Изобретение относится к триазоловым соединениям, их получению и использованию в качестве противогрибковых средств или промежуточных соединений для изготовления противогрибковых лекарственных трав, а также агрохимикатов.
Существует много соединений, которые могут быть отнесены к противогрибковым агентам.
Известны производные триазола, обладающие противогрибковой активностью (выложенные заявки Японии 189173/83, 98072/84). Однако из этих публикаций непонятно, являются ли данные соединения достаточно эффективными в качестве лекарственных средств, если рассматривать их с точки зрения их противогрибковой активности, побочных эффектов в абсорбирующей способности.
Что касается существующих стандартных противогрибковых средств, то они не являются достаточно эффективными и обладают определенными недостатками, например побочные эффекты.
Для устранения указанных недостатков в качестве лекарственных средств желательно использовать соединения, обладающие более сильной антигрибковой активностью и большей надежностью.
На фиг.1 показана порошковая рентгенограмма гидрохлорида соединения 43; на фиг.2 то же, гидробромида соединения 43.
Настоящее изобретение относится:
1. К соединению формулы
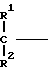 A (I) где Ro, R1 и R2 являются одинаковыми или различными и представляют собой атом водорода или алкильную группу;
A (I) где Ro, R1 и R2 являются одинаковыми или различными и представляют собой атом водорода или алкильную группу;
А группа формулы
-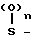 X R3 где Х химическая связь или группа формулы
X R3 где Х химическая связь или группа формулы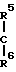 - X1- где Х1 химическая связь или алкиленовая группа с 1-5 атомами углерода, которая может содержать атом серы или кислорода в качестве составляющих атомов; R5 и R6 являются одинаковыми или различными и означают атом водорода или низшую алкильную группу;
- X1- где Х1 химическая связь или алкиленовая группа с 1-5 атомами углерода, которая может содержать атом серы или кислорода в качестве составляющих атомов; R5 и R6 являются одинаковыми или различными и означают атом водорода или низшую алкильную группу;
R3 ароматическая гетероциклическая группа, которая может быть замещенной; n означает 0, 1 или 2,
или А является группой формулы
-S-R4, где R4 атом водорода, алканоильная группа, если Х и Х1 в радикале А являются химической связью или А является группой формулы
-S-R4, где R4 указан выше,
то R0 или R1 низшая алкильная группа, или к соли описанного соединения формулы I.
2. К способу получения соединения формулы I.
3. К противогрибковым агентам, содержащим соединение формулы I.
Соединение настоящего изобретения может быть также представлено общей формулой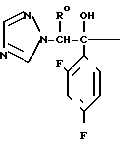
 x____R3 (I') где R0,R1,R2,R3,x,n определены выше,
x____R3 (I') где R0,R1,R2,R3,x,n определены выше,
и общей формулой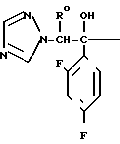
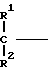 S____R4 (I'') где R0,R1,R2 и R4 определены выше.
S____R4 (I'') где R0,R1,R2 и R4 определены выше.
В соединениях (I) низшими алкильными группами, представленными R0,R1 или R2, являются С1-3 алкильные группы с прямой или разветвленной цепью, такие как метил, этил, пропил и изопропил; или R1 и R2 в сочетании представляют собой низшие алкиленовые группы, например этилен, пропилен, предпочтительными являются соединения, в которых R1 метильная группа, а R0 и R2 атомы водорода.
Если в соединениях формулы I Х в А представляет собой группу формулы - X1- то предпочтительно, чтобы R5 представлял собой атом водорода или низшую алкильную группу, например метил, этил, пропил, или чтобы R6 представлял собой атом водорода или метил, а Х представлял собой алкиленовую группу с 1-5 атомами углерода, которая может содержать в качестве составляющих атомов атомы серы или кислорода и примерами которой могут служить следующие группы:
- X1- то предпочтительно, чтобы R5 представлял собой атом водорода или низшую алкильную группу, например метил, этил, пропил, или чтобы R6 представлял собой атом водорода или метил, а Х представлял собой алкиленовую группу с 1-5 атомами углерода, которая может содержать в качестве составляющих атомов атомы серы или кислорода и примерами которой могут служить следующие группы:
CH2, CH2CH2,  ,
,  ,
,  H2CH2
H2CH2
CH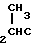 H2, CH2CH2CH2CH2, CH2CH2CH2CH2CH2 СН2S, CH2SCH2, CH2SCH2S, CH2SCH2CH2, CH2SCH2CH2S, CH2SCH2CH2CH2, CH2CH2S, CH2CH2SCH2, CH2CH2SCH2S, CH2CH2SCH2CH2,
H2, CH2CH2CH2CH2, CH2CH2CH2CH2CH2 СН2S, CH2SCH2, CH2SCH2S, CH2SCH2CH2, CH2SCH2CH2S, CH2SCH2CH2CH2, CH2CH2S, CH2CH2SCH2, CH2CH2SCH2S, CH2CH2SCH2CH2,
CH CH2, CH2CH2CH2S, CH2CH2CH2SCH2
CH2, CH2CH2CH2S, CH2CH2CH2SCH2 CH2OCH2CH2S, CH2SCH2CH2O, CH2OCH2CH2O, CH2OCH2CH2, CH2OCH2CH2CH2, CH2CH2CH2O.
CH2OCH2CH2S, CH2SCH2CH2O, CH2OCH2CH2O, CH2OCH2CH2, CH2OCH2CH2CH2, CH2CH2CH2O.
Если Х является алкиленовой группой, содержащей атомы серы в качестве составляющих атомов, то указанные атомы серы могут быть оксидированы с образованием сульфоксида или сульфона. Если эти группы, имеющие химические связи с обеих сторон, имеют на конце атом серы или кислорода, то эти атомы связаны с R3.
Если в соединениях формулы I А представляет собой
- X R3 то ароматическая гетероциклическая группа, которая может быть замещенной, может быть также конденсированной с ядром, имеющим 5-7 членов. При этом примерами таких конденсированных ароматических гетероциклических групп могут служить 1-бензимидазолил, 2-бензимидазолил, 5Н-6,7-дигидропиролло[1,2-a] имидазол-2-ил, 5Н-6,7-дигидропиролло[1,2-c]имидазол-3-ил, 2-имидазо[1,2-a]пиримидинил, 5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-ил, 2-имидазо[1,2-a]пиридинил, 3-имидазо [1,5-a]пиразинил, 2-имидазо[1,2-a]пиразинил, 6-имидазо[1,2-b] пиридазинил, 2-имидазо[1,2-b] пиридазинил, 3-имидазо[1,2-b] пиридазинил, 5-имидазо[1,5-a]пиридинил, 6-имидазо[1,5-a]пиридинил, 7-имида- зо[1,5-b] пиридазинил, 2-бензотиазолил, 4,5,6,7-тетра- гидробензотиазол-2-ил, 4Н-5,6-дигидроциклопента[d] тиазол-2-ил, 4Н-5,6,7,8-тетрагидроциклогепта [d] тиазол-2-ил, хинолил, изохинолил, хиназолинил, индолизинил и индолил.
X R3 то ароматическая гетероциклическая группа, которая может быть замещенной, может быть также конденсированной с ядром, имеющим 5-7 членов. При этом примерами таких конденсированных ароматических гетероциклических групп могут служить 1-бензимидазолил, 2-бензимидазолил, 5Н-6,7-дигидропиролло[1,2-a] имидазол-2-ил, 5Н-6,7-дигидропиролло[1,2-c]имидазол-3-ил, 2-имидазо[1,2-a]пиримидинил, 5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-ил, 2-имидазо[1,2-a]пиридинил, 3-имидазо [1,5-a]пиразинил, 2-имидазо[1,2-a]пиразинил, 6-имидазо[1,2-b] пиридазинил, 2-имидазо[1,2-b] пиридазинил, 3-имидазо[1,2-b] пиридазинил, 5-имидазо[1,5-a]пиридинил, 6-имидазо[1,5-a]пиридинил, 7-имида- зо[1,5-b] пиридазинил, 2-бензотиазолил, 4,5,6,7-тетра- гидробензотиазол-2-ил, 4Н-5,6-дигидроциклопента[d] тиазол-2-ил, 4Н-5,6,7,8-тетрагидроциклогепта [d] тиазол-2-ил, хинолил, изохинолил, хиназолинил, индолизинил и индолил.
Примерами неконденсированных гетероциклических групп в ароматических гетероциклических группах, представленных радикалом R3, которые могут быть замещенными, являются 1-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, 1-(Н)-1,2,4-три- азолил, 3-(4Н)-1,2,4-триазолил, 3-(1Н)-1,2,4-триазолил, 5-(1Н)= 1,2,4-триазолил, 4-(4Н)-1,2,4-триазолил, 1,2,3-триазолил, 1-пиразолил, 3-пиразолил, 4-пиразолил, 4-пиридил, 2-пиридил, 3-пиридил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,3,4-тиадиазол-2-ил, 2-тиенил, 2-фурил, 1-пирролил, 2-пиразинил, 3-пиримизинил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 3-озоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, 1-тетразолил, 5-тетразолил и 2-оксо-1,3-диоксол-4-ил.
Заместителями, которые могут иметь конденсированные или неконденсированные ароматические гетероциклические группы, являются атомы галогена, амино-, гидрокси-, алкильные, алкениловые, арильные, аралкильные, галогенированные алкильные, алкилтио-, циклоалкильные и циклоалкилалкильные группы, причем указанными атомами галогена могут быть атомы хлора, фтора, брома и иода.
Указанными алкильными группами, имеющими предпочтительно 1-4 атома углерода, являются группы с прямой или разветвленной цепью, такие как метил, этил, н-пропил, изопропил, бутил, изобутил, вторбутил и третбутил.
Указанными алкенильными группами, имеющими 2-4 атомов углерода, являются винил, аллил и 1,3-бутадиенил.
Указанной арильной группой могут быть фенил и нафтил.
Указанной аралкильной группой могут быть бензил, фенетил и фенилпропил.
Примерами указанной галогенированной алкильной группой являются алкильные группы, имеющие 1-4 атома углерода, каждый из которых является замещенным 1-5 атомами галогена, фторометил, дифторометил, трифторометил, фтороэтил, дифтороэтил, трифтороэтил, дифторопропил и тетрафторопропил.
Примерами указанной алкилтиогруппы, имеющей 1-4 атома углерода, могут служить метилтио-, этилтио-, пропилтио- и бутилтиогруппы, причем в указанных алкилтиогруппах атом серы может быть окислен с образованием сульфоксида или сульфона.
Примерами циклоалкильной группы, имеющей 3-6 атомов углерода, являются циклопропил, циклобутил, циклопентил, циклогексил.
Примерами циклоаралкильных групп, имеющих 1-4 атома углерода, каждая из которых является замещенной циклоалкильными группами, имеющими 3-6 атомов углерода, являются циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил, циклопропилэтил, циклопропилбутил.
Если в соединениях формулы I А представляет собой -S-R4, то алканоильной группой, представленной R4, является ацильная группа, происходящая от карбоновых кислот, например С2-5-алканоильная группа, такая как пропионил, бутурил, изобутурил, валерил и ацетил, и арил С1-3-алканоильная группа, такая как фенилацетил и фенилпропионил. Предпочтительной ацильной группой является группа, которая обладает способностью гидролизоваться в организме.
Конкретные соединения настоящего изобретения представлены в табл.1-4.
Если соединения (I) имеют один или несколько асимметрических атомов углерода, то предлагаемое изобретение включает в себя стереоизомеры R- и S-конфигураций и их смеси. Предпочтительными соединениями являются соединения (I), имеющие в качестве R1 метильную группу, а в качестве R2атом водорода, абсолютной конфигурацией которых является R-конфигурация.
Соединения (I) могут быть получены также в виде солей, например соли неорганических кислот, такие как гидрохлориды, гидробромиды, сульфаты, нитраты и фосфаты и соли органических кислот, такие как ацетаты, тартраты, цитраты, фумараты, малеаты, толуолсульфонаты и метансульфонаты.
Если в формуле (I') соединений настоящего изобретения n является 0, то указанные соединения могут быть получены, например, с помощью реакции соединения формулы N-
N-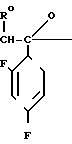
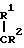 (II)
(II)
с соединением общей формулы
HS X R3. (III)
Указанную реакцию обычно проводят в присутствии воды или органического растворителя, например, ацетонитрила, диметилформамида, диметилсульфоксида, тетрагидрофурана, метилового спирта, этилового спирта, которые могут быть использованы как отдельно, так и в сочетании друг с другом, или без какого-либо растворителя при температуре от -20 до 150оС. В целях ускорения реакции в реакционную систему может быть добавлено основание, например, такое как карбонат калия, гидроксид калия, гидроксид натрия, гидрид натрия, метилат натрия, этилат натрия или фторид тетрабутиламмония.
Если в формуле (I) соединений настоящего изобретения n является 0, то такие соединения могут быть также получены, например, с помощью реакции соединения формулы N
N
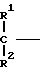 SH (IV) с соединением общей формулы
SH (IV) с соединением общей формулы
W X R3, (V) где Х и R3 определены выше; W атом галогена или группа формулы R3-SO2-O-, где R3 низший С1-4-алкил, трифторoметил, фенил или р-толуол.
Обычно указанную реакцию проводят в присутствии воды или органического растворителя, например ацетонитрила, диоксана, диметилформамида, диметилсульфоксида, тетрагидрофурана, метилового спирта, этилового спирта, которые могут быть использованы как отдельно, так и в сочетании друг с другом, или без какого-либо растворителя при температуре от -20 до 150оС. В целях ускорения реакции в реакционную систему может быть добавлено основание, например, такое как карбонат калия, гидроксид калия, гидроксид натрия, гидрид натрия, метилат натрия, этилат натрия или фторид тетрабутиламмония.
Если в формуле (I") настоящего изобретения R4 является алканоильной группой, то такие соединения могут быть получены с помощью реакции соединения формулы N
N
 SH с соединением формулы
SH с соединением формулы
R4' W', (VI) где R4' является алканоильной группой; W' атом галогена или -О-R4'', где R4'' ацильная группа.
Обычно указанную реакцию проводят в воде или органическом растворителе, например метиленхлориде, хлороформе, этилацетате, бензоле, диоксане, тетрагидрофуране, которые могут быть использованы как отдельно, так и в сочетании друг с другом, при температуре от -20 до 100оС. В целях ускорения реакции в реакционную систему может быть добавлено неорганическое основание, такое как карбонат калия, гидрокарбонат натрия или гидроокись натрия, или органическое основание, такое как триэтиламин, пиридин, или пиколин.
Если в формуле (I') соединений настоящего изобретения n является 1 или 2, то такие соединения могут быть получены, например, с помощью соединения формулы N
N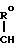
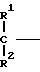 SH
SH
Реакцию окисления обычно проводят в присутствии воды или органического растворителя, например метиленхлориде, хлороформе, изопропиловом спирте, бензоле уксусной кислоте, которые могут быть использованы как отдельно, так и в сочетании друг с другом, при температуре от -20 до 50оС с оксидантом, например m-хлоропербензойной кислотой, перуксусной кислотой, перекисью водорода, бензоилпероксидом. Количество оксиданта, эквивалентного соединению (8), может быть соответственно скорректировано так, что соединение формулы (I'), в котором n равно 1, и соединение, в котором n равно 2, могут быть получены отдельно или в смеси. Реакционная температура и время прохождения реакции могут быть откорректированы так, что соединения формулы (I'), в которых n равно 1 и 2, могут быть получены отдельно или в смеси. Для указанной реакции предпочтительным оксидантом является m-хлорбензойная кислота.
Соединение (I") настоящего изобретения, где R4-водород, может быть также получено с помощью реакции разблокирования соединения формулы N
N 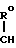
 S ___ Rp (XXIX)
S ___ Rp (XXIX)
где R0, R1 и R2 определены выше; Rр бензил, n-метоксибензил, n-метилбензил или тритил. Указанная реакция разблокирования может быть проведена, например, путем воздействия кислоты, такой как фтороводород, трифторуксусная кислота, трифторметансульфоновая кислота и т.п. на субстратное соединение (XXIX) в присутствии или отсутствия анизола или триазола, путем воздействия металлического натрия на соединение XXIX в жидком аммиаке или путем обработки субстратного соединения тяжелым металлом, например нитратом серебра, ацетатом ртути, трифторoацетатом ртути и т.п. с последующей реакцией этого соединения с меркаптосоединением, например сероводородом, β-меркаптоэтанолом и т.п. Указанная реакция разблокирования обычно протекают в присутствии или отсутствии органического растворителя, например уксусной кислоты, метиленхлорида, хлороформа, трифторуксусной кислоты и т.п. при температуре приблизительно от -10 до 60о.
Соединение (I), полученное в результате указанных реакций, может быть выделено из реакционной смеси с помощью стандартных процедур очистки, таких как экстрагирование, концентрирование, нейтрализация, фильтрация, перекристаллизация, колоночная хроматография и тонкослойная хроматография.
Соединение (I) может существовать по меньшей мере в двух стереомерах. Указанные изомеры, а также их смеси, если необходимо, могут быть получены отдельно. Это может быть осуществлено, например, с помощью описанной соответствующей реакции конкретного изомера исходного соединения (II), (III), (V), (VI), (VII), (VIII) или (XXIX), в результате которой может быть получен соответствующий изомер соединения (I). Если реакционный продукт является смесью двух или нескольких изомеров, он может быть фракционирован в соответствующие изомеры путем стандартной техники разделения или фракционирования, такой как образование соли с оптически активной кислотой, например камфоросульфоновой кислотой, винной кислотой и т.п. хроматография нескольких типов, фракционирование, перекристаллизация и т.п.
Физиологически приемлемые соли соединения (I) могут быть получены путем добавления указанных неорганических кислот и органических кислот.
Среди синтетических промежуточных соединений (II), используемых в настоящем изобретении, соединениe (IX), где R0 и R2 являются водородом, может быть получено способом, который проиллюстрирован в приведенной ниже реакционной схеме.

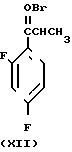
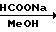

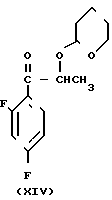
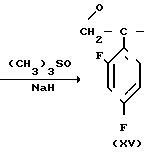
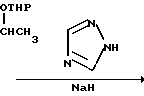
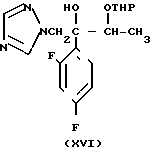
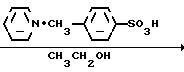
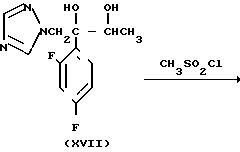



Способ получения соединения (IX) подробно описан ниже. Так, например, 2,4-дифторoбензол (Х) подвергают реакции Фриделя-Крафтса с пропионилхлоридом с получением соединения (XI), которое затем обрабатывают бромидом, в результате чего получают бромид (XII). Указанное соединение (XII) может быть также получено с помощью реакции Фриделя-Крафтса соединения (Х) и 2-бромопропионилхлорида. Реакция (XII) (XIII) является реакцией гидролиза, которая может быть легко осуществлена в присутствии формата натрия в метаноле. С помощью обычной реакции тетрагидропиранилирования соединения (XIII) получают соединение (XIV). Если указанное соединение (XIV) подвергают реакции с триметилсульфоксонийиодидом в присутствии гидрида натрия, то в результате получают соединение (XV), в котором ТНР является 2-(2Н)-3,4,5,6-тетрагидропиранилом. Реакция (XV)-(XVI) является реакцией размыкания эпоксикольца с помощью триазолнатриевой соли, образованной из триазола и гидрида натрия, и может быть легко осуществлена в диметилформамиде при 60-90оС. При воздействии n-толуолсульфонатом пиридиния на соединение (XVI) в этаноле имеет место реакция разблокирования с образованием соединения (XVII). В результате взаимодействия этого соединения (XVII) с метансульфонилхлоридом получают соединение (XVIII), которое затем обрабатывают основанием, например метоксидом натрия, в целях получения соединения (IX).
В описанном методе синтеза соединения (XVII), (XVIII) и (IX) получают в виде смеси двух диастереоизомеров. По желанию каждое соединение из (XVII), (XVIII) и (IX) может быть фракционировано в диастереомеры компонент путем фракционированной перекристаллизации, хроматографии или т. п. либо перед соответствующей реакцией каждое из предшествующих соединений (XV) и (XVI) могут быть фракционированы путем фракционированной перекристаллизации, хроматографии или т. п. что приводит к получению диастереомеров (XVII), (XVIII) и (IX). Кроме того, диастереомер (XVIII) или (IX) может быть получен путем использования соответствующего диастереомера (XVII) или (XVIII).
Можно также использовать оптически активный сложный эфир (R)-молочной кислоты [(R)-(XIX)] или сложный эфир (S)-молочной кислоты [(S) (XIX)] в качестве исходного материала для синтеза соответствующего оптически активного соединения (XIV)[(R) (XIV)] или (S) (XIV)] в соответствии с реакционной схемой, приведенной ниже, а затем с помощью реакции в соответствии с реакционной схемой, приведенной выше, с последующим (если необходимо) разделением диастереоизомеров, в результате получают необязательно активные соединения (XVII) [(2R, 3R) (XVII), (2R, 3S) (XVII), (2S, 3S) (XVII)] (XVIII), [(2S, 3RS) (XVIII) (2R, 3S (XVIII), (2S, 3S) (XVIII), (2S, 3R) (XVIII)] и (IX) [(2R, 3R) (IX), (2R, 3S) (IX), (2S, 3S) (IX), (2S, 3R) (IX)]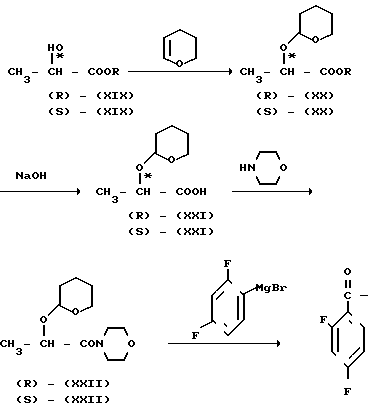
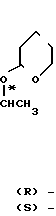
 где заместитель R является С1-4-алкильной группой. Реакция сложного эфира (R)-молочной кислоты с (2Н)-3,4-дигидрофураном в присутствии n-толуолсульфоновой кислоты дает соединение (R)-(XX). Указанную реакцию проводят в растворителе, таком как метиленхлорид, хлороформ и т.п. в основном при температуре от -10 до 30оС. Реакция (R) (XX) ->> (R) (XXI) является обычной гидролизной реакцией, которая легко осуществляется в присутствии основания, например гидроокиси натрия, гидроокиси калия и т.п. в воде или в органическом растворителе, например метаноле, этаноле, иди в их смеси при температуре в пределах 0 40оС. Реакция конденсации между полученным соединением (R) (XXI) и морфолином может быть предпочтительно проведена в присутствии стандартного дегидратирующего агента конденсации в целях получения соединения (R) (XXII). Дегидрирующим агентом конденсации, используемым в указанной реакции, является, например, дициклогексилкарбодиимид, карбонилдимидазол, диэтилцианофосфат и т. п. В качестве растворителя может быть использован, например, тетрагидрофуран, диоксан или ацетонитрил. Указанную реакцию обычно проводят при температуре от -10оС до 40оС. Реакция (R) (XXII) ->> (R) (XIV) является реакцией Гриньяра и может быть осуществлена с помощью реакции соединения (R) (XXIII) с бромидом 2,4-дифторофенилмагния, полученным из 2,4-дифторобромбензола и металлического магния, в органическом растворителе, например тетрагидрофуране, этиловом эфире и т.п. и при температуре от -10 до 40оС. Амидное соединение (R) (XXII), используемое в реакции Гриньяра, вместо морфолиногруппы может иметь другую циклическую аминогруппу, например, 1-пирролидинил, пиперидино и т.п. или вторичную аминогруппу, например диметиламино, диэтиламино, дибутиламино и т.п. и указанные амиды могут быть синтезированы путем конденсации соответствующих аминов с (R) (XXI). Кроме того, реакция сложного эфира (S)-молочной кислоты, проводимая тем же способом, как описано выше, [(S) (XIX) ->> (S) (XX) ->> (S) (XXI) ->> (S) (XXII) ->> (S) (XIV)] приводит к получению (S) (XIV).
где заместитель R является С1-4-алкильной группой. Реакция сложного эфира (R)-молочной кислоты с (2Н)-3,4-дигидрофураном в присутствии n-толуолсульфоновой кислоты дает соединение (R)-(XX). Указанную реакцию проводят в растворителе, таком как метиленхлорид, хлороформ и т.п. в основном при температуре от -10 до 30оС. Реакция (R) (XX) ->> (R) (XXI) является обычной гидролизной реакцией, которая легко осуществляется в присутствии основания, например гидроокиси натрия, гидроокиси калия и т.п. в воде или в органическом растворителе, например метаноле, этаноле, иди в их смеси при температуре в пределах 0 40оС. Реакция конденсации между полученным соединением (R) (XXI) и морфолином может быть предпочтительно проведена в присутствии стандартного дегидратирующего агента конденсации в целях получения соединения (R) (XXII). Дегидрирующим агентом конденсации, используемым в указанной реакции, является, например, дициклогексилкарбодиимид, карбонилдимидазол, диэтилцианофосфат и т. п. В качестве растворителя может быть использован, например, тетрагидрофуран, диоксан или ацетонитрил. Указанную реакцию обычно проводят при температуре от -10оС до 40оС. Реакция (R) (XXII) ->> (R) (XIV) является реакцией Гриньяра и может быть осуществлена с помощью реакции соединения (R) (XXIII) с бромидом 2,4-дифторофенилмагния, полученным из 2,4-дифторобромбензола и металлического магния, в органическом растворителе, например тетрагидрофуране, этиловом эфире и т.п. и при температуре от -10 до 40оС. Амидное соединение (R) (XXII), используемое в реакции Гриньяра, вместо морфолиногруппы может иметь другую циклическую аминогруппу, например, 1-пирролидинил, пиперидино и т.п. или вторичную аминогруппу, например диметиламино, диэтиламино, дибутиламино и т.п. и указанные амиды могут быть синтезированы путем конденсации соответствующих аминов с (R) (XXI). Кроме того, реакция сложного эфира (S)-молочной кислоты, проводимая тем же способом, как описано выше, [(S) (XIX) ->> (S) (XX) ->> (S) (XXI) ->> (S) (XXII) ->> (S) (XIV)] приводит к получению (S) (XIV).
В синтезе оптически активного соединения согласно настоящему изобретению может быть синтезировано также оптически активное промежуточное соединение (XXII) в соответствии со следующей реакционной схемой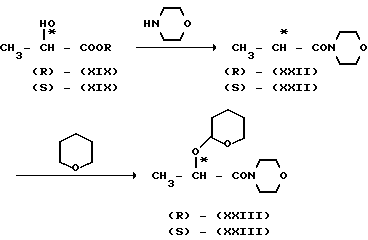 где заместитель R имеет значения, определенные выше. Реакция конверсии сложного эфира (R)-молочной кислоты (R) (XIX) в амид (R) (XXIII) может быть проведена предпочтительно путем нагревания соединения (R) (XIX) и морфолина в присутствии или в отсутствии растворителя, например бензола, толуола и т.п. при 60 100оС. Реакция соединения (R) (XXIII) ->> соединение (R) (XXII) может быть проведена тем же способом, что и реакция (R) (XIX) ->> (R) (XX). В указанной реакционной схеме использование циклического амина, не являющегося морфолином, например пирролидина, пиперидина и т.п. или вторичного амина, например диметиламина, диэтиламина, дибутиламина и т.п. вместо морфолина при прочих равных условиях позволяет получить соединение, в котором морфолиногруппа является замененной соответствующей циклической аминогруппой или вторичной аминогруппой. Указанное соединение может быть использовано так же, как и соединение (R) (XXII), в реакции Гриньяра с бромидом 2,4-дифторофенилмагния. Кроме того, с помощью реакции сложного эфира (S)-молочной кислоты (S) (XIX), аналогичной описанной, получают соединение (S) (XXII) по схеме [(S) (XIX) ->> (S) (XIII) ->> (S) (XXII)]
где заместитель R имеет значения, определенные выше. Реакция конверсии сложного эфира (R)-молочной кислоты (R) (XIX) в амид (R) (XXIII) может быть проведена предпочтительно путем нагревания соединения (R) (XIX) и морфолина в присутствии или в отсутствии растворителя, например бензола, толуола и т.п. при 60 100оС. Реакция соединения (R) (XXIII) ->> соединение (R) (XXII) может быть проведена тем же способом, что и реакция (R) (XIX) ->> (R) (XX). В указанной реакционной схеме использование циклического амина, не являющегося морфолином, например пирролидина, пиперидина и т.п. или вторичного амина, например диметиламина, диэтиламина, дибутиламина и т.п. вместо морфолина при прочих равных условиях позволяет получить соединение, в котором морфолиногруппа является замененной соответствующей циклической аминогруппой или вторичной аминогруппой. Указанное соединение может быть использовано так же, как и соединение (R) (XXII), в реакции Гриньяра с бромидом 2,4-дифторофенилмагния. Кроме того, с помощью реакции сложного эфира (S)-молочной кислоты (S) (XIX), аналогичной описанной, получают соединение (S) (XXII) по схеме [(S) (XIX) ->> (S) (XIII) ->> (S) (XXII)]
Оптически активное промежуточное соединение (R) (XIV) может быть получено с использованием производного (R)-молочной кислоты (R) (XXIV) в качестве исходного материала в соответствии со следующей реакционной схемой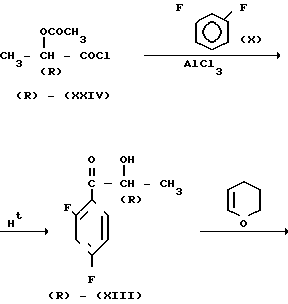
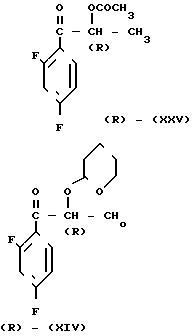
Использование соответствующих диастереомеров соединений (XVII), (XVIII) и (IX) или их оптически активных форм, полученных указанным способом, в качестве промежуточных соединений позволяет получить соответствующий диастереомер соединения (I) или его оптически активной формы в зависимости от обстоятельств.
Что касается синтетического промежуточного соединения (II), то соединение (XXVI), в котором Ro метил, а R1 и R2 водород, может быть получено, например, способом, проиллюстрированным приведенной ниже схемой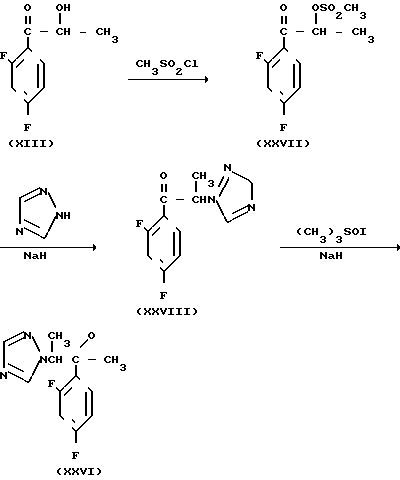
Синтетические промежуточные соединения (XXIX) настоящего изобретения могут быть получены, например, следующим образом N-
N- R2
R2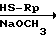 (XXIX)
(XXIX)
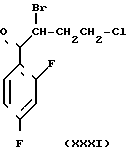
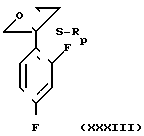
Среди синтетических промежуточных соединений (XXIX) настоящего изобретения соединение (XXX), в котором R0 водород, а R1 и R2 вместе взятые представляют собой этиленовую группу, может быть получено, например, способом, проиллюстрированным в следующей реакционной схеме
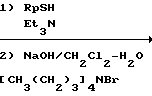

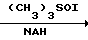
 N__CH
N__CH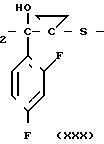 Rp
Rp
Если в соединениях формулы (I") настоящего изобретения R4 является атомом водорода, то такие соединения могут быть получены, например, с помощью реакции соединения формулы
Az  -
-  C
C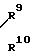 (XXIV) (XXXIV) где R7, R8, R9, R10 и R11 являются одинаковыми или различными и представляют собой атом водорода или остаток углеводорода, который может быть замещенным; А триазолил, имидазолил, с соединением формулы
(XXIV) (XXXIV) где R7, R8, R9, R10 и R11 являются одинаковыми или различными и представляют собой атом водорода или остаток углеводорода, который может быть замещенным; А триазолил, имидазолил, с соединением формулы
HS CH2 CH где по меньшей мере один из Z и Y является цианогруппой или карбоксильной группой, которая может быть эстерифицированной или амидированной, а другой является атомом водорода, низшей алкильной группой или аминогруппой, которая может быть ацилированной.
где по меньшей мере один из Z и Y является цианогруппой или карбоксильной группой, которая может быть эстерифицированной или амидированной, а другой является атомом водорода, низшей алкильной группой или аминогруппой, которая может быть ацилированной.
Указанная реакция может быть проведена в одну стадию, используемые в этой реакции реагенты являются недорогостоящими и удобными в обращении, а целевые соединения общей формулы (I") могут быть получены в большом количестве; таким образом, описанный способ может быть с успехом использован в промышленном производстве.
Реакцию между соединением общей формулы (XXXIV) и соединением общей формулы (IV) желательно проводить в присутствии органического или неорганического основания, примерами которого могут служить гидроокись натрия, гидроокись калия, карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия, метилат натрия, этилат натрия, метилат калия, этилат калия, третбутилат калия, гидрид натрия, гидрид калия, бутиллитий, диизопропиламид лития, триэтиламин, N-метилморфолин, диметиламинопиридин, лютидин, фторид тетрабутиламмония, гидроксид тетрабутиламмония; гидроксид N-бензилтриметиламмония, 1,8-диазабицикло [5,4,0] унд-7-цен; 1,5-диазабицикло [4,3,0] нон-5-ен, 1,4-диазабицикло [2,2,2] октан, причем предпочтительным основанием являются гидрид натрия и метилат натрия.
В формуле (XXXIV), описанной выше, триазолом, обозначенным AZ, может быть 1,2,4-триазол.
Углеводородные остатки, обозначенные в формуле (XXXIV) R7, R8, R9, R10 и R11 могут быть замещенными и представляют собой алкильные, циклоалкильные, алкенильные и арильные группы.
Примерами указанных алкильных групп, имеющих 1-12 атомов углерода каждая, могут служить группы с прямой или разветвленной цепью, такие как метил, этил, пропил, бутил, гептил, окстил, нонил, децил и додецил.
Указанными циклоалкильными группами могут быть группы, содержащие 3-7 атомов углерода, например, такие как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Указанными алкенильными группами могут быть группы, включающие 2-6 атомов углерода, например, такие как аллил, винил, 1,3-бутадиенил, изопренил и 2,4-пентадиенил.
Указанными арильными группами могут быть фенил, нафтил, бифенил, антрил и инденил.
Указанные углеводородные остатки могут иметь заместителей, таких как гидроксильная группа, карбоксильные группы, которые могут быть эстерифицированы, например карбокси, этоксикарбонил, метоксикарбонил, бутоксикарбонил, аминогруппы, ациламиногруппы, например ацетиламино, пропиониламино, бутуриламино, алкиламиногруппы, например метиламино, диметиламино, диэтиламино, дибутиламино, алкоксигруппы, например метокси, этокси, бутокси, атомы галогена, например фтора, хлора, брома, оксо-, тио-, меркапто-, например метилтио, этилтио, бутилтио, цианогруппы, кроме этого, алкильные циклоалкильные, алкенильные и арильные группы, описанные выше. Указанные углеводородные остатки могут иметь от 1 до 3 заместителей, которые являются либо одинаковыми, либо различными.
Низшими С1-4-алкильными группами, представленными Х и Y в формуле (IV), могут быть метил, пропил, этил, бутил, изопропил и третбутил. Примерами аминогрупп, представленных Х или Y, которые могут быть ацилированными, являются ацетиламино, бензоиламино, тозиламино и мелиламино.
Примерами карбоксильных групп, представленных Х и Y в формуле (IV), которые могут быть эстерифицированы или амидированы, являются карбокси, метоксикарбонил, этоксикарбонил, бутоксикарбонил, карбамоил, диметиламинокарбонил, диэтиламинокарбонил, морфолинокарбонил, пиперидинкарбонил, и 1-пирролидинилкарбонил.
Каждое из меркаптосоединений, представленных общей формулой (IX), имеет один или несколько асимметрических атомов и два или более стереоизомеров. Предлагаемое изобретение относится к способу получения всех стереоизомеров.
Способ может быть осуществлен следующим образом.
К соответствующему растворителю, например метанолу, этанолу, изопропиловому спирту, диметилформамиду, диметилсульфоксиду, ацетону, толуолу, бензолу, этилацетату, диоксану, тетрагидрофурану и воде, которые могут быть использованы отдельно или в сочетании друг с другом, добавляют соединение общей формулы (XXXIV), а затем один или более эквивалентов, предпочтительно 2-10 эквивалентов, указанного основания по отношению к количеству соединения (XXXIV). Затем добавляют один или более эквивалентов, предпочтительно 2-20 эквивалентов, соединения общей формулы (IV) по отношению к количеству соединения (XXXIV). Полученную в результате смесь выдерживают при температуре от -10 до 100оС, предпочтительно от 0 до 80оС, в целях проведения реакции. После завершения реакции получают соединение общей формулы (IX) путем выделения известным способом per se, например путем концентрации, нейтрализации, экстрагирования, перекристаллизации, фильтрации или различными методами хроматографии.
Наиболее предпочтительным исходным соединением (IV) способа настоящего изобретения является соединение, в котором Х представляет собой метоксикарбонильную группу, а Y является атомом водорода (метиловый сложный эфир 3-меркаптопропионовой кислоты).
Предпочтительными исходными соединениями (XXXIV) являются соединения, в которых AZ 1,2,4-триазолил; R7, R8, R9 и R10 атомы водорода или низшие алкильные группы, R11 замещенная арильная группа, причем особенно предпочтительными являются соединения, в которых R7, R8и R9 атомы водорода; R10 метильная группа, R11 2,4-дифторфенил.
Среди исходных соединений формулы (XXXIV) соединения (IX), в которых AZ 1,2,4-триазолил; R7, R8 и R9 атомы водорода; R10 метил; R11 2,4-дифторoфенил, могут быть синтезированы, например, в соответствии с указанными схемами.
Противогрибковую активность соединений (I) оценивали следующим образом: лист диска фильтровальной бумаги (8 мм в диаметре), изготовленный Тоуо Seisakusho, пропитанный путем погружения в 1000 мкг/мл раствора соединения (I) в метаноле, помещали на агаровую чашку и инкубировали при 28оС в течение 2 дн. после чего вокруг диска фильтровальной бумаги измеряли диаметр зоны ингибирования роста. При этом использовали следующие культуральные среды:
А дрожжевая агаровая среда с азотным основанием;
В дрожжевая агаровая среда с азотным основанием (рН 7,0);
С агаровая среда Сабурода.
Противогрибковый спектр соединений (I) представлен в табл.5.
Противогрибковые активности соединений (I) против грибка Candida albicans представлены в табл.6 и 7.
Предотвращающее заражение действие соединений I, оцениваемое в экспериментальном инфецировании мышей, показано в табл.8 и 9.
Способ 1 (табл. 8): мышам штамма Crj:CDF1 в возрасте 5 недель вводили внутривенно минимальную летальную дозу Candida albicans. Испытуемое соединение вводили дважды: через 0 и 2 ч после инфецирования. Эффективность лекарственного средства, выраженная в ED50-величинах, рассчитывали по методу Рида-Менча (на основании подсчета степени выживаемости через 7 дн. после инфецирования). ED50-величины рассчитывались с учетом полной дозы, введенной в указанных двух случаях.
Способ 2 табл. 9: мышам (штамм Crj CDF1, возраст 5 недель) внутривенно вводили пороговую летальную дозу Candida albicans. Испытуемое соединение вводили сразу после инфецирования. Эффективность лекарственного средства, выраженная в ED50-величинах, рассчитывали по методу Рида-Менча по степени выживаемости через 7 дн. после инфецирования.
ED50-величины рассчитывались на основании полной дозы, введенной в двух случаях.
Соединения настоящего изобретения обладают низкой токсичностью и высокой противогрибковой активностью с широким спектром противогрибкового действия и могут быть использованы для предупреждения и лечения грибковых инфекций человека, домашних животных и птиц.
При лечении человека соединения настоящего изобретения могут быть введены перорально или парентерально в виде фармацевтических композиций, изготовленных в стандартных лекарственных формах, таких как порошки, гранулы, таблетки, капсулы и инъекции, путем смешивания активных соединений с фармацевтически приемлемыми носителями, наполнителями или разбавителями. Доза активного соединения может варьироваться в зависимости от степени заражения и способа введения, например пероральная доза для взрослого, зараженного кандидой, составляет от 0,1 до 100 мг/кг в день, предпочтительно от 1 до 50 мг/кг в день.
Соединение настоящего изобретения также может быть использовано в качестве противогрибкового препарата при наружном применении. Например, слизистые и кожные оболочки могут быть стерилизованы и дезинфицированы путем нанесения на них мази, содержащей от 0,1 до 100 мг, предпочтительно от 1 до 50 мг соединения настоящего изобретения на грамм.
Ниже приводятся сравнительные примеры и примеры, подробно иллюстрирующие осуществление настоящего изобретения.
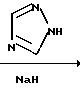
С р а в н и т е л ь н ы й п р и м е р 1. Смесь бензимидазола (4,72 г, 40 мМ), бромохлорэтана (3,4 мл, 41 мМ), карбоната калия (5,52 г, 40 мМ) и диметилформамида (60 мл) перемешивали при комнатной температуре в течение 20 ч. К этой реакционной смеси добавляли этилацетат и воду, а затем экстрагировали этилацетатом. Органический слой промывали водой, а затем насыщенным водным раствором хлорида натрия и высушивали безводным сульфатом натрия. После чего растворитель отгоняли, а остаток хроматографировали на силикагеле (3,0 х 40 см), элюируя смесью этилацетата и гексана (2:1), в результате чего получали 1-(2-хлороэтил)бензимидазол (5,56 гб, 38%) в виде бесцветного маслянистого вещества.
1Н-ЯМР (CDCl3) δ ppm: 3,78 (2Н, т. I 6 Гц); 4,45 (2Н, т, I 6 Гц); 7,19-7,40 (3Н, м); 7,76 7,95 (1Н, м); 7,90 (1Н, с).
К раствору тритилмеркаптана (9,7 г, 35 мМ) в этаноле (90 мл) добавляли при 0оС раствор метоксида натрия и метанола (28% 7,1 мл). К реакционной смеси добавляли 1-(2-хлорэтил) бензимидазол (6,32 г, 35 мМ) и нагревали в течение 2 ч с обратным холодильником. Нерастворившиеся вещества отфильтровывали, а фильтрат концентрировали при пониженном давлении. Полученный концентрат хроматографировали на колонках с силикагелем (3,0 х 40 см, этилацетат: гексан 1:1 ->> 2:1) и получали 1-2-тритилтиоэтил бензимидазол (5,6 г, 38%).
1Н-ЯМР (CDCl3) δ ppm: 2,69 (2Н, т, I 6,5 Гц); 3,76 (2Н, т, I 6,5 Гц); 6,90-7,90 (21Н, м).
К раствору 1-(2-тритилтиоэтил)-бензимидазол (7,0 г, 16,6 мМ) в смеси метанола (50 мл) и хлороформа (80 мл) добавляли пиридин (1,32 мл, 16,3 мМ), а затем нитрат серебра (2,9 г, 17,1 мМ). Полученную смесь перемешивали в течение 3 ч при комнатной температуре. Осадок выделяли и промывали метанолом, а затем этиловым эфиром, в результате чего получали серебряную соль 1-(2-меркаптоэтил) бензимидазола (4,73 г, 99%). Указанную серебряную соль суспендировали (4,73 г, 16,6 мМ) в дихлорметане (250 мл) и барботировали с сероводородом при 0оС в течение 1 ч. Преципитаты отфильтровывали, а фильтрат концентрировали и в результате получали 1-(2-меркаптоэтил)бензимидазол (2,35 г, 79%) в виде бесцветного порошка (2,35 г, 79%).
1Н-ЯМР (DMCO-d6-CDCl3) δ ppm: 2,25 (1Н, т, I 7,5 Гц); 3,07 (2Н, кв, I 7,5 Гц); 4,73 (2Н, т, I 7,5 Гц); 7,40-8,10 (4Н, м); 9,69 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 2. Смесь 1Н-1,2,4-триазола (10,3 г), 1,3-оксатиолан-2-она (5,2 г) и толуола (100 мл) нагревали в колбе с обратным холодильником в течение 4 дн. Затем растворитель отгоняли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида натрия (50 мл), а затем экстрагировали 4 раза метиленхлоридом (50 мл). Экстракт высушивали безводным сульфатом натрия и подвергали дистилляции при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат ->> этилацетат: ацетон 2:1) и получали 2-(1,2,4-триазол-1-ил) этантиол (2,5 г) в виде бесцветного маслянистого вещества.
1Н-ЯМР (CDCl3) δ ppm: 1,35 (1Н, т, I 8,8 Гц); 3,01 (2Н, м); 4,37 (2Н, т, I 6,6 Гц); 7,99 (1Н, с); 8,18 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 3. К имидазолу (2,16 г, 32 мМ) добавляли метансульфон (1,03 мл, 16 мМ) и полученную смесь перемешивали в течение 5 мин. Затем к реакционной смеси добавляли этиленсульфид (1,04 мл, 18 мМ) и перемешивали при 55оС в течение 17 ч. После этого реакционную смесь хроматографировали на колонках с силикагелем (3,0 х 30, этилацетат: метанол 10:1) и получали 2-(1-имидазолио) этантиол (0,85 г, 38%) в виде маслянистого вещества бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ ppm: 1,75 (1Н, т, I 8г Гц); 2,84 (2Н, т, I 7,5 Гц); 4,13 (2Н, т, I 7,5 Гц); 7,10 (2Н, с); 7,51 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 4. Смесь имидазола (13,6 г, 0,2 M), бромохлорэтана (16,6 мл, 0,2 М), карбоната калия (0,2 М) и диметилформамида (100 мл) перемешивали в течение 20 ч при комнатной температуре. Нерастворившиеся вещества отфильтровывали, а фильтрат концентрировали при пониженном давлении, в результате чего получали 1-(2-хлороэтил)имидазол в виде жидкого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ ppm: 3,76 (2Н, т, I 6 Гц); 4,29 (2Н, т, I 6 Гц); 7,01 (1Н, с); 7,07 (1Н, с); 7,57 (1Н, с).
1-(2-хлороэтил)имидазол растворяли в этаноле (30 мл), а затем к раствору добавляли 1,2-этандитиол (25 мл) и метоксид натрия (28%-ный метаноловый раствор, 16,6 мл), полученную смесь нагревали в колбе с обратным холодильником в течение 30 мин. Нерастворившиеся вещества отфильтровывали, а фильтрат концентрировали при пониженном давлении. Затем к концентрату добавляли дихлорметан (300 мл). Органический слой промывали водой, а затем насыщенным водным раствором хлорида натрия и высушивали сульфатом натрия. Растворитель отгоняли, а остаток хроматографировали на колонках с силикагелем (3,0 х 40 см, этилацетат ->> этилацетат: метанол 10:1), в результате чего получали 2-[2-(1-имидазолил)этилтио] этандиол (3,0 г) в виде маслянистого вещества бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ ppm: 1,70 (1Н, т, I 7,8 Гц); 2,55-2,70 (4Н, м); 2,89 (2Н, т, I 6,8 Гц); 4,15 (2Н, т, I 6,8 Гц); 6,97 (1Н, с); 7,08 (1Н, с); 7,55 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 5. К этанолу (100 мл), содержащему этандитиол (5 мл) и 28%-ный метаноловый раствор метилата натрия (11,5 г), добавляли гидрохлорид 2-хлорометил-1-метилимидазола (2 г). Полученную смесь перемешивали в течение 15 мин. Затем этанол отгоняли при пониженном давлении. Остаток нейтрализовали с помощью 5 н. водного раствора соляной кислоты (9,5 мл), а затем подвергали дистилляции при пониженном давлении. Полученный остаток хроматографировали на силикагеле (3,5 х 15 см), элюируя смесью метанола и этилацетата (5:95). Целевую фракцию концентрировали и получали 2-(1-метил-2-имидазолилтио)этантиол (1,4 г) в виде бесцветного маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 1,62 (1Н, т, I 7,8 Гц); 2,55-2,80 (4Н, м); 3,69 (3Н, с); 3,81 (2Н, с); 6,87 (1Н, д, I 1,4 Гц); 6,91 (1Н, д, I 1,4 Гц).
С р а в н и т е л ь н ы й п р и м е р 6. К раствору ацетона (40 мл), содержащему 2-меркапто-1-метилимидазол (4 г) и безводный карбонат калия (20 г), добавляли по каплям и при охлаждении льдом 1-бромо-2-хлороэтан (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч, а затем к ней добавляли метиленхлорид (40 мл) и смесь фильтровали. Фильтрат концентрировали при пониженном давлении и получали в результате 2-(2-хлороэтилтио)-1-метилимидазол (6,2 г) в виде бесцветного маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 3,35 (2Н, т, I 7,0 Гц); 3,63 (3Н, с); 3,75 (2Н, т, I 7,0 Гц); 6,94 (1Н, д, I 1,2 Гц); 7,05 (1Н, д, I 1,2 Гц).
С р а в н и т е л ь н ы й п р и м е р 7. В диметилформамиде (100 мл) растворяли 1H-1,2,4-триазол (13,8 г) и 1-бромо-2-хлороэтан (28,7 г). К полученному раствору добавляли карбонат калия (27,6 г) и смесь перемешивали в течение 4 дн. Растворитель отгоняли при пониженном давлении. К остатку добавляли дихлорометан (100 мл). Нерастворившиеся вещества отфильтровывали, фильтрат концентрировали и получали 1-(2-хлороэтил)-1Н-1,2,4-триазол (23,3 г).
1Н-ЯМР (200 МГц, CDCl3) δ: 3,90 (2Н, т, I 5,7 Гц); 4,51 (2Н, т, I 5,7 Гц); 7,99 (1Н, с); 8,17, с).
Полученное хлорэтиловое соединение (2,8г) и 1,2-этандитиол (4,2 г) растворяли в этаноле (30 мл), а затем к этому раствору добавляли 28%-ный тетраоловый раствор метилата натрия (3,6 мл) и полученную смесь нагревали в колбе с обратным холодильником в течение 30 мин. Растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (элюент: этилацетат) и в результате получали 2-[2-(1Н-1,2,4-триазол-1-ил)этилтио]этантиол (2,03 г) в виде бесцветного маслянистого вещества.
1H-ЯМР (CDCl3) δ: 1,67 (1Н, т, Н); 2,62 (4Н, м); 3,02 (2Н, т, I 6,5 Гц); 4,36 (2Н, т, I 6,5 Гц); 7,97 (1Н, с); 8,14 (1Н, с).
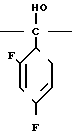
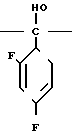
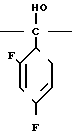
С р а в н и т е л ь н ы й п р и м е р 8. К раствору диметилформамида (160 мл), 2-(2,4-дифторофенил)-2-(1Н-1,2,4-триазол-1-ил-метил)оксирана (8 г) и сложного метилового эфира 3-меркаптопропионовой кислоты добавляли, охлаждая льдом, 60%-ный гидрид натрия (4 г). Полученную смесь перемешивали в течение 15 мин, а затем к этой смеси по каплям добавляли 1н. водный раствор соляной кислоты (101 мл) для доведения рН до 7, после чего диметилформамид и воду отгоняли. К остатку добавляли воду (20 мл), а затем экстрагировали этилацетатом (50 мл х 3). Экстракт промывали насыщенным водным раствором хлорида натрия, затем высушивали безводным сульфатом натрия, а растворитель отгоняли. Остаток хроматографировали на силикагеле (6,0 х 9,0 см), элюируя смесью этилацетата и гексана (3:1). Целевую фракцию концентрировали, добавляли диэтиловый эфир и получали 2-(2,4-дифторфенил)-3-меркапто-1- (1Н-1,2,4-триазол-1-ил) пропан-2-ол (6,44 г) в виде бесцветных игольчатых кристаллов с т.пл. 112-113оС.
Элементный анализ для C11H11F2N3OS.
Вычислено, С 48,70; Н 4,09; N 15,49.
Найдено, С 48,96; Н 4,11; N 15,62.
1Н-ЯМР (CDCl3) δ: 1,37 (1Н, дд, I 6,80 Гц, 10,8 Гц); 2,84 (1Н, дд, I 10,8 Гц, 13,8 Гц); 3,27 (1Н, дд, I 6,80 Гц, 13,8 Гц); 4,53 (1Н, с); 4,72 (2Н, с); 6,74-6,88 (2Н, м); 7,42-7,55 (1Н, м); 7,83 (1Н, с); 8,00 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 9. К ацетоновому раствору (50 мл), содержащему 2-меркапто-1-метилтетразол (4 г) и безводный карбонат калия (20 г), добавляли 1-бромо-2-хлороэтaн (5 мл), охлаждая при этом льдом. Полученную смесь перемешивали при комнатной температуре в течение 90 мин, а затем добавляли метиленхлорид (50 мл). Далее смесь фильтровали, фильтрат концентрировали при пониженном давлении и получали 2-(хлороэтилтио)-1-метилтетразол (6 г) в виде маслянистого вещества желтого цвета.
1Н-ЯМР (CDCl3) δ: 3,69 (2Н, т, I 6,8 Гц); 3,92 (2Н, т, I 6,8 Гц); 3,96 (3Н, с).
С р а в н и т е л ь н ы й п р и м е р 10. Метаноловый раствор (2 мл), содержащий (2RS, 3RS)-2-(2,4-дифторофенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил-метил)-оксиран (0,05 г), метил 3-меркаптопропионат (0,11 мл) и 28%-ный раствор метилата натрия и метанола, нагревали в колбе с обратным холодильником в течение 1 ч. Затем реакционную смесь охлаждали и добавляли воду (10 мл). Смесь нейтрализовали 1н. водным раствором соляной кислоты, после чего экстрагировали метиленхлоридом (5,0 мл х 2 раза). Раствор экстракта высушивали безводным сульфатом натрия, а затем растворитель отгоняли.
Остаток хроматографировали на силикагеле (2,5 см х 5 см), элюируя смесью этилацетата и гексана (1:2). Целевую фракцию концентрировали, а затем добавляли этилацетат, в результате чего получали (2RS, 3RS)-2-(2,4-дифторофенил)-3-меркапто-1-[(1H)-1, 2,4-триазол-1-ил] 2-бутанол (0,03 г) в виде бесцветных игольчатых кристаллов.
1Н-ЯМР (CDCl3) δ: 1,17 (3Н, д, I 7,0 Гц); 1,96 (1Н, д, I 10,2 Гц); 3,45 (1Н, д, кв, I 7,0 Гц, 10,2 Гц); 4,77 (1Н, с); 4,82 (1Н, д, I 14,4 Гц); 5,01 (1Н, д, I 14,4 Гц); 5,01 (1Н, д, I 14,4 Гц); 6,70-6,81 (2Н, м); 7,33-7,45 (1Н, м); 7,79 (1Н, с); 7,80 (1Н, с).
Полученный продукт перекристаллизовывали из этилацетата и получали бесцветное вещество в виде призмообразных кристаллов, т.пл. 145-147оС.
С р а в н и т е л ь н ы й п р и м е р 11. К этанолу (20 мл), содержащему 28% -ный раствор метанола и метилата натрия, добавляли при 80оС этаноловый раствор (20 мл) 2-хлорoметилимидазо 1,2-а пиридина гидрохлорид (2 г). Полученную смесь нагревали в колбе с обратным холодильником в течение 2,5 ч. Затем реакционную смесь охлаждали и добавляли разбавленную соляную кислоту в целях доведения рН до 1, после чего трижды промывали толуолом (30 мл х 3). После этого к водному слою добавляли водный раствор гидроокиси натрия для доведения рН до 10 и затем экстрагировали 3 раза метиленхлоридом (30 мл х 3). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрат хроматографировали на силикагеле (3 см х 15 см), элюируя смесью этилацетата и гексана (2:1). Целевую фракцию концентрировали при пониженном давлении и получали 2-(4-метоксибензилтиометилимидазо [1,2-a] пиридин (2,5 г) в виде бесцветного маслянистого продукта.
1Н-ЯМР (CDCl3) δ: 3,73 (2Н, с); 3,78 (2Н, с); 3,78 (2Н, с); 3,79 (3Н, с); 6,75 (1Н, м); 6,83 (2Н, д, I 6,6 Гц); 7,13 (1Н, м); 7,25 (2Н, д, I 6,6 Гц), 7,46 (1Н, с); 7,55 (1Н, д, I 9,0 Гц); 8,04 (1Н, дд, I 1,0 Гц, 5,6 Гц).
К смеси полученного продукта (2,5 г), анизола (20 мл) и трифторуксусной кислоты (50 мл) добавляли ацетат серебра [II] (3,2 г), а затем перемешивали при комнатной температуре в течение 4 ч. После этого реакционную смесь концентрировали при пониженном давлении. В концентрат добавляли петролейный эфир. Полученный в результате бесцветный порошок отделяли, собирали путем фильтрации и промывали диэтиловым эфиром. Полученный порошок (5 г) суспендировали в N, N-диметилформамиде (60 мл). В суспензию вдували сероводород до тех пор, пока она не станет черной. Затем реакционную смесь барботировали с газообразным азотом в целях удаления избыточного сероводорода, а затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (2 см х 15 см), элюируя смесью метанола и метиленхлорида (1: 19). Целевую фракцию концентрировали при пониженном давлении. К концентрату добавляли метанол и метиленхлорид и в результате получали 2-меркаптометилимидазо [1,2-a]пиридин (1,3 г) в качестве бесцветных игольчатых кристаллов, т.пл. 168-178оС.
1Н ЯМР (DMCO-d6) δ: 3,30 (1Н, т, I 8,0 Гц); 4,00 (2Н, д, I 8,0 Гц); 7,36 (1Н, дт. I 1,8 Гц, 6,6 Гц); 7,75-7,89 (2Н, м); 8,16 (1Н, с); 8,82 (1Н, д, I 7,2 Гц).
С р а в н и т е л ь н ы й п р и м е р 12. К суспензии имидазол-2-карбоксиальдегида (2,5 г) в N,N-диметилформамиде (25 мл) добавляли 60%-ный гидрид натрия в масле (1,2 г) при комнатной температуре, полученную смесь перемешивали в течение 30 мин. К этому раствору при комнатной температуре добавляли 2,2,3,3-тетрафторопропил-n-толуолсульфонат (11,2 г) и смесь перемешивали при 110оС в течение 2,5 ч. Затем смесь охлаждали, добавляли воду (100 мл) и толуол (30 мл) для экстрагирования. Затем водный слой еще раз экстрагировали толуолом 3 раза (30 мл х 3). Слои толуола объединяли, промывали насыщенным водным хлоридом натрия (30 мл) и высушивали безводным сульфатом натрия, после чего растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (3,0 см х 10 см), элюируя смесью этилацетата и гексана (1: 1). Целевую фракцию концентрировали и получали 1-(2,2,3,3-тетрафторопропил)имидазол-2-карбоксиальдегид (2,7 г) в виде бесцветных пластинок с т.пл. 51-54оС.
1Н-ЯМР (CDCl3) δ: 5,15 (2Н, т, I 12,6 Гц); 5,92 (1Н, т, т, I 54 Гц, 2,6 Гц); 7,27 (1Н, с); 7,38 (1Н, с); 9,84 (1Н, с).
Элементный анализ для C7H6F4N2O.
Вычислено, С 40,01; Н 2,99; N 13,33.
Найдено, С 39,68; Н 2,86; N 13,11.
К раствору 1-(2,2,3,3-тетрафторопропил)-имидазол-2-карбоксиальдегида (1,5 г) в метаноле (15 мл) добавляли борогидрид натрия (0,08г г) при 0оС, полученную смесь перемешивали при 0оС в течение 40 мин. После этого к смеси добавляли насыщенный водный раствор хлорида натрия (5 мл) и смесь опять перемешивали в течение 50 мин, а затем экстрагировали этилацетатом (30 мл х 4 раза). Органический слой высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. После добавления к остатку этилацетата и гексана получали 2-гидроксиметил-1-(2,2,3,3-тетрафторопропил)имидазол (1,5 г) в виде бесцветных игольчатых кристаллов, т.пл. 91-92оС.
Элементный анализ для C7H8F4N2O.
Вычислено, С 39,63; Н 3,80; N 13,20.
Найдено, С 39,79; Н 3,78; N 13,20.
1Н-ЯМР (CDCl3) δ: 4,68 (2Н, т, I 12,4 Гц); 4,68 (2Н, с); 5,88 (1Н, т,т, I 53,2 Гц, 2,6 Гц); 6,93 (1Н, с); 6,96 (1Н, с).
К тионилхлориду (7 мл) постепенно добавляли 2-гидроксиметил-1-(2,23,3-тетрафторoпропил)имидазол (0,7 г) при 0оС и полученную смесь нагревали в колбе с обратным холодильником в течение 45 мин. Затем смесь концентрировали при пониженном давлении, добавляли диэтиловый эфир и осажденные кристаллы собирали фильтрацией. Эти кристаллы растворяли в этаноле и перекристаллизовывали из диэтилового эфира, в результате чего получали 2-хлорометил-1-(2,2,3,3-тетрафторопропил) имидазолгидрохлорид (0,9 г) в виде бесцветных игольчатых кристаллов с т.пл. 104-107оС.
Элементный анализ для C7H8Cl2F4N2.
Вычислено, С 31,48; Н 3,02; N 10,49.
Найдено, С 31,74; Н 2,94; N 10,44.
1Н-ЯМР (DMCO-d6) δ: 5,13 (2Н, с); 5,17 (2Н, т, I 16,2 Гц); 6,76 (1Н, т, т. I 51,8 Гц, 5 Гц); 7,66 (1Н, д, I 1,8 Гц); 7,69 (1Н, шир.с).
С р а в н и т е л ь н ы й п р и м е р 13. Смесь 1Н-1,2,4-триазола (20 г) и параформальдегида (9 г) нагревали при 170оС в течение 1,5 ч, а затем к этой смеси добавляли еще 9 г параформальдегида. Полученную смесь нагревали при 170оС в течение 1,5 ч, а затем подвергали дистилляции при пониженном давлении с целью удаления оставшегося триазола. Остаток охлаждали и добавляли диметилформамид (150 мл). Затем при охлаждении льдом к смеси добавляли третбутилдиметилхлорид (25 г), после чего смесь перемешивали при комнатной температуре в течение 1,25 ч. Реакционную смесь концентрировали при пониженном давлении, добавляли водный насыщенный раствор бикарбоната натрия (100 мл) и экстрагировали метиленхлоридом (30 мл х 3 раза). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрат хроматографировали на силикагеле (5 х 20 см), элюируя смесью этилацетата и гексана (3:1). Целевую фракцию концентрировали и получали в результате 3-третбутилдиметилсилоксиметил- 1н-1,2,4-триазол (15 г) в виде бесцветного маслянистого продукта.
1Н-ЯМР (CDCl3) δ: 0,13 (6Н, с), 0,93 (9Н, с); 4,94 (2Н, с); 8,03 (1Н, с).
Раствор 3-третбутилдиметилсилоксиметил-1Н-1,2,4-триазола (8 г) в N,N-диметилформамиде (20 мл) по каплям добавляли при 0оС к смеси 60% гидрида натрия в масле (1,5 г), метилиодида (2,8 мл) и N,N-диметилформамида (80 мл) в течение 10 мин. Полученную смесь перемешивали в течение 10 мин, после чего к этой смеси добавляли воду (300 мл) и экстрагировали этилацетатом (100 мл х 3 раза). Органический слой промывали насыщенным водным раствором хлорида натрия, высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрат хроматографировали на силикагеле, элюируя сначала смесью этилацетата и гексана (1:1), а затем этилацетатом. Целевые фракции концентрировали при пониженном давлении и получали 5-третбутилдиметилсилоксиметил-1-метил- 1Н-1,2,4-триазол (4,4 г), 3-третбутилдиметилсилоксиметил-1-метил-1Н-1,2,4-триазол (2,4 г) и 3-третбутилдиметилсилоксиметил-4-метил-4Н-1,2,4-триазол (0,5 г).
5-третбутилдиметилсилоксиметил-1-ме- тил-1Н-1,2,4-триазол бесцветное маслянистое вещество.
1H-ЯМР (CDCl3) δ: 0,09 (6Н, с); 0,90 (9Н, с); 3,96 (3Н, с); 4,85 (2Н, с); 7,79 (1Н, с);
3-третбутилдиметилсилоксиметил-1-ме- тил-1Н-1,2,4-триазол бесцветное маслянистое вещество.
1Н-ЯМР (CDCl3) δ: 0,13 (6Н, с); 0,93 (9Н, с); 3,90 (3Н, с); 4,77 (2Н, с); 7,97 (1Н, с).
2-третбутилдиметилсилоксиметил-4-ме- тил-4Н-1,2,4-триазол бесцветные игольчатые кристаллы.
1Н-ЯМР (CDCl3) δ: 0,09 (6Н, с); 0,89 (9Н, с); 3,76 (3Н, с); 4,90 (2Н, с); 8,08 (1Н, с).
Т.пл. (кристаллизованного из диэтилового эфира) 94-95оС.
Смесь 5-третбутилдиметилсилоксиметил-1-метил-1Н-1,2,4-триазола (3 г), этанола (20 мл), 5н.водного раствора гидроокиси натрия (4 мл) и метанола (30 мл) перемешивали в течение 24 ч при комнатной температуре. Затем реакционную смесь концентрировали при пониженном давлении, а концентрат хроматографировали на силикагеле (3,0 х 15 см), элюируя смесью метанола и метиленхлорида (1:9). Целевую фракцию концентрировали при пониженном давлении, в результате чего получали 5-гидроксиметил-1-метил-1Н-1,2,4-триазол (1,2 г) в виде бесцветного твердого продукта.
1Н-ЯМР (CDCl3) δ: 3,95 (3Н, с); 4,76 (2Н, шир.с), 5,33 (1Н, шир.с), 7,78 (1Н, с).
К тионилхлориду (8 мл) постепенно при 0оС добавляли 5-гидроксиметил-1-метил-1Н-1,2,4-триазол (0,8 г) и смесь нагревали в колбе с обратным холодильником в течение 3 ч. Полученную реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли диэтиловый эфир и полученный продукт в виде порошка собирали путем фильтpации. Затем этот продукт растворяли в этаноле, добавляли диэтиловый эфир для кристаллизации и получали 5-хлоро-1-метил-1Н-1,2,4-триазол гидрохлорида (1,1 г) в виде бесцветных кристаллических пластинок с т.пл. 77-78оС.
Элементный анализ для C4H7Cl2N3 х х 1/2H2O.
Вычислено, С 27,14; Н 4,55; N 23,74.
Найдено, С 27,60; Н 3,98; N 23,66.
1Н-ЯМР (DMCO-d6) δ: 3,92 (3Н, с); 5,01 (2Н, с); 8,01 (1Н, с).
Смесь 3-третбутилдиметилсилоксиметил-1-метил-1Н-1,2,4-триазола (2 г), этанола (10 мл), метанола (20 мл) и 5н.водного раствора гидроокиси натрия (2,6 мл) перемешивали при комнатной температуре в течение 27 ч, а затем перемешивали еще 21 ч при 45оС. Реакционную смесь концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (2 х 10 см), элюируя смесью метанола и метиленхлорида (1:9). Целевую фракцию концентрировали при пониженном давлении, в результате получали 3-гидроксиметил-1-метил-1Н-1,2,4-триазол (1 г) в виде бесцветных игольчатых кристаллов.
1Н-ЯМР (CDCl3) δ: 3,91 (3Н, с); 4,75 (2Н, д, I 4,6 Гц); 4,06 (1Н, шир. с), 8,02 (1Н, с).
Т.пл. 72-75оС.
К тионилхлориду (8 мл) постепенно при 0оС добавляли 3-гидрокси-1-метил-1Н-1,2,4-триазол (0,6 г), полученную смесь нагревали в колбе с обратным холодильником в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении, после чего к смеси добавляли диэтиловый эфир. Полученный порошок кристаллизовали из смеси этанола и диэтилового эфира и получали 3-хлорометил-1-метил-1Н-1,2,4-триазол гидрохлорида (1 г) в виде бесцветных игольчатых кристаллов с т.пл. 69-70оС.
Элементный анализ для C4H7Cl2N3.
Вычислено, С 28,59; Н 4,20; N 25,01.
Найдено, С 28,16; Н 4,08; N 24,51.
1Н ЯМР (DMCO-d6) δ: 3,87 (3Н, с); 4,72 (2Н, с); 8,57 (1Н, с).
Смесь 3-третбутилдиметилсилоксиметил-4-метил-4Н-1,2,4-триазола (0,4 г), этанола (12 мл) и 5н. гидроокиси (0,53 мл) перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь концентрировали при пониженном давлении и полученный концентрат хроматографировали на силикагеле (2 х 10 см), элюируя смесью метанола и метиленхлорида (1:4). Целевую фракцию концентрировали при пониженном давлении и получали 3-гидроксиметил-4-метил-4Н-1,2,4-триазол (0,2 г) в виде бесцветного твердого вещества.
1Н-ЯМР (DMCO-d6) δ: 3,66 (3Н, с); 4,59 (2Н, д, I 5,6 Гц); 5,52 (1Н, т, I 5,6 Гц), 8,40 (1Н, с).
К тионилхлориду (2 мл) добавляли (при 0оС) 3-гидроксиметил-4-метил-4-1,2,4-триазол (0,15 г) и полученную смесь нагревали в колбе с обратным холодильником в течение 1,5 ч. Затем смесь концентрировали при пониженном давлении, после чего добавляли диэтиловый эфир. Полученные кристаллы собирали путем фильтрации и растворяли в этаноле, а затем к этому раствору добавляли в целях перекристаллизации диэтиловый эфир, в результате чего получали 3-хлорометил-4-метил-4Н-1,2,4-триазол гидрохлорида (0,22 г) в виде бесцветных призмообразных кристаллов с т.пл. 96-97оС.
Элементный анализ для C4H7Cl2N3.
Вычислено, С 28,59; Н 4,20; N 25,01.
Найдено, С 28,70; Н 4,18; N 24,91.
1Н-ЯМР (DMCO-d6) δ: 3,81 (3Н, с); 5,11 (2Н, с); 9,26 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 14. 3-Гидроксиметил-5-меркапто-4-метил-4Н-1, 2,4-триазол (1,5 г) добавляли при 0оС в течение 1 ч к смеси концентрированной азотной кислоты (d 1,38, 2,3 мл), воды (6 мл) и нитрита натрия (0,005 г). Реакционную смесь нагревали до комнатной температуры, после чего ее выдерживали при этой температуре в течение 1 ч, а затем добавляли водный раствор гидроокиси натрия в целях доведения рН до 8. Воду отгоняли при пониженном давлении, а остаток хроматографировали (2,5 см х 10 см), элюируя смесью метанола и метиленхлорида (1:4). Целевую фракцию концентрировали, а концентрат перекристаллизовывали из этилацетата, в результате чего получали 3-гидрокси-4-метил-4Н-1,2,4-триазол (1,1 г) в виде бесцветных игольчатых кристаллов.
1Н-ЯМР (DMCO-d6) δ: 3,66 (3Н, с); 4,59 (2Н, g, I 5,6 Гц); 5,52 (1Н, т, I 5,6 Гц); 8,40 (1Н, с).
Т.пл. 81-82оС.
С р а в н и т е л ь н ы й п р и м е р 15. Смесь 6,7-дигидро-5Н-пирроло [1,2-c] имидазола (7 г) и параформальдегида (4 г) нагревали при 160оС, после чего через 20 мин добавляли параформальдегид (2 г), а еще через 20 мин добавляли 1 г параформальдегида и полученную в результате смесь нагревали в течение 45 мин. Затем реакционную смесь хроматографировали (4 х 15 см) на силикагеле, элюируя смесью метанола и метиленхлорида (1:9). Целевую фракцию концентрировали. К концентрату добавляли этанол и диэтиловый эфир и в результате получали 6,7-дигидро-3-гидроксиметил-5Н-пирроло[1,2-c]имидазол (3,4 г) в виде бесцветных игольчатых кристаллов с т.пл. 110-120оС.
Элементный анализ для C7H10N2O.
Вычислено, С 60,85; Н 7,29; N 20,27.
Найдено, С 61,08; Н 7,30; N 20,27.
1Н-ЯМР (CDCl3) δ: 2,5-2,7 (2Н, м); 2,81 (2Н, т, I 7,4 Гц); 4,01 (2Н, т, I 7,0 Гц); 4,56 (2Н, с); 6,2 (1Н, шир.с); 6,54 (1Н, с).
К тиенилхлориду (0,8 мл) при 0оС постепенно добавляли 6,7-дигидро-3-гидроксиметил-5-пирроло [1,2-c)имидазола (0,8 г) и полученную смесь нагревали с обратным холодильником в течение 40 мин, после чего тионилхлорид отгоняли при пониженном давлении. К остатку добавляли диэтиловый эфир и полученное твердое вещество собирали путем фильтрации. Затем это вещество растворяли в этаноле и после добавления диэтилового эфира получали 3-хлорометил-6,7-дигидро-5Н-пирроло [1,2-c]имидазол гидрохлорида (0,7 г) в виде игольчатых кристаллов бледно-желтого цвета, т.пл. 120-140оС.
Элементный анализ для C7H10Cl2N2.
Вычислено, С 43,55; Н 5,22; N 14,51.
Найдено, С 43,74; Н 5,21; N 14,31.
1Н-ЯМР (DMCO-d6) δ: 2,5-2,7 (2Н, м); 2,96 (2Н, т, I 6,8 Гц); 4,28 (2Н, т, I 7,0 Гц); 5,16 (2Н, с); 7,43 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 16. Смесь 2-ацетокситиоацетамида (10 г), 2-хлороциклопентанона (10,6 г) и диметилформамида (100 мл) перемешивали в течение 24 ч при 80оС, затем реакционную смесь охлаждали и выливали в ледяную воду (500 мл), после чего дважды экстрагировали этилацетатом (200 мл). Экстракт дважды промывали водой (100 мл) и высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (2,5 х 50 см), элюируя смесью этилацетата и гексана (3:7). Целевую фракцию концентрировали и получали в результате 2-ацетоксиметил-5,6-дигидро-4Н-циклопента- тиазол (5 г) в виде маслянистого продукта желтого цвета.
К полученному продукту (5 г) добавляли 5н. гидроксид натрия (10 мл), смесь перемешивали при 80оС в течение 2 ч. Затем реакционную смесь охлаждали, нейтрализовали с помощью 2н. соляной кислоты и экстрагировали этилацетатом (200 мл). Экстракт промывали водой (50 мл) и высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (2,5 х 30 см), элюируя смесью ацетата и дихлорометана (3: 2). Целевую фракцию концентрировали и получали 2-гидроксиметил-5,6-дигидро-4Н-циклопентатиазол (3,5 г) в виде маслянистого продукта желтого цвета.
1Н-ЯМР (CDCl3) δ: 2,40-2,59 (4Н, м); 2,74-2,99 (4Н, м); 3,49 (1Н, шир. с); 4,85 (2Н, с).
Полученный продукт (0,17 г) растворяли в метиленхлориде (4 мл); к полученному раствору по капле добавляли тионилхлорид (1,52 мл), после чего смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель отгоняли при пониженном давлении и получали 2-хлорометил-5,6-дигидро-4Н-циклопентатиазол гидрохлорида (0,22 г) в виде маслянистого продукта красноватого цвета.
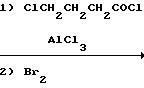
С р а в н и т е л ь н ы й п р и м е р 17. К смеси m-дифторобензола (75 мл) и безводного хлорида алюминия (115 г) добавляли по каплям, перемешивали при этом, в течение 50 мин 2-бромопропионилхлорид (100 г). Смесь перемешивали затем на масляной бане при 50-55оС в течение 2 ч. Затем реакционную смесь охлаждали и добавляли метиленхлорид (500 мл). Далее полученный раствор, перемешивали при этом, добавляли в ограниченных количествах в ледяную воду (1,5 л). Затем слой метиленхлорида отделяли, а водный слой дважды экстрагировали метиленхлоридом (100 мл). После этого метиленхлоридные слои объединяли и промывали водой 500 мл, а затем высушивали безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и получали 2-бромо-2',4'-дифторопропиофенон (142,5 г) в виде маслянистого продукта желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,90 (3Н); 5,25 (1Н); 6,85-7,06 (2Н); 7,93-8,05 (1Н).
С р а в н и т е л ь н ы й п р и м е р 18. К смеси m-дифторобензола (100 мл) и безводного хлорида алюминия (114 г) добавляли по каплям, в течение 45 мин, и перемешивали при этом, пропионилхлорид (66 мл). Затем смесь перемешивали на масляной бане при 50-55оС в течение 2 ч. После этого реакционную смесь охлаждали и добавляли в нее метиленхлорид (300 мл). Полученный раствор порциями добавляли в ледяную воду (1 л), при этом перемешивая. Затем слои метиленхлорида объединяли, промывали водой (200 мл) и высушивали безводным сульфатом магния. Растворитель отгоняли и получали 2', 4'-дифторопропиофенон (111,4 г) в виде маслянистого продукта желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,17-1,24 (3Н); 2,92-3,05 (2Н); 6,82-7,02 (2Н); 7,89-8,03 (1Н).
С р а в н и т е л ь н ы й п р и м е р 19. В метиленхлориде (300 мл) растворяли 2', 4'-дифторопропиофенон (55 г), а затем по каплям в течение 30 мин, при этом перемешивая, добавляли бром (50 г). Смесь перемешивали при комнатной температуре в течение 30 мин. Затем к этой реакционной смеси добавляли воду (200 мл), а слой метиленхлорида промывали 3 раза и высушивали безводным сульфатом магния. Растворитель отгоняли и получали 2-бромо-2', 4'-дифторопропиофенон (77 г) в виде маслянистого продукта бледно-желтого цвета.
С р а в н и т е л ь н ы й п р и м е р 20. 2-Бромо-2', 4'-дифторопропиофенон (141 г) растворяли в метаноле (1100 г), к раствору добавляли формат натрия (176,2 г) и полученную смесь перемешивали при 50оС в течение 2 дн. Затем метанол отгоняли при пониженном давлении. Остаток экстрагировали путем добавления этилацетата (700 мл) и воды (500 мл). Слой этилацетата высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток кристаллизовали из гексана (200 мл) и получали 2',4'-дифторо-2-гидроксипропиофенон (50,5 г) в виде бесцветных призмообразных кристаллов с т.пл. 49-51оС.
ЯМР (CDCl3) δ: 1,40, 1,41 (3Н, д х 2, I 7 Гц); 3,74 (1Н, д, I 6,2 Гц); 4,96-5,11 (1Н, м); 6,87-7,27 (2Н, м); 7,69-8,09 (1Н, м).
С р а в н и т е л ь н ы й п р и м е р 21. 2', 4'-Дифторо-2-гидроксипропиофенон (61 г) растворяли в метиленхлориде (500 мл), а затем добавляли, охлаждая льдом, гидрат n-толуолсульфоновой кислоты (1 г). К полученной смеси при охлаждении льдом и перемешивании в течение 10 мин добавляли 3,4-дигидро-2Н-пиран (41,4 г). Смесь перемешивали при охлаждении льдом в течение 1 ч, а затем к этой смеси добавляли 5%-ный водный раствор бикарбоната натрия (240 мл). Смесь перемешивали 10 мин при охлаждении льдом. Слой метиленхлорида отделяли и высушивали безводным сульфатом магния, а растворитель отгоняли. Остаточный маслянистый продукт очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (5:1), в результате получали 2', 4'-дифторо-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропиофенона (86,5 г) в виде маслянистого продукта желтого цвета.
С р а в н и т е л ь н ы й п р и м е р 22. К диметилсульфоксиду (650 мл) добавляли при комнатной температуре порциями в течение 10 мин 60% гидрата натрия в масле (15,2 г). Смесь перемешивали при комнатной температуре в течение 10 мин, а затем к этой смеси порциями в течение 1 ч добавляли йодид триметилсульфоксония (83,7 г). К полученной смеси по каплям в течение 1 ч добавляли раствор 2', 4'-дифторо-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропиофенона (86,5 г) в диметилсульфоксиде (150 мл). Смесь перемешивали в течение 3 ч при комнатной температуре, а затем реакционную смесь выливали в ледяную воду (1,5 л) и 5 раз экстрагировали этилацетатом (300 мл). Слой этилацетата промывали 4 раза водой (300 мл), а затем высушивали безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и получали 2-[1-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)этил] -2-(2,4- дифторофенил)оксиран (83,4 г) в виде маслянистого продукта желтого цвета.
С р а в н и т е л ь н ы й п р и м е р 23. К N,N-диметилформамиду (700 мл) при комнатной температуре и перемешивании в течение 10 мин порциями добавляли 60%-ный гидрид натрия в масле (35,2 г). Смесь перемешивали в течение 5 мин при комнатной температуре, а затем к ней добавляли порциями в течение 20 мин при комнатной температуре 1Н-1,2,4-триазол (25 г). Реакционную смесь охлаждали льдом и к смеси, примешивая, добавляли в течение 30 мин 1Н-1,2,4-триазол (44,6 г), после чего смесь перемешивали при комнатной температуре еще 10 мин. К полученной смеси в течение 5 мин по каплям добавляли 2-[1-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) этил] -2-(2,4-дифторофенил) оксиран (83,4 г) и смесь перемешивали при 90оС в течение 4 ч. Затем реакционную смесь охлаждали, выливали в ледяную воду (1,5 л) и экстрагировали этилацетатом (500 мл) 4 раза. Слой этилацетата промывали 3 раза водой (300 мл) и высушивали безводным сульфатом магния. Затем растворитель отгоняли и получали маслянистый продукт светло-желтого цвета. Этот продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и ацетона (4: 1 ->> 1:1). Полученный маслянистый продукт кристаллизовали из гексана и получали 2-(2,4-дифторофенил)-3-(3,4,5,6-тетрагидро-2Н-пиран-2-ил)окси- 1-(1Н- 1,2,4-триазол-1-ил)-2-бутанол (51,4 г) в виде бесцветного порошка с т.пл. 93-95оС.
Элементный анализ для C17H21F2N3O3.
Вычислено, С 57,78; Н 5,99; N 11,89.
Найдено, С 57,82; Н 6,04; N 11,77.
С р а в н и т е л ь н ы й п р и м е р 24. В этаноле (500 мл) растворяли 2-(2,4-дифторофенил)-3-(3,4,5,6-тетрагидро-2Н-пиран-2-ил)окси-1- (1Н-1,2,4-триазол-1-ил)-2-бутанол (51,4 г), затем к полученному раствору добавляли n-толуолсульфонат пиридиния (13,2 г) и смесь перемешивали при 55оС в течение 6 ч. После этого к полученной смеси добавляли еще 2 г n-толуолсульфонат пиридиния и смесь перемешивали при 55оС в течение 1,5 ч. Затем смесь охлаждали, а растворитель отгоняли при пониженном давлении. К остатку добавляли этилацетат (900 мл) и смесь промывали 3 раза водой (50 мл). Слой этилацетата высушивали безводным сульфатом магния, а растворитель отгоняли. К остатку добавляли этилацетат (50 мл) и этиловый эфир (100 мл). Осажденные кристаллы собирали фильтрацией и получали чистое соединение (99% чистоты): (2RS, 3RS)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триа- зол-1-ил)-2,3-бутандиол (29 г) в виде диастеромера, т.пл. 154-156оС.
1Н-ЯМР (CDCl3) δ: 0,93 (3Н, д, I 6,2 Гц); 4,26-4,39 (1Н, м); 4,82 (2Н, с); 6,71-6,83 (2Н, м); 7,35-7,51 (1Н, м); 7,84 (1Н, с); 7,87 (1Н, с).
Элементный анализ для C12H13F2N3O2.
Вычислено, С 53,53; Н 4,87; N 15,61.
Найдено, С 53,35; Н 4,90; N 15,49.
С р а в н и т е л ь н ы й п р и м е р 25. В смеси этилацетата (200 мл) и метиленхлорида (50 мл) растворяли (2RS, 3RS)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил)-2, 3-бутандиол (11 г), к полученной смеси добавляли при охлаждении льдом триэтиламин (6,21 мл). К этой смеси в течение 3 мин по каплям добавляли метансульфонилхлорид (3,46 мл), при этом перемешивая и охлаждая льдом, а затем перемешивали при комнатной температуре в течение 45 мин. К реакционной смеси добавляли воду (100 мл), затем органический слой отделяли, промывали водой и высушивали безводным сульфатом натрия, после чего растворитель отгоняли при пониженном давлении и получали (2RS, 3RS)-2-(2,4-дифторофенил)-3-метансульфонилокси-1-(1Н-1,2,4-триазол-1- ил)-2-бутанол в виде маслянистого продукта. Затем продукт растворяли в метаноле (200 мл) и к раствору при охлаждении льдом добавляли 28%-ный метаноловый раствор метилата натрия (8,84 г), после чего смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель отгоняли при пониженном давлении. К остатку добавляли этилацетат (200 мл) и воду (100 мл) для экстрагирования. Слой этилацетата промывали водой и высушивали безводным сульфатом натрия, а растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и метиленхлорида (4:1), после кристаллизации из гексана получали (2RS, 3RS)-2-(2,4-дифторофенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил)метилоксиран (8,3 г) в виде одного диастереомера бесцветных кристаллов, т.пл. 66-68оС.
1Н-ЯМР (CDCl3) δ: 1,65 (3Н, д, I 5,6 Гц); 3,20 (1Н, кв, I 5,6 Гц); 4,42 (1Н, д, I 14,6 Гц); 4,89 (1Н, д, I 14,6 Гц); 6,68-6,83 (2Н, м), 6,93-7,08 (1Н, м); 7,82 (1Н, с); 7,97 (1Н, с).
Элементный анализ для C12H11F2N3O.
Вычислено, С 57,37г; Н 4,41; N 16,73.
Найдено, С 57,31. Н 4,44; N 16,62.
С р а в н и т е л ь н ы й п р и м е р 26. В дихлорметане (200 мл) растворяли (S)-(-)-этиллактат (35,4 г), к раствору, охлаждая льдом, добавляли гидрат n-толуолсульфоновой кислоты (570 мг). Затем к смеси по каплям в течение 30 мин добавляли 3,4-дигидро-2Н-пиран (30,2 г) и полученную смесь размешивали при охлаждении льдом в течение 1 ч. К реакционной смеси добавляли 5% -ный водный раствор бикарбоната натрия (50 мл), энергично перемешивая, после чего органический слой отделяли. Затем органический слой промывали 5% -ным водным раствором бикарбоната натрия и высушивали безводным сульфатом магния, после чего растворитель отгоняли при пониженном давлении и получали этил (2S)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)пропионат (61 г) в виде маслянистого продукта желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,28 (3Н, т, I 7,0 Гц); 1,40-1,46 (3Н, д, I 6,8 Гц); 1,40-2,00 (6Н, м); 3,40-3,60 (2Н, м), 3,80-4,00 (2Н, м); 4,10-4,44 (3Н, м); 4,68-4,76 (1Н, м).
Сравнительный пример 27. В этаноле (450 мл) растворяли этил (2S)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионат и к раствору при охлаждении льдом добавляли 2н. раствор гидроокиси натрия (150 мл), полученную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали льдом, а затем к этой смеси добавляли при 27оС водный раствор уксусной кислоты (100 мл) и 3 раза экстрагировали дихлорометаном (200 мл). Слои дихлорометана объединяли, промывали дважды насыщенным раствором и солевым раствором (100 мл) и высушивали безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и получали (2S)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионовую кислоту (29,5 г) в виде бесцветного воскообразного продукта.
К (2S)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионовой кислоте (29,5 г) добавляли безводный тетрагидрофуран (250 мл). Перемешивая при комнатной температуре, к смеси в течение 10 мин добавляли 1,1-карбонил-диимидазол (33,1 г). Полученную смесь перемешивали при комнатной температуре в течение 30 мин, охлаждая льдом, а затем по капле в течение 15 мин добавляли морфолин (34,8 г). Полученную смесь перемешивали в течение 15 мин в ледяной бане, а затем концентрировали при пониженном давлении. Концентрат растворяли в дихлорметане (300 мл), промывали насыщенным водным раствором хлорида натрия (50 мл) и высушивали безводным сульфатом магния, а растворитель отгоняли. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:4), и получали N-[(2S)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси]пропионил морфолин (17,5 г).
1Н-ЯМР (CDCl3) δ: 1,39-1,44 (3Н, д, I 6,8 Гц); 1,45-1,96 (6Н, м); 3,40-3,95 (10Н, м); 4,52, 4,68 (1Н, кв. I 6,8 Гц); 4,60 (1Н, м).
ИК (пленка): 2945, 2855, 1662, 1650, 1460, 1435, 1370, 1270, 1230, 1110, 1020, 980 см-1.
С р а в н и т е л ь н ы й п р и м е р 28. В безводном тетрагидрофуране (40 мл) растворяли 1-бромо-2,4-дифторобензол (7,72 г). К раствору добавляли при комнатной температуре магний (хлопья, 972 мг) и очень небольшое количество йода, затем смесь энергично перемешивали приблизительно в течение 2 ч, в результате чего получали 1М раствора бромида 2,4-дифторофенилмагния. Из этого раствора брали порцию в 9,5 мл, в ней разбавляли 9,5 мл безводного тетрагидрофурана, а затем этот раствор по каплям добавляли к безводному раствору тетрагидрофурана (25 мл) и N-[(2S)-2-(3,4,5-тетрагидро-2Н-пиран-2-илокси)пропионил] морфолина (2,26 г) в течение 25 мин при температуре (-30) (-20)оС. По окончании добавления температуру смеси повышали до 20оС в течение 1 ч. Затем смесь перемешивали при 20оС еще в течение 2 ч. После этого реакционную смесь охлаждали льдом и к этой смеси добавляли насыщенный водный раствор хлорида аммония (20 мл) с последующим экстрагированием этилацетатом (100 мл). Экстракт высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (10:1), и получали в результате (2S)-2', 4'-дифторо-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропиофенон (1,02 г) в виде маслянистого продукта бледно-желтого цвета.
ИК (пленка): 3075, 2950, 2875, 1695, 1605, 1500, 1422, 1370, 1265, 1230, 1132, 1090, 1030, 970, 848 см-1.
Оптическую чистоту этого соединения определяли следующим способом.
В этаноле (3 мл) растворяли (2S)-2',4'-дифторо-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропиофенон (95 мг) и к полученному раствору добавляли n-толуолсульфонат пиридиния (21 мг), после чего смесь перемешивали при 55оС в течение 1 ч. Растворитель отгоняли при пониженном давлении, а остаток растворяли в этилацетате (20 мл) и высушивали безводным сульфатом магния, после чего растворитель отгоняли. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (5:1), в результате чего получали (2S)-2', 4'-дифторо-2-гидроксипропиофенон (40 мг) в виде маслянистого продукта бледно-желтого цвета.
Полученный продукт анализировали с помощью высоко разрешающей жидкостной хроматографии (подвижная фаза: гексан, изопропиловый спирт 9:1), используя колонку для разделения оптического изомера (CHIRALCEL 0,46 см х 25 см, изготовлена Daicel Chemical Industries, Ltd]
0,46 см х 25 см, изготовлена Daicel Chemical Industries, Ltd]
Избыток энантиомера составлял 98,4%
1Н-ЯМР (CDCl3) δ: 1,41 (3Н, дд, I 7,0 Гц); I 1,6 Гц); 3,74 (1Н, д); 5,01 (1Н, м); 6,86-7,08 (2Н, м); 7,96-8,08 (1Н, м).
ИК (пленка): 3450, 1690, 1612, 1500, 1430, 1270, 1145, 1100, 1030, 980, 858 см-1.
С р а в н и т е л ь н ы й п р и м е р 29. К диметилсульфоксиду (5 мл) добавляли 60% -ный гидрид натрия в масле (85 мг), а затем йодид триметилсульфоксония (0,49 г), перемешивая при этом при 15оС, смесь перемешивали еще 15 мин при комнатной температуре. Реакционную смесь охлаждали льдом и к этой смеси добавляли диметилсульфоксидный раствор [1.2 мл] (2S)-2', 4'-дифторо-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионфенона (0,5 г), после чего полученную смесь перемешивали при комнатной температуре в течение 2 ч. Затем к реакционной смеси добавляли воду (10 мл) и этилацетат (50 мл) и полученную в результате смесь встряхивали. Органический слой промывали насыщенным водным раствором хлорида натрия 2 раза (10 мл), высушивали безводным сульфатом магния. Растворитель отгоняли при пониженном давлении, а остаток очищали с помощью хроматографии на силикагеле (элюент: смесь гексана и этилацетата 10: 1), в результате чего получали 2-[(1S)-1-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)этил] -2-(2, 4-дифторофенил) оксиран (0,4 г) в виде маслянистого продукта бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,10-1,26 (3Н, м); 1,40-1,95 (6Н, м); 2,83 (1Н, м); 3,05, 3,32 (1Н, д, I 5,2 Гц); 3,42-3,59 (1Н, м); 3,76-4,12 (2Н, м); 4,74-4,96 (1Н, м); 6,72-6,95 (2Н, м); 7,32-7,60 (1Н, м).
С р а в н и т е л ь н ы й п р и м е р 30. В диметилформамиде (5 мл) диспергировали 60% -ный гидрид натрия в масле (0,23 г), к этой дисперсии добавляли, охлаждая при этом льдом, триазол (0,59 г), после чего смесь перемешивали в течение 15 мин. К полученной смеси добавляли диметилформамидный раствор (1 мл) 2-[(1S)-1-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) этил]-2-(2,4-дифторофенил) оксиран (0,4 г) и нагревали при 80оС в течение 3 ч. Затем реакционную смесь охлаждали и добавляли холодную воду (1 мл) и этилацетат (50 мл), затем смесь встряхивали. Водный слой дважды экстрагировали этилацетатом. Слои этилацетата комбинировали, промывали насыщенным раствором водного хлорида натрия и высушивали безводным сульфатом натрия, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью дихлорметана, этилацетата и ацетона (6:1:1). Полученный воскообразный продукт дважды переосаждали из этилацетата и гексана, в результате чего получали (3S)-2-(2,4-дифторофенил)-3-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)-1-(1Н-1,2 4-т- 2-бутанол (0,22 г) в виде бесцветного воскообразного продукта.
1Н-ЯМР (CDCl3) δ: 0,99, 1,12 (3Н, д, I 6,4 Гц); 1,40-2,00 (6Н, м); 3,40-3,65 (1Н, м); 3,80-4,06 (1Н, м); 4,25-4,45 (1Н, м); 4,31 (1Н, с); 4,63 (1Н, д, I 14,2 Гц); 4,71 (1Н, м); 4,90 (1Н, д, I 14,2 Гц); 6,65-6,85 (2Н, м); 7,35-7,50 (1Н, м); 7,72, 7,73 (1Н, с); 7,93, 7,96 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 31. В этаноле (4 мл) растворяли (3S)-2-(2,4- дифторофенил)-3-(3,4,5,6-тетрагидро-2Н-пиран-2-ил)окси-1- (1Н-1,2,4-триазол-1-ил)-2-бутанол (0,22 г) и n-толуолсульфонат пиридиния (47 мг), полученный раствор перемешивали при 55оС в течение 2 ч. Затем к раствору добавляли еще 10 мг n-толуолсульфоната пиридиния и полученный раствор перемешивали при 55оС еще 2 ч. Затем реакционную смесь охлаждали, а растворитель отгоняли. К остатку добавляли этилацетат (30 мл) и смесь промывали насыщенным водным раствором хлорида натрия (10 мл). Слой этилацетата высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. К остатку добавляли этиловый эфир, а осаждающиеся кристаллы собирали фильтрацией, в результате чего получали (2S, 3S)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол (60 мг), т.пл. 115-117оС.
1Н-ЯМР (CDCl3) δ: 0,97 (3Н, д, I 6,4 Гц); 4,32 (1Н, м); 4,82 (2Н, с); 6,69-6,82 (2Н, м); 7,35-7,48 (1Н, м); 7,83 (1Н, с); 7,84 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 32. В смеси этилацетата (2 мл) и дихлорометана (0,5 мл) растворяли (2S, 3S)-2-(2,4-дифторофенил- 1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол (58 г). К полученному раствору добавляли, охлаждая при этом льдом, триэтиламин (66 мл) и метансульфонилхлорид (37 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Затем к этой реакционной смеси добавляли этилацетат (30 мл), полученную смесь промывали водой, высушивали безводным сульфатом магния и концентрировали, в результате чего получали (2S, 3S)-2-(2,4-дифторфенил)-3-метан- сульфонилокси-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол в виде маслянистого продукта. Этот продукт растворяли в метаноле (2 мл), после чего к нему добавляли при охлаждении льдом 28% метилата натрия (116 мл) и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Затем к реакционной смеси добавляли дихлорметан (30 мл), полученную смесь промывали водой и высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата и дихлорметана (4:1), в результате чего после кристаллизации из гексана получали (2S, 3S)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-три- азол-1-ил) метилоксиран (42 мл) в виде бесцветных игольчатых кристаллов, т.пл. 89-90оС.
[α]D23 7,8о (с 1,0 в МеОН).
1Н-ЯМР (CDCl3) δ: 1,65 (3Н, д, I 5,6 Гц); 3,20 (1Н, кв. I 5,5 Гц); 4,43 (1Н, д, I 14,6 Гц); 4,88 (1Н, д, I 14,6 Гц); 6,68-6,83 (2Н, м); 6,93-7,08 (1Н, м); 7,82 (1Н, с); 7,96 (1Н, с).
Элементный анализ для C12H11F2N3O.
Вычислено, С 57,37; Н 4,41; N 16,73.
Найдено, С 56,98; Н 4,40; N 16,53.
Полученный продукт анализировали с помощью высокоразрешающей жидкостной хроматографии (подвижная фаза: гексан изопропанол 9:1), используя колонку для разделения оптических изомеров [CHIRALCEL 0,46 см х 25 см, изготовлена Daicel Chemical Industries, Ltd. Избыток энантиомера составлял 97,9%
0,46 см х 25 см, изготовлена Daicel Chemical Industries, Ltd. Избыток энантиомера составлял 97,9%
С р а в н и т е л ь н ы й п р и м е р 33. Метил (R)-(+)-лактат (25 г) растворяли в дихлорoметане (250 мл), к раствору при охлаждении льдом добавляли гидрат n-толуол- сульфоновой кислоты (456 мг). Затем к смеси по каплям в течение 30 мин добавляли 3,4-дигидро-2Н-пиран (24,2 г) и полученную смесь перемешивали при охлаждении льдом в течение 1 ч. После этого к реакционной смеси добавляли 5%-ный водный раствор бикарбоната натрия (50 мл), смесь энергично перемешивали с последующим отделением органического слоя. Органический слой промывали 5%-ным водным раствором бикарбоната натрия, высушивали безводным сульфатом магния с последующей отгонкой растворителя при пониженном давлении и получали метил (2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)пропионат (42,7 г) в виде маслянистого продукта желтого цвета.
С р а в н и т е л ь н ы й п р и м е р 34. В этаноле (510 мл) растворяли метил (2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионат (42,7 г), к полученному раствору, охлаждая льдом, добавляли раствор гидроокиси натрия (170 мл), после чего смесь перемешивали при комнатной температуре в течение 1 ч. Затем реакционную смесь охлаждали льдом, добавляли в нее 26%-ный водный раствор уксусной кислоты (120 мл) и 3 раза экстрагировали дихлорметаном (200 мл). Слои дихлорметана комбинировали, промывали насыщенным водным раствором хлорида натрия (100 мл) 2 раза и высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении, в результате чего получали (2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионовую кислоту (32 г) в виде бесцветного воскообразного продукта.
К (2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионовой кислоте (32 г) добавляли безводный тетрагидрофуран (250 мл). Затем к смеси порциями добавляли 1,1'-карбонил-диимидазол (35,8 г) в течение 10 мин. Полученную смесь перемешивали при комнатной температуре в течение 30 мин, а затем после охлаждения льдом к ней по каплям в течение 15 мин добавляли морфолин (38,3 г), после чего смесь перемешивали в ледяной бане еще 15 мин. Далее реакционную смесь концентрировали при пониженном давлении и концентрат растворяли в дихлорметане (300 мл). Раствор промывали насыщенным водным раствором хлорида натрия (50 мл) и высушивали безводным сульфатом магния, а растворитель отгоняли. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:4), в результате чего получали N-[(2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)пропионил]морфолин (25,7 г).
1Н-ЯМР (CDCl3) δ: 1,39 (1,44, 3Н, д, I 6,8 Гц); 1,45-1,96 (6Н, м); 3,40-3,95 (10Н, м); 4,52, 4,68 (1Н, кв, I 6,8 Гц); 4,59-4,65 (1Н, м).
ИК (пленка): 2945, 2855, 1662, 1650, 1462, 1438, 1370, 1270, 1230, 1112, 1030, 980 см-1.
С р а в н и т е л ь н ы й п р и м е р 35. В безводном тетрагидрофуране (50 мл) растворяли 1-бромо-2,4-дифторобензол (9,69 г). Затем к раствору при комнатной температуре добавляли магний (1,22 г) и небольшое количество йода, после чего полученную смесь энергично перемешивали в течение 2 ч и получали 1М раствор 2,4-дифторофенилмагний бромида. Этот раствор разбавляли 50 мл безводного тетрагидрофурана и добавляли к безводному тетрагидрофурановому раствору (125 мл) N-[(2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропионил] морфолина (12,7 г) при температуре (-30)-(-20)оС в течение 45 мин. По окончании добавления температуру смеси повышали до 20оС в течение 1 ч. Смесь перемешивали при этой температуре в течение 1 ч. Затем реакционную смесь охлаждали льдом и добавляли к ней насыщенный водный раствор хлорида аммония (40 мл) с последующей экстракцией этилацетатом (300 мл). Экстракт высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (10; 1), и получали в результате (2R)-2', 4'-дифторо-2-(3,4,5,6- тетрагидро-2Н-пиран-2-илокси) пропиофенон (5,03 г) в виде маслянистого продукта бледно-желтого цвета.
ИК (пленка): 3075, 2950, 2875, 1695, 1605, 1500, 1422, 1370, 1266, 1235, 1138, 1090, 1030, 970, 850 см-1.
Оптическую чистоту этого соединения определяли следующим способом.
В этаноле (3 мл) растворяли (2R)-2', 4'-дифторо-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропиофенон (121 мг) и к полученному раствору добавляли n-толуолсульфонат пиридиния (25 мг), после чего смесь перемешивали при 55оС в течение 1 ч. Растворитель отгоняли при пониженном давлении, а остаток растворяли в этилацетате (20 мл). Раствор промывали водой и высушивали безводным сульфатом магния, а растворитель отгоняли. Остаток высушивали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (5:1), в результате чего получали (2R)-2',4'-дифторо-2-гидроксипропиофенон (62 мг) в виде маслянистого продукта бледно-желтого цвета.
[d] + 68,7 (с 1,8 в хлороформе).
Этот продукт анализировали с помощью жидкостной хроматографии высокого разрешения (подвижная фаза: гексан изопропанол 9:1), используя колонку для разделения оптических изомеров [CHIRALCEL 0,46 см х 25 см, изготовлена Daicel Chemical Industries, Ltd]
Энантиомер с (2R)-конфигурацией не обнаружили.
1Н-ЯМР (CDCl3) δ: 1,41 (3Н, д, I 7,0 Гц, I 1,6 Гц); 3,74 (1Н, д, I 7,0 Гц); 5,01 (1Н, м); 6,86-7,08 (2Н, м); 7,96-8,08 (1Н, м).
ИК (пленка): 3450, 1690, 1610, 1500, 1430, 1268, 1140, 1095, 1030, 980, 855 см-1.
С р а в н и т е л ь н ы й п р и м е р 36. К диметилсульфоксиду (50 мл) добавляли 60%-ный гидрид натрия (0,833 г) в минеральном масле. В полученную смесь, перемешивая при 15оС, добавляли триметилсульфоксонийиодид (4,80 г), после чего смесь перемешивали при комнатной температуре в течение 15 мин. Реакционную смесь охлаждали льдом и добавляли к ней раствор 10 мл (2R)-2', 4'-дифторo-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси) пропиофенон (4,90 г). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь выливали в ледяную воду (120 мл) и экстрагировали этилацетатом (150, 100, 100 мл). Слои этилацетата объединяли и промывали водой (50 мл) и насыщенным водным раствором хлорида натрия (50 мл), а затем высушивали безводным сульфатом магния. Растворитель отгоняли при пониженном давлении, а остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (10:1), в результате чего получали 2-[(1R)-1-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)этил] -2-(2,4-дифторофенил)- оксиран (4,7 г) в виде маслянистого продукта бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,10-1,30 (3Н, м); 1,40-1,95 (6Н, м); 2,83 (1Н, м); 3,05, 3,32 (1Н, д, I 5,2 Гц); 3,42-3,60 (1Н, м); 3,76-4,14 (2Н, м); 4,76, 4,93 (1Н, м); 6,72-6,95 (2Н, м); 7,32-7,60 (1Н, м).
ИК (пленка): 2950, 1618, 1600, 1510, 1425, 1270, 1140, 1120, 1075, 1020, 990, 985, 850 см-1.
С р а в н и т е л ь н ы й п р и м е р 37. В диметилформамиде (50 мл) диспергировали 60% -ный гидрид натрия (2,64 г) в минеральном масле, после чего в эту дисперсию, охлаждая льдом, добавляли триазол (6,84 г) и смесь перемешивали в течение 15 мин. Затем к этой смеси добавляли раствор 10 мл 2-[(1R)-1-(3,4,5,6-тетрагидро-2Н-пиран-2-ил- окси)этил]-2-(2,4-дифторофенил) оксирана (4,7 г) в диметилформамиде. Полученную смесь нагревали при 80оС в течение 3 ч. Реакционную смесь охлаждали, выливали в холодную воду (200 мл) и 3 раза экстрагировали этилацетатом (150 мл). Слои этилацетата комбинировали, промывали водой (100 мл х 3) и насыщенным водным раствором хлорида натрия (100 мл), высушивали безводным сульфатом натрия, а растворитель отгоняли. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью дихлорметана, этилацетата и ацетона (6:1:1), и получали (3R)-2-2,4-дифторофенил-3-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)-1- (1Н-1,2,4-триазол-1-ил)-2-бутанол (4,4 г) в виде бесцветного вязкого маслянистого вещества.
1Н-ЯМР (CFCl3) δ: 0,99-1,12 (3Н, д, I 6,4 Гц); 1,40-2,00 (6Н, м); 3,40-3,65 (1Н, м); 3,80-4,06 (1Н, м), 4,25-4,45 (1Н, м); 4,29 (1Н, с); 4,62 (1Н, д, I 14,2 Гц); 4,71 (1Н, м); 4,90 (1Н, д, I 14,2 Гц); 6,65-6,83 (2Н, м); 7,35-7,50 (1Н, м); 7,71, 7,72 (1Н, с); 7,91, 7,94 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 38. В этаноле (50 мл) растворяли (3R)-2-(2,4-дифторофенил)-3-(3,4,5,6-тетрагидро-2Н-пиран-2-ил)окси-1- (1Н-1,2,4-триазол-1-ил)-2-бутанол (4,4 г) и n-толуолсульфонат пиридиния (0,93 г). Раствор перемешивали при 55оС в течение 2 ч, а затем к раствору добавляли 0,20 г n-толуолсульфоната пиридиния и перемешивали при 55оС еще 2 ч. Затем реакционную смесь охлаждали, а растворитель отгоняли. К остатку добавляли этилацетат (250 мл) и промывали водой (50 мл), а затем насыщенным водным раствором хлорида натрия (50 мл). Слой этилацетата высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. К остатку добавляли этиловый эфир и осаждавшиеся кристаллы собирали путем фильтрации, в результате чего получали (2R, 3R)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триа- зол- 1-ил)-2,3-бутандиол (1,37 г), т.пл. 115-117оС.
[α]D -80,3оС (с 1,0 в метаноле).
1Н-ЯМР (CDCl3) δ: 0,97 (3Н, д, I 6,4 Гц); 4,33 (1Н, м); 4,82 (2Н, с); 6,69-6,82 (2Н, м); 7,35-7,48 (1Н, м); 7,84 (1Н, c); 7,85 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 39. В смеси этилацетата (40 мл) и дихлорометана (10 мл) растворяли (2R, 3R)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутан- диол (1,25 г). К раствору при охлаждении льдом добавляли триэтиламин (0,84 мл) и метансульфонилхлорид (0,48 мл), после чего смесь перемешивали при комнатной температуре в течение 30 мин. Затем к реакционной смеси добавляли этилацетат (50 мл), полученную смесь промывали водой, высушивали безводным сульфатом магния и концентрировали, в результате чего получали (2R, 3R)-2-(2,4-дифторoфенил)-3-метансульфонилок- си-1-(1Н-1,2,4-триазол-1-ил)-2 -бутанол в виде маслянистого продукта. Этот продукт растворяли в метаноле (40 мл), а затем к нему добавляли 28%-ный метаноловый раствор метилата натрия (1,16 мл) при охлаждении льдом. Смесь перемешивали при комнатной температуре в течение 30 мин. После этого реакционную смесь концентрировали при пониженном давлении до объема 10 мл. К концентрату добавляли этилацетат (100 мл), полученную смесь промывали водой и высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата и дихлорметана (4:1), и после перекристаллизации из смеси этилацетата и гексана получали (2R,3S)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол- 1-ил)метил оксиран (520 мг) в виде бесцветных игольчатых кристаллов, т.пл. 89-90оС.
[α]23 -8,3 (с 1,0 в МеОН).
1Н-ЯМР (CDCl3) δ: 1,65 (3Н, д, I 5,6 Гц); 3,20 (1Н, кв, 5,6 Гц); 4,43 (1Н, д, I 14,6 Гц); 4,88 (1Н, д, I 14,6 Гц); 6,68-6,83 (2Н, м); 6,93-7,08 (1Н, м); 7,82 (1Н, с); 7,97 (1Н, с).
Элементный анализ для C12H11F2N3O.
Вычислено, С 57,37; Н 4,41; N 16,73.
Найдено, С 57,27; Н 4,43; N 16,83.
Полученный продукт анализировали с помощью жидкостной хроматографии высокого разрешения (подвижная фаза: гексан изопропиловый спирт 9:1), используя при этом колонку для разделения оптических изомеров [CHIRALCEL 0,46 см х 25 см, изготовлена Daicel Chemical Industries, Ltd. Избыток энантиомера составлял 99,2%
С р а в н и т е л ь н ы й п р и м е р 40. Смесь метил (R)-лактата (104 г) и морфолина (260 мл) нагревали при 85оС в течение 60 ч. Реакционную смесь концентрировали при пониженном давлении, а концентрат очищали с помощью хроматографии на силикагеле (800 г силикагель, элюент: гексан этилацетат 1:1 ->> этилацетат), в результате чего получали N-[(2R)-2-гидроксипропионил]морфолин (141 г) в качестве маслянистого продукта бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ:1,34 (3Н, д, I 6,6 Гц); 3,43 (2Н, т, I 4,8 Гц); 3,55-3,80 (6Н, м); 3,79 (1Н, д); 4,45 (1Н, м).
[α]D +0,98 (с 5,24 в CHCl3).
С р а в н и т е л ь н ы й п р и м е р 41. К раствору дихлорoметана (500 мл) и N-[(2R)-2-гидроксипропионил] морфолина (141 г) добавляли моногидрат n-толуолсульфоновой кислоты (1,67 г). Затем к смеси по каплям в течение 30 мин добавляли, охлаждая при этом льдом, 3,4-дигидро-2Н-пиран (89,3 г) и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь промывали 5%-ным водным раствором бикарбоната натрия (150 мл х 2), высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (силикагель: 800 г, элюент: гексан этилацетат 8:1 ->> этилацетат) и получали N-[(2R)-2-(3,4,5,6-тетрагидро-2Н-пиран-2-илокси)пропионил] морфолин (184 г) в виде маслянистого продукта бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,39, 1,44 (3Н, д, I 6,8 Гц); 1,40-1,95 (6Н, м); 3,40-3,95 (10Н, м); 4,48-4,75 (2Н, м).
[α]D +34,9о (с 6,3 в CHCl3).
С р а в н и т е л ь н ы й п р и м е р 42. Маточный раствор, полученный после перекристаллизации (2RS, 3RS)-2-(2,4-дифторoфенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиола (см. сравнительный пример 24), подвергали дистилляции при пониженном давлении. Остаток очищали с помощью колоночной хроматографии, элюируя смесью этилацетата и метанола (30:1), и получали (2RS, 3RS)-2-(2,4-дифторoфенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол (в виде первой фракции элюата), после перекристаллизации которого из метанола получали бесцветные призмообразные кристаллы (1,9 г) в качестве диастереомера с чистотой 98% и т.пл. 146-148оС.
1Н-ЯМР (CDCl3) δ: 1,26 (3Н, д, I 5,8 Гц), 2,41-2,52 (1Н, м); 3,92-4,07 (1Н, м); 4,57 (1Н, д, I 14 Гц); 5,03 (1Н, с); 5,04 (1Н, д, I 14 Гц); 6,68-6,87 (2Н, м); 7,50-7,68 (1Н, м); 7,79 (1Н, с); 8,05 (1Н, с).
Элементный анализ для C12H13F2O2.
Вычислено, С 55,53; Н 4,87; N 15,61.
Найдено, С 53,70; Н 4,97; N 15,59.
С р а в н и т е л ь н ы й п р и м е р 43. Полученное в сравнительном примере 42 соединение (2RS, 3RS)-2-(2,4-дифторoфенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бу- тадиол (1,1 г) способом, описанным в сравнительном примере 25, подвергали взаимодействию с метансульфонилхлоридом (0,35 мл), после обработки 28%-ным метаноловым раствором метилата натрия получали (2RS, 3RS)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил)метилоксиран. Полученный продукт очищали с помощью колоночной хроматографии, элюируя смесью этилацетата и метиленхлорида (4:1), и получали маслянистый продукт желтого цвета (0,8г г).
1Н-ЯМР (CDCl3) δ: 1,06 (3Н, д, I 5,4 Гц); 3,18 (2Н, кв. I 5,4, 10,7 Гц); 4,42 (1Н, д, I 15 Гц); 4,81 (1Н, д, I 15 Гц); 6,76-6,92 (2Н, м); 7,07-7,20 (1Н, м); 7,86 (1Н, с); 8,07 (1Н, с).
С р а в н и т е л ь н ы е п р и м е р ы 44-50. Способом, аналогичным описанному в сравнительном примере 12, имидазол-2-карбоксиальдегид подвергали взаимодействию с алкилирующим агентом, указанным в табл.10 (стадия 1), а затем без очистки продукт подвергали реакции восстановления с борогидридом натрия (стадия 2) с последующим хлорированием с помощью тионилхлорида (стадия 3), в результате чего получали производное 2-хлорометилимидазол.
Сравнительный пример 51. К этаноловому раствору (100 мл) 3-(n-метоксибензилтио)-пропиональдегида (5,7 г) добавляли 40%-ный водный раствор глиоксала (4,3 г), а затем 30%-ный водный раствор аммония (4,1 мл) при -10оС. Смесь перемешивали при комнатной температуре в течение 1 ч, после чего к ней добавляли 40% -ный водный раствор глиоксала (4 мл) и 30%-ный водный раствор аммония (4,1 мл), полученную смесь перемешивали еще 1 ч. Затем реакционную смесь концентрировали при пониженном давлении. Концентрат подкисляли соляной кислотой и промывали метиленхлоридом (30 мл х 2), рН водного слоя доводили до 8 водным раствором гидроокиси натрия, а затем экстрагировали метиленхлоридом (30 мл х 3). Раствор высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. К концентрату добавляли смесь этанола и этилацетата, в результате чего получали 2-[2-(n-метоксибензилтио)этил]-имидазол (2,7 г) в виде кристаллов. Маточный раствор хроматографировали на силикагеле (колонка 3 см х 15 см), элюируя смесью метанола и метиленхлорида (1: 9). Целевую фракцию концентрировали при пониженном давлении и получали 1,1 г продукта в виде кристаллов. Т.пл. 116-118оС (бесцветные пластинки).
1Н-ЯМР (DMCO-d6) δ: 2,68 (2Н, т, I 7,0 Гц); 2,85 (2Н, т, I 7,0 Гц); 3,65 (2Н, с); 3,73 (3Н, с); 6,87 (2Н, д, I 8,8 Гц); 7,23 (2Н, д, I 8,8 Гц); 6,7-7,1 (2Н, шир.с); 11,73 (1Н, шир.с).
Элементный анализ для C13H16N2OS.
Вычислено, С 62,87; Н 6,49; N 11,28.
Найдено, С 62,86; Н 6,45; N 11,29.
К раствору (15 мл) 2-[2-(n-метоксибензилтио)этил]имидазола (1,5 г) в диметилформамиде добавляли 60% -ный гидрид натрия (0,29 г) в минеральном масле при 0оС. Затем через 15 мин к смеси добавляли метилйодид (0,41 мл) при -20оС, после чего смесь размешивали в течение 10 мин. Реакционную смесь выливали в воду (60 мл) и экстрагировали метиленхлоридом (20 мл х 3). Органический слой промывали водой (20 мл), высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Концентрат хроматографировали на колонках с силикагелем (3 см х 15 см), элюируя смесью метанола и метиленхлорида (5,95). Целевую фракцию концентрировали при пониженном давлении и получали 1-метил-2-[2-(n-метоксибензилтио)этил]имида- зол (1,6 г) в виде бесцветного маслянистого продукта.
1Н-ЯМР (CDCl3) δ: 2,8-2,95 (4Н, м); 3,54 (3Н, с); 3,65 (2Н, с); 3,79 (3Н, с); 6,78 (1Н, д, I 1,4 Гц); 6,54 (2Н, д, I 8,6 Гц); 6,94 (1Н, д, I 1,4 Гц); 7,24 (2Н, д, I 8,6 Гц).
В смеси трифторуксусной кислоты (25 мл) и анизола (10 мл) растворяли 1-метил-2-[2-(n-метоксибензилтио)этил]имидазол (1,4 г). К этому раствору при 0оС добавляли ацетат ртути (1,9 г) и полученную смесь размешивали в течение 1 ч 45 мин. Затем реакционную смесь концентрировали при пониженном давлении. К концентрату добавляли петролейный эфир (50 мл). Надосадочную жидкость удаляли. К осадку добавляли диэтиловый эфир (30 мл) и после фильтрации получали белый порошок (2,6 г). Этот порошок (1 г) растворяли в N,N-диметилформамиде (5 мл) и в этот раствор барботировали сероводород при 0оС в течение 10 мин. Затем в целях удаления избыточного количества сероводорода в реакционную смесь барботировали азот. Нерастворившиеся вещества отфильтровывали и фильтрат концентрировали при пониженном давлении, в результате чего получали 1-метил-2-(2-меркаптоэтил)имидазол (0,5 г) в виде бесцветного маслянистого продукта.
1Н-ЯМР (DMCO-d6) δ: 2,74 (1Н, т, I 7,0 Гц); 2,88 (2Н, д, I 7,0 Гц, д, I 12 Гц); 3,1-3,4 (2Н, м); 3,82 (3Н, с); 7,61 (1Н, д, I 2 Гц); 7,63 (1Н, д, I 2 Гц).
С р а в н и т е л ь н ы й п р и м е р 52. К раствору метанола (31 мл), содержащему 37% -ный водный раствор формальдегида (8,9 г) и 40%-ный раствор гликозала (16 г), добавляли, охлаждая при этом льдом, метаноловый раствор (5 мл), содержащий циклопропиламин (6,3 г) и 28%-ный водный раствор аммония (7,6 г), в течение 25 мин. Полученную смесь размешивали при 0оС в течение 1 ч, а затем концентрировали при пониженном давлении до получения объема около 10 мл. Нерастворившиеся вещества отфильтровывали. К фильтрату добавляли воду (200 мл). Водный раствор промывали гексаном (100 мл х 4), а затем смесью гексана (50 мл) и диэтилового эфира (30 мл). Полученный водный раствор насыщали хлоридом натрия и экстрагировали этилацетатом (100 мл х 8). Слой этилацетата промывали насыщенным водным раствором хлорида натрия (50 мл), высушивали безводным сульфатом магния и после отгонки растворителя при пониженном давлении получали сырое соединение 7-циклопропилимидазола (4,2 г) в виде бесцветного порошка. Смесь этого сырого продукта (3,5 г) и параформальдегида (3 г) нагревали при 170оС в течение 30 мин, а затем к ней добавляли еще 2 г параформальдегида. Полученную смесь нагревали при 170оС в течение 20 мин. К этой смеси добавляли еще 2 г параформальдегида, а затем снова нагревали при 170оС в течение 20 мин. После этого реакционную смесь охлаждали льдом и растворяли в метаноле (20 мл), к полученному раствору добавляли насыщенный водный раствор хлорида натрия (20 мл) с последующим экстрагированием этилацетатом (40 мл х 2). Слой этилацетата высушивали безводным сульфатом магния и концентрировали при пониженном давлении. Концентрат хроматографировали на колонках с силикагелем (3 х 13 см), элюируя смесью метанола и дихлорметана (1:9), а целевую фракцию концентрировали при пониженном давлении. К концентрату добавляли смесь этилацетата и диэтилового эфира, в результате чего получали 1-циклопропил-2-гидроксиметилимидазол (0,7 г) в виде бесцветных кристаллов. Полученное соединение (0,7 г) добавляли к тионилхлориду (7 мл) при 0оС, а затем смесь размешивали в течение 5 мин. Далее указанную реакционную смесь нагревали в колбе с обратным холодильником в течение 15 мин. Избыток тионилхлорида отгоняли при пониженном давлении. К остатку добавляли диэтиловый эфир, а полученный порошок собирали фильтрацией. После перекристаллизации из смеси этанола и диэтилового эфира получали 1-циклопропил-2-хлорометилимидазол гидрохлорида (0,75 г).
1-Циклопропил-2-гидроксиметилимида- зол, т. пл. 90-95оС, бесцветные игольчатые кристаллы.
1Н-ЯМР (CDCl3) δ: 0,9-1,2 (4Н, м); 3,25-3,40 (1Н, м); 4,76 (2Н, с); 5,9 (1Н, шир.с); 6,86 (2Н, с).
Элементный анализ для C7H10N2O.
Вычислено, С 60,85; Н 7,29; N 20,27.
Найдено, С 60,83; Н 7,29; N 20,24.
1-Циклопропил-2-хлорoметилимидазол гидрохлорид, т. пл. 100-101оС бесцветные призмообразные кристаллы.
1Н-ЯМР (DMCO-d6) δ: 1,1-1,3 (4Н, м); 3,65-3,80 (1Н, м), 5,21 (2Н, с), 7,72 (1Н, д, I 2,0 Гц); 7,80 (1Н, д, I 2,0 Гц).
Элементный анализ для C7H9ClN2 HCl.
Вычислено, С 43,55; Н 5,22; N 14,51.
Найдено, С 43,61; Н 5,22; N 14,37.
С р а в н и т е л ь н ы й п р и м е р 53. К метаноловому раствору (50 мл) гидразина гликолевой кислоты (5 г) добавляли изотиоцианат циклопропила (5,6 г) при 20оС. Смесь перемешивали в течение 1 ч, после чего к ней добавляли воду (30 мл) при 0оС. Затем к полученной смеси по каплям добавляли 5н. водный раствор NaOH (11 мл), поднимали температуру раствора до 20оС и перемешивали при этой температуре в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении до получения объема около 10 мл. Концентрат разбавляли этанолом (100 мл), после чего к нему по каплям, охлаждая льдом, добавляли 5н. водный раствор HCl (11 мл). Нерастворившиеся вещества отфильтровывали, а фильтрат концентрировали досуха при пониженном давлении, в результате чего получали сырые кристаллы (6,4 г) 4-циклопропил-5-гидроксиметил-3-меркапто- 4Н-1,2,4-триазола. Это соединение (3 г) добавляли в смесь концентрированной азотной кислоты (d 1,38 4,6 мл), воды (12 мл) и нитрита натрия (10 мг) при 60оС. Затем добавляли 1 мл концентрированной азотной кислоты. Когда часть азотной кислоты взаимодействовала с небольшим количеством триазолового соединения на внутренней стенке реакционного сосуда, начиналась реакция и температура достигала 90-100оС, после чего реакция завершалась. Затем реакционную смесь охлаждали льдом, нейтрализовали водным раствором NaOH и концентрировали при пониженном давлении. Концентрат хроматографировали на силикагеле (3 х 10 см), элюируя смесью метанола и дихлорэтана (1:4). Целевую фракцию концентрировали и получали 4-циклопропил-3-гидроксиметил-4Н-1,2,4- триазол (2 г). Этот продукт (1 г) добавляли к тионилхлориду (10 мл) при 0оС, после чего смесь нагревали с обратным холодильником в течение 20 мин. Смесь охлаждали, а избыток тионилхлорида отгоняли при пониженном давлении. К остатку добавляли диэтиловый эфир, а полученный порошок собирали фильтрацией. Этот продукт перекристаллизовывали из смеси этанола и этилацетата и получали 4-циклопропил-3-хлорометил-1,2,4-триазол гидрохлорид (1,34 г).
4-Циклопропил-5-гидроксиметил-3-ме- ркапто-4Н-1,2,4-триазол, т. пл. 159-160оС, бесцветные призмообразные кристаллы.
1Н-ЯМР (DMCO-d6) δ: 1,0-1,2 (4Н, м); 2,9-3,0 (1Н, м); 3,37 (1Н, шир.с); 4,51 (2Н, с); 5,60 (1Н, шир.с).
Элементный анализ для C6H9N3OS.
Вычислено, С 42,09; Н 5,30; N 24,54.
Найдено, С 42,11; Н 5,34; N 24,50.
4-Циклопропил-3-гидроксиметил-4Н-1, 2,4-триазол.
1Н-ЯМР (DMCO-d6) δ: 1,01 (4Н, д, I 5,4 Гц); 3,3-3,5 (1Н, м); 4,64 (2Н, д, I 5,8 Гц); 5,50 (1Н, т, I 5,8 Гц); 8,42 (1Н, с).
Масс-спектр (m/z): 140 (МН)+.
4-Циклопропил-3-хлорометил-1,2,4-три- азол гидрохлорид, т.пл. 60-65оС, бесцветные игольчатые кристаллы.
1Н-ЯМР (DMCO-d6) δ: 1,0-1,3 (4Н, м); 3,5-3,7 (1Н, м); 5,12 (2Н, с); 9,51 (1Н, с).
Элементный анализ для C6H8ClN3 HCl x x 0,5 H2O.
Вычислено, С 35,49; Н 4,96; N 20,69.
Найдено, С 35,86; Н 4,55; N 20,30.
С р а в н и т е л ь н ы й п р и м е р 54. Смесь этилбромопирувата (6,82 г), 2,2,2-трифторотиоацетамида (4,52 г) и этанола (30 мл) нагревали с обратным холодильником в течение 3 ч. Затем реакционную смесь охлаждали, а этанол отгоняли при пониженном давлении. К остатку добавляли воду (40 мл) и экстрагировали этилацетатом (60 мл х 2). Раствор экстракта промывали водой (40 мл) и высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (2,5 х 45 см), элюируя смесью этилацетата и гексана (1:5). Целевую фракцию концентрировали и получали этил 2-трифторметил-4-тиазолкарбоксилат (2,2 г) в виде игольчатых кристаллов бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,43 (3Н, т, I 7 Гц); 4,48 (2Н, кв, I 7 Гц); 8,40 (1Н, с).
К безводному эфиру (40 мл) добавляли алюмогидрид лития (0,33 г). К этой смеси по каплям добавляли безводный эфирный раствор (20 мл) этил 2-трифторметил-4-тиазолкарбоксилат (2 г). Смесь размешивали при комнатной температуре в течение 3 ч. Затем к реакционной смеси по каплям добавляли, охлаждая льдом, воду (20 мл) в целях разложения избыточного количества восстанавливающего агента. К полученной реакционной смеси добавляли этилацетат (50 мл) и воду (25 мл) и экстрагировали этилацетатом. Органический слой промывали водой (25 мл), а затем насыщенным водным раствором хлорида натрия (25 мл) и высушивали сульфатом магния. Растворитель отгоняли, а остаток хроматографировали на силикагеле (2,5 х 30 см), элюируя этилацетатом и гексаном (3:1). Целевую фракцию концентрировали и получали 2-трифторметил-4-гидроксиметилтиазол (1,1 г) в виде маслянистого бледно-желтого продукта.
1Н-ЯМР (CDCl3) δ: 2,50 (1Н, с); 4,87 (2Н, д, I 5 Гц); 7,50 (1Н, с).
В хлороформе (10 мл) растворяли 2-трифторметил-4-гидроксиметилтиазол (1 г) и к полученному раствору добавляли по каплям тионилхлорид (2 мл). Затем смесь нагревали с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали, а избыточное количество тионилхлорида отгоняли при пониженном давлении. К остатку добавляли дихлорметан (30 мл), затем промывали последовательно водным раствором бикарбоната натрия (20 мл) и водой (20 мл) и высушивали сульфатом магния. Растворитель отгоняли, а остаток хроматографировали на силикагеле (2,5 х 20 см), элюируя смесью этилацетата и гексана (1:3). Целевую фракцию концентрировали и получали 2-трифторметил-4-хлорметилтиазол (0,66 г) в виде красноватого маслянистого продукта.
1Н-ЯМР (CDCl3) δ: 4,62 (2Н, с); 7,57 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 55. Смесь этилборопирувата (3,6 г), циклопропантиоамида (2,2 г) и этанола (30 мл) нагревали с обратным холодильником в течение 2 ч. Затем реакционную смесь охлаждали, а этанол отгоняли при пониженном давлении. К остатку добавляли воду (30 мл) и дважды экстрагировали этилацетатом (50 мл). Pаствор экстракта промывали водой (30 мл) и высушивали сульфатом магния. Растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (2,5 х 30 см), элюируя смесью этилацетата и гексана (1:3). Целевую фракцию концентрировали и получали 2-циклопропил-4-тиазолкарбоксилат (0,84 г) в виде игольчатых кристаллов бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ: 1,01-1,24 (4Н, м); 1,40 (3Н, т, I 7 Гц); 2,32-2,50 (1Н, м); 4,41 (2Н, кв, I 7 Гц); 7,95 (1Н, с).
К смеси алюмогидрида лития (0,32 г) и безводного эфира (40 мл) добавляли по каплям безводный эфирный раствор (10 мл) этил-2-циклопропил-4-тиазолкарбоксилат (1,65 г) и полученную смесь размешивали при комнатной температуре в течение 1 ч. Затем к реакционной смеси по каплям добавляли воду (20 мл), охлаждая льдом, для разложения избыточного количества восстанавливающего агента. К полученной смеси добавляли этилацетат (50 мл) и воду (25 мл) и экстрагировали этилацетатом. Органический слой промывали водой (25 мл) и насыщенным водным раствором хлорида натрия (25 мл), а затем высушивали сульфатом магния. Растворитель отгоняли, а остаток хроматографировали на силикагеле (2,5 х 30 см), элюируя смесью этилацетата и гексана (2:1). Целевую фракцию концентpировали и получали 2-циклопропил-4-гидроксиметилтиазол (0,94 г) в виде игольчатых кристаллов бледно-желтого цвета.
1Н-ЯМР (CDCl3) δ: 0,98-1,18 (4Н, м); 2,24-2,39 (2Н, м); 3,20 (1Н, с); 4,70 (2Н, д, I 5 Гц); 6,93 (1Н, с).
В хлороформе (10 мл) растворяли 2-циклопропил-4-гидроксиметилтиазол (0,9 г), а затем к этому раствору добавляли по каплям, охлаждая при этом льдом, тионилхлорид (0,68г мл), после чего смесь размешивали при комнатной температуре в течение 30 мин. Избыток тионилхлорида отгоняли при пониженном давлении. К остатку добавляли диэтиловый эфир и получали 2-циклопропил-4-хлорметилтиазол гидрохлорида (1,1 г) в виде коричневого порошка, т.пл. 108-110оС.
1Н-ЯМР (DMCO-d6) δ: 0,93-1,18 (4Н, м); 2,32-2,48 (1Н, м); 4,72 (2Н, с); 7,47 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 56. Маточный раствор, полученный после перекристаллизации (2R, 3R)-2-(2,4-дифторoфе-нил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиола, как описано в сравнительном примере 31, концентрировали при пониженном давлении. Концентрат очищали хроматографией на силикагеле (элюент:этилацетат), в результате чего получали (2S, 3R)-2-(2,4-дифторометил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол в качестве первой фракции элюата.
Т.пл. 154-156оС.
1Н-ЯМР (CDCl3) δ: 1,27 (3Н, дд, I 6,4 Гц, I 1,6 Гц); 2,44 (1Н, д, ОН); 3,99 (1Н, м); 4,56 (1Н, дд, I 14 Гц, I 1,6 Гц); 5,05 (1Н, дд, I14 Гц); 1,6 Гц); 6,65-6,86 (2Н, м); 7,50-7,62 (1Н, м); 7,80 (1Н, с); 8,05 (1Н, с).
ИК (КВr), см-1: 3400, 1615, 1500, 1420, 1275, 1200, 1135.
С р а в н и т е л ь н ы й п р и м е р 57. В этилацетате (40 мл) растворяли (2S, 3R)-2-(2,4-дифторoфенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол (2,5 г). К раствору при охлаждении льдом добавляли триэтиламин 1,82 мл и метансульфонилхлорид (1,51 г). Смесь размешивали при комнатной температуре в течение 30 мин. Затем к реакционной смеси добавляли этилацетат (40 мл), после чего смесь промывали водой, высушивали безводным сульфатом магния и после концентрирования получали (2S, 3R)-2-(2,4-дифторофенил)-3-метансульфонилокси-1-(1Н, 1,2,4-триазол-1-ил)-2-бутанол в виде маслянистого продукта. Этот продукт растворяли в метаноле (40 мл), а затем при охлаждении льдом к нему добавляли 28%-ный метаноловый раствор метилата натрия (2,04 г). Полученную смесь размешивали при комнатной температуре в течение 30 мин и концентрировали при пониженном давлении. К концентрату добавляли этилацетат (100 мл), смесь промывали водой и высушивали безводным сульфатом магния. Растворитель отгоняли при пониженном давлении, а остаток очищали на силикагеле, элюируя смесью этилацетата и дихлорметана (4:1), в результате чего получали (2S, 3S)-2-(2,4-дифторофенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил)метил оксиран (1,62 г) в виде бесцветного маслянистого продукта. При замораживании этот продукт становился бесцветным твердым веществом с т.пл. 41-43оС.
[α]23 + 5,8о (с 1,0 в метаноле).
ЯМР (CDCl3) δ: 1,06 (3Н, д, I 5,4 Гц), 3,18 (1Н, кв, I 5,4 Гц); 4,42 (1Н, д, I 15 Гц); 4,80 (1Н, д, 15 Гц); 6,76-6,90 (2Н, м); 7,07-7,20 (1Н, м); 7,85 (1Н, с); 8,06 (1Н, с).
ИК(КВr), см-1: 3150, 1615, 1595, 1502, 1420, 1270, 1130.
С р а в н и т е л ь н ы й п р и м е р 58. Маточный раствор, полученный после перекристаллизации (2S, 3S)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиола, как описано в сравнительном примере 38, концентрировали при пониженном давлении. Концентрат очищали с помощью хроматографии на силикагеле (элюент: этилацетат) и получали (2R, 3S)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол в виде первой фракции элюата.
Т.пл. 156-157оС.
1Н-ЯМР (CDCl3) δ: 1,27 (3Н, дд, I 6,4 Гц, I 1,6 Гц); 2,42 (1Н, д, ОН); 3,99 (1Н, м); 4,57 (1Н, дд, I 14 Гц, I 1,6 Гц); 5,05 (1Н, дд, I 14 Гц, 1,6 Гц); 6,67-6,86 (2Н, м); 7,50-7,62 (1Н, м); 7,80 (1Н, с); 8,04 (1Н, с).
ИК (КВr), см-1: 3350, 1615, 1510, 1420, 1275, 1200, 1130.
С р а в н и т е л ь н ы й п р и м е р 59. В этилацетате (4 мл) растворяли (2R, 3S)-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил)-2,3-бутандиол (0,18 г). К полученному раствору добавляли триэтиламин (0,1 мл) и метансульфонилхлорид (84 мг), охлаждая при этом льдом. Смесь размешивали при комнатной температуре в течение 30 мин, после чего к ней добавляли этилацетат (10 мл). Реакционную смесь промывали водой, высушивали безводным сульфатом магния и после концентрирования получали (2R, 3S)-2-(2,4-дифторoфенил)-3-метансульфонилок- си-1-(1Н-1,2,4-триазол-1-ил) -2-бутанол в виде маслянистого продукта. Этот продукт растворяли в метаноле (6 мл), к полученному раствору добавляли, охлаждая при этом льдом, 5,6%-ный метаноловый раствор метилата натрия. Смесь размешивали при комнатной температуре в течение 30 мин, а затем концентрировали при пониженном давлении. К концентрату добавляли этилацетат (30 мл), промывали водой и высушивали безводным сульфатом магния. Растворитель отгоняли, а остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:2), в результате чего получали (2R, 3R)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил)метил оксиран (0,15 г) в виде бесцветного маслянистого продукта.
ЯМР (CDCl3) δ: 1,06 (3Н, д, I 5,4 Гц); 3,18 (1Н, кв, I 5,4 Гц); 4,42 (1Н, д, I 15 Гц); 4,80 (1Н, д, I 15 Гц); 6,76-6,90 (2Н, м); 7,07-7,20 (1Н, м); 7,85 (1Н, с); 8,06 (1Н, с).
ИК (пленка), см-1: 1615, 1595, 1505, 1420, 1270, 1140.
С р а в н и т е л ь н ы й п р и м е р 60. 2',4'-Дифторо-2-гидроксипропиофенон (2,8 г) растворяли в метиленхлориде (28 мл), а затем к этому раствору добавляли при 0оС триэтиламин (2,5 мл) и метансульфонилхлорид (1,3 мл). После размешивания в течение 15 мин смесь промывали водой (30 мл), а органический слой высушивали и концентрировали при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (3 см х 10 см), элюируя метилен- хлоридом. Целевую фракцию концентрировали и получали 2',4'-дифтор-2-метансульфонилоксипропиофенон (3 г) в виде бесцветного маслянистого продукта. Этот продукт (3 г) растворяли в 30 мл N,N-диметилформамида, после чего к этому раствору добавляли 1Н-1,2,4-триазол (0,94 г) и затем 60%-ную суспензию гидрида натрия в масле (0,5 г) при -10оС. После размешивания при 0оС в течение 50 мин реакционную смесь добавляли в смесь этилацетата (100 мл) и воды (200 мл) для экстрагирования. Водный слой экстрагировали этилацетатом (100 мл), слои этилацетата комбинировали, промывали насыщенным водным раствором хлорида натрия (30 мл х 2), высушивали безводным сульфатом магния и концентрировали при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (3 х 10 см), элюируя этилацетатом. Целевую фракцию концентрировали и получали 2', 4'-дифторо-2-(1Н-1,2,4-триазол-1-ил) пропиофенон (1,5 г) в виде бесцветного маслянистого продукта. Этот продукт (1,24 г) добавляли к смеси диметилсульфоксида (30 мл), 60%-ной суспензии гидрида натрия в масле (0,25 г) и иодида триметилсульфоксония (1,38 г) при 10оС, после чего температуру повышали до 25оС. Через 2 ч реакционную смесь добавляли к смеси диэтилового эфира (100 мл) и воды (150 мл), слой диэтилового эфира отделяли, а водный слой экстрагировали диэтиловым эфиром (100 мл х 2). Органические слои объединяли, промывали насыщенным водным раствором хлорида натрия (50 мл х 2), высушивали безводным сульфатом магния и концентрировали при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (3 х 10 см) и элюировали смесью этилацетата и гексана (3:1), в результате чего получали 2-(2,4-дифторoфенил)-2-1-(1Н-1,2,4-триазол-1-ил)этил-оксиран в виде бесцветного маслянистого продукта (0,87 г).
1Н-ЯМР (CDCl3) δ: 1,61 (3Н, д, I 7,0 Гц); 1,62 (3Н, д, I= 6,0 Гц); 2,64 (1Н, д, I 4,6 Гц); 2,81 (1Н, д, I 5,6 Гц); 2,87 (1Н, д, I 4,6 Гц); 3,18 (1Н, д, I 4,6 Гц); 4,92 (2Н, кв. I 7,0 Гц); 6,7-7,2 (6Н, м); 7,87 (1Н, с); 7,94 (1Н, с); 8,04 (1Н, с); 8,12 (1Н, с).
С р а в н и т е л ь н ы й п р и м е р 61. К раствору 2-(2,4-дифторофенил)-2-[(1R)-1-(3,4,5,6- тетрагидро-2Н-пиран-2-илокси)этил] оксирана (1,7 г) и имидазола (0,49 г) в N,N-диметилформамиде (17 мл) добавляли порциями 60% маслянистого гидрида натрия в минеральном масле (0,29 г), при этом постоянно перемешивая при 20оС. Через 5 мин смесь нагревали при 70оС в течение 3 ч. Реакционную смесь охлаждали, выливали в воду (50 мл) и экстрагировали этилацетатом (20 мл х 3). Слои этилацетата объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл), высушивали безводным сульфатом магния и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (3 см х 15 см), элюируя смесью метанола и этилацетата (5:95). Целевую фракцию концентрировали при пониженном давлении и получали (3R)-2-(2,4-дифторофенил)-1-(1-имидазоил)-3-(3,4,5,6-тетрагидро- 2Н- пиран-2-илокси)-2-бутанол (1,8 г) в виде бесцветного сиропа.
1Н-ЯМР (CDCl3) δ: 0,92 (1,03 3Н, д, I 6,2 Гц, I 6,4 Гц); 1,5-2,0 (6Н, м); 3,5-4,8 (6Н, м); 6,6-7,5 (6Н, м).
ИК (чистый), см-1: 3300, 2900, 1650, 1650, 1600, 1490.
МСВИ (масс-спектроскопия вторичных ионов m/z):353 (МН+).
В этаноле (8,5 мл) растворяли (3R)-2-(2,4-дифторoфенил)-1-(1-имидазолил)-3-(3,4,5,6-те- трагидро- 2Н-пиран-2-илокси)-2-бутанол (1,7 г), к полученному раствору добавляли при 0оС трифторуксусную кислоту (8,5 мл). Через 10 мин температуру доводили до 20оС, а смесь оставляли на 1 ч. Затем реакционную смесь концентрировали при пониженном давлении, а остаток хроматографировали на силикагеле (2 х 10 см), элюируя смесью метанола и метиленхлорида (1: 9). Целевую фракцию концентрировали и получали (3)-2-(2,4-дифторoфенил) -1-(1-имидазолил)-2,3-бутандиол (1,7 г).
1Н-ЯМР (DMCO-d6) δ: 0,83, 1,03 (3Н, д, I 6,2 Гц, I 6,4 Гц); 4,15-4,35 (1Н, м); 4,62 (1Н, д, I 14,2 Гц); 4,71 (1Н, д, I 14,2 Гц); 5,5 (1Н, шир.с); 5,71 (1Н, с); 6,9-7,0 (1Н, м); 7,15-8,8 (5Н, м).
ИК (чистый), см-1: 3300, 1660, 1495, 1410, 1190, 1120.
МСВИ (m/z): 269 (МН+).
К раствору метиленхлорида (60 мл), содержащему (3R)-2-(2,4-дифторoфенил)-1-(1-имидазол)-2,3-бутандиол (1,7 г), триэтиламин (0,88 мл) и тетрагидрофуран (2 мл), добавляли по каплям при 0оС метансульфонилхлорид (0,5 мл), при этом постоянно перемешивая. Через 10 мин температуру доводили до 20оС и смесь перемешивали еще 50 мин. Затем добавляли метансульфонилхлорид (0,50 мл) и триэтиламин (0,88 мл), после этого реакционную смесь размешивали в течение 1 ч, выливали в воду (100 мл) и экстрагировали метиленхлоридом (30 мл х 3). Слои этилацетата объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл), высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (3 см х 15 см), элюируя этилацетатом. Целевую фракцию концентрировали при пониженном давлении. К остатку добавляли изопропиловый эфир-н-гексан, с помощью которого кристаллизовали (2R, 3S)-2-(2,4-дифторoфенил)-2-(1-имидазолил)метил-3- метилоксиран (0,14 г) в виде призмообразных бесцветных кристаллов с т.пл. 73-76оС.
С р а в н и т е л ь н ы й п р и м е р 62. К N,N-диметилформамидному раствору (35 мл), содержащему (2R, 3S)-3-(2,4-дифторoфенил)-3,4- эпокси-2-бутилметансульфонат (3,5 г) и имидазол (1,2 г), добавляли 60%-ный гидрид натрия в минеральном масле (0,7 г), постоянно перемешивая при 0оС. Через 10 мин температуру доводили до 20оС и смесь размешивали в течение 20 ч. Затем реакционную смесь выливали в воду (100 мл) и экстрагировали этилацетатом (30 мл х 4). Слои этилацетата объединяли, промывали насыщенным водным раствором хлорида натрия (30 мл), высушивали безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (3 х 15 см), элюируя смесью метанола и этилацетата (5:95). Целевую фракцию концентрировали при пониженном давлении, остаток кристаллизовали путем добавления изопропилового эфира и н-гексана и получали (2R, 3S)-2-(2,4-дифторoфе-нил)-2-(1-имидазоил)метил-3-метилоксиран (1,7 г) в виде бесцветных призмообразных кристаллов, т.пл. 73-76оС.
1Н-ЯМР (CDCl3) δ: 1,61 (3Н, д, I 5,6 Гц); 3,16 (1Н, кв, 5,6 Гц); 4,13 (1Н, д, I 14,8 Гц); 4,63 (1Н, д, I 14,8 Гц); 6,66-6,78 (2Н, м); 6,83 (1Н, с); 6,94 (1Н, с); 6,92-7,04 (1Н, м); 7,29 (1Н, с).
Элементный анализ для C13H12F2N2O x x 1/4 H2O.
Вычислено, С 61,29; Н 4,95; N 11,00.
Найдено, С 61,46; Н 4,73; N 10,89.
ИК (КВr), см-1: 1600, 1585, 1495, 1415, 1270, 1260, 1210, 1110, 1090.
П р и м е р 1. В диметилформамиде (20 мл) растворяли 2-(2,4-дифторoфенил)-2-(1Н-1,2,4, -триазол-1-ил-метил) оксиран (1,2 г) и 1-(2-меркаптоэтил)бензимидазол (1,1 г). К этому раствору добавляли, охлаждая при этом льдом, 60%-ный гидрид натрия в масле (0,36 г) и полученную смесь размешивали в течение 90 мин. Затем диметилформамид отгоняли при пониженном давлении, остаток разбавляли водой (50 мл) и экстрагировали этилацетатом. Экстракт промывали водой, а затем насыщенным водным раствором хлорида натрия и высушивали MgSO4, а растворитель отгоняли. Остаток хроматографировали на силикагеле (2,5 х 40 см), элюируя смесью этилацетата, ацетона и метанола (6:2: 1). Целевую фракцию концентрировали и получали соединение 1 (1,1 г) в виде бесцветного порошка.
Элементный анализ для C20H19F2N5OS x x 0,5 H2О.
Вычислено, С 56,59; Н 4,75; N 16,50.
Найдено, С 57,07; Н 4,96; N 15,98.
1Н-ЯМР (CDCl3) δ: 2,81-3,12 (4Н, м); 4,30-4,37 (2Н, м); 4,64 (2Н, с); 6,74-6,82 (2Н, м); 7,28-7,50 (5Н, м); 7,71-7,83 (1Н, шир.с); 7,81 (1Н, с); 7,88 (1Н, с); 7,93 (1Н, с).
МСВИ (m/z): 416 (М+Н)+.
П р и м е р 2. Смесь 2-(1Н-1,2,4-триазол-1-ил)этантиола (4,5 г), 2-(2,4-дифторoфенил)-2-(1Н-1,2,4-триазол-1-ил-метил) окси- ранметасульфоната (4 г), карбоната калия (6,5 г) и диметилформамида (20 мл) размешивали при 70оС в течение 1 ч. После охлаждения реакционную смесь разбавляли водой (150 мл) и насыщенным хлоридом натрия и экстрагировали этилацетатом (150 мл х 3). Экстракт промывали водой (50 мл х 3) и высушивали сульфатом натрия, а растворитель отгоняли, в результате чего получали маслянистое вещество желтого цвета. Полученный продукт очищали хроматографией на силикагеле, элюируя смесью этилацетата, ацетона и метанола (6:2:1), полученный маслянистый продукт кристаллизовали из смеси этилового эфира и этилацетата, в результате чего получали соединение 2 (0,85 г) в виде бесцветных призмообразных кристаллов.
Т.пл. 114-115оС.
Элементный анализ для C15H16F2N6OS.
Вычислено, С 49,17; Н 4,40; N 22,94.
Найдено, С 48,93; Н 4,40; N 22,87.
П р и м е р 3. В метиленхлориде (10 мл) растворяли соединение 2 (0,2 г) и, размешивая при комнатной температуре, добавляли m-хлоробензойную кислоту (чистота 85% 0,29 г). Затем смесь размешивали при комнатной температуре в течение 16 ч. Смесь промывали 5%-ным водным раствором бикарбоната натрия, органический слой высушивали MgSO4 и концентрировали при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (1,5 x х 15 см), элюируя смесью этилацетата, ацетона и метанола (8:2:1). Целевую фракцию концентрировали и получали соединение 3 (0,12 г) в виде бесцветного порошка.
1Н-ЯМР (CDCl3) δ: 3,36-3,79 (4Н, м); 4,63-4,79 (3Н, м); 4,89 (1Н, д, I 14,2 Гц); 5,61 (1Н, с); 6,78-6,92 (2Н, м); 7,38-7,53 (1Н, м); 7,86 (1Н, с); 7,91-7,95 (2Н, м); 8,18 (1Н, с).
МСВИ (m/z): 399 (М+Н)+.
П р и м е р 4. В метиленхлориде (15 мл) растворяли соединение 2 (0,3 г) и, перемешивая при охлаждении льдом, добавляли m-хлоробензойную кислоту (чистота 85% 0,22 г). Смесь размешивали, охлаждая при этом льдом, и промывали 5%-ным водным раствором бикарбоната натрия. Органический слой высушивали MgSO4 и концентрировали при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (1,5 х 20 см), элюируя смесью этилацетата, ацетона и метанола (5:4:1). Целевую фракцию концентрировали и получали соединение 4 (0,07 г) в виде бесцветного порошка.
1Н-ЯМР (CDCl3) δ: 2,87-3,48 (4Н, м); 4,36-4,72 (4Н, м); 5,48 (1Н, шир. с); 6,81-7,02 (2Н, м); 7,59-7,75 (1Н, м); 7,85-7,99 (2Н, м); 8,11 (1Н, с); 8,15 (1Н, с).
П р и м е р 5. Смесь 2-(1-имидазолил) этантиола (4,5 г), 2-(2,4-дихлорoфенил-2-(1Н-1,2,4-триазол-1-ил-метил) оксиранметансульфоната (4 г), карбоната калия (6,5 г) и диметилформамида (30 мл) размешивали при 70оС в течение 1 ч. После охлаждения реакционную смесь разбавляли водой (100 мл), насыщали хлоридом натрия и экстрагировали этилацетатом (100 мл х 3). Экстракт промывали водой (30 мл х 3) и высушивали безводным сульфатом натрия, а растворитель отгоняли, в результате чего получали бесцветное маслообразное вещество. Этот продукт очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата, ацетона в метаноле (6:2:1), и получaли маслянистый продукт, после кристаллизации которого из этилового эфира получали соединение 5 (1,28 г) в виде бесцветного порошка с т.пл. 103-105оС.
Элементный анализ для C16H17F2N5OS.
Вычислено, С 52,59; Н 4,69; N 19,17.
Найдено, С 52,32; Н 4,69; N 19,03.
П р и м е р 6. Способом, аналогичным описанному в примере 3, соединение 5 (0,3 г) окисляли с помощью m-хлоробензойной кислоты, в результате чего получали соединение 6 (0,17 г, 51%) в виде бесцветного порошка.
1Н-ЯМР (CDCl3) δ: 3,51-3,73 (4Н, м); 4,37-4,43 (2Н, м); 4,59 (1Н, д, I 14,2 Гц); 4,80 (1Н, д, I 14,2 Гц); 6,44 (1Н, шир.с); 6,80-6,99 (4Н, м); 7,40-7,54 (2Н, м); 7,81 (1Н, с); 8,11 (1Н, с).
МСВИ (m/z): 398 (М+Н)+.
П р и м е р ы 7-12. Способом, аналогичным описанному в примере 1, проводили реакцию взаимодействия 2-(2,4-дифторoфенил)-2-(1Н-1,2,4-триазол-1-ил-метил) оксирана (эпоксисоединение в табл.11) с различными тиолами, приведенными в табл.11, в результате чего получали соединения 7,8,9,10,11,17,19 и 21.
П р и м е р 13. Смесь (2RS, 3SR)-2-(2,4-дифторoфенил)-3-метил-2- (1Н-1,2,4-триазол-1-ил-метил) оксирана (0,5 г), 2-меркапто-5,6- дигидро-4Н-циклопентатиазола (0,38 г) и 1М тетрабутиламмония фторида (2,2 мл) в этаноле (15 мл) нагревали в колбе с обратным холодильником в течение 4 ч. Затем этанол отгоняли при пониженном давлении, а остаток разбавляли водой (25 мл) и экстрагировали этилацетатом. Экстракт промывали водой и насыщенным водным раствором хлорида натрия и высушивали MgSO4, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (2,5 х 40 см), элюируя смесью этилацетата и н-гексана (3:2). Целевую фракцию концентрировали, к остатку добавляли смесь эфира и гексана (2:1) и получали соединениe 12 (0,34 г) в виде бесцветных призмообразных кристаллов. Т.пл. 70-72оС.
Элементный анализ для C18H18F2N4OS.
Вычислено, С 52,93; Н 4,44; N 13,72.
Найдено, С 52,65; Н 4,38; N 13,70.
1Н-ЯМР (CDCl3) δ: 1,22 (3Н, д, I 7,2 Гц); 2,43-2,62 (2Н, м); 2,87-2,99 (2Н, м); 4,02 (1Н, кв, I 7,2 Гц); 4,98 (2Н, с); 6,64-6,82 (2Н, м); 7,08 (1Н, с); 7,39-7,52 (1Н, м); 7,67 (1Н, с); 8,06 (1Н, с).
П р и м е р 14. Смесь (2RS, 3SR)-2-(2,4-дифторoметил)-3-метил-2- (1Н-1,2,4-триазол-1-ил-метил) оксирана (0,5 г), 1-(2-меркаптоэтил)- 1Н-1,2,3-триазола (0,31 г) и 1М тетрабутиламмония фторида (2,2 мл) в этаноле (20 мл) нагревали в колбе с обратным холодильником в течение 12 ч. Затем этанол отгоняли при пониженном давлении, а остаток разбавляли водой (25 мл) и экстрагировали этилацетатом. Экстракт промывали водой и насыщенным водным раствором хлорида натрия и высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (2,5 х 40 см), элюируя смесью этилацетата, ацетона и метанола (10: 2:1). Целевую фракцию концентрировали и получали соединение 13 (0,4 г) в виде маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 1,11 (3Н, д, I 7,2 Гц); 3,09-3,38 (3Н, м); 4,37-4,59 (2Н, м); 4,73 (1Н, д, I 14 Гц); 4,93 (1Н, д, I 14 Гц); 5,03 (1Н, шир.с); 6,69-6,78 (2Н, м); 7,29-7,41 (1Н, м), 7,78 (1Н, с); 7,79 (1Н, с); 8,02 (1Н, с); 8,19 (1Н, с).
После обработки этого продукта (0,14 г) хлороводородом в этилацетате получали гидрохлорид указанного соединения (0,11 г) в виде бесцветного порошка.
Т.пл. 179-181оС.
Элементный анализ для C16H18F2N6OS x x 2HCl ˙ 0,5 H2O.
Вычислено, С 41,57; Н 4,58; N 18,18.
Найдено, С 41,87; Н 4,41; N 18,45.
П р и м е р 15. Способом, аналогичным описанному в примере 3, соединение 13 (0,2 г) окисляли с помощью m-хлоропербензойной кислоты (0,28 г), в результате чего получали соединение 14 (0,8 г, 37%) в виде бесцветного порошка.
1Н-ЯМР (CDCl3) δ: 1,22 (3Н, д, I 7,2 Гц); 3,56-4,08 (3Н, м); 4,76-4,82 (2Н, м); 4,91 (1Н, д, I 14,2 Гц); 5,36 (1Н, д, I 14,2 Гц); 5,36 (1Н, д, I 14,2 Гц); 5,71 (1Н, с); 6,71-6,81 (2Н, м); 7,18-7,32 (1Н, м); 7,75 (1Н, с); 7,77 (1Н, с); 8,00 (1Н, с); 8,25 (1Н, с).
МСВИ (m/z): 413 (М+Н+).
П р и м е р 16. Смесь раствора (2RS, 3SR)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1, 2,4-триазол-1-ил-метил) оксирана (0,6 г), пиридин-4-метантиола (0,45 г) и 28% метоксида натрия и метанола (0,55 г) в этаноле (12 мл) нагревали в колбе с обратным холодильником в течение 1 ч. Затем этанол отгоняли при пониженном давлении, а остаток хроматографировали на колонках с силикагелем (2,5 х 15 см), элюируя смесью метанола и этилацетата (5:95). Целевую фракцию концентрировали, а к остатку добавляли смесь этилацетата и диэтилового эфира, в результате чего получали соединение 15 (0,67 г) в виде бесцветных игольчатых кристаллов.
Т.пл. 98-99оС.
Элементный анализ для C18H18F2N4OS.
Вычислено, С 57,43; Н 4,82; N 14,88.
Найдено, С 57,12; Н 4,70; N 14,78.
1Н-ЯМР (CDCl3) δ: 1,14 (3Н, д, I 6,8 Гц); 3,16 (1Н, кв, 6,80 Гц); 3,80 (1Н, д, I 13,8 Гц); 3,92 (1Н, д, I 13,8 Гц); 4,57 (1Н, д, I 14,2 Гц); 4,97 (1Н, д, I 14,2 Гц); 5,04 (1Н, с); 6,64-6,77 (2Н, м); 7,28-7,40 (1Н, м); 7,31 (2Н, д, I 6,0 Гц); 7,74 (1Н, с); 7,76 (1Н, с); 8,59 (2Н, д, I 6,0 Гц).
П р и м е р ы 17-19. Способом, аналогичным описанному в примере 16, приводили реакцию взаимодействия (2RS- 3SR)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1, 2,4-триазол-1-ил-метил) оксирана (метилэпоксисоединение в табл.12) с различными тиолами, приведенными в табл. 12, в результате чего получали соединения 16, 22 и 23.
П р и м е р 20. Смесь раствора 2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-три- азол-1-ил)пропан-2-ол (1,2 г), 2-(2-хлороэтилтио)-1-метилимидазол (1,2 г) и 28% метоксида натрия-метанола (0,94 г) в этаноле (30 мл) нагревали в колбе с обратным холодильником в течение 2 ч. Затем этанол отгоняли при пониженном давлении и остаток хроматографировали на силикагеле (3,5 х 15 см), элюируя смесью метанола и этилацетата (5:95). Целевую фракцию концентрировали, а к остатку добавляли дихлорметан и диэтиловый эфир, в результате чего получали соединение 18 (0,9 г) в виде бесцветных игольчатых кристаллов. Т.пл. 92-93оС.
Элементный анализ для C17H19F2N5OS2.
Вычислено, С 49,62; Н 4,65; N 17,02.
Найдено, С 49,45; Н 4,71; N 16,84.
1Н-ЯМР (CDCl3) δ: 2,89 (2Н, шир. т, I 7,4 Гц); 3,22-3,30 (2Н, м); 3,20 (1Н, д, I 14,8 Гц); 3,39 (1Н, д, I 14,8 Гц); 3,57 (3Н, с); 4,65 (1Н, д, I 14,2 Гц); 4,75 (1Н, д, I 14,2 Гц); 6,19 (1Н, с); 6,74-6,85 (2Н, м); 6,92 (1Н, с); 6,98 (1Н, с); 7,47-7,60 (1Н, м); 7,76 (1Н, с); 8,07 (1Н, с).
П р и м е р ы 21 и 22. Способом, аналогичным описанному в примере 20, проводили реакцию взаимодействия 2-(2,4-дифторoфенил)-3- меркапто-1-(1Н-1,2,4-триазол-1-ил) пропан-2-ола, (представленного в табл.13 как производное тиола, с хлоpпроизводным, показанным в табл.13, в результате чего получали соединения 20 и 24.
П р и м е р 23. В диэтиловом эфире (30 мл) растворяли соединение 16 (0,52 г), полученное в соответствии с описанием примера 17, после чего к этому раствору добавляли хлороводород в этилацетате. Полученную смесь оставляли отстаиваться и надосадочную жидкость удаляли путем декантации. Остаток разбавляли диэтиловым эфиром (30 мл), отстаивали и надосадочную жидкость удаляли путем декантации. Остаток растворяли в этаноле (10 мл), а затем к нему добавляли этилацетат (100 мл), полученную смесь выдерживали в течение дня для кристаллизации. Полученные кристаллы собирали фильтрацией и высушивали при пониженном давлении, в результате чего получали соединение 16-гидрохлорид (0,49 г). Т.пл. 114-116оС.
Элементный анализ для C17H19F2N5OS x x 2HCl ˙ H2O.
Вычислено, С 43,41; Н 4,93; N 14,89.
Найдено, С 43,76; Н 4,72; N 14,96.
1Н-ЯМР (DMCO-d6) δ: 1,05 (3Н, д, I 6,6 Гц); 3,43 (1Н, кв, I 6,6 Гц); 3,90 (3Н, с); 4,32 (1Н, д, I 15,4 Гц); 4,45 (1Н, д, I 15,4 Гц); 4,56 (1Н, д, I 14,4 Гц); 4,86 (1Н, д, I 14,4 Гц); 6,95 (1Н, м); 7,10-7,40 (2Н, м); 7,68 (1Н, с); 7,73 (1Н, с); 7,82 (1Н, с); 8,54 (1Н, с).
П р и м е р 24. Смесь (2RS- 3SR)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол- 1-ил) метилоксирана (0,3 г), 2-метил-1,3,4-тиадиазол-5-тиола (0,19 г) и 1М фторида тетрабутиламмония (1,3 мл) в этаноле (10 мл) нагревали в колбе с обратным холодильником в течение 4,5 ч. Затем этанол отгоняли при пониженном давлении, а остаток разбавляли водой (15 мл) и экстрагировали этилацетатом.
Экстракт промывали водой и насыщенным водным раствором хлорида натрия и высушивали сульфатом магния. Затем растворитель отгоняли при пониженном давлении, а остаток хроматографировали на колонках с силикагелем (2,5 х 30 см), элюируя смесью этилацетата и дихлорметана (3:2). Целевую фракцию концентрировали и к остатку добавляли смесь эфира и изопропилового эфира (1:2), в результате чего получали соединение 26 (0,07 г) в виде бесцветных призмообразных кристаллов. Т.пл. 130-131оС.
Элементный анализ для C15H15F2N5OS.
Вычислено, С 46,99; Н 3,94; N 18,26.
Найдено, С 46,77; Н 3,86; N 18,06.
1Н-ЯМР (CDCl3) δ: 1,29 (3Н, д, I 7 Гц); 2,77 (3Н, с); 4,62 (1Н, кв, I 7 Гц); 4,89 (1Н, д, I 13,8 Гц); 5,12 (1Н, д, I 13,8 Гц); 5,97 (1Н, с); 6,72-6,86 (2Н, м); 7,41-7,52 (1Н, м), 7,75 (1Н, с), 7,91 (1Н, с).
П р и м е р 25. Смесь (2RS, 3SR)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил)метилоксирана (0,25 г), 2-имидазо [1,2-a] пиридинметантиола (0,25 г), 28% метоксида натрия и метанола (0,25 мл) и этанола (7,5 мл) нагревали с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении и остаток разбавляли метиленхлоридом (20 мл) и водой (20 мл), а затем экстрагировали метиленхлоридом. Слой метиленхлорида высушивали безводным сульфатом натрия и концентрировали при пониженном давлении, а остаток хроматографировали на силикагеле (2,5 х 7,0 см), элюируя этилацетатом. Целевую фракцию концентрировали и получали соединение 27 (0,25 г) в виде сиропа. Этот сироп (0,19 г) растворяли в диэтиловом эфире (30 мл), а затем к нему добавляли хлороводород в этилацетате. Полученную смесь отстаивали и надосадочную жидкость удаляли путем декантации. Остаток разбавляли 30 мл диэтилового эфира, а надосадочную жидкость удаляли путем декантации. К остатку добавляли смесь диэтилового эфира и этанола для кристаллизации и получали соединение 27 гидрохлорид (0,17 г). Т.пл. 123-125оС.
Элементный анализ для C20H19F2N5OS x x 2HCl ˙ 1/2H2O.
Вычислено, С 48,30; Н 4,46; N 14,08.
Найдено, С 48,38; Н 4,44; N 13,92.
1Н-ЯМР (DMCO-d6) δ: 1,08 (3Н, д, I 8,2 Гц); 3,43 (1Н, кв, I 8,2 Гц); 4,22 (1Н, д, I 14,4 Гц); 4,32 (1Н, д, I 14,4 Гц); 4,68 (1Н, д, I 14,6 Гц); 4,99 (1Н, д, I 14,6 Гц); 6,92 (1Н, м); 7,12 (1Н, м), 7,29 (1Н, м), 7,50 (1Н, м); 7,77 (1Н, с), 7,97 (2Н, м); 8,37 (1Н, с); 8,64 (1Н, с); 8,98 (1Н, д, I 6,6 Гц).
П р и м е р 26. К раствору (2RS, 3SR)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4- триазол-1-ил)-2-бутанола (0,30 г) в этаноле (3 мл) добавляли 28% метоксид натрия-этанол (0,98 мл) при комнатной температуре. После добавления 1-метил-5-хлорометилимидазола гидрохлорида (0,48 г) смесь размешивали в течение 10 мин. Эту реакционную смесь разбавляли диэтиловым эфиром (30 мл) и водой (30 мл) и экстрагировали диэтиловым эфиром (30 мл х 3). Слои диэтилового эфира объединяли, промывали насыщенным водным раствором хлорида натрия (20 мл), высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли диэтиловый эфир и получали (2RS, 3SR)-2-(2,4-дифторoфенил)-3-(1-метилимидазол-5-ил)метилтио-1-(1Н-1, 2,4- триазол-1-ил)-2-бутанол соединение 29 (0,30 г) в виде кристаллов. Эти кристаллы перекристаллизовывали из смеси этанола и диэтилового эфира, в результате чего получали бесцветные игольчатые кристаллы (0,27 г). Т.пл. 159-160оС.
Элементный анализ для C17H19F2N5OS.
Вычислено, С 53,81; Н 5,05; N 18,46.
Найдено, С 53,40; Н 5,23; N 18,22.
1Н-ЯМР (CDCl3) δ: 1,13 (3Н, д, I 7,0 Гц); 3,20 (1Н, кв, I 7,0 Гц); 3,73 (3Н, с); 3,82 (1Н, д, I 14,8 Гц); 3,91 (1Н, д, I 14,8 Гц); 4,45 (1Н, д, I 14,2 Гц); 4,83 (1Н, д, I 14,2 Гц); 5,00 (1Н, шир. с); 6,72 (2Н, м); 7,01 (1Н, с); 7,35 (1Н, м); 7,48 (1Н, с); 7,72 (1Н, с); 7,77 (1Н, с).
П р и м е р ы 27-37. Способом, аналогичным описанному в примере 26, проводили реакцию взаимодействия (2RS, 3SR)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол, обозначенного в табл. 14 метилтиоловым производным, с хлорсоединениями, приведенными в табл.14, в результате чего получали соединения 25, 28 и 30-38.
П р и м е р 38. Смесь (2RS, 3SR)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол- 1-ил)метилоксирана (7 г), метил 3-меркаптопропионата (30,8 мл) и 28% метоксида натрия-метанола (19,6 мл) в метаноле (210 мл) нагревали в колбе с обратным холодильником в течение 2 ч. Затем добавляли 28% метоксида натрия-метанолa (9,8 мл) и смесь нагревали с обратным холодильником еще 1 ч. После этого добавляли метил 3-меркаптопропионат и смесь нагревали с обратным холодильником еще 2 ч. Затем реакционную смесь охлаждали, разбавляли водой (100 мл), нейтрализовали 5%-ным водным раствором фосфорной кислоты и экстрагировали метиленхлоридом (200 мл х 2). Экстракт высушивали безводным сульфатом натрия, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (4 х 50 см), элюируя смесью этилацетата и гексана (3:1), и целевую фракцию концентрировали. К остатку добавляли эфир и получали соединение 39 (5,5 г) в виде бесцветных игольчатых кристаллов.
1Н-ЯМР (CDCl3) δ: 1,17 (3Н, д, I 7,0 Гц); 1,96 (1Н, д, I 10,2 Гц); 3,45 (1Н, д, кв. I 7,0 Гц, I 10,2 Гц); 4,77 (1Н, с); 4,82 (1Н, д, I 14,4 Гц); 5,01 (1Н, д, I 14,4 Гц); 6,70-6,81 (2Н, м); 7,33-7,45 (1Н, м); 7,79 (1Н, с); 7,80 (1Н, с). Т.пл. 145-147оС.
П р и м е р 39. К дихлорoметану (5 мл) добавляли (2R, 3R)-2-(2,4-дифторoфенил)-3-меркапто-1-(1H-1,2,4-триазол-1-ил)-2-бута- нол (0,3 г), а затем при охлаждении льдом добавляли триэтиламин (0,16 мл). После этого к смеси добавляли по каплям ацетилхлорид (0,082 мл) и смесь размешивали при комнатной температуре в течение 30 мин. Затем растворитель отгоняли при пониженном давлении, а остаток хроматографировали на силикагеле (2,5 х 20 см), элюируя смесью этилацетата и гексана (2:1). Целевую фракцию концентрировали, к остатку добавляли гексан, в результате чего получали соединение 40 (0,2 г) в виде бесцветных игольчатых кристаллов. Т.пл. 73-75оС.
Элементный анализ C14H15F2N3O2.
Вычислено, С 51,37; Н 4,62; N 12,84.
Найдено, С 51,19; Н 4,53; N 12,84.
1Н-ЯМР (CDCl3) δ: 1,10 (3Н, д, I 7,2 Гц); 2,42 (2Н, с); 4,29 (1Н, кв, I 7,2 Гц); 4,67 (1H, д, I 15,2 Гц); 4,91 (1Н, д, I 15,2 Гц); 5,09 (1Н, с); 6,69-6,88 (2Н, м); 7,26-7,43 (1Н, м); 7,77 (1Н, с); 7,78 (1Н, с).
П р и м е р 40. Смесь (2S, 3R)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил) метилоксирана (30 ил), метил 3-меркаптопропионата (0,09 мл) и 28% метоксида натрия-метанола (0,08 мл) в метаноле (2 мл) нагревали в колбе с обратным холодильником в течение 2 ч, после чего добавляли смесь 28% метоксида натрия и метанола (0,04 мл) и полученную в результате смесь нагревали с обратным холодильником еще в течение 1 ч. Затем добавляли метил 3-меркаптопропионат (0,04 мл) и смесь нагревали с обратным холодильником еще в течение 2 ч. Реакционную смесь охлаждали, разбавляли водой (2 мл), нейтрализовали 5%-ным водным раствором фосфорной кислоты и экстрагировали метиленхлоридом (3 мл х 2). Экстракт высушивали сульфатом натрия, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на силикагеле (1 х 5 см), элюируя смесью этилацетата и гексана (3:1). Целевую фракцию концентрировали, а остаток кристаллизовали из смеси этилацетата и изопропилового эфира, в результате чего получали (2S, 3S)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2, 4-триазол-1-ил)-2-бутанол (соединение 42 11 г) в виде бесцветных призмообразных кристаллов.
[α]D25+ 55,7o (с 1,0, метанол).
1Н-ЯМР (CDCl3) δ: 1,17 (3Н, д, I 6,8 Гц); 1,96 (1Н, д, I 10,4 Гц); 3,39-3,54 (1Н, м); 4,75 (1Н, с); 4,81 (1Н, д, I 14,4 Гц); 5,01 (1Н, д, I 14,4 Гц); 6,69-6,81 (2Н, м); 7,33-7,46 (1Н, м); 7,79 (1Н, с); 7,80 (1Н, с). T.пл. 175-178оС.
Для определения избытка энантиомера полученный продукт S ацетилировали (соединение 44 в примере 43) и анализировали с помощью жидкостной хроматографии высокого разрешения, используя хиральную колонку (Chiralcel OF, 0,46 см х 25 см, Daicel Chemical, подвижная фаза: гексанизопропиловый спирт 7: 3). При скорости потока 1 мл/мин соединение 44 имело в основном один пик при времени удерживания 10 мин и избыток энантиомера составлял 97,4% Соответствующее рацемическое соединение (соединение 40 в примере 39) имело два пика в отношении 1:1 при времени удерживания 10 и 17 мин при тех же условиях.
П р и м е р 41. В метаноле (10 мл) растворяли (2R, 3S)-2-(2,4-дифторoфенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил)метилокси- ран (0,40 г), метил 3-меркаптопропионат (1,42 мл) и 28%-ный метоксид натрия метанол (1,25 мл). Полученный раствор нагревали с обратным холодильником. После чего через 2 ч, а затем через 3,5 ч добавляли метил 3-меркаптопропионат (0,53 и 0,32 мл), а через 2,5 мин добавляли 28%-ный метоксид натрия метанол (0,63 мл). Через 4,5 ч после начала нагревания масляную баню удаляли, а реакционную смесь охлаждали, нейтрализовали 1н. соляной кислотой (9,6 мл) и экстрагировали дихлорметаном (100 мл). Экстракт промывали насыщенным водным раствором хлорида натрия (20 мл) и высушивали безводным сульфатом натрия, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:3). Целевую фракцию концентрировали и полученные кристаллы собирали и промывали изопропиловым эфиром, в результате чего получали (2R, 3R)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-три- азол-1-ил)-2-бутанол (соединение 43, 0,22 г) в виде бесцветных игольчатых кристаллов. Т.пл. 176-178оС.
[α]D25-56,8о (с 0,7, метанол).
Элементный анализ для C12H13F2N3OS
Вычислено, С 50,52; Н 4,59; N 14,73.
Найдено, С 50,81; Н 4,64; N 14,64.
1Н-ЯМР (CDCl3) δ: 1,17 (3Н, д, I 7,0 Гц); 1,96 (1Н, д, I 10,2 Гц); 3,45 (1Н, м); 4,76 (1Н, с); 4,82 (1Н, д, I 14,4 Гц); 5,01 (1Н, д, I 14,4 Гц); 6,74 (2Н, м); 7,33-7,45 (1Н, м); 7,79 (2Н, с).
В целях определения избытка энантиомера полученный продукт S ацетилировали (соединение 45 в примере 44) и анализировали с помощью жидкостной хроматографии высокого разрешения, используя хиральную колонку (Chiralce OF, 0,46 х 25 см, Daicel Chemical, подвижная фаза: гексанизопропиловый спирт 7,3). При скорости потока 1 мл/мин соединение 45 имело в основном один пик при времени удерживания 17 мин, а избыток энантиомера составлял 99,7% Соответствующее рацемическое соединение (соединение 40 в примере 39) имело два пика в отношении (1:1) при времени удерживания 10 и 17 мин при тех же условиях.
OF, 0,46 х 25 см, Daicel Chemical, подвижная фаза: гексанизопропиловый спирт 7,3). При скорости потока 1 мл/мин соединение 45 имело в основном один пик при времени удерживания 17 мин, а избыток энантиомера составлял 99,7% Соответствующее рацемическое соединение (соединение 40 в примере 39) имело два пика в отношении (1:1) при времени удерживания 10 и 17 мин при тех же условиях.
П р и м е р 42. К дихлорoметану (1 мл) добавляли (2S, 3S)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бута- нол (3 мг), а затем добавляли при охлаждении льдом дихлорметановый раствор триэтиламина (10% 16 мкл). После этого добавляли дихлорметановый раствор ацетилхлорида (10% 8,2 мкл) и полученную смесь размешивали при комнатной температуре в течение 30 мин. Затем растворитель отгоняли при пониженном давлении, а остаток хроматографировали на силикагеле (1 х 1 см), элюируя смесью этилацетата и гексана (2: 1). Целевую фракцию концентрировали и получали (2S, 3S)-3-ацетилтио-2-(2,4-дифторофенил)-1-(1Н-1,2,4-триазол-1-ил) -2-бутанол (соединение 44, 1,2 мг) в виде бесцветного твердого вещества.
П р и м е р 43. В дихлорметане (1,5 мл) растворяли (2R, 3R)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бута-нол (60 мг), после чего к раствору, охлаждая льдом, добавляли триэтиламин (33 мл) и ацетилхлорид (13 мл). Затем смесь размешивали при комнатной температуре в течение 30 мин, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1: 2), и концентрировали целевую фракцию, в результате чего получали (2R, 3R)-3-ацетилтио-2-(2,4- дифторoфенил)-1-(1Н-1,2,4-триазол-1-ил)-2- бутанол (соединение 45, 59 мг) в виде бесцветного твердого вещества.
1Н-ЯМР (CDCl3) δ: 1,11 (3Н, д, I 7,2 Гц); 2,42 (3Н, с); 4,31 (1Н, д, I 7,2 Гц); 4,67 (1Н, д, I 14,4 Гц); 4,92 (1Н, д, I 14,4 Гц); 5,11 (1Н, д, I 1,8 Гц); 6,69-6,88 (2Н, м); 7,27-7,43 (1Н, м); 7,78 (2Н, с).
Избыток энантиомера этого продукта составлял 99,7%
П р и м е р ы 44-53. Способом, аналогичным описанному в примере 26, проводили реакцию взаимодействия (2RS, 3SR)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил-2-бутанол)[(2R 3R)-метиловое производное в табл.15] с хлоросоединениями, приведенными в табл.15, в результате чего получали соединения 46-49, 51-55 и 59.
П р и м е р 54. Смесь (2RS, 3RS)-2-(2,4-дифторoфенил)-1-метил-2-(1Н-триазол-1-ил-метил)оксиран (0,18 г), 1-метил-2-(2-меркаптоэтил)имидазола (0,1 г), 28% -ный метоксид натрия-метанола (0,20 мл) и этанола (5,4 мл) нагревали в колбе с обратным холодильником в течение 1 ч. Затем реакционную смесь разбавляли водой (10 мл) и экстрагировали диэтиловым эфиром (20 мл х 3). Экстракт промывали насыщенным водным раствором хлорида натрия (10 мл), высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли диэтиловый эфир и получали сырые кристаллы (2RS, 3RS)-2-(2,4-дифторoфенил)-3-[2-(1-метилимидазол-2-ил)этил] тио- 1-(1Н- 1,2,4-триазол-1-ил)-2-бутанол (0,088 г). Кристаллы перекристаллизовывали из этилацетата и получали 0,050 г соединения (соединение 50). Т.пл. 175-176оС.
1Н-ЯМР (CDCl3) δ: 1,20 (3Н, д, I 6,6 Гц); 2,7-3,6 (5Н, м), 3,60 (3Н, с); 4,71 (1Н, д, I14,2 Гц); 5,03 (1Н, д, I 14,2 Гц); 6,87-6,77 (2Н, м); 6,87 (1Н, с); 7,08 (1Н, с); 7,43-7,55 (1Н, м); 7,69 (1Н, с); 7,82 (1Н, с); 8,03 (1Н, с).
Элементный анализ для C18H21F2N5OS.
Вычислено, С 54,95; Н 5,38; N 17,80.
Найдено, С 55,04; Н 5,39; N 17,62.
П р и м е р 55. К раствору (2RS, 3RS)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (0,060 г), 4-бромометил-5-метил-2-оксо-1,3-диоксол (0,049 г) в N,N-диметилформамиде (2 мл) добавляли безводный карбонат (0,2 г) при -20оС и полученную смесь размешивали в течение 10 мин. К реакционной смеси добавляли этилацетат (10 мл) и воду (10 мл) и органический слой отделяли. Затем смесь экстрагировали этилацетатом (10 мл х 2). Органические слои объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл), высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (1 х 5 см), элюировали смесью этилацетата и гексана (1:1). Целевую фракцию концентpировали, а остаток кристаллизовали из смеси хлороформа и диэтилового эфира, в результате чего получали (2RS, 3RS)-2-(2,4-дифторoфенил)-3-(5-метил-2-оксо-1,3-диоксол-4-ил) метилтио-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (соединение 56, 0,065 г), т.пл. 173-174оС.
1Н-ЯМР (CDCl3) δ: 1,20 (3Н, д, I 7,0 Гц); 2,15 (3Н, с); 3,32 (1Н, кв, I 7,0 Гц); 3,67 (1Н, д, I 15,4 Гц); 3,77 (1Н, д, I 15,4 Гц); 4,83 (1Н, д, I 14,2 Гц); 5,04 (1Н, д, I 14,2 Гц); 5,11 (1Н, д, I 1,6 Гц); 6,68-6,79 (2Н, м); 7,30-7,42 (1Н, м); 7,79 (1Н, с); 7,80 (1Н, с).
Элементный анализ для C17H17F2N3O4S.
Вычислено, С 51,38; Н 4,31; N 10,57.
Найдено, С 51,39; Н 4,30; N 10,55.
П р и м е р 56. В 1,2-дихлорoэтане (5 мл) растворяли N-(2-пиразинилметил)хлорoаце-тамид (0,5 г), а затем добавляли оксихлорид фосфора (5 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 40 мин, а затем концентрировали при пониженном давлении. Остаток растворяли в метаноле (1 мл) и добавляли диэтиловый эфир, в результате чего получали неочищенный 3-хлорометилимидазо [1,5-a] пиразин гидрохлорида в виде порошка (0,46 г). Этот порошок (0,18 г) добавляли к смеси (2RS, 3RS)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н, 2,4-триазол-1-ил)-2-бутанола (0,25 г), этанола (2,5 мл) и 28% -ного метоксида натрия и метанола (0,36 мл) при комнатной температуре. Полученную смесь размешивали в течение 7 мин.
Затем реакционную смесь выливали в воду (20 мл) и экстрагировали этилацетатом (20 мл х 3). Органические слои объединяли, промывали насыщенным водным раствором хлорида натрия (10 мл), высушивали безводным сульфатом магния и концентpировали при пониженном давлении. Остаток хроматографировали (2 х 8 см), элюируя метанолом-метиленхлоридом (5:95). Целевые фракции объединяли, а затем хроматографировали на силикагеле (2 х 8 см), элюируя смесью метиленхлорида и этилацетата (1:2). Целевые фракции объединяли и концентрировали. К остатку добавляли смесь диэтилового эфира и гексана, в результате чего получали (2RS, 3RS)-2-(2,4-дифторфенил)-3- (имидазо[1,5-a]пирадин-3-ил)метилтио-1- (1Н-1,2,4-триазол-1-ил)-2-бутанол (соединение 57, 0,02 г) в виде бесцветного порошка.
1Н-ЯМР (CDCl3) δ: 1,14 (3Н, д, I 7,0 Гц), 3,34 (1Н, кв, I 7,0 Гц); 4,28 (1Н, д, I 15 Гц); 4,38 (1Н, д, I 15 Гц); 4,49 (1Н, д, I 14,4 Гц); 4,86 (1Н, д, I 14,4 Гц); 5,56 (1Н, шир.с); 6,66-6,77 (2Н, м); 7,27-7,44 (1Н, м); 7,62 (1Н, д, I 5,0 Гц); 7,74 (1Н, с); 7,77 (1H, с); 7,80 (1Н, с); 7,86 (1Н, д, I 5,0 Гц); 9,01 (1Н, д, I 1,4 Гц).
МСВИ (m/z): 417 (МН+).
П р и м е р 57. Метаноловый раствор (25 мл), содержащий (2RS, 3RS)-2-(2,4-дифторофенил)-3-метил-2-(1Н-1,2,4-триазол-1-ил) метилоксиран (1,1 г), метил 3-меркаптопропионат (2,5 мл), 28%-ный метоксид натрия-метанола (2,4 мл), нагревали с обратным холодильником в течение 3 ч. После добавления 28% -ного метоксида натрия-метанола (1,2 мл) смесь нагревали с обратным холодильником в течение 2 ч. После охлаждения реакционную смесь разбавляли водой (25 мл), нейтрализовали 5%-ным водным раствором фосфорной кислоты и экстрагировали метиленхлоридом (25 мл х 3). Экстракт высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (4 х 50 см), элюируя смесью этилацетата и гексана (3: 1). Целевую фракцию концентрировали, к остатку добавляли эфир и получали соединение 60 (0,5 г) в виде бесцветных игольчатых кристаллов.
Полученный продукт (0,5 г) перекристаллизовывали из этилацетата (40 мл), в результате чего получали бесцветные призмообразные кристаллы (2 г) соединения 60. Т.пл. 107-109оС.
1Н-ЯМР (CDCl3) δ: 1,47 (3Н, д, I Гц); 2,11 (1Н, д, I 8,4 Гц); 3,61 (1Н, кв, I 7 Гц); 6,42 (1Н, д, I 14,2 Гц); 4,71 (1Н, д, I 14,2 Гц); 5,84 (1Н, с); 6,81-6,92 (1Н, м); 6,99-7,15 (1Н, м); 7,21-7,37 (1Н, м); 7,65 (1Н, м); 8,23 (1Н, с).
Элементный анализ для C12H13F2N3OS.
Вычислено, С 50,52; Н 4,59; N 14,73.
Найдено, С 50,31; Н 4,59; N 14,60.
П р и м е р 58. К дихлорометану (5 мл) добавляли (2RS, 3RS)-2-(2,4-дифторфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бута- нол (0,35 г), а затем добавляли, охлаждая льдом, триэтиламин (0,18 мл). Далее к смеси по каплям добавляли ацетилхлорид (0,1 мл) и полученную смесь размешивали при комнатной температуре в течение 30 мин. После этого растворитель отгоняли при пониженном давлении, а остаток хроматографировали на силикагеле (2,5 х 20 см), элюируя смесью этилацетата и гексана (2:1). Целевую фракцию концентрировали, а к остатку добавляли гексан, в результате чего получали соединение 61 (0,22 г) в виде бесцветных игольчатых кристаллов. Т.пл. 128-130оС.
1Н-ЯМР (CDCl3) δ: 1,57 (3Н, д, I 7 Гц); 2,14 (3Н, с); 4,22 (1Н, кв, I 7 Гц); 4,55 (1Н, д, I 14 Гц); 4,99 (1Н, д, I 14 Гц); 5,13 (1Н, с); 6,62-6,79 (2Н, м); 7,31-7,42 (1Н, м); 7,77 (1Н, с); 7,87 (1Н, с).
Элементный анализ для C14H15F2N3O2S.
Вычислено, С 51,37; Н 4,62; N 12,84.
Найдено, С 51,32; Н 4,61; N 12,71.
П р и м е р 59. В дихлорометане (5 мл) растворяли (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бута- нол (143 мг), а затем охлаждая льдом, добавляли триэтиламин (0,076 мл) и изобутурилхлорид (58,6 мг). Смесь перемешивали при комнатной температуре в течение 15 мин, после чего растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1: 2). Целевую фракцию концентрировали и получали (2R, 3R)-2-(2,4-дифторофенил)-3-изобутурилтио-1-(1Н-1,2,4-триазол-1-ил)-2-бута- нол (соединение 62, 140 мг) в виде бесцветного сиропа. Этот продукт обрабатывали 4н, соляной кислотой в этилацетате, в результате чего получали гидрохлорид соединения 62 (156 мг) в виде бесцветного порошка. Т.пл. 129-138оС.
1Н-ЯМР (CDCl3) δ: 1,03 (3Н, д, I 7,0 Гц); 1,18 (6Н, дд, I 6,8 Гц, I 4,0 Гц); 2,84 (1Н, м); 4,33 (1Н, кв, I 7,0 Гц), 4,67 (2Н, с); 6,92 (1Н, м); 7,08-7,30 (2Н, м), 7,76 (1Н, с); 8,42 (1Н, с).
П р и м е р 60. Метил-3-меркаптопропионата (0,88 мл) добавляли к N,N-диметилформамиду (14 мл), содержащую 60%-ный гидрид натрия в масле (0,32 г), охлаждая при этом льдом. Затем через 5 мин в течение 5 мин, охлаждая льдом, добавляли раствор 2-(2,4-дифторофенил)-2-[1-(1Н-1,2,4-триазол-1-ил)этил] оксиран (0,67 г) в N,N-диметилформамиде (3,5 мл). Через 15 мин реакционную смесь выливали в воду (150 мл), нейтрализовали соляной кислотой и экстрагировали этилацетатом (50 мл х 3). Органические слои объединяли, промывали насыщенным водным раствором хлорида натрия (30 мл х 2), высушивали сульфатом натрия и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (3 х 10 см), элюируя смесью этилацетата и гексана (1:1). Целевую фракцию концентрировали и к остатку добавляли диэтиловый эфир, в результате чего получали неочищенные кристаллы (0,25 г) 2-(2,4-дифторофенил)-1-меркап- то-3-[(1Н-1,2,4-триазол-1-ил)-2-бутанола] низкой полярности, соединение 116, диастереомер В. Кристаллы перекристаллизовывали из смеси хлороформа и диэтилового эфира и получали 0,21 г соединения 116. Маточный раствор полученных неочищенных кристаллов хроматографировали на силикагеле (2 х 12 см), элюируя смесью этилацетата и гексана (1:1). Целевую фракцию концентрировали и к остатку добавляли диэтиловый эфир и гексан, в результате чего получали сырые кристаллы (0,25 г) 2-(2,4-дифторофенил)-1-меркапто-3-[(1Н-1,2,4-триазол-1-ил)-2-бутанола] высокой полярности; соединение 115, диастереомер А. Эти кристаллы перекристаллизовывали из смеси хлороформа и диэтилового эфира и получали кристаллы соединения 115 (0,611 г).
Соединение 115 (диастереомер А).
Т.пл. 112-117оС, бесцветные призмообразные кристаллы.
1Н-ЯМР (CDCl3) δ: 1,19 (1Н, т, I 8,2 Гц; 1,69 (3Н, д, I 7,0 Гц); 3,05 (1Н, д, I 8,2 Гц, д, I 14,0 Гц); 3,31 (1Н, д, I 8,2 Гц, д, I 14 Гц); 4,55 (1Н, с); 5,06 (1Н, кв, I 7,0 Гц); 6,68-6,79 (2Н, м); 7,23-7,35 (1Н, м); 7,73 (1Н, с); 7,88 (1Н, с).
Соединение 116 (диастереомер В).
Т.пл. 183-184оС.
1Н-ЯМР (CDCl3) δ: 0,93 (1Н, д, I 6,8 Гц, д, I 10,2 Гц); 1,36 (3Н, д, I 7,0 Гц); 2,18 (1Н, д, I 14 Гц, д, I 10,2 Гц); 3,28 (1Н, д, I 6,8 Гц, д, I 14 Гц); 4,19 (1Н, с); 5,09 (1Н, кв, I 7,0 Гц); 6,80-7,04 (2Н, м); 7,67-7,79 (1Н, м); 7,99 (1Н, с); 8,29 (1Н, с).
П р и м е р 61. К этанолу (25 мл) добавляли (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (0,9 г) и 4-хлорометил-2-метилтиазол гидрохлорид (0,7 г), а затем, постоянно перемешивая при комнатной температуре, добавляли 28% -ную смесь этоксида натрия и этанола (1,3 мл). После этого смесь размешивали при комнатной температуре в течение 30 мин, а затем добавляли этилацетат (100 мл) и воду (100 мл). Слой этилацетата отделяли, а водный слой экстрагировали этилацетатом (50 мл). Слои этилацетата объединяли, промывали водой (50 мл), высушивали (MgSO4) и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (2,9 х 100 см), элюируя смесью этилацетата и ацетона (4:1). Целевую фракцию концентрировали и получали соединение 107 (0,8 г) в виде бесцветного маслянистого вещества.
1Н-ЯМР (CDCl3) δ: 1,24 (3Н, д, I 7,2 Гц); 2,79 (3Н, с); 3,49 (1Н, кв, I 7,2 Гц); 3,82 (1Н, д, I 14,6 Гц); 4,06 (1Н, д, I 14,6 Гц); 4,68 (1Н, д, I 14,6 Гц); 5,08 (1Н, д, I 14,6 Гц); 6,25 (1Н, с); 6,69-6,78 (2Н, м); 6,93 (1Н, с); 7,36-7,50 (1Н, м); 7,72 (1Н, с); 7,83 (1Н, с).
Этот продукт (0,8 г) обрабатывали 4 н. HCl-этилацетатом и получали гидрохлорид соединения в виде бесцветных кристаллов (0,64 г). Т.пл. 165-167оС.
[α]D23 83,1о (с 1,0, метанол).
Элементный анализ для C17H18F2N4OS2 х х 2HCl ˙ 1/2 H2O.
Вычислено, С 42,68; H 4,42; N 11,71.
Найдено, С 42,64; Н 4,37; N 11,59.
П р и м е р 62. В этаноле (7,5 мл) растворяли (2R, 3R)-2-(2,4-дифторoфенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (0,74 г), а затем, примешивая при комнатной температуре, добавляли 28%-ный метоксид натрия-метанол (1,06 мл) и 2-хлорoметил-1-цикло- пропилимидазол гидрохлорид (0,5 г). Смесь перемешивали при комнатной температуре в течение 10 мин, затем разбавляли водой (20 мл) и экстрагировали этиловым эфиром (20 мл х 3). Экстракт промывали насыщенным водным раствором хлорида натрия, высушивали сульфатом магния и концентрировали при пониженном давлении.
Остаток очищали с помощью хроматографирования на силикагеле, элюируя смесью: этилацетат этилацетат-метанол= 95:5 и кристаллизовали из этилового эфира, в результате чего получали соединение 110 (0,82 г) в виде бесцветных призмообразных кристаллов.
[α]D23 119,5о (с 1,0, метанол).
Т.пл. 127-128оС.
Элементный анализ для C19H21F2N5O1S1.
Вычислено, С 56,28; Н 5,22; N 17,27.
Найдено, С 56,34; Н 5,26; N 17,14.
1Н-ЯМР (CDCl3) δ: 0,95-1,17 (4Н, м); 1,26 (3Н, д, I 7,0 Гц); 3,20-3,33 (1Н, м); 3,60 (1Н, кв, I 7,0 Гц); 3,94 (1Н, д, I 15,4 Гц); 4,03 (1Н, д, I 15,4 Гц); 4,62 (1Н, д, I 14,2 Гц); 4,88 (1Н, д, I 14,2 Гц); 6,67-6,80 (2Н, м); 6,87 (1Н, с); 6,93 (1Н, с); 7,40-7,53 (1Н, м); 7,65 (1Н, с); 7,68 (1Н, с); 8,03 (1Н, с).
П р и м е р 63. В N,N-диметилформамиде (30 мл) растворяли (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутан (1 г) и 4-бромометил-5-метил-1,3-диоксол-2-он (0,81 г), а затем, перемешивая при комнатной температуре, добавляли карбонат калия (3 г). После этого смесь размешивали в течение 7 мин, а затем добавляли воду (100 мл) и экстрагировали этилацетатом (50 мл х 3). Органические слои объединяли, промывали насыщенным водным раствором хлорида (30 мл х 2), высушивали сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1 ->> 2:1), и полученное маслянистое вещество кристаллизовали из смеси хлороформа и этилацетата, в результате чего получали соединение 111 (1 г) в виде бесцветных игольчатых кристаллов. Т.пл. 133-134оС.
[α]D23 77г,8о (с 1,0, метанол).
Элементный анализ для C17H17F2N3O4S1.
Вычислено, С 51,38; Н 4,31; N 10,57.
Найдено, С 51,20; Н 4,35; N 10,52.
1Н-ЯМР (CDCl3) δ: 1,20 (3Н, д, I 7,2 Гц); 2,15 (3Н, с); 3,32 (1Н, кв, I 7,2 Гц); 3,67 (1Н, д, I 15,2 Гц); 3,77 (1Н, д, I 15,2 Гц); 4,83 (1Н, д, I 14,2 Гц); 5,04 (1Н, д, I 14,2 Гц); 5,11 (1Н, д, I 1,4 Гц); 6,68-6,79 (2Н, м); 7,30-7,42 (1Н, м); 7,79 (1Н, с); 7,80 (1Н, с).
П р и м е р 64. (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (0,25 г) растворяли в этаноле (2,5 мл), а затем, перемешивая при комнатной температуре, добавляли 28%-ный метоксид натрия-метанол (0,36 мл) и 3-хлорометил-4-циклопропил-4Н-1,2,4-триазол гидрохлорид (0,17 г). Смесь размешивали в течение 10 мин, а затем разбавляли насыщенным водным раствором хлорида натрия (3 мл) и экстрагировали этилацетатом (10 мл х 3). Органические слои объединяли и высушивали сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на колонках с силикагелем, элюируя смесью метиленхлорида и метанола (98: 2 ->> 1:9), в результате чего получали соединение 114 (0,29 г) в виде бесцветного маслянистого продукта.
1Н-ЯМР (CDCl3) δ: 1,01-1,1 (4Н, м); 1,18 (3Н, д, I 6,8 Гц); 3,28-3,40 (1Н, м); 3,57 (1Н, кв, I 6,8 Гц); 4,04 (1Н, д, I 15 Гц); 4,16 (1Н, д, I 15 Гц); 4,64 (1Н, д, I 14,2 Гц); 4,84 (1Н, д, I 14,2 Гц); 5,58 (1Н, с); 6,66-6,80 (2Н, м); 7,30-7,43 (1Н, м); 7,73 (1Н, с); 7,86 (1Н, с); 8,12 (1Н, с).
Полученный продукт растворяли в этилацетате, а затем добавляли смесь хлороводорода и этилацетата. Осадок кристаллизовали из смеси этанола и этилацетата и получали дигидрохлорид соединения 114 (0,31 г) в виде бесцветного порошка.
[α]D23 75,6о (с 1,0, метанол).
П р и м е р ы 65-75. Способом, аналогичным описанному в примере 64, проводили реакцию взаимодействия (2R, 3R)-2-(2,4- дифторофенил)-3-меркапто-1-(1Н-1,2,4-три- азол-1-ил)-2-бутанола, обозначенного в табл.16 2R, 3R-метилтиол, производн. с хлоросоединениями, приведенными в табл.16, в результате чего получали соединения 78, 97, 99, 108, 112, 121-125.
П р и м е р 76. В метаноле (25 мл) растворяли (2S, 3S)-2-(2,4-дифторофенил)-3-метил-2-[(1Н)-1,2,4-триазол-1-ил)метил] окси- ран (1 г), метил-3-меркаптопропионат (3,5 мл) и 29%-ный метоксид натрия-метанол (3,07 г). Полученный раствор нагревали с обратным холодильником в масляной бане. Через 2 и 3 ч добавляли метил-3-метилпропионат (1,75 мл) и 28%-ный метоксид натрия-метанол (1,5 г). После нагревания в течение 4 ч масляную баню удаляли, а реакционную смесь нейтрализовали холодной 1н. соляной кислотой (32 мл) и экстрагировали дихлорэтаном (200 мл). Экстракт промывали насыщенным водным раствором хлорида натрия, высушивали безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:3). Целевую фракцию концентрировали и полученные кристаллы перекристаллизовывали из смеси этилацетата и гексана, в результате чего получали (2S, 3S)-2-(2,4-дифторo- фенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (соединение 117, 472 мг) в виде бесцветных игольчатых кристаллов. Т. пл. 141-144оС.
[α]D25 + 61,1о (с 1,0, в метаноле).
Элементный анализ для C12H13F2N3OS.
Вычислено, С 50,52; Н 4,59; N 14,73.
Найдено, С 50,51; Н 4,59; N 14,49.
1H-ЯМР (CDCl3) δ: 1,52 (3Н, д, I 7 Гц); 1,54 (1Н, д, I 6 Гц); 3,69 (1Н, м); 4,56 (1Н, с); 4,62 (1Н, д, I 14 Гц); 4,94 (1Н, дд, I 14 Гц, I 1,8 Гц); 6,68-6,81 (2Н, м); 7,30-7,43 (1Н, м); 7,73 (1Н, с); 7,95 (1Н, с).
ИК (KBr), см-1: 3260, 1615, 1500, 1420, 1260, 1200, 1125.
П р и м е р 77. В трифтороуксусной кислоте (75 мл) растворяли [1-(1,2,4-дифторофенил)-1-гидрокси-2-(1Н-1,2,4-триазол-1-ил)-этил-1-(4-меток сибециклопропан (4,26 г), анизол (26 мл) и ацетат ртути [II] (3,58 г). Полученный раствор размешивали при комнатнойе температуре в течение 2 ч. Затем реакционную смесь концентрировали при пониженном давлении, а остаток разбавляли петролейным эфиром (50 мл), а надосадочную жидкость удаляли. К остатку добавляли эфир (30 мл), в результате чего получали ртутное соединение в виде бесцветного порошка. Порошок отбирали фильтрацией и промывали небольшим количеством (10 мл) эфира. Полученный продукт (6,2 г) суспендировали в дихлорметане (150 мл) и в суспензию при комнатной температуре барботировали сероводород в течение 30 мин. Полученный осадок отфильтровывали и фильтрат концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (2,5 х 40 мл), элюируя смесью этилацетата и гексана (3:1). Целевую фракцию концентрировали и остаток обрабатывали изопропиловым эфиром, в результате чего получали соединение 120 (2 г) в виде бесцветных игольчатых кристаллов. Т.пл. 92-94оС.
1Н-ЯМР (CDCl3) δ: 0,72-1,28 (4Н, м); 2,26 (1Н, с); 4,80 (1Н, дд, I 1,6 Гц, 14 Гц); 5,19 (1Н, шир.с); 5,31 (1Н, дд, I 1,6, 14 Гц); 6,63-6,88 (2Н, м); 7,54-7,66 (1Н, м); 7,81 (1Н, с); 8,14 (1Н, дд, I 1,6 Гц).
Элементный анализ для C13H13F2N3OS.
Вычислено, С 52,52; Н 4,41; N 14,13.
Найдено, С 52,41; Н 4,45; N 13,97.
П р и м е р 78. В метаноле (25 мл) растворяли (2R, 3R)-2-(2,4-дифторoфенил)-3-метил-2-[(1Н-1,2,4-триазол-1-ил)-метил] окси- ран (0,98 г), метил-3-меркаптопропионат (3,3 мл) и 28%-ный метоксид натрия-метанол (3 г). Полученный раствор нагревали с обратным холодильником в масляной бане. Через 2 а затем через 3 ч добавляли соответственно метил-3-метилпропионат (0,83 мл) и 28%-ный метоксид натрия-метанол (0,75 г). После нагревания в течение 4 ч масляную баню удаляли, а реакционную смесь нейтрализовали холодной 1н. HCl (23 мл) и экстрагировали дихлорметаном (200 мл). Экстракт промывали насыщенным водным раствором хлорида натрия и высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:3). Целевую фракцию концентрировали и полученные кристаллы перекристаллизовывали из этилацетата и гексана, в результате чего получали (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанол (соединение 118, 461 мг) в виде бесцветных игольчатых кристаллов. Т, пл. 141-143оС.
[α]D25 63,4о (с 1,0, метанол).
Элементный анализ C12H13F2N3OS.
Вычислено, С 50,52; Н 4,59; N 14,73.
Найдено, С 50,51; Н 4,68; N 14,53.
1Н-ЯМР (CDCl3) δ: 1,52 (3Н, д, I 7,0 Гц); 1,54 (1Н, д, I 6 Гц); 3,69 (1Н, м); 4,55 (1Н, с); 4,62 (1Н, д, I 14 Гц); 4,93 (1Н, дд, I 14 Гц, I 1,8 Гц); 6,68-6,82 (2Н, м); 7,29-7,45 (1Н, м); 7,72 (1Н, с); 7,95 (1Н, с).
ИК (KBr), см-1: 3260, 1615, 1500, 1420, 1200, 1120.
П р и м е р 79. В трифтороуксусной кислоте (20 мл) растворяли 3-(4-метоксибензилтио)-2-(2,4-дифторофенил)-3-метил- 1- (1Н-1,2,4-триазол -1-ил)-2-бутанол (1,23 г), анизол (7,5 мл) и ацетат ртути [II](1,03 г). Полученный раствор размешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь концентрировали при пониженном давлении, остаток разбавляли петролейным эфиром (20 мл), а слой надосадочной жидкости удаляли. К остатку добавляли эфир (20 мл). Полученный бесцветный порошок собирали фильтрацией, промывали небольшим количеством (5 мл) эфира и получали ртутное соединение (1,6 г). Этот продукт (1,6 г) суспендировали в дихлорометане (50 мл) и в суспензию, при комнатной температуре в течение 20 мин барботировали хлороводород. Затем осадок отфильтровывали, в фильтрат в течение 15 мин барботировали газ N2, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (2,5 х 20 см), элюируя смесью этилацетата и гексана (3:1). Целевую фракцию концентрировали, к остатку добавляли изопропиловый эфир, в результате чего получали (соединение 119, 0,6 г) в виде бесцветных игольчатых кристаллов. Т.пл. 95-96оС.
1Н-ЯМР (CDCl3) δ: 1,39 (3Н, с); 1,42-1,48 (3Н, м); 2,39 (1Н, с); 4,93 (1Н, д,д, I 2,6 Гц, I 14 Гц); 5,32 (1Н, дд, I 2,6 Гц, 14 Гц); 5,44 (1Н, с); 6,59-6,85 (2Н, м); 7,62-7,71 (1Н, м); 7,75 (1Н, с); 8,08 (1Н, д, I 2,6 Гц).
Элементный анализ C13H15F2N3OS.
Вычислено, С 52,16; Н 5,05; N 14,04.
Найдено, С 52,13; Н 5,10; N 14,03.
П р и м е р ы 80-85. Способом, аналогичным описанному в примере 64, проводили реакцию взаимодействия (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2- бутанола (в табл.17 обозначен 2R, 3R-метилтиоловым производным) с хлоросоединениями, приведенными в табл.17, в результате чего получали соединения 90, 91, 126-129.
П р и м е р 86. В этилацетате (20 мл) растворяли (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бу- танол (соединение 43, 0,2 г). Полученный раствор концентрировали до объема 10 мл при пониженном давлении. Затем концентрат оставляли при комнатной температуре, в результате чего получали соединение 43 в виде бесцветных призмообразных кристаллов. Анализ с помощью рентгеновской кристаллографии показал следующие результаты: Кристаллографические данные Формула C12H13N3OF2 Формульная масса 285,31 Кристаллографическая система Орторомбическая Размеры ячейки а 10,754 (1) 
b 13,771 (2) 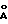
с 9,069 (1) 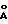 Объем ячейки 1343,2 (3)
Объем ячейки 1343,2 (3)  Пространственная группа Р212121 Число формульных единиц в элементарной ячейке 4 Вычисленная плотность 1,411 г/см3 Излучение Мо-Ка (λ=0,71069
Пространственная группа Р212121 Число формульных единиц в элементарной ячейке 4 Вычисленная плотность 1,411 г/см3 Излучение Мо-Ка (λ=0,71069  ) Конечная Р-величина 0,041
) Конечная Р-величина 0,041
П р и м е р 87. В этилацетате (10 мл) растворяли (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бута- нол (соединение 43, 0,2 г), а затем к полученному раствору добавляли 4н. HCl-этилацетат (1 мл). Смесь концентрировали при пониженном давлении до получения объема около 1 мл, в результате чего получали кристаллы. Затем добавляли этиловый эфир (2 мл), смесь фильтровали и получили гидрохлорид соединения 43 (0,15 г) в виде игольчатых кристаллов. Т.пл. 155-162оС.
Элементный анализ C12H13F2N3OS ˙ HCl.
Вычислено, С 44,79; Н 4,39; N 13,06.
Найдено, С 44,77; Н 4,48; N 12,85.
На фиг.1 представлена порошковая рентгенограмма.
П р и м е р 88. В этилацетате (20 мл) растворяли (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бута- нол (соединение 43, 0,48 г), а затем к полученному раствору добавляли 25%-ную смесь бромоводорода и уксусной кислоты (0,7 мл). После этого смесь разбавляли 100 мл гексана и полученный порошок собирали путем фильтрации, а затем растворяли в этилацетате (10 мл). После добавления этилового эфира (20 мл) раствор выдерживали и полученные кристаллы собирали путем фильтрации (0,35 г, гидробромид соединения 43) в виде пластинчатых кристаллов. Т.пл. 180-175оС.
Элементный анализ С12H13F2N3OS ˙ HBr.
Вычислено, С 39,36; Н 3,85; N 11,47.
Найдено, С 39,28; Н 3,85; N 11,32.
На фиг.2 представлена порошковая рентгенограмма соединения.
П р и м е р ы 89-93. Способом, аналогичным описанному в примере 64, проводили реакцию взаимодействия (2R, 3R)-2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол-1-ил)-2-бутанола, в табл.18 обозначенного 2R, 3R-метилтиоловым производным, с хлоросоединениями, приведенными в табл.18, в результате чего получали соединения 95, 101, 102, 130, 131.
П р и м е р 94. К раствору 2-(2,4-дифторофенил)-2-(1Н-1,2,4-триазол-1-ил-метил) оксирана (8 г) и метилового сложного эфира 3-меркаптопропионовой кислоты (11,2 мл) в диметилформамиде (160 мл) добавляли 60%-ную суспензию гидрида натрия в масле (4 г). Полученную смесь размешивали в течение 15 мин. Затем для доведения рН до 7 по каплям добавляли 1 н. водный раствор соляной кислоты (101 мл), а диметилформамид и воду выпаривали при пониженном давлении. К остатку добавляли 20 мл воды и экстрагировали этилацетатом (50 мл х 3). Экстракт промывали насыщенным водным раствором хлорида натрия, высушивали безводным сульфатом натрия, а затем выпаривали при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (6,0 х 9,0 см), элюируя смесью этилацетата и гексана (3:1). Целевую фракцию концентрировали и к остатку добавляли диэтиловый эфир. В результате чего получали 2-(2,4-дифторофенил)-3-меркапто-1-(1Н-1,2,4-триазол- 1-ил)пропан-2-ол (6,44 г) в виде бесцветных игольчатых кристаллов. Т.пл. 112-113оС.
Элементный анализ C11H11F2N3OS.
Вычислено, С 48,70; Р 4,09; N 15,49.
Найдено, С 48,96; Н 4,11; N 15,62.
П р и м е р 95. Раствор метанола (75 мл), содержащий (2R, 3S)-2-(2,4-дифторофенил)-2-(1-имидазолил)-метил-3-метилоксиран (2,5 г), метил-3-меркаптопропионат (5,5 мл) и 28%-ный раствор метоксида натрия и метанола (8,1 мл) нагревали с обратным холодильником в течение 1,5 ч. Затем к раствору добавляли 1-меркаптопропионат (5,5 мл) и 28%-ный метоксид натрия-метанол (8,1 мл) и полученную смесь нагревали с обратным холодильником еще 2 ч. Затем реакционную смесь охлаждали льдом, нейтрализовали 5н. соляной кислотой (16 мл), разбавляли насыщенным водным раствором хлорида натрия (100 мл) и экстрагировали этилацетатом (200 мл х 3). Экстракт высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток растворяли в метиленхлориде (40 мл), а затем к нему добавляли 1н. водный раствор гидроокиси натрия (8 мл) и воду (100 мл). Далее водный слой экстрагировали метиленхлоридом (40 мл х 3). Слои метиленхлорида объединяли и экстрагировали 5 раз водой (30 мл), содержащей 1н, соляную кислоту (8 мл). Водные слои объединяли, нейтрализовали гидроокисью натрия и экстрагировали метиленхлоридом (40 мл х 4). Экстракт высушивали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонках с силикагелем (4 см х 15 см), используя в качестве элюента смесь метанола и метиленхлорида (5:95). Целевую фракцию концентрировали и после добавления к остатку диэтилового эфира получали (2R, 3R)-2-(2,4-дифторофенил)-1-(1-имидазолил)-3-меркапто-2-бутанол (0,84 г) в виде бесцветных призмообразных кристаллов.
1Н-ЯМР (CDCl3) δ 1,12 (3Н, д, I 7,0 Гц); 1,69 (1Н, д, I 6,0 Гц); 3,68 (1Н, м); 4,45 (1Н, дд, I 1,4 Гц, 14,2 Гц); 4,59 (1Н, д, I 14,2 Гц); 6,57 (1Н, с); 6,71 (1Н, с); 6,7-6,85 (1Н, м); 7,28 (1Н, с); 7,3-7,5 (1Н, м).
Т.пл. 125-135оС.
Элементный анализ C13H14F2N2OS.
Вычислено, С 54,92; Н 4,96; N 9,85.
Найдено, С 54,94; Н 5,10; N 9,62.
ИК (КВr), см-1: 3000, 1610, 1590, 1500, 1420, 1260, 1200, 1130.
Полученное соединение растворяли в диэтиловом эфире и добавляли к нему смесь соляной кислоты и диэтилового эфира. Полученный порошок перекристаллизовывали из смеси этанола и диэтилового эфира, в результате чего получали гидрохлорид в виде призмообразных кристаллов.
1Н-ЯМР (DMCO-d6) δ:1,06 (3Н, д, I 6,8 Гц); 2,95 (1Н, д, I=9,2 Гц); 3,63 (1Н, м); 4,65 (1Н, д, I 14,6 Гц); 4,92 (1Н, д, I 14,6 Гц); 6,31 (1Н, с); 6,98 (1Н, д, I 2,8 Гц, 8,8 Гц); 7,20-7,37 (2Н, м); 7,29 (1Н, с); 7,45 (1Н, с); 8,85 (1Н, с).
Элементный анализ C13H15F2N2OS ˙ ˙ HCl ˙ 1/2H2O.
Вычислено, С 47,34; Н 4,89; N 8,49.
Найдено, С 46,74; Н 4,43; N 8,44.
ИК (КВr), см-1: 3270, 3000, 1600, 1490, 1410, 1260, 1120.
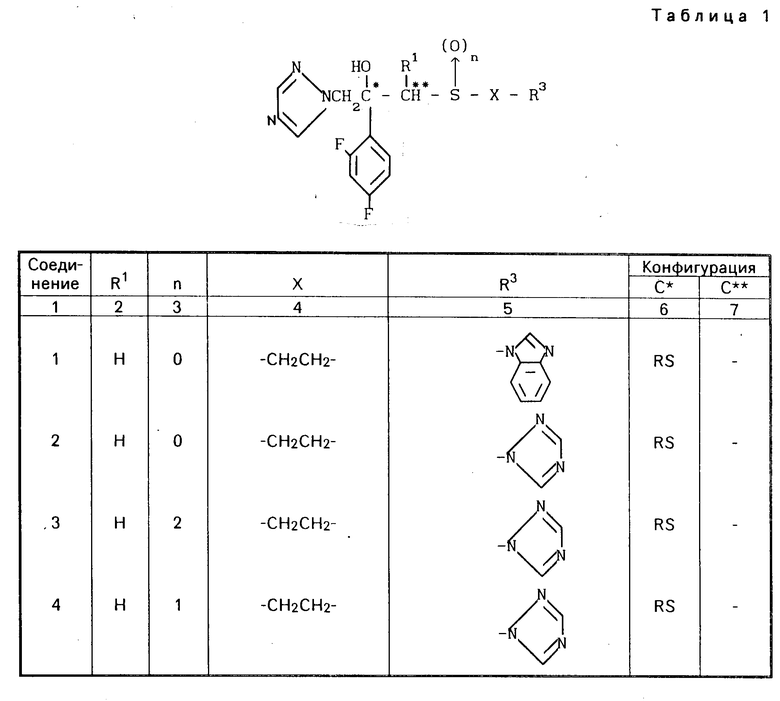
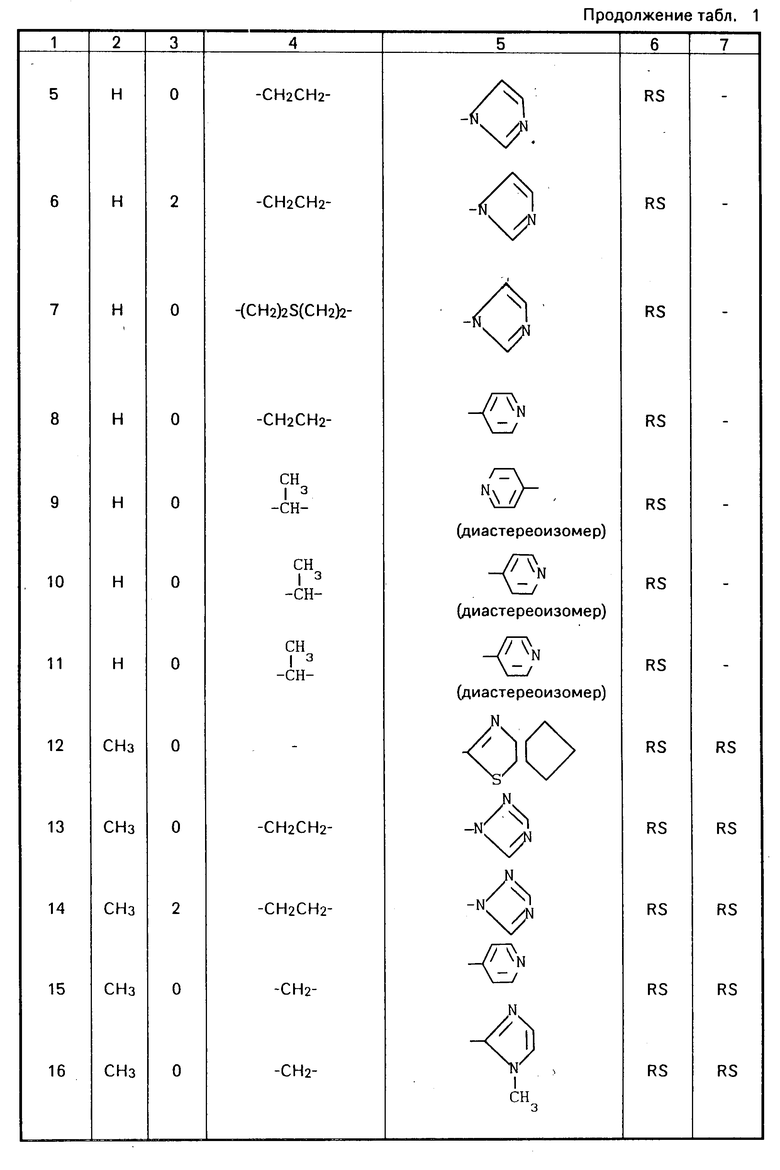
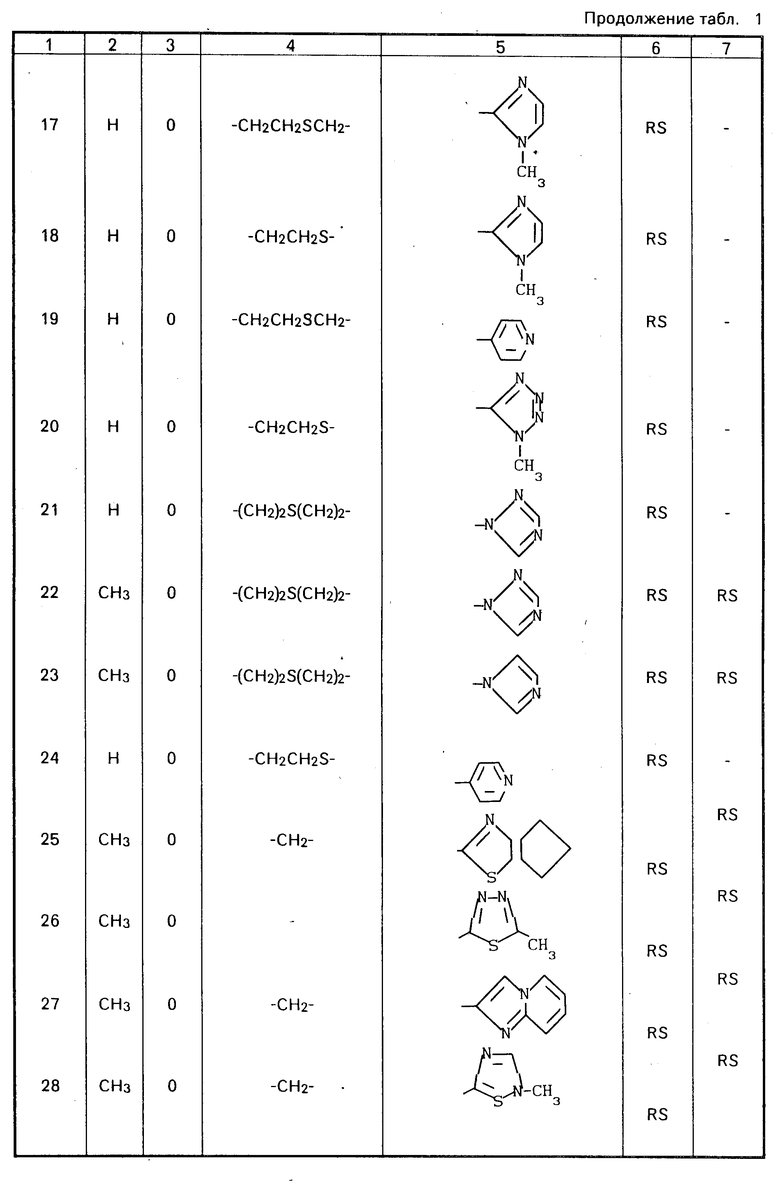

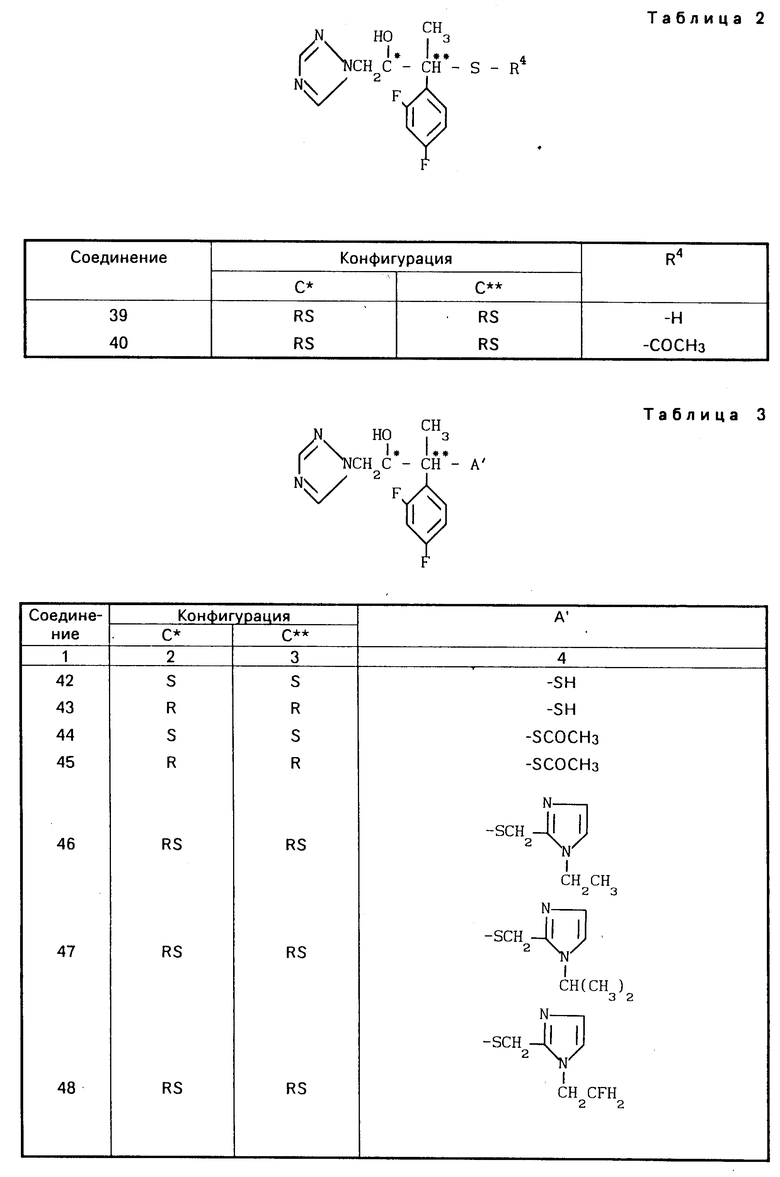
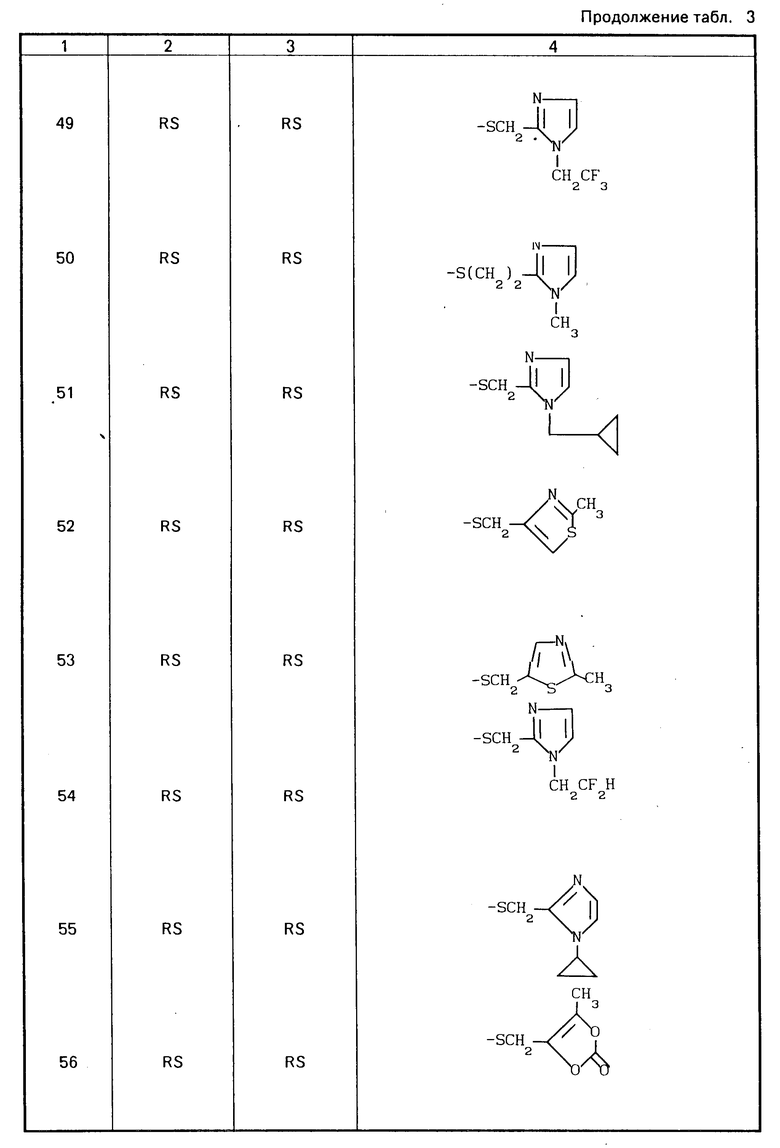
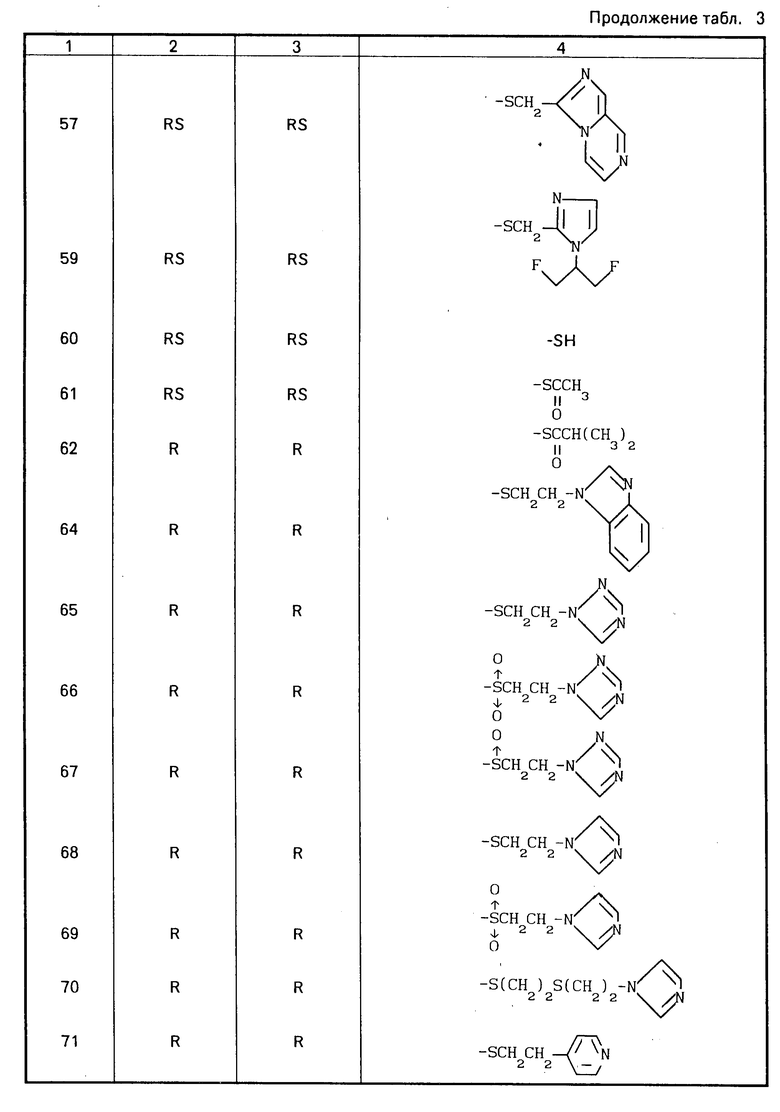
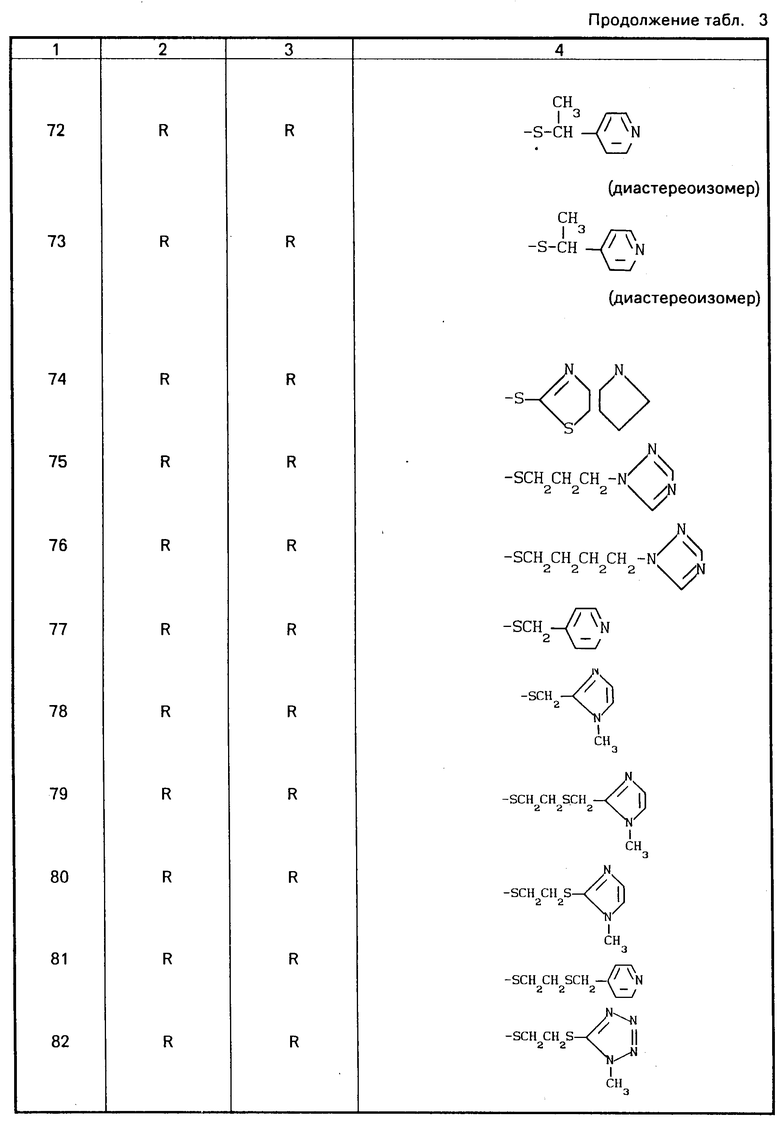
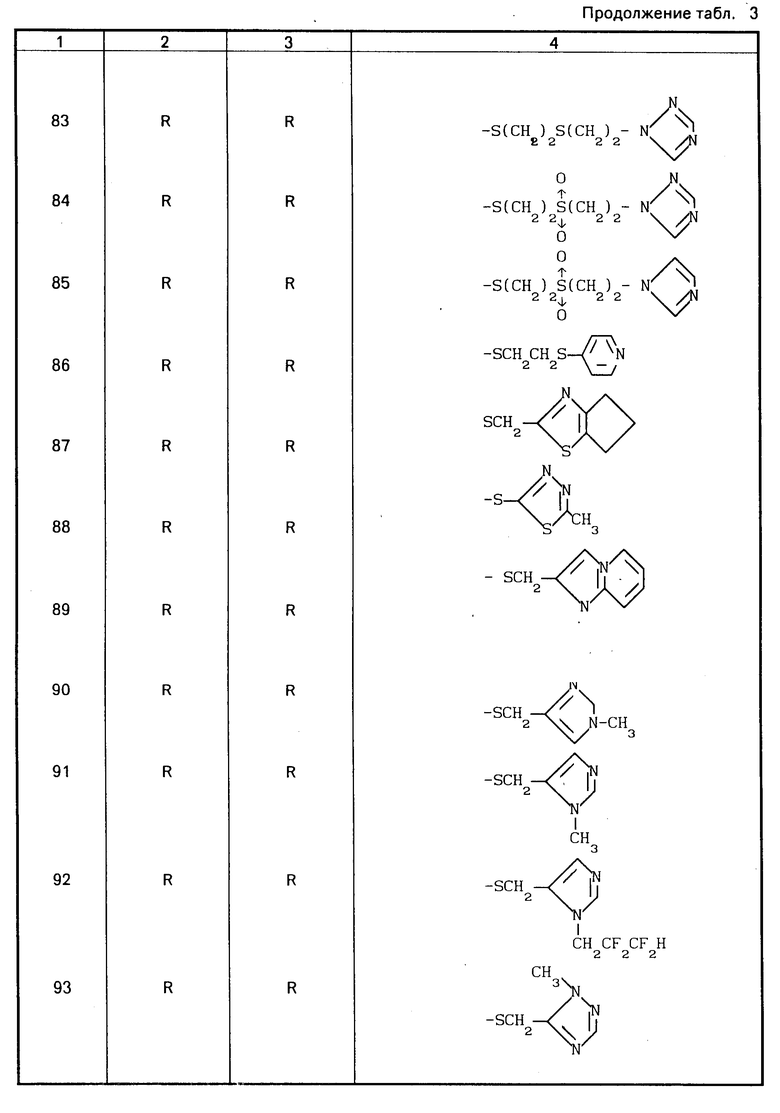
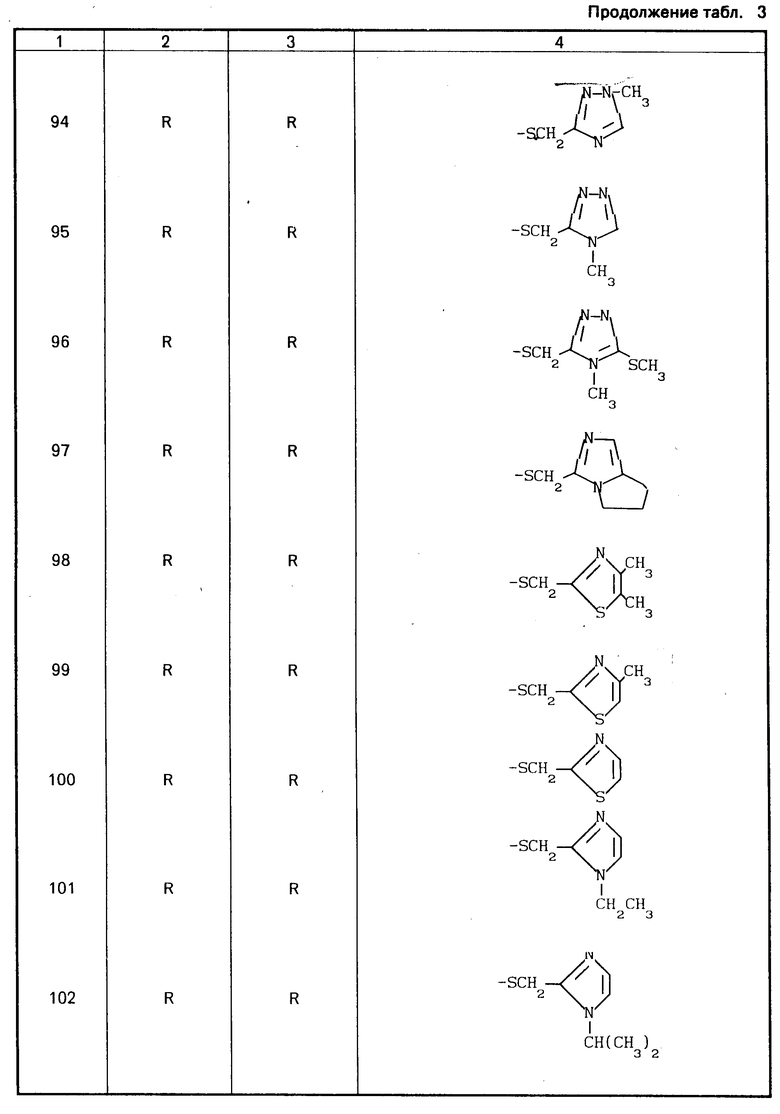
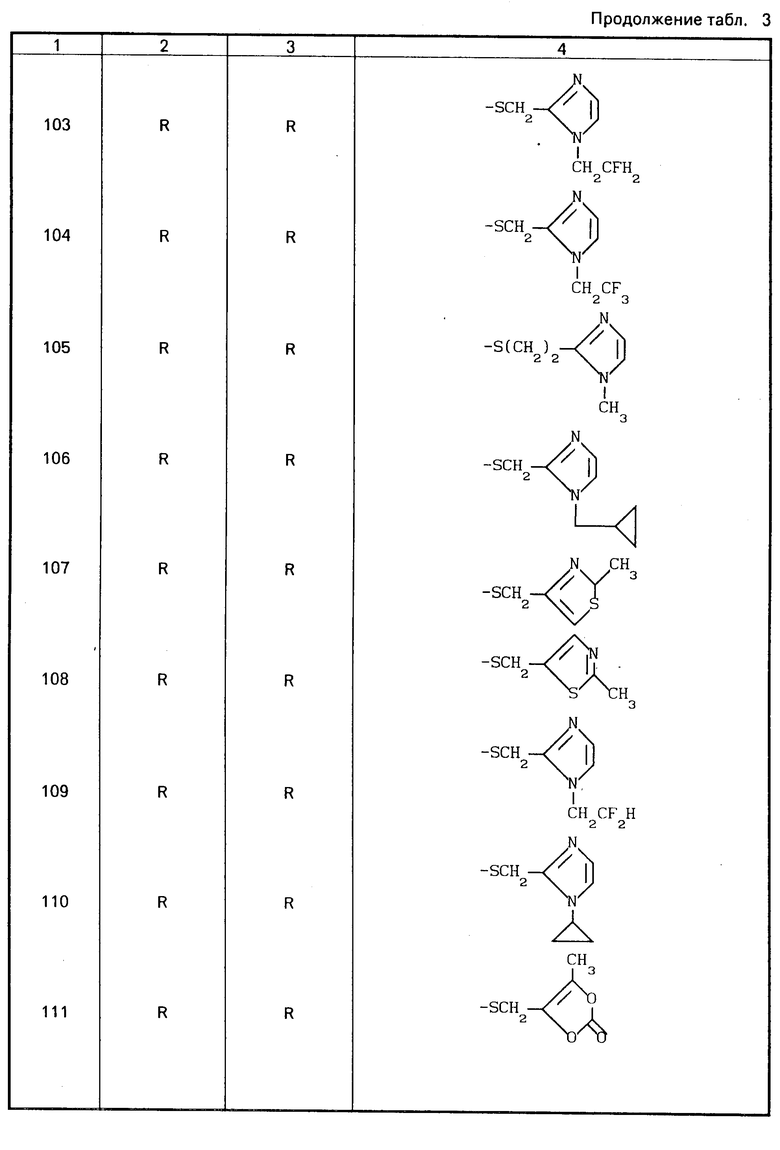
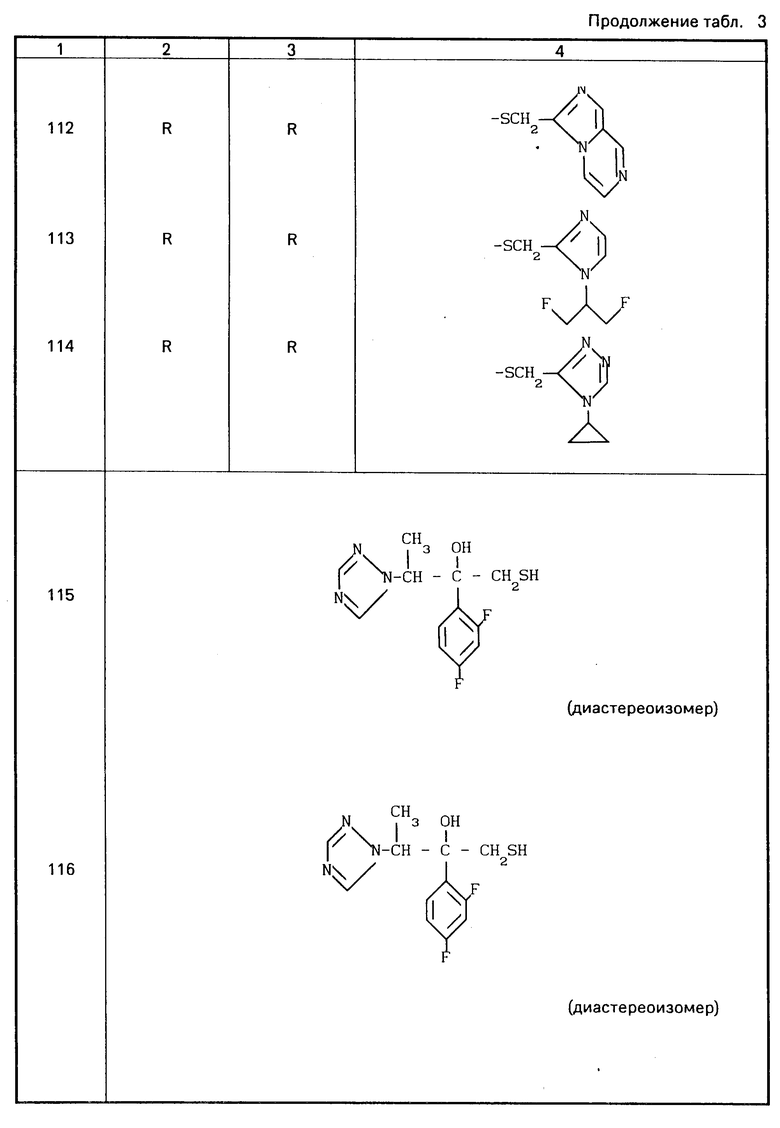
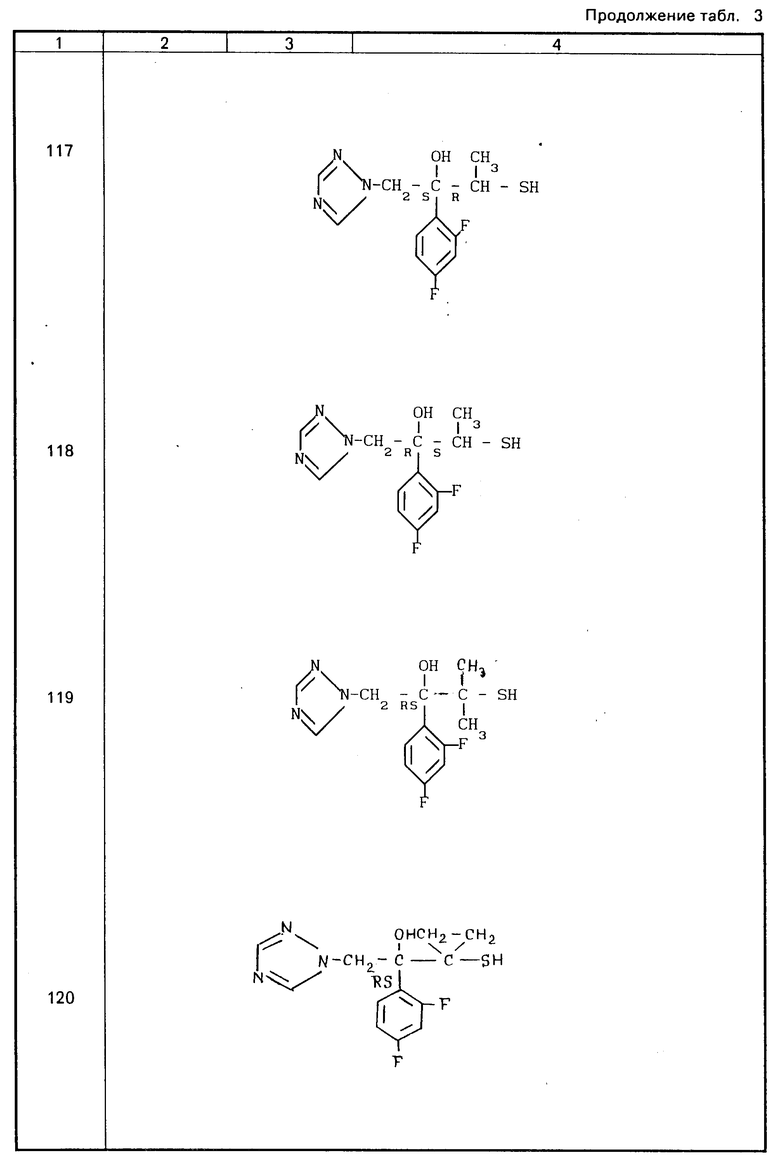

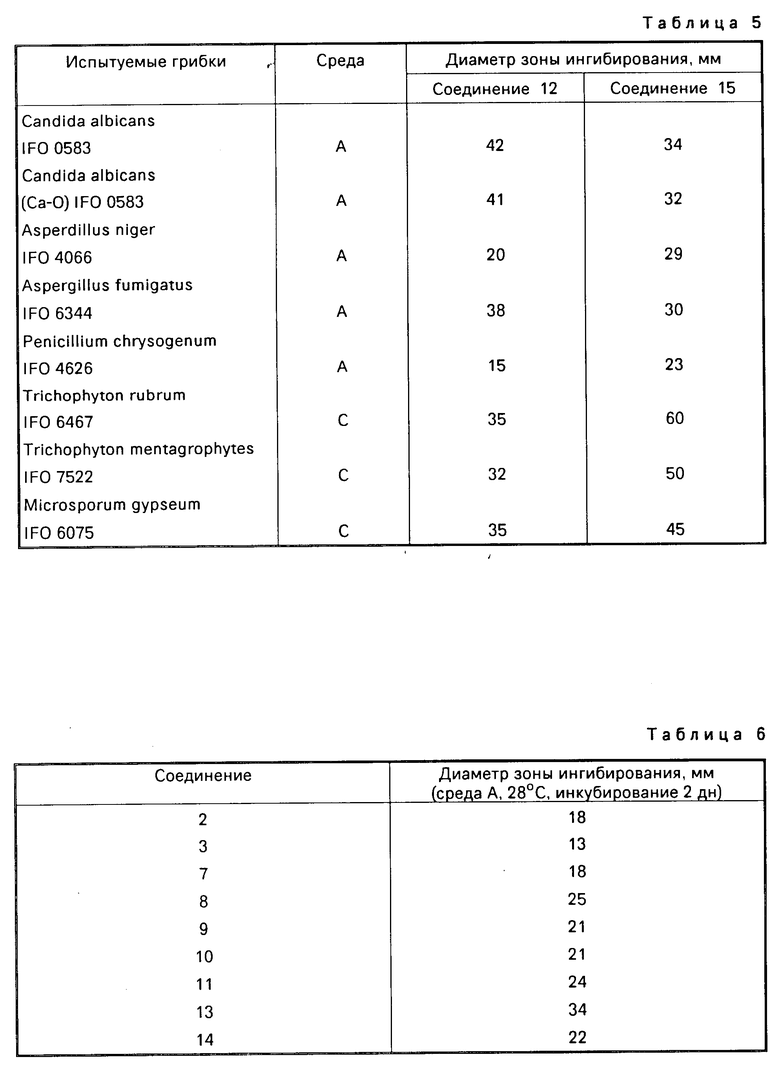






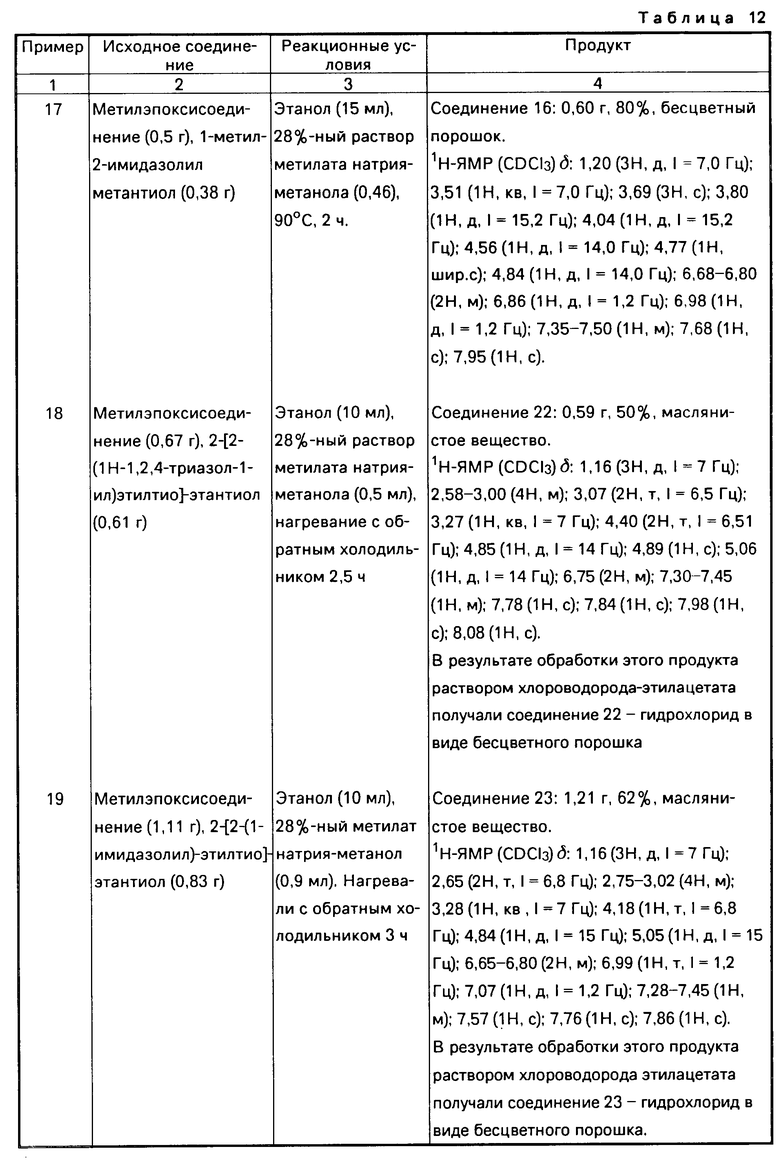
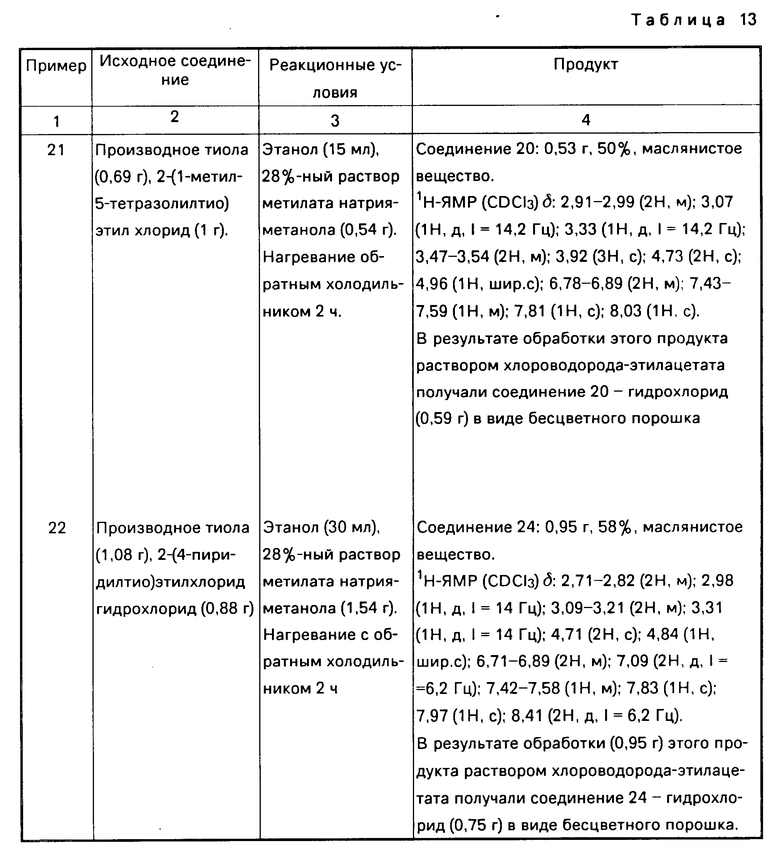

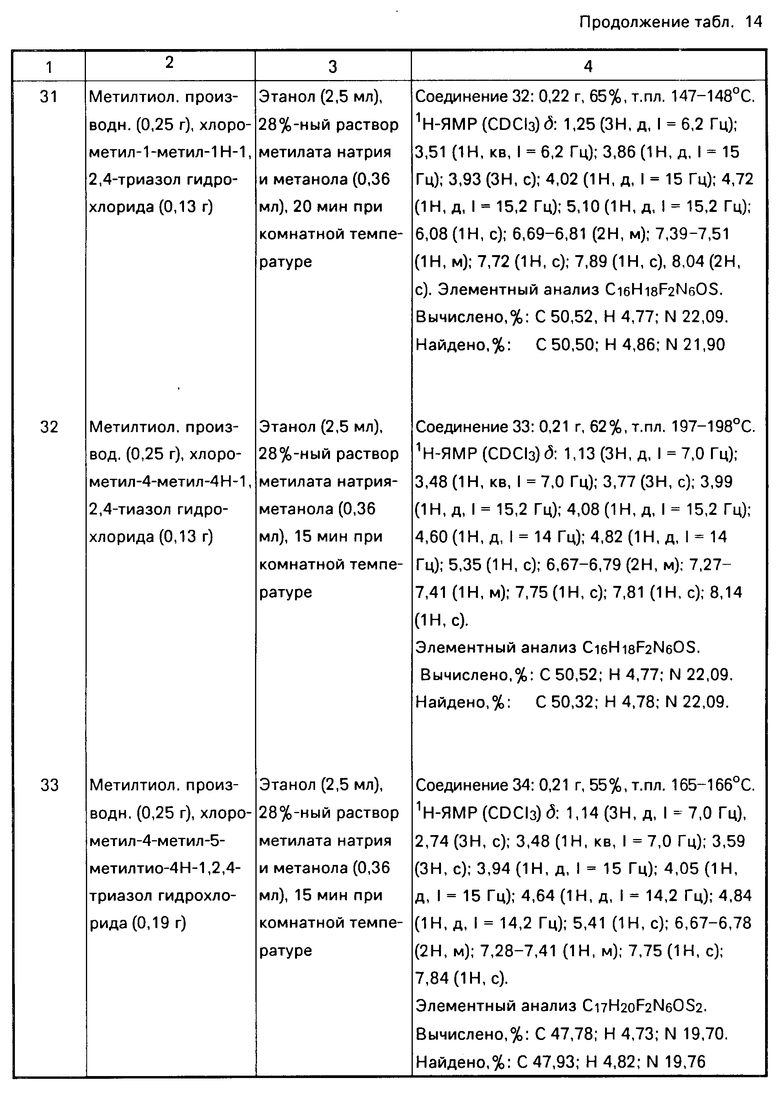

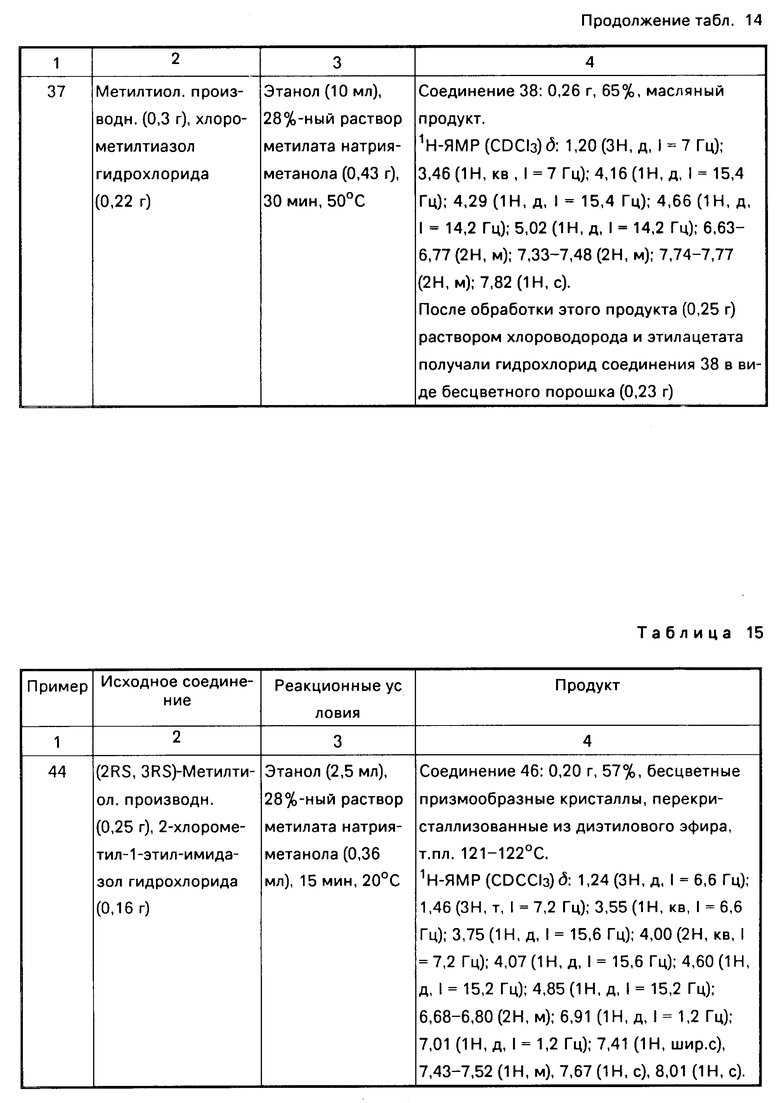




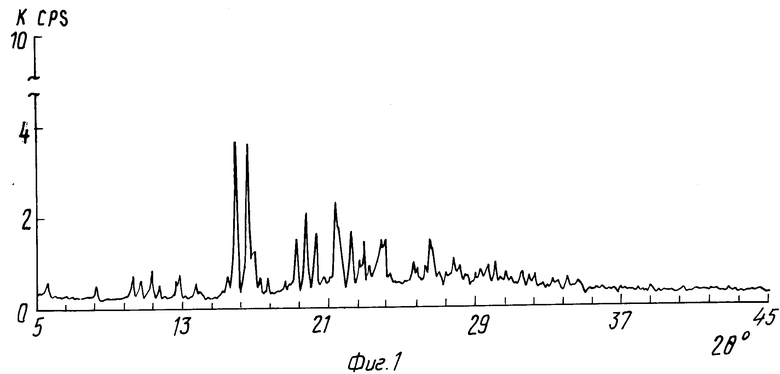

Использование: в качестве противогрибковых средств или промежуточных продуктов для их синтеза. Сущность изобретения: продукт производное 1,2,4-триазола ф-лы I, где R°, R1 и R2- одинаковы или различны, водород или низшая алкильная группа; А-группа ф-лы II, где Х химическая связь или группа ф-лы III, где X1 химическая связь или C1-C5 -алкиленовая группа, которая может содержать атом серы; R5 и R6 одинаковы или различны, водород или низшая алкильная группа; R3 ароматический гетероцикл, в котором гетероатом это атом азота, серы или кислорода и который может быть замещен низшим алкилом (незамещенным или фторзамещенным), C3 -циклоалкилом, амино, низшей алкилмеркаптогруппой или оксогруппой; n 0,1 или 2; или А-группа ф-лы S-R4, где R4 указано выше, Ro или R1 низшая алкильная группа. Реагент 1: соединение ф-лы II, R°, R1 и R2 указаны выше. Реагент 2: HS-X-R3, где Х и R3 указаны выше. Условия реакции: при температуре от 20 до 150°С с окислением при необходимости продукта реакции. 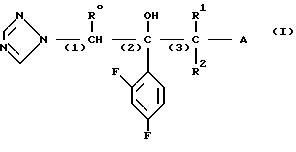
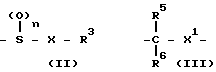
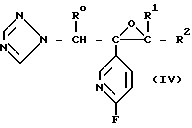 2 с.п. ф-лы, 2 ил. 18 табл.
2 с.п. ф-лы, 2 ил. 18 табл.
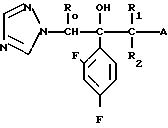
где R0, R1 и R2, одинаковые или различные, водород или низшая алкильная группа;
A группа общей формулы II
где X химическая связь или группа общей формулы III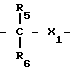
где X1 химическая связь или С1 С5-алкиленовая группа, которая может содержать атом серы в числе составляющих атомов;
R5 и R6, одинаковые или различные, водород или низшая алкильная группа;
R3 ароматическая гетероциклическая группа, в которой гетероатомом является атом азота, серы или кислорода и которая может быть замещенной низшим алкилом (незамещенным или фторзамещенный), С3-циклоалкилом, амино, низшей алкилмеркаптогруппой или оксогруппой;
n 0,1 или 2,
или
A группа общей формулы IV
S R4,
где R4 водород или низшая алканоильная группа,
причем когда X или X1 в радикале A является химической связью или A группа общей формулы IV, где R4 имеет указанные значения, R0 или R1 низшая алкильная группа;
оптически инертные или имеющие R- или S-конфигурации C-2 и C-3 асимметричных центров, или их соли, обладающие фунгицидной активностью.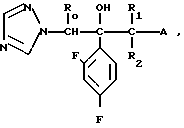
где R0, R1 и R2, одинаковые или различные, водород или низшая алкильная группа;
A группа общей формулы II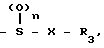
где X химическая связь или группа общей формулы III
где X1 химическая связь или С1 С5-алкиленовая группа, которая может содержать атом серы или кислорода в качестве составляющих атомов;
R5 и R6, одинаковые или различные, водород или низшая алкильная группа;
R3 ароматическая гетероциклическая группа, в которой гетероатомом является атом азота, серы или кислорода и которая может быть замещенной незамещенным или фторзамещенным низшим алкилом, С3-циклоалкилом, амино, низшей алкилмеркаптогруппой или оксогруппой;
n=0,1 или 2;
или
A группа общей формулы IV
S R4,
где R4 водород или низшая алканоильная группа,
причем когда X или X1 в A являются химической связью или A группа общей формулы IV, где R4 имеет указанные значения, определенные выше, R0 или R1 низшая алкильная группа,
оптически инертных или имеющих R- или S-конфигурацию C-2 и C-3 асимметричных центров, или его соли, отличающийся тем, что соединение общей формулы V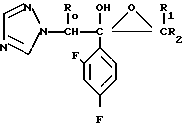
где R0, R1 и R2 имеют указанные значения,
подвергают взаимодействию с соединением общей формулы VI
HS X R3,
где X и R3 имеют указанные значения,
при температуре от -20 до 150oС, и если необходимо, подвергают продукт реакции окислению.
Приоритет по признакам:
26.09.89 при R0 водород; R1 и R2 водород, низший алкил; A группа общей формулы
где X C1 C5-алкиленовая группа, которая в качестве заместителя может содержать серу; R3 ароматический гетероцикл, в котором гетероатомом является атом азота, серы или кислорода и который может быть замещен низшим алкилом (незамещенным или фторзамещенным), С3-циклоалкилом, амино, низшей алкилмеркаптогруппой или оксогруппой, n 0,1 или 2.
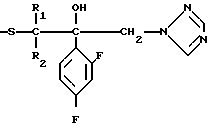
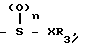

| АВТОМАТИЧЕСКИЙ ЛЕНТОЧНЫЙ ВЕСОВОЙ ДОЗАТОР НЕПРЕРЫВНОГО ДЕЙСТВИЯ ДЛЯ СЫПУЧИХ МАТЕРИАЛОВ | 0 |
|
SU189173A1 |
