Область техники
Настоящее изобретение относится к жидким водным фармацевтическим композициям, содержащим полипептиды фактора VII, и к способам получения и применения таких композиций, а также к контейнерам, содержащим такие композиции, и к применению таких композиций для лечения синдрома, зависимого от фактора VII. Более конкретно, изобретение относится к жидким композициям, стабилизированным против химической и/или физической деградации.
Предшествующий уровень техники
Было идентифицировано множество факторов, вовлеченных в процесс свертывания крови, включая фактор VII (FVII), представляющий собой гликопротеин плазмы. Коагуляция инициируется образованием комплекса между тканевым фактором (TF), подвергающимся воздействию циркулирующей крови после повреждения стенки сосуда, и FVIIa, который присутствует в кровотоке в количестве, соответствующем примерно 1% общей массы белка FVII. FVII существует в плазме главным образом в виде одноцепочечного зимогена, который расщепляется FXa на его двухцепочечную активированную форму FVIIa. Рекомбинантный активированный фактор VIIa (rFVIIa) был разработан как прогемостатический агент. Введение rFVIIa обеспечивает быстрый и высокоэффективный прогемостатический ответ у гемофилических субъектов с кровотечениями, которых невозможно лечить другими продуктами факторов свертывания крови вследствие образования антител. С помощью FVIIa можно успешно лечить также кровотечение у субъектов с дефицитом фактора VII или у субъектов, имеющих нормальную систему коагуляции, но подверженных чрезмерным кровотечениям.
Желательно иметь формы для введения фактора VIIa, подходящие как для хранения, так и для доставки. Идеально, когда лекарственный продукт хранят и вводят в виде жидкости. В качестве альтернативы лекарственный продукт лиофилизируют, т.е. высушивают сублимацией, и затем растворяют, добавляя подходящий разбавитель перед тем, как он будет использоваться пациентом. Идеально, когда лекарственный продукт имеет стабильность, достаточную для хранения в течение длительного периода времени, т.е. более шести месяцев.
Решение либо сохранять готовый лекарственный препарат в виде жидкости, либо лиофилизировать его обычно основано на стабильности белкового лекарства в этих формах. На стабильность белка могут оказывать влияние, в частности, такие факторы, как ионная сила, рН, температура, повторные циклы замораживания/оттаивания и воздействие сил трения. Активный белок может быть утрачен в результате физической нестабильности, включая денатурацию и агрегирование (образование как растворимых, так и нерастворимых агрегатов), а также химической нестабильности, включая, например, гидролиз, деамидирование и окисление, которые приведены лишь в качестве примера. Для общего обзора стабильности белковых фармацевтических средств можно сослаться, например, на Manning, et al., Pharmaceutical Research 6:903-918 (1989).
Хотя возможное возникновение белковой нестабильности широко признано, невозможно предсказать конкретные проблемы нестабильности конкретного белка. Любой из видов этой нестабильности может приводить к образованию побочного белкового продукта или производного, имеющего сниженную активность, повышенную токсичность и/или повышенную иммуногенность. Действительно, осаждение белка может приводить к тромбозу, неоднородности лекарственной формы и количества, а также к засорению шприцов. Кроме того, посттрансляционные модификации, такие как, например, гамма-карбоксилирование некоторых остатков глутаминовой кислоты на N-конце и добавление боковых углеводных цепей, обеспечивают потенциальные сайты, которые могут оказаться чувствительными к модификации при хранении. Также специфически для фактора VIIa, который является сериновой протеазой, может происходить фрагментация в результате автокатализа (ферментативная деградация). Таким образом, безопасность и эффективность любой композиции белка напрямую связаны с его стабильностью. Поддержание стабильности в жидкой форме, как правило, отличается от поддержания стабильности в лиофилизированной форме из-за значительно большего потенциала молекулярного движения и, следовательно, повышенной вероятности молекулярных взаимодействий. Поддержание стабильности в концентрированной форме также отличается от упомянутой выше стабильности благодаря склонности к образованию агрегатов при повышенных концентрациях белка.
При разработке жидкой композиции учитывают многие факторы. Для достижения кратковременной, т.е. менее шести месяцев, стабильности жидкости, как правило, следует предотвращать грубые структурные изменения, такие как денатурация и агрегирование. Эти процессы описаны в литературе для целого ряда белков, и существуют многие примеры стабилизирующих агентов. Общеизвестно, что агент, эффективный для стабилизации одного белка, фактически дестабилизирует другие белки. После того как белок был стабилизирован против грубых структурных изменений, разработка жидкой композиции для достижения долговременной стабильности (например более шести месяцев) следует дополнительно стабилизировать белок от типов деградации, специфических для этого белка. Более специфические типы деградации могут включать в себя, например, изменение положения дисульфидной связи, окисление некоторых остатков, деамидирование, циклизацию. Хотя не всегда возможно точно определить конкретные виды деградации, разработаны анализы для мониторинга едва уловимых изменений, для того чтобы контролировать способность конкретных наполнителей однозначно стабилизировать интересующий белок.
Желательно, чтобы рН композиции находилось в физиологически подходящем диапазоне после инъекции/инфузии, иначе это может привести к боли и дискомфорту для пациента.
Общий обзор белковых композиций представлен, например, в Cleland et al.: The development of stable protein compositions: A closer look at protein aggregation, deamidation and oxidation, Critical Reviews in Therapeutic Drug Carrier Systems 1993, 10(4): 307-377; и Wang et ai., Parenteral compositions of proteins and peptides: Stability and stabilizers, Journal of Parenteral Science and Technology 1988 (Supplement), 42 (2S).
Фактор VIIa подвергается нескольким путям деградации, в особенности агрегации (димеризации), окислению и автолитическому расщеплению (укорочению пептидного скелета или "деградации тяжелой цепи"). Кроме того, может наблюдаться осаждение. Многие из этих реакций могут быть существенно замедлены удалением воды из белка. Однако разработка водной композиции для фактора VIIa имеет преимущества устранения ошибок разведения, тем самым увеличивая точность дозирования, а также упрощая применение продукта в клинике, улучшая, таким образом, соблюдение пациентом режима и схемы лечения. Идеально, когда композиции фактора VIIa должны быть стабильны в течение более 6 месяцев в широком диапазоне концентраций белка. Это дает возможность приспособиться к методам введения. Как правило, более высококонцентрированные формы позволяют вводить меньшие объемы, что весьма желательно с точки зрения пациентов. Жидкие композиции могут иметь многие преимущества над лиофилизированными продуктами, что касается легкости введения и применения.
В настоящее время единственная, имеющаяся в продаже композиция, содержащая полученный рекомбинантными методами полипептид FVII, представляет собой лиофилизированный продукт фактора FVIIa, который растворяют перед использованием; она содержит относительно низкую концентрацию фактора VIIa, например приблизительно 0,6 мг/мл. Ампула (1,2 мг) NovoSeven® (Novo Nordisk A/S, Дания) содержит 1,2 мг рекомбинантного фактора VIIa человека, 5,84 мг NaCl, 2,94 мг CaCl2, 2 Н2O, 2,64 мг глицилглицина (GlyGly), 0,14 мг полисорбата 80 и 60,0 мг маннита; разводится до рН 5,5 2,0 мл воды для инъекции (WFI). После разведения белковый раствор является стабильным для применения в течение 24 часов. Таким образом, никаких жидких, готовых для применения или концентрированных продуктов фактора VII в настоящее время нет в продаже.
Соответственно, задача этого изобретения состоит в том, чтобы предложить жидкую водную фармацевтическую композицию, содержащую полипептид фактора VII, в которой снижено образование продуктов химической и/или физической деградации, таких как продукты ферментативной деградации или автокатализа.
Раскрытие изобретения
Авторы настоящего изобретения обнаружили, что фактор VII или его аналоги ("полипептиды фактора VII"), будучи приготовлены в виде жидких водных фармацевтических композиций вместе с по меньшей мере одним стабилизирующим агентом (3), содержащим фрагмент -C(=N-Z1-R1)-NH-Z2-R2, проявляют улучшенную стабильность и, таким образом, делают возможным длительное хранение перед фактическим применением.
Таким образом, один аспект настоящего изобретения касается жидкой водной фармацевтической композиции, содержащей:
по меньшей мере 0,01 мг/мл полипептида фактора VII (1);
буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0; и
по меньшей мере один стабилизирующий агент (3), содержащий фрагмент -C(=N-Z1-R1)-NH-Z2-R2, где
Z1 и Z2 независимо выбраны из группы, состоящей из -О-, -S-, -NRH- и одинарной связи, где RH выбран из группы, состоящей из водорода, C1-4-алкила, арила и арилметила, и R1 и R2 независимо выбраны из группы, состоящей из водорода, возможно замещенного C1-6-алкила, возможно замещенного C2-6-алкенила, возможно замещенного арила, возможно замещенного гетероциклила, или
Z2 и R2 такие, как определено выше, и -C=N-Z1-R1 образует часть гетероциклического кольца, или
Z1 и R1 такие, как определено выше, и -C-NH-Z2-R2 образует часть гетероциклического кольца, или
-C(=N-Z1-R1)-NH-Z2-R2 образует гетероциклическое кольцо, где -Z1-R1-R2-Z2-представляет собой бирадикал.
Второй аспект настоящего изобретения касается способа получения жидкой водной фармацевтической композиции полипептида фактора VII, включающего стадию получения раствора полипептида фактора VII (1) в концентрации по меньшей мере 0,01 мг/мл, где раствор содержит буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0; и по меньшей мере один стабилизирующий агент (3), содержащий фрагмент -C(=N-Z1-R1)-NH-Z2-R2, где
Z1 и Z2 независимо выбраны из группы, состоящей из -О-, -S-, -NRH- и одинарной связи, где RH выбран из группы, состоящей из водорода, C1-4-алкила, арила и арилметила, и R1 и R2 независимо выбраны из группы, состоящей из водорода, возможно замещенного С1-6-алкила, возможно замещенного C2-6-алкенила, возможно замещенного арила, возможно замещенного гетероциклила, или
Z2 и R2 такие, как определено выше, и -C=N-Z1-R1 образует часть гетероциклического кольца, или
Z1 и R1 такие, как определено выше, и -C-NH-Z2-R2 образует часть гетероциклического кольца, или
-C(=N-Z1-R1)-NH-Z2-R2 образует гетероциклическое кольцо, где -Z1-R1-R2-Z2-представляет собой бирадикал.
Третий аспект настоящего изобретения касается жидкой водной фармацевтической композиции для применения в качестве лекарства.
Четвертый аспект настоящего изобретения касается применения жидкой водной фармацевтической композиции для изготовления лекарства для лечения синдрома, зависимого от фактора VII.
Пятый аспект настоящего изобретения касается способа лечения синдрома, зависимого от фактора VII, включающего введение субъекту, нуждающемуся в этом, эффективного количества жидкой водной фармацевтической композиции.
Шестой аспект настоящего изобретения касается герметичного контейнера, содержащего жидкую водную фармацевтическую композицию и возможно инертный газ.
Сведения, подтверждающие возможность осуществления изобретения
Как упомянуто выше, настоящее изобретение состоит в разработке новой стабилизированной жидкой водной фармацевтической композиции, содержащей полипептид фактора VII. Более конкретно, жидкая водная фармацевтическая композиция содержит:
по меньшей мере 0,01 мг/мл полипептида фактора VII (1);
буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0; и
по меньшей мере один стабилизирующий агент (3), содержащий фрагмент -C(=N-Z1-R1)-NH-Z2-R2, где
Z1 и Z2 независимо выбраны из группы, состоящей из -О-, -S-, -NRH- и одинарной связи, где RH выбран из группы, состоящей из водорода, С1-4-алкила, арила и арилметила, и R1 и R2 независимо выбраны из группы, состоящей из водорода, возможно замещенного C1-6-алкила, возможно замещенного С2-6-алкенила, возможно замещенного арила, возможно замещенного гетероциклила, или
Z2 и R2 такие, как определено выше, и -C=N-Z1-R1 образует часть гетероциклического кольца, или
Z1 и R1 такие, как определено выше, и -C-NH-Z2-R2 образует часть гетероциклического кольца, или
-C(=N-Z1-R1)-NH-Z2-R2 образует гетероциклическое кольцо, где -Z1-R1-R2-Z2-представляет собой бирадикал.
Термин "C1-6-алкил" означает ациклические и циклические насыщенные углеводородные радикалы, которые имеют 1-6 атомов углерода и могут быть линейными или разветвленными. Конкретные примеры представляют собой метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропилметил, н-пентил, изопентил, н-гексил и т.д. Аналогично, термин "С1-4-алкил" охватывает ациклические и циклические насыщенные углеводородные остатки, которые имеют 1-4 атома углерода и могут быть линейными или разветвленными.
Аналогично, термин "С2-6-алкенил" означает ациклические и циклические насыщенные углеводородные радикалы, которые имеют 2-6 атомов углерода и содержат одну ненасыщенную связь, которые могут быть линейными или разветвленными. Примеры С2-6-алкенильных групп представляют собой винил, аллил, бут-1-ен-1-ил, бут-2-ен-1-ил, пент-1-ен-1-ил, и гекс-1-ен-1-ил.
Термин "возможно замещенный" в связи с C1-6-алкильными и C2-6-алкенильными группами предназначен для обозначения того, что рассматриваемая группа может быть замещена одной или несколькими, предпочтительно одной-тремя группами, выбранными из группы, состоящей из гидрокси, C1-6-алкокси (т.е. C1-6-алкил-окси), С2-6-алкенилокси, оксо (образующей кето- или альдегидную функциональную группу), арила, арилокси, арилкарбонила, гетероциклила, гетероциклилокси, гетероциклилкарбонила, амино, моно- и ди(С1-6-алкил)амино, галогена, где любой арил и гетероцикпил могут быть замещены, как в частности описано ниже для возможно замещенного арила и гетероциклила.
"Галоген" включает фтор, хлор, бром и иод.
Используемый в данном описании термин "арил" предназначен для обозначения полностью или частично ароматического карбоциклического кольца или кольцевой системы, таких как фенил, нафтил, 1,2,3,4-тетрагидронафтил, антрацил, фенантрацил, пиренил, бензопиренил, флуоренил и ксантенил, среди которых предпочтительным примером является фенил.
Термин "гетероциклил" предназначен для обозначения насыщенного, частично ненасыщенного, частично ароматического или полностью ароматического карбоциклического кольца или кольцевой системы, где один или более атомов углерода замещены гетероатомами, например атомами азота (=N- или -NH), серы (-S-) и/или кислорода (-O-). Примеры таких гетероциклильных групп представляют собой оксазолил, оксазолинил, оксазолидинил, изоксазолил, изоксазолинил, изоксазолидинил, оксадиазолил, оксадиазолинил, оксадиазолидинил, тиазолил, изотиазолил, пирролил, пирролинил, пирролидинил, имидазолил, имидазолинил, имидазолидинил, пиразолил, пиридинил, пиразинил, пиридазинил, пиперидинил, кумарил, фурил, хинолил, бензотиазолил, бензотриазолил, бензодиазолил, бензоксозолил, диазолил, диазолинил, диазолидинил, триазолил, триазолинил, триазолидинил, тетразол и т.д. Предпочтительные гетероциклильные группы представляют собой 5-, 6- или 7-членные моноциклические группы, такие как изоксазолил, изоксазолинил, оксадиазолил, оксадиазолинил, пирролил, пирролинил, диазолил, диазолинил, триазолил, триазолинил, имидазолил, имидазолинил и т.д.
Термин "гетероциклическое кольцо" предназначен для обозначения кольца, соответствующего тем, которые определены термином "гетероциклил".
В связи с терминами "арил", "гетероциклил" и "гетероциклическое кольцо", термин "возможно замещенный" предназначен для обозначения того, что рассматриваемая группа может быть замещена одной или несколькими, предпочтительно одной-тремя группами, выбранными из гидрокси (которая, когда присутствует в енольной системе, может быть представлена в таутомерной кетоформе), C1-6-алкила, С2-6-алкенила, фенила, бензила, C1-6-алкокси, оксо (которая может быть представлена в таутомерной енольной форме), карбокси, С1-6-алкоксикарбонила, C1-6-алкилкарбонила, амино, моно- и ди(С1-6-алкил)амино, дигалоген-С1-4-алкила, тригалоген-С1-4-алкила и галогена. Наиболее типичные примеры заместителей представляют собой гидроксил, C1-4-алкил, фенил, бензил, C1-4-алкокси, оксо, амино, моно- и диметиламино и галоген.
Кроме того, что R1 и R2 независимо могут быть выбраны из группы, состоящей из водорода, возможно замещенного C1-6-алкила, возможно замещенного С2-6-алкенила, возможно замещенного арила, возможно замещенного гетероциклила, также возможно, что часть фрагмента -C(=N-Z1-R1)-NH-Z2-R2 может быть частью гетероциклического кольца, тогда как другая часть фрагмента имеет значение, определенное соответственно для Z1, Z2, R1 и R2. В некоторых воплощениях изобретения группа -C=N-Z1-R1 может образовывать часть гетероциклического кольца, выбранного из группы, состоящей из 1,2-диазольного кольца, изоксазольного кольца, 1,2,4-триазольного кольца, и 1,2,4-оксадиазольного кольца, или группа -C-NH-Z2-R2 может образовывать часть гетероциклического кольца, выбранного из группы, состоящей из 1,2-диазолинового кольца, изоксазолинового кольца, 1,2,4-триазолинового кольца и 1,2,4-оксадиазолинового кольца. Такие гетероциклические кольца могут быть замещены, как описано выше.
В некоторых воплощениях по меньшей мере один из R1 и R2 представляет собой водород, например, оба представляют собой водород. Кроме того, в другом воплощении, которое может быть объединено с воплощениями, упомянутыми выше, по меньшей мере один из Z1 и Z2 представляет собой одинарную связь, например, оба представляют собой одинарную связь. В конкретных воплощениях R1 и R2 оба представляют собой водород, и Z1 и Z2 оба представляют собой одинарную связь.
Предполагают, что фрагмент -C(=N-Z1-R1)-NH-Z2-R2 является особенно важным для стабилизирующего эффекта стабилизирующего агента (3). В частности, предполагают, что фрагмент -C(=N-Z1-R1)-NH-Z2-R2 имитирует аргининовую группировку субстрата для полипептида фактора VII.
В более конкретных воплощениях стабилизирующий агент (3) представляет собой по меньшей мере один агент, выбранный из группы, состоящей из амидиновых соединений, содержащих фрагмент -C-C(=N-Z1-R1)-NH-Z2-R2, и гуанидиновых соединений, содержащих фрагмент >N-C(=N-Z1-R1)-NH-Z2-R2.
В некоторых воплощениях стабилизирующий агент (3) представляет собой по меньшей мере одно амидиновое соединение, выбранное из группы, состоящей из бензамидинов, содержащих фрагмент -C6H4-C(=N-Z1-R1)-NH-Z2-R2, где C6H4 означает возможно замещенное бензольное кольцо, из которых бензамидин (R1 и R2 представляют собой водород, и Z1 и Z2 представляют собой одинарную связь) представляет конкретное воплощение (смотри Экспериментальную часть).
В других конкретных воплощениях изобретения бензамидины содержат фрагмент >N-C6H4-C(=N-Z1-R1)-NH-Z2-R2, где C6H4 означает возможно замещенное бензольное кольцо, т.е. о-аминобензамидин, м-аминобензамидин или п-аминобензамидин, из которых п-аминобензамидин является в настоящее время наиболее предпочтительным.
Кроме того, иллюстративные примеры п-аминобензамидинов представляют собой соединения, раскрытые в ЕР 1162194 А1 (Aventis), в частности, определенные в пп.1-6 формулы изобретения и в параграфах [0009]-[0052], и в ЕР 1270551 А1, в частности, в пп.1 и 2 формулы изобретения и параграфах [0010]-[0032].
В другом воплощении стабилизирующий агент (3) представляет собой по меньшей мере одно гуанидиновое соединение, выбранное из группы, состоящей из гуанидиновых соединений, содержащих фрагмент -CH2-NH-C(=N-Z1-R1)-NH-Z2-R2. Примеры гуанидиновых соединений представляют собой соединения, выбранные из группы, состоящей из аргинина, производных аргинина и пептидов из 2-5 аминокислотных остатков, включающих по меньшей мере один остаток аргинина. Конкретным воплощением является аргинин (смотри Экспериментальную часть).
Термин "производные аргинина" предназначен для обозначения гомологов аргинина с функционапными группами на N-конце (например, N-метилированных и N-ацилированных производных аргинина (например, ацетилированных производных)), с функционалными группами на С-конце (например, С-амидированных, С-алкиламидированных и С-алкилированных производных) и их комбинаций.
Как указано выше, один ключевой фрагмент стабилизирующих агентов представляет собой -C(=N-Z1-R1)-NH-Z2-R2. Другие части стабилизирующего агента также могут быть важными, в особенности, что касается оптимизации стабилизирующего эффекта и переносимости пациентом. Как правило, стабилизирующий агент имеет формулу Y-C(=N-Z1-R1)-NH-Z2-R2, где Y представляет собой органический радикал. Радикал Y выбирают таким, чтобы улучшить эффективность стабилизирующего эффекта. Кроме того, радикал Y может содержать один или более дополнительных фрагментов формулы -C(=N-Z1-R1)-NН-Z2-R2.
Молекулярная масса стабилизирующего агента как правило составляет не более 1000 Да, например не более 500 Да.
Соединения согласно настоящему изобретению могут иметь один или более чем один асимметрический центр, и, если не указано иначе, подразумевается, что стереоизомеры (оптические изомеры) в виде выделенных, чистых или частично очищенных стереоизомеров или их рацемических смесей включены в объем данного изобретения.
Концентрация стабилизирующего агента (или агентов) (3) как правило составляет по меньшей мере 1 мкМ. Желательная (или необходимая) концентрация как правило зависит от выбранного стабилизирующего агента (или агентов), более конкретно от аффинности связывания выбранного стабилизирующего агента с полипептидом фактора VII.
В различных воплощениях стабилизирующий агент (3) присутствует в концентрации по меньшей мере 5 мкМ, по меньшей мере 10 мкМ, по меньшей мере 20 мкМ, по меньшей мере 50 мкМ, по меньшей мере 100 мкМ, по меньшей мере 150 мкМ, по меньшей мере 250 мкМ, по меньшей мере 500 мкМ, по меньшей мере 1 мМ, по меньшей мере 2 мМ, по меньшей мере 4 мМ, по меньшей мере 5 мМ, по меньшей мере 8 мМ, по меньшей мере 9 мМ, по меньшей мере 10 мМ, по меньшей мере 15 мМ, по меньшей мере 20 мМ, такой, которая находится, например, в диапазоне 1-10000 мкМ, 10-10000 мкМ, 20-10000 мкМ, 50-10000 мкМ, 10-5000 мкМ, 10-2000 мкМ, 20-5000 мкМ, 20-2000 мкМ, 50-5000 мкМ, 0,1-100 мМ, 0,1-75 мМ, 0,1-50 мМ, 0,1-10 мМ, 0,2-75 мМ, 0,2-50 мМ, 0,2-20 мМ, 0,5-75 мМ или 0,5-50 мМ.
В одном воплощении стабилизирующий агент (3) представляет собой бензамидин, и концентрация указанного агента составляет по меньшей мере 1 мМ, например, по меньшей мере 2 мМ, хотя предполагается, что замещенные бензамидины могут быть более эффективными, поэтому они могут быть добавлены в меньших концентрациях.
В одном воплощении стабилизирующий агент (3) не является бензамидином.
В одном воплощении стабилизирующий агент (3) представляет собой аргинин, и концентрация указанного агента составляет по меньшей мере 10 мМ, например, по меньшей мере 50 мМ.
В другом воплощении стабилизирующий агент (3) представляет собой п-аминобензамидин, и концентрация указанного агента составляет по меньшей мере 0,001 мМ.
В другом воплощении стабилизирующий агент (3) представляет собой S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамид формулы
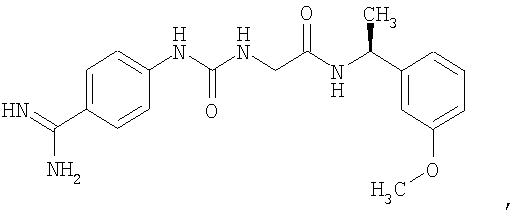
и концентрация указанного агента составляет по меньшей мере 0,001 мМ.
В другом воплощении стабилизирующий агент (3) представляет собой S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-(1-фенилэтил)-ацетамид формулы
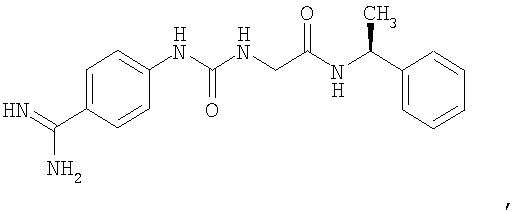
и концентрация указанного агента составляет по меньшей мере 0,001 мМ.
В другом воплощении стабилизирующий агент (3) представляет собой N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамид формулы
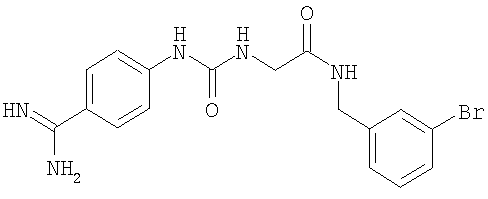
и концентрация указанного агента составляет по меньшей мере 0,001 мМ. В различных воплощениях молярное соотношение между стабилизирующим агентом (3) и полипептидом FVII (агент (3):FVII) составляет: более 0,1, более 0,5, более 1, более 2, более 5, более 10, более 25, более 100, более 250, более 1000, более 2500 или более 5000, такое, которое находится, например, в диапазоне 0,1-10000, 0,1-5000, 0,1-2500, 0,1-1000, 0,1-250, 0,1-100, 0,1-25, 0,1-10, 0,5-10000, 0,5-5000, 0,5-2500, 0,5-1000, 0,5-250, 0,5-100, 0,5-25, 0,5-10, 1-10000, 1-5000, 1-2500, 1-1000, 1-250, 1-100; 1-25; 1-10, 10-10000, 10-5000, 10-250, 1000-10000 или 1000-5000.
Желательная концентрация как правило зависит от выбранного стабилизирующего агента (или агентов), более конкретно от аффинности связывания выбранного агента с полипептидом фактора VII.
Биологический эффект фармацевтической композиции определяется главным образом присутствием полипептида фактора VII, хотя другие активные ингредиенты могут быть включены в комбинации с полипептидом фактора VII.
Используемый в данном описании термин "полипептид фактора VII" охватывает фактор VII дикого типа (т.е. полипептид, имеющий аминокислотную последовательность, раскрытую в патенте США № 4784950), а также варианты фактора VII, проявляющие по существу такую же или улучшенную биологическую активность по сравнению с фактором VII дикого типа. Термин "фактор VII" предназначен для обозначения полипептидов фактора VII в их нерасщепленной (зимогенной) форме, а также полипептидов, которые были протеолитически обработаны с получением их соответствующих биоактивнных форм, которые могут быть обозначены как фактор VIIa. Как правило, фактор VII расщепляется между остатками 152 и 153 с получением фактора VIIa. Термин "полипептид фактора VII" также охватывает полипептиды, включая варианты, в которых биологическая активность фактора VIIa была по существу модифицирована или в некоторой степени уменьшена по сравнению с активностью фактора VIIa дикого типа. Эти полипептиды включают в себя, но не ограничены ими, фактор VII или фактор VIIa, в которые были введены специфические изменения аминокислотной последовательности, которые модифицируют или нарушают биоактивность данного полипептида.
Биологическая активность фактора VIIa в свертывании крови обусловлена его способностью (1) связываться с тканевым фактором (TF) и (2) катализировать протеолитическое расщепление фактора IX или фактора Х с получением активированного фактора IX или Х (Фактора IXa или Ха соответственно).
Для целей изобретения биологическая активность полипептидов фактора VII ("биологическая активность фактора VII") может быть количественно определена путем определения способности препарата стимулировать свертывание крови (см. Анализ 4, представленный в данном описании). В этом анализе биологическая активность выражается как уменьшение времени свертывания крови относительно контрольного образца и переводится в "единицы фактора VII" путем сравнения с объединенным сывороточным стандартом человека, содержащим 1 ед./мл активности фактора VII. Альтернативно, биологическая активность фактора VIIa может быть количественно определена путем (1) определения способности фактора VIIa или полипептида, родственного фактору VII, продуцировать активированный фактор Х (фактор Ха) в системе, включающей TF, погруженный в липидную мембрану, и фактор Х (Persson et al., J. Biol. Chem. 272:19919-19924, 1997); (2) определения степени гидролиза фактора Х в водной системе ("Анализ протеолиза in vitro", смотри Анализ 2 ниже); (3) определения степени физического связывания фактора VIIa или полипептида, родственного фактору VII, с TF с использованием прибора на основе поверхностного плазменного резонанса (Persson, FEBS Letts. 413:359-363, 1997); (4) определения степени гидролиза синтетического субстрата фактором VIIa и/или полипептидом, родственным фактору VII ("Анализ гидролиза in vitro", смотри Анализ 1 ниже); или (5) определения продукции тромбина in vitro в TF-независимой системе (смотри Анализ 3 ниже).
Варианты фактора VII, имеющие по существу такую же или улучшенную биологическую активность по сравнению с фактором VIIa дикого типа, охватывают варианты, которые проявляют по меньшей мере примерно 25%, например, по меньшей мере примерно 50%, по меньшей мере примерно 75% или по меньшей мере примерно 90% специфической активности фактора VIIa, который продуцировался в таком же типе клеток, при тестировании в одном или более чем одном анализе свертывания крови (Анализ 4), анализе протеолиза (Анализ 2) или анализе связывания TF, как описано выше. Варианты фактора VII, имеющие в значительной степени сниженную биологическую активность относительно фактора VIIa дикого типа, представляют собой варианты, которые проявляют менее примерно 25%, такую как, например, менее примерно 10% или менее примерно 5% специфической активности фактора VIIa дикого типа, который продуцировался в таком же типе клеток, при тестировании в одном или более анализе свертывания крови (Анализ 4), анализе протеолиза (Анализ 2) или анализе связывания TF, как описано выше. Варианты фактора VII, имеющие по существу модифицированную биологическую активность относительно фактора VII дикого типа, включают в себя, без ограничения, варианты фактора VII, которые проявляют TF-независимую протеолитическую активность в отношении фактора X, и варианты, которые связывают TF, но не расщепляют фактор X.
Варианты фактора VII, независимо от того, проявляют они по существу такую же или лучшую биоактивность, чем фактор VII дикого типа, или наоборот проявляют по существу модифицированную или уменьшенную биоактивность относительно фактора VII дикого типа, включают в себя, без ограничения, полипептиды, имеющие аминокислотную последовательность, которая отличается от последовательности фактора VII дикого типа вставкой, делецией или заменой одной или более чем одной аминокислоты.
Не ограничивающие примеры вариантов фактора VII, имеющие по существу такую же биологическую активность, как и фактор VII дикого типа, включают в себя S52A-FVIIa, S60А-FVIIa (Lino et al., Arch. Biochem. Biophys. 352: 182-192, 1998); варианты FVIIa, проявляющие повышенную протеолитическую стабильность, как раскрыто в патенте США № 5580560; фактор VIIa, который был протеолитически расщеплен между остатками 290 и 291 или между остатками 315 и 316 (Mollerup et al., Biotechnol. Bioeng. 48:501-505, 1995); окисленные формы фактора VIIa (Kornfelt et al., Arch. Biochem. Biophys. 363:43-54, 1999); варианты FVII, раскрытые в PCT/DK02/00189; и варианты FVII, проявляющие повышенную протеолитическую стабильность, как раскрыто в WO 02/38162 (Scripps Research Institute); варианты FVII, имеющие модифицированный Gla-домен и демонстрирующие повышенное мембранное связывание, как раскрыто в WO 99/20767 (University of Minnesota); и варианты FVII, раскрытые в WO 01/58935 (Maxygen ApS).
He ограничивающие примеры вариантов фактора VII, имеющих повышенную биологическую активность по сравнению с FVIIa дикого типа, включают варианты FVII, раскрытые в WO 01/83725, WO 02/22776, WO 02/077218, WO 03/27147, WO 03/37932; WO 02/38162 (Scripps Research Institute); и варианты FVIIa с повышенной активностью, раскрытые в JP 2001061479 (Chemo-Sero-Therapeutic Res Inst).
He ограничивающие примеры вариантов фактора VII, имеющих по существу уменьшенную или модифицированную биологическую активность относительно фактора VII дикого типа, включают в себя R152E-FVIIa (Wildgoose et al., Biochem 29:3413-3420, 1990).
Примеры полипептидов фактора VII включают в себя, но не ограничены ими, фактор VII дикого типа, L305V-FVII, L305V/M306D/D309S-FVII, L305I-FVII, L305T-FVII, F374P-FVII, V158T/M298Q-FVII, V158D/E296V/M298Q-FVII, K337A-FVII, M298Q-FVII, V158D/M298Q-FVII, L305V/K337A-FVII, V158D/E296V/M298Q/L305V-FVII, V158D/E296V/M298Q/K337A-FVII, V158D/E296V/M298Q/L305V/K337A-FVII, К157А-FVII, E296V-FVII, E296V/M298Q-FVII, V158D/E296V-FVII, V158D/M298K-FVII и S336G-FVII, L305V/K337A-FVII, L305V/V158D-FVII, L305V/E296V-FVII, L305V/M298Q-FVII, L305V/V158T-FVII, L305V/K337A/V158T-FVII, L305V/K337A/M298Q-FVII, L305V/K337A/E296V-FVII, L305V/K337A/V158D-FVII, L305V/V158D/M298Q-FVII, L305V/V158D/E296V-FVII, L305V/V158T/M298Q-FVII, L305V/V158T/E296V-FVII, L305V/E296V/M298Q-FVII, L305V/V158D/E296V/M298Q-FVII, L305V/V158T/E296V/M298Q-FVII, L305V/V158T/K337A/M298Q-FVII, L305V/V158T/E296V/K337A-FVII, L305V/V158D/K337A/M298Q-FVII, L305V/V158D/E296V/K337A-FVII, L305V/V158D/E296V/M298Q/K337A-FVII, L305V/V158T/E296V/M298Q/K337A-FVII, S314E/K316H-FVII, S314E/K316Q-FVII, S314E/L305V-FVII, S314E/K337A-FVII, S314E/V158D-FVII, S314E/E296V-FVII, S314E/M298Q-FVII, S314E/V158T-FVII, K316H/L305V-FVII, K316H/K337A-FVII, K316H/V158D-FVII, K316H/E296V-FVII, K316H/M298Q-FVII, K316H/V158T-FVII, K316Q/L305V-FV1I, K316Q/K337A-FVII, K316Q/V158D-FVII, K316Q/E296V-FVII, K316Q/M298Q-FVII, K316Q/V158T-FVII, S314E/L305V/K337A-FVII, S314E/L305V/V158D-FVII, S314E/L305V/E296V-FVII, S314E/L305V/M298Q-FVII, S314E/L305V/V158T-FVII, S314E/L305V/K337A/V158T-FVII, S314E/L305V/K337A/M298Q-FVII, S314E/L305V/K337A/E296V-FVII, S314E/L305V/K337A/V158D-FVII, S314E/L305V/V158D/M298Q-FVII, S314E/L305V/V158D/E296V-FVII, S314E/L305V/V158T/M298Q-FVII, S314E/L305V/V158T/E296V-FVII, S314E/L305V/E296V/M298Q-FVII, S314E/L305V/V158D/E296V/M298Q-FVII, S314E/L305V/V158T/E296V/M298Q-FVII, S314E/L305V/V158T/K337A/M298Q-FVII,S314E/L305V/V158T/E296V/K337A-FVII, S314E/L305V/V158D/K337A/M298Q-FVII, S314E/L305V/V158D/E296V/K337A-FVII, S314E/L305V/V158D/E296V/M298Q/K337A-FVII, S314E/L305V/V158T/E296V/M298Q/K337A-FVII,K316H/L305V/K337A-FVII, K316H/L305V/V158D-FVII, K316H/L305V/E296V-FVII, K316H/L305V/M298Q-FVII, K316H/L305V/V158T-FVII, K316H/L305V/K337A/V158T-FVII, K316H/L305V/K337A/M298Q-FVII, K316H/L305V/K337A/E296V-FVII, K316H/L305V/K337A/V158D-FVII, K316H/L305V/V158D/M298Q-FVII, K316H/L305V/V158D/E296V-FVII, K316H/L305V/V158T/M298Q-FVII, K316H/L305V/V158T/E296V-FVII, K316H/L305V/E296V/M298Q-FVII, K316H/L305V/V158D/E296V/M298Q-FVII, K316H/L305V/V158T/E296V/M298Q-FVII, K316H/L305V/V158T/K337A/M298Q-FVII, K316H/L305V/V158T/E296V/K337A-FVII, K316H/L305V/V158D/K337A/M298Q-FVII,K316H/L305V/V158D/E296V/K337A-FVII, K316H/L305V/V158D/E296V/M298Q/K337A-FVII, K316H/L305V/V158T/E296V/M298Q/K337A-FVII, K316Q/L305V/K337A-FVII, K316Q/L305V/V158D-FVII, K316Q/L305V/E296V-FVII, K316Q/L305V/M298Q-FVII, K316Q/L305V/V158T-FVII, K316Q/L305V/K337A/V158T-FVII, K316Q/L305V/K337A/M298Q-FVII, K316Q/L305V/K337A/E296V-FVII, K316Q/L305V/K337A/V158D-FVII, K316Q/L305V/V158D/M298Q-FVII, K316Q/L305V/V158D/E296V-FVII, K316Q/L305V/V158T/M298Q-FVII, K316Q/L305V/V158T/E296V-FVII, K316Q/L305V/E296V/M298Q-FVII, K316Q/L305V/V158D/E296V/M298Q-FVII, K316Q/L305V/V158T/E296V/M298Q-FVII, K316Q/L305V/V158T/K337A/M298Q-FVII, K316Q/L305V/V158T/E296V/K337A-FVII, K316Q/L305V/V158D/K337A/M298Q-FVII, K316Q/L305V/V158D/E296V/K337A-FVII, K316Q/L305V/V158D/E296V/M298Q/K337A-FVII, К316Q/L305V/V158T/E296V/M298Q/K337A-FVII, F374Y/K337A-FVII, F374Y/V158D-FVII, F374Y/E296V-FVII, F374Y/M298Q-FVII, F374Y/V158T-FVII, F374Y/S314E-FVII, F374Y/L305V-FVII, F374Y/L305V/K337A-FVII, F374Y/L305V/V158D-FVII, F374Y/L305V/E296V-FVII, F374Y/L305V/M298Q-FVII, F374Y/L305V/V158T-FVII, F374Y/L305V/S314E-FVII, F374Y/K337A/S314E-FVII, F374Y/K337A/V158T-FVII, F374Y/K337A/M298Q-FVII,F374Y/K337A/E296V-FVII, F374Y/K337A/V158D-FVII, F374Y/V158D/S314E-FVII, F374Y/V158D/M298Q-FVII, F374Y/V158D/E296V-FVII, F374Y/V158T/S314E-FVII, F374Y/V158T/M298Q-FVII, F374Y/V158T/E296V-FVII, F374Y/E296V/S314E-FVII,F374Y/S314E/M298Q-FVII, F374Y/E296V/M298Q-FVII, F374Y/L305V/K337A/V158D-FVII, F374Y/L305V/K337A/E296V-FVII, F374Y/L305V/K337A/M298Q-FVII, F374Y/L305V/K337A/V158T-FVII, F374Y/L305V/K337A/S314E-FVII, F374Y/L305V/V158D/E296V-FVII, F374Y/L305V/V158D/M298Q-FVII, F374Y/L305V/V158D/S314E-FVII, F374Y/L305V/E296V/M298Q-FVII, F374Y/L305V/E296V/V158T-FVII, F374Y/L305V/E296V/S314E-FVII,F374Y/L305V/M298Q/V158T-FVII, F374Y/L305V/M298Q/S314E-FVII, F374Y/L305V/V158T/S314E-FV11, F374Y/K337A/S314E/V158T-FVII, F374Y/K337A/S314E/M298Q-FVII, F374Y/K337A/S314E/E296V-FVII, F374Y/K337A/S314E/V158D-FVII, F374Y/K337A/V158T/M298Q-FVII, F374Y/K337A/V158T/E296V-FVII, F374Y/K337A/M298Q/E296V-FVH, F374Y/K337A/M298Q/V158D-FVII, F374Y/K337A/E296V/V158D-FVII, F374Y/V158D/S314E/M298Q-FVII, F374Y/V158D/S314E/E296V-FVII, F374Y/V158D/M298Q/E296V-FVII, F374Y/V158T/S314E/E296V-FVII, F374Y/V158T/S314E/M298Q-FVII, F374Y/V158T/M298Q/E296V-FVII, F374Y/E296V/S314E/M298Q-FVII, F374Y/L305V/M298Q/K337A/S314E-FVII, F374Y/L305V/E296V/K337A/S314E-FVII, F374Y/E296V/M298Q/K337A/S314E-FVII, F374Y/L305V/E296V/M298Q/K337A-FVII, F374Y/L305V/E296V/M298Q/S314E-FVII, F374Y/V158D/E296V/M298Q/K337A-FVII, F374Y/V158D/E296V/M298Q/S314E-FVII, F374Y/L305V/V158D/K337A/S314E-FVII, F374Y/V158D/M298Q/K337A/S314E-FVII, F374Y/V158D/E296V/K337A/S314E-FVII, F374Y/L305V/V158D/E296V/M298Q-FVII, F374Y/L305V/V158D/M298Q/K337A-FVII, F374Y/L305V/V158D/E296V/K337A-FVII, F374Y/L305V/V158D/M298Q/S314E-FVII, F374Y/L305V/V158D/E296V/S314E-FVII, F374Y/V158T/E296V/M298Q/K337A-FVII, F374Y/V158T/E296V/M298Q/S314E-FVII, F374Y/L305V/V158T/K337A/S314E-FVII, F374Y/V158T/M298Q/K337A/S314E-FVII, F374Y/V158T/E296V/K337A/S314E-FVII, F374Y/L305V/V158T/E296V/M298Q-FVII, F374Y/L305V/V158T/M298Q/K337A-FVII, F374Y/L305V/V158T/E296V/K337A-FVII, F374Y/L305V/V158T/M298Q/S314E-FVII, F374Y/L305V/V158T/E296V/S314E-FVII,
F374Y/E296V/M298Q/K337A/V158T/S314E-FVII, F374Y/V158D/E296V/M298Q/K337A/S314E-FVII,
F374Y/L305V/V158D/E296V/M298Q/S314E-FVII,
F374Y/L305V/E296V/M298Q/V158T/S314E-FVII,
F374Y/L305V/E296V/M298Q/K337A/V158T-FVII,
F374Y/L305V/E296V/K337A/V158T/S314E-FVII,
F374Y/L305V/M298Q/K337A/V158T/S314E-FVII,
F374Y/L305V/V158D/E296V/M298Q/K337A-FVII,
F374Y/L305V/V158D/E296V/K337A/S314E-FVII,
F374Y/L305V/V158D/M298Q/K337A/S314E-FVII,
F374Y/L305V/E296V/M298Q/K337A/V158T/S314E-FVII,
F374Y/L305V/V158D/E296V/M298Q/K337A/S314E-FVII, S52A-фактор VII, S60A-фактор VII; Р152Е-фактор VII, S344A-фактор VII, фактор VIIa без домена Gla; и P11Q/K33E-FVII, T106N-FVII, K143N/N145T-FVII, V253N-FVII, R290N/A292T-FVII, G291N-FVII, R315N/V317T-FVII, K143N/N145T/R315N/V317T-FVII; и FVII, имеющий замены, вставки или делеции в аминокислотной последовательности от 233 Тhr до 240 Asn; FVII, имеющий замены, вставки или делеции в аминокислотной последовательности от 304 Аrg до 329 Cys, и FVII, имеющий замены, делеции или вставки в аминокислотной последовательности Ilе 153-Аrg 223.
В некоторых воплощениях полипептид фактора VII представляет собой фактор VIIa человека (hFVIIa), предпочтительно рекомбинантно полученный фактор VIIa человека (rhVIIa).
В других воплощениях полипептид фактора VII представляет собой вариант последовательности фактора VII.
В некоторых воплощениях полипептид фактора VII имеет гликозилирование, отличное от человеческого фактора VII дикого типа.
В различных воплощениях, например таких, где полипептид фактора VII представляет собой полипептид, родственный фактору VII, или вариант последовательности фактора VII, соотношение между активностью полипептида фактора VII и активностью нативного фактора VIIa человека (FVIIa дикого типа) составляет по меньшей мере примерно 1,25, предпочтительно по меньшей мере примерно 2,0 или 4,0, наиболее предпочтительно по меньшей мере примерно 8,0, при тестировании в "Анализе протеолиза in vitro" (Анализ 2), как описано в настоящем описании.
В некоторых воплощениях полипептиды фактора VII представляют собой полипептиды, родственные фактору VII, в частности варианты, где соотношение между активностью указанного полипептида фактора VII и активностью нативного фактора VIIa человека (FVIIa дикого типа) составляет по меньшей мере примерно 1,25 при тестировании в "Анализе гидролиза in vitro" (смотри Анализ 1 ниже); в других воплощениях это соотношение составляет по меньшей мере примерно 2,0; в дополнительных воплощениях это соотношение составляет по меньшей мере примерно 4,0.
В фармацевтической композиции часто желательно, чтобы концентрация активного ингредиента была такой, чтобы введение единичной дозы не вызывало нежелательного дискомфорта у пациента. Таким образом, единичная доза более чем примерно 2-10 мл часто не желательна. Поэтому для цели настоящего изобретения концентрация полипептида фактора VII составляет по меньшей мере 0,01 мг/мл. В различных воплощениях полипептид фактора VII присутствует в концентрации 0,01-20 мг/мл; 0,1-20 мг/мл; 0,1-15 мг/мл; 0,1-10 мг/мл; 0,5-5,0 мг/мл; 0,6-4,0 мг/мл; 1,0-4,0 мг/мл; 0,1-5 мг/мл; 0,1-4,0 мг/мл; 0,1-2 мг/мл или 0,1-1,5 мг/мл.
Концентрация фактора VIIa соответственно выражается как мг/мл или как МЕ/мл, причем 1 мг обычно означает 43000-56000 ME или более.
Для того чтобы сделать жидкую водную фармацевтическую композицию полезной для непосредственного парентерального введения млекопитающему, такому как человек, обычно необходимо, чтобы значение рН композиции сохранялось в некоторых пределах, таких как от примерно 4,0 до примерно 9,0. Для обеспечения подходящего значения рН в заданных условиях, фармацевтическая композиция также содержит буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0.
Термин "буферный агент" включает такие агенты или комбинации агентов, которые поддерживают рН раствора в приемлемом диапазоне от примерно 4,0 до примерно 9,0.
В одном воплощении буферный агент (2) представляет собой по меньшей мере один компонент, выбранный из групп, состоящих из кислот и солей MES (2-(4-морфолинил)-этансульфоновая кислота), PIPES (1,4-пиперазиндиэтансульфоновая кислота), ACES (N-(2-ацетамидо)-2-аминоэтансульфоновая кислота), BES (2-[бис(2-гидроксиэтил)амино]-этансульфоновая кислота), TES (2-[трис(гидроксиметил)-метиламино]-1-этансульфоновая кислота), HEPES (2-[4-(2-гидроксиэтил)-1-пиперазинил]-этансульфоновая кислота), TRIS (трис(гидроксиметил)-аминометан), гистидина (например, L-гистидина), имидазола, глицина, глицилглицина, глицинамида, фосфорной кислоты (например, фосфата натрия или калия), уксусной кислоты (например, ацетата аммония, натрия или кальция), молочной кислоты, глутаровой кислоты, лимонной кислоты (например, цитрата натрия или калия), винной кислоты, яблочной кислоты, малеиновой кислоты и янтарной кислоты. Следует понимать, что буферный агент может содержать смесь двух или более компонентов, где данная смесь способна обеспечивать значение рН в указанном диапазоне. В качестве примеров могут быть упомянуты уксусная кислота и ацетат натрия и т.д.
Концентрацию буферного агента выбирают таким образом, чтобы сохранить предпочтительное значение рН раствора. В различных воплощениях концентрация буферного агента составляет 1-100 мМ; 1-50 мМ; 1-25 мМ или 2-20 мМ.
В одном воплощении рН композиции составляет от примерно 4,0 до примерно 9,0; от 5,0 до примерно 9,0; от примерно 5,0 до примерно 8,0; например, от примерно 5,0 до примерно 7,5; от примерно 5,0 до примерно 7,0; от примерно 5,0 до примерно 6,5; от примерно 5,0 до примерно 6,0; от примерно 5,5 до примерно 7,0; от примерно 5,5 до примерно 6,5; от примерно 6,0 до примерно 7,0; от примерно 6,0 до примерно 6,5; от примерно 6,3 до примерно 6,7, или от примерно 5,2 до примерно 5,7.
Кроме трех обязательных компонентов жидкая водная фармацевтическая композиция может содержать дополнительные компоненты, полезные для получения композиции, приготовления препарата, стабильности или введения композиции.
Поэтому фармацевтическая композиция может также содержать неионное поверхностно-активное вещество. Поверхностно-активные вещества (также известные как детергенты), как правило, включают такие агенты, которые защищают белок от напряжений, индуцированных поверхностью раздела воздух/раствор, и напряжений, индуцированных на границе раствора/поверхности (например, приводящих к агрегации белка).
Типичные неионные поверхностно-активные вещества представляют собой полисорбаты, полоксамеры, полиоксиэтиленалкиловые эфиры, блок-сополимеры полиэтилен/полипропилен, полиэтиленгликоль (ПЭГ), полиоксиэтиленстеараты и полиоксиэтиленкасторовые масла.
Иллюстративные примеры неионных поверхностно-активных веществ представляют собой Tween®, полисорбат 20, полисорбат 80, Brij-35 (полиоксиэтилендодециловый эфир), полоксамер 188, полоксамер 407, ПЭГ8000, полиолы Pluronic®, полиокси-23-лауриловый эфир, Myrj 49 и Кремофор А.
В одном воплощении неионное поверхностно-активное вещество присутствует в количестве 0,005-2,0 мас.%.
Кроме того, композиция может дополнительно содержать агент (5), модифицирующий тоничность.
Используемый в данном описании термин "агент, модифицирующий тоничность" включает агенты, которые вносят вклад в осмотическое давление раствора. Агент (5), модифицирующий тоничность, включает в себя по меньшей мере один агент, выбранный из группы, состоящей из нейтральных солей, аминокислот, пептидов из 2-5 аминокислотных остатков, моносахаридов, дисахаридов, полисахаридов и сахарных спиртов. В некоторых воплощениях композиция содержит два или более таких агентов в комбинации.
Под "нейтральной солью" подразумевают соль, которая не является ни кислой, ни щелочной при растворении в водном растворе.
В одном воплощении по меньшей мере один агент (5), модифицирующий тоничность, представляет собой нейтральную соль, выбранную из групп, состоящих из солей натрия, солей калия, солей кальция и солей магния, таких как хлорид натрия, хлорид калия, хлорид кальция, ацетат кальция, глюконат кальция, левулат кальция, хлорид магния, ацетат магния, глюконат магния и левулат магния.
В еще одном воплощении агент (5), модифицирующий тоничность, включает хлорид натрия в комбинации с по меньшей мере одним агентом, выбранным из группы, состоящей из хлорида кальция, ацетата кальция, хлорида магния и ацетата магния.
В еще одном воплощении агент (5), модифицирующий тоничность, представляет собой по меньшей мере агент, выбранный из группы, состоящей из хлорида натрия, хлорида кальция, сахарозы, глюкозы и маннита.
В различных воплощениях агент (5), модифицирующий тоничность, присутствует в концентрации по меньшей мере 1 мМ, по меньшей мере 5 мМ, по меньшей мере 10 мМ, по меньшей мере 20 мМ, по меньшей мере 50 мМ, по меньшей мере 100 мМ, по меньшей мере 200 мМ, по меньшей мере 400 мМ, по меньшей мере 800 мМ, по меньшей мере 1000 мМ, по меньшей мере 1200 мМ, по меньшей мере 1500 мМ, по меньшей мере 1800 мМ, по меньшей мере 2000 мМ или по меньшей мере 2200 мМ.
В одной группе воплощений, агент (5), модифицирующий тоничность, присутствует в концентрации 5-2200 мМ, такой как 25-2200 мМ, 50-2200 мМ, 100-2200 мМ, 200-2200 мМ, 400-2200 мМ, 600-2200 мМ, 800-2200 мМ, 1000-2200 мМ, 1200-2200 мМ, 1400-2200 мМ, 1600-2200 мМ, 1800-2200 мМ или 2000-2200 мМ; 5-1800 мМ, 25-1800 мМ, 50-1800 мМ, 100-1800 мМ, 200-1800 мМ, 400-1800 мМ, 600-1800 мМ, 800-1800 мМ, 1000-1800 мМ, 1200-1800 мМ, 1400-1800 мМ, 1600-1800 мМ; 5-1500 мМ, 25-1400 мМ, 50-1500 мМ, 100-1500 мМ, 200-1500 мМ, 400-1500 мМ, 600-1500 мМ, 800-1500 мМ, 1000-1500 мМ, 1200-1500 мМ; 5-1200 мМ, 25-1200 мМ, 50-1200 мМ, 100-1200 мМ, 200-1200 мМ, 400-1200 мМ, 600-1200 мМ или 800-1200 мМ.
В одном воплощении изобретения по меньшей мере один агент (5), модифицирующий тоничность, представляет собой агент (5/а), модифицирующий ионную силу.
Используемый в данном описании термин "агент, модифицирующий ионную силу", включает в себя агенты, которые вносят вклад в ионную силу раствора. Эти агенты включают, но не ограничиваются этим, нейтральные соли, аминокислоты, пептиды из 2-5 аминокислотных остатков. В некоторых воплощениях композиция содержит два или более таких агентов в комбинации.
Не ограничивающие примеры агентов, модифицирующих ионную силу (5/а), представляют собой нейтральные соли, такие как хлорид натрия, хлорид калия, хлорид кальция и хлорид магния. В одном воплощении этот агент (5/а) представляет собой хлорид натрия.
Термин "ионная сила" означает ионную силу раствора (µ), которую определяют уравнением: µ=1/2∑([i](Zi 2)), где и означает ионную силу, [i] означает миллимолярную концентрацию иона, и Zi представляет собой заряд (+ или -) этого иона (см., например, Solomon, Journal of Chemical Education, 78(12):1691-92, 2001; James Fritz and George Schenk: Quantitative Analytical Chemistry, 1979).
В различных воплощениях изобретения ионная сила композиции составляет по меньшей мере 50 мМ, например, по меньшей мере 75 мМ, по меньшей мере 100 мМ, по меньшей мере 150 мМ, по меньшей мере 200 мМ, по меньшей мере 250 мМ, по меньшей мере 400 мМ, по меньшей мере 500 мМ, по меньшей мере 650 мМ, по меньшей мере 800 мМ, по меньшей мере 1000 мМ, по меньшей мере 1200 мМ, по меньшей мере 1600 мМ, по меньшей мере 2000 мМ, по меньшей мере 2400 мМ, по меньшей мере 2800 мМ или по меньшей мере 3200 мМ.
В некоторых конкретных воплощениях общая концентрация агента (5), модифицирующего тоничность, и агента (5/а), модифицирующего ионную силу, находится в диапазоне 1-1000 мМ, таком как 1-500 мМ, 1-300 мМ, 10-200 мМ или 20-150 мМ; или таком как 100-1000 мМ, 200-800 мМ или 500-800 мМ, причем другие ингредиенты могут оказывать влияние на тоничность и ионную силу.
В одном воплощении композиция является изотонической; в другом воплощении она является гипертонической.
Термин "изотонический" означает "изотонический с сывороткой", т.е. при примерно 300±50 миллиосмоль/кг. Подразумевается, что тоничность является мерой осмотического давления раствора перед введением. Термин "гипертонический" предназначен для обозначения уровней осмотического давления выше физиологического уровня сыворотки, таких как уровни выше 300±50 миллиосмоль/кг.
Кроме того, конкретное воплощение настоящего изобретения относится к комбинации стабилизирующего агента (3) с довольно высокой концентрацией агента (5/а), модифицирующего ионную силу. В одном его воплощении агент (5/а), модифицирующий ионную силу, выбран из группы, состоящей из солей натрия, солей кальция и солей магния. В этом воплощении агент (5/а), модифицирующий ионную силу, т.е. соль натрия, соль кальция и/или соль магния, присутствует в концентрации 15-1500 мМ, такой как 15-1000 мМ, 25-1000 мМ, 50-1000 мМ, 100-1000 мМ, 200-1000 мМ, 300-1000 мМ, 400-1000 мМ, 500-1000 мМ, 600-1000 мМ, 700-1000 мМ; 15-800 мМ, 25-800 мМ, 50-800 мМ, 100-800 мМ, 200-800 мМ, 300-800 мМ, 400-800 мМ, 500-800 мМ; 15-600 мМ, 25-600 мМ, 50-600 мМ, 100-600 мМ, 200-600 мМ, 300-600 мМ; 15-400 мМ, 25-400 мМ, 50-400 мМ или 100-400 мМ.
В этих воплощениях соль натрия может представлять собой хлорид натрия, соль кальция может быть выбрана из группы, состоящей из хлорида кальция, ацетата кальция, глюконата кальция и левулата кальция, и соль магния может быть выбрана из группы, состоящей из хлорида магния, ацетата магния, глюконата магния, левулата магния и солей магния сильных кислот. В более конкретном воплощении соль кальция и/или соль магния используют в комбинации с хлоридом натрия.
В одном воплощении композиция содержит один или более чем один агент, модифицирующий ионную силу, выбранный из группы, состоящей из солей кальция (Са2+) и солей магния (Мg2+), например одну или более чем одну соль, выбранную из группы, состоящей из хлорида кальция, ацетата кальция, глюконата кальция, левулата кальция, хлорида магния, ацетата магния, сульфата магния, глюконата магния, левулата магния, солей магния сильных кислот.
В одном воплощении кальций (Са2+) и/или магний (Мg2+) присутствует в концентрации по меньшей мере примерно 0,1 мкМ, такой как, например, по меньшей мере примерно 0,5 мкМ, по меньшей мере примерно 1 мкМ, по меньшей мере примерно 5 мкМ, по меньшей мере примерно 10 мкМ, по меньшей мере примерно 50 мкМ, по меньшей мере примерно 100 мкМ, по меньшей мере примерно 1 мМ, по меньшей мере примерно 2 мМ, по меньшей мере примерно 5 мМ или по меньшей мере примерно 10 мМ. В конкретном воплощении композиция содержит по меньшей мере 2 мМ Са2+.
В различных воплощениях молярное соотношение между ионами кальция (Са2+) и/или магния (Мg2+) и полипептидом FVII составляет: 0,001-750; 0,001-250; 0,001-100; 0,001-10; 0,001-1,0; 0,001-0,5; 0,5-750; 0,5-250; 0,5-100; 0,5-10; 0,5-1,0; 0,001-0,4999; 0,005-0,050.
В одном воплощении настоящего изобретения молярное соотношение некомплексного кальция (Са2+) и/или магния (Мg2+) c полипептидом фактора VII составляет менее 0,5, например, находится в диапазоне 0,001-0,499, таком как 0,005-0,050, или в диапазоне 0,000-0,499, таком как в диапазоне 0,000-0,050, или примерно 0,000. В одном воплощении настоящего изобретения молярное соотношение некомплексного кальция (Са2+) c полипептидом фактора VII составляет менее 0,5, например, в диапазоне 0,001-0,499, таком как 0,005-0,050, или в диапазоне 0,000-0,499, таком как в диапазоне 0,000-0,050, или примерно 0,000.
Используемый в данном описании термин "концентрация некомплексных ионов кальция и/или магния" предназначен для обозначения различия между общей концентрацией ионов кальция и/или магния и концентрацией кальция и/или магния, связанных хелатообразующими агентами кальция/магния. В этом отношении полипептид фактора VII не рассматривается в качестве "хелатообразующего агента кальция/магния", хотя предполагается, что кальций и/или магний связываются или становятся связанными с полипептидом фактора VII в определенных условиях.
В другом воплощении молярное соотношение некомплексных ионов кальция и/или магния с полипептидом фактора VII составляет более 0,5. В другом воплощении молярное соотношение некомплексных ионов кальция с полипептидом фактора VII составляет более 0,5.
Для того чтобы получить низкое относительное соотношение между ионами кальция и/или магния (Са2+) и полипептидом фактора VII, может быть необходимо или желательно удалить избыток ионов кальция и/или магния, например путем приведения композиции в контакт с ионообменным материалом в условиях, подходящих для удаления Са2+ и/или Мg2+, или добавить хелатообразующий агент кальция/магния для того, чтобы связать (образовать комплекс) избыток ионов кальция и/или магния. Это особенно важно, когда соотношение между ионами кальция и/или магния и полипептидом фактора VII в растворе из стадии способа, предшествующей стадии приготовления препарата, превышает предел, указанный выше. Примеры "кальциевых/магниевых хелатообразующих агентов" включают EDTA (этилендиаминтетрауксусную кислоту), лимонную кислоту, NTA (нитрилтрехуксусную кислоту), DTPA (диэтилентриаминпентауксусную кислоту), винную кислоту, молочную кислоту, яблочную кислоту, янтарную кислоту, HIMDA (N-(2-гидроксиэтил)-иминодиуксусную кислоту), ADA (N-(2-ацетамидо)-иминодиуксусную кислоту) и аналогичные соединения.
В еще одном воплощении композиция дополнительно содержит антиоксидант (6). В различных воплощениях антиоксидант выбран из группы, состоящей из L-метионина, D-метионина, аналогов метионина, метионинсодержащих пептидов, гомологов метионина, аскорбиновой кислоты, цистеина, гомоцистеина, глутатиона, цистина и цистатионина. В предпочтительном воплощении антиоксидант представляет собой L-метионин.
Концентрация антиоксиданта составляет как правило 0,1-5,0 мг/мл, например, 0,1-4,0 мг/мл, 0,1-3,0 мг/мл, 0,1-2,0 мг/мл или 0,5-2,0 мг/мл.
В особых воплощениях композиция не содержит антиоксидант; вместо этого чувствительность полипептида фактора VII к окислению контролируется путем исключения атмосферного воздуха. Применение антиоксиданта, конечно, может быть также объединено с исключением атмосферного воздуха.
Таким образом, в настоящем изобретении также предложен герметичный контейнер (например ампула или картридж (такой как картридж для ручки-аппликатора)), содержащий жидкую водную фармацевтическую композицию, как определено в данном описании, и возможно инертный газ.
Инертный газ может быть выбран из группы, состоящей из азота, аргона и т.д. Контейнер (например, ампула или картридж) как правило выполнены из стекла или пластика, в частности стекла, возможно, закрытого резиновой мембраной или другим укупорочным средством, делающим возможным проникновение с сохранением целостности фармацевтической композиции. В особом воплощении композиция не содержит консервант (7). В еще одном воплощении контейнер представляет собой ампулу или картридж, вложенные в герметичный пакет, например герметичный пластиковый пакет, такой как пакет из ламинированного материала (например пластиковый пакет, покрытый металлом (таким как алюминий)).
Кроме обязательных компонентов, неионного поверхностно-активного вещества (4), агента, модифицирующего тоничность (5), и возможного антиоксиданта (6), фармацевтическая композиция может дополнительно содержать консервант (7).
Консервант может быть включен в композицию для замедления микробного роста, в результате чего обеспечивется возможность "многократно использовать" упаковку для полипептидов фактора VII. Примеры консервантов включают фенол, бензиловый спирт, орто-крезол, мета-крезол, пара-крезол, метилпарабен, пропилпарабен, бензалкония хлорид и бензэтония хлорид. Консервант обычно включают в концентрации 0,1-20 мг/мл в зависимости от диапазона рН и типа консерванта.
Кроме того, композиция может также включать один или более агентов, способных ингибировать деамидирование и изомеризацию.
В одном воплощении жидкая водная фармацевтическая композиция содержит:
0,1-20 мг/мл полипептида фактора VII (1);
буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0;
по меньшей мере один стабилизирующий агент (3), содержащий фрагмент -C6H4-C(=N-Z1-R1)-NH-Z2-R2, в концентрации по меньшей мере 5 мкМ;
неионное поверхностно-активное вещество (4); и
по меньшей мере один агент (5), модифицирующий тоничность, в концентрации по меньшей мере 5 мМ.
В другом воплощении жидкая водная фармацевтическая композиция содержит:
0,1-10 мг/мл полипептида фактора VII (1);
буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0;
по меньшей мере один стабилизирующий агент (3), содержащий фрагмент -CH2-NH-C(=N-Z1-R1)-NH-Z2-R2, в концентрации по меньшей мере 500 мкМ;
неионное поверхностно-активное вещество (4); и
по меньшей мере один агент (5), модифицирующий тоничность (5), в концентрации по меньшей мере 5 мМ.
Предполагают, что используемые в данном описании значения рН, определенные как "примерно", означают ±0,1, например, примерно рН 8,0 включает рН 8,0±0,1.
Процентные отношения представляют собой отношения «масса/масса» как при ссылке на твердые вещества, растворенные в растворе, так и на жидкости, смешанные в растворы. Например, для Tween® это означает отношение массы 100%-ного исходного раствора к массе раствора.
Композиции по настоящему изобретению применимы в качестве стабильных и предпочтительно готовых для применения композиций полипептидов фактора VII. Кроме того, предполагают, что принципы, рекомендации и конкретные воплощения, предложенные в данном описании, в равной степени применимы для хранения полипептидов фактора VII в больших количествах, mutatis mutandis (с соответствующими изменениями). Композиции как правило стабильны в течение по меньшей мере шести месяцев, предпочтительно вплоть до 36 месяцев; когда хранятся при температурах от 2°С до 8°С. Композиции являются химически и/или физически стабильными, в частности химически стабильными, когда хранятся в течение по меньшей мере 6 месяцев при температурах от 2°С до 8°С.
Термин "стабильный" предназначен для обозначения того, что (1) после хранения в течение 6 месяцев при температуре от 2°С до 8°С композиция сохраняет по меньшей мере 50% своей исходной биологической активности, как измерено посредством одностадийного анализа свертывания крови, как описано в Анализе 4 настоящего описания, или что (2) после хранения в течение 6 месяцев при температуре от 2°С до 8°С увеличение в содержании продуктов деградации тяжелых цепей составляет не более 40% (мас./мас.) от исходного содержания полипептида фактора VII.
Термин "исходное содержание" относится к количеству полипептидов фактора VII, добавленных к композиции при изготовлении этой композиции.
В одном воплощении стабильная композиция сохраняет по меньшей мере 70%, например, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90% или по меньшей мере 95% своей исходной биологической активности после хранения в течение 6 месяцев при температуре от 2 до 8°С.
В различных воплощениях изобретения стабильная композиция дополнительно сохраняет по меньшей мере 50% своей исходной биологической активности, как измерено посредством одностадийного анализа свертывания крови, как описано в Анализе 4 настоящего описания, после хранения в течение по меньшей мере 30 дней, например 60 дней или 90 дней.
В различных воплощениях увеличение в содержании продуктов деградации тяжелых цепей в стабильных композициях составляет не более примерно 30% (мас./мас.), не более примерно 25% (мас./мас.), не более примерно 20% (мас./мас.), не более примерно 15% (мас./мас.), не более примерно 10% (мас./мас.), не более примерно 5% (мас./мас.) или не более примерно 3% (мас./мас.) от исходного содержания полипептида фактора VII.
С целью определения содержания продуктов деградации тяжелых цепей, ВЭЖХ с обращенной фазой осуществляли на колонке 4,5×250 мм с бутилсвязанным силикагелем с размером частиц 5 мкм и размером пор 300 Å. Температура колонки: 70°С. А-буфер: 0,1% (об./об.) трифторуксусной кислоты. В-буфер: 0,09% (об./об.) трифторуксусной кислоты, 80% (об./об.) ацетонитрила. Колонку элюировали линейным градиентом от Х до (Х+13)% В в течении 30 минут. Х устанавливали таким образом, чтобы FVIIa элюировался с временем удерживания приблизительно 26 минут. Скорость потока: 1,0 мл/мин. Детекция: 214 нм. Нагрузка: 25 мкг FVIIa.
Термин "физическая стабильность" полипептидов фактора VII относится к образованию нерастворимых и/или растворимых агрегатов в виде димерных, олигомерных и полимерных форм полипептидов фактора VII, а также к любой структурной деформации и денатурации молекулы. Физически стабильная композиция охватывает композиции, которые остаются визуально прозрачными. Физическая стабильность композиций часто оценивается посредством визуального контроля и помутнения композиции после хранения при различных температурах в течение различных периодов времени. Визуальный контроль композиций осуществляют в резко сфокусированном свете с темным фоном. Композиция рассматривается как физически нестабильная, когда она демонстрирует видимое помутнение.
Термин "химическая стабильность" предназначен для обозначения появления любого химического изменения в полипептидах фактора VII при хранении в растворе в ускоренных условиях. Примеры представляют собой гидролиз, деамидирование и окисление, а также ферментативную деградацию, являющуюся результатом образования фрагментов полипептидов фактора VII. В частности, серосодержащие аминокислоты склонны к окислению с образованием соответствующих сульфоксидов.
Термин "химически стабильная" предназначен для обозначения композиции, которая сохраняет по меньшей мере 50% своей исходной биологической активности после хранения в течение 6 месяцев при температуре от 2 до 8°С, как измерено посредством одностадийного анализа свертывания крови (Анализ 4).
В следующем аспекте изобретения также предложен способ получения жидкой водной фармацевтической композиции полипептида фактора VII, включающий стадию получения полипептида фактора VII в концентрации по меньшей мере 0,01 мг/мл (1) в растворе, содержащем буферный агент (2), подходящий для поддержания рН в диапазоне от примерно 4,0 до примерно 9,0; и по меньшей мере один стабилизирующий агент (3), содержащий фрагмент -C(=N-Z1-R1)-NH-Z2-R2, где
Z1 и Z2 независимо выбраны из группы, состоящей из -О-, -S-, -NRH- и одинарной связи, где RH выбран из группы, состоящей из водорода, C1-4-алкила, арила и арилметила, и R1 и R2 независимо выбраны из группы, состоящей из водорода, возможно замещенного С1-6-алкила, возможно замещенного С2-6-алкенила, возможно замещенного арила, возможно замещенного гетероциклила, или
Z2 и R2 - такие, как определено выше, и -C=N-Z1-R1 образует часть гетероциклического кольца, или
Z1 и R1 - такие, как определено выше, и -C-NH-Z2-R2 образует часть гетероциклического кольца, или
-C(=N-Z1-R1)-NH-Z2-R2 образует гетероциклическое кольцо, где -Z1-R1-R2-Z2-представляет собой бирадикал.
Способы применения
Как будет понятно, жидкие водные фармацевтические композиции, определенные в данном описании, могут быть использованы в области медицины. Поэтому в настоящем изобретении, в частности, предложены жидкие водные фармацевтические композиции, определенные в данном описании, для применения в качестве лекарства, более конкретно для применения в качестве лекарства для лечения синдрома, зависимого от фактора VII.
Поэтому в настоящем изобретении также предложено применение жидкой водной фармацевтической композиции, как определено в данном описании, для изготовления лекарства для лечения синдрома, зависимого от фактора VII, а также способ лечения синдрома, зависимого от фактора VII, включающий введение субъекту, нуждающемуся в этом, эффективного количества жидкой водной фармацевтической композиции, как определено в данном описании.
Препараты по настоящему изобретению могут быть использованы для лечения любого синдрома, зависимого от фактора VII, таких как, например, расстройства, связанные с кровотечениями, включая расстройства, вызванные дефицитом факторов свертывания крови (например, гемофилию А, гемофилию В, дефицит фактора коагуляции XI, дефицит фактора коагуляции VII), тромбоцитопенией или болезнью фон Виллебранда, или ингибиторами фактора свертывания крови и внутри мозговым кровоизлиянием, или чрезмерным кровотечением по любой причине. Препараты можно также вводить пациентам в сочетании с хирургической или другой травмой или пациентам, получающим антикоагулянтную терапию.
Термин "эффективное количество" означает эффективную дозу, определяемую квалифицированным практикующим врачом, который может определить дозировки для достижения желательного ответа. Факторы для рассмотрения дозы будут включать в себя эффективность, биодоступность, желательные фармакокинетические/фармакодинамические профили, условия лечения, факторы, связанные с пациентом (например, масса, общее состояние здоровья, возраст и т.д.), наличие совместно вводимых лекарств (например, антикоагулянтов), время введения или другие факторы, известные специалисту в области медицины.
Термин "лечение" определяется как помощь и уход за субъектом, например млекопитающим, в частности человеком, с целью противостояния заболеванию, состоянию или расстройству, и включает в себя введение полипептида фактора VII с целью предупреждения появления симптомов или осложнений, или облегчения симптомов или осложнений, или устранения данного заболевания, состояния или расстройства. Фармацевтические композиции по настоящему изобретению, содержащие полипептид фактора VII, могут быть введены субъектам, нуждающимся в таком лечении, парентерально. Парентеральное введение может быть осуществлено подкожной, внутримышечной или внутривенной инъекцией посредством шприца, возможно шприца-ручки. Альтернативно, парентеральное введение может быть осуществлено посредством инфузионного насоса.
В важных воплощениях фармацевтическая композиция адаптирована для подкожной, внутримышечной или внутривенной инъекции в соответствии со способами, известными в данной области.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Основные методы
Анализы, подходящие для определения биологической активности полипептидов фактора VII
Полипептиды фактора VII, применяемые согласно настоящему изобретению, могут быть выбраны с помощью подходящих анализов, которые можно осуществлять в виде простых предварительных тестов in vitro. Соответственно, в настоящем описании раскрыт простой тест (названный "Анализ гидролиза in vitro") на активность полипептидов фактора VII.
Анализ гидролиза in vitro (Анализ 1)
Нативный (дикого типа) фактор VIIa и полипептид фактора VII (оба в дальнейшем обозначаются как "фактор VIIa") могут быть проанализированы на специфические активности. Они могут быть также проанализированы параллельно для прямого сравнения их специфических активностей. Этот анализ проводят в титрационном микропланшете (MaxiSorp, Nunc, Denmark). Хромогенный субстрат D-Ile-Pro-Arg-п-нитроанилид (S-2288, Chromogenix, Sweden) в конечной концентрации 1 мМ добавляют к фактору VIIa (конечная концентрация 100 нМ) в 50 мМ HEPES, рН 7,4, содержащем 0,1 М NaCl, 5 мМ CaCl2 и 1 мг/мл бычьего сывороточного альбумина. Поглощение при 405 нм измеряют непрерывно в планшет-ридере SpectraMaxTM 340 (Molecular Devices, USA). Поглощение, развивающееся в течение 20-минутной инкубации, после вычитания поглощения в контрольной лунке, не содержащей фермент, используют для вычисления соотношения между активностями полипептида фактора VII и фактора VIIa дикого типа:
Соотношение=(полипептид фактора VII при А405 нм)/(фактор VIIa дикого типа при А405 нм).
На основании этого могут быть идентифицированы полипептиды фактора VII с активностью, меньшей, чем сопоставимой с или более высокой, чем активность нативного фактора VIIa, такие как, например, полипептиды фактора VII, где соотношение между активностью полипептида фактора VII и активностью нативного фактора VII (FVII дикого типа) составляет примерно 1,0 в сравнении с выше чем 1,0.
Активность полипептидов фактора VII может быть также измерена с использованием физиологического субстрата, такого как фактор Х ("Анализ протеолиза in vitro"), соответственно при концентрации 100-1000 нМ, где образующийся фактор Ха измеряют после добавления подходящего хромогенного субстрата (например S-2765). Кроме того, анализ активности можно проводить при физиологической температуре.
Анализ протеолиза in vitro (Анализ 2)
Нативный (дикого типа) фактор VIIa и полипептид фактора VII (оба в дальнейшем обозначаются как "фактор VIIa") проанализированы параллельно для прямого сравнения их специфических активностей. Анализ проводят в титрационном микропланшете (MaxiSorp, Nunc, Denmark). Фактор VIIa (10 нМ) и фактор Х (0,8 мкМ) в 100 мкл 50 мМ HEPES, рН 7,4, содержащего 0,1 М NaCl, 5 мМ CaCl2 и 1 мг/мл бычьего сывороточного альбумина, инкубируют в течение 15 мин. Расщепление фактора Х затем останавливают путем добавления 50 мкл 50 мМ HEPES, рН 7,4, содержащего 0,1 М NaCl, 20 мМ EDTA и 1 мг/мл бычьего сывороточного альбумина. Количество образующегося фактора Ха измеряют путем добавления хромогенного субстрата Z-D-Arg-Gly-Аrg-п-нитроанилида (S-2765, Chromogenix, Sweden) в конечной концентрации 0,5 мМ. Поглощение при 405 нм измеряют непрерывно в планшет-ридере SpectraMaxTM 340 (Molecular Devices, USA). Поглощение, развивающееся в течение 10 минут, после вычитания поглощения в контрольной лунке, не содержащей FVIIa, используют для вычисления соотношения между протеолитическими активностями полипептида фактора VII и фактора VIIa дикого типа:
Соотношение=(полипептид фактора VII при А405 нм)/(фактор VIIa дикого типа при А405 нм).
На основании этого могут быть идентифицированы полипептиды фактора VII с активностью, меньшей, чем сопоставимой с или более высокой, чем активность нативного фактора VIIa, такие как, например, полипептиды фактора VII, где соотношение между активностью полипептида фактора VII и активностью нативного фактора VII (FVII дикого типа) составляет примерно 1,0 в сравнении с выше чем 1,0.
Анализ образования тромбина (Анализ 3)
Способность фактора VIIa или полипептидов фактора VII образовывать тромбин может быть также измерена в анализе (Анализ 3), включающем все релевантные факторы коагуляции и ингибиторы в физиологических концентрациях (минус фактор VIII при имитации условий гемофилии А) и активированные тромбоциты (как описано на стр.543 в Monroe et al. (1997) Brit. J. Haematol. 99, 542-547, которое включено таким образом в данное описание в виде ссылки).
Одностадийный анализ коагуляции (анализ свертывания крови) (Анализ 4) Полипептиды фактора VII могут быть также проанализированы в отношении специфических активностей ("коагуляционная активность") с использованием одностадийного анализа свертывания крови (Анализ 4). Для этой цели тестируемый образец разбавляют в 50 мМ PIPES-буфера (рН 7,5), 0,1% БСА и 40 мкл инкубируют с 40 мкл плазмы с дефицитом фактора VII и 80 мкл человеческого рекомбинантного тканевого фактора, содержащего 10 мМ Са2+ и синтетические фосфолипиды. Время коагуляции (время свертывания) измеряют и сравнивают со стандартной кривой, используя ссылочный стандарт в параллельном анализе.
Получение и очистка полипептидов фактора VII
Человеческий очищенный фактор VIIa, подходящий для применения в настоящем изобретении, предпочтительно получают технологией рекомбинантной ДНК, например, как описано Наgеn с соавт. в Proc.Natl.Acad.Sci. USA 83: 2412-2416, 1986, или как описано в европейском патенте №0200421 (ZymoGenetics, inc.).
Фактор VII может быть также получен способами, описанными Broze и Majerus в J.Biol.Chem. 255 (4): 1242-1247, 1980, и Hedner и Kisiel в J.Clin. Invest. 71: 1836-1841, 1983. Эти способы дают фактор VII без детектируемых количеств других факторов свертывания крови. Еще более очищенный препарат фактора VII может быть получен путем включения дополнительной гель-фильтрации в качестве конечной стадии очистки. Фактор VII затем превращают в активированный фактор VIIa известными способами, например с помощью некоторых различных плазматических белков, таких как фактор XIIa, IXa или Ха. Альтернативно, как описано Bjoern с соавт. (Research Disclosure, 269 September 1986, pp.564-565), фактор VII может быть активирован путем пропускания его через ионообменную хроматографическую колонку, такую как Mono Q® (Pharmacia fine Chemicals) или тому подобное, или путем самоактивации в растворе.
Полипептиды, родственные фактору VII, могут быть получены модификацией фактора VII дикого типа или рекомбинантной технологией. Полипептиды, родственные фактору VII, с измененной аминокислотной последовательностью, при сравнении с фактором VII дикого типа, могут быть получены путем модификации последовательности нуклеиновой кислоты, кодирующей фактор VII дикого типа, либо путем изменения аминокислотных кодонов, либо путем удаления некоторых аминокислотных кодонов в нуклеиновой кислоте, кодирующей природный фактор VII, известными способами, например сайт-специфическим мутагенезом.
Специалистам в данной области будет очевидно, что замены могут быть осуществлены за пределами областей, ответственных за функционирование молекулы фактора VIIa, и все же давать активный полипептид. Аминокислотные остатки, существенные для активности полипептида фактора VII, и поэтому предпочтительно не подвергнутые замене, могут быть идентифицированы согласно процедурам, известным в данной области, таким как сайт-специфический мутагенез или аланин-сканирующий мутагенез (смотри, например, Cunningham and Wells, 1989, Science 244: 1081-1085). В последней методике мутации вводят по любому положительно заряженному остатку в молекуле, и полученные мутантные молекулы тестируют на коагулянтную, соответственно перекрестно-сшивающую активность для идентификации аминокислотных остатков, которые являются критическими для активности данной молекулы. Сайты субстрат-ферментного взаимодействия могут быть также определены путем анализа трехмерной структуры, как определено такими методами, как ядерный магнитный резонансный анализ, кристаллография или фотоаффинное мечение (смотри, например, de Vos et al., 1992, Science 255: 306-312; Smith et al., 1992, Journal of Molecular Biology 224: 899-904; Wlodaver et al., 1992, FEBS Letters 309: 59-64).
Введение мутации в последовательность нуклеиновой кислоты для замены одного нуклеотида другим нуклеотидом может быть осуществлено сайт-специфическим мутагенезом с использованием любого из способов, известных в данной области. Особенно полезной является процедура, в которой используют суперскрученный двунитевой ДНК-вектор с интересующей вставкой и два синтетических праймера, содержащих желательную мутацию. Олигонуклеотидные праймеры, каждый комплементарный противоположным нитям вектора, удлиняются во время термоциклирования посредством Pfu ДНК-полимеразы. После введения праймеров образуется мутантная плазмида, содержащая разбросанные ники (однонитевые разрывы). После термоциклирования продукт обрабатывают Dpnl, которая является специфической для метилированной и полуметилированной ДНК, для того чтобы переваривать родительскую ДНК-матрицу и селектировать содержащую мутацию синтезированную ДНК. Другие процедуры, известные в данной области для создания, идентификации и выделения вариантов, также могут быть использованы, такие как, например, методы "перетасовки" генов (gene shuffling) или фагового дисплея.
Отделение полипептидов от клеток, из которых они происходят, может быть достигнуто любым способом, известным в данной области, включая, без ограничения, удаление клеточной культуральной среды, содержащей желательный продукт, из адгезивной клеточной культуры; центрифугирование или фильтрацию для удаления неприкрепившихся клеток; и тому подобное.
Возможно, полипептиды фактора VII могут быть дополнительно очищены. Очистка может быть достигнута с использованием любого способа, известного в данной области, включая, без ограничения, аффинную хроматографию, такую как, например, колонка с антителом против фактора VII (смотри, например, Wakabayashi et al, J. Biol. Chem. 261:11097, 1986; и Thim et al., Biochem. 27:7785, 1988); гидрофобную хроматографию; ионообменную хроматографию; гель-фильтрацию; электрофоретические процедуры (например, препаративное изоэлектрофокусирование (ИЭФ), селективную растворимость (например, осаждение сульфатом аммония) или экстракцию и тому подобное. Смотри в основном Scopes, Protein Purification, Springer-Verlag, New York, 1982; и Protein Purification, J.C.Janson and Lars Ryden, editors, VCH Publishers, New York, 1989. После очистки препарат предпочтительно содержит менее 10 мас.%, более предпочтительно менее 5% и наиболее предпочтительно менее 1% полипептидов, не являющихся фактором VII, происходящих из клетки-хозяина.
Полипептиды фактора VII могут быть активированы протеолитическим расщеплением с использованием фактора XIIа или других протеаз, имеющих трипсинподобную специфичность, таких как, например, фактор IХа, калликреин, фактор Ха и тромбин. Смотри, например, Osterud et al., Biochem. 11:2853 (1972); Thomas, патент США № 4456591; и Hedner et al., J. Clin. Invest. 71:1836 (1983). Альтернативно, полипептиды фактора VII могут быть активированы путем пропускания их через ионообменную хроматографическую колонку, такую как Mono Q® (Pharmacia), или тому подобное, или путем самоактивации в растворе. Полученный активированный полипептид фактора VII может быть затем приготовлен в виде препарата и введен, как описано в настоящей заявке.
Следующие примеры иллюстрируют осуществление изобретения. Эти примеры включены только для иллюстративных целей и не предназначены для ограничения объема заявленного изобретения каким-либо образом.
Примеры осуществления изобретения
В примерах, приведенных ниже, содержание продуктов деградации тяжелых цепей определяют посредством ОФ-ВЭЖХ, как описано ниже:
ВЭЖХ с обращенной фазой осуществляли на колонке 4,5×250 мм с бутилсвязанным силикагелем с размером частиц 5 мкм и размером пор 300Å. Температура колонки: 70°С. А-буфер: 0,1% (об./об.) трифторуксусной кислоты. В-буфер: 0,09% (об./об.) трифторуксусной кислоты, 80% (об./об.) ацетонитрила. Колонку элюировали линейным градиентом от Х до (Х+13)% В в течение 30 минут. Х устанавливали таким образом, чтобы FVIIa элюировался с временем удерживания приблизительно 26 минут. Скорость потока: 1,0 мл/мин. Детекция: 214 нм. Нагрузка: 25 мкг FVIIa.
В примерах ниже коагуляционную активность измеряют с использованием одностадийного анализа свертывания крови по существу, как описано в Анализе 4 настоящего описания.
Пример 1
Для исследования влияния бензамидина на стабильность rFVIIa готовили следующие препараты:
Препарат 1
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ ацетата натрия
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мМ бензамидина
рН 6,5
Препарат 2
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ ацетата натрия
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мМ бензамидина
рН 7,0
Препарат 3
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ ацетата натрия
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 6,5
Препарат 4
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ ацетата натрия
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 7,0
Препараты готовили путем добавления 10 мМ гистидина, 10 мМ ацетата натрия и 50 мМ бензамидина (только для препаратов 1 и 2) к 1,0 мг/мл основного раствора rFVIIa, уже содержащего глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно доводили до 6,5 и 7,0 соответственно 1 М гидроксидом натрия и 1 М соляной кислотой.
Препараты хранили при температуре 5°С и 30°С и проводили анализы образования продуктов деградации тяжелых цепей в моменты времени, показанные в Таблице (Таблица 1).
Образование продуктов деградации тяжелых цепей (ДТЖ) в препаратах бензамидина.
Как можно видеть из Таблицы 1, после 6 месяцев хранения при 5°С увеличение в содержании продуктов деградации тяжелых цепей в контрольных препаратах (3 и 4) составляло 34,0% и 57,7% соответственно, тогда как увеличение в содержании продуктов деградации тяжелых цепей в иллюстративных композициях (1 и 2) составляло только 1,4% и 4,2% соответственно. После 14 месяцев хранения при 5°С увеличение в содержании продуктов деградации тяжелых цепей в иллюстративных композициях (1 и 2) составляло только 3,0% и 8,1% соответственно.
Содержание продуктов деградации тяжелых цепей определяют посредством ОФ-ВЭЖХ, как описано ниже:
ВЭЖХ с обращенной фазой осуществляли на колонке 4,5×250 мм с бутилсвязанным силикагелем с размером частиц 5 мкм и размером пор 300 Å. Температура колонки: 70°С. А-буфер: 0,1% (об./об.) трифторуксусной кислоты, В-буфер: 0,09% (об./об.) трифторуксусной кислоты, 80% (об./об.) ацетонитрила. Колонку элюировали линейным градиентом от Х до (Х+13)% В через 30 минут. Х устанавливали таким образом, чтобы, что FVIIa элюировался с временем удержания приблизительно 26 минут. Скорость потока: 1,0 мл/мин. Детекция: 214 нм. Нагрузка: 25 мкг FVIIa.
Пример 2
Для исследования влияния аргинина на стабильность rFVIIa готовили следующие растворы:
Препарат 5
1,0 мг/мл rFVIIa
25 мМ HEPES
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
pН 7,5
Препарат 6
1,0 мг/мл rFVIIa
25 мМ HEPES
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
200 мМ аргинина
рН 7,5
Препарат 7
1,0 мг/мл rFVIIa
20 мМ гистидина
20 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 7,0
Препарат 8
1,0 мг/мл rFVIIa
50 мМ гистидина
50 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
200 мМ аргинина
рН 7,0
Препарат 9
1,0 мг/мл rFVIIa
50 мМ гистидина
50 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
400 мМ аргинина
рН 7,0
Препараты готовили путем добавления 25 мМ HEPES (растворы 5 и 6) и аргинина (растворы 6, 8 и 9) к 1,0 мг/мл основного раствора rFVIIa, уже содержащего глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно подводили 1 М гидроксидом натрия и 1 М соляной кислотой.
Препараты хранили при температуре от 5°С до 30°С и анализы образования продуктов деградации тяжелых цепей проводили как в Примере 1 в моменты времени, показанные в Таблице 2.
Образование продуктов деградации тяжелых цепей (ДТЖ) в препаратах аргинина
н/а: не анализировали
Пример 3
Исследовали приготовление следующих жидких водных фармацевтических композиций:
А)
Б)
В)
Г)
Фармацевтические композиции А-Г могут быть впоследствии перенесены в стерильные ампулы или картриджи, промытые азотом или аргоном, и могут быть затем упакованы в герметичные, покрытые слоем алюминия, пластиковые пакеты.
Пример 4
Для исследования влияния бензамидина на стабильность rFVIIa готовили следующие препараты:
Препарат 1
1 1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 6,5
Препарат 2
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мМ бензамидина
рН 6,5
Препарат 3
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
pН 7,5
Препарат 4
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мМ бензамидина
pН 7,5
Препараты готовили путем добавления 10 мМ гистидина и 50 мМ бензамидина (только для препаратов 2 и 4) к 1,0 мг/мл основного раствора rFVIIa, уже содержащего глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно доводили до 6,5 и 7,5 соответственно 1 М гидроксидом натрия и 1 М соляной кислотой.
Препараты хранили при температуре 5°С, и проводили анализы коагуляционной активности в моменты времени, показанные в Таблице 1:
Коагуляционная активность (МЕ/мл)
Пример 5
Для исследования влияния различных стабилизаторов на стабильность rFVIIa приготавливали следующие препараты:
Препарат 1
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 6,5
Препарат 2
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
5 мМ п-аминобензамидина
рН 6,5
Препарат 3
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
0,5 мМ 8-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 6,5
Препарат 4
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 7,5
Препарат 5
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
5 мМ п-аминобензамидина
рН 7,5
Препарат 6
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
0,5 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 7,5
Препараты готовили путем добавления 10 мМ гистидина, 5 мМ п-аминобензамидина (для препаратов 2 и 5) и 0,5 мМ S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида (для препаратов 3 и 6) к 1,0 мг/мл основного раствора rFVIIa, уже содержащего глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно доводили до 6,5 и 7,5 соответственно 1 М гидроксидом натрия и 1 М соляной кислотой.
Препараты хранили при температуре 5°С и проводили анализы коагуляционной активности (Таблица 1) и деградации тяжелых цепей (Таблица 2) в моменты времени, показанные в Таблицах:
Коагуляционная активность (МЕ/мл)
Содержание продуктов деградации тяжелой цепи (%)
Пример 6
Для исследования влияния бензамидина и N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамида HCl на стабильность rFVIIa готовили следующие препараты:
Препарат 1
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 6,5
Препарат 2
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мМ бензамидина
рН 6,5
Препарат 3
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мкМ N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамида HCl
рН 6,5
Препарат 4
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
500 мкМ N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамида HCl
рН 6,5
Препарат 5
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
pН 7,5
Препарат 6
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мМ бензамидина
pH 7,5
Препарат 7
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
50 мкМ N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамида HCl
pН 7,5
Препарат 8
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
500 мкМ N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамида HCl
pН 7,5
Препараты готовили путем добавления 10 мМ гистидина, 50 мМ бензамидина (для препаратов 2 и 6) и 50/500 мкМ N-(3-бромбензил)-2-[3-(4-карбамимидоилфенил)-уреидо]-ацетамида HCl (для препарата 3, 4 и 7,8 к 1,0 мг/мл основного раствора rFVIIa, уже содержащего глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно доводили до 6,5 и 7,5 соответственно 1 М гидроксидом натрия и 1 М соляной кислотой.
Препараты хранили при температуре 5°С и 30°С, и проводили анализы коагуляционной активности в моменты времени, показанные в Таблице 1:
Коагуляционная активность (МЕ/мл)
Пример 7
Для исследования влияния различных стабилизаторов на стабильность rFVIIa приготавливали следующие препараты:
Препарат 1
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 6,5
Препарат 2
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
5 мМ п-аминобензамидина
рН 6,5
Препарат 3
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
0,05 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 6,5
Препарат 4
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
0,5 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 6,5
Препарат 5
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
рН 7,5
Препарат 6
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
5 мМ п-аминобензамидина
рН 7,5
Препарат 7
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
0,05 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 7,5
Препарат 8
1,0 мг/мл rFVIIa
10 мМ гистидина
10 мМ глицилглицина
50 мМ хлорида натрия
10 мМ хлорида кальция
0,5 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 7,5
Препараты готовили путем добавления 10 мМ гистидина, 5 мМ п-аминобензамидина (для препаратов 2 и 6) и 0,05/0,5 мМ S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида (для препаратов 3, 7 и 4, 8) к 1,0 мг/мл основного раствора rFVIIa, уже содержащего глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно доводили до 6,5 и 7,5 соответственно 1 М гидроксидом натрия и 1 М соляной кислотой.
Препараты хранили при температуре 5°С, и проводили анализы коагуляционной активности (Таблица 1) и деградации тяжелых цепей (Таблица 2) в моменты времени, показанные в Таблицах:
Коагуляционная активность (МЕ/мл)
Содержание продуктов деградации тяжелой цепи (%)
Пример 8
Для исследования влияния S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида на стабильность rFVIIa приготавливали следующие препараты:
Препарат 1
1,0 мг/мл rFVIIa
50 мМ хлорида натрия
10 мМ хлорида кальция
10 мМ глицилглицина
20 мМ гистидина
0,5 мг/мл метионина
5 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 6,0
Препарат 2
1,0 мг/мл rFVIIa
50 мМ хлорида натрия
10 мМ хлорида кальция
10 мМ глицилглицина
20 мМ гистидина
0,5 мг/мл метионина
5 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 6,5
Препарат 3
1,0 мг/мл rFVIIa
50 мМ хлорида натрия
10 мМ хлорида кальция
10 мМ глицилглицина
20 мМ гистидина
0,5 мг/мл метионина
5 мМ
S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида
рН 7,5
Препараты готовили путем добавления 20 мМ гистидина, 5 мМ S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида и 0,5 мг/мл метионина к раствору rFVIIa, уже содержащему глицилглицин, хлорид натрия и хлорид кальция в вышеупомянутых концентрациях. рН окончательно доводили до 6,0, 6,5 и 7,5, соответственно, 1 М гидроксидом натрия/соляной кислотой.
Препараты хранили при температуре 25°С, и анализы коагуляционной активности (Таблица 1) и деградации тяжелых цепей (Таблица 2) проводили в моменты времени, показанные в Таблицах:
Коагуляционная активность (МЕ/мл)
Ссылочные препараты без S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамида демонстрируют следующую коагуляционную активность.
Коагуляционная активность (МЕ/мл) для контрольных растворов
Изобретение относится к медицине и касается жидких водных фармацевтических композиций, содержащих полипептиды фактора VII вместе с стабилизирующим агентом, способа получения и применения таких композиций, а также контейнера, содержащего такую композицию, и применения таких композиций для лечения синдрома, зависимого от фактора VII. Изобретение обеспечивает получение жидкой водной фармацевтической композиции, содержащей полипептид фактора VII, в которой снижено образование продуктов химической и/или физической деградации, таких как продукты ферментативной деградации или автокатализа. 5 н. и 19 з.п. ф-лы, 10 табл.
1. Жидкая водная фармацевтическая композиция, адаптированная для парентерального введения, содержащая:
по меньшей мере 0,01 мг/мл полипептида фактора VII;
буферный агент, подходящий для поддержания рН в диапазоне от примерно 5,0 до примерно 9,0; и
по меньшей мере один стабилизирующий агент, содержащий фрагмент -C(=NH)-NH2.
2. Композиция по п.1, где стабилизирующий агент представляет собой по меньшей мере одно амидиновое соединение, выбранное из группы, состоящей из бензамидинов, содержащих фрагмент -С6Н4-С(=NН)-NН2, где С6Н4 означает возможно замещенное бензольное кольцо.
3. Композиция по п.2, где бензамидины содержат фрагмент >N-C6H4-C(=NH)-NH2, где С6Н4 означает возможно замещенное бензольное кольцо.
4. Композиция по п.1, где молекулярная масса стабилизирующего агента составляет не более 1000 Да.
5. Композиция по п.1, где концентрация стабилизирующего агента составляет по меньшей мере 1 мкМ.
6. Композиция по п.1, где стабилизирующий агент представляет собой п-аминобензамидин, и концентрация указанного агента составляет по меньшей мере 0,001 мМ.
7. Композиция по п.1, где стабилизирующий агент представляет собой S-2-[3-(4-карбамимидоилфенил)-уреидо]-N-[1-(3-метоксифенил)-этил]-ацетамид, и концентрация указанного агента составляет по меньшей мере 0,001 мМ.
8. Композиция по п.1, где полипептид фактора VII представляет собой фактор VIIa человека.
9. Композиция по п.1, где полипептид фактора VII представляет собой вариант последовательности фактора VII.
10. Композиция по п.1, дополнительно содержащая неионное поверхностно-активное вещество.
11. Композиция по п.10, где неионное поверхностно-активное вещество представляет собой по меньшей мере одно поверхностно-активное вещество, выбранное из группы, состоящей из полисорбатов, полоксамеров, полиоксиэтиленалкиловых зфиров, блок-сополимеров этилена/полипропилена и полиэтиленгликоля (ПЭГ).
12. Композиция по п.1, дополнительно содержащая агент, модифицирующий тоничность.
13. Композиция по п.12, где агент, модифицирующий тоничность, представляет собой по меньшей мере один агент, выбранный из группы, состоящей из нейтральных солей, аминокислот, пептидов из 2-5 аминокислотных остатков, моносахаридов, дисахаридов, полисахаридов и сахарных спиртов.
14. Композиция по п.12 или 13, где агент, модифицирующий тоничность, присутствует в концентрации по меньшей мере 1 мМ.
15. Композиция по п.1, дополнительно содержащая антиоксидант.
16. Композиция по п.15, где антиоксидант выбран из L-метионина, D-метионина, аналогов метионина, метионинсодержащих пептидов, гомологов метионина, аскорбиновой кислоты, цистеина, гомоцистеина, глутатиона, цистина и цистатионина.
17. Композиция по п.15 или 16, где антиоксидант присутствует в концентрации 0,1-5,0 мг/мл.
18. Композиция по п.1, дополнительно содержащая консервант.
19. Композиция по п.18, где консервант выбран из группы, состоящей из фенола, бензилового спирта, орто-крезола, мета-крезола, пapa-крезола, метилпарабена, пропилпарабена, бензалкония хлорида и бензаэтония хлорида.
20. Композиция по п.1, адаптированная для подкожной, внутримышечной или внутривенной инъекции.
21. Способ получения жидкой водной фармацевтической композиции полипептида фактора VII, адаптированной для парентерального введения, включающий получение раствора полипептида фактора VII в концентрации по меньшей мере 0,01 мг/мл, содержащего:
буферный агент, подходящий для поддержания рН в диапазоне от примерно 5,0 до примерно 9,0; и
по меньшей мере один стабилизирующий агент, включающий фрагмент -C(=NH)-NH2.
22. Применение жидкой водной фармацевтической композиции, охарактеризованной в любом из пп.1-20, для изготовления лекарственного средства для лечения синдрома, зависимого от фактора VII.
23. Способ лечения синдрома, зависимого от фактора VII, включающий введение субъекту, нуждающемуся в этом, эффективного количества жидкой водной фармацевтической композиции, охарактеризованной в любом из пп.1-20.
24. Герметичный контейнер, содержащий жидкую водную фармацевтическую композицию, охарактеризованную в любом из пп.1-20, и возможно инертный газ.
| РАЗБРАСЫВАТЕЛЬ УДОБРЕНИЙ | 0 |
|
SU182943A1 |
| DE 10131404 (A1), 11.07.2002 | |||
| Sichler K | |||
| et al | |||
| "Crystal structures of uninhibited factor VIIa link its cofactor and substrate-assisted activation to specific interactions" | |||
| J | |||
| Mol Biol | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| JP 62195335 (A), 28.08.1987 | |||
| US 2003109446 (A1), 12.06.2003 | |||
| ИЗМЕРИТЕЛЬ СКО^РОСТИ ИЗЛ\ЕНЕНИЯ ДАВЛЕНИ51'' ^' г?ПШ"и1?^г1'^1СНА1! ^-44ЬЛМОТЕКЛ | 0 |
|
SU336548A1 |

