Область техники, к которой относится изобретение
Настоящее изобретение относится, в целом, к способу жидкофазного каталитического окисления ароматического соединения. Один из аспектов настоящего изобретения относится к частичному окислению диалкилароматического соединения (например, пара-ксилола) для получения сырой ароматической дикарбоновой кислоты (например, сырой терефталевой кислоты), которая после этого может подвергаться очистке и разделению. Другой аспект настоящего изобретения относится к усовершенствованной барботажной колонне реакторного типа, которая обеспечивает более эффективный и экономичный способ жидкофазного окисления.
Уровень техники
Жидкофазные реакции окисления используются во множестве существующих промышленных процессов. Например, жидкофазное окисление используется в настоящее время для окисления альдегидов до кислот (например, пропиональдегида до пропионовой кислоты), для окисления циклогексана до адипиновой кислоты и для окисления алкилароматических соединений до спиртов, кислот или дикислот. Особенно важный промышленный способ окисления в последней категории (окисление алкилароматических соединений) представляет собой жидкофазное каталитическое частичное окисление пара-ксилола до терефталевой кислоты. Терефталевая кислота является важным соединением с множеством применений. Первичное использование терефталевой кислоты представляет собой использование в качестве исходного материала при получении полиэтилентерефталата (PET). PET является хорошо известным пластиком, используемым в больших количествах в мире для изготовления таких продуктов, как бутылки, волокна и упаковка.
В типичном способе жидкофазного окисления, включая частичное окисление пара-ксилола до терефталевой кислоты, жидкофазный поток исходных материалов и газофазный поток окислителя вводятся в реактор и образуют многофазную реакционную среду в реакторе. Жидкофазный поток исходных материалов, введенный в реактор, содержит, по меньшей мере, одно окисляемое органическое соединение (например, пара-ксилол), в то время как газофазный поток окислителя содержит молекулярный кислород. По меньшей мере, часть молекулярного кислорода, введенного в реактор в виде газа, растворяется в жидкой фазе реакционной среды, обеспечивая доступность кислорода для жидкофазной реакции. Если жидкая фаза многофазной реакционной среды содержит недостаточную концентрацию молекулярного кислорода (например, если определенные части реакционной среды являются "обедненными кислородом"), нежелательные побочные реакции могут образовывать примеси и/или целевые реакции могут замедляться по скорости. Если жидкая фаза реакционной среды содержит слишком мало окисляемого соединения, скорость реакции может быть нежелательно медленной. Кроме того, если жидкая фаза реакционной среды содержит избыточную концентрацию окисляемого соединения, дополнительные нежелательные побочные реакции могут образовывать примеси.
Обычные реакторы жидкофазного окисления снабжаются средствами перемешивания для перемешивания многофазной реакционной среды, содержащейся в них. Перемешивание реакционной среды производится в попытке облегчить растворение молекулярного кислорода в жидкой фазе реакционной среды, поддержать относительно однородные концентрации растворенного кислорода в жидкой фазе реакционной среды и поддержать относительно однородные концентрации окисляемого органического соединения в жидкой фазе реакционной среды.
Перемешивание реакционной среды, подвергающейся жидкофазному окислению, часто обеспечивается с помощью механических средств перемешивания в емкостях, таких, например, как проточные реакторы смешения (CSTR). Хотя CSTR могут обеспечить тщательное перемешивание реакционной среды, CSTR имеют ряд недостатков. Например, CSTR имеют относительно высокую капитальную стоимость из-за необходимости в них в дорогостоящих двигателях, непроницаемых для текучих сред подшипниках и приводных валах и/или сложных механизмах перемешивания. Кроме того, вращающиеся и/или осциллирующие механические компоненты обычных CSTR требуют регулярного обслуживания. Затраты труда и время остановки, связанные с таким обслуживанием, увеличивают стоимость работы CSTR. Однако даже при регулярном обслуживании системы механического перемешивания, используемые в CSTR, склонны к механическим отказам и могут потребовать замены через относительно короткие периоды времени.
Барботажные колонны реакторного типа обеспечивают привлекательную альтернативу CSTR и другим механически перемешиваемым реакторам окисления. Барботажные колонны реакторного типа обеспечивают перемешивание реакционной среды, не требуя дорогостоящего и ненадежного механического оборудования. Барботажные колонны реакторного типа, как правило, содержат вытянутую снизу вверх реакционную зону, в которой содержится реакционная среда. Перемешивание реакционной среды в реакционной зоне обеспечивается, прежде всего, естественным всплыванием газовых пузырьков, восходящих через жидкую фазу реакционной среды. Это перемешивание под действием естественного всплывания, обеспечиваемое в барботажных колоннах реакторного типа, уменьшает капитальные затраты и затраты на обслуживание по сравнению с механически перемешиваемыми реакторами. Кроме того, отсутствие, по существу, подвижных механических деталей, ассоциируемое с барботажно-реакторными колоннами, обеспечивает систему окисления, которая менее склонна к механическому отказу, чем механически перемешиваемые реакторы.
Когда жидкофазное частичное окисление пара-ксилола осуществляют в обычном реакторе окисления (CSTR или барботажной колонне), продукт, извлекаемый из реактора, как правило, представляет собой суспензию, содержащую сырую терефталевую кислоту (CTA) и маточную жидкость. CTA содержит относительно высокие уровни примесей (например, 4-карбоксибензальдегида, пара-толуиловой кислоты, флуоренонов и других окрашенных частиц), которые делают ее непригодной в качестве исходных материалов для получения PET. Таким образом, CTA, получаемая в обычных реакторах окисления, как правило, подвергается очистке, которая преобразует CTA в очищенную терефталевую кислоту (PTA), пригодную для получения PET.
Один из типичных способов очистки для преобразования CTA в PTA включает в себя следующие стадии: (1) замену маточной жидкости CTA-содержащей суспензии водой, (2) нагрев суспензии CTA/вода для растворения CTA в воде, (3) каталитическое гидрирование раствора CTA/вода для преобразования примесей в более желательные и/или легкоотделяемые соединения, (4) осаждение полученной PTA из гидрированного раствора посредством множества стадий кристаллизации и (5) выделение кристаллизованной PTA из остающихся жидкостей. Несмотря на эффективность этот тип обычного способа очистки может быть очень дорогостоящим. Отдельные факторы, вносящие вклад в высокую стоимость обычных способов очистки CTA, включают в себя, например, тепловую энергию, необходимую для облегчения растворения CTA в воде, катализатор, необходимый для гидрирования, поток водорода, необходимый для гидрирования, потери выхода, вызываемые гидрированием части терефталевой кислоты, и множество емкостей, необходимое для многостадийной кристаллизации. Таким образом, было бы желательным получение продукта CTA, который мог бы очищаться без необходимости в облегчаемом нагревом растворении в воде, гидрировании и/или многостадийной кристаллизации.
Цели изобретения
Следовательно, целью настоящего изобретения является создание более эффективного и экономичного реактора и способа жидкофазного окисления.
Другой целью настоящего изобретения является создание более эффективного и экономичного реактора и способа жидкофазного каталитического частичного окисления пара-ксилола до терефталевой кислоты.
Еще одной целью настоящего изобретения является создание барботажной колонны реакторного типа, которая облегчает улучшенные жидкофазные реакции окисления при уменьшении образования примесей.
Еще одной целью настоящего изобретения является создание более эффективной и экономичной системы получения чистой терефталевой кислоты (PTA) посредством жидкофазного окисления пара-ксилола с получением сырой терефталевой кислоты (CTA), а впоследствии, очистки CTA до PTA.
Кроме того, целью настоящего изобретения является создание барботажной колонны реакторного типа для окисления пара-ксилола и получения продукта CTA, который можно очищать без необходимости в облегчаемом нагревом растворении CTA в воде, гидрировании растворенной CTA и/или многостадийной кристаллизации гидрированной PTA.
Необходимо отметить, что рамки настоящего изобретения, как определяется в прилагаемой формуле изобретения, не ограничиваются способами или устройствами, которые могут реализовать все цели, перечисленные выше. Скорее, рамки заявляемого изобретения могут охватывать разнообразные системы, которые не достигают всех перечисленных выше целей или какой-либо из них. Дополнительные цели и преимущества настоящего изобретения будут легко понятны специалисту в данной области при просмотре следующего далее подробного описания и прилагаемых чертежей.
Сущность изобретения
Один из вариантов осуществления настоящего изобретения относится к способу, включающему в себя следующие стадии: (a) введения в основном газофазного потока окислителя, содержащего молекулярный кислород, в реакционную зону барботажной колонны реакторного типа; (b) введения в основном жидкофазного потока исходных материалов, содержащего пара-ксилол, в реакционную зону через множество входных отверстий, где реакционная зона имеет максимальный диаметр (D), где, по меньшей мере, два из входных отверстий отделены по вертикали друг от друга, по меньшей мере, примерно на 0,5 D, где, по меньшей мере, часть потока исходных материалов поступает в реакционную зону при поверхностной скорости на входе, по меньшей мере, примерно 5 м/с; и (c) окисления, по меньшей мере, части пара-ксилола в жидкой фазе многофазной реакционной среды, содержащейся в реакционной зоне, с получением при этом сырой терефталевой кислоты, где реакционная среда имеет максимальную высоту (H), максимальную ширину (W) и отношение H:W равно, по меньшей мере, примерно 3:1.
Другой вариант осуществления настоящего изобретения относится к способу получения терефталевой кислоты, включающему в себя следующие стадии: (a) введения в основном газофазного потока окислителя, содержащего молекулярный кислород, в реакционную зону барботажной колонны реакторного типа; (b) введения в основном жидкофазного потока исходных материалов, содержащего пара-ксилол, в реакционную зону через множество входных отверстий, где реакционная зона имеет максимальный диаметр (D), где, по меньшей мере, два входных отверстия отдалены по вертикали друг от друга, по меньшей мере, примерно на 0,5 D, где, по меньшей мере, часть потока исходных материалов поступает в реакционную зону при поверхностной скорости на входе, по меньшей мере, примерно 5 м/с, где, по меньшей мере, часть реакционной зоны определяется одной или несколькими расположенными вертикально боковыми стенками реактора, где, по меньшей мере, примерно 25 мас.% пара-ксилола поступает в реакционную зону в одном или нескольких положениях, отдаленных внутрь, по меньшей мере, на 0,05 D от расположенных вертикально боковых стенок; (c) окисления, по меньшей мере, части пара-ксилола в жидкой фазе трехфазной реакционной среды, содержащейся в реакционной зоне, с получением при этом частиц сырой терефталевой кислоты, где реакционная среда имеет максимальную высоту (H), максимальную ширину (W), и отношение H:W равно, по меньшей мере, примерно 3:1; и (d) окисления, по меньшей мере, части частиц сырой терефталевой кислоты в реакторе вторичного окисления с получением при этом более чистой терефталевой кислоты.
Еще один вариант осуществления настоящего изобретения относится к барботажной колонне реакторного типа для взаимодействия в основном жидкофазного потока с в основном газофазным потоком. Барботажная колонна реакторного типа содержит корпус, множество отверстий для жидкости и множество отверстий для газа. Корпус определяет удлиненную реакционную зону, простирающуюся вдоль в целом расположенной вертикально центральной оси оболочки. Реакционная зона имеет максимальную длину (L), измеренную параллельно оси оболочки, максимальный диаметр (D), измеренный перпендикулярно оси оболочки, и отношение L:D находится в пределах примерно от 6:1 примерно до 30:1. Множество отверстий для жидкости вводят жидкофазный поток в реакционную зону. По меньшей мере, два отверстия для жидкости отдалены аксиально друг от друга, по меньшей мере, примерно на 0,5 D. Множество отверстий для газа вводят газофазный поток в реакционную зону. Реакционная зона предоставляет первый и второй противоположные края, отдаленные друг от друга на максимальную длину (L). Большая часть общей открытой площади, определяемой всеми отверстиями для газа, располагается в пределах примерно 0,25 D от первого уровня реакционной зоны.
Краткое описание чертежей
Предпочтительные варианты осуществления настоящего изобретения описываются подробно ниже со ссылкой на прилагаемые фигуры чертежей, в которых
фиг.1 представляет собой вид сбоку реактора окисления, сконструированного в соответствии с одним из вариантов осуществления настоящего изобретения, в частности, иллюстрирующий введение потоков исходных материалов, окислителя и флегмы в реактор, присутствие многофазной реакционной среды в реакторе и извлечение газа и суспензии из верхней и нижней части реактора, соответственно;
фиг.2 представляет собой увеличенный вид в разрезе сбоку нижней части барботажной колонны реакторного типа по линии 2-2 на фиг.3, в частности, иллюстрирующий расположение и конфигурацию устройства распределения окислителя, используемого для введения потока окислителя в реактор;
фиг.3 представляет собой вид сверху устройства распределения окислителя на фиг.2, в частности, иллюстрирующий отверстия для окислителя в верхней части устройства распределения окислителя;
фиг.4 представляет собой вид снизу устройства распределения окислителя на фиг.2, в частности, иллюстрирующий отверстия для окислителя в нижней части устройства распределения окислителя;
фиг.5 представляет собой вид в разрезе сбоку устройства распределения окислителя по линии 5-5 на фиг.3, в частности, иллюстрирующий ориентацию отверстий для окислителя в верхней и нижней части устройства распределения окислителя;
фиг.6 представляет собой увеличенный вид сбоку нижней части барботажной колонны реакторного типа, в частности, иллюстрирующий систему для введения потока исходных материалов в реактор во множестве положений, отделенных друг от друга некоторым расстоянием по вертикали;
фиг.7 представляет собой вид сверху в разрезе по линии 7-7 на фиг.6, в частности, иллюстрирующий, как система введения исходных материалов, показанная на фиг.6, распределяет поток исходных материалов внутри, в предпочтительной радиальной зоне ввода (FZ) и в более чем одном азимутальном квадранте (Q1, Q2, Q3, Q4);
фиг.8 представляет собой вид сверху в разрезе, подобный фиг.7, но иллюстрирующий альтернативные средства для высвобождения потока исходных материалов в реакторе с использованием байонетных труб, каждая из которых имеет множество малых входных отверстий;
фиг.9 представляет собой изометрический вид альтернативной системы для введения потока исходных материалов в реакционную зону во множестве положений, отделенных друг от друга некоторым расстоянием по вертикали, без необходимости во множестве проходов через стенку емкости, в частности, иллюстрирующий, что система распределения исходных материалов может, по меньшей мере, частично поддерживаться на устройстве распределения окислителя;
фиг.10 представляет собой вид сбоку системы распределения исходных материалов с одним прохождением через стенку и устройства распределения окислителя, иллюстрируемых на фиг.9;
фиг.11 представляет собой вид сверху в разрезе по линии 11-11 на фиг.10, дополнительно иллюстрирующий систему распределения исходных материалов с одним прохождением, поддерживаемую на устройстве распределения окислителя;
фиг.12 представляет собой изометрический вид альтернативного устройства распределения окислителя, имеющего все отверстия для окислителя, расположенные в нижней части кольцевого элемента;
фиг.13 представляет собой вид сверху альтернативного устройства распределения окислителя фиг.12;
фиг.14 представляет собой вид снизу альтернативного устройства распределения окислителя на фиг.12, в частности, иллюстрирующий расположение отверстий в нижней части для введения потока окислителя в реакционную зону;
фиг.15 представляет собой вид в разрезе сбоку устройства распределения окислителя по линии 15-15 на фиг.13, в частности, иллюстрирующий ориентацию нижних отверстий для окислителя;
фиг.16 представляет собой вид сбоку барботажной колонны реакторного типа, снабженной внутренней деаэрационной емкостью вблизи нижнего выхода реактора;
фиг.17 представляет собой увеличенный вид в разрезе сбоку нижней части барботажной колонны реакторного типа на фиг.16 по линии 17-17 на фиг.18, в частности, иллюстрирующий конфигурацию внутренней деаэрационной емкости, расположенной на нижнем выходе барботажной колонны реакторного типа;
фиг.18 представляет собой вид сверху в разрезе по линии 18-18 на фиг.16, в частности, иллюстрирующий гаситель вихрей, расположенный в деаэрационной емкости;
фиг.19 представляет собой вид сбоку барботажной колонны реакторного типа, снабженной наружной деаэрационной емкостью, иллюстрирующей способ, в котором часть деаэрированной суспензии, покидающей нижнюю часть деаэрационной емкости, может использоваться для прочистки отводной линии, соединенной с нижней частью реактора;
фиг.20 представляет собой вид сбоку барботажной колонны реакторного типа, снабженной гибридной внутренней/наружной деаэрационной емкостью для отделения газовой фазы реакционной среды, извлеченной из приподнятого бокового положения в реакторе;
фиг.21 представляет собой вид сбоку барботажной колонны реакторного типа, снабженной альтернативной гибридной деаэрационной емкостью вблизи нижней части реактора;
фиг.22 представляет собой увеличенный вид сбоку в разрезе нижней части барботажной колонны реакторного типа на фиг.21, в частности, иллюстрирующий использование альтернативного устройства распределения окислителя, использующего входные проходы, которые принимают поток окислителя через днище реактора;
фиг.23 представляет собой увеличенный вид сбоку в разрезе, подобный фиг.22, в частности, иллюстрирующий альтернативные средства для введения потока окислителя в реактор через множество отверстий в днище реактора, необязательно использующие пластины отбойников для более однородного распределения потока окислителя в реакторе;
фиг.24 представляет собой вид сбоку барботажной колонны реакторного типа, использующей внутренний проход для потока, для улучшения диспергирования окисляемого соединения посредством рециркулирования части реакционной среды из верхней части реактора в нижнюю часть реактора;
фиг.25 представляет собой вид сбоку барботажной колонны реакторного типа, использующей наружный проход для потока, для улучшения диспергирования окисляемого соединения посредством рециркулирования части реакционной среды из верхней части реактора в нижнюю;
фиг.26 представляет собой вид в разрезе сбоку горизонтального эжектора, который может использоваться для улучшения диспергирования окисляемого соединения в реакторе окисления, в частности, иллюстрирующий эжектор, который использует поступающие жидкие исходные материалы для нагнетания реакционной среды в эжектор и высвобождает смесь исходных материалов и реакционной среды в реакционной зоне при высокой скорости;
фиг.27 представляет собой вид в разрезе сбоку вертикального эжектора, который может использоваться для улучшения диспергирования окисляемого соединения в реакторе окисления, в частности, иллюстрирующий эжектор, который объединяет жидкие исходные материалы и входной газ и использует объединенную двухфазную текучую среду для нагнетания реакционной среды в эжектор и высвобождения смеси жидких исходных материалов, входного газа и реакционной среды в реакционной зоне при высокой скорости;
фиг.28 представляет собой вид сбоку барботажной колонны реакторного типа, содержащей многофазную реакционную среду, в частности, иллюстрирующий реакционную среду, которая является теоретически распределенной на 30 горизонтальных слоях равного объема для количественного определения градиентов параметров в реакционной среде;
фиг.29 представляет собой вид сбоку барботажной колонны реакторного типа, содержащей многофазную реакционную среду, в частности, иллюстрирующий первый и второй дискретные 20% сплошные объемы реакционной среды, которая имеет по существу различные концентрации кислорода и/или скорости расходования кислорода;
фиг.30 представляет собой вид сбоку двух расположенных друг над другом реакционных емкостей, с необязательным механическим перемешиванием или без него, содержащих многофазную реакционную среду, в частности, иллюстрирующий, что емкости содержат дискретные 20% сплошные объемы реакционной среды, имеющей по существу различные концентрации кислорода и/или скорости потребления кислорода;
фиг.31 представляет собой вид сбоку трех расположенных бок о бок реакционных емкостей, с необязательным механическим перемешиванием или без него, содержащих многофазную реакционную среду, в частности, иллюстрирующий, что емкости содержат дискретные 20% сплошные объемы реакционной среды, имеющей по существу различные концентрации кислорода и/или скорости потребления кислорода;
фиг.32A и 32B представляют собой увеличенные виды частиц сырой терефталевой кислоты (CTA), полученных в соответствии с одним из вариантов осуществления настоящего изобретения, в частности, иллюстрирующие, что каждая частица CTA представляет собой частицу с низкой плотностью, высокой площадью поверхности, состоящую из множества непрочно связанных субчастиц CTA;
фиг.33A и 33B представляют собой увеличенные виды получаемой обычно CTA, в частности, иллюстрирующие, что обычная частица CTA имеет больший размер частицы, более низкую плотность и более низкую площадь поверхности, чем частицы CTA по настоящему изобретению на фиг.32A и 32B;
фиг.34 представляет собой упрощенную блок-схему способа, известного из литературы, получения очищенной терефталевой кислоты (PTA); и
фиг.35 представляет собой упрощенную блок-схему способа получения PTA в соответствии с одним из вариантов осуществления настоящего изобретения.
Подробное описание
Один из вариантов осуществления настоящего изобретения относится к жидкофазному частичному окислению окисляемого соединения. Такое окисление предпочтительно осуществляют в жидкой фазе многофазной реакционной среды, содержащейся в одном или нескольких реакторах смешения. Пригодные для использования реакторы смешения включают в себя, например, реактора, перемешиваемые барботированием (например, барботажные колонны реакторного типа), механически перемешиваемые реактора (например, проточные реакторы смешения) и реакторы, перемешиваемые потоком (например, струйные реакторы). В одном из вариантов осуществления настоящего изобретения жидкофазное окисление осуществляют в одной барботажной колонне реакторного типа.
Как здесь используется, термин "барботажная колонна реакторного типа" должен обозначать реактор для облегчения химических реакций в многофазной реакционной среде, где перемешивание реакционной среды обеспечивается, прежде всего, посредством перемещения снизу вверх газовых пузырьков через реакционную среду. Как здесь используется, термин "перемешивание" должен обозначать работу, диссипируемую в реакционной среде, вызывая поток текучей среды и/или перемешивание. Как здесь используется, термины "большинство", "в основном" и "преобладающе" должны обозначать больше чем 50%. Как здесь используется, термин "механическое перемешивание" должен обозначать перемешивание реакционной среды, вызываемое физическим перемещением жесткого или гибкого элемента (элементов) по отношению к реакционной среде или внутри нее. Например, механическое перемешивание может обеспечиваться посредством вращения, колебания и/или вибрации внутренних мешалок, лопастей, вибраторов или акустических диафрагм, расположенных в реакционной среде. Как здесь используется, термин "перемешивание потоком" должен обозначать перемешивание реакционной среды, вызываемое высокоскоростной инжекцией и/или рециркуляцией одной или нескольких текучих сред в реакционную среду. Например, перемешивание потоком может обеспечиваться соплами, эжекторами и/или эжекционными устройствами.
В предпочтительном варианте осуществления настоящего изобретения меньше, примерно, чем 40% перемешивания реакционной среды в барботажной колонне реакторного типа во время окисления обеспечивается посредством механического перемешивания и/или перемешивания потоком, более предпочтительно меньше, примерно, чем 20% перемешивания обеспечивается посредством механического перемешивания и/или перемешивания потоком, наиболее предпочтительно меньше, чем 5% перемешивания обеспечивается посредством механического перемешивания и/или перемешивания потоком. Предпочтительно величина механического перемешивания и/или перемешивания потоком, придаваемого многофазной реакционной среде во время окисления, меньше, примерно, чем 3 кВт/м3 реакционной среды, более предпочтительно меньше, примерно, чем 2 кВт/м3, наиболее предпочтительно меньше, чем 1 кВт/м3.
Обращаясь теперь к фиг.1, предпочтительная барботажная колонна реакторного типа 20 иллюстрируется как содержащая корпус 22, имеющий реакционную секцию 24 и секцию отделения 26. Реакционная секция 24 определяет внутреннюю реакционную зону 28, в то время как секция отделения 26 определяет внутреннюю зону отделения 30. В основном жидкофазный поток исходных материалов вводится в реакционную зону 28 через входы для исходных материалов 32a, b, c, d. В основном газофазный поток окислителя вводится в реакционную зону 28 через устройство распределения окислителя 34, расположенное в нижней части реакционной зоны 28. Жидкофазный поток исходных материалов и газофазный поток окислителя совместно образуют многофазную реакционную среду 36 в пределах реакционной зоны 28. Многофазная реакционная среда 36 содержит жидкую фазу и газовую фазу. Более предпочтительно многофазная реакционная среда 36 содержит трехфазную среду, имеющую твердофазный, жидкофазный и газофазный компоненты. Твердофазный компонент реакционной среды 36 предпочтительно осаждается в пределах реакционной зоны 28 в результате реакции окисления, осуществляемой в жидкой фазе реакционной среды 36. Барботажная колонна реакторного типа 20 содержит выход для суспензии 38, расположенный вблизи нижней части реакционной зоны 28, и выход для газа 40, расположенный вблизи верхней части зоны отделения 30. Выходящий поток суспензии, содержащий жидкофазный и твердофазный компоненты реакционной среды 36, извлекается из реакционной зоны 28 через выход для суспензии 38, в то время как преимущественно газообразный выходящий поток извлекается из зоны отделения 30 через выход для газа 40.
Жидкофазный поток исходных материалов, вводимый в барботажную колонну реакторного типа 20 через входы для исходных материалов 32a, b, c, d, предпочтительно содержит окисляемое соединение, растворитель и систему катализаторов.
Окисляемое соединение, присутствующее в жидкофазном потоке исходных материалов, предпочтительно содержит, по меньшей мере, одну углеводородную группу. Более предпочтительно окисляемое соединение представляет собой ароматическое соединение. Еще более предпочтительно окисляемое соединение представляет собой ароматическое соединение, по меньшей мере, с одной присоединенной углеводородной группой или, по меньшей мере, одной присоединенной замещенной углеводородной группой или, по меньшей мере, одним присоединенным гетероатомом или, по меньшей мере, одной присоединенной функциональной группой карбоновой кислоты (-COOH). Еще более предпочтительно окисляемое соединение представляет собой ароматическое соединение, по меньшей мере, с одной присоединенной углеводородной группой или, по меньшей мере, одной присоединенной замещенной углеводородной группой, при этом каждая присоединенная группа содержит от 1 до 5 атомов углерода. Еще более предпочтительно окисляемое соединение представляет собой ароматическое соединение, имеющее точно две присоединенных группы, при этом каждая присоединенная группа содержит точно один атом углерода и состоит из метильных групп и/или замещенных метильных групп, и/или, самое большее, одной группы карбоновой кислоты. Еще более предпочтительно окисляемое соединение представляет собой пара-ксилол, мета-ксилол, пара-толуальдегид, мета-толуальдегид, пара-толуиловую кислоту, мета-толуиловую кислоту и/или ацетальдегид. Наиболее предпочтительно окисляемое соединение представляет собой пара-ксилол.
"Углеводородная группа", как здесь определено, представляет собой, по меньшей мере, один атом углерода, который соединен только с атомами водорода или с другими атомами углерода. "Замещенная углеводородная группа", как здесь определено, представляет собой, по меньшей мере, один атом углерода, соединенный, по меньшей мере, с одним гетероатомом и, по меньшей мере, с одним атомом водорода. "Гетероатомы", как здесь определено, представляют собой все атомы, иные, чем атомы углерода и водорода. Ароматические соединения, как здесь определено, содержат ароматическое кольцо, предпочтительно имеющее, по меньшей мере, 6 атомов углерода, еще более предпочтительно имеющее только атомы углерода в качестве части кольца. Соответствующие примеры таких ароматических колец включают в себя, но не ограничиваясь этим, бензол, бифенил, терфенил, нафталин и другие конденсированные ароматические кольца на основе углерода.
Соответствующие примеры окисляемого соединения включают в себя алифатические углеводороды (например, алканы, разветвленные алканы, циклические алканы, алифатические алкены, разветвленные алкены и циклические алкены); алифатические альдегиды (например, ацетальдегид, пропиональдегид, изобутиральдегид и н-бутиральдегид); алифатические спирты (например, этанол, изопропанол, н-пропанол, н-бутанол и изобутанол); алифатические кетоны (например, диметилкетон, этилметилкетон, диэтилкетон и изопропилметилкетон); сложные алифатические эфиры (например, метилформиат, метилацетат, этилацетат); алифатические пероксиды, перкислоты и гидропероксиды (например, трет-бутилгидропероксид, перуксусную кислоту и ди-трет-бутилгидропероксид); алифатические соединения с группами, которые представляют собой сочетания указанных выше алифатических видов плюс другие гетероатомы (например, алифатические соединения, содержащие один или несколько молекулярных сегментов углеводородов, альдегидов, спиртов, кетонов, сложных эфиров, пероксидов, перкислот и/или гидропероксидов в сочетании с натрием, бромом, кобальтом, марганцем и цирконием); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы с одной или несколькими присоединенными углеводородными группами (например, толуол, этилбензол, изопропилбензол, н-пропилбензол, неопентилбензол, пара-ксилол, мета-ксилол, орто-ксилол, все изомеры триметилбензолов, все изомеры тетраметилбензолов, пентаметилбензол, гексаметилбензол, все изомеры этилметилбензолов, все изомеры диэтилбензолов, все изомеры этилдиметилбензолов, все изомеры диметилнафталинов, все изомеры этилметилнафталинов, все изомеры диэтилнафталинов, все изомеры диметилбифенилов, все изомеры этилметилбифенилов и все изомеры диэтилбифенилов, стильбен, и он же с одной или несколькими присоединенными углеводородными группами, флуорен, и он же с одной или несколькими присоединенными углеводородными группами, антрацен, и он же с одной или несколькими присоединенными углеводородными группами, и дифенилэтан, и он же с одной или несколькими присоединенными углеводородными группами); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы с одной или несколькими присоединенными углеводородными группами и/или с одним или несколькими присоединенными гетероатомами, которые могут соединяться с другими атомами или группами атомов (например, фенолом, всеми изомерами метилфенолов, всеми изомерами диметилфенолов, всеми изомерами нафтолов, простым бензилметиловым эфиром, всеми изомерами бромфенолов, бромбензолом, всеми изомерами бромтолуолов, включая альфа-бромтолуол, дибромбензолом, кобальт нафтенатом и всеми изомерами бромбифенилов); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы с одной или несколькими присоединенными углеводородными группами и/или одним или несколькими присоединенными гетероатомами и/или с одной или несколькими присоединенными замещенными углеводородными группами (например, бензальдегидом, всеми изомерами бромбензальдегидов, всеми изомерами бромированных толуальдегидов, включая все изомеры альфа-бромтолуальдегидов, всеми изомерами гидроксибензальдегидов, всеми изомерами бромгидроксибензальдегидов, всеми изомерами бензолдикарбоксальдегидов, всеми изомерами бензолтрикарбоксальдегидов, пара-толуальдегидом, мета-толуальдегидом, орто-толуальдегидом, всеми изомерами толуолдикарбоксальдегидов, всеми изомерами толуолтрикарбоксальдегидов, всеми изомерами толуолтетракарбоксальдегидов, всеми изомерами диметилбензолдикарбоксальдегидов, всеми изомерами диметилбензолтрикарбоксальдегидов, всеми изомерами диметилбензолтетракарбоксальдегидов, всеми изомерами триметилбензолтрикарбоксальдегидов, всеми изомерами этилтолуальдегидов, всеми изомерами триметилбензолдикарбоксальдегидов, тетраметилбензолдикарбоксальдегидом, гидроксиметилбензолом, всеми изомерами гидроксиметилтолуолов, всеми изомерами гидроксиметилбромтолуолов, всеми изомерами гидроксиметилтолуальдегидов, всеми изомерами гидроксиметилбромтолуальдегидов, бензилгидропероксидом, бензоилгидропероксидом, всеми изомерами толилметилгидропероксидов и всеми изомерами метилфенолметилгидропероксидов); различные бензольные кольца, нафталиновые кольца, бифенилы, терфенилы и другие ароматические группы с одной или несколькими присоединенными выбранными группами, выбранные группы означают углеводородные группы, и/или присоединенные гетероатомы, и/или замещенные углеводородные группы, и/или группы карбоновых кислот, и/или группы перкислот (например, бензойной кислоты, пара-толуиловой кислоты, мета-толуиловой кислоты, орто-толуиловой кислоты, всех изомеров этилбензойных кислот, всех изомеров пропилбензойных кислот, всех изомеров бутилбензойных кислот, всех изомеров пентилбензойных кислот, всех изомеров диметилбензойных кислот, всех изомеров этилметилбензойных кислот, всех изомеров триметилбензойных кислот, всех изомеров тетраметилбензойных кислот, всех изомеров пентаметилбензойных кислот, всех изомеров диэтилбензойных кислот, всех изомеров бензолдикарбоновых кислот, всех изомеров бензолтрикарбоновых кислот, всех изомеров метилбензолдикарбоновых кислот, всех изомеров диметилбензолдикарбоновых кислот, всех изомеров метилбензолтрикарбоновых кислот, всех изомеров бромбензойных кислот, всех изомеров дибромбензойных кислот, всех изомеров бромтолуиловых кислот, включая альфа-бромтолуиловые кислоты, толилуксусной кислоты, всех изомеров гидроксибензойных кислот, всех изомеров гидроксиметилбензойных кислот, всех изомеров гидрокситолуиловых кислот, всех изомеров гидроксиметилтолуиловых кислот, всех изомеров гидроксиметилбензолдикарбоновых кислот, всех изомеров гидроксибромбензойных кислот, всех изомеров гидроксибромтолуиловых кислот, всех изомеров гидроксиметилбромбензойных кислот, всех изомеров карбоксибензальдегидов, всех изомеров дикарбоксибензальдегидов, пербензойной кислоты, всех изомеров гидропероксиметилбензойных кислот, всех изомеров гидропероксиметилгидроксибензойных кислот, всех изомеров гидропероксикарбонилбензойных кислот, всех изомеров гидропероксикарбонилтолуолов, всех изомеров метилбифенилкарбоновых кислот, всех изомеров диметилбифенилкарбоновых кислот, всех изомеров метилбифенилдикарбоновых кислот, всех изомеров бифенилтрикарбоновых кислот, всех изомеров стильбена с одной или несколькими присоединенными выбранными группами, всех изомеров флуоренона с одной или несколькими присоединенными выбранными группами, всех изомеров нафталина с одной или несколькими присоединенными выбранными группами, бензила, всех изомеров бензила с одной или несколькими присоединенными выбранными группами, бензофенона, всех изомеров бензофенона с одной или несколькими присоединенными выбранными группами, антрахинона, всех изомеров антрахинона с одной или несколькими присоединенными выбранными группами, всех изомеров дифенилэтана с одной или несколькими присоединенными выбранными группами, бензокумарина и всех изомеров бензокумарина с одной или несколькими присоединенными выбранными группами).
Если окисляемое соединение, присутствующее в жидкофазном потоке исходных материалов, представляет собой твердое соединение при нормальных условиях (т.е. является твердым при стандартной температуре и давлении), предпочтительно, чтобы окисляемое соединение по существу растворялось в растворителе, когда вводится в реакционную зону 28. Предпочтительно, чтобы температура кипения окисляемого соединения при атмосферном давлении составляла, по меньшей мере, примерно 50°C. Более предпочтительно температура кипения окисляемого соединения находится в пределах примерно от 80 примерно до 400°C, а наиболее предпочтительно в пределах от 125 до 155°C. Количество окисляемого соединения, присутствующее в жидкофазных исходных материалах, предпочтительно находится в пределах примерно от 2 примерно до 40 мас.%, более предпочтительно в пределах примерно от 4 примерно до 20 мас.%, наиболее предпочтительно в пределах от 6 до 15 мас.%.
Теперь отметим, что окисляемое соединение, присутствующее в жидкофазных исходных материалах, может содержать сочетание из двух или более различных окисляемых химических соединений. Эти два или более различных химических материалов могут вводиться смешанные в жидкофазном потоке исходных материалов, или могут вводиться по отдельности во множестве потоков исходных материалов. Например, окисляемое соединение, содержащее пара-ксилол, мета-ксилол, пара-толуальдегид, пара-толуиловую кислоту и ацетальдегид, может вводиться в реактор через один вход или множество отдельных входов.
Растворитель, присутствующий в жидкофазном потоке исходных материалов, предпочтительно содержит компонент кислоты и компонент воды. Растворитель предпочтительно присутствует в жидкофазном потоке исходных материалов при концентрации в пределах примерно от 60 примерно до 98 мас.%, более предпочтительно в пределах примерно от 80 примерно до 96 мас.%, наиболее предпочтительно в пределах от 85 до 94 мас.%. Компонент кислоты растворителя предпочтительно представляет собой в основном органическую низкомолекулярную монокарбоновую кислоту, имеющую 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно компонент кислоты растворителя представляет собой, в основном, уксусную кислоту. Предпочтительно компонент кислоты составляет, по меньшей мере, примерно 75 мас.% растворителя, более предпочтительно, по меньшей мере, примерно 80 мас.% растворителя, наиболее предпочтительно от 85 до 98 мас.% растворителя, остаток представляет собой в основном воду. Растворитель, вводимый в барботажную колонну реакторного типа 20, может содержать малые количества примесей, таких, например, как пара-толуальдегид, терефтальдегид, 4-карбоксибензальдегид (4-CBA), бензойную кислоту, пара-толуиловую кислоту, пара-толуиловый альдегид, альфа-бром-пара-толуиловую кислоту, изофталевую кислоту, фталевую кислоту, тримеллитовую кислоту, полиароматические соединения и/или суспендированные частицы. Предпочтительно, чтобы общее количество примесей в растворителе, вводимом в барботажную колонну реакторного типа 20, было меньше, примерно, чем 3 мас.%.
Система катализаторов, присутствующая в жидкофазном потоке исходных материалов, предпочтительно представляет собой гомогенную, жидкофазную систему катализаторов, которая может облегчать окисление (включая частичное окисление) окисляемого соединения. Более предпочтительно система катализаторов содержит, по меньшей мере, один переходный металл переменной валентности. Еще более предпочтительно переходный металл переменной валентности включает в себя кобальт. Еще более предпочтительно система катализаторов включает в себя кобальт и бром. Наиболее предпочтительно система катализаторов включает в себя кобальт, бром и марганец.
Когда в системе катализаторов присутствует кобальт, предпочтительно, чтобы количество кобальта, присутствующего в жидкофазном потоке исходных материалов, было таким, чтобы концентрация кобальта в жидкой фазе реакционной среды 36 поддерживалась в пределах примерно от 300 примерно до 6000 м.д. мас., более предпочтительно в пределах примерно от 700 примерно до 4200 м.д. мас., наиболее предпочтительно в пределах от 1200 до 3000 м.д. мас. Когда бром присутствует в системе катализаторов, предпочтительно, чтобы количество брома, присутствующего в жидкофазном потоке исходных материалов, было таким, чтобы концентрация брома в жидкой фазе реакционной среды 36 поддерживалось в пределах примерно от 300 примерно до 5000 м.д. мас., более предпочтительно в пределах примерно от 600 примерно до 4000 м.д. мас., наиболее предпочтительно в пределах от 900 до 3000 м.д. мас. Когда в системе катализаторов присутствует марганец, предпочтительно, чтобы количество марганца, присутствующего в жидкофазном потоке исходных материалов, было таким, чтобы его концентрация в жидкой фазе реакционной среды 36 поддерживалась в пределах примерно от 20 примерно до 1000 м.д. мас., более предпочтительно в пределах примерно от 40 примерно до 500 м.д. мас., наиболее предпочтительно в пределах от 50 до 200 м.д. мас.
Концентрации кобальта, брома и/или марганца в жидкой фазе реакционной среды 36, приведенные выше, выражаются как усредненные по времени и по объему. Как здесь используется, термин "усредненный по времени" должен обозначать усреднение, по меньшей мере, по 10 измерениям, осуществляемым через равные промежутки времени за непрерывный период, по меньшей мере, 100 с. Как здесь используется, термин "усредненный по объему" должен обозначать усреднение, по меньшей мере, по 10 измерениям, осуществляемым на однородных 3-мерных промежутках в определенном объеме.
Массовое отношение кобальта к брому (Co:Br) в системе катализаторов, вводимых в реакционную зону 28, предпочтительно находится в пределах примерно от 0,25:1 примерно до 4:1, более предпочтительно в пределах примерно от 0,5:1 примерно до 3:1, наиболее предпочтительно в пределах от 0,75:1 до 2:1. Массовое отношение кобальта к марганцу (Co:Mn) в системе катализаторов, вводимых в реакционную зону 28, предпочтительно находится в пределах примерно от 0,3:1 примерно до 40:1, более предпочтительно в пределах примерно от 5:1 примерно до 30:1, наиболее предпочтительно в пределах от 10:1 до 25:1.
Жидкофазный поток исходных материалов, вводимый в барботажную колонну реакторного типа 20, может содержать малые количества примесей, таких, например, как толуол, этилбензол, пара-толуальдегид, терефтальдегид, 4-карбоксибензальдегид (4-CBA), бензойная кислота, пара-толуиловая кислота, пара-толуиловый альдегид, альфа-бром-пара-толуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или суспендированные частицы. Когда барботажная колонна реакторного типа 20 используется для получения терефталевой кислоты, мета-ксилол и орто-ксилол также рассматриваются как примеси. Предпочтительно, чтобы общее количество примесей в жидкофазном потоке исходных материалов, вводимом в барботажную колонну реакторного типа 20, было меньше, примерно, чем 3 мас.%.
Хотя фиг.1 иллюстрирует вариант осуществления, где окисляемое соединение, растворитель и система катализаторов смешиваются вместе и вводятся в барботажную колонну реакторного типа 20 как единый поток исходных материалов, в альтернативном варианте осуществления настоящего изобретения окисляемое соединение, растворитель и катализатор могут в барботажную колонну реакторного типа 20 вводиться раздельно. Например, является возможным введение чистого потока пара-ксилола в барботажную колонну реакторного типа 20 через вход, отдельный от входа (входов) для растворителя и катализаторов.
В основном газофазный поток окислителя, вводимый в барботажную колонну реакторного типа 20 через устройство распределения окислителя 34, содержит молекулярный кислород (O2). Предпочтительно поток окислителя содержит в пределах примерно от 5 примерно до 40% молярных молекулярного кислорода, более предпочтительно в пределах примерно от 15 примерно до 30% молярных молекулярного кислорода, а наиболее предпочтительно в пределах от 18 до 24% молярных молекулярного кислорода. Предпочтительно, чтобы оставшаяся часть потока окислителя состояла в основном из газа или газов, таких как азот, которые являются инертными для окисления. Более предпочтительно поток окислителя состоит по существу из молекулярного кислорода и азота. Наиболее предпочтительно поток окислителя представляет собой сухой воздух, который содержит примерно 21% молярный молекулярного кислорода и примерно от 78 примерно до 81% молярных азота. В альтернативном варианте осуществления настоящего изобретения поток окислителя может содержать по существу чистый кислород.
Обращаясь опять к фиг.1, барботажная колонна реакторного типа 20 предпочтительно снабжена распределителем флегмы 42, расположенным над верхней поверхностью 44 реакционной среды 36. Распределитель флегмы 42 работает, вводя капли в основном жидкофазного потока флегмы в зону отделения 30 с помощью любых средств образования капель, известных в данной области. Более предпочтительно распределитель флегмы 42 производит спрей из капель, направленный вниз по направлению к верхней поверхности 44 реакционной среды 36. Предпочтительно это направленный вниз спрей из капель оказывает воздействие (т.е. перекрывает и влияет), по меньшей мере, примерно на 50% максимальной площади горизонтального поперечного сечения зоны отделения 30. Более предпочтительно спрей капель оказывает воздействие, по меньшей мере, примерно на 75% максимальной площади горизонтального поперечного сечения зоны отделения 30. Наиболее предпочтительно спрей капель оказывает воздействие, по меньшей мере, на 90% максимальной площади горизонтального поперечного сечения зоны отделения 30. Этот направленный вниз спрей жидкой флегмы может помочь в подавлении пенообразования на верхней поверхности 44 реакционной среды 36 или над ней и может также помочь при отделении любых капель жидкости или суспензии, захваченных в движущемся снизу вверх газе, который протекает по направлению к выходу для газа 40. Кроме того, жидкая флегма может служить для уменьшения количества частиц и потенциально осаждающихся соединений (например, растворенной бензойной кислоты, пара-толуиловой кислоты, 4-CBA, терефталевой кислоты и каталитических солей металлов), выходящих в газовом выходящем потоке, извлекаемом из зоны отделения 30 через выход для газа 40. В дополнение к этому, введение капель флегмы в зону отделения 30 может, посредством дистилляционного воздействия, использоваться для регулировки состава газового выходящего потока, извлекаемого через выход для газа 40.
Поток жидкой флегмы, вводимый в барботажную колонну реакторного типа 20 через распределитель флегмы 42, предпочтительно имеет примерно такой же состав, как и компонент растворителя жидкофазного потока исходных материалов, вводимого в барботажную колонну реакторного типа 20 через входы для исходных материалов 32a, b, c, d. Таким образом, предпочтительно, чтобы поток жидкой флегмы содержал компонент кислоты и воду. Компонент кислоты потока флегмы предпочтительно представляет собой низкомолекулярную органическую монокарбоновую кислоту, имеющую 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно компонент кислоты потока флегмы представляет собой уксусную кислоту. Предпочтительно компонент кислоты составляет, по меньшей мере, примерно 75 мас.% потока флегмы, более предпочтительно, по меньшей мере, примерно 80 мас.% потока флегмы, а наиболее предпочтительно от 85 до 98 мас.% потока флегмы, при этом остаток представляет собой воду. Поскольку поток флегмы, как правило, имеет по существу такой же состав, как и растворитель в жидкофазном потоке исходных материалов, когда это описание относится к "общему количеству растворителя", вводимому в реактор, такое "общее количество растворителя" должно включать в себя как часть потока исходных материалов в потоке флегмы, так и часть их в растворителе.
Во время жидкофазного окисления в барботажной колонне реакторного типа 20, предпочтительно, чтобы потоки исходных материалов, окислителя и флегмы по существу непрерывно вводились в реакционную зону 28, в то время как выходящие потоки газа и суспензии по существу непрерывно извлекаются из реакционной зоны 28. Как здесь используется, термин "по существу непрерывно" должен обозначать в течение периода, по меньшей мере, 10 часов с перерывами, меньшими, чем 10 минут. Во время окисления предпочтительно, чтобы окисляемое соединение (например, пара-ксилол) по существу непрерывно вводилось в реакционную зону 28 со скоростью, по меньшей мере, примерно 8000 кг/час, более предпочтительно со скоростью в пределах примерно от 13000 примерно до 80000 кг/час, еще более предпочтительно в пределах примерно от 18000 примерно до 50000 кг/час и наиболее предпочтительно в пределах от 22000 до 30000 кг/час. Хотя, как правило, предпочтительно, чтобы скорости потока для поступающих потоков исходных материалов, окислителя и флегмы были по существу стационарными, сейчас отметим, что один из вариантов осуществления настоящего изобретения предполагает импульсный режим поступающего потока исходных материалов, окислителя и/или флегмы для улучшения перемешивания и массопередачи. Когда поступающие потоки исходных материалов, окислителя и/или флегмы вводятся импульсным образом, предпочтительно, чтобы их скорости потока изменялись в пределах примерно от 0 примерно до 500% от стационарных скоростей потоков, упоминаемых здесь, более предпочтительно в пределах примерно от 30 примерно до 200% от стационарных скоростей потоков, упоминаемых здесь, наиболее предпочтительно в пределах от 80 до 120% от стационарных скоростей потоков, упоминаемых здесь.
Усредненная по пространству и времени скорость реакции (STR) в барботажной колонне реакторного типа окисления 20 определяется как масса окисляемого соединения, вводимого на единицу объема реакционной среды 36 на единицу времени (например, килограммы пара-ксилола, вводимого на кубический метр в час). При обычном использовании количество окисляемого соединения, не преобразованного в продукт, должно, как правило, вычитаться из количества окисляемого соединения в потоке исходных материалов перед вычислением STR. Однако степени преобразования и выходы, как правило, для многих окисляемых соединений, предпочтительных здесь (например, пара-ксилола), являются высокими, и здесь является обычным определение термина, как сформулировано выше. По причинам капитальных затрат и стоимости работы, среди прочего, как правило, предпочтительно, чтобы реакция осуществлялась с высокими STR. Однако осуществление реакции при все более высоких STR может повлиять на качество или выход частичного окисления. Барботажная колонна реакторного типа 20 является особенно пригодной для использования, когда STR окисляемого соединения (например, пара-ксилола) находится в пределах примерно от 25 кг/м3×ч примерно до 400 кг/м3×ч, более предпочтительно в пределах примерно от 30 кг/м3×ч примерно до 250 кг/м3×ч, еще более предпочтительно примерно от 35 кг/м3×ч примерно до 150 кг/м3×ч, а наиболее предпочтительно в пределах от 40 кг/м3×ч до 100 кг/м3×ч.
STR по кислороду в барботажной колонне реакторного типа окисления 20 определяется как масса молекулярного кислорода, потребляемая на единицу объема реакционной среды 36 в единицу времени (например, килограммы молекулярного кислорода, потребляемого на кубический метр в час). По причинам капитальных затрат и расходования растворителя на окисление, среди прочего, является, как правило, предпочтительным, чтобы реакция осуществлялась при больших STR по кислороду. Однако осуществление реакции при все более высоких STR по кислороду может понизить качество или выход частичного окисления. Не ограничиваясь теорией, видно, что это, возможно, связано со скоростью переноса молекулярного кислорода из газовой фазы в жидкую на площади поверхности границы раздела и, следовательно, в объем жидкости. Слишком высокая STR по кислороду, возможно, приводит к слишком низкому содержанию растворенного кислорода в объеме жидкой фазы реакционной среды.
Общая усредненная STR по кислороду определяется здесь как масса всего кислорода, потребленного во всем объеме реакционной среды 36 в единицу времени (например, килограммы молекулярного кислорода, потребленные на кубический метр в час). Барботажная колонна реакторного типа 20 является особенно пригодной для использования, когда общая усредненная STR по кислороду находится в пределах примерно от 25 кг/м3×ч примерно до 400 кг/м3×ч, более предпочтительно в пределах примерно от 30 кг/м3×ч примерно до 250 кг/м3×ч, еще более предпочтительно примерно от 35 кг/м3×ч примерно до 150 кг/м3×ч, наиболее предпочтительно в пределах от 40 кг/м3×ч до 100 кг/м3×ч.
Во время окисления в барботажной колонне реакторного типа 20, предпочтительно, чтобы отношение массовой скорости потока общего количества растворителя (как из потока исходных материалов, так и из потока флегмы) к массовой скорости потока окисляемого соединения, поступающего в реакционную зону 28, поддерживалось в пределах примерно от 2:1 примерно до 50:1, более предпочтительно в пределах примерно от 5:1 примерно до 40:1, наиболее предпочтительно в пределах от 7,5:1 до 25:1. Предпочтительно отношение массовой скорости потока растворителя, вводимого как часть потока исходных материалов, к массовой скорости потока растворителя, вводимого как часть потока флегмы, поддерживается в пределах примерно от 0,5:1 до полного отсутствия потока флегмы, более предпочтительно в пределах примерно от 0,5:1 примерно до 4:1, еще более предпочтительно в пределах примерно от 1:1 примерно до 2:1, наиболее предпочтительно в пределах от 1,25:1 до 1,5:1.
Во время жидкофазного окисления в барботажной колонне реакторного типа 20, предпочтительно, чтобы поток окислителя вводился в барботажную колонну реакторного типа 20 в количестве, которое обеспечивает молекулярный кислород, несколько превосходящий стехиометрическую потребность в нем. Величина избытка молекулярного кислорода, необходимого для наилучших результатов с конкретным окисляемым соединением, влияет на общие экономические показатели жидкофазного окисления. Во время жидкофазного окисления в барботажной колонне реакторного типа 20, предпочтительно, чтобы отношение массовой скорости потока для потока окислителя к массовой скорости потока окисляемого органического соединения (например, пара-ксилола), поступающего в реактор 20, поддерживалось в пределах примерно от 0,5:1 примерно до 20:1, более предпочтительно в пределах примерно от 1:1 примерно до 10:1, наиболее предпочтительно в пределах от 2:1 до 6:1.
Обращаясь опять к фиг.1, потоки исходных материалов, окислителя и флегмы, вводимые в барботажную колонну реакторного типа 20, совместно образуют, по меньшей мере, часть многофазной реакционной среды 36. Реакционная среда 36 представляет собой предпочтительно трехфазную среду, содержащую твердую фазу, жидкую фазу и газовую фазу. Как рассмотрено выше, окисление окисляемого соединения (например, пара-ксилола) имеет место в основном в жидкой фазе реакционной среды 36. Таким образом, жидкая фаза реакционной среды 36 содержит растворенный кислород и окисляемое соединение. Экзотермическая природа реакции окисления, которая имеет место в барботажной колонне реакторного типа 20, вызывает кипение/испарение части растворителя (например, уксусной кислоты и воды), вводимого через входы для исходных материалов 32a, b, c, d. Таким образом, газовая фаза реакционной среды 36 в реакторе 20 формируется в основном из испаренного растворителя и нерастворенной, непрореагировавшей части потока окислителя. Определенные, известные из литературы реакторы окисления используют теплообменные трубы/ребра для нагрева или охлаждения реакционной среды. Однако такие структуры теплообмена могут быть нежелательными в реакторе по настоящему изобретению и в способе, описываемом здесь. Таким образом, предпочтительно, чтобы барботажная колонна реакторного типа 20 по существу не содержала никаких поверхностей, которые вступают в контакт с реакционной средой 36 и демонстрируют усредненный по времени тепловой поток, больший, чем 30000 Вт/м2.
Концентрация растворенного кислорода в жидкой фазе реакционной среды 36 представляет собой результат динамического равновесия между скоростью массопередачи из газовой фазы и скоростью его расходования по реакциям в жидкой фазе (т.е. она не регулируется просто парциальным давлением молекулярного кислорода в поступающей газовой фазе, хотя оно и является одним из факторов для скорости подачи растворенного кислорода, и оно не влияет на предельную верхнюю концентрацию растворенного кислорода). Количество растворенного кислорода изменяется локально, будучи более высоким вблизи границы раздела пузырьков. В целом, количество растворенного кислорода зависит от соотношения факторов накопления и расходования в различных областях реакционной среды 36. Количество растворенного кислорода в данный момент времени зависит от однородности смешивания газа и жидкости относительно скоростей химического расходования. При конструировании, для соответствующего согласования подачи и потребности в растворенном кислороде в жидкой фазе реакционной среды 36, предпочтительно, чтобы усредненная по времени и по объему концентрация кислорода в жидкой фазе реакционной среды 36 поддерживалась примерно выше 1 м.д. молярной, более предпочтительно в пределах примерно от 4 примерно до 1000 м.д. молярных, еще более предпочтительно в пределах примерно от 8 примерно до 500 м.д. молярных, наиболее предпочтительно в пределах от 12 до 120 м.д. молярных.
Жидкофазная реакция окисления, осуществляемая в барботажной колонне реакторного типа 20, предпочтительно представляет собой реакцию осаждения, которая образует твердые продукты. Более предпочтительно жидкофазное окисление, осуществляемое в барботажной колонне реакторного типа 20, заставляет, по меньшей мере, примерно 10 мас.% окисляемого соединения (например, пара-ксилола), вводимого в реакционную зону 28, образовывать твердое соединение (например, частицы сырой терефталевой кислоты) в реакционной среде 36. Еще более предпочтительно жидкофазное окисление заставляет, по меньшей мере, примерно 50 мас.% окисляемого соединения образовывать твердые соединения в реакционной среде 36. Наиболее предпочтительно жидкофазное окисление заставляет, по меньшей мере, 90 мас.% окисляемого соединения образовывать твердые соединения в реакционной среде 36. Предпочтительно, чтобы общее количество твердых продуктов в реакционной среде 36 было большим, примерно, чем 3 мас.%, усредненное по времени и по объему. Более предпочтительно общее количество твердых продуктов в реакционной среде 36 поддерживается в пределах примерно от 5 примерно до 40 мас.%, еще более предпочтительно в пределах примерно от 10 примерно до 35 мас.%, наиболее предпочтительно в пределах от 15 до 30 мас.%. Предпочтительно, чтобы существенная часть продукта окисления (например, терефталевой кислоты), получаемого в барботажной колонне реакторного типа 20, присутствовала в реакционной среде 36 в виде твердых продуктов, вместо того, чтобы оставаться растворенной в жидкой фазе реакционной среды 36. Количество твердофазного продукта окисления, присутствующего в реакционной среде 36, предпочтительно составляет, по меньшей мере, примерно 25 мас.% от общего количества продукта окисления (твердой и жидкой фазы) в реакционной среде 36, более предпочтительно, по меньшей мере, примерно 75 мас.% от общего количества продукта окисления в реакционной среде 36, наиболее предпочтительно, по меньшей мере, 95 мас.% от общего количества продукта окисления в реакционной среде 36. Численные диапазоны, приведенные выше для количества твердых продуктов в реакционной среде 36, относятся к стационарной, по существу, работе барботажной колонны 20 в течение по существу непрерывного периода времени, но не к пуску, остановке или работе в неоптимальном режиме барботажной колонны реакторного типа 20. Количество твердых продуктов в реакционной среде 36 определяется гравиметрически. При этом в гравиметрическом способе представительный образец суспензии извлекается из реакционной среды и взвешивается. При условиях, которые эффективно поддерживают общее распределение твердые продукты - жидкость, присутствующее в реакционной среде, свободная жидкость удаляется из части твердых продуктов посредством седиментации или фильтрации, эффективно, без потерь осажденных твердых продуктов и с менее, примерно, чем 10% от начальной массы жидкости, остающимися в части твердых продуктов. Оставшаяся жидкость на твердых продуктах выпаривается досуха, эффективно, без сублимации твердых продуктов. Оставшаяся часть твердых продуктов взвешивается. Отношение массы части твердых продуктов к массе исходной части суспензии представляет собой долю твердых продуктов, как правило, выражаемую в процентах.
Реакция осаждения, осуществляемая в барботажной колонне реакторного типа 20, может вызвать образование осадка (т.е. отложение твердых продуктов) на поверхности определенных жестких структур, которые вступают в контакт с реакционной средой 36. Таким образом, в одном из вариантов осуществления настоящего изобретения предпочтительно, чтобы барботажная колонна реакторного типа 20 по существу не содержала внутренних структур теплообмена, перемешивания или дефлекторов в реакционной зоне 28, поскольку такие структуры были бы склонны к образованию осадка. Если внутренние структуры присутствуют в реакционной зоне 28, является желательным исключение внутренних структур, имеющих наружные поверхности, которые имеют значительную величину площади плоской поверхности, обращенной вверх, поскольку такие обращенные вверх плоские поверхности были бы в высшей степени склонными к образованию осадка. Таким образом, если какие-либо внутренние структуры присутствуют в реакционной зоне 28 предпочтительно, чтобы меньше, примерно, чем 20% от общей площади открытой наружной поверхности, обращенной вверх, у таких внутренних структур формировались бы посредством по существу плоских поверхностей, наклоненных под углом меньше, примерно, чем 15°, от горизонтали.
Обращаясь опять к фиг.1, физическая конфигурация барботажной колонны реакторного типа 20 помогает обеспечить оптимизированное окисление окисляемого соединения (например, пара-ксилола) при минимальном образовании примесей. Предпочтительно, чтобы удлиненная реакционная секция 24 корпуса 22 содержала по существу цилиндрический главный корпус 46 и днище 48. Верхний уровень реакционной зоны 28 определяется горизонтальной плоскостью 50, простирающейся по верхней части цилиндрического главного корпуса 46. Нижний уровень 52 реакционной зоны 28 определяется самой нижней внутренней поверхностью днища 48. Как правило, нижний уровень 52 реакционной зоны 28 располагается вблизи отверстия для выхода суспензии 38. Таким образом, удлиненная реакционная зона 28, определенная внутри барботажной колонны реакторного типа 20, имеет максимальную длину L, измеряемую от верхнего края 50 до нижнего края 52 реакционной зоны 28 вдоль продольной оси цилиндрического главного корпуса 46. Длина L реакционной зоны 28 предпочтительно находится в пределах примерно от 10 примерно до 100 м, более предпочтительно в пределах примерно от 20 примерно до 75 м, наиболее предпочтительно в пределах от 25 до 50 м. Реакционная зона 28 имеет максимальный диаметр (ширину) D, который, как правило, равен максимальному внутреннему диаметру цилиндрического главного корпуса 46. Максимальный диаметр D реакционной зоны 28 предпочтительно находится в пределах примерно от 1 примерно до 12 м, более предпочтительно в пределах примерно от 2 примерно до 10 м, еще более предпочтительно в пределах примерно от 3,1 примерно до 9 м, наиболее предпочтительно в пределах от 4 до 8 м. В предпочтительном варианте осуществления настоящего изобретения реакционная зона 28 имеет отношение длины к диаметру L:D в пределах примерно от 6:1 примерно до 30:1. Еще более предпочтительно реакционная зона 28 имеет отношение L:D в пределах примерно от 8:1 примерно до 20:1. Наиболее предпочтительно реакционная зона 28 имеет отношение L:D в пределах от 9:1 до 15:1.
Как обсуждалось выше, реакционная зона 28 барботажной колонны реакторного типа 20 принимает многофазную реакционную среду 36. Реакционная среда 36 имеет нижний уровень, совпадающий с нижним уровнем 52 реакционной зоны 28, и верхний уровень, расположенный на верхней поверхности 44. Верхняя поверхность 44 реакционной среды 36 определяется вдоль горизонтальной плоскости, которая пересекает реакционную зону 28 в вертикальном положении, где содержимое реакционной зоны 28 переходит из сплошного газообразного состояния в сплошное жидкофазное состояние. Верхняя поверхность 44 предпочтительно располагается в вертикальном положении, где локальное усредненное по времени содержание газа тонкого горизонтального слоя содержимого реакционной зоны 28 равно 0,9.
Реакционная среда 36 имеет максимальную высоту H, измеренную между ее верхним и нижним краями. Максимальная ширина W реакционной среды 36, как правило, равна максимальному диаметру D цилиндрического главного корпуса 46. Во время жидкофазного окисления в барботажной колонне реакторного типа 20, предпочтительно, чтобы H поддерживалось как примерно от 60 примерно до 120% от L, более предпочтительно примерно как от 80 примерно до 110% от L, наиболее предпочтительно как от 85 до 100% от L. В предпочтительном варианте осуществления настоящего изобретения реакционная среда 36 имеет отношение высоты к ширине H:W больше, примерно, чем 3:1. Более предпочтительно реакционная среда 36 имеет отношение H:W в пределах примерно от 7:1 примерно до 25:1. Еще более предпочтительно реакционная среда 36 имеет отношение H:W в пределах примерно от 8:1 примерно до 20:1. Наиболее предпочтительно реакционная среда 36 имеет отношение H:W в пределах от 9:1 до 15:1. В одном из вариантов осуществления настоящего изобретения L=H и D=W, так что различные размеры или отношения, приведенные здесь для L и D, применяются также к H и W, и наоборот.
Относительно высокие отношения L:D и H:W, предусмотренные в соответствии с одним из вариантов осуществления настоящего изобретения, могут вносить вклад в несколько важных преимуществ системы по настоящему изобретению. Как обсуждается более подробно ниже, обнаружено, что более высокие отношения L:D и H:W, а также некоторые другие особенности, обсуждаемые ниже, могут способствовать созданию градиентов по высоте концентраций молекулярного кислорода и/или окисляемого соединения (например, пара-ксилола) в реакционной среде 36. В противоположность общепринятой концепции, которая склоняется к хорошо перемешиваемой реакционной среде с относительно однородными концентрациями в ней, обнаружено, что вертикальное ступенчатое изменение концентрации кислорода и/или окисляемого соединения способствует более эффективной и экономичной реакции окисления. Сведение к минимуму концентраций кислорода и окисляемого соединения вблизи верхней части реакционной среды 36 может помочь предотвратить потери непрореагировавшего кислорода и непрореагировавшего окисляемого соединения через верхний выход для газа 40. Однако если концентрация окисляемого соединения и непрореагировавшего кислорода являются низкими по всей реакционной среде 36, тогда скорость и/или селективность окисления понижаются. Таким образом, предпочтительно, чтобы концентрации молекулярного кислорода и/или окисляемого соединения были значительно выше вблизи нижней части реакционной среды 36, чем вблизи верхней части реакционной среды 36.
В дополнение к этому, высокие отношения L:D и H:W делают давление в нижней части реакционной среды 36 существенно более высоким, чем давление в верхней части реакционной среды 36. Этот градиент давления по высоте является результатом большой высоты и плотности реакционной среды 36. Одно из преимуществ этого градиента давления по высоте заключается в том, что повышенное давление в нижней части емкости обеспечивает большую растворимость кислорода и больший массоперенос, чем те, которые достигались бы в противном случае при сравнимых температурах и давлениях в верхней части колонны в более низких реакторах. Таким образом, реакция окисления может осуществляться при более низких температурах, чем потребовались бы в более низкой емкости. Когда барботажная колонна реакторного типа 20 используется для частичного окисления пара-ксилола до сырой терефталевой кислоты (CTA), способность ее к работе при более низких температурах реакции, при тех же или лучших скоростях массопередачи кислорода, имеет ряд преимуществ. Например, низкотемпературное окисление пара-ксилола уменьшает количество растворителя, сжигаемого во время реакции. Как обсуждается более подробно ниже, низкотемпературное окисление также благоприятствует образованию малых, имеющих высокую площадь поверхности, непрочно связанных, легко растворяющихся частиц CTA, которые могут подвергаться более экономичным технологиям очистки, чем большие, имеющие низкую площадь поверхности, плотные частицы CTA, получаемые с помощью обычных высокотемпературных способов окисления.
Во время окисления в реакторе 20 предпочтительно, чтобы усредненная по времени и по объему температура реакционной среды 36 поддерживалась в пределах примерно от 125 примерно до 200°C, более предпочтительно в пределах примерно от 140 примерно до 180°C, наиболее предпочтительно в пределах от 150 до 170°C. Давление в верхней части колонны над реакционной средой 36 предпочтительно поддерживается в пределах примерно от 1 примерно до 20 бар в датчике (бар), более предпочтительно в пределах примерно от 2 примерно до 12 бар, наиболее предпочтительно в пределах от 4 до 8 бар. Предпочтительно разность давлений между верхней частью реакционной среды 36 и нижней частью реакционной среды 36 находится в пределах примерно от 0,4 примерно до 5 бар, более предпочтительно разность давлений находится в пределах примерно от 0,7 примерно до 3 бар, наиболее предпочтительно разность давлений составляет от 1 до 2 бар. Хотя, как правило, предпочтительно, чтобы давление в верхней части колонны над реакционной средой 36 поддерживалось при относительно постоянном значении, один из вариантов осуществления настоящего изобретения предполагает импульсный режим давления в верхней части колонны для облегчения улучшенного перемешивания и/или массопередачи в реакционной среде 36. Когда давление в верхней части колонны пульсирует, предпочтительно, чтобы импульсные режимы давления находились в пределах между примерно 60 и примерно 140% от стационарного давления в верхней части колонны, упоминаемого здесь, более предпочтительно в пределах между примерно 85 и примерно 115% стационарного давления в верхней части колонны, упоминаемого здесь, наиболее предпочтительно в пределах между 95 и 105% стационарного давления в верхней части колонны, упоминаемого здесь.
Дополнительное преимущество высокого отношения L:D реакционной зоны 28 заключается в том, что это может вносить вклад в увеличение средней поверхностной скорости реакционной среды 36. Термин "поверхностная скорость" и "поверхностная скорость газа", как здесь используется по отношению к реакционной среде 36, должен обозначать объемную скорость потока газовой фазы реакционной среды 36 на некоторой высоте в реакторе, разделенную на площадь горизонтального поперечного сечения реактора на этой высоте. Увеличенная поверхностная скорость, обеспечиваемая высоким отношением L:D реакционной зоны 28, может ускорять локальное перемешивание и увеличивать содержание газа в реакционной среде 36. Усредненные по времени поверхностные скорости реакционной среды 36 на одной четвертой высоты, на половине высоты и/или на трех четвертях высоты реакционной среды 36 предпочтительно больше, примерно, чем 0,3 м/с, более предпочтительно находятся в пределах примерно от 0,8 примерно до 5 м/с, еще более предпочтительно в пределах примерно от 0,9 примерно до 4 м/с, наиболее предпочтительно в пределах от 1 до 3 м/с.
Обращаясь опять к фиг.1, секция отделения 26 барботажной колонны реакторного типа 20 представляет собой просто расширенную часть корпуса 22, расположенную непосредственно над реакционной секцией 24. Секция отделения 26 уменьшает скорость проходящей снизу вверх газовой фазы в барботажной колонне реакторного типа 20, когда газовая фаза поднимается выше верхней поверхности 44 реакционной среды 36 и достигает выхода для газа 40. Это уменьшение скорости движения снизу вверх газовой фазы помогает облегчить удаление захваченных жидких и/или твердых продуктов в проходящей снизу вверх газовой фазе и тем самым уменьшает нежелательные потери определенных компонентов, присутствующих в жидкой фазе реакционной среды 36.
Секция отделения 26 предпочтительно содержит в целом расходящуюся коническую переходную стенку 54, в целом цилиндрическую широкую боковую стенку 56 и крышку 58. Узкий нижний край переходной стенки 54 соединен с верхней частью цилиндрического главного корпуса 46 реакционной секции 24. Широкий верхний край переходной стенки 54 соединен с нижней частью широкой боковой стенки 56. Предпочтительно, чтобы переходная стенка 54 простиралась вверх и наружу от ее узкого нижнего края под углом в пределах примерно от 10 примерно до 70° от вертикали, более предпочтительно в пределах примерно 15 примерно до 50° от вертикали, а наиболее предпочтительно в пределах от 15 до 45° от вертикали. Широкая боковая стенка 56 имеет максимальный диаметр X, который, как правило, больше, чем максимальный диаметр D реакционной секции 24, хотя, когда верхняя часть реакционной секции 24 имеет диаметр, меньший, чем максимальный диаметр реакционной секции 24 в целом, тогда X может реально быть меньшим, чем D. В предпочтительном варианте осуществления настоящего изобретения отношение диаметра широкой боковой стенки 56 к максимальному диаметру реакционной секции 24 X:D находится в пределах примерно от 0,8:1 примерно до 4:1, наиболее предпочтительно в пределах от 1,1:1 до 2:1. Крышка 58 соединена с верхней частью широкой боковой стенки 56. Крышка 58 предпочтительно представляет собой, в целом, эллиптический элемент крышки, определяющий центральное отверстие, которое позволяет газу уходить из зоны отделения 30 через выход для газа 40. Альтернативно, крышка 58 может иметь любую форму, включая коническую. Зона отделения 30 имеет максимальную высоту Y, измеренную от верхней части 50 реакционной зоны 28 до самой верхней части зоны отделения 30. Отношение длины реакционной зоны 28 к высоте зоны отделения 30 L:Y предпочтительно находится в пределах примерно от 2:1 примерно до 24:1, более предпочтительно в пределах примерно от 3:1 примерно до 20:1, наиболее предпочтительно в пределах от 4:1 до 16:1.
Обращаясь к фиг.1-5, теперь будут обсуждаться более подробно положение и конфигурация устройства распределения окислителя 34. Фиг.2 и 3 показывают, что устройство распределения окислителя 34 может содержать кольцевой элемент 60, диаметральный элемент 62 и пару проходов 64a, b для поступления окислителя. Удобно, чтобы эти проходы 64a, b для поступления окислителя могли входить в емкость несколько выше кольцевого элемента 60, а потом поворачивать вниз, как показано на фиг.2 и 3. Альтернативно, проход 64a, b для поступления окислителя может входить в емкость ниже кольцевого элемента 60 или примерно в той же горизонтальной плоскости, что и кольцевой элемент 60. Каждый проход 64a, b для поступления окислителя содержит первый край, соединенный с соответствующим входом для окислителя 66a, b, сформированный в корпусе 22, и второй край, соединенный с сообщением текучих сред с кольцевым элементом 60. Кольцевой элемент 60 предпочтительно формируется из проходов, более предпочтительно из множества прямых секций проходов, наиболее предпочтительно из множества прямых трубных секций, жестко соединенных друг с другом, с образованием трубчатого многоугольного кольца. Предпочтительно кольцевой элемент 60 формируется, по меньшей мере, из 3 прямых трубных секций, более предпочтительно из 6-10 трубных секций, а наиболее предпочтительно из 8 трубных секций. Соответственно, когда кольцевой элемент 60 формируется из 8 трубных секций, он имеет, в целом, восьмиугольную конфигурацию. Диаметральный элемент 62 предпочтительно формируется по существу из прямой трубной секции, которая соединена с сообщением текучих сред с противоположными трубными секциями кольцевого элемента 60 и простирается по диагонали между ними. Трубная секция, используемая для диаметрального элемента 62, предпочтительно имеет по существу такой же диаметр, как и трубные секции, используемые для формирования кольцевого элемента 60. Предпочтительно, чтобы трубные секции, которые составляют проходы 64a, b для поступления окислителя, кольцевой элемент 60 и диаметральный элемент 62, имели номинальный диаметр, больший, примерно, чем 0,1 м, более предпочтительно находящийся в пределах примерно от 0,2 примерно до 2 м, а наиболее предпочтительно в пределах от 0,25 до 1 м. Как лучше всего, вероятно, иллюстрируется на фиг.3, кольцевой элемент 60 и диаметральный элемент 62, каждый, предоставляют множество верхних отверстий 68 для окислителя, для высвобождения потока окислителя снизу вверх в реакционную зону 28. Как лучше всего, вероятно, иллюстрируется на фиг.4, кольцевой элемент 60 и/или диаметральный элемент 62 могут предоставлять одно или несколько нижних отверстий 70 для окислителя, для высвобождения потока окислителя вниз, в реакционную зону 28. Нижние отверстия 70 для окислителя могут также использоваться для высвобождения жидкости и/или твердых продуктов, которые могли бы поступать в кольцевой элемент 60 и/или диаметральный элемент 62. Для предотвращения осаждения твердых продуктов внутри устройства распределения окислителя 34 поток жидкости может непрерывно или периодически проходить через устройства распределения 34, смывая любые аккумулированные твердые продукты.
Обращаясь опять к фиг.1-4, во время окисления в барботажной колонне реакторного типа 20 потоки окислителя нагнетаются через входы для окислителя 66a, b и в проходы 64a, b для поступления окислителя, соответственно. Затем потоки окислителя переносятся через проходы 64a, b для поступления окислителя в кольцевой элемент 60. Когда поток окислителя поступает в кольцевой элемент 60, поток окислителя распределяется по внутренним объемам кольцевого элемента 60 и диаметрального элемента 62. Затем поток окислителя принудительно выводится из устройства распределения окислителя 34 и в реакционную зону 28 через верхние и нижние отверстия для окислителя 68, 70 кольцевого элемента 60 и диаметрального элемента 62.
Выходы верхних отверстий 68 для окислителя отделены друг от друга латерально на некоторое расстояние и располагаются по существу на одной и той же высоте в реакционной зоне 28. Таким образом, выходы верхних отверстий 68 для окислителя располагаются в целом вдоль по существу горизонтальной плоскости, определенной верхней частью устройства распределения окислителя 34. Выходы нижних отверстий 70 для окислителя отделены друг от друга латерально на некоторое расстояние и располагаются по существу на одной и той же высоте в реакционной зоне 28. Таким образом, выходы нижних отверстий 70 для окислителя располагаются в целом вдоль по существу горизонтальной плоскости, определенной нижней частью устройства распределения окислителя 34.
В одном из вариантов осуществления настоящего изобретения устройство распределения окислителя 34 имеет, по меньшей мере, примерно 20 верхних отверстий 68 для окислителя, сформированных в нем. Более предпочтительно устройство распределения окислителя 34 имеет в пределах примерно от 40 примерно до 800 верхних отверстий для окислителя, сформированных в нем. Наиболее предпочтительно устройство распределения окислителя 34 имеет в пределах от 60 до 400 верхних отверстий 68 для окислителя, сформированных в нем. Устройство распределения окислителя 34 предпочтительно имеет, по меньшей мере, примерно 1 нижнее отверстие 70 для окислителя, сформированное в нем. Более предпочтительно устройство распределения окислителя 34 имеет в пределах примерно от 2 примерно до 40 нижних отверстий для окислителя 70, сформированных в нем. Наиболее предпочтительно устройство распределения окислителя 34 имеет в пределах от 8 до 20 нижних отверстий 70 для окислителя, сформированных в нем. Отношение количества верхних отверстий 68 для окислителя к нижним отверстиям 70 для окислителя в устройстве распределения окислителя 34 предпочтительно находится в пределах примерно от 2:1 примерно до 100:1, более предпочтительно в пределах примерно от 5:1 примерно до 25:1, наиболее предпочтительно в пределах от 8:1 до 15:1. Диаметры по существу всех верхних и нижних отверстий 68, 70 для окислителя предпочтительно являются по существу одинаковыми, так что отношение объемной скорости потока окислителя из верхних и нижних отверстий 68, 70 является по существу таким же, как и отношения, приведенные выше, для относительного количества верхних и нижних отверстий 68, 70 для окислителя.
Фиг.5 иллюстрирует направление высвобождения окислителя из верхних и нижних отверстий 68, 70 для окислителя. При упоминании верхних отверстий 68 для окислителя, предпочтительно, чтобы, по меньшей мере, часть верхних отверстий 68 для окислителя, для высвобождения потока окислителя находилась под углом A, т.е. под углом к вертикали. Предпочтительно, чтобы процент верхних отверстий для окислителя 68, которые отклоняются от вертикали на угол A, находился в пределах примерно от 30 примерно до 90%, более предпочтительно в пределах примерно от 50 примерно до 80%, еще более предпочтительно в пределах от 60 до 75%, а наиболее предпочтительно составлял примерно 67%. Угол A предпочтительно находится в пределах примерно от 5 примерно до 60°, более предпочтительно в пределах примерно от 10 примерно до 45°, а наиболее предпочтительно в пределах от 15 до 30°. Относительно нижних отверстий 70 для окислителя, предпочтительно, чтобы по существу все нижние отверстия 70 для окислителя располагались вблизи самой нижней части кольцевого элемента 60 и/или диаметрального элемента 62. Таким образом, любые жидкие и/или твердые продукты, которые могут ненамеренно поступать в устройство распределения окислителя 34, могут легко высвобождаться из устройства распределения окислителя 34 через нижние отверстия 70 для окислителя. Предпочтительно нижние отверстия для окислителя 70 высвобождают поток окислителя вниз, по существу под вертикальным углом. Для целей настоящего описания верхнее отверстие для окислителя может представлять собой любое отверстие, которое высвобождает поток окислителя в направлении в целом снизу вверх (т.е. под углом выше горизонтали), и нижнее отверстие для окислителя может представлять собой любое отверстие, которое высвобождает поток окислителя в направлении в целом сверху вниз (т.е. под углом ниже горизонтали).
Во многих обычных барботажных колоннах реакторного типа, содержащих многофазную реакционную среду, по существу вся реакционная среда, находящаяся ниже устройства распределения окислителя (или другого механизма введения потока окислителя в реакционную зону), имеет очень низкое значение содержания газа. Как известно в данной области, "содержание газа" представляет собой просто объемную долю многофазной среды, которая находится в газообразном состоянии. Зоны с низким содержанием газа в среде могут также упоминаться как "неаэрированные" зоны. Во многих обычных суспензионных барботажных колоннах реакторного типа значительная часть общего объема реакционной среды располагается ниже устройства распределения окислителя (или другого механизма для введения потока окислителя в реакционную зону). Таким образом, значительная часть реакционной среды, присутствующей в нижней части обычных барботажных колонн реакторного типа, является неаэрированной.
Обнаружено, что сведение к минимуму количества неаэрированных зон в реакционной среде, подвергающейся окислению в барботажной колонне реакторного типа, может свести к минимуму образование определенных типов нежелательных примесей. Неаэрированные зоны реакционной среды содержат относительно мало пузырьков окислителя. Этот малый объем пузырьков окислителя уменьшает количество молекулярного кислорода, доступного для растворения в жидкой фазе реакционной среды. Таким образом, жидкая фаза в неаэрированной зоне реакционной среды имеет относительно низкую концентрацию молекулярного кислорода. Эти обедненные кислородом, неаэрированные зоны реакционной среды имеют тенденцию к ускорению нежелательных побочных реакций, вместо желательной реакции окисления. Например, когда пара-ксилол частично окисляется с образованием терефталевой кислоты, недостаточная доступность кислорода в жидкой фазе реакционной среды может вызывать образование нежелательно высоких количеств бензойной кислоты и связанных ароматических колец, в заметных количествах содержащих в высшей степени нежелательные окрашенные молекулы, известные как флуореноны и антрахиноны.
В соответствии с одним из вариантов осуществления настоящего изобретения жидкофазное окисление осуществляют в барботажной колонне реакторного типа, сконфигурированной и работающей таким способом, что объемная доля реакционной среды с низкими значениями содержания газа сводится к минимуму. Это сведение к минимуму неаэрированных зон может количественно определяться посредством теоретического разделения всего объема реакционной среды на 2000 дискретных горизонтальных слоев одинакового объема. За исключением самого верхнего и самого нижнего горизонтальных слоев, каждый горизонтальный слой представляет собой дискретный объем, ограниченный на его сторонах боковой стенкой реактора и ограниченный сверху и снизу воображаемыми горизонтальными плоскостями. Самый высокий горизонтальный слой ограничен снизу воображаемой горизонтальной плоскостью, а сверху - верхней поверхностью реакционной среды. Самый нижний горизонтальный слой ограничен сверху воображаемой горизонтальной плоскостью, а снизу - нижним уровнем емкости. Когда реакционная среда теоретически разделяется на 2000 дискретных горизонтальных слоев равного объема, может быть определено усредненное по времени и по объему содержание газа каждого горизонтального слоя. Когда используется этот способ количественного определения количества неаэрированных зон, предпочтительно, чтобы количество горизонтальных слоев, имеющих усредненное по времени и усредненное по объему содержание газа, меньшее, чем 0,1, было меньшим, чем 30, более предпочтительно меньшим, чем 15, еще более предпочтительно меньшим, чем 6, еще более предпочтительно меньшим, чем 4, а наиболее предпочтительно меньшим, чем 2. Предпочтительно, чтобы количество горизонтальных слоев, имеющих содержание газа, меньшее, чем 0,2, было меньшим, чем 80, более предпочтительно меньшим, чем 40, еще более предпочтительно меньшим, чем 20, еще более предпочтительно меньшим, чем 12, а наиболее предпочтительно меньшим, чем 5. Предпочтительно, чтобы количество горизонтальных слоев, имеющих содержание газа, меньшее, чем 0,3, было меньшим, чем 120, более предпочтительно меньшим, чем 80, еще более предпочтительно меньшим, чем 40, еще более предпочтительно меньшим, чем 20, а наиболее предпочтительно меньшим, чем 15.
Обращаясь опять к фиг.1 и 2, обнаружено, что расположение устройства распределения окислителя 34 ниже в реакционной зоне 28 обеспечивает ряд преимуществ, включая уменьшение количества неаэрированных зон в реакционной среде 36. При данной высоте H реакционной среды 36, длине L реакционной зоны 28 и максимальном диаметре D реакционной зоны 28 предпочтительно, чтобы большая часть (т.е. >50 мас.%) потока окислителя вводилась в реакционную зону 28 в пределах примерно 0,025 H, 0,022 L и/или 0,25 D от нижнего края 52 реакционной зоны 28. Более предпочтительно большая часть потока окислителя вводится в реакционную зону 28 в пределах примерно 0,02 H, 0,018 L и/или 0,2 D от нижнего края 52 реакционной зоны 28. Наиболее предпочтительно большая часть потока окислителя вводится в реакционную зону 28 в пределах 0,015 H, 0,013 L и/или 0,15 D от нижнего края 52 реакционной зоны 28.
В варианте осуществления, иллюстрируемом на фиг.2, вертикальное расстояние Y1 между нижним краем 52 реакционной зоны 28 и выходом верхних отверстий 68 для окислителя устройства распределения окислителя 34 является меньшим, примерно, чем 0,25 H, 0,022 L и/или 0,25 D, так что по существу весь поток окислителя поступает в реакционную зону 28 в пределах примерно 0,25 H, 0,022 L и/или 0,25 D от нижнего края 52 реакционной зоны 28. Более предпочтительно Y1 меньше, примерно, чем 0,02 H, 0,018 L и/или 0,2 D. Наиболее предпочтительно Y1 меньше, чем 0,015 H, 0,013 L и/или 0,15 D, но больше, чем 0,005 H, 0,004 L и/или 0,06 D. Фиг.2 иллюстрирует линию начала изгиба 72, в положении, где нижний край цилиндрического главного корпуса 46 корпуса 22 соединяется с верхним краем эллиптического днища 48 корпуса 22. Альтернативно, днище 48 может иметь любую форму, включая коническую, и линия начала изгиба по-прежнему определяется как нижний уровень цилиндрической части главного корпуса 46. Вертикальное расстояние Y2 между линией начала изгиба 72 и верхней частью устройства распределения окислителя 34 предпочтительно равно, по меньшей мере, примерно 0,0012 H, 0,001 L и/или 0,01 D; более предпочтительно, по меньшей мере, примерно 0,005 H, 0,004 L и/или 0,05 D; а наиболее предпочтительно, по меньшей мере, 0,01 H, 0,008 L и/или 0,1 D. Вертикальное расстояние Y3 между нижним краем 52 реакционной зоны 28 и выходом нижних отверстий 70 для окислителя устройства распределения окислителя 34 предпочтительно меньше, примерно, чем 0,015 H, 0,013 L и/или 0,15 D; более предпочтительно меньше, примерно, чем 0,012 H, 0,01 L и/или 0,1 D; а наиболее предпочтительно меньше, чем 0,01 H, 0,008 L и/или 0,075 D, но больше, чем 0,003 H, 0,002 L и/или 0,025 D.
В предпочтительном варианте осуществления настоящего изобретения, отверстия, которые высвобождают поток окислителя и поток исходных материалов в реакционной зоне, конфигурируются таким образом, что количество (по массе) потока окислителя или исходных материалов, высвобождаемого из отверстия, является прямо пропорциональным открытой площади отверстия. Таким образом, например, если 50% общей открытой площади, определяемой всеми отверстиями для окислителя, располагается в пределах 0,15 D от нижней части реакционной зоны, тогда 50 мас.% потока окислителя поступает в реакционную зону в пределах 0,15 D от нижней части реакционной зоны, и наоборот.
В дополнение к преимуществам, обеспечиваемым посредством сведения к минимуму неаэрированных зон (т.е. зон с низким содержанием газа) в реакционной среде 36, обнаружено, что окисление может быть улучшено посредством доведения до максимума содержания газа во всей реакционной среде 36. Реакционная среда 36 предпочтительно имеет усредненное по времени и усредненное по объему содержание газа, по меньшей мере, примерно 0,4, более предпочтительно в пределах примерно от 0,6 примерно до 0,9, а наиболее предпочтительно в пределах от 0,65 до 0,85. Несколько физических и рабочих атрибутов барботажной колонны реакторного типа 20 вносят вклад в высокое содержание газа, обсуждаемое выше. Например, для данного размера реактора и скорости потока для потока окислителя, высокое отношение L:D реакционной зоны 28 дает меньший диаметр, который увеличивает поверхностную скорость в реакционной среде 36, что, в свою очередь, увеличивает содержание газа. В дополнение к этому, реальный диаметр барботажной колонны и отношение L:D, как известно, влияют на среднее содержание газа даже для данной постоянной поверхностной скорости. В дополнение к этому, сведение к минимуму неаэрированных зон, в особенности в нижней части реакционной зоны 28, вносит вклад в увеличение значения содержания газа. Кроме того, давление в верхней части колонны и механическая конфигурация барботажной колонны реакторного типа могут влиять на стабильность работы при высоких поверхностных скоростях и значениях содержания газа, описываемых здесь.
Более того, авторы обнаружили важность работы с оптимизированным давлением в верхней части колонны для получения увеличенного содержания газа и увеличенной массопередачи. Может показаться, что работа при более низком избыточном давлении, которое уменьшает растворимость молекулярного кислорода в соответствии с законом Генри, уменьшала бы скорость массопередачи молекулярного кислорода из газа в жидкость. В механически перемешиваемой емкости так обычно и происходит, поскольку уровни аэрации и скорости массопередачи определяются конструкцией мешалки и давлением в верхней части колонны. Однако в барботажной колонне реакторного типа в соответствии с предпочтительным вариантом осуществления настоящего изобретения обнаружено, как использовать более низкое давление в верхней части колонны для создания данной массы газофазного потока окислителя, занимая больший объем, увеличивая поверхностную скорость в реакционной среде 36 и, в свою очередь, увеличивая содержание газа и скорость переноса молекулярного кислорода.
Равновесие между коалесценцией и разрушением пузырьков представляет собой исключительно сложное явление, приводя, с одной стороны, к тенденции пенообразования, которое уменьшает внутренние скорости циркуляции жидкой фазы и которое может потребовать очень значительных зон отделения, а с другой стороны, к тенденции образования нескольких очень больших пузырьков, которые дают более низкое содержание газа и более низкую скорость массопередачи из потока окислителя в жидкую фазу. Относительно жидкой фазы, ее состав, плотность, вязкость и поверхностное натяжение, среди других факторов, как известно, взаимодействуют очень сложным способом, с получением очень сложных результатов даже в отсутствие твердой фазы. Например, лабораторные исследователи находят полезным квалифицировать, представляет ли собой "вода" водопроводную воду, дистиллированную воду или деионизованную воду, когда они описывают и оценивают наблюдения даже для простых барботажных колонн вода-воздух. Для сложных смесей в жидкой фазе и при добавлении твердой фазы степень сложности дополнительно возрастает. Нерегулярности поверхности индивидуальных частиц твердых продуктов, средний размер твердых продуктов, распределение размеров частиц, количество твердых продуктов относительно жидкой фазы и способность жидкости смачивать поверхность твердых продуктов, среди других вещей, все они являются важными при их взаимодействии с жидкой фазой и потоком окислителя при установлении того, какое поведение пузырьков и какие структуры потока естественной конвекции получатся в результате.
Таким образом, способность барботажной колонны реакторного типа функционировать с пользой при высоких поверхностных скоростях и высоких содержаниях газа, описанных здесь, зависит, например, от правильного выбора: (1) состава жидкой фазы реакционной среды; (2) количества и типа осаждаемых твердых продуктов, оба они могут регулироваться условиями реакции; (3) величины потока окислителя, вводимого в реактор; (4) давления в верхней части колонны, которое оказывает воздействие на объемную скорость потока, для потока окислителя, стабильность пузырьков и, через баланс энергии, на температуру реакции; (5) самой температуры реакции, которая влияет на свойства текучей среды, свойства осаждаемых твердых продуктов и удельный объем потока окислителя; и (6) геометрии и механических деталей реакционной емкости, включая отношение L:D.
Обращаясь опять к фиг.1, обнаружено, что улучшение распределения окисляемого соединения (например, пара-ксилола) в реакционной среде 36 может быть обеспечено посредством введения жидкофазного потока исходных материалов в реакционную зону 28 во множестве положений, отделенных друг от друга некоторым расстоянием по высоте. Предпочтительно жидкофазный поток исходных материалов вводится в реакционную зону 28 посредством, по меньшей мере, 3 входных отверстий, более предпочтительно, по меньшей мере, 4 входных отверстий. Как здесь используется, термин "входные отверстия" должен обозначать отверстия, где жидкофазный поток исходных материалов высвобождается в реакционную зону 28 для смешивания с реакционной средой 36. Предпочтительно, чтобы, по меньшей мере, 2 из входных отверстий были отделены друг от друга некоторым расстоянием по вертикали, по меньшей мере, примерно 0,5 D, более предпочтительно, по меньшей мере, примерно 1,5 D, а наиболее предпочтительно, по меньшей мере, 3 D. Однако предпочтительно, чтобы самое высокое входное отверстие было отделено некоторым расстоянием по вертикали от самого нижнего отверстия для окислителя, не большим, примерно, чем 0,75 H, 0,65 L и/или 8 D; более предпочтительно не большим, примерно, чем 0,5 H, 0,4 L и/или 5 D; наиболее предпочтительно не большим, чем 0,4 H, 0,35 L и/или 4 D.
Хотя является желательным введение жидкофазного потока исходных материалов во множестве положений по высоте, также обнаружено, что улучшенное распределение окисляемого соединения в реакционной среде 36 обеспечивается, если большая часть жидкофазного потока исходных материалов вводится в нижней половине реакционной среды 36 и/или реакционной зоны 28. Предпочтительно, по меньшей мере, примерно 75 мас.% жидкофазного потока исходных материалов вводится в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Наиболее предпочтительно, по меньшей мере, 90 мас.% жидкофазного потока исходных материалов вводится в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. В дополнение к этому, предпочтительно, чтобы, по меньшей мере, примерно 30 мас.% жидкофазного потока исходных материалов вводилось в реакционную зону 28 в пределах примерно 1,5 D от самого нижнего положения по высоте, где поток окислителя вводится в реакционную зону 28. Это самое нижнее положение по высоте, где поток окислителя вводится в реакционную зону 28, как правило, находится в нижней части устройства распределения окислителя; однако предпочтительным вариантом осуществления настоящего изобретения рассматривается множество альтернативных конфигураций для введения потока окислителя в реакционную зону 28. Предпочтительно, по меньшей мере, примерно 50 мас.% жидкофазных исходных материалов вводится в пределах примерно 2,5 D от самого нижнего положения по высоте, где поток окислителя вводится в реакционную зону 28. Предпочтительно, по меньшей мере, примерно 75 мас.% жидкофазного потока исходных материалов вводится в пределах примерно 5 D от самого нижнего положения по высоте, где поток окислителя вводится в реакционную зону 28.
Каждое входное отверстие определяет открытую площадь, через которую высвобождаются исходные материалы. Предпочтительно, чтобы, по меньшей мере, примерно 30% общей открытой площади всех входов для исходных материалов располагалось в пределах примерно 1,5 D от самого нижнего положения по высоте, где поток окислителя вводится в реакционную зону 28. Предпочтительно, по меньшей мере, примерно 50% общей открытой площади всех входов для исходных материалов располагается в пределах примерно 2,5 D от самого нижнего положения по высоте, где поток окислителя вводится в реакционную зону 28. Предпочтительно, по меньшей мере, примерно 75% общей открытой площади всех входов для исходных материалов располагается в пределах примерно 5 D от самого нижнего положения по высоте, где поток окислителя вводится в реакционную зону 28.
Обращаясь опять к фиг.1, в одном из вариантов осуществления настоящего изобретения входы для исходных материалов 32a, b, c, d представляют собой просто ряд расположенных по высоте отверстий вдоль одной стороны корпуса 22. Эти входные отверстия предпочтительно имеют по существу сходные диаметры, меньшие, примерно, чем 7 см, более предпочтительно в пределах примерно от 0,25 примерно до 5 см, а наиболее предпочтительно в пределах от 0,4 до 2 см. Барботажная колонна реакторного типа 20 предпочтительно снабжается системой для контроля скорости потока, для жидкофазного потока исходных материалов из каждого входного отверстия. Такая система контроля потока предпочтительно содержит индивидуальный клапан контроля потока 74a, b, c, d для каждого соответствующего входа для исходных материалов 32a, b, c, d. В дополнение к этому предпочтительно, чтобы барботажная колонна реакторного типа 20 снабжалась системой контроля потока, которая дает возможность для введения, по меньшей мере, части жидкофазного потока исходных материалов в реакционную зону 28 с повышенной входной поверхностной скоростью, по меньшей мере, примерно 2 м/с, более предпочтительно, по меньшей мере, примерно 5 м/с, еще более предпочтительно, по меньшей мере, примерно 6 м/с, а наиболее предпочтительно в пределах от 8 до 20 м/с. Как здесь используется, термин "входная поверхностная скорость" обозначает усредненную по времени объемную скорость потока для потока исходных материалов из входного отверстия, деленную на площадь входного отверстия. Предпочтительно, по меньшей мере, примерно 50 мас.% потока исходных материалов вводится в реакционную зону 28 при повышенной входной поверхностной скорости. Наиболее предпочтительно по существу почти весь поток исходных материалов вводится в реакционную зону 28 при повышенной входной поверхностной скорости.
Обращаясь теперь к фиг.6-7, иллюстрируется альтернативная система для введения жидкофазного потока исходных материалов в реакционную зону 28. В этом варианте осуществления поток исходных материалов вводится в реакционную зону 28 на четырех различных уровнях высоты. Каждый уровень высоты снабжается соответствующей системой распределения исходных материалов 76a, b, c, d. Каждая система распределения исходных материалов 76 содержит главный проход 78 для исходных материалов и коллектор 80. Каждый коллектор 80 снабжается, по меньшей мере, двумя выходами 82, 84, соединенными с соответствующими вставными проходами 86, 88, которые простираются в реакционную зону 28 корпуса 22. Каждый вставной проход 86, 88 предоставляет соответствующее входное отверстие 87, 89 для высвобождения потока исходных материалов в реакционную зону 28. Входные отверстия 87, 89 предпочтительно имеют по существу сходные диаметры, меньшие, примерно, чем 7 см, более предпочтительно находящиеся в пределах примерно от 0,25 примерно до 5 см, наиболее предпочтительно в пределах от 0,4 до 2 см. Предпочтительно, чтобы входные отверстия 87, 89 каждой системы распределения исходных материалов 76a, b, c, d были диаметрально противоположными, с тем, чтобы ввести поток исходных материалов в реакционную зону 28 в противоположных направлениях. Кроме того, предпочтительно, чтобы диаметрально противоположные входные отверстия 86, 88 соседних систем распределения исходных материалов 76 были ориентированы под углом поворота 90° по отношению друг к другу. При работе жидкофазный поток исходных материалов загружается в главный проход для исходных материалов 78 и впоследствии поступает в коллектор 80. Коллектор 80 распределяет поток исходных материалов равномерно для одновременного введения на противоположных сторонах реактора 20 через входные отверстия 87, 89.
Фиг.8 иллюстрирует альтернативную конфигурацию, где каждая система распределения исходных материалов 76 снабжена байонетными трубами 90, 92 вместо вставных проходов 86, 88 (показанных на фиг.7). Байонетные трубы 90, 92 выступают в реакционную зону 28 и содержат множество малых входных отверстий 94, 96 для высвобождения жидкофазных исходных материалов в реакционную зону 28. Предпочтительно, чтобы малые входные отверстия 94, 96 байонетных труб 90, 92 имели по существу одинаковые диаметры, меньшие, примерно, чем 50 мм, более предпочтительно примерно от 2 примерно до 25 мм, наиболее предпочтительно от 4 до 15 мм.
Фиг.9-11 иллюстрирует альтернативную систему распределения исходных материалов 100. Система распределения исходных материалов 100 вводит жидкофазный поток исходных материалов во множестве положений, отделенных друг от друга некоторым расстоянием по высоте и отделенных друг от друга некоторым латеральным расстоянием, без необходимости во множестве проходов в боковой стенке барботажной колонны реакторного типа 20. Система введения исходных материалов 100, как правило, содержит один входной проход 102, коллектор 104, множество расположенных вертикально распределительных труб 106, механизм латеральной поддержки 108 и механизм вертикальной поддержки 110. Вход 102 проходит через боковую стенку главного корпуса 46 корпуса 22. Вход 102 соединяется с сообщением текучих сред с коллектором 104. Коллектор 104 равномерно распределяет поток исходных материалов, принимаемый из входа 102, среди распределительных труб 106, расположенных по высоте. Каждая распределительная труба 106 имеет множество отделенных друг от друга некоторым расстоянием по высоте входных отверстий 112a, b, c, d для высвобождения потока исходных материалов в реакционную зону 28. Механизм латеральной поддержки 108 соединен с каждой распределительной трубой 106 и затрудняет относительное латеральное перемещение распределительных труб 106. Механизм вертикальной поддержки 110 предпочтительно соединяется с механизмом латеральной поддержки 108 и с верхней частью устройства распределения окислителя 34. Механизм вертикальной поддержки 110 по существу затрудняет перемещение распределительных труб 106 по высоте в реакционной зоне 28. Предпочтительно, чтобы входные отверстия 112 имели по существу одинаковые диаметры, меньшие, примерно, чем 50 мм, более предпочтительно примерно от 2 примерно до 25 мм, наиболее предпочтительно от 4 до 15 мм. Вертикальное расстояние между соседними входными отверстиями 112 системы распределения исходных материалов 100, иллюстрируемой на фиг.9-11, может быть по существу таким же, как описано выше, при упоминании системы распределения исходных материалов фиг.1.
Обнаружено, что структуры потока реакционной среды во множестве барботажных колонн реакторного типа могут сделать возможным неравномерное азимутальное распределение окисляемого соединения в реакционной среде, особенно, когда окисляемое соединение в основном вводится вдоль одной стороны реакционной среды. Как здесь используется, термин "азимутальный" должен обозначать угол или расстояние вокруг расположенной вертикально продольной оси реакционной зоны. Как здесь используется, "расположенный вертикально" должен обозначать в пределах 45° от вертикали. В одном из вариантов осуществления настоящего изобретения поток исходных материалов, содержащий окисляемое соединение (например, пара-ксилол), вводится в реакционную зону через множество отделенных друг от друга некоторым азимутальным расстоянием входных отверстий. Эти отделенные друг от друга некоторым азимутальным расстоянием входные отверстия могут помочь предотвратить возникновение областей высоких и низких концентраций окисляемого соединения в реакционной среде. Различные системы введения исходных материалов, иллюстрируемые на фиг.6-11, представляют собой примеры систем, которые обеспечивают правильное азимутальное расстояние между входными отверстиями.
Обращаясь опять к фиг.7, для количественного определения азимутально распределенного введения жидкофазного потока исходных материалов в реакционную среду реакционная среда может быть теоретически разделена на четыре расположенных вертикально азимутальных квадранта Q1, Q2, Q3, Q4 приблизительно равного объема. Эти азимутальные квадранты Q1, Q2, Q3, Q4 определяются посредством пары воображаемых пересекающихся перпендикулярных вертикальных плоскостей P1, P2, простирающихся через максимальный вертикальный размер и максимальный радиальный размер реакционной среды. Когда реакционная среда содержится в цилиндрической емкости, линия пересечения воображаемых пересекающихся вертикальных плоскостей P1, P2 будет приблизительно совпадать с вертикальной центральной линией цилиндра и каждый азимутальный квадрант Q1, Q2, Q3, Q4 будет представлять собой, в целом, клинообразный вертикальный объем, имеющий высоту, равную высоте реакционной среды. Предпочтительно, чтобы существенная часть окисляемого соединения высвобождалась в реакционную среду через входные отверстия, расположенные, по меньшей мере, в двух различных азимутальных квадрантах.
В предпочтительном варианте осуществления настоящего изобретения не более, примерно, чем 80 мас.% окисляемого соединения высвобождается в реакционную среду через входные отверстия, которые могут располагаться в одном азимутальном квадранте. Более предпочтительно не более, примерно, чем 60 мас.% окисляемого соединения высвобождается в реакционную среду через входные отверстия, которые могут располагаться в одном азимутальном квадранте. Наиболее предпочтительно не более чем 40 мас.% окисляемого соединения высвобождается в реакционную среду через входные отверстия, которые могут располагаться в одном азимутальном квадранте. Эти параметры для азимутального распределения окисляемого соединения измеряются, когда азимутальные квадранты азимутально ориентируются таким образом, что максимально возможное количество окисляемого соединения высвобождается в один из азимутальных квадрантов. Например, если весь поток исходных материалов высвобождается в реакционную среду через два входных отверстия, которые азимутально отделены друг от друга на 89°, с целью определения азимутального распределения в четырех азимутальных квадрантах, 100 мас.% потока исходных материалов высвобождается в реакционную среду в одном азимутальном квадранте, поскольку азимутальные квадранты могут азимутально ориентироваться таким образом, что оба входных отверстия располагаются в одном азимутальном квадранте.
В дополнение к преимуществам, связанным с правильным азимутальным расстоянием между входными отверстиями, также обнаружено, что правильное радиальное расстояние между входными отверстиями в барботажной колонне реакторного типа также может быть важным. Предпочтительно, чтобы существенная часть окисляемого соединения, вводимого в реакционную среду, высвобождалась через входные отверстия, которые радиально распределены внутрь от боковой стенки корпуса. Таким образом, в одном из вариантов осуществления настоящего изобретения существенная часть окисляемого соединения поступает в реакционную зону через входные отверстия, расположенные в "предпочтительной радиальной входной зоне", которая распределена внутрь от расположенных вертикально боковых стенок, определяющих реакционную зону.
Обращаясь опять к фиг.7, предпочтительная радиальная входная зона FZ может принимать форму теоретического расположенного вертикально цилиндра, с центром в реакционной зоне 28, и имеющего наружный диаметр D0 в 0,9 D, где D представляет собой диаметр реакционной зоны 28. Таким образом, определяется наружный кольцевой зазор OA, имеющий толщину 0,05 D между предпочтительной радиальной входной зоной FZ и внутри боковых стенок, определяющих реакционную зону 28. Предпочтительно, чтобы или малое количество окисляемого соединения вводилось в реакционную зону 28 через входные отверстия, расположенные в этом наружном кольцевом зазоре OA, или оно вообще не вводилось.
В другом варианте осуществления предпочтительно, чтобы малое количество окисляемого соединения вводилось в центр реакционной зоны 28, или оно вообще не вводилось. Таким образом, как иллюстрируется на фиг.8, предпочтительная радиальная входная зона FZ может принимать форму теоретического расположенного вертикально кольцевого зазора, с центром в реакционной зоне 28, имеющего наружный диаметр D0 0,9 D и имеющего внутренний диаметр Di 0,2 D. Таким образом, в настоящем варианте осуществления внутренний цилиндр IC, имеющий диаметр 0,2 D, "вырезается" из центра предпочтительной радиальной входной зоны FZ. Предпочтительно, чтобы малое количество окисляемого соединения вводилось в реакционную зону 28 через входные отверстия, расположенные в этом внутреннем цилиндре IC, или оно вообще не вводилось.
В предпочтительном варианте осуществления настоящего изобретения существенная часть окисляемого соединения вводится в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной входной зоне, независимо от того, имеет ли предпочтительная радиальная входная зона цилиндрическую или кольцевую форму, описанную выше. Более предпочтительно, по меньшей мере, примерно 25 мас.% окисляемого соединения высвобождается в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной входной зоне. Еще более предпочтительно, по меньшей мере, примерно 50 мас.% окисляемого соединения высвобождается в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной входной зоне. Наиболее предпочтительно, по меньшей мере, 75 мас.% окисляемого соединения высвобождается в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной входной зоне.
Хотя теоретические азимутальные квадранты и теоретическая предпочтительная радиальная входная зона, иллюстрируемые на фиг.7 и 8, описываются со ссылками на распределение жидкофазного потока исходных материалов, обнаружено, что правильное азимутальное и радиальное распределение газофазного потока окислителя может также обеспечить определенные преимущества. Таким образом, в одном из вариантов осуществления настоящего изобретения описание азимутального и радиального распределения жидкофазного потока исходных материалов, приведенное выше, также применимо к способу, которым газофазный поток окислителя вводится в реакционную среду 36.
Обращаясь теперь к фиг.12-15, иллюстрируется альтернативное устройство распределения окислителя 200 как содержащее в целом кольцевой элемент 202 и пару проходов 204, 206 для поступления окислителя. Устройство распределения окислителя 200 на фиг.12-15 является сходным с устройством для распределения окислителя 34 на фиг.1-11 при следующих трех основных отличиях: (1) устройство распределения окислителя 200 не содержит диагонального диаметрального элемента; (2) верхняя часть кольцевого элемента 202 не имеет отверстий для высвобождения окислителя в направлении снизу вверх и (3) устройство распределения окислителя 200 имеет гораздо больше отверстий в нижней части кольцевого элемента 202.
Как, вероятно, лучше всего иллюстрируется на фиг.14 и 15, нижняя часть кольца 202 устройства распределения окислителя предоставляет множество отверстий 208 для окислителя. Отверстия 208 для окислителя предпочтительно конфигурируются таким образом, что, по меньшей мере, примерно 1% общей открытой площади, определяемой отверстиями 208 для окислителя, располагается ниже центральной линии 210 (фиг.15) кольцевого элемента 202, где центральная линия 210 располагается на высоте объемного центроида кольцевого элемента 202. Более предпочтительно, по меньшей мере, примерно 5% общей открытой площади, определяемой всеми отверстиями отверстия 208 для окислителя, располагается ниже центральной линии 210, при этом, по меньшей мере, примерно 2% общей открытой площади определяются отверстиями 208, которые высвобождают поток окислителя в направлении, в целом, вниз, в пределах примерно 30° от вертикали. Еще более предпочтительно, по меньшей мере, примерно 20% общей открытой площади, определяемой всеми отверстиями 208 для окислителя, располагается ниже центральной линии 210, при этом, по меньшей мере, примерно 10% общей открытой площади определяется отверстиями 208, которые высвобождают поток окислителя в направлении, в целом, вниз, в пределах 30° от вертикали. Наиболее предпочтительно, по меньшей мере, примерно 75% общей открытой площади, определяемой всеми отверстиями 208 для окислителя, располагается ниже центральной линии 210, при этом, по меньшей мере, примерно 40% общей открытой площади определяется отверстиями 208, которые высвобождают поток окислителя в направлении, в целом, вниз, в пределах 30° от вертикали. Доля общей открытой площади, определяемой всеми отверстиями для окислителя 208, которые располагаются выше центральной линии 210, предпочтительно является меньшей, примерно, чем 75%, более предпочтительно меньшей, примерно, чем 50%, еще более предпочтительно меньшей, примерно, чем 25%, наиболее предпочтительно меньшей, чем 5%.
Как иллюстрируется на фиг.14 и 15, отверстия 208 для окислителя содержат направленные вниз отверстия 208a и наклонные отверстия 208b. Направленные вниз отверстия 208a конфигурируются для высвобождения потока окислителя, в целом, вниз, под углом в пределах примерно 30° от вертикали, более предпочтительно в пределах примерно 15° от вертикали, наиболее предпочтительно в пределах 5° от вертикали. Наклонные отверстия 208b конфигурируются для высвобождения потока окислителя, в целом, наружу и вниз, под углом A, который находится в пределах примерно от 15 примерно до 75° от вертикали, более предпочтительно угол A находится в пределах примерно от 30 примерно до 60° от вертикали, наиболее предпочтительно угол A находится в пределах от 40 до 50° от вертикали.
Предпочтительно, чтобы по существу все отверстия 208 для окислителя имели приблизительно одинаковый диаметр. Диаметр отверстий для окислителя 208 предпочтительно находится в пределах примерно от 2 примерно до 300 мм, более предпочтительно в пределах примерно от 4 примерно до 120 мм, а наиболее предпочтительно в пределах от 8 до 60 мм. Общее количество отверстий 208 для окислителя в кольцевом элементе 202 выбирается, чтобы удовлетворить критериям малого перепада давления, подробно описанным ниже. Предпочтительно общее количество отверстий 208 для окислителя, сформированных в кольцевом элементе 202, равно, по меньшей мере, примерно 10, более предпочтительно общее количество отверстий 208 для окислителя находится в пределах примерно от 20 примерно до 200, а наиболее предпочтительно общее количество отверстий 208 для окислителя находится в пределах от 40 до 100.
Хотя фиг.12-15 иллюстрируют очень специфичную конфигурацию устройства распределения окислителя 200, заметим теперь, что для достижения преимуществ, описанных здесь, могут использоваться разнообразные конфигурации устройства распределения окислителя. Например, устройство распределения окислителя необязательно должно иметь конфигурацию восьмиугольного кольцевого элемента, иллюстрируемую на фиг.12-13. Скорее, является возможным, чтобы устройство распределения окислителя был сформировано из любой конфигурации прохода (проходов) для потока, которые используют множества отделенных друг от друга некоторым расстоянием отверстий для высвобождения потока окислителя. Размер, количество и направление высвобождения отверстий для окислителя в проходе для потока предпочтительно находятся в пределах, сформулированных выше. Кроме того, устройство распределения окислителя предпочтительно конфигурируется для обеспечения азимутального и радиального распределения молекулярного кислорода, описанного выше.
Независимо от конкретной конфигурации устройства распределения окислителя предпочтительно, чтобы устройство распределения окислителя было физически сконфигурировано и работало способом, который сводит к минимуму перепад давления, связанный с высвобождением потока окислителя из прохода (проходов) для потока, через отверстия для окислителя, и в реакционную зону. Такой перепад давления вычисляется как усредненное по времени статическое давление потока окислителя внутри прохода для потока на входах для окислителя 66a, b устройства распределения окислителя минус усредненное по времени статическое давление в реакционной зоне на высоте, где половина потока окислителя вводится выше этого положения по высоте и половина потока окислителя вводится ниже этого положения по высоте. В предпочтительном варианте осуществления настоящего изобретения усредненный по времени перепад давления, связанный с высвобождением потока окислителя из устройства распределения окислителя, является меньшим, примерно, чем 0,3 МПа, более предпочтительно меньшим, примерно, чем 0,2 МПа, еще более предпочтительно меньшим, примерно, чем 0,1 МПа, а наиболее предпочтительно меньшим, чем 0,05 МПа. При предпочтительных рабочих условиях барботажной колонны реакторного типа, описанных здесь, давление потока окислителя внутри прохода (проходов) для потока в устройстве, для распределения окислителя, предпочтительно находится в пределах примерно от 0,35 примерно до 1 МПа, более предпочтительно в пределах примерно от 0,45 примерно до 0,85 МПа, а наиболее предпочтительно в пределах от 0,5 до 0,7 МПа.
Как указывалось ранее со ссылками на конфигурацию устройства распределения окислителя, иллюстрируемую на фиг.2-5, может быть желательным непрерывно или периодически прочищать устройство распределения окислителя с помощью жидкости (например, уксусной кислоты, воды и/или пара-ксилола), чтобы предотвратить образование осадка в устройстве распределения окислителя из твердых продуктов. Когда используется такая прочистка жидкостью, предпочтительно, чтобы эффективное количество жидкости (т.е. не только малое количество жидких капель, которые могли бы естественным образом присутствовать в потоке окислителя) проходило через устройство распределения окислителя и из отверстий для окислителя, по меньшей мере, в течение одного периода, большего, чем одна минута, ежедневно. Когда жидкость непрерывно или периодически высвобождается из устройства распределения окислителя, предпочтительно, чтобы усредненное по времени отношение массовой скорости потока жидкости через устройство распределения окислителя к массовой скорости потока молекулярного кислорода через устройство распределения окислителя находилось в пределах примерно от 0,05:1 примерно до 30:1, или в пределах примерно от 0,1:1 примерно до 2:1, или даже в пределах от 0,2:1 до 1:1.
В одном из вариантов осуществления настоящего изобретения значительная часть окисляемого соединения (например, пара-ксилола) может вводиться в реакционную зону через устройство распределения окислителя. В такой конфигурации предпочтительно, чтобы окисляемое соединение и молекулярный кислород высвобождались из устройства распределения окислителя через одни и те же отверстия в устройстве распределения окислителя. Как отмечено выше, окисляемое соединение, как правило, представляет собой жидкость при STP (при стандартных температуре и давлении). Следовательно, в настоящем варианте осуществления двухфазный поток может высвобождаться из устройства распределения окислителя, при этом жидкая фаза содержит окисляемое соединение и газовая фаза содержит молекулярный кислород. Необходимо заметить, однако, что, по меньшей мере, часть окисляемого соединения может находиться в газообразном состоянии, когда высвобождается из устройства распределения окислителя. В одном из вариантов осуществления жидкая фаза, высвобождающаяся из устройства распределения окислителя, формируется преобладающе из окисляемого соединения. В другом варианте осуществления жидкая фаза, высвобождающаяся из устройства распределения окислителя, имеет по существу такой же состав, как поток исходных материалов, описанный выше. Когда жидкая фаза, высвобождающаяся из устройства распределения окислителя, имеет по существу такой же состав, как поток исходных материалов, такая жидкая фаза может содержать растворитель и/или систему катализаторов в количествах и при отношениях, описанных выше при упоминании состава потока исходных материалов.
В одном из вариантов осуществления настоящего изобретения предпочтительно, чтобы, по меньшей мере, примерно 10 мас.% всего окисляемого соединения, вводимого в реакционную зону, вводилось через устройство распределения окислителя, более предпочтительно, по меньшей мере, примерно 40 мас.% окисляемого соединения вводилось в реакционную зону через устройство распределения окислителя, наиболее предпочтительно, по меньшей мере, 80 мас.% окисляемого соединения вводилось в реакционную зону через устройство распределения окислителя. Когда все окисляемое соединение или его часть вводится в реакционную зону через устройство распределения окислителя, предпочтительно, чтобы, по меньшей мере, примерно 10 мас.% всего молекулярного кислорода, вводимого в реакционную зону, вводилось через то же устройство распределения окислителя, более предпочтительно, по меньшей мере, примерно 40 мас.% окисляемого соединения вводилось в реакционную зону через то же устройство распределения окислителя, наиболее предпочтительно, по меньшей мере, 80 мас.% окисляемого соединения вводилось в реакционную зону через то же устройство распределения окислителя. Когда значительная часть окисляемого соединения вводится в реакционную зону через устройство распределения окислителя, предпочтительно, чтобы одно или несколько устройств, отслеживающих температуру (например, термопар), располагалось в устройстве распределения окислителя. Эти температурные сенсоры могут использоваться, чтобы помочь удостовериться в том, что температура в устройстве распределения окислителя не становится опасно высокой.
Обращаясь теперь к фиг.16-18, барботажная колонна реакторного типа 20 иллюстрируется как содержащая внутреннюю деаэрационную емкость 300, расположенную в нижней части реакционной зоны 28 вблизи выхода 38 для суспензии. Обнаружено, что побочные реакции, формирующие примеси, осуществляются при относительно высокой скорости во время деаэрации реакционной среды 36. Как здесь используется, "деаэрация" должна означать отделение газовой фазы из многофазной реакционной среды. Когда реакционная среда 36 является сильно аэрированной (содержание газа >0,3), образование примесей является минимальным. Когда реакционная среда 36 является сильно неаэрированной (содержание газа <0,01), образование примесей также является минимальным. Однако, когда реакционная среда является частично аэрированной (содержание газа 0,01-0,3), нежелательные побочные реакции облегчаются и образуется больше примесей. Деаэрационная емкость 300 решает эту и другие проблемы посредством сведения к минимуму объема реакционной среды 36 в частично аэрированном состоянии и посредством сведения к минимуму времени, которое отнимает деаэрация реакционной среды 36. По существу деаэрированная суспензия получается из нижней части деаэрационной емкости 300 и покидает реактор 20 через выход 38 для суспензии. По существу деаэрированная суспензия предпочтительно содержит меньше, примерно, чем 5% объемных газовой фазы, более предпочтительно меньше, примерно, чем 2% объемных газовой фазы, наиболее предпочтительно меньше, чем 1% объемный газовой фазы.
На фиг.16 барботажная колонна реакторного типа 20 иллюстрируется как содержащая контроллер уровня 302 и клапан контроля потока 304. Контроллер уровня 302 и клапан контроля потока 304 совместно поддерживают реакционную среду 36 по существу при постоянной высоте в реакционной зоне 28. Контроллер уровня 302 работает, отслеживая (например, посредством отслеживания уровня по разнице давлений или посредством ядерного отслеживания уровня) высоту верхнего уровня поверхности 44 реакционной среды 36 и образуя контрольный сигнал 306, чувствительный к высоте реакционной среды 36. Клапан контроля потока 304 принимает контрольный сигнал 306 и регулирует скорость потока суспензии через проход для выхода суспензии 308. Таким образом, скорость потока суспензии из выхода для суспензии 38 может изменяться в пределах между максимальной объемной скоростью потока суспензии (Fmax), когда высота реакционной среды 36 является слишком большой, и минимальной объемной скоростью потока суспензии (Fmin), когда высота реакционной среды 36 является слишком низкой.
Для удаления твердофазного продукта окисления из реакционной зоны 28 порция должна сначала пройти через деаэрационную емкость 300. Деаэрационная емкость 300 обеспечивает внутренний объем с низкой турбулентностью, который дает возможность газовой фазе реакционной среды 36 естественным образом подняться из жидкой и твердых фаз реакционной среды 36, когда жидкость и твердые продукты протекают вниз по направлению к выходу для суспензии 38. Подъем газовой фазы из жидкой и твердой фаз вызывается естественным всплыванием газовой фазы в жидкой и твердой фазах. Когда используется деаэрационная емкость 300, переход реакционной среды 36 от полностью аэрированной, трехфазной среды к полностью деаэрированной, двухфазной суспензии является быстрым и эффективным.
Обращаясь теперь к фиг.17 и 18, деаэрационная емкость 300 содержит, в целом, расположенную вертикально боковую стенку 308, определяющую внутри себя деаэрационную зону 312. Предпочтительно боковая стенка 308 простирается снизу вверх в пределах примерно 30° от вертикали, более предпочтительно в пределах примерно 10° от вертикали. Наиболее предпочтительно боковая стенка 308 является по существу вертикальной. Деаэрационная зона 312 является отдельной от реакционной зоны 28 и имеет высоту h и диаметр d. Верхний уровень 310 боковой стенки 308 является открытым, таким образом, чтобы принимать реакционную среду из реакционной зоны 28 во внутренний объем 312. Нижний уровень боковой стенки 308 является соединенным с сообщением текучих сред с выходом для суспензии 38 через переходную секцию 314. В определенных случаях, например, когда отверстие выхода для суспензии 38 большее или когда диаметр d боковой стенки 308 маленький, переходная секция 314 может быть устранена. Как, вероятно, лучше всего иллюстрируется на фиг.18, деаэрационная емкость 300 может также содержать гаситель вихрей 316, расположенный в деаэрационной зоне 312. Гаситель вихрей 316 может иметь любую структуру, работающую для замедления образования вихрей, когда твердая и жидкая фазы протекают вниз по направлению к выходу для суспензии 38.
Чтобы сделать возможным соответствующее отделение газовой фазы от твердой и жидкой фаз в деаэрационной емкости 300, высота h и горизонтальная площадь поперечного сечения внутренней деаэрационной зоны 312 тщательно подбираются. Высота h и горизонтальная площадь поперечного сечения внутренней деаэрационной зоны 312 должны обеспечивать достаточное расстояние и время, так что даже когда извлекается максимальное количество суспензии (т.е. когда суспензия извлекается при Fmax), по существу весь объем газовых пузырьков может подняться из твердой и жидкой фаз до того, как газовые пузырьки достигнут нижнего выхода деаэрационной емкости 300. Таким образом, предпочтительно, чтобы площадь поперечного сечения деаэрационной зоны 312 была такой, чтобы максимальная скорость движения вниз (Vdmax) жидкой и твердых фаз через деаэрационную зону 312 была существенно меньшей, чем скорость естественного подъема (Vu) пузырьков газовой фазы через жидкую и твердую фазы. Максимальная скорость движения вниз (Vdmax) жидкой и твердой фаз через деаэрационную зону 312 осуществляется при максимальной объемной скорости потока суспензии (Fmax), обсуждаемой выше. Естественная скорость подъема (Vu) пузырьков газа через жидкую и твердую фазу изменяется в зависимости от размера пузырьков; однако естественная скорость подъема (Vu0,5) газовых пузырьков диаметром 0,5 см через жидкую и твердую фазы может использоваться в качестве порогового значения, поскольку по существу весь объем пузырьков, изначально, в реакционной среде 36 будет большим, чем 0,5 см. Предпочтительно площадь поперечного сечения деаэрационной зоны 312 является такой, что Vdmax является меньшей, примерно, чем 75% от Vu0,5, более предпочтительно Vdmax является меньшей, примерно, чем 40% от Vu0,5, наиболее предпочтительно Vdmax является меньшей, чем 20% от Vu0,5.
Скорость движения вниз жидкой и твердой фаз в деаэрационной зоне 312 деаэрационной емкости 300 вычисляется как объемная скорость потока деаэрированной суспензии через выход для суспензии 38, деленная на минимальную площадь поперечного сечения деаэрационной зоны 312. Скорость движения вниз жидкой и твердой фаз в деаэрационной зоне 312 деаэрационной емкости 300 предпочтительно меньше, примерно, чем 50 см/с, более предпочтительно меньше, примерно, чем 30 см/с, а наиболее предпочтительно меньше, чем 10 см/с.
Отметим сейчас, что, хотя расположенная вертикально боковая стенка 308 деаэрационной емкости 300 имеет цилиндрическую конфигурацию, боковая стенка 308 может включать в себя множество боковых стенок, которые образуют множество конфигураций (например, треугольную, квадратную или овальную), постольку поскольку стенки определяют внутренний объем, имеющий соответствующий объем, площадь поперечного сечения, ширину d и высоту h. В предпочтительном варианте осуществления настоящего изобретения d находится в пределах примерно от 0,2 примерно до 2 м, более предпочтительно в пределах примерно от 0,3 примерно до 1,5 м, а наиболее предпочтительно в пределах от 0,4 до 1,2 м. В предпочтительном варианте осуществления настоящего изобретения h находится в пределах примерно от 0,3 м примерно до 5 м, более предпочтительно в пределах примерно от 0,5 примерно до 3 м, а наиболее предпочтительно в пределах от 0,75 до 2 м.
В предпочтительном варианте осуществления настоящего изобретения боковая стенка 308 является по существу вертикальной, так что горизонтальная площадь поперечного сечения деаэрационной зоны 312 является по существу постоянной по всей высоте h деаэрационной зоны 312. Предпочтительно максимальная горизонтальная площадь поперечного сечения деаэрационной зоны 312 меньше, примерно, чем 25% от максимальной горизонтальной площади поперечного сечения реакционной зоны 28. Более предпочтительно максимальная горизонтальная площадь поперечного сечения деаэрационной зоны 312 находится в пределах примерно от 0,1 примерно до 10% от максимальной горизонтальной площади поперечного сечения реакционной зоны 28. Наиболее предпочтительно максимальная горизонтальная площадь поперечного сечения деаэрационной зоны 312 находится в пределах от 0,25 до 4% от максимальной горизонтальной площади поперечного сечения реакционной зоны 28. Предпочтительно максимальная горизонтальная площадь поперечного сечения деаэрационной зоны 312 находится в пределах от примерно 0,02 примерно до 3 м2, более предпочтительно в пределах примерно от 0,05 примерно до 2 м2, наиболее предпочтительно в пределах от 0,1 до 1,2 м2. Объем деаэрационной зоны 312 предпочтительно меньше, примерно, чем 5% от общего объема реакционной среды 36 или реакционной зоны 28. Более предпочтительно объем деаэрационной зоны 312 находится в пределах от примерно 0,01 примерно до 2% от общего объема реакционной среды 36 или реакционной зоны 28. Наиболее предпочтительно объем деаэрационной зоны 312 находится в пределах от 0,05 примерно до 1% от общего объема реакционной среды 36 или реакционной зоны 28. Объем деаэрационной зоны 312 предпочтительно меньше, примерно, чем 2 м3, более предпочтительно находится в пределах примерно от 0,01 примерно до 1 м3, наиболее предпочтительно находится в пределах от 0,05 до 0,5 м3.
Обращаясь теперь к фиг.19, барботажная колонна реакторного типа 20 иллюстрируется как содержащая наружную деаэрационную емкость 400. В этой конфигурации аэрированная реакционная среда 36 извлекается из реакционной зоны 28 через приподнятое отверстие на боку корпуса 22. Извлеченная аэрированная среда переносится в наружную деаэрационную емкость 400 через выходной проход 402 для отделения газовой фазы от твердой и жидкой фаз. Отделенная газовая фаза покидает деаэрационную емкость 400 через проход 404, в то время как по существу деаэрированная суспензия покидает деаэрационную емкость 400 через проход 406.
На фиг.19 выходной проход 402 показан как являющийся приблизительно прямым, горизонтальным и ортогональным корпусу 22. Это просто одна из удобных конфигураций; и выходной проход 402 может быть отличным в любом отношении, при условии, что он пригодным для использования образом соединяет барботажную колонну реакторного типа 20 с наружной деаэрационной емкостью 400. Обращаясь к проходу 404, является полезным, чтобы этот проход присоединялся сверху деаэрационной емкости 400 или вблизи ее верхней части для надежного контроля проблем, связанных с застойным газовым карманом, содержащим окисляемое соединение и окислитель. Более того, проходы 402 и 404 могут пригодным для использования образом содержать средства изоляции потоков, такие как клапаны.
Когда реакционная среда 36 извлекается из реактора 20 через приподнятый выход, как показано на фиг.19, предпочтительно, чтобы барботажная колонна реакторного типа 20 была снабжена нижним выходом 408 вблизи нижней части 52 реакционной зоны 28. Нижний выход 408 и нижний проход 410, соединенный с ним, может использоваться для запасного (т.е. пустого) реактора 20 во время выключений. Предпочтительно один или несколько нижних выходов 408 предусматриваются в нижней трети высоты реакционной среды 36, более предпочтительно в нижней четверти реакционной среды 36, а наиболее предпочтительно в самой нижней точке реакционной зоны 28.
Для приподнятой системы извлечения суспензии и деаэрации, показанной на фиг.19, нижний проход 410 и выход 408 не используются для извлечения суспензии из реакционной зоны 28 во время окисления. В данной области известно, что твердые продукты имеют тенденцию к оседанию под действием силы тяжести в неаэрированных и не перемешиваемых другим образом частях суспензии, включая застойные зоны прохождения потока. Кроме того, осажденные твердые продукты (например, терефталевая кислота) могут иметь тенденцию к отверждению в виде больших агломератов посредством продолжения осаждения и/или кристаллической реорганизации. Таким образом, для предотвращения забивания нижнего прохода для потока 410 часть деаэрированной суспензии из нижней части деаэрационной емкости 400 может использоваться для непрерывной или периодической прочистки нижнего выхода 410 во время нормальной работы реактора 20. Предпочтительные способы осуществления такой прочистки суспензией выхода 410 представляют собой периодическое открывание клапана 412 в выходе 410 и предоставление возможности части деаэрированной суспензии для протекания через выход 410 и в реакционную зону 28 через нижнее отверстие 408. Даже когда клапан 412 является полностью или частично открытым, только часть деаэрированной суспензии протекает через нижний выход 410 и назад в реакционную зону 28. Оставшаяся часть деаэрированной суспензии, не используемая для прочистки нижнего прохода 410, переносится через проход 414 из реактора 20 для дополнительной последующей переработки (например, очистки).
Во время нормальной работы барботажной колонны реакторного типа 20 в течение достаточного отрезка времени (например, >100 часов) предпочтительно, чтобы количество деаэрированной суспензии, используемой для прочистки нижнего выхода 410, было меньшим, чем 50 мас.% от общего количества деаэрированной суспензии, получаемой из нижней части деаэрационной емкости 400, более предпочтительно меньшим, примерно, чем 20 мас.%, а наиболее предпочтительно меньшим, чем 5 мас.%. Кроме того, предпочтительно, чтобы в течение достаточного отрезка времени средняя массовая скорость потока деаэрированной суспензии, используемой для прочистки нижнего выхода 410, была меньше, примерно, чем умноженная на 4 средняя массовая скорость потока окисляемого соединения в реакционной зоне 28, более предпочтительно меньше, примерно, чем умноженная на 2 средняя массовая скорость потока окисляемого соединения в реакционной зоне 28, еще более предпочтительно меньше, чем средняя массовая скорость потока окисляемого соединения в реакционной зоне 28, а наиболее предпочтительно меньше, чем 0,5 от средней массовой скорости потока окисляемого соединения в реакционной зоне 28.
Обращаясь опять к фиг.19, деаэрационная емкость 400 содержит расположенную по существу вертикально предпочтительно цилиндрическую боковую стенку 416, определяющую деаэрационную зону 418. Деаэрационная зона 418 имеет диаметр d и высоту h. Высота h измеряется как вертикальное расстояние между положением, где аэрированная реакционная среда поступает в деаэрационную емкость 400, и нижней частью боковой стенки 416. Высота h, диаметр d, площадь и объем деаэрационной зоны 418 предпочтительно являются по существу такими же, как описано выше, при упоминании деаэрационной зоны 312 деаэрационной емкости 300, иллюстрируемой на фиг.16-18. В дополнение к этому, деаэрационная емкость 400 содержит верхнюю секцию 420, сформированную посредством выступающей боковой стенки 416 выше деаэрационной зоны 418. Верхняя секция 420 деаэрационной емкости 400 может иметь любую высоту, хотя предпочтительно она простирается вверх до уровня реакционной среды 36 в реакционной зоне 28 или выше. Верхняя секция 420 обеспечивает то, что газовая фаза имеет пространство для соответствующего отделения от жидкой и твердой фаз до выхода из деаэрационной емкости 400 через проход 404. Теперь отметим, что хотя проход 404 иллюстрируется как возвращающий отделенную газовую фазу в зону отделения реактора 20, выход 404 альтернативно может соединяться с корпусом 22 на любой высоте, выше выхода 402. Необязательно, выход 404 может соединяться с проходом для выхода газа 40, так что отделенная газовая фаза из деаэрационной емкости 400 объединяется с удаленным из верхней части колонны потоком пара в проходе 40 и направляется далее для дополнительной переработки.
Обращаясь теперь к фиг.20, барботажная колонна реакторного типа 20 иллюстрируется как содержащая гибридную внутреннюю-наружную деаэрационную емкость 500. В этой конфигурации, часть реакционной среды 36 извлекается из реакционной зоны 28 через относительно приподнятое отверстие 502 в боковой стенке корпуса 22. Затем извлеченная реакционная среда 36 переносится через коленчатый проход 504 относительно большого диаметра и поступает в верхнюю часть деаэрационной емкости 500. На фиг.20 коленчатый проход 504 показан как присоединяющийся ортогонально к боковой стенке корпуса 22 и содержащий плавный поворот под углом примерно 90°. Это всего лишь одна из удобных конфигураций; и коленчатый проход 504 может быть отличным в любом отношении, при условии, что он соединяет пригодным для использования способом барботажную колонну реакторного типа 20 с наружной деаэрационной емкостью 500, как описано. Кроме того, коленчатый проход 504 может содержать пригодным для использования способом средства для изоляции потока, такие как клапаны.
В деаэрационной емкости 500 газовая фаза движется снизу вверх, в то время как твердая и жидкая фазы двигаются сверху вниз. Движущаяся снизу вверх газовая фаза может повторно поступать в коленчатый проход 504, а затем уходить через отверстие 502 назад в реакционную зону 28. Таким образом, в отверстии 502 может осуществляться противоток поступающей реакционной среды 36 и выходящего отделенного газа. Деаэрированная суспензия покидает деаэрационную емкость 500 через проход 506. Деаэрационная емкость 500 содержит расположенную по существу вертикально предпочтительно цилиндрическую боковую стенку 508, определяющую деаэрационную зону 510. Деаэрационная зона 510 имеет высоту h и диаметр d. Предпочтительно, чтобы приподнятое отверстие 502 и коленчатый проход 504 имели диаметр такой же или больший, чем диаметр d деаэрационной зоны 510. Высота h, диаметр d, площадь и объем деаэрационной зоны 510 предпочтительно являются по существу таким же, как описано выше при упоминании деаэрационной зоны 312 деаэрационной емкости 300, иллюстрируемой на фиг.16-18.
Фиг.19 и 20 иллюстрируют вариант осуществления барботажной колонны реакторного типа 20, где твердый продукт (например, сырая терефталевая кислота), получаемый в реакционной зоне 28, извлекается из реакционной зоны 28 через приподнятый по высоте выход. Извлечение аэрированной реакционной среды 36 из приподнятого по высоте положения над нижней частью барботажной колонны реакторного типа 20 может помочь предотвратить аккумуляцию и застой плохо аэрированной реакционной среды 36 в нижней части 52 реакционной зоны 28. В соответствии с другими аспектами настоящего изобретения концентрации кислорода и окисляемого соединения (например, пара-ксилола) в реакционной среде 36 вблизи верхней части реакционной среды 36 предпочтительно являются более низкими, чем вблизи нижней части. Таким образом, извлечение реакционной среды 36 в приподнятом по высоте положении может увеличить выход посредством понижения количества непрореагировавших реагентов, извлекаемых из реактора 20. Также температура реакционной среды 36 значительно изменяется в вертикальном направлении, когда барботажная колонна реакторного типа 20 работает при высоких STR и градиентах химического состава, как описано здесь. В таких условиях температура реакционной среды 36, как правило, будет иметь локальные минимумы вблизи нижнего уровня поверхности и верхнего уровня поверхности реакционной зоны 28. Вблизи нижнего уровня поверхности минимум связан с испарением растворителя вблизи области, где принимается окислитель или его часть. Вблизи верхнего уровня поверхности минимум опять же вызывается испарением растворителя, хотя и вызванным здесь уменьшением давления в реакционной среде. В дополнение к этому, другие локальные минимумы могут возникать между верхним и нижним уровнями поверхности, когда в реакционную среду принимаются дополнительные исходные материалы или окислитель. Таким образом, имеется один или несколько максимумов температуры, возникающих под действием экзотермического тепла реакций окисления, между нижним краем и верхним краем реакционной зоны 28. Извлечение реакционной среды 36 в приподнятом положении с повышенной температурой может быть особенно преимущественным, когда последующая переработка осуществляется при более высоких температурах, поскольку стоимость энергии, связанной с нагревом извлеченной среды для последующей переработки, уменьшается.
Таким образом, в предпочтительном варианте осуществления настоящего изобретения и, особенно, когда последующая переработка осуществляется при более высоких температурах, реакционная среда 36 извлекается из барботажной колонны реакторного типа 20 через приподнятый по высоте выход (выходы), расположенный выше положения (положений) поступления в реакционную зону 28, по меньшей мере, 50 мас.% жидкофазного потока исходных материалов и/или газофазного потока окислителя. Более предпочтительно реакционная среда 36 извлекается из барботажной колонны реакторного типа 20 через приподнятый по высоте выход (выходы), расположенный по высоте выше положения (положений), где поступают в реакционную зону 28 по существу весь жидкофазный поток исходных материалов и/или газофазный поток окислителя. Предпочтительно, по меньшей мере, 50 мас.% твердофазных и жидкофазных компонентов, извлекаемых из барботажной колонны реакторного типа 20, извлекаются через приподнятый по высоте выход (выходы). Более предпочтительно по существу все твердофазные и жидкофазные компоненты, извлекаемые из барботажной колонны реакторного типа 20, извлекаются через приподнятый по высоте выход (выходы). Предпочтительно приподнятый по высоте выход (выходы) располагается, по меньшей мере, примерно на 1 D выше нижнего уровня 52 реакционной зоны 28. Более предпочтительно приподнятый по высоте выход (выходы) располагается, по меньшей мере, примерно на 2 D выше нижнего уровня 52 реакционной зоны 28. Наиболее предпочтительно приподнятый по высоте выход (выходы) располагается, по меньшей мере, на 3 D выше нижнего уровня 52 реакционной зоны 28. При данной высоте H реакционной среды 36 предпочтительно, чтобы приподнятый по высоте выход (выходы) располагался вертикально в пределах между примерно 0,2 H и примерно 0,8 H, более предпочтительно между примерно 0,3 H и примерно 0,7 H, наиболее предпочтительно между 0,4 H и 0,6 H. Кроме того, предпочтительно, чтобы температура реакционной среды 36 на приподнятом по высоте выходе из реакционной зоны 28 была, по меньшей мере, на 1°C больше, чем температура реакционной среды 36 вблизи нижнего уровня 52 реакционной зоны 28. Более предпочтительно температура реакционной среды 36 на приподнятом по высоте выходе из реакционной зоны 28 выше, в пределах примерно от 1,5 примерно до 16°C, чем температура реакционной среды 36 вблизи нижнего уровня 52 реакционной зоны 28. Наиболее предпочтительно температура реакционной среды 36 на приподнятом по высоте выходе из реакционной зоны 28 выше, в пределах от 2 до 12°C, чем температура реакционной среды 36 на нижнем краю 52 реакционной зоны 28.
Обращаясь теперь к фиг.21, барботажная колонна реакторного типа 20 иллюстрируется как содержащая альтернативную гибридную деаэрационную емкость 600, расположенную в нижней части реактора 20. При этой конфигурации аэрированная реакционная среда 36 извлекается из реакционной зоны 28 через относительно большое отверстие 602 в низу 52 корпуса 22. Отверстие 602 определяет открытый верхний уровень деаэрационной емкости 600. В деаэрационной емкости 600 газовая фаза движется снизу вверх, в то время как твердая и жидкая фазы движутся сверху вниз. Движущаяся снизу вверх газовая фаза может повторно поступать в реакционную зону 28 через отверстие 602. Таким образом, в отверстии 602 может осуществляться противоток поступающей реакционной среды 36 и уходящего отделенного газа. Деаэрированная суспензия покидает деаэрационную емкость 600 через проход 604. Деаэрационная емкость 600 содержит расположенную по существу вертикально предпочтительно цилиндрическую боковую стенку 606, определяющую деаэрационную зону 608. Деаэрационная зона 608 имеет высоту h и диаметр d. Предпочтительно, чтобы отверстие 602 имело диаметр такой же, как диаметр d деаэрационной зоны 608, или больший. Высота h, диаметр d, площадь и объем деаэрационной зоны 608 предпочтительно являются по существу такими же, как описано выше при упоминании деаэрационной зоны 312 деаэрационной емкости 300, иллюстрируемой на фиг.16-18.
Обращаясь теперь к фиг.22, барботажная колонна реакторного типа 20 фиг.21 иллюстрируется как содержащая альтернативное устройство распределения окислителя 620. Устройство распределения окислителя 620 содержит кольцевой элемент 622 и пару входных проходов 624, 626. Кольцевой элемент 622 предпочтительно имеет по существу такую же конфигурацию, как кольцевой элемент 202, описанный выше при упоминании фиг.12-15. Входные проходы 624, 626 простираются снизу вверх через отверстия в днище 48 корпуса 22 и обеспечивают поток окислителя к кольцевому элементу 622.
Обращаясь теперь к фиг.23, барботажная колонна реакторного типа 20 фиг.21 иллюстрируется как содержащая средства для введения потока окислителя в реакционную зону 28, не содержащие устройств распределения. В конфигурации фиг.23 поток окислителя доставляется в реактор 20 через проходы для окислителя 630, 632. Проходы для окислителя 630, 632 соединяются с соответствующими отверстиями для окислителя 634, 636 в днище 48 корпуса 22. Поток окислителя вводится непосредственно в реакционную зону 28 через отверстия для окислителя 634, 636. Необязательные пластины дефлекторов 638, 640 могут предусматриваться для отклонения направления потока, для потока окислителя, после того как он изначально поступает в реакционную зону 28.
Как рассмотрено выше, предпочтительно, чтобы реактор окисления сконфигурировался и работал способом, который устраняет зоны с высокой концентрацией окисляемого соединения в реакционной среде, поскольку такие зоны могут приводить к образованию примесей. Один из способов улучшения начального диспергирования окисляемого соединения (например, пара-ксилола) в реакционной среде заключается в разбавлении окисляемого соединения жидкостью. Жидкость, используемая для разбавления окисляемого соединения, может происходить от части реакционной среды, расположенной на значительном расстоянии от положения (положений), где окисляемое соединение вводится в реакционную зону. Эта жидкость из удаленной части реакционной среды может циркулировать в положение вблизи от положения поступления окисляемого соединения через проход для потока, который располагается внутри и/или снаружи главной реакционной емкости.
Фиг.24 и 25 иллюстрируют два предпочтительных способа циркуляции жидкости от удаленной части реакционной среды к положению вблизи входа окисляемого соединения, использующие внутренний (фиг.24) или наружный (фиг.25) проход. Предпочтительно длина прохода для потока от его входа (т.е. отверстия (отверстий), где жидкость поступает в проход) до его выхода (т.е. отверстия (отверстий), где жидкость высвобождается из прохода) больше, примерно, чем 1 м, более предпочтительно больше, примерно, чем 3 м, еще более предпочтительно больше, примерно, чем 6 м, а наиболее предпочтительно больше, чем 9 м. Однако реальная длина прохода становится менее важной, если жидкость получают из отдельной емкости, вероятно, расположенной непосредственно над емкостью, в которую изначально высвобождаются исходные материалы окисляемого соединения, или рядом с ней. Жидкость из любой отдельной емкости, содержащая, по меньшей мере, часть реакционной среды, представляет собой предпочтительный источник для начального разбавления окисляемого соединения.
Предпочтительно, чтобы жидкость, протекающая через проход, каким бы не был ее источник, имела более низкую постоянную концентрацию окисляемого соединения, чем реакционная среда в непосредственной близости, по меньшей мере, с одним выходом из прохода. Кроме того, предпочтительно, чтобы жидкость, протекающая через проход, имела концентрацию окисляемого соединения в жидкой фазе примерно ниже 100000 м.д. мас., более предпочтительно примерно ниже 10000 м.д. мас., еще более предпочтительно примерно ниже 1000 м.д. мас., а наиболее предпочтительно ниже 100 м.д. мас., где концентрации измеряются до добавления в проход малой добавки исходных материалов окисляемого соединения и любых необязательных, отдельных исходных материалов растворителя. Когда измерения осуществляют после добавления малой добавки исходных материалов окисляемого соединения и необязательных исходных материалов растворителя, предпочтительно, чтобы объединенный жидкий поток, поступающий в реакционную среду, имел концентрацию окисляемого соединения в жидкой фазе примерно ниже 300000 м.д. мас., более предпочтительно примерно ниже 50000 м.д. мас., а наиболее предпочтительно ниже 10000 м.д. мас.
Является желательным поддерживать поток через проход при достаточно низкой скорости, так, чтобы циркулирующая жидкость не подавляла желательный общий градиент окисляемого соединения в реакционной среде. В этом отношении предпочтительно, чтобы отношение массы жидкой фазы в реакционной зоне, в которую сначала высвобождается малая добавка окисляемого соединения, к массовой скорости потока жидкости, протекающей через проход, было большим, примерно, чем 0,3 мин, более предпочтительно большим, примерно, чем 1 мин, еще более предпочтительно находилось в пределах примерно между 2 мин и примерно 120 мин, а наиболее предпочтительно в пределах между 3 мин и 60 мин.
Существует множество средств для того, чтобы заставить жидкость протекать через проход. Предпочтительные средства включают в себя силу тяжести, эжекторы всех типов, использующие либо газ, либо жидкость, либо как то, так и другое в качестве движущей текучей среды, и механические насосы всех типов. Когда используется эжектор, один из вариантов осуществления настоящего изобретения использует в качестве движущей текучей среды, по меньшей мере, одну текучую среду, выбранную из группы, состоящей из исходных материалов окисляемого соединения (жидкости или газа), исходных материалов окислителя (газа), исходных материалов растворителя (жидкости) и прокачиваемого источника реакционной среды (суспензии). Другой вариант осуществления использует в качестве движущей текучей среды, по меньшей мере, две текучих среды, выбранных из группы, состоящей из исходных материалов окисляемого соединения, исходных материалов окислителя и исходных материалов растворителя. Еще один вариант осуществления использует в качестве движущей текучей среды сочетание исходных материалов окисляемого соединения, исходных материалов окислителя и исходных материалов растворителя.
Соответствующий диаметр или диаметры циркуляционного прохода могут изменяться в соответствии с количеством и свойствами материала, который переносится, энергии, доступной для принудительного движения потока, и соображениями капитальных затрат. Предпочтительно, чтобы минимальный диаметр для такого прохода был большим, примерно, чем 0,02 м, более предпочтительно находился в пределах примерно между 0,06 м и примерно 2 м, наиболее предпочтительно между 0,12 и 0,8 м.
Как отмечалось выше, является желательным контролировать поток через проход в определенных предпочтительных пределах. Имеется множество средств, известных в данной области, для осуществления этого контроля посредством подбора соответствующей фиксированной геометрии во время конструирования прохода для потока. Другой предпочтительный вариант осуществления представляет собой использование геометрии, которая изменяется во время работы, явным образом включающей в себя клапаны всех сортов и описаний, включая как приводимые в действие вручную, так и приводимые в действие приводом, с помощью любых средств, включая контроль с помощью петель обратной связи от отслеживающего элемента или без них. Другие предпочтительные средства контроля потока жидкости разбавления заключаются в изменении поступления энергии между входом и выходом из прохода. Предпочтительные средства включают в себя изменения скорости потока одной или нескольких движущих текучих сред для эжектора, изменение поступления энергии для привода насоса и разности плотностей или разности высот, когда используется гравитационная сила. Эти предпочтительные средства могут использоваться также во всех сочетаниях.
Проход, используемый для циркуляции жидкости из реакционной среды, может быть любого типа, известного в данной области. Один из вариантов осуществления использует проход, сконструированный полностью или частично с использованием обычных трубных материалов. Другой вариант осуществления использует проход, сконструированный полностью или частично с использованием стенки реакционной емкости в качестве одной из деталей прохода. Проход может конструироваться как полностью заключенный внутри границ реакционной емкости (фиг.24), или же он может конструироваться полностью снаружи реакционной емкости (фиг.25), или он может содержать секции как внутри, так и снаружи реакционной емкости.
Авторы предполагают, что, в особенности, в реакторах больших размеров может быть желательным иметь множество проходов и различные их конструкции для перемещения жидкости через проход. Кроме того, может быть желательным создание множества выходов во множестве положений на одном или на всех проходах. Особенности конструкции будут балансировать желательный общий градиент постоянных концентраций окисляемого соединения с желаемым начальным разбавлением исходных материалов окисляемого соединения в соответствии с другими аспектами настоящего изобретения.
Фиг. как 24, так и 25 иллюстрируют конструкции, которые используют деаэрационную емкость, соединенную с проходом. Эта деаэрационная емкость обеспечивает то, что часть реакционной среды, используемая для разбавления поступающего окисляемого соединения, представляет собой по существу деаэрированную суспензию. Теперь отметим, однако, что жидкость или суспензия, используемая для разбавления поступающего окисляемого соединения, может находиться в аэрированной форме, а также в деаэрированной форме.
Использование жидкости, протекающей через проход для обеспечения разбавления исходных материалов окисляемого соединения, является особенно полезным в барботажных колоннах реакторного типа. Кроме того, в барботажных колоннах реакторного типа может быть достигнут хороший выигрыш для начального разбавления исходных материалов окисляемого соединения, даже без добавления исходных материалов окисляемого соединения непосредственно в проход, при условии, что выход из прохода расположен достаточно близко к положению добавления окисляемого соединения, в таком варианте осуществления предпочтительно, чтобы выход из прохода находился в пределах примерно 27 диаметров выхода из прохода от ближайшего положения добавления для окисляемого соединения, более предпочтительно в пределах примерно 9 диаметров выхода из прохода, еще более предпочтительно в пределах примерно 3 диаметров выхода из прохода, а наиболее предпочтительно в пределах 1 диаметра выхода из прохода.
Обнаружено также, что эжекторы потока могут быть пригодными для начального разбавления окисляемого соединения в барботажных колоннах окисления в соответствии с одним из вариантов осуществления настоящего изобретения, даже без использования проходов для получения жидкости разбавления из удаленной части реакционной среды. В таких случаях эжектор располагается в реакционной среде и имеет открытый путь из реакционной среды в горловину эжектора, где низкое давление вовлекает в расположенную рядом реакционную среду. Примеры двух возможных конфигураций эжектора иллюстрируются на фиг.26 и 27. В предпочтительном варианте осуществления этих эжекторов ближайшее положение поступающего окисляемого соединения находится в пределах примерно 4 м, более предпочтительно в пределах примерно 1 м, а наиболее предпочтительно 0,3 м от горловины эжектора. В другом варианте осуществления окисляемое соединение вводится под давлением в качестве движущей текучей среды. Еще в одном варианте осуществления либо растворитель, либо окислитель вводится под давлением в качестве дополнительной движущей текучей среды вместе с окисляемым соединением. Еще в одном варианте осуществления как растворитель, так и окислитель вводятся под давлением в качестве дополнительной движущей текучей среды вместе с окисляемым соединением.
Авторы предполагают, что, в особенности в реакторах больших размеров, может быть желательным иметь множество эжекторов и различные их конструкции, расположенные в различных положениях в реакционной среде. Особенности конструкций будут балансировать желаемый общий градиент постоянных концентраций окисляемого соединения с желаемым начальным разбавлением исходных материалов окисляемого соединения, в соответствии с другими аспектами настоящего изобретения. В дополнение к этому, авторы предполагают, что струи выходного потока из эжектора могут ориентироваться в любом направлении. Когда используется множество эжекторов, каждый эжектор может ориентироваться по отдельности, опять же, в любом направлении.
Как рассмотрено выше, определенные физические и рабочие особенности барботажной колонны реакторного типа 20, описанные выше, при упоминании фиг.1-27, обеспечивают градиенты по высоте давления, температуры и концентраций реагентов (т.е. кислорода и окисляемого соединения) в реакционной среде 36. Как описывается выше, эти градиенты по высоте могут обеспечить более эффективный и экономичный способ окисления, по сравнению с обычными способами окисления, которые хорошо воспринимают хорошо перемешанную реакционную среду с относительно однородными давлением, температурой и концентрациями реагента в ней. Градиенты кислорода по высоте окисляемого соединения (например, пара-ксилола) и температуры, которые становятся возможными посредством использования системы окисления в соответствии с вариантом осуществления настоящего изобретения, теперь будут обсуждаться более подробно.
Обращаясь теперь к фиг.28, для количественного определения градиентов концентраций реагентов, существующих в реакционной среде 36 во время окисления в барботажной колонне реакторного типа 20, весь объем реакционной среды 36 может теоретически быть разделен на 30 дискретных горизонтальных слоев равного объема. Фиг.28 иллюстрирует концепцию деления реакционной среды 36 на 30 дискретных горизонтальных слоев равного объема. За исключением самого высокого и самого нижнего горизонтальных слоев, каждый горизонтальный слой представляет собой дискретный объем, ограниченный сверху и снизу воображаемыми горизонтальными плоскостями и ограниченный на своих сторонах стенкой реактора 20. Самый высокий горизонтальный слой ограничен на своей нижней части воображаемой горизонтальной плоскостью, а на своей верхней части - верхней поверхностью реакционной среды 36. Самый нижний горизонтальный слой ограничен на своей верхней части воображаемой горизонтальной плоскостью, а на своей нижней части - нижней частью корпуса. После того как реакционная среда 36 теоретически разделяется на 30 дискретных горизонтальных слоев равного объема, затем может быть определена усредненная по времени и усредненная по объему концентрация каждого горизонтального слоя. Индивидуальный горизонтальный слой, имеющий максимальную концентрацию из всех 30 горизонтальных слоев, может идентифицироваться как "горизонтальный слой C-max". Индивидуальный горизонтальный слой, расположенный выше горизонтального слоя C-max и имеющий минимальную концентрацию из всех горизонтальных слоев, расположенных выше горизонтального слоя C-max, может идентифицироваться как "горизонтальный слой C-min". Градиент концентраций по высоте затем может быть вычислен как отношение концентрации в горизонтальном слое C-max к концентрации в горизонтальном слое C-min.
Относительно количественного определения градиента концентрации кислорода, когда реакционная среда 36 теоретически разделяется на 30 дискретных горизонтальных слоев равного объема, горизонтальный слой O2-max идентифицируется как имеющий максимальную концентрацию кислорода из всех 30 горизонтальных слоев, а горизонтальный слой O2-min идентифицируется как имеющий минимальную концентрацию кислорода из горизонтальных слоев, расположенных выше горизонтальных слоев O2-max. Концентрации кислорода горизонтальных слоев измеряются в газовой фазе реакционной среды 36 как усредненная по времени и усредненная по объему молярная концентрация во влажном состоянии. Предпочтительно, чтобы отношение концентраций кислорода горизонтального слоя O2-max к концентрации кислорода в горизонтальном слое O2-min находилась в пределах примерно от 2:1 примерно до 25:1, более предпочтительно в пределах примерно от 3:1 примерно до 15:1, а наиболее предпочтительно в пределах от 4:1 до 10:1.
Как правило, горизонтальный слой O2-max будет располагаться вблизи нижней части реакционной среды 36, в то время как горизонтальный слой O2-min будет располагаться вблизи верхней части реакционной среды 36. Предпочтительно горизонтальный слой O2-min представляет собой один из 5 самых верхних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Наиболее предпочтительно горизонтальный слой O2-min представляет собой самый верхний слой из 30 дискретных горизонтальных слоев, как иллюстрируется на фиг.28. Предпочтительно горизонтальный слой O2-max представляет собой один из 10 самых нижних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Наиболее предпочтительно горизонтальный слой O2-max представляет собой один из 5 самых нижних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Например, фиг.28 иллюстрирует горизонтальный слой O2-max как третий горизонтальный слой от нижней части реактора 20. Предпочтительно, чтобы расстояние по высоте между горизонтальными слоями O2-min и O2-max составляло, по меньшей мере, примерно 2 W, более предпочтительно, по меньшей мере, примерно 4 W, а наиболее предпочтительно, по меньшей мере, 6 W. Предпочтительно, чтобы расстояние по высоте между горизонтальными слоями O2-min и O2-max составляло, по меньшей мере, примерно 0,2 H, более предпочтительно, по меньшей мере, примерно 0,4 H, а наиболее предпочтительно, по меньшей мере, 0,6 H.
Усредненная по времени и по объему концентрация кислорода во влажном состоянии для горизонтального слоя O2-min предпочтительно находится в пределах примерно от 0,1 примерно до 3% молярных, более предпочтительно в пределах примерно от 0,3 примерно до 2% молярных, а наиболее предпочтительно в пределах от 0,5 до 1,5% молярного. Усредненная по времени и по объему концентрация кислорода горизонтального слоя O2-max предпочтительно находится в пределах примерно от 4 примерно до 20% молярных, более предпочтительно в пределах примерно от 5 примерно до 15% молярных, а наиболее предпочтительно в пределах от 6 до 12% молярных. Усредненная по времени концентрация кислорода, в сухом состоянии, в газообразном выходящем потоке, высвобождаемом из реактора 20 через выход для газа 40, предпочтительно находится в пределах примерно от 0,5 примерно до 9% молярных, более предпочтительно в пределах примерно от 1 примерно до 7% молярных, а наиболее предпочтительно в пределах от 1,5 до 5% молярных.
Поскольку концентрация кислорода затухает так заметно по направлению к верхней части реакционной среды 36, является желательным, чтобы потребность в кислороде в верхней части реакционной среды 36 уменьшалась. Это пониженная потребность в кислороде вблизи верхней части реакционной среды 36 может достигаться посредством создания градиента по высоте концентрации окисляемого соединения (например, пара-ксилола), где минимальная концентрация окисляемого соединения находится вблизи верхней части реакционной среды 36.
Относительно количественного определения градиента концентрации окисляемого соединения (например, пара-ксилола), когда реакционная среда 36 теоретически разделяется на 30 дискретных горизонтальных слоев равного объема, горизонтальный слой OC-max идентифицируется как имеющий максимальную концентрацию окисляемого соединения из всех 30 горизонтальных слоев и горизонтальный слой OC-min идентифицируется как имеющий минимальную концентрацию окисляемого соединения, из горизонтальных слоев, расположенных выше горизонтального слоя OC-max. Концентрации окисляемого соединения горизонтальных слоев измеряются в жидкой фазе по отношению к усредненной по времени и по объему массовой доле. Предпочтительно, чтобы отношение концентрации окисляемого соединения горизонтального слоя OC-max к концентрации окисляемого соединения горизонтального слоя OC-min было большим, примерно, чем 5:1, более предпочтительно большим, примерно, чем 10:1, еще более предпочтительно большим, примерно, чем 20:1, наиболее предпочтительно находилось в пределах от 40:1 до 1000:1.
Как правило, горизонтальный слой OC-max будет располагаться вблизи нижней части реакционной среды 36, в то время как горизонтальный слой OC-min будет располагаться вблизи верхней части реакционной среды 36. Предпочтительно горизонтальный слой OC-min представляет собой один из 5 самых верхних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Наиболее предпочтительно горизонтальный слой OC-min представляет собой самый верхний из 30 дискретных горизонтальных слоев, как иллюстрируется на фиг.28. Предпочтительно горизонтальный слой OC-max представляет собой один из 10 самых нижних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Наиболее предпочтительно горизонтальный слой OC-max представляет собой один из 5 самых нижних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Например, фиг.28 иллюстрирует горизонтальный слой OC-max как пятый горизонтальный слой от нижней части реактора 20. Предпочтительно, чтобы вертикальное расстояние между горизонтальными слоями OC-min и OC-max составляло, по меньшей мере, примерно 2 W, где W представляет собой максимальную ширину реакционной среды 36. Более предпочтительно вертикальное расстояние между горизонтальными слоями OC-min и OC-max составляет, по меньшей мере, примерно 4 W, а наиболее предпочтительно, по меньшей мере, 6 W. Для данной высоты H реакционной среды 36 предпочтительно, чтобы вертикальное расстояние между горизонтальными слоями OC-min и OC-max составляло, по меньшей мере, примерно 0,2 H, более предпочтительно, по меньшей мере, примерно 0,4 H, а наиболее предпочтительно, по меньшей мере, 0,6 H.
Усредненная по времени и по объему концентрация окисляемого соединения (например, пара-ксилола) в жидкой фазе горизонтального слоя OC-min предпочтительно является меньшей, примерно, чем 5000 м.д. мас., более предпочтительно меньшей, примерно, чем 2000 м.д. мас., еще более предпочтительно меньшей, примерно, чем 400 м.д. мас., наиболее предпочтительно находится в пределах от 1 м.д. мас. до 100 м.д. мас. Усредненная по времени и по объему концентрация окисляемого соединения в жидкой фазе горизонтального слоя OC-max предпочтительно находится в пределах примерно от 100 м.д. мас. примерно до 10000 м.д. мас., более предпочтительно в пределах примерно от 200 м.д. мас. примерно до 5000 м.д. мас., наиболее предпочтительно в пределах от 500 м.д. мас. до 3000 м.д. мас.
Хотя предпочтительно, чтобы барботажная колонна реакторного типа 20 обеспечивала градиенты концентрации окисляемого соединения по высоте, является также предпочтительным, чтобы объемный процент реакционной среды 36, имеющий концентрацию окисляемого соединения в жидкой фазе выше 1000 м.д. мас., сводился к минимуму. Предпочтительно усредненный по времени объемный процент реакционной среды 36, имеющий концентрацию окисляемого соединения в жидкой фазе выше 1000 м.д. мас., меньше, примерно, чем 9%, более предпочтительно меньше, примерно, чем 6%, наиболее предпочтительно меньше, чем 3%. Предпочтительно усредненный по времени объемный процент реакционной среды 36, имеющий концентрацию окисляемого соединения в жидкой фазе выше 2500 м.д. мас., меньше, примерно, чем 1,5%, более предпочтительно меньше, примерно, чем 1%, наиболее предпочтительно меньше, чем 0,5%. Предпочтительно усредненный по времени объемный процент реакционной среды 36, имеющий концентрацию окисляемого соединения в жидкой фазе выше 10000 м.д. мас., меньше, примерно, чем 0,3%, более предпочтительно меньше, примерно, чем 0,1%, наиболее предпочтительно меньше, чем 0,03%. Предпочтительно усредненный по времени объем процент реакционной среды 36, имеющий концентрацию окисляемого соединения в жидкой фазе выше 25000 м.д. мас., меньше, примерно, чем 0,03%, более предпочтительно меньше, примерно, чем 0,015%, наиболее предпочтительно меньше, чем 0,007%. Авторы отмечают, что объем реакционной среды 36, имеющий повышенные уровни окисляемого соединения, не должен обязательно лежать в едином сплошном объеме. Во многих случаях хаотические структуры потока в емкости барботажной колонны реакторного типа образуют одновременно две или несколько сплошных, но отделенных друг от друга частей реакционной среды 36, имеющих повышенные уровни окисляемого соединения. В каждом случае использования усреднения по времени все такие сплошные, но отделенные друг от друга объемы, большие, чем 0,0001% объемный от реакционной среды в целом, складываются вместе для определения общего объема, имеющего повышенные уровни концентрации окисляемого соединения в жидкой фазе.
В дополнение к градиентам концентрации кислорода и окисляемого соединения, обсуждаемым выше, предпочтительно, чтобы в реакционной среде 36 существовал градиент температуры. Обращаясь опять к фиг.28, этот градиент температуры может быть количественно определен способом, подобным градиентам концентрации, посредством теоретического разделения реакционной среды 36 на 30 дискретных горизонтальных слоев равного объема и измерения усредненной по времени и по объему температуры каждого слоя. Горизонтальный слой с самый низкой температурой из самых нижних 15 горизонтальных слоев затем может быть идентифицирован как горизонтальный слой T-мин, и горизонтальный слой, расположенный выше горизонтального слоя T-мин, и имеющий температуру, максимальную среди всех слоев выше горизонтального слоя T-мин, может затем быть идентифицирован как "горизонтальный слой T-макс". Предпочтительно, чтобы температура горизонтального слоя T-макс была, по меньшей мере, примерно на 1°C выше, чем температура горизонтального слоя T-мин. Более предпочтительно температура горизонтального слоя T-макс выше, в пределах примерно от 1,25 примерно до 12°C, чем температура горизонтального слоя T-мин. Наиболее предпочтительно температура горизонтального слоя T-макс выше, в пределах от 2 до 8°C, чем температура горизонтального слоя T-мин. Температура горизонтального слоя T-макс предпочтительно находится в пределах примерно от 125 примерно до 200°C, более предпочтительно в пределах примерно от 140 примерно до 180°C, наиболее предпочтительно в пределах от 150 до 170°C
Как правило, горизонтальный слой T-макс будет располагаться вблизи центра реакционной среды 36, в то время как горизонтальный слой T-мин будет располагаться вблизи нижней части реакционной среды 36. Предпочтительно горизонтальный слой T-мин представляет собой один из 10 самых нижних горизонтальных слоев, из 15 самых нижних горизонтальных слоев. Наиболее предпочтительно горизонтальный слой T-мин представляет собой один из 5 самых нижних горизонтальных слоев, из 15 самых нижних горизонтальных слоев. Например, фиг.28 иллюстрирует горизонтальный слой T-мин как второй горизонтальный слой снизу в реакторе 20. Предпочтительно горизонтальный слой T-макс представляет собой один из 20 средних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Наиболее предпочтительно горизонтальный слой T-мин представляет собой один из 14 средних горизонтальных слоев, из 30 дискретных горизонтальных слоев. Например, фиг.28 иллюстрирует горизонтальный слой T-макс как двадцатый горизонтальный слой снизу реактора 20 (т.е. один из средних 10 горизонтальных слоев). Предпочтительно, чтобы вертикальное расстояние между горизонтальными слоями T-мин и T-макс составляло, по меньшей мере, примерно 2 W, более предпочтительно, по меньшей мере, примерно 4 W, наиболее предпочтительно, по меньшей мере, 6 W. Предпочтительно, чтобы вертикальное расстояние между горизонтальными слоями T-мин и T-макс составляло, по меньшей мере, примерно 0,2 H, более предпочтительно, по меньшей мере, примерно 0,4 H, наиболее предпочтительно, по меньшей мере, 0,6 H.
Как обсуждалось выше, когда имеется градиент по высоте температуры в реакционной среде 36, может быть преимущественным извлечение реакционной среды 36 в приподнятом положении, где температура реакционной среды является самой высокой, в особенности, когда извлеченный продукт подвергается последующей дополнительной переработке при более высокой температуре. Таким образом, когда реакционная среда 36 извлекается из реакционной зоны 28 через один или несколько приподнятых выходов, как иллюстрируется на фиг.19 и 20, предпочтительно, чтобы приподнятый по высоте выход (выходы) располагался вблизи горизонтального слоя T-макс. Предпочтительно приподнятый по высоте выход располагается в пределах 10 горизонтальных слоев от горизонтального слоя T-макс, более предпочтительно в пределах 5 горизонтальных слоев от горизонтального слоя T-макс, наиболее предпочтительно в пределах 2 горизонтальных слоев от горизонтального слоя T-макс.
Теперь отметим, что многие особенности настоящего изобретения, описываемые здесь, могут использоваться в системах с множеством реакторов окисления - а не только в системах, использующих один реактор окисления. В дополнение к этому, определенные особенности настоящего изобретения, описываемые здесь, могут использоваться в механически перемешиваемых и/или перемешиваемых потоком реакторах окисления - а не только в барботажных перемешиваемых реакторах (т.е. барботажных колоннах реакторного типа). Например, авторы обнаружили определенные преимущества, связанные со ступенчатым изменением/плавным изменением концентрации кислорода и/или скорости потребления кислорода в реакционной среде. Преимущества, реализуемые посредством ступенчатого изменения концентрации/потребления кислорода в реакционной среде, могут быть реализованы независимо от того, содержится ли весь объем реакционной среды в одной емкости или во множестве емкостей. Кроме того, преимущества, реализуемые посредством ступенчатого изменения концентрации/потребления кислорода в реакционной среде, могут быть реализованы независимо от того, является ли реакционная емкость (емкости) перемешиваемой механически, перемешиваемой потоком и/или барботажно-перемешиваемой.
Один из способов количественного определения уровня ступенчатого изменения концентрации и/или скорости расходования кислорода в реакционной среде заключается в сравнении двух или нескольких отдельных 20% сплошных объемов реакционной среды. Эти 20% сплошные объемы не должны обязательно быть определены посредством любой конкретной формы. Однако каждый 20% сплошной объем должен быть сформирован из сплошного отдельного объема реакционной среды (т.е. каждый объем является "сплошным") и 20% сплошные объемы не должны перекрываться друг с другом (т.е. объемы являются "отдельными"). Фиг.29-31 иллюстрируют, что эти отдельные 20% сплошные объемы могут располагаться в одном и том же реакторе (фиг.29) или во множестве реакторов (фиг.30 и 31). Необходимо отметить, что реакторы, иллюстрируемые на фиг.29-31, могут представлять собой перемешиваемые механически, перемешиваемые потоком и/или барботажно-перемешиваемые реакторы. В одном из вариантов осуществления предпочтительно, чтобы реакторы, иллюстрируемые на фиг.29-31, представляли собой барботажно-перемешиваемые реакторы (т.е. барботажные колонны реакторного типа).
Обращаясь теперь к фиг.29, реактор 20 иллюстрируется как содержащий реакционную среду 36. Реакционная среда 36 включает в себя первый отдельный 20% сплошной объем 37 и второй отдельный 20% сплошной объем 39.
Обращаясь теперь к фиг.30, система из множества реакторов иллюстрируется как содержащая первый реактор 720a и второй реактор 720b. Реакторы 720a, b вместе содержат общий объем реакционной среды 736. Первый реактор 720a содержит первую часть реакционной среды 736a, в то время как второй реактор 720b содержит вторую часть реакционной среды 736b. Первый отдельный 20% сплошной объем 737 реакционной среды 736 показан как являющийся определенным внутри первого реактора 720a, в то время как второй отдельный 20% сплошной объем 739 реакционной среды 736 показан как являющийся определенным внутри второго реактора 720b.
Обращаясь теперь к фиг.31, система из множества реакторов иллюстрируется как содержащая первый реактор 820a, второй реактор 820b и третий реактор 820c. Реакторы 820a, b, c вместе содержат общий объем реакционной среды 836. Первый реактор 820a содержит первую часть реакционной среды 836a; второй реактор 820b содержит вторую часть реакционной среды 836b; и третий реактор 820c содержит третью часть реакционной среды 836c. Первый отдельный 20% сплошной объем 837 реакционной среды 836 показан как являющийся определенным внутри первого реактора 820a; второй отдельный 20% сплошной объем 839 реакционной среды 836 показан как являющийся определенным внутри второго реактора 820b; и третий отдельный 20% сплошной объем 841 реакционной среды 836 показан как являющийся определенным внутри третьего реактора 820c.
Ступенчатое изменение доступности кислорода в реакционной среде может быть количественно определено посредством упоминания 20% сплошного объема реакционной среды, имеющего наиболее избыточную молярную фракцию кислорода в газовой фазе, и посредством упоминания 20% сплошного объема реакционной среды, имеющего наиболее истощенную молярную фракцию кислорода в газовой фазе. В газовой фазе отдельного 20% сплошного объема реакционной среды, содержащего самую высокую концентрацию кислорода в газовой фазе, усредненная по времени и по объему концентрация кислорода, во влажном состоянии, предпочтительно находится в пределах примерно от 3 примерно до 18% молярных, более предпочтительно в пределах примерно от 3,5 примерно до 14% молярных, наиболее предпочтительно в пределах от 4 до 10% молярных. В газовой фазе отдельного 20% сплошного объема реакционной среды, содержащего самую низкую концентрацию кислорода в газовой фазе, усредненная по времени и по объему концентрация кислорода, во влажном состоянии, предпочтительно находится в пределах примерно от 0,3 примерно до 5% молярных, более предпочтительно в пределах примерно от 0,6 примерно до 4% молярных, наиболее предпочтительно в пределах от 0,9 до 3% молярных. Более того отношение усредненной по времени и по объему концентрации кислорода, во влажном состоянии, в наиболее избыточном 20% сплошном объеме реакционной среды, по сравнению с наиболее истощенным 20% сплошным объемом реакционной среды, предпочтительно находится в пределах примерно от 1,5:1 примерно до 20:1, более предпочтительно в пределах примерно от 2:1 примерно до 12:1, наиболее предпочтительно в пределах от 3:1 до 9:1.
Ступенчатое изменение скорости потребления кислорода в реакционной среде может быть количественно определено в терминах STR по кислороду, изначально описанной выше. STR по кислороду описана ранее в общем смысле (т.е. с точки зрения средней STR по кислороду реакционной среды в целом); однако STR по кислороду может также быть рассмотрена в локальном смысле (т.е. для части реакционной среды), для количественного определения ступенчатого изменения скорости потребления кислорода в реакционной среде.
Авторы обнаружили, что является очень пригодным для использования вызывать изменение STR по кислороду в реакционной среде в общей гармонии с желательными градиентами, описываемыми здесь по отношению к давлению в реакционной среде и к молярной доле молекулярного кислорода в газовой фазе реакционной среды. Таким образом, предпочтительно, чтобы отношение STR по кислороду первого отдельного 20% сплошного объема реакционной среды по сравнению с STR по кислороду второго отдельного 20% сплошного объема реакционной среды находилось в пределах примерно от 1,5:1 примерно до 20:1, более предпочтительно в пределах примерно от 2:1 примерно до 12:1, наиболее предпочтительно в пределах от 3:1 до 9:1. В одном из вариантов осуществления "первый отдельный 20% сплошной объем" располагается ближе, чем "второй отдельный 20% сплошной объем", к положению, где молекулярный кислород изначально вводится в реакционную среду. Эти большие градиенты STR по кислороду являются желательными, независимо от того, содержится ли реакционная среда частичного окисления в барботажной колонне реакторного типа окисления или в любом другом типе реакционной емкости, в которой создаются градиенты давления и/или молярной доли молекулярного кислорода в газовой фазе реакционной среды (например, в емкости с механическим перемешиванием, имеющей множество вертикально расположенных перемешиваемых зон, получаемых посредством использования множества импеллеров, имеющих сильный радиальный поток, возможно расширяемый посредством, в целом, горизонтальных узлов дефлекторов, с восходящим потоком окислителя, как правило, снизу вверх от входа вблизи нижней части реакционной емкости, несмотря на то что значительное обратное перемешивание потока окислителя может происходить в пределах каждой вертикально расположенной зоны перемешивания и что некоторое обратное перемешивание потока окислителя может происходить между соседними расположенными вертикально зонами перемешивания). Т.е., когда имеется градиент давления и/или молярной доли молекулярного кислорода в газовой фазе реакционной среды, как обнаружили авторы, является желательным создание подобного градиента химического расходования растворенного кислорода с помощью средств, описываемых здесь.
Предпочтительные средства для принудительного изменения локальной STR по кислороду представляют собой осуществление контроля положений введения окисляемого соединения и контроль перемешивания жидкофазной реакционной среды для контроля градиентов концентрации окисляемого соединения в соответствии с другими описаниями настоящего изобретения. Другие пригодные для использования средства принудительного изменения локальной STR по кислороду включают в себя принудительное изменение реакционной активности посредством принудительного локального изменения температуры и посредством локального изменения смеси катализатора и компонентов растворителя (например, посредством введения дополнительного газа, чтобы охлаждать испарением в конкретной части реакционной среды, и посредством добавления потока растворителя, содержащего более высокое количество воды, для уменьшения активности в конкретной части реакционной среды).
Как обсуждается выше при упоминании фиг.30 и 31, реакция частичного окисления может быть осуществлена пригодным для использования образом во множестве реакционных емкостей, где, по меньшей мере, часть, предпочтительно, по меньшей мере, 25%, более предпочтительно, по меньшей мере, 50%, наиболее предпочтительно, по меньшей мере, 75% молекулярного кислорода, покидающего первую реакционную емкость, переносится в одну или несколько последующих реакционных емкостей для потребления дополнительной малой добавки, предпочтительно более чем 10%, более предпочтительно более чем 20%, наиболее предпочтительно более чем 40% молекулярного кислорода, покидающего первую/первую по ходу процесса реакционную емкость. Когда используется такой последовательный поток молекулярного кислорода из одного реактора в другие, является желательным, чтобы первая реакционная емкость работала при более высокой интенсивности реакции, чем, по меньшей мере, одна из последующих реакционных емкостей, предпочтительно при отношении усредненной по емкости STR по кислороду внутри первой реакционной емкости к усредненной по емкости STR по кислороду в последующей реакционной емкости в пределах примерно от 1,5:1 примерно до 20:1, более предпочтительно в пределах примерно от 2:1 примерно до 12:1, наиболее предпочтительно в пределах от 3:1 до 9:1.
Как обсуждалось выше, все типы первой реакционной емкости (например, барботажная колонна, механически перемешиваемая, с обратным перемешиванием, с внутренним ступенчатым изменением, с поршневым потоком, и так далее) и все типы последовательных реакционных емкостей, которые могут быть или не быть типов, иных, чем первая реакционная емкость, являются пригодными для использования, для последовательного протекания молекулярного кислорода в последующие реакционные емкости в соответствии с настоящим изобретением. Средства принудительного уменьшения усредненной по емкости STR по кислороду, для уменьшения в пределах последующих реакционных емкостей, пригодным для использования образом включают в себя уменьшение температуры, уменьшение концентрации окисляемого соединения и уменьшение реакционной активности конкретной смеси каталитических компонентов и растворителя (например, уменьшение концентрации кобальта, увеличение концентрации воды и добавление такого каталитического ингибитора, как малые количества ионной меди).
При протекании из первой реакционной емкости в последующую реакционную емкость поток окислителя может обрабатываться с помощью любых средств, известных в данной области, таких как сжатие или понижение давления, охлаждение или нагрев и удаление массы или добавление массы любого количества или любого типа. Однако использование уменьшения усредненной по емкости STR по кислороду в последующей реакционной емкости является конкретно пригодным для использования, когда абсолютное давление в верхней части первой реакционной емкости меньше, примерно, чем 2,0 МПа, более предпочтительно меньше, примерно, чем 1,6 МПа, наиболее предпочтительно меньше, чем 1,2 МПа. Более того использование уменьшения усредненной по емкости STR по кислороду в последующей реакционной емкости является конкретно пригодным для использования, когда отношение абсолютного давления в верхней части первой реакционной емкости, по сравнению с абсолютным давлением в верхней части, по меньшей мере, одной последующей реакционной емкости находится в пределах примерно от 0,5:1 до 6:1, более предпочтительно в пределах примерно от 0,6:1 примерно до 4:1, наиболее предпочтительно в пределах от 0,7:1 до 2:1. Понижение давления в последующей емкости ниже этих нижних пределов излишне понижает доступность молекулярного кислорода, а увеличение давления выше этих верхних пределов является все более дорогостоящим, по сравнению с использованием подачи свежего окислителя.
Когда используется последовательный поток молекулярного кислорода в последующую реакционную емкость, имеющую пониженную усредненную по емкости STR по кислороду, потоки свежих исходных материалов и окисляемого соединения, растворителя и окислителя могут протекать в последующую реакционную емкость и/или в первую реакционную емкость. Потоки жидкой фазы и твердой фазы, если они присутствуют, реакционной среды могут протекать в любом направлении между реакционными емкостями. Вся газовая фаза или ее часть, покидающая первую реакционную емкость и поступающая в последующую реакционную емкость, может протекать отдельно или перемешиваться с частью жидкой фазы или твердой фазы, если они присутствуют, реакционной среды из первой реакционной емкости. Протекающий поток продукта, содержащего жидкую фазу и твердую фазу, если они присутствуют, может извлекаться из реакционной среды в любой реакционной емкости в системе.
Обращаясь опять к фиг.1-31, окисление предпочтительно осуществляют в барботажной колонне реакторного типа 20 при условиях, которые являются заметно иными, в соответствии с предпочтительными вариантами осуществления, описанными здесь, чем обычные реакторы окисления. Когда барботажная колонна реакторного типа 20 используется для осуществления жидкофазного частичного окисления пара-ксилола до сырой терефталевой кислоты (CTA) в соответствии с предпочтительными вариантами осуществления, описанными здесь, пространственные профили локальной интенсивности реакции, локальной интенсивности испарения и локальной температуры в сочетании со структурами потока жидкости в реакционной среде и предпочтительными относительно низкими температурами окисления вносят вклад в образование частиц CTA, имеющих уникальные и преимущественные свойства.
Фиг.32A и 32B иллюстрируют основные частицы CTA, полученные в соответствии с одним из вариантов осуществления настоящего изобретения. Фиг.32A показывает основные частицы CTA при 500-кратном увеличении, в то время как фиг.32B увеличивает одну из основных частиц CTA и показывает эту частицу при 2000-кратном увеличении. Как, вероятно, лучше всего иллюстрируется на фиг.32B, каждая основная частица CTA, как правило, формируется из большого количества малых агломерированных субчастиц CTA, при этом получается основная частица CTA с относительно большой площадью поверхности, высокой пористостью, низкой плотностью и хорошей растворимостью. Основные частицы CTA, как правило, имеют средний размер частиц в пределах примерно от 20 примерно до 150 мкм, более предпочтительно в пределах примерно от 30 примерно до 120 мкм, а наиболее предпочтительно в пределах от 40 до 90 мкм. Субчастицы CTA, как правило, имеют средний размер частиц в пределах примерно от 0,5 примерно до 30 мкм, более предпочтительно примерно от 1 примерно до 15 мкм, а наиболее предпочтительно в пределах от 2 до 5 мкм. Относительно высокая площадь поверхности основных частиц CTA, иллюстрируемых на фиг.32A и 32B, может количественно определяться с использованием способа измерения площади поверхности Браунауэра-Эмметта-Теллера (БЭТ). Предпочтительно основные частиц CTA имеют среднюю поверхность БЭТ, по меньшей мере, примерно 0,6 м2/г. Более предпочтительно основные частиц CTA имеют среднюю площадь поверхности по БЭТ в пределах примерно от 0,8 примерно до 4 м2/г. Наиболее предпочтительно основные частиц CTA имеют среднюю площадь поверхности по БЭТ в пределах от 0,9 до 2 м2/г. Физические свойства (например, размер частиц, площадь поверхности по БЭТ, пористость и растворимость) основных частиц CTA, сформированных посредством оптимизированного способа окисления предпочтительного варианта осуществления настоящего изобретения, делают возможной очистку частиц CTA посредством более эффективных и/или экономичных способов, как описано более подробно ниже в связи с фиг.35.
Значения среднего размера частиц, приведенные выше, определяют с использованием микроскопии поляризованного света и анализа изображения. Оборудование, используемое при анализе размеров частиц, включает в себя оптический микроскоп Nikon E800 с объективом 4× Plan Flour N.A. 0.13, цифровую камеру Spot RT™ и персональный компьютер, использующий программное обеспечение для анализа изображений Image Pro Plus™ V4.5.0,19. Способ анализа размеров частиц включает в себя следующие главные стадии: (1) диспергирования порошков CTA в минеральном масле; (2) приготовления препарата дисперсии между предметным и покровным стеклом микроскопа; (3) исследования объекта с использованием микроскопии поляризованного света (условия скрещенных поляризаторов - частицы появляются как яркие объекты на черном фоне); (4) регистрации различных изображений для препарата каждого образца (размер поля=3×2,25 мм; размер пикселя=1,84 мкм/пиксель); (5) осуществления анализа изображений с помощью программного обеспечения Image Pro Plus™; (6) переноса измерений частиц в таблицу и (7) осуществления статистической характеризации данных. Стадия (5) "осуществление анализа изображений с помощью программного обеспечения Image Pro Plus™" включает в себя подстадии: (a) установки порога изображения для детектирования белых частиц на темном фоне; (b) создания двоичного изображения; (c) запуска однопроходного открытого фильтра для отфильтровывания пиксельного шума; (d) измерения всех частиц в изображении и (e) регистрации среднего диаметра, измеренного для каждой частицы. Программное обеспечение Image Pro Plus™ определяет средний диаметр отдельных частиц как среднечисленную длину диаметров частицы, измеренную на интервалах 2° и при прохождении через центроид частицы. Стадия 7 "осуществления статистической характеризации в таблице" включает в себя вычисление взвешенного по объему среднего размера частиц следующим образом. Объем каждой из n частиц в образце вычисляется, как если бы они были сферическими, используя выражение pi/6·di^3; умножая объем каждой частицы на ее диаметр, с получением выражения pi/6·di^4; суммируя для всех частиц в образце значения pi/6·di^4; суммируя объемы всех частиц в образце и вычисляя взвешенный по объему диаметр частиц как сумму выражений (pi/6·di^4) для всех n частиц в образце, деленную на сумму выражений (pi/6·di^3) для всех n частиц в образце. Как здесь используется, "средний размер частицы" относится к взвешенному по объему среднему размеру частицы, определенному в соответствии с описанным выше способом исследований; и он также упоминается как D (4, 3).
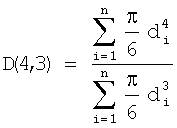
В дополнение к этому, стадия 7 включает в себя нахождение размеров частиц, для которых различные доли общего объема образца являются меньшими. Например, D (v, 0,1) представляет собой размер частиц, для которого 10% от общего объема образца являются меньшими, а 90% являются большими; D (v, 0,5) представляет собой размер частиц, для которого половина объема образца являются большими, а половина меньшими; D (v, 0,9) представляет собой размер частиц, для которого 90% от общего объема образца являются меньшими; и так далее. В дополнение к этому, стадия 7 включает в себя вычисление значения D (v, 0,9) минус D (v, 0,1), которое определяется здесь как "разброс размеров частиц"; и стадия 7 включает в себя вычисление значения разброса размеров частиц, деленного на D (4, 3), которое определяется здесь как "относительный разброс размеров частиц".
Кроме того, предпочтительно, чтобы D (v, 0,1) частиц CTA, как измерено выше, находился в пределах примерно от 5 примерно до 65 мкм, более предпочтительно в пределах примерно от 15 примерно до 55 мкм, а наиболее предпочтительно в пределах от 25 до 45 мкм. Предпочтительно, чтобы D (v, 0,5) частиц CTA, как измерено выше, находился в пределах примерно от 10 примерно до 90 мкм, более предпочтительно в пределах примерно от 20 примерно до 80 мкм, а наиболее предпочтительно в пределах от 30 до 70 мкм. Предпочтительно, чтобы D (v, 0,9) частиц CTA, как измерено выше, находился в пределах примерно от 30 примерно до 150 мкм, более предпочтительно в пределах примерно от 40 примерно до 130 мкм, а наиболее предпочтительно в пределах от 50 до 110 мкм. Предпочтительно, чтобы относительный разброс размеров частиц находился в пределах примерно от 0,5 примерно до 2,0, более предпочтительно в пределах примерно от 0,6 примерно до 1,5, а наиболее предпочтительно в пределах от 0,7 до 1,3.
Значения площади поверхности по БЭТ, приведенные выше, измеряются на Micromeritics ASAP2000 (доступном от Micromeritics Instrument Corporation of Norcross, GA). На первой стадии процесса измерения от 2 до 4 граммов образца частиц взвешивают и сушат в вакууме при 50°C. Затем образец помещают в газовый коллектор для анализа и охлаждают до 77K. Изотерму адсорбции азота измеряют, минимум, при пяти равновесных давлениях посредством экспонирования образца для известных объемов газообразного азота и измерения уменьшения давления. Равновесные давления находятся примерно в пределах P/P0=0,01-0,20, где P представляет собой равновесное давление, и P0 представляет собой давление паров жидкого азота при 77K. Полученная изотерма затем может быть представлена в виде графика в соответствии со следующим уравнением БЭТ:
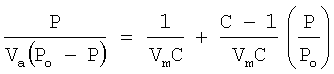
где Va представляет собой объем газа, адсорбированного образцом при P, Vm представляет собой объем газа, необходимый для покрытия всей поверхности образца монослоем газа, и C - это константа. Из этого графика определяют Vm и C. Затем Vm преобразуется в площадь поверхности с использованием площади поперечного сечения азота при 77K посредством:
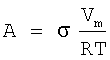
где σ представляет собой площадь поперечного сечения азота при 77K, T равно 77°K и R представляет собой газовую константу.
Как ясно из указанного выше, CTA, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, демонстрирует превосходные свойства растворения, по сравнению с обычной CTA, полученной другими способами. Эта повышенная скорость растворения делает возможной очистку CTA по настоящему изобретению посредством более эффективных и/или более продуктивных способов очистки. Следующее далее описание описывает способ, с помощью которого может количественно определяться скорость растворения CTA.
Скорость растворения известного количества твердых продуктов в известном количестве растворителя в перемешиваемой смеси может измеряться посредством различных протоколов. Как здесь используется, способ измерения, называемый "исследование временной зависимости растворения", определяется следующим образом. В течение исследования временной зависимости растворения используют давление окружающей среды примерно 0,1 МПа. Температура окружающей среды, используемая в течение исследования временной зависимости растворения, составляет примерно 22°C. Кроме того, твердые продукты, растворитель и все устройство для растворения являются полностью уравновешенными термически при этой температуре до начала исследований, и нет заметного нагрева или охлаждения стакана или его содержимого в период времени растворения. Порция растворителя из свежего тетрагидрофурана, аналитического качества для ВЭЖХ (чистота >99,9%), далее THF, насчитывающая 250 г, помещается в чистый высокий 400-мл стеклянный стакан KIMAX (Kimble® part number 14020, Kimble/Kontes, Vineland, NJ), который является неизолированным, имеет гладкие стенки и, в целом, цилиндрическую форму. Покрытый тефлоном брусок магнитной мешалки (VWR part number 58948-230, длиной примерно 1 дюйм, диаметром 3/8-дюйма, восьмиугольного поперечного сечения, VWR International, West Chester, PA 19380) помещают в стакан, где он естественным образом оседает на дно. Образец перемешивают с использованием магнитного перемешивающего устройства Variomag® multipoint 15 (H&P Labortechnik AG, Oberschleissheim, Germany), при его настройке на 800 об/мин. Это перемешивание начинается не более чем за 5 минут до добавления твердых продуктов и продолжается непрерывно в течение, по меньшей мере, 30 мин после добавления твердых продуктов. Твердый образец из частиц сырой или очищенной TPA в количестве 250 мг взвешивают на не прилипающем к образцу поддоне для взвешивания. В начальный момент времени, обозначаемый как t=0, взвешенные твердые продукты высыпают, все вместе, в перемешиваемый THF и одновременно с этим запускают таймер. Приготовленный соответствующим образом THF очень быстро смачивает твердые продукты и в пределах 5 с образует разбавленную, хорошо перемешанную суспензию. Впоследствии, образцы этой смеси получают в следующие моменты времени, измеренные в минутах от t=0: 0,08, 0,25, 0,50, 0,75, 1,00, 1,50, 2,00, 2,50, 3,00, 4,00, 5,00, 6,00, 8,00, 10,00, 15,00 и 30,00. Каждый малый образец извлекают из разбавленной, хорошо перемешанной смеси с использованием нового одноразового шприца (Becton, Dickinson and Co, 5 milliliter, REF 30163, Franklin Lakes, NJ 07417). Непосредственно при извлечении из стакана приблизительно 2 мл прозрачного образца жидкости быстро высвобождают через новый неиспользованный шприцевой фильтр (диаметр 25 мм, 0,45 мкм, Gelnan GHP Acrodisc GF®, Pall Corporation, East Hills, NY 11548) в новый меченый стеклянный флакон для образца. Продолжительность каждого заполнения шприца, замены фильтра и высвобождения во флакон для образца является заметно меньшей, примерно, чем 5 с, и этот интервал, соответственно, начинается и заканчивается в пределах примерно 3 с в любую сторону от каждого целевого времени отбора образца. В пределах примерно пяти минут каждого заполнения флаконы для образцов закрываются и поддерживаются приблизительно при постоянной температуре до осуществления следующего химического анализа. После того как конечный образец отбирается в момент времени 30 мин после времени t=0, все шестнадцать образцов анализируются на количество растворенной TPA с использованием способа ВЭЖХ-DAD (матричный диодный детектор), в целом, как описывается в другом месте в настоящем описании. Однако при настоящем исследовании как калибровочные стандарты, так и регистрируемые результаты приводятся как миллиграммы растворенной TPA на грамм растворителя THF (далее "м.д. в THF"). Например, если все 250 мг твердых продуктов представляют собой очень чистую TPA и если все это количество полностью растворяется в 250 г растворителя THF до отбора конкретного образца, правильно измеренная концентрация составляла бы примерно 1000 м.д. в THF.
Когда CTA в соответствии с настоящим изобретением подвергается исследованию временной зависимости растворения, описанному выше, предпочтительно, чтобы образец, отобранный через одну минуту после t=0, растворялся до концентрации, по меньшей мере, примерно 500 м.д. в THF, более предпочтительно, по меньшей мере, 600 м.д. в THF. Для образца, отобранного через две минуты после t=0, предпочтительно, чтобы CTA в соответствии с настоящим изобретением растворялась до концентрации, по меньшей мере, примерно 700 м.д. в THF, более предпочтительно, по меньшей мере, 750 м.д. в THF. Для образца, отобранного через четыре минуты после t=0, предпочтительно, чтобы CTA в соответствии с настоящим изобретением растворялась до концентрации, по меньшей мере, примерно 840 м.д. в THF, более предпочтительно, по меньшей мере, 880 м.д. в THF.
Авторы обнаружили, что относительно простая модель отрицательного экспоненциального роста является пригодной для описания временной зависимости полного набора данных от полного исследования временной зависимости растворения, несмотря на сложность отбора образцов частиц и процесса растворения. Форма уравнения, далее "модели временной зависимости растворения", является следующей:
S=A+B·(1-exp(-C·t)), где
t = время в единицах минут;
S = растворимость, в единицах м.д. в THF, в момент времени t;
exp = экспоненциальная функция на основе натурального логарифма 2;
A, B = константы регрессии в единицах м.д. в THF, где A относится по большей части к быстрому растворению частиц меньших размеров при очень коротких временах и где сумма A+B относится по большей части к общему количеству растворения вблизи конца указанного периода исследования; и
C = временная постоянная регрессии в единицах обратных минут.
Константы регрессии подбирают, сводя к минимуму сумму среднеквадратичной ошибки между точками реальных данных и соответствующими значениями модели, этот способ обычно называется подгонка "наименьших квадратов". Предпочтительный пакет программного обеспечения для осуществления этой регрессии данных представляет собой JMP Release 5.1.2 (SAS Institute Inc., JMP Software, SAS Campus Drive, Cary, NC 27513).
Когда CTA в соответствии с настоящим изобретением исследуют с помощью исследования временной зависимости растворения и подгоняют к модели временной зависимости растворения, описанной выше, предпочтительно, чтобы CTA имела временную константу C, большую, примерно, чем 0,5 мин-1, более предпочтительно большую, примерно, чем 0,6 мин-1, а наиболее предпочтительно большую, чем 0,7 мин-1.
Фиг.33A и 33B иллюстрируют обычную частицу CTA, полученную с помощью обычного высокотемпературного способа окисления в проточном реакторе смешения (CSTR). Фиг.33A показывает обычную частицу CTA при 500-кратном увеличении, в то время как фиг.33B увеличивает и показывает частицу CTA при 2000-кратном увеличении. Визуальное сравнение частиц CTA по настоящему изобретению, иллюстрируемых на фиг.324A и 32B, и обычной частицы CTA, иллюстрируемой на фиг.33A и 33B, показывает, что обычная частица CTA имеет более высокую плотность, более низкую площадь поверхности, более низкую пористость и больший размер частиц, чем частица CTA по настоящему изобретению. Фактически, обычная CTA, представленная на фиг.33A и 33B, имеет средний размер частиц примерно 205 мкм и площадь поверхности по БЭТ примерно 0,57 м2/г.
Фиг.34 иллюстрирует обычный способ получения очищенной терефталевой кислоты (PTA). В обычном способе получения PTA пара-ксилол частично окисляется в высокотемпературном реакторе окисления 700 с механическим перемешиванием. Суспензия, содержащая CTA, извлекается из реактора 700, а затем очищается в системе очистки 702. Продукт PTA из системы очистки 702 вводится в систему разделения 706 для выделения и сушки частиц PTA. Система очистки 702 соответствует большей части затрат, связанных с получением частиц PTA посредством обычных способов. Система очистки 702, как правило, содержит систему добавления/замены воды 708, систему растворения 710, систему гидрирования 712 и три отдельных емкости кристаллизации 704a, b, c. В системе добавления/замены воды 708 существенная часть маточной жидкости заменяется водой. После добавления воды суспензия вода/CTA вводится в систему растворения 710, где смесь вода/CTA нагревается до тех пор, пока частицы CTA не растворятся полностью в воде. После растворения CTA раствор CTA в воде подвергается гидрированию в системе гидрирования 712. Гидрированный выходящий поток из системы гидрирования 712 затем подвергают воздействию трех ступеней кристаллизации в емкостях кристаллизации 704a, b, c, с последующим выделением PTA в системе разделения 706.
Фиг.35 иллюстрирует улучшенный способ получения PTA с использованием барботажной колонны реакторного типа окисления 800, конфигурированной в соответствии с вариантом осуществления настоящего изобретения. Исходная суспензия, содержащая твердые частицы CTA и жидкую маточную жидкость, извлекается из реактора 800. Как правило, исходная суспензия может содержать в пределах примерно от 10 примерно до 50 мас.% твердых частиц CTA, при этом жидкий остаток представляет собой маточную жидкость. Твердые частицы CTA, присутствующие в исходной суспензии, как правило, содержат, по меньшей мере, примерно 400 м.д. мас. 4-карбоксибензальдегида (4-CBA), чаще, по меньшей мере, примерно 800 м.д. мас. 4-CBA, чаще всего в пределах от 1000 до 15000 м.д. мас. 4-CBA. Исходная суспензия, извлеченная из реактора 800, вводится в систему очистки 802 для уменьшения концентрации 4-CBA и других примесей, присутствующих в CTA. Более чистая/очищенная суспензия получается из системы очистки 802 и подвергается разделению и сушке в системе разделения 804, для получения при этом твердых частиц более чистой терефталевой кислоты, содержащих менее, примерно, чем 400 м.д. мас. 4-CBA, более предпочтительно менее, примерно, чем 250 м.д. мас. 4-CBA, а наиболее предпочтительно в пределах от 10 до 200 м.д. мас. 4-CBA.
Система очистки 802 системы получения PTA, иллюстрируемой на фиг.35, обеспечивает ряд преимуществ по сравнению с системой очистки 802 известной из литературы системы, иллюстрируемой на фиг.34. Предпочтительно система очистки 802, в целом, содержит систему замены маточной жидкости 806, дигестер 808 и отдельный кристаллизатор 810. В системе замены маточной жидкости 806, по меньшей мере, примерно 50 мас.% маточной жидкости, присутствующей в исходной суспензии, заменяется свежим замещающим растворителем, с получением при этом суспензии с замененным растворителем, содержащим частицы CTA и замещающий растворитель. Суспензия с замененным растворителем, покидающая систему замены маточной жидкости 806, вводится в дигестер (или реактор вторичного окисления) 808. В дигестере 808 реакция вторичного окисления осуществляется при несколько более высоких температурах, чем используются в начальной/первичной реакции окисления, осуществляемой в барботажной колонне реакторного типа 800. Как обсуждалось выше, высокая площадь поверхности, малый размер частиц и низкая плотность частиц CTA, получаемых в реакторе 800, делает определенные примеси, захваченные в частицах CTA, доступными для окисления в дигестере 808 без необходимости в полном растворении частиц CTA в дигестере 808. Таким образом, температура в дигестере 808 может быть ниже, чем во многих сходных способах, известных из литературы. Вторичное окисление, осуществляемое в дигестере 808, предпочтительно уменьшает концентрацию 4-CBA в CTA, по меньшей мере, на 200 м.д. мас., более предпочтительно, по меньшей мере, примерно на 400 м.д. мас., наиболее предпочтительно в пределах от 600 до 6000 м.д. мас. Предпочтительно температура вторичного окисления в дигестере 808, по меньшей мере, примерно на 10°C выше, чем температура первичного окисления в барботажной колонне реакторного типа 800, более предпочтительно выше в пределах примерно от 20 примерно до 80°C, чем температура первичного окисления в реакторе 800, а наиболее предпочтительно выше в пределах от 30 до 50°C, чем температура первичного окисления в реакторе 800. Температура вторичного окисления предпочтительно находится в пределах примерно от 160 примерно до 240°C, более предпочтительно в пределах примерно от 180 примерно до 220°C, а наиболее предпочтительно в пределах от 190 до 210°C. Очищенный продукт из дигестера 808 требует только одной стадии кристаллизации в кристаллизаторе 810 перед разделением в системе разделения 804. Пригодные для использования технологии вторичного окисления/дигерирования обсуждаются более подробно в публикации заявки на патент США №2005/0065373, полное описание которой явным образом включается сюда в качестве ссылки.
Терефталевая кислота (например, PTA), полученная посредством системы, иллюстрируемой на фиг.35, предпочтительно состоит из частиц PTA, имеющих средний размер частиц, по меньшей мере, примерно 40 мкм, более предпочтительно в пределах примерно от 50 примерно до 2000 мкм, а наиболее предпочтительно в пределах от 60 до 200 мкм. Частицы PTA предпочтительно имеют среднюю площадь поверхности по БЭТ, меньшую, примерно, чем 0,25 м2/г, более предпочтительно находящуюся в пределах примерно от 0,005 примерно до 0,2 м2/г, а наиболее предпочтительно в пределах от 0,01 до 0,18 м2/г. PTA, полученная посредством системы, иллюстрируемой на фиг.35, является пригодной для использования в качестве исходных материалов при получении PET. Как правило, PET получают этерификацией терефталевой кислоты с помощью этиленгликоля с последующей поликонденсацией. Предпочтительно терефталевая кислота, полученная посредством одного из вариантов осуществления настоящего изобретения, используется в качестве исходных материалов для способа получения PET в трубчатом реакторе, описанном в заявке на патент США, серийный №10/013318, зарегистрированной 7 декабря 2001 года, полное описание которой включается сюда в качестве ссылки.
Частицы CTA с предпочтительной морфологией, описанной здесь, являются особенно пригодными для использования в описанном выше способе окислительного дигерирования для понижения содержания 4-CBA. В дополнение к этому, эти предпочтительные частицы CTA обеспечивают преимущества в широком диапазоне других, следующих далее процессов, включающих в себя растворение и/или химическую реакцию частиц. Эти дополнительные следующие далее процессы включают в себя, но не ограничиваясь этим, реакцию, по меньшей мере, с одним гидроксилсодержащим соединением, с образованием сложноэфирных соединений, в частности реакцию CTA с метанолом, с образованием диметилтерефталата и сложных эфиров примесей; реакцию, по меньшей мере, с одним диолом, с образованием сложноэфирных мономерных и/или полимерных соединений, в частности реакцию CTA с этиленгликолем, с образованием полиэтилентерефталата (PET); и полное или частичное растворение в растворителях, включая, но не ограничиваясь этим, воду, уксусную кислоту и N-метил-2-пирролидон, которые могут включать в себя дополнительную переработку, включая, но не ограничиваясь этим, повторное осаждение более чистой терефталевой кислоты и/или селективное химическое восстановление карбонильных групп, иных, чем группы карбоновых кислот. В явном виде является включенным, по существу, растворение CTA в растворителе, содержащем воду, объединенное с частичным гидрированием, которое уменьшает количество альдегидов, в частности 4-CBA, флуоренонов, фенонов и/или антрахинонов.
Авторы также предполагают, что частицы CTA, имеющие предпочтительные свойства, описанные здесь, могут быть получены из частиц CTA, не совпадающих по предпочтительным свойствам с теми, которые описаны здесь (несовпадающих частиц CTA), с помощью средств, включающих в себя, но не ограничиваясь этим, механическое перемешивание несовпадающих частиц CTA и полное или частичное растворение несовпадающих частиц CTA, с последующим полным или частичным повторным осаждением.
В соответствии с одним из вариантов осуществления настоящего изобретения предусматривается способ частичного окисления окисляемого ароматического соединения до одного или нескольких типов ароматической карбоновой кислоты, где чистота части растворителя исходных материалов (т.е. "исходных материалов растворителя") и чистота части окисляемого соединения исходных материалов (т.е. "исходных материалов окисляемого соединения") контролируются в определенных пределах, описанных ниже. Вместе с другими вариантами осуществления настоящего изобретения это делает возможным контроль чистоты жидкой фазы и твердой фазы, если она присутствует, и объединенной фазы суспензии (т.е. твердые продукты плюс жидкость) реакционной среды в определенных предпочтительных пределах, указанных ниже.
Относительно исходных материалов растворителя, является известным окисление окисляемого ароматического соединения (соединений), с получением ароматической карбоновой кислоты, где исходные материалы растворителя, вводимые в реакционную среду, представляют собой смесь уксусной кислоты аналитической чистоты и воды, как часто используется при работе в лабораторном масштабе и пилотном масштабе. Подобным же образом, является известным осуществление окисления окисляемого ароматического соединения до ароматической карбоновой кислоты, где растворитель, покидающий реакционную среду, отделяется от полученной ароматической карбоновой кислоты, а затем рециклируется назад в реакционную среду как исходный материал растворителя, прежде всего, по причинам затрат на производство. Это рециклирование растворителя вызывает накопление со временем определенных примесей исходных материалов и побочных продуктов способа в рециклируемом растворителе. В данной области известны различные средства, чтобы помочь в очистке рециклированного растворителя перед повторным введением в реакционную среду. Как правило, более высокий уровень очистки рециклированнного растворителя приводит к значительно более высоким затратам на производство, чем более низкий уровень очистки с помощью подобных средств. Один из вариантов осуществления настоящего изобретения относится к пониманию и определению предпочтительных пределов большого количества примесей в исходных материалах растворителя, многие из которых до сих пор рассматриваются по большей части как вредные, для нахождения оптимального баланса между общими затратами на производство и общей чистотой продукта.
"Рециклируемые исходные материалы растворителя" определяются здесь как исходные материалы растворителя, содержащие, по меньшей мере, примерно 5 мас.% массы, которая ранее прошла через реакционную среду, содержащую одно или несколько окисляемых ароматических соединений, подвергающихся частичному окислению. По причинам запасов растворителя и времени протекания в производственной установке предпочтительно, чтобы порции рециклированного растворителя проходили через реакционную среду, по меньшей мере, раз в рабочий день, более предпочтительно, по меньшей мере, раз в день в течение, по меньшей мере, семи последовательных рабочих дней, а наиболее предпочтительно, по меньшей мере, раз в день в течение, по меньшей мере, 30 последовательных рабочих дней. По экономическим причинам предпочтительно, чтобы, по меньшей мере, примерно 20 мас.% исходных материалов растворителя в реакционной среде по настоящему изобретению представляли собой рециклированный растворитель, более предпочтительно, по меньшей мере, примерно 40 мас.%, еще более предпочтительно, по меньшей мере, примерно 80 мас.%, а наиболее предпочтительно, по меньшей мере, 90 мас.%.
Авторы обнаружили, что по причинам реакционной активности и по соображениям металлических примесей, остающихся в продукте окисления, концентрации выбранных металлов переменной валентности в рециклированных исходных материалах растворителя предпочтительно находятся в пределах, указанных непосредственно ниже. Концентрация железа в рециклированном растворителе предпочтительно примерно ниже 150 м.д. мас., более предпочтительно примерно ниже 40 м.д. мас., а наиболее предпочтительно находится в пределах между 0 и 8 м.д. мас. Концентрация никеля в рециклированном растворителе предпочтительно примерно ниже 150 м.д. мас., более предпочтительно примерно ниже 40 м.д. мас., а наиболее предпочтительно находится в пределах между 0 и 8 м.д. мас. Концентрация хрома в рециклированном растворителе предпочтительно примерно ниже 150 м.д. мас., более предпочтительно примерно ниже 40 м.д. мас., а наиболее предпочтительно находится в пределах между 0 и 8 м.д. мас. Концентрация молибдена в рециклированном растворителе предпочтительно примерно ниже 75 м.д. мас., более предпочтительно примерно ниже 20 м.д. мас., а наиболее предпочтительно находится в пределах между 0 и 4 м.д. мас. Концентрация титана в рециклированном растворителе предпочтительно примерно ниже 75 м.д. мас., более предпочтительно примерно ниже 20 м.д. мас., а наиболее предпочтительно находится в пределах между 0 и 4 м.д. мас. Концентрация меди в рециклированном растворителе предпочтительно примерно ниже 20 м.д. мас., более предпочтительно примерно ниже 4 м.д. мас., а наиболее предпочтительно находится в пределах между 0 и 1 м.д. мас. Другие металлические примеси, как правило, также присутствуют в рециклированном растворителе, как правило, изменяясь на более низких уровнях по отношению к одному или нескольким из перечисленных выше металлов. Контроль перечисленных выше металлов в предпочтительных пределах будет поддерживать другие металлические примеси на соответствующих уровнях.
Эти металлы могут возникать как примеси в любом из поступающих в способ исходных материалов (например, в поступающем окисляемом соединении, растворителе, окислителе и соединениях катализаторов). Альтернативно, металлы могут возникать как продукты коррозии из любой из установок, вступающих в контакт с реакционной средой и/или вступающих в контакт с рециклированным растворителем. Средства для контроля металлов в описанных пределах концентраций включают в себя соответствующее качество и мониторинг чистоты различных исходных материалов и правильное использование материалов конструкции, включая, но не ограничиваясь этим, множество коммерческих сортов титана и нержавеющих сталей, включая сорта, известные как дуплексные нержавеющие стали и высокомолибденовые нержавеющие стали.
Авторы также обнаружили предпочтительные пределы для выбранных ароматических соединений в рециклируемом растворителе. Они включают в себя как осажденные, так и растворенные ароматические соединения в рециклированном растворителе.
Неожиданно, даже осажденный продукт (например, TPA) от частичного окисления пара-ксилола представляет собой примесь, содержанием которой нужно управлять в рециклируемом растворителе. Поскольку неожиданно возникают предпочтительные пределы для уровней твердых продуктов в реакционной среде, любой осажденный продукт в исходных материалах растворителя непосредственно вычитается из количества окисляемого соединения, которое может вводиться вместе с ним. Кроме того, введение осажденных твердых продуктов TPA в рециклируемом растворителе при повышенных уровнях, как обнаружено, отрицательно влияет на характер частиц, образующихся в осаждающей среде окисления, приводя к нежелательному характеру следующих далее операций (например, фильтрования продукта, промывки растворителем, окислительного дигерирования сырого продукта, растворения сырого продукта для дополнительной переработки, и так далее). Другая нежелательная характеристика осажденных твердых продуктов в исходных материалах рециклируемого растворителя заключается в том, что они часто содержат очень высокие уровни осажденных примесей, по сравнению с концентрациями примесей в объеме твердых продуктов в суспензиях TPA, из которых получают большую часть рециклированного растворителя. Возможно, повышенные уровни примесей, наблюдаемые в твердых продуктах, суспендированных в рециклированном фильтрате, могут быть связаны со временами зародышеобразования для осаждения определенных примесей из рециклируемого растворителя и/или с охлаждением рециклируемого растворителя, либо намеренным, либо вызванным потерями в окружающей среде. Например, концентрации сильно окрашенного и нежелательного 2,6-дикарбоксифлуоренона наблюдаются при гораздо более высоких уровнях в твердых продуктах, присутствующих в рециклированном растворителе, при 80°C, чем наблюдается в твердых продуктах TPA, выделенных из рециклируемого растворителя при 160°C. Подобным же образом, концентрации изофталевой кислоты наблюдаются при гораздо более высоких уровнях в твердых продуктах, присутствующих в рециклируемом растворителе, по сравнению с уровнями, наблюдаемыми в твердых продуктах TPA из реакционной среды. Как именно конкретные осажденные примеси, захваченные в рециклируемом растворителе, ведут себя, когда повторно вводятся в реакционную среду, видимо, варьируется. Это зависит, вероятно, от относительной растворимости примеси в жидкой фазе реакционной среды, вероятно, от того, как осажденная примесь распределяется по слоям внутри осажденных твердых продуктов, и вероятно, от локальной скорости осаждения TPA, когда твердый продукт сначала повторно вводится в реакционную среду. Таким образом, авторы обнаружили, что полезно контролировать уровень определенных примесей в рециклируемом растворителе, как описано ниже, вне зависимости от того, присутствуют ли эти примеси в рециклируемом растворителе в растворенной форме или представляют собой захваченные в нем частицы.
Количество осажденных твердых продуктов, присутствующих в рециклированном фильтрате, определяется гравиметрически следующим образом. Представительный образец извлекается из растворителя, подаваемого в реакционную среду, в то время как растворитель протекает в проходе по направлению к реакционной среде. Пригодный для использования размер образца составляет примерно 100 г, заключенные в стеклянный контейнер, имеющий примерно 250 мл внутреннего объема. Перед высвобождением до атмосферного давления, но при этом непрерывно протекая по направлению к контейнеру с образцом, рециклируемый фильтрат охлаждается до менее чем 100°C; это охлаждение осуществляется для ограничения испарения растворителя в течение короткого интервала до герметизации в замкнутом стеклянном контейнере. После захвата образца при атмосферном давлении стеклянный контейнер немедленно герметизируется. Затем образцу дают возможность для охлаждения примерно до 20°C, в то время как он окружен воздухом примерно при 20°C и без принудительной конвекции. После достижения примерно 20°C образец выдерживается в этом состоянии в течение, по меньшей мере, примерно 2 часов. Затем герметизированный контейнер энергично встряхивают до тех пор, пока не будет получено визуально однородное распределение твердых продуктов. Непосредственно после этого в контейнер для образца добавляют магнитный перемешиватель и вращают при достаточной скорости для поддержания эффективно однородного распределения твердых продуктов. 10-миллилитровую аликвоту перемешанной жидкости с суспендированными твердыми продуктами извлекают посредством пипетки и взвешивают. Затем объем жидкой фазы из этой аликвоты отделяют вакуумным фильтрованием, по-прежнему, примерно при 20°C, и эффективно, без потерь твердых продуктов. Влажные твердые продукты, отфильтрованные из этой аликвоты, затем сушат, эффективно, без сублимации твердых продуктов, и эти высушенные твердые продукты взвешивают. Отношение массы высушенных твердых продуктов к массе исходной аликвоты суспензии представляет собой долю твердых продуктов, как правило, выражаемую в процентах и упоминаемую здесь как содержание осажденных твердых продуктов в рециклированном фильтрате при 20°C.
Авторы обнаружили, что ароматические соединения, растворенные в жидкой фазе реакционной среды и содержащие ароматические карбоновые кислоты, в которых нет неароматических углеводородных групп (например, изофталевая кислота, бензойная кислота, фталевая кислота, 2,5,4'-трикарбоксибифенил), являются неожиданно вредными компонентами. Хотя эти соединения имеют гораздо более низкую химическую активность в рассматриваемой реакционной среде, по сравнению с окисляемыми соединениями, имеющими неароматические углеводородные группы, авторы обнаружили, что эти соединения, тем не менее, подвергаются многочисленным вредным превращениям. Таким образом, является преимущественным контроль содержания этих соединений в предпочтительных пределах, в жидкой фазе реакционной среды. Это приводит к появлению предпочтительных пределов выбранных соединений в исходных материалах рециклируемого растворителя, а также предпочтительных пределов выбранных предшественников в исходных материалах окисляемого ароматического соединения.
Например, при жидкофазном частичном окислении пара-ксилола до терефталевой кислоты (TPA) авторы обнаружили, что сильноокрашенная и нежелательная примесь 2,7-дикарбоксифлуоренон (2,7-DCF) по существу не детектируется в реакционной среде и в пробе продукта, когда мета-замещенные ароматические соединения находятся в реакционной среде на очень низких уровнях. Авторы обнаружили, что когда примесь изофталевой кислоты присутствует в исходных материалах растворителя при увеличивающихся уровнях, образование 2,7-DCF возрастает почти прямопропорционально. Авторы также обнаружили, что когда примесь мета-ксилола присутствует в исходных материалах пара-ксилола, образование 2,7-DCF опять возрастает почти прямопропорционально. Кроме того, даже если исходные материалы растворителя и исходные материалы окисляемого соединения свободны от мета-замещенных ароматических соединений, авторы обнаружили, что некоторое количество изофталевой кислоты образуется во время типичного частичного окисления очень чистого пара-ксилола, в частности, когда бензойная кислота присутствует в жидкой фазе реакционной среды. Это самопроизвольно образуемая изофталевая кислота может, благодаря ее более высокой растворимости, чем у TPA, в растворителе, содержащем уксусную кислоту и воду, аккумулироваться со временем в промышленных установках, использующих рециклируемый растворитель. Таким образом, количество изофталевой кислоты в исходных материалах растворителя, количество мета-ксилола в исходных материалах окисляемого ароматического соединения и скорость самопроизвольного образования изофталевой кислоты в реакционной среде, все они соответствующим образом рассматриваются в балансе друг с другом и в балансе с любыми реакциями, которые потребляют изофталевую кислоту. Изофталевая кислота, как обнаружено, подвергается дополнительным реакциям с ее потреблением, помимо образования 2,7-DCF, как описано ниже. В дополнение к этому, авторы обнаружили, что имеются другие предметы для рассмотрения, когда устанавливаются соответствующие пределы для мета-замещенных ароматических частиц при частичном окислении пара-ксилола до TPA. Другие сильно окрашенные и нежелательные примеси, такие как 2,6-дикарбоксифлуоренон (2,6-DCF), видимо, сильно связаны с растворенными, пара-замещенными ароматическими частицами, которые всегда присутствуют вместе с исходными материалами пара-ксилола для жидкофазного окисления. Таким образом, подавление 2,7-DCF лучше всего рассматривать в перспективе с уровнем других окрашенных примесей, которые производятся.
Например, при жидкофазном частичном окислении пара-ксилола до TPA авторы обнаружили, что образование тримеллитовой кислоты увеличивается, когда в реакционной среде увеличиваются уровни изофталевой кислоты и фталевой кислоты. Тримеллитовая кислота представляет собой трехфункциональную карбоновую кислоту, приводящую к разветвлению полимерных цепей во время получения PET из TPA. Во многих применениях PET уровни ветвления должны контролироваться при низких уровнях, и, следовательно, тримеллитовая кислота должна контролироваться при низких уровнях в очищенной TPA. Кроме того, что они приводят к получению тримеллитовой кислоты, присутствие мета-замещенных и орто-замещенных частиц в реакционной среде также приводит к появлению других трикарбоновых кислот (например, 1,3,5-трикарбоксибензола). Кроме того, повышенное присутствие трикарбоновых кислот в реакционной среде увеличивает величину образования тетракарбоновой кислоты (например, 1,2,4,5-тетракарбоксибензола). Контроль общего получения всех ароматических карбоновых кислот, имеющих более двух групп карбоновых кислот, является одним из факторов при установлении предпочтительных уровней мета-замещенных и орто-замещенных частиц в исходных материалах рециклируемого растворителя, в исходных материалах окисляемого соединения и в реакционной среде в соответствии с настоящим изобретением.
Например, при жидкофазном частичном окислении пара-ксилола до TPA авторы обнаружили, что повышенные уровни в жидкой фазе реакционной среды нескольких растворенных ароматических карбоновых кислот, у которых нет неароматических углеводородных групп, непосредственно приводят к увеличению образования моноокиси углерода и двуокиси углерода. Это увеличенное образование окислов углерода представляет собой потери выхода как по окислителю, так и по окисляемому соединению, последнее связано с тем, что многие получаемые совместно ароматические карбоновые кислоты, которые, с одной стороны, могут рассматриваться как примеси, с другой стороны, также имеют промышленную ценность. Таким образом, соответствующее удаление относительно растворимых карбоновых кислот, у которых нет неароматических углеводородных групп, из рециклируемого растворителя имеет экономическую ценность при предотвращении потерь выхода окисляемого ароматического соединения и окислителя, в дополнение к подавлению образования очень нежелательных примесей, таких как различные флуореноны и тримеллитовая кислота.
Например, при жидкофазном частичном окислении пара-ксилола до TPA авторы обнаружили, что образование 2,5,4'-трикарбоксибифенила, видимо, является неизбежным. 2,5,4'-трикарбоксибифенил представляет собой ароматическую трикарбоновую кислоту, сформированную посредством соединения двух ароматических колец, вероятно, посредством связывания растворенных пара-замещенных ароматических частиц с арильным радикалом, вероятно, с арильным радикалом, сформированным посредством декарбоксилирования или декарбонилирования пара-замещенных ароматических частиц. К счастью, 2,5,4'-трикарбоксибифенил обычно образуется при более низких уровнях, чем тримеллитовая кислота, и не приводит обычно к значительному увеличению проблем с ветвлением полимерных молекул во время получения PET. Однако авторы обнаружили, что повышенные уровни 2,5,4'-трикарбоксибифенила в реакционной среде, содержащей окисление алкилароматических соединений в соответствии с предпочтительными вариантами осуществления настоящего изобретения, приводят к повышению уровней сильно окрашенного и нежелательного 2,6-DCF. Повышенный уровень 2,6-DCF возможно возникает из 2,5,4'-трикарбоксибифенила посредством замыкания кольца с потерей молекулы воды, хотя точный механизм реакции не известен с определенностью. Если 2,5,4'-трикарбоксибифенил, который является более растворимым в растворителе, содержащем уксусную кислоту и воду, чем TPA, получает возможность для накопления до слишком высокого уровня в рециклируемом растворителе, скорости преобразования в 2,6-DCF могут стать неприемлемо высокими.
Например, при жидкофазном частичном окислении пара-ксилола до TPA авторы обнаружили, что ароматические карбоновые кислоты, у которых нет неароматических углеводородных групп (например, изофталевая кислота), как правило, приводят к мягкому подавлению химической активности реакционной среды, когда они присутствуют в жидкой фазе в достаточной концентрации.
Например, при жидкофазном частичном окислении пара-ксилола до TPA авторы обнаружили, что осаждение очень часто является неидеальным (т.е. неравновесным) по отношению к относительным концентрациям различных химических частиц в твердой фазе и в жидкой фазе. Вероятно, это связано с тем, что скорость осаждения является очень быстрой при пространственно-временных скоростях реакций, предпочтительных здесь, приводя к неидеальному совместному осаждению примесей или даже окклюзии. Таким образом, если желательным является ограничение концентрации определенных примесей (например, тримеллитовой кислоты и 2,6-DCF) в сырой TPA, из-за конфигурации работы следующих далее установок, предпочтителен контроль их концентрации в исходных материалах растворителя, а также скорости их образования в реакционной среде.
Например, авторы обнаружили, что бензофеноновые соединения (например, 4,4'-дикарбоксибензофенон и 2,5,4'-трикарбоксибензофенон), полученные во время частичного окисления пара-ксилола, имеют нежелательные воздействия в реакционной среде для получения PET, даже несмотря на то что бензофеноновые соединения не являются сильно окрашенными в TPA сами по себе, как флуореноны и антрахиноны. Соответственно, является желательным ограничение присутствия бензофенонов и выбор предшественников в рециклируемом растворителе и в исходных материалах окисляемого соединения. Кроме того, авторы обнаружили, что присутствие повышенных уровней бензойной кислоты, либо поступившей в рециклированном растворителе, либо образовавшейся в реакционной среде, приводит к повышению скоростей образования 4,4'-дикарбоксибензофенона.
В качестве обзора, авторы обнаружили и достаточно определили количественно неожиданный ряд реакций для ароматических соединений, у которых нет неароматических углеводородных групп, которые присутствуют при жидкофазном частичном окислении пара-ксилола до TPA. Рассматривая как раз отдельный случай бензойной кислоты, авторы обнаружили, что повышенные уровни бензойной кислоты в реакционной среде в некоторых вариантах осуществления настоящего изобретения приводят к сильному увеличению образования сильно окрашенной и нежелательной 9-флуоренон-2-карбоновой кислоты, к сильному повышению уровней 4,4'-дикарбоксибифенила, к повышению уровней 4,4'-дикарбоксибензофенона, к мягкому подавлению химической активности предполагаемого окисления пара-ксилола и к повышению уровней окислов углерода, и к соответствующим потерям выхода. Авторы обнаружили, что повышение уровней бензойной кислоты в реакционной среде также приводит к увеличению образования изофталевой кислоты и фталевой кислоты, уровни которых желательно контролировать в низких пределах, в соответствии со сходными аспектами настоящего изобретения. Количество и важность реакций, включающих в себя бензойную кислоту, являются, вероятно, еще более неожиданными, поскольку некоторые авторы недавних изобретений предполагают использование бензойной кислоты вместо уксусной кислоты в качестве первичного компонента растворителя (см., например, патент США №6562997). В дополнение к этому, авторы настоящего изобретения наблюдают, что бензойная кислота самопроизвольно образуется во время окисления пара-ксилола при скоростях, которые являются заведомо важными по отношению к ее образованию из примесей, таких как толуол и этилбензол, обычно находящихся в исходных материалах окисляемого соединения, содержащих пара-ксилол промышленной чистоты.
С другой стороны, авторы обнаружили небольшую пользу от дополнительного регулирования состава рециклируемого растворителя относительно присутствия окисляемого ароматического соединения и относительно присутствия ароматических промежуточных продуктов реакции, которые как содержат неароматические углеводородные группы, так и являются также относительно растворимыми в рециклируемом растворителе. Как правило, эти соединения либо вводятся, либо образуются в реакционной среде с долями, существенно большими, чем их присутствие в рециклируемом растворителе; и скорость расходования этих соединений в реакционной среде является достаточно большой, при сохранении одной или нескольких неароматических углеводородных групп, для ограничения соответствующим образом их накопления в рециклируемом растворителе. Например, во время частичного окисления пара-ксилола в многофазной реакционной среде пара-ксилол испаряется до ограниченной степени вместе с большими количествами растворителя. Когда этот испаренный растворитель покидает реактор как часть входящего газа и конденсируется для извлечения в качестве рециклируемого растворителя, существенная часть испаренного пара-ксилола также конденсируется в нем. Не является необходимым ограничение концентрации этого пара-ксилола в рециклируемом растворителе. Например, если растворитель отделяется от твердых продуктов при выходе суспензии из реакционной среды окисления пара-ксилола, этот извлеченный растворитель будет содержать концентрацию растворенной пара-толуиловой кислоты, сходную с той, которая присутствует в точке удаления из реакционной среды. Хотя может быть важным ограничение постоянной концентрации пара-толуиловой кислоты в жидкой фазе реакционной среды, см. ниже, не является необходимым отдельное регулирование пара-толуиловой кислоты в этой части рециклируемого растворителя из-за ее относительно хорошей растворимости и ее низкой массовой скорости потока по отношению к образованию пара-толуиловой кислоты в реакционной среде. Подобным же образом, авторы обнаружили мало причин для ограничения в рециклируемом растворителе концентраций ароматических соединений с метильными заместителями (например, толуиловых кислот), ароматических альдегидов (например, терефтальдегида), ароматических соединений с гидроксиметильными заместителями (например, 4-гидроксиметилбензойной кислоты) и бромированных ароматических соединений, сохраняющих, по меньшей мере, одну неароматическую углеводородную группу (например, альфа-бром-пара-толуиловой кислоты), ниже тех, которые изначально находятся в жидкой фазе, уходящей из реакционной среды, возникающей при частичном окислении ксилола в соответствии с предпочтительными вариантами осуществления настоящего изобретения. Неожиданно, авторы также обнаружили, что также нет необходимости в регулировании в рециклируемом растворителе концентрации выбранных фенолов, образующихся собственно во время частичного окисления ксилола, поскольку эти соединения образуются и разрушаются в реакционной среде при гораздо больших долях, чем их присутствие в рециклируемом растворителе. Например, авторы обнаружили, что 4-гидроксибензойная кислота имеет относительно малое воздействие на химическую активность в предпочтительных вариантах осуществления настоящего изобретения, когда совместно вводится при долях более 2 г 4-гидроксибензойной кислоты на 1 кг пара-ксилола, гораздо больших, чем естественное присутствие в рециклируемом растворителе, несмотря на это, другие сообщают о ней как о важном яде в подобной реакционной среде (см., например, W. Partenheimer, Catalysis Today 23 (1995) p.81).
Таким образом, имеются многочисленные реакции и многочисленные соображения при установлении предпочтительных пределов различных ароматических примесей в исходных материалах растворителя, как теперь описывается. Эти данные формулируются с точки зрения агрегированных средневзвешенных составов всех потоков растворителей, которые вводятся в реакционную среду в течение заданного периода времени, предпочтительно одного дня, более предпочтительно одного часа, а наиболее предпочтительно одной минуты. Например, если исходные материалы одного растворителя протекают по существу непрерывно с содержанием 40 м.д. мас. изофталевой кислоты при скорости потока 7 кг/мин, исходные материалы второго растворителя протекают по существу непрерывно с составом 2000 м.д. мас. изофталевой кислоты при скорости потока 10 кг/мин и нет других потоков исходных материалов растворителей, поступающих в реакционную среду, тогда агрегированный средневзвешенный состав исходных материалов растворителя вычисляется как (40·7+2000·10)/(7+10)=1,193 м.д. мас. изофталевой кислоты. Заметим, что масса любых исходных материалов окисляемого соединения или любых исходных материалов окислителя, которые, вероятно, смешиваются с исходными материалами растворителя до поступления в реакционную среду, не рассматривается при вычислении агрегированной средневзвешенной композиции исходных материалов растворителя.
Таблица 1, ниже, перечисляет предпочтительные значения для определенных компонентов в исходных материалах растворителя, вводимых в реакционную среду. Компоненты исходных материалов растворителей, перечисленные в таблице 1, представляют собой следующие соединения: 4-карбоксибензальдегид (4-CBA), 4,4'-дикарбоксистильбен (4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-DCF), 2,7-дикарбоксифлуоренон (2,7-DCF), 3,5-дикарбоксифлуоренон (3,5-DCF), 9-флуоренон-2-карбоновую кислоту (9F-2CA), 9-флуоренон-4-карбоновую кислоту (9F-4CA), флуореноны в целом, включая другие флуореноны, не перечисленные отдельно (флуореноны в целом), 4,4'-дикарбоксибифенил (4,4'-DCB), 2,5,4'-трикарбоксибифенил (2,5,4'-TCB), фталевую кислоту (PA), изофталевую кислоту (IPA), бензойную кислоту (BA), тримеллитовую кислоту (TMA), 2,6-дикарбоксибензокумарин (2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-DCBP), 2,5,4'-трикарбоксибензофенон (2,5,4'-TCBP), терефталевую кислоту (TPA), осажденные твердые продукты при 20°C и ароматические карбоновые кислоты в целом, у которых нет неароматических углеводородных групп. Таблица 1, ниже, приводит предпочтительные количества этих примесей в CTA, полученной в соответствии с вариантом осуществления настоящего изобретения.
Множество других ароматических примесей, как правило, также присутствуют в рециклируемом растворителе, как правило, изменяясь при еще более низких уровнях и/или в пропорции к одному или нескольким описанным ароматическим соединениям. Способы контроля описанных ароматических соединений в предпочтительных пределах будут, как правило, поддерживать другие ароматические примеси при соответствующих уровнях.
Когда в реакционной среде используется бром, большое количество ионных и органических форм брома, как известно, существуют в динамическом равновесии. Эти различные формы брома имеют различные характеристики стабильности, когда они покидают реакционную среду и проходят через различные операции установки, относящиеся к рециклируемому растворителю. Например, альфа-бром-пара-толуиловая кислота может существовать как таковая при некоторых условиях или может быстро гидролизоваться при других условиях с образованием 4-гидроксиметилбензойной кислоты и бромистого водорода. В настоящем изобретении предпочтительно, чтобы, по меньшей мере, примерно 40 мас.%, более предпочтительным, чтобы, по меньшей мере, примерно 60 мас.%, а наиболее предпочтительным, чтобы, по меньшей мере, примерно 80 мас.% от общей массы брома, присутствующего в агрегированных исходных материалах растворителя, поступающих в реакционную среду, находились в одной или нескольких из следующих химических форм: ионный бром, альфа-бром-пара-толуиловая кислота и бромуксусная кислота.
Хотя важность и ценность контроля агрегированной средневзвешенной чистоты исходных материалов растворителя в описанных желательных пределах по настоящему изобретению не были до этого обнаружены и/или описаны, соответствующие средства для контроля чистоты исходных материалов растворителя могут быть собраны из различных способов, уже известных в данной области. Во-первых, любой растворитель, испаряемый из реакционной среды, как правило, имеет соответствующую чистоту, при условии, что жидкость или твердые продукты из реакционной среды не захватываются вместе с испаряемым растворителем. Введение капель флегмы растворителя в пространстве для отделения уходящего газа над реакционной средой, как описано здесь, соответствующим образом ограничивает такой захват; и рециклируемый растворитель соответствующей чистоты по отношению к ароматическому соединению может конденсироваться из такого уходящего газа. Во-вторых, более сложная и дорогостоящая очистка исходных материалов рециклируемого растворителя, как правило, относится к растворителю, отобранному из реакционной среды в жидкой форме, и к растворителю, который впоследствии вступает в контакт с жидкой и/или твердой фазами реакционной среды, извлекаемыми из реакционной емкости (например, к рециклируемому растворителю, полученному из фильтра, в котором твердые продукты концентрируются и/или промываются, к рециклируемому растворителю, получаемому из центрифуги, в которой твердые продукты концентрируются и/или промываются, к рециклируемому растворителю, отбираемому из операции кристаллизации, и так далее). Однако в данной области также известны средства для осуществления необходимой очистки этих потоков рециклируемого растворителя с использованием одного или нескольких из предыдущих описаний. Относительно контроля осажденных твердых продуктов в рециклируемом растворителе, чтобы они находились в указанных пределах, соответствующие средства контроля включают в себя, но не ограничиваясь этим, гравиметрическую седиментацию, механическое фильтрование с использованием фильтровальной ткани на роторных ленточных фильтрах и роторных барабанных фильтрах, механическое фильтрование с использованием стационарной среды фильтра в емкостях высокого давления, гидроциклонах и центрифугах. Относительно контроля растворенных ароматических частиц в рециклируемом растворителе, чтобы они находились в указанных пределах, средства контроля включают в себя, но не ограничиваясь этим, те, которые описаны в патенте США №4939297 и в публикации заявки на патент США №2005-0038288, включенных сюда в качестве ссылок. Однако ни одно из этих предыдущих изобретений не обнаруживает и не описывает предпочтительных уровней чистоты в агрегированных исходных материалах растворителя, как описывается здесь. Вместо этого эти предыдущие изобретения всего лишь предусматривают средства для очистки выбранных и частичных потоков рециклируемого растворителя без рассмотрения оптимальных значений композиции агрегированных средневзвешенных исходных материалов растворителя, вводимых в реакционную среду по настоящему изобретению.
Обращаясь теперь к чистоте исходных материалов окисляемого соединения, известно, что определенные уровни изофталевой кислоты, фталевой кислоты и бензойной кислоты присутствуют и являются допустимыми при низких уровнях в очищенной TPA, используемой для получения полимера. Кроме того, известно, что эти частицы являются относительно более растворимыми во многих растворителях и могут преимущественно удаляться из очищенной TPA посредством способов кристаллизации. Однако из одного из вариантов осуществления настоящего изобретения, описанных здесь, теперь известно, что контроль уровня нескольких относительно растворимых ароматических частиц, включая явным образом изофталевую кислоту, фталевую кислоту и бензойную кислоту, в жидкой фазе реакционной среды является неожиданно важным для контроля уровня полициклических и окрашенных ароматических соединений, образующихся в реакционной среде, для контроля соединений с более чем 2 функциональными группами карбоновых кислот на молекулу, для контроля реакционной активности в реакционной среде частичного окисления и для контроля потерь выхода окислителя и ароматического соединения.
В данной области известно, что изофталевая кислота, фталевая кислота и бензойная кислота образуются в реакционной среде следующим образом. Примесь исходных материалов мета-ксилола окисляется с хорошим преобразованием и выходом до IPA. Примесь исходных материалов орто-ксилола окисляется с хорошим преобразованием и выходом до фталевой кислоты. Примеси исходных материалов этилбензола и толуола окисляются с хорошим преобразованием и выходом до бензойной кислоты. Однако авторы наблюдают, что значительные количества изофталевой кислоты, фталевой кислоты и бензойной кислоты также образуются в реакционной среде, содержащей пара-ксилол, с помощью средств, иных, чем окисление мета-ксилола, орто-ксилола, этилбензола и толуола. Эти другие действительные химические маршруты, возможно, включают в себя декарбонилирование, декарбоксилирование, перегруппировку переходных состояний и дополнение метильных и карбонильных радикалов до ароматических колец.
При определении предпочтительных пределов примесей в исходных материалах окисляемого соединения важными являются многие факторы. Любая примесь в исходных материалах, вероятно, будет вызывать непосредственные потери выхода и затраты на очистку продукта, если требования к чистоте окисленного продукта являются достаточно высокими (например, в реакционной среде для частичного окисления пара-ксилола толуол и этилбензол, как правило, находящиеся в пара-ксилоле промышленной чистоты, приводят к образованию бензойной кислоты, и эта бензойная кислота по большей части удаляется из большинства промышленных сортов TPA). Когда продукт частичного окисления примеси исходных материалов участвует в дополнительных реакциях, факторы, иные, чем просто потери выхода и удаление, становятся важными, когда рассматривают, какими должны быть затраты на очистку исходных материалов (например, в реакционной среде для частичного окисления пара-ксилола этилбензол приводит к образованию бензойной кислоты, а бензойная кислота впоследствии приводит к образованию сильно окрашенной 9-флуоренон-2-карбоновой кислоты, изофталевой кислоты, фталевой кислоты и к увеличению количества окислов углерода, среди прочего). Когда реакционная среда самопроизвольно образует дополнительные количества примеси с помощью химических механизмов, не связанных непосредственно с примесями исходных материалов, анализ становится еще более сложным (например, в реакционной среде для частичного окисления пара-ксилола бензойная кислота также самопроизвольно образуется из самого пара-ксилола). В дополнение к этому, последующая переработка сырого продукта окисления может повлиять на соображения относительно предпочтительной чистоты исходных материалов. Например, стоимость удаления до соответствующих уровней непосредственной примеси (бензойной кислоты) и последующих примесей (изофталевой кислоты, фталевой кислоты, 9-флуоренон-2-карбоновой кислоты, и тому подобное) может быть одной и той же, может отличаться от остальных и может отличаться от требований удаления по большей части не связанной с ней примеси (например, продукта неполного окисления 4-CBA при окислении пара-ксилола до TPA).
Описанные далее пределы чистоты исходных материалов для пара-ксилола являются предпочтительными, когда пара-ксилол вводится вместе с растворителем и окислителем в реакционную среду для частичного окисления с получением TPA. Эти пределы являются более предпочтительными в способе получения TPA, имеющем стадии последующего окисления для удаления из реакционной среды примесей, иных, чем окислитель и растворитель (например, металлических катализаторов). Эти пределы являются еще более предпочтительными в способах получения TPA, которые удаляют дополнительную 4-CBA из CTA (например, посредством преобразования CTA до диметилтерефталата, плюс сложные эфиры примесей, и последующего отделения сложного метилового эфира 4-CBA посредством дистилляции, посредством способов окислительного дигерирования, для превращения 4-CBA до TPA, посредством способов гидрирования для превращения 4-CBA в пара-толуиловую кислоту, которая затем отделяется посредством способов фракционной кристаллизации). Эти пределы являются наиболее предпочтительными в способах получения TPA, которые удаляют дополнительную 4-CBA из CTA посредством способов окислительного дигерирования для преобразования 4-CBA в TPA.
Используя новое знание предпочтительных пределов рециклирования ароматических соединений и относительных количеств ароматических соединений, образующихся непосредственно от окисления примесей исходных материалов, по сравнению с другими действительными химическими маршрутами, обнаружены улучшенные пределы для примесей, для загрязнения пара-ксилола, представляющего собой исходные материалы, вводимые в способы частичного окисления для получения TPA. Таблица 2, ниже, приводит предпочтительные значения для количества мета-ксилола, орто-ксилола и этилбензола+толуола в исходных материалах пара-ксилола.
Специалисты в данной области заметят теперь, что указанные выше примеси в загрязненном пара-ксилоле могут оказывать свое наибольшее воздействие на реакционную среду после того, как продукты их частичного окисления аккумулируются в рециклируемом растворителе. Например, введение верхнего количества из наиболее предпочтительного диапазона мета-ксилола, 400 м.д. мас., непосредственно приведет к получению примерно 200 м.д. мас. изофталевой кислоты в жидкой фазе реакционной среды, если работать примерно с 33 мас.% твердых продуктов в реакционной среде. Это сравнимо с поступлением от верхнего количества из наиболее предпочтительного диапазона для изофталевой кислоты в рециклируемом растворителе 400 м.д. мас., что, после того как дается возможность для охлаждения реакционной среды с помощью испарения типичного растворителя, приводит к получению примерно 1200 м.д. мас. изофталевой кислоты в жидкой фазе реакционной среды. Таким образом, со временем происходит накопление продуктов частичного окисления в рециклируемом растворителе, которое представляет собой наибольшее вероятное воздействие примесей мета-ксилола, орто-ксилола, этилбензола и толуола в исходных материалах загрязненного пара-ксилола. Соответственно, предпочтительно, чтобы указанные выше диапазоны примесей в исходных материалах загрязненного пара-ксилола поддерживались в течение, по меньшей мере, половины дня, в каждый день работы любой реакционный среды частичного окисления в конкретной промышленной установке, более предпочтительно в течение, по меньшей мере, трех четвертей каждого дня в течение, по меньшей мере, семи последовательных дней работы, а наиболее предпочтительно, когда средневзвешенные параметры составов исходных материалов загрязненного пара-ксилола находятся в предпочтительных пределах в течение, по меньшей мере, 30 последовательных дней работы.
Средства для получения загрязненного пара-ксилола предпочтительной чистоты уже известны в данной области и включают в себя, но не ограничиваясь этим, дистилляцию, способы фракционной кристаллизации при температурах ниже температуры окружающей среды и способы с молекулярными ситами, с использованием адсорбции, селективной по размеру пор. Однако предпочтительные пределы чистоты, указанные здесь, являются, в их самой высшей форме, более требовательными и дорогостоящими, чем то, что характерно для осуществления промышленными поставщиками пара-ксилола; и еще в низшей форме, предпочтительные пределы устраняют излишне дорогостоящую очистку пара-ксилола, предназначенного для введения в реакционную среду частичного окисления, посредством обнаружения и описания того, где объединенные воздействия самопроизвольного образования примесей из самого пара-ксилола и реакций с потреблением примесей в реакционной среде становятся более важными, чем скорости введения примесей в загрязненном пара-ксилоле.
Когда поток исходных материалов, содержащий ксилол, содержит выбранные примеси, такие как этилбензол и/или толуол, окисление этих примесей может образовывать бензойную кислоту. Как здесь используется, термин "образуемая примесями бензойная кислота" должен обозначать бензойную кислоту, полученную из любого источника, иного, чем ксилол, во время окисления ксилола.
Как описывается здесь, часть бензойной кислоты, образующейся во время окисления ксилола, получается из самого ксилола. Это получение бензойной кислоты из ксилола является явным образом дополнительным по отношению к любой части получения бензойной кислоты, которая может представлять собой бензойную кислоту, образуемую примесями. Не желая ограничиваться теорией, предполагается, что бензойная кислота получается из ксилола в реакционной среде, когда различные промежуточные продукты окисления ксилола спонтанно декарбонилируются (теряют моноокись углерода) или декарбоксилируются (теряют двуокись углерода), производя при этом арильные радикалы. Эти арильные радикалы могут затем извлекать атом водорода из любого из множества доступных источников в реакционной среде и образовывать самопроизвольно образуемую бензойную кислоту. В качестве химического механизма термин "самопроизвольно образуемая бензойная кислота", как здесь используется, должен обозначать бензойную кислоту, полученную из ксилола во время окисления ксилола.
Как также описывается здесь, когда пара-ксилол окисляется с получением терефталевой кислоты (TPA), получение самопроизвольно образуемой бензойной кислоты вызывает потери выхода по пара-ксилолу и потери выхода по окислителю. В дополнение к этому, присутствие самопроизвольно образуемой бензойной кислоты в жидкой фазе реакционной среды коррелирует с усилением множества нежелательных побочных реакций, в явном виде включающим в себя образование сильно окрашенных соединений, называемых моно-карбоксифлуоренонами. Самопроизвольно образуемая бензойная кислота также вносит вклад в нежелательное накопление бензойной кислоты в рециклированном фильтрате, который дополнительно поднимает концентрацию бензойной кислоты в жидкой фазе реакционной среды. Таким образом, образование самопроизвольно образуемой бензойной кислоты желательно свести к минимуму, но это также рассматривается соответствующим образом одновременно с образуемой примесями бензойной кислотой, с факторами, влияющими на потребление бензойной кислоты, с факторами, относящимися к другим вопросам селективности реакции, и с общей экономикой.
Авторы обнаружили, что самопроизвольное образование бензойной кислоты может контролироваться на низких уровнях посредством соответствующего выбора, например, температуры, распределения ксилола и доступности кислорода в реакционной среде во время окисления. Не желая ограничиваться теорией, более низкие температуры и улучшение доступности кислорода, видимо, подавляют скорости декарбонилирования и/или декарбоксилирования, таким образом, устраняя аспект потери выхода самопроизвольно образуемой бензойной кислоты. Достаточная доступность кислорода, видимо, направляет арильные радикалы по направлению к другим, более вредным продуктам, в частности гидроксибензойным кислотам. Распределение ксилола в реакционной среде может также влиять на баланс между преобразованием арильного радикала в бензойную кислоту или в гидроксибензойные кислоты. Какими бы не были химические механизмы, авторы обнаружили условия реакции, которые, хотя и достаточно мягкие для уменьшения получения бензойной кислоты, являются достаточно жесткими для окисления высокой доли получаемой гидроксибензойной кислоты до моноокиси углерода и/или двуокиси углерода, которые легко удаляются из продукта окисления.
В предпочтительном варианте осуществления настоящего изобретения реактор окисления конфигурируется и работает таким образом, что образование самопроизвольно образуемой бензойной кислоты сводится к минимуму и окисление гидроксибензойных кислот до моноокиси углерода и/или двуокиси углерода доводится до максимума. Когда реактор окисления используется для окисления пара-ксилола до терефталевой кислоты, предпочтительно, чтобы пара-ксилол восполнялся, по меньшей мере, примерно на 50 мас.% от общего ксилола в потоке исходных материалов, вводимом в реактор. Более предпочтительно пара-ксилол восполняется, по меньшей мере, примерно на 75 мас.% от общего ксилола в потоке исходных материалов. Еще более предпочтительно пара-ксилол восполняется, по меньшей мере, на 95 мас.% от общего ксилола в потоке исходных материалов. Наиболее предпочтительно пара-ксилол восполняется по существу на все количество ксилола в потоке исходных материалов.
Когда реактор используется для окисления пара-ксилола до терефталевой кислоты, предпочтительно, чтобы скорость образования терефталевой кислоты доводилась до максимума, в то время как скорость образования самопроизвольно образуемой бензойной кислоты доводится до минимума. Предпочтительно отношение скорости образования (по массе) терефталевой кислоты к скорости образования (по массе) самопроизвольно образуемой бензойной кислоты составляет, по меньшей мере, примерно 500:1, более предпочтительно, по меньшей мере, примерно 1000:1, а наиболее предпочтительно, по меньшей мере, 1500:1. Как будет видно ниже, скорость образования самопроизвольно образуемой бензойной кислоты предпочтительно измеряется, когда концентрация бензойной кислоты в жидкой фазе реакционной среды ниже 2000 м.д. мас., более предпочтительно ниже 1000 м.д. мас., а наиболее предпочтительно ниже 500 м.д. мас., поскольку эти низкие концентрации подавляют до соответствующих низких скоростей реакции, которые преобразуют бензойную кислоту в другие соединения.
При объединении самопроизвольно образуемой бензойной кислоты и образуемой примесями бензойной кислоты отношение скорости образования (по массе) терефталевой кислоты к скорости образования (по массе) бензойной кислоты в целом предпочтительно составляет, по меньшей мере, примерно 400:1, более предпочтительно, по меньшей мере, примерно 700:1, а наиболее предпочтительно, по меньшей мере, 1100:1. Как будет видно ниже, общая скорость образования самопроизвольно образуемой бензойной кислоты плюс бензойной кислоты, образуемой примесями, предпочтительно измеряется, когда концентрация бензойной кислоты в жидкой фазе реакционной среды ниже 2000 м.д. мас., более предпочтительно ниже 1000 м.д. мас., а наиболее предпочтительно ниже 500 м.д. мас., поскольку эти низкие концентрации подавляют до соответствующих низких скоростей реакции, которые преобразуют бензойную кислоту в другие соединения.
Как здесь описывается, повышенная концентрация бензойной кислоты в жидкой фазе реакционной среды приводит к увеличению образования множества других ароматических соединений, несколько из которых являются вредными примесями в TPA; и, как описано здесь, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к увеличению образования газообразных окислов углерода, образование которых представляет собой потери выхода по окислителю и по ароматическим соединениям, и/или растворителю. Кроме того, теперь описывается, что авторы обнаружили, что значительная часть этого увеличения образования других ароматических соединений и окислов углерода происходит из-за реакций, которые преобразуют некоторые из молекул бензойной кислоты сами по себе, в противоположность бензойной кислоте, катализирующей другие реакции, без расходования ее самой. Соответственно, "образование бензойной кислоты в целом" определяется здесь как усредненная по времени масса всей бензойной кислоты, покидающей реакционную среду, минус усредненная по времени масса всей бензойной кислоты, поступающей в реакционную среду за тот же период времени. Это образование бензойной кислоты в целом часто является положительным благодаря скоростям образования бензойной кислоты, образуемой примесями, и самопроизвольно образуемой бензойной кислоты. Однако авторы обнаружили, что скорость преобразования бензойной кислоты в окислы углерода и в несколько других соединений, видимо, увеличивается приблизительно линейно, когда концентрация бензойной кислоты увеличивается в жидкой фазе реакционной среды, при измерении, когда другие условия реакции, включая температуру, доступность кислорода, STR и реакционную активность, поддерживаются, соответственно, постоянными. Таким образом, когда концентрация бензойной кислоты в жидкой фазе реакционной среды является достаточно большой, вероятно, из-за повышенной концентрации бензойной кислоты в рециклируемом растворителе, тогда преобразование молекул бензойной кислоты в другие соединения, включая окислы углерода, может стать равным или большим, чем химическое образование новых молекул бензойной кислоты. В этом случае образование бензойной кислоты в целом может балансировать вблизи нуля или даже быть отрицательным. Авторы обнаружили, что когда образование бензойной кислоты в целом является положительным, тогда отношение скорости образования (по массе) терефталевой кислоты в реакционной среде по сравнению со скоростью образования бензойной кислоты в целом в реакционной среде предпочтительно примерно выше 700:1, более предпочтительно примерно выше 1100:1, а наиболее предпочтительно выше 4000:1. Авторы обнаружили, что когда образование бензойной кислоты в целом является отрицательным, отношение скорости образования (по массе) терефталевой кислоты в реакционной среде по сравнению со скоростью образования бензойной кислоты в целом в реакционной среде предпочтительно примерно выше 200:(-1), более предпочтительно примерно выше 1000:(-1), а наиболее предпочтительно выше 5000:(-1).
Авторы также обнаружили предпочтительные пределы для состава суспензии (жидкость + твердые продукты), извлекаемой из реакционной среды, и для твердой части суспензии CTA. Составы предпочтительной суспензии и предпочтительной CTA являются неожиданно превосходными и пригодными для использования. Например, очищенная TPA, полученная из этой предпочтительной CTA посредством окислительного дигерирования, имеет достаточно низкий уровень общих примесей и окрашенных примесей, так что очищенная TPA является пригодной, без гидрирования дополнительной 4-CBA и/или окрашенных примесей, для широкого диапазона применений в волокнах из PET и в применениях для упаковки из PET. Например, предпочтительный состав суспензии обеспечивает жидкую фазу реакционной среды, в которой концентрация важных примесей является относительно низкой, и это значительно уменьшает образование других, еще более нежелательных примесей, как здесь описывается. В дополнение к этому, предпочтительный состав суспензии значительно помогает дальнейшей переработке жидкости из суспензии, для получения соответствующего чистого рециклированного растворителя, в соответствии с другими вариантами осуществления настоящего изобретения.
CTA, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, содержит меньше примесей, выбранных типов, чем CTA, полученная с помощью обычных способов и устройств, в частности, тех, которые используют рециклируемый растворитель. Примеси, которые могут присутствовать в CTA, включают в себя следующие: 4-карбоксибензальдегид (4-CBA), 4,4'-дикарбоксистильбен (4,4'-DCS), 2,6-дикарбоксиантрахинон (2,6-DCA), 2,6-дикарбоксифлуоренон (2,6-DCF), 2,7-дикарбоксифлуоренон (2,7-DCF), 3,5-дикарбоксифлуоренон (3,5-DCF), 9-флуоренон-2-карбоновую кислоту (9F-2CA), 9-флуоренон-4-карбоновую кислоту (9F-4CA), 4,4'-дикарбоксибифенил (4,4'-DCB), 2,5,4'-трикарбоксибифенил (2,5,4'-TCB), фталевую кислоту (PA), изофталевую кислоту (IPA), бензойную кислоту (BA), тримеллитовую кислоту (TMA), пара-толуиловую кислоту (PTAC), 2,6-дикарбоксибензокумарин (2,6-DCBC), 4,4'-дикарбоксибензил (4,4'-DCBZ), 4,4'-дикарбоксибензофенон (4,4'-DCBP), 2,5,4'-трикарбоксибензофенон (2,5,4'-TCBP). Таблица 3, ниже, приводит предпочтительные количества этих примесей в CTA, полученной в соответствии с одним из вариантов осуществления настоящего изобретения.
В дополнение к этому, предпочтительно, чтобы CTA, полученная по одному из вариантов осуществления настоящего изобретения, имела пониженное содержание цвета по отношению к CTA, полученной посредством обычных способов и устройств, а именно тех, которые используют рециклируемый растворитель. Таким образом, предпочтительно, чтобы CTA, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, имела процент пропускания на 340 нанометрах (нм), по меньшей мере, примерно 25%, более предпочтительно, по меньшей мере, примерно 50%, а наиболее предпочтительно, по меньшей мере, 60%. Кроме того, предпочтительно, чтобы CTA, полученная в соответствии с одним из вариантов осуществления настоящего изобретения, имела процент пропускания на 400 нанометрах (нм), по меньшей мере, примерно 88%, более предпочтительно, по меньшей мере, примерно 90%, а наиболее предпочтительно, по меньшей мере, 92%.
Исследование для процента пропускания предусматривает измерение концентрации окрашенных, поглощающих свет примесей, присутствующих в TPA или CTA. Как здесь используется, исследование относится к измерениям, осуществляемым на порции раствора, полученного посредством растворения 2,00 г сухой твердой TPA или CTA в 20,0 мл диметилсульфоксида (DMSO), аналитического или еще лучшего качества. Часть этого раствора затем помещают в проточную полумикроскопическую ячейку Hellma, PN 176.700, которая изготовлена из кварца и имеет путь света 1,0 см и объем 0,39 мл (Hellma USA, 80 Skyline Drive, Plainview, NY 11803). Спектрофотометр с диодной матрицей Agilent 8453 используют для измерения пропускания различных длин волн света через эту заполненную проточную ячейку (Agilent Technologies, 395 Page Mill Road, Palo Alto, CA 94303). После соответствующей корректировки на поглощение от фона, включая, но не ограничиваясь этим, используемую ячейку и растворитель, результаты по проценту пропускания, характеризующие долю падающего света, которая проходит через раствор, регистрируются непосредственно машиной. Значения процента пропускания на длинах волн света 340 нм и 400 нм являются особенно пригодными для использования с целью выявления различий чистой TPA от многих примесей, как правило, находящихся в ней.
Предпочтительные пределы различных ароматических примесей в фазе суспензии (твердые продукты + жидкость) реакционной среды приводятся ниже в таблице 4.
(м.д. мас.)
Эти предпочтительные составы для суспензии воплощают предпочтительный состав жидкой фазы реакционной среды, в то же время полезным образом устраняя экспериментальные трудности, относящиеся к осаждению дополнительных компонентов жидкой фазы из реакционной среды в компоненты твердой фазы во время отбора образца из реакционной среды, разделения жидкости и твердых продуктов и к изменению аналитических условий.
Множество других ароматических примесей, как правило, также присутствуют в фазе суспензии реакционной среды и в CTA из реакционной среды, как правило, изменяясь на более низких уровнях и/или в пропорции к одному или нескольким описанных ароматических соединений. Контроль описанных ароматических соединений в предпочтительных пределах будет поддерживать другие ароматические примеси на соответствующих уровнях. Эти преимущественные составы для фазы суспензии в реакционной среде и для твердой CTA, отобранной непосредственно из суспензии, делают возможной работу с вариантами осуществления настоящего изобретения, описанными здесь, для частичного окисления пара-ксилола до TPA.
Измерение концентрации компонентов с низкими уровнями в растворителе, рециклируемом растворителе, CTA, суспензии из реакционной среды и PTA осуществляются с использованием способов жидкостной хроматографии. Теперь описываются два взаимозаменяемых варианта осуществления.
Способ, упоминаемый здесь как ВЭЖХ-DAD, включает в себя жидкостную хроматографию высокого давления (ВЭЖХ), объединенную с матричным диодным детектором (DAD), для обеспечения разделения и количественного определения различных молекулярных частиц внутри данного образца. Прибор, используемый при этом измерении, представляет собой модель 1100 HPLC, снабженную DAD, поставляемую Agilent Technologies (Palo Alto, CA), хотя и другие пригодные для использования приборы являются коммерчески доступными также и от других поставщиков. Как известно в данной области, как время элюирования, так и отклик детектора калибруются с использованием известных соединений, присутствующих в известных количествах, эти соединения и их количества находятся в соответствии с теми, которые встречаются в реальных неизвестных образцах.
Способ, упоминаемый здесь как ВЭЖХ-МС, включает в себя жидкостную хроматографию высокого давления (ВЭЖХ), соединенную с масс-спектрометрией (МС), для обеспечения разделения, идентификации и количественного определения различных молекулярных частиц внутри данного образца. Приборы, используемые при этом измерении, представляют собой Alliance HPLC и ZQ MS, поставляемые Waters Corp. (Milford, MA), хотя и другие пригодные для использования приборы являются коммерчески доступными также и от других поставщиков. Как известно в данной области, как время элюирования, так и отклик детектора калибруются с использованием известных соединений, присутствующих в известных количествах, эти соединения и их количества находятся в соответствии с теми, которые встречаются в реальных неизвестных образцах.
Другой вариант осуществления настоящего изобретения относится к частичному окислению ароматического окисляемого соединения при соответствующем балансе подавления вредных ароматических примесей, с одной стороны, по сравнению с образованием двуокиси углерода и моноокиси углерода, совместно, окислов углерода (COx), с другой. Эти окислы углерода, как правило, покидают реакционную емкость в уходящем газе, и они соответствуют деструктивным потерям растворителя и окисляемого соединения, включая, в конечном счете, предпочтительные окисленные производные (например, уксусную кислоту, пара-ксилол и TPA). Авторы обнаружили нижние границы для образования окислов углерода, ниже которых, видимо, высокое образование вредных ароматических примесей, как описано ниже, и низкий общий уровень преобразования неизбежно являются слишком плохими, чтобы иметь экономическое применение. Авторы также обнаружили верхние пределы границы окислов углерода, выше которых образование окислов углерода продолжает увеличиваться при малой дополнительной ценности, обеспечиваемой уменьшением образования вредных ароматических примесей.
Авторы обнаружили, что снижение жидкофазных концентраций исходных материалов ароматического окисляемого соединения и ароматических промежуточных частиц в реакционной среде приводит к более низким скоростям образования для вредных примесей во время частичного окисления ароматического окисляемого соединения. Эти вредные примеси включают в себя связанные ароматические кольца и/или ароматические молекулы, содержащие большее, чем желательное, количество групп карбоновых кислот (например, при окислении пара-ксилола вредные примеси включают в себя 2,6-дикарбоксиантрахинон, 2,6-дикарбоксифлуоренон, тримеллитовую кислоту, 2,5,4'-трикарбоксибифенил и 2,5,4'-бензофенон). Ароматические промежуточные частицы включают в себя ароматические соединения, выпадающие из исходных материалов окисляемого ароматического соединения и все еще удерживающие неароматические углеводородные группы (например, при окислении пара-ксилола ароматические промежуточные частицы включают в себя пара-толуальдегид, терефтальдегид, пара-толуиловую кислоту, 4-CBA, 4-гидроксиметилбензойную кислоту и альфа-бром-пара-толуиловую кислоту). Исходные материалы окисляемого ароматического соединения и ароматические промежуточные частицы, содержащие неароматические углеводородные группы, когда они присутствуют в жидкой фазе реакционной среды, видимо, приводят к появлению вредных примесей способом, подобным тому, который уже описывался здесь, для растворенных ароматических частиц, у которых нет неароматических углеводородных групп (например, изофталевой кислоты).
Настраиваясь против этой необходимости в более высокой реакционной активности для подавления образования вредных ароматических примесей во время частичного окисления окисляемого ароматического соединения, авторы обнаружили, что нежелательный сопутствующий результат представляет собой увеличение образования окислов углерода. Важно заметить, что эти окислы углерода представляют собой потери выхода окисляемого соединения и окислителя, а не только растворителя. На самом деле, значительная, а иногда главная доля окислов углерода происходит от окисляемого соединения и его производных, а не из растворителя; и часто окисляемое соединение стоит больше на единицу углерода, чем растворитель. Кроме того, важно заметить, что желаемый продукт карбоновой кислоты (например, TPA) также подвергается сверхокислению до окислов углерода, когда присутствует в жидкой фазе реакционной среды.
Важно также заметить, что настоящее изобретение относится к реакциям в жидкой фазе реакционной среды и к концентрациям реагентов в ней. Это контрастирует с некоторыми предыдущими изобретениями, которые относятся непосредственно к образованию ароматического соединения в осажденной твердой форме, удерживающего неароматические углеводородные группы. Конкретно, для частичного окисления пара-ксилола до TPA, определенные предыдущие изобретения относятся к количеству 4-CBA, осажденному в твердой фазе CTA. Однако авторы настоящего изобретения обнаружили разброс, больший, чем два к одному, для отношения 4-CBA в твердой фазе к 4-CBA в жидкой фазе, используя такие же условия температуры, давления, катализа, состава растворителя и пространственно-временной скорости реакции пара-ксилола, в зависимости от того, осуществляется ли частичное окисление в хорошо перемешиваемом автоклаве или в реакционной среде со ступенчатым изменением концентрации кислорода и пара-ксилола в соответствии с настоящим изобретением. Кроме того, авторы настоящего изобретения обнаружили, что отношение 4-CBA в твердой фазе к 4-CBA в жидкой фазе может также изменяться более чем на два к одному либо в хорошо перемешиваемой реакционной среде, либо в реакционной среде со ступенчатым изменением концентрации, в зависимости от пространственно-временной скорости реакции пара-ксилола, при сходных в остальном условиях температуры, давления, катализа и состава растворителя. В дополнение к этому, 4-CBA в твердой фазе CTA, видимо, не вносит вклад в образование вредных примесей, а 4-CBA в твердой фазе может извлекаться и окисляться до TPA просто и с высоким выходом (например, посредством окислительного дигерирования суспензии CTA, как здесь описывается); в то время как удаление вредных примесей является гораздо более сложным и дорогостоящим, чем удаление твердой фазы 4-CBA, и образование окислов углерода представляет собой постоянную потерю выхода. Таким образом, важно отличать, что данный аспект настоящего изобретения относится к жидкофазным составам в реакционной среде.
Происходят ли они из растворителя или окисляемого соединения, но авторы обнаружили, что при превращениях промышленного значения образование окислов углерода сильно связано с уровнем общей реакционной активности, несмотря на широкий разброс для каждого конкретного сочетания температуры, металлов, галогенов, температуры, кислотности реакционной среды, как измеряется посредством pH, концентрации воды, используемых для получения уровня общей реакционной активности. Авторы находят полезным для частичного окисления ксилола оценивать уровень общей реакционной активности с использованием жидкофазной концентрации толуиловых кислот на серединной высоте реакционной среды, в нижней части реакционной среды и в верхней части реакционной среды.
Таким образом, возникает важное одновременное уравновешивание для сведения к минимуму образования вредных примесей посредством увеличения реакционной активности и еще для сведения к минимуму образования окислов углерода посредством снижения реакционной активности. Т.е. если общее образование окислов углерода подавляется до слишком низкого значения, тогда образуются избыточные уровни вредных примесей, и наоборот.
Кроме того, авторы настоящего изобретения обнаружили, что растворимость и относительная реакционная способность желаемой карбоновой кислоты (например, TPA) и присутствие других растворенных ароматических частиц, у которых нет неароматических углеводородных групп, вносят очень важный фактор в это уравновешивание окислов углерода, по сравнению с вредными примесями. Желаемый продукт карбоновой кислоты обычно растворяют в жидкой фазе реакционной среды, даже если он присутствует также и в твердой форме. Например, при температурах в предпочтительных пределах TPA является растворимой в реакционной среде, содержащей уксусную кислоту и воду, при уровнях, находящихся в пределах примерно от одной тысячи м.д. мас. до более чем 1 мас.%, при настоящем растворимость увеличивается, когда увеличивается температура. Несмотря на то что имеются различия в скоростях реакции по отношению к образованию различных вредных примесей из исходных материалов окисляемого ароматического соединения (например, пара-ксилола), из ароматических промежуточных продуктов реакции (например, пара-толуиловой кислоты), из желаемого продукта ароматической карбоновой кислоты (например, TPA) и из ароматических частиц, у которых нет неароматических углеводородных групп (например, изофталевой кислоты), присутствие и реакционная способность последних двух групп устанавливает область выгодности уменьшения, по отношению к дальнейшему подавлению первых двух групп исходных материалов окисляемого ароматического соединения и ароматических промежуточных продуктов реакции. Например, при частичном окислении пара-ксилола до TPA, если в жидкой фазе реакционной среды при данных условиях растворенная TPA доходит до 7000 м.д. мас., растворенная бензойная кислота доходит до 8000 м.д. мас., растворенная изофталевая кислота доходит до 6000 м.д. мас. и растворенная фталевая кислота доходит до 2000 м.д. мас., тогда значение для дальнейшего снижения концентрации вредных соединений в целом начинает понижаться, когда реакционная активность увеличивается, с подавлением жидкофазной концентрации пара-толуиловой кислоты и 4-CBA ниже подобных уровней. Т.е. присутствие и концентрация в жидкой фазе реакционной среды ароматических частиц, у которых нет неароматических углеводородных групп, очень мало изменяется с увеличением реакционной активности, и их присутствие служит для расширения вверх области выгодности уменьшения, для снижения концентрации промежуточных продуктов реакции для подавления образования вредных примесей.
Таким образом, один из вариантов осуществления настоящего изобретения предусматривает предпочтительные диапазоны окислов углерода, ограниченные снизу низкой реакционной активностью и избыточным образованием вредных примесей, а сверху избыточными потерями углерода, но при уровнях более низких, чем те, которые ранее обнаруживались и описывались как пригодные промышленно. Соответственно, образование окислов углерода предпочтительно контролируется следующим образом. Отношение образующихся молей окислов углерода в целом, по отношению к молям вводимого окисляемого ароматического соединения, предпочтительно больше, примерно, чем 0,02:1, более предпочтительно больше, примерно, чем 0,04:1, еще более предпочтительно больше, примерно, чем 0,05:1, а наиболее предпочтительно больше, чем 0,06:1. В то же самое время отношение образующихся молей окислов углерода в целом к молям вводимого окисляемого ароматического соединения предпочтительно меньше, примерно, чем 0,24:1, более предпочтительно меньше, примерно, чем 0,22:1, еще более предпочтительно меньше, примерно, чем 0,19:1, а наиболее предпочтительно меньше, чем 0,15:1. Отношение молей образующейся двуокиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно больше, примерно, чем 0,01:1, более предпочтительно больше, примерно, чем 0,03:1, еще более предпочтительно больше, примерно, чем 0,04:1, а наиболее предпочтительно больше, чем 0,05:1. В то же самое время отношение молей образующейся двуокиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно меньше, примерно, чем 0,21:1, более предпочтительно меньше, примерно, чем 0,19:1, еще более предпочтительно меньше, примерно, чем 0,16:1, а наиболее предпочтительно меньше, чем 0,11. Отношение молей образующейся моноокиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно больше, примерно, чем 0,005:1, более предпочтительно больше, примерно, чем 0,010:1, еще более предпочтительно больше, примерно, чем 0,015:1, а наиболее предпочтительно больше, чем 0,020:1. В то же самое время отношение молей образующейся моноокиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно составляет меньше, примерно, чем 0,09:1, более предпочтительно меньше, примерно, чем 0,07:1, еще более предпочтительно меньше, примерно, чем 0,05:1, а наиболее предпочтительно меньше, чем 0,04:1.
Содержание двуокиси углерода в сухом уходящем газе из реактора окисления предпочтительно больше, примерно, чем 0,10% молярных, более предпочтительно больше, примерно, чем 0,20% молярного, еще более предпочтительно больше, примерно, чем 0,25% молярного, а наиболее предпочтительно больше, чем 0,30% молярного. В то же самое время содержание двуокиси углерода в сухом уходящем газе из реактора окисления предпочтительно меньше, примерно, чем 1,5% молярных, более предпочтительно меньше, примерно, чем 1,2% молярного, еще более предпочтительно меньше, примерно, чем 0,9% молярного, а наиболее предпочтительно меньше, чем 0,8% молярного. Содержание моноокиси углерода в сухом уходящем газе из реактора окисления предпочтительно больше, примерно, чем 0,05% молярного, более предпочтительно больше, примерно, чем 0,10% молярного, еще более предпочтительно больше, чем 0,15, а наиболее предпочтительно больше, чем 0,18% молярного. В то же самое время содержание моноокиси углерода в сухом уходящем газе из реактора окисления предпочтительно меньше, примерно, чем 0,60% молярного, более предпочтительно меньше, примерно, чем 0,50% молярного, еще более предпочтительно меньше, примерно, чем 0,35% молярного, а наиболее предпочтительно меньше, чем 0,28% молярного.
Авторы настоящего изобретения обнаружили, что важный фактор для уменьшения образования окислов углерода до этих предпочтительных пределов представляет собой улучшение чистоты рециклируемого фильтрата и исходных материалов окисляемого соединения для понижения концентрации ароматических соединений, у которых нет неароматических углеводородных групп, в соответствии с описаниями настоящего изобретения - это одновременно уменьшает образование окислов углерода и вредных примесей. Другой фактор представляет собой улучшение распределения пара-ксилола и окислителя в реакционной емкости в соответствии с описаниями настоящего изобретения. Другие факторы, делающие возможными указанные выше предпочтительные уровни окислов углерода, представляют собой работу с градиентами в реакционной среде, как здесь описано, давления, температуры, концентрации окисляемого соединения в жидкой фазе и окислителя в газовой фазе. Другие факторы, делающие возможным указанные выше предпочтительные уровни окислов углерода, представляют собой работу в пределах описаний, приведенных здесь, предпочтительных для пространственно-временной скорости реакции, давления, температуры, состава растворителя, состава катализатора и механической геометрии реакционной емкости.
Важное преимущество работы в предпочтительных пределах образования окислов углерода заключается в том, что использование молекулярного кислорода может быть уменьшено, хотя и не до стехиометрических значений. Несмотря на хорошее ступенчатое изменение концентрации окислителя и окисляемого соединения в соответствии с настоящим изобретением избыток кислорода должен поддерживаться выше стехиометрического значения, как вычисляется только лишь для исходных материалов окисляемого соединения, чтобы сделать возможными некоторые потери на окислы углерода и обеспечить избыток молекулярного кислорода для контроля образования вредных примесей. Конкретно, для случая, где ксилол представляет собой исходные материалы окисляемого соединения, отношение в исходных материалах массы молекулярного кислорода к массе ксилола предпочтительно больше, примерно, чем 0,91:1,00, более предпочтительно больше, примерно, чем 0,95:1,00, а наиболее предпочтительно больше, чем 0,99:1,00. В то же самое время отношение в исходных материалах массы молекулярного кислорода к массе ксилола предпочтительно меньше, примерно, чем 1,20:1,00, более предпочтительно меньше, примерно, чем 1,12:1,00, а наиболее предпочтительно меньше, чем 1,06:1,00. Конкретно для исходных материалов ксилола, усредненное по времени содержание молекулярного кислорода в сухом уходящем газе из реактора окисления предпочтительно больше, примерно, чем 0,1% молярного, более предпочтительно больше, примерно, чем 1% молярный, а наиболее предпочтительно больше, чем 1,5% молярного. В то же самое время усредненное по времени содержание молекулярного кислорода в сухом уходящем газе из реактора окисления предпочтительно меньше, примерно, чем 6% молярных, более предпочтительно меньше, примерно, чем 4% молярных, а наиболее предпочтительно меньше, чем 3% молярных.
Другое важное преимущество от работы в предпочтительных пределах образования окиси углерода заключается в том, что меньше ароматического соединения превращается в окислы углерода и другие, менее ценные формы. Это преимущество оценивается с использованием суммы молей всех ароматических соединений, покидающих реакционную среду, деленной на сумму молей всех ароматических соединений, поступающих в реакционную среду за непрерывный период времени, предпочтительно за один час, более предпочтительно один день, а наиболее предпочтительно 30 последовательных дней. Это отношение далее упоминается как "молярное отношение выживания" для ароматических соединений при прохождении через реакционную среду и выражается как численный процент. Если все поступающие ароматические соединения покидают реакционную среду как ароматические соединения, хотя, по большей части, в окисленных формах поступающих ароматических соединений, тогда молярное отношение выживания имеет свое максимальное значение 100%. Если точно 1 из каждых 100 поступающих ароматических молекул преобразуется в окислы углерода и/или другие неароматические молекулы (например, уксусную кислоту) в то время, когда она проходит через реакционную среду, тогда молярное отношение выживания равно 99%. Конкретно для случая, когда ксилол представляет собой главный исходный материал окисляемого ароматического соединения, молярное отношение выживания для ароматических соединений через реакционную среду предпочтительно больше, примерно, чем 98%, более предпочтительно больше, примерно, чем 98,5%, а наиболее предпочтительно меньше, чем 99,0%. В то же самое время, и для того, чтобы присутствовала достаточная общая реакционная активность, молярное отношение выживания для ароматических соединений при прохождении через реакционную среду предпочтительно является меньшим, примерно, чем 99,9%, более предпочтительно меньшим, примерно, чем 99,8%, а наиболее предпочтительно меньшим, чем 99,7%, когда ксилол представляет собой главный исходный материал окисляемого ароматического соединения.
Другой аспект настоящего изобретения включает в себя получение метилацетата в реакционной среде, содержащей уксусную кислоту и одно или несколько окисляемых ароматических соединений. Этот метилацетат является относительно летучим, по сравнению с водой и уксусной кислотой, и, таким образом, стремится следовать за уходящим газом, если только не используется дополнительное охлаждение или другая работа установки для его извлечения и/или его разрушения перед высвобождением уходящего газа назад в окружающую среду. Таким образом образование метилацетата представляет собой затраты на работу, а также капитальные затраты. Вероятно, метилацетат образуется посредством, сначала, объединения метильного радикала, вероятно, от разложения уксусной кислоты, с кислородом, с получением метилгидропероксида, посредством последующего разложения с образованием метанола и посредством конечного взаимодействия полученного метанола с оставшейся уксусной кислотой, с образованием метилацетата. Каким бы ни был химический путь, авторы обнаружили, что, когда скорость образования метилацетата является слишком низкой, тогда и образование окислов углерода также является слишком низким, а образование вредных ароматических примесей является слишком высоким. Если скорость образования метилацетата является слишком высокой, тогда образование окислов углерода также является излишне высоким, приводя к потерям выхода растворителя, окисляемого соединения и окислителя. Когда используются предпочтительные варианты осуществления, описываемые здесь, отношение образования молей образующегося метилацетата к молям вводимого окисляемого ароматического соединения предпочтительно больше, примерно, чем 0,005:1, более предпочтительно больше, чем примерно 0,010:1, а наиболее предпочтительно больше, чем 0,020:1. В то же самое время отношение образования молей образующегося метилацетата к молям вводимого окисляемого ароматического соединения предпочтительно меньше, примерно, чем 0,09:1, более предпочтительно меньше, примерно, чем 0,07:1, еще более предпочтительно меньше, примерно, чем 0,05:1, а наиболее предпочтительно меньше, чем 0,04:1.
Настоящее изобретение может дополнительно иллюстрироваться посредством следующих далее примеров предпочтительных вариантов его осуществления, хотя будет понятно, что эти примеры включаются исключительно для целей иллюстрации и не предназначены для ограничения объема притязаний настоящего изобретения, если только этого не указано конкретно.
Пример 1
Это рабочий пример промышленного окисления пара-ксилола в барботажной колонне реакторного типа. Этот пример демонстрирует, например, что имеются большие градиенты концентрации по высоте пара-ксилола, когда используются соответствующие геометрические и технологические условия в соответствии с аспектами настоящего изобретения.
Настоящий пример использует промышленную емкость барботажной колонны окисления, имеющую приблизительно вертикальный по существу цилиндрический корпус с внутренним диаметром примерно 2,44 м. Высота барботажной колонны окисления составляет примерно 32 м, от нижней линии начала изгиба (TL) до верхней TL. Емкость соединяется с эллиптическими днищем и крышкой, примерно 2:1, в верхней и нижней частях цилиндра. Рабочий уровень составляет примерно 25 м реакционной среды над нижней TL. Скорость поступления исходных материалов пара-ксилола промышленной чистоты является эффективно постоянной при скорости примерно 81 кг/мин, он поступает в реакционную емкость через круговое отверстие, расположенное в стенке цилиндрической секции на высоте примерно 4,35 м над нижней TL. Внутренний диаметр указанного отверстия в стенке составляет примерно 0,076 м. Фильтрованный растворитель вводится при эффективно постоянной скорости примерно 777 кг/мин. Неотмеряемая доля этого фильтрованного растворителя, устанавливаемая размерами прохода и перепадами давления как равная примерно 20 кг/мин, вводится в качестве жидкости для промывки устройства распределения окислителя. Оставшаяся часть отфильтрованного растворителя, примерно 757 кг/мин, вводится совместно с пара-ксилолом промышленной чистоты. Объединенный жидкофазный поток исходных материалов отфильтрованного растворителя и пара-ксилола промышленной чистоты, таким образом, составляет примерно 838 кг/мин, давая поверхностную скорость потока на входе через указанное отверстие в стенке примерно 3 м/с. Этот отфильтрованный растворитель получают из системы рециклирования установки, и он содержит более, примерно, чем 97 мас.% уксусной кислоты и воды. Концентрация компонентов катализатора в отфильтрованном растворителе является такой, что состав в жидкой фазе реакционной среды представляет собой примерно 1777 м.д. мас. кобальта, примерно 1518 м.д. мас. брома и примерно 107 м.д. мас. марганца. Отдельный поток флегмы растворителя вводится в виде капель в зону отделения газа над рабочим уровнем реакционной среды при эффективно постоянной скорости примерно 572 кг/мин. Эта флегма растворителя содержит более, примерно, чем 99 мас.% уксусной кислоты и воды; и флегму растворителя получают из отдельной системы рециклирования установки, при этом флегма не содержит значительных уровней компонентов катализатора. Объединенное содержание воды исходных материалов отфильтрованного растворителя и исходных материалов флегмы растворителя является таким, что концентрация воды в жидкой фазе реакционной среды составляет примерно 6,0 мас.%. Окислитель представляет собой сжатый воздух, который вводится при эффективно постоянной скорости примерно 384 кг/мин через устройство распределения окислителя, подобное тому, которое иллюстрируется на фиг.2-5. Это устройство распределения окислителя содержит скошенный проход для потока, который представляет собой приблизительно равносторонний восьмиугольник с диаметральным элементом, соединяющим одну из сторон с противоположной стороной и проходящим через вертикальную ось симметрии реакционной емкости. Скошенный проход для потока изготавливается из номинально 12-дюймовых трубных компонентов Schedule 10S. Ширина восьмиугольника от центроида на одной стороне прохода для потока до центроида на противоположной стороне составляет примерно 1,83 м. Восьмиугольник лежит приблизительно горизонтально, и средняя высота восьмиугольного прохода составляет примерно 0,11 м над нижней TL реакционной емкости. Устройство распределения окислителя содержит 75 круговых отверстий, которые имеют диаметр примерно 0,025 м. Отверстия располагаются приблизительно однородно по восьмиугольнику и диаметральному элементу, располагаясь вблизи верхней части указанной 12-дюймовой трубы. Имеется одно круговое отверстие с диаметром примерно 0,012 м вблизи нижней части одной только стороны восьмиугольного прохода. Рабочее давление газа в верхней части реакционной емкости постоянно составляет примерно 0,52 МПа в датчике. Реакция осуществляется по существу адиабатическим образом, так что тепло реакции повышает температуру поступающих исходных материалов и испаряет большую часть поступающего растворителя. Измеренная вблизи серединной высоты реакционной среды рабочая температура составляет примерно 160°C. Уходящая суспензия, содержащая сырую терефталевую кислоту (CTA), удаляется вблизи нижней части эллиптического днища реакционной емкость при эффективно постоянной скорости. Скорость потока уходящей суспензии составляет примерно 408 кг/мин.
Образцы суспензии из реакционной среды получают с трех уровней высоты в реакционной емкости, как описано ниже. При определении концентрации различных частиц в различных положениях, в реакционной среде, необходимо принимать во внимание стохастическую природу системы, отбирая достаточное количество образцов для определения усредненной по времени величины с достаточным разрешением.
Один набор из пяти образцов получают из прохода для уходящей суспензии вблизи нижней части эллиптического днища реакционной емкости. Другой набор из пяти образцов получают из отверстия в стенке, расположенного на высоте примерно 12,4 м над нижней TL реакционной емкости. Третий набор из пяти образцов получают из отверстия в стенке, расположенного на высоте примерно 17,2 м над нижней TL реакционной емкости.
Все образцы суспензии анализируют с помощью способа калиброванной газовой хроматографии (ГХ) на состав пара-ксилола и пара-толуальдегида в жидкой фазе. Таблица 5, ниже, показывает среднее значение для пяти результатов, которые получают на трех различных уровнях в колонне. Результаты приводятся как массовые доли анализируемого вещества на миллион массовых долей (м.д. мас.) жидкой фазы.
Эти результаты показывают большие градиенты, возникающие по высоте, для локальных концентраций пара-ксилола и пара-толуальдегида. Например, градиент концентрации пара-ксилола, наблюдаемый в данных таблицы 5, составляет более 20:1 (455:21). Эти результаты демонстрируют, что внутреннее перемешивание текучих сред поступающих исходных материалов пара-ксилола в барботажной колонне значительно медленнее, чем внутренние скорости реакции. До меньшей степени, градиенты по высоте также наблюдаются для концентраций других родственных ароматических реагирующих частиц в реакционной среде (например, пара-толуиловой кислоты и 4-карбоксибензальдегида).
Как демонстрируется в следующих далее примерах, подробные численные модели показывают, что реальный диапазон концентрации пара-ксилола в жидкой фазе реакционной среды настоящего примера намного превосходит 100:1. Даже без использования строго численной модели специалисты в данной области заметят, что реальная максимальная концентрация пара-ксилола возникает в области, расположенной вблизи той, где исходные материалы пара-ксилола вводятся в реакционную емкость барботажной колонны через стенку емкости. Эта высота максимальной концентрации пара-ксилола составляет примерно 4,35 м над нижней TL, между положениями отбора образцов примерно на 12,4 м и в нижней части потока. Подобным же образом реальная минимальная концентрация пара-ксилола, вероятно, устанавливается в верхней части реакционной среды или очень близко от нее, примерно на 25 м, намного выше самого высокого уровня высоты, с которого отбираются указанные выше образцы.
Концентрации пара-ксилола и других окисляемых соединений могут измеряться для других положений в реакционной среде посредством использования соответствующих механических устройств для отбора образцов, в любом положении по вертикали или горизонтали, в реакционной среде. Необязательно, концентрации для положений, откуда образцы физически не берутся и не анализируются химически, могут вычисляться с разумной точностью, используя компьютерные модели достаточной сложности, для соответствия очень сложным структурам потоков текучих сред, кинетике химических реакций, балансу энергии, равновесиям пар-жидкость-твердое тело и скоростям обмена между фазами.
Примеры 2-5
Примеры 2-5 представляют собой численные модели барботажных колонн реакторного типа, либо идентичных реактору примера 1, либо в целом подобных ему, с указанными усовершенствованиями. Численное моделирование динамики текучих сред (CFD), осуществляемое для образования примеров 2-5, осуществляют в соответствии со способом моделирования, описанным в ожидающей совместного решения заявке на патент США, серийный №60/594774, озаглавленной "Modeling of Liquid-Phase Oxidation", полное описание которой в явном виде включается сюда в качестве ссылки.
В примерах 2-5 моделирование CFD осуществляют с использованием CFX release 5.7 (ANSYS, Inc. 275 Technology Drive, Canonsburg, PA 15317). Примеры 2-5 содержат свыше примерно 100000 дискретных пространственных ячеек вычисления, каждый. Шаги изменения времени, используемые в примерах 2-5, меньше, чем 0,1 с. Множество размеров пузырьков в диапазоне диаметров примерно от 0,005 примерно до 0,20 м, как показано, являются пригодными для настройки модели CFD для аппроксимации вблизи среднего содержания пузырьков, оцениваемого посредством измерения разности давлений, вертикальному профилю содержания пузырьков, оцениваемому посредством гамма-сканирования, и горизонтальным профилям содержания пузырьков, оцениваемым посредством анализа изображений компьютерной томографии (CT). Для выбора соответствующих размеров и популяций пузырьков в моделях CFD примеров 2-5 реальные рабочие данные установок получают для барботажных колонн с суспензией, с цилиндрическими внутренними диаметрами примерно 2,44 м и примерно 3,05 м, работающих с реакционной средой вблизи соответствующей композиции и условий способа, как описано ниже. Эталонные данные для общего содержания пузырьков получают с использованием разностей давлений, измеренных от положения вблизи основания емкостей и до уходящего газа из верхней части колонны. Эталонные данные для вертикального профиля содержания пузырьков получают с использованием гамма-испускающего радиоактивного источника и пошагового способа детектирования снаружи реакционной емкости с шагами в пределах примерно от 0,05 м примерно до 0,3 м. Эталонные данные для горизонтальных профилей содержания пузырьков получают посредством анализа изображений CT, осуществляемых на решетке девять на девять в горизонтальной плоскости работающей барботажной колонны, с использованием гамма-испускающего радиоактивного источника и способа детектирования. Т.е. источник располагается на заданной высоте в девяти различных положениях, расположенных примерно на равных расстояниях по периметру барботажной колонны. Для каждого положения источника гамма-излучения количество гамма-излучения, проходящее через реакционную емкость и реакционную среду, детектируется в девяти различных положениях, расположенных примерно на одинаковых расстояниях друг от друга по периметру барботажной колонны. Различные математические модели применяются затем к этим дискретным данным для получения оценок разброса содержания пузырьков в реакционной среде для указанной высоты. Множество горизонтальных изображений CT получают в два различных дня, для двух различных уровней высоты и для двух различных скоростей поступления исходных материалов пара-ксилола, сжатого воздуха, и тому подобное.
Модель химической реакции расходования пара-ксилола в этой окружающей среде настраивается по профилям реагентов для пара-ксилола, как найдено в примере 1, вместе с другими данными для сходных температур, давлений, интенсивностей реакции, катализа, концентрации воды, и так далее, от исследований как в промышленном, так и в пилотном масштабе. В качестве показательной аппроксимации временная псевдоконстанта первого порядка для затухания реакционного индикатора пара-ксилола равна примерно 0,2 сек-1 примерно для 160°C и для средних, примерно, условий реакционной среды, используемых в примерах 2-4.
Важно, что модели CFD полей потока, полученные в примерах 2-4, дают крупномасштабные флуктуации в виде групп пузырьков и всплесков жидкости, которые в целом согласуются с наблюдаемым низкочастотным волнообразным движением в рабочей реакционной емкости барботажной колонны.
Пример 2
Настоящий пример развивает вычисления, относящиеся к механической конфигурации примера 1, и устанавливает сравнительную основу для примеров 3 и 4. В настоящем примере механическая конфигурация барботажной колонны реакторного типа идентична примеру 1, она имеет круглое входное отверстие диаметром 0,076 м сквозь стенку реакционной емкости для потока исходных материалов, содержащих пара-ксилол и отфильтрованный растворитель. Скорость поступления исходных материалов пара-ксилола составляет примерно 1,84 кг/с - выше, чем в примере 1. Скорость поступления исходных материалов отфильтрованного растворителя, вводимого в тесной смеси с пара-ксилолом, равна примерно 18,4 кг/с. Поверхностная скорость объединенного потока пара-ксилола плюс отфильтрованного растворителя, поступающего через отверстие в стенке, таким образом, составляет примерно 4 м/с. Скорость поступления исходных материалов флегмы растворителя в пространство для отделения газа в верхней части колонны равна 12,8 кг/с. Скорость поступления исходных материалов сжатого воздуха через устройство распределения окислителя составляет примерно 9 кг/с. Содержание твердых продуктов реакционной суспензии составляет примерно 31 мас.%. Суспензия продукта извлекается из центра днища реакционной емкости с использованием эффективно постоянной скорости для поддержания приблизительно постоянного уровня реакционной среды примерно 25 м. Среднее содержание газа для серединной высоты реакционной среды составляет примерно 55%, при усреднении по площади, усреднении по времени, где отрезок усреднения по времени составляет, по меньшей мере, примерно 100 с времени в модели CFD. Давление в верхней части колонны над реакционной средой равно примерно 0,50 МПа в датчике. Температура равна примерно 160°C, измеренная вблизи серединной высоты реакционной среды. Содержание воды и кобальта, брома и марганца в жидкой части реакционной среды является по существу таким же, как в примере 1.
Пример 3
Настоящий пример развивает вычисления, относящиеся к улучшению диспергирования исходных материалов пара-ксилола посредством повышения поверхностной скорости жидкофазных исходных материалов, содержащих пара-ксилол, в их точке поступления в реакционную среду в соответствии с одним из аспектов настоящего изобретения. В этом примере механическая конфигурация барботажной колонны реакторного типа идентична примеру 2, за исключением того, что отверстие в стенке, через которое поступают жидкофазные исходные материалы, содержащие пара-ксилол, уменьшается до кругового диаметра 0,025 м. Скорость поступления исходных материалов пара-ксилола и другие условия способа являются такими же, как в примере 2, за исключением того, что поверхностная скорость объединенного жидкофазного потока исходных материалов пара-ксилола плюс отфильтрованного растворителя, поступающих через отверстие в стенке, составляет теперь примерно 36 м/с.
Вычисления модели CFD усредненных по времени частей реакционной среды с концентрацией реакционного индикатора пара-ксилола в жидкой фазе, превышающей различные пороги, представлены ниже в таблице 6. Объем реакционной среды с очень высокой концентрацией реакционного индикатора пара-ксилола в жидкой фазе уменьшается при работе с более высокими входными скоростями жидкофазного потока исходных материалов, содержащих пара-ксилол, в соответствии с настоящим изобретением. Уменьшение областей с высокой концентрацией пара-ксилола является важным для ограничения нежелательных реакций связывания, как потому, что концентрации многих растворимых ароматических частиц являются в ней повышенными, так и потому, что такие концентрации приводят к локально высокому расходованию растворенного молекулярного кислорода и поэтому приводят к локальному подавлению постоянных концентраций растворенного молекулярного кислорода.
с концентрацией pX
Пример 4
Настоящий пример развивает вычисления для усовершенствованных механических средств для введения окислителя и пара-ксилола в барботажную колонну реакторного типа. Этот пример осуществляют в такой же барботажной колонне реакторного типа, как используется в примерах 1-3. Однако реактор модифицируется относительно способа, которым как окислитель, так и пара-ксилол поступают в реакционную среду. При обсуждении примера 4 внимание направляется, во-первых, на модифицированное устройство для поступления пара-ксилола в реакционную среду, при этом уменьшая зоны высоких концентраций пара-ксилола. Во-вторых, внимание направляется на модифицированное устройство для поступления окислителя в реакционную среду, тем самым, уменьшая зоны, которые плохо аэрируются. Это не предполагает того, что две эти модификации являются совершенно независимыми по своим результатам, но просто представляют собой постадийное представление.
Количество реакционной среды с очень высокими концентрациями в жидкой фазе реакционного индикатора пара-ксилола уменьшается в примере 4 посредством использования системы распределения жидкофазных исходных материалов, в целом, как показано на фиг.9-11. Удобно, чтобы эта система распределения жидкофазных исходных материалов имела четыре прохода для потоков, удобно, стоящих приблизительно вертикально. Каждый из этих четырех проходов для потоков находится примерно на расстоянии 0,75 м от вертикальной оси симметрии барботажной колонны. Удобно изготовлять эти четыре прохода для потока из номинально 1,5-дюймовых труб Schedule 10S. Удобно, чтобы нижний уровень каждой стойки в настоящем примере имел конически сходящуюся секцию с внутренним углом, измеряемым между противоположными сторонами конуса, который составляет, удобно, примерно 24°; однако другие формы также являются пригодными для закрывания заднего края прохода для потока (например, коническая крышка с другим внутренним углом, крышка в виде плоской пластины, крышка в виде колпачка из трубы, крышка в форме клина и так далее). Каждый из этих четырех проходов для потока имеет в целом девять отверстий, при этом каждое из них имеет круговой диаметр примерно 0,0063 м. Самое нижнее из девяти отверстий в каждом проходе находится в нижней части нижней конической секции. Для каждого прохода это самое нижнее отверстие располагается примерно на 0,4 м выше нижней TL реакционной емкости. Измеряя всегда от этого нижнего края обрезанной нижней части конической секции, следующие три отверстия в каждом проходе находятся на высоте примерно 0,3 м, следующие три отверстия находятся на высоте примерно 1,6 м, и два самых верхних отверстия находятся на высоте примерно 2,7 м. Таким образом, вертикальное расстояние от самого нижнего отверстия до самого высокого отверстия в каждом проходе составляет примерно 2,7 м или примерно 1,1 D. Линейное (не вертикальное) расстояние наибольшего удаления для отверстия, от самого нижнего отверстия в одном вертикальном проходе до самого верхнего отверстия вертикального прохода, диагонально противоположного, составляет примерно 3,44 м или примерно 1,4 D. Для каждого уровня отверстия распределены примерно равномерно по окружности каждого прохода для потока. Удобно, чтобы подающий проход для подачи исходных материалов окисляемого соединения и растворителя в верхнюю часть четырех приблизительно вертикальных проходов был расположен примерно горизонтально на высоте примерно 3,60 м над нижней TL реакционной емкости. Удобно, чтобы подающий проход был изготовлен из номинально 3-дюймовых трубных компонентов Schedule 10S. Имеется соответствующая механическая жесткая связь внутри узла и механическая жесткая связь между узлом и устройством распределения окислителя и реакционной емкостью для противостояния как статическим, так и динамическим силам, возникающим как при нормальной, так и при чрезвычайной работе.
Хотя они не рассчитываются в настоящем примере, являются возможными множество других конструкций для этой системы распределения жидкофазных исходных материалов. Например, проходы для потоков жидкости могут иметь большие или меньшие размеры, или поперечные сечения, иные, чем приблизительно круговые, или количество, отличное от четырех. Например, каждый из четырех по существу вертикальных проходов может запитываться независимо посредством проходов для потока, по отдельности пересекающих удерживающую давление стенку реакционной емкости. Например, соединение для подачи поступающего пара-ксилола и исходных материалов растворителя может входить вблизи серединной высоты или вблизи более низкой высоты, или на любой высоте, или на множестве уровней высоты приблизительно вертикальных проходов. Например, подающие проходы могут быть приблизительно вертикальными, с распределительными отверстиями, располагающимися в приблизительно горизонтальных проходах, или оба направления потоков могут быть наклонными или нелинейными или неортогональными. Например, отверстия могут располагаться по разному, в радиальном, азимутальном или вертикальном направлении, по отношению к реакционной среде. Например, может использоваться большее или меньшее количество отверстий, и/или отверстия различных форм, и/или отверстия со смешанными размерами и/или смешанными формами. Например, вместо выходных отверстий могут использоваться выходные сопла. Например, одно или несколько устройств для отклонения потока могут находиться вне прохода для потока вблизи выходных отверстий и на пути текучих сред при выходе в реакционную среду.
В зависимости от характера и содержания твердых продуктов, если они имеются, от объединенных исходных материалов пара-ксилола и растворителя или реакционной среды и в зависимости от пуска, остановки и других рабочих процедур, используемых в реальных рабочих операциях, может возникнуть необходимость в прочистке системы распределения жидкофазных исходных материалов от твердых продуктов внутри нее. Хотя это не рассчитывается в настоящем примере, отверстие для прочистки может полезным образом быть большим, чем отверстия однородных размеров, показанные в настоящем примере. Отверстие на нижнем краю каждой из четырех приблизительно вертикальных стоек является особенно пригодным для прочистки от твердых продуктов, хотя оно и не является единственным возможным средством. Более сложные механические устройства, такие как узлы заслонок, контрольные клапаны, клапаны для выпуска избыточного потока, электрические клапаны и тому подобное, могут использоваться либо для предотвращения накопления твердых продуктов, либо для высвобождения аккумулируемых твердых продуктов из внутреннего пространства системы распределения жидкофазных исходных материалов.
Теперь внимание направлено на устройство распределения окислителя, которое является в целом таким, как показано на фиг.12-15. Удобно, чтобы кольцевой элемент устройства распределения окислителя содержал скошенный проход для потока, который, удобно и приблизительно, представляет собой восьмиугольник с равными сторонами без диаметрального элемента. Скошенный проход для потока удобно изготавливать из номинально 10-дюймовых труб Schedule 10S. Ширина восьмиугольника, от центроида на одной стороне прохода для потока до центроида на противоположной стороне, составляет примерно 1,12 м. Удобно, чтобы восьмиугольная секция лежала приблизительно горизонтально и серединная высота восьмиугольной секции была примерно на 0,24 м ниже нижней TL реакционной емкости. Это представляет собой резкий контраст с кольцевым элементом устройства распределения окислителя примеров 1-3, уровни которых центрируются выше нижней TL реакционной емкости. Восьмиугольная часть прохода перфорируется 64 примерно круговыми отверстиями, каждое диаметром примерно 0,030 м, расположенными приблизительно на равных расстояниях вокруг прохода. Примерно половина отверстий расположена вокруг прохода, в положениях, которые находятся под углом примерно 45° ниже горизонтали, измеряя от каждого отверстия до ближайшего центроида поперечного сечения прохода для потока. Примерно половина отверстий расположена вокруг прохода в положениях, которые находятся примерно в нижней части прохода для потока (т.е. под углом примерно 90° ниже горизонтали, измеряя от каждого отверстия до ближайшего центроида поперечного сечения прохода для потока). Авторы опять отмечают, в дополнение к примечаниям для входного распределителя жидкой фазы, что является возможным множество других конкретных конструкций устройства распределения окислителя, попадающих в рамки нескольких аспектов настоящего изобретения. Например, стенку, удерживающую давление, могут пересекать больше или меньше чем два подающих прохода. Например, подающие проходы устройства распределения окислителя могут конструироваться не содержащими кольцевого элемента. Например, могут присутствовать более одного кольцевого элемента, и любой кольцевой элемент может иметь не 8 сторон или может иметь несимметричные стороны. Например, конструкция может получать предпочтительный перепад давлений или предпочтительное качество аэрирования, или предпочтительную природу без образования осадка, используя при этом различное количество или размер, или размеры, или расположение отверстий или выходов из прохода. Например, конструкция может включать различные диаметры проходов в предпочтительных пределах. Например, конструкция может достигать природы без образования осадка посредством использования промывки жидкостью.
В этом примере реакционная среда извлекается с эффективно постоянной скоростью сбоку реакционной емкости на высоте примерно 14 м через отверстие в стенке, которое имеет внутренний круговой диаметр примерно 0,076 м. Извлекаемая реакционная среда разделяется на суспензию продукта, содержащую сырую терефталевую кислоту, и уходящий газ, используя наружную деаэрационную емкость, которая описывается полностью в примере 6. Отделенный уходящий газ из наружной деаэрационной емкости переносится посредством прохода, присоединяясь к главному потоку уходящего газа, покидающего верхнюю часть реакционной емкости.
Способы моделирования CFD настоящего примера являются по существу такими же, как для примеров 2 и 3, с этими исключениями. Пространственное разделение на ячейки модифицируется, как принято и известно в данной области, для рассматриваемого устройства распределения поступающего окислителя, для распределения поступающего окисляемого соединения и для удаления суспензии продукта из боковой стенки реакционной емкости, примерно на 14 м выше нижней TL.
Для оценки результатов модели CFD относительно распределения реакционного индикатора пара-ксилола используются такие же способы, как в примерах 2 и 3. А именно определяются усредненные по времени доли реакционной среды с концентрацией реакционного индикатора пара-ксилола в жидкой фазе, превышающей различные пороги. Для простоты сравнения, результаты этого примера представлены выше в таблице 6. Эти результаты показывают, что улучшение распределения реакционного индикатора пара-ксилола в этом примере реально вызывает небольшое увеличение количества реакционной среды при более чем 1000 м.д. мас., но более вредные пороговые уровни 2500 м.д. мас., 10000 м.д. мас. и 25000 м.д. мас. уменьшаются. Эти усовершенствования обеспечиваются, например, более высокими входными скоростями исходных материалов вместе с улучшенным вертикальным, радиальным и азимутальным позиционированием и распределением введения пара-ксилола в реакционную среду.
Теперь, обращаясь к качеству аэрации реакционной среды, способ 2000 горизонтальных слоев равного субобъема используется для оценки количества плохо аэрируемого объема в реакционной среде примеров 2-4. Начиная с самой нижней части реакционной среды, а именно с нижней части эллиптического днища, в этом примере реакционная среда разделяется на 2000 равных субобъемов, с использованием теоретических горизонтальных плоскостей. Для каждого из приращений времени модели CFD, в пределах каждого из указанных 2000 равных субобъемов, количество суспензии и количество газа определяются и используются для вычисления среднего содержания газа в нем. Чтобы сделать возможной стохастическую природу процесса и его модели CFD, выходной параметр модели CFD усредняется по времени, по временам модели, имеющим продолжительность, по меньшей мере, примерно 100 с, с получением усредненных по времени значений содержания газа в каждом из 2000 равных субобъемов.
После определения усредненного по времени содержания газа для каждого из 2000 равных субобъемов эти значения сравниваются с пороговыми значениями, описанными здесь. Для каждого порога учитывается общее количество негодных субобъемов, которые не превосходят указанное пороговое значение. Таблица 7, ниже, показывает количество, из 2000 горизонтальных слоев равного объема реакционной среды, с усредненным по времени содержанием газа ниже 10% объемных, ниже 20% объемных и ниже 30% объемных, как для примера 2, так и примера 4. Пример 4 является значительно улучшенным, по сравнению с примером 2.
При сравнении рассчитанных примеров 2 и 4 заметно также, что исходные материалы пара-ксилола примера 4 высвобождаются в реакционной среде ниже и ближе к поступающему потоку окислителя, чем в примере 2.
Примеры 5 и 6
Примеры 5 и 6 представляют собой рабочие примеры, демонстрирующие в промышленной барботажной колонне окисления важность сведения к минимуму областей плохой аэрации, усовершенствования способа введения исходных материалов пара-ксилола промышленной чистоты для лучшего вертикального, азимутального и радиального диспергирования и понижения уровня входа для исходных материалов пара-ксилола промышленной чистоты, чтобы он был ближе к точке наивысшей доступности молекулярного кислорода, в соответствии с описаниями настоящего изобретения. А дополнение к этому эти примеры демонстрируют выигрыш в выходе реакции, когда имеется приподнятый по высоте выход для суспензии.
Имеется множество различных соединений примесей, как правило, получаемых посредством связывания ароматических колец во время частичного окисления пара-ксилола. Один из них представляет собой 4,4'-дикарбоксистильбен. Это соединение имеет гораздо более высокое поглощение света, чем имеет терефталевая кислота, и оно сильно уменьшает оптическое пропускание предполагаемого продукта. В дополнение к этому, 4,4'-дикарбоксистильбен представляет собой удобную примесь для использования при мониторинге качества непрерывного окисления, поскольку он распределяется селективно в твердую фазу реакционной среды; следовательно, как правило, очень малое количество 4,4'-дикарбоксистильбена присутствует в потоках рециклированного растворителя реакционных емкостей промышленных барботажных колонн, описанных в примерах 5 и 6. В примерах 5 и 6 концентрации 4,4'-дикарбоксистильбена измеряют с помощью аналитического метода, использующего ВЭЖХ-МС, калиброванную с помощью соответствующей эталонной смеси, содержащей растворитель и известные количества нескольких анализируемых веществ, в частности, содержащей известное количество 4,4'-дикарбоксистильбена. Аналитический метод ВЭЖХ-МС описан выше в разделе «Подробное описание».
Пример 5
Барботажная колонна реакторного типа, используемая в настоящем примере, имеет по существу такую же механическую конфигурацию, как и реактор в примерах 1 и 2. Реактор находится при условиях способа, сравнимых с примером 6, и обеспечивает основу для сравнения. Рабочий уровень составляет примерно 25 м реакционной среды. Исходные материалы пара-ксилола промышленной чистоты при эффективно постоянной скорости примерно 81 кг/мин. Отфильтрованный растворитель вводится при эффективно постоянной скорости примерно 793 кг/мин. Неотмеряемая доля этого, устанавливаемая размерами прохода и перепадами давления как равная примерно 20 кг/мин вводится в качестве жидкости для промывки устройства распределения окислителя. Оставшаяся часть отфильтрованного растворителя, примерно 773 кг/мин, вводится тесно смешанной с пара-ксилолом промышленной чистоты. Объединенный жидкофазный поток отфильтрованного растворителя и пара-ксилола промышленной чистоты, таким образом, составляет примерно 854 кг/мин. Этот отфильтрованный растворитель получают из системы рециклирования установки, и он содержит более, примерно, чем 97 мас.% уксусной кислоты и воды. Концентрация компонентов катализатора в отфильтрованном растворителе является такой, что состав в жидкой фазе реакционной среды представляет собой примерно 2,158 м.д. мас. кобальта, примерно 1,911 м.д. мас. брома и примерно 118 м.д. мас. марганца. Отдельный поток флегмы растворителя вводится в виде капель в зону отделения газа над рабочим уровнем реакционной среды при эффективно постоянной скорости примерно 546 кг/мин. Эта флегма растворителя содержит более, примерно, чем 99 мас.% уксусной кислоты и воды; и флегму растворителя получают из отдельной системы рециклирования установки, она не содержит значительных уровней компонентов катализатора. Объединенное содержание воды исходных материалов отфильтрованного растворителя и исходных материалов флегмы растворителя является таким, что концентрация воды в жидкой фазе реакционной среды составляет примерно 5,8 мас.%. Окислитель представляет собой сжатый воздух, который вводится при эффективно постоянной скорости примерно 352 кг/мин. Рабочее давление газа в верхней части реакционной емкости постоянно составляет примерно 0,42 МПа в датчике. Реакционная емкость работает, по существу, в адиабатическом режиме, так что тепло реакции повышает температуру поступающих исходных материалов и испаряет большую часть поступающего растворителя. Измеренная вблизи серединной высоты реакционной среды рабочая температура составляет примерно 154,6°C. Уходящая суспензия, содержащая сырую терефталевую кислоту (CTA), удаляется вблизи нижней части эллиптического днища реакционной емкости при эффективно постоянной скорости примерно 428 кг/мин.
В настоящем примере отношение скорости получения нежелательного 4,4'-дикарбоксистильбена к скорости получения желаемой терефталевой кислоты измеряют с помощью ВЭЖХ-МС на трех отдельных образцах продукта суспензии как примерно 8,6, 9,1 и 9,2 м.д. мас., таким образом, в среднем, примерно 9,0 м.д. мас. Концентрация пара-ксилола в жидкой фазе уходящей суспензии измеряется посредством калиброванной ГХ на трех отдельных образцах продукта суспензии как примерно 777, 539 и 618 м.д. мас., таким образом, в среднем примерно 645 м.д. мас. Концентрация пара-толуальдегида в жидкой фазе уходящей суспензии измеряется посредством калиброванной ГХ на указанных отдельных образцах продукта суспензии, как примерно 1,055, 961 и 977 м.д. мас., таким образом, в среднем, примерно 998 м.д. мас.
Пример 6
Барботажная колонна реакторного типа настоящего примера соответствует механической конфигурации, разработанной в рассчитанном примере 4. Реактор настоящего примера содержит усовершенствования относительно высоты, скорости, количества и распределения входов для исходных материалов пара-ксилола, таким образом, обеспечивая улучшенное распределение исходных материалов пара-ксилола и улучшенное ступенчатое изменение концентрации относительно молекулярного кислорода. Кроме того, он содержит усовершенствования в качестве аэрации в реакционной среде посредством использования усовершенствованного устройства распределения окислителя и относительно высоты и способа удаления и деаэрации суспензии, покидающей реакционную среду. По сравнению с примером 5 видны важные усовершенствования относительно выхода пара-ксилола и видны важные уменьшения относительно образования примесей.
Реактор настоящего примера имеет улучшенную механическую конфигурацию, как описано в модели CFD примера 4. Рабочий уровень составляет примерно 25 м реакционной среды. Поступление исходных материалов пара-ксилола промышленной чистоты является эффективно постоянным при скорости примерно 81 кг/мин. Отфильтрованный растворитель вводится совместно с пара-ксилолом промышленной чистоты при эффективно постоянной скорости примерно 744 кг/мин. Объединенный поток отфильтрованного растворителя и исходных материалов пара-ксилола промышленной чистоты, таким образом, составляет примерно 825 кг/мин. Этот отфильтрованный растворитель происходит из такой же системы рециклирования установки и по существу имеет такой же состав, как в примере 5. Концентрация компонентов катализатора в отфильтрованном растворителе является такой, что состав в жидкой фазе реакционной среды представляет собой примерно 1996 м.д. мас. кобальта, примерно 1693 м.д. мас. брома и примерно 108 м.д. мас. марганца. Отдельный поток флегмы растворителя вводится в виде капель в зону отделения газа над рабочим уровнем реакционной среды при эффективно постоянной скорости примерно 573 кг/мин. Эта флегма растворителя содержит более, примерно, чем 99 мас.% уксусной кислоты и воды; и флегму растворителя получают из отдельной системы рециклирования установки, он не содержит значительных уровней компонентов катализатора. Объединенное содержание воды исходных материалов отфильтрованного растворителя и исходных материалов флегмы растворителя является таким, что концентрация воды в жидкой фазе реакционной среды составляет примерно 5,7 мас.%. Окислитель представляет собой сжатый воздух, который вводится при эффективно постоянной скорости примерно 329 кг/мин. Рабочее давление газа в верхней части реакционной емкости постоянно составляет примерно 0,41 МПа в датчике. Реакционная емкость работает по существу адиабатическим образом, так что тепло реакции повышает температуру поступающих исходных материалов и испаряет большую часть поступающего растворителя. Измеренная вблизи серединной высоты реакционной среды рабочая температура составляет примерно 153,3°C.
Реакционную среду извлекают сбоку реакционной емкости на высоте примерно 14 м, через отверстие в стенке, которое имеет внутренний круговой диаметр примерно 0,076 м. Извлеченная реакционная среда переносится через горизонтальный по существу проход, изготовленный из номинально 3-дюймовых труб Schedule 10S, к боковой стенке по существу вертикальной наружной деаэрационной емкости. Наружная деаэрационная емкость имеет внутренний круговой диаметр примерно 0,315 м и конструируется в основном из номинально 12-дюймовой трубы Schedule 10S. Горизонтальная площадь поперечного сечения внутри наружной деаэрационной емкости составляет, таким образом, примерно 0,0779 м2. Это сравнивается с площадью горизонтального поперечного сечения внутри реакционной емкости примерно 4,67 м2 для высоты, где извлекается реакционная среда. Таким образом, отношение меньшей площади горизонтального поперечного сечения к большей площади равно примерно 0,017.
Наружная деаэрационная емкость простирается вниз от высоты ввода реакционной среды примерно на 1,52 м до перехода внизу к диаметру, согласующемуся с нижним выходным проходом для потока. Эффективно постоянная скорость потока равна примерно 433 кг/мин для деаэрированной по существу суспензии, содержащей сырую терефталевую кислоту, выходящей из нижней части наружной деаэрационной емкости. Таким образом, по существу деаэрированная суспензия на более низких уровнях высоты номинально 12-дюймовой деаэрационной емкости имеет поверхностную скорость движения вниз, которая равна примерно 0,093 м/с; и в этой уходящей суспензии нет вредного захвата окислителя. Уходящая суспензия переносится вперед посредством прохода для потока, изготовленного из номинально 3-дюймовых труб Schedule 10S, для соединения со следующим далее технологическим оборудованием. В настоящем примере средства для контроля скорости потока извлеченной реакционной среды располагаются в потоке, покидающем нижнюю часть деаэрационной емкости, хотя и другие положения для контроля являются возможными и пригодными для использования.
Наружная деаэрационная емкость простирается выше высоты, на которой поступает реакционная среда, примерно на 14 м, до перехода от номинально 12-дюймового размера трубы, с уменьшением диаметра для согласования с верхним выходным проходом для потока, изготовленным из номинально 2-дюймовых труб Schedule 10S. Отделенный уходящий газ из наружной деаэрационной емкости переносится через этот номинально 2-дюймовый проход, присоединяясь к главному потоку уходящего газа, покидающего верхнюю часть реакционной емкости.
В настоящем примере отношение скорости получения нежелательного 4,4'-дикарбоксистильбена к скорости получения желаемой терефталевой кислоты измеряется посредством ВЭЖХ-МС на трех отдельных образцах продукта суспензии, как примерно 2,3, 2,7 и 3,2 м.д. мас., в среднем примерно 2,7 м.д. мас. Это значительно меньше по сравнению с примером 5. Концентрация пара-ксилола в жидкой фазе суспензии, уходящей из приподнятого бокового выхода, измеряется посредством калиброванной ГХ на трех отдельных образцах продукта суспензии как примерно 86, 87 и 91 м.д. мас., в среднем, примерно 88 м.д. мас. Концентрация пара-толуальдегида в жидкой фазе уходящей суспензии измеряется посредством калиброванной ГХ на указанных отдельных образцах продукта суспензии как примерно 467, 442 и 423 м.д. мас., в среднем, примерно 444 м.д. мас. Это представляет собой улучшение преобразования и выхода в потоке извлеченной суспензии по сравнению с примером 5.
Примеры 7-10
Примеры 7-10 представляют собой рассчитанные примеры, относящиеся, в частности, к начальному диспергированию пара-ксилола в реакционной среде, но демонстрирующие также и другие аспекты настоящего изобретения.
Пример 7
Настоящий пример относится к введению испаренного пара-ксилола. В этом примере исходные материалы пара-ксилола нагреваются и испаряются перед поступлением в реакционную среду. Это помогает начальному диспергированию пара-ксилола. Это обеспечивает увеличение поступающих объемов и облегчает увеличение скоростей. Кроме того, это замедляет перенос поступающего пара-ксилола в объем жидкой фазы и заставляет исходные материалы пара-ксилола двигаться в направлении реакционноспособной жидкой фазы в лучшей гармонии с газообразными исходными материалами молекулярного кислорода.
В настоящем примере емкость барботажной колонны окисления имеет вертикальный цилиндрический корпус с внутренним диаметром 2,44 м. Высота емкости барботажной колонны окисления равна 32 м, от нижней линии начала изгиба (TL) до верхней TL. Емкость соединяется с эллиптическими днищем и крышкой, 2:1, в верхней и нижней части цилиндра. Рабочий уровень составляет примерно 25 м реакционной среды над нижней TL. Исходные материалы отфильтрованного растворителя, который отделяют от пара-ксилола, поступают при скорости 18,4 кг/с через круговое входное отверстие диаметром 0,076 м через стенку реакционной емкости на высоте 4,35 м над нижней TL. Скорость поступления исходных материалов флегмы растворителя в зону отделения газа выше рабочего уровня реакционной среды, равна примерно 14,3 кг/с. Скорость поступления исходных материалов сжатого воздуха равна примерно 9 кг/с, через устройство распределения окислителя, по существу такое же, как в примерах 4 и 6. Суспензию, содержащую примерно 31 мас.% твердых продуктов, извлекают из реакционной среды через боковую выходную стойку, по существу такую же, как в примерах 4 и 6. Давление в верхней части колонны над реакционной средой равно примерно 0,50 МПа в датчике. Содержание воды и кобальта, брома и марганца в жидкой части реакционной среды является по существу таким же, как в примере 4.
Скорость поступления исходных материалов пара-ксилола равна 1,84 кг/с. Перед высвобождением в реакционной среде поток исходных материалов жидкофазного пара-ксилола подвергается повышению давления, а затем испаряется при давлении примерно 0,69 МПа в датчике посредством нагрева от температуры хранения примерно 40°C, до температуры примерно 233°C. Это требует поступления тепла, примерно, 1,3 МДж/с, в поток исходных материалов пара-ксилола. Теплообменник, использующий пар при давлении 4 МПа, используется для этой цели, но любой другой источник энергии с достаточной температурой будет в такой же степени удовлетворительным, включая избыток тепла от технологических текучих сред. Это поступление тепла представляет собой примерно 5% от тепла реакции преобразования пара-ксилола в терефталевую кислоту. Удаление этой дополнительной тепловой нагрузки заставляет температуру реакционной среды несколько увеличиться при постоянном давлении, по сравнению с исходными материалами жидкого пара-ксилола. (См. пример 8). Температура составляет примерно 162°C, измеренная вблизи серединной высоты реакционной среды. Необязательно, давление может понижаться, с уменьшением температуры реакции до 160°C, измеренной вблизи серединной высоты реакционной среды.
Объемный поток испаренного пара-ксилола составляет примерно 0,084 м3/с. Этот поток поступает в реакционную емкость на высоте 0,1 м над нижней TL емкости через 3 прохода, соединенные параллельно. Рядом с реакционной емкостью каждый проход изготавливается из номинально 1,5-дюймовых труб и присоединяется к круговому отверстию такого же диаметра в стенке емкости. 3 отверстия в стенке располагаются по горизонтали, на азимутальном расстоянии друг от друга 120°. Поверхностная скорость каждого поступающего потока пара-ксилола составляет приблизительно 21 м/с, и поступающий пара-ксилол диспергируется в реакционной среде, в то же время, когда он растворяется в реакционноспособной жидкой фазе, где находятся в основном частицы катализатора.
Пример 8
Настоящий пример относится к введению частично испаренного пара-ксилола. В этом рассчитанном примере исходные материалы пара-ксилола частично испаряются посредством смешивания с подающимся окислителем перед поступлением в реакционную среду. Это помогает начальному диспергированию пара-ксилола. Это обеспечивает увеличение поступающих объемов и облегчает увеличение скоростей; и это разбавляет концентрацию пара-ксилола. Кроме того, это замедляет перенос поступающего пара-ксилола в объем жидкой фазы и заставляет исходные материалы пара-ксилола двигаться в направлении реакционноспособной жидкой фазы в лучшей гармонии с газообразными исходными материалами молекулярного кислорода.
В настоящем примере емкость барботажной колонны окисления имеет вертикальный цилиндрический корпус с внутренним диаметром 2,44 м. Высота емкости барботажной колонны окисления равна 32 м от нижней линии начала изгиба (TL) до верхней TL. Емкость соединяется с эллиптическими днищем и крышкой, 2:1, в верхней и нижней части цилиндра. Рабочий уровень составляет примерно 25 м реакционной среды над нижней TL. Исходные материалы отфильтрованного растворителя, который отделяется от пара-ксилола, поступают при скорости 18,4 кг/с через круговое входное отверстие диаметром 0,076 м, через стенку реакционной емкости на высоте 4,35 м над нижней TL. Скорость поступления исходных материалов флегмы растворителя в зону отделения газа над рабочим уровнем реакционной среды равна примерно 12,8 кг/с. Скорость поступления исходных материалов сжатого воздуха равна примерно 9 кг/с, через устройство распределения окислителя, сходное с устройством в примерах 4 и 6, но модифицированное, как отмечено ниже. Суспензия, содержащая примерно 31 мас.% твердых продуктов, извлекается из реакционной среды через боковую выходную стойку, по существу, такую же, как в примерах 4 и 6. Давление в верхней части колонны над реакционной средой равно примерно 0,50 МПа в датчике. Содержание воды и кобальта, брома и марганца в жидкой части реакционной среды по существу такое же, как в примере 4.
Скорость поступления исходных материалов пара-ксилола опять равна 1,84 кг/с. Он протекает в виде жидкости через проходы во внутреннее пространство устройства распределения окислителя, где жидкость высвобождается в сжатый воздух в 4 положениях, с использованием сопел для распыления жидкости, как известно в данной области. Необязательно, проходы для жидкости с открытыми краями или газо-жидкостные распылительные сопла могут использоваться в точке, где жидкость поступает в устройство распределения окислителя. Для обеспечения безопасности 4 сенсора температуры располагаются внутри устройства распределения окислителя. Эти сенсоры температуры соединяются с устройствами подачи сигнала тревоги и блокировочными устройствами для отключения подачи окислителя и пара-ксилола, если детектируются высокие температуры. При подаче сжатого воздуха примерно при 80°C, благодаря теплоте сжатия, без последующего охлаждения на конечной стадии сжатия, и при подаче исходных материалов пара-ксилола примерно при 40°C испаряется приблизительно 17 мас.% пара-ксилола, при давлении, преобладающем внутри устройства распределения окислителя. Оставшийся жидкий пара-ксилол переносится в реакционную среду, при этом газ в двухфазном потоке перемешивается с газом при скоростях, достигающих скорости газового потока. В дополнение к этому, указанная остающаяся жидкость помогает прочищать устройство распределения окислителя от любых твердых продуктов, которые в него попали, в соответствии с аспектами настоящего изобретения.
Температура равна примерно 160°C, измеренная вблизи серединной высоты реакционной среды. Поскольку никакой дополнительной энергии не добавляется в какой-либо поток исходных материалов, она примерно такая же, как в примерах 4 и 6.
Необязательно, либо исходные материалы сжатого воздуха, либо исходные материалы пара-ксилола могут предварительно нагреваться перед смешиванием в устройстве распределения окислителя, для увеличения доли пара-ксилола, который поступает в реакционную среду в виде пара. Например, поступление тепла, в 300 кДж/с, в пара-ксилол повышает его температуру примерно до 124°C и увеличивает долю испаренного пара-ксилола примерно до 33%. Например, поступление тепла, 600 кДж/с, в сжатый воздух повышает его температуру примерно до 146°C и увеличивает долю испаренного пара-ксилола примерно до 54%. В обоих случаях для нагрева требуется более низкое качество энергии, чем в примере 7. Фактически, избыточное тепло от уходящего газа из реакционной среды может использоваться в качестве всего источника тепла или его части. Однако когда к исходным материалам добавляется некоторое количество энергии, температура реакционной среды будет слегка увеличиваться, устанавливаясь, при данном давлении, потоках и составах фаз, в пределах между 160°C и 162°C, измеренная посредине высоты. Необязательно, давление может регулироваться для регулировки температуры. В дополнение к этому, когда некоторое количество энергии добавляется к исходным материалам, количество растворителя, поступающего в реакционную емкость, регулируется, когда желательным является удерживание приблизительно постоянной доли твердых продуктов. Например, в примерах 7 и 8 поток флегмы растворителя изменяется в пределах между примерно 12,8 и примерно 14,3 кг/с, в зависимости от величины добавляемой энергии, чтобы поддерживать приблизительно постоянное содержание твердых продуктов около 31 мас.%.
Пример 9
Настоящий пример относится к введению пара-ксилола вдали от стенки реакционной емкости с использованием эжектора для жидкости. В этом рассчитанном примере начальное диспергирование жидких исходных материалов пара-ксилола улучшается посредством использования эжектора, использующего поток жидкости в качестве движущей силы. Реактор настоящего примера имеет такую же механическую конфигурацию и граничные условия способа, как в примере 4, с исключениями, описанными ниже. Смешанный жидкофазный поток пара-ксилола плюс отфильтрованного растворителя поступает через стенку реакционной емкости на такой же высоте через такой же номинально 3-дюймовый проход для потока. Однако вместо внутренней системы распределения жидкофазных исходных материалов примера 4 смешанные жидкофазные исходные материалы высвобождаются в реакционной среде в виде движущей текучей среды в проточном эжекторе, как известно в данной области и как показано на диаграмме на фиг.26. Эжектор конструируется для разности давлений 0,1 МПа на движущей текучей среде. Эжектор располагается и ориентируется с соплом для потока, выходящим вертикально вверх, вдоль аксиальной центральной линии реакционной емкости, на высоте примерно 4,5 м над нижней TL. Объем реакционной среды, эжектируемой и смешиваемой с движущей жидкостью, изменяется со временем в зависимости от стохастических событий возникновения групп пузырьков в барботажной колонне на входе эжекции. Однако усредненный по времени эжектируемый поток больше, чем поток движущей текучей среды, таким образом, обеспечивая более быстрое разбавление поступающего пара-ксилола. Последующее смешивание и химическая реакция осуществляются в соответствии с обычными стохастическими явлениями в барботажной колонне.
Пример 10
Настоящий пример относится к введению пара-ксилола вдали от стенки реакционной емкости с использованием эжектора для газа и жидкости. В этом рассчитанном примере начальное диспергирование исходных материалов пара-ксилола улучшается посредством использования эжектора, использующего поток газа в качестве движущей силы. Реактор настоящего примера имеет такую же механическую конфигурацию и граничные условия способа, как в примере 4, с исключениями, описанными ниже. Как восьмиугольное устройство распределения окислителя, так и система распределения жидкофазных исходных материалов удаляются. Вместо этого поступающий поток окислителя и смешанные жидкофазные исходные материалы пара-ксилола плюс отфильтрованного растворителя переносятся через раздельные проходы во внутреннюю часть реакционной емкости. Там оба потока объединяются в качестве движущих текучих сред на входе проточного эжектора, как известно в данной области и как показано на диаграмме на фиг.27. Эжектор расположен по вертикали вдоль осевой центральной линии реакционной емкости. Он располагается с выходом, направленным вниз и расположенным на 0,2 м ниже нижней линии начала изгиба реакционной емкости. Эжектор конструируется для разности давлений 0,1 МПа на движущих текучих средах. Два сенсора температуры располагаются вблизи положения, где исходные материалы сжатого воздуха и пара-ксилола первый раз объединяются. Эти сенсоры температуры соединяются с тревожными устройствами и блокировочными устройствами для отключения подачи окислителя и пара-ксилола, если детектируются высокие температуры.
Объем эжектируемой реакционной среды увеличивается, по сравнению с примером 9, и начальное разбавление поступающего пара-ксилола дополнительно улучшается. В дополнение к этому, жидкофазная часть реакционной среды с самыми высокими локальными концентрациями пара-ксилола подвергается еще более непосредственному ступенчатому изменению концентрации, по сравнению с газофазной частью с самой высокой концентрацией молекулярного кислорода. Последующее перемешивание и химическая реакция осуществляются в соответствии с обычными стохастическими явлениями в барботажной колонне.
Примеры 11-13
Примеры 11-13 представляют собой рассчитанные примеры, относящиеся конкретно к использованию потоков жидкости из реакционной среды в проходах для облегчения начального диспергирования пара-ксилола в реакционной среде, но демонстрируют также и другие аспекты настоящего изобретения.
Пример 11
Настоящий пример относится к использованию прохода для потока внутри реакционной емкости для переноса жидкости с целью облегчения начального диспергирования поступающего пара-ксилола. Реактор настоящего примера имеет такую же механическую конфигурацию и граничные условия способа, как в примере 4, с исключениями, описанными ниже. Упоминается диаграмма на фиг.24. Смешанный жидкофазный поток пара-ксилола плюс отфильтрованного растворителя поступает через стенку корпуса, через номинально 3-дюймовый проход для потока, подобный примеру 4. Однако внутренняя система распределения жидкофазных исходных материалов примера 4 удаляется, и указанный смешанный поток жидкости, вместо этого, высвобождаются в проход для потока. Проход для потока имеет круговой внутренний диаметр примерно 0,15 м для большей части его длины, включая его нижнее окончание, которое находится на 1 м выше нижней TL емкости. Проход для потока поднимается вертикально до общей высоты 21 м над нижней TL емкости. На высоте 20 м над нижней TL емкости проход для потока расширяется, чтобы он имел внутреннюю площадь поперечного сечения 0,5 м2, когда он поднимается в высоту еще на 1 м. Эта верхняя секция с большим диаметром указанного прохода для потока может рассматриваться как внутренняя деаэрационная емкость, и она реально формируется, частично используя стенку корпуса. Весь проход для потока располагается внутри реакционной емкости. На верхнем входе в проход для потока реакционная среда сильно обеднена пара-ксилолом и пара-толуальдегидом, хотя имеются заметные концентрации паратолуиловой кислоты и 4-карбоксибензальдегида. Реакционная среда, поступающая в верхнюю часть указанного прохода для потока, по существу деаэрируется, создавая более плотную среду внутри указанного прохода для потока, чем в остальной части корпуса. Суспензия в проходе для потока движется вниз со скоростью, согласно оценкам, примерно 150 кг/с, в этой точке перепад давления в потоке, интегрируемый по всей длине указанного прохода для потока, приходит в равновесие с разностью плотностей внутри и снаружи, проинтегрированной по всей длине указанного прохода для потока. Из этого направленного вниз потока суспензии примерно 104 кг/с представляет собой жидкость, составляющую примерно 69 мас.%. Поток исходных материалов тесно перемешанного пара-ксилола и отфильтрованного растворителя, в целом, примерно 20,2 кг/с поступает в указанный проход для потока примерно на 5 м выше нижней TL. Затем эта смесь проходит вниз по проходу для потока дополнительно 4 м, примерно 27 диаметров прохода, менее чем за 1 с, и становится соответствующим образом перемешанной. Концентрация пара-ксилола, таким образом, полезным образом уменьшается примерно до 15000 м.д. мас. перед высвобождением в главном корпусе с реакционной средой в барботажной колонне. Последующее перемешивание и химическая реакция осуществляются в соответствии с обычными стохастическими явлениями в барботажной колонне.
Пример 12
Настоящий пример относится к использованию прохода для потока вне реакционной емкости для переноса жидкости с целью облегчения начального диспергирования поступающего пара-ксилола. Реактор настоящего примера имеет такую же механическую конфигурацию и граничные условия способа, как в примере 11, с исключениями, описанными ниже, и с упоминанием диаграммы на фиг.25. Внутренний проход для потока удаляется и заменяется наружным проходом для потока. Секция прохода, соединяющая реакционную емкость с наружной деаэрационной секцией, имеет внутренний круговой диаметр 0,30 м и располагается на 20 м выше нижней TL. Внутренний круговой диаметр наружной деаэрационной секции равен 1 м, а его высота равна 2 м. Внутренний круговой диаметр прохода для потока ниже деаэрационной секции равен 0,20 м, делая возможными большие потоки, с использованием примерно такой же доступной высоты крышки. Сенсор потока и контрольный клапан для потока включаются в проход для потока с целью контроля скорости потока в желаемом диапазоне. Например, контроль потока настраивается, чтобы сделать возможным перенос суспензии 150 кг/с, такой же, как, согласно оценкам, осуществляется через внутренний проход для потока примера 11. Смешанный жидкофазный поток пара-ксилола и отфильтрованного растворителя поступает в наружный проход для потока примерно на 5 м выше нижней TL реакционной емкости. Выход наружного прохода для потока соединяется с днищем реакционной емкости. Таким образом, концентрация пара-ксилола опять полезным образом уменьшается примерно до 15000 м.д. мас. перед высвобождением в главном корпусе с реакционной средой в барботажной колонне. Последующее перемешивание и химическая реакция осуществляются в соответствии с обычными стохастическими явлениями в барботажной колонне. Извлечение продукта суспензии для последующей переработки осуществляется посредством ответвления от указанного прохода для потока ниже секции деаэрации и выше добавления жидкофазного потока пара-ксилола и отфильтрованного растворителя, таким образом, устраняя необходимость в отдельной системе для удаления и деаэрации суспензии.
Пример 13
Настоящий пример относится к использованию прохода для потока, состоящего из секций как наружных, так и внутренних по отношению к реакционной емкости для переноса жидкости с целью облегчения начального диспергирования поступающего пара-ксилола. Этот рассчитанный пример является идентичным примеру 12, за исключением того, что второе ответвление вне наружного прохода для потока располагается примерно на 3 м выше нижней TL реакционной емкости, которая находится ниже точки добавления смешанного жидкофазного потока пара-ксилола и отфильтрованного растворителя. Второе ответвление прохода для потока также имеет внутренний круговой диаметр 0,20 м. Отдельный клапан для контроля потока располагается во втором ответвлении прохода для потока, опять же, для регулировки потока. Разветвленный проход для потока проходит через боковую стенку реакционной емкости на 3 м выше нижней TL, и разветвленный проход для потока простирается внутри стенок реакционной емкости на 0,4 м. Таким образом, разветвленный проход содержит секции как наружные, так и внутренние по отношению к реакционной емкости. Поток может поступать в реакционную емкость через любой проход, либо из выхода прохода в днище, либо из выхода прохода внутри боковой стенки, или через оба выхода, и при любом отношении.
Настоящее изобретение описывается подробно с конкретными ссылками на его предпочтительные варианты осуществления, но будет понятно, что вариации и модификация могут осуществляться в пределах сущности и объема настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2393146C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382758C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382759C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ В БАРБОТАЖНОЙ КОЛОННЕ РЕАКТОРНОГО ТИПА | 2005 |
|
RU2363534C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388745C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2363535C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2006 |
|
RU2435753C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2384563C2 |
| СОСТАВ СЫРОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2388744C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2381212C2 |
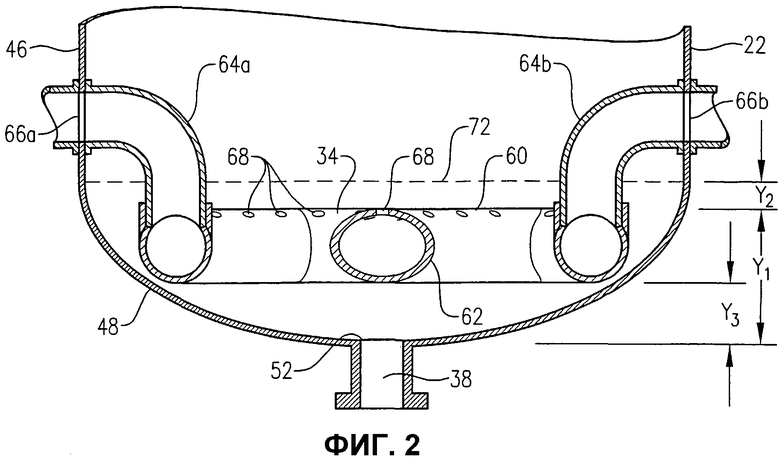
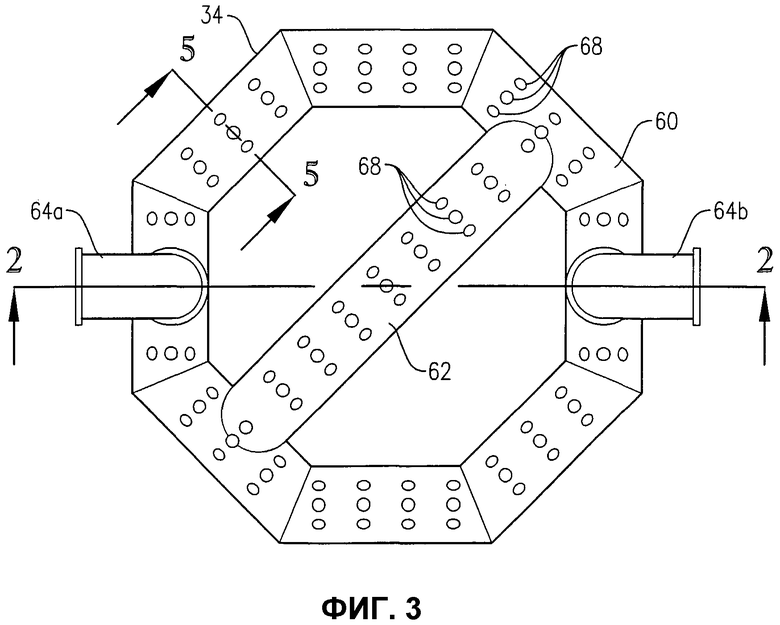
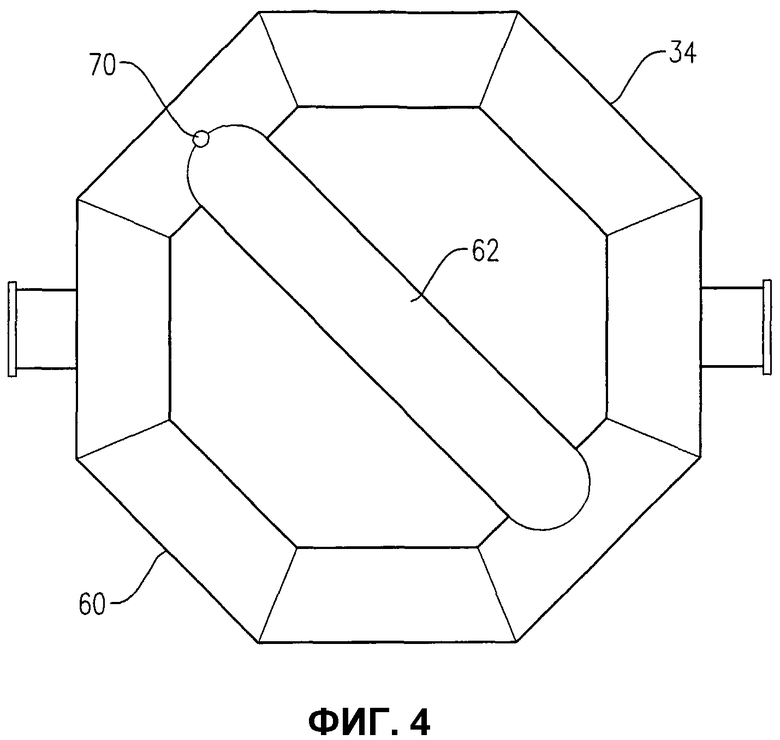
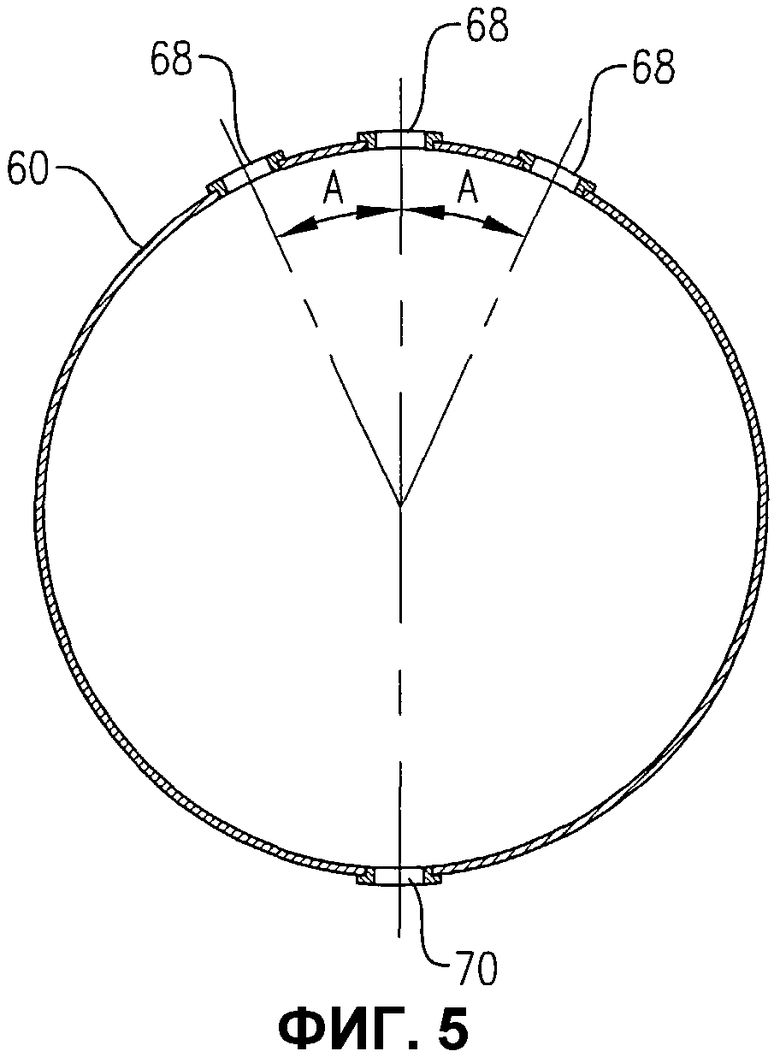
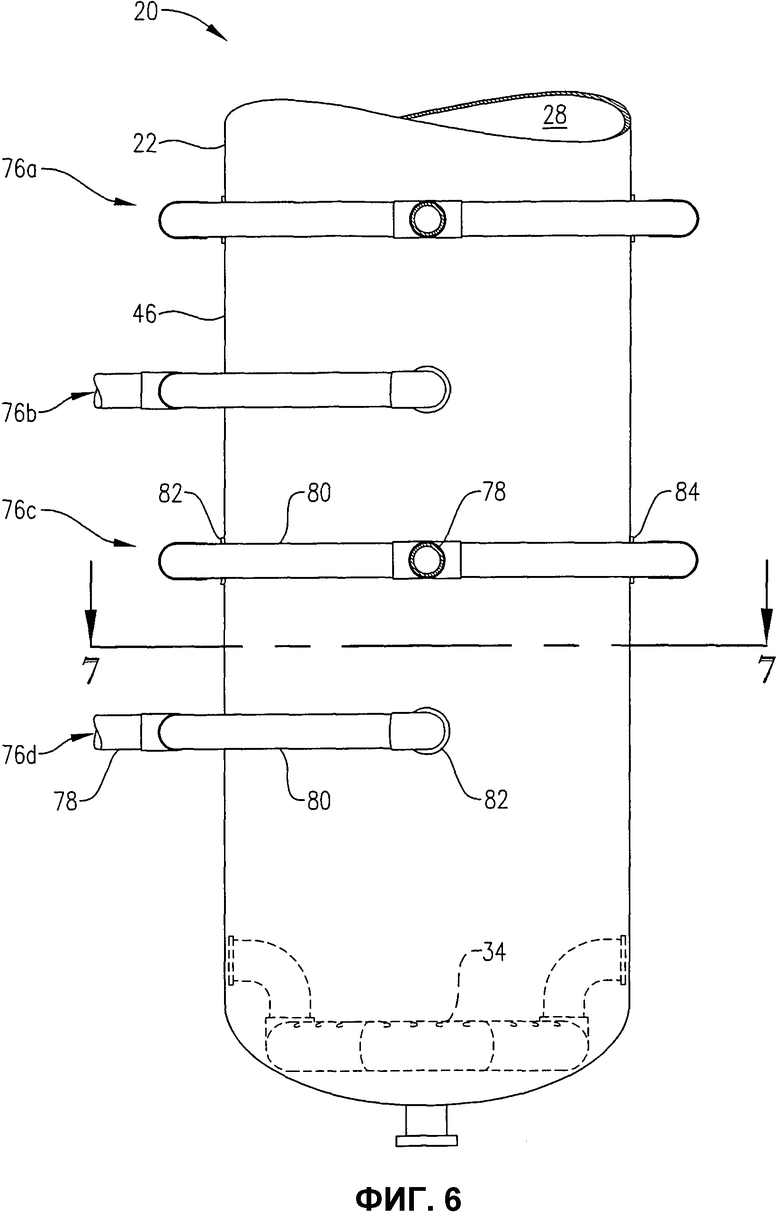
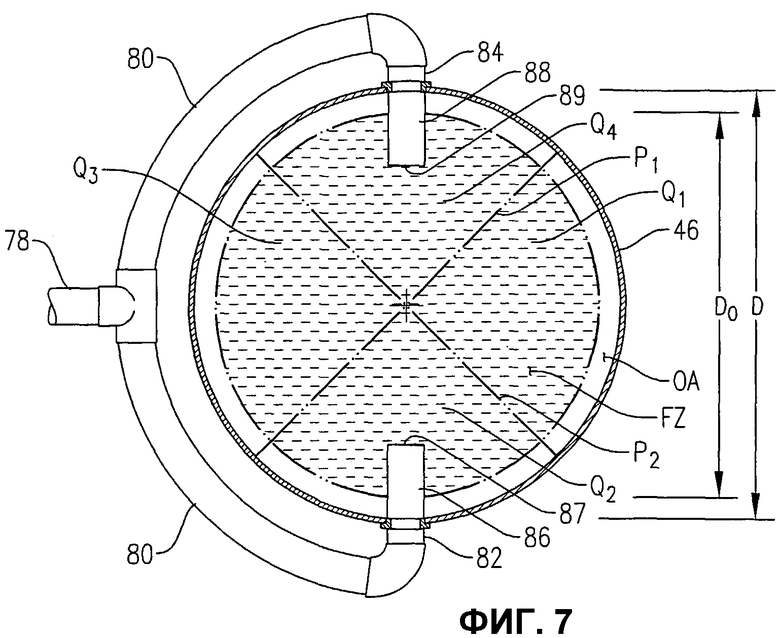
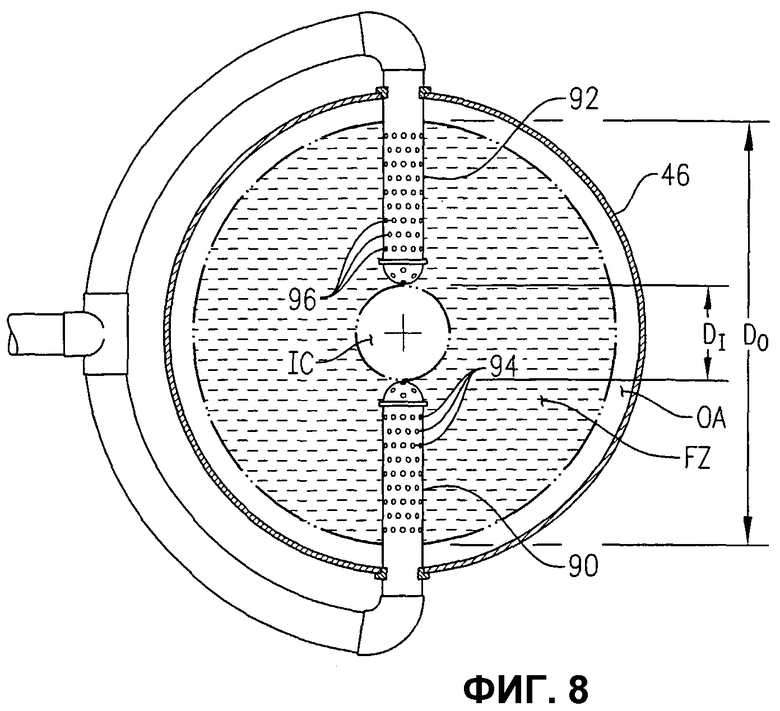
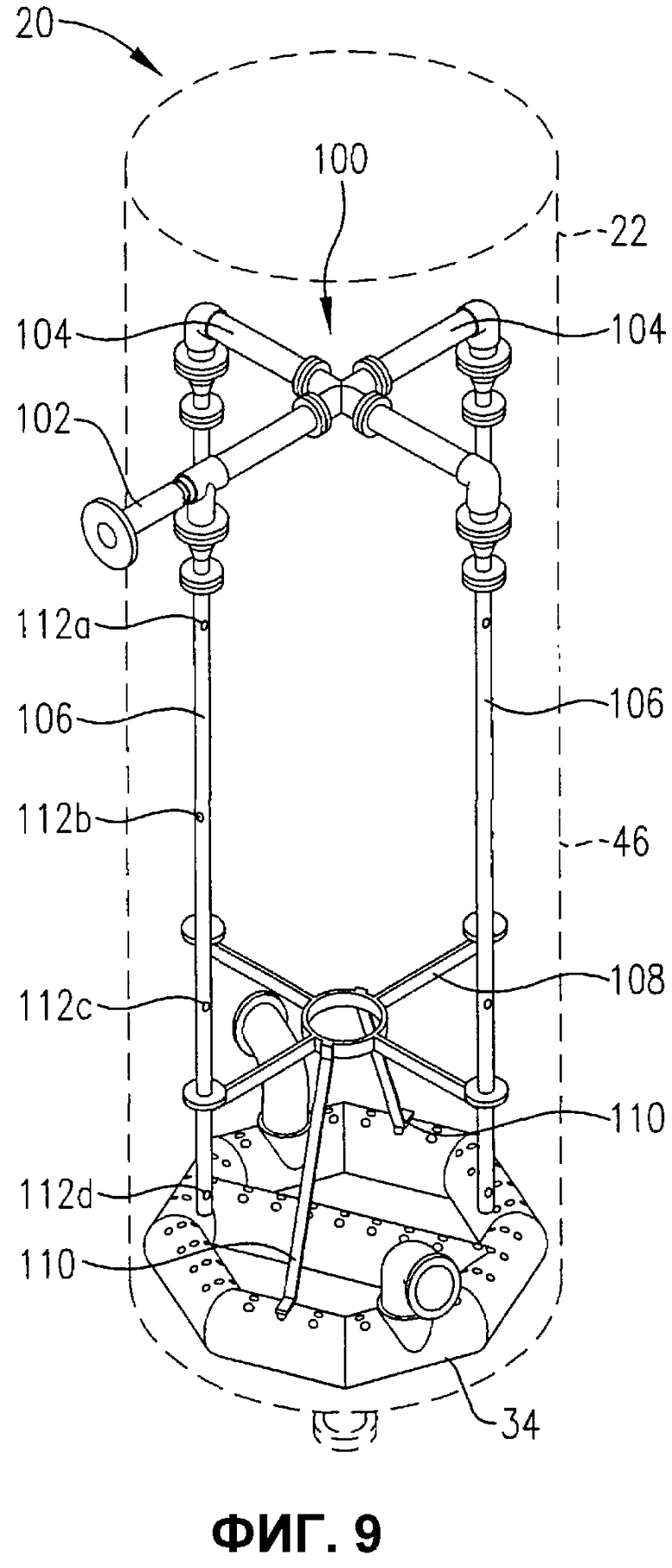
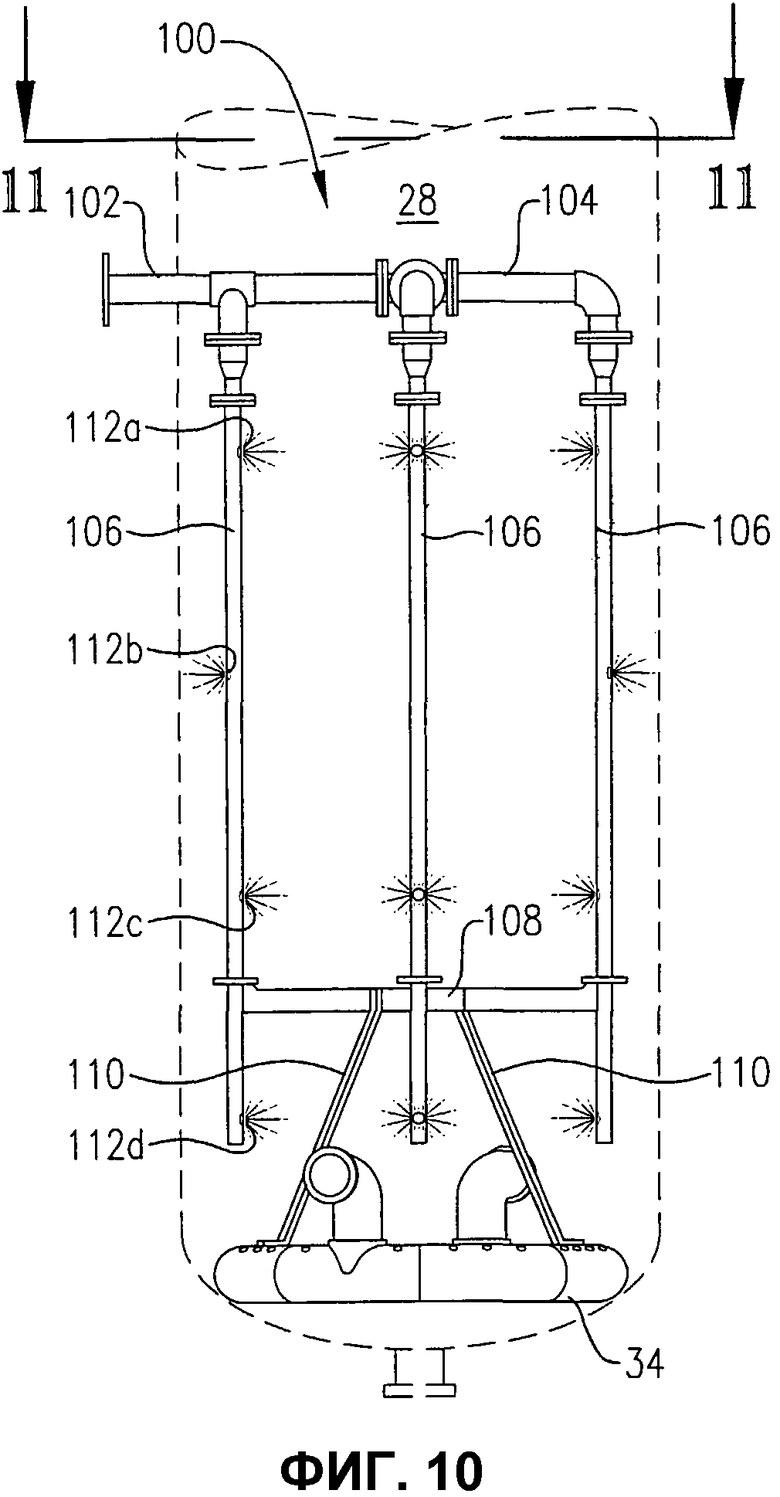
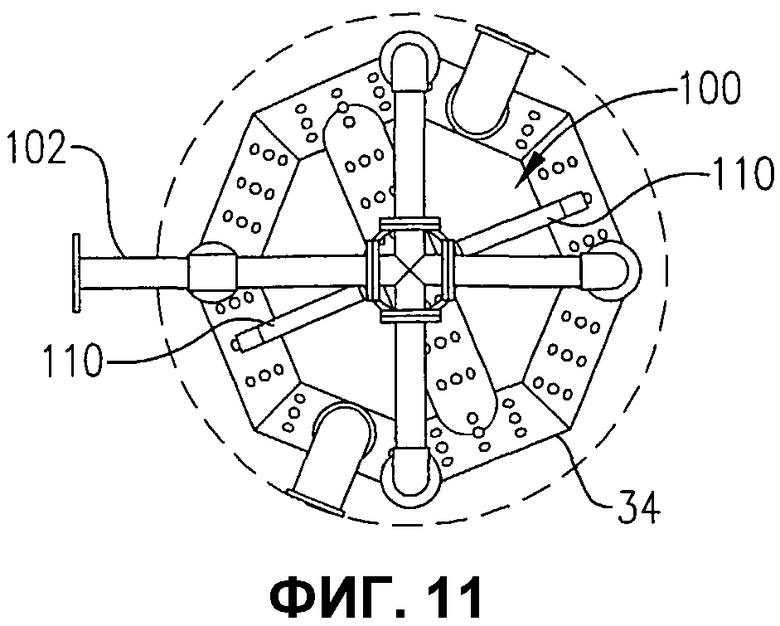
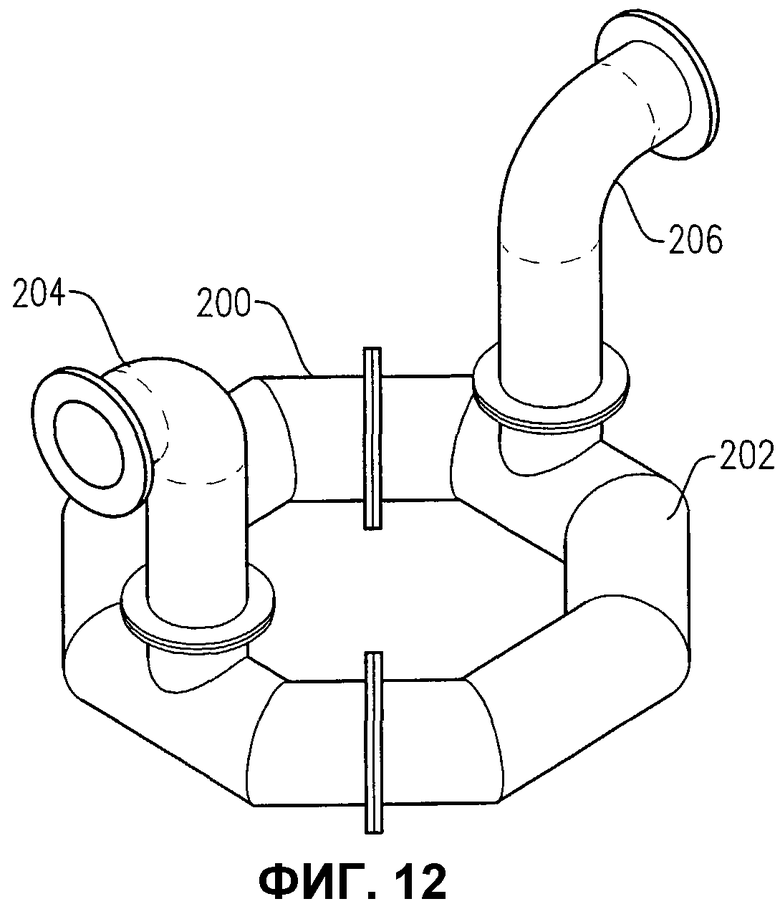
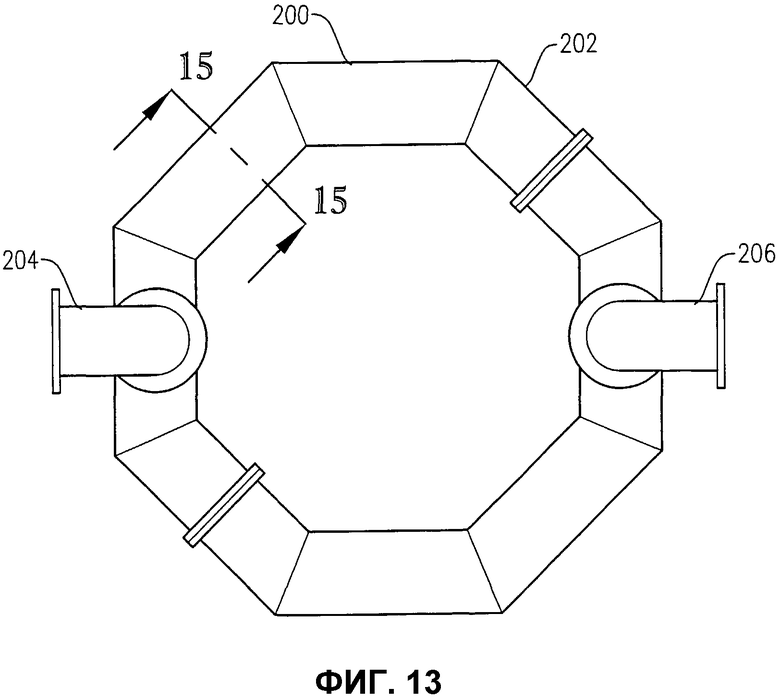
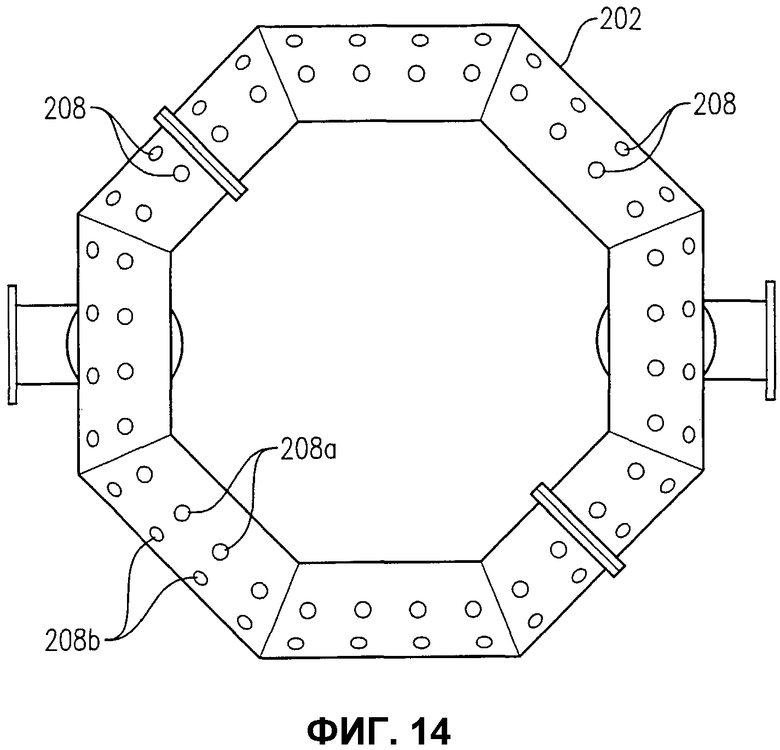
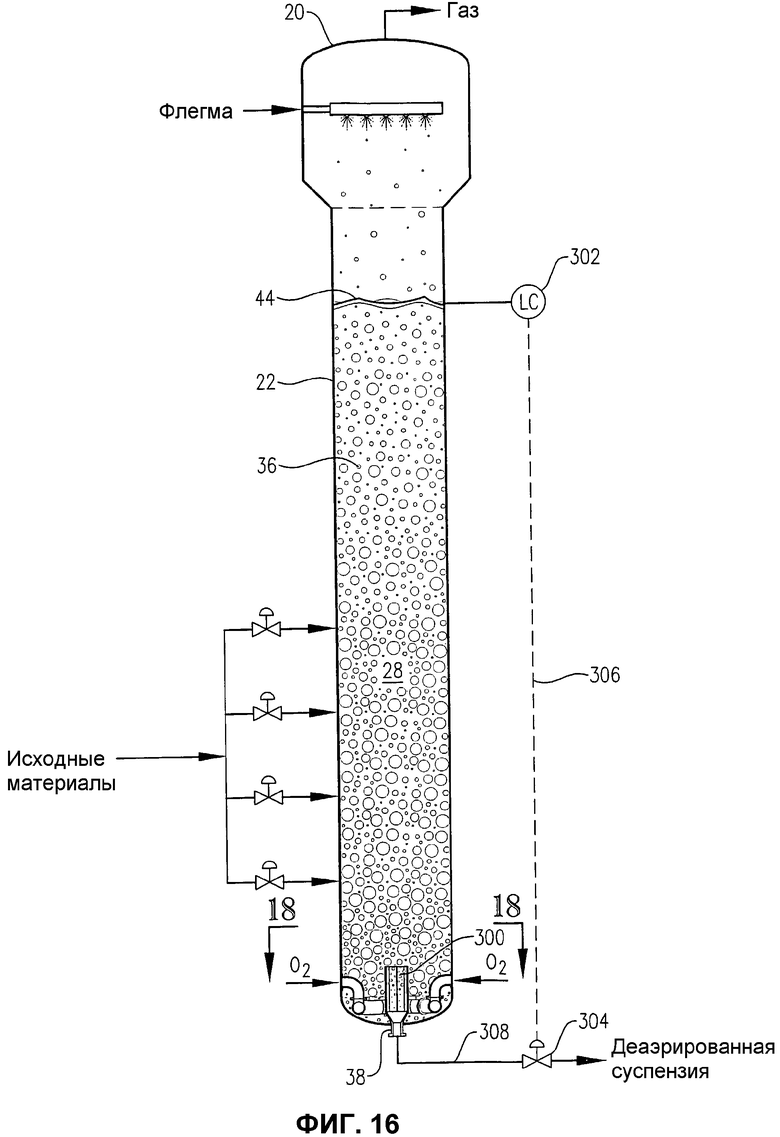
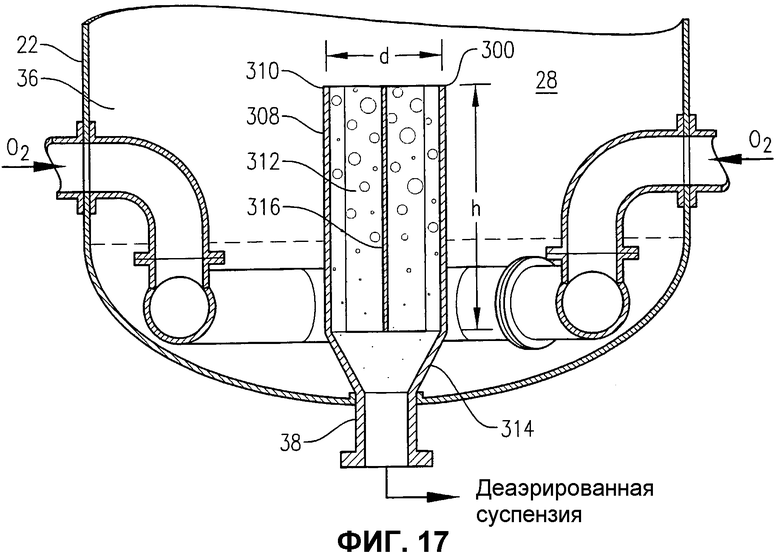
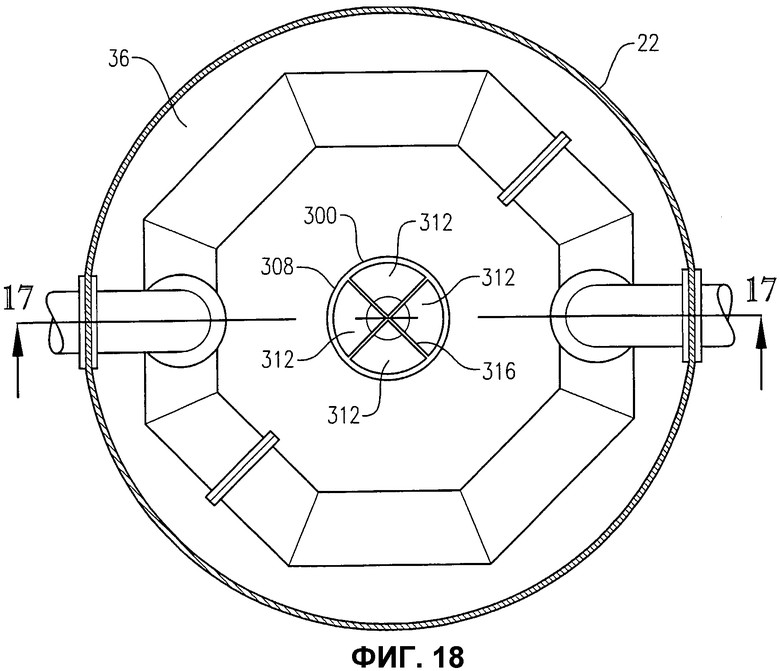
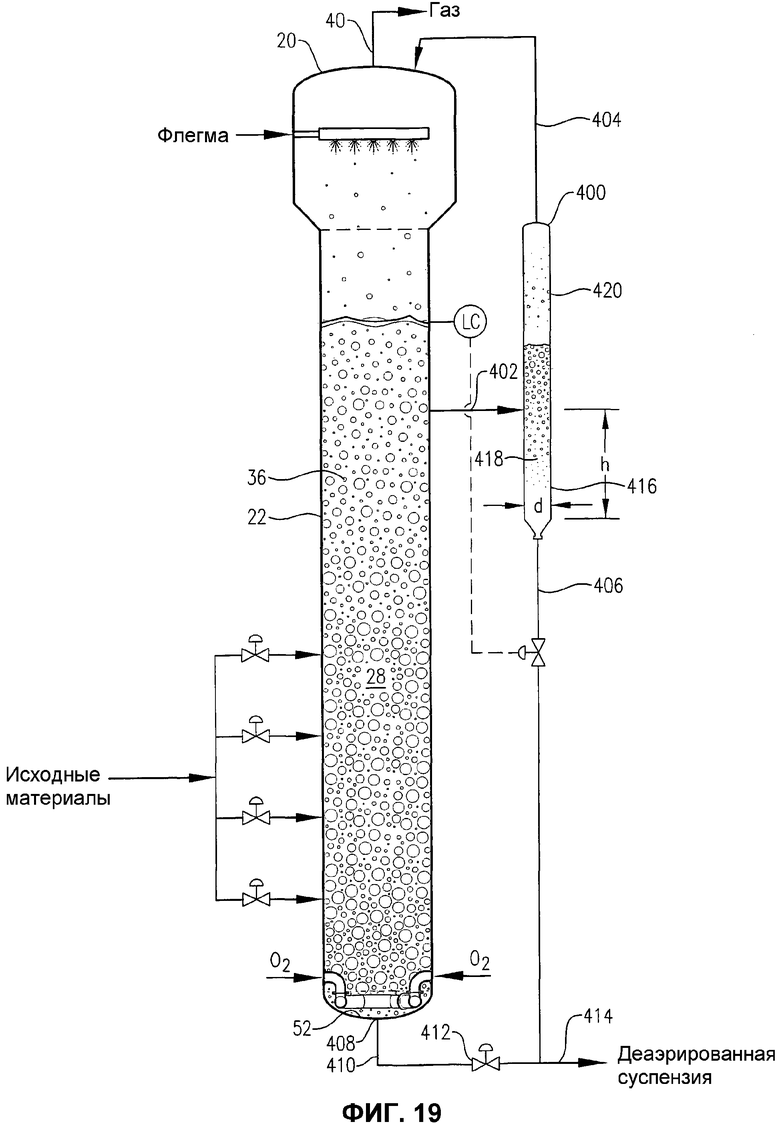
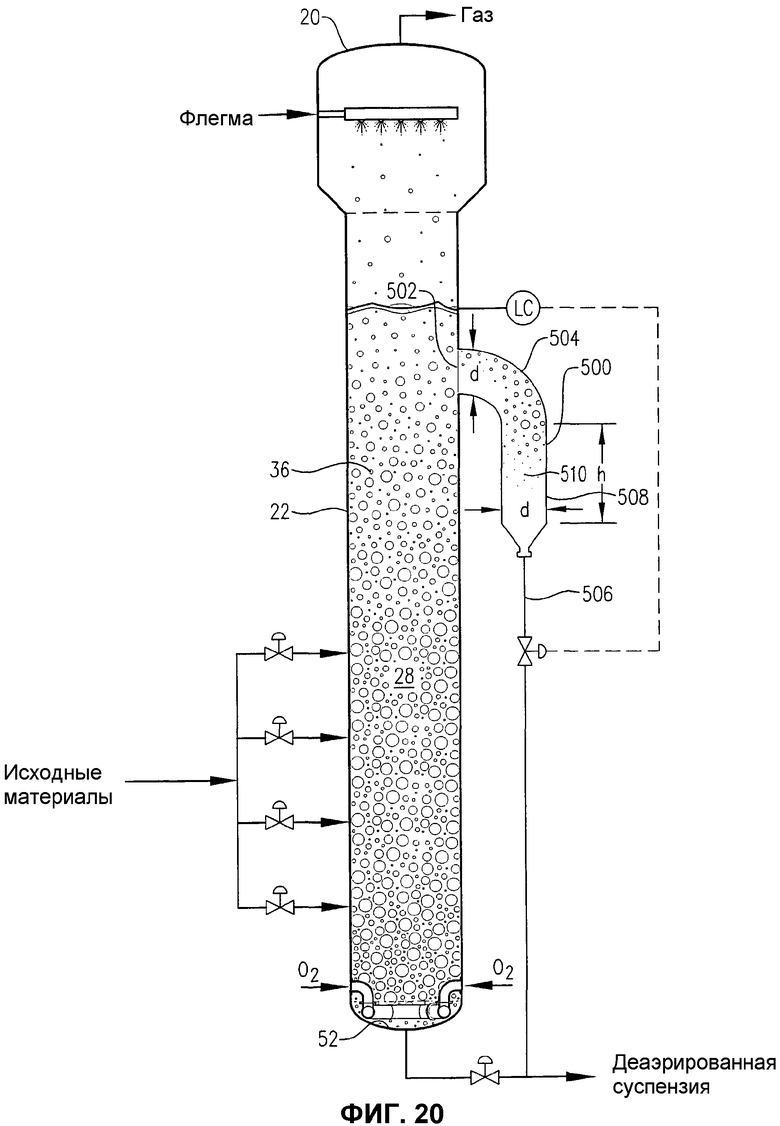
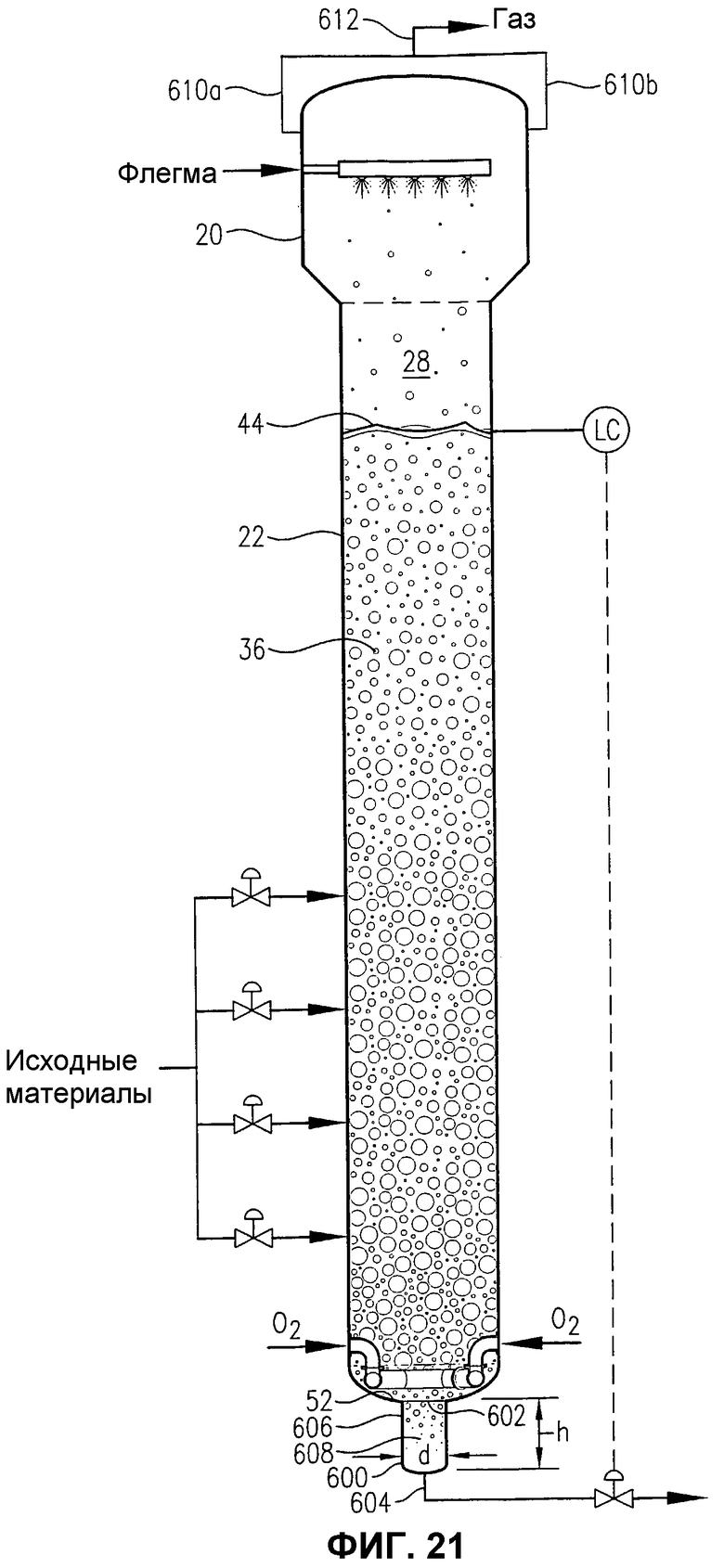
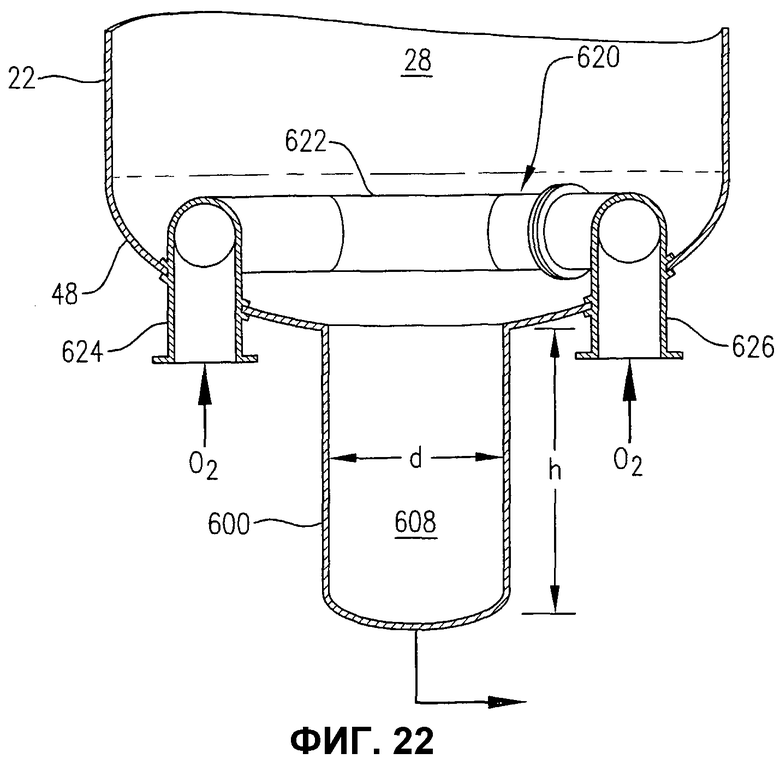
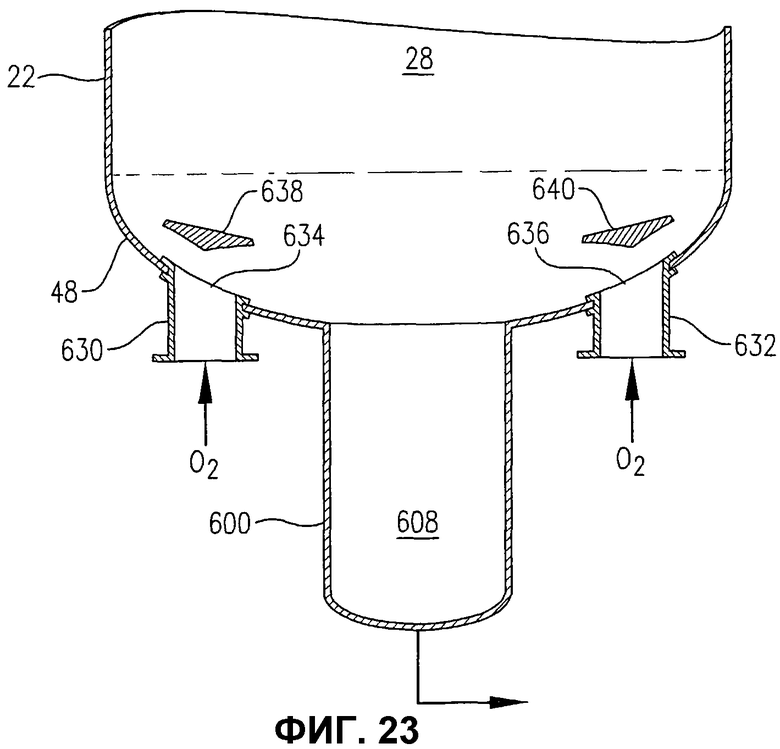
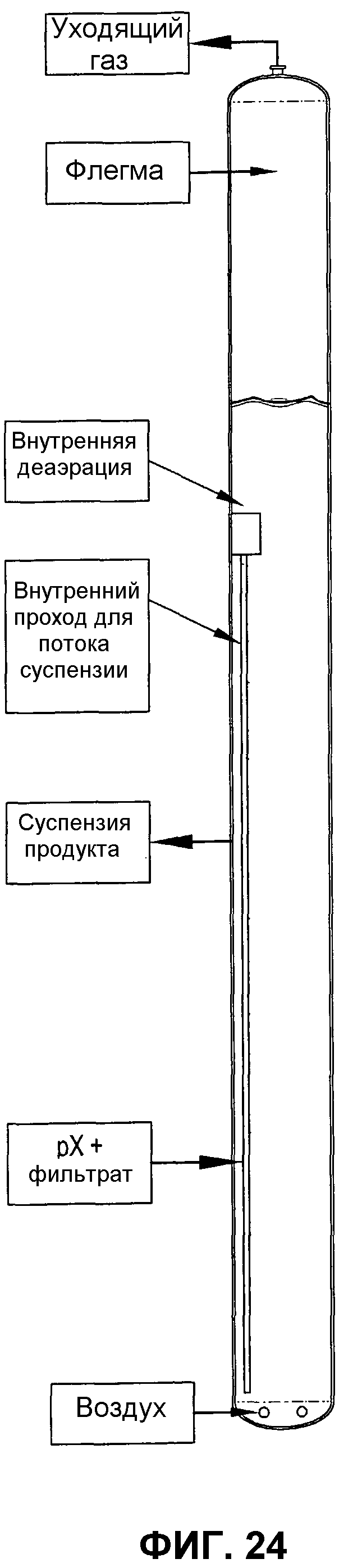
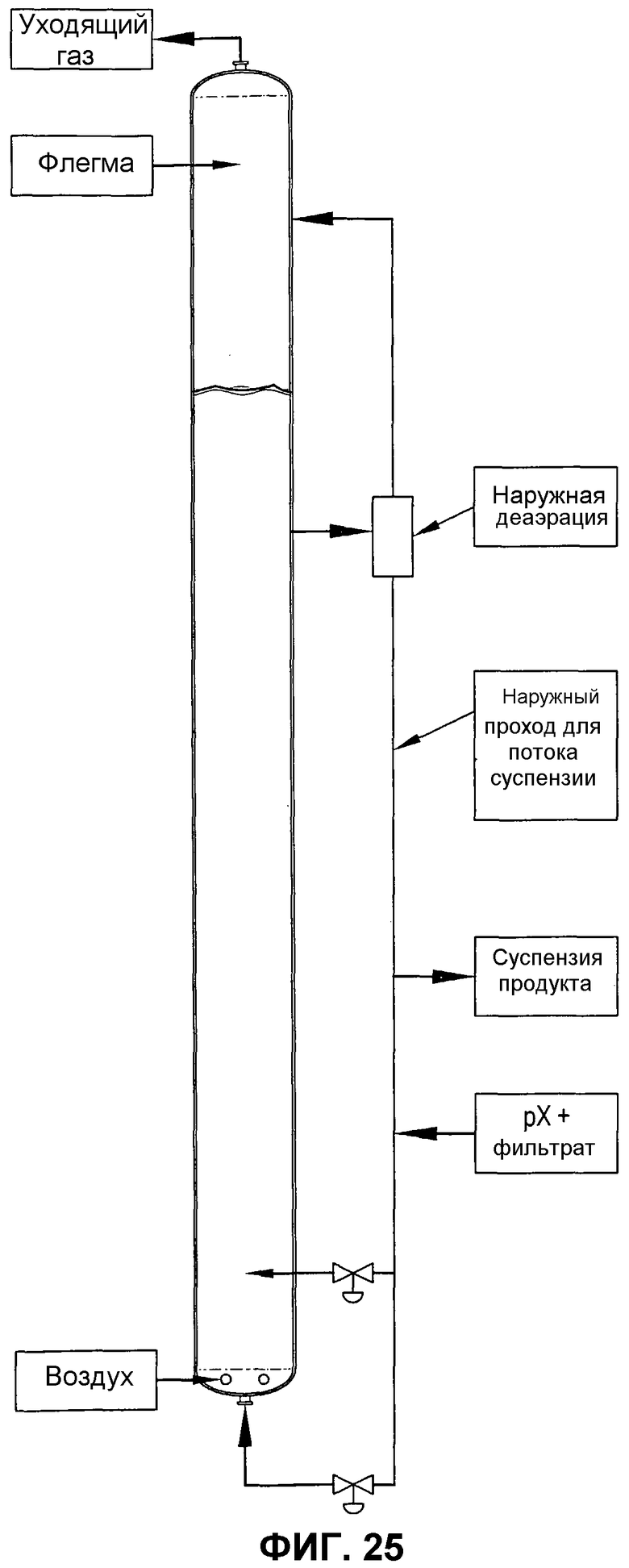
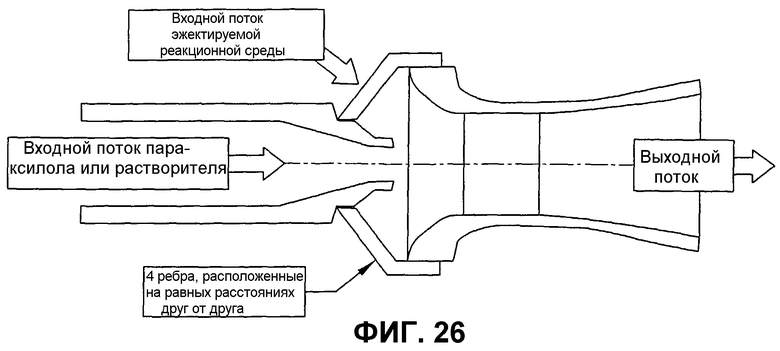
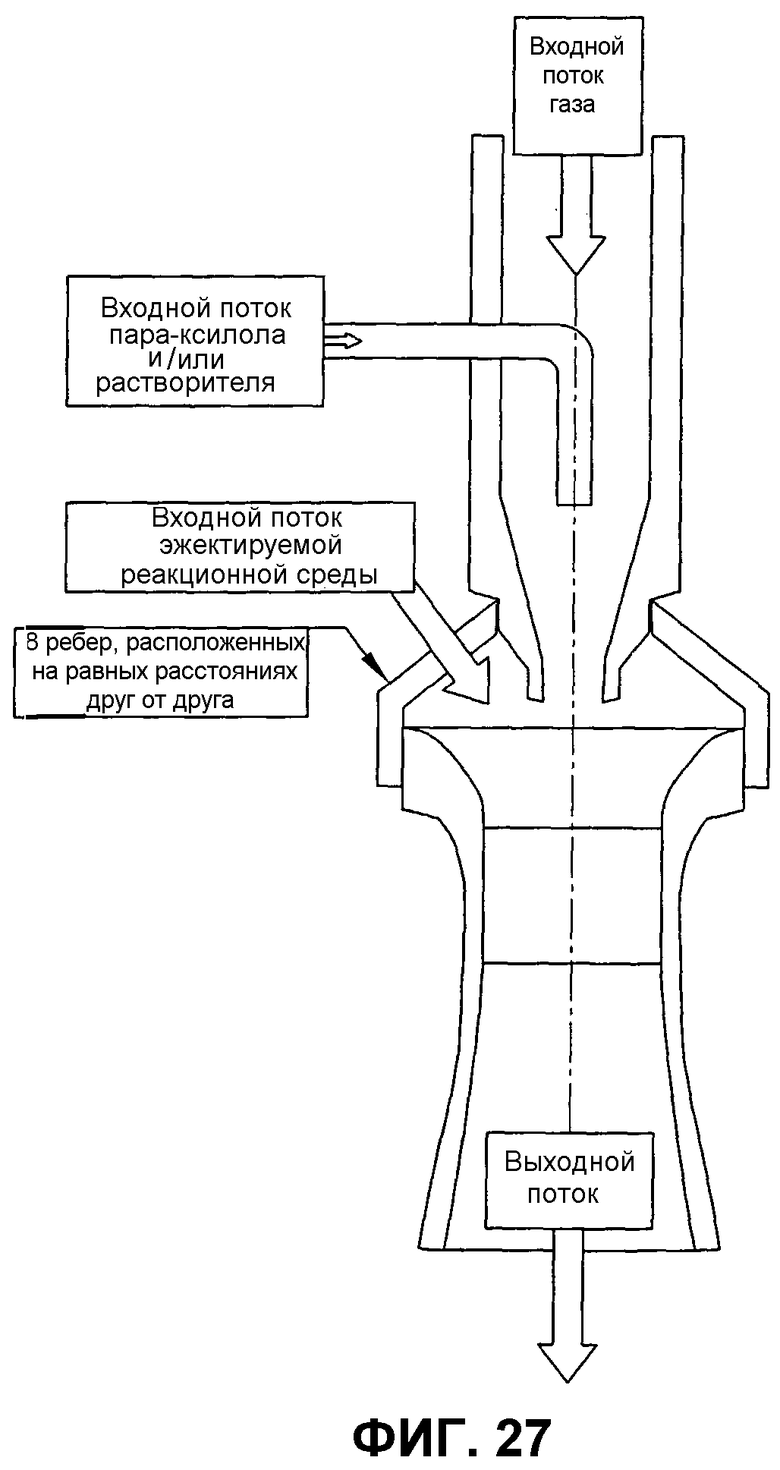
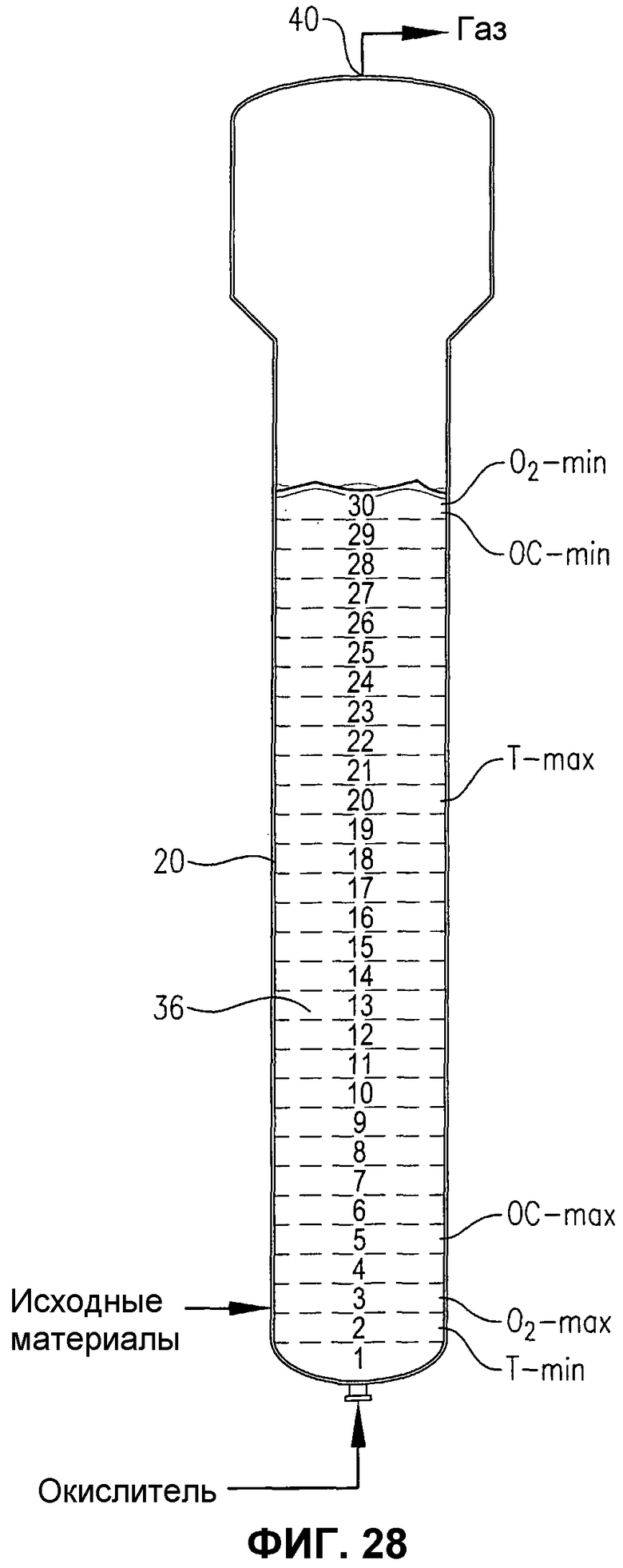
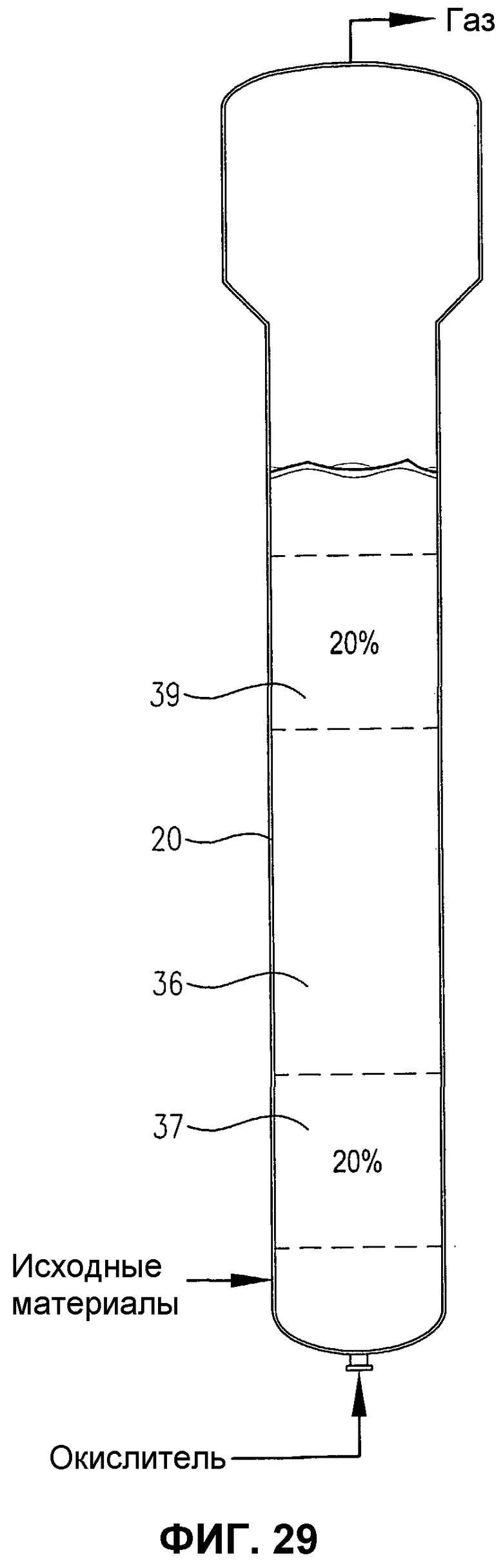
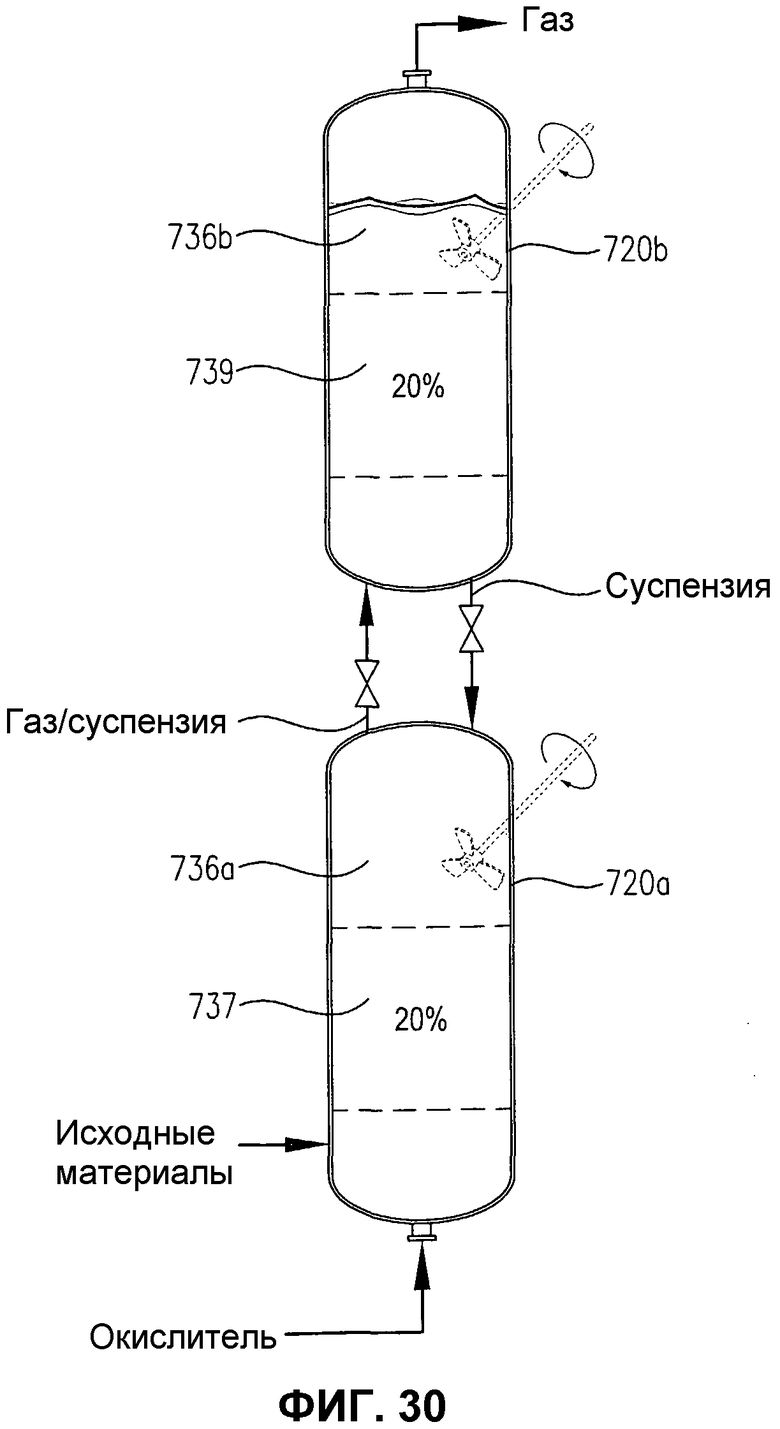
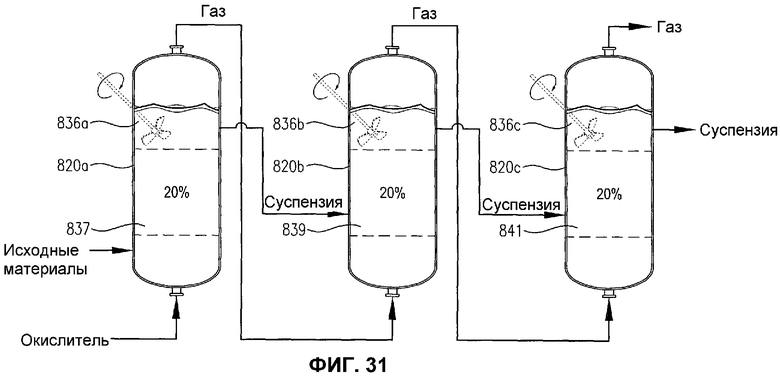




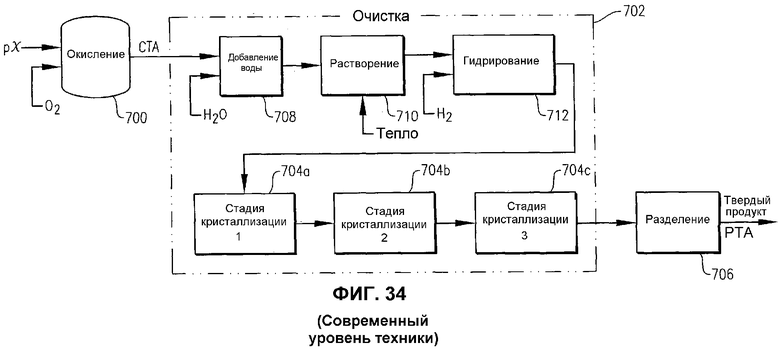
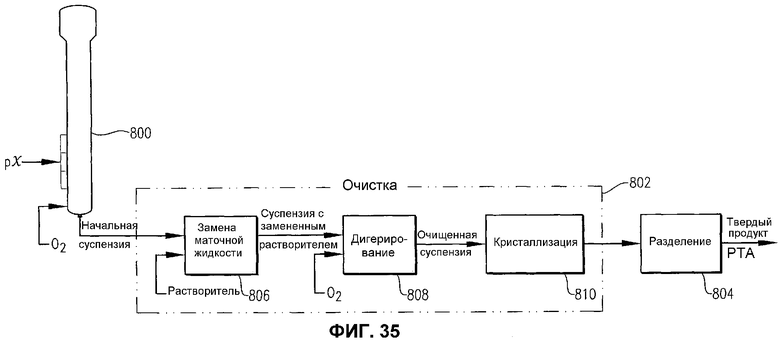
Группа изобретений относится к окислению пара-ксилола с получением сырой терефталевой кислоты в барботажной колонне реакторного типа, которые обеспечивают более эффективное и экономичное жидкофазное окисление окисляемого соединения при относительно низких температурах. В реакционную зону с максимальным диаметром D барботажной колонны реакторного типа вводят преимущественно газофазный поток окислителя, содержащий молекулярный кислород, и преимущественно жидкофазный поток исходных материалов, содержащих пара-ксилол, и, по меньшей мере, часть пара-ксилола окисляют в жидкой фазе многофазной реакционной среды, содержащейся в реакционной зоне, с получением сырой терефталевой кислоты. Исходные материалы вводят в реакционную зону через множество входных отверстий, по меньшей мере, два из которых отдалены друг от друга по вертикали на, по меньшей мере, примерно 0,5 D. По меньшей мере, часть реакционной зоны определена одной или несколькими расположенными вертикально боковыми стенками барботажной колонны, при этом, по меньшей мере, примерно 25 мас.% пара-ксилола подают в упомянутую реакционную зону в одном или нескольких местах, расположенных внутрь от вертикальных боковых стенок на расстоянии по меньшей мере 0,05 D. Окисление ведут в объеме реакционной среды с максимальной высотой Н, максимальной шириной W и отношением H:W, равным, по меньшей мере, примерно 3:1. 2 н. и 29 з.п. ф-лы, 35 ил., 7 табл.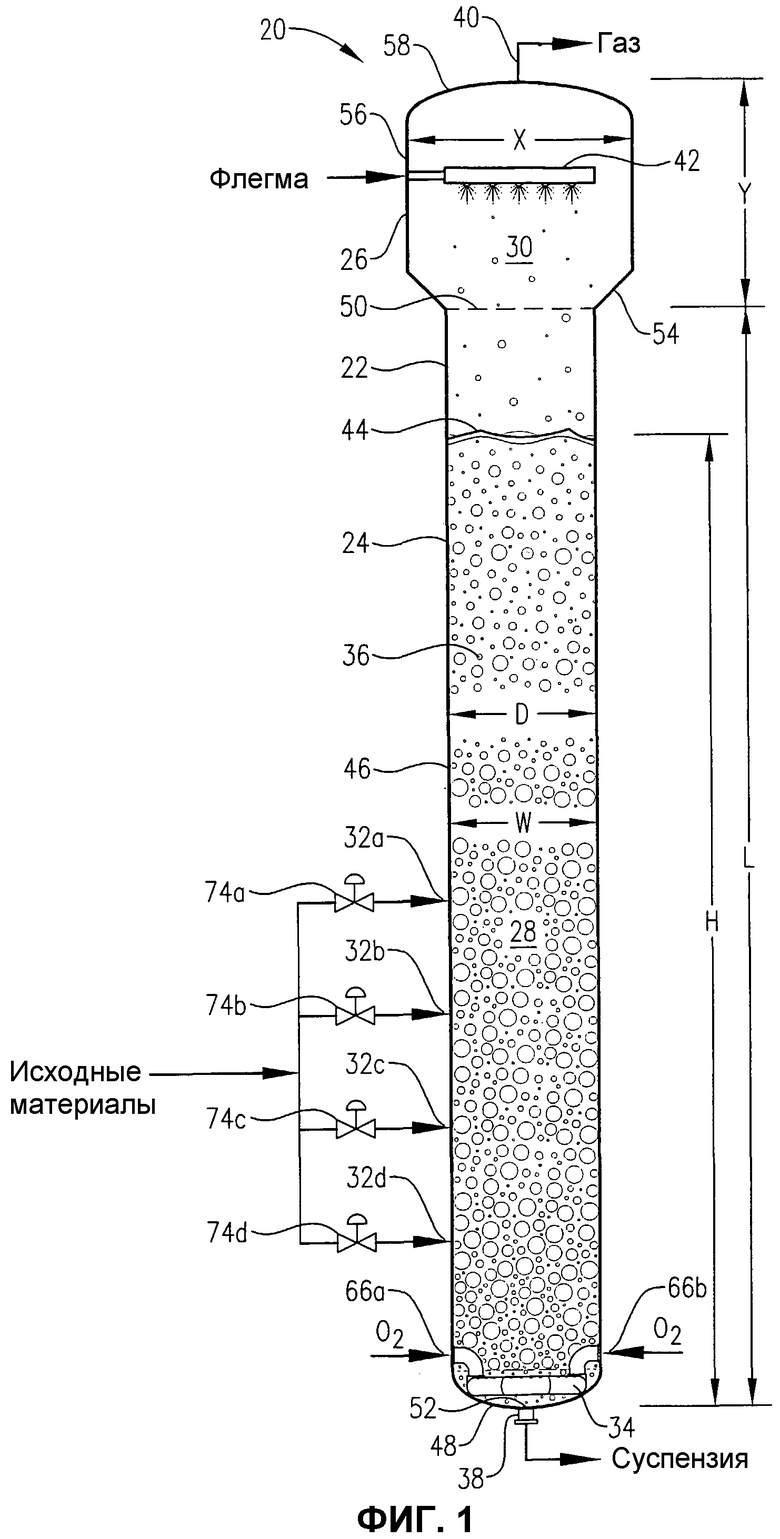
1. Способ окисления параксилола с получением сырой терефталевой кислоты, включающий введение в реакционную зону с максимальным диаметром D барботажной колонны реакторного типа преимущественно газофазного потока окислителя, содержащего молекулярный кислород, и преимущественно жидкофазного потока исходных материалов, содержащих параксилол, и окисление, по меньшей мере, части параксилола в жидкой фазе многофазной реакционной среды, содержащейся в реакционной зоне, с получением сырой терефталевой кислоты, отличающийся тем, что исходные материалы вводят в реакционную зону через множество входных отверстий, по меньшей мере два из которых отдалены друг от друга по вертикали на, по меньшей мере, примерно 0,5 D, при этом, по меньшей мере, часть реакционной зоны определена одной или несколькими расположенными вертикально боковыми стенками барботажной колонны, а, по меньшей мере, примерно 25 мас.% параксилола подают в упомянутую реакционную зону в одном или нескольких местах, расположенных внутрь от вертикальных боковых стенок на расстоянии по меньшей мере 0,05 D, причем окисление ведут в объеме реакционной среды с максимальной высотой Н, максимальной шириной W и соотношением H:W, по меньшей мере, примерно 3:1.
2. Способ по п.1, отличающийся тем, что, по меньшей мере, часть потока исходных материалов подают в реакционную зону с поверхностной скоростью на входе, по меньшей мере, примерно 5 м/с.
3. Способ по п.1, отличающийся тем, что, по меньшей мере, два входных отверстия для ввода исходных материалов отдалены друг от друга по вертикали на, по меньшей мере, примерно 1,5 D и, по меньшей мере, примерно 30 мас.% параксилола подают в реакционную зону на расстоянии примерно 1,5 D от самого нижнего положения ввода молекулярного кислорода в реакционную зону, при этом, по меньшей мере, примерно 50 мас.% параксилола подают в реакционную зону в одном или нескольких местах, расположенных внутрь от вертикальных боковых стенок на расстоянии, по меньшей мере, 0,05 D при соотношении H:W от примерно 7:1 до примерно 25:1.
4. Способ по п.1, отличающийся тем, что большая часть молекулярного кислорода поступает в реакционную зону в пределах примерно 0,25 W от нижней части реакционной зоны.
5. Способ по п.1, отличающийся тем, что большую часть молекулярного кислорода подают в реакционную зону на расстоянии примерно 0,2 W и примерно 0,02 Н от нижней части реакционной зоны.
6. Способ по п.1, отличающийся тем, что извлекают, по меньшей мере, часть реакционной среды из реакционной зоны через приподнятый по высоте выход, расположенный выше нижней части реакционной зоны.
7. Способ по п.6, отличающийся тем, что приподнятый по высоте выход расположен, по меньшей мере, примерно на 2 D выше нижней части реакционной зоны.
8. Способ по п.1, отличающийся тем, что газовую фазу реакционной среды подают с усредненной по времени поверхностной скоростью, по меньшей мере, примерно 0,3 м/с на половинной высоте реакционной зоны.
9. Способ по п.1, отличающийся тем, что подают исходные материалы, содержащие от примерно 60 до примерно 98 мас.% растворителя и от примерно 2 до примерно 40 мас.% параксилола.
10. Способ по п.9, отличающийся тем, что растворитель содержит уксусную кислоту.
11. Способ по п.1, отличающийся тем, что поток окислителя содержит меньше примерно 50 моль% молекулярного кислорода.
12. Способ по п.1, отличающийся тем, что окисление осуществляют в присутствии системы катализаторов, содержащей кобальт.
13. Способ по п.12, отличающийся тем, что система катализаторов дополнительно содержит бром и марганец.
14. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования по меньшей мере примерно 10 мас.% параксилола твердых частиц сырой терефталевой кислоты в реакционной среде.
15. Способ по п.1, отличающийся тем, что окисление осуществляют таким образом, что при теоретическом разделении всего объема реакционной среды на 30 горизонтальных слоев равного объема горизонтальный слой O2-max имеет максимальную концентрацию кислорода из всех горизонтальных слоев, а горизонтальный слой O2-min имеет минимальную концентрацию кислорода из всех горизонтальных слоев, расположенных выше горизонтального слоя O2-max, при этом концентрацию кислорода в молях измеряют в газовой фазе реакционной среды путем усреднения ее по времени и по объему во влажном состоянии, а отношение концентрации кислорода горизонтального слоя O2-max к концентрации кислорода горизонтального слоя O2-min поддерживают, по меньшей мере, примерно 2:1.
16. Способ по п.1, отличающийся тем, что окисление осуществляют таким образом, что при теоретическом разделении всего объема реакционной среды на 30 горизонтальных слоев равного объема горизонтальный слой рХ-max имеет максимальную концентрацию параксилола из всех горизонтальных слоев, а горизонтальный слой pX-min имеет минимальную концентрацию параксилола из всех горизонтальных слоев, расположенных выше горизонтального слоя рХ-max, при этом концентрацию параксилола по массе измеряют в жидкой фазе реакционной среды путем усреднения ее по времени и по объему, а отношение концентрации пара-ксилола горизонтального слоя рХ-max к концентрации пара-ксилола горизонтального слоя pX-min поддерживают, по меньшей мере, примерно 5:1.
17. Способ по п.1, отличающийся тем, что поток исходных материалов вводят в реакционную зону таким образом, что при теоретическом разделении реакционной зоны на 4 вертикальных квадранта равного объема посредством пары пересекающихся вертикальных плоскостей не более примерно 80 мас.% указанного параксилола подают в реакционную зону в один из вертикальных квадрантов.
18. Способ по п.1, отличающийся тем, что давление в нижней части реакционной среды поддерживают, по меньшей мере, на примерно 0,4 бар больше, чем давление в верхней части реакционной среды.
19. Способ по п.1, отличающийся тем, что, по меньшей мере, часть терефталевой кислоты дополнительно окисляют в реакторе вторичного окисления.
20. Способ по п.19, отличающийся тем, что окисление в реакторе вторичного окисления осуществляют при средней температуре, по меньшей мере, примерно на 10°С больше, чем окисление в реакторе начального окисления.
21. Способ по п.19, отличающийся тем, что окисление в реакторе вторичного окисления осуществляют при средней температуре, превышающей на примерно от 20 до примерно 80°С среднюю температуру реактора начального окисления, при этом окисление в реакторе начального окисления осуществляют при средней температуре от примерно 140 до примерно 180°С, а окисление в реакторе вторичного окисления осуществляют при средней температуре от примерно 180 до примерно 220°С.
22. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования в реакционной среде твердых частиц сырой терефталевой кислоты, при этом представительный образец упомянутых частиц дополнительно содержит меньше примерно 12 млн-1 4,4-дикарбоксистильбена, и/или меньше примерно 800 млн-1 изофталевой кислоты, и/или меньше примерно 100 млн-1 2,6-дикарбоксифлуоренона, и/или имеет коэффициент пропускания на 340 нм (%Т340) больше примерно 25%.
23. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования в реакционной среде твердых частиц сырой терефталевой кислоты, при этом упомянутые частицы характеризуются растворимостью в тетрагидрофуране до концентрации, по меньшей мере, примерно 500 млн-1 за одну минуту.
24. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования в реакционной среде твердых частиц сырой терефталевой кислоты, при этом упомянутые частицы характеризуются временной константой регрессии "С", большей примерно 0,5 мин-1.
25. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования в реакционной среде твердых частиц сырой терефталевой кислоты, при этом упомянутые частицы имеют среднюю площадь поверхности по БЭТ больше примерно 0,6 м2/г.
26. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования в реакционной среде твердых частиц сырой терефталевой кислоты, при этом упомянутые частицы имеют средний размер от примерно 20 до примерно 150 мкм.
27. Способ по п.1, отличающийся тем, что окисление проводят с обеспечением образования в реакционной среде твердых частиц сырой терефталевой кислоты, при этом 90% упомянутых частиц имеют размер от примерно 30 до примерно 150 мкм.
28. Барботажная колонна реакторного типа для окисления преимущественно жидкофазного потока преимущественно газофазным потоком, содержащая корпус, определяющий удлиненную реакционную зону, простирающуюся вдоль расположенной в целом вертикально центральной оси корпуса, множество отверстий для введения жидкофазного потока в реакционную зону и множество отверстий для введения газофазного потока в реакционную зону, отличающаяся тем, что реакционная зона имеет максимальную длину L, измеряемую параллельно оси корпуса, и максимальный диаметр D, измеряемый перпендикулярно оси корпуса, при отношении L:D от примерно 6:1 до примерно 30:1, по меньшей мере два отверстия для введения жидкофазного потока отделены друг от друга аксиально на, по меньшей мере, примерно 0,5 D, при этом, по меньшей мере, часть реакционной зоны определена одной или несколькими расположенными вертикально боковыми стенками корпуса и, по меньшей мере, примерно 25% от общей открытой площади отверстий для введения жидкофазного потока расположены внутрь от вертикальных боковых стенок на расстоянии по меньшей мере 0,05 D, a реакционная зона имеет первый и второй противоположные уровни, расположенные на максимальной длине L друг от друга, причем большая часть общей открытой площади отверстий для введения газофазного потока расположена на расстоянии примерно 0,25 D от первого уровня реакционной зоны.
29. Барботажная колонна по п.28, отличающаяся тем, что отношение L:D составляет от примерно 8:1 до примерно 20:1, при этом, по меньшей мере, два входных отверстия для введения жидкофазного потока аксиально отделены друг от друга, по меньшей мере, примерно на 1,5 D, большая часть общей открытой площади отверстий для введения газофазного потока расположена на расстоянии примерно 0,2 D от первого уровня реакционной зоны, а по меньшей мере, примерно 30% от общей открытой площади отверстий для введения жидкофазного потока расположены на расстоянии примерно 1,5 D от отверстия для введения газофазного потока, расположенного ближе всех к первому уровню реакционной зоны.
30. Барботажная колонна по п.28, отличающаяся тем, что в реакторе выполнен приподнятый выход, аксиально отделенный, по меньшей мере, на примерно 1 D от первого уровня реакционной зоны.
31. Барботажная колонна по п.28, отличающаяся тем, что при теоретическом разделении реакционной зоны на 4 вертикальных квадранта равного объема посредством пары пересекающихся вертикальных плоскостей не более примерно 80% общей открытой площади отверстий для введения жидкофазного потока расположены в одном из вертикальных квадрантов.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| EP 0847800 A2,17.06.1996 | |||
| US 6375921 B1, 23.04.2002 | |||
| СПОСОБ ПОЛУЧЕНИЯ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1994 |
|
RU2079480C1 |
| Реактор жидкофазного окисления | 1981 |
|
SU1012967A1 |

