Настоящее изобретение относится к соединению формулы I
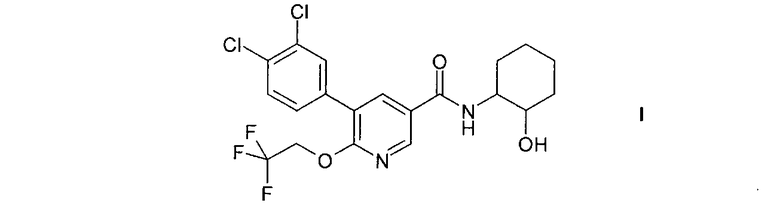
и к его изомерным формам и фармацевтически приемлемым солям, к их получению, содержащим их фармацевтическим композициям и к их применению в качестве лекарственных средств. Соединение формулы I, его изомерные формы и фармацевтически приемлемые соли являются особенно подходящими для применения в качестве средств, повышающих концентрацию ЛВП холестерина.
Атеросклероз и связанная с ним ишемическая болезнь сердца являются основной причиной смерти в промышленно-развитых странах. Показано, что риск возникновения ишемической болезни сердца хорошо коррелирует с концентрациями некоторых липидов в плазме. Липиды переносятся в крови липопротеинами. Общая структура липопротеинов представляет собой ядро, образованное нейтральными липидами (триглицерид и холестериловый эфир) и оболочку, состоящую из полярных липидов (фосфолипидов и неэтерифицированного холестерина). Существуют 3 разных класса липопротеинов плазмы, обладающих разными содержаниями липидов в ядре: липопротеин низкой плотности (ЛНП), который обогащен холестериловым эфиром (ХЭ); липопротеин высокой плотности (ЛВП), который также обогащен холестериловым эфиром (ХЭ); и липопротеин очень низкой плотности (ЛОНП), который обогащен триглицеридом (ТГ). Разные липопротеины можно разделить с помощью флотации, основанной на различиях их плотностей или размеров.
Высокие концентрации ЛНП-холестерина (ЛНП-Х) и триглицеридов положительно коррелируют, а высокие концентрации ЛВП-холестерина (ЛВП-Х) отрицательно коррелируют с риском развития сердечно-сосудистых заболеваний.
Отсутствуют полностью удовлетворительные терапевтические средства, приводящие к повышению концентрации ЛВП. Ниацин может значительно повысить концентрацию ЛВП, но он плохо переносится, что ухудшает соблюдение режима лечения. Фибраты и ингибиторы HMG СоА редуктазы приводят лишь к умеренному повышению концентрации ЛВП (10-12%). Вследствие этого для медицины все еще необходимо хорошо переносимое средство, которое может значительно повысить концентрацию ЛВП в плазме.
Таким образом, средства, повышающие концентрацию ЛВП холестерина, можно применять в качестве лекарственных средств для лечения и/или профилактики атеросклероза, заболевания периферических сосудов, дислипидемии, гипербеталипопротеинемии, гипоальфалипопротеинемии, гиперхолестеринемии, гипертриглицеридемии, семейной гиперхолестеринемии, сердечно-сосудистых нарушений, стенокардии, ишемии, ишемической болезни сердца, удара, инфаркта миокарда, реперфузионного поражения, рестеноза после ангиопластики, гипертензии и сосудистых осложнений диабета, ожирения или эндотоксикоза.
Кроме того, средства, повышающие концентрацию ЛВП холестерина, можно использовать в комбинации с другим соединением, где такое соединение представляет собой ингибитор HMG-CoA редуктазы, ингибитор микросомного белка-переносчика триглицеридов (МТР)/секреции АроВ, активатор PPAR, ингибитор повторного поглощения желчных кислот, ингибитор белка-переносчика холестерилового эфира (СЕТР), ингибитор поглощения холестерина, ингибитор синтеза холестерина, фибрат, ниацин, препараты, содержащие ниацин или другие агонисты НМ74а, ионообменную смолу, антиоксидант, ингибитор АСАТ или вещество, усиливающее экскрецию желчных кислот.
Поэтому объектом настоящего изобретения является получение соединения, которое является активным средством, повышающим концентрацию ЛВП холестерина. Согласно изобретению было установлено, что соединение формулы I, предлагаемое в настоящем изобретении, является особенно подходящим для лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП холестерина, т.е. соединение формулы I является особенно подходящим для лечения и/или предупреждения дислипидемии, атеросклероза и сердечно-сосудистых заболеваний. Объектом настоящего изобретения также является получение соединения, которое в терапевтически активных концентрациях, которые повышают концентрации ЛВП, не взаимодействует с рецептором СВ1. Это обусловлено тем, что лиганды рецептора СВ1 могут ухудшать терапевтическую применимость средств, повышающих концентрацию ЛВП холестерина, поскольку и агонисты, и антагонисты рецептора СВ1 могут привести к побочным эффектам.
Соединения с общими структурными элементами раскрыты в качестве антагонистов рецептора СВ1 (WO 2006/106054) и смешанных антагонистов рецептора CB1/средств, повышающих концентрацию ЛВП холестерина (WO 2008/040651).
Если не указано иное, приведенные ниже определения предназначены для иллюстрации и определения значения и объема различных терминов, использующихся для описания настоящего изобретения.
"Изомерными формами" являются все формы соединения, которые обладают идентичными молекулярными формулами, но различаются по природе или последовательности связывания своих атомов или по расположению своих атомов в пространстве. Предпочтительно, если изомерные формы различаются по расположению своих атомов в пространстве и их также можно назвать "стереоизомерами". Стереоизомеры, которые не являются зеркальными отображениями друг друга, называются "диастереоизомерами" и стереоизомеры, которые не являются налагающимися друг на друга зеркальными отображениями, называются "энантиомерами", а иногда и оптическими изомерами. Атом углерода, обладающий четырьмя разными заместителями, называется "хиральным центром".
Термин "фармацевтически приемлемые соли" означает такие соли, которые сохраняют биологическую эффективность и характеристики свободных оснований или свободных кислот и которые не являются нежелательными в биологическом или другом отношении. Эти соли образуются с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., предпочтительно хлористоводородная кислота, и органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, салициловая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т.п. Таким образов, предпочтительные "фармацевтически приемлемые соли" включают ацетат, бромид, хлорид, формиат, фумарат, малеат, мезилат, нитрат, оксалат, фосфат, сульфат, тартрат и тозилат соединений формулы I. Кроме того, фармацевтически приемлемые соли можно получить путем добавления неорганического основания или органического основания к свободной кислоте. Соли, образованные из неорганического основания, включают, но не ограничиваются только соли натрия, калия, лития, аммония, кальция, магния и т.п. Соли, образованные из органических оснований, включают, но не ограничиваются только ими соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, диэтиламин, лизин, аргинин, N-этилпиперидин, пиперидин, пиперазин и т.п. Соединение формулы I также может содержаться в форме цвиттерионов или в форме гидратов. Особенно предпочтительными фармацевтически приемлемыми солями соединений формулы I являются гидрохлориды.
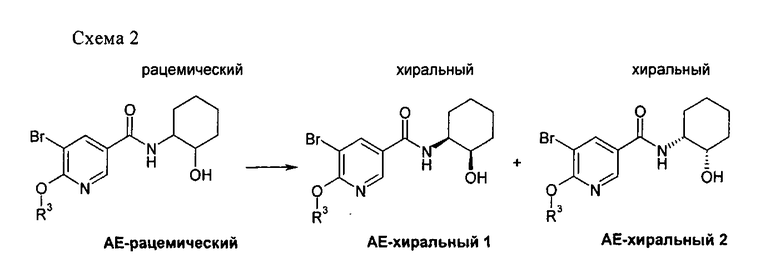
Предпочтительным объектом настоящего изобретения является 5-(3,4-дихлорфенил)-N-((1R,2)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид, т.е. соединение формулы I в изомерной форме 1а.
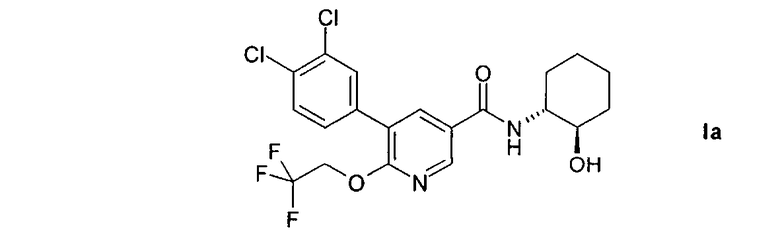
Настоящее изобретение также относится к 5-(3,4-дихлорфенил)-N-((1R,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамиду и его фармацевтически приемлемым солям.
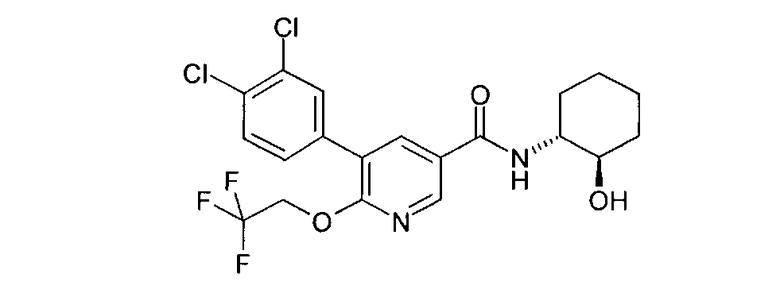
Другим предпочтительным объектом настоящего изобретения является 5-(3,4-дихлорфенил)-N-((1S,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид, т.е. соединение формулы I в изомерной форме Ib.
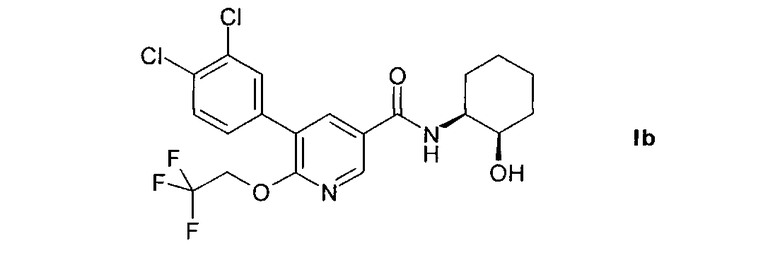
Настоящее изобретение также относится к 5-(3,4-дихлорфенил)-N-((1S,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамиду и его фармацевтически приемлемым солям.
Соединение формулы I можно получить способом, который включает введение в реакцию сочетания соединения формулы
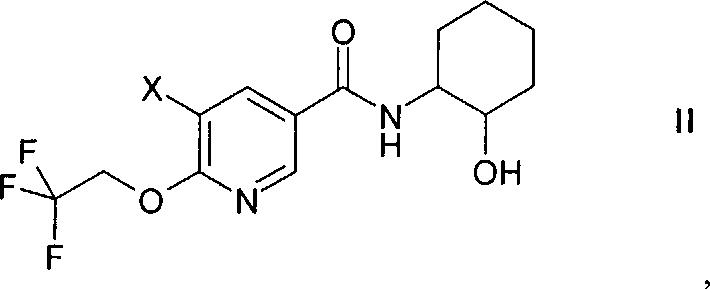
в которой X обозначает галоген, с содержащим металл арилом формулы

в которой М обозначает бороновую кислоту или сложный эфир бороновой кислоты, в присутствии содержащего Pd катализатора в щелочной среде,
и необязательно разделение изомеров на хиральной колонке ВЭЖХ,
и если необходимо, превращение полученного соединения формулы I в его фармацевтически приемлемую соль.
Содержащим металл арил, предпочтительно представляет собой арилбороновую кислоту или сложный эфир арилбороновой кислоты. Содержащий палладий катализатор предпочтительно представляет собой смесь ацетат палладия(II)/трифенилфосфин или комплекс хлорид палладия(II)-dppf, который используют в присутствии основания, предпочтительно триэтиламина или карбоната натрия. X обозначает галоген, более предпочтительно, если X обозначает бром или йод.
Синтез соединений общей структуры I можно провести по приведенным ниже схемам 1 и 2.
По методике, представленной на схеме 1, соединение АА (5-бром-6-хлор-3-пиридинкарбоновая кислота, CAS RN 29241-62-1) можно использовать в качестве исходного вещества. Соединение АА имеется в продаже, или его альтернативно можно получить с помощью последовательности нескольких реакций из 6-гидрокси-3-пиридинкарбоновой кислоты по описанным в литературе методикам.
Соединение АС можно получить из соединения АА по реакции с подходящим замещенным первичным или вторичным спиртом формулы АВ в присутствии основания, например гидроксида калия, в инертном растворителе, например диметилсульфоксиде, при температурах от комнатной температуры до температуры кипения растворителя, предпочтительно при комнатной температуре.
Соединение АЕ можно получить по реакции сочетания АС с соответствующим амином формулы AD с помощью подходящих реакций образования амидной связи. Эти реакции известны в данной области техники. Например, для проведения такого превращения можно использовать реагенты сочетания, такие как N,N'-карбонилдиимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (EDCI), 1-[бис(диметиламино)-метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид гексафторфосфат (HATU), 1-гидрокси-1,2,3-бензотриазол (НОВТ) и 0-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийтетрафторборат (TBTU). Обычной методикой является использование, например, TBTU и основания, например основания Хюнига (N-этилдиизопропиламин) в инертном растворителе, таком как например, диметилформамид, при комнатной температуре.
На следующей стадии соединения формулы I получают по реакции сочетания подходящего замещенного содержащего металл арила формулы AF, предпочтительно арилбороновой кислоты или сложного эфира арилбороновой кислоты, с АЕ в присутствии подходящего катализатора, предпочтительно содержащего палладий катализатора и более предпочтительно смесей ацетат палладия(II)/трифенилфосфин или комплексов хлорид палладия(II)-dppf (1,1'-бис(дифенилфосфино)ферроцен) и основания, предпочтительно триэтиламина или карбоната натрия в инертном растворителе, таком как диметилформамид или толуол.
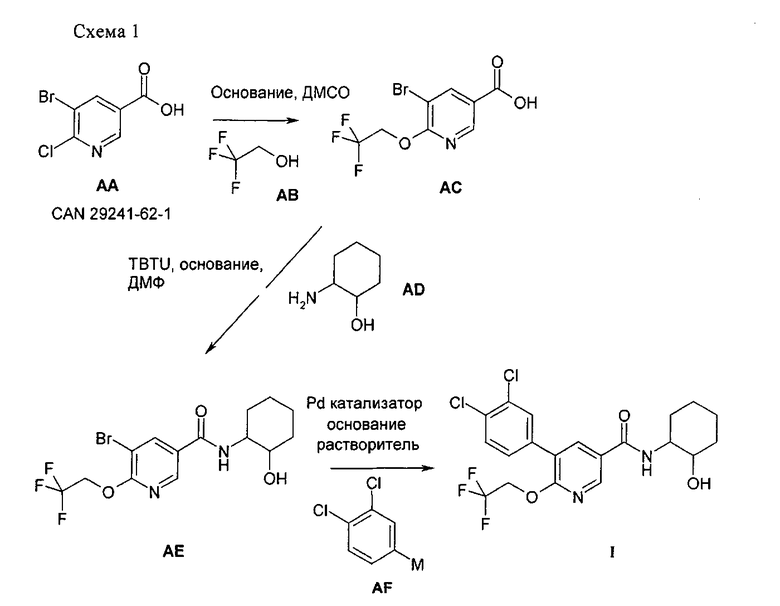
Соединения формулы АЕ или соединения I, которые получают в соответствии со схемой 1, в зависимости от природы амина AD могут содержать один или большее количество хиральных центров. Хиральные соединения АЕ-хиральный или I-хиральный можно получить по различным методикам, известным в данной области техники, таким как синтез из хиральных предшественников или методики хирального разделения. Хиральное разделение на хиральных колонках ВЭЖХ предпочтительно проводить для соединения, которое лучше растворимо в подвижной фазе. Соединения формулы АЕ обычно растворимы в смесях гептан/спирт лучше, чем соединения формулы I. Выделение соединений АЕ-хиральный 1 и АЕ-хиральный 2 из АЕ-рацемического можно провести в соответствии со схемой 2 с использованием подходящей хиральной колонки ВЭЖХ, такой как ChiralPak AD® или использующих аналогичные неподвижные фазы в периодическом режиме или по технологии псевдоожиженного слоя с использованием подходящих подвижных фаз, таких как смеси гептан/изопропанол.
Как отмечено выше, соединения формулы I, предлагаемые в настоящем изобретении, можно применять в качестве лекарственных средств для лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП холестерина. Примерами таких заболеваний являются атеросклероз, заболевание периферических сосудов, дислипидемия, гипербеталипопротеинемия, гипоальфалипопротеинемия, гиперхолестеринемия, гипертриглицеридемия, семейная гиперхолестеринемия, сердечно-сосудистые заболевания, такие как стенокардия, ишемия, ишемическая болезнь сердца, удар, инфаркт миокарда, реперфузионное поражение, рестеноз после ангиопластики, гипертензия и сосудистые осложнения диабета, ожирение или эндотоксикоз. Предпочтительным является применение в качестве лекарственного средства для лечения и/или предупреждения дислипидемии, атеросклероза и сердечно-сосудистых заболеваний.
Поэтому настоящее изобретение также относится к фармацевтическим композициям, включающим соединение, определенное выше, и фармацевтически приемлемый носитель и/или вспомогательное вещество, которые применимы для лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП холестерина.
Таким образом, настоящее изобретение относится к фармацевтической композиции, определенной выше, предназначенной для лечения и/или профилактики атеросклероза, заболевания периферических сосудов, дислипидемии, гипербеталипопротеинемии, гипоальфалипопротеинемии, гиперхолестеринемии, гипертриглицеридемии, семейной гиперхолестеринемии, сердечно-сосудистых заболеваний, таких как стенокардия, ишемия, ишемическая болезнь сердца, удар, инфаркт миокарда, реперфузионное поражение, рестеноз после ангиопластики, гипертензия и сосудистые осложнения диабета, ожирения или эндотоксикоза.
В другом варианте осуществления настоящее изобретение относится к способу лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП холестерина, способ включает введение нуждающемуся в нем пациенту соединения формулы I в терапевтически эффективном количестве. Примерами таких заболеваний являются атеросклероз, заболевание периферических сосудов, дислипидемия, гипербеталипопротеинемия, гипоальфалипопротеинемия, гиперхолестеринемия, гипертриглицеридемия, семейная гиперхолестеринемия, сердечно-сосудистые заболевания, такие как стенокардия, ишемия, ишемическая болезнь сердца, удар, инфаркт миокарда, реперфузионное поражение, рестеноз после ангиопластики, гипертензия и сосудистые осложнения диабета, ожирение или эндотоксикоз. Предпочтительным является способ лечения и/или профилактики дислипидемии, атеросклероза и сердечно-сосудистых заболеваний.
Кроме того, настоящее изобретение относится к применению соединений формулы I, определенных выше, для приготовления лекарственного средства, предназначенного для лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП. Примерами таких заболеваний являются атеросклероз, заболевание периферических сосудов, дислипидемия, гипербеталипопротеинемия, гипоальфалипопротеинемия, гиперхолестеринемия, гипертриглицеридемия, семейная гиперхолестеринемия, сердечно-сосудистые нарушения, такие как стенокардия, ишемия, ишемическая болезнь сердца, удар, инфаркт миокарда, реперфузионное поражение, рестеноз после ангиопластики, гипертензия и сосудистые осложнения диабета, ожирение или эндотоксикоз. Предпочтительным является применение соединений формулы I, определенных выше, для приготовления лекарственных средств, предназначенных для лечения и/или профилактики дислипидемии, атеросклероза и сердечно-сосудистых заболеваний.
Кроме того, средства, повышающие концентрацию ЛВП, формулы I применимы в комбинации или совместно с другим соединением, где такое соединение выбрано из группы, включающей ингибитор HMG-CoA редуктазы, ингибитор микросомного белка-переносчика триглицеридов (МТР)/секреции АроВ, активатор PPAR, ингибитор белка-переносчика холестерилового эфира (СЕТР), ингибитор повторного поглощения желчных кислот, ингибитор поглощения холестерина, ингибитор синтеза холестерина, фибрат, ниацин, препарат, содержащий ниацин или другие агонисты НМ74а, ионообменную смолу, антиоксидант, ингибитор АСАТ или вещество, усиливающее экскрецию желчных кислот
Поэтому настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы I, определенное выше, в комбинации или совместно с соединением, выбранным из группы, включающей ингибитор HMG-CoA редуктазы, ингибитор микросомного белка-переносчика триглицеридов (МТР)/секреции АроВ, активатор PPAR, ингибитор белка-переносчика холестерилового эфира (СЕТР), ингибитор повторного поглощения желчных кислот, ингибитор поглощения холестерина, ингибитор синтеза холестерина, фибрат, ниацин, препарат, содержащий ниацин или другие агонисты НМ74а, ионообменную смолу, антиоксидант, ингибитор АСАТ или вещество, усиливающее экскрецию желчных кислот, а также фармацевтически приемлемый носитель и/или вспомогательное вещество.
Настоящее изобретение также относится к применению соединений формулы I, определенных выше, в комбинации или совместно с соединением, выбранным из группы, включающей ингибитор HMG-CoA редуктазы, ингибитор микросомного белка-переносчика триглицеридов (МТР)/секреции АроВ, активатор PPAR, ингибитор белка-переносчика холестерилового эфира (СЕТР), ингибитор повторного поглощения желчных кислот, ингибитор поглощения холестерина, ингибитор синтеза холестерина, фибрат, ниацин, препарат, содержащий ниацин или другие агонисты НМ74а, ионообменную смолу, антиоксидант, ингибитор АСАТ или вещество, усиливающее экскрецию желчных кислот, для приготовления лекарственного средства, предназначенного для лечения и/или профилактики заболеваний, таких как атеросклероз, заболевание периферических сосудов, дислипидемия,
гипербеталипопротеинемия, гипоальфалипопротеинемия, гиперхолестеринемия, гипертриглицеридемия, семейная гиперхолестеринемия, сердечно-сосудистые нарушения, стенокардия, ишемия, ишемическая болезнь сердца, удар, инфаркт миокарда, реперфузионное поражение, рестеноз после ангиопластики, гипертензия и сосудистые осложнения диабета, ожирение или эндотоксикоз.
Настоящее изобретение также относится к способу лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП холестерина, который включает введение соединения формулы I в терапевтически эффективном количестве в комбинации или совместно с терапевтически эффективным количеством соединения, выбранного из группы, включающей ингибитор HMG-CoA редуктазы, ингибитор микросомного белка-переносчика триглицеридов (МТР)/секреции АроВ, активатор PPAR, ингибитор белка-переносчика холестерилового эфира (СЕТР), ингибитор повторного поглощения желчных кислот, ингибитор поглощения холестерина, ингибитор синтеза холестерина, фибрат, ниацин, препарат, содержащий ниацин или другие агонисты НМ74а, ионообменную смолу, антиоксидант, ингибитор АСАТ или вещество, усиливающее экскрецию желчных кислот.
Соединения формулы I и/или их фармацевтически приемлемые соли можно использовать в форме фармацевтических композиций для энтерального, парентерального или местного введения. Их можно вводить, например, перорально, например, в форме таблеток, таблеток с покрытием, драже, капсул из твердого или мягкого желатина, растворов, эмульсий, суспензий, перорально, например в полость рта, ректально, например, в форме суппозиториев, парентерально, например, в форме растворов для инъекции или растворов для вливания, предназначенных для внутримышечной, внутривенной или подножной инъекции, или местно, например, в форме мазей, кремов или масел. Предпочтительным является пероральное введение.
Приготовление фармацевтических композиций можно проводить по технологиям, которые известны любому специалисту в данной области техники, путем внесения описанных соединений формулы I и/или их фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически ценными веществами, в галеновые препаративные формы вместе с подходящими нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими носителями и при необходимости с обычными фармацевтическими вспомогательными веществами.
Подходящими носителями являются не только неорганические носители, но и органические носители. Так, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновые кислоты или их соли и т.п. можно использовать в качестве носителей для таблеток, таблеток с покрытием, драже и капсул из твердого желатина. Для капсул из мягкого желатина подходящими носителями являются, например, растительные масла, воски, жиры и полужидкие и жидкие полиолы (однако в зависимости от природы активного вещества в случае капсул из мягкого желатина носитель может не потребоваться). Носителями, подходящими для приготовления растворов и сиропов, являются, например, вода, полиолы, сахароза, инвертный сахар и т.п. Носителями, подходящими для растворов для инъекций, являются, например, вода, спирты, полиолы, глицерин и растительные масла. Носителями, подходящими для суппозиториев, являются, например, натуральные или отвержденные масла, воски, жиры, полужидкие и жидкие полиолы. Носителями, подходящими для препаратов, предназначенных для местного применения, являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воски, жидкие парафины, жидкие жирные спирты, стерины, полиэтиленгликоли и производные целлюлозы.
В качестве фармацевтических вспомогательных веществ рассматриваются обычные стабилизаторы, консерванты, смачивающие и эмульгирующие агенты, агенты, улучшающие консистенцию, агенты, улучшающие вкус, соли для регулирования осмотического давления, буферные вещества, солюбилизаторы, красители и маскирующие агенты и антиоксиданты.
Терапевтически эффективное количество или дозировка соединений формулы I может меняться в широких пределах в зависимости от подвергающегося лечению заболевания, возраста и индивидуального состояния пациента и пути введения и, разумеется, в каждом конкретном случае будет меняться в соответствии с индивидуальными потребностями. Для взрослых пациентов рассматривается суточная доза, составляющая примерно от 1 до 100 мг, предпочтительно примерно от 1 до 50 мг. В зависимости от тяжести заболевания и точного фармакокинетического профиля соединение можно вводить в виде одной или нескольких дозированных единиц в сутки, например от 1 до 3 дозированных единиц.
Фармацевтические композиции обычно содержат примерно 1-100 мг, предпочтительно 5-50 мг соединения формулы I.
В приведенных ниже примерах описаны исследования, которые проводили для определения активности соединений формулы I и в особенности их ценных фармакологических характеристик.
Примеры
МС - масс-спектрометрия; ЭУ - электронный удар; ИОР - ионное распыление, соответствует ИЭР (электрораспыление); данные ЯМР приведены в частях на миллион (δ) относительно внутреннего стандарта тетраметилсилана и для сравнения использован сигнал дейтерия растворителя (d6-ДМСО если не указано иное); константы спин-спинового взаимодействия (J) приведены в герцах, tпл - температура плавления; tкип - температура кипения; ВЭЖХ - высокоэффективная жидкостная хроматография, Rt - время удерживания, ТСХ - тонкослойная хроматография, КТ - комнатная температура, TBTU - О-(бензотриазол-1-ил)-N,N',N'-тетраметилуронийтетрафторборат; ДМФ - диметилформамид, ДМСО - диметилсульфоксид, ТГФ - тетрагидрофуран, CAN - регистрационный номер С AS.
Пример 1
Влияние на концентрацию липидов в плазме у хомяков Эффективность соединений для модулирования концентрации липидов в плазме определяли у хомяков после 5 дней ежедневного введения соединений. Во всех исследованиях использовали самцов хомяков в возрасте 6-8 недель. После недельной акклиматизации у животных после 4-часового голодания отбирали пробы крови для определения концентрации липидов в плазме. Затем животных распределяли на группы на основании значений концентрации ЛВП холестерина. Соединения вводили через желудочный зонд один раз в день в течение 5 дней. Контрольным животным вводили только разбавитель. Для определения концентрации липидов в плазме пробы крови у хомяков отбирали на пятый день после 4-часового голодания через 2 часа после введения последней дозы. Общий холестерин, ЛВП холестерин, ЛНП холестерин и триглицериды определяли с помощью колориметрических ферментных анализов (Roche Diagnostic GmbH, Mannheim, Germany). ЛВП холестерин также определяли после селективного осаждения ЛВП из плазмы, проводимого по стандартным методикам.
Пример 2
Сродство к рецепторам СВ1 и СВ2
Сродство соединений, предлагаемых в настоящем изобретении, к каннабиноидным рецепторам определяли с использованием препаратов мембран клеток почек эмбриона человека (ПЭЧ), в которые каннабиноидный рецептор СВ1 человека временно трансфицирован с использованием системы вируса лихорадки леса Семлики вместе с [3Н]-СР-55,940 в качестве радиолиганда. После инкубации свежеприготовленного препарата мембран клеток с лигандом [3Н] с добавлением или без добавления соединений, предлагаемых в настоящем изобретении, разделение связанного и свободного лигандов проводили фильтрованием через стекловолоконные фильтры. Радиоактивность фильтра измеряли по сцинтилляционной методике
Сродство соединений, предлагаемых в настоящем изобретении, к каннабиноидным рецепторам СВ2 определяли с использованием препаратов мембран клеток почек эмбриона человека (ПЭЧ), в которые каннабиноидный рецептор СВ2 человека временно трансфицирован с использованием системы вируса лихорадки леса Семлики вместе с [3Н]-СР-55,940 в качестве радиолиганда. После инкубации свежеприготовленного препарата мембран клеток с лигандом [3Н] с добавлением или без добавления соединений, предлагаемых в настоящем изобретении, разделение связанного и свободного лигандов проводили фильтрованием через стекловолоконные фильтры. Радиоактивность фильтра измеряли по сцинтилляционной методике
Значения Ki рассчитывали по значениям IC50 по уравнению Чена-Прусова.
трифторэтокси)никотинамид
гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамид
Пример 3
Получение 5-(3,4-дихлорфенил)-N-((1R,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамида
а) 5-Бром-6-(2,2,2-трифторэтокси)никотиновая кислота
5-Бром-6-хлор-3-пиридинкарбоновую кислоту (68,0 г, 0,288 моля, CAN 29241-62-1) растворяли в ДМСО (1000 мл). К этому раствору при перемешивании добавляли гидроксид калия (48,25 г, 0,86 моля) и после 10 мин перемешивания при комнатной температуре добавляли 2,2,2-трифторэтанол (26,9 мл, 0,374 моля). Смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли воду (1000 мл) и концентрированную хлористоводородную кислоту (107 мл, 1280 ммоля, 37%) и суспензию энергично перемешивали в течение 4 ч. Осадок фильтровали, промывали водой (4×100 мл) и сушили в вакууме в течение ночи и получали искомое соединение (80,4 г) в виде почти белого твердого вещества; МС (ЭУ) 299, 301 (М)+.
b) 5-Бром-N-((1R,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид
5-Бром-6-(2,2,2-трифторэтокси)-никотиновую кислоту (50,0 г, 0,166 моля) растворяли в ДМФ (600 мл). К раствору добавляли TBTU (58,9 г, 0,183 моля), N,N-диизопропилэтиламин (142,6 мл, 0,83 моля) и (JR,2R)-2-аминоциклогексанол (21,1 г, 0,183 моля). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. Растворитель выпаривали в вакууме, остаток растворяли в смеси этилацетата (1200 мл) и ТГФ (300 мл). Раствор дважды промывали водой (700 мл) и водные фазы экстрагировали этилацетатом (600 мл). Органические фазы объединяли, сушили над MgSO4 и концентрировали до объема, равного примерно 900 мл. При перемешивании и охлаждении до 0°С осаждался продукт. Фильтрование, промывка смесью этилацетат/н-гептан (1:1) и сушка в вакууме давали искомое соединение (53,1 г) в виде белого твердого вещества; МС (ИОР) 397, 399 (М)+.
c) 5-(3,4-Дихлорфенил)-N-((1R,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамид
5-Бром-N-((1R,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид (59,8 г, 151 ммоля) растворяли в толуоле (2500 мл) и ДМФ (200 мл). К этому раствору при перемешивании добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) CH2Cl2 (6,15 г, 7,5 ммоля), 3,4-дихлорфенилбороновую кислоту (30,2 г, 158 ммоля) и раствор карбоната натрия (2М, 150 мл). Эту смесь нагревали при 90°С в течение 2 ч, охлаждали до комнатной температуры и фильтровали через диатомовую землю. Осадок на фильтре тщательно промывали этилацетатом (3000 мл). Фильтраты объединяли, дважды промывали водой (2×2000 мл) и водные фазы экстрагировали этилацетатом (2×1500 мл). Органические фазы объединяли, сушили над MgSO4 и летучие вещества удаляли в вакууме. Остаток очищали фильтрованием через диоксид кремния (500 г) с промывкой этилацетатом. Растворитель удаляли и остаток растирали с диэтиловым эфиром и получали после сушки в вакууме искомое соединение (45,6 г) в виде сероватого твердого вещества; МС 463,079, 465,077 (М+Н)+.
Пример 4
Получение 5-(3,4-дихлорфенил)-N-((1S,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамида
a) 5-Бром-N-((1SR,2RS)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид
5-Бром-6-(2,2,2-трифторэтокси)никотиновую кислоту (75,0 г, 0,25 моля) растворяли в ДМФ (850 мл). К раствору добавляли TBTU (91,0 г, 0,275 моля), N,N-диизопропилэтиламин (214 мл, 1,25 моля) и (!SR,2RS)-2-аминоциклогексанолгидрохлорид (41,7 г, 0,275 моля). Реакционную смесь перемешивали в течение 1,5 ч при комнатной температуре. Растворитель выпаривали в вакууме, остаток подвергали распределению между этилацетатом (2500 мл) и 1 н. раствором гидроксида натрия (2000 мл), водную фазу отделяли, еще раз экстрагировали этилацетатом (1000 мл) и органические фазы 2 раза промывали водой (2×1500 мл). Органические фазы объединяли, сушили над MgSO4 и концентрировали до объема, равного примерно 900 мл. При перемешивании и охлаждении до 0°С осаждался продукт. Фильтрование, промывка смесью этилацетат/н-гептан (1:1) и сушка в вакууме давала искомое соединение (81,1 г) в виде белого твердого вещества; МС (ИОР) 397, 399 (М)+.
b) 5-Бром-N-((1S,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид
5-Бром-N-((1SR,2RS)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид (91,3 г, 0,23 моля) обрабатывали с помощью препаративной ВЭЖХ на колонке ChiralPak AD® (колонка 250×110 мм) с использованием смеси н-гептан/изопропанол 85/15 в качестве подвижной фазы. Обеспечивалось основное разделение и искомое соединение (43,6 г) выделяли в виде бесцветного твердого вещества из фракции, соответствующей первому пику; МС (ИОР) 395,2, 397,2 (М-Н); дисперсия оптического вращения (589 нМ, 20°С, СНС13) -21,6°.
с) 5-(3,4-Дихлорфенил)-N-((1S,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)никотинамид
5-Бром-N-((1S,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид (42,0 г, 106 ммоля) растворяли в толуоле (1900 мл) и ДМФ (100 мл). К этому раствору при перемешивании добавляли [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) CH2Cl2 (0,9 г, 1,06 ммоля), 3,4-дихлорфенилбороновую кислоту (20,2 г, 106 ммоля) и раствор карбоната натрия (2М, 106 мл). Эту смесь нагревали при 90°С в течение 2 ч, охлаждали до комнатной температуры и подвергали распределению между этилацетатом (1000 мл) и водой (2000 мл), водную фазу отделяли, еще дважды экстрагировали этилацетатом (2×1000 мл) и органические фазы промывали один раз водой и один раз рассолом (по 1000 мл каждого). Органические фазы объединяли, сушили над MgSO4 и летучие вещества удаляли в вакууме. Остаток растворяли в диэтиловом эфире (500 мл) и фильтровали через диатомовую землю. Искомое соединение осаждалось при проводимом по каплям добавлении н-гептана (500 мл) к раствору в диэтиловом эфире, его отфильтровывали и сушили в вакууме и получали 33,3 г искомого соединения в виде почти белого твердого вещества; МС 463,079 (М+Н)+.
Пример 5
Таблетки с пленочным покрытием, содержащие указанные ниже ингредиенты, можно приготовить обычным образом:
Активный ингредиент просеивают и смешивают с микрокристаллической целлюлозой и смесь гранулируют с использованием водного раствора поливинилпирролидона. Затем гранулят смешивают с натриевой солью гликолята крахмала и стеаратом магния, прессуют и получают ядра массой 120 или 350 мг соответственно. Ядра лакируют водным раствором/суспензией указанного выше пленочного покрытия.
Пример 6
Капсулы, содержащие указанные ниже ингредиенты, можно приготовить обычным образом:
Компоненты просеивают, смешивают и помещают в капсулы размера 2.
Пример 7
Растворы для инъекции могут обладать следующим составом:
Активный ингредиент растворяют в смеси полиэтиленгликоля 400 и воды для инъекции (часть). Значение рН устанавливают равным 5,0 путем добавления уксусной кислоты. Объем доводят до 1,0 мл путем добавления оставшегося количества воды. Раствор фильтруют, помещают во флаконы с соответствующим избытком и стерилизуют.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3-ПИРИДИНКАРБОКСАМИДА И 2-ПИРАЗИНКАРБОКСАМИДА В КАЧЕСТВЕ АГЕНТОВ, ПОВЫШАЮЩИХ УРОВЕНЬ ЛВП-ХОЛЕСТЕРИНА | 2007 |
|
RU2454405C2 |
| N-ПИРИДИН-3-ИЛ ИЛИ N-ПИРАЗИН-2-ИЛ КАРБОКСАМИДЫ В КАЧЕСТВЕ АГЕНТОВ, ПОВЫШАЮЩИХ УРОВЕНЬ ХОЛЕСТЕРИНА ЛПВП | 2011 |
|
RU2540069C2 |
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ТЕТРАГИДРОПИРОЛЛО[3,2-c]ПИРИДИН-4-ОНОВЫХ ПРОИЗВОДНЫХ ДЛЯ ЛЕЧЕНИЯ ОЖИРЕНИЯ, ПСИХИАТРИЧЕСКИХ И НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2006 |
|
RU2415856C2 |
| ПРОИЗВОДНЫЕ ПИРИДИН-3-КАРБОКСАМИДА В КАЧЕСТВЕ ОБРАТНЫХ АГОНИСТОВ СВ1 | 2006 |
|
RU2404164C2 |
| СПОСОБ ЛЕЧЕНИЯ АТЕРОСКЛЕРОЗА У СУБЪЕКТОВ С ВЫСОКИМ УРОВНЕМ ТРИГЛИЦЕРИДОВ | 2012 |
|
RU2609200C2 |
| СОСТАВЫ, ОБЛАДАЮЩИЕ ЛИПИДОПОНИЖАЮЩИМИ СВОЙСТВАМИ | 2006 |
|
RU2428973C2 |
| КОНЪЮГАТЫ ЖИРНЫХ КИСЛОТ И НИАЦИНА И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2569063C2 |
| 4-ЦИКЛОАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВ | 2005 |
|
RU2393151C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ1 | 2005 |
|
RU2377238C2 |
| НИАЦИНОВЫЕ МИМЕТИКИ И СПОСОБ ИХ ИСПОЛЬЗОВАНИЯ | 2011 |
|
RU2588133C2 |
Изобретение относится к области органической химии, а именно к соединению формулы I, к его изомерным формам и фармацевтически приемлемым солям. Также изобретение относится к фармацевтической композиции на основе соединения формулы I. Технический результат: получено новое соединение, повышающее концентрацию ЛВП холестерина. 2 н. и 4 з.п. ф-лы, 2 табл., 7 пр.
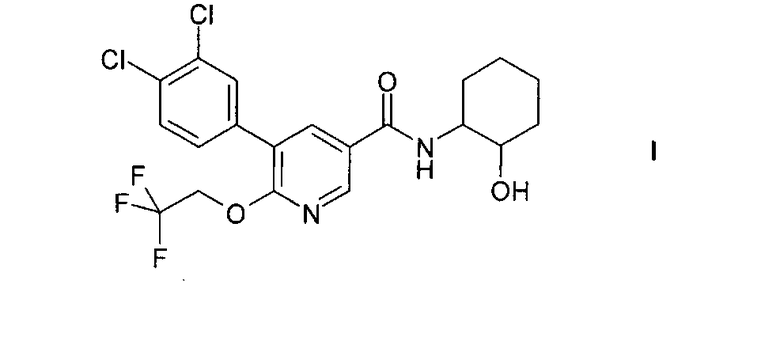
1. Соединение формулы
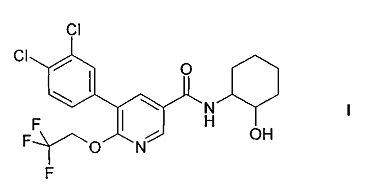
и его изомерные формы и фармацевтически приемлемые соли.
2. Соединение формулы I по п.1, которое представляет собой 5-(3,4-дихлорфенил)-N-((1R,2R)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид.
3. Соединение формулы I по п.1, которое представляет собой 5-(3,4-дихлорфенил)-N-((1S,2S)-2-гидроксициклогексил)-6-(2,2,2-трифторэтокси)-никотинамид.
4. Фармацевтическая композиция, обладающая активностью повышения концентрации ЛВП холестерина в плазме, включающая соединение формулы I по любому из пп.1-3 и фармацевтически приемлемый носитель и/или вспомогательное вещество.
5. Фармацевтическая композиция по п.4, предназначенная для лечения и/или профилактики заболеваний, которые можно лечить средствами, повышающими концентрацию ЛВП холестерина.
6. Соединение формулы I по любому из пп.1-3, предназначенное для применения в качестве лекарственного средства, обладающего активностью повышения концентрации ЛВП холестерина в плазме.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| RU 2003105805 A, 10.08.2004 | |||

