Изобретение относится к новым производным пролина, и, более конкретно к отдельным формам нового производного 1-замещенного N-[2-метил-1-(трифторацетил)- пропил]пирролидин-2-карбоксамида, которые являются ингибиторами эластазы лейкоцитов человека (ЭЛЧ), известной также как эластаза нейтрофилов человека (ЭНЧ), которые имеют важное значение, например, как средства научно-исследовательской работы в фармакологических, диагностических и связанных с ними исследованиях и при лечении заболеваний млекопитающих, к которым причастна ЭЛЧ. Например, ЭЛЧ является причиной патогенеза острого респираторного дистресс-синдрома (ARDS), ревматоидного артрита, атеросклероза, эмфиземы легких и других воспалительных заболеваний, включая воспалительные заболевания дыхательных путей, характеризуемые повышенной и аномальной секрецией в дыхательных путях, такие как острый и хронический бронхит и муковисцидоз. ЭЛЧ также является причиной некоторых сосудистых заболеваний и сопутствующих им состояний (и используется при их лечении), в которых принимают участие нейтрофилы или к которым причастны нейтрофилы, например кровотечение, связанное с острым лейкозом, не являющимся лимфолейкозом, а также реперфузионной травмы, связанной, например, с ишемией миокарда, и сопутствующими состояниями, связанными с заболеванием коронарной артерии, таким как стенокардия и инфаркт, цереброваскулярной ишемией, как например преходящее нарушение мозгового кровообращения или внезапный приступ нарушения мозгового кровообращения, периферическим обтурирующим сосудистым заболеванием, таким как перемежающаяся хромота и критическая ишемия конечности, венозной недостаточностью, такой как венозная гипертензия, варикозные вены и образование венозных язв, а также с ослабленными состояниями с реперфузией, такими как состояния, связанные с восстановительной хирургией на сосудах, тромболизом и пластической операцией на сосудах. Изобретение относится также к способам лечения одного или нескольких из этих состояний заболевания и к применению одной или нескольких отдельных форм нового производного при изготовлении лекарственного средства для применения в случае одного или нескольких указанных состояний. Изобретение, кроме того, относится к фармацевтическим композициям, содержащим одну или несколько отдельных форм нового производного в качестве активного ингредиента, а также к способам получения отдельных форм нового производного, к новым промежуточным продуктам, используемым в указанных способах, и к способам получения указанных промежуточных продуктов.
В связи с очевидной ролью эластазы лейкоцитов человека в последние годы имел место значительный объем исследовательских работ, направленных на разработку ингибиторов ЭЛЧ. В патенте США 4910190 описан ряд близких по структуре пептидоильных производных трифторметана, которые являются ингибиторами эластазы лейкоцитов человека. В настоящее время заявителями обнаружено, что определенные формы нового производного 1-замещенного N-[2-метил-1-(трифторацетил) пропил]пирролидин-2-карбоксамида формулы I (представлена в конце описания) являются неожиданно мощными ингибиторами ЭЛЧ.
Согласно одному аспекту изобретения предлагается соединение (S)-1-[(S)-2-(метоксикарбониламино)-3-метил-бутирил]-N-[2- метил-1-(трифторацетил) пропил] пирролидин-2-карбоксамид, или его сольват, оба в форме диастереомерной смеси (S)-1-[(S)-2- (метоксикарбониламино)-3-метилбутирил]-N-[(S)-2-метил-1- (трифторацетил) пропил]пирролидин-2-карбоксамида (или его сольвата) и (S)-1-[(S)-2-(метоксикарбониламино)-3-метилбутирил]-N-[(R)-2- метил-1-(трифторацетил) пропил]пирролидин-2-карбоксамида (или его сольвата) и в форме существенно или практически чистого диастереоизомера (S)-1-[(S)-2-(метоксикарбониламино)-3- метилбутирил] -N-[(S)-2-метил-1-трифторацетил)пропил] пирролидин-2-карбоксамида (или его сольвата).
Следует учесть, что соединение формулы I имеет три хиральных центра (обозначенных в формуле I как * и #) и, следовательно, может существовать в восьми различных стереомерных формах или в виде диастереомерной смеси двух или нескольких из этих форм. Например, соединение (S)-1-[(S)-2-(метоксикарбониламино)-3-метилбутирил]-N- [2-метил-1-(трифторацетил) пропил] пирролидин-2-карбоксамид является соединением формулы 1, в котором два хиральных центра, обозначенных как *, имеют S-конфигурацию, а третий хиральный центр, обозначенный как #, имеет RS-конфигурацию. Следовательно, соединение является диастереомерной смесью, содержащей диастереоизомер с хиральными центрами, обозначенными * и #, имеющими все S-конфигурацию, то есть (S)-1-[(S)-2-(метоксикарбониламино)-3-метилбутирил]-N- [(S)- 2-метил-1-(трифторацетил) пропил] пирролидин-2-карбоксамид (именуемый далее "SSS-диастереоизомер" формулы I, и который может также быть представлен как показано формулой Ia, приведенной в конце описания, в которой изображенная жирно связь обозначает связь, выступающую над плоскостью бумаги), и диастереизомер с хиральными центрами, обозначенными *, имеющими S-конфигурацию, и центром, обозначенным #, имеющим R-конфигурацию, то есть (S)-1-[(S)-2- (метоксикарбониламино)-3-метилбутирил]-N-[(R)-2-метил-1- (трифторацетил)пропил] -пирролидин-2-карбоксамид (именуемый далее "SSR-диастереоизомер" формулы I), или их сольваты. Такая диастереомерная смесь включает, например, смесь, содержащую приблизительно равные количества SSS- и SSR-диастереоизомеров, т.е. соотношение SSS:SSR составляет около 1:1. Например, получены диастереомерные смеси, содержащие SSS- и SSR-диастереоизомеры в соотношениях 53:47 и 47:53 (SSS:SSR). Отдельные формы соединения формулы I, которые являются предпочтительными, представляют собой диастереомерные смеси, которые обогащены SSS-диастереоизомером, т.е. соотношение SSS:SSR составляет более 1:1. Особенно предпочтительная форма соединения представляет собой существенно или практически чистый SSS-диастереоизомер, то есть SSS-диастереоизомер, содержащий менее 5% (более конкретно, менее 3% и предпочтительно менее 2%) других диастереоизомеров.
Следует учесть, что SSS-диастереоизомер формулы I может также образовывать диастереомерную смесь с одной или несколькими другими формами формулы I, например может быть получен (S)-1- [2-(метоксикарбониламино)-3-метилбутирил] -N-[(S)-2-метил-1- (трифторацетил) пропил] пирролидин-2-карбоксамид (диастереомерная смесь SSS- и RSS-форм формулы I) или I-[(S)-2-(метоксикарбониламино) -3-метилбутирил] -N-[(S)-2-метил-1- (трифторацетил) пропил] пирролидин-2-карбоксамид (диастереомерная смесь SSS- и SRS-форм формулы I). Эти конкретные диастереомерные смеси и другие диастереомерные смеси, содержащие около 50% или более SSS-диастереоизомера, совместно с одним или несколькими другими возможными диастереоизомерами с различными конфигурациями при хиральных центрах, обозначенных в формуле I как * и #, являются поэтому следующими аспектами настоящего изобретения.
Диастереомерная смесь SSS- и SSR-диастереоизомеров может существовать в аморфной, некристаллической форме или в кристаллической форме в зависимости от соотношения SSS: SSR присутствующих диастереоизомеров. Предпочтительная диастереомерная смесь представляет собой смесь, которая может быть выделена в кристаллической форме, которая особенно подходит при изготовлении соединения или составов на его основе, со степенью чистоты и однородностью, требуемыми для аттестации установленного образца. Следует учесть, что крайне сложно получить соединение, представляющее собой единственный диастереоизомер, полностью свободный от других возможных диастереомерных форм, в особенности соединение, которое имеет три хиральных центра. Поэтому настоящее изобретение включает кристаллическую форму SSS-диастереоизомера формулы I или его сольвата, который содержит другие возможные диастереомерные формы с различными конфигурациями при хиральных центрах, обозначенных в формуле I как * и # . Установлено, что может быть получена кристаллическая диастереомерная смесь SSS- и SSR-диастереоизомеров, или их гидратов, которая представляет собой по существу или практически диастереомерную смесь SSS- и SSR-диастереоизомеров в соотношении (SSS:SSR) 65:35 или более, т.е. она содержит 35% или менее SSR-диастереоизомера. Поэтому настоящее изобретение включает кристаллическую форму соединения формулы I или его сольват с содержанием SSS-диастереоизомера по меньшей мере 65%. Предпочтительно кристаллическая диастереомерная смесь имеет, например, соотношение SSS:SSR, которое составляет 80: 20 или более, например 95:5 или более, и в особенности 98,5:1,5 или более. Особенно предпочтительной формой соединения согласно изобретению является кристаллический SSS-диастереоизомер, который является существенно или практически чистым, т.е. он содержит менее 5% других диастереоизомеров, например менее 5% SSR-диастереоизомера, предпочтительно менее 3% SSR-диастереоизомера и более предпочтительно менее 2% SSR-диастереоизомера.
Аморфная или кристаллическая диастереомерная смесь SSS- и SSR-форм, либо существенно, либо практически чистый диастереоизомер, существует в форме, которая является существенно или практически свободной от растворителя (которая именуется ниже "кетонная форма" и которая иллюстрируется формулой Ia для чистого SSS-диастереоизомера), или в виде сольватированной, например, гидратированной формы, или в виде смеси кетонной и сольватированной (гидратированной) форм. Гидратированная форма может существовать, например, в виде гем-диола трифторкетонного радикала, то есть в виде соединения формулы Ib (представлена в конце описания) для существенно или практически чистого SSS-диастереоизомера, или в виде соединения формулы Ic (представлена в конце описания), или в форме, которая включает молекулу воды в качестве части кристаллической решетки, или смеси таких форм. Соединения формулы Ib или Ic, кроме того, могут быть, например, гидратированными.
Следует учесть, что степень гидратации диастереомерной смеси либо существенно, либо практически чистого SSS-диастереоизомера может быть выражена как отношение количества гидратной формы к количеству кетонной формы. Например, выделена аморфная, некристаллическая диастереомерная смесь SSS- и SSR-форм, в которой отношение гидратированной формы к кетонной форме изменяется, например, от около 30:70 (т.е. смесь, обогащенная кетонной формой) до около 95:5 или более (т.е. смесь по существу или практически в гидратированной форме), включая такие соотношения, как около 50:50 и около 60:40. Например, были получены кристаллические формы, которые имеют отношение SSS: SSR около 95:5 одновременно с отношением гидрат:кетон около 80:20, и которые имеют отношение SSS:SSR около 65:35 или более (как например 98,5:1,5) и находятся по существу или практически в гидратированной форме. Были также получены кристаллические гидраты существенно или практически чистого SSS-диастереоизомера, содержащего приблизительно 4,1% (по весу) и 7,8% (по весу) воды. Такие отдельные формы являются дальнейшими аспектами изобретения. Кроме того, следует учесть, что настоящее изобретение охватывает также любой кеталь или гемикеталь (или их смеси) диастереомерной смеси или формы SSS-диастереоизомера, или их сольвата, в отношении тех из них, которые превращаются в гем-диол in vivo, например, путем гидролиза или ферментативного расщепления (и у которых остаток является фармацевтически приемлемым). Настоящее изобретение включает также любой таутомер или пролекарство SSS-диастереоизомера или его сольвата.
Следует учесть, что соединение формулы Ib может обозначаться как гем-диольная форма соединения формулы Ia или при помощи химического наименования (S)-1-[(S)-2- (метоксикарбониламино)-3- (метилбутирил)] -N-[(S)-2-метил-1- (2,2,2-трифтор-1,1-дигидроксиэтил) пропил]пирролидин-2- карбоксамид. Следует также учесть, что альтернативным наименованием для соединения формулы Ia является метил-N-[(1S)-1- ((2S)-2-[N-((1S)-2-метил-1- (2,2,2-трифторацетил)-пропил) карбамоил]пирролидин-1-илкарбонил) -2-метилпропил]-карбамат, а альтернативным наименованием для соединения формулы Ib является метил-N-[(S)-1-((2S)-2-[N-((S)-3,3,3-трифтор-2,2-дигидрокси-1- изопропилпропил) карбамоил-пирролидин-1-илкарбонил) -2-метилпропил]карбамат.
Температура плавления кристаллического SSS-диастереоизомера, содержащего SSR-диастереоизомер, обычно зависит от уровня содержания присутствующего SSR-диастереоизомера и от степени сольватации (гидратации). Она может быть определена при помощи стандартных методик, хорошо известных в технологии, например при помощи дифференциальной сканирующей калориметрии (ДСК).
Предпочтительно кристаллический SSS-диастереоизомер находится в гидратированной форме. Например, были обнаружены гидратированные формы SSS-диастереоизомера, которые обладают таким положительным свойством, что они не являются гигроскопичными, например форма А и форма В, которые описаны ниже. Таким образом, предпочтительная форма SSS-диастереоизомера представляет собой кристаллическую форму, содержащую менее 5% (предпочтительно менее 3% и особенно предпочтительно менее 2%) SSR-диастереоизомера, и находится существенно или практически в гидратированной форме. Установлено, что такие кристаллические гидратированные формы, например форма A и форма B, обладают хорошей биологической пригодностью и хорошей растворимостью в водном буфере, и оба эти свойства являются положительными.
Особенно предпочтительная кристаллическая форма SSS-диастереоизомера формулы I, когда он является существенно или практически чистым и находится в гидратированной форме, имеет порошковую рентгенограмму, включающую два главных характерных пика приблизительно при 2θ = 10,8 и 11,4o. Эта форма (называемая в настоящем описании форма A) содержит приблизительно 4,1% воды. Порошковая рентгенограмма включает также относительно менее интенсивные характерные пики, встречающиеся приблизительно при 2θ = 15,4, 16,8, 18,2, 18,6, 20,6, 21,6, 21,9, 22,8 и 25,0o. Спектр порошковой дифракции рентгеновских лучей (XDS) типичного образца этой формы показан на фиг. 1 и 2, где на фиг. 2 показаны менее интенсивные пики в увеличенном масштабе. Дополнительные физические данные дают основание предполагать, что эта кристаллическая форма находится по существу или практически в форме диола формулы Ib.
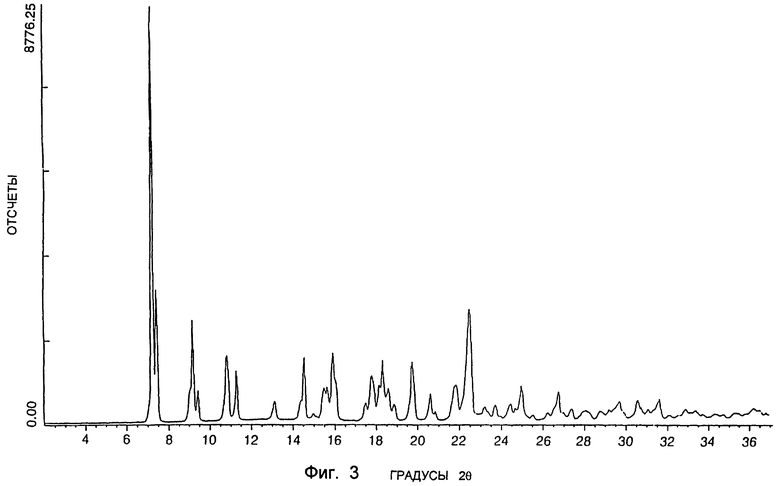
Следующая предпочтительная кристаллическая форма SSS-диастереоизомера формулы I, когда он является существенно или практически чистым и находится в гидратированной форме, имеет порошковую рентгенограмму, включающую главный характерный пик приблизительно при 2θ = 7,2°. Эта форма (называемая в настоящем описании форма В) содержит приблизительно 7,8% по весу (например, 7,3-8,3% по весу) воды. Порошковая рентгенограмма включает также относительно менее интенсивные характерные пики, встречающиеся приблизительно при 2θ = 7,4, 9,0, 10,8, 11,3, 14,5, 15,9, 17,8, 18,1, 19,7 и 22,5o. Спектр порошковой дифракции рентгеновских лучей типичного образца этой формы показан на фиг. 3. Дополнительные физические данные дают основание предполагать, что эта кристаллическая форма является существенно или практически моногидратом диола формулы Ib.
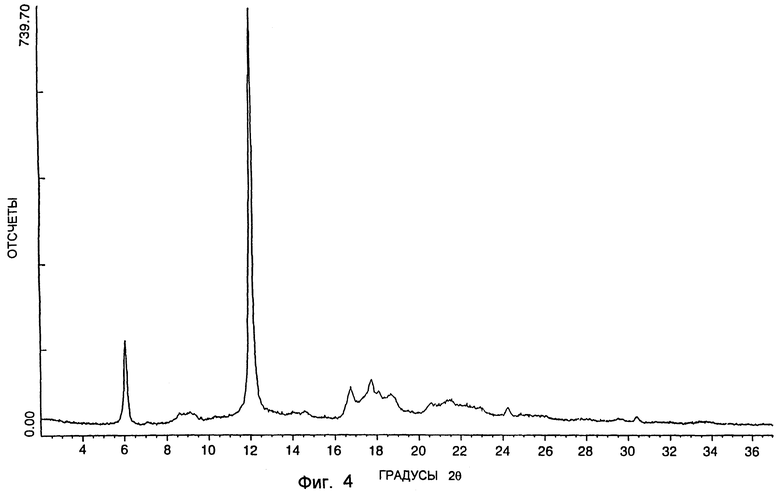
Когда он является существенно или практически чистым и существенно или практически свободным от растворителя (т.е. находится в "кетонной" форме), SSS-диастереоизомер формулы I имеет порошковую рентгенограмму, включающую главный характерный пик приблизительно при 2θ = 12,1. Эта рентгенограмма включает также относительно менее интенсивные пики, встречающиеся приблизительно при 2θ = 6,0, 16,8 и 17,7o. Спектр XDS типичного образца в "кетонной" форме показан на фиг. 4.
Спектры порошковой дифракции рентгеновских лучей определяли, например, используя рентгеновский дифрактометр Scintag XDS-2000 с твердофазным детектором фотонов EC& G, серия GLP (германий), управляемый компьютером Microvax, и используя программное обеспечение Diffraction Management System, поставляемое фирмой Scintag Inc., Sunnydale, California, USA. Использовавшаяся рентгеновская трубка представляла собой трубку Cu K-альфа с длиной волны  при 45 кВ и 40 мА. Приемные щели были установлены на расстоянии 2 и 4 мм, а щели, осуществляющие дивергенцию, были установлены на расстоянии 0,2 и 0,5 мм по отношению к пути падающего луча. Спектры были получены в режиме непрерывного сканирования с приращением обтюратора (а chopper increment) 0,02. Каждый образец экспонировали при скорости 1 градус угла 2-тета в минуту (время прохода составляло 38 минут) и снимали показания от 2 до 40 градусов 2-тета с получением кривой параметра кристаллической решетки в зависимости от интенсивности для этого интервала.
при 45 кВ и 40 мА. Приемные щели были установлены на расстоянии 2 и 4 мм, а щели, осуществляющие дивергенцию, были установлены на расстоянии 0,2 и 0,5 мм по отношению к пути падающего луча. Спектры были получены в режиме непрерывного сканирования с приращением обтюратора (а chopper increment) 0,02. Каждый образец экспонировали при скорости 1 градус угла 2-тета в минуту (время прохода составляло 38 минут) и снимали показания от 2 до 40 градусов 2-тета с получением кривой параметра кристаллической решетки в зависимости от интенсивности для этого интервала.
Для проведения дифракционного анализа образцы упаковывали в круглые чашки для образцов из алюминиевого сплава диаметром 25 мм и высотой 2 мм. Порошкообразный образец помещали в чашку таким образом, чтобы вещество оказывалось в избытке по сравнению с объемом чашки, а затем выравнивали по краю чашки при помощи предметного стекла микроскопа. В качестве внешнего стандарта использовали кремний типа NBS 640b.
В альтернативном варианте использовали рентгеновский дифрактометр Siemens D5000, записывающий дифрактограмму в θ - θ режиме в интервале от 2 до 40 градусов 2-тета, с экспозицией в 4 секунды на приращение 2θ в 0,02o.
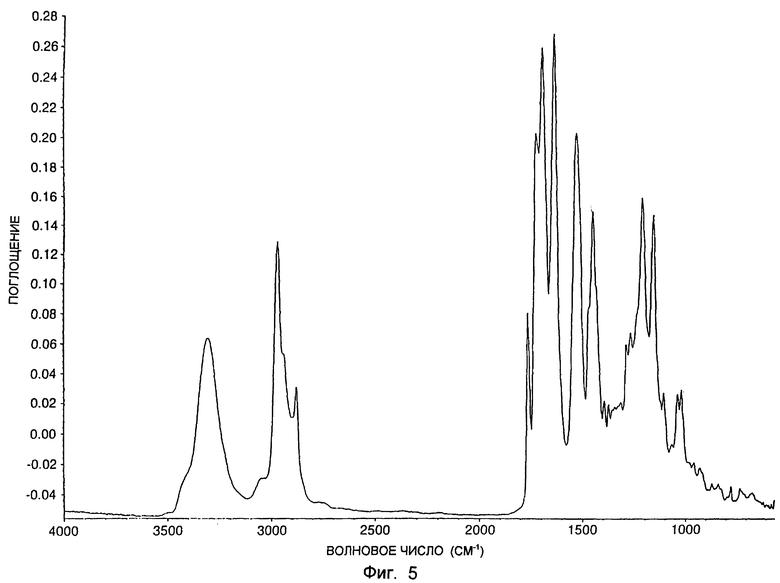
Для типичного образца формы А был получен инфракрасный спектр. Инфракрасный спектр был получен при помощи методики заливки растворителя, хорошо известной в технологии, из ацетонитрильных заливок образца в соляное окно для анализа путем прямого пропускания. Инфракрасный спектр определяли в диапазоне волновых чисел от 4000 до 400 см-1. Инфракрасный спектр показан на фиг. 5. Спектр на фиг. 5 включает резкие пики приблизительно при 2968, 1762, 1721, 1690, 1632, 1525, 1447, 1207 и 1154 см-1.
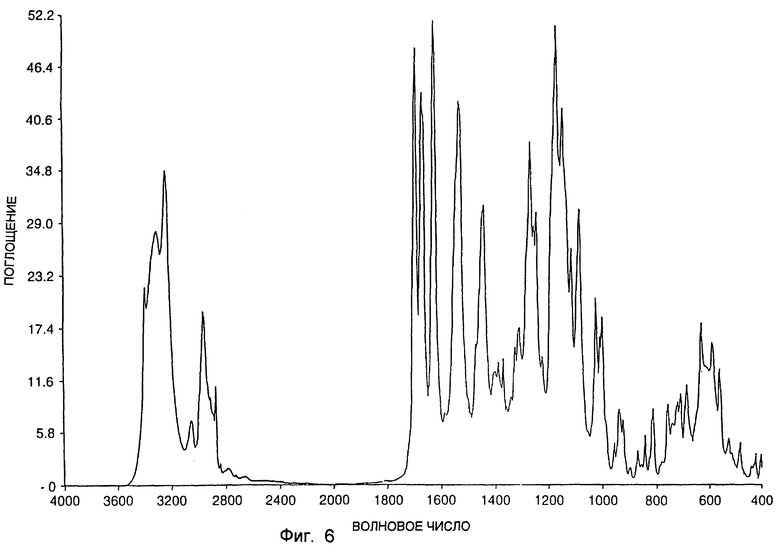
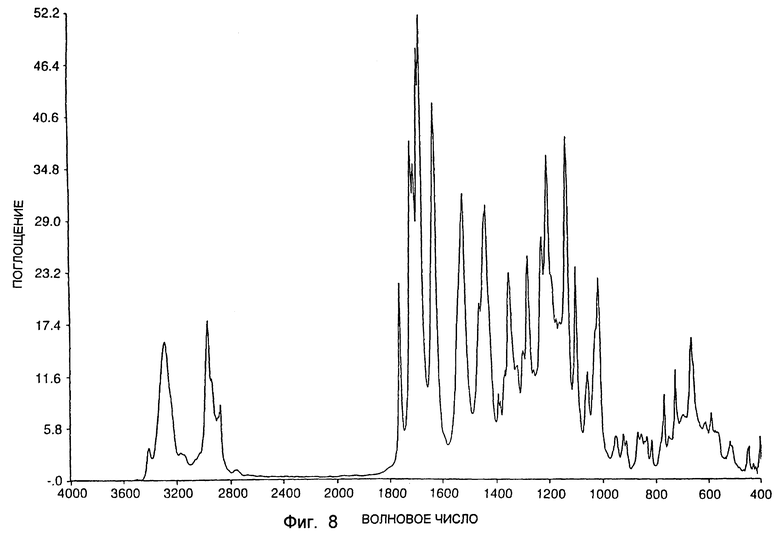
Инфракрасный спектр был получен также для типичного образца формы A с использованием спектрометра Nicolet 20SXC FTIR. Спектр получали, используя 2% дисперсию образца в бромистом калии. Инфракрасный спектр показан на фиг. 6. Спектр на фиг. 6 включает резкие пики приблизительно при 3402, 3321, 3252, 3060, 2967, 2878, 1699, 1674, 1629, 1535, 1532, 1446, 1271, 1258, 1249, 1175, 1152, 1118, 1089, 1029, 1013, 1004, 635, 593 и 567 см-1. При использовании аналогичных условий был получен инфракрасный спектр для типичного образца формы В. Инфракрасный спектр показан на фиг. 7. Спектр на фиг. 7 включает резкие пики приблизительно при 3428, 3304, 2971, 2875, 1708, 1682, 1637, 1556, 1518, 1470, 1449, 1428, 1316, 1310, 1277, 1265, 1236, 1196, 1175, 1144, 1120, 1081, 1036, 1005, 928, 818, 790 и 727 см-1. При использовании аналогичных условий был получен инфракрасный спектр для типичного образца SSS-диастереоизомера в существенно "кетонной" форме. Инфракрасный спектр показан на фиг. 8. Спектр на фиг. 8 включает резкие пики приблизительно при 3415, 3300, 2967, 2876, 1764, 1723, 1711, 1695, 1686, 1634, 1527, 1445, 1356, 1286, 1234, 1213, 1139, 1105, 1061, 1020, 774, 774, 732 и 671 см-1.
Следует учесть, что значения 2θ на порошковых рентгенограммах и длины волн инфракрасных спектров могут слегка изменяться от одного прибора к другому, и, таким образом, количественно определенные значения не должны рассматриваться как абсолютные. Например, два характерных специфичных пика, которые встречаются приблизительно при 2θ = 10,8 и 11,4o, для типичного образца формы А в случае использования рентгеновского дифрактометра Scintag XDS-2000 встречаются приблизительно при 2θ = 10,6 и 11,2o соответственно в случае использования рентгеновского дифрактометра Siemens D-5000 (с менее интенсивными пиками, также встречающимися при пропорционально более низких относительных значениях 2θ).
Следует учесть, что атомы водорода гидроксильных групп форм, имеющих формулу Ib или Ic (или их гидрата), являются кислотными и что, следовательно, такие соединения могут образовывать кристаллические фарамацевтически приемлемые соли при использовании стандартных методик, например, с основаниями, дающими физиологически приемлемые катионы, например солями щелочного металла (такого как натрий или калий), щелочно-земельного металла или органического амина. Поэтому изобретение включает кристаллические фармацевтически приемлемые соли форм формулы Ib и Ic или их гидратов.
Различные формы соединения формулы I или их сольваты (гидраты) могут быть получены, например, при помощи следующих процессов, которые являются, далее, отдельными аспектами изобретения.
Некристаллическая (аморфная) диастереомерная смесь SSS- и SSR-диастереоизомеров может быть получена окислением соединения формулы II (представлена в конце описания) подходящим окислителем.
Подходящий окислитель представляет собой известный в технологии окислитель для превращения гидроксильной группы в кетонную группу. Подходящие окислители и условия включают, например, использование оксалилхлорида, диметилсульфоксида и третичного амина; использование уксусного ангидрида и диметилсульфоксида; использование пиридинового комплекса с трехокисью хрома в дихлорметане; использование реагента, содержащего гипервалентный йод, такого как 1,1,1-триацетокси-2,1-бензоксидол-3(3H)-он с трифторуксусной кислотой в дихлорметане; использование избытка диметилсульфоксида и растворимого в воде карбодиимида в присутствии дихлоруксусной кислоты; или перманганта щелочного металла в щелочном водном растворе, такого как щелочной водный раствор перманганата калия или перманганата натрия. Особенно подходящими окислителями являются два последних из названных, в особенности щелочной водный раствор перманганата калия или натрия, например смесь гидроксида натрия и перманганата калия или натрия.
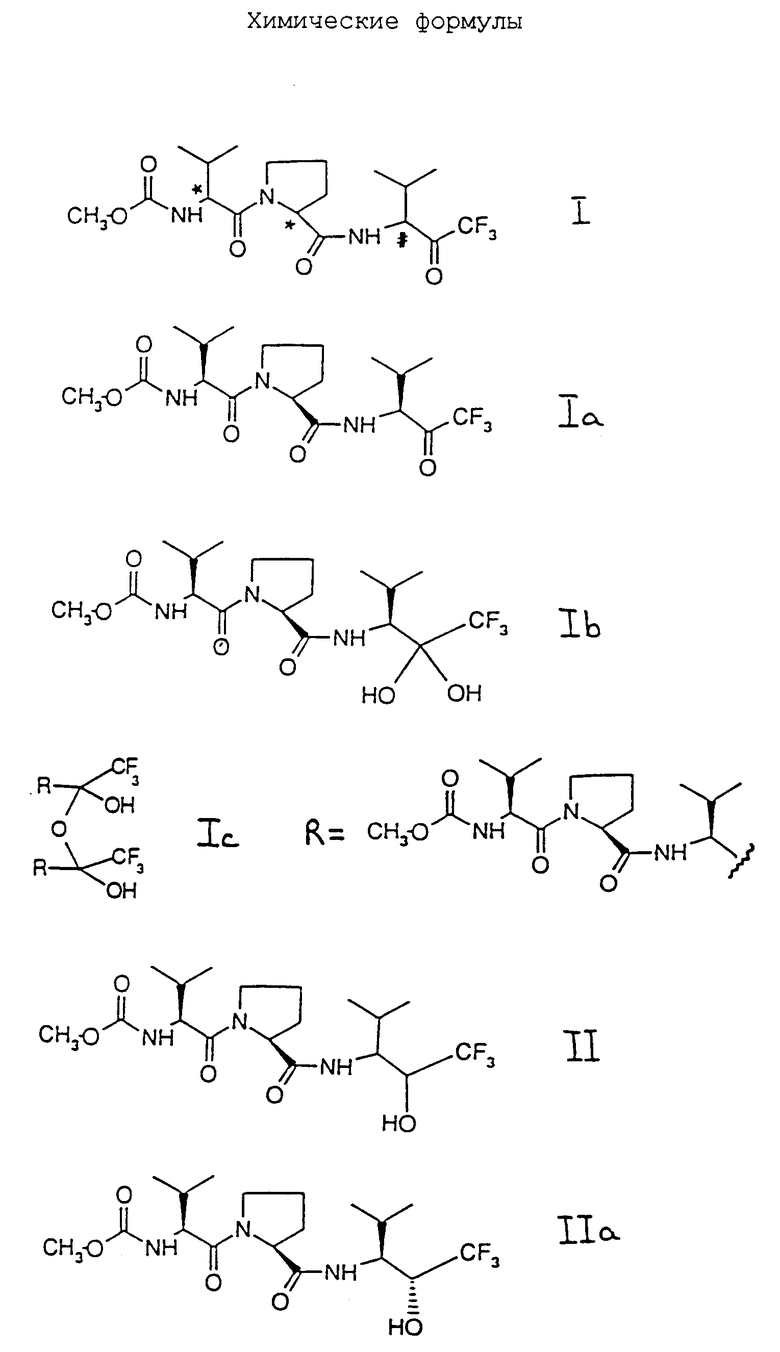
Соединение формулы II может быть получено, например, как показано на схемах 1 и 2, приведенных в конце описания, с использованием стандартных методик или как проиллюстрировано в примерах. Стадии с (а) по (d) схемы 1 могут быть проведены как описано в патенте США 5194588 или Европейском патенте 189305. Стадию (е) проводят с использованием стандартных методик получения карбамата из первичного амина, например, с использованием метилгалогенформиата, такого как метилхлорформиат, в присутствии подходящего основания, такого как триэтиламин или N-метилморфолин, и в подходящем растворителе или разбавителе, например хлорированном углеводороде (таком как дихлорметан или хлороформ) или в растворителе типа простого эфира (таком как тетрагидрофуран или диоксан) и при температуре, например, в диапазоне от -10oC до 50oC, например от 0oC до 30oC. Стадии реакции согласно схеме 2 включают стандартные стадии защиты (стадия (10)), снятия защиты или избирательного снятия защиты (стадии (1), (3), (6), (8), (9) и (12)), сочетания (стадии (4), (5), (13) и (14)) и образования карбамата (стадии (2), (7) и (11)), хорошо известные в технологии.
Следует учесть, что диастереомерные смеси SSS- и RSS-диастереоизомеров и SSS- и SRS-диастереоизомеров могут быть получены с использованием аналогичных методик с соответствующим выбором L- или DL-валина или пролина (или их защищенных производных) в качестве исходных веществ и использованием (2R, 3S)-3-амино-4-метил-1,1,1-трифтор-2-пентанола на соответствующих стадиях сочетания.
Существенно или практически чистый SSS-диастереоизомер может быть получен, например, путем окисления (S)-1-[(S)-2-(метоксикарбониламино)-3-(метилбутирил)] - N-[(S)-2-метил-1-((R)-2,2,2-трифтор-1-гидроксиэтил)пропил] пирролидин-2-карбоксамида (формулы IIa, приведенной в конце описания) подходящим окислителем, таким как один из окислителей, описанных выше. Исходный спирт может быть получен как показано на схеме 2, приведенной в конце описания.
Кристаллические формы SSS-диастереоизомера, содержащие 35% или менее SSR-диастереоизомера, могут быть получены из некристаллической (аморфной) диастереомерной смеси SSS- и SSR-диастереоизомеров, содержащей SSS- и SSR-диастереоизомеры в приблизительно равных количествах (т.е. отношение около 1: 1, обычно 53:47 или 47:53), путем кристаллизации из подходящего неполярного растворителя, такого как смесь метил-трет-бутилового простого эфира и гексана, предпочтительно содержащего небольшое количество воды и необязательно содержащего небольшое количество соляной кислоты, например 0-0,2 мольных эквивалента 36% соляной кислоты, и 1-2,1 мольных эквивалента воды. Установлено, что является предпочтительным добавлять водную соляную кислоту к растворителю, из которого проводится кристаллизация, в том случае, когда используется некристаллическая диастереомерная смесь с отношением SSS:SSR, составляющим 47: 53. Чтобы инициировать кристаллизацию, является предпочтительным внесение затравки кристаллического SSS-диастереоизомера. Кристаллический продукт обычно выделяется в виде смеси гидратированной и кетонной формы, обычно в соотношении около 80:20 (гидрат:кетон) или более. Гидратированная форма или смесь кетонной и гидратированной форм может быть преобразована в существенно или практически "кетонную" форму путем сушки в вакуумной печи (например, при температуре около 50oC). Однако такая кетонная форма является гигроскопичной.
Существенно или практически чистые кристаллические формы SSS-диастереоизомера могут быть получены перекристаллизацией или повторной перекристаллизацией кристаллических форм SSS-диастереоизомера, содержащих SSR-диастереоизомер. Растворители или смеси растворителей, которые могут использоваться для этого, включают, например, бутилацетат, бутилацетат/гексан, ацетон/вода, ацетон/гексан, ацетон/фракция нефти с температурой кипения 100-120oC, 1,2-диметоксиэтан/гексан, 1,2-диметоксиэтан/вода/гексан, этилацетат/вода/гексан, этилацетат/гексан, вода, дибутиловый эфир/гексан, дихлорметан/гексан, 1,2-диметоксиэтан/вода, метанол/толуол, метил-трет-бутиловый эфир/гексан, изопропанол/гексан и тетрагидрофуран/гексан. Для получения формы A предпочтительными являются первые десять растворителей или смесей растворителей из перечисленных выше. Особенно пригодными растворителями или смесями растворителей для получения формы В являются 1,2-диметоксиэтан/вода и вода/метанол, хотя эта форма может быть также получена в случае использования смеси этилацетат/вода/гексан. В понятие "гексан", когда оно используется в настоящей работе, включаются изомеры гексана (как например изогексан) или их смеси.
Существенно или практически чистые кристаллические формы SSS-диастереоизомера могут быть также получены кристаллизацией существенно или практически чистого SSS-диастереоизомера, выделенного в некристаллической форме (например, путем окисления соединения формулы IIa), такой как масло, с использованием растворителей или смесей растворителей, подобных тем, что описаны выше, особенно смеси этилацетата, воды и гексана.
Кроме того, форма A может быть также получена из формы В путем перекристаллизации, например, как показано в примере 9. В дополнение к этому кристаллическая "кетонная" форма (которая является гигроскопичной) может быть получена из формы А, например, как показано в примере 10.
Получение кеталя или геми-кеталя из кетона хорошо известно в технологии.
3-амино-4-метил-1,1,1- трифтор-2-пентанол может быть получен как описано в патенте США N 4910190 или как показано в примерах.
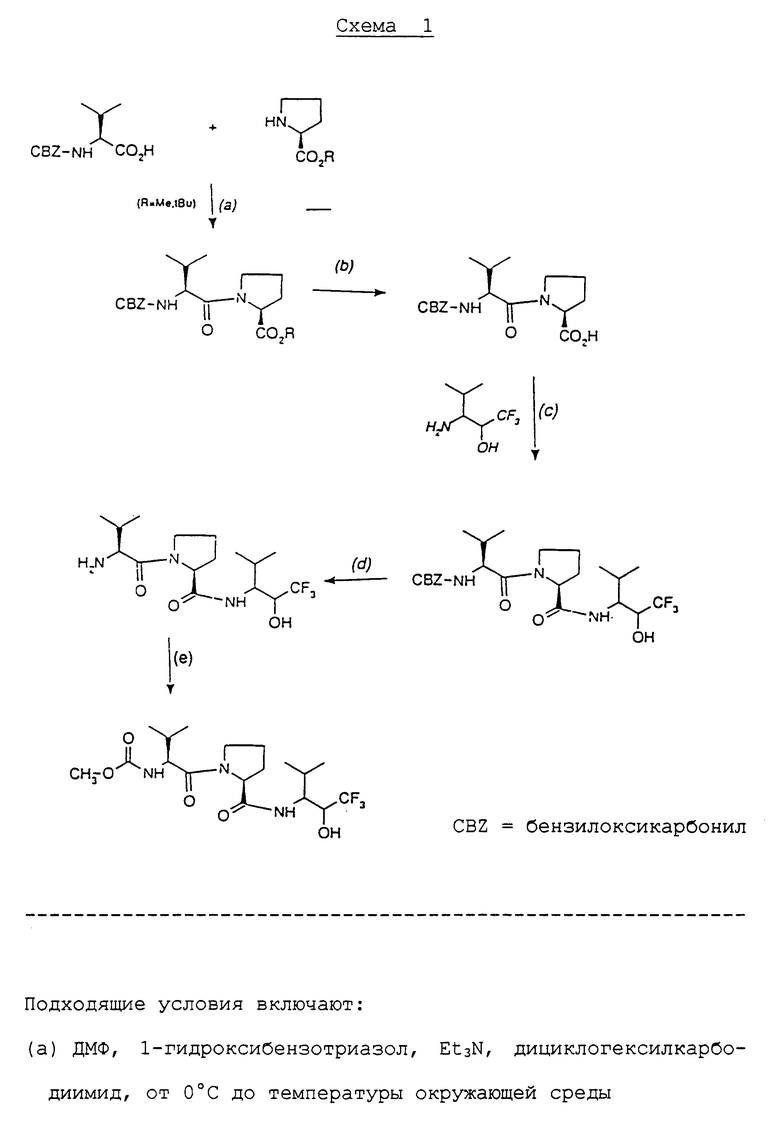
Особенно удобным является способ получения (2R,3S)-3-амино-4-метил-1,1,1-трифтор-2-пентанола, который является следующим аспектом изобретения и включает (как показано на схеме 3, приведенной в конце описания):
(1) взаимодействие (2RS,3SR)-3-амино-4- метил-1,1,1-трифтор-2-пентанола или его соли с трифосгеном или диметилкарбонатом в присутствии подходящего основания с получением (4RS,5SR)-4-изопропил-5-трифторметилоксазолидин-2-она; с последующим
(2) взаимодействием (4RS,5SR)-4-изопропил-5- трифторметил-оксазолидин-2-она или его соли со щелочным металлом с (-)-ментилхлорформиатом с получением (4RS,5SR)-4-изопропил-3- [(1R,3R,4S)-3-п-ментил-оксикарбонил]-5-трифторметил- оксазолидин-2-она, и выделением изомера (4S,5R)-4-изопропил-3- [(1R, 3R,4S)-3-п-ментил-оксикарбонил]-5-трифтор- метилоксазолидин-2-он; с последующим
(3) гидролизом изомера (4S, 5R)-4-изопролпил-3-[(1R,3R,4S)-3-п-ментилоксикарбонил]-5- трифторметилоксазолидин-2-он в щелочной среде с получением (2R,3S)-3-амино-4-метил-1,1,1-трифтор-2-пентанола.
На стадии (1) подходящим основанием является водный раствор гидроксида щелочного металла, например гидроксида натрия или калия. Реакция в целом проводится в подходящем инертном растворителе или разбавителе, например углеводороде, таком как толуол. Реакция является экзотермической и, таким образом, реакция в целом проводится в условиях внешнего охлаждения с поддержанием температуры приблизительно от 0oC до 50oC, например приблизительно при температуре окружающей среды.
На стадии (2) реакция проводится в подходящем растворителе или разбавителе, например в растворителе, имеющем эфирный характер, таком как тетрагидрофуран. Удобным образом перед добавлением (-)- ментилхлорформиата оксазолидинон преобразуют в его соль с щелочным металлом, например, используя бутиллитий при температуре около -78oC. В процессе обработки желаемый (4S,5R)-изомер кристаллизуется из смеси изомеров и собирается при помощи фильтрования.
На стадии (3) подходящие условия включают, например, использование водного раствора гидроксида щелочного металла (такого как гидроксид калия или натрия) в растворителе или разбавителе эфирного характера, таком как диоксан, при температуре в диапазоне, например, 60-130oC (таком, как например 90-120oC).
Полезность соединения согласно изобретению может быть продемонстрирована при помощи стандартных испытаний и клинических исследований, включая таковые, описанные ниже.
Количественные измерения торможения
Способность соединения согласно изобретению (или его отдельной формы) действовать в качестве ингибитора эластазы лейкоцитов человека (ЭЛЧ) на пептидный субстрат метокси-сукцинил-аланил-аланил-пролил-валин-п- нитроанилид с низким молекулярным весом определяли как описано в патенте США 4910190. Способность соединения оценивали путем получения кинетической оценки константы диссоциации, Ki, комплекса, образуемого в результате взаимодействия ингибитора с ЭЛЧ. Было найдено, что соединение из примера 1 имеет Ki, равную 36 нМ. Было найдено, что соединение из примера 2 имеет Ki, равную 9 нМ.
Модель повреждения легкого в острой форме
Модели эмфиземы на животных включали внутритрахеальное (i.t.) введение эластолитической протеиназы, чтобы вызвать медленно прогрессирующее поражение легких разрушительного характера. Обычно это поражение оценивали через промежуток времени, составляющий от нескольких недель до нескольких месяцев после исходного кровоизлияния. Однако эти протеазы вызывают также поражение, которое проявляется в первые несколько часов. Раннее поражение вначале является геморрагическим, прогрессирует в воспалительное поражение к концу первых 24 часов и устраняется в течение первой недели после кровоизлияния. Чтобы использовать преимущество этого раннего поражения, может быть использована следующая модель.
Хомячков вначале слегка анестезируют при помощи бревитала (Brevital). Затем непосредственно в трахею вводят физиологический раствор с фосфатным буфером (PBS) с pH 7,4, либо сам по себе, либо содержащий эластазу лейкоцитов человека (ЭЛЧ). Через двадцать четыре часа животных умерщвляют и легкие удаляют и тщательно срезают посторонние ткани. После определения массы сырых легких их (легкие) промывают PBS и определяют общее количество выделенных лаважируемых (способных к удалению из тканей и органов при промывании) красных и белых клеток. Значения масс сырых легких, общего количества лаважируемых красных клеток и общего количества лаважируемых белых клеток возрастают в зависимости от дозы последующего введения ЭЛЧ. Соединения, которые являются эффективными ингибиторами эластазы, могут предотвратить или уменьшить степень тяжести вызванного ферментом поражения, приводя к более низкому весу сырого легкого и снижая значения общего количества лаважируемых клеток, как красных, так и белых, по отношению к введению одной только ЭЛЧ. Провести оценку соединений можно путем их введения внутритрахеально в виде растворов или суспензий в физиологическом растворе с фосфатным буфером, либо одновременно, либо в различные моменты времени с тестовым введением ЭЛЧ (400 мкг), или путем их дозированного введения внутривенно или перорально в виде растворов в различные моменты времени перед тестовым введением ЭЛЧ (100 мкг), чтобы определить их полезность в предотвращении вызванного ЭЛЧ поражения. Раствор соединения согласно изобретению (или его отдельной формы) может быть удобным образом приготовлен с использованием 10% полиэтиленгликоля 400 в PBS.
Испытание на кровотечение в острой форме
Это испытание основывается на наблюдении только за количественной характеристикой кровоизлияния в легкое после внутритрахеального введения эластазы нейтрофилов человека (ЭНЧ). Количественную характеристику кровоизлияния определяют путем разрушения эритроцитов, выделенных из использовавшейся промывной жидкости после лаважа легких, и проведения сравнения с разведением цельной крови хомячка. Порядок проверки, подобный порядку, описанному в работе Fletcher et al., American Review of Respiratory Disease (1990), 141, 672-677, состоит в следующем. Соединения, для которых показано, что они являются ингибиторами ЭНЧ in vitro, удобным образом готовят для дозированного введения как описано выше для модели повреждения легкого в острой форме. Самцов сирийских хомячков (голодавших в течение 16-128 часов перед использованием) слегка анестезируют при помощи бревитал-натрия (30 мг/кг внутрибрюшинно). Затем производят дозированное введение хомячкам соединений внутривенно или перорально за заданное время, как например 30 или 90 минут, до внутритрахеального введения ЭНЧ в 300 мкл физиологического раствора с фосфатным буфером с pH 7,4 из расчета 50 мкг/животное. Через четыре часа после введения фермента животных умерщвляют при помощи избыточной дозы пентобарбитал-натрия, вскрывают грудную клетку и удаляют легкие и сердце и очищают легкие от побочного материала. Иссеченные легкие промывают тремя сменами по 2 мл PBS через трахеотомическую трубку. Выделенные промывки объединяют, регистрируют их объемы (около 5 мл) и промывки сохраняют при 4oC до проведения анализа. Для расчета количества крови в каждом образце оттаявшие промывки и образец цельной крови хомячка обрабатывают ультразвуком, чтобы разрушить эритроциты, и соответствующим образом разбавляют в отдельных лунках 96-луночного титрационного микропланшета. Оптические плотности промывок и образцов крови, подвергшихся разрушению эритроцитов, определяют при 540 нм. Отношения (мкл эквивалентов крови)/(мл промывки) определяют путем сравнения оптической плотности испытуемых образцов с оптической плотностью калибровочной кривой, полученной на основе цельной крови хомячков. Общее количество выделенных эквивалентов крови в мкл определяют путем умножения объема выделенной промывки на отношение (мкл эквивалентов крови)/(мл промывки) для каждого образца. Результаты даются в виде % торможения вызванного ЭНЧ кровотечения по отношению к контрольным образцам, обработанным физиологическим раствором с фосфатным буфером, для испытуемого соединения, взятого в определенной дозе и с определенным временем введения до введения ЭНЧ. Было найдено, что для соединения согласно примеру 1 ED50 (эффективная доза, обеспечивающая торможение на 50%) составляет 4,5 мг/кг при пероральном дозированном введении. Для соединения согласно примеру 2 было найдено, что ED50 составляет 1,9 мг/кг при пероральном дозированном введении и 0,6 мг/кг при внутривенном дозированном введении.
Не наблюдалось явной токсичности в тех случаях, когда соединение согласно изобретению вводили в указанных выше испытаниях in vivo.
Следует понимать, что выводы об активности соединения в модели повреждения легкого в острой форме или в испытании на кровотечение в острой форме не ограничиваются эмфиземой, но скорее, что испытание дает доказательство общего ингибирования ЭЛЧ in vivo.
Согласно следующей особенности изобретения предусматривается фармацевтическая композиция, включающая фармацевтически эффективное количество соединения согласно изобретению (либо его отдельной формы) или его сольвата и фармацевтически приемлемый разбавитель или носитель. Как отмечено выше, другой особенностью изобретения является способ использования соединения согласно изобретению (либо его отдельной формы) или его сольвата при лечении заболеваний или состояний млекопитающего, в особенности человека, к которым причастна ЭЛЧ, таких как заболевания или состояния, описанные выше, и, в частности, острого и хронического бронхита, эмфиземы легких, реперфузионной травмы, респираторного дистресс-синдрома у взрослых, муковисцидоза или периферического сосудистого заболевания (такого как критическая ишемия конечности или перемежающаяся хромота).
Соединение согласно изобретению (или его отдельная форма) может вводиться теплокровному животному, в частности человеку, которое (который) нуждается в лечении заболевания, к которому причастна ЭЛЧ, в форме стандартной фармацевтической композиции, например, как в целом описано в патенте США 4910190. Один способ введения может осуществляться посредством порошкообразного или жидкого аэрозоля. В виде порошкообразного аэрозоля соединение согласно изобретению (или его отдельная форма) может вводиться тем же способом, что и хромолин-натрий, через устройство типа турбоингалятора "Spinhaler" (товарный знак), производимого фирмой Fisons Corp. of Bedford, Massachusets, с нормой приблизительно от 0,1 до 50 мг на капсулу, причем среднему человеку вводится от 1 до 8 капсул в день. Каждая капсула, предназначенная для использования в турбоингаляторе, содержит требуемое количество соединения согласно изобретению (или его отдельной формы), а остальное содержимое массой 20 мг на капсулу представляет собой фармацевтически приемлемый носитель, такой как лактоза. В виде жидкого аэрозоля соединение согласно изобретению (или его отдельная форма) может вводиться с использованием распылителя (аэрозольного ингалятора), такого как, например, распылитель "Retec" (товарный знак), в котором раствор распыляется сжатым воздухом. Аэрозоль может вводиться, например, по норме от одного до приблизительно восьми раз в день следующим образом: распылитель заполняют раствором соединения (или его отдельной формы), например 3,5 мл раствора, содержащего 10 мг/мл; раствор в распылителе распыляют сжатым воздухом; и пациент дышит нормальным образом (дыхательный объем) в течение восьми минут с распылителем во рту.
В альтернативном варианте режим введения может быть парентеральным, включая подкожное отложение, при посредстве осмотического насоса или предпочтительно пероральным. Соединение согласно изобретению (или его отдельная форма) может быть удобным образом приготовлена в виде пероральной или парентеральной лекарственной формы путем смешивания приблизительно от 10 до 250 мг на единицу лекарственной формы со стандартным носителем, эксципиентом, связующим веществом, консервантом, стабилизатором, вкусовой добавкой или тому подобное, как предусматривается общепринятой фармацевтической практикой, например, как описано в патенте США 3755340. Для парентерального введения следует проводить внутривенную, внутримышечную или подкожную инъекцию объемом от 1 до 10 мл, содержащую приблизительно от 0,02 мг до 10 мг на кг массы тела соединения согласно изобретению (или его отдельной формы), 3 или 4 раза в день. Инъекция должна содержать соединение согласно изобретению (или его отдельную форму) в водном изотоническом стерильном растворе или суспензии, необязательно с консервантом, таким как фенол, или с солюбилизатором, таким как этилендиаминтетрауксусная кислота (ЭДТК). Для парентерального введения или использования в виде аэрозоля может быть приготовлен водный состав, например, путем растворения соединения (или его отдельной формы) в 5-10% полиэтиленгликоле 400 в физиологическом растворе с фосфатным буфером, за которым следует асептическая фильтрация и хранение в стерильных условиях с использованием стандартных методик.
Обычно соединение согласно изобретению (или его отдельная форма) может быть введено человеку в дневной дозе в диапазоне, например, от 5 до 100 мг соединения (или его отдельной формы) в виде аэрозоля, или от 50 до 1000 мг внутривенно или перорально, или в их сочетании. Однако понятно, что может быть необходимо изменить дозу вводимого соединения (или его отдельной формы) в соответствии с хорошо известной медицинской практикой, принимая во внимание природу и тяжесть заболевания, лечение которого производится, сопутствующую терапию и возраст, вес и пол пациента, подвергающегося лечению. Одновременно следует учесть, что могут также использоваться обычно эквивалентные количества сольватированной (например, гидратированной) формы соединения. Порядок введения ингибитора ЭЛЧ и оценки их действия на пациентах описаны в Европейских патентных заявках с номерами публикаций 458535, 458536, 458537 и 463811 для лечения или предотвращения муковисцидоза, острого респираторного дистресс-синдрома, бронхита и кровотечения, связанного с острым лейкозом, не относящимся
к лимфолейкозу, или его терапией соответственно; и соединение согласно изобретению (или его отдельная форма) может использоваться подобным образом или предпочтительно использоваться путем перорального введения для лечения этих заболеваний и состояний либо само по себе, либо в сочетании с другим терапевтическим средством, обычно показанным для лечения конкретного состояния. Для терапевтического лечения или профилактики у млекопитающего сосудистого заболевания или связанного с ним состояния, в котором участвуют нейтрофилы или к которому причастны нейтрофилы, соединение согласно изобретению (или его отдельная форма) может быть удобным образом введено пероральным или парентеральным путем либо само по себе, либо одновременно или последовательно с другими терапевтически активными средствами, обычно вводимыми при таком состоянии. Полезность соединения согласно изобретению (или его отдельной формы) в таком лечении сосудистых заболеваний и связанных с ними состояний может быть продемонстрирована с использованием методик, описанных в International Patent Application, Publication N WO 92/22309.
Различные аспекты изобретения далее проиллюстрированы следующими, не являющимися ограничивающими примерами, в которых, если не указано иначе:
(1) температуры даны в градусах по Цельсию (oC); операции проводили при комнатной температуре или температуре окружающей среды, то есть при температуре в интервале 18-25oC;
(2) органические растворители сушили над безводным сульфатом магния; выпаривание растворителя проводили при помощи роторного испарителя под пониженным давлением (600-4000 Па; 4,5-30 мм рт.ст.) при температуре бани до 60oC;
(3) хроматография обозначает флеш-хроматографию (метод Стилла (Still)), проводимую на сорбенте Merck Kieselgel (Art 9835 from E.Merck, Darmstadt, Germany), элюирование, в котором использовались как ступенчатые, так и пологие градиенты, обозначено заключенным в скобки термином "градиентное", за которым следуют начальные и конечные соотношения растворителей; тонкослойную хроматографию (ТСХ) проводили на пластинках с силикагелем, например на пластинках GHLF со слоем силикагеля в 0,25 мм (Art 21521 from Analtech, Newark, DE, USA);
(4) обычно за протеканием реакции следовала тонкослойная хроматография, и времена реакций даны только для иллюстрации;
(5) температуры плавления являются нескорректированными, и обозначение (разл. ) указывает на разложение; данные температуры плавления являются температурами плавления, полученными для веществ, приготовленных согласно описанию; полиморфизм может приводить к выделению в некоторых препаратах веществ с различными температурами плавления;
(6) конечные продукты имели удовлетворительные спектры ядерного магнитного резонанса (ЯМР); и в тех случаях, когда они исследовались при помощи жидкостной хроматографии высокого разрешения, являлись существенно чистыми;
(7) выходы даны только для иллюстрации и не являются обязательными значениями для выходов, которые могут быть получены при тщательной разработке процесса; если требовалось больше вещества, его приготовление повторяли;
(8) данные ЯМР, когда они даны, представлены в виде значений дельта для главных диагностических протонов, данных в частях на миллион (млн-1), по отношению к тетраметилсилану (ТМС) как встроенному стандарту, определенному при 250 МГц с использованием ДМСО-d6 в качестве растворителя; используются стандартные сокращения для формы сигналов; для спектров AB приводятся непосредственно наблюдавшиеся сдвиги;
(9) химические символы имеют их обычные значения; используются единицы и символы СИ;
(10) пониженные давления даны как абсолютные давления в паскалях (Па); повышенные давления даны как избыточные давления в паскалях (Па);
(11) соотношения растворителей даны в виде отношений объем/объем (об/об);
(12) съемку масс-спектров проводили с энергией электронов в 70 электрон-вольт в режиме химической ионизации с использованием зонда прямого экспонирования; причем указанная ионизация вызывалась электронным ударом (EI) или бомбардировкой быстрыми атомами (FAB); обычно приводятся только пики, которые указывают на массу вещества-источника; и
(13) чтобы установить соотношение SSS:SSR диастереоизомеров формулы I в выделенном веществе, использовали жидкостную хроматографию высокого разрешения с применением колонки 25 см х 4,6 мм с обращенной фазой SUPELCO LC-18 и смеси вода:ацетонитрил (70:30) в качестве элюента. Скорость течения составляла 0,1 мл/мин, объем ввода составлял 20 мкл при помощи клапана, и длина волны детектирования составляла 205 нм. Время задержки для SSS-диастереоизомера было около 9,9 минут, а время задержки для SSR-диастереоизомера составляло около 11,7 минут.
Пример 1
Гидрохлорид 1-(3-диметиламинопропил)-3- этилкарбодиимида (1,84 г) добавляли к раствору (S)-1-[(S)-2- (метокси-карбониламино) -3-метилбутирил]-N-[2-метил-1-(2,2,2- трифтор-1-гидроксиэтил) пропил]пирролидин-2-карбоксамида (0,41 г), растворенного в диметилсульфоксиде (ДМСО; 5 мл) и толуоле (5 мл), после чего по каплям добавляли дихлоруксусную кислоту (0,32 мл). Полученный раствор перемешивали при 20oC в течение 2 часов. Затем раствор выливали в этилацетат (200 мл) и последовательно промывали 1 М соляной кислотой, водой и солевым раствором. Органический раствор сушили (MgSO4) и упаривали в вакууме. Остаток очищали при помощи флеш-хроматографии (градиентное элюирование; метанол: метиленхлорид от 3:97 до 5:95) с получением (S)-1-[(S)-2-(метоксикарбониламино)-3- метилбутирил]-N-[2-метил-1- (2,2,2-трифторацетил) пропил] пирролидин-2-карбоксамида (0,27 г) в виде белой пены (в виде смеси кетонной и гидратированной форм); ТСХ, Rf=0,4 (метанол:дихлорметан 2,5: 97,5); 1H ЯМР (ДМСО/D2O): 4,44 (м, 1H), 4,00 (м, 2H), 3,72 (м, 1H), 3,51 (м, 4H), 2,02-1,75 (м, 6H), 0,95-0,78 (м, 12H); элементный анализ для C18H28F3N3O5•0,3H2O: рассчитано: C 50,42; H 6,72; N 9,80; найдено: C 50,31; H 6,28; N 9,56.
Исходное вещество (S)-1-[(S)-2- (метоксикарбониламино)-3-метилбутирил] -N-[2-метил-1-(2,2,2- трифтор-1-гидрокси-этил) пропил] пирролидин-2-карбоксамид было получено следующим образом.
Метилхлорформиат (0,12 мл) добавляли к раствору (2RS),(3SR)-L-валил-N-[3- (4-метил-1,1,1-трифтор-1- гидроксипентил)] -L-пролинамида (полученного как описано в патенте США 5194588) (0,5 г) и триэтиламина (0,57 мл) в дихлорметане (13,6 мл) при 0oC. Раствор перемешивали в течение 0,5 часа и затем выливали в этилацетат (100 мл). Органический раствор последовательно промывали насыщенным водным раствором бикарбоната натрия, водой и солевым раствором. Раствор сушили (MgSO4) и упаривали в вакууме. Остаток очищали при помощи флеш-хроматографии (градиентное элюирование; метанол:метиленхлорид от 5:95 до 7:93) с получением (S)-1-[(S)-2-(метоксикарбониламино)-3-метил- бутирил] -N-[2-метил-1- (2,2,2-трифтор-1-гидроксиэтил)- пропил]пирролидин-2-карбоксамида (0,51 г); ТСХ, Rf=0,2 (метанол:метиленхлорид 5:95); MS: m/z = 426 (М+1).
Пример 2
Раствор перманганата калия (16,6 г) в воде (100 мл) добавляли по каплям к имеющему температуру 0oC раствору (S)-1- [(S)-2-(метоксикарбониламино) -3-метилбутирил] -N-[(S)-2- метил-1-((R)-2,2,2- трифтор-1-гидроксиэтил) пропил] -пирролидин- 2-карбоксамида (15 г) в смеси трет-бутилового спирта (175 мл), воды (100 мл) и 0,6 М раствора гидроксида натрия (175 мл). Раствор перемешивали в течение 2 часов и затем гасили путем добавления метанола (70 мл) с последующим перемешиванием в течение 1 часа. Раствор фильтровали через диатомовую землю и фильтрат подкисляли до pH 2, используя 1 М соляную кислоту, и насыщали хлоридом натрия. Продукт экстрагировали эфиром (5 х 100 мл) и растворитель удаляли в вакууме. Полученное масло хроматографировали (метанол:дихлорметан 5:95) и растворитель удаляли с получением масла. К перемешиваемому раствору масла в этилацетате (который был предварительно насыщен водой) (40 мл) добавляли гексан (40 мл) и перемешивание продолжали в течение 24 часов, на протяжении которых образовывался кристаллический осадок. Добавляли другую порцию гексана (40 мл) и твердое вещество собирали и сушили в вакууме (40oC) с получением (S)-1-[(S)-2-(метоксикарбониламино)- 3-метилбутирил] -N-[(S)-2- метил-1-(2,2,2-трифторацетил) пропил]пирролидин-2-карбоксамида (9,45 г) в виде белого кристаллического твердого вещества (в виде, по существу или практически, формы A); 1H ЯМР (300 МГц, ДМСО/D2O): 4,42 (м, 1H), 4,02 (д, 1H), 3,73 (м, 1H), 3,59 (м, 1H), 3,54 (с, 3H), 2,23 (м, 1H), 2,00-1,76 (м, 6H), 0,91 (м, 6H), 0,85 (д, 3H), 0,80 (д, 3H); элементный анализ для C18H28F3N3O5•H2O: рассчитано: C 48,97; H 6,85; N 9,51; найдено: C 49,02; H 6,80; N 9,66 (спектр дифракции рентгеновских лучей показан на фиг. 1).
Исходное вещество (S)-1-[(S)-2-(метоксикарбониламино)-3-метилбутирил]-N- [(S)-2-метил-1-((R)-2,2,2-трифтор-1-гидроксиэтил) пропил] пирролидин-2-карбоксамид было получено следующим образом.
(1) Трет-бутиловый эфир N-[(фенилметокси)карбонил]-L- валил-L-пролина (905 г) растворяли в этаноле (4 литра) и добавляли 10% палладий на угле (20 г). Реакционную смесь встряхивали в атмосфере водорода (345 кПа) в течение 12 часов и затем катализатор удаляли при помощи фильтрования через диатомовую землю. Фильтрат упаривали в вакууме и остаток дважды выпаривали с толуолом (1 литр) с получением трет-бутилового эфира L-валил-L-пролина в виде масла (628 г); ТСХ, Rf=0,2, ацетон:гексан 20:80; MS: m/z=271 (M+1).
(2) Растворы карбоната натрия (110,5 г) в воде (1,5 литра) и трет-бутилового эфира L-валил-L-пролина в тетрагидрофуране (ТГФ; 1 л) объединяли и охлаждали до 0oC. Смесь разбавляли эфиром (400 мл) и по каплям добавляли метилхлорформиат (39,4 г). Затем реакционной смеси давали нагреться до температуры окружающей среды в течение 2 часов. Слои разделяли и органическую фазу промывали дважды 1 М соляной кислотой, а затем насыщенным водным раствором бикарбоната натрия и солевым раствором. Водную фазу экстрагировали эфиром. Все органические фазы объединяли и сушили (MgSO4) и растворитель удаляли с получением трет-бутилового эфира N-(метоксикарбонил)- L-валил-L-пролина (125,9 г); 1H ЯМР (300 МГц, ДМСО/d4-трифторуксусная кислота): 4,23 (дд, 1H), 4,06 (д, 1H), 3,78 (м, 1H), 3,57 (м, 1H), 3,55 (с, 3H), 2,16 (м, 1H), 1,95 (м, 1H), 1,80 (м, 1H), 1,42 (с, 9H), 0,94 (м, 6H); MS: m/z = 329 (М+1).
(3) К раствору трет-бутилового эфира N-(метокси-карбонил)-L-валил-L-пролина (813 г) в толуоле (3 литра) добавляли ионообменную смолу Amberlyst-15 (190 г). Реакционную смесь нагревали до 120oC, чтобы отогнать воду, присутствующую в смоле, в виде азеотропа вода/толуол. Собиралось приблизительно 400 мл дистиллята. Затем нагревание продолжали с обратным холодильником в течение 1,5 часов. Реакционную смесь охлаждали до 60oC и смолу удаляли фильтрованием. Фильтрат экстрагировали 1 М NaOH (2,5 литра), а затем насыщенным водным раствором бикарбоната натрия. Объединенные щелочные экстракты экстрагировали смесью ТГФ/этилацетат (1:1, 1 литр) и затем охлаждали на ледяной бане. Водный раствор подкисляли до pH 1,5, используя холодную 3 М соляную кислоту (1 литр) и экстрагировали дважды смесью ТГФ/этилацетат (1:1, 1,5 литра и 1 литр). Экстракты объединяли и промывали солевым раствором, сушили (MgSO4) и растворитель удаляли путем выпаривания. Полученное вещество растворяли в эфире (1 литр) и давали ему кристаллизоваться при 0oC в течение 48 часов. Полученное твердое вещество собирали при помощи фильтрования, промывали холодным эфиром и сушили в вакууме с получением N-(метоксикарбонил)-L-валил-L-пролина (373 г); 1H ЯМР (300 МГц, ДМСО): 12,4 (с, 1H), 7,37 (д, 1H), 4,25 (дд, 1H), 4,00 (т, 1H), 3,79 (м, 1H), 3,55 (м, 1H), 3,51 (с, 3H), 2,11 (м, 1H), 1,85 (м, 4H), 0,91 (д, 3H), 0,87 (д, 3H); MS: m/z = 273 (М+1).
(4) N-Метилморфолин (8,5 мл) добавляли к раствору N- (метоксикарбонил)-L-валил-L-пролина (12,5 г) в ТГФ (150 мл) и раствор охлаждали до -15oC на бане из смеси льда с ацетоном. Добавляли по каплям изобутилхлорформиат (6,6 мл) и смесь перемешивали в течение 1 часа. Добавляли вторую порцию N-метилморфолина (8,5 мл), а затем - гемиоксалат (2R), (3S)-3-амино- 4-метил-1,1,1-трифтор-2-пентанола (10 г). Затем реакционную смесь перемешивали в течение 12 часов, давая ей в это время нагреться до комнатной температуры. Реакционную смесь разбавляли эфиром (500 мл) и последовательно промывали насыщенным водным раствором бикарбоната натрия, 1 М HCl и солевым раствором. Водные слои экстрагировали эфиром и все органические фазы объединяли и сушили (MgSO4). Раствор фильтровали и растворитель удаляли путем выпаривания. Полученное вещество фильтровали через силикагель, используя эфир в качестве элюента. Эфирные фракции, содержащие продукт, объединяли и растворитель удаляли путем выпаривания. Продукт сушили в вакууме с получением (S)-1-[(S)-2- (метоксикарбониламино)-3-метилбутирил]-N-[(S)-2-метил-1-((R)- 2,2,2-трифтор-1-гидроксиэтил) пропил] пирролидин-2-карбоксамида (16,1 г); 1H ЯМР (300 МГц, ДМСО): 7,61 (д, 1H), 7,28 (д, 1H), 6,44 (д, 1H), 4,44 (м, 1H), 4,05 (м, 1H), 3,98 (м, 1H), 3,75 (м, 2H), 3,55 (м, 1H), 3,50 (с, 3H), 1,83 (м, 6H), 0,90 (д, 3H), 0,86 (д, 3H); MS: m/z = 426.
Гемиоксалат (2R),(3S)-3-амино-4-метил-1,1,1-трифтор-2-пентанола, использовавшийся на стадии (4), был получен следующим образом.
(1) Трифосген (23 г) добавляли единой порцией к хорошо перемешиваемой смеси гемиоксалата (2RS), (3SR)-3- амино-4-метил-1,1,1-трифтор-2-пентанола (50 г) в толуоле (250 мл) и 2 М раствора гидроксида натрия (350 мл). Реакция начинала выделять тепло, и ее помещали на ледяную баню. Через 0,5 часа реакционную смесь нагревали до 25oC, и тонкослойная хроматография указала на присутствие значительного количества не вступившего в реакцию амина. pH раствора вновь доводили до значения около 12, используя 50% раствор гидроксида натрия. Добавляли дополнительную порцию трифосгена (8 г) и раствор перемешивали в течение 1 часа. pH реакционной смеси снижали до pH 7, используя 1 М соляную кислоту, и дважды экстрагировали эфиром. Объединенные эфирные слои промывали водой, солевым раствором и сушили (MgSO4). Растворитель удаляли путем выпаривания с получением масла, которое при стоянии кристаллизовалось. Получившееся твердое вещество собирали при помощи фильтрования и промывали смесью эфир: гексан (1:1) с получением 27 г (4RS),(5SR)-4-изопропил-5-трифторметил- оксазолидин-2-она в виде белого твердого вещества, Т.пл. 71-72oC; 1H ЯМР (300 МГц, ДМСО): 8,45 (с, 1H), 5,11 (м, 1H), 3,61 (м, 1H), 1,72 (м, 1H), 0,86 (д, 6H).
(2) н-Бутиллитий (20 мл 10 М раствора в гексане) добавляли к раствору (4RS), (5SR)-4-изопропил-5-трифторметилоксазолидин-2-она (35,8 г) в ТГФ (600 мл) при -78oC, с последующим перемешиванием в течение 0,5 часа. Добавляли (-)-ментилхлорформиат (41 мл, свежеперегнанный), после чего продолжали перемешивание при -78oC в течение 0,5 часа. Раствор нагревали до 25oC и реакцию гасили добавлением насыщенного водного раствора бикарбоната натрия. Продукт экстрагировали эфиром и промывали водой и солевым раствором. Раствор сушили (MgSO4) и растворитель удаляли в вакууме. Получившееся масло кристаллизовалось при стоянии, образуя твердое вещество, которое собирали при помощи фильтрования. Твердое вещество промывали смесью эфир:гексан (1:1) и сушили с получением (4S), (5R)-4-изопропил-3-[(1R,3R,4S) -3-п-ментилоксикарбонил]-5-трифторметилоксазолидин-2-она (23,15 г); Т.пл. 138-140oC; 1H ЯМР (300 МГц, ДМСО): 5,51 (дд, 1H), 4,68 (м, 1H), 4,26 (м, 1H), 2,27 (м, 1H), 1,94 (д, 1H), 1,78 (м, 1H), 1,62 (д, 2H), 1,42 (м, 2H), 1,01 (дд, 2H), 0,95-0,84 (м, 24H), 0,71 (д, 3H); 19F ЯМР (376,5 МГц, ДМСО): -76,9910; 99% d.e. (Дополнительный выход продукта в количестве 4,3 г (99% d.e.) был получен из маточной жидкости). [Замечание: (4R),(5S)-изомер имеет Т.пл. 80-82oC и 19F ЯМР (376,5 МГц, ДМСО): -77,0019.]
(3) Раствор (4S),(5R)-4-изопропил-3-[(1R,3R,4S)-3-п-ментилоксикарбонил] -5- трифторметилоксазолидин-2-она (27 г) в диоксане (70 мл) и 50% раствор гидроксида калия (80 мл) нагревали при 100oC в течение 2 дней. Реакционную смесь охлаждали, разбавляли эфиром (400 мл) и отделяли органический слой. pH водного раствора доводили до 9 (исходно составлял около 14), используя 6 М соляную кислоту. Водный слой трижды экстрагировали эфиром (300 мл). Органические фазы объединяли, сушили (MgSO4) и добавляли к хорошо перемешиваемому раствору дигидрата щавелевой кислоты (4,5 г) в ацетонитриле (100 мл). Твердое вещество, которое выпадало в осадок, собирали при помощи фильтрования, промывали эфиром и сушили в вакууме (60oC) с получением 15,9 г белого твердого вещества. Твердое вещество растирали с эфиром (300 мл), собирали при помощи фильтрования и сушили с получением (2R),(3S)-3-амино-4-метил-1,1,1-трифтор- 2-пентанола, выделенного в форме его соли гемиоксалата (13,4 г, выход 88%), в виде твердого вещества белого цвета, Т.пл. 184-186oC; 1H ЯМР (300 МГц, ДМСО): 5,71 (ушир.с, 3H), 4,08 (ддд, 1H), 2,88 (м, 1H), 1,81 (м, 1H), 0,92 (м, 6H); элементный анализ для C6H12F3NO•0,5C2H2O4: рассчитано: C 38,89; H 6,06; N 6,48; найдено: C 38,75; H 5,95; N 6,47.
(2RS), (3SR)-3-Амино-4-метил-1,1,1-трифтор-2-пентанол, использовавшийся на стадии (1), получали как описано в патенте США 4910190.
Пример 3
Используя методику окисления, подобную методике, описанной в примере 2, но с использованием (S)-1-[(S)-2- (метоксикарбониламино)-3-метилбутирил]-N-[2-метил-1-(2,2,2- трифтор-1-гидроксиэтил)пропил]пирролидин-2-карбоксамида и добавлением раствора перманганата калия при 5-10oC, а затем перемешиванием при 10oC в течение одного часа перед обработкой метанолом получали (после обработки при помощи экстракции трет-бутилметиловым эфиром с последующим промыванием солевым раствором и упариванием в вакууме) (S)-1-[(S)-2-(метоксикарбониламино)- 3-метилбутирил] -N-[2-метил-1-(2,2,2-трифторацетил) пропил] - пирролидин-2-карбоксамид в виде смолы (с выходом 75%); соотношение SSS: SSR составляет 53:47; соотношение гидрат:кетон 1:1; спектр 1H ЯМР подобен спектру продукта из примера 1. [При использовании аналогичной методики, однако с добавлением раствора перманганата калия при температуре окружающей среды вместо 5-10oC получали продукт с соотношением SSS:SSR, составляющим 47:53.]
Исходное вещество (S)-1-[(S)-2-(метоксикарбониламино)-3- метилбутирил] -N-[2-метил-1-(2,2,2-трифтор-1-гидрокси-этил) пропил] пирролидин-2-карбоксамид было получено в виде масла (с выходом 55%) при использовании методики, аналогичной методике, описанной в примере 2, часть (4), но с применением 3-амино-4- метил-1,1,1-трифтор-2-пентанола (в виде смеси диастереоизомеров), который, в свою очередь, получали как описано в патенте США 4910190 или следующим образом.
(1) Раствор мочевины (72 г) в диметилформамиде (810 мл) добавляли к нитриту натрия (90 г), перемешивали в течение 10 минут, а затем охлаждали до 15oC. В течение 30 минут добавляли изобутилйодид (97,2 мл) и реакционной смеси давали перемешиваться при температуре окружающей среды в течение 20 часов. Смесь вновь охлаждали до 15oC и медленно добавляли воду (810 мл). Смесь перемешивали в течение 5 минут при температуре окружающей среды и затем дважды экстрагировали метил-трет-бутиловым эфиром. Объединенные органические экстракты дважды промывали 20% раствором тиосульфата натрия и упаривали в вакууме с получением 2-метил-1-нитропропана (39 г), который использовали без дальнейшей очистки.
(2) Молекулярные сита 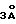 (27,04 г) нагревали при 120oC в вакууме в течение 20 часов и добавляли к раствору 2-метил-1-нитропропана (13,0 г) в метил-трет-бутиловом эфире (420 мл). Смесь перемешивали в течение 5 минут, добавляли карбонат калия (64,5 г) и смесь перемешивали в течение последующих 30 минут. Смесь охлаждали до 15oC и в течение 30 минут добавляли флуоральгидрат (22,0 г). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов, затем охлаждали до 15oC и добавляли воду (270 мл). После перемешивания в течение 5 минут при температуре окружающей среды органическую фазу отделяли и промывали 10% водным раствором карбоната калия, 2 М раствором соляной кислоты и водой. Затем растворитель удаляли при помощи выпаривания под пониженным давлением при температуре ниже 40oC и масло отгоняли в виде безводного азеотропа с изопропиловым спиртом при температуре ниже 50oC с получением 4-метил-3-нитро-1,1,1- трифтор-2-пентанола (21,3 г) в виде масла, которое использовали без дальнейшей очистки.
(27,04 г) нагревали при 120oC в вакууме в течение 20 часов и добавляли к раствору 2-метил-1-нитропропана (13,0 г) в метил-трет-бутиловом эфире (420 мл). Смесь перемешивали в течение 5 минут, добавляли карбонат калия (64,5 г) и смесь перемешивали в течение последующих 30 минут. Смесь охлаждали до 15oC и в течение 30 минут добавляли флуоральгидрат (22,0 г). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов, затем охлаждали до 15oC и добавляли воду (270 мл). После перемешивания в течение 5 минут при температуре окружающей среды органическую фазу отделяли и промывали 10% водным раствором карбоната калия, 2 М раствором соляной кислоты и водой. Затем растворитель удаляли при помощи выпаривания под пониженным давлением при температуре ниже 40oC и масло отгоняли в виде безводного азеотропа с изопропиловым спиртом при температуре ниже 50oC с получением 4-метил-3-нитро-1,1,1- трифтор-2-пентанола (21,3 г) в виде масла, которое использовали без дальнейшей очистки.
(3) Раствор 4-метил-3-нитро-1,1,1- трифтор-2-пентанола (17,1 г) в изопропаноле (115 мл) и уксусной кислоте (0,43 мл) гидрировали на 10% палладии на угле (2,4 г) при давлении 350 кПа до тех пор, пока не завершалось поглощение водорода. Катализатор удаляли при помощи фильтрования через диатомовую землю и фильтровальную лепешку промывали изопропанолом. Фильтрат упаривали в вакууме до тех пор, пока изопропанол больше не отгонялся, и остаток растворяли в ацетонитриле (40 мл). Добавляли при перемешивании раствор щавелевой кислоты (3,94 г) в ацетонитриле (80 мл) и смесь охлаждали при 5oC. Продукт, который кристаллизовался, собирали при помощи фильтрования, промывали холодным ацетонитрилом и сушили при 50oC с получением 4-метил-3-нитро-1,1,1-трифтор-2-пентанола в форме его соли со щавелевой кислотой (оксалата) (9,08 г).
Пример 4
Гексан (13 мл) добавляли к раствору (S)-1-[(S)-2- (метоксикарбониламино)-3-метилбутирил] -N-[2-метил-1-(2,2,2- трифторацетил)пропил]пирролидин-2-карбоксамида (0,85 г; SSS:SSR 53:47; гидрат:кетон 1: 1) в трет-бутилметиловом эфире (8,5 мл) до тех пор, пока продолжалось помутнение. Затем раствор нагревали с получением прозрачного раствора, вносили затравку существенно чистого кристаллического SSS-диастереоизомера и давали отстояться. Кристаллизовалось твердое вещество белого цвета, которое собирали при помощи фильтрования с получением (S)-1-[(S)-2- (метоксикарбонил-амино)-3- метилбутирил]-N-[(S)-2-метил -1-(2,2,2-трифторацетил) пропил] пирролидин-2-карбоксамида в виде кристаллического твердого вещества с выходом 30%, SSS: SSR 95:5; гидрат:кетон 80:20; спектр ЯМР подобен спектру продукта из примера 2.
Пример 5
Используя методику, подобную методике, описанной в примере 4, но исходя из диастереомерной смеси с SSS:SSR 53:47 (1,73 г) и гидрат:кетон 95:5 и добавляя к растворителю, из которого проводится кристаллизация, 36% (массовых) соляную кислоту (0,06 мл) и воду (0,04 мл) перед добавлением гексана, получали с выходом 22% кристаллический SSS-диастереоизомер с SSS:SSR 98,5:1,5 и существенно или практически в гидратной форме.
Пример 6
Используя методику, подобную методике, описанной в примере 5, но исключая соляную кислоту, получали кристаллическую диастереомерную смесь с SSS:SSR 65:35 и находящуюся существенно или практически в гидратной форме.
Пример 7
Используя методику, подобную методике, описанной в примере 5, но исходя из диастереомерной смеси с SSS:SSR 47:53 и гидрат:кетон 60:40, получали с выходом 18% кристаллический SSS-диастереоизомер с SSS:SSR 98,5:1,5 и находящуюся существенно или практически в гидратной форме.
Пример 8
Продукт из примера 2 (5 г) растворяли в 1,2- диметоксиэтане (6 мл) при слабом нагревании. К раствору осторожно добавляли воду (5 мл) с получением прозрачного раствора. Раствору давали остыть до температуры окружающей среды, вносили затравку существенно чистого кристаллического SSS-диастереоизомера и давали отстояться в течение 16 часов. Кристаллическую массу, которая образовалась на дне сосуда, осторожно размельчали и собирали при помощи вакуумного фильтрования. Кристаллический продукт промывали смесью 1,2-диметоксиэтана и воды и давали сохнуть в токе воздуха в течение 16 часов с получением кристаллического SSS-диастереоизомера (содержащего менее чем 2% SSR-диастереоизомера) в виде по существу или практически формы B с содержанием воды 7,3% массовых; (спектр дифракции рентгеновских лучей показан на фиг. 3). [Используя подобную методику, но применяя в качестве исходного вещества перекристаллизованную форму A, получали форму B с содержанием воды 7,7% массовых.]
Пример 9
Продукт из примера 8 (4,78 г) растворяли в этилацетате (14,7 мл) при нагревании до 60oC в инертной атмосфере. Медленно добавляли гексан (22 мл) и раствору давали остыть до 22oC. Кристаллический продукт собирали при помощи фильтрования и промывали гексаном (10 мл), затем давали сохнуть в токе воздуха с получением кристаллического существенно чистого SSS-диастереоизомера (содержащего менее чем 2% SSR-диастереоизомера) в виде по существу или практически формы A с содержанием воды 4,1% массовых.
Пример 10
Продукт из примера 2 (1 г) растворяли в циклогексане (20 мл) и раствор перегоняли при атмосферном давлении при 80oC, уменьшая объем до 7 мл. Затем прозрачному раствору давали остыть до 24oC. Суспендированное твердое вещество собирали при помощи фильтрования под пониженным давлением, проводимого в токе сухого азота, и сушили в эксикаторе в вакууме в присутствии пятиокиси фосфора. Таким образом получали кристаллический SSS-диастереоизомер (содержащий менее чем 2% SSR-диастереоизомера) в виде по существу или практически "кетонной" формы; (спектр дифракции рентгеновских лучей показан на фиг. 4).





Описываются новые производные пролина общей формулы I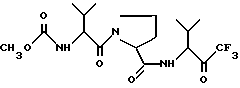
или его сольватированная форма, или его кеталь, или геми-кеталь в виде диастереомерной смеси, содержащей 50% или более диастереоизомера формулы Ia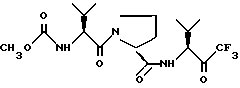
или его сольватированной формы, или его кеталя, или геми-кеталя. Соединения являются ингибиторами эластазы лейкоцитов человека. 6 с. и 10 з.п. ф-лы, 8 ил.
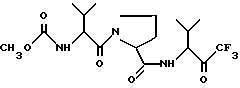
или его сольватированная форма, или его кеталь, или геми-кеталь в виде диастереомерной смеси, содержащей 50% или более диастереоизомера формулы Ia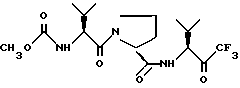
или его сольватированной формы, или его кеталя, или геми-кеталя.
или его гидратированная форма, или его фармацевтически приемлемая соль.
подходящим окислителем.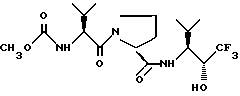
10. Способ по п. 9 с последующим образованием кристаллов из раствора продукта в растворителе, выбранном из бутилацетата, смеси бутилацетата и гексана, смеси ацетона и воды, смеси ацетона и гексана, смеси ацетона и петролейного эфира с температурой кипения 100 - 120oC, смеси 1,2-диметоксиэтана и гексана, смеси 1,2-диметоксиэтана, воды и гексана, смеси этилацетата и гексана, смеси этилацетата, гексана и воды, воды, смеси дибутилового эфира и гексана, смеси дихлорметана и гексана, смеси метанола и толуола, смеси метил-трет-бутилового эфира и гексана, смеси изопропанола и гексана и смеси тетрагидрофурана и гексана.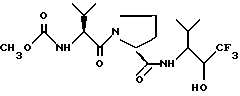
подходящим окислительным агентом с последующим образованием кристаллов из продукта в растворителе, выбранном из бутилацетата, смеси бутилацетата и гексана, смеси ацетона и воды, смеси ацетона и гексана, смеси ацетона и петролейного эфира с температурой кипения 100 - 120oC, смеси 1,2-диметоксиэтана и гексана, смеси 1,2-диметоксиэтана, воды и гексана, смеси этилацетата и гексана, смеси этилацетата, гексана и воды, воды, смеси дибутилового эфира и гексана, смеси дихлорметана и гексана, смеси метанола и толуола, смеси изопропанола и гексана и смеси тетрагидрофурана и гексана.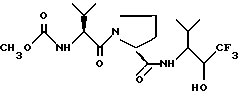
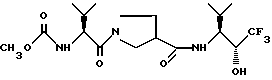
15. Способ получения промежуточного продукта (2R, 3S)-3-амино-4-метил-1,1,1-трифтор-2-пентанола, или его соли, который заключается во: (1) взаимодействии (4RS, 5SR)-4-изопропил-5-трифторметилоксазолидин-2-она или его соли с щелочным металлом с (-)-ментилхлорформиатом с получением (4RS, 5SR)-4-изопропил-3-[(1R, 3R, 4S] -3-п-ментил-оксикарбонил]-5-трифторметилоксазолидин-2-она; (2) выделении изомера (4S, 5R)-4-изопропил-3-[(1R, 3R, 4S)-3-п-ментилоксикарбонил] -5-трифторметилоксазолидин-2-он и (3) гидролизе изомера (4S, 5R)-4-изопропил-3-[(1R, 3R, 4S)-3-п-ментилоксикарбонил]-5-трифторметилоксазолидин-2-он в щелочной среде с получением (2R, 3S)-3-амино-4-метил-1,1,1-трифтор-2-пентанола.
| Способ получения производных 1, 2, 5, 6-тетрагидропиридина или их гидрохлоридов, или бензолсульфонатов | 1988 |
|
SU1678203A3 |
| US 4643991 A, 17.02.1987 | |||
| US 4910190 A, 20.03.1990 | |||
| EP 433811 A2, 20.06.1991 | |||
| Шихта для изготовления огнеупорного материала | 1973 |
|
SU458535A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
