Изобретение относится к способам получения порошков фосфатов кальция, которые могут быть использованы для производства биодеградируемых медицинских материалов, стимулирующих восстановление дефектов костной ткани.
Фосфаты кальция с мольным соотношением Са/Р, равным 1, могут служить основой для создания биодеградируемых материалов. Такими фосфатами являются брушит СаНРO4*2Н2O, монетит СаНРO4 и пирофосфат Са2Р2O7 (ПФК) [1]. И если гидроксиапатит (ГАП) и некоторые другие сложные фосфаты являются конечными продуктами процесса биологической минерализации, то ПФК является одним из промежуточных продуктов в этом процессе. Спеченный ПФК в лучшей степени способствует образованию костных клеток, чем ГАП. Пирофосфат-ионы принимают участие в регуляции многих важных биологических процессов и существенно влияют на образование и рост костного минерала [2].
Известен способ получения порошка ПФК твердофазным методом из карбоната кальция СаСО3 и гидрофосфата аммония (NH4)2HPO4. Керамику из этого порошка изготавливают спеканием при 1150°С [3]. Порошки, полученные в результате твердофазной реакции обычно обладают низкой активностью к спеканию, а микроструктура такого материала далека от совершенства.
Известны различные способы получения ПФК с использованием высокотемпературной обработки продукта (брушита или монетита), полученного в результате взаимодействия фосфорной кислоты или растворимых ортофосфатов аммония, калия, натрия и хорошо растворимых солей кальция (нитрата, хлорида, ацетата) [4, 5, 6] или трудно растворимых (гидроксида или сульфата кальция) [7]. Частицы ПФК наследуют после термообработки пластинчатую форму частиц брушита или монетита [8]. Частицы пластинчатой формы препятствуют уплотнению материала при формовании и спекании, что приводит к получению пористого материала с крупными зернами и плотностью не выше 85% [9].
Известен способ [9], в котором керамический биодеградируемый материал на основе ПФК получают из порошка монетита, синтезированного в результате взаимодействия водных растворов соли кальция (Са(NO3)2) и гидрофосфата аммония ((NH4)2HPO4). Полученный порошок прессуют, а заготовки затем обжигают. Недостатком данного керамического биодеградируемого материала является размер кристаллов, достигающий 100 мкм, а также наличие внутри и межкристаллической пористости. Формирование крупнокристаллической пористой микроструктуры также связано с пластинчатой формой частиц и необходимостью использовать более высокие температуры обжига. Однако температура обжига не может быть больше 1300°С (Т пл. ПФК) и не должна превышать 1150°С (Т перехода β-ПФК в α-ПФК, сопровождающегося значительными объемными изменениями). Именно поэтому при получении керамики на основе ПФК стремятся снизить температуру обжига, например, введением добавок с низкой Т плавления (СаС12 или Na4P2O7).
Для получения сферических частиц ПФК применяют пиролиз при распылении жидкости [10, 11], представляющей собой растворенный в разбавленной азотной кислоте СаНРO4. После распыления образуются сферические частицы монетита (СаНРO4), которые затем при термообработке преобразуются в ПФК в соответствии с реакцией (1). Реализация метода, использующего распыление, требует применения специального оборудования, а образующиеся при этом частицы - пористые.
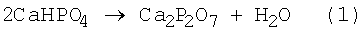
Наиболее близким к предполагаемому изобретению является способ получения порошка гидратированного ПФК, который может быть получен в виде ди- или тетрагидрата [12], который включает взаимодействие растворимой соли кальция (CaCl2, Са(NО3)2 и растворимого пирофосфата (Na4P2O7), остаривание, фильтрование. Термообработка гидратированного ПФК, которая использована в работе [12] как термический анализ, приводит к дегидратации и получению ПФК. Следует отметить, что при получении активных к спеканию оксидных порошков достаточно часто предварительно получают порошок нерастворимого соединения (оксалата, гидроксида или гидратированного соединения), который дезагрегируют, а потом переводят в оксидную форму при термообработке [13]. Таким образом получают порошки Аl2О3 или ZrO2 из Аl(ОН)3 или Zr(OH)4, а также ПФК из брушита или монетита (реакция 1).
Недостатком данного способа [12] является применение длительного (до 7 дней) остаривания, которое неизбежно способствует увеличению размера частиц, которые достигают 20-40 мкм. Выращенные таким образом кристаллы имеют призматическую форму. Поученные кристаллы использованы в публикации [12] для уточнения структуры и модификации гидратированного ПФК.
Целью настоящего изобретения было получение активного к спеканию порошка ПФК, состоящего из частиц изометричной формы.
Поставленная цель была достигнута настоящим изобретением.
В способе получения активного к спеканию порошка ПФК используют взаимодействие водных растворов соли кальция и растворимого ПФК, остаривание осадка в маточном растворе, фильтрование, термообработку, дезагрегацию. Согласно изобретению для получения активного к спеканию порошка ПФК с размером частиц не более 200 нм остаривание проводят в течение 30-60 мин, а термообработку в интервале 300-600°С в течение 2-4 часов.
Растворимые соли кальция (нитрата Са(NO3)2, хлорида СаСl2, ацетата Са(СН3СОО)2) берут в количествах, позволяющих получить водные растворы с концентрацией 0,1-2М. Растворимые пирофосфаты (пирофосфат аммония (NH4)4P2O7, пирофосфат натрия Na4P2O7, пирофосфат калия К4P2O7) берут в количествах, позволяющих получать водные растворы с концентрацией 0,05-1M. При проведении синтеза в соответствии с реакцией (2) используют равные объемы растворов соли кальция и растворимого прирофосфата. При проведении синтеза раствор соли кальция приливают к раствору растворимого пирофосфата.
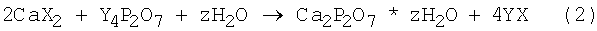
Где X=NO3 1-, Cl1-, СН3СOO1-; Y=NH4 1+, Na1+, K1+, 2≤z≤4
В результате взаимодействия образуется гелеобразный осадок, рентгенограмма которого представлена на чертеже (фигура 1). Использование растворов с концентрациями растворов ниже 0,1М для соли кальция и 0,05М для растворимого пирофосфата аммония или щелочных металлов (Na, К) приводят к невысокому выходу продукта. Использование растворов с концентрациями растворов выше 2М для соли кальция и 1М для растворимого пирофосфата аммония или щелочных металлов (Na, К) приводят к получению осадка, перемешивание которого затруднено. Полученный гелеобразный осадок гидратированного ПФК выдерживают в маточном растворе 30-60 минут. Выдерживание осадка менее 30 минут снижает выход продукта. Осадок остается аморфным при выдерживании в маточном растворе в течение 3 суток, а затем кристаллизуется с образованием смеси 2 - и 4-водного ПФК, а частицы теряют изометричную форму и приобретают призматическую форму, что сопровождается увеличением размера частиц. Однако выдерживание осадка дольше 60 минут организационно и экономически не целесообразно.
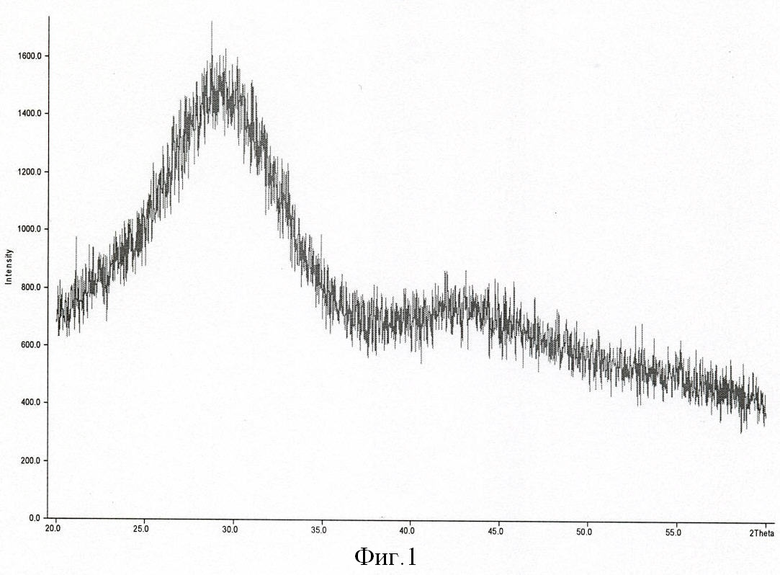
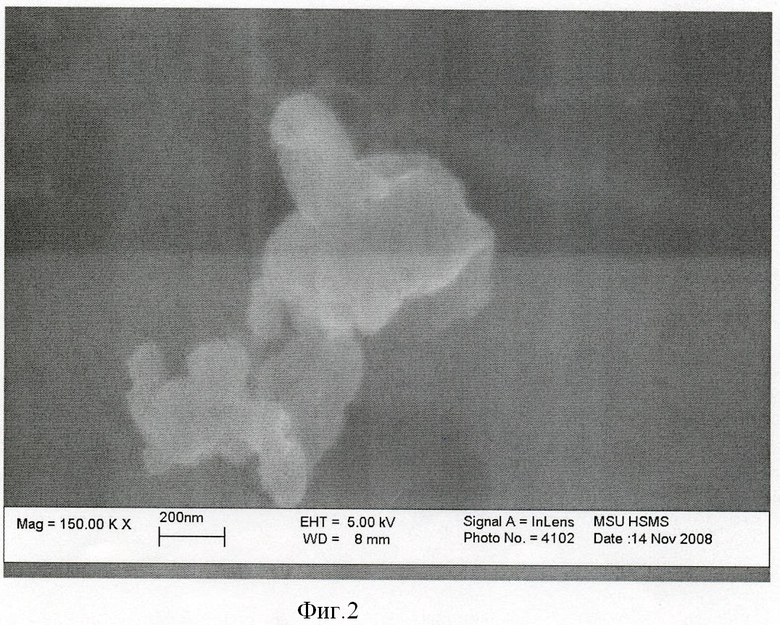
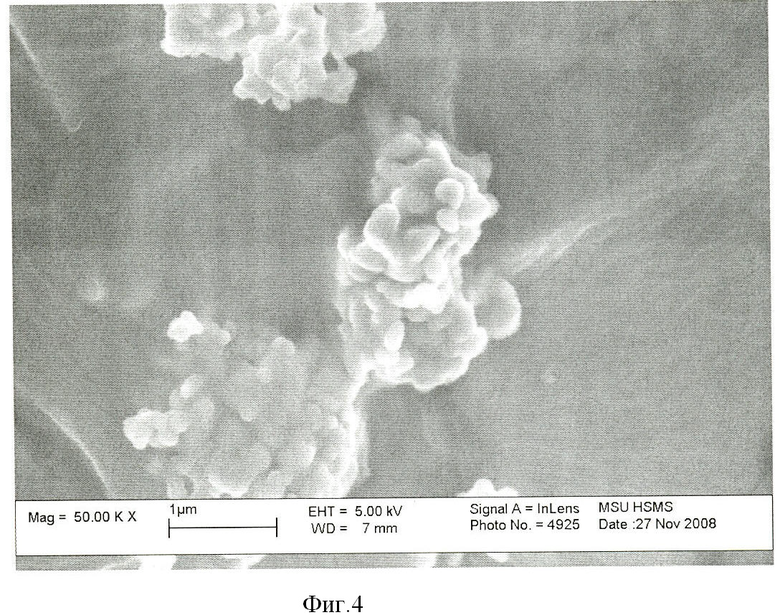
Полученный осадок фильтруют с использованием вакуума, а затем сушат в тонком слое на воздухе при комнатной температуре. Полученный продукт дезагрегируют в ацетоне в шаровой планетарной мельнице. При получении гидратированного ПФК, содержащего в качестве сопутствующего продукта хлориды или ацетаты калия или натрия, высушенный порошок промывают дистиллированной водой для удаления указанных солей. Высушенный порошок, не содержащий сопутствующих продуктов, подвергают дополнительной дезагрегации. Микрофотография частиц после синтеза представлена на чертеже (фигура 2). Размер частиц в агрегате не превышает 200 нм. Дезагрегированный порошок сушат, а затем обрабатывают в течение 2-4 часов при 300-600°С. При высокотемпературной обработке происходит разложение гидратированного ПФК в соответствии с реакцией (3). Обработка порошка ниже температуры 300°С менее 2 часов недостаточна для полной дегидратации аморфного ПФК, тогда как обработка порошка при температуре выше 600°С более 4 часов приводит к огрублению порошка, т.е. образованию более крупных частиц вследствие спекания. Данные РФА порошка после термообработки (фигура 3) и микрофотография частиц порошка (фигура 4) свидетельствуют о том, что заявленный способ позволяет получить частицы ПФК изометричной формы. При термообработке при 300-600°С в течение 2-4 часов размер первичных частиц сохраняется и не превышает 200 нм.
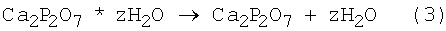
Порошок после термообработки дезагрегигируют в шаровой планетарной мельнице в ацетоне. После сушки из полученного порошка прессуют образцы размером 10×5×3 мм, которые обжигают в при температуре в интервале 800-1000°С. Усадка для всех образцов лежит в интервале 18-22%. Для сравнения, при обжиге в указанном интервале температур образцы из порошков, синтезированных твердофазным методом; из порошков ПФК, полученных в результате термической конверсии брушита или монетита, синтезированных осаждением из растворов; а также из порошков ПФК, синтезированных из растворимой соли кальция и растворимого пирофосфата с остариванием в течение недели, обладали усадкой, не превышающей 1-2%. Столь значительная усадка (18-22%) при относительно низких температурах обжига (до 1000°С) для керамики на основе порошка, полученного в соответствии со способом, предлагаемым настоящим изобретением, свидетельствует о высокой активности данного порошка ПФК к спеканию.
Изобретение иллюстрируется чертежами и примерами:
Фигуры:
Фигура 1. Данные рентгенофазового анализа для порошка ПФК, синтезированного из растворимой соли кальция и растворимого пирофосфата.
Фигура 2. Электронно-микроскопическая фотография частиц порошка после синтеза.
Фигура 3. Данные рентгенофазового анализа для порошка ПФК после термообработки.
Фигура 4. Электронно-микроскопическая фотография частиц порошка после термообработки.
Пример 1.
Синтез гидратированного аморфного ПФК проводят в соответствии с реакцией (4), используя 500 мл водного 1 М раствора нитрата кальция и 500 мл водного 0,5 М раствора пирофосфата аммония по реакции (4).
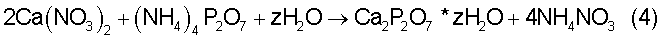
Полученный гелеобразный осадок выдерживают в маточном растворе в течение 45 минут, а затем фильтруют для отделения осадка от маточного раствора. Полученный продукт сушат, а затем дезагрегируют в шаровой планетарной мельнице в ацетоне. После сушки порошок прокаливают при 450°С в течение 3 часов. Дегидратированный порошок дезагрегируют в шаровой планетарной мельнице в ацетоне. Из полученного порошка прессуют образцы размером 10×5×3 мм. Плотность образцов после прессования составляет 45-48%. Отпрессованные образцы обжигают при 900°С в течение 3 часов. Относительная линейная усадка образцов после обжига составляет 18-22%.
Аналогично были синтезированы порошки ПФК из хлорида кальция и растворимого пирофосфата аммония, а также из ацетата кальция и растворимого пирофосфтат аммония.
Пример 2.
Синтез гидратированного аморфного ПФК проводят в соответствии с реакцией (5), используя 500 мл водного 1М раствора хлорида кальция и 500 мл водного 0,5М раствора пирофосфата натрия по реакции (4).
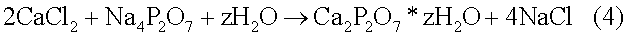
Полученный гелеобразный осадок выдерживают в маточном растворе в течение 45 минут, а затем фильтруют для отделения осадка от маточного раствора. Полученный продукт сушат, а затем дезагрегируют в шаровой планетарной мельнице в ацетоне. Полученный осадок промывают дистиллированной водой для удаления сопутствующего продукта реакции NaCl. Порошок после промывания сушат, а затем дезагрегируют в шаровой планетарной мельнице в ацетоне. После сушки порошок прокаливают при 450°С в течение 3 часов. Дегидратированный порошок дезагрегируют в шаровой планетарной мельнице в ацетоне. Из полученного порошка прессуют образцы размером 10×5×3 мм. Плотность образцов после прессования составляет 45-48%. Отпрессованные образцы обжигают при 900°С в течение 3 часов. Относительная линейная усадка образцов после обжига составляет 18-22%.
Аналогично были синтезированы порошки ПФК из нитрата кальция и растворимого пирофосфата натрия, из нитрата кальция и растворимого пирофосфата калия, из ацетата кальция и растворимого пирофосфтат натрия, из ацетата кальция и растворимого пирофосфтат калия, а также из хлорида кальция и растворимого пирофосфата калия.
Из таблицы следует, что при указанных условиях получения порошков ПФК из растворимых солей кальция и растворимых пирофосфатов усадка после обжига при относительно невысоких температурах обжига 800-1000°С составляет 18-22%, что свидетельствует о высокой активности порошков к спеканию.

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КЕРАМИЧЕСКОГО БИОДЕГРАДИРУЕМОГО МАТЕРИАЛА, СОСТОЯЩЕГО ИЗ ПИРОФОСФАТА КАЛЬЦИЯ И ТРИКАЛЬЦИЙФОСФАТА | 2008 |
|
RU2391316C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА ПИРОФОСФАТА КАЛЬЦИЯ | 2016 |
|
RU2629079C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТОГО КЕРАМИЧЕСКОГО МАТЕРИАЛА НА ОСНОВЕ ПИРОФОСФАТА КАЛЬЦИЯ | 2012 |
|
RU2531377C2 |
| СПОСОБ ПОДГОТОВКИ ШИХТЫ ДЛЯ ПОЛУЧЕНИЯ КЕРАМИЧЕСКОГО БИОДЕГРАДИРУЕМОГО МАТЕРИАЛА | 2010 |
|
RU2456253C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТОГО ПИРОФОСФАТА КАЛЬЦИЯ | 2012 |
|
RU2537615C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКА БРУШИТА | 2009 |
|
RU2431599C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЕРАМИЧЕСКОГО БИОДЕГРАДИРУЕМОГО МАТЕРИАЛА НА ОСНОВЕ ПИРОФОСФАТА КАЛЬЦИЯ | 2008 |
|
RU2392006C2 |
| Способ получения окрашенного однофазного пирофосфата кальция | 2019 |
|
RU2714188C1 |
| СПОСОБ ПОЛУЧЕНИЯ КЕРАМИЧЕСКОГО КОМПОЗИЦИОННОГО БИОДЕГРАДИРУЕМОГО МАТЕРИАЛА НА ОСНОВЕ ДВОЙНОГО ФОСФАТА КАЛИЯ КАЛЬЦИЯ | 2008 |
|
RU2395303C1 |
| СПОСОБ ПОДГОТОВКИ ШИХТЫ ДЛЯ ПОЛУЧЕНИЯ БИОКЕРАМИКИ | 2009 |
|
RU2431627C2 |
Изобретение относится к области химии. Активный к спеканию порошок пирофосфата кальция получают путем взаимодействия водных растворов соли кальция и растворимого пирофосфата, остаривания полученного осадка в маточном растворе в течение 30-60 мин, фильтрования, термообработки в интервале 300-600°С в течение 2-4 часов и дезагрегации. Изобретение позволяет получать порошок пирофосфата кальция, состоящий из частиц изометричной формы размером 100-200 нм, активный к спеканию. 4 ил., 1 табл.
Способ получения активного к спеканию порошка пирофосфата кальция, включающий взаимодействие водных растворов соли кальция и растворимого пирофосфата, остаривание осадка в маточном растворе, фильтрование, термообработку, дезагрегацию, отличающийся тем, что остаривание порошка проводят в течение 30-60 мин, а термообработку - в интервале 300-600°С в течение 2-4 ч.
| MARGARET R | |||
| CHRISTOFFERSEN et al, Growth and precipitation of a monoclinic calcium pyrophosphate tetrahydrate indicating auto-inhibition at pH 7, Journal of Crystal Growth, 2000, Volume 212, Issues 3-4, Pages 500-506 | |||
| Способ получения двузамещенных пирофосфатов щелочных металлов | 1982 |
|
SU1057416A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРОФОСФАТА НАТРИЯ | 2000 |
|
RU2170210C1 |
| US 5676917 A, 14.10.1997 | |||
| US 4721615 A, 26.01.1988 | |||
| GB 1304218 A, 24.01.1973 | |||
| GB 1304217 A, 24.01.1973 | |||
| US 3635660 A, 18.01.1972. | |||

