Область техники, к которой относится изобретение
Настоящее изобретение относится к новой комбинированной терапии, включающей вориконазол.
Уровень техники
(2R,3S)-2-(2,4-дифторфенил)-3-(5-фтор-4-пиримидинил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол, известный также как вориконазол, описан в ЕР-А-440372; смотри, в частности, пример 7. Вориконазол имеет следующую структуру:
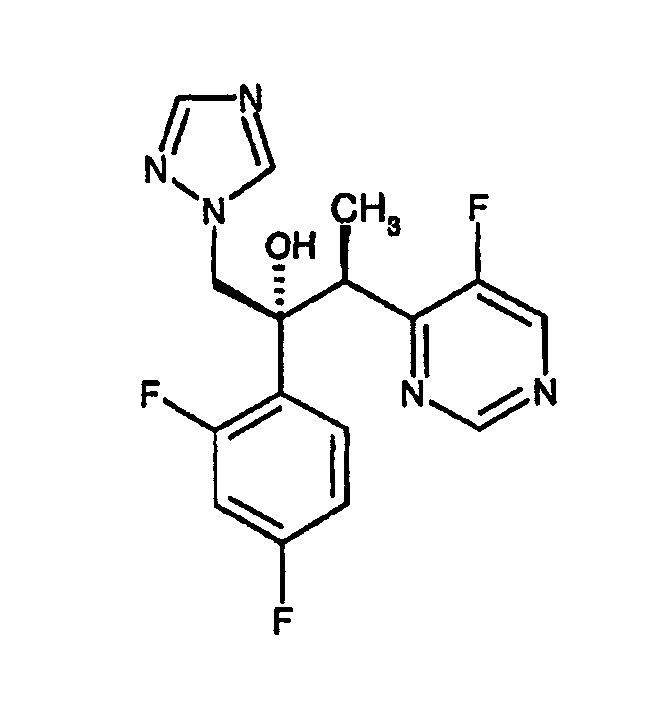
и является пригодным для лечения грибковых инфекций.
Фармакокинетика вориконазола характеризуется насыщающим метаболизмом, что приводит к нелинейным увеличениям воздействия при возрастающих уровнях доз. Кроме того, воздействие лекарственного средства на различных пациентов значительно варьирует. Вориконазол метаболизируется изоферментами CYP2C19, CYP2C9 и CYP3A4 системы цитохрома Р450. Главный циркулирующий в крови метаболит, структура которого приводится ниже, получается в результате N-окисления.
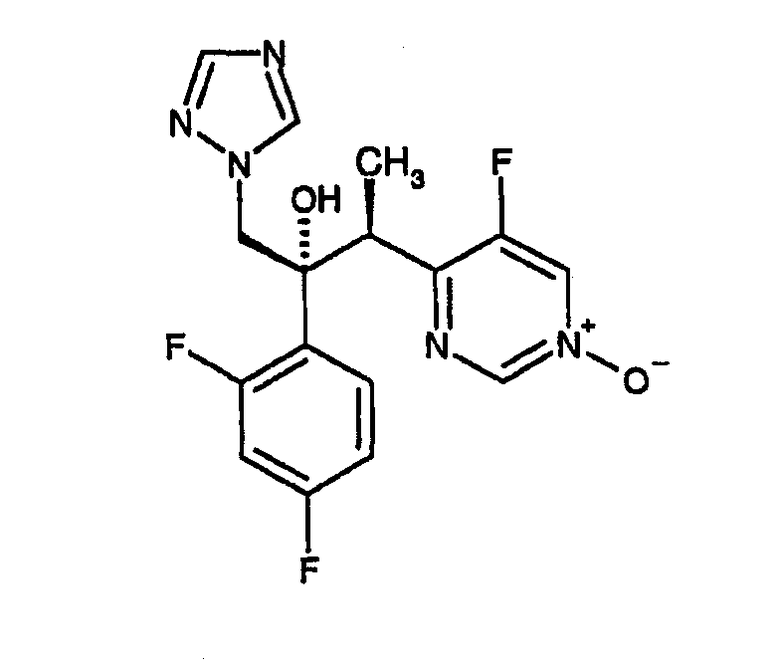
Заявители установили, что метаболизм вориконазола зависит в большой степени от генотипа субъектов, подвергаемых лечению. Один генотип метаболизирует вориконазол в значительной степени, что приводит к быстрому клиренсу вориконазола из организма и, как следствие, к низким уровням вориконазола в плазме крови (в пределах приблизительно от 0,6 до 1,4 мкг/мл). В данном описании указанный генотип будет называться «экстенсивными метаболизаторами». Второй генотип можно охарактеризовать как слабый метаболизатор вориконазола: при данном генотипе клиренс вориконазола происходит гораздо медленнее и, следовательно, его уровни в организме остаются более высокими (в пределах приблизительно от 3,5 до 5,5 мкг/мл). В данном описании указанный генотип будет называться «слабыми метаболизаторами». Генотипирование, как полагают, происходит по рецессивному гену: гомозиготные экстенсивные метаболизаторы составляют приблизительно до 73% популяции белой расы и 35% популяции японцев; гетерозиготные экстенсивные метаболизаторы составляют приблизительно до 25% популяции белой расы и 46% популяции японцев. Напротив, слабые метаболизаторы составляют лишь приблизительно до 2% популяции белой расы и приблизительно 19% популяции японцев.
В настоящее время понятно, что вариабельность генотипов с точки зрения метаболизма вориконазола зависит от того, в какой степени в организме присутствует фермент 2С19 системы цитохрома Р450 (далее в настоящем документе называется CYP2C19): фермент CYP2C19 присутствует у экстенсивных метаболизаторов, в то время как у слабых метаболизаторов имеет место недостаток функционального фермента. Смотри, например, M. de Morais, G. Wilkinson, J. Blaisdell et al. J. Biol. Chem. (1994), 269, 15419-15422 (целиком включенный в настоящий документ в качестве ссылки).
На практике вориконазол вводят как экстенсивным, так и слабым метаболизаторам, без подбора дозы. Однако, необходимость для вориконазола присутствовать в плазме крови в количествах, достаточных для получения терапевтического эффекта у экстенсивных метаболизаторов, требует более высокой дозы лекарственного средства: обычная рекомендованная суточная доза составляет 400 мг (200 мг два раза в день). Указанная доза у слабых метаболизаторов приводит к более выраженному системному воздействию лекарственного средства, которое может приводить к появлению нежелательных побочных эффектов. Кроме того, быстрый клиренс лекарственного средства у экстенсивных метаболизаторов требует вводить соединение два раза в день, чтобы поддерживать его уровни в плазме крови в течение дня и оказывать терапевтическое действие. Необходимость принимать лекарственное средство дважды в день порождает проблемы с выполнением пациентом схемы и режима лечения, если вориконазол он вводит себе самостоятельно.
M. Ghannoum, N. Isham, M. Hossain and D. Sheehan, Int. J. Infect. Dis. (2002), Vol. 6 Supp. 2, 2S50 описали комбинацию in vitro вориконазола и противогрибковых агентов, включая амфотерицин В, AbelcetTM, 5-фторцитозин и флуконазол, и изучали их механистический синергизм против ряда организмов. Сообщается, что комбинации вориконазола с флуконазолом в 39% случаев является аддитивными и в 61% случаев - индифферентными.
M. Ghannoum, N. Isham and D. Sheehan, Abstracts of the Interscience Conference on Antimicrobial Agents and Chemotherapy (2002) 42, 385, также описывают комбинацию in vitro вориконазола с амфотерицином В, AbelcetTM, флуконазолом, микафунгином, равуконазолом и каспофунгином. Сообщается, что комбинации вориконазола с флуконазолом в 100% случаев являются индифферентными, т.е., никакого синергизма не наблюдается.
H.J. Scherpbier, M.I. Hilhorst and T.W. Kuijpers, Clin. Infect. Dis. (2003) 37, 828, описывают лечение пациента со СПИДом с использованием комбинации антиретровирусных лекарственных средств и сообщают о взаимодействии между ингибиторами протеиназ и вориконазолом, когда последний был добавлен к терапии пациента для лечения кандидоза пищевода. Взаимодействие затрагивало нарушение функции печени и повышение концентраций в плазме лопинавира, невирапина и ампренавира. Концентрации в плазме вориконазола у пациентов не измерялись.
N. Wood, K. Tan, L. Purkins, G. Layton, J. Hamlin, D. Kleinermans and D. Nichols, Br. J. Clin. Pharmacol. (2003) 56, 56 описывают исследование с целью определения влияния ингибитора протонного насоса. Омепразола, ингибитора CYP2C19, на устойчивое состояние фармакокинетики вориконазола. Исследование установило, что омепразол не оказывал клинически значимого действия на воздействие вориконазола, что предполагало отсутствие необходимости коррекции дозы вориконазола для пациентов, которым начали лечение омепразолом. Было бы желательным разработать такое лечение вориконазолом, которое бы устраняло и уменьшало вариабельность его действия на различных субъектов. Помимо этого, было бы желательным разработать такое лечение вориконазолом, при котором можно было бы уменьшить вводимую дозу вориконазола. Кроме того, было бы желательным разработать такое лечение вориконазолом, при котором клиренс лекарственного средства из плазмы был бы уменьшен, что позволило бы вводить вориконазол один раз в день.
Заявители неожиданно установили, что ингибирование фермента CYP2C19 путем совместного введения вориконазола со вторым, другим противогрибковым лекарственным средством, способным ингибировать активность CYP2C19, заметно уменьшает метаболизм у экстенсивных метаболизаторов вориконазола, что делает фармакокинетический профиль у подобных лиц приблизительно таким же, как у слабых метаболизаторов. Указанные результаты заметно уменьшают разброс между субъектами и между терапевтическими уровнями в плазме, которые достигались значительно менее высокими дозами вориконазола. Кроме того, результатом явился также уменьшенный клиренс вориконазола из организма, в результате чего вориконазол присутствовал в течение дня в плазме в концентрациях, достаточных для достижения терапевтического эффекта при введении один раз в день.
Сущность изобретения
Изобретение в первом аспекте относится к терапевтической комбинации, включающей вориконазол и противогрибковый ингибитор CYP2C19 в конкретных количествах или массовых соотношениях, определенных более подробно далее в настоящем документе.
Изобретение во втором аспекте относится также к фармацевтической композиции, включающей терапевтически эффективное количество вориконазола и противогрибкового ингибитора CYP2C19, вместе с фармацевтически приемлемым носителем или разбавителем.
Изобретение в третьем аспекте относится также к дозированной лекарственной форме, включающей терапевтически эффективное количество вориконазола и терапевтически эффективное количество противогрибкового ингибитора CYP2C19.
Изобретение в четвертом аспекте относится также к набору, включающему множество отдельных контейнеров, в котором по меньшей мере один контейнер содержит вориконазол и по меньшей мере один другой контейнер содержит противогрибковый ингибитор CYP2C19.
Изобретение в пятом аспекте относится дополнительно к применению указанных выше комбинации, композиции, набора или дозированной лекарственной формы для изготовления лекарственного средства для лечения грибковой инфекции у млекопитающего.
Изобретение в шестом аспекте относится также к способу лечения грибковой инфекции у млекопитающего, включающему введение млекопитающему, которое нуждается в указанном лечении, эффективного количества указанных выше комбинации, композиции, набора или дозированной лекарственной формы.
Далее в настоящем документе, терапевтическая комбинация, фармацевтическая композиция, дозированная лекарственная форма, набор, применение и способ по настоящему изобретению будут обобщенно называться «комбинацией по настоящему изобретению».
Краткое описание рисунков
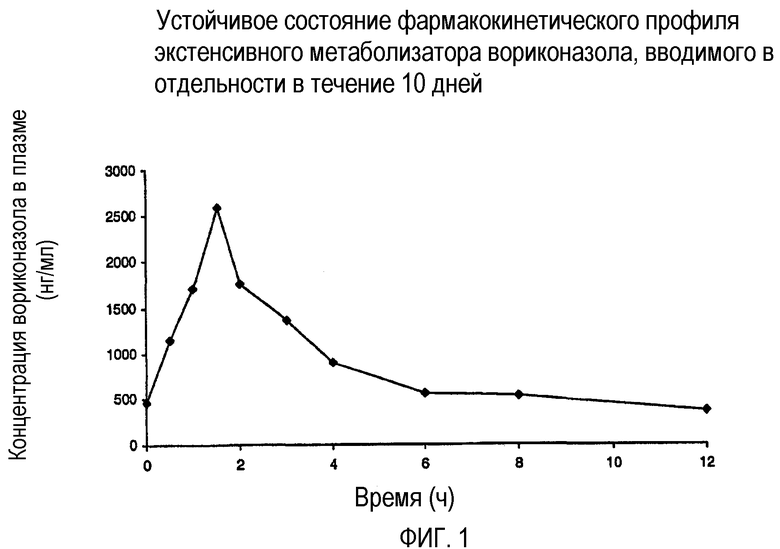
Фиг. 1 иллюстрирует фармакокинетический профиль экстенсивного метаболизатора вориконазола при введении в отдельности.
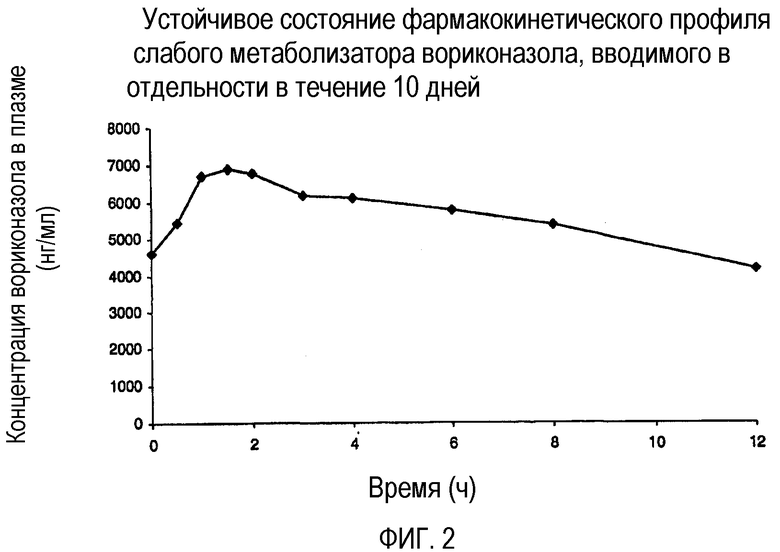
Фиг. 2 иллюстрирует фармакокинетический профиль слабого метаболизатора вориконазола при введении в отдельности.
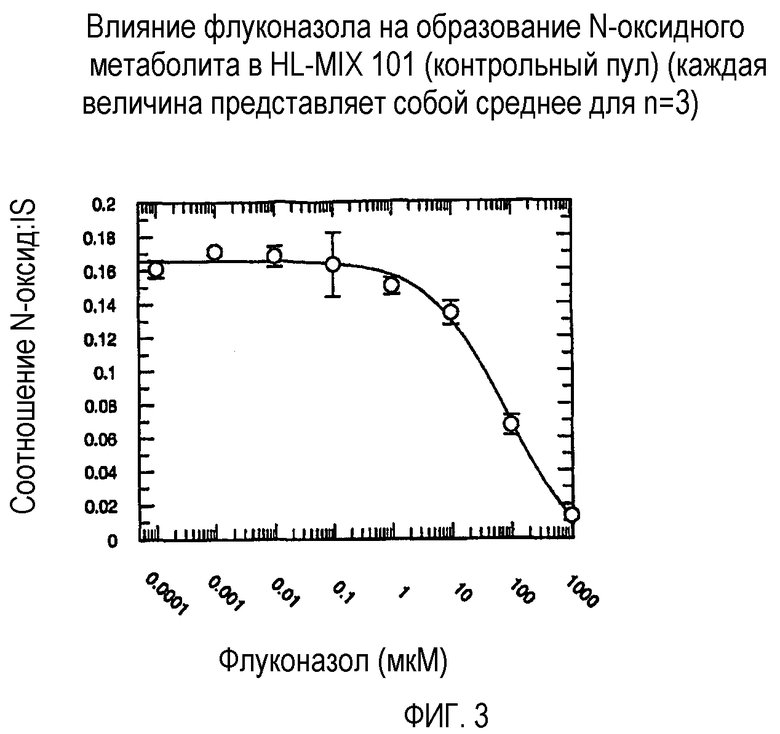
Фиг. 3 иллюстрирует влияние флуконазола на образование N-оксидного метаболита in vitro в человеческих микросомах печени HL-MIX 101 (контрольный пул).
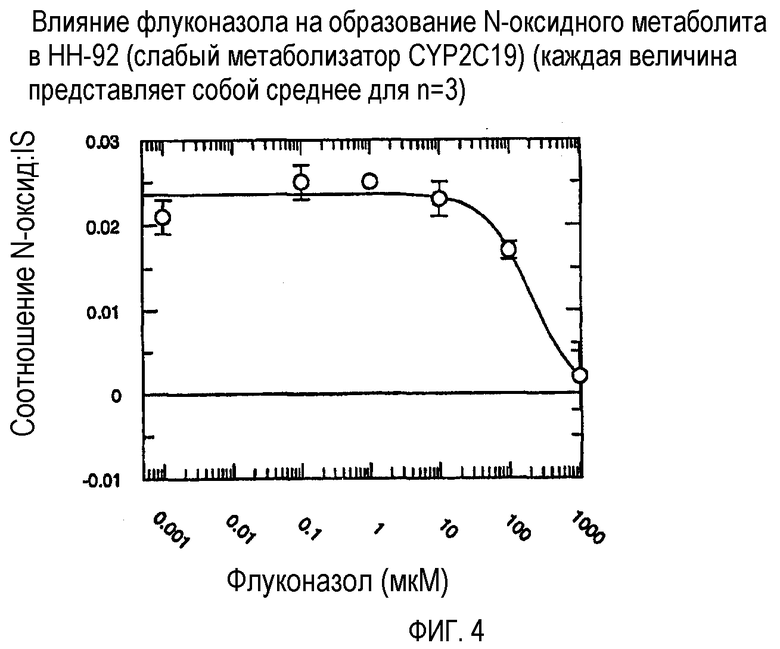
Фиг. 4 иллюстрирует влияние флуконазола на образование N-оксидного метаболита in vitro в человеческих микросомах печени HH-92 (слабый метаболизатор CYP2C19).
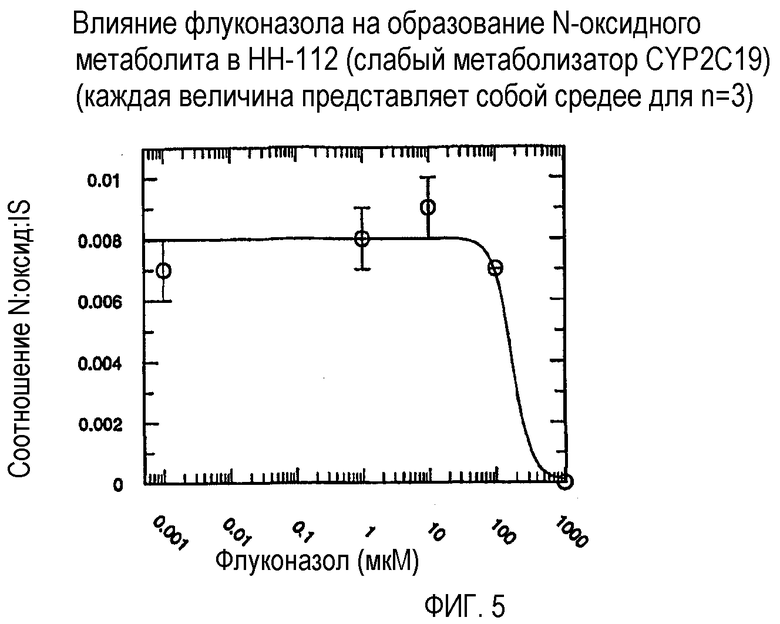
Фиг. 5 иллюстрирует влияние флуконазола на образование N-оксидного метаболита in vitro в человеческих микросомах печени HH-122 (слабый метаболизатор CYP2C19).
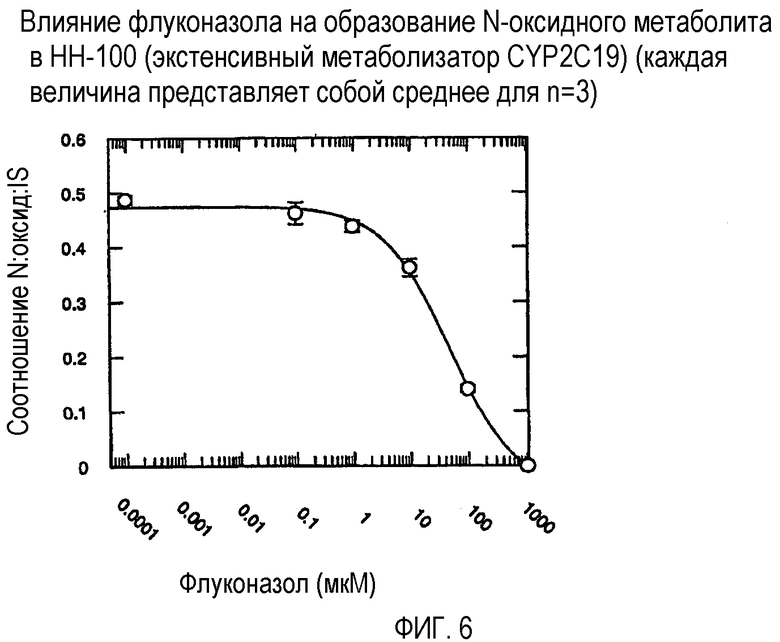
Фиг. 6 иллюстрирует влияние флуконазола на образование N-оксидного метаболита in vitro в человеческих микросомах печени HH-100 (экстенсивный метаболизатор CYP2C19).
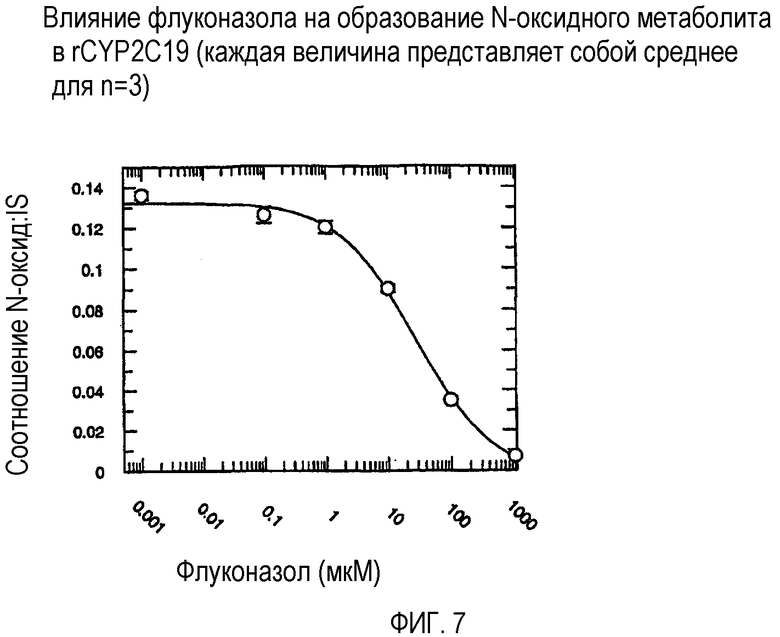
Фиг. 7 иллюстрирует влияние флуконазола на образование N-оксидного метаболита в rCYP2C19.
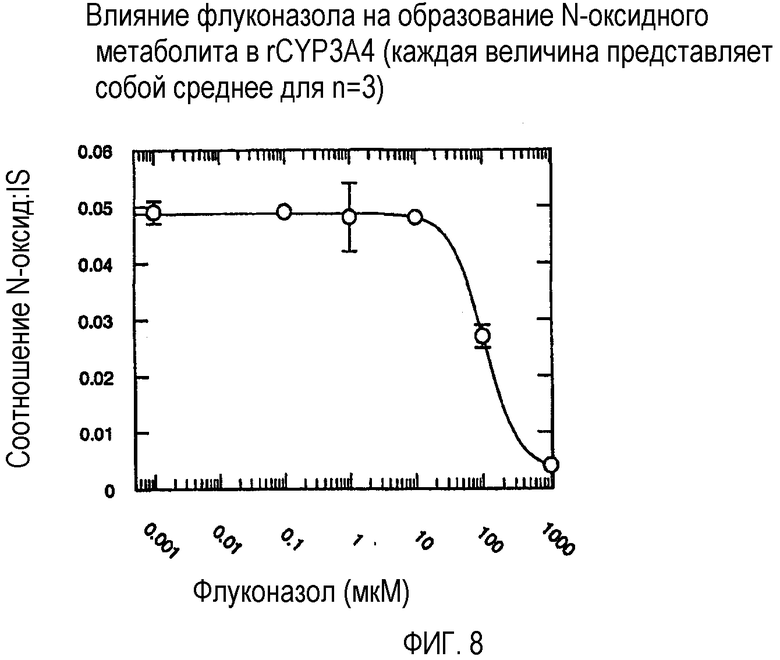
Фиг. 8 иллюстрирует влияние флуконазола на образование N-оксидного метаболита в rCYP3А4.
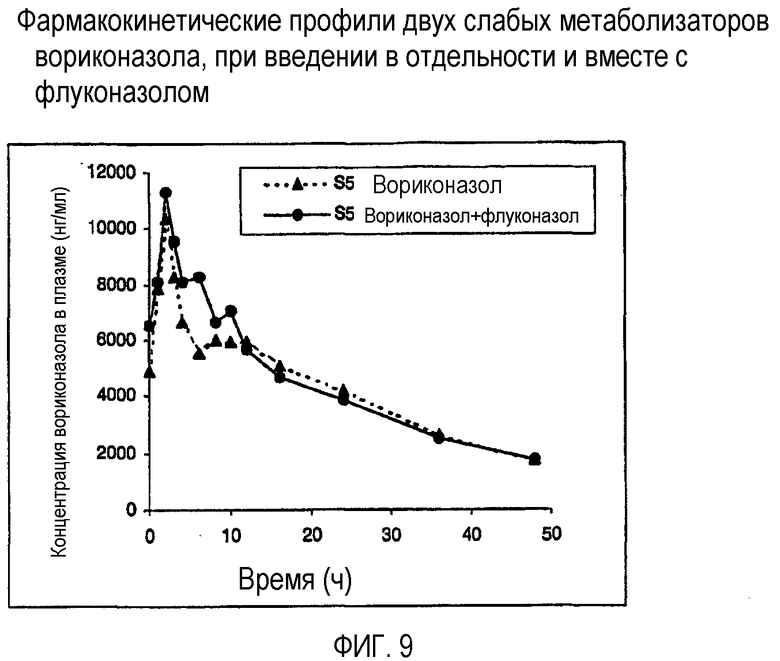
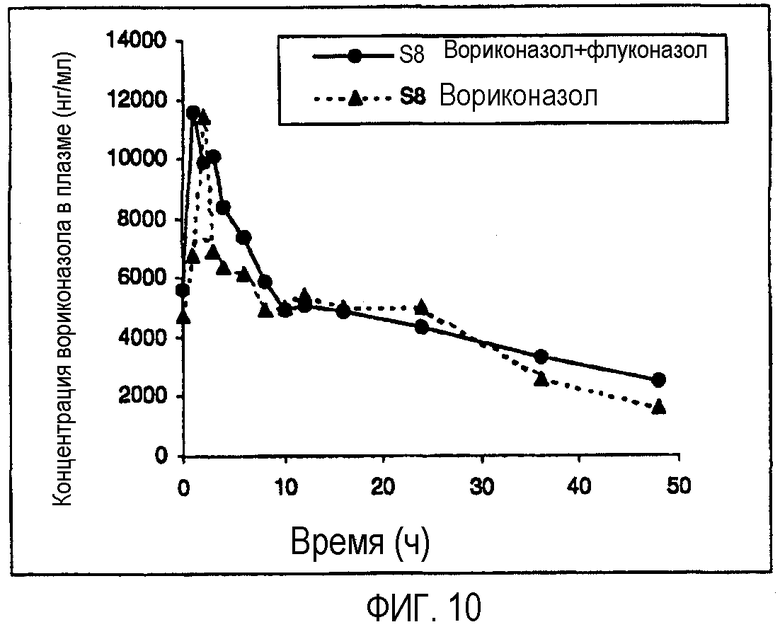
Фиг. 9 и 10 представляют собой фармакокинетический профиль двух слабых метаболизаторов вориконазола при введении по отдельности и вместе с флуконазолом.
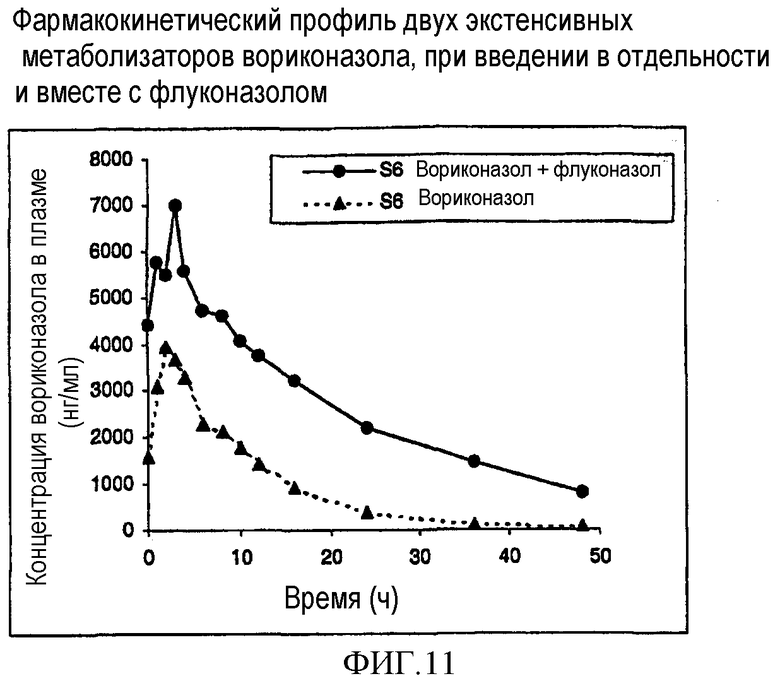
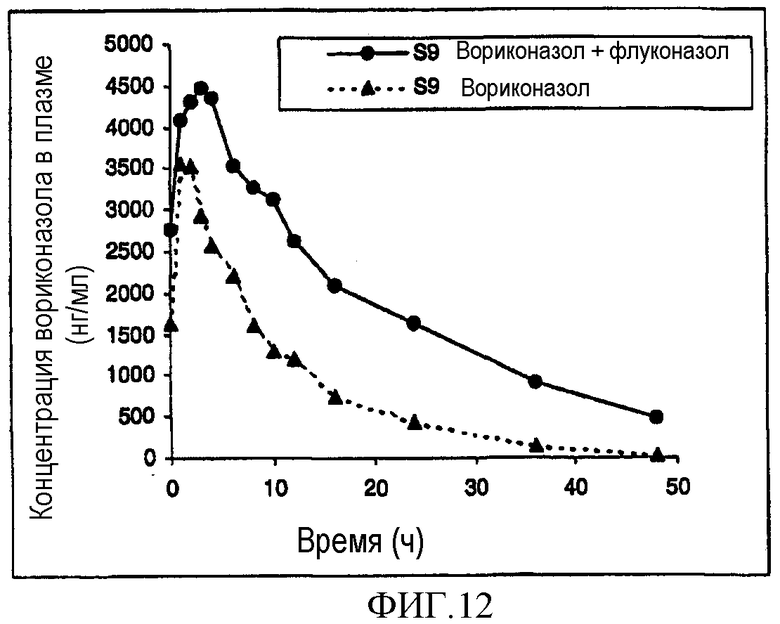
Фиг. 11 и 12 представляют собой фармакокинетический профиль двух экстенсивных метаболизаторов вориконазола при введении по отдельности и вместе с флуконазолом.
Подробное описание предпочтительных вариантов осуществления настоящего изобретения
Одной частью комбинации по настоящему изобретению является вориконазол. Вориконазол описан в ЕР-А-440372 (целиком включенном в настоящий документ в качестве ссылки); смотри, в частности, пример 7. Как описано более подробно ниже, вориконазол можно вводить в форме свободного основания или в форме его соли, сольвата или пролекарства.
Другой частью комбинации по настоящему изобретению является противогрибковый ингибитор CYP2C19. Точная природа противогрибкового агента специальным образом не ограничивается, при условии, что он обладает противогрибковой активностью и способен действовать в качестве ингибитора фермента CYP2C19 млекопитающего. В настоящем описании «ингибитор CYP2C19» подразумевает любое соединение, способное ингибировать действие фермента CYP2C19 млекопитающего, как описано в обзоре Desta et al. (2992) Clinical Pharmacokinetics 41(12), 913-958 (целиком включенном в настоящий документ в качестве ссылки). Величина KI менее 10 мкм предпочтительно требуется для гарантированного ингибирования метаболизма вориконазола при использовании обычных терапевтических доз. Стандартный in vitro тест для определения KI использует (S)-мефенитоин в качестве зондового субстрата и измерения 4'-гидроксилирования - смотри Meier et al. (1985) Anal. Biochem. 151, 286-291 (целиком включено в настоящий документ в качестве ссылки).
Примеры противогрибковых ингибиторов CYP2C19 включают флуконазол, который имеет KI 2 мкМ - см. L.C. Winkers, C.J. Wurden, E. Storch et al., Drug Metab. Dispos. (1996) 24, 610-4. Другие примеры ингибиторов CYP2C19 описаны в Desta et al. (2002) (ссылка приводилась выше).
Предпочтительно, чтобы противогрибковый ингибитор CYP2C19 был способен селективно ингибировать метаболизм вориконазола ферментом CYP2C19 по сравнению с ферментом CYP3A4. Селективность в отношении изоферментов можно измерить посредством относительной ингибирующей активностью против (S)-мефенитоин-4-гидроксилазы и тестостерон-6-бета-гидроксилазы: измерение активности последней описано у Funa and Imaoka (1987) Biochem. Biophys. Acta. 926, 349-358 (целиком включено в настоящий документ в качестве ссылки). Более конкретно, селективность можно продемонстрировать влиянием на N-окисление вориконазола с использованием концентраций субстрата 25 и 2500 мкМ, для оценки влияния на CYP2C19 (высокоаффинный фермент) и 3А4 (низкоаффинный фермент), соответственно. Методология измерения N-окисления вориконазола изоферментами системы цитохрома Р450 описана у Hyland, Jones and Smith (2003) Drug Metabolism and Disposition, 31(5), 540-547 (целиком включено в настоящий документ в качестве ссылки). Точный уровень требующейся селективности зависит от противогрибкового ингибитора CYP2C19 и его фармакокинетики и вариабельности между субъектами: однако, заявители предпочитают, чтобы противогрибковый ингибитор CYP2C19 проявлял селективность в отношении CYP2C19 по сравнению с CYP3A4 (по измерению их относительных величин IC50 в указанной выше статье Hyland et al.) в 2-10 и, предпочтительно, в 3-6 раз больше. Не желая ограничиваться теорией, полагают, что селективное ингибирование CYP2C19, превосходящее CYP3А4, позволяет уменьшить вариабельность между субъектами и достигать терапевтических уровней в плазме при более низких дозах вориконазола, в то же время уменьшая возможные нежелательные побочные эффекты в популяции пациентов при современной рекомендованной схеме лечения. При низких концентрациях CYP2C19 имеется преимущественный путь клиренса, в то время как при более высоких концентрациях CYP3A4 путь метаболизма становится основным, ненасыщаемым, путем клиренса вориконазола.
Предпочтительно, противогрибковый ингибитор CYP2C19 имеет фармакокинетический полупериод существования, подходящий для того, чтобы позволить комбинации вориконазол/противогрибковый ингибитор CYP2C19 поддерживать уровни в плазме в течение дня и достигать терапевтического эффекта при введении один раз в день. Подходящие полупериоды существования варьируют от 6 до 72 часов, предпочтительно, от 12 до 48 часов, и, более предпочтительно, от 18 до 36 часов.
Предпочтительно, чтобы противогрибковый ингибитор CYP2C19 экскретировался из организма преимущественно в виде неизмененного лекарственного средства. Не желая ограничиваться теорией, полагают, что противогрибковый ингибитор CYP2C19, который экскретируется из организма преимущественно в виде неизмененного лекарственного средства, позволяет уменьшить вариабельность между субъектами как для противогрибкового ингибитора CYP2C19, так и для вориконазола. Предпочтительно, от 50 до 99%, более предпочтительно, от 70 до 80% и, наиболее предпочтительно, приблизительно 75% лекарственного средства экскретируется из организма неизмененным.
Противогрибковый ингибитор CYP2C19 обладает противогрибковой активностью. Примеры ингибиторов CYP2C19, которые также являются противогрибковыми агентами, включают азолы, такие как флуконазол. В результате появляется преимущество, которое выражается в том, что комбинация не оказывает никакого другого действия, кроме противогрибкового. Второй противогрибковый агент предпочтительно должен добавлять эффект к противогрибковой активности и может быть, предпочтительно, разрешен для безопасного применения в той же самой популяции пациентов. Суммарный противогрибковый эффект зависит, разумеется, от конкретной инфекции, по поводу которой осуществляют лечение, введенной дозы вориконазола и противогрибкового ингибитора CYP2C19, и возраста, пола, массы тела и состояния пациента, который подвергается лечению.
Особенно предпочтительным противогрибковым ингибитором CYP2C19 является флуконазол.
Комбинация по настоящему изобретению включает вориконазол и противогрибковый ингибитор CYP2C19. Вориконазол и противогрибковый ингибитор CYP2C19 можно вводить в форме свободной кислоты или основания, или в форме их фармацевтически приемлемой соли, сольвата или пролекарства.
Фармацевтически приемлемые соли вориконазола и противогрибкового ингибитора CYP2C19 включают их кислотно-аддитивные и основные соли.
Подходящие кислотно-аддитивные соли образуются из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, аспартат, бензоат, бесилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, эдисилат, эсилат, формиат, фумарат, глуцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидроиодид/иодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат, фосфат/дигидрофосфат, сахарат, стеарата, сукцинат, тартрат, тозилат и трифторацетат.
Подходящие основные соли образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка.
Обзор подходящих солей смотри в «Handbook of Pharmaceutical Salts: Properties, Selection, and Use», Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Фармацевтически приемлемую соль вориконазола или противогрибкового ингибитора CYP2C19 можно легко получить смешиванием растворов вориконазола или противогрибкового ингибитора CYP2C19 и желаемой кислоты или основания должным образом. Соль может выпадать в осадок из раствора, и ее можно собирать фильтрованием или получать выпариванием растворителя. Степень ионизации соли может варьировать от полностью ионизированной до почти неионизированной.
Вориконазол и противогрибковый ингибитор CYP2C19 могут существовать как в несольватированной, так и в сольватированной формах. Термин «сольват» используется в настоящем документе для описания молекулярного комплекса, включающего в себя вориконазол и/или противогрибковый ингибитор CYP2C19 и одну или более молекул фармацевтически приемлемого растворителя, например, этанола. Термин «гидрат» используется, когда указанным растворителем является вода.
Далее в настоящем документе все ссылки на вориконазол и/или противогрибковый ингибитор CYP2C19 включают в себя ссылки на их соли, сольваты и комплексы и на сольваты и комплексы их солей.
Комбинация по настоящему изобретению включает вориконазол и противогрибковый ингибитор CYP2C19, как определено ранее в настоящем документе, их полиморфы и пролекарства.
Как было заявлено, изобретение включает все полиморфы вориконазола и/или противогрибкового ингибитора CYP2C19, как определено ранее в настоящем документе.
Также в объем настоящего изобретения входят так называемые «пролекарства» вориконазола и/или противогрибкового ингибитора CYP2C19. Так, некоторые производные вориконазола и/или противогрибкового ингибитора CYP2C19, которые сами по себе могут иметь низкую фармакологическую активность или не иметь ее вовсе, при введении внутрь организма или при нанесении снаружи на организм, могут превращаться в соединения, обладающие желательной активностью, например, посредством гидролитического расщепления. Указанные производные называются «пролекарствами». Дополнительную информацию по использованию пролекарств можно найти в «Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Sympisium Series (T. Higuchi and W. Stella) and «Bioreversible Carriers in Drug Desighn», Pergamon Press, 1987 (ed. E.B. Roche, American Pharmaceutical Association).
Пролекарства в соответствии с настоящим изобретением могут быть, например, произведены заменой соответствующих функциональных групп, имеющихся в вориконазоле или противогрибковом ингибиторе CYP2C19, определенными частями, известными специалистам как «про-части», как описано, например, в «Design of Prodrugs», H. Bundgaard (Elsevier, 1985).
Вориконазол содержит спиртовую функциональную группу (-ОН), следовательно, некоторые примеры пролекарств в соответствии с изобретением могут включать его сложный или простой эфир, например, путем замены водорода фосфорилированием, с получением (2R,3S)-2-(2,4-дифторфенил)-3-(5-фтор-4-пиримидинил)-1-(1Н-1,2,4-триазол-1-ил)-2-бутил дигидрофосфата, как описано в WO 97/28169 (целиком включено в настоящий документ в качестве ссылки); см., в частности, пример 3.
Подобно этому, когда противогрибковым ингибитором CYP2C19 является флуконазол, данное соединение также содержит спиртовую функциональную группу (-ОН), следовательно, некоторые примеры пролекарств в соответствии с изобретением могут включать его сложный или простой эфир, например, путем замены водорода фосфорилированием, с получением 2-(2,4-дифторфенил)-1,3-бис(1Н-1,2,4-триазол-1-ил)-2-пропил дигидрофосфата, как описано в WO 97/28169; (целиком включено в настоящий документ в качестве ссылки); смотри, в частности, пример 1, или его фармацевтически приемлемой соли, особенно, динатриевой соли (Prodif®). Другие примеры замещающих групп в соответствии с указанными примерами, и примеры других типов пролекарств можно найти в упомянутых выше ссылках.
Противогрибковый ингибитор CYP2C19 может содержать один или более асимметричных атомов углерода и может, следовательно, существовать в виде двух или более стереоизомеров. В случае, когда противогрибковый ингибитор CYP2C19 содержит алкенильную или алкениленовую группу, возможными являются геометрические цис/транс (или Z/E) изомеры. В случае, когда соединение содержит, например, группу кето или оксим или ароматическую часть, может наблюдаться таутомерная изомерия ("таутомеризация"). Из этого следует, что одно соединение может демонстрировать более одного типа изомерии.
В объем настоящего изобретения включены все стереоизомеры, геометрические изомеры и таутомерные формы противогрибкового ингибитора CYP2C19, включая соединения, демонстрирующие более одного типа изомерии, и смеси одного или более из них. Также включаются кислотно-аддитивные или основные соли, в которых противоион является оптически активным, например D-лактат или L-лизин, или рацемические, например DL-тартрат или DL-аргинин.
Цис/транс изомеры могут быть разделены с использованием обычных методик, хорошо известных специалистам, например с использованием хроматографии или фракционной кристаллизации.
Обычные методики для получения/изоляции отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника, или разделение рацемата (или рацемата соли или производного), с использованием, например, хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ).
Альтернативно, рацемат (или рацемический предшественник) может взаимодействовать с подходящим оптически активным соединением, например спиртом, или, в случае, когда противогрибковый ингибитор CYP2C19 содержит кислую или основную часть, с кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученную диастереоизомерную смесь можно разделить хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера конвертировать в соответствующий чистый энантиомер (энантиомеры) способами, хорошо известными специалистам.
Хиральные соединения, используемые в комбинации по настоящему изобретению (и их хиральные предшественники), могут быть получены в энантимерно обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметричной смоле с мобильной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащей от 0 до 50% изопропанола, обычно, от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата позволяет получить обогащенную смесь.
Стереоизомерные конгломераты можно разделить обычными методиками, известными специалистами; смотри, например, «Stereochemistry of Organic Compounds», E.L. Ellel (Wiley, New York, 1994).
Комбинация по настоящему изобретению включает количество противогрибкового ингибитора CYP2C19, эффективное в качестве противогрибкового агента и эффективного для ингибирования действия фермента CYP2C19, предпочтительно, для селективного ингибирования указанного фермента по сравнению с ферментом CYP3A4. Точная вводимая доза зависит от различных факторов, таких как возраст, пол, масса тела и состояния пациента, которому осуществляют лечение. Однако, заявители предпочитают, чтобы вводимая доза составляла приблизительно от 5 до 500 мг, более предпочтительно, приблизительно от 10 до 250 мг, еще более предпочтительно, приблизительно от 50 мг до 200 мг, и, наиболее предпочтительно, приблизительно от 75 до 125 мг. Предпочтительно, противогрибковый ингибитор CYP2C19 представляет собой флуконазол.
Комбинация по настоящему изобретению включает количество вориконазола, эффективное в качестве противогрибкового агента. Точная вводимая доза зависит от различных факторов, таких как возраст, пол, масса тела и состояния пациента, которому осуществляют лечение. Однако заявители предпочитают, чтобы вводимая доза составляла приблизительно от 5 до 600 мг, предпочтительно, приблизительно от 10 до 500 мг, более предпочтительно, приблизительно от 20 мг до 300 мг, и, наиболее предпочтительно, приблизительно от 25 до 250 мг. Доза около 200 мг является особенно предпочтительной: это сообщает значительное преимущество настоящему изобретению в том, что дозу вориконазола можно уменьшить наполовину, от ее обычного уровня 400 мг, сводя к минимуму, таким образом, нежелательные побочные эффекты.
В предпочтительных вариантах осуществления настоящего изобретения комбинацию по настоящему изобретению следует вводить пациенту один раз в день. Это имеет особенные преимущества с точки зрения соблюдения пациентом режима и схемы лечения. Однако введение два, три или четыре раза в день также находится в пределах объема настоящего изобретения, хотя и в этом случае доза на одно введение будет дополнительно уменьшаться.
Массовое соотношение, в соответствии с которым вводятся компоненты комбинации по настоящему изобретению, варьирует в зависимости от различных факторов, таких как возраст, пол, масса тела и состояние, по поводу которого пациент проходит лечение. Однако заявители предпочитают, чтобы вориконазол и противогрибковый ингибитор CYP2C19 вводились в массовом соотношении в пределах приблизительно от 1:4 до 6:1, предпочтительно, приблизительно от 1:2 до 3:1, и, более предпочтительно, приблизительно от 3:2 до 5:2. Предпочтительно, противогрибковым ингибитором CYP2C19 является флуконазол.
Грибковые инфекции, которые можно лечить комбинацией по настоящему изобретению, широко описаны в литературе, включая ЕР-А-440372, и включают местные инфекции, инфекции слизистых оболочек, такие как вагинальный кандидоз, пищеводный и орофарингеальный кандидоз, и системные инфекции. Кроме того, комбинацию по настоящему изобретению можно использовать для лечения аллергических реакций, таких как аллергический риносинусит. Грибковые инфекции, которые можно лечить комбинацией по настоящему изобретению, включают инфекции, вызываемые, inter alia, Candida spp, Trichophyton spp, Microsporium spp, Epidermophyton floccosum, Cryptococcus neoformans, Aspergillus spp, Fusarium spp, Scedosporium spp, Coccidioides immitis, Paracoccidioides brasillensis, Histoplasma spp, Blastomyces dermatiditis, Alternaria spp, Exophiala spp, Fonsecaea pedrosol, Penicillium marneffei, Phialophora spp или Paecilomyces lilacinus. Следует понимать, что ссылка на лечения включает также и профилактику, а также облегчение уже имеющихся симптомов.
В комбинации по настоящему изобретению вориконазол и противогрибковый ингибитор CYP2C19 можно вводить, с точки зрения форм дозирования, как по отдельности, так и в сочетании друг с другом; а с точки зрения времени их введения, как одновременно, так и последовательно. Таким образом, введение вориконазола можно осуществлять до, одновременно или после введения противогрибкового ингибитора CYP2C19. Время между введением может варьировать в пределах 24-часового интервала.
Единичная дозированная форма по настоящему изобретению представляет собой лекарственную форму, в которой присутствуют вориконазол и противогрибковый ингибитор CYP2C19. Она может представлять собой твердую композицию для перорального введения, такую как таблетка, капсула, содержащая множество частиц, жидкость или порошок, таблетка в форме ромба (включая наполненную жидкостью), жевательная таблетка, гель, твердый раствор, липосома, пленка (включая мукоадгезивную), овула, спрей или жидкую композицию, или композиции для парентерального введения (обычно водный раствор, который может содержать наполнители, как определено ниже в настоящем документе), или композицию для местного нанесения на кожу или слизистую оболочку (то есть, дермально или трансдермально), такую как гидрогель, лосьон, раствор, крем, мазь, распыляемый порошок, повязка, пенка, кожный пластырь, тонкая пластинка, имплантат, губка, волокно, бандаж или микроэмульсия. Предпочтительно, единичная дозированная форма по настоящему изобретению представляет собой таблетку или капсулу, особенно таблетку, содержащую вориконазол и противогрибковый ингибитор CYP2C19.
Соединения комбинации по настоящему изобретению, предназначенные для фармацевтического применения, можно вводить в форме кристаллических или аморфных продуктов. Их можно получать, например, в форме твердых пробок, порошков или пленок, такими способами как осаждение, кристаллизация, лиофильная сушка, распылительная сушка или сушка выпариванием. Для данной цели можно использовать также микроволновую сушку или сушку с использованием радиоволн.
Обычно композицию по настоящему изобретению будут вводить в форме композиции в сочетании с одним или более фармацевтически приемлемым эксципиентом. Термин «эксципиент» используется в настоящем документе для описания любого ингредиента, не являющегося соединениями по настоящему изобретению. Выбор эксципиента в большой степени будет зависеть от таких факторов, как конкретный способ введения, влияние эксципиента на растворимость и стабильность и от природы лекарственной формы.
Фармацевтические композиции, подходящие для доставки соединений по настоящему изобретению, и способы из изготовления будут очевидны для специалистов в данной области. Указанные композиции и способы их изготовления можно найти, например, в «Remington's Pharmaceutical Sciences», 19-е издание (Mack Publishing Company, 1995).
Соединения по настоящему изобретению можно вводить перорально. Пероральное введение может включать проглатывание, таким образом, что соединения проникают в желудочно-кишечный тракт, или буккальное, или сублингвальное введение, посредством которых соединения проникают в кровь непосредственно из ротовой полости.
Композиции, подходящие для перорального введения включают твердые композиции, такие как таблетки, капсулы. Содержащие множество частиц жидкости или порошки, таблетки в форме ромба (включая наполненные жидкостью), жевательные таблетки, мульти- и наночастицы, гели, твердый раствор, липосому, пленки (включая мукоадгезивные), овулы, спреи и жидкие композиции.
Жидкие композиции включают суспензии, растворы, сиропы и эликсиры. Указанные композиции можно использовать как наполнители в мягких или твердых капсулах и обычно включают в себя носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более эмульгирующих агентов и/или суспендирующих агентов. Жидкие композиции можно также изготавливать повторным растворением твердого вещества, например, из пакетика.
Соединения по настоящему изобретению можно использовать также в виде быстрорастворимых, быстрораспадающихся лекарственных форм, таких, которые описаны в Expert Opinion in Therapeutic Patents, 11 (6), 981-986, Liang and Chen (2001).
В лекарственных формах в виде таблеток, в зависимости от дозы, лекарственное средство может составлять от 1 мас.% до 80 мас.% лекарственной формы, более обычно, от 5 мас.% до 60 мас.% лекарственной формы. Помимо лекарственного средства, таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают гликолят натрий-крахмал, натрий-карбоксиметилцеллюлозу, кальций-карбоксиметилцеллюлозу, натрий-кроскармеллозу, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшим алкилом, крахмал, предварительно желатинизированный крахмал и альгинат натрия. Обычно разрыхлитель будет включать от 1 мас.% до 25 мас.%, предпочтительно, от 5 мас.% до 20 мас.% лекарственной формы.
Связующие агенты обычно используют для придания свойств сцепления таблеточной композиции. Подходящие связующие агенты включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические смолы, поливинилпирролидон, предварительно желатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, моногидрат, полученный распылительной сушкой, безводный и т.п.), маннит, ксилит, декстрозу, сахарозу, сорбит, микрокристаллическую целлюлозу, крахмал и дигидрат двухкальциевого фосфата.
Таблетки могут также, необязательно, включать поверхностно-активные агенты, такие как лаурилсульфат натрия и полисорбат 80, и агенты, обеспечивающие скольжение, такие как диоксид кремния и тальк. Если поверхностно-активные агенты присутствуют, то они могут составлять от 0,2 мас.% до 5 мас.% таблетки, а агенты, обеспечивающие скольжение, могут составлять от 0,2 мас.% до 1 мас.% таблетки.
Таблетки также обычно содержат лубриканты, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния и лаурилсульфатом натрия. Лубриканты обычно составляют от 0,25 мас.% до 10 мас.%, предпочтительно, от 0,5 мас.% до 3 мас.% таблетки.
Другие возможные ингредиенты включают антиоксиданты, красители, корригенты, консерванты и агенты, маскирующие вкус.
Примеры таблеток содержат приблизительно до 80% лекарственного средства, приблизительно от 10 мас.% до 90 мас.% связующего агента, приблизительно от 0 мас.% до 85 мас.% разбавителя, приблизительно от 2 мас.% до 10 мас.% разрыхлителя и приблизительно от 0,25 мас.% до 10 мас.% смазывающего агента.
Таблетировочные смеси можно прессовать непосредственно или с использованием роллера для формирования таблеток. Таблетировочные смеси или части смесей можно, альтернативно гранулировать влажным способом, сухим способом или способом расплава, замораживанием расплава или экструдированием перед таблетированием. Конечная композиция может включать один или более слоев и может иметь или не иметь покрытия; она может быть даже инкапсулированной.
Изготовление таблеток обсуждается в «Pharmaceutical Dosage Forms: Tablets, Vol. 1», H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-8247-6918-X).
Твердые композиции для перорального введения могут быть изготовлены для немедленного и/или модифицированного высвобождения. Композиции для модифицированного высвобождения включают замедленное, отсроченное, импульсное, контролируемое, нацеленное и запрограммированное высвобождение.
Подходящие композиции с модифицированным высвобождением для целей настоящего изобретения описаны в патенте США № 6106864. Подробности других подходящих методик высвобождения, таких как высокоэнергетические дисперсии и осмотические и имеющие покрытие частицы, можно найти в Verma et al., Pharmaceutical Technology On-line, 25(2), 1-14 (2001). Использование жевательной резинки для достижения контролируемого высвобождения описано в WO 00/35298.
Соединения по настоящему изобретению можно также вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие средства для парентерального введения включают внутривенный, внутриартериальный, интраперитонеальный, интратекальный, интравентрикулярный, интрауретральный, интрастернальный, интракраниальный, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) инъекторы, безыгольные инъекторы и инфузионные технологии. Примером безыгольного инъектора является PowderjectTM.
Парентеральные композиции обычно представляют собой водные растворы, которые могут содержать наполнители, такие как соли, углеводы и буферные агенты (предпочтительно, с рН от 3 до 9), но, для некоторых видов применения, они могут быть более удобно изготовлены в форме стерильного неводного раствора или в виде порошка, высушенного для использования в сочетании с подходящим носителем, таким как стерильная апирогенная вода.
Изготовление парентеральных композиций в стерильных условиях, например, лиофилизацией, может быть легко осуществлено с использованием стандартных фармацевтических методик, хорошо известных специалистам в данной области.
Растворимость вориконазола и противогрибкового ингибитора CYP2C19, используемых при изготовлении парентеральных растворов, может быть повышена с использованием соответствующих методик изготовления, таких как инкорпорация агентов, усиливающих растворимость. Композиции для использования в безыгольных инъекторах включают соединение по настоящему изобретению в порошкообразной форме в сочетании с подходящим носителем, таким как стерильная апирогенная вода. Например, растворимость вориконазола в воде можно повысить добавлением к нему одного или более полоксамеров, как описано в патентной заявке UK № 0327390.1 (включена в настоящий документ в качестве ссылки). Альтернативно растворимость вориконазола в воде можно повысить добавлением к нему сульфобутилового эфира циклодекстрина, такого, который описан в WO 91/11172 и WO 94/025518 (включены в настоящий документ в качестве ссылки). Композиция вориконазола с сульфобутиловым эфиром циклодекстрина описана в WO 98/58677 (включена в настоящий документ в качестве ссылки).
Композиции для парентерального введения могут быть изготовлены для немедленного и/или модифицированного/контролируемого высвобождения. Композиции для контролируемого/модифицированного высвобождения включают замедленное, отсроченное, импульсное, контролируемое, нацеленное и запрограммированное высвобождение. Таким образом, соединения по настоящему изобретению могут быть изготовлены в виде твердого вещества, полутвердого вещества или тиксотропной жидкости для введения в качестве имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры указанных композиций включают покрытые лекарственным средством стенты и PGLA микросферы.
Соединения по настоящему изобретению также можно наносить местно на кожу или слизистую оболочку, т.е., дермально или трансдермально. Типичные композиции для указанной цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, распыляемые порошки, повязки, пенки, пленки, кожные пластыри, тонкие пластинки, имплантаты, губки, волокна, бандажи и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Можно инкорпорировать усилители проникновения - см., например, J. Pharm. Sci., 88(10), 955-958, Finnin and Morgan (октябрь 1999 г.). Местное введение также может достигаться с использованием пластыря, такого как чрескожный ионофоретический пластырь.
Другие средства местного введения включают доставку путем электропорации, ионофореза, фонофореза, сонофореза и с помощью микроигольных или безыгольных (например, PowderjectTM, BiojectTM и т.п.) инъекторов.
Композиции для местного введения могут быть изготовлены для немедленного и/или модифицированного/контролируемого высвобождения. Композиции для контролируемого/модифицированного высвобождения включают замедленное, отсроченное, импульсное, контролируемое, нацеленное и запрограммированное высвобождение. В объем настоящего изобретения входят также композиции, содержащие вориконазол и противогрибковый ингибитор CYP2C19, которые удобно скомбинированы в форме набора, подходящего для совместного введения композиций.
Таким образом, набор по настоящему изобретению включает две или более отдельных фармацевтических композиций, по меньшей мере одна из которых содержит вориконазол, а другая содержит противогрибковый ингибитор CYP2C19, и средства для отдельного отпуска указанных композиций, такие как контейнер, бутылка с делениями или пакет из фольги с делениями. Примером указанного набора является известная блистерная упаковка, используемая для упаковки таблеток, капсул и т.п.
Набор по настоящему изобретению является особенно удобным для введения различных лекарственных форм, например пероральной и парентеральной, для введения отдельных композиций с различными интервалами или для дозирования отдельных композиций относительно друг друга. Для облегчения пациенту соблюдения режима и схемы лечения, набор обычно включает инструкции по введению и может снабжаться так называемыми памятками.
Примеры
Пример 1
Ингибирование активности цитохрома Р450 в человеческих микросомах печени флуконазолом
Данные в таблице 1, ниже, были определены, согласно методике, описанной Meier et al. (1985) Anal. Biochem. 151, 286-291, и в Funa and Imaoka (1987) Biochem. Biophys. Acta. 926, 349-358.
Как следует из представленных выше данных, флуконазол демонстрирует селективное ингибирование фермента CYP2C19 по сравнению с CYP3А4.
Пример 2
In vitro метаболизм вориконазола в человеческих микросомах печени и rCYP2C19 и rCYP3А4: Селективность ингибирования фермента флуконазолом
Следующая инкубационная смесь (конечные концентрации) использовали во всех анализах, описанных в данном исследовании; 50 мМ буфера фосфата калия (рН 7,4), 5 мМ MgCl2, 5 мМ изолимонной кислоты, 1 ЕД/мл дегидрогеназы изолимонной кислоты. Восстановительные эквиваленты, требующиеся для метаболизма Р450, обеспечивались NADPH, регенерация которого осуществлялась с использованием дегидрогеназы изолимонной кислоты/изолимонной кислоты.
Ход исследований
Инкубировали следующие препараты человеческих микросом печени; контрольная серия HL-MIX-101 (приготовленная из пула от 60 доноров); два донора генотипировали как слабых метаболизаторовCYP2C19 (НН-92, НН-112) и экстенсивного метаболизатора CYP2C19 с высокой активностью CYP2C19 (НН-100). Относительные активности CYP2C19 и CYP3A4 показаны в таблице 2, ниже:
Характеристики человеческих микросом печени
(мг/мл)
(пмоль/мл)
(нмоль/мин/мг белка)
(нмоль/мин/мг белка)
Дополнительную инкубацию производили с микросомами, приготовленными из клеток насекомых, трансфектированными рекомбинантными CYP2C19 и CYP3A4 (BDGENTEST® суперсомы и бакулосомы Panvera соответственно).
Ряд предварительных исследований потребовался для того, чтобы убедиться в том, что образование N-оксидного метаболита было линейным в течение периода инкубации, с использованием упомянутой выше реакционной смеси. Человеческие микросомы печени (1 мг/мл) предварительно инкубировали в присутствии субстрата вориконазола (24 мкМ конечная концентрация) перед добавлением NADPH. Следует заметить, что для исследований с rCYP2C19 и rCYP3A4, предварительные инкубации были выполнены с NADPH, а реакцию инициировали добавлением субстрата вориконазола. Аликвоты (1 мл) реакционной смеси собирали в течение периода времени (0-60 мин) и добавляли к 4 мл дихлорметана, содержавшего 50 мкл внутреннего стандарта (2-(2,4-дифторфенил)-3-(4-пиримидил)-1-(1Н-1,2,4-триазол-1-ил)-2-бутанола; 15 мкг/мл). Образцы смешивали на ротационной мешалке в течение 10 минут, а затем центрифугировали на 3000 об./мин в течение 5 мин (4°С). Верхние водные слои удаляли и сливали. Нижний органический слой выпаривали до сухости при 37°С под потоком N2. Образцы затем разводили в 100 мкл подвижной фазы А (смотри ниже) и 25 мкл инъекцировали на ВЭЖХ.
Образцы анализировали на Agilent 1 100 Series UV-HPLC. Хроматографическое разделение осуществляли с использованием колонки Hichrom 100-5C18 (150 х 4,6 мм), при скорости потока 1 мл/мин с UV-λ на 254 нм. Использовали следующие подвижные фазы: (А) 0,1 М фосфат аммония в 70:30 Н2О:MeOH (рН доводили до 7,0, перед добавлением MeOH) и (В) MeOH, с использованием следующего градиента; 0-2 мин (0% В), 2-20 (0-100% В), 20-23 (100% В), 23,1 (0% В). Колонке позволяли вновь уравновеситься в течение 7 мин до следующей инъекции. Типичное время удерживания составляло 7,9 мин для флуконазола; 11,7 мин для N-оксидного метаболита; 13,1 мин для внутреннего стандарта и 14,4 мин для вориконазола.
Ингибирующий потенциал флуконазола против N-окисления вориконазола изучали на человеческих микросомах печени и препаратах rCYP2C19 и rCYP3A4. Конечные изучавшиеся концентрации флуконазола составляли 1, 0,1, 1, 10, 100 и 1000 мкМ.
Человеческие микросомы печени инкубировали при 1 мг/мл в течение 60 мин, rCYP2C19 при 10 пмоль CYP/мл в течение 60 мин, и rCYP3А4 при 1000 пмоль CYP/мл в течение 20 мин. Для каждого матрикса аликвоты (4 мл) реакционной смеси предварительно нагревали до 37°С на водяной бане, с последующим добавлением 200 мкл NADPH (20 мМ) и 40 мкл соответствующих растворов флуконазола (0-100 мМ). Реакции инициировали добавлением 50 мкл вориконазола (2 мМ). К концу времени инкубации аликвоты (n=3) отбирали из каждой инкубационной смеси и добавляли в пробирки, содержавшие 4 мл дихлорметана и 50 мкл внутреннего стандарта (2-(2,4-дифторфенил)-3-(4-пиримидил)-1-(1Н-1,2,4-триазол-1-ил)-2-бутанола; 15 мкг/мл). Экстрагирование и анализ осуществляли как описано выше.
При всех заявленных концентрациях образование N-оксидного метаболита было линейным до 60 мин в человеческих микросомах печени и в rCYP2C19. В rCYP3А4 линейность определялась в течение 20 мин.
Ингибирующий потенциал (IC50) флуконазола в отношении метаболизма вориконазола до его N-оксидного метаболита изучали в пуле человеческих микросом печени (HL-MIX-101), а также от отдельных доноров, генотипированных как слабые (HH-92 и НН-112) или экстенсивные метаболизаторы (НН-100). Кроме того, РМ были разделены также на имеющие высокие и низкие метаболические способности в отношении. Для того, чтобы лучше определить селективность флуконазола в отношении ингибирования CYP3А4- или CYP2C19-опосредованного метаболизма вориконазола, изучали также rCYP3А4 или rCYP2C19. Результаты суммированы на фиг.3-8 и сопоставлены в таблице 3.
Определение 1С50 для флуконазола в человеческих микросомах
печени (CY2С19 PM & EM) и rCYP2C19 и rCYP3A4
В случае человеческих микросом печени слабого метаболизатора CYP2C19 флуконазол является слабым ингибитором N-окисления вориконазола (IC50˜175 мкМ и 214 мкМ). Более мощное ингибирование наблюдалось в CYP2C19 микросомах экстенсивного метаболизатора (IC50˜50 мкМ). Эксперименты с использованием rCYP2C19 и rCYP3А4 показали, что флуконазол является более мощным ингибитором CYP2C19-опосредованного N-окисления (IC50˜29 мкМ), по сравнению с CYP3А4 (IC50˜106 мкМ).
Общим открытием указанных экспериментов выявляется то, что селективность флуконазола в 3-4 раза превышает ингибирование CYP2C19-опосредованного N-окисления вориконазола по сравнению с CYP3А4-опосредованным N-окислением.
Пример 3
Клинические испытания
Проводилось исследование с целью изучения влияния совместного введения флуконазола на устойчивое состояние фармакокинетики вориконазола у здоровых субъектов мужского пола и с целью оценки безопасности и переносимости совместного введения флуконазола и вориконазола. Десять здоровых субъектов в возрасте 21-55 лет были набраны для участия в испытаниях, чтобы гарантировать завершение исследования по меньшей мере 8 субъектами, включая двух слабых метаболизаторов CYP2C19. Два вида лечения были следующими:
1. Ударная доза вориконазола 400 мг два раза в день перорально в 1 день, затем 200 мг два раза в день перорально на 2-3 дни и однократная 200 мг доза перорально утром 4 дня.
2. Ударная доза вориконазола 400 мг два раза в день перорально в 1 день, затем 200 мг два раза в день перорально на 2-3 дни и однократная 200 мг доза перорально утром 4 дня, плюс однократная доза 400 мг флуконазола перорально, с утренней дозой вориконазола в 1 день и однократные дозы 200 мг флуконазола перорально один раз в день утром 2-5 дней.
Субъекты были отобраны слепым способом для каждого вида лечения, как в отношении последовательности введения вориконазола, с последующим введением вориконазола плюс флуконазол, так и в отношении последовательности вориконазол плюс флуконазол, с последующим введением вориконазола.
Образцы крови отбирали на 4, 5 и 6 дни каждого периода лечения для определения концентраций вориконазола в плазме для расчета фармакокинетических параметров Cmax, Tmax, AUCT, AUCt и AUC на 4 день каждого периода лечения. Для субъектов, классифицированных как экстенсивные метаболизаторы CYP2C19, данные параметры затем использовали для сравнения вориконазола в отдельности с вориконазолом плюс флуконазол. Те же параметры для субъектов, классифицированных как слабые метаболизаторы CYP2C19, использовали также для сравнения данных, полученных для экстенсивных метаболизаторов.
Результаты показаны на фиг.9 и 10 (слабые метаболизаторы) и 11 и 12 (экстенсивные метаболизаторы). Фиг.9 и 10 ясно показывают, что флуконазол оказывал незначительное влияние или вовсе не оказывал влияния на фармакокинетику вориконазола у слабых метаболизаторов.
Однако фиг.11 и 12 показывают влияние 200 мг флуконазола один раз в день на стандартную дозу вориконазола 200 мг два раза в день у экстенсивного метаболизатора. Следует отметить, что через 24 часа после последней дозы уровни вориконазола сохранялись >2000 нг/мл, что считается избыточной концентрацией в плазме, требующейся для эффективности. Кроме того, полупериод существования вориконазола у экстенсивных метаболизаторов увеличивался до >18 часов.
Это показывает, что совместное введение вориконазола с флуконазолом приводит к уменьшению клиренса вориконазола из организма у экстенсивного метаболизатора. Это делает возможным присутствие вориконазола в плазме крови в течение дня в концентрациях, достаточных для достижения терапевтического эффекта у экстенсивного метаболизатора при введении один раз в день.
Кроме того, данные показывают, что посредством совместного введения вориконазола с флуконазолом уровни вориконазола в плазме могут достигаться при значительно сниженных дозах вориконазола; можно предвидеть снижение в два раза при введении один раз в день и снижение в четыре раза при введении два раза в день. Помимо этого данные показывают, что можно достигать значительного уменьшения вариабельности в популяции пациентов.
Данные показывают, что терапевтические уровни вориконазола в плазме могут достигаться при значительно сниженных дозах вориконазола. Это может позволить слабым метаболизаторам или гетерозиготным экстенсивным метаболизаторам уменьшить вводимые дозы и уровни в крови вориконазола в 2-4 раза. В свою очередь, достигаемое уменьшение системного воздействия может минимизировать любые нежелательные побочные эффекты.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ РИНОСИНУСИТА | 2004 |
|
RU2410083C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ПРОИЗВОДНОЕ ФЕНИЛАМИДИНА, И СПОСОБ ПРИМЕНЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ В КОМБИНАЦИИ С ПРОТИВОГРИБКОВЫМ СРЕДСТВОМ | 2007 |
|
RU2429843C2 |
| ИДЕНТИФИКАЦИЯ РЕАКЦИИ ПАЦИЕНТОВ НА ВВЕДЕНИЕ МОДУЛЯТОРА РЕЦЕПТОРОВ S1P | 2013 |
|
RU2785736C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2021 |
|
RU2824066C1 |
| ИСПОЛЬЗОВАНИЕ СОЕДИНЕНИЙ ПРИ ЛЕЧЕНИИ ГРИБКОВЫХ ИНФЕКЦИЙ | 2021 |
|
RU2805930C1 |
| НОВАЯ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ЛЕЧЕНИЯ ГРИБКОВОЙ ИНФЕКЦИИ | 2010 |
|
RU2481100C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ И СПОСОБ ПРИМЕНЕНИЯ ПРОТИВОГРИБКОВОГО СРЕДСТВА В КОМБИНАЦИИ | 2006 |
|
RU2396955C2 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 2017 |
|
RU2760681C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ АЛЛЕРГИЧЕСКИХ И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ КОЖИ | 2009 |
|
RU2440108C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ГОШЕ | 2018 |
|
RU2781242C2 |
Настоящее изобретение относится к области лекарственных средств, в частности к терапевтической комбинации, включающей вориконазол и противогрибковый ингибитор CYP2C19 флуконазол. Кроме того, изобретение относится к композиции, набору, единичной дозированной формы, включающих указанные компоненты, а также к способу лечения грибковой инфекции. Технический результат - снижение метаболизма у экстенсивных метаболизаторов вориканозола, а также синергетический эффект, проявляющийся при введении указанной комбинации. 7 н.п. ф-лы, 12 ил., 3 табл.
| GHANNOUM М | |||
| et al | |||
| In vitro evaluated of voriconazole in combination with antifungals | |||
| International journal of infection diseases, vol.6, 02.06.2002, p.S50-S51 | |||
| ERNST EJ et al | |||
| Rates and extents of antifungal activities of amphotericin B, flucytosine, fluconazole, and voriconazole against Candida lusitaniae determined by microdilution, Etest, and |

