Изобретение относится к улучшенному способу производства промежуточных продуктов, полезных в получении толтеродина, фезотеродина и других фармацевтически полезных соединений. В изобретении также предложены улучшенные способы получения таких фармацевтически полезных соединений с использованием этих промежуточных продуктов.
Толтеродин {2-[(1R)-3-[бис(1-метилэтил)амино]-1-фенилпропил]-4-метилфенол или альтернативно (+)-N,N-диизопропил-3-(2-гидрокси-5-метилфенил)-3-фенилпропиламин},
2124
представляет собой антагонист мускариновых рецепторов для лечения гиперактивного мочевого пузыря, включающего недержание мочи. Он был одобрен для продажи (в виде тартрата) в 1997 и в последующие годы был выпущен на многие рынки под товарными знаками DETROL и DETRUSITOL. Тартрат толтеродина раскрыт в международной патентной заявке WO 89/06644 (смотри в частности Пример 22 и пункт 7).
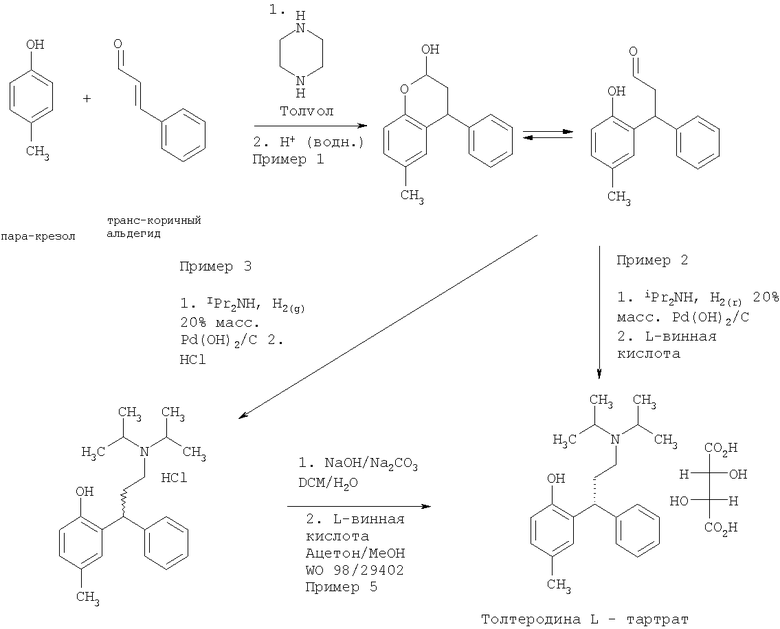
В WO 98/29402 раскрыт способ получения толтеродина, включающий конденсирование пара-крезола (а) с коричной кислотой (б), затем восстановление полученного лактона (в) восстановителем, таким как диизобутилалюминия гидрид (DIBAL), натрия бис(2-метоксиэтокси)алюминия гидрид или лития три-трет-бутоксиалюминогидрид, с получением соответствующего бензопиран-2-ольного соединения (г). Бензопиран-2-ольное соединение (г) затем может быть превращено в рацемический гидрохлорид толтеродина (д) посредством восстановительного аминирования с диизопропиламином с последующим добавлением водной соляной кислоты. Наконец, толтеродина L-тартрат получают путем нейтрализации солянокислой соли (д) NaOH/NaHCO3 и последующего разделения с использованием L-винной кислоты. Способ представлен на Схеме 1
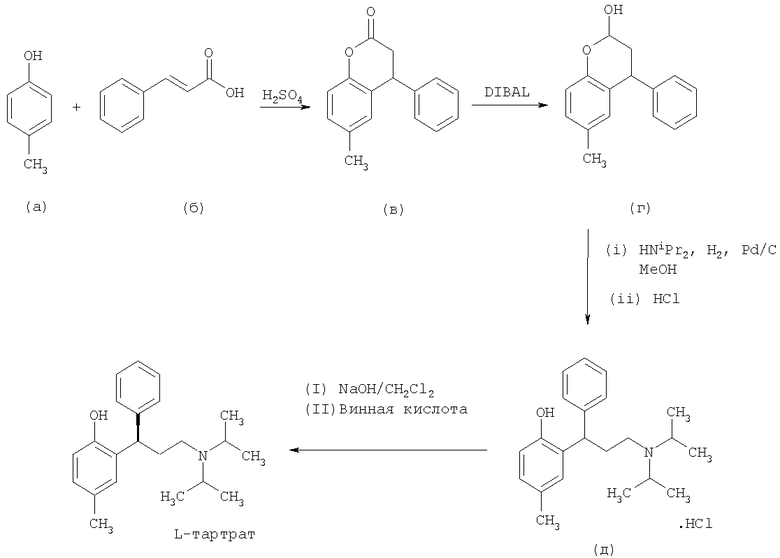
Видно, что в способе, раскрытом в WO 98/29402, бензопиран-2-ольное соединение (г) получают за две стадии и этот способ включает использование относительно дорогостоящего восстановителя (DIBAL).
В WO 01/49649 описано восстановительное аминирование энантиомеров вышеуказанного соединения (г) с получением толтеродина и его энантиомера. Энантиомеры соединения (г) получают посредством энантиоселективных взаимодействий. Это также относится к таким же способам, применяемым к аналогичным соединениям, в частности аналогам толтеродина, в которых метильная группа в фенольном кольце заменена 5-гидроксиметильной группой.
В патентной заявке US 2003/0236438 (MacMillan et al.) раскрыто применение относительно сложных хиральных имидазолидиноновых катализаторов [например, (2S,5S)-5-бензил-2-трет-бутил-3-метилимидазолидин-4-она] для осуществления энантиоселективных реакций 1,4-присоединения между анилиновыми нуклеофилами и α,β-ненасыщенными альдегидами (данная работа описана также в MacMillan et al., J. Am. Chem. Soc, 2002, 124, 7894-7895). Пример 2 из патентной заявки US 2003/0236438 представляет собой типичную описанную реакцию:
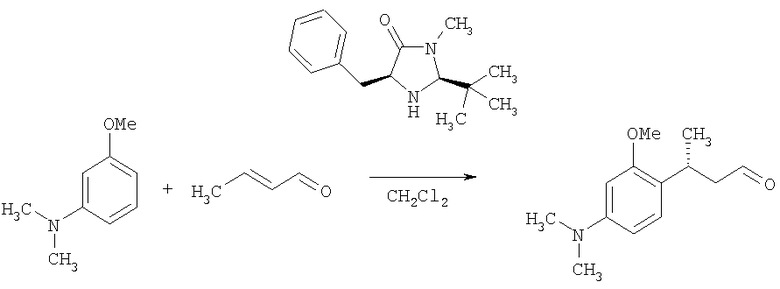
Видно, что атом углерода ароматического кольца в пара-положении относительно аминогруппы связывается с альфа-бета ненасыщенным альдегидом.
В Jurd (Journal of Heterocylic Chemistry, том 28 (4), 1991, стр.983-986) раскрыто взаимодействие 3,4-метилендиоксифенола, морфолина и коричного альдегида в метаноле с получением 2-морфолинил-4-фенилбензопиранов.
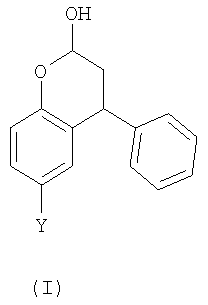
Неожиданно было установлено, что бензопиран-2-ольное соединение (г) из Схемы 1 может быть получено в однореакторном взаимодействии, начиная из пара-крезола (а). Также могут быть получены аналогичные соединения. Таким образом, согласно первому аспекту настоящего изобретения, предложен способ получения соединения формулы (I),
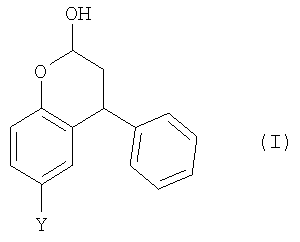
где Y выбран из СН3, СН2ОН, СН2СН2ОН, CH2Br и Br; включающий стадии:
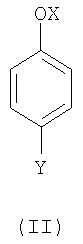
(1) взаимодействия соединения формулы (II),
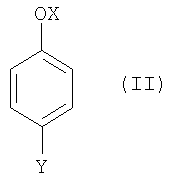
где
OX представляет собой гидрокси или O-М+, где М+ представляет собой катион, выбранный из Li+, Na+и К+, и
Y такой, как определено выше;
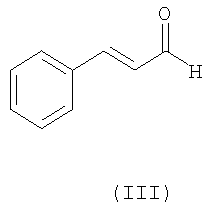
с транс-циннамальдегидом (III),
 ;
;
в присутствии вторичного аминного соединения; затем
(2) обработки продукта с предыдущей стадии кислотой с получением соединения формулы (I).
Под "вторичным аминным соединением" авторы изобретения подразумевают органическое соединение, которое содержит по меньшей мере одну вторичную аминную группу, то есть соединение формулы:
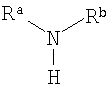
где Ra и Rb не являются водородом. Предпочтительно, каждый из Ra и Rb связан с атомом азота посредством группы СН2, например они независимо представляют собой С1-6алкил или вместе образуют 4- или 5-членную алкильную цепь, в которой один атом углерода возможно заменен O или N.
Предпочтительными воплощениями первого аспекта данного изобретения являются такие, в которых:
(а) ОХ представляет собой гидрокси;
(б) Y представляет собой СН3 или СН2ОН;
(в) вторичное аминное соединение является ахиральным;
(г) вторичное аминное соединение содержит две вторичные аминные группы, например пиперазин (данный катализатор обеспечивает особенно высокие выходы продукта);
(д) если вторичное аминное соединение содержит две вторичные аминные группы, то 0,5-1,25 моль-эквивалентов этого вторичного аминного соединения используют на стадии (1);
(е) альтернативно, вторичное аминное соединение содержит одну вторичную аминную группу, и, более предпочтительно, вторичное аминное соединение представляет собой морфолин, дибутиламин, дибензиламин, 1,1,3,3-тетраметилгуанидин, диэтиламин, диизопропиламин, пиперидин или N-(С1-6алкил)пиперазин. N-Метилпиперазин особенно предпочтителен, так как он обеспечивает хорошие выходы, исходное вещество [смотри формулу (VI) ниже] легко гидролизуется в соответствующее лактольное соединение формулы (I), и неочищенное соединение формулы (1) имеет улучшенную чистоту, что облегчает кристаллизацию;
(з) если вторичное аминное соединение содержит одну вторичную аминогруппу, то на стадии (1) используют 1-5, более предпочтительно 1-2,5 моль-эквивалентов вторичного аминного соединения;
(и) кислота, применяемая на стадии (2), представляет собой водную соляную кислоту (предпочтительно в концентрации не более 2 М), хотя следующие водные кислоты в соответствующих концентрациях также обеспечивают хорошие результаты: лимонная кислота, уксусная кислота, оксалиновая кислота, трифторуксусная кислота, малеиновая кислота, фумаровая кислота, салициловая кислота, транс-коричная кислота, бензойная кислота, камфорсульфокислота и толуолсульфокислота;
(к) взаимодействие на стадии (1) осуществляют в органическом растворителе, выбранном из толуола, ксилола, N-бутилацетата, трет-амилового спирта, диоксана и дибутилового эфира, наиболее предпочтительно в толуоле (который обеспечивает особенно высокие выходы);
(л) взаимодействие на стадии (1) осуществляют при температуре в пределах от 80°С до температуры дефлегмации растворителя;
(м) взаимодействие на стадии (1) осуществляют при условиях, при которых удаляется вода из реакционной системы (например, в условиях Дина-Старка, когда вода, полученная при реакции, конденсируется в боковом конденсаторе так, что она не возвращается в реакционную смесь и может быть отведена, если это требуется); и
(н) взаимодействие на стадии (1) осуществляют при давлении, равном или приблизительно равном давлению окружающей среды (например, можно использовать незначительно повышенное давление атмосферы азота, особенно в промышленном масштабе).
Особенно предпочтительно чтобы, когда Y представляет собой СН2ОН, тогда вторичное аминное соединение представляло собой N-метилпиперазин.
Вторичное аминное соединение предпочтительно содержит два основных атома азота. Такие соединения образуют исходные продукты [смотри формулу (VI) ниже], которые легко гидролизуются в соединения формулы (I).
Согласно второму аспекту изобретения предложен способ получения соединения формулы (IV),
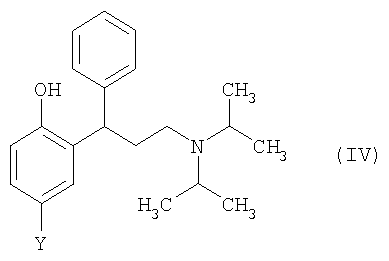
где Y выбран из СН3, СН2ОН, СН2СН2ОН, CH2Br и Br, или его соли, включающий:
(а) получение соединения формулы (I), как определено выше, с использованием способа согласно первому аспекту изобретения; затем
(б) восстановительное аминирование соединения формулы (I) диизопропиламином;
(в) и, если требуется, превращение полученного соединения в соль.
Во втором аспекте изобретения Y предпочтительно представляет собой СН3 или СН2ОН. Если Y представляет собой СН3, то соединение формулы (IV) может быть обработано L-винной кислотой на стадии (в) с получением толтеродина L-тартрата [например, R-(+)-толтеродина L-тартрата]. Если предполагается применение соединения формулы (IV) в качестве лекарственного средства, то солевая форма, полученная в данном втором аспекте изобретения, предпочтительно является фармацевтически приемлемой. Однако это не существенно, если соединение будет подвергаться дополнительной обработке.
Восстановительное аминирование соединения формулы (I) может включать обработку диизопропиламином в подходящем растворителе, таком как метанол (который является предпочтительным) или трет-амиловый спирт или их смеси, затем гидрирование в присутствии катализатора, такого как Pd/C или Pd(OH)2/C.
В одном воплощении соединение формулы (IV) может быть обработано водной кислотой, какой как соляная кислота, с получением соответствующей солянокислой соли. Рацемическое соединение может быть превращено в соответствующий (R)-энантиомер L-тартратной соли путем нейтрализации солянокислой соли в присутствии основания, такого как смесь гидроксида натрия и карбоната натрия, с последующим отделением с L-винной кислотой. В одном воплощении получают толтеродина L-тартрат [то есть R-(+)-толтеродина L-тартрат].
В альтернативном воплощении (R)-энантиомер L-тартратной соли соединения формулы (IV) может быть получен непосредственно после восстановительного аминирования соединения формулы (I) без образования солянокислой соли. Например, в одном воплощении продукт стадии восстановительного аминирования может быть обработан растворителем, таким как ацетон и L-винная кислота с получением L-тартратной соли. Если Y представляет собой СН3, то получают толтеродина L-тартрат [например, R-(+)-толтеродина L-тартрат].
Согласно третьему аспекту в изобретении предложен способ получения фезотеродина,
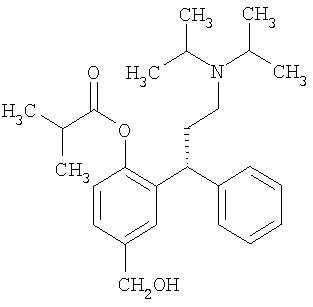
или его фармацевтически приемлемой соли, который включает:
(а) получение соединения формулы (IV), как определено выше, в котором Y представляет собой СН2ОН, с использованием способа, описанного выше;
(б) разделение продукта со стадии (а) с получением (R)-энантиомера;
(в) ацилирование фенольной гидроксигруппы продукта со стадии (б) с получением соответствующего эфира изомасляной кислоты;
(г) и, когда это желательно или необходимо, превращение полученного соединения в фармацевтически приемлемую соль.
Фезотеродин, имеющий химическое название 2-[(1R)-3-[бис(1-метилэтил)амино]-1-фенилпропил]-4-гидроксиметилфенила изобутират или альтернативно R-(+)-изомасляной кислоты 2-(3-диизопропиламино-1-фенилпропил)-4-гидроксиметилфениловый эфир, описан в ЕР 1077912 (смотри страницу 32 строка 5 и формула 4, 3-е соединение). Он показан для лечения гиперактивного мочевого пузыря.
В данном третьем аспекте разделение предпочтительно осуществляют посредством фракционной кристаллизации с хиральной кислотой, предпочтительно (R)-(-)-ацетокси(фенил)уксусной кислотой.
Ацилирующий агент предпочтительно представляет собой изобутирилхлорид.
Соединение формулы (I) может существовать в форме с открытым кольцом, хотя предполагается, что оно существует главным образом в форме с замкнутым кольцом (лактол). Кроме того, предполагается, что в способе согласно первому аспекту изобретения получают смесь диастереоизомеров лактола:
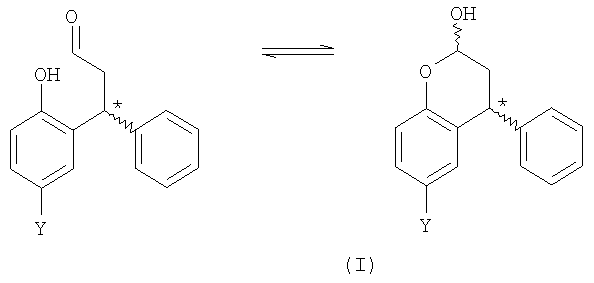
и R- и S-энантиомеры хирального центра, отмеченного выше звездочкой, присутствуют в эквивалентных количествах. Получение всех таких таутомерных и стереоизомерных форм охватывается настоящим изобретением.
Когда пиперазин применяют на стадии (1) первого аспекта изобретения, то реакция протекает через поддающееся отделению промежуточное соединение формулы (V),
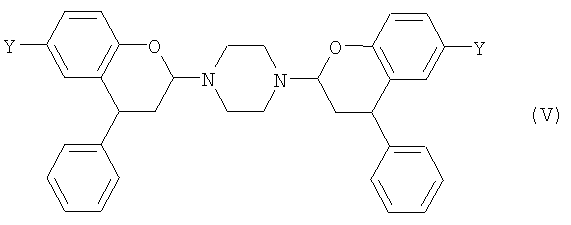
где Y выбран из СН3, СН2ОН, СН2СН2ОН, CH2Br и Br. Такие соединения предлагаются согласно четвертому аспекту настоящего изобретения. Предпочтительно, Y представляет собой СН3.
Если на стадии (1) первого аспекта изобретения применяют N-метилпиперазин, то взаимодействие происходит через промежуточное соединение формулы (VI)
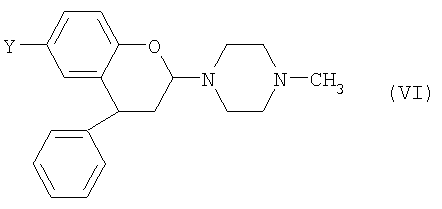
где Y выбран из СН3, СН2ОН, СН2СН2ОН, CH2Br и Br. Такие соединения предлагаются согласно пятому аспекту настоящего изобретения. Предпочтительно, Y представляет собой СН2ОН.
В данном изобретении дополнительно предлагается соединение формулы (I),
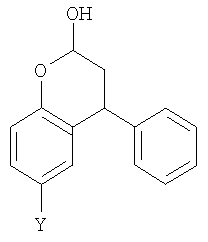
где Y выбран из СН2СН2ОН, CH2Br и Br.
Способ по изобретению отличается от патентной заявки US 2003/0236438 (смотри выше) тем, что ни один из реагентов не является анилиновым соединением, и в соединении формулы (II) по изобретению не присутствует дополнительная сильно активирующая или сильно электронодонорная группа (такая как метокси в Примере 2 патентной заявки US 2003/0236438). Кроме того, аминные катализаторы, используемые в настоящем изобретении, являются более простыми (например, они не должны быть хиральными) и, следовательно, более дешевыми.
Способ по изобретению отличается от указанного выше в Jurd (Journal of Heterocylic Chemistry, том 28 (4), 1991, стр.983-986) тем, что ни один из реагентов не является анилиновым соединением, и в соединении формулы (II) по изобретению не присутствует дополнительная сильно активированная или сильно электронодонорная группа, такая как алкокси или гидрокси.
Дополнительным преимуществом изобретения является то, что как часть способа получения толтеродина, по сравнению со способом, описанным в WO 98/29402, исключен ряд реакций и технологических стадий, что приводит к снижению затрат. Кроме того, в этом способе избегают применения дорогих восстановителей, таких как диизобутилалюминий гидрид (DIBAL), натрий бис(2-метоксиэтокси)алюминий гидрид или литий три-трет-бутоксиалюмогидрид, которые также трудно утилизировать.
Дополнительным преимуществом изобретения является то, что как часть способа получения фезотеродина, по сравнению со способами, описанными в уровне техники, исключен ряд реакций и технологических стадий, что приводит к снижению затрат. Кроме того, в этом способе избегают применения опасных и экологически нежелательных реагентов, которые трудно утилизировать.
Изобретение проиллюстрировано следующими примерами, в которых могут использоваться следующие сокращения:
BuOH = бутанол
DEA = диэтиламин
DMA = диметилацетамид
DMF = диметилформамид
DMPU = 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидон
DMSO = диметилсульфоксид
EDTA = этилендиаминтетрауксусная кислота
э.и. = энантиомерный избыток
EtOAc = этилацетат
EtOH = этанол
h = час
IPA = изопропиловый спирт
LC-MS = жидкостная хроматография - масс-спектрометрия
LOD = потери при высушивании
МеОН = метанол
min = минута
n-BuOH = н-бутанол
фунт/кв.дюйм = фунтов на квадратный дюйм
TFA = трифторуксусная кислота
THF = тетрагидрофуран
tlc = тонкослойная хроматография
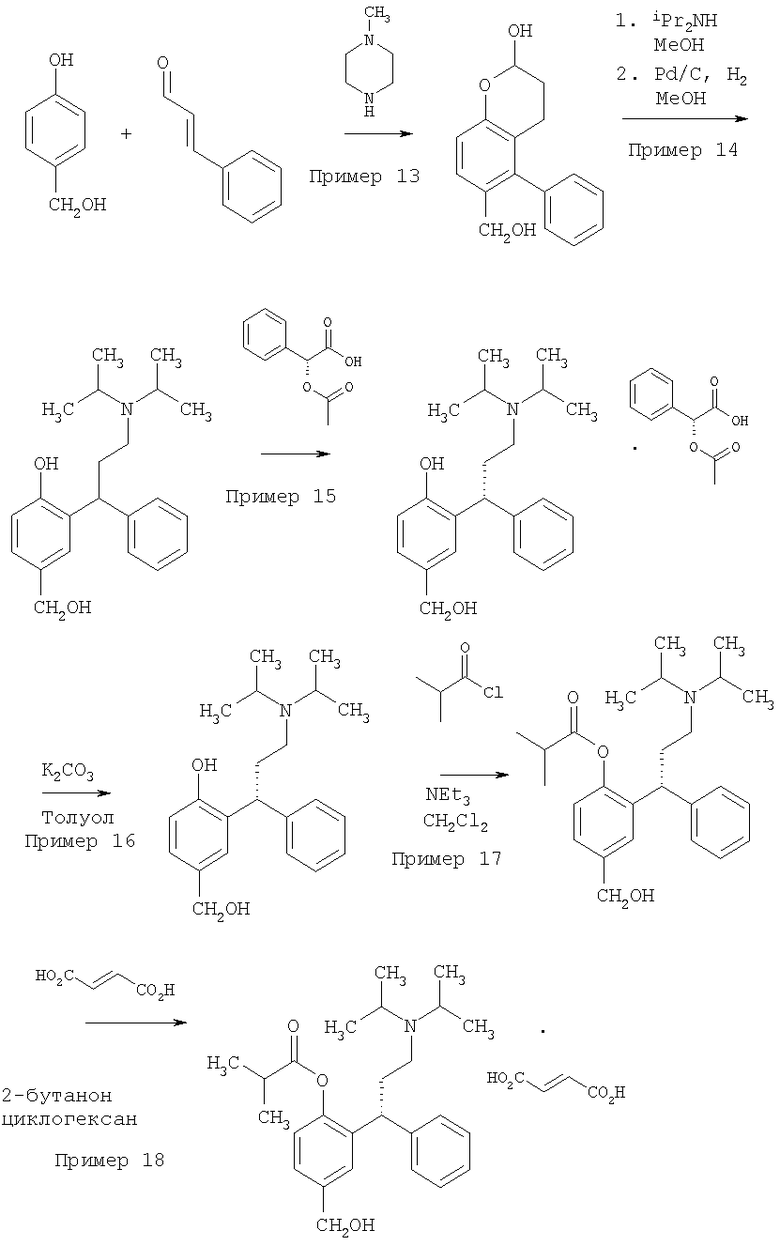
Пример 1
Синтез 3.4-дигидро-6-метил-4-фенил-2Н-бензопиран-2-ола
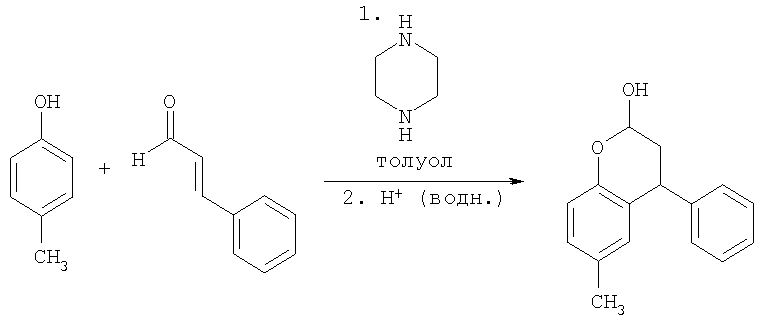
Пара-крезол (150 г, 1,387 моль) перемешивали с пиперазином (72 г, 0,832 моль, 0,6 экв.) в толуоле (1.5 л, 10 мл/г) и затем нагревали до кипении с обратным холодильником в условиях Дина-Старка в течение по меньшей мере 30 минут для удаления воды с получением прозрачного светло-желтого раствора. Затем добавляли транс-циннамальдегид (262 мл, 275 г, 2,081 моль, 1,5 экв.) в течение 2 часов, поддерживая реакционную смесь при кипении с обратным холодильником в условиях Дина-Старка. Когда добавление было закончено, нагревание реакционной смеси продолжали с обратным холодильником в условиях Дина-Старка в течение еще 4 часов. Черный раствор оставляли охлаждаться до 80°С и затем медленно гасили в течение 45 минут раствором 0,67 М HCl (водн.) (750 мл, 0,601 моль, 1,3 экв.). Затем двухфазный раствор интенсивно перемешивали в течение по меньшей мере 12 часов при температуре 75-80°С. Затем перемешивание прекращали, и смесь оставляли охлаждаться до комнатной температуры, и фазы разделяли. Затем раствор толуола промывали 1 М HCl (водн.) (750 мл, 5 мл/г), затем водой (3×750 мл, 5 мл/г). 3,4-Дигидро-6-метил-4-фенил-2Н-бензопиран-2-ол не отделяли, но использовали вместо раствора толуола непосредственно на стадии восстановительного аминирования (Пример 2).
Пример 2
Синтез толтеродина L-тартрата
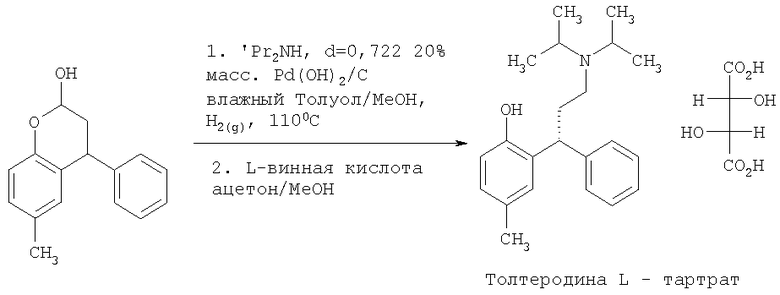
Раствор толуола, содержащий неочищенный 3,4-дигидро-6-метил-4-фенил-2Н-бензопиран-2-ол из Примера 1 (теоретически=333,3 г в 1,5 л толуола), разбавляли метанолом (750 мл, 5 мл/г), затем добавляли диизопропиламин (583 мл, 421 г, 4,161 моль, 3 экв.). Черный раствор затем гидрировали над влажным 20% мac. Pd(OH)2/C (10% мас., 33 г) при давлении 621×103 Нм-2 (90 фунт/кв. дюйм) и 110°С в течение 48 часов. Образец отбирали для анализа.
Реакционную смесь фильтровали через Arbocel™ (вспомогательный фильтрующий материал) для удаления остатков катализатора и затем нагревали до кипения с обратным холодильником, и весь диизопропиламин и метанол удаляли дистилляцией и замещали толуолом, получая конечный объем 10 мл/г. Затем черный раствор охлаждали до 25°С, добавляли ацетон (750 л, 5 мл/г), и затем раствор нагревали до 55-60°С. Добавляли раствор L-винной кислоты (312 г, 2,081 моль, 1,5 экв.) в метаноле (1,05 л, 7 мл/г) в течение 30 минут, поддерживая температуру 55-60°С. Затем полученную суспензию оставляли охлаждаться до комнатной температуры и перемешивали в течение 12 часов. Суспензию фильтровали, промывали ацетоном (2×600 мл, 4 мл/г), затем сушили в вакууме при 50°С в течение 12 часов с получением указанного в заголовке соединения в виде не совсем белого твердого вещества [159,2 г, 48% (24% от пара-крезола)]. Ахиральная чистота составляла 100% (нет определяемых примесей) и хиральная чистота составляла 91,4% э.и.
Пример 3
Синтез рацемического толтеродина гидрохлорида
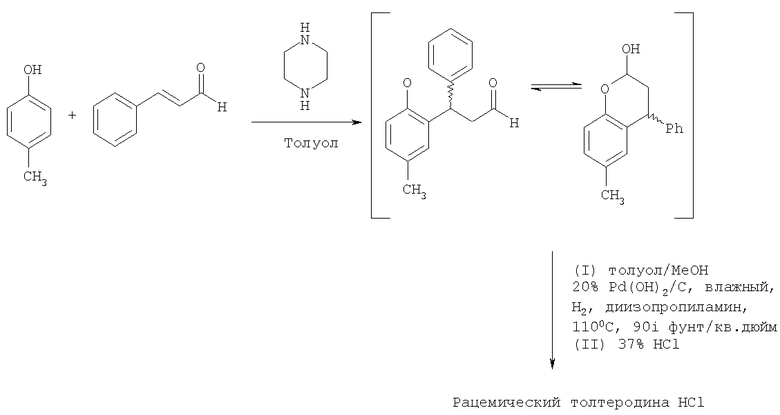
Стадия А. Получение 3,4-дигидро-6-метил-4-фенил-2Н-бензопиран-2-ола
К раствору пара-крезола (25 г, 0,231 моль, 1 экв.), пиперазина (11,94 г, 0,139 моль, 0,6 экв.) и толуола (250 мл, 10 мл/г) при нагревании с обратным холодильником в условиях Дина-Старка добавляли циннамальдегид (45,83 г, 44 мл, 0,347 моль, 1,5 экв.) в течение 2 часов, и реакционную смесь контролировали при помощи HPLC на присутствие пара-крезола. По завершении (2-3 часа) смесь охлаждали до 80°С и медленно добавляли раствор конц. HCl (25 мл, 0,301 моль, 1,3 экв.) в воде (100 мл, 5 мл/г) и нагревали при 80-90°С в течение по меньшей мере 5 часов. Полученный раствор оставляли охлаждаться до комнатной температуры, и фазы разделяли. Раствор толуола промывали 1 М HCl (125 мл, 5 мл/г), затем водой (3×125 мл). Полученный органический слой переносили на стадию восстановительного аминирования (Стадия Б) в виде неочищенной смеси.
Стадия Б. Получение рацемического толтеродина гидрохлорида
К неочищенному раствору со Стадии А добавляли метанол (125 мл, 5 мл/г крезола) и диизопропиламин (92 мл, 0,693 моль, 3 экв.). Затем смесь гидрировали над влажным 20% мас. Pd(OH)2/C (5,6 г, теоретически 10% мас. бензопиран-2-ола) при 110°С при давлении водорода 586×103 Нм-2 (85 фунт/кв.дюйм). Развитие реакции контролировали с помощью HPLC (завершение обычно происходит между 16 и 24 ч). После завершения смесь охлаждали, продували азотом, фильтровали и промывали толуолом (2×25 мл). Затем фильтрат азеотропно перегоняли с толуолом для удаления до конца всего метанола и диизопропиламина с конечным объемом, соответствующим 10 мл/г крезола. Затем раствор перемешивали при 50-60°С и добавляли 37% HCl (19,3 мл, 0,231 моль, 1,0 экв. c.f крезол), вызывая осаждение рацемического толтеродина гидрохлорида. Суспензию охлаждали до 25°С и перемешивали в течение 2 часов, затем фильтровали и промывали толуолом (2×50 мл). Затем рацемический толтеродина гидрохлорид сушили под вакуумом при 50°С. Выход составлял 52,7 г, 63% от пара-крезола с ахиральной чистотой 97%.
Синтез толтеродина L-тартрата согласно способам Примеров 1-3 показан на Схеме 2.
Пример 4
Влияние аминного катализатора и растворителя на выход 3.4-дигидро-6-метил-4-фенил-2Н-бензопиран-2-ола
Взаимодействие из Примера 1 повторяли, но варьируя аминный катализатор и растворитель. Используемая температура составляла приблизительно 100°С или температуру дефлегмации любого растворителя, если она ниже (если не указано иное). Условия Дина-Старка не использовали, если не указано (посредством *). Выходы представлены в следующей таблице.
Пример 5
1.4-бис-(6-метил-4-фенилхроман-2-ил)пиперазин
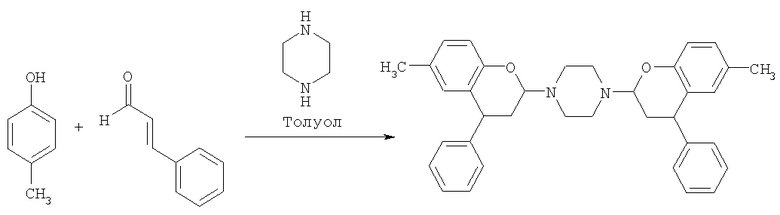
Получение указанного в заголовке соединения осуществляли, используя методику из Примера 1, за исключением гашения водной кислотой. Вместо этого после завершения реакции смесь оставляли охлаждаться до температуры окружающей среды, что приводило к образованию коричневой суспензии. В результате фильтрации данной суспензии получали коричневое твердое вещество, подтверждение структуры которого в присутствии 1Н и 13С дает NMR. Температура плавления: 241°С.
Пример 6
Получение 6-(2-гидроксиэтил)-4-фенилхроман-2-ола
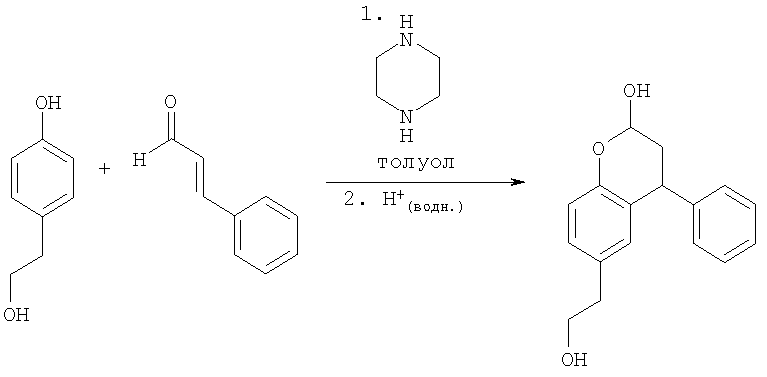
К раствору 4-гидроксифенетилового спирта (Tyrosol) (5,0 г, 36 ммоль, 1 экв.), пиперазина (1,87 г, 22 ммоль, 0,6 экв.) и толуола (50 мл) при кипении с обратным холодильником в атмосфере N2 и в условиях Дина-Старка добавляли циннамальдегид (6,4 мл, 51 ммоль, 1,4 экв.), и реакционную смесь поддерживали при нагревании в течение 17 ч. Реакционную смесь охлаждали до 80°С и гасили водной HCl (0,7 молярная, 1,3 экв.), затем перемешивали при нагревании в течение 18 ч. Двухфазную смесь оставляли охлаждаться до температуры окружающей среды, разделяли, органическую фазу промывали водной HCl, водную и органическую фазу концентрировали до черного остатка при пониженном давлении. В результате флэш-хроматографии, элюируя смесью 20% EtOAc/гептан, получали указанное в заголовке соединение в виде основного компонента приблизительно 80% чистого желтого масла, Rf=0,37 (50% EtOAc/гептан), и структуру подтверждали 1Н NMR и LC-MS (М+1=271).
Пример 7
Альтернативное получение 6-(2-гидроксиэтил)-4-фенилхроман-2-ола с использованием N-метилпиперазина
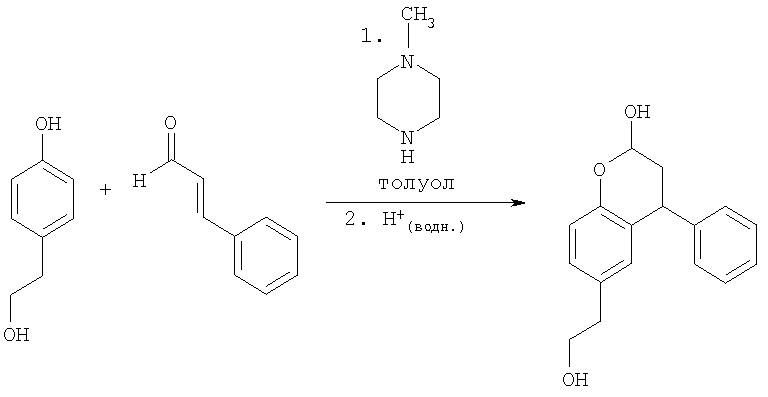
К раствору 4-гидроксифенетилового спирта (Tyrosol) (25,0 г, 181 ммоль, 1 экв.), N-метилпиперазина (54,4 г, 543 ммоль, 3 экв.) и толуола (200 мл) при кипении с обратным холодильником в атмосфере N2 и условиях Дина-Старка добавляли циннамальдегид (35,9 г мл, 272 ммоль, 1,5 экв.) в течение 2 часов, и реакционную смесь поддерживали при кипении с обратным холодильником в течение 17 ч. Реакционную смесь охлаждали до 50°С и гасили водной HCl (2 М, 375 мл, ~4 экв.). Двухфазную смесь оставляли охлаждаться до температуры окружающей среды, разбавляли этилацетатом (250 мл), и органическую фазу отделяли. Органическую фазу промывали водной HCl (250 мл), бикарбонатом калия (1 М, 250 мл), сушили над сульфатом магния и упаривали при пониженном давлении с получением черного масла (50,0 г, предположительно количественно).
Пример 8
Получение 2-[3-(диизопропиламино)-1-фенилпропил]-4-(2-гидроксиэтил)-фенола гидрохлорида
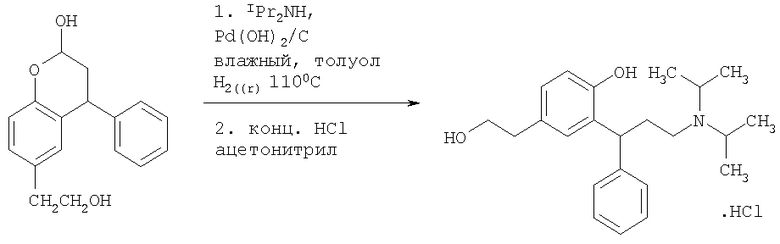
Смесь 6-(2-гидроксиэтил)-4-фенилхроман-2-ола (Пример 7, 30 г, 111 ммоль, 1 экв.), диизопропиламина (33,7 г, 333 ммоль, 3 экв.) и гидроксида палладия на угле [50% влажный катализатор (50% от массы составляет вода), 6 г, 0,2 экв.] в толуоле (120 мл) гидрировали при давлении водорода 621×103 Нм-2 (90 фунт/кв.дюйм) при 110°С. Реакционную смесь охлаждали до комнатной температуры и фильтровали через Arbocel и упаривали при пониженном давлении. Полученное масло растворяли в ацетонитриле (200 мл) и добавляли концентрированную соляную кислоту (11,6 мл, 1,05 экв.). Смесь перегоняли при атмосферном давлении, удаляя приблизительно 100 мл ацетонитрила, и отогнанный растворитель заменяли свежим ацетонитрилом. Смесь оставляли охлаждаться и кристаллизоваться в течение ночи. Продукт фильтровали и промывали небольшой порцией ацетонитрила и сушили в течение ночи in vacuo при 50°С с получением указанного в заголовке соединения в виде белого твердого вещества (26,7 г, 68,1 ммоль, 61%).
Пример 9
Получение 2-[3-(диизопропиламино)-1-фенилпропил]-4-(2-гидроксиэтил)-фенола
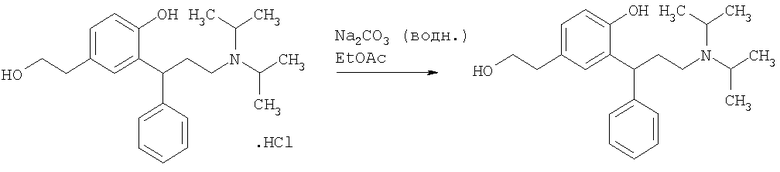
Водный натрия бикарбонат (165 мл) добавляли к смеси HCl соли (Пример 8, 16,5 г, 42,1 ммоль, 1 экв.) в этилацетате (165 мл), и смесь перемешивали в течение 1 часа. Фазы разделяли, и органическую фазу промывали водой (195 мл), сушили над сульфатом магния, упаривали при пониженном давлении с получением указанного в заголовке соединения в виде масла, содержащего ~25% мас./мас., этилацетата (14,6 г всего, 11,03 г указанного в заголовке соединения, 31 ммоль, 74%).
Пример 10
Получение соли (R)-2-[3-(диизопропиламин)-1-фенилпропил]-4-(2-гидроксиэтил)фенол (S)-2-феноксипропионовой кислоты
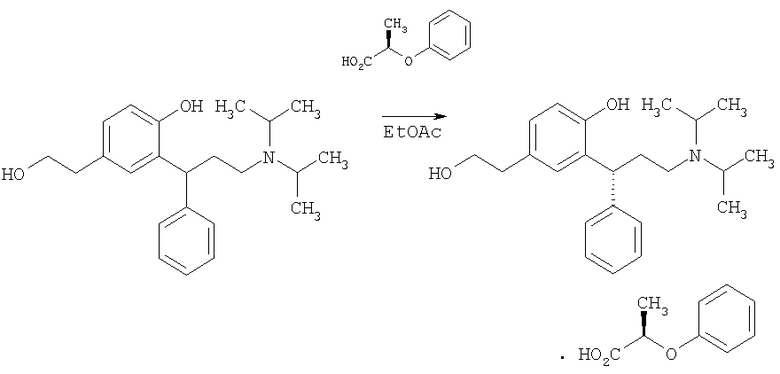
(S)-2-Феноксипропионовую кислоту (3,40 г, 20,5 ммоль, 1 экв.) добавляли к раствору 2-[3-(диизопропиламино)-1-фенилпропил]-4-(2-гидроксиэтил)фенола (Пример 9; 7,28 г, 20,5 ммоль, 1 экв.) в этилацетате. Смесь нагревали при 80°С в течение 2 суток, после чего смесь охлаждали до комнатной температуры, фильтровали и промывали этилацетатом и сушили in vacuo при 50°С в течение ночи с получением указанного в заголовке соединения в виде белого твердого вещества (3,9 г, 7,48 ммоль, 37% выход, 94% э.и.).
Энантиомерный избыток определяли путем превращения соли в свободное основание при помощи гидроксида натрия и выполнения нормально-фазовой хиральной HPLC-хроматографии (Chiral Рак AS-H колонка, элюирование гексаном (89,8%), IPA (10%), DEA (0,1%), TFA (0,1%) при 1 мл/мин).
Указанное в заголовке соединение может быть полезным в качестве исходного вещества для получения Примера 5 в одновременно находящейся на рассмотрении международной патентной заявке PCT/IB07/000619. Соответствующая указанному в заголовке соединению солянокислая соль описана как в примере 12 в WO 98/43942.
Пример
Синтез 3,4-дигидро-6-бром-4-фенил-2Н-бензопиран-2-ола

4-Бромфенол (2,0 г, 11,6 ммоль) перемешивали с N-метилпиперазином (3,48 г, 34,8 ммоль, 3 экв.) в толуоле (30 мл, 15 мл/г) и нагревали при кипении с обратным холодильником в условиях Дина-Старка. При достижении начала дефлегмации добавляли транс-циннамальдегид (2,2 г, 17,4 ммоль, 1,5 экв.) в течение 2 часов. После того как добавление закончилось, продолжали нагревание реакционной смеси при кипении с обратным холодильником в условиях Дина-Старка в течение 3 часов. Темный раствор охлаждали до 25°С и разбавляли этилацетатом (20 мл, 10 мл/г) и гасили 2 М HCl (30 мл, 15 мл/г). Фазы разделяли, и верхний органический слой промывали дополнительно 2 М HCl (20 мл, 10 мл/г) и 1 М раствором гидрокарбоната натрия (20 мл, 10 мл/г). Органическую фазу сушили (MgSO4), фильтровали и концентрировали с получением темно-окрашенного масла (4,2 г, 11,6 ммоль, предположительно количественно).
Пример 12
Синтез соли 2-[3-(диизопропиламино)-1-фенилпропил]-4-бромфенола
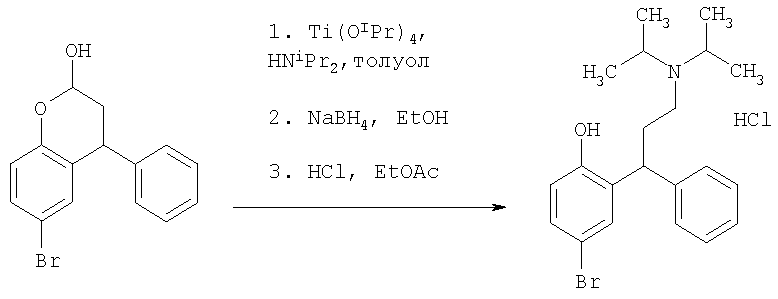
Неочищенный 3,4-гидро-6-бром-4-фенил-2Н-бензопиран-2-ол (Пример 11, 2,0 г, 6,55 ммоль) растворяли в толуоле (20 мл, 10 мл/г), и к данному раствору добавляли тетраизопропилат титана (5,84 мл, 3 экв.) и диизопропиламин (1,0 мл, 1,1 экв.). Реакционную смесь охлаждали до 0-5°С и добавляли боргидрид натрия (0,75 г, 3 экв.) по частям в течение 15 минут. Этанол вводили по каплям в течение 15 минут и перемешивали при 0-5°С в течение еще 2 часов. Реакционную смесь гасили водой (20 мл), этилацетатом (50 мл) и концентрированным раствором аммиака (20 мл). Суспензию фильтровали через Celite, и фазы разделяли. Органический слой промывали водой (50 мл), сушили (MgSO4), фильтровали и концентрировали с получением свободного основания в виде коричневого масла. Его растворяли в этилацетате (50 мл) и добавляли 5 М HCl (2 мл). Избыток кислоты и воды азеотропно перегоняли со свежим этилацетатом (2×50 мл), и полученное твердое вещество гранулировали в свежем этилацетате (20 мл) в течение 48 часов. Твердое вещество собирали посредством фильтрации, промывали этилацетатом (10 мл) и сушили при 50°С под вакуумом в течение 4 часов. Указанное в заголовке соединение получали в виде белого твердого вещества (1,12 г, 40% от 4-бромфенола).
Бензилокси-аналог указанного в заголовке соединения описан как в примере 1(e) в WO 94/11337. Указанное в заголовке соединение после разделения также может быть полезным в качестве исходного вещества в получении по Примеру 3 в одновременно находящейся на рассмотрении международной патентной заявке PCT/IB07/000619.
Пример 13
Синтез (2-гидрокси-4-фенил-3,4-дигидро-2Н-хромен-6-ил)метанола
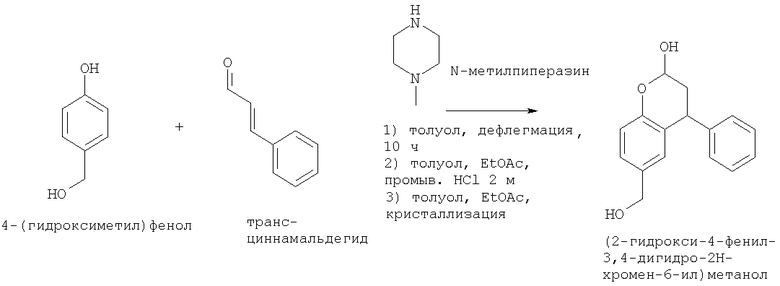
4-(гидроксиметил)фенол (2,515 кг, 20,26 мол, 1 экв.) перемешивали с N-метилпиперазином (5,06 кг, 50,52 мол, 2,5 экв.) в толуоле (17,74 кг, 20,5 л, 8,15 мл/г) и затем нагревали до кипения. Затем добавляли транс-циннамальдегид (3,35 кг, 25,35 мол, 1,25 экв.) в течение 2 часов, поддерживая реакционную смесь при кипении с обратным холодильником. Линию подачи промывали толуолом (0,9 кг, 0,35 мл/г). После окончания добавления реакционную смесь продолжали нагревать с обратным холодильником в течение 19 ч. Затем некоторое количество толуола отгоняли, уменьшая объем до приблизительно 18,5 л. Затем смесь оставляли охлаждаться до комнатной температуры и добавляли EtOAc (13,5 кг 15 л, 6 мл/г). Органическую фазу промывали 2 М HCl (46,4 кг, 46,4 л, 18,5 мл/г). Фазы разделяли, и добавляли этилацетат (27,1 кг, 30 л, 12 мл/г) для разбавления органического слоя. Органическую фазу промывали 1 М HCl (17,75 кг, 17,75 л 7,1 мл/г), 5% мас./мас. NaHCO3 (17,5 л, 7 мл/г) и водой (25 л, 10 мл/г). Фазы разделяли и добавляли к органическому слою толуола (6,5 кг, 7,5 л, 3 мл/г), и смесь перегоняли до приблизительно 8 л. Загружали дополнительно толуол (7 кг), затем этилацетат (3,9 л). Смесь нагревали до кипения, затем охлаждали до 22°С со скоростью 1°С/минуту, затем перемешивали в течение 20 часов. Суспензию охлаждали до 2°С и гранулировали в течение 2 ч. Суспензию фильтровали, и остаток промывали холодным толуолом (2×4,3 кг, 5 л). Полученное светло-коричневое твердое вещество сушили в вакууме в течение 68 часов при 40°С, с получением 2,76 кг продукта (2-гидрокси-4-фенил-3,4-дигидро-2H-хромен-6-ил)метанола (выход 53,4%), который использовали в следующем примере без очистки:
1Н NMR 300 МГц d6 DMSO δ м.д. (миллионный доли) (смесь изомеров, 10,1): 1.95-2.10 (m, 2Н), 2.15-2.35 (m, минорный изомер), 3.25-3.35 (m, 1Н), 4.15-4.35 (m, 3Н), 4.80-4.95 (m, 1Н), 5.35-5.45 (m, минорный изомер), 5.46-5.55 (m, 1Н), 6.51-6.54 (m, минорный изомер), 6.58-6.63 (m, 1Н), 6.75 (d J=8,2 Гц, 1Н), 6.98-7.40 (m, 6Н).
Пример 14
Синтез 2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенола

(2-Гидрокси-4-фенил-3,4-дигидро-2H-хромен-6-ил)метанол (Пример 13, 830 г, 3,24 моль, 1,0 экв.) перемешивали в метаноле (4150 мл, 5,0 мл/г). Затем с помощью капельной воронки в течение 15 минут добавляли диизопропиламин (1362 мл, 9,72 моль, 3,0 экв.). Затем полученный раствор перемешивали в течение одного часа в атмосфере азота.
Катализатор Pd-ESCAT 142 [(5% Pd/C паста, влажность приблизительно 50% воды) 83 г, 10% мас./мас.] добавляли в виде суспензии в метаноле (2075 мл, 2,5 мл/г). Систему продували водородом, затем смесь гидрировали при 115 фунт/кв.дюйм (793×103 Нм-2, 7,92 бар) при температуре 40°С в течение 20 часов.
Смесь продували азотом и фильтровали через Arbocel™ (вспомогательный фильтрующий материал). Фильтровальную подушку с остатком промывали метанолом (2×1660 мл, 2×2,0 мл/г).
Из-за ограничений со стороны оборудования, вышеуказанную процедуру выполняли два раза в масштабе 830 и 840 г.
Затем три фильтрата и их соответствующие промывные воды с фильтровальных подушек комбинировали с получением одного раствора, эквивалентного одной реакции в масштабе 2,50 кг. Конечный объем фиксировали как исходный целевой объем в следующей методике дистилляции:
- Диизопропиламин (2500 мл, 1,0 мл/г) и трет-амиловый спирт (10000 мл, 4,0 мл/г) добавляли к реакционной смеси. Затем выполняли вакуум-дистилляцию (установка вакуума 100 мбар (104 Па)), чтобы отдистиллировать с уменьшением объема до целевого, фиксированного ранее.
- Диизопропиламин (2500 мл, 1,0 мл/г) и трет-амиловый спирт (10000 мл, 4,0 мл/г) добавляли к реакционной смеси. Затем выполняли вакуум-дистилляцию (установка вакуума 100 мбар (104 Па)), чтобы отдистиллировать с уменьшением объема до целевого, фиксированного ранее.
- Диизопропиламин (2500 мл, 1,0 мл/г) и трет-амиловый спирт (10000 мл, 4,0 мл/г) добавляли к реакционной смеси. Затем выполняли вакуум-дистилляцию (установка вакуума 100 мбар (104 Па)), чтобы отдистиллировать с уменьшением объема до целевого, фиксированного ранее.
- Трет-амиловый спирт (12500 мл, 5,0 мл/г) добавляли к реакционной смеси. Затем выполняли вакуум-дистилляцию (установка вакуума 100 мбар), чтобы отдистиллировать с уменьшением объема до 12500 мл.
- Трет-амиловый спирт (12500 мл, 5,0 мл/г) добавили к реакционной смеси. Затем выполняли вакуум-дистилляцию (установка вакуума 100 мбар), чтобы отдистиллировать с уменьшением объема до 12500 мл. Трет-амиловый спирт (12500 мл, 5,0 мл/г) добавляли к реакционной смеси с получением конечного объема 25 л.
Раствор неочищенного продукта 2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенола в трет-амиловом спирте использовали на следующей стадии без дополнительной очистки. HPLC-анализ (площадь/площадь) показал 93,3% продукта, плюс: 4,2% исходного вещества и другие примеси в количестве 1,4% и 0,4%. Количественный HPLC-анализ показал неочищенный раствор, содержащий 2950 г продукта (89% выход).
Пример 15
Синтез (R)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенол(R)-ацетокси(фенил)ацетата
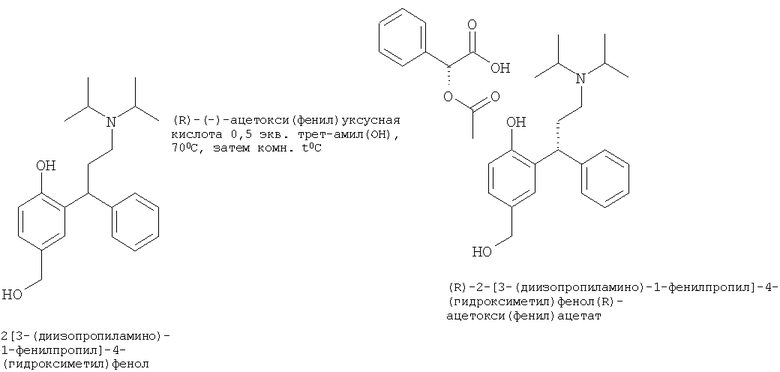
Трет-амиловый спирт (19,2 л) добавляли к предыдущему раствору 2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенола (из Примера 14) в трет-амиловом спирте (25 л, содержащий 2,95 кг, 8,64 моль, 1 экв.) с получением конечного объема 442 л. Этот раствор нагревали до 70°С. В отдельном резервуаре при 50°С получали раствор (R)-(-)-ацетокси(фенил)уксусной кислоты (0,839 кг, 4,32 мол, 0,5 экв.) в трет-амиловом спирте (14,8 л), затем, когда вся кислота растворилась, его охлаждали до комнатной температуры. Этот раствор затем добавляли к раствору 2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенола в трет-амиловом спирте в течение одного часа. В полученный раствор затем вводили затравку продукта (0,03 кг, 1% мас., предварительно полученную аналогичным методом, но в меньшем масштабе). Суспензию охлаждали до 60°С в течение 2 часов и затем до 25°С в течение еще 3 часов. Смесь перемешивали при 25°С в течение еще 12 часов. Суспензию фильтровали, и остаток дважды ресуспендировали с трет-амиловым спиртом (2×29,5 л, 2×10 мл/г) и тщательно отделяли от жидкости. Белое твердое вещество сушили при пониженном давлении при 40°С в течение 12 часов с получением 2,04 кг (R)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенола (R)-ацетокси(фенил)ацетата (выход 37,8%, скорректированный с учетом 14,3% мас./мас. трет-амилового спирта (определенного посредством LOD анализа) с 99% э.и. при помощи хиральной HPLC.
HPLC-способ мониторинга э.и:
Колонка: Chiralpak AS-H
Скорость потока: 1 мл/мин
Подвижная фаза: гептан 92,5/этанол 7,5/диэтиламин 0,12/трифторуксусная кислота 0,18
Температура: 35°С
Разрешение: 220 нм
Время удерживания:
(R)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенол 15 мин
(S)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенол 18,4 мин
Пример 16
Синтез (R)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенола
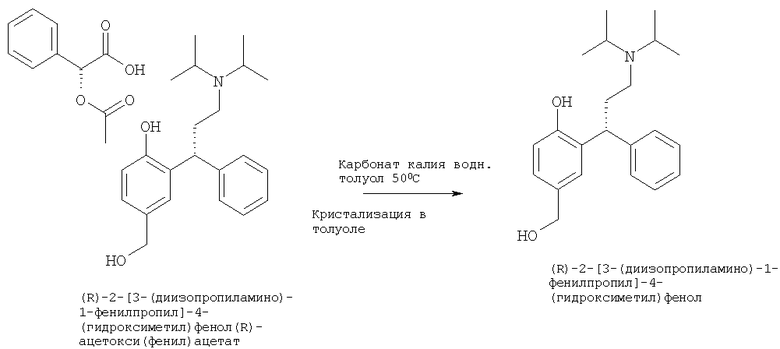
(R)-2-[3-(диизопропиламино)1-фенилпропил]-4-(гидроксиметил)фенола (R)-ацетокси(фенил)ацетат (Пример 15, 1,75 кг, 3,27 мол, 1 экв.) суспендировали в толуоле (15,2 кг, 10 мл/г) и нагревали до 50°С. Загружали 10% водный раствор К2СО3 (1,75 кг K2CO3 растворяли в 17,5 л очищенной воды, 10 мл/г). Смесь интенсивно перемешивали при 50°С в течение 30 минут. Две фазы раствора разделяли при 50°С. Органическую фазу промывали очищенной водой (1,75 кг, 1 мл/г) при 50°С. Фазы разделяли при 50°С, и объем толуола уменьшали до 3 мл/г (5,5 л) посредством дистилляции. Кристаллизацию проводили путем понижения температуры до 62°С и затем охлаждения до 40°С в течение 40 минут. Партию выдерживали при 40°С в течение 30 минут и затем вводили затравку, используя (R)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)фенол (0,01 кг, полученный предварительно с использованием аналогичного метода в меньшем масштабе). Партию перемешивали в течение еще 1 часа при 40°С и затем охлаждали до 20°С в течение 3,5 часов. Партию гранулировали при 20°С в течение 10 часов. Затем суспензию охлаждали до 2°С в течение 1 часа и гранулировали при 2°С в течение 1 часа (смотри ниже температурный профиль). Суспензию фильтровали, и остаток промывали холодным толуолом (1,5 кг, 1 мл/г). Влажный продукт (0,933 кг, % высушивания определяли анализом LOD) представлял собой белое кристаллическое твердое вещество.
Затем выполняли ресуспендирование толуола. Толуол (2,42 кг, 2,6 мл/г (на основе оценки высушивания)) охлаждали до 3°С, и влажный продукт загружали и перемешивали при 3°С в течение 15 минут. Суспензию фильтровали, и остаток промывали холодным толуолом (1,6 кг, 1,5 мл/г (на основе оценки высушивания)). Влажный продукт сушили in vacuo при 45°С с выходом (R)-2-[3-(диизопропиламино)-1-фенилпропил]-4-(гидроксиметил)-фенола (0,74 кг, 2,17 мол) с выходом 66,7% в виде белого кристаллического вещества. HPLC показывает чистоту >99,6%, и хиральная HPLC показывает >99% э.и.
Пример 17
Получение (R)-(+)-изомасляной кислоты 2-[3-(диизопропиламино)-1-фенилпропил]-(гидроксиметил) фенилового эфира
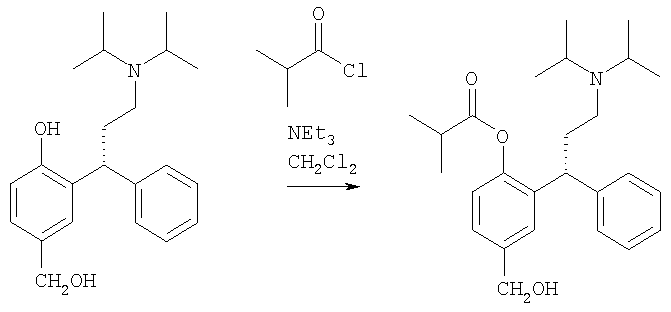
Указанное в заголовке соединение получают из соединения Примера 16, используя способ из патента US 6858650 (смотри раздел 5, строка 16). В качестве альтернативы такое взаимодействие может быть выполнено без добавления извне нейтрализующего кислоту основания - смотри патент US 6858650, колонка 10, строки 32-40.
Пример 18
Получение (R)-(+)-изомасляной кислоты 2-[3-(диизопропиламино)-фенилпропил]-4-(гидроксиметил)Фенилового эфира гидрофумарата
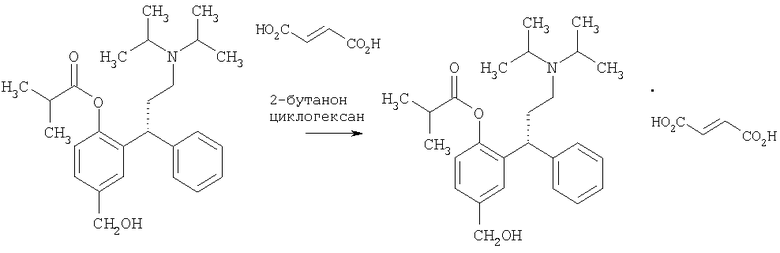
Указанное в заголовке соединение получают из соединения Примера 17, используя методику патента US 6858650 (смотри раздел 6, колонка 16).
Пример 19
Синтез [2-(4-метилпиперазин-1-ил)-4-фенил-2Н-хроман-6-ил]-метанола
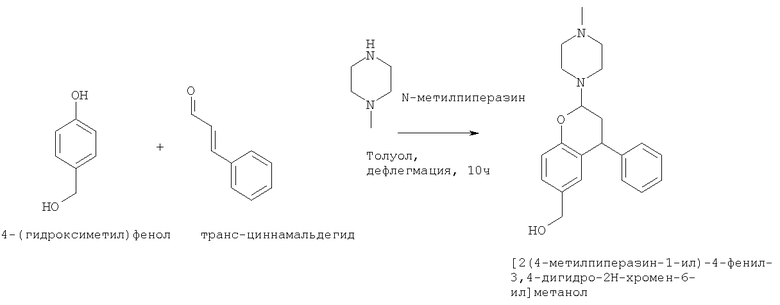
Транс-циннамальдегид (66,5 г, 0,66 мол, 1,25 экв.) разбавляли толуолом (100 мл, 2 мл/г на основе 4-(гидроксиметил)фенола), и дважды промывали насыщенным раствором натрия гидрокарбоната (2×100 мл) и один раз водой (100 мл). Такой толуольный раствор циннамальдегида затем добавляли в течение 2 часов к смеси 4-(гидроксиметил)фенола (50 г, 0,40 мол, 1 экв.) и N-метилпиперазина (113 мл, 1,0 мол, 2,5 экв.) в толуоле (350 мл, 7 мл/г), нагревали до кипения с обратным холодильником в условиях Дина-Старка. Как только добавление заканчивалось, реакционную смесь продолжали нагревать до кипения с обратным холодильником в условиях Дина-Старка в течение 10 ч. Затем смесь охлаждали до комнатной температуры, и образец упаривали досуха для аналитических целей при пониженном давлении. Темное масло содержит неочищенный [2-(4-метилпиперазин-1-ил)-4-фенил-2Н-хроман-6-ил]метанол (смесь диастереоизомеров) и примеси.
EI-GC-MS (Agilent), колонка: ZB-5HT, температурная программа: 50°С (0,5 мин), 20°С /мин до 320°С (минут), получили: RT=24,4 мин, MW: 338.
1Н NMR (DMSO) (неочищенная смесь) 300 мГц δ (м.д.): 7.40-7.00 (28Н, m); 6.89 (1Н, d, J=2,0 Гц); 6.81 (1Н, d, J=8,3 Гц); 6.75 (1Н, d, J=8,2 Гц); 6.54 (1Н, d, J=1,0 Гц); 4.87 (1Н, d, J=10,2 Гц); 4.45 (1Н, d, J=10,0 Гц); 4.40-4.20 (3Н, m); 4.36 (2Н, s); 4.23 (1,8Н, s); 2.86 (4Н, m); 2.80-2.50 (14Н, m); 2.50-2.00 (50Н, m), включая 2.31 (s); 2.17 (s); 2.14 (s); 2.1.1 (s).
Синтез фезотеродина гидрофумарата согласно способам из Примеров 13-18 представлен на Схеме 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 5-ГИДРОКСИМЕТИЛ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВ, СПОСОБ ПОЛУЧЕНИЯ 5-АМИНОМЕТИЛ ЗАМЕЩЕННЫХ ОКСАЗОЛИДИНОНОВЫХ АМИНОВ, ОКСАЗОЛИДИНОНСУЛЬФОНАТ | 1997 |
|
RU2176643C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3,3-ДИАРИЛПРОПИЛАМИНОВ (ВАРИАНТЫ) И СОЕДИНЕНИЯ (ВАРИАНТЫ) | 2001 |
|
RU2270188C2 |
| ЗАМЕЩЕННЫЕ АМИНОМАСЛЯНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ НЕПРИЛИЗИНА | 2012 |
|
RU2604522C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТОЛТЕРОДИНА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1997 |
|
RU2176246C2 |
| ВЫСОКОЧИСТЫЕ СЛОЖНЫЕ 3,3-ДИФЕНИЛПРОПИЛАМИНОМОНОЭФИРЫ | 2004 |
|
RU2394019C2 |
| КОМБИНАЦИЯ ИНГИБИТОРА ВИЧ-ПРОТЕАЗЫ С ДРУГИМИ АНТИВИРУСНЫМИ СОЕДИНЕНИЯМИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1994 |
|
RU2139052C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛПЕНТАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ВИЧ-ПРОТЕАЗЫ | 1992 |
|
RU2131416C1 |
| ПРОИЗВОДСТВО СОЕДИНЕНИЙ И КОМПОЗИЦИЙ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2019 |
|
RU2797951C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ИНДОЛОПИРРОЛОКАРБАЗОЛА | 2003 |
|
RU2337105C2 |
| ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2261248C2 |
В данном изобретении предлагается способ получения соединения формулы (I), где Y выбран из СН3, СН2ОН, СН2СН2ОН, CH2Br и Br; включающий стадии: (1) взаимодействия соединения формулы (II), где ОХ представляет собой гидрокси или O-M+, где М+ представляет собой катион, выбранный из Li+, Na+ и К+, и Y такой, как определено выше; с транс-циннамальдегидом (III), в присутствии вторичного аминного соединения; затем (2) обработки продукта с предыдущей стадии кислотой с получением соединения формулы (I). Вышеуказанный способ можно использовать для получения толтеродина и фезотеродина, которые полезны в лечении гиперактивного мочевого пузыря. Заявлены также соединения формулы V, VI и VII. 6 н. и 19 з.п. ф-лы, 1 табл.
1. Способ получения соединения формулы (I)
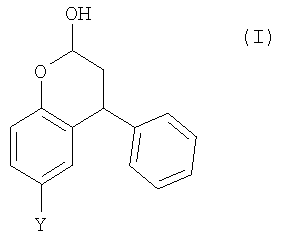
где Y выбран из СН3, СН2ОН, СН2СН2ОН, CH2Br и Br, включающий стадии:
(1) взаимодействия соединения формулы (II)
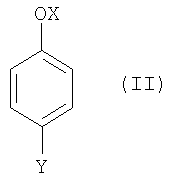
где ОХ представляет собой гидрокси или O-M+, где M+ представляет собой катион, выбранный из Li+, Na+ и K+, и
Y такой, как определено выше,
с трансциннамальдегидом (III)
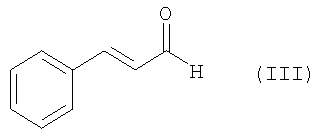 ;
;
в присутствии вторичного аминного соединения; затем
(2) обработки продукта с предыдущей стадии кислотой с получением соединения формулы (I).
2. Способ по п.1, где ОХ представляет собой гидрокси.
3. Способ по п.1, где Y представляет собой СН3 или СН2ОН.
4. Способ по п.1, где вторичное аминное соединение является ахиральным.
5. Способ по п.1, где вторичное аминное соединение содержит две вторичные аминные группы.
6. Способ по п.1, где вторичное аминное соединение представляет собой пиперазин.
7. Способ по п.5 или 6, где на стадии (1) используют 0,5-1,25 моль-экв. вторичного аминного соединения.
8. Способ по п.1, где вторичное аминное соединение содержит одну вторичную аминогруппу.
9. Способ по любому из пп.1-4 и 8, где вторичное аминное соединение представляет собой морфолин, дибутиламин, дибензиламин, 1,1,3,3-тетраметилгуанидин, диэтиламин, диизопропиламин, пиперидин или N-(C1-6алкил)пиперазин.
10. Способ по п.9, где вторичное аминное соединение представляет собой N-метилпиперазин.
11. Способ по п.8, где на стадии (1) используют 1-5 моль-экв. вторичного аминного соединения.
12. Способ по п.1, где кислота, используемая на стадии (2), представляет собой водную соляную кислоту.
13. Способ по п.1, где взаимодействие на стадии (1) осуществляют в органическом растворителе, выбранном из толуола, ксилола, N-бутилацетата, трет-амилового спирта, диоксана и дибутилового эфира.
14. Способ по п.13, где растворитель представляет собой толуол.
15. Способ по п.1, где взаимодействие на стадии (1) осуществляют при температуре от 80°С до температуры дефлегмации растворителя.
16. Способ по п.1, где взаимодействие на стадии (1) осуществляют при условиях, при которых из реакционной системы удаляется вода.
17. Способ по п.1, где взаимодействие на стадии (1) осуществляют при давлении, равном или приблизительно равном давлению окружающей среды.
18. Способ получения соединения формулы (IV)
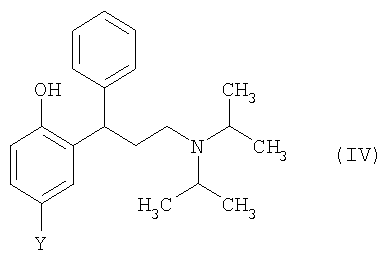
где Y такой, как определено в п.1, или его соли, включающий:
(а) получение соединения формулы (I) по п.1 с использованием способа по любому из пп.1-17; затем
(б) восстановительное аминирование соединения формулы (I) диизопропиламином
(в) и, при необходимости, превращение полученного соединения в соль.
19. Способ по п.18, где Y представляет собой СН3 или СН2ОН.
20. Способ по п.19, где Y представляет собой СН3, соединение формулы (IV) обрабатывают L-винной кислотой на стадии (в) и получают толтеродина L-тартрат.
21. Способ получения фезотеродина,
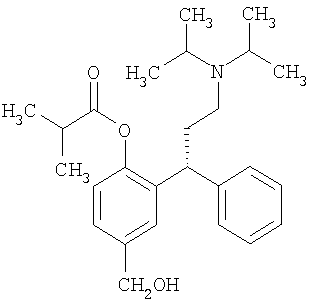
или его фармацевтически приемлемой соли, который включает:
(а) получение соединения формулы (IV), как определено в п.18, где Y представляет собой СН2ОН, с использованием способа по п.18;
(б) разделение продукта со стадии (а) с получением (R)-энантиомера;
(в) ацилирование фенольной гидроксигруппы продукта со стадии (б) с получением соответствующего сложного эфира изомасляной кислоты
(г) и, при желании или необходимости, превращение полученного соединения в фармацевтически приемлемую соль.
22. Способ по п.21, где вторичное аминное соединение, используемое для получения соединения формулы I, представляет собой N-метилпиперазин.
23. Соединение формулы (V)
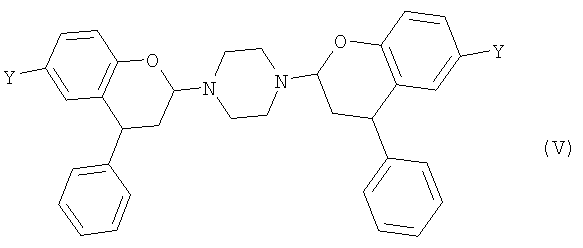
или его соль, где Y такой, как определено в п.1.
24. Соединение формулы (VI)
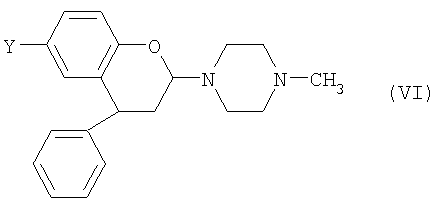
или его соль, где Y такой, как определено в п.1.
25. Соединение формулы (VII)
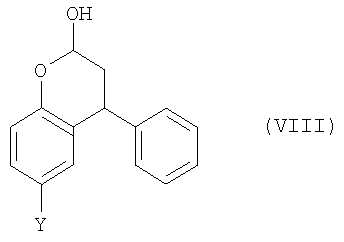
где Y выбран из CH2CH2OH, CH2Br и Br.
| Jurd L., Heterocyclic J., 1991, v.28, p.983-986 | |||
| WO 9829402 A1, 09.07.1998 | |||
| EP 1254890 A1, 06.11.2002 | |||
| US 6858650 В1, 22.02.2005 | |||
| US 20040209916 A1, 21.10.2004 | |||
| СПОСОБ ПОЛУЧЕНИЯ ТОЛТЕРОДИНА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1997 |
|
RU2176246C2 |
| 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1996 |
|
RU2178409C2 |

