Настоящее изобретение применимо в медицине. Более конкретно, настоящее изобретение относится к эффективному способу получения в промышленном масштабе соединения, применимого в медицине.
Полученное в соответствии со способом по настоящему изобретению производное индолопирролокарбазола, представленное формулой (I):
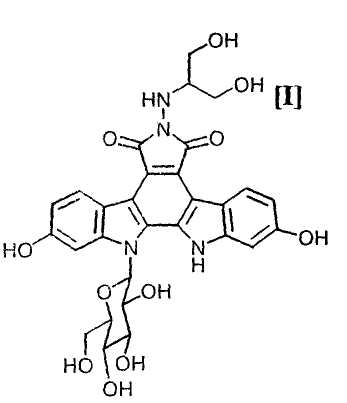
обладает противоопухолевой активностью, и соединение было клинически протестировано (Mitsuru Ohkubo et al., Bioorganic & Medicinal Chemistry Letters, vol. 9, pages 3307-3312, 1999).
Способы получения соединения по настоящему изобретению были раскрыты в WO 95/30682 и WO 01/62769.
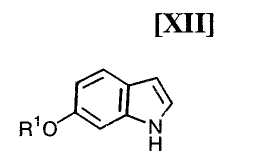
Кроме того, в Organic Synthesis Collective Volumes, vol. 7, page 34 раскрыт способ получения производного индолопирролокарбазола, представленного формулой (XII):
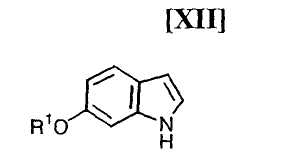
в которой R1 представляет собой гидроксизащитную группу.
В дополнение, известна (патент США № 5105012) реакция гидрирования с применением соединения родия, в которой для восстановления нитрогруппы производного нитробензола в кислотном растворителе, таком как уксусная кислота, в качестве катализатора применяется большое количество железного порошка.
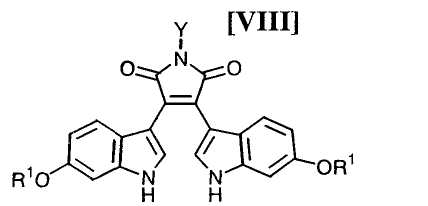
Кроме того, в WO 95-30682 раскрыт способ получения бис-индольного соединения, представленного формулой (VIII):
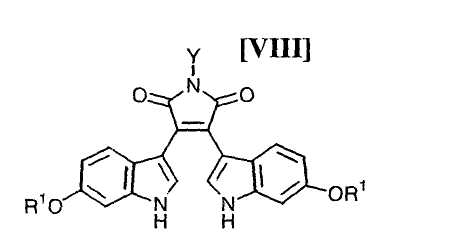
в которой R1 представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную или аралкильную группу.
Настоящее изобретение относится к устранению нежелательных аспектов в общепринятых способах получения представленного формулой (I) производного индолопирролокарбазола, применимого в качестве лекарственного средства. Другими словами, настоящее изобретение относится к способу получения, в который не включена стадия с низким выходом продукта и в соответствии с которым не применяется реагент, производство которого связано с высоким риском и который вызывает серьезное загрязнение окружающей среды.
В известном способе получения индольного соединения (Organic synthesis Collective volumes, vol. 7, page 34) стадию восстановления осуществляют с применением гидразина в присутствии катализатора никеля Ренея. Однако этот способ нежелателен для промышленного производства, так как гидразин обладает высокой взрывоопасностью. Кроме того, в связи с тем, что необходимо большое количество катализатора никеля Ренея, наблюдается серьезное загрязнение окружающей среды полученными в процессе производства жидкими отходами, что указывает на нежелательность применения этого способа в крупномасштабном промышленном производстве.
Кроме того, выход в известных способах получения бис-индольного соединения является невысоким, а потому данные процессы являются экономически неэффективными.
С другой стороны, известна (патент США № 5105012) реакция гидрирования с применением соединения родия, в которой для восстановления нитрогруппы производного нитробензола в кислотной среде, такой как уксусная кислота, в качестве катализатора применяется большое количество железного порошка. В этом случае, в связи с тем, что реакцию гидрирования осуществляют в кислотных условиях, этот способ неприменим для нестабильных в кислотных условиях веществ.
Авторы настоящего изобретения провели тщательное изучение способа получения производного индолопирролокарбазола формулы (I) и обнаружили следующее (i-v):
(i) новый способ получения производного индолопирролокарбазола формулы (I), при котором не наблюдается серьезного загрязнения окружающей среды полученными в процессе производства жидкими отходами, который является исключительно экономически выгодным и производство которого может осуществляться безопасно и высокой воспроизводимостью в качестве промышленного способа производства;
(ii) безопасный новый способ получения производного индола формулы (XII);
(iii) новый и экономически более совершенный способ получения производного бис-индола формулы (VIII);
(iv) новый катализатор гидрирования, который безопасен, не вызывает серьезного загрязнения окружающей среды обработанными жидкими отходами и может применяться не только в кислотных, но и в других условиях, и
(v) способ получения соединения (VII), в котором регулирование стадий является легким, а образование цианистого водорода в качестве побочного продукта в реакции замыкания цикла с применением 1,2-дихлор-5,6-дициано-1,4-бензохинона может быть предотвращено.
Авторы настоящего изобретения провели дальнейшие исследования и в результате создали настоящее изобретение.
А именно настоящее изобретение относится к новому способу получения производного индолопирролокарбазола формулы (I), к новому способу получения производного индола, к новому способу получения производного бис-индола и к новому катализатору гидрирования, который включает следующие (1)-(24).
(1) Способ получения производного индолопирролокарбазола представленного формулой (I), который включает следующие стадии:
(i): стадию взаимодействия соединения формулы (XIII)
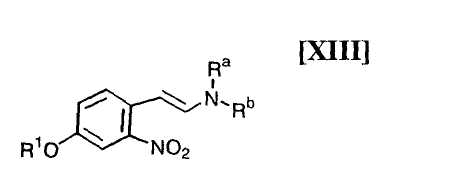
в которой R1 представляет собой гидроксизащитную группу, а каждый Ra и Rb независимо представляет собой C1-C7-алкильную группу, или Ra и Rb могут быть объединены вместе с образованием C3-C6-алкиленовой группы, или его соли, с газообразным водородом в присутствии соединения родия и соединения металлас получением индольного соединения формулы (XII):
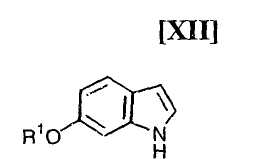
в котором значения R1 определены выше в этом документе, или его соли;
(ii): стадию осуществления взаимодействия полученного индольного соединения формулы (XII) или его соли с магнийхлоридом формулы (XI):
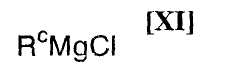
в которой Rc представляет собой C1-C7-алкильную группу, фенильную группу, винильную группу или аллильную группу; или с соединением магния формулы (X):
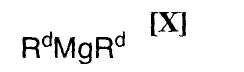
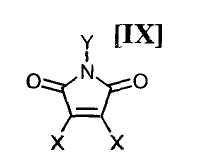
в которой Rd представляет собой C1-C7-алкильную группу или фенильную группу, или с его солью, или со смесью магнийхлорида (XI) и соединения магния (X), с последующим осуществлением взаимодействия полученного продукта с малеимидным соединением формулы (IX):
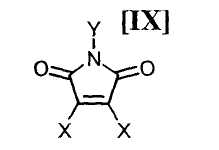
в которой X представляет собой атом галогена, и Y представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную группу или C7-C12-аралкильную группу, с получением бис-индольного соединения формулы (VIII):
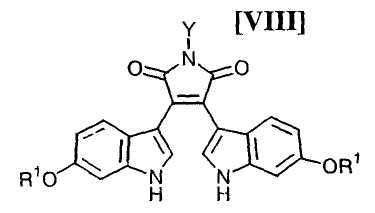
в которой значения для каждого R1 и Y определены выше, или его соли;
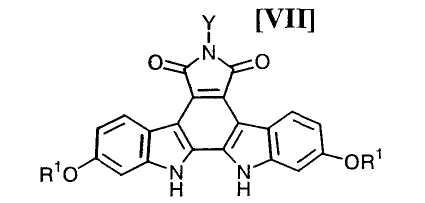
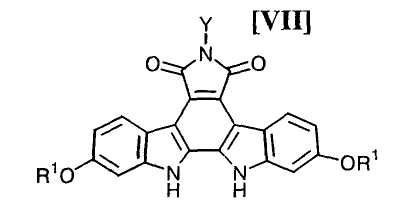
(iii): стадию осуществления реакции замыкания цепи в полученном бис-индольном соединении (VIII) или его соли с получением соединения формулы (VII):
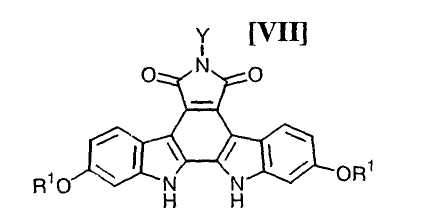
в которой значения для каждого R1 и Y определены выше, или его соли;
(iv): стадию сочетания полученного соединения (VII) или его соли с активированным производным глюкозы формулы (VI):
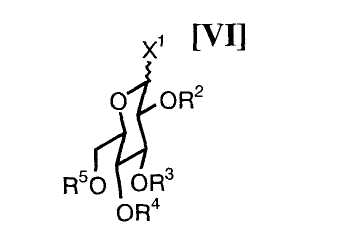
в которой каждый R2, R3, R4 и R5 представляет собой гидроксизащитную группу, и X1 представляет собой атом галогена, с получением соединения формулы (V):
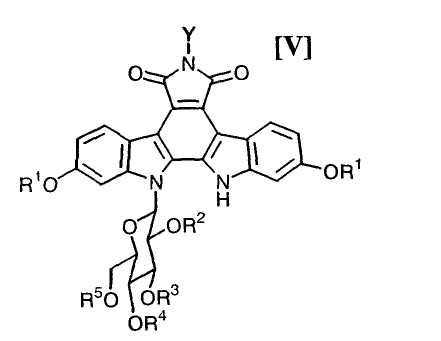
в которой значения для каждого R1, R2, R3, R4, R5 и Y определены выше, или его соли;
(v): стадию обработки полученного соединения (V) или его соли основанием с получением соединения формулы (IV):
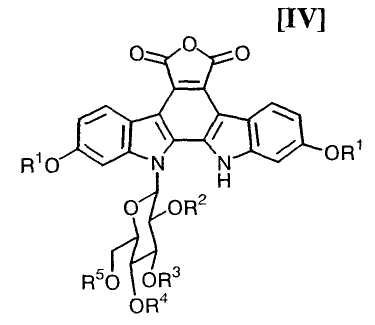
в которой значения для каждого R1, R2, R3, R4 и R5 определены выше, или его соли;
(vi): стадию взаимодействия соединения (IV) или его соли с соединением формулы (III):
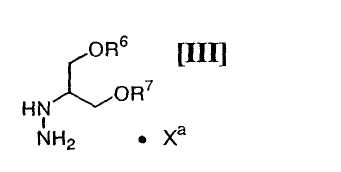
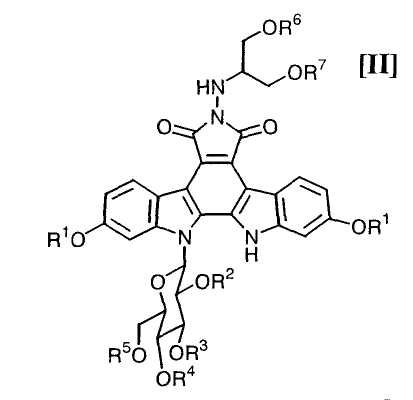
в которой каждый R6 и R7 представляет собой гидроксизащитную группу, и Xa представляет собой молекулу кислоты, с получением соединения формулы (II):
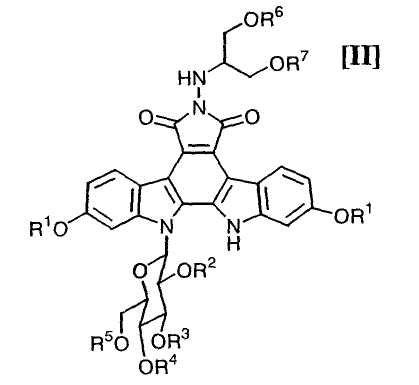
в которой значения для каждого R1, R2, R3, R4, R5, R6 и R7 определены выше, или его соли; и
(vii): стадию снятия защитных групп с полученного соединения (II) или его соли с получением производного индолопирролокарбазола формулы (I):
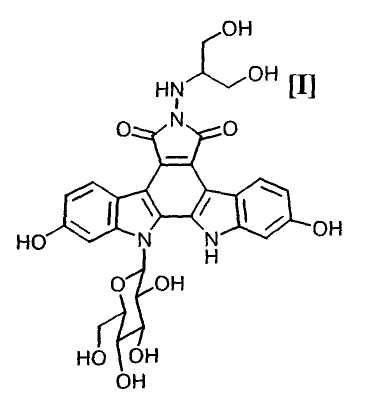
или его соли;
(2) способ в соответствии с приведенным выше пунктом (1), в котором соединение родия представляет собой родий/углерод, родий/оксид алюминия, родий/карбонат кальция или родий/сульфат бария;
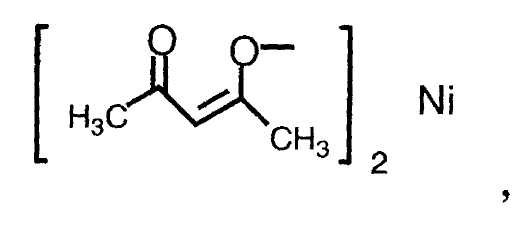
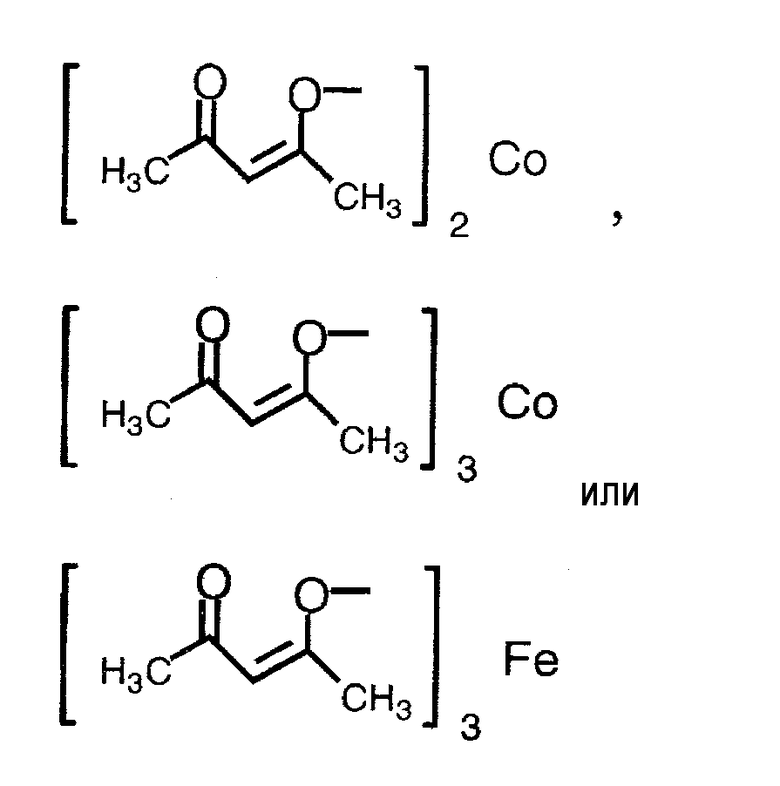
(3) способ в соответствии с приведенным выше пунктом (1), в котором соединение металлапредставляет собой соединение никеля(II), соединение железа(II), соединение железа(III), соединение кобальта(II) или соединение кобальта(III);
(4) способ в соответствии с приведенным выше пунктом (3), в котором соединение никеля(II), соединение железа(II), соединение железа(III), соединение кобальта(II) или соединение кобальта(III) представляют собой NiBr2, Ni(NO3)2, Ni(OCOCH3)2, FeBr3, FeCl2, FeSO4, FeCl3,FeCl3-SiO2, Fe(OCOCH3)2, фумарат Fe(II), CoBr2, CoCl2,
(5) способ в соответствии с приведенным выше пунктом (1), в котором каждый R1, R2, R3, R4, R5, R6 и R7 представляет собой бензильную группу;
(6) способ в соответствии с приведенным выше пунктом (1), в котором магнийхлорид формулы (XI) представляет собой этилмагнийхлорид, изопропилмагнийхлорид или н-бутилмагнийхлорид;
(7) способ в соответствии с приведенным выше пунктом (1), в котором соединение магния формулы (X) представляет собой ди(н-бутил)магний, ди(втор-бутил)магний, (н-бутил)(втор-бутил)магний, диметилмагний или диэтилмагний;
(8) способ в соответствии с приведенным выше пунктом (1), в котором малеимидное соединение формулы (IX) представляет собой малеимидное соединение, представленное формулой (IX-a):
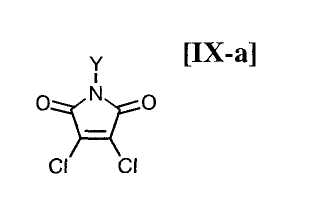
в которой Y представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную группу или аралкильную группу;
(9) способ в соответствии с приведенным выше пунктом (1), в котором Y представляет собой метильную группу;
(10) способ в соответствии с приведенным выше пунктом (1), в котором Xa представляет собой щавелевую кислоту;
(11) способ в соответствии с приведенным выше пунктом (1), в котором сочетание осуществляют в присутствии катализатора межфазного переноса, такого как Aliquat 336;
(12) способ получения индольного соединения или его соли, который включает получение индольного соединения, представленного формулой (XII):
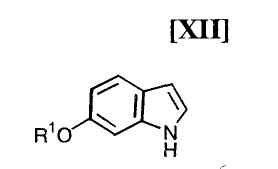
в которой R1 представляет собой гидроксизащитную группу, или его соли путем осуществления взаимодействия соединения, представленного формулой (XIII):
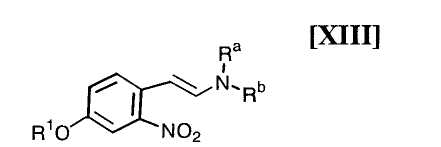
в которой значение R1 определено выше, и каждый Ra и Rb независимо представляет собой C1-C7-алкильную группу, или Ra и Rb могут быть объединены вместе с образованием C3-C6-алкиленовой группы, с газообразным водородом в присутствии соединения родия и соединения металла;
(13) способ в соответствии с приведенным выше пунктом (12), который включает осуществление взаимодействия соединения, представленного формулой (XIII):
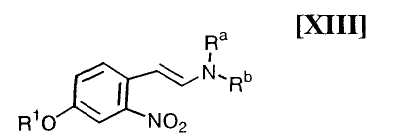
в которой R1 представляет собой гидроксизащитную группу, и каждый Ra и Rb независимо представляет собой C1-C7-алкильную группу, или Ra и Rb могут быть объединены вместе с образованием C3-C6-алкиленовой группы, или его соли, с газообразным водородом в присутствии соединения родия и соединения металла, и обработку полученного неочищенного продукта силикагелем;
(14) способ получения бис-индольного соединения или его соли, который включает осуществление взаимодействия индольного соединения формулы (XII):
в которой R1 представляет собой гидроксизащитную группу, или его соли с магнийхлоридом формулы (XI):
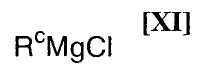
в которой Rc представляет собой C1-C7-алкильную группу, фенильную группу, винильную группу или аллильную группу; или с соединением магния формулы (X):
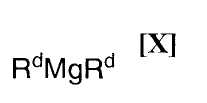
в которой Rd представляет собой C1-C7-алкильную группу или фенильную группу, или с его солью, или со смесью магнийхлорида формулы (XI) и соединения магния формулы (X), в инертном растворителе, с последующим осуществлением взаимодействия полученного продукта с малеимидным соединением формулы (IX):
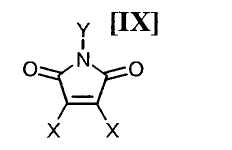
в которой X представляет собой атом галогена, и Y представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную группу или C7-C12-аралкильную группу, предпочтительно в инертном растворителе с получением бис-индольного соединения формулы (VIII):
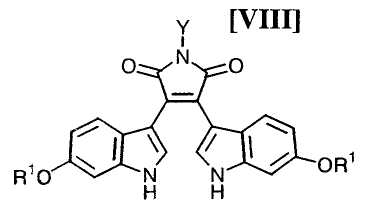
в которой значения для каждого R1 и Y определены выше, или его соли;
(15) способ в соответствии с приведенным выше пунктом (14), в котором малеимидное соединение формулы (IX) представляет собой малеимидное соединение, представленное формулой (IX-a):
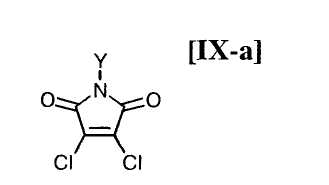
в которой Y представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную группу или C7-C12-аралкильную группу;
(16) способ получения соединения, представленного формулой (VII):
в которой R1 представляет собой гидроксизащитную группу, и Y представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную группу или C7-C12-аралкильную группу, или его соли, который включает обработку бис-индольного соединения, представленного формулой (VIII):
в которой значения для каждого R1 и Y определены выше, или его соли, с 2,3-дихлор-5,6-дициано-1,4-бензохиноном в неполярном растворителе в реакции замыкания цепи;
(17) способ в соответствии с приведенным выше пунктом (16), в котором неполярный растворитель представляет собой бензол, толуол, ксилол (орто-, мета- или пара-), этилбензол или 1,2,4-триметилбензол;
(18) применяемый в реакции гидрирования катализатор содержит соединение родия и соединение металла;
(19) катализатор в соответствии с приведенным выше пунктом (18) дополнительно содержит амин;
(20) катализатор в соответствии с приведенным выше пунктом (18) или (19), в котором соединение родия представляет собой родий/углерод, родий/оксид алюминия, родий/карбонат кальция или родий/сульфат бария;
(21) катализатор в соответствии с приведенным выше пунктом (18) или (19), в котором соединение металлапредставляет собой соединение никеля(II), соединение железа(II), соединение железа(III), соединение кобальта(II) или соединение кобальта(III);
(22) катализатор в соответствии с приведенным выше пунктом (19), в котором амин представляет собой вторичный амин или третичный амин;
(23) катализатор в соответствии с приведенным выше пунктом (19), в котором амин представляет собой пирролидин, пиперидин, диметиламин, диэтиламин, диизопропиламин, дибутиламин, триметиламин, триэтиламин или трибутиламин; и
(24) катализатор в соответствии с приведенным выше пунктом (21), в котором соединение никеля(II), соединение железа(II), соединение железа(III), соединение кобальта(II) или соединение кобальта(III) представляют собой NiBr2, Ni(NO3)2, Ni(OCOCH3)2, FeBr3, FeCl2, FeSO4, FeCl3,FeCl3-SiO2, Fe(OCOCH3)2, фумарат Fe(II), CoBr2, CoCl2,
 .
.
Способом в соответствии с изобретением стало возможным получать соединение, которое может безопасно, просто и эффективно применяться в медицине в качестве противоопухолевого средства.
НАИЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет проиллюстрировано ниже более подробно. Прежде всего, будут разъяснены термины, используемые в описании настоящего изобретения.
Примеры «C1-C7-алкильной группы» включают неразветвленную или разветвленную алкильную группу, такую как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил и гептил, среди которых предпочтительным является метил, этил, пропил, изопропил или бутил, и более предпочтительным является метил, этил, пропил, бутил или гептил.
Примеры «C3-C6-алкиленовой группы» включают неразветвленную алкиленовую группу, такую как триметилен, тетраметилен, пентаметилен и гексаметилен, среди которых предпочтительным является тетраметилен или пентаметилен.
Примеры «C7-C12-аралкильной группы» включают C7-C12-аралкильную группу, такую как бензил, 1-нафтилметил и 2-нафтилметил, среди которых предпочтительным является бензил.
Примеры «кислотной молекулы» включают протонную кислоту, такую как соляная кислота, серная кислота, азотная кислота, уксусная кислота, метилсульфоновая кислота, пара-толуолсульфоновая кислота, щавелевая кислота, пропионовая кислота, муравьиная кислота и бензойная кислота, среди которых предпочтительной является щавелевая кислота.
Примеры «гидроксизащитных групп» включают группы, защищающие гидроксигруппы, такие как бензильная, толильная, пара-нитробензильная, пара-метоксибензильная и бензилоксиметильная группы, среди которых предпочтительной является бензильная группа.
Термин «соединение родия» относится к содержащему атом родия катализатору, обычно к родиевому катализатору на носителе, и его предпочтительные примеры включают родий/углерод, родий/оксид алюминия, родий/карбонат кальция или родий/сульфат бария.
Примеры «атома галогена» включают хлор, йод и бром.
Термин «соединение металла» не включает соединение родия и относится к катализатору, который вместе с соединением родия способствует реакции восстановления, и его примеры включают соединение никеля(II), соединение железа(II), соединение железа(III), соединение кобальта(II) и соединение кобальта(III), предпочтительно NiBr2, Ni(NO3)2, Ni(OCOCH3)2, FeBr3, FeCl2, FeSO4, FeCl3, FeCl3-SiO2, Fe(OCOCH3)2, фумарат Fe(II), CoBr2, CoCl2,
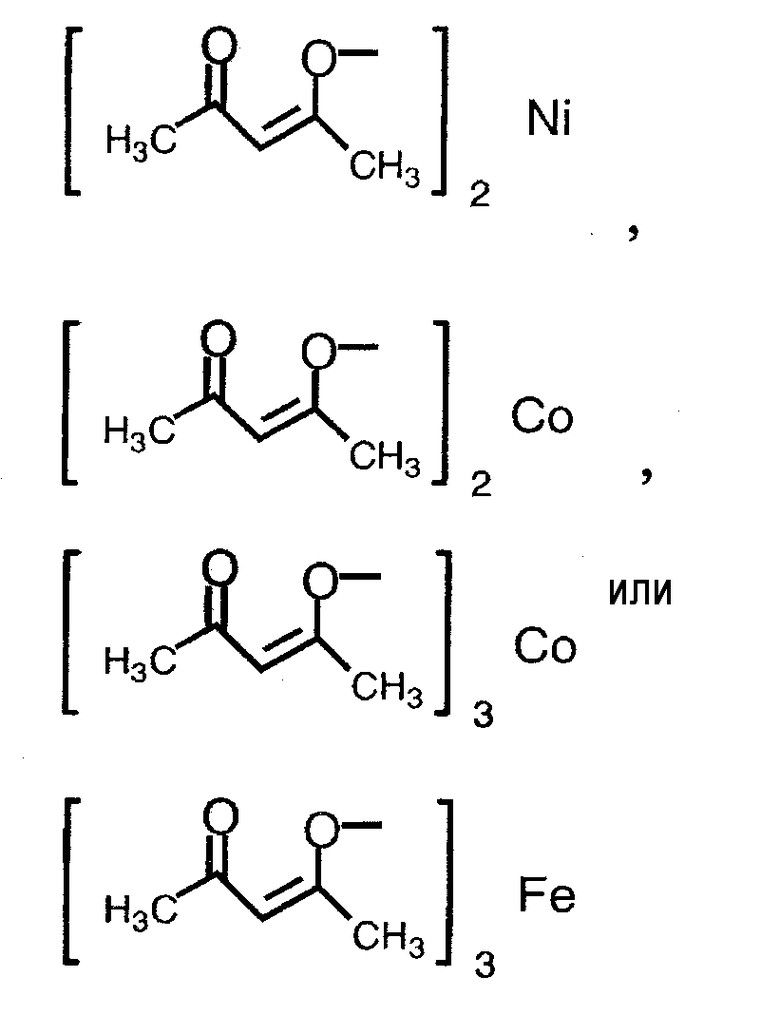 .
.
Термин «катализатор межфазного переноса» относится к катализатору, который способствует взаимодействию между гидрофобным (олеофильным) органическим соединением и гидрофильным органическим соединением в двухфазной системе, состоящей из жировой фазы и водной фазы, и его примеры включают соединение формулы (XIV):
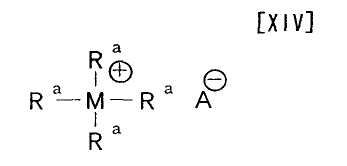
в которой каждый Ra независимо представляет собой водород, бензил или C1-C18-углеводород; M представляет собой атом азота или атом фосфора; и A представляет собой гидроксигруппу, атом фтора, атом брома, атом хлора, атом йода, цианогруппу, HSO4, CH3SO3 или PhCH2COO, и трис(2-(2-метоксиэтокси)этил)амин, и его предпочтительные примеры включают трикаприлметиламмонийхлорид, трис(2-(2-метоксиэтокси)этил)амин, бензилтриэтиламмонийхлорид и гидросульфат трибутиламмония. Конкретные примеры соединения формулы (XIV) включают трикаприлметиламмонийхлорид, и тому подобное.
Термин «соль» обычно относится к аддитивной соли кислоты, и предпочтительной является фармацевтически приемлемая соль. Примеры кислоты и аддитивные соли кислоты включают неорганическую кислоту, такую как соляная кислота и серная кислота, и органическую кислоту, такую как уксусная кислота и щавелевая кислота.
Примеры «амина» включают первичный амин, вторичный амин и третичный амин, и более конкретно включают амин, такой как пирролидин, пиперидин, диметиламин, диэтиламин, диизопропиламин, дибутиламин, триметиламин, триэтиламин и трибутиламин, предпочтительно вторичный амин или третичный амин, и более предпочтительно пирролидин.
Термин «обработка силикагелем» относится к процессу фильтрации неочищенного продукта, растворенного в растворителе, через наполненную силикагелем колонку или через фильтр, поверхность которого покрыта силикагелем.
Предпочтительный способ получения по настоящему изобретению будет проиллюстрирован ниже подробно.
Стадия получения индольного соединения формулы (XII):
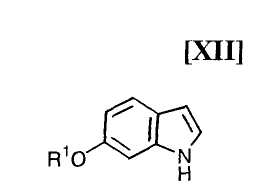
в которой R1 представляет собой гидроксизащитную группу, путем осуществления взаимодействия соединения (XIII):
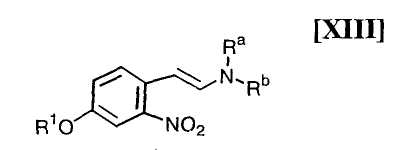
в которой значение R1 определено выше, и каждый Ra и Rb независимо представляет собой C1-C7-алкильную группу, или Ra и Rb могут быть объединены вместе с образованием C3-C6-алкиленовой группы, с газообразным водородом в присутствии соединения родия и соединения металла, выполняют так, что соединение (XIII) взаимодействует с газообразным водородом при давлении в 1-5 атм. в присутствии приблизительно от 0,5 мол.% до 30 мол.% соединения родия и приблизительно от 1 мол.% до 100 мол.% соединения металла по отношению к 1 молю соединения (XIII), в инертном растворителе при температуре приблизительно от -20°C до 80°C в течение приблизительно от 1 до 120 часов.
Примеры инертного растворителя, который может применяться на указанной выше стадии, включают тетрагидрофуран, диэтиловый эфир, трет-бутилметиловый эфир, диизопропиловый эфир, дибутиловый эфир, метанол, этанол, изопропанол, пропанол, ацетон, этилацетат, изопропилацетат и циклопентилметиловый эфир, или смесь этих растворителей, среди которых предпочтительным является тетрагидрофуран, циклопентилметиловый эфир или трет-бутилметиловый эфир.
Соединение родия, которое может применяться на указанной выше стадии, может представлять собой любое соединение, содержащее в составе молекулы, по крайней мере, один атом родия, и его примеры предпочтительно включают содержащий 1-10% родия родий/углерод, содержащий 1-10% родия родий/оксид алюминия, содержащий 1-10% родия родий/карбонат кальция или содержащий 1-10% родия родий/сульфат бария, и более предпочтительно представляет собой родий/углерод.
Примеры соединения металла, которое может применяться на указанной выше стадии, включают соединение никеля(II), соединение железа(II), соединение железа(III), соединение кобальта(II) и соединение кобальта(III), предпочтительно NiBr2, Ni(NO3)2, Ni(OCOCH3)2, FeBr3, FeCl2, FeSO4, FeCl3, FeCl3-SiO2, Fe(OCOCH3)2, фумарат Fe(II), CoBr2, CoI2, CoCl2,
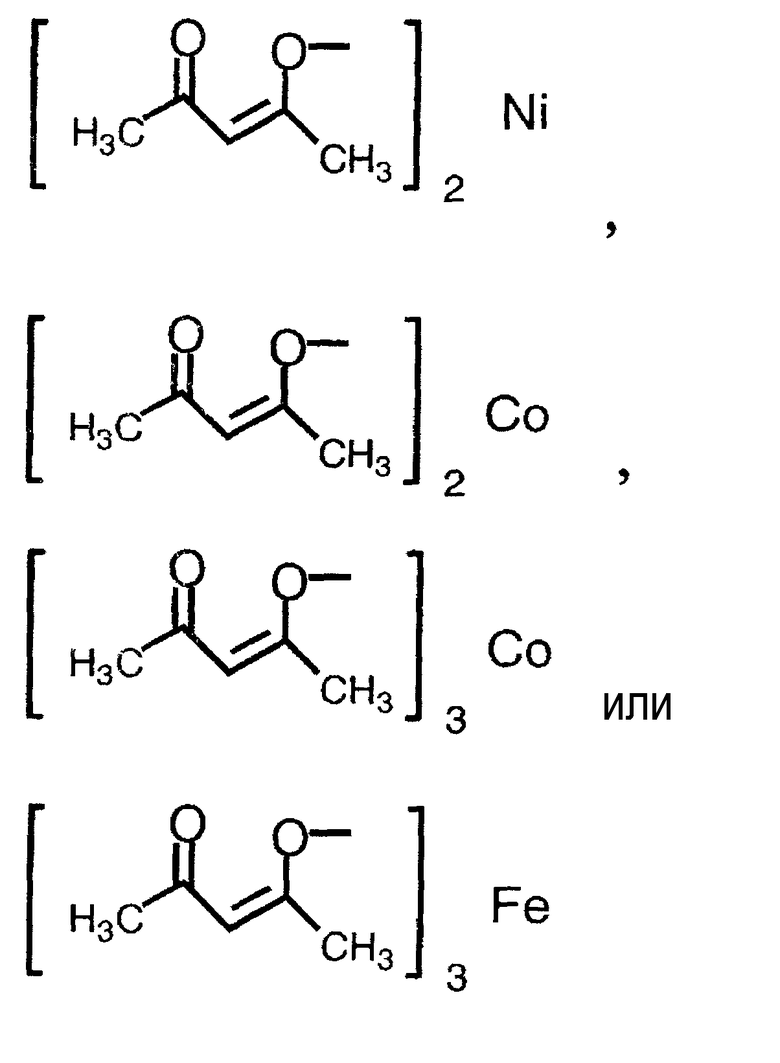
Применяемые на этой стадии исходные соединения могут быть получены в соответствии со способом, описанным, например, в Organic Synthesis Collective Volumes, vol. 7, page 34, или в соответствии со сходным с ним способом.
На указанной выше стадии взаимодействие предпочтительно осуществляют в присутствии амина в дополнение к соединению родия и соединению металла. Дополнительное присутствие амина в химической реакции может увеличивать скорость и выход реакции. С применением амина может быть резко увеличена скорость реакции. Амин включает первичный амин, вторичный амин или третичный амин, и более конкретно включает амин, такой как пирролидин, пиперидин, диметиламин, диэтиламин, диизопропиламин, дибутиламин, триметиламин, триэтиламин и трибутиламин, предпочтительно вторичный амин или третичный амин, и более предпочтительно пирролидин.
Амин обычно используют в количестве приблизительно от 0,01 до 10 эквивалентов по отношению к подвергаемому гидрированию материалу (например, соединению (XIII)). Более того, в случае подбора подходящего реагента, генерирующего амин в реакционном растворе при постоянном гидрировании в соответствии с реакцией по настоящему изобретению, дополнительное добавление амина к реакционному раствору не требуется.
К полученному на этой стадии реакционному раствору (суспензии) добавляют водный раствор аммиака и насыщенный солевой раствор, предпочтительно их суспензию, перемешивают смесь приблизительно в течение одного часа и фильтруют для разделения твердого вещества, а затем остаток промывают растворителем, таким как бензол, толуол или ксилол. Фильтрат и промывную жидкость объединяют, затем последовательно промывают водным раствором лимонной кислоты, 5% водным бикарбонатом натрия и насыщенным солевым раствором, а затем упаривают в вакууме досуха. После растворения полученного соединения формулы (XII) или его соли в растворителе, таком как бензол, толуол или ксилол, и тому подобное, раствор наносят на колонку, заполненную силикагелем с тем же самым что и у соединения (XII) весом, или на фильтр, чья поверхность покрыта упомянутым силикагелем, и подвергают воздействию давлением инертного газа, такого как азот. Образующиеся на стадии реакции примеси, такие как окрашенные вещества, могут быть эффективно удалены посредством очистки на силикагеле. Степень чистоты соединения (XII) повышают посредством способа очистки по настоящему изобретению, и поэтому химические реакции и очистка продуктов на последующих стадиях могут проводиться эффективными с точки зрения промышленного производства способами без применения особого способа.
Затем на стадии получения бис-индольного соединения формулы (VIII):
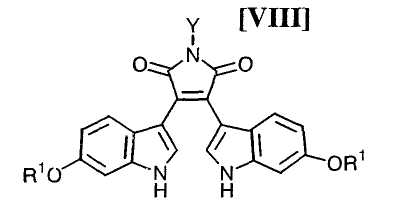
в которой значения для каждого R1 и Y определены выше, или его соли, взаимодействие полученного выше индольного соединения формулы (XII):
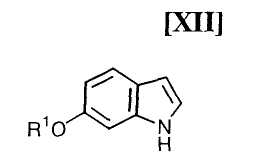
в которой значения для R1 определены выше, или его соли, с магнийхлоридом формулы (XI):
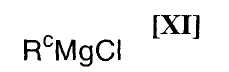
в которой Rc представляет собой C1-C7-алкильную группу, фенильную группу, винильную группу или аллильную группу; с соединением магния формулы (X):
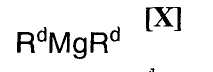
в которой Rd представляет собой C1-C7-алкильную группу или фенильную группу, или с его солью, или со смесью магнийхлорида формулы (XI) и соединения магния формулы (X), в вышеупомянутом инертном растворителе, с последующим осуществлением взаимодействия полученного продукта с малеимидным соединением формулы (IX):
в которой X представляет собой атом галогена, и Y представляет собой атом водорода, C1-C7-алкильную группу, фенильную группу, бензилоксиметильную группу или C7-C12-аралкильную группу, в вышеупомянутом инертном растворителе предпочтительно осуществляют в соответствии со следующими способами 1), 2) или 3).
1) При температуре приблизительно от 30°C до 120°C в течение приблизительно от 0,5 до 24 часов в инертном растворителе осуществляют взаимодействие приблизительно от 2 до 4 моль индольного соединения (XII) и приблизительно от 2 до 4 моль магнийхлорида (XI) по отношению к 1 молю малеимидного соединения (IX).
2) При температуре приблизительно от 30°C до 120°C в течение приблизительно от 0,5 до 24 часов в инертном растворителе осуществляют взаимодействие приблизительно от 2 до 4 моль индольного соединения (XII) и приблизительно от 2 до 4 моль соединения магния (X) по отношению к 1 молю малеимидного соединения (IX).
3) При температуре приблизительно от 30°C до 120°C в течение приблизительно от 0,5 до 24 часов в инертном растворителе осуществляют взаимодействие приблизительно от 2 до 4 моль индольного соединения (XII) и приблизительно от 0,8 до 4 моль смеси, содержащей магнийхлорид (XI) и соединение магния (X), по отношению к 1 молю малеимидного соединения (IX).
Предпочтительные примеры применяемого в способах 1), 2) и 3) растворителя включают толуол и смесь толуола и тетрагидрофурана.
Примеры используемого на приведенной выше стадии магнийхлорида формулы (XI) включают алкилмагнийхлориды, такие как метилмагнийхлорид, этилмагнийхлорид, н-пропилмагнийхлорид, изопропилмагнийхлорид, н-бутилмагнийхлорид, втор-бутилмагнийхлорид, изобутилмагнийхлорид, трет-бутилмагнийхлорид, н-пентилмагнийхлорид, н-гексилмагнийхлорид, фенилмагнийхлорид, винилмагнийхлорид и аллилмагнийхлорид, или их смесь.
Примеры применяемого на приведенной выше стадии соединения магния формулы (X) включают диметилмагний, диэтилмагний, ди(н-пропил)магний, диизопропилмагний, ди(н-бутил)магний, ди(втор-бутил)магний, диизобутилмагний, ди(трет-бутил)магний, ди(н-пентил)магний, ди(н-гексил)магний, (н-бутил)(втор-бутил)магний, (метил)(втор-бутил)магний, (этил)(втор-бутил)магний, (метил)(н-бутил)магний, (этил)(н-бутил)магний, (метил)(трет-бутил)магний, (этил)(трет-бутил)магний, (н-пропил)(н-бутил)магний, (н-пропил)(втор-бутил)магний, (н-пропил)(изопропил)магний, (н-бутил)(изопропил)магний, (втор-бутил)(изопропил)магний, (изобутил)(изопропил)магний, (н-пропил)(изобутил)магний и дифенилмагний, или их смесь.
Затем, на стадии получения соединения формулы (VII):
в которой значения для каждого R1 и Y определены выше, или его соли, реакцию замыкания цепи полученного выше бис-индольного соединения, представленного формулой (VIII):
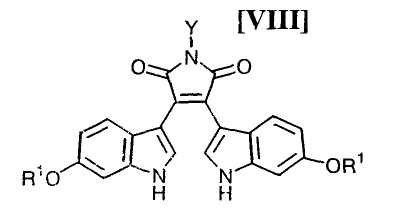
в которой значения для каждого R1 и Y определены выше, или его соли, предпочтительно осуществляют в соответствии со следующими способами 1) и 2).
1) Соединение формулы (VIII) или его соль обрабатывают в инертном растворителе при температуре приблизительно от 20°C до 200°C в течение приблизительно от 1 минуты до 5 суток, например, 2,3-дихлор-5,6-дициано-1,4-бензохиноном (DDQ), палладиевым реагентом, таким как PdCl2 и Pd(OAc)2, или медным реагентом, таким как CuCl2, в количестве приблизительно от 1 до 10 молярных эквивалентов по отношению к 1 моль соединения (VIII) или его соли.
Растворителем, который может применяться на стадии 1), может служить любой общеизвестный инертный растворитель, и его примеры включают полярные растворители, такие как тетрагидрофуран, метанол, этанол, N,N-диметилформамид, диметилсульфоксид, N-метилпирролидон и N,N-диметилацетамид, и неполярный растворитель, такой как бензол, толуол, ксилол (орто-, мета- или пара-), этилбензол и 1,2,4-триметилбензол. В случае применения 2,3-дихлор-5,6-дициано-1,4-бензохинона при использовании полярного растворителя во время химической реакции или при обработке образуется цианистый водород, в то время как при использовании неполярного растворителя образование цианистого водорода подавлено, что благоприятно для ступенчатого контроля.
2) Соединение формулы (VIII) или его соль обрабатывают в инертном растворителе реагентом, состоящим из приблизительно 0,01-1,0 эквивалента нанесенного на углерод, оксид алюминия, карбонат кальция, сульфат бария или силикагель катализатора, содержащего металл переменной валентности (например, палладий, платину и т.д.), по отношению к 1 моль соединения (VIII) при температуре приблизительно от 20°C до 200°C в течение приблизительно от 1 минуты до 5 суток при давлении от 1 до 5 атм. окислителя, выбираемого из группы, состоящей из кислорода, воздуха, этилена и ацетилена.
Примеры инертного растворителя, который может применяться на стадии 2), включают толуол, тетрагидрофуран, метанол, этанол, диметилформамид, диметилсульфоксид, N-метилпирролидон и диметилацетамид.
Затем, на стадии получения соединения формулы (V):
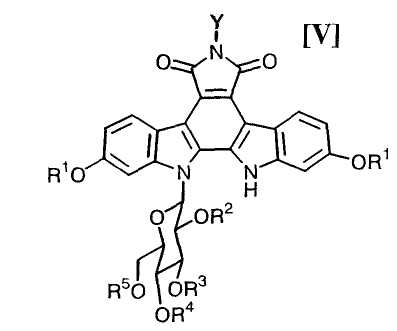
в которой значения для каждого R1 и Y определены выше, и каждый R2, R3, R4 и R5 представляет собой гидроксизащитную группу, или его соли, сочетание полученного выше соединения формулы (VII)
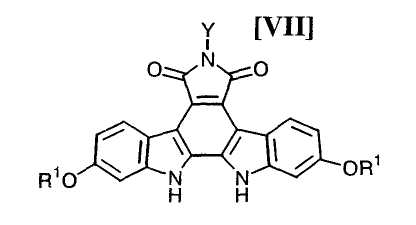
в которой значения для каждого R1 и Y определены выше, или его соли, или его соли, с активированным производным глюкозы формулы (VI):
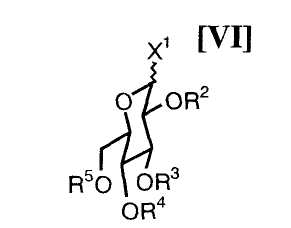
в которой значения для каждого R2, R3, R4 и R5 определены выше, и X1 представляет собой атом галогена, с предпочтительным использованием системы, содержащей основание в водном растворителе и катализатор межфазного переноса в инертном органическом растворителе, может быть осуществлено следующим образом.
Активированное производное глюкозы (VI):
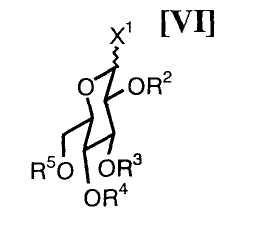
в котором каждый R2, R3, R4 и R5 представляет собой гидроксизащитную группу, и X1 представляет собой атом галогена, может быть получено путем осуществления взаимодействия производного глюкозы (VIa):
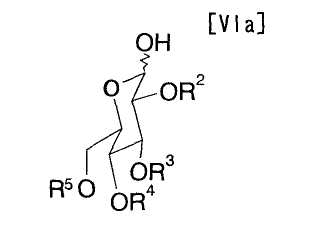
в которой каждый R2, R3, R4 и R5 представляет собой гидроксизащитную группу, например, с галогенангидридом, сульфонилхлоридом или йодтрифенилфосфином при температуре приблизительно от -50°C до 200°C, предпочтительно приблизительно от -10°C до 30°C, предпочтительно в инертном растворителе.
Примеры галогенангидрида, применяемого на вышеупомянутой стадии, включают SOCl2, POCl3, SOBr3, POBr3, PBr3 и оксалилхлорид, среди которых предпочтительным является SOCl2 или оксалилхлорид, а более предпочтительным - SOCl2.
Примеры инертного растворителя, применяемого на вышеупомянутой стадии, включают углеводород, такой как толуол, ксилол, гептан и гексан; нитрил, такой как ацетонитрил; и простой эфир, такой как трет-бутилметиловый эфир и тетрагидрофуран; галогенированный углеводород, такой как метиленхлорид, тетрахлорид углерода, хлороформ, трифтортолуол и дихлорбензол; и кетон, такой как метилизобутиловый кетон и ацетон, среди которых предпочтительным является трет-бутилметиловый простой эфир или тетрагидрофуран, а более предпочтительным - трет-бутилметиловый простой эфир.
Что касается производного глюкозы (VIa), то могут использоваться коммерчески доступные продукты.
Полученное выше активированное производное глюкозы формулы (VI) сочетают с соединением формулы (VII):
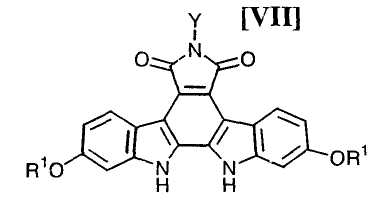
в которой значения для каждого R1 и Y определены выше, или с его солью, с использованием системы, содержащей основание в водном растворителе и катализатор межфазного переноса в инертном органическом растворителе, обычно при температуре от -50°C до 200°C, предпочтительно от 0°C до 40°C.
Примером водного растворителя, применяемого на вышеупомянутой стадии, является вода.
Примеры основания, применяемого на вышеупомянутой стадии, включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид цезия, среди которых предпочтительным является гидроксид натрия или гидроксид калия. Концентрация применяемого основания составляет приблизительно от 5 мас.% до 95 мас.%, предпочтительно приблизительно от 45 мас.% до 50 мас.%.
Примеры инертного растворителя, применяемого на вышеупомянутой стадии, включают углеводород, такой как толуол, ксилол, гептан и гексан; нитрил, такой как ацетонитрил; и простой эфир, такой как трет-бутилметиловый эфир и тетрагидрофуран; галогенированный углеводород, такой как метиленхлорид, тетрахлорид углерода, хлороформ, трифтортолуол и дихлорбензол; кетон, такой как метилизобутиловый кетон и ацетон; и неионный растворитель, такой как N,N-диметилформамид и 1-метил-2-пирролидинон, среди которых предпочтительным является трет-бутилметиловый эфир, метиленхлорид или трифтортолуол.
Примеры катализатора межфазного переноса, применяемого на вышеупомянутой стадии, включают соединение формулы (XIV):
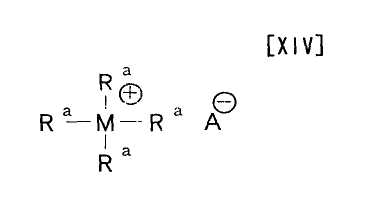
в которой каждый Ra независимо представляет собой водород, бензил или C1-C18-углеводород; M представляет собой атом азота или атом фосфора; и A представляет собой гидроксигруппу, атом фтора, атом брома, атом хлора, атом йода, цианогруппу, HSO4, CH3SO3 или PhCH2COO, и трис(2-(2-метоксиэтокси)этил)амин, и его предпочтительные примеры включают трикаприлметиламмонийхлорид, трис(2-(2-метоксиэтокси)этил)амин, бензилтриэтиламмонийхлорид и гидросульфат трибутиламмония.
Следующую стадию обработки полученного выше соединения формулы (V):
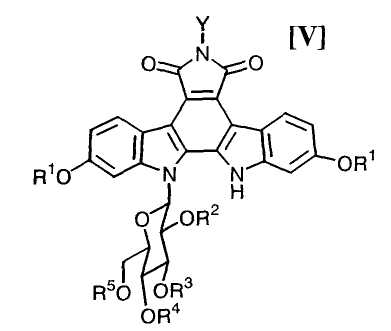
в которой значения для каждого R1, R2, R3, R4, R5 и Y определены выше, или его соли, основанием предпочтительно в инертном растворителе с получением соединения, представленного формулой (IV):
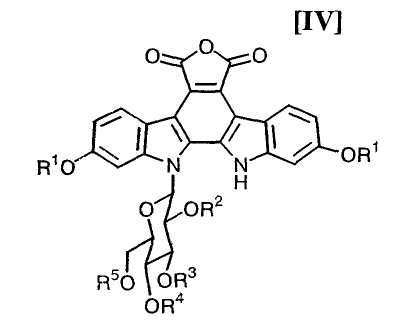
в которой значения для каждого R1, R2, R3, R4 и R5 определены выше, или его соли, обычно выполняют с применением основания в количестве приблизительно от 50 до 100 моль, предпочтительно приблизительно от 50 до 70 моль, по отношению к 1 моль соединения (V) или его соли, предпочтительно в инертном растворителе, который не оказывает отрицательного эффекта на химическую реакцию.
Примеры упомянутого выше инертного растворителя включают спирты, такие как метанол, этанол, изопропанол, трет-бутанол, диметилсульфоксид, и смешанный из них растворитель, среди которых предпочтительным является метанол, этанол или изопропанол.
Примеры упомянутого выше основания включают основание, такое как гидроксид натрия, гидроксид калия, метоксид натрия, метоксид калия, трет-бутоксид натрия и трет-бутоксид калия, среди которых предпочтительным является гидроксид натрия, гидроксид калия или метоксид натрия.
Температура реакции обычно составляет от комнатной температуры приблизительно до 60°C, предпочтительно приблизительно от 30°C до 50°C, а длительность химической реакции обычно составляет приблизительно от 1 часа до 1 суток, предпочтительно приблизительно от 3 часов до 10 часов.
Далее, стадию получения соединения формулы (II):
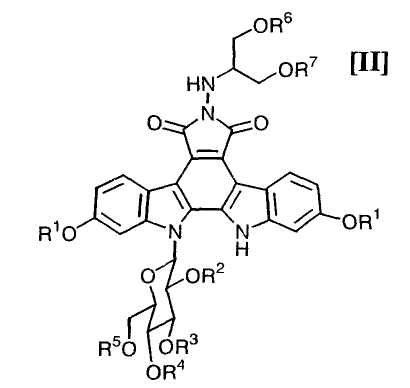
в которой значения для каждого R1, R2, R3, R4 и R5 определены выше, и каждый R6 и R7 представляет собой гидроксизащитную группу, или его соли, путем осуществления взаимодействия полученного выше соединения формулы (IV):
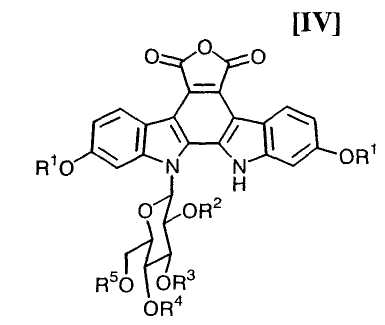
в которой значения для каждого R1, R2, R3, R4 и R5 определены выше, или его соли, с соединением формулы (III):
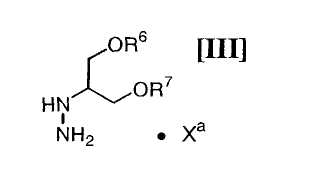
в которой значения для каждого R6 и R7 определены выше, и Xa представляет собой молекулу кислоты, обычно осуществляют с применением соединения (III) в количестве приблизительно от 1 моль до 3,0 моль, предпочтительно приблизительно от 1,0 до 1,5 моль, по отношению к 1 моль соединения формулы (IV), или его соли, в инертном растворителе, который не оказывает отрицательного влияния на химическую реакцию.
Упомянутая выше стадия может выполняться в присутствии акцептора кислоты или одновременно акцептора кислоты и десикканта.
Количество применяемого акцептора кислоты составляет приблизительно от 0,1 до 100 моль, предпочтительно приблизительно от 0,1 до 2 моль, по отношению к 1 моль соединения формулы (IV) или его соли. Количество применяемого десикканта составляет приблизительно от 0,1 до 100 моль, более предпочтительно приблизительно от 0,1 до 2 моль, по отношению к 1 моль соединения формулы (IV) или его соли.
Примеры упомянутого выше инертного растворителя включают N,N-диметилформамид, N,N-диметилацетамид, тетрагидрофуран, диметилсульфоксид и N-метилпирролидон, и их смесь, среди которых предпочтительным является N,N-диметилформамид, N,N-диметилацетамид или N-метилпирролидон.
Температура реакции обычно составляет от комнатной температуры приблизительно до 90°C, предпочтительно от 30°C до 70°C, а длительность химической реакции обычно составляет приблизительно от 1 часа до 1 суток, предпочтительно приблизительно от 1 часа до 3 часов.
Примеры акцептора кислоты включают этилдиметиламин, триэтиламин, изопропилдиэтиламин, диизопропилэтиламин, трибутиламин, пиридин, 2,6-лутидин, 2,6-трет-бутилпиридин, 2,4,6-коллидин, 1,8-диазабицикло[5.4.0]нон-5-ен (DBU), 1,5-диазабицикло[4.3.0]ундец-7-ен (DBN), диизопропиламин, N,N-диметиланилин, 1,4-диазабицикло[2.2.2]октан (DABCO) и N-метилморфолин, среди которых более предпочтительными являются низшие алкиламины, такие как триэтиламин, диизопропилэтиламин, трибутиламин или диизопропиламин, а более предпочтительным является триэтиламин.
Примеры десикканта включают сульфат магния, сульфат натрия, молекулярное сито, HC(O-изо-Pr)3, HC(O-Et)3, НС(О-СН3)3 и (СН3)2С(ОСН3)2, среди которых более предпочтительным является сульфат магния, сульфат натрия или молекулярное сито, а более предпочтительным является сульфат магния.
Далее, на стадии получения индолопирролокарбазольного производного формулы (I):
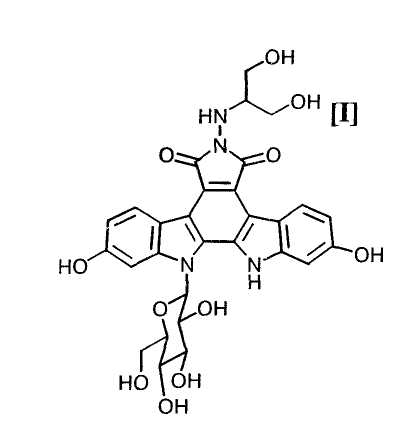
или его соли путем снятия защитных групп с соединения формулы (II):
в которой значения для каждого R1, R2, R3, R4, R5, R6 и R7 определены выше, или его соли, в случае осуществления реакции путем каталитического восстановления, катализатор включает, например, палладий-на-угле и никель Ренея. Такие катализаторы могут представлять собой известные катализаторы.
При каталитическом восстановлении предпочтительное давление водорода обычно составляет от нормального давления до 3 атм., и удельное отношение количества применяемого катализатора к массе исходного соединения (II) обычно составляет приблизительно от 1/100 до 1, предпочтительно приблизительно от 1/100 до 1/10.
Примеры применяемого в реакции растворителя включают растворители, смешанные из спиртовых растворителей, например, метанола, этанола, изопропанола, бутанола и тетрагидрофурана, среди которых предпочтительным является смешанный растворитель, включающий изопропанол и тетрагидрофуран (50:50).
Температура реакции обычно составляет от -30°C до 60°C, предпочтительно от 0°C до 50°C, а продолжительность химической реакции обычно составляет приблизительно от мгновенного протекания реакции приблизительно до 7 суток, предпочтительно от мгновенного протекания реакции приблизительно до 24 часов.
Способ очистки полученного соединения формулы (I) или его соли может осуществляться следующим образом.
Полученный реакционный раствор фильтруют и корректируют pH фильтрата до значения приблизительно от 1,5 приблизительно до 6,5, более предпочтительно приблизительно до 2,5.
Для доведения концентрации соединения формулы (I) в растворе до значения приблизительно от 10 мл/г приблизительно до 20 мл/г, предпочтительно приблизительно от 12 мл/г приблизительно до 18 мл/г, более предпочтительно приблизительно до 15 мл/г к полученному раствору добавляют приблизительно от 10% приблизительно до 30%, предпочтительно приблизительно от 15% приблизительно до 25%, более предпочтительно приблизительно 20% водный раствор спирта.
Полученный таким образом раствор нагревают до температуры приблизительно от 50°C приблизительно до 100°C, предпочтительно приблизительно до 70°C.
К раствору добавляют спирт в количестве, равном двум третям от объема раствора.
Полученный раствор выдерживают при температуре приблизительно от 50°C до 100°C, предпочтительно приблизительно при 70°C, и фильтруют, собирая выпадающие в осадок кристаллы.
В указанном выше процессе фильтрации содержание воды в суспензии кристаллов доводят до значения приблизительно от 1 приблизительно до 10 мас./об.%.
Пример применяемого на указанной выше стадии спирта включает C1-C5-алифатический спирт, предпочтительно метанол, этанол, пропанол, изопропанол, бутанол, втор-бутанол, изобутанол, пентанол, изопентанол, и более предпочтительно изопропанол.
Примеры применяемых для корректировки значения pH оснований включают триэтиламин, диизопропилэтиламин, трибутиламин, пиридин, 2,6-лутидин, 2,4,6-коллидин, 1,8-диазабицикло[5.4.0]нон-5-ен (DBU), 1,5-диазабицикло[4.3.0]ундец-7-ен (DBN), диизопропиламин, N,N-диметиланилин, 1,4-диазабицикло[2.2.2]октан (DABCO) или N-метилморфолин, среди которых более предпочтительными являются низшие алкиламины, такие как триэтиламин, диизопропилэтиламин, трибутиламин или диизопропиламин, а более предпочтительным является триэтиламин.
Соединения, полученные на каждом из указанных выше стадий, могут быть очищены и, при необходимости, выделены по отдельности или в комбинации посредством известных per se в данной области техники способов, таких как колоночная хроматография на силикагеле или адсорбирующей смоле, жидкостная хроматография, тонкослойная хроматография, экстракция растворителями и перекристаллизация/переосаждение.
Настоящее изобретение также относится к используемому катализатору гидрирования, содержащему упомянутое выше соединение родия и упомянутое выше соединение металла.
Этот катализатор применяется для восстановления соединений и содержит упомянутое выше соединение родия и соединение металла. В соответствии с настоящим изобретением предполагается совместное присутствие соединения родия и металла или их смесь. Поэтому, кроме соединения родия и соединения металла, в катализаторе по настоящему изобретению может содержаться, например, растворитель. Реакция восстановления, в которой используется катализатор по настоящему изобретению, не должна ограничиваться реакцией восстановления в описанной выше стадии (1), хотя восстановление нитрогрупп до аминогрупп и восстановление алкенильных групп или алкинильных групп до соответствующих алкильных групп является предпочтительным. Катализатор по настоящему изобретению обладает избирательным действием в случае его применения для восстановления нитрогрупп до аминогрупп и для восстановления алкенильных групп или алкинильных групп до соответствующих алкильных групп, что является очень выгодным для промышленного применения. В качестве примера упомянутого избирательного действия может быть приведен тот заметный факт, что при восстановлении нитрогрупп до аминогрупп и при восстановлении алкенильных групп или алкинильных групп до соответствующих алкильных групп, даже если в подвергаемом восстановлению материале, содержащем, кроме нитрогруппы, алкенильной или алкинильной групп, другие функциональные группы, такие как бензилоксигруппа, карбонильная группа, например альдегид или кетон, или галоген, причем восстановление отличных от нитрогруппы, алкенила или алкинила функциональных групп по существу не происходитилиподавляется, или восстановление нитрогрупп до аминогрупп и восстановление алкенильных групп или алкинильных групп до соответствующих алкильных групп является преимущественным по сравнению с восстановлением других, отличных от нитрогруппы, алкенила или алкинила, функциональных групп. Более того, также показана способность катализатора по настоящему изобретению ускорять восстановление нитрогрупп до аминогрупп и восстановление алкенильных групп или алкинильных групп до соответствующих алкильных групп. Конкретно, в случае осуществления каталитического восстановления с применением родиевого катализатора и катализатора, содержащего в качестве соединения металла соль железа, соль никеля или соль кобальта, восстановление функциональных групп, таких как бензилэфирные группы, арилгалогениды, альдегиды или кетоны, по существу отсутствуетилиподавлено, а потому становится возможным избирательное восстановление нитрогрупп до аминогрупп и восстановление алкенильных групп или алкинильных групп до соответствующих алкильных групп. Следовательно, в случае применения катализатора по настоящему изобретению в реакции восстановления не требуется проведение применяемых в традиционных реакциях восстановлениясложных стадий, включающих защиту функциональных групп защитными группами, последующий процесс восстановления и снятие защитных групп.
В настоящем изобретении является предпочтительным, чтобы дополнительно к упомянутым выше соединениям родия и металла упомянутый выше катализатор содержал амин, как упомянуто выше. При применении катализатора, содержащего амин по настоящему изобретению, в отличие от катализатора, не содержащего амин, скорость реакции резко увеличивается, скорость восстановления легко восстанавливаемых групп, таких как бензилэфирная группа, снижается, и может достигаться избирательное восстановление нитрогруппы, алкенила или алкинила, улучшая тем самым выход продукта восстановления.
ПРИМЕРЫ
Настоящее изобретение подробно описывается посредством примеров и сравнительных примеров, но не ограничивается ими.
Пример 1
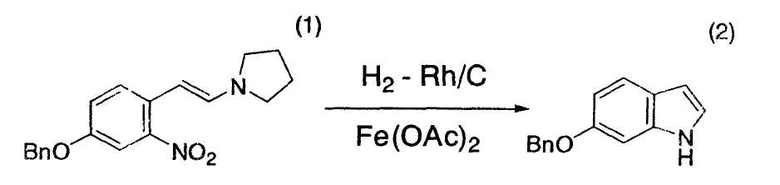
(Bn - бензильная группа (здесь и далее в тексте); Rh/C - порошкообразный родий/угольный порошок; Ac - ацетильная группа)
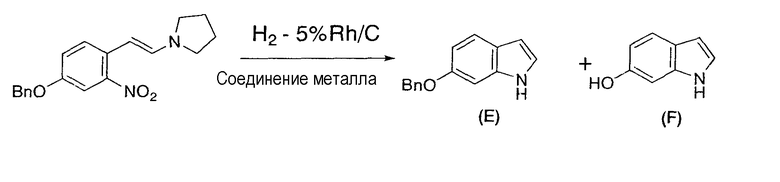
3-Бензилокси-6-(2-пирролидинилвинил)нитробензол (1) (5,00 г, 15,4 ммоль), порошкообразный 5% родий/угольный порошок (952 мг, 0,462 ммоль), ацетат железа (II) (536 мг, 3,08 ммоль) и тетрагидрофуран (50 мл) помещали в атмосфере азота в трехгорлую колбу емкостью 100 мл, оснащенную магнитной мешалкой и термометром. Полученную суспензию перемешивали при 22°C-25°C в течение 24 часов в атмосфере водорода и затем перемешивали в течение ночи в атмосфере азота. К суспензии добавляли 28% водный раствор аммиака (50 г) и 5% соляной раствор (20 мл) и перемешивали смесь в течение одного часа, затем фильтровали для отделения твердого вещества. Сухой остаток промывали толуолом (100 мл) и объединяли предыдущий фильтрат и промывную жидкость. Раствор последовательно промывали 10% водной лимонной кислотой (50 г), 5% водным бикарбонатом натрия (50 г) и 20% солевым раствором (50 г) и упаривали в вакууме досуха. Остаток растворяли в толуоле (приблизительно 100 мл) и фильтровали на покрытом силикагелем (5 г) фильтре. Силикагель промывали толуолом и полученный бесцветный раствор (152,15 г) анализировали высокоэффективной жидкостной хроматографией, которая свидетельствовала о том, что выход целевого соединения (2) составлял 91% (партия 3,15 г).
1H-ЯМР (500 МГц, ДМСО-d6,  ч/млн): 10,90 (ушир.с, 1H), 7,49 (д, J=7,5 Гц, 1H), 7,44 (д, J=8,6 Гц, 1H), 7,40 (дд, J=7,5, 7,5 Гц, 1H), 7,33 (дд, J=7,5, 7,5 Гц, 1H), 7,21 (ушир.дд, J=2,4, 2,4 Гц, 1H), 7,01 (ушир.м, 1H), 6,77 (дд, J=1,8, 8,6 Гц, 1H), 6,36 (ушир.м, 1H), 5,12 (с, 2H).
ч/млн): 10,90 (ушир.с, 1H), 7,49 (д, J=7,5 Гц, 1H), 7,44 (д, J=8,6 Гц, 1H), 7,40 (дд, J=7,5, 7,5 Гц, 1H), 7,33 (дд, J=7,5, 7,5 Гц, 1H), 7,21 (ушир.дд, J=2,4, 2,4 Гц, 1H), 7,01 (ушир.м, 1H), 6,77 (дд, J=1,8, 8,6 Гц, 1H), 6,36 (ушир.м, 1H), 5,12 (с, 2H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 154,7, 138,0, 136,8, 128,7, 128,0, 127,9, 124,4, 122,5, 120,9, 110,1, 101,3, 96,3, 69,9.
Примеры 2-18
Выполняли операции, сходные с приведенными в примере 1, используя родий/угольный порошок (Rh/C) и представленные в следующей таблице добавки вместо ацетата железа(II).
(используемое количество)
(10 мол.%)
В данной таблице "acac" означает группу, представленную следующей формулой, а Ac означает ацетил.
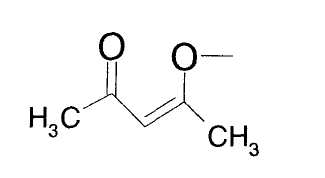
Пример 19
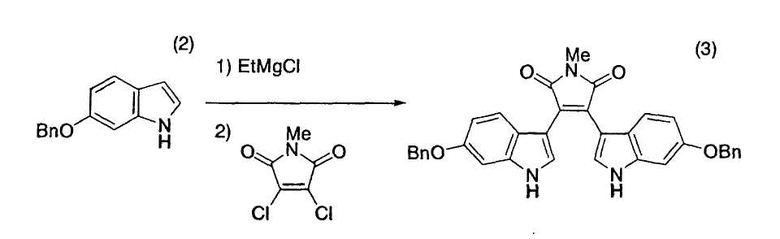
[Et означает этил и Me означает метил (здесь и далее в этом документе)]
6-Бензилоксииндол (2) (2,00 г, 8,96 ммоль), тетрагидрофуран (2,70 мл) и толуол (15,2 мл) помещали в атмосфере азота в трехгорлую колбу емкостью 50 мл, оснащенную магнитной мешалкой, холодильником Димрота и термометром. Смесь нагревали до 33°C, добавляли 2,00M этилмагнийхлорид/диэтиловый эфир (4,48 мл, 8,96 ммоль) в течение 7 минут, смесь нагревали до 55°C-60°C и перемешивали при 55°C-60°C в течение одного часа. N-Метил-1,2-дихлормалеимид (730 мг, 4,06 ммоль) растворяли в толуоле (4,4 мл), добавляли в течение 10 минут раствор к полученной смеси и промывали толуолом (1 мл) емкость с N-метил-1,2-дихлормалеимидом. После добавления промывной жидкости смесь перемешивали при 55°C-60°C в течение 20 минут. Реакционную смесь дополнительно нагревали до 100°C-108°C, перемешивали при 100°C-108°C в течение 12 часов и оставляли охлаждаться до комнатной температуры и перемешиваться в течение ночи. Реакционную смесь нагревали до 80°C и добавляли к смеси толуол (15,2 мл) и 13% водный хлорид аммония (17 мл). Полученную смесь охлаждали до комнатной температуры с получением суспензии, которую фильтровали с получением красного твердого вещества. Твердое вещество последовательно промывали толуолом (20 мл), толуолом/водой (отношение смеси 1:1, 20 мл) и метанолом (20 мл×2). Твердое вещество сушили в вакууме в течение ночи при комнатной температуре с получением целевого бис-индола (3) с выходом 84% (партия 1,89 г).
1H-ЯМР (500 МГц, ДМСО-d6, ч/млн): 11,50 (с, 2H), 7,63 (д, J=2,3 Гц, 2H), 7,42 (д, J=7,3 Гц, 2H), 7,37 (дд, J=7,3, 7,3 Гц, 2H), 7,30 (дд, J=7,3, 7,3 Гц, 2H), 6,97 (д, J=2,1 Гц, 2H), 6,72 (д, J=8,8 Гц, 2H), 6,41 (дд, J=2,1, 8,8 Гц, 2H), 5,04 (с, 4H), 3,03 (с, 3H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 172,2, 155,0, 137,7, 137,1, 128,7, 128,5, 128,1, 128,0, 127,1, 122,0, 120,1, 110,4, 106,1, 96,3, 69,7, 24,3.
Рассчитанная т.пл.˜240°C (с разложением).
Пример 20
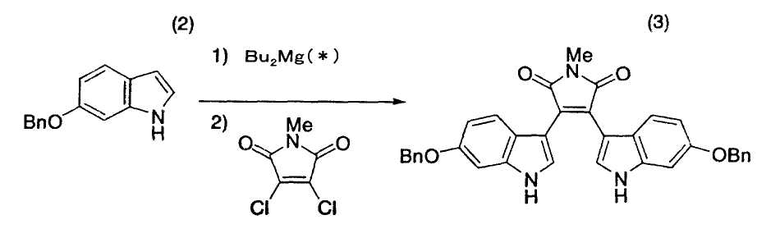
* - смесь ди(н-бутил)магния, ди(втор-бутил)магния и (н-бутил)(втор-бутил)магния
6-Бензилоксииндол (2) (2,00 г, 8,96 ммоль), тетрагидрофуран (2,64 мл) и толуол (15,2 мл) помещали в атмосфере азота в трехгорлую колбу емкостью 50 мл, оснащенную магнитной мешалкой, холодильником Димрота и термометром. Смесь нагревали до 38°C, добавляли в течение 10 минут 0,90M дибутилмагнийхлорид/гептан (4,96 мл, 4,47 ммоль), причем гептан содержал смесь ди(н-бутил)магния, ди(втор-бутил)магния и (н-бутил)(втор-бутил)магния, и перемешивали смесь при 55°C-60°C в течение одного часа. N-Метил-1,2-дихлормалеимид (730 мг, 4,06 ммоль) растворяли в толуоле (4,4 мл), добавляли в течение 10 минут раствор к полученной выше смеси и промывали толуолом (1 мл) емкость с N-метил-1,2-дихлормалеимидом. После добавления промывной жидкости смесь нагревали до 55°C-60°C, перемешивали при 55°C-60°C с получением твердого вещества, которое растворяли до гомогенного состояния добавлением тетрагидрофурана (1,5 мл). Раствор перемешивали при 55°C-60°C в течение 30 минут, затем нагревали до 98°C-100°C, перемешивали при 98°C-100°C в течение 12 часов и оставляли охлаждаться до комнатной температуры и перемешиваться в течение ночи. После нагревания реакционной смеси до 90°C к реакционной смеси добавляли 13% водный хлорид аммония (17 мл) и охлаждали смесь до комнатной температуры с получением суспензии, которую фильтровали с получением красного твердого вещества, которое последовательно промывали толуолом (20 мл), толуолом/водой (отношение смеси 1:1, 20 мл) и метанолом (20 мл×2). Твердое вещество сушили в вакууме в течение ночи при комнатной температуре с получением целевого бис-индола (3) с выходом 82% (партия 1,85 г).
1H-ЯМР (500 МГц, ДМСО-d6,  ч/млн): 11,50 (с, 2H), 7,63 (д, J=2,3 Гц, 2H), 7,42 (д, J=7,3 Гц, 2H), 7,37 (дд, J=7,3, 7,3 Гц, 2H), 7,30 (дд, J=7,3, 7,3 Гц, 2H), 6,97 (д, J=2,1 Гц, 2H), 6,72 (д, J=8,8 Гц, 2H), 6,41 (дд, J=2,1, 8,8 Гц, 2H), 5,04 (с, 4H), 3,03 (с, 3H).
ч/млн): 11,50 (с, 2H), 7,63 (д, J=2,3 Гц, 2H), 7,42 (д, J=7,3 Гц, 2H), 7,37 (дд, J=7,3, 7,3 Гц, 2H), 7,30 (дд, J=7,3, 7,3 Гц, 2H), 6,97 (д, J=2,1 Гц, 2H), 6,72 (д, J=8,8 Гц, 2H), 6,41 (дд, J=2,1, 8,8 Гц, 2H), 5,04 (с, 4H), 3,03 (с, 3H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 172,2, 155,0, 137,7, 137,1, 128,7, 128,5, 128,1, 128,0, 127,1, 122,0, 120,1, 110,4, 106,1, 96,3, 69,7, 24,3.
Рассчитанная т.пл.˜240°C (с разложением).
Пример сравнения 2
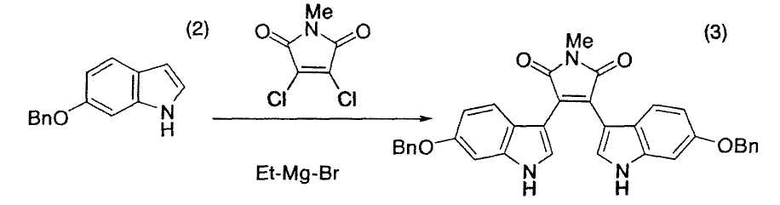
[Bn - бензил; Me - метил]
6-Бензилоксииндол (2) (2,00 г, 8,96 ммоль), тетрагидрофуран (2,64 мл) и толуол (15,2 мл) помещали в атмосфере азота в трехгорлую колбу емкостью 50 мл, оснащенную магнитной мешалкой, холодильником Димрота и термометром. Смесь нагревали до 38°C, добавляли в течение 7 минут 2,82M этилмагнийбромид/диэтиловый эфир (3,12 мл, 8,82 ммоль), нагревали до 55°C-60°C и перемешивали при 55°C-60°C в течение одного часа. N-Метил-1,2-дихлормалеимид (730 мг, 4,06 ммоль) растворяли в толуоле (4,4 мл), добавляли в течение 7 минут раствор к полученной выше смеси и промывали толуолом (1 мл) емкость с N-метил-1,2-дихлормалеимидом. После добавления промывной жидкости смесь перемешивали при 55°C-60°C в течение 30 минут, затем нагревали до 100°C-107°C, перемешивали при 100°C-107°C в течение 12 часов и оставляли охлаждаться до комнатной температуры и перемешиваться в течение ночи. После нагревания реакционной смеси до 80°C к реакционной смеси добавляли толуол (15,2 мл) и 13% водный хлорид аммония (17 мл) и охлаждали смесь до комнатной температуры с получением суспензии. Суспензию фильтровали с получением красного твердого вещества, которое последовательно промывали толуолом (20 мл), толуолом/водой (1:1, 20 мл) и метанолом (20 мл×2). Твердое вещество сушили в вакууме в течение ночи при комнатной температуре с получением целевого бис-индола (3) с выходом 70% (партия 1,73 г).
1H-ЯМР (500 МГц, ДМСО-d6, ч/млн): 11,50 (с, 2H), 7,63 (д, J=2,3 Гц, 2H), 7,42 (д, J=7,3 Гц, 2H), 7,37 (дд, J=7,3, 7,3 Гц, 2H), 7,30 (дд, J=7,3, 7,3 Гц, 2H), 6,97 (д, J=2,1 Гц, 2H), 6,72 (д, J=8,8 Гц, 2H), 6,41 (дд, J=2,1, 8,8 Гц, 2H), 5,04 (с, 4H), 3,03 (с, 3H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 172,2, 155,0, 137,7, 137,1, 128,7, 128,5, 128,1, 128,0, 127,1, 122,0, 120,1, 110,4, 106,1, 96,3, 69,7, 24,3.
Рассчитанная т.пл.˜240°C (с разложением).
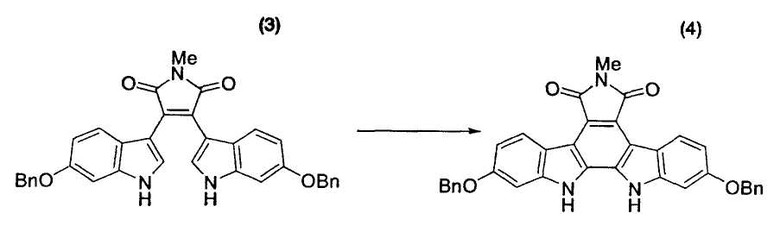
Пример 21
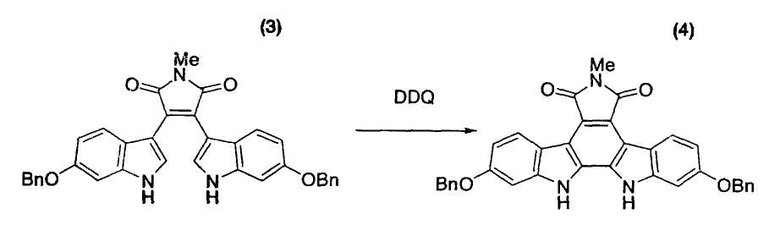
Бис-индольное соединение (3) (3,00 г, 5,42 ммоль) и толуол (75,3 мл) помещали в атмосфере азота в трехгорлую колбу емкостью 300 мл, оснащенную магнитной мешалкой, холодильником Димрота и термометром, и нагревали смесь до 110°C. К полученной смеси в течение 15 минут при 107°C-110°C добавляли раствор 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) (1,37 г, 6,03 ммоль) в толуоле (48,0 мл). После промывания содержащей DDQ емкости толуолом (12 мл) к реакционной смеси также добавляли промывную жидкость. Смесь перемешивали при 108°C-110°C в течение одного часа и подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о факте исчезновения исходного материала. Реакционный раствор охлаждали до 71°C и в течение 3 часов при 60°C-71°C добавляли метанол (134 мл) [этап измерения A]. Смесь охлаждали до комнатной температуры и перемешивали в течение ночи [этап измерения B]. Реакционный раствор фильтровали и промывали метанолом (15 мл×2) с получением коричневого твердого вещества (2876 мг), которое суспендировали в N,N-диметилформамиде (54 мл). Суспензию нагревали и перемешивали при 95°C-105°C в течение одного часа, охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционный раствор фильтровали и промывали метанолом (15 мл×2) с получением желтого твердого вещества (3018 мг), которое суспендировали в диметилсульфоксиде (28,3 мл). Суспензию нагревали до 60°C-70°C для растворения полученного выше твердого вещества. Последовательно добавляли метанол (13,3 мл) и небольшое количество охарактеризованного выше соединения в виде затравочного кристалла и перемешивали смесь в течение одного часа для выдерживания суспензии. После добавления метанола (42,4 мл) в течение 2 часов смесь охлаждали до комнатной температуры и затем перемешивали в течение ночи. Реакционную смесь фильтровали, промывали метанолом (15 мл×2), сушили в вакууме при 60°C в течение ночи с получением целевого индолкарбазольного производного (4) в виде желтых кристаллов с выходом 87% (партия 2589 мг).
1H-ЯМР (500 МГц, ДМСО-d6, ч/млн): 11,26 (с, 2H), 8,69 (д, J=8,7 Гц, 2H), 7,54 (д, J=7,3 Гц, 2H), 7,43 (дд, J=7,3, 7,3 Гц, 2H), 7,37 (дд, J=7,3, 7,3 Гц, 2H), 7,27 (д, J=2,1 Гц, 2H), 6,72 (д, J=8,8 Гц, 2H), 6,96 (дд, J=2,1, 8,7 Гц, 2H), 5,22 (с, 4H), 2,96 (с, 3H).
Рассчитанная т.пл.˜342°C (с разложением).
Условия проведения ВЭЖХ-анализа: разделительная колонка YMC AM-303 250×4,6 мм; температура при измерении - 40°C; детектирование на длине волны 220 нм; количество наносимого образца - 10 мкл; подвижная фаза: MeCN/0,1% фосфорная кислота=(t=0, 65:35; t=20, 90:30); скорость потока - 1 мл/мин.
Пример 22
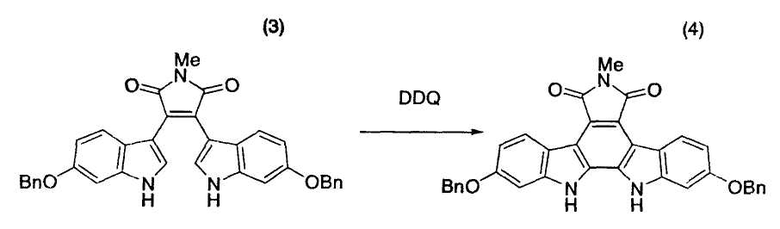
Толуол (75,3 мл) и бис-индольное соединение (3) (3,00 г, 5,42 ммоль) помещали в атмосфере азота в трехгорлую колбу емкостью 300 мл, оснащенную магнитной мешалкой, холодильником Димрота и термометром, и нагревали смесь до 70°C. К полученной смеси в течение одного часа добавляли раствор 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) (1,29 г, 5,69 ммоль) в N,N-диметилформамиде (24,0 мл). После промывания содержащей DDQ емкости N,N-диметилформамидом (6,0 мл) к реакционной смеси также добавляли промывную жидкость. Смесь перемешивали при 70°C в течение одного часа [этап измерения C] и подвергали анализу по методу высокоэффективной жидкостной хроматографии, который указывал на факт исчезновения исходного материала. После добавления метанола (134 мл) в течение 2 часов реакционный раствор перемешивали при 70°C в течение одного часа, охлаждали до 25°C и дополнительно перемешивали при той же самой температуре в течение ночи. Реакционную смесь фильтровали и промывали метанолом (15 мл×2) с получением желтого твердого вещества, которое сушили в вакууме в течение ночи при 25°C с получением целевого индолкарбазольного производного (4) в виде неочищенных желтых кристаллов (партия 3,08 г).
Пример 23
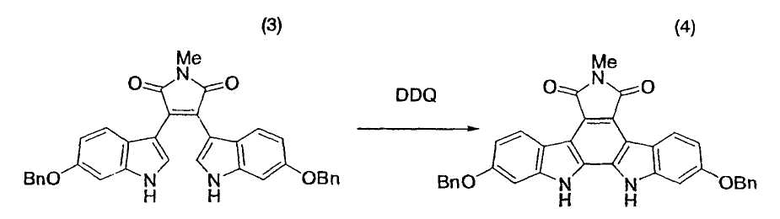
Толуол (28,6 кг) и бис-индольное соединение (3) (1,50 кг, 2,71 моль) помещали в атмосфере азота в реакционный сосуд емкостью 80 л и промывали внутреннюю стенку сосуда толуолом (3,9 кг). После добавления промывной жидкости полученную суспензию нагревали до 110°C и добавляли к полученной смеси в течение 1 часа раствор 2,3-дихлор-5,6-дициано-1,4-бензохинона (DDQ) (0,65 кг, 2,86 моль) в толуоле (20,8 л). После промывания содержащей DDQ емкости толуолом (5,2 кг) к реакционному раствору также добавляли промывную жидкость. Смесь перемешивали при 110°C в течение одного часа и подвергали анализу по методу высокоэффективной жидкостной хроматографии, который указывал на факт исчезновения исходного материала. Смесь охлаждали до 25°C в течение получаса [этап измерения D] и перемешивали при той же самой температуре в течение одного часа. Реакционную смесь фильтровали и промывали толуолом (13 кг) с получением коричневого твердого вещества (2,07 кг), которое сушили в вакууме при 60°C в течение ночи. Твердое вещество суспендировали в N,N-диметилформамиде (25,4 кг) и перемешивали суспензию при 100°C-105°C в течение одного часа. Суспензию охлаждали до 25°C в течение одного часа и перемешивали в течение ночи [этап измерения E]. Реакционный раствор фильтровали, промывали N,N-диметилформамидом (9,8 кг) и метанолом (8,2 кг) и сушили при 60°C в течение ночи с получением желтого твердого вещества (1,51 кг). Желтое твердое вещество суспендировали в диметилсульфоксиде (16,7 кг). Суспензию нагревали до 60°C для растворения полученного выше твердого вещества. К суспензии последовательно добавляли метанол (5,6 кг) и затравочный кристалл (8,0 г) целевого индолкарбазольного производного (4). Суспензию перемешивали при 60°C-65°C в течение одного часа для вызревания кристаллов. Затем, после добавления метанола (18,0 кг) в течение 2 часов, смесь перемешивали при той же самой температуре в течение одного часа, охлаждали до 25°C и перемешивали в течение ночи. Реакционную смесь фильтровали, промывали метанолом (12 кг), сушили в вакууме при 60°C в течение ночи с получением целевого индолкарбазольного производного (4) в виде желтых кристаллов (1,29 кг, выход 86%). Представленный в скобках термин «этап измерения» означает этап, на котором проводили измерение цианистого водорода, и результаты измерения цианистого водорода представлены в следующих тестовых примерах.
ЯМР-спектр и ИК-спектр целевого соединения (4) в этом примере согласуется с таковыми для целевого соединения примера 21.
Тестовые примеры
Измерение цианистого водорода
Способ измерения:
Во время обработки выпускаемый из реакционного сосуда газообразный азот вводили в 0,05 н. раствор гидроксида натрия (на 1 г бис-индольного соединения (3) использовали 7 мл 0,05 н. раствора гидроксида натрия). Что касается полученного раствора гидроксида натрия, цианид-ион измеряли с применением ион-индикаторной бумаги (CN-) производства ADVANTEC.
Пример 24
Бис-индольное соединение (3) (500 мг, 0,903 ммоль), порошкообразный 5% палладий/угольный порошок (384 мг, 0,181 ммоль) и толуол (22 мл) помещали на воздухе в баклажановидную колбу емкостью 50 мл, оснащенную магнитной мешалкой. Смесь нагревали в масляной бане при 105°C, перемешивали при той же самой температуре в течение 4 суток, охлаждали и упаривали досуха. После добавления к остатку диметилсульфоксида (40 мл) нерастворимые материалы удаляли путем фильтрации. Фильтрат подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о получении целевого соединения (4) в виде раствора в диметилсульфоксиде с выходом 66% (330 мг).
ЯМР-спектр и ИК-спектр целевого соединения (4) в этом примере согласуется с таковыми для целевого соединения примера 21.
Пример 25
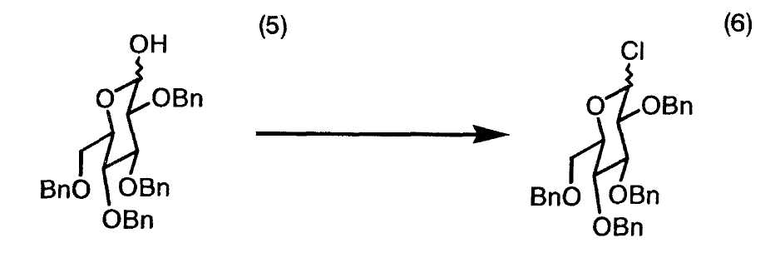
2,3,4,6-O-Тетрабензил-D-глюкопиранозу (5) (100,00 г, 185 ммоль) растворяли при 23°C в N,N-диметилформальдегиде (360 мл). Раствор охлаждали до 9°C и в течение 15 минут добавляли к нему тионилхлорид (16,2 мл, 222 ммоль), в результате чего температура повышалась до 20°C. Полученный раствор нагревали до 30°C и выдерживали в течение одного часа. Раствор охлаждали до -10°C и добавляли 10 мас.% раствор гидроксида калия (приблизительно 150 мл), поддерживая температуру ниже 0°C. Раствор нагревали до 22°C и разделяли на органический слой и водный слой. Водный слой экстрагировали трет-бутилметиловым эфиром (300 мл×1). Полученный ранее органический слой и трет-бутилметилэфирный экстракт объединяли, и последовательно промывали раствор насыщенным солевым раствором (150 мл×1) и водой (200 мл×1). Раствор концентрировали в вакууме до объема 350 мл, содержащего 1-хлор-2,3,4,6-тетрабензил-1-дезокси-D-глюкопиранозу (6). Неочищенный концентрат служил в качестве исходного материала для нужд примера 26.
Пример 26
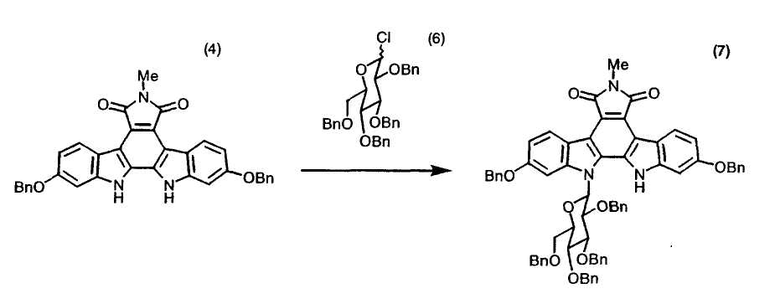
Полученное в примере 21 индолкарбазольное производное (4) (72,00 г, 131 ммоль) растворяли в трет-бутилметиловом эфире (600 мл). Раствор перемешивали при 23°C в течение 10 минут и к этому раствору добавляли раствор (350 мл), содержащий 1-хлор-2,3,4,6-тетрабензил-1-дезокси-D-глюкопиранозу (6), полученную в примере 25. Раствор перемешивали в течение 10 минут и добавляли 45 мас.% раствор гидроксида калия (300 мл). Раствор перемешивали в течение 10 минут и постепенно в течение 22 минут добавляли 40 мас.% Aliquat (зарегистрированный товарный знак) 336 (наименование продукта) в трет-бутилметиловом эфире (полученный путем растворения Aliquat 336 (72 г) в трет-бутилметиловом эфире (110 г)). Смесь перемешивали при 23°C в течение 6 часов и добавляли к ней воду (350 мл). Смесь перемешивали в течение 5 минут и разделяли на органический слой и водный слой. Водный слой экстрагировали трет-бутилметиловым эфиром (300 мл×1). Трет-бутилметилэфирный слой и органический слой объединяли, промывали 10 мас.% водной лимонной кислотой (300 мл×1), а затем водой (300 мл×1). Органический раствор перемешивали при 22°C в течение ночи для выпадения в осадок кристаллов целевого индолокарбазольного соединения (7). Полученную суспензию концентрировали при атмосферном давлении до объема приблизительно 625 мл и охлаждали до 23°C. К суспензии постепенно в течение одного часа добавляли метанол (225 мл), охлаждали суспензию до -5°C и перемешивали в течение 45 минут с получением кристаллов. Кристаллы собирали путем фильтрации, промывали холодным метанолом/трет-бутилметиловым эфиром (1:1) (400 мл×2) и сушили в вакууме при 25°C-40°C.
Анализ полученных кристаллов по методу высокоэффективной жидкостной хроматографии показал, что содержание целевого индолокарбазольного соединения (7) составило 99%.
Использованный в этом примере Aliquat (зарегистрированный товарный знак) 336 производства Aldrich Chemical Co., Inc. представляет собой трикаприлметиламмонийхлорид.
Пример 27
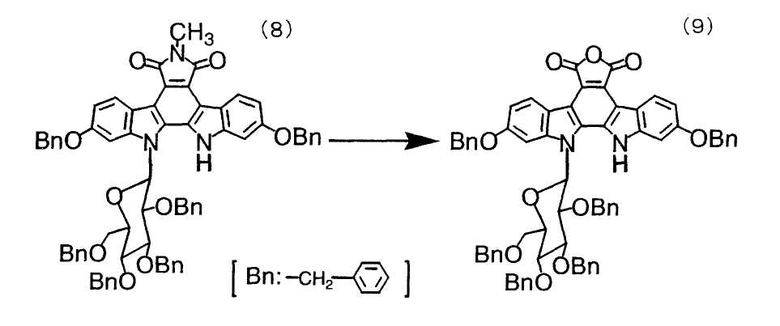
Этанол (36 мл) добавляли в четырехгорлую колбу емкостью 300 мл, оснащенную мешалкой и термометром, и добавляли к нему при перемешивании 12,13-дигидро-2,10-дибензилокси-13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]пирроло[3,4-c]карбазол-6-метил-5,7(6H)-дион (8) (670 мг, 0,62 ммоль). Смесь перемешивали при комнатной температуре в течение одного часа и при той же самой температуре в течение 20 минут добавляли по каплям 5 н. водный гидроксид калия (8 мл). Смесь перемешивали при температуре смеси 60°C в течение 4 часов и при комнатной температуре в течение ночи с получением коричневого раствора, к которому добавляли толуол (20 мл). После добавления по каплям при той же самой температуре в течение 30 минут 1,0 н. соляной кислоты (62 мл) для корректировки значения pH до 2,6 получали желтый раствор. К нему добавляли тетрагидрофуран (10 мл) и перемешивали смесь в течение 6 часов. Водный слой (нижний слой) отделяли, последовательно промывали органический слой очищенной водой (10 мл×2) и насыщенным солевым раствором (10 мл), сушили над безводным сульфатом натрия (5 г) и фильтровали. Фильтрат концентрировали в вакууме с получением 12,13-дигидро-2,10-дибензилокси-13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]карбазол-5,6-дикарбонового ангидрида (0,63 г) в виде маслянистого желтого остатка с выходом 85%.
1H-ЯМР (270 МГц, CDCl3), (ч/млн): 10,79 (1H, с), 9,04 (1H, д, J=9,2 Гц), 8,95 (1H, д, J=9,6 Гц), 7,26 (32H, м), 6,17 (2H, д, J=7,3 Гц), 5,85 (1H, д, J=8,2 Гц), 4,89 (10H, м), 4,32 (1H, т, J=8,9 Гц), 3,96 (6H, м), 3,13 (1H, д, J=10,2 Гц).
Пример 28
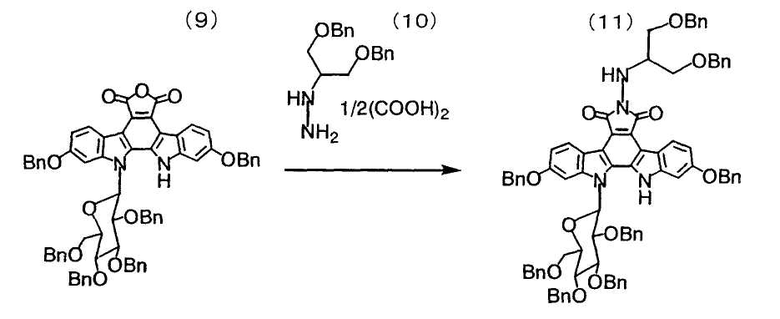
Смесь полученного в примере 27 12,13-дигидро-2,10-дибензилокси-13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]карбазол-5,6-дикарбонового ангидрида (9) (1,50 г, 1,41 ммоль), гемиоксалата N-(1-бензилоксиметил-2-бензилоксиэтил)гидразина (10) (609 мг, 1,84 ммоль) и N,N-диметилацетамида (14 мл) подвергали дегазации и замещали атмосферу азотом. Смесь нагревали до 62°C и по каплям добавляли триэтиламин (0,26 мл, 1,84 ммоль). После перемешивания при той же самой температуре в течение 3 часов реакционную смесь охлаждали до комнатной температуры и добавляли к ней метил-трет-бутиловыйэфир (10 мл) и воду (7 мл). Органический слой отделяли, промывали водой, сушили над сульфатом натрия и фильтровали. Растворитель удаляли путем выпаривания в вакууме с получением целевого соединения (11).
1H-ЯМР (270 МГц, CDCl3, ч/млн): 10,63 (1H, ушир.с), 9,24 (1H, ушир.д, J=9,6 Гц), 9,16 (1H, ушир.д, J=9,6 Гц), 7,50-6,84 (42H, м), 6,20 (2H, ушир.д, J=7,6 Гц), 5,84 (1H, д, J=8,6 Гц), 5,33 (1H, ушир.д, J=3,0 Гц), 5,21 (1H, д, J=12,2 Гц), 5,19 (1H, д, J=11,9 Гц), 5,16 (1H, д, J=12,2 Гц), 5,08 (1H, д, J=11,9 Гц), 5,08 (1H, д, J=10,9 Гц), 4,96 (1H, д, J=10,9 Гц), 4,89 (1H, д, J=10,9 Гц), 4,85 (1H, д, J=10,9 Гц), 4,72 (1H, д, J=12,9 Гц), 4,68 (1H, д, J=12,9 Гц), 4,62-4,48 (4H, м), 4,33 (1H, дд, J=9,6, 9,6 Гц), 4,06-3,77 (7H, м), 3,72 (4H, д, J=5,6 Гц), 3,04 (1H, д, J=9,9 Гц).
1H-ЯМР (68 МГц, CDCl3, ч/млн): 168,8, 168,7, 159,4, 159,3, 143,2, 142,9, 138,0, 137,9, 137,6, 136,9, 136,8, 136,6, 136,0, 130,2, 128,7, 128,6, 128,5, 128,4, 128,3, 128,2, 128,2, 128,1, 128,0, 127,9, 127,8, 127,7, 127,6, 127,5, 127,4, 127,3, 126,9, 126,6, 119,4, 119,1, 118,0, 116,9, 116,7, 116,1, 110,4, 96,7, 96,3, 85,8, 84,7, 80,9, 77,4, 77,2, 76,0, 75,9, 75,4, 74,9, 73,9, 73,3, 73,2, 70,7, 70,4, 69,9, 69,8, 66,7, 58,7, 49,4, 30,9, 27,0.
Пример 29
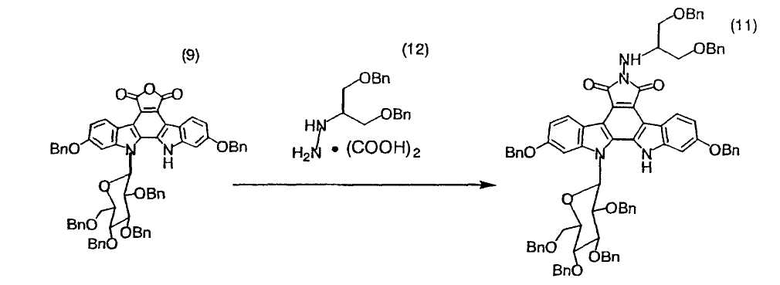
Смесь полученного в примере 27 12,13-дигидро-2,10-дибензилокси-13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]карбазол-5,6-дикарбонового ангидрида (9) (1,30 г, 1,23 ммоль), монооксалата N-(1-бензилоксиметил-2-бензилоксиэтил)гидразина (599 мг, 1,59 ммоль) и N,N-диметилацетамида (12,3 мл) подвергали дегазации. Смесь нагревали до 45°C в атмосфере азота и по каплям добавляли триэтиламин (34,1 мкл, 0,25 ммоль). Реакционную смесь перемешивали при той же самой температуре в течение 16 часов и охлаждали до комнатной температуры. После добавления метил-трет-бутиловогоэфира (25 мл) и воды (6,1 мл) органический слой отделяли, промывали водой (5,2 мл×4), сушили над сульфатом натрия и фильтровали. Фильтрат подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о получении целевого соединения (11) (1,50 г) в виде раствора с выходом 92%.
Так как ЯМР-спектр и ИК-спектр полученного в этом примере целевого соединения (11) согласовывался с таковыми для целевого соединения примера 28, соединение (11) было охарактеризовано как 12,13-дигидро-2,10-дибензилокси-6-N-(1-бензилоксиметил-2-бензилоксиэтиламино)13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]пирроло[3,4-c]карбазол-6-метил-5,7(6H)-дион.
Пример 30
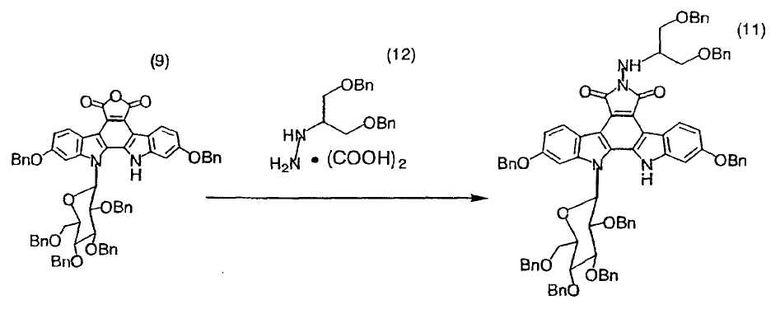
Смесь полученного в примере 27 12,13-дигидро-2,10-дибензилокси-13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]карбазол-5,6-дикарбонового ангидрида (9) (1,30 г, 1,23 ммоль), монооксалата N-(1-бензилоксиметил-2-бензилоксиэтил)гидразина (599 мг, 1,59 ммоль), магнийсульфата (1,48 г, 12,3 ммоль) и N,N-диметилацетамида (12,3 мл) подвергали дегазации. Смесь нагревали до 45°C в атмосфере азота и по каплям добавляли триэтиламин (446 мкл, 3,20 ммоль). Реакционную смесь перемешивали при той же самой температуре в течение 10 часов и охлаждали до комнатной температуры. После добавления метил-трет-бутиловогоэфира (25 мл) и воды (6,1 мл) корректировали значение pH водного слоя до pH 3,5 добавлением 2н HCl (1,34 мл). Органический слой отделяли, четырежды промывали водой (5,2 мл), сушили над сульфатом натрия и фильтровали. Фильтрат подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о получении целевого соединения (11) (1,50 г) в виде раствора с выходом 92%.
Так как ЯМР-спектр и ИК-спектр полученного в этом примере целевого соединения (11) согласовывался с таковыми для целевого соединения примера 28, соединение (11) было охарактеризовано как 12,13-дигидро-2,10-дибензилокси-6-N-(1-бензилоксиметил-2-бензилоксиэтиламино)-13-(β-D-2,3,4,6-тетра-O-бензилглюкопиранозил)-5H-индоло[2,3-a]пирроло[3,4-c]карбазол-5,7(6H)-дион.
Пример 31
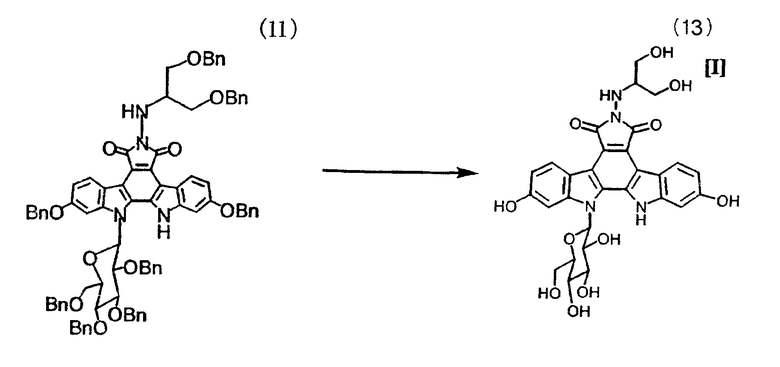
10% палладий/углерод (50 масс./масс.%, 112 г) помещали в аппарат для гидрирования и добавляли к нему 12-β-D-(2,3,4,6-тетра-O-бензилглюкопиранозил)-12,13-дигидро-2,10-дибензилокси-6-[[(2-бензилокси-1-(бензилоксиметил)этил)амино]-5H-индоло[2,3-a]пирроло[3,4-c]карбазол-5,7(6H)-дион (11) в тетрагидрофуране (175 г/л, 6,4 л, 1,12 кг), изопропанол (7,9 л) и 3 н. HCl (224 мл). Смесь гидрировали при давлении водорода 40 Па/дюйм2 при энергичном перемешивании при 40°C в течение 4-14 часов до тех пор, пока теоретическое поглощение водорода не составило 110%. Реакционный раствор охлаждали до 25°C и фильтровали с применением Solka Floc (зарегистрированный товарный знак) для того, чтобы собрать твердые вещества, такие как катализатор. Эти твердые вещества промывали изопропанолом/тетрагидрофураном (3:2) (3 л×1). Фильтрат и промывную жидкость объединяли, корректировали значение pH раствора до pH 2,5 добавлением 1M триэтиламина/изопропанола (приблизительно 600 мл). После добавления воды (4,0 л) раствор концентрировали при атмосферном давлении до объема 7,5 л, затем концентрировали при добавлении изопропанола/воды (4:1) (6,5 л) и окончательно концентрировали до содержания воды 20 мас./об.%, дополнительно добавляя изопропанол (приблизительно 9 л) и поддерживая приблизительный объем 7,5 л. Концентрат выдерживали при 70°C и добавляли к нему суспензию затравочных кристаллов (5 г) в изопропаноле (50 мл). Смесь выдерживали при 70°C в течение одного часа и в течение 1,5 часа добавляли изопропанол (5,0 л). Полученную смесь выдерживали при 70°C в течение 9-24 часов для выпадения кристаллов в осадок. Полученную суспензию концентрировали при атмосферном давлении, добавляя изопропанол (17 л) до достижения в концентрате содержания воды 3 мас./об.%. Суспензию выдерживали при 70°C в течение 3-6 часов, охлаждали до 22°C и выдерживали при той же самой температуре в течение одного часа. Суспензию фильтровали с получением на фильтре осадка, который последовательно промывали изопропанолом (2,5 л) и метанолом (1,5 л). Осадок с фильтра сушили в вакууме при 38°C в течение 6 часов с получением оранжевых кристаллов с выходом 80% или выше (содержание: 99% или выше). Так как ЯМР-спектр и ИК-спектр полученных в этом примере оранжевых кристаллов согласовывался с таковыми для соединения примера 6 в WO 95/30682, полученное в этом примере целевое соединение (13) было охарактеризовано как 12,13-дигидро-2,10-дигидрокси-6-N-(1-гидроксиметил-2-гидроксиэтиламино)-13-(β-D-глюкопиранозил)-5H-индоло[2,3-a]пирроло[3,4-c]карбазол-5,7(6H)-дион.
Условия проведения анализа по методу высокоэффективной жидкостной хроматографии: разделительная колонка YMC ODS-AQ (250×4,6 мм); скорость потока - 1,5 мл/мин; детектирование на длине волны 228 нм; подвижная фаза: A=0,1% H3PO4/вода, B=ацетонитрил; количество наносимого образца - 10 мкл; температура при измерении - 25°C.
Пример 32
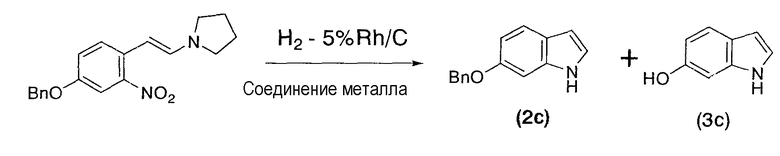
3-Бензилокси-6-(2-пирролидинилвинил)нитробензол (1,00 г, 3,08 мл), 5% родий/угольный порошок (63,5 мг, 0,0309 ммоль), ацетат железа(II) (5,6 мг, 0,0309 ммоль) в качестве соединения металла и тетрагидрофуран (20 мл) помещали в атмосфере азота в баклажановидную колбу емкостью 30 мл, оснащенную магнитной мешалкой, а затем замещали газообразный азот газообразным водородом. Полученную суспензию перемешивали при комнатной температуре в течение 41 часов в атмосфере водорода, затем замещали газообразный водород газообразным азотом и перемешивали в течение ночи в атмосфере азота. Реакционную смесь фильтровали для получения твердого вещества, которое промывали тетрагидрофураном. Фильтрат и промывную жидкость объединяли с получением коричневого раствора (79,83 г). Раствор подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о получении целевого 6-бензилоксииндола (2c) (661 мг, выход 96%) и попутно образовавшегося 6-гидроксииндола (3c) (2 мг, выход 0,6%).
6-Бензилоксииндол:
1H-ЯМР (500 МГц, ДМСО-d6, ч/млн): 10,90 (ушир.с, 1H), 7,49 (д, J=7,5 Гц, 1H), 7,44 (д, J=8,6 Гц, 1H), 7,40 (дд, J=7,5, 7,5 Гц, 1H), 7,33 (дд, J=7,5, 7,5 Гц, 1H), 7,21 (ушир.дд, J=2,4, 2,4 Гц, 1H), 7,01 (ушир.м, 1H), 6,77 (дд, J=1,8, 8,6 Гц, 1H), 6,36 (ушир.м, 1H), 5,12 (с, 2H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 154,7, 138,0, 136,8, 128,7, 128,0, 127,9, 124,4, 122,5, 120,9, 110,1, 101,3, 96,3, 69,9.
6-Гидроксииндол:
1H-ЯМР (500 МГц, ДМСО-d6, ч/млн): 10,68 (ушир.с, 1H), 8,88 (ушир.с, 1H), 7,33 (д, J=8,4 Гц, 1H), 7,12 (м, 1H), 6,79 (м, 1H), 6,56 (дд, J=8,4, 1,5 Гц, 1H), 6,30 (м, 1H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 153,78, 137,90, 124,00, 121,90, 121,17, 110,37, 101,75, 97,36.
Примеры 33-35
Следуя методике, сходной с описанной в примере 32, 2-(2-пирролидинилвинил)нитробензол гидрировали в присутствии катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок и ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла. Индол получали с высоким выходом, как представлено ниже в таблице 3.
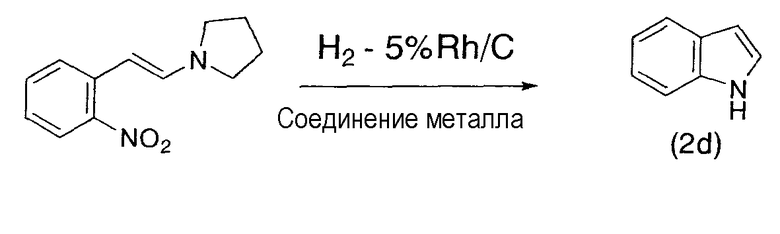
Индол:
Продукт (2d) был охарактеризован в результате сравнения со спектрами различных коммерчески доступных продуктов.
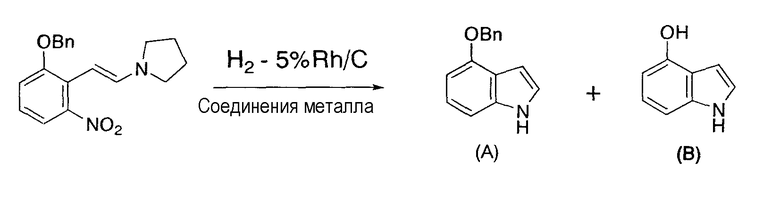
Примеры 36-38
Следуя методике, сходной с описанной в примере 32, 3-бензилокси-2-(2-пирролидинилвинил)нитробензол в качестве исходного материала восстанавливали в присутствии катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок и ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла.
Результаты представлены в следующей таблице 4, и по сравнению с примером сравнения 3 в случае применения восстановителя по настоящему изобретению 4-бензилоксииндол получали с более высоким выходом, а 4-гидроксииндол побочно образовывался с меньшим выходом.
(1 мол.%)
(20 мол.%)
(5 мол.%)
4-Бензилоксииндол:
1H-ЯМР (500 МГц, CDCl3, ч/млн): 8,12 (ушир.с, 1H), 7,49 (ушир.д, J=7,5 Гц, 1H), 7,44 (ушир.т, J=7,5 Гц, 1H), 7,37 (м, 1H), 7,14 (дд, J=8,0, 7,8 Гц, 1H), 7,10 (м, 1H), 7,04 (д, J=8,0 Гц, 1H), 6,77 (м, 1H), 6,64 (д, J=7,7 Гц, 1H), 5,28 (с, 2H).
13C-ЯМР (126 МГц, CDCl3, ч/млн): 152,88, 137,94, 137,66, 128,81, 128,05, 127,66, 123,00, 119,23, 105,02, 101,46, 100,40, 70,27.
4-Гидроксииндол:
1H-ЯМР (500 МГц, CDCl3, ч/млн): 8,19 (ушир.с, 1H), 7,13 (м, 1H), 7,06 (дд, J=8,0, 7,6 Гц, 1H), 7,01 (д, J=8,0 Гц, 1H), 6,62 (м, 1H), 6,54 (д, J=7,6 Гц, 1H), 5,22 (ушир.с, 1H).
13C-ЯМР (126 МГц, CDCl3, ч/млн): 149,08, 137,86, 123,18, 123,03, 117,64, 104,37, 104,29, 98,91.
Примеры 39-41
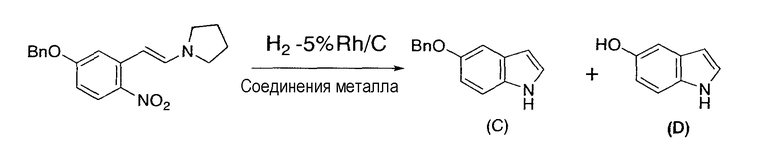
Следуя методике, сходной с описанной в примере 32, 4-бензилокси-2-(2-пирролидинилвинил)нитробензол в качестве исходного материала восстанавливали в присутствии катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок и ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла.
Результаты представлены в следующей таблице 5, и по сравнению с примером сравнения 4 в случае применения восстановителя по настоящему изобретению 5-бензилоксииндол получали с более высоким выходом, а 5-гидроксииндол побочно образовывался с меньшим выходом.
(мол.%)
(1 мол.%)
(20 мол.%)
(5 мол.%)
5-Бензилоксииндол:
1H-ЯМР (500 МГц, CDCl3, ч/млн): 7,92 (ушир.с, 1H), 7,44 (ушир.д, J=7,5 Гц, 1H), 7,36-7,26 (м, 2H), 7,17-7,15 (м, 2H), 7,03 (м, 1H), 6,92 (дд, J=8,8, 2,4 Гц, 1H), 6,42 (м, 1H), 5,06 (с, 2H).
13C-ЯМР (126 МГц, CDCl3, ч/млн): 153,64, 137,99, 131,45, 128,82, 128,53, 128,08, 127,89, 125,30, 113,29, 112,06, 104,32, 102,59, 71,26.
5-Гидроксииндол:
1H-ЯМР (500 МГц, ДМСО-d6, ч/млн): 10,76 (ушир.с, 1H), 8,61 (ушир.с, 1H), 7,24 (м, 1H), 7,21 (д, J=8,6 Гц, 1H), 6,89 (ушир.д, J=2,0 Гц, 1H), 6,65 (дд, J=8,6, 2,0 Гц, 1H), 6,27 (м, 1H).
13C-ЯМР (126 МГц, ДМСО-d6, ч/млн): 151,42, 131,38, 129,30, 126,37, 112,50, 112,20, 104,76, 101,12.
Примеры 42-44
Следуя методике, сходной с описанной в примере 32, 5-бензилокси-2-(2-пирролидинилвинил)нитробензол в качестве исходного материала восстанавливали в присутствии катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок и ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла.
Результаты представлены в следующей таблице 6, и по сравнению с примером сравнения 5 в случае применения восстановителя по настоящему изобретению 6-бензилоксииндол получали с более высоким выходом, а 6-гидроксииндол побочно образовывался с меньшим выходом.
(1 мол.%)
(20 мол.%)
(5 мол.%)
Пример 45
Нитробензол (379 мг, 3,08 ммоль), бензилфениловый эфир (567 мг, 3,08 ммоль) в качестве субстрата, 5% родий/угольный порошок (63,5 мг, 0,0309 ммоль), ацетат железа(II) (5,6 мг, 0,0309 ммоль) в качестве соединения металла и тетрагидрофуран (20 мл) помещали в атмосфере азота в баклажановидную колбу емкостью 30 мл, оснащенную магнитной мешалкой. После замещения газообразного азота газообразным водородом полученную суспензию перемешивали при комнатной температуре в течение 16 часов в атмосфере водорода и после замены газообразного водорода газообразным азотом, полученную суспензию перемешивали в течение ночи в атмосфере азота. После удаления сухого остатка путем фильтрации остаток промывали тетрагидрофураном. Фильтрат и промывную жидкость объединяли с получением коричневого раствора. Раствор подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о том, что анилин (A'), бензилфениловый эфир в качестве извлекаемого субстрата и фенол (B') в качестве восстановленного субстрата получали с выходом 86% (порция 247 мг), с извлечением 89% (порция 503 мг) и с выходом 1% (порция 4 мг), соответственно.
Каждый компонент полученного раствора был охарактеризован в результате сравнения со спектрами различных коммерчески доступных продуктов.
Примеры 46-48
Следуя методике, сходной с описанной в примере 45, реакцию восстановления осуществляли с применением катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок, ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла, и бензилфенилового эфира в качестве субстрата. Результаты представлены в следующей таблице 7.
Анилин и восстановленный субстрат (фенол) были охарактеризованы в результате сравнения со спектрами различных коммерчески доступных продуктов.
(1 мол.%)
(5 мол.%)
Выход соединения (A') представляет собой выход анилина, а выход соединения (B') представляет собой выход фенола.
Примеры 49-51
Следуя методике, сходной с описанной в примере 45, реакцию восстановления осуществляли с применением катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок, ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла, и хлорбензола в качестве субстрата. Результаты представлены ниже в таблице 8.
Анилин и восстановленный субстрат (бензол) были охарактеризованы в результате сравнения со спектрами различных коммерчески доступных продуктов.
(20 мол.%)
(1 мол.%)
(5 мол.%)
Выход соединения (C') представляет собой выход анилина, а выход соединения (D') представляет собой выход бензола.
Примеры 52-54
Следуя методике, сходной с описанной в примере 45, реакцию восстановления осуществляли с применением катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок, ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла, и бензальдегида в качестве субстрата. Результаты представлены ниже в таблице 9.
Анилин и восстановленный субстрат (бензиловый спирт) были охарактеризованы в результате сравнения со спектрами различных коммерчески доступных продуктов.
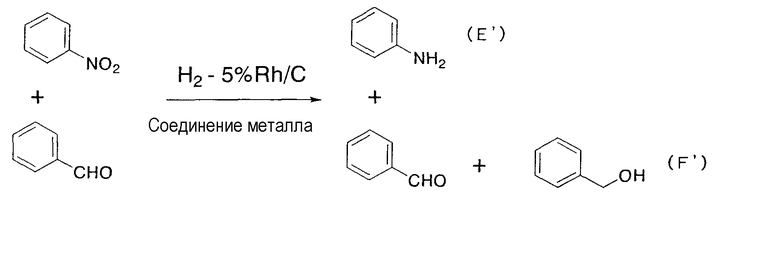
(20 мол.%)
(1 мол.%)
Выход соединения (E') представляет собой выход анилина, а выход соединения (F') представляет собой выход бензилового спирта.
Пример 55
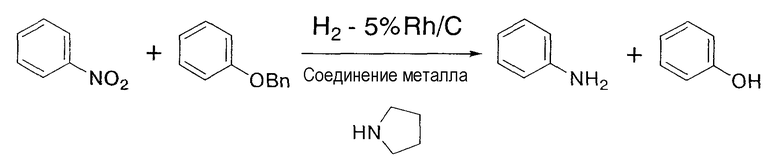
Нитробензол (379 мг, 3,08 ммоль), бензилфениловый эфир (567 мг, 3,08 ммоль), пирролидин (0,257 мл, 3,08 ммоль), 5% родий/угольный порошок (63,5 мг, 0,0309 ммоль), ацетат железа(II) (5,6 мг, 0,0309 ммоль) в качестве соединения металла и тетрагидрофуран (20 мл) помещали в атмосфере азота в баклажановидную колбу емкостью 30 мл, оснащенную магнитной мешалкой. После замещения газообразного азота газообразным водородом полученную суспензию перемешивали при комнатной температуре в течение 5 часов в атмосфере водорода и перемешивали в течение ночи в атмосфере азота. После удаления сухого остатка путем фильтрации остаток промывали тетрагидрофураном. Фильтрат и промывную жидкость объединяли с получением коричневого раствора. Раствор подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о получении анилина с выходом 88% (порция 252 мг) и побочно образовавшегося фенола, полученного путем восстановления бензилфенилового эфира, с выходом 0,4% (порция 1 мг).
Примеры 56-58
Следуя методике, сходной с описанной в примере 55, реакцию восстановления осуществляли с применением катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок, ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла, и пирролидин в качестве основания. Результаты представлены ниже в таблице 10.
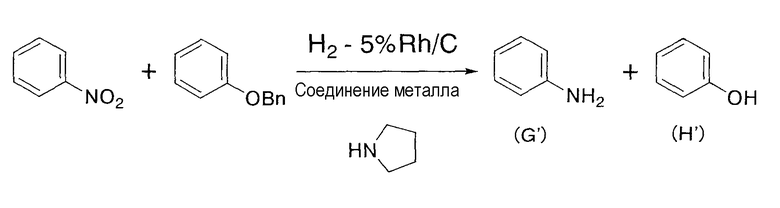
(20 мол.%) Пирролидин
(1 экв.)
(1 мол.%) Пирролидин
(1 экв.)
(5 мол.%) Пирролидин
(1 экв.)
Пирролидин
(1 экв.)
Пирролидин
(1 экв.)
Пирролидин
(1 экв.)
Выход соединения (G') представляет собой выход анилина, а выход соединения (H') представляет собой выход фенола. Сокращение "экв." означает эквивалент.
Полученные выше результаты подтвердили, что избирательность каталитического восстановления функциональных групп может быть улучшена путем дополнительного введения амина в катализатор по настоящему изобретению.
Пример 59
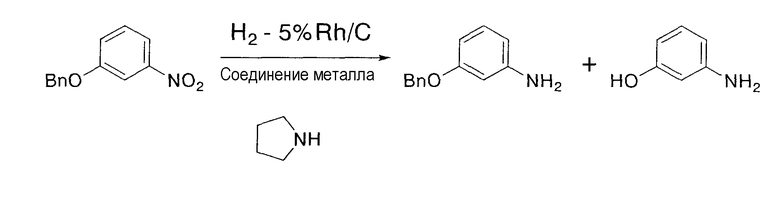
3-Бензилоксинитробензол (706 мг, 3,08 ммоль), пирролидин (0,257 мл, 3,08 ммоль), 5% родий/угольный порошок (190 мг, 0,0924 ммоль), гексагидрат нитрата никеля(II) (179 мг, 0,616 ммоль) и тетрагидрофуран (20 мл) помещали в атмосфере азота в баклажановидную колбу емкостью 30 мл, оснащенную магнитной мешалкой. После замещения газообразного азота газообразным водородом полученную суспензию перемешивали при комнатной температуре в течение 2,5 часов в атмосфере водорода и замещали атмосферу водорода атмосферой азота. Полученное твердое вещество собирали путем фильтрации и промывали тетрагидрофураном. Фильтрат и промывную жидкость объединяли с получением коричневого раствора. Раствор подвергали анализу по методу высокоэффективной жидкостной хроматографии, который свидетельствовал о получении 3-бензилоксианилина с выходом 92% (порция 565 мг) и 3-гидроксианилина с выходом 1% (порция 3 мг).
Примеры 60-66
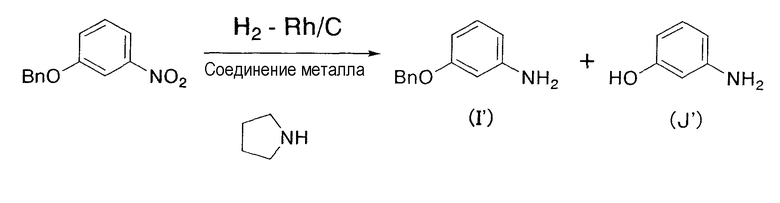
Следуя методике, сходной с описанной в примере 59, реакцию восстановления осуществляли с применением ацетата железа(II), нитрата никеля(II) или ацетилацетоната кобальта(III) в качестве соединения металла. Результаты представлены ниже в таблице 11.
(1 мол.%)
(1 мол.%)
(5 мол.%)
(5 мол.%)
15
16
17
Выход соединения (I') представляет собой выход (%) 3-бензилоксианилина; выход соединения (J') представляет собой выход 3-гидроксианилина.
Результаты примеров 60-66 подтвердили, что в случае добавления амина, такого как пирролидин, к каталитической восстановительной системе по настоящему изобретению, содержащей катализатор родия на носителе и соли железа, соли никеля или соли кобальта в качестве соединения металла, наблюдается резкое возрастание скорости реакции, снижение вероятности восстановления способных к такому восстановлению функциональных групп, таких как бензилэфирная группа, избирательное восстановление нитрогруппы, и возрастание выхода.
Примеры 67-68
Следуя методике, сходной с описанной в примере 45, реакцию восстановления осуществляли с применением катализатора по настоящему изобретению, содержащего 5% родий/угольный порошок и ацетат железа(II), нитрат никеля(II) или ацетилацетонат кобальта(III) в качестве соединения металла, и стирола (K') в качестве субстрата. Результаты представлены в следующей таблице 12.
Анилин и восстановленный субстрат (этилбензол (L')) были охарактеризованы в результате сравнения со спектрами различных коммерчески доступных продуктов.
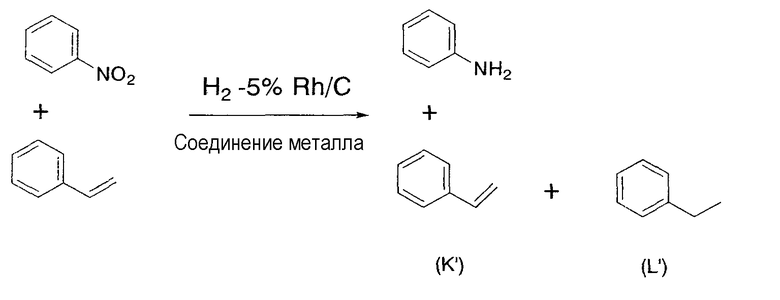
(1 мол.%)
(5 мол.%)
Выход соединения (K') представляет собой выход анилина, а выход соединения (L') представляет собой выход этилбензола.
В полученных выше результатах подтверждено, что в случае осуществления каталитического восстановления с применением катализатора по настоящему изобретению восстановление нитрогруппы и винильной группы происходит без восстановления бензилэфирной группы, заместителей хлора и альдегидной группы.
Поэтому, в случае осуществления каталитического восстановления с применением катализатора по настоящему изобретению, т.е. катализатора, содержащего катализатор родия на носителе и соли железа, соли никеля или соли кобальта в качестве соединения металла, восстановление нитрогруппы, алкенильной или алкинильной группы может быть избирательным без восстановления функциональных групп, таких как бензилэфирная группа, арилгалогениды, альдегиды, кетоны и т.д.
ПРИМЕНЯЕМОСТЬ В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Целевое соединение (I) по настоящему изобретению применимо в качестве лекарственного средства, и настоящее изобретение относится к выгодному в промышленном масштабе способу производства указанного соединения. Дополнительно, катализатор по настоящему изобретению может применяться в качестве катализатора в различных реакциях восстановления, делая возможным избирательное восстановление.
Настоящее изобретение относится к способу получения производного индолопирролокарбазола формулы (I) или его фармацевтически приемлемой соли, обладающего противоопухолевой активностью. Изобретение относится также к способу получения индольного соединения формулы (XII), в которой R1 - гидроксизащитная группа, или его фармацевтически приемлемой соли, отличающемуся тем, что осуществляют взаимодействие соединения формулы (XIII), в которой R1 определено выше, Ra и Rb независимо представляют собой С1-С7-алкил, или Ra и Rb вместе образуют С3-С6-алкиленовую группу, или его фармацевтически приемлемой соли, с газообразным водородом при 1-5 атм. в присутствии катализатора гидрирования (предложенного также в качестве нового), состоящего из соединения родия, соединения металла и, необязательно, амина, в инертном растворителе при комнатной температуре, причем соединение родия представляет собой 1-10% родий на угле, окиси алюминия, карбонате кальция или сульфате бария, и соединение металла представляет собой соль никеля (II), железа (II), железа (III), кобальта (II) или кобальта (III). Предложен также способ получения бис-индольного соединения формулы (VIII), в которой R1 - гидроксизащитная группа; Y - водород, С1-С7-алкил, фенил, бензилоксиметил или С7-С12-аралкил, или его фармацевтически приемлемой соли, заключающийся во взаимодействии индольного соединения формулы (XII), в которой R1 - гидроксизащитная группа, или его фармацевтически приемлемой соли с этилмагнийхлоридом или бутилмагнийхлоридом, или с соединением магния формулы (X), в которой Rd - бутил, в инертном растворителе, с последующим осуществлением взаимодействия полученного продукта с малеимидным соединением формулы (IX), в которой X - галоген, a Y определен выше, в инертном растворителе. 4 н. и 11 з.п. ф-лы, 12 табл.
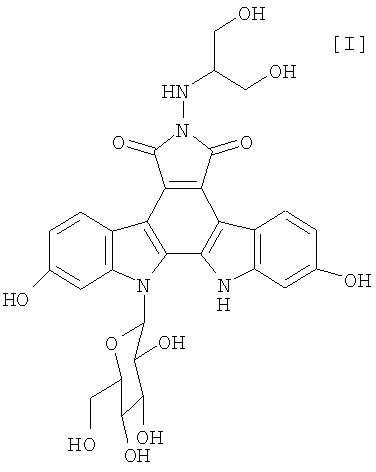
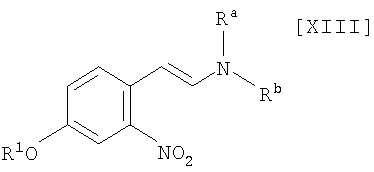
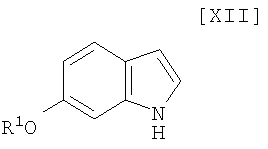
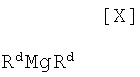
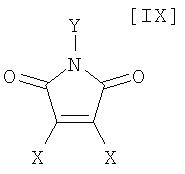
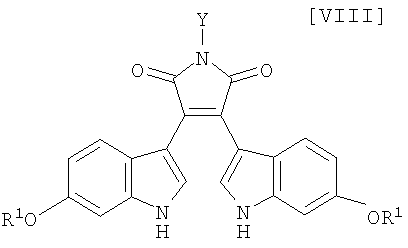
(i): стадию, в которой соединение формулы (XIII)
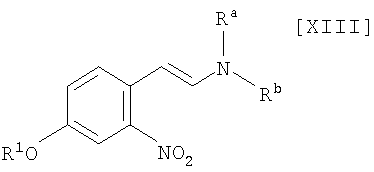
в которой R1 представляет собой гидроксизащитную группу, а каждый из Ra и Rb независимо представляет собой С1-С7-алкильную группу, или Ra иRb могут быть объединены вместе с образованием С3-С6-алкиленовой группы, или его фармацевтически приемлемую соль, подвергают взаимодействию с газообразным водородом при 1-5 атм в присутствии соединения родия, соединения металла и, необязательно, амина, в инертном растворителе при комнатной температуре,
причем соединение родия представляет собой 1-10% родий на угле, родий на окиси алюминия, родий на карбонате кальция или родий на сульфате бария, и
соединение металла представляет собой соль никеля (II), железа (II), железа (III), кобальта (II) или кобальта (III),
с получением индольного соединения формулы (XII):
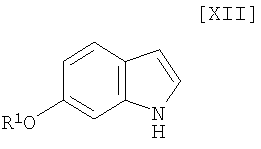
в котором значения R1 определены выше, или его фармацевтически приемлемой соли;
(ii): стадию взаимодействия полученного индольного соединения формулы (XII) или его фармацевтически приемлемой соли с магнийхлоридом формулы (XI):
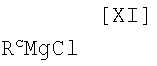
в которой Rc представляет собой этил или бутил; или с соединением магния формулы (X):
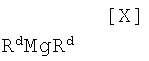
в которой Rd представляет собой бутил, в инертном растворителе, с последующим добавлением к полученной смеси малеимидного соединения формулы (IX):
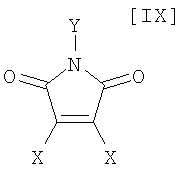
в которой X представляет собой атом галогена, и Y представляет собой атом водорода, С1-С7-алкильную группу, фенильную группу, бензилоксиметильную группу или С7-С12-аралкильную группу, с получением бис-индольного соединения формулы (VIII):
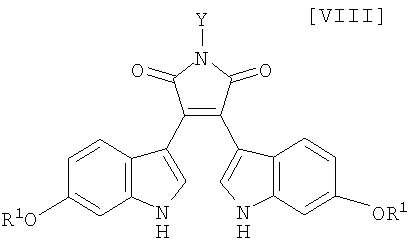
в которой значения для каждого R1 и Y определены выше, или его фармацевтически приемлемой соли;
(iii): стадию осуществления реакции замыкания цепи в полученном бис-индольном соединении (VIII) или его фармацевтически приемлемой соли с получением соединения формулы (VII):
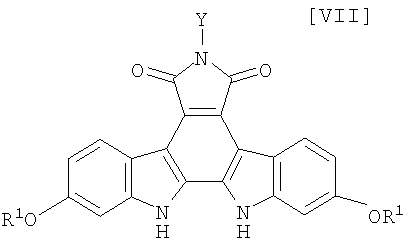
в которой значения для каждого R1 и Y определены выше, или его фармацевтически приемлемой соли;
(iv): стадию сочетания полученного соединения (VII) или его фармацевтически приемлемой соли с активированным производным глюкозы формулы (VI):
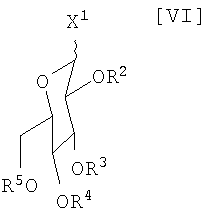
в которой каждый из R2, R3, R4 и R5 представляет собой гидроксизащитную группу, и X1 представляет собой атом галогена, с получением соединения формулы (V):
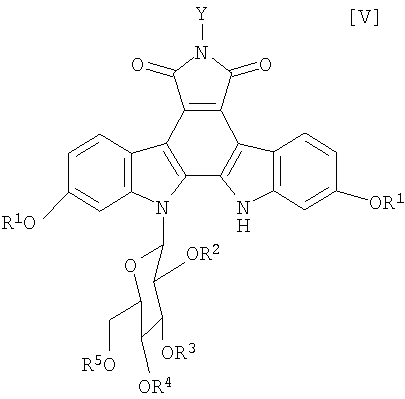
в которой значения для каждого R1, R2, R3, R4, R5 и Y определены выше, или его фармацевтически приемлемой соли;
(v): стадию обработки полученного соединения (V) или его фармацевтически приемлемой соли неорганическим основанием в инертном растворителе с получением соединения формулы (IV):
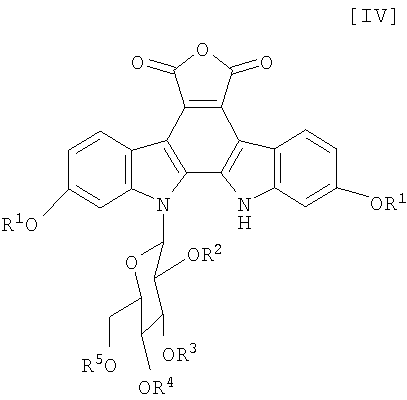
в которой значения для каждого из R1, R2, R3, R4 и R5 определены выше, или его фармацевтически приемлемой соли;
(vi): стадию взаимодействия соединения (IV) или его фармацевтически приемлемой соли с соединением формулы (III):
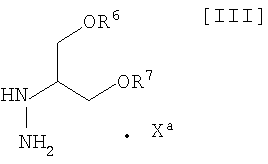
в которой каждый из R6 и R7 представляет собой гидроксизащитную группу, и Ха представляет собой молекулу кислоты, с получением соединения формулы (II):
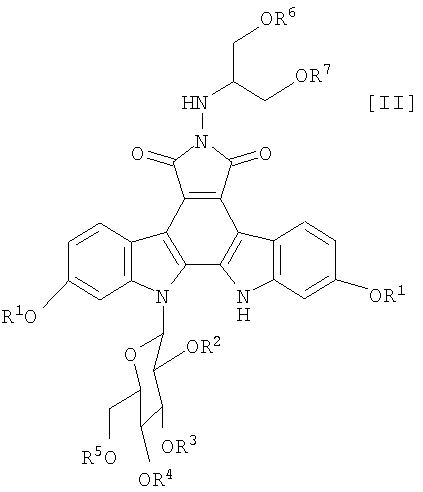
в которой значения для каждого R1, R2, R3, R4, R5, R6 и R7 определены выше, или его фармацевтически приемлемой соли; и
(vii): стадию удаления гидроксизащитных групп с полученного соединения (II) или его фармацевтически приемлемой соли с получением производного индолопирролокарбазола формулы (I):
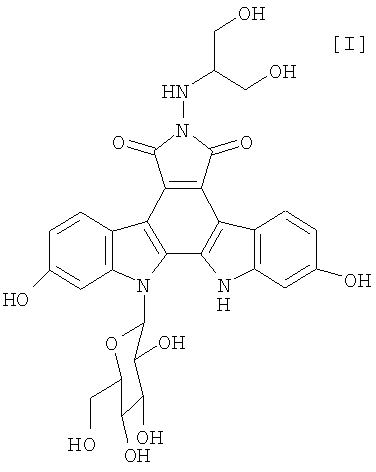
или его фармацевтически приемлемой соли.
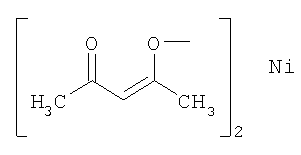 ,
,
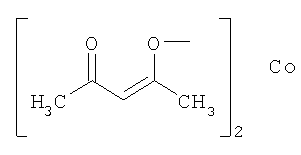 ,
,
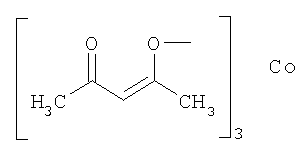 или
или
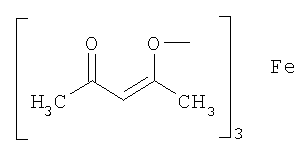 .
.
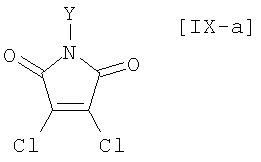
в которой Y представляет собой атом водорода, С1-С7-алкильную группу, фенильную группу, бензилоксиметильную группу или C7-C12-аралкильную группу.
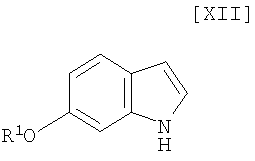
в которой R1 представляет собой гидроксизащитную группу, или его фармацевтически приемлемой соли, отличающийся тем, что осуществляют взаимодействие соединения, представленного формулой (XIII):
в которой значение R1 определено выше, и каждый из Ra и Rb независимо представляет собой С1-С7-алкильную группу, или Ra и Rb могут быть объединены вместе с образованием С3-С6-алкиленовой группы, или его фармацевтически приемлемой соли, с газообразным водородом при 1-5 атм в присутствии соединения родия и соединения металла и, необязательно, амина, в инертном растворителе при комнатной температуре,
причем соединение родия представляет собой 1-10% родий на угле, родий на окиси алюминия, родий на карбонате кальция или родий на сульфате бария, и
соединение металла представляет собой соль никеля (II), соль железа (II), соль железа (III), соль кобальта (II) или соль кобальта (III).
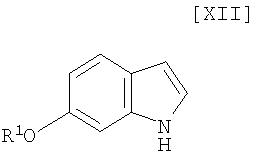
в которой R1 представляет собой гидроксизащитную группу, или его фармацевтически приемлемой соли с магнийхлоридом формулы (XI):
в которой Rc представляет собой этил или бутил, или с соединением магния формулы (X):
в которой Rd представляет собой бутил, в инертном растворителе, с последующим осуществлением взаимодействия полученного продукта с малеимидным соединением формулы (IX)
в которой X представляет собой атом галогена, и Y представляет собой атом водорода, С1-С7-алкильную группу, фенильную группу, бензилоксиметильную группу или С7-С12-аралкильную группу, в инертном растворителе с получением бис-индольного соединения формулы (VIII):
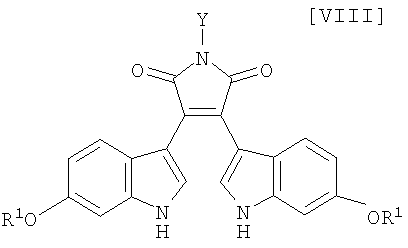
в которой значения для каждого R1 и Y определены выше, или его фармацевтически приемлемой соли.
в которой Y представляет собой атом водорода, С1-С7-алкильную группу, фенильную группу, бензилоксиметильную группу или С7-С12-аралкильную группу.
где соединение родия представляет собой 1-10% родий на угле, родий на окиси алюминия, родий на карбонате кальция или родий на сульфате бария, и
соединение металла представляет собой соль никеля (II), соль железа (II), соль железа (III), соль кобальта (II) или соль кобальта (III).
,
,
или
.
| US 5804564 A, 08.09.1998 | |||
| EP 760375 A1, 05.03.1997 | |||
| КОМПОЗИЦИЯ ДЛЯ ПРИГОТОВЛЕНИЯ ТЕСТА ДЛЯ ХЛЕБА ПШЕНИЧНОГО "ДАРЫ МОРЯ" | 2009 |
|
RU2399209C1 |
| Центробежный пульсатор | 1940 |
|
SU61036A1 |
| Устройство для измерения параметров диэлектрических материалов | 1985 |
|
SU1276966A1 |
| BIOSCIENCE, BIOTECHNOLOGY AND BIOCHEMISTRY, 2000, 64(4), 808-815 | |||
| Способ получения парафиновых и олефиновых углеводородов | 1972 |
|
SU496712A3 |
| Устройство для дражирования семян сельскохозяйственных культур | 1986 |
|
SU1400529A1 |
| КАТАЛИЗАТОР ДЛЯ ГИДРИРОВАНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ НА ОСНОВЕ РОДИЯ | 1971 |
|
SU428772A1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОФЕНИЛАЛКИЛОВЫХЭФИРОВ | 0 |
|
SU352459A1 |

