Область техники, к которой относится изобретение
Настоящее изобретение касается способа получения 3-замещенных 2-амино-5-цианобензойных кислот и их производных.
Предпосылки создания изобретения
Получение определенных 2-амино-5-цианобензойных кислот и их применение в качестве промежуточных соединений для получения соответствующих инсектицидных диамидов цианоантраниловой кислоты раскрыто (см., например, схему 9 в патентной публикации PCT WO 2004/067528; схему 9 и пример 2, стадия A в патентной публикации PCT WO 2006/068669; и схему 15 и пример 6, стадия B в патентной публикации PCT WO 2006/062978).
Тем не менее, продолжает существовать необходимость в новых или усовершенствованных способах, пригодных для быстрого и экономичного получения 2-амино-5-цианобензойных кислот и их производных.
Сущность изобретения
Настоящее изобретение направлено на способ получения соединения формулы 1
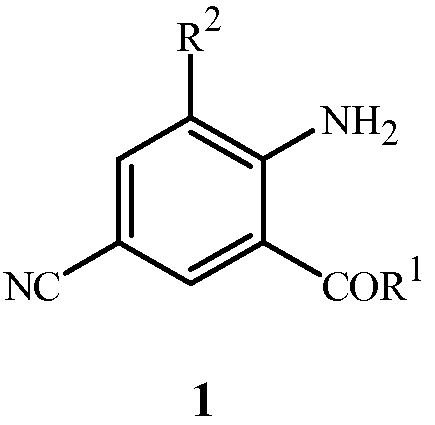
где
R1 представляет собой NHR3 или OR4;
R2 представляет собой CH3 или Cl;
R3 представляет собой H, C1-C4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил; и
R4 представляет собой H или C1-C4алкил;
включающий приведение в контакт (1) соединения формулы 2
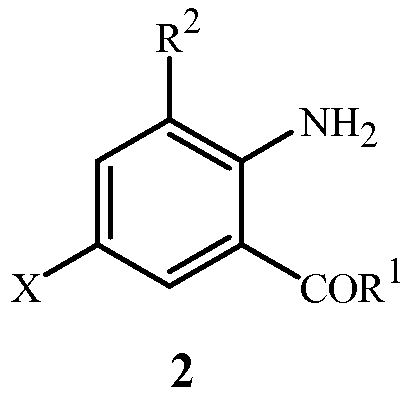
где X представляет собой Br или Cl;
с (2) реагентом на основе цианида металла, (3) реагентом на основе соли меди(I), (4) реагентом на основе соли йодистоводородной кислоты и (5) по меньшей мере одним соединением формулы 3
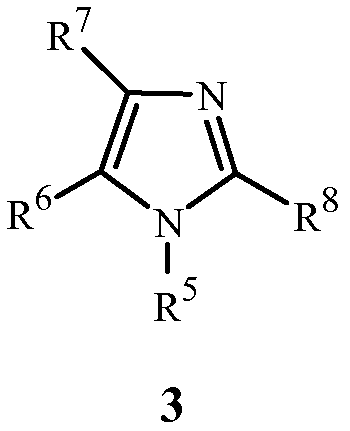
где
R5 представляет собой H, фенил или бензил; или C1-C12алкил, необязательно замещенный NR9R10;
каждый R6, R7 и R8 представляет собой независимо H, C1-C12алкил, фенил или бензил; или
R6 и R7 вместе образуют -CH=CH-CH=CH-; и
R9 и R10 вместе образуют -CH=N-CH=CH-, необязательно имеющий до 3 заместителей, независимо выбранных из C1-C12алкила;
при условии, что когда X представляет собой Cl, то R2 представляет собой метил.
Настоящее изобретение также относится к способу получения соединения формулы 4
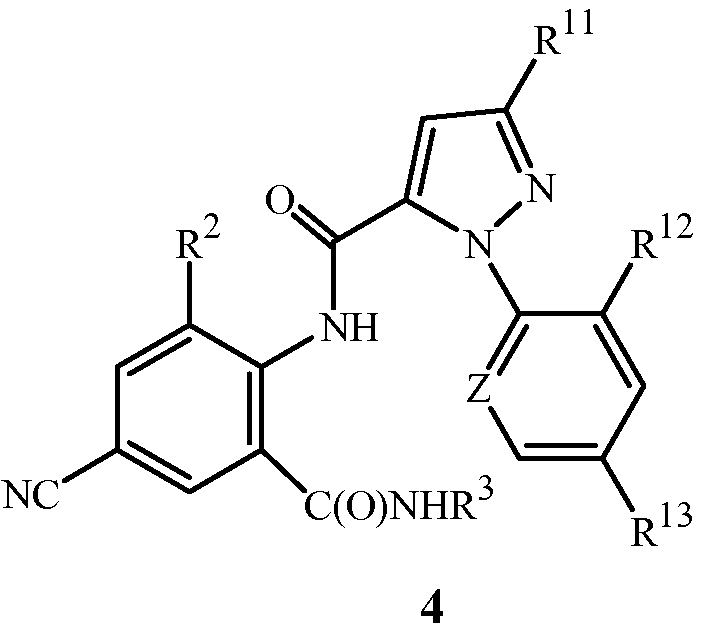 ,
,
где
R2 представляет собой CH3 или Cl;
R3 представляет собой H, C1-C4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил;
Z представляет собой CR14 или N;
R11 представляет собой Cl, Br, CF3, OCF2H или OCH2CF3;
R12 представляет собой F, Cl или Br;
R13 представляет собой H, F или Cl; и
R14 представляет собой H, F, Cl или Br;
с использованием соединения формулы 1. Данный способ отличается тем, что (a) соединение формулы 1 получают из соединения формулы 2 способом, описанным выше, или (b) в качестве указанного соединения формулы 1 используют соединение формулы 1, полученное способом, описанным выше.
Подробное описание изобретения
Как используется в данном описании, выражения “содержит”, “содержащий”, “включает”, “включающий”, “имеет”, “имеющий” или любая другая их вариация, предназначены для того, чтобы охватить неисключительное включение. Например, композиция, процесс, способ, изделие или устройство, которые содержат перечень элементов, необязательно ограничены только перечисленными элементами, но могут включать другие элементы, специально не перечисленные или присущие такой композиции, процессу, способу, изделию или устройству. Кроме того, если точно не указано иное, “или” относится к включающему "или", но не к исключающему его. Например, условие A или B удовлетворяет любому из следующего: A является верным (или присутствует), и B является ложным (или отсутствует), A является ложным (или отсутствует), и B является верным (или присутствует), и оба A и B являются верными (или присутствуют).
Также подразумевается, что формы единственного числа, предшествующие элементу или компоненту по настоящему изобретению, не является ограничивающим в отношении числа примеров (т.е. экземпляров) элемента или компонента. Тем самым, формы единственного числа следует понимать, как включающие один или по меньшей мере один, и форма единственного числа элемента или компонента также включает множественное число, если только, несомненно, не подразумевается, что число является единственным.
Как используется в данном описании, следующие обозначения следует применять, если особо не указано иное. Выражение "необязательно замещенный" используется взаимозаменяемо с фразой “замещенный или незамещенный” или с выражением “(не)замещенный”. Если не указано иное, когда присутствует более одного заместителя в группе, каждое замещение является независимым от другого. Также группа, обозначенная как необязательно замещенная заместителем, является замещенной нулем или одним примером указанного заместителя, если только не указан верхний предел.
В некоторых случаях в данном описании соотношения перечисляются как отдельные числа, которые относятся к числу 1; например, соотношение 4 означает 4:1.
Как используется в данном описании, выражение “эквивалент цианида” и связанные выражения, такие как “эквивалентное соотношение цианида”, когда оно относится к соединению, содержащему одну или более цианидных групп, относится к количеству ионов цианида (CN-) на моль содержащего цианид соединения. В частности, “эквивалент(ы) цианида” означает число молей иона цианида, которое может быть обеспечено молем содержащего цианид соединения (т.е. источника цианида) для преобразования соединения формулы 2 в соединение формулы 1 согласно данному способу. Например, реагент на основе гексацианоферрата(II) имеет шесть молей ионов цианида на моль гексацианоферрата(II); поэтому, если эквивалентное соотношение цианида в реагенте на основе гексацианоферрата(II) относительно другого реагента (например, соединения формулы 2) составляет 1:1, то мольное соотношение будет 0,167:1. Более того, способ настоящего изобретения включает применение реагента (2) (т.е. реагента на основе цианида металла), который может включать одно или более содержащих цианид соединений (например, один или более цианидов металла). В случаях, где реагент (2) содержит более чем одно содержащее цианид соединение, “эквиваленты цианида”, обеспеченные реагентом (2), представляют собой сумму количества эквивалентов цианида (т.е. молей CN-), обеспеченных каждым из содержащих цианид соединений в реагенте (2). Например, для конкретной реакции, реагент (2) может состоять из комбинации цианида натрия (NaCN) и цианида меди(I) (CuCN); поэтому эквивалентное соотношение цианида относительно другого реагента (например, соединение формулы 2) представляет собой сумму числа эквивалентов цианида, обеспеченных цианидом натрия и цианидом меди(I), относительно числа молей другого реагента. Поскольку как цианид натрия, так и цианид меди(I) содержат один моль иона цианида на моль (т.е. молекулярная масса по формуле соединения) соединения, то эквивалентное соотношение цианида относительно другого реагента представляет собой также сумму числа молей цианида натрия и цианида меди(I) относительно числа молей другого реагента.
В приведенных выше перечислениях выражение “алкил” включает неразветвленный или разветвленный алкил, такой как метил, этил, н-пропил, изопропил или различные изомеры бутила, пентила или гексила.
Выражение “циклопропилциклопропил” означает замещение циклопропила другим кольцом циклопропила. Примеры “циклопропилциклопропила” включают 1,1'-бициклопропил-1-ил, 1,1'-бициклопропил-2-ил и различные цис- и транс-изомеры циклопропилциклопропила, такие как (1R,2S)-1,1'-бициклопропил-2-ил и (1R,2R)-1,1'-бициклопропил-2-ил.
Как используется в данном описании, выражение “лиганд” относится к органической молекуле, содержащей по меньшей мере одну пару электронов, доступную для координации с атомом металла (в данном случае атомом меди). Выражение “бидентатный лиганд” относится к органической молекуле, содержащей по меньшей мере две пары электронов, которые доступны для координации с атомом металла (например, атомом меди).
Радикал на основе углерода относится к одновалентному молекулярному компоненту, содержащему атом углерода, который соединяет радикал с остатком химической структуры посредством одинарной связи. Радикалы на основе углерода могут необязательно содержать насыщенные, ненасыщенные и ароматические группы, цепи, кольца и системы колец, и гетероатомы. Хотя радикалы на основе углерода не подвергаются какому-либо конкретному ограничению в размере, в контексте настоящего изобретения они обычно содержат от 1 до 16 атомов углерода и от 0 до 3 гетероатомов. Следует отметить радикалы на основе углерода, выбранные из C1-C4алкила, C1-C2галогеналкила и фенила, необязательно замещенные 1-3 заместителями, выбранными из C1-C3алкила, галогена и нитро.
Способ настоящего изобретения включает реагент (2) (т.е. реагент на основе цианида металла), реагент (3) (т.е. реагент на основе соли меди(I)) и реагент (4) (т.е. реагент на основе соли йодистоводородной кислоты). Реагент (2) альтернативно и эквивалентно описывается как по меньшей мере один цианид металла, так как реагент на основе цианида металла содержит один или более цианидов металла. Реагент (3) альтернативно и эквивалентно описывается как по меньшей мере одна соль меди(I), так как реагент на основе соли меди(I) содержит одну или несколько солей меди(I). Реагент (4) альтернативно и эквивалентно описывается как по меньшей мере одна соль йодистоводородной кислоты, так как реагент на основе соли йодистоводородной кислоты содержит одну или несколько солей йодистоводородной кислоты. Более того, число молей реагента на основе цианида металла относится к числу молей цианида, содержащегося в реагенте (как описано выше). Число молей реагента на основе соли меди(I) относится к числу молей меди(I), содержащейся в реагенте. Число молей реагента на основе соли йодистоводородной кислоты относится к числу молей йодида, содержащегося в реагенте.
Согласно изложенному в данном описании, выражение “карбоновая кислота” означает органическое химическое соединение, содержащее по меньшей мере одну функциональную группу карбоновой кислоты (т.е. -C(O)OH). Выражение “карбоновая кислота” не включает соединение угольной кислоты (т.е. HOC(O)OH). Карбоновые кислоты включают, например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, хлоруксусную кислоту, бензойную кислоту, малеиновую кислоту и лимонную кислоту. Выражение “эффективный pKa” относится к pKa функциональной группы карбоновой кислоты, или если соединение имеет более чем одну функциональную группу карбоновой кислоты, то “эффективный pKa” относится к pKa наиболее кислой функциональной группы карбоновой кислоты. Согласно изложенному в данном описании, “эффективный pH” неводного вещества или смеси, такой как реакционная смесь, определяется смешиванием аликвоты вещества или смеси с примерно 5-20 объемами воды и затем измерением pH полученной водной смеси (например, с помощью pH-метра). Согласно изложенному в данном описании, “по существу безводное” вещество означает, что вещество содержит не более чем примерно 1% воды по массе. Химическое название “изатиновый ангидрид” является другим названием, соответствующим действующему названию по Химической реферативной службе “2H-3,1-бензоксазин-2,4(1H)-дион”.
Варианты осуществления настоящего изобретения включают:
Вариант осуществления A1. Способ, описанный в разделе «Сущность изобретения», для получения соединения формулы 1, включающий приведение в контакт реагента (1) (т.е. соединения формулы 2) с реагентом (2) (т.е. реагентом на основе цианида металла), реагентом (3) (т.е. реагентом на основе соли меди(I)), реагентом (4) (т.е. реагентом на основе соли йодистоводородной кислоты) и реагентом (5) (т.е. по меньшей мере одним соединением формулы 3).
Вариант осуществления A2. Способ варианта осуществления A1, где R1 представляет собой NHR3.
Вариант осуществления A3. Способ варианта осуществления A1 или A2, где R3 представляет собой C1-C4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил.
Вариант осуществления A4. Способ варианта осуществления A3, где R3 представляет собой C1-C4алкил или циклопропилметил.
Вариант осуществления A5. Способ варианта осуществления A4, где R3 представляет собой метил.
Вариант осуществления A6. Способ любого из вариантов осуществления A1-A5, где R2 представляет собой метил.
Вариант осуществления A7. Способ любого из вариантов осуществления A1-A6, где X представляет собой Br.
Вариант осуществления A8. Способ любого из вариантов осуществления A1-A7, где реагент (1) содержит 2-амино-5-бром-N,3-диметилбензамид.
Вариант осуществления A9. Способ любого из вариантов осуществления A1-A8, где реагент (2) содержит один или более цианидов металлов, выбранных из группы, содержащей цианиды щелочных металлов, гексацианоферраты(II) щелочных металлов и цианид меди(I).
Вариант осуществления A10. Способ варианта осуществления A9, где реагент (2) содержит один или более цианидов, выбранных из группы, содержащей цианиды щелочных металлов и гексацианоферраты(II) щелочных металлов.
Вариант осуществления A11. Способ варианта осуществления A10, где реагент (2) содержит один или более цианидов металлов, выбранных из группы, содержащей цианид натрия, цианид калия, гексацианоферрат(II) калия и гексацианоферрат(II) натрия.
Вариант осуществления A12. Способ варианта осуществления A11, где реагент (2) содержит один или более цианидов металлов, выбранных из группы, содержащей цианид натрия, цианид калия и гексацианоферрат(II) калия.
Вариант осуществления A13. Способ варианта осуществления A12, где реагент (2) содержит цианид натрия или гексацианоферрат(II) калия.
Вариант осуществления A14. Способ варианта осуществления A13, где реагент (2) содержит цианид натрия.
Вариант осуществления A15. Способ любого из вариантов осуществления A1-A14, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет по меньшей мере примерно 1.
Вариант осуществления A16. Способ варианта осуществления A15, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет по меньшей мере примерно 1,15.
Вариант осуществления A17. Способ варианта осуществления A16, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет по меньшей мере примерно 1,25.
Вариант осуществления A17a. Способ варианта осуществления A17, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет по меньшей мере примерно 1,4.
Вариант осуществления A18. Способ любого из вариантов осуществления A1-A17a, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет не более чем примерно 2,1.
Вариант осуществления A19. Способ варианта осуществления A19, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет не более чем примерно 1,55.
Вариант осуществления A19a. Способ варианта осуществления A19, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет не более чем примерно 1,5.
Вариант осуществления A20. Способ варианта осуществления A19a, где эквивалентное соотношение цианида реагента (2) к реагенту (1) составляет не более чем примерно 1,4.
Вариант осуществления A21. Способ любого из вариантов осуществления A1-A20, где R5 представляет собой H; или C1-C6алкил, необязательно замещенный NR9R10.
Вариант осуществления A22. Способ варианта осуществления 21, где R5 представляет собой H, метил, этил, н-пропил или н-бутил; или C1-C6алкил, замещенный NR9R10.
Вариант осуществления A23. Способ варианта осуществления A22, где R5 представляет собой метил или н-бутил.
Вариант осуществления A24. Способ любого из вариантов осуществления A1-A22, где R9 и R10 вместе образуют -CH=N-CH=CH-.
Вариант осуществления A25. Способ любого из вариантов осуществления A1-A24, где каждый R6, R7 и R8, когда взят отдельно (т.е. R6 и R7 не взяты вместе), представляет собой независимо H или C1-C3алкил.
Вариант осуществления A26. Способ варианта осуществления A25, где каждый R6, R7 и R8, когда взят отдельно, представляет собой независимо H или метил.
Вариант осуществления A27. Способ варианта осуществления A26, где каждый R6, R7 и R8, когда взят отдельно, представляет собой H.
Вариант осуществления A27a. Способ любого из вариантов осуществления A1-A27, где каждый R6, R7 и R8 взяты отдельно.
Вариант осуществления A28. Способ любого из вариантов осуществления A1-A27a, где реагент (5) содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-этил-1H-имидазол, 1-пропил-1H-имидазол, 1-бутил-1H-имидазол, 1-пентил-1H-имидазол, 1-гексил-1H-имидазол, 4-метилимидазол, 1,1'-(1,4-бутандиил)бис-1H-имидазол, 1,1'-(1,5-пентандиил)бис-1H-имидазол и 1,1'-(1,6-гександиил)бис-1H-имидазол.
Вариант осуществления A28a. Способ варианта осуществления A28, где реагент (5) содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-этил-1H-имидазол, 1-пропил-1H-имидазол, 1-бутил-1H-имидазол, 4-метилимидазол, 1,1'-(1,4-бутандиил)бис-1H-имидазол и 1,1'-(1,6-гександиил)бис-1H-имидазол.
Вариант осуществления A28b. Способ варианта осуществления A28a, где реагент (5) содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-бутил-1H-имидазол, 4-метилимидазол и 1,1'-(1,6-гександиил)бис-1H-имидазол.
Вариант осуществления A29. Способ любого из вариантов осуществления A1-A28b, где реагент (5) содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-этил-1H-имидазол, 1-пропил-1H-имидазол, 1-бутил-1H-имидазол, 1-пентил-1H-имидазол, 1-гексил-1H-имидазол, 1,1'-(1,4-бутандиил)бис-1H-имидазол, 1,1'-(1,5-пентандиил)бис-1H-имидазол и 1,1'-(1,6-гександиил)бис-1H-имидазол.
Вариант осуществления A30. Способ варианта осуществления A29, где реагент (5) содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-этил-1H-имидазол, 1-бутил-1H-имидазол и 1,1'-(1,4-бутандиил)бис-1H-имидазол.
Вариант осуществления A31. Способ варианта осуществления A28b или A30, где реагент (5) содержит 1-метил-1H-имидазол или 1-бутил-1H-имидазол.
Вариант осуществления A32. Способ варианта осуществления A31, где реагент (5) содержит 1-метил-1H-имидазол.
Вариант осуществления A33. Способ варианта осуществления A31, где реагент (5) содержит 1-бутил-1H-имидазол.
Вариант осуществления A34. Способ любого из вариантов осуществления A1-A33, где мольное соотношение реагента (5) к реагенту (3) (на основании содержания меди(I)) составляет по меньшей мере примерно 1.
Вариант осуществления A34a. Способ варианта осуществления A34, где мольное соотношение реагента (5) к реагенту (3) (на основании содержания меди(I)) составляет по меньшей мере примерно 1,5.
Вариант осуществления A35. Способ варианта осуществления A34a, где мольное соотношение реагента (5) к реагенту (3) составляет по меньшей мере примерно 2.
Вариант осуществления A36. Способ варианта осуществления A35, где мольное соотношение реагента (5) к реагенту (3) составляет по меньшей мере примерно 2,5.
Вариант осуществления A37. Способ варианта осуществления A36, где мольное соотношение реагента (5) к реагенту (3) составляет по меньшей мере примерно 3.
Вариант осуществления A38. Способ варианта осуществления A37, где мольное соотношение реагента (5) к реагенту (3) составляет по меньшей мере примерно 4.
Вариант осуществления A39. Способ любого из вариантов осуществления A1-A38, где мольное соотношение реагента (5) к реагенту (3) (на основании содержания меди(I)) составляет не более чем примерно 10.
Вариант осуществления A40. Способ варианта осуществления A39, где мольное соотношение реагента (5) к реагенту (3) составляет не более чем примерно 6.
Вариант осуществления A41. Способ варианта осуществления A40, где мольное соотношение реагента (5) к реагенту (3) составляет не более чем примерно 5,5.
Вариант осуществления A42. Способ варианта осуществления A41, где мольное соотношение реагента (5) к реагенту (3) составляет не более чем примерно 5.
Вариант осуществления A43. Способ любого из вариантов осуществления A1-A42, где мольное соотношение реагента (3) (на основании содержания меди(I)) к реагенту (1) составляет по меньшей мере примерно 0,01.
Вариант осуществления A44. Способ варианта осуществления A43, где мольное соотношение реагента (3) к реагенту (1) составляет по меньшей мере примерно 0,1.
Вариант осуществления A45. Способ варианта осуществления A44, где мольное соотношение реагента (3) к реагенту (1) составляет по меньшей мере примерно 0,15.
Вариант осуществления A45a. Способ варианта осуществления A45, где мольное соотношение реагента (3) к реагенту (1) составляет по меньшей мере примерно 0,3, когда X представляет собой Cl.
Вариант осуществления A46. Способ любого из вариантов осуществления A1-A45a, где мольное соотношение реагента (3) (на основании содержания меди(I)) к реагенту (1) составляет менее чем примерно 1.
Вариант осуществления A47. Способ любого из вариантов осуществления A1-A46, где мольное соотношение реагента (3) (на основании содержания меди(I)) к реагенту (1) составляет не более чем примерно 0,99.
Вариант осуществления A48. Способ варианта осуществления A47, где мольное соотношение реагента (3) к реагенту (1) составляет не более чем примерно 0,5.
Вариант осуществления A49. Способ варианта осуществления A48, где мольное соотношение реагента (3) к реагенту (1) составляет не более чем примерно 0,4.
Вариант осуществления A50. Способ варианта осуществления A49, где мольное соотношение реагента (3) к реагенту (1) составляет не более чем примерно 0,3.
Вариант осуществления A51. Способ варианта осуществления A50, где мольное соотношение реагента (3) к реагенту (1) составляет не более чем примерно 0,25, когда X представляет собой Br.
Вариант осуществления A52. Способ варианта осуществления A51, где мольное соотношение реагента (3) к реагенту (1) составляет не более чем примерно 0,2, когда X представляет собой Br.
Вариант осуществления A53. Способ любого из вариантов осуществления A1-A52, где мольное соотношение реагента (4) к реагенту (1) составляет по меньшей мере примерно 0,001.
Вариант осуществления A54. Способ варианта осуществления A53, где мольное соотношение реагента (4) к реагенту (1) составляет по меньшей мере примерно 0,05.
Вариант осуществления A55. Способ варианта осуществления A54, где мольное соотношение реагента (4) к реагенту (1) составляет по меньшей мере примерно 0,1.
Вариант осуществления A56. Способ варианта осуществления A55, где мольное соотношение реагента (4) к реагенту (1) составляет по меньшей мере примерно 0,15.
Вариант осуществления A57. Способ любого из вариантов осуществления A1-A56, где мольное соотношение реагента (4) к реагенту (1) составляет менее чем примерно 1.
Вариант осуществления A58. Способ любого из вариантов осуществления A1-A57, где мольное соотношение реагента (4) к реагенту (1) составляет не более чем примерно 0,5.
Вариант осуществления A59. Способ варианта осуществления A58, где мольное соотношение реагента (4) к реагенту (1) составляет не более чем примерно 0,4.
Вариант осуществления A60. Способ варианта осуществления A59, где мольное соотношение реагента (4) к реагенту (1) составляет не более чем примерно 0,3.
Вариант осуществления A61. Способ варианта осуществления 60, где мольное соотношение реагента (4) к реагенту (1) составляет не более чем примерно 0,2.
Вариант осуществления A62. Способ любого из вариантов осуществления A1-A61, где реагент (3) и реагент (4) содержит йодид меди(I).
Вариант осуществления A63. Способ любого из вариантов осуществления A1-A62, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт в присутствии подходящего органического растворителя.
Вариант осуществления A64. Способ любого из вариантов осуществления A1-A63, где реагент (1) приводят в контакт с подходящим органическим растворителем с образованием смеси, и затем реагент (2), реагент (3), реагент (4) и реагент (5) последовательно добавляют в смесь.
Вариант осуществления A65. Способ любого из вариантов осуществления A63 и A64, где подходящий органический растворитель включает один или более растворителей, выбранных из группы, включающей галогенированные и негалогенированные алифатические и ароматические углеводороды.
Вариант осуществления A66. Способ варианта осуществления A65, где подходящий органический растворитель включает один или более растворителей, выбранных из группы, включающей ксилолы, толуол, хлорбензол, метоксибензол (также известный как анизол), 1,2,4-триметилбензол, 1,3,5-триметилбензол (также известный как мезитилен), этилбензол, (1-метилэтил)бензол (также известный как кумол), C1-C3алкил-замещенные нафталины (например, 1-метилнафталин, 2-метилнафталин, 1,5-диметилнафталин, 2,6-диметилнафталин и 1,3-диметилнафталин), ShellSol A100 (смесь C9-C10 ароматических углеводородов) и ShellSol A150 (смесь C10-C11 ароматических углеводородов).
Вариант осуществления A67. Способ варианта осуществления A66, где подходящий органический растворитель включает один или более растворителей, выбранных из группы, включающей ксилолы, толуол, хлорбензол, анизол, 1,2,4-триметилбензол, мезитилен, 1-метилнафталин, ShellSol A100 и ShellSol A150.
Вариант осуществления A67a. Способ варианта осуществления A66, где подходящий органический растворитель включает один или более растворителей, выбранных из группы, содержащей ксилолы, толуол, 1,2,4-триметилбензол, мезитилен и 1-метилнафталин.
Вариант осуществления A68. Способ варианта осуществления A67 или A67a, где подходящий органический растворитель включает ксилолы, толуол, 1-метилнафталин или мезитилен.
Вариант осуществления A69. Способ варианта осуществления A68, где подходящий органический растворитель включает ксилолы, толуол или мезитилен.
Вариант осуществления A69a. Способ варианта осуществления A68, где подходящий органический растворитель включает 1-метилнафталин или мезитилен.
Вариант осуществления A70. Способ варианта осуществления A69, где подходящий органический растворитель включает ксилолы.
Вариант осуществления A71. Способ варианта осуществления A69, где подходящий органический растворитель включает толуол.
Вариант осуществления A72. Способ любого из вариантов осуществления A69 и A69a, где подходящий органический растворитель включает мезитилен.
Вариант осуществления A73. Способ любого из вариантов осуществления A63-A72, где соотношение объема подходящего органического растворителя к массе реагента (1) составляет по меньшей мере примерно 2 мл/г.
Вариант осуществления A74. Способ варианта осуществления A73, где соотношение объема подходящего органического растворителя к массе реагента (1) составляет по меньшей мере примерно 3 мл/г.
Вариант осуществления A75. Способ варианта осуществления A74, где соотношение объема подходящего органического растворителя к массе реагента (1) составляет по меньшей мере примерно 4 мл/г.
Вариант осуществления A76. Способ любого из вариантов осуществления A63-A75, где соотношение объема подходящего органического растворителя к реагенту (1) составляет не более чем примерно 10 мл/г.
Вариант осуществления A77. Способ варианта осуществления A76, где соотношение объема подходящего органического растворителя к массе реагента (1) составляет не более чем примерно 6 мл/г.
Вариант осуществления A78. Способ варианта осуществления A77, где соотношение объема подходящего органического растворителя к массе реагента (1) составляет не более чем примерно 5 мл/г.
Вариант осуществления A79. Способ любого из вариантов осуществления A1-A78, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт в присутствии подходящего органического растворителя с образованием смеси, давление указанной выше смеси увеличивают выше атмосферного давления и температуру смеси увеличивают выше нормальной точки кипения растворителя (т.е. точки кипения при давлении 100 кПа или 14,5 фунт/дюйм2 (абс.)).
Вариант осуществления A80. Способ варианта осуществления A79, где подходящий органический растворитель включает ксилолы, толуол или анизол.
Вариант осуществления A81a. Способ варианта осуществления A80, где подходящий органический растворитель включает ксилолы или толуол.
Вариант осуществления A82. Способ любого из вариантов осуществления A1-A81a, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре не более чем примерно 200°C.
Вариант осуществления A83. Способ варианта осуществления A82, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре не более чем примерно 180°C.
Вариант осуществления A84. Способ варианта осуществления A83, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре не более чем примерно 170°C.
Вариант осуществления A85. Способ варианта осуществления A84, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре не более чем примерно 165°C.
Вариант осуществления A86. Способ любого из вариантов осуществления A1-A85, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре более чем примерно 115°C.
Вариант осуществления A87. Способ варианта осуществления A86, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре более чем примерно 145°C.
Вариант осуществления A88. Способ варианта осуществления A87, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре более чем примерно 155°C.
Вариант осуществления A89. Способ варианта осуществления A88, где реагент (1), реагент (2), реагент (3), реагент (4) и реагент (5) приводят в контакт с подходящим органическим растворителем при температуре более чем примерно 160°C.
Вариант осуществления A90. Способ варианта осуществления A1, где X представляет собой Br, и соединение формулы 1 получают в виде твердого вещества, включающий приведение в контакт реагента (1) с подходящим органическим растворителем с образованием смеси, и затем последовательно добавление реагента (2), реагента (3), реагента (4) и реагента (5) к смеси, поддержание температуры смеси примерно от 145 до 180°C в течение примерно от 5 до примерно 8 часов, охлаждение смеси до примерно 0-50°C, добавление воды к смеси, необязательно добавление медь-координирующего агента к смеси, необязательно перемешивание в течение примерно от 1 до примерно 24 часов, и затем восстановление соединения формулы 1 в виде твердого вещества из смеси.
Вариант осуществления A91. Способ варианта осуществления A1, где X представляет собой Cl и соединение формулы 1 получают в виде твердого вещества, включающий приведение в контакт реагента (1) с подходящим органическим растворителем с образованием смеси, и затем последовательно добавление реагента (2), реагента (3), реагента (4) и реагента (5) к смеси, поддержание температуры смеси примерно от 150 до 200°C в течение примерно от 5 до примерно 24 часов, охлаждение смеси примерно до 0-50°C, добавление воды к смеси, необязательно добавление медь-координирующего агента к смеси, необязательно перемешивание в течение примерно 1-примерно 24 часов, и затем восстановление соединения формулы 1 в виде твердого вещества из смеси.
Вариант осуществления B1. Способ, описанный в разделе «Сущность изобретения», для получения соединения формулы 4, с использованием соединения формулы 1, полученного из соединения формулы 2.
Вариант осуществления B2. Способ варианта осуществления B1, где соединение формулы 1 получают из соединения формулы 2 способом любого из вариантов осуществления A1-A91.
Вариант осуществления B3. Способ варианта осуществления B1 или B2, где Z представляет собой N.
Вариант осуществления B4. Способ варианта осуществления B1 или B2, где Z представляет собой CH.
Вариант осуществления B5. Способ любого из вариантов осуществления B1-B4, где R3 представляет собой C1-C4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил.
Вариант осуществления B6. Способ варианта осуществления B5, где R3 представляет собой C1-C4алкил или циклопропилметил.
Вариант осуществления B7. Способ варианта осуществления B6, где R3 представляет собой метил.
Вариант осуществления B8. Способ любого из вариантов осуществления B1-B7, где R2 представляет собой метил.
Вариант осуществления B9. Способ любого из вариантов осуществления B1-B8, где R11 представляет собой Br.
Вариант осуществления B10. Способ любого из вариантов осуществления B1-B9, где R12 представляет собой Cl.
Вариант осуществления B11. Способ любого из вариантов осуществления B1-B10, где R13 представляет собой H.
Вариант осуществления B12. Способ любого из вариантов осуществления A1-A91 или B1-B11, где соединение формулы 1 представляет собой 2-амино-5-циано-N,3-диметилбензамид.
Вариант осуществления C1. Способ, описанный в разделе «Сущность изобретения» или любом из вариантов осуществления A1-A91 или B1-B12, где R5 представляет собой H; или C1-C12алкил, необязательно замещенный NR9R10, если не дано более точное определение.
Вариант осуществления C2. Способ, описанный в разделе «Сущность изобретения» или любом из вариантов осуществления A1-A91, B1-B12 или C1, где каждый R6, R7 и R8 представляет собой независимо H или C1-C12алкил, если не дано более точное определение.
Варианты осуществления настоящего изобретения могут быть комбинированы любым образом. Следует отметить способ любого из вариантов осуществления A1-A90 или B1-B12, где X представляет собой Br. Также следует отметить способ любого из вариантов осуществления A1-A89, A91 или B1-B12, где X представляет собой Cl.
На следующих схемах 1-8 обозначения R1, R2, R3, R4, R5, R6, R7, R8, R11, R12, R13, X и Z в соединении Формул 1-10 являются такими, как определено выше в разделе «Сущность изобретения» и описании вариантов осуществления, если не указано иное. Формулы 1a, 1b и 1c являются подклассом формулы 1. Формула 2a является подклассом формулы 2.
Как показано на схеме 1, в способе настоящего изобретения соединение формулы 1 получают путем приведения в контакт соединения формулы 2 по меньшей мере с одним цианидом металла (т.е. реагентом на основе цианида металла), по меньшей мере одной солью меди(I) (т.e. реагентом на основе соли меди(I)), по меньшей мере одной солью йодистоводородной кислоты (т.е. реагентом на основе соли йодистоводородной кислоты) и по меньшей мере одним соединением формулы 3.
Схема 1
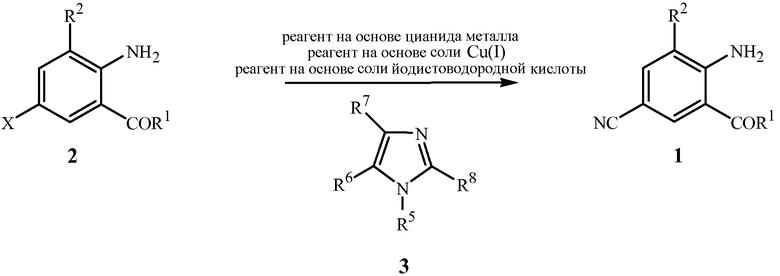
В данном способе реагент на основе цианида металла, в частности, содержит по меньшей мере одно соединение, выбранное из группы, состоящей из цианидов щелочных металлов и гексацианоферратов(II) щелочных металлов и цианида меди(I). Подходящие цианиды щелочных металлов включают соединения формулы M1CN, где M1 означает щелочной металл, такой как натрий или калий. Подходящие гексацианоферраты(II) щелочных металлов включают, например, гексацианоферрат(II) калия и гексацианоферрат(II) натрия, оба из которых являются коммерчески доступными по низкой цене, являются нетоксичными, удобными в обращении, и имеют шесть ионов цианида, доступных для переноса на соединения формулы 2. Самые высокие выходы соединений формулы 1 обычно достигаются, когда используется реагент на основе цианида металла, включающий цианид натрия. Как правило, эквивалентное соотношение цианида реагента на основе цианида металла относительно соединения формулы 2 составляет примерно от 1 до примерно 2,1, и более конкретно примерно от 1,15 до примерно 1,55. Тем не менее, использование бόльших количеств реагента на основе цианида металла может быть преимущественным при удалении меди в ходе выделения соединений формулы 1. Цианиды щелочных металлов, такие как цианид натрия, являются особенно подходящими в качестве медь-координирующих агентов для облегчения удаления меди в ходе выделения соединений формулы 1. Когда дополнительные количества реагента на основе цианида металла, включающего цианид щелочного металла (например, цианид натрия), включены в реакционную смесь для облегчения последующего удаления меди, эквивалентное соотношение реагента на основе цианида металла относительно соединения формулы 2 составляет обычно примерно от 1,4 до примерно 2,1 или даже выше. Когда используется цианид щелочного металла, то может быть полезным снижение размера частиц цианида щелочного металла с помощью стандартных способов, таких как растирание или размалывание перед добавлением цианида щелочного металла к реакционной смеси. Как правило, цианид щелочного металла, который был растерт или размолот, является особенно предпочтительным, когда используется только стехиометрическое или немного больше количество цианида щелочного металла. Напротив, когда цианид щелочного металла используется в большом избытке, таком как количество, достаточное не только для стадии цианирования, но также и для последующего удаления меди из реакционной смеси (т.е. примерно 1,4-2,1 относительно формулы 2), растирание или размалывание цианида щелочного металла может обеспечить небольшую пользу, по сравнению с использованием цианида щелочного металла, не растертого или размолотого перед добавлением к реакционной смеси.
В способе схемы 1 реагент на основе соли меди(I), как полагают, действует как источник химических частиц, которые катализируют преобразование соединений формулы 2 в соединения формулы 1. Подходящие реагенты соли меди(I) включают одно или более соединений, выбранных из группы, включающей соли меди(I), такие как йодид меди(I), бромид меди(I), хлорид меди(I), цианид меди(I) и трифлат меди(I) (CuOSO2CF3). Мольное соотношение реагента на основе соли меди(I) (на основе Cu(I)) к соединению формулы 2 составляет примерно от 0,01 до примерно 1, обычно примерно от 0,01 до примерно 0,99, и более конкретно примерно от 0,1 до примерно 0,4. Когда X представляет собой Br, то оптимальные результаты обычно получают от мольных соотношений примерно от 0,1 до примерно 0,3 реагента на основе соли меди(I) к соединению формулы 2. Так как соединения формулы 2, где X представляет собой Cl, как правило, менее реакционноспособны, чем соответствующие соединения формулы 2, в реакции схемы 1 обычно используют большее количество меди(I) для того, чтобы вызвать реакцию, когда X представляет собой Cl. Таким образом, когда X представляет собой Cl, обычно используют мольные соотношения примерно от 0,3 до примерно 0,4 реагента на основе соли меди(I) к соединению формулы 2.
Вне связи с какой-либо конкретной теорией, полагают, что в условиях данного способа 5-бром или -хлор производное формулы 2 по меньшей мере частично превращается в соответствующее 5-йод производное в присутствии соли йодистоводородной кислоты. Подходящие реагенты соли йодистоводородной кислоты включают одно или более соединений, выбранных из группы, включающей соли йодистоводородной кислоты четвертичного аммония, щелочных и щелочноземельных металлов, таких как йодид меди(I), йодид натрия, йодид калия, йодид цинка, йодид лития, йодид кальция, йодид тетрабутиламмония и йодид тетраметиламмония. Мольное соотношение соли йодистоводородной кислоты к соединению формулы 2 составляет примерно от 0,001 до примерно 1, и более конкретно примерно от 0,05 до примерно 0,4, и наиболее конкретно примерно от 0,1 до примерно 0,4.
В способе схемы 1 самые высокие выходы соединений формулы 1 с оптимальными скоростями реакции часто получают, когда в качестве источника реагента на основе соли меди(I) и реагента на основе соли йодистоводородной кислоты используют йодид меди(I) (CuI). Когда йодид меди(I) (CuI) используют в данном способе, как правило, мольное соотношение составляет примерно от 0,1 до примерно 0,4 относительно соединения формулы 2. В некоторых случаях может быть полезно использовать йодид меди(I) в комбинации с другим реагентом на основе соли йодистоводородной кислоты, таким как йодид натрия, йодид калия, йодид цинка, йодид тетрабутиламмония или йодид тетраметиламмония. Польза комбинирования йодида меди(I) с другим реагентом на основе соли йодистоводородной кислоты зависит от конкретных условий реакции и субстрата. Обычно оптимальные выходы соединений формулы 1 могут быть получены из данного способа просто с использованием йодида меди(I) в качестве единственного источника реагента на основе соли йодистоводородной кислоты.
Соединения формулы 3 действуют в качестве лигандов в способе схемы 1. Могут использоваться как монодентатные хелатирующие лиганды, содержащие необязательно замещенное имидазольное кольцо, так и бидентатные хелатирующие лиганды, содержащие 2 необязательно замещенных имидазольных кольца. Обнаружено, что такие лиганды ускоряют скорость преобразования соединений формулы 2 в соединения формулы 1. Вне связи с какой-либо конкретной теорией, полагают, что лиганды облегчают реакцию путем увеличения растворимости, реакционной способности и/или стабильности активных каталитических видов меди(I) посредством образования комплекса медь-лиганд. Соединения формулы 3, включающие имидазол и широкое разнообразие имидазол-замещенных производных, могут быть использованы в качестве лигандов в данном способе. Обычно лиганды формулы 3 включают соединения, где R5, R6, R7 и R8 представляют собой независимо H или C1-C4алкил, такие как 1-метил-1H-имидазол, 1-этил-1H-имидазол, 1-пропил-1H-имидазол, 1-бутил-1H-имидазол, 1-пентил-1H-имидазол, 1-гексил-1H-имидазол и 4-метилимидазол. Также подходящими являются бис(имидазолил)алканы (т.е., где R5 представляет собой C1-C12алкил, замещенный NR9R10), такие как 1,1'-(1,4-бутандиил)бис-1H-имидазол, 1,1'-(1,5-пентандиил)бис-1H-имидазол и 1,1'-(1,6-гександиил)бис-1H-имидазол. В способе схемы 1 обычно самые высокие выходы соединений формулы 1 и наиболее благоприятные скорости реакции достигаются с использованием одного или нескольких из следующих коммерчески доступных лигандов: 1-метил-1H-имидазол, 1-бутил-1H-имидазол, 4-метилимидазол и 1,1'-(1,6-гександиил)бис-1H-имидазол. Мольное соотношение соединений формулы 3 к реагенту на основе соли меди(I) составляет обычно примерно от 1 до примерно 10. Так как мольные соотношения больше чем 1 могут зачастую ускорять реакцию, тогда как соотношения выше 6, в общем, оказывают небольшую дополнительную пользу при возрастающих затратах, то предпочтительным является соотношение примерно от 1,5 до примерно 6.
Реакцию схемы 1 обычно проводят в подходящем органическом растворителе. Могут использоваться разнообразные растворители для образования подходящего растворителя для данного способа. Как правило, способ наиболее удовлетворительно проводят, используя растворители, в которых соединения формулы 2 предпочтительно полностью или по меньшей мере значительно растворяются, и реагент на основе цианида металла имеет низкую растворимость в объеме используемых растворителей и при температурах реакции. Примеры подходящих растворителей включают галогенированные и негалогенированные алифатические и ароматические углеводороды, такие как ксилолы, толуол, хлорбензол, метоксибензол (также известный как анизол), 1,2,4-триметилбензол, 1,3,5-триметилбензол (также известный как мезитилен), этилбензол, (1-метилэтил)бензол (также известный как кумол), C1-C3алкил-замещенные нафталины (например, 1-метилнафталин, 2-метилнафталин, 1,5-диметилнафталин, 2,6-диметилнафталин и 1,3-диметилнафталин) и смеси ароматических растворителей, которые являются твердыми, например, от Shell Chemical под торговым названием ShellSol, в частности, ShellSol A100 (смесь C9-C10 ароматических углеводородов) и ShellSol A150 (смесь C10-C11 ароматических углеводородов), включая смеси указанных выше растворителей. Способ наиболее удовлетворительно проводят, используя растворитель, который предусматривает температуры реакции примерно от 150 до 180°C. Этого можно достигнуть с использованием растворителя с нормальной точкой кипения (т.е. точкой кипения при давлении 100 кПа) в пределах или выше этого диапазона или путем работы при повышенном давлении с низкокипящим растворителем, таким как ксилолы или толуол. Растворители ксилолы или толуол являются подходящими растворителями, так как высокие выходы соединений формулы 1 обычно получают, когда используют такие растворители, особенно когда данный способ осуществляют при повышенном давлении. При использовании ксилолов в качестве растворителя может быть использован отдельный изомер (т.е. о-ксилол, м-ксилол или п-ксилол), но использование смеси изомеров ксилолов является коммерчески предпочтительным, так как это обеспечивает в равной степени хорошие результаты при более низкой стоимости. Способ также традиционно проводят, используя растворитель с нормальной точкой кипения в диапазоне примерно от 150 до 180°C, такой как 1,3,5-триметилбензол, 1-метилнафталин, смеси C9-C11 ароматических растворителей или их смеси. В частности, обнаружено, что использование растворителя (т.е. с нормальной точкой кипения в диапазоне примерно от 150 до 180°C), включающего 1-метилнафталин или 1,3,5-триметилбензол, приводит к высоким выходам соединений формулы 1. Объем органического растворителя относительно массы соединения формулы 2 составляет обычно примерно от 2 мл/г до примерно 10 мл/г. Количество растворителя более чем 2 мл/г может облегчить перемешивание реакционной смеси, но большие количества растворителя могут замедлять реакцию, а также увеличивать стоимость; поэтому обычно объем растворителя к массе соединения формулы 2 составляет примерно от 2 мл/г до примерно 5 мл/г, и более конкретно, примерно от 2 мл/г до 4 мл/г. Растворитель может быть добавлен различными способами и в различные промежутки времени в ходе реакции, а именно: в одной загрузке в начале последовательности реакций, или порциями в ходе последовательности реакций, или периодически в процессе добавления одного или более реагентов. Например, один или несколько реагентов могут быть диспергированы, растворены или частично растворены в подходящем органическом растворителе и затем добавлены к смеси, содержащей другие реагенты и дополнительное количество подходящего органического растворителя.
В данном способе порядок, в котором комбинируют реагенты, не является критическим для результата реакции. Один порядок комбинации, например, включает комбинирование соединения формулы 2 с подходящим органическим растворителем с образованием смеси, и затем последовательное добавление реагента на основе цианида металла, реагента на основе соли меди(I), реагента на основе соли йодистоводородной кислоты и по меньшей мере одного соединения формулы 3 к смеси. Альтернативно, в некоторых случаях предпочтительным является растворение по меньшей мере одного соединения формулы 3 и реагента на основе соли меди(I) в подходящем органическом растворителе и добавление полученного раствора к смеси, содержащей соединение формулы 2, реагент на основе цианида металла, реагент на основе соли йодистоводородной кислоты и подходящий органический растворитель. Или, альтернативно, по меньшей мере одно соединение формулы 3 может быть растворено в подходящем органическом растворителе и добавлено к смеси, содержащей соединение формулы 2, реагент на основе цианида металла, реагент на основе соли меди(I), реагент на основе соли йодистоводородной кислоты и подходящий органический растворитель. Для такого способа добавления, обычно подходящий органический растворитель (т.е. растворяющее соединение или смесь растворяющих соединений), используемый для растворения соединения(й) формулы 3 и реагента на основе соли меди(I), является таким же подходящим органическим растворителем, используемым при образовании смеси, содержащей компоненты реакции. Разнообразие других порядков добавления также являются подходящими для данного способа.
Способ схемы 1 предпочтительно проводят в бескислородной среде, хотя это не является существенным для успешного результата реакции. Обнаружено, что снижение присутствия атмосферного кислорода в реакционном сосуде до и в процессе добавления реагентов и поддержание бескислородной среды в ходе протекания реакции является предпочтительным. Могут использоваться стандартные методики получения бескислородной среды, включая, например, создание вакуума в реакционном сосуде с использованием вакуумного насоса и затем накачивание до атмосферного давления инертного газа (например, азота или аргона). Такой способ можно повторять два или более раз для дальнейшего снижения присутствия кислорода в реакционном сосуде. Альтернативно, реакционный сосуд может продуваться инертным газом, и затем положительное давление инертного газа можно поддерживать в течение реакции.
Данный способ обычно осуществляют при температурах примерно от 115 до 200°C и более конкретно примерно от 145 до 200°C. Температуры примерно от 150 до 180°C часто приводят к самому высокому выходу и чистоте с наиболее благоприятными скоростями реакции; например, в большинстве случаев соединения формулы 1 получают с выходами более чем 95% или более, примерно за 5-8 часов.
Продукт формулы 1 можно выделить стандартными методиками, известными в данной области, включая фильтрование, экстракцию, выпаривание и кристаллизацию. Например, реакционную среду можно разбавить примерно 2-8 частями по массе воды относительно соединения формулы 2 для растворения неорганических солей, которые присутствуют в реакционной среде. Так как соединения формулы 1 являются обычно твердыми веществами при температуре окружающей среды и, как правило, трудно растворимы в растворителе реакции, они наиболее легко выделяются с помощью фильтрации, с последующей промывкой водой и необязательно органическим растворителем (например, ксилолами, толуолом, 1,3,5-триметилбензолом). Если соединения формулы 1 растворимы в растворителе реакции, их наиболее традиционно выделяют разбавлением реакционной среды водой, чтобы растворить неорганические соли, затем отделить органическую фазу, необязательно с последующей промывкой водой, для удаления остаточных количеств солей и/или цианидов металлов, и затем удаляют растворитель дистилляцией или выпариванием при пониженном давлении. В некоторых случаях может быть предпочтительным добавление водорастворимого медь-координирующего агента для оптимизации удаления меди перед выделением соединений формулы 1. Подходящие медь-координирующие агенты включают, например, 2,2'-тиодиэтанол, этилендиамин, N,N'-диметилэтилендиамин, N,N,N',N'-тетраметилэтилендиамин и цианиды щелочных металлов. Особенно подходящими для удаления меди являются этилендиамин и цианиды щелочных металлов. Если цианид щелочного металла (например, цианид натрия) используется в данном способе в качестве медь-координирующего агента, обычно примерно от 0,3 до примерно 0,6 молей относительно соединений формулы 2 являются подходящими для снижения количества остаточной меди в соединениях формулы 1. Такое количество цианида натрия может быть добавлено, когда добавлен реагент на основе цианида металла (т.е. в процессе реакции цианирования, как обсуждается выше) или по завершению реакции и перед выделением соединений формулы 1. Для первого способа добавления цианид щелочного металла добавляют в безводной форме, и для второго способа его добавляют либо в безводной форме, либо в виде водного раствора. Соединения формулы 1 далее могут быть очищены перекристаллизацией из приемлемого органического растворителя. Примеры приемлемых растворителей включают метанол, этанол, изопропанол, н-пропанол, толуол, ксилолы и хлорбензол. Способ схемы 1 проиллюстрирован в примерах 1-21 ниже. Примеры 3 и 4 иллюстрируют способ схемы 1, включая обработку реакционной смеси этилендиамином перед выделением соединения формулы 1.
Особенности данного способа обеспечивают эффективные методы, используя недорогие реагенты для производства производных 3-замещенной 2-амино-5-цианобензойной кислоты формулы 1 с высокими выходами (обычно 95% или больше на моль используемого соединения формулы 2) примерно за 5-8 часов. Следует особо отметить, что данный способ может быть использован для достижения необыкновенно высоких выходов соединений формулы 1 с исключительной чистотой, несмотря на то, что данные соединения, также как и исходные соединения формулы 2, содержат амино-заместители и в некоторых случаях амидные заместители, которые могут потенциально участвовать в побочных реакциях.
Исходные соединения формулы 2 могут быть получены с помощью ряда способов, известных в данной области. Как показано на схеме 2, согласно одному способу соединения формулы 2 могут быть получены путем галогенирования соединения формулы 5 с использованием различных реагентов, известных в литературе, включая бром, хлор, сульфурилхлорид, N-хлорсукцинимид (NCS), N-бромсукцинимид (NBS) и галогенирующие агенты, такие как смеси, включающие пероксид водорода и гидрогалогенид. Для ознакомления со ссылками, описывающими приведенные способы, см. патентные публикации РСТ WO 1998/16503 (схема 4 и пример 132), WO 2006/068669 (схема 11), WO 2003/015519 (схема 4 и пример 1, стадия A) и WO 2006/062978 (схема 15; пример 4, стадия B и пример 5, стадия B).
Схема 2
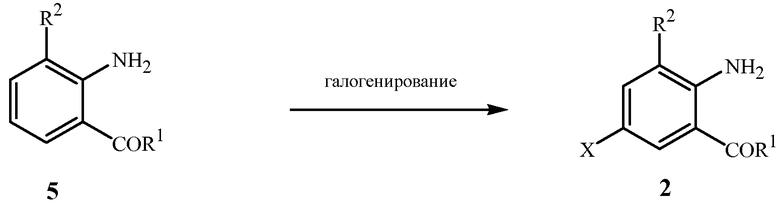
Другой способ получения соединения формулы 2, где X представляет собой Br, и R1 представляет собой NHR3, включает бромирование соединений формулы 5 путем обработки газом, содержащим бром, как проиллюстрировано в методике справочного примера 1 (справочный пример 1 также показан в патентной публикации PCT WO 2008/082502).
Соединения формулы 2a (формулы 2, где R1 представляет собой NHR3) также могут быть получены путем приведения в контакт изатинового (isatoic) ангидрида формулы 6 с алкиламином формулы 7 в присутствии карбоновой кислоты, как показано на схеме 3.
Схема 3
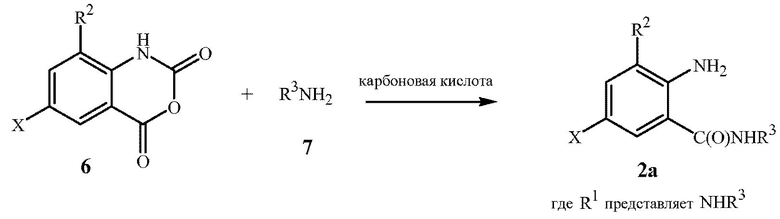
Так как амины, такие как соединение формулы 7, являются основаниями, то в отсутствие карбоновой кислоты, смесь соединений формул 6 и 7 будет основной (т.е. эффективный pH>7). Карбоновая кислота действует как буфер для уменьшения эффективного pH реакционной смеси. Может быть использовано широкое разнообразие карбоновых кислот, так как единственным требованием по меньшей мере для одной группы карбоновой кислоты является обеспечение кислотности. Могут присутствовать другие функциональные группы, и более чем одна группа карбоновой кислоты может присутствовать на молекуле карбоновой кислоты. Как правило, карбоновая кислота имеет эффективный pKa в диапазоне примерно от 2 до примерно 5. Карбоновые кислоты включают, например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, хлоруксусную кислоту, бензойную кислоту, фталевую кислоту, малеиновую кислоту, винную кислоту и лимонную кислоту. Ввиду стоимости, недорогие карбоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота и бензойная кислота, являются предпочтительными. Уксусная кислота, которая коммерчески доступна по низкой цене в ее безводной форме (известной как “ледяная уксусная кислота”) является особенно предпочтительной.
Комбинация карбоновой кислоты с основным амином формулы 7 образует аминную соль карбоновой кислоты. Такая аминная соль может быть получена перед добавлением изатинового ангидрида соединения формулы 6, или аминная соль может быть образована in situ путем отмеренного добавления амина формулы 7 в смесь соединения формулы 6 и карбоновой кислоты. Для любого из способов добавления поддержание эффективного pH смеси в ходе реакции примерно от 3 до примерно 7 является в общем оптимальным.
Так как эффективный pH смеси является результатом буферного эффекта карбоновой кислоты в комбинации с амином формулы 7, то эффективный pH может быть отрегулирован согласно эффективному pKa карбоновой кислоты путем регулирования молярного соотношения карбоновой кислоты к амину формулы 7. Обычно молярное количество амина формулы 7 относительно карбоновой кислоты находятся в диапазоне примерно от 0,8 до примерно 3. Более конкретно, когда способ комбинации включает отмеренное добавление амина формулы 7 в смесь изатинового ангидрида соединения формулы 6 и карбоновой кислоты, то молярное соотношение амина формулы 7 к карбоновой кислоте составляет предпочтительно примерно от 0,95 до примерно 3. Когда способ комбинации включает образование аминной соли перед добавлением соединения формулы 6, то молярное соотношение амина формулы 7 к карбоновой кислоте составляет предпочтительно примерно от 0,8 до примерно 1,05; поскольку используется почти эквимолярное соотношение (например, примерно от 0,95 до примерно 1,05) амина формулы 7 к карбоновой кислоте, то аминная соль, которая образуется таким образом, обычно используется в соотношении примерно от 1,1 до примерно 5 молярных эквивалентов относительно соединения формулы 6. Для оптимального преобразования молярное соотношение амина формулы 7 к изатиновому ангидриду соединения формулы 6 должно быть по меньшей мере 1,0, хотя предпочтительно, чтобы молярное соотношение составляло примерно от 1,1 до примерно 1,5 для эффективности и экономичности, вне зависимости от того, как смешивают компоненты. Молярное количество амина формулы 7 относительно соединения формулы 6 может быть значительно больше, чем 1,5, особенно когда используется почти эквимолярное соотношение (например, примерно от 0,95 до примерно 1,05) амина к кислоте.
Самый высокий выход продукта и чистота достигаются, когда реакционная среда является по существу безводной. Реакционная среда, таким образом, образуется из по существу безводного соединения формул 6 и 7 и карбоновой кислоты. Предпочтительно реакционная среда и образующие материалы содержат примерно 5% или меньше, более предпочтительно примерно 1% или меньше, и наиболее предпочтительно примерно 0,1% воды или меньше (по массе). Если карбоновая кислота представляет собой уксусную кислоту, то предпочтительно в форме ледяной уксусной кислоты.
Реакцию схемы 3 обычно проводят в жидкой фазе. Во многих случаях реакция может быть осуществлена без растворителя, отличного от соединений формул 2a, 6 и 7 и карбоновой кислоты. Но предпочтительная методика включает использование растворителя, который может суспендировать и по меньшей мере частично растворять реагенты. Предпочтительными растворителями являются такие, которые являются инертными в отношении компонентов реакции и имеют диэлектрическую постоянную, равную примерно 5 или больше, такие как алкилнитрилы, сложные эфиры, простые эфиры и кетоны. Предпочтительно, растворитель должен быть по существу безводным, чтобы облегчить получение по существу безводной реакционной среды. Массовое отношение растворителя к соединению формулы 6 обычно составляет примерно от 1 до примерно 20 и предпочтительно примерно 5 в целях эффективности и экономичности.
Диоксид углерода образуется как побочный продукт реакции схемы 3. Основная часть образованного диоксида углерода выделяется из реакционной среды в виде газа. Добавление соединения формулы 6 в реакционную среду, содержащую амин формулы 7, или добавление амина формулы 7 в реакционную среду, содержащую соединение формулы 6, предпочтительно проводят при такой скорости и температурах, чтобы облегчить контроль выделения диоксида углерода. Температура реакционной среды составляет, как правило, примерно от 5 до 75°C, более конкретно, примерно от 35 до 55°C.
Продукт формулы 2a может быть выделен стандартными методами, известными в данной области, включающими регулирование pH, экстракцию, выпаривание, кристаллизацию и хроматографию. Например, реакционная среда может быть разбавлена примерно 3-15 частями по массе воды относительно исходного соединения формулы 6, pH можно необязательно регулировать либо кислотой, либо основанием для оптимизации удаления либо кислотных, либо основных примесей, водную фазу можно необязательно отделять, и основную часть органического растворителя можно удалять путем дистилляции или выпаривания при пониженном давлении. Так как соединения формулы 2a обычно являются кристаллическими твердыми веществами при температуре окружающей среды, то оно могут быть легко выделены фильтрованием, необязательно с последующей промывкой водой и затем сушкой.
Как показано на схеме 4, изатиновые ангидриды формулы 6 можно получить из антраниловых кислот формулы 2b (формулы 2, где R1 представляет собой OR4, и R4 представляет собой H, которые могут быть получены способом схемы 2) путем реакции циклизации, включая обработку антраниловых кислот фосгеном или эквивалентом фосгена, таким как трифосген или алкилхлорформиат (например, метилхлорформиат), в подходящем растворителе, таком как толуол или тетрагидрофуран. Способ описан в патентной публикации PCT WO 2006/068669, включая специфический пример, относящийся к схеме 4. Также см. Coppola, Synthesis 1980, 505 и Fabis et al., Tetrahedron 1998, 10789.
Схема 4
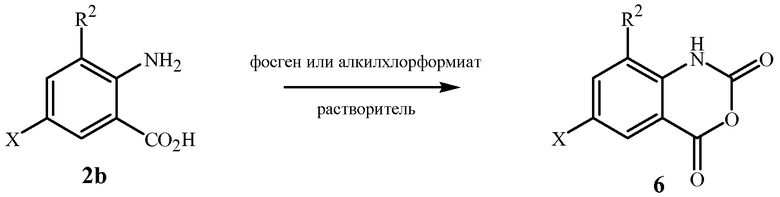
Соединения формулы 3 являются коммерчески доступными, и в литературе по синтезу описано множество общих способов получения имидазолов; см., например, Grimmett, Science of Synthesis 2002, 12, 325-528 и процитированные там ссылки. Бис(имидазолил)алканы формулы 3, такие как 1,1'-(1,4-бутандиил)бис-1H-имидазол, 1,1'-(1,5-пентандиил)бис-1H-имидазол, 1,1'-(1,6-гександиил)бис-1H-имидазол и подобные, могут быть получены путем взаимодействия соответствующего дигалогеналкана (например, 1,4-дибромбутана, 1,5-дибромпентана, 1,6-дибромгексана) с 2 эквивалентами необязательно замещенного имидазола в присутствии основания, согласно общим методикам, описанным Diez-Barra et al., Heterocycles 1992, 34(7), 1365-1373, Torres et al., Journal of Heterocyclic Chemistry 1988, 25(3), 771-782, Sato, et al., Heterocycles 2003, 60(4), 779-784, и Luo et al., Heterocycles 1995, 41(7), 1421-1424.
В другом аспекте настоящего изобретения соединения формулы 1, полученные способом схемы 1, могут быть использованы в качестве промежуточных соединений для получения соединений формулы 4. Соединения формулы 4 полезны в качестве инсектицидов, как описано, например, в патентных публикациях PCT WO 2003/015518 и WO 2006/055922.
где
R2 представляет собой CH3 или Cl;
R3 представляет собой H, C1-C4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил;
Z представляет собой CR14 или N;
R11 представляет собой Cl, Br, CF3, OCF2H или OCH2CF3;
R12 представляет собой F, Cl или Br;
R13 представляет собой H, F или Cl; и
R14 представляет собой H, F, Cl или Br;
Возможны различные способы получения соединения формулы 4 из соединения формулы 1. Как показано на схеме 5, один такой способ включает сочетание соединения формулы 1a (формула 1, где R1 представляет собой OR4, и R4 представляет собой H) и пиразол-5-карбоновой кислоты формулы 8, с получением в результате цианобензоксазинона формулы 9. Последующее взаимодействие цианобензоксазинона с амином формулы 7 обеспечивает соединение формулы 4. Условия для первой стадии включают последовательное добавление метансульфонилхлорида в присутствии третичного амина, такого как триэтиламин или пиридин, к пиразолу формулы 8, с последующим добавлением соединения формулы 1a, с последующим вторым добавлением третичного амина и метансульфонилхлорида. Реакция может протекать в чистом виде или в различных подходящих растворителях, включая тетрагидрофуран, диэтиловый эфир, диоксан, толуол, дихлорметан или хлороформ, при оптимальных температурах, лежащих в диапазоне от комнатной температуры до температуры кипения растворителя с обратным холодильником. Вторая стадия, взаимодействие бензоксазинонов с аминами с получением антраниламидов, хорошо описана в химической литературе. Для общего обзора химии бензоксазинонов см. Jakobsen et al., Biorganic and Medicinal Chemistry 2000, 8, 2095-2103 и процитированные там ссылки, и G.M. Coppola, J. Heterocyclic Chemistry 1999, 36, 563-588. См. также патентную заявку PCT WO 2004/067528, в которой сообщается об общем способе, показанном на схеме 5, включая экспериментальные примеры, относящиеся к схеме 5.
Схема 5
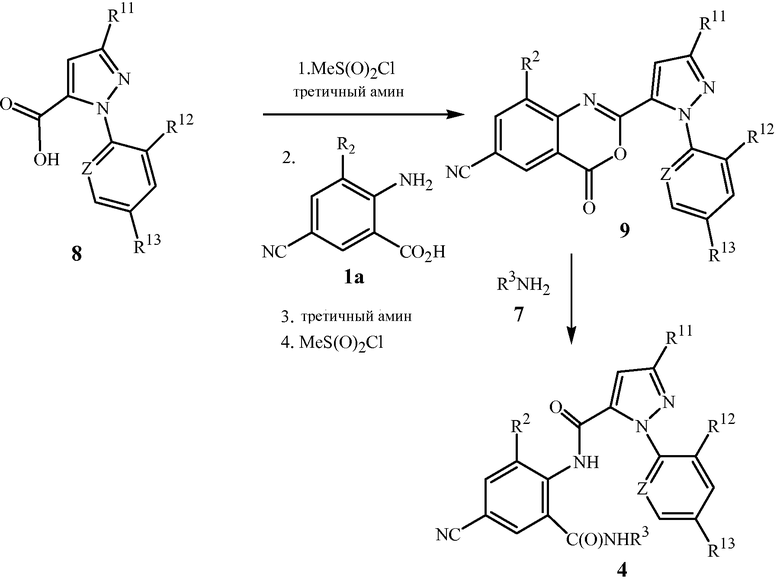
Другой способ получения соединений формулы 4 показан на схеме 6. В данном способе соединение формулы 4 получают путем объединения соединения формулы 1b (формула 1, где R1 представляет собой NHR3), пиразола формулы 8 и сульфонилхлорида согласно общему способу, о котором сообщается в патентной публикации PCT WO 2006/062978, которая тем самым включена в данное описание в своей полноте посредством ссылки.
Схема 6
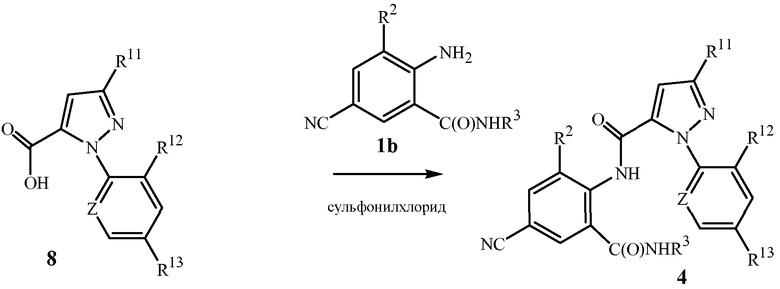
Как описано в WO 2006/062978, для данного преобразования возможны различные условия реакции. Обычно сульфонилхлорид добавляют к смеси соединений формул 1b и 8 в присутствии растворителя и основания. Сульфонилхлориды в общем имеют формулу RS(O)2Cl, где R представляет собой радикал на основе углерода. Обычно для данного способа R представляет собой C1-C4алкил, C1-C2галогеналкил или фенил, необязательно замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, C1-C3алкила и нитро. Коммерчески доступные сульфонилхлориды включают метансульфонилхлорид (R представляет собой CH3), пропансульфонилхлорид (R представляет собой (CH2)2CH3), бензолсульфонилхлорид (R представляет собой фенил) и п-толуолсульфонилхлорид (R представляет собой 4-метилфенил). Метансульфонилхлорид следует особо отметить с точки зрения более низкой стоимости, простоты добавления и/или меньшего количества отходов. По меньшей мере один молярный эквивалент сульфонилхлорида на моль соединения формулы 8 стехиометрически необходим для завершения преобразования. Обычно молярное соотношение сульфонилхлорида к соединению формулы 8 составляет не более примерно 2,5, более конкретно, не более примерно 1,4.
Соединение формулы 4 образуется, когда исходные соединения формул 1b, 8 и сульфонилхлорид приводят в контакт друг с другом в комбинированной жидкой фазе, в которой каждый является по меньшей мере частично растворимым. Поскольку исходные вещества формул 1b и 8 обычно являются твердыми веществами при средних температурах окружающей среды, то способ наиболее удовлетворительно осуществляют, используя растворитель, в котором исходные соединения имеют значительную растворимость. Таким образом, способ, как правило, осуществляют в жидкой фазе, включающей растворитель. В некоторых случаях карбоновая кислота формулы 8 может иметь только слабую растворимость, но ее основно-аддитивная соль может иметь большую растворимость в растворителе. Подходящие растворители для данного способа включают нитрилы, такие как ацетонитрил и пропионитрил; сложные эфиры, такие как метилацетат, этилацетат и бутилацетат; кетоны, такие как ацетон, метилэтилкетон (MEK) и метилбутилкетон; галогеналканы, такие как дихлорметан и трихлорметан; простые эфиры, такие как этиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран (ТГФ) и п-диоксан; ароматические углеводороды, такие как бензол, толуол, хлорбензол и дихлорбензол; третичные амины, такие как триалкиламины, диалкиланилины и необязательно замещенные пиридины; и смеси вышеперечисленного. Растворители, которые следует отметить, включают ацетонитрил, пропионитрил, этилацетат, ацетон, MEK, дихлорметан, метил-трет-бутиловый эфир, ТГФ, п-диоксан, толуол и хлорбензол. Растворителем, который следует отметить особо, является ацетонитрил, так как он часто обеспечивает продукты с превосходным выходом и/или чистотой.
Так как при реакции по данному способу образуется хлористый водород в качестве побочного продукта, который может иным способом связываться с основными центрами соединений формул 1b, 4 и 8, то способ наиболее удовлетворительно осуществляют в присутствии по меньшей мере одного дополнительного основания. Основание может также облегчить структурное взаимодействие карбоновой кислоты с сульфонилхлорида и антраниламидом. При взаимодействии добавленного основания с карбоновой кислотой формулы 8 образуется соль, которая имеет большую растворимость, чем карбоновая кислота в реакционной среде. Хотя основание может быть добавлено одновременно, последовательно или даже после добавления сульфонилхлорида, основание обычно добавляют перед добавлением сульфонилхлорида. Некоторые растворители, такие как третичные амины, также служат в качестве оснований, и когда они используются в качестве растворителей, то они будут в большом стехиометрическом избытке в качестве основания. Когда основание не используется как растворитель, то номинальное мольное соотношение основания к сульфонилхлориду составляет обычно примерно от 2,0 до примерно 2,2, и предпочтительно составляет примерно от 2,1 до примерно 2,2. Предпочтительные основания представляют собой третичные амины, включая замещенные пиридины. Более предпочтительные основания включают 2-пиколин, 3-пиколин, 2,6-лутидин и пиридин. В качестве основания следует особо отметить 3-пиколин, так как его соли с карбоновыми кислотами формулы 8 являются часто высоко растворимыми в растворителях, таких как ацетонитрил. Способ схемы 6 проиллюстрирован в примере 22 ниже.
Соединения формулы 4 могут быть выделены из реакционных смесей способами, известными специалистам в данной области, включая кристаллизацию, фильтрование и экстракцию. Как раскрывается в WO 2006/062978, в некоторых случаях в условиях реакции сочетания схемы 6 соединения формулы 4 могут частично циклизоваться с образованием производных иминобензоксазина формулы 10, как показано ниже на схеме 7.
Схема 7
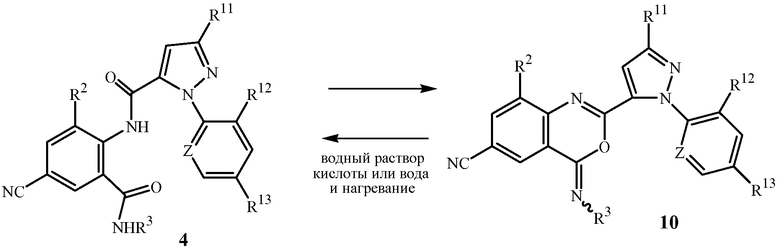
Как обсуждается в WO 2006/062978, в этих случаях часто предпочтительным является преобразование соединений иминобензоксазина формулы 10 обратно в амиды формулы 4 перед выделения. Такое преобразование может быть осуществлено путем обработки реакционной смеси водным раствором кислоты (например, водным раствором хлористоводородной кислоты); или путем выделения смеси соединений формулы 10 и формулы 4, и затем обработки смеси водным раствором кислоты, необязательно в присутствии подходящего органического растворителя (например, ацетонитрила). В WO 2006/062978 раскрыты конкретные примеры, относящиеся к способу схемы 6, включая примеры, иллюстрирующие обработку реакционной смеси водным раствором кислоты перед выделением соединений формулы 4. Пример 22 ниже иллюстрирует способ схемы 6, включающий обработку реакционной смеси водным раствором хлористоводородной кислоты перед выделением соединения формулы 4.
Альтернативно, соединения формулы 10 могут быть преобразованы обратно в соединения формулы 4 перед выделением путем приведения в контакт реакционной смеси с водой и нагревания. Обычно, преобразование соединений формулы 10 в соединения формулы 4 может достигаться путем добавления воды примерно от 2 до 6 частей по массе относительно массы исходного соединения формулы 1 и затем нагревания примерно от 45 до примерно 65°C. Преобразование соединения формулы 10 в соединение формулы 4 обычно завершается за 1 час или меньше. Справочный пример 2 ниже иллюстрирует способ схемы 6, включающий обработку реакционной смеси водой и нагревание перед выделением соединения формулы 4.
Пиразол-5-карбоновые кислоты формулы 8 могут быть получены из 5-оксо-3-пиразолидинкарбоксилатов путем обработки галогенирующим агентом с получением 3-галоген-4,5-дигидро-1H-пиразол-5-карбоксилатов, которые могут быть впоследствии обработаны окисляющим агентом, чтобы получить сложные эфиры формулы 8. Сложные эфиры могут быть затем преобразованы в кислоты (т.е. соединение формулы 8). Галогенирующие агенты, которые могут использоваться, включают, например, оксигалогениды фосфора, тригалогениды фосфора, пентагалогениды фосфора, тионилхлорид, дигалогентриалкилфосфораны, дигалогендифенилфосфораны, оксалилхлорид и фосген. Окисляющими агентами могут быть, например, пероксид водорода, органические пероксиды, персульфат калия, персульфат натрия, персульфат аммония, моноперсульфат калия (например, Oxone®) или перманганат калия. См. патентные публикации РСТ WO 2003/016283, WO 2004/087689 и WO 2004/011453 для описания способов галогенирования и окисления, и методик получения исходных 5-оксо-3-пиразолидинкарбоксилатов. Для преобразования сложных эфиров в карбоновые кислоты могут быть использованы различные способы, описанные в химической литературе, включающие нуклеофильное отщепление в безводных условиях или гидролиз, включающий использование либо кислот, либо оснований (см. T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2-е изд., John Wiley & Sons, Inc., Нью Йорк, 1991, с. 224-269 для обзора способов). Гидролитические способы, катализируемые основанием, являются предпочтительными для получения карбоновых кислот формулы 8 из соответствующих сложных эфиров. Подходящие основания включают гидроксиды щелочных металлов (такие как гидроксиды лития, натрия или калия). Например, сложные эфиры могут быть растворенными в смеси воды и спирта, такого как метанол. При обработке гидроксидом натрия или гидроксидом калия, сложные эфиры омыляются с получением соли натриевой или калиевой карбоновой кислоты. Закисление сильной кислотой, такой как хлористоводородная кислота или серная кислота, дает карбоновые кислоты. В патентной публикации PCT WO 2003/016283 представлен соответствующий экспериментальный пример, иллюстрирующий способ гидролиза, катализируемый основанием, для преобразования сложного эфира в кислоту.
Альтернативно, пиразол-5-карбоновые кислоты формулы 8 могут быть получены из 4,5-дигидро-5-гидрокси-1H-пиразол-5-карбоксилатов путем реакции дегидратации, катализируемой кислотой, с получением сложных эфиров, которые затем можно превратить в кислоты формулы 8. Конкретные условия реакции включают обработку 4,5-дигидро-5-гидрокси-1H-пиразол-5-карбоксилатов кислотой, например, хлористоводородной кислотой, в органическом растворителе, таком как уксусная кислота, при температурах примерно от 0 до 100°C. Способ описан в патентной публикации PCT WO 2003/016282. Преобразование сложных эфиров в кислоты может быть выполнено с использованием способов, описанных выше. Также в WO 2003/016282 представлен соответствующий экспериментальный пример для преобразования сложного эфира в кислоту.
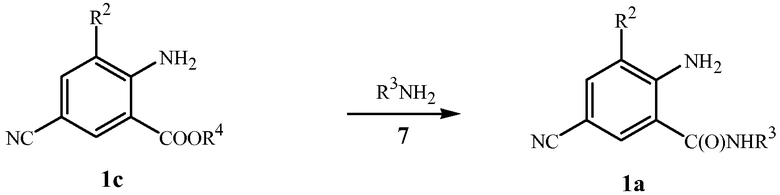
Антраниловые амиды формулы 1a также могут быть получены из соответствующих кислот или сложных эфиров формулы 1c (формула 1, где R1 представляет собой OR4, и R4 представляет собой H или C1-C4алкил) как показано ниже на схеме 8. Получение амидов из карбоновых кислот обычно включает добавление агента сочетания (например, тетрахлорида кремния или, альтернативно, дициклогексилкарбодиимида, или 1-этил-3-(3-диметиламинопропил)карбодиимида, часто в присутствии 1-гидроксибензотриазола). Получение антраниловых амидов из антраниловых кислот раскрыто M.J. Kornet, в Journal of Heterocyclic Chemistry 1992, 29(1), 103-5; публикации PCT WO 01/66519-A2; T. Asano et al., Bioorganic & Medicinal Chemistry Letters 2004, 14(9), 2299-2302; H.L. Birch et al., Bioorganic & Medicinal Chemistry Letters 2005, 15(23), 5335-5339; и D. Kim et al., Bioorganic & Medicinal Chemistry Letters 2005, 15(8), 2129-2134. T. Asano et al. также сообщают о получении антраниламида из антраниловой кислоты через промежуточное соединение N-защищенный анилин или через промежуточное соединение 4H-3,1-бензоксазин-2,4(1H)-дион (изатиновый ангидрид). Образование амидов из сложных эфиров часто включает нагревание сложного эфира с соответствующим амином в полярном растворителе, таком как этиленгликоль. Методика, пригодная для преобразования антраниловых сложных эфиров в антраниламиды, описана в патентной публикации PCT WO 2006/062978. Также E.B. Skibo et al., Journal of Medicinal Chemistry 2002, 45(25), 5543-5555 раскрывает получение антраниламида из соответствующего антранилового сложного эфира с использованием катализатора цианида натрия.
Схема 8
Способы схем 5 и 6 являются иллюстративными только для двух из многих способов преобразования соединения формулы 1 в карбоксамидное соединения формулы 4. В данной области техники известно широкое разнообразие общих способов получения карбоксамидов из карбоновых кислот и аминов. Для общего обзора см. M. North, Contemporary Org. Synth. 1995, 2, 269-287. Конкретные способы включают приведение в контакт соединения формулы 1b с соединением формулы 8 в присутствии дегидратирующего агента сочетания, такого как 1,1'-карбонилдиимидазол, бис(2-оксо-3-оксазолидинил)фосфинхлорид или гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония, или связанный с полимером аналогичный агент, такой как связанный с полимером дициклогексилкарбодиимид, обычно в инертном растворителе, таком как дихлорметан или N,N-диметилформамид, как в общем раскрыто в патентной публикации PCT WO 2003/15518. Также в WO 2003/15518 раскрывается способ получения ацилхлоридного эквивалента соединения формулы 8, путем приведения в контакт с тионилхлоридом или оксалилхлоридом в присутствии каталитического количества N,N-диметилформамида, и затем приведение в контакт полученного хлорангидрида с соединением формулы 1b в присутствии акцептора кислоты, такого как аминное основание (например, триэтиламин, N,N-диизопропилэтиламин, пиридин и аналоги на полимерной подложке) или гидроксид, или карбонат (например, NaOH, KOH, Na2CO3, K2CO3), обычно в инертном растворителе, таком как тетрагидрофуран, 1,4-диоксан, этиловый эфир или дихлорметан. Продукты соединений формулы 4 могут быть выделены из реакционных смесей способами, известными специалистам в данной области техники, включающими кристаллизацию, фильтрование и экстракцию.
Без дальнейшего уточнения, полагают, что специалист в данной области техники, используя приведенное выше описание, может применять настоящее изобретение в его полном объеме. Последующие примеры, поэтому, расцениваются только как иллюстративные и не ограничивающие каким-либо образом настоящее изобретение. Последующие примеры иллюстрируют методики синтеза, и исходное вещество для каждого примера может быть необязательно получено с помощью конкретного предварительного опыта, чья методика описана в других примерах. В примерах 13-16 реакционные смеси анализируют с помощью ВЭЖХ с обращенной фазой (высокоэффективная жидкостная хроматография) (HP Zorbax® Eclipse XDB-C8, производства Agilent Technologies, 3,5 мкм, 4,6 мм×75 мм). Система растворителей представляла собой растворитель A: вода с pH, доведенным до 2,5 путем добавления фосфорной кислоты, и растворитель B: ацетонитрил (градиент, начинающийся на 0 минутах с 81% растворителя A и 19% растворителя B, и растворитель B повышался до 87% в пределах 27 минут; скорость потока составляла 1,5 мл/мин). В примерах 17-21 реакционные смеси анализировали с помощью ВЭЖХ с обращенной фазой (HP Zorbax® SB-Phenyl, производства Agilent Technologies, 3,5 мкм, 4,6 мм×15 мм). Система растворителей представляла собой растворитель A: вода с pH, доведенным до 3,0 путем добавления фосфорной кислоты, и растворитель B: ацетонитрил (градиент, начинающийся на 0 минутах с 83% растворителя A и 17% растворителя B, и растворитель B повышался до 95% в пределах 15 минут; скорость потока составляла 1,5 мл/мин). Спектры 1H ЯМР (ядерно-магнитный резонанс) регистрировались в миллионных долях (м.д.) сдвига от тетраметилсилана; с означает синглет, д означает дублет, м означает мультиплет, ушир.с означает широкий синглет, и ушир.д означает широкий дублет.
Справочный пример 1
Получение 2-амино-5-бром-N,3-диметилбензамида (соединение формулы 2)
В 1000-мл колбу, оснащенную механической мешалкой, термопарой, конденсатором и фторполимерной трубкой Teflon (1/16" (0,16) см внутренний диаметр×1/8" (0,32 см) внешний диаметр) (расположенной так, что конец трубки погружен ниже поверхности реакционной смеси), загружали уксусной кислотой (226 мл). Раствор водного гидроксида натрия (50%, 25 г) в воде (85 г) добавляли в течение 15 минут и затем добавляли 2-амино-N,3-диметилбензамид (50 г, 0,305 моль) (см. патентную публикацию PCT WO 2006/062978 для способа получения) и смесь нагревали при 55°C. Двугорлую 200-мл колбу, на одно горлышко которой насаживали погружаемую трубку Teflon®, наполняли жидким бромом (50,1 г), и другое горлышко соединяли с трубкой Teflon® на 1000-мл колбе. Газообразный азот затем пропускали через трубку, погруженную ниже поверхности жидкого брома, со скоростью примерно 0,012 м3 (0,4 куб. фут) в час в течение 2,5 часов, на протяжении которых весь бром испарялся, и пары брома, захваченные газообразным азотом, истекали из двугорлой 200-мл колбы и попадали в реакционную смесь через трубку Teflon®. Температуру реакции поддерживали примерно при 55°C в ходе добавления паров брома и в течение 30 минут после этого, затем охлаждали до 45°C и перемешивали в течение ночи. Раствор водного гидроксида натрия (50%, 52 г) в воде (88 мл) добавляли к реакционной смеси со скоростью 0,8 мл/мин. После того, как было добавлено примерно 10% от общего объема раствора гидроксида натрия, добавление прекращали и реакционную смесь перемешивали в течение 1 часа при 45°C. Через 1 час оставшийся раствор гидроксида натрия добавляли со скоростью 0,8 мл/мин. После того, как добавление завершали, реакционную смесь перемешивали в течение 30 минут при 45°C, затем охлаждали до 10°C и перемешивали в течение 1 часа. Смесь фильтровали, собранное твердое вещество промывали метанолом (130 мл) и водой (260 мл) и затем сушили до постоянного веса в вакуумной печи при 45°C, с получением указанного в заголовке соединения в виде твердого вещества (67 г, 99,4% чистоты по данным ВЭЖХ, 90% выход), которое плавилось при 133-135°C.
1H ЯМР (ДМСО (диметилсульфоксид)-d6) δ 8,30 (м, 1H), 7,49 (д, 1H), 7,22 (д, 1H), 6,35 (ушир.с, 2H), 2,70 (д, 3H), 2,06 (с, 3H).
Пример 1
Получение 2-амино-5-циано-N,3-диметилбензамида (соединение формулы 1)
В 100-мл трехгорлую колбу, оснащенную механической мешалкой, термометром и конденсатором, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99,1% чистота, 5,0 г, 0,02 моль) и 1-метилнафталин (20 г), поддерживая поток азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (1,25 г, 0,024 моль, допуская 95% чистоту), йодид меди(I) (0,57 г, 0,003 моль, 98% чистоты) и 1-бутил-1H-имидазол (2,15 г, 0,017 моль, 98% чистоты). Смесь нагревали при 158-167°C в течение 5 часов и охлаждали в течение ночи. Воду (20 мл) добавляли по каплям к реакционной смеси в течение 5 минут при перемешивании. После перемешивания в течение дополнительного 1 часа, реакционную смесь фильтровали и собранное твердое вещество промывали водой (3×10 мл), ксилолами (10 мл) и сушили до постоянного веса в вакуумной печи при 80°C, с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (3,4 г).
1H ЯМР (ДМСО-d6) δ 8,40 (ушир.с, 1H), 7,79 (ушир.с, 1H), 7,41 (ушир.с, 1H), 7,18 (ушир.с, 2H), 2,72 (д, 3H), 2,08 (с, 3H).
Пример 2
Второе получение 2-амино-5-циано-N,3-диметилбензамида
В 100-мл трехгорлую колбу, оснащенную механической мешалкой, термометром и конденсатором, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (5,0 г, 0,020 моль, 99,1% чистоты) и 1-метилнафталин (20 г), поддерживая поток азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (1,25 г, 0,024 моль, допуская 95% чистоту), йодид меди(I) (0,57 г, 0,0030 моль, 98% чистоты) и 1-метил-1H-имидазол (1,40 г, 0,017 моль, 99%). Смесь нагревали при 160-165°C в течение 6 часов, затем переносили в 200-мл колбу и охлаждали в течение ночи. Воду (20 мл) добавляли по каплям к реакционной смеси в течение 5 минут при перемешивании. После перемешивания в течение дополнительных 2 часов реакционную смесь фильтровали, собранное твердое вещество промывали водой (3×10 мл) и ксилолами (10 мл) и сушили до постоянного веса в вакуумной печи при 80°C, с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (3,85 г).
1H ЯМР (ДМСО-d6) δ 8,40 (ушир.с, 1H), 7,80 (ушир.с, 1H), 7,40 (ушир.с, 1H), 7,15 (ушир.с, 2H), 2,72 (д, 3H), 2,07 (с, 3H).
Пример 3
Третье получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (6,2 г, 0,121 моль, допуская 95% чистоты), йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-бутил-1H-имидазол (10,8 г, 0,085 моль, 98% чистоты), после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 7 часов, пропуская через скруббер. После охлаждения в течение ночи добавляли воду (100 мл) к реакционной смеси и перемешивание продолжали в течение 1 часа. Этилендиамин (0,09 г, примерно 0,015 моль, 99% чистоты) добавляли к смеси, и перемешивание продолжали в течение дополнительных 15 минут. Смесь фильтровали и собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (18,3 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.д, 1H), 7,82 (д, 1H), 7,44 (ушир.с, 1H), 7,17 (ушир.с, 2H), 2,75 (д, 3H), 2,10 (с, 3H).
Пример 4
Четвертое получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (6,2 г, 0,121 моль, допуская, 95% чистоты), йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-бутил-1H-имидазол (5,4 г, 0,0425 моль, 98% чистоты), после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 6 часов, пропуская через скруббер. После охлаждения в течение ночи, к реакционной смеси добавляли воду (100 мл) и перемешивание продолжали в течение 1 часа. К реакционной смеси добавляли этилендиамин (0,09 г, примерно 0,015 моль, 99% чистоты) и перемешивание продолжали в течение дополнительных 3,5 часов. Смесь фильтровали, собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (19,0 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.д, 1H), 7,83 (д, 1H), 7,44 (ушир.с, 1H), 7,19 (ушир.с, 2H), 2,74 (д, 3H), 2,10 (с, 3H).
Пример 5
Пятое получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (7,7 г, 0,15 моль, допуская 95% чистоты), йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-бутил-1H-имидазол (10,8 г, 0,085 моль, 98% чистоты), после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 7 часов, пропуская через скруббер. После охлаждения в течение ночи, к реакционной смеси добавляли воду (100 мл) и перемешивание продолжали в течение 1 часа. Смесь фильтровали, собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (16,9 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.д, 1H), 7,82 (д, 1H), 7,44 (ушир.с, 1H), 7,19 (ушир.с, 2H), 2,74 (д, 3H), 2,10 (с, 3H).
Пример 6
Шестое получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (7,7 г, 0,15 моль, допуская 95% чистоты), йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-бутил-1H-имидазол (7,6 г, 0,06 моль, 98% чистоты), после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 6,75 часов, пропуская через скруббер. После охлаждения до 25°C к реакционной смеси добавляли воду (100 мл) и перемешивание продолжали в течение 1 часа. Смесь фильтровали, собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (17,7 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.д, 1H), 7,82 (д, 1H), 7,44 (ушир.с, 1H), 7,19 (ушир.с, 2H), 2,74 (д, 3H), 2,10 (с, 3H).
Пример 7
Седьмое получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (7,7 г, 0,15 моль, допуская 95% чистоты) и йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-этил-1H-имидазол (5,9 г, 0,06 моль, 98% чистоты), после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 8 часов, пропуская через скруббер. После охлаждения в течение ночи добавляли воду (100 мл) и перемешивание продолжали в течение 1 часа. Смесь фильтровали, собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (18,0 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.д, 1H), 7,82 (д, 1H), 7,44 (ушир.с, 1H), 7,19 (ушир.с, 2H), 2,74 (д, 3H), 2,11 (с, 3H).
Пример 8
Восьмое получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Смесь перемешивали при комнатной температуре и добавляли цианид натрия (измельченный в порошок непосредственно перед использованием) (7,7 г, 0,15 моль, допуская 95% чистоты), йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-бутил-1H-имидазол (9,5 г, 0,075 моль, 98% чистоты), после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 6 часов, пропуская через скруббер. После охлаждения в течение ночи к реакционной смеси добавляли воду (100 мл) и перемешивание продолжали в течение 1 часа. Смесь фильтровали, собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (17,0 г).
Пример 9
Девятое получение 2-амино-5-циано-N,3-диметилбензамида
В 500-мл четырехгорлую колбу, оснащенную магнитной мешалкой, термопарой и конденсатором, боковой капельной воронкой и скруббером с гидроксидом натрия/гипохлоритом натрия, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (24,6 г, 0,10 моль, 99% чистоты) и мезитилен (100 г), поддерживая атмосферу азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре, и гранулированный цианид натрия (Alfa Aesar®, 7,7 г, 0,15 моль, допуская 95% чистоту), йодид меди(I) (2,9 г, 0,015 моль, 98% чистоты) и 1-бутил-1H-имидазол (10,8 г, 0,085 моль, 98% чистоты) добавляли к реакционной смеси, после чего приемную линию для азота прикрепляли непосредственно к реакционной колбе. Реакционную смесь нагревали при кипячении с обратным холодильником (примерно 166-170°C) в течение 6 часов, пропуская через скруббер, и затем охлаждали в течение ночи. К реакционной смеси добавляли воду (100 мл) и перемешивание продолжали в течение 45 минут. Больше воды (25 мл) добавляли к реакционной смеси и перемешивание продолжали в течение дополнительных 15 минут. Смесь фильтровали, собранное твердое вещество промывали водой (3×50 мл), мезитиленом (50 г) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (16,8 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.д, 1H), 7,82 (д, 1H), 7,44 (ушир.с, 1H), 7,18 (ушир.с, 2H), 2,74 (д, 3H), 2,10 (с, 3H).
Пример 10
Десятое получение 2-амино-5-циано-N,3-диметилбензамида
В 100-мл трехгорлую колбу, оснащенную магнитной мешалкой, термометром и конденсатором, загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 5,0 г, 0,02 моль) и мезитилен (20 г), поддерживая поток азота через газоприемную линию, соединенную с конденсатором. Реакционную смесь перемешивали при комнатной температуре, и добавляли порошок цианида натрия (CyPlus®, 1,40 г, 0,027 моль, допуская 95% чистоты), цианид меди(I) (0,27 г, 0,003 моль, 99% чистоты), йодид натрия (0,45 г, 0,003 моль, 99% чистоты) и 1-бутил-1H-имидазол (1,9 г, 0,015 моль, 98% чистоты). Смесь нагревали при кипячении с обратным холодильником (примерно 167-168°C) в течение 6 часов и затем охлаждали в течение ночи. Воду (20 мл) добавляли по каплям к реакционной смеси в течение 5 минут при перемешивании. После перемешивания в течение дополнительного 1 часа реакционную смесь фильтровали, собранное твердое вещество промывали последовательно водой (3×10 мл), мезитиленом (10 мл) и сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (3,4 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.с, 1H), 7,82 (ушир.с, 1H), 7,44 (ушир.с, 1H), 7,18 (ушир.с, 2H), 2,74 (д, 3H), 2,10 (с, 3H).
Пример 11
Одиннадцатое получение 2-амино-5-циано-N,3-диметилбензамида
100 мл реактор (HP реакторная установка высокого давления, разработанная Hastelloy® C и произведенная HEL, Inc., Lawrenceville, Нью Джерси) оснащали механической мешалкой (разработанной Hastelloy® C) с двойной турбинной мешалкой (нижняя турбина, которая накачивает, и верхняя турбина, которая откачивает). Реактор продували азотом, затем выдерживали в атмосфере азота и загружали последовательно 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 7,4 г, 0,03 моль), порошок цианида натрия (CyPlus®, 2,3 г, 0,045 моль, допуская 95% чистоту), йодид меди(I) (98% чистоты, 0,87 г, 0,0045 моль), ксилолы (30 г) и 1-бутил-1H-имидазол (3,2 г, 0,0255 моль, 98% чистоты). Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли пять раз. Перемешивание начинали при 300 об/мин, и реактор затем испытывали на герметичность путем нагнетания до 690 кПа в течение 20 минут. Из реактора выпускали газ, выпускное отверстие закрывали, и реакционную смесь нагревали при 170°C в течение 6 часов. Реакционную смесь охлаждали до 20-25°C, из реактора выпускали газ и отстаивали в течение выходных. Реакционную смесь переносили в 250-мл колбу Эрленмейера, и добавляли воду (30 г). После перемешивания в течение 1 часа, реакционную смесь фильтровали, собранное твердое вещество промывали последовательно водой (3×15 мл) и ксилолами (15 мл), сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (5,2 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.с, 1H), 7,82 (ушир.с, 1H), 7,44 (ушир.с, 1H), 7,18 (ушир.с, 2H), 2,74 (д, 3H), 2,10 (с, 3H).
Пример 12
Двенадцатое получение 2-амино-5-циано-N,3-диметилбензамида
100 мл реактор (HP реакторная установка высокого давления, разработанная Hastelloy® C и произведенная HEL, Inc.) оснащали механической мешалкой (разработанной Hastelloy® C) с двойной турбинной мешалкой (нижняя турбина, которая накачивает, и верхняя турбина, которая откачивает). Реактор продували азотом, затем выдерживали в атмосфере азота и загружали последовательно 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 7,4 г, 0,03 моль), порошок цианида натрия (CyPlus®, 2,3 г, 0,045 моль, допуская 95% чистоту), йодид меди(I) (98% чистоты, 0,87 г, 0,0045 моль), толуол (30 г) и 1-бутил-1H-имидазол (3,2 г, 0,0255 моль, 98% чистоты). Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли пять раз. Перемешивание начинали при 300 об/мин, и реактор затем испытывали на герметичность путем нагнетания до 690 кПа в течение 20 минут. Из реактора выпускали газ, выпускное отверстие закрывали, и реакционную смесь нагревали при 170°C в течение 6 часов. Реакционную смесь охлаждали до 20-25°C, из реактора выпускали газ и отстаивали в течение выходных. Затем реакционную смесь переносили в 250-мл колбу Эрленмейера и добавляли воду (30 г). После перемешивания в течение 1 часа реакционную смесь фильтровали, собранное твердое вещество промывали последовательно водой (3×15 мл) и толуолом (15 мл), сушили до постоянного веса в вакуумной печи при 55°C, с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (5,0 г).
1H ЯМР (ДМСО-d6) δ 8,44 (ушир.с, 1H), 7,83 (ушир.с, 1H), 7,44 (ушир.с, 1H), 7,19 (ушир.с, 2H), 2,75 (д, 3H), 2,11 (с, 3H).
Пример 13
Тринадцатое получение 2-амино-5-циано-N,3-диметилбензамида
100 мл реактор (HP реакторная установка высокого давления, разработанная Hastelloy® C и произведенная HEL, Inc.) оснащали механической мешалкой (разработанной Hastelloy® C) с двойной турбинной мешалкой (нижняя турбина, которая накачивает, и верхняя турбина, которая откачивает). Реактор продували азотом, затем выдерживали в атмосфере азота и загружали последовательно 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 7,4 г, 0,03 моль), порошок цианида натрия (CyPlus®, 2,3 г, 0,045 моль, допуская 95% чистоту), йодид меди(I) (98% чистоты, 0,87 г, 0,0045 моль) и ксилолы (20 г). Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Перемешивание начинали при 300 об/мин, и реактор затем испытывали на герметичность путем нагнетания до 690 кПа в течение 20 минут. Из реактора выпускали газ до атмосферного давления, и раствор 1-метилимидазола (1,86 г, 0,0255 моль, 99% чистоты) в ксилолах (10 мл) добавляли к реакционной смеси. Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Выпускное отверстие реактора закрывали, и смесь нагревали при 170°C в течение 8 часов. Затем реакционную смесь охлаждали до 20-25°C, из реактора выпускали газ и отстаивали в течение ночи. Затем реакционную смесь разбавляли N,N-диметилформамидом (ДМФА) (общая масса реакционной смеси после разбавления ДМФА составляла 100 г) и анализировали ВЭЖХ, которая показала 98% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 14
Четырнадцатое получение 2-амино-5-циано-N,3-диметилбензамида
100 мл реактор (HP реакторная установка высокого давления, разработанная Hastelloy® C и произведенная HEL, Inc.) оснащали механической мешалкой (разработанной Hastelloy® C) с двойной турбинной мешалкой (нижняя турбина, которая накачивает, и верхняя турбина, которая откачивает). Реактор продували азотом, затем выдерживали в атмосфере азота и загружали последовательно 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 7,4 г, 0,03 моль), порошок цианида натрия (CyPlus®, 2,3 г, 0,045 моль, допуская 95% чистоту), йодид меди(I) (98% чистоты, 0,87 г, 0,0045 моль) и толуол (20 г). Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Перемешивание начинали при 300 об/мин, и реактор затем испытывали на герметичность путем нагнетания до 690 кПа в течение 20 минут. Из реактора выпускали газ до атмосферного давления, и раствор 1-метилимидазола (1,86 г, 0,0255 моль, 99% чистоты) в толуоле (10 мл) добавляли к реакционной смеси. Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Выпускное отверстие реактора закрывали, и смесь нагревали при 170°C в течение 8 часов. Реакционную смесь охлаждали до 20-25°C, из реактора выпускали газ и отстаивали в течение ночи. Затем реакционную смесь разбавляли ДМФА (общая масса реакционной смеси после разбавления ДМФА составляла 100 г) и анализировали ВЭЖХ, которая показала 99% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 15
Пятнадцатое получение 2-амино-5-циано-N,3-диметилбензамида
100 мл реактор (HP реакторная установка высокого давления, разработанная Hastelloy® C и произведенная HEL, Inc.) оснащали механической мешалкой (разработанной Hastelloy® C) с двойной турбинной мешалкой (нижняя турбина, которая накачивает, и верхняя турбина, которая откачивает). Реактор продували азотом, затем выдерживали в атмосфере азота и загружали последовательно 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 7,4 г, 0,03 моль), порошок цианида натрия (CyPlus®, 2,3 г, 0,045 моль, допуская 95% чистоту), йодид меди(I) (98% чистоты, 0,87 г, 0,0045 моль) и ксилолы (20 г). Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Перемешивание начинали при 300 об/мин, и реактор затем испытывали на герметичность путем нагнетания до 690 кПа в течение 20 минут. Из реактора выпускали газ до атмосферного давления и раствор 4-метилимидазола (1,86 г, 0,0255 моль, 98% чистоты) в ксилолах (10 мл) добавляли к реакционной смеси. Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Выпускное отверстие реактора закрывали, и смесь нагревали при 170°C в течение 12 часов. Реакционную смесь охлаждали до 20-25°C, из реактора выпускали газ и отстаивали в течение ночи. Затем реакционную смесь разбавляли ДМФА (общая масса реакционной смеси после разбавления ДМФА составляла 100 г) и анализировали ВЭЖХ, которая показала 88% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 16
Шестнадцатое получение 2-амино-5-циано-N,3-диметилбензамида
100 мл реактор (HP реакторная установка высокого давления, разработанная Hastelloy® C и произведенная HEL, Inc.) оснащали механической мешалкой (разработанной Hastelloy® C) с двойной турбинной мешалкой (нижняя турбина, которая накачивает, и верхняя турбина, которая откачивает). Реактор продували азотом, затем выдерживали в атмосфере азота и загружали последовательно 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 7,4 г, 0,03 моль), порошок цианида натрия (CyPlus®, 2,3 г, 0,045 моль, допуская 95% чистоту), йодид меди(I) (98% чистоты, 0,87 г, 0,0045 моль), 1,6-бис(имидазол-1-ил)гексан (2,5 г, 0,011 моль) и ксилолы (30 г). Реактор накачивали до 345 кПа азотом и затем выпускали газ. Процедуру нагнетания/выпускания азота повторяли два раза. Перемешивание начинали при 300 об/мин, и реактор затем испытывали на герметичность путем нагнетания до 690 кПа в течение 20 минут. Из реактора выпускали газ до атмосферного давления, выпускное отверстие закрывали и реакционную смесь нагревали при 170°C в течение 12 часов. Реакционную смесь охлаждали до 20-25°C, из реактора выпускали газ и отстаивали в течение ночи. Затем реакционную смесь разбавляли ДМФА (общая масса реакционной смеси после разбавления ДМФА составляла 100 г) и анализировали ВЭЖХ, которая показала 93% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 17
Семнадцатое получение 2-амино-5-циано-N,3-диметилбензамида
125-мл реактор (Advantage Series® 3400 Process Chemistry Workstation, продаваемый Biotage) оснащали механической мешалкой (ось сконструирована Hastelloy® C с полиэфирэфиркетоновым (PEEK) вкладышем мешалки и PEEK турбинным смесителем), температурным датчиком (разработанным Hastelloy® C) и дефлегматором. Реактор загружали 2-амино-5-бром-N,3-диметилбензамид (полученный способом справочного примера 1) (99% чистоты, 9,8 г, 0,04 моль), йодид меди(I) (98% чистоты, 1,17 г, 0,006 моль) и гранулированный цианид натрия (Alfa Aesar®, 3,09 г, 0,060 моль, допуская 95% чистоту). Реактор продували азотом через газоприемную линию, соединенную с конденсатором, затем раствор 1-бутил-1H-имидазола (0,76 г, 0,006 моль, 98% чистоты) в мезитилене (40 г) добавляли к реакционной смеси. Смеситель включали при 300 об/мин, и смесь нагревали при кипячении с обратным холодильником в течение 6 часов. Реакционную смесь охлаждали до 20°C и отстаивали в течение ночи. Затем реакционную смесь разбавляли ДМФА (общая масса реакционной смеси после разбавления ДМФА составляла 130 г) и анализировали ВЭЖХ, которая показала 97% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 18
Восемнадцатое получение 2-амино-5-циано-N,3-диметилбензамида
Указанное соединение получали по методике примера 17, используя другое количество 1-бутил-1H-имидазола (1,52 г, 0,012 моль). Анализ реакционной смеси с помощью ВЭЖХ показал 97% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 19
Девятнадцатое получение 2-амино-5-циано-N,3-диметилбензамида
Указанное соединение получали по методике примера 17, используя другое количество 1-бутил-1H-имидазола (2,27 г, 0,018 моль). Анализ реакционной смеси с помощью ВЭЖХ показал 97% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 20
Двадцатое получение 2-амино-5-циано-N,3-диметилбензамида
Указанное соединение получали по методике примера 17, используя другое количество 1-бутил-1H-имидазола (3,03 г, 0,024 моль). Анализ реакционной смеси с помощью ВЭЖХ показал 99% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 21
Двадцать первое получение 2-амино-5-циано-N,3-диметилбензамида
Указанное соединение получали по методике примера 17, используя другое количество 1-бутил-1H-имидазола (3,82 г, 0,030 моль). Анализ реакционной смеси с помощью ВЭЖХ показал 98% преобразование 2-амино-5-бром-N,3-диметилбензамида, причем 2-амино-5-циано-N,3-диметилбензамид являлся основным продуктом.
Пример 22
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1H-пиразол-5-карбоксамида
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоновой кислоты (см. патентную публикацию PCT WO 2003/015519 для способа получения) (3,02 г, 0,010 моль) и 2-амино-5-циано-N,3-диметилбензамида (полученного способом примера 9, за исключением того, что 2-амино-5-бром-N,3-диметилбензамид получали из коммерческого источника и использовали порошок цианида натрия от CyPlus®) (1,99 г, 0,0105 моль) в ацетонитриле (16 мл) добавляли 3-пиколин (2,92 мл, 0,030 моль). Реакционную смесь охлаждали до 15-20°C и затем по каплям добавляли метансульфонилхлорид (8,2 г, 0,071 моль). После перемешивания в течение 2 часов при 20°C, к реакционной смеси по каплям добавляли воду (7,5 мл), поддерживая температуру при 20-25°C. Через 15 минут добавляли концентрированную хлористоводородную кислоту (0,50 мл) и реакционную смесь перемешивали в течение 1 часа при 20°C. Смесь фильтровали, собранные твердые вещества промывали смесью ацетонитрил-вода (3:1 смесь, 2×2 мл) и водой (2×2 мл), и сушили в атмосфере азота, с получением указанного в заголовке соединения (4,86 г) в виде не совсем белого твердого вещества, которое плавилось при 206-208°C.
1H ЯМР (ДМСО-d6) δ 10,52 (ушир.с, 1H), 8,50 (дд, 1H), 8,36 (м, 1H), 8,17 (дд, 1H), 7,88 (д, 1H), 7,76 (д, 1H), 7,62 (м, 1H), 7,41 (с, 1H), 2,66 (д, 3H), 2,21 (с, 3H).
Справочный пример 2
Получение 3-бром-1-(3-хлор-2-пиридинил)-N-[4-циано-2-метил-6-[(метиламино)карбонил]фенил]-1H-пиразол-5-карбоксамида (соединение формулы 4)
К смеси 3-бром-1-(3-хлор-2-пиридинил)-1H-пиразол-5-карбоновой кислоты (см. патентную публикацию PCT WO 2003/015519 для способа получения) (15 г, 0,049 моль, 97,4% чистоты) и 2-амино-5-циано-N,3-диметилбензамида (см. патентную публикацию PCT WO 2006/62978 для способа получения) (10,0 г, 0,0525 моль) в ацетонитриле (80 мл) добавляли 3-пиколин (13,9 г, 0,148 моль). Реакционную смесь охлаждали до 15-20°C и затем по каплям добавляли метансульфонилхлорид (8,2 г, 0,071 моль). Через 1 час к реакционной смеси по каплям добавляли воду (37,3 г), поддерживая температуру при 15-20°C. Смесь нагревали при 45-50°C в течение 30 минут и затем охлаждали до 15-25°C в течение 1 часа. Смесь фильтровали, собранные твердые вещества промывали смесью ацетонитрил-вода (смесью приблизительно 5:1, 2×10 мл) и ацетонитрилом (2×10 мл) и затем сушили в атмосфере азота, с получением указанного в заголовке соединения (24,0 г, 93,6% скорректированный выход на основе анализа 91,6%) в виде не совсем белого твердого вещества.
1H ЯМР (ДМСО-d6) δ 10,53 (ушир.с, 1H), 8,49 (дд, 1H), 8,36 (м, 1H), 8,16 (дд, 1H), 7,87 (д, 1H), 7,76 (д, 1H), 7,60 (м, 1H), 7,41 (с, 1H), 2,67 (д, 3H), 2,21 (с, 3H).
Таблица 1 иллюстрирует конкретные преобразования для получения соединений формулы 1 согласно способу по настоящему изобретению. Для таких превращений реагент на основе соли меди(I) и реагент на основе соли йодистоводородной кислоты представляют собой йодид меди(I). В таблице 1 и последующих таблицах: трет означает третичный, втор означает вторичный, н означает нормальный, изо означает изо, ц означает цикло, Me означает метил, Et означает этил, Pr означает пропил, и Bu означает бутил. Объединения групп сокращаются подобным образом; например, “ц-PrCH2” означает циклопропилметил.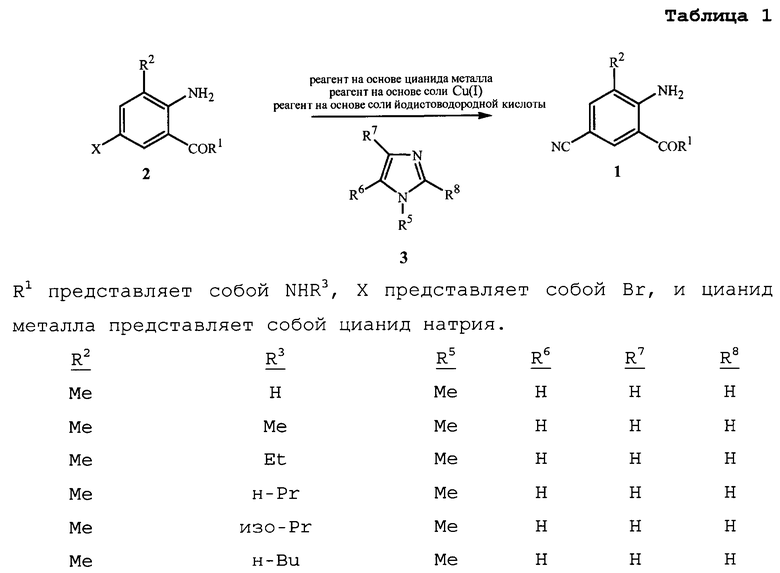





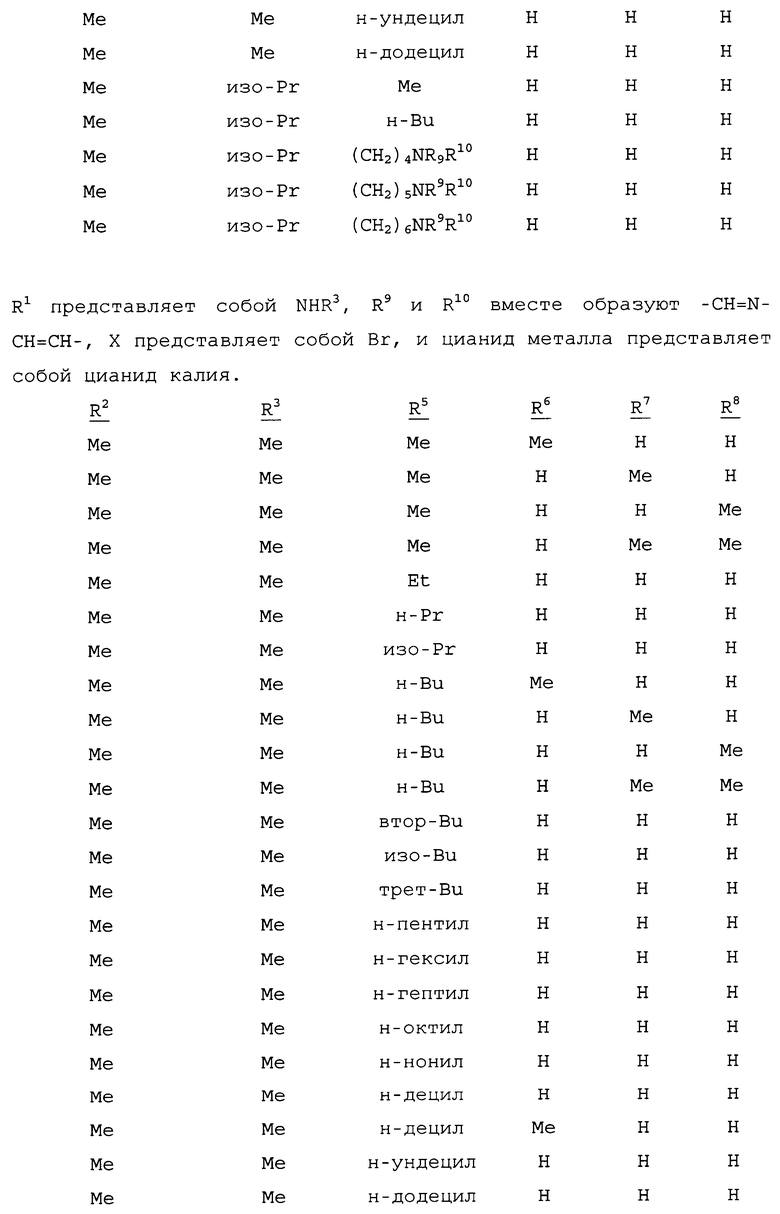
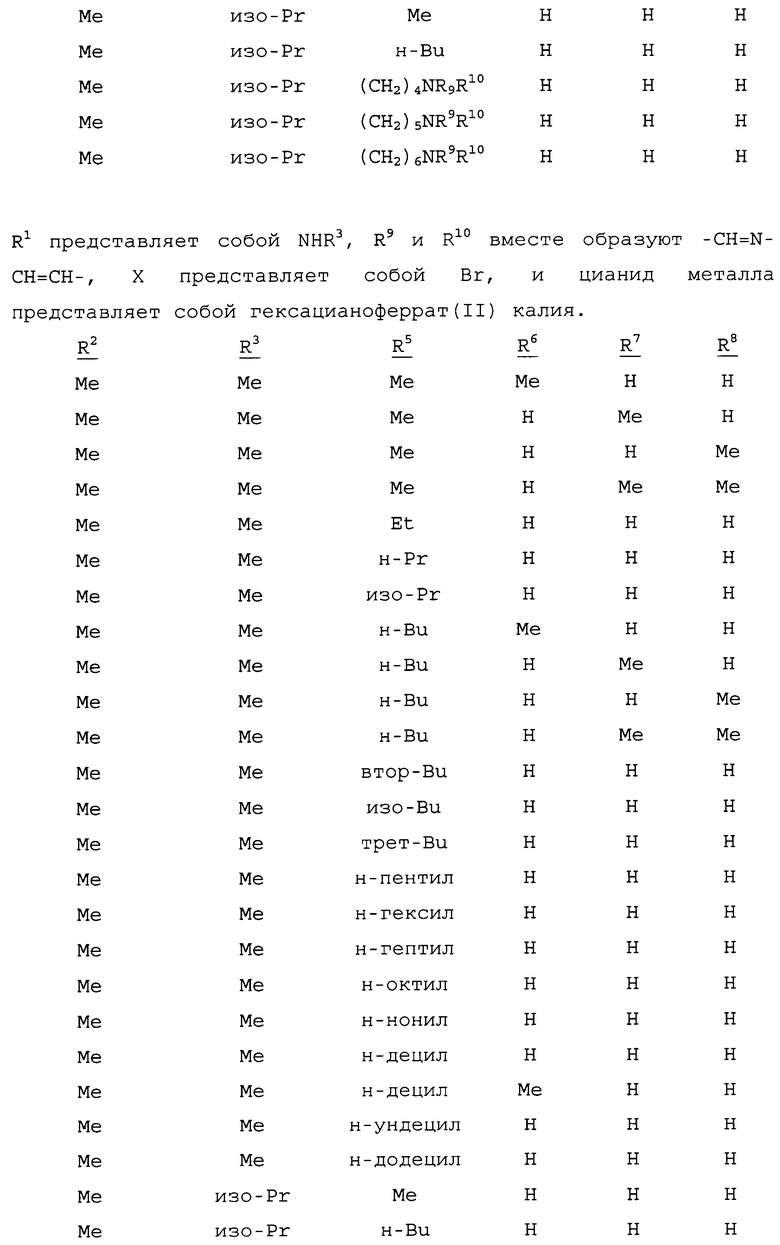

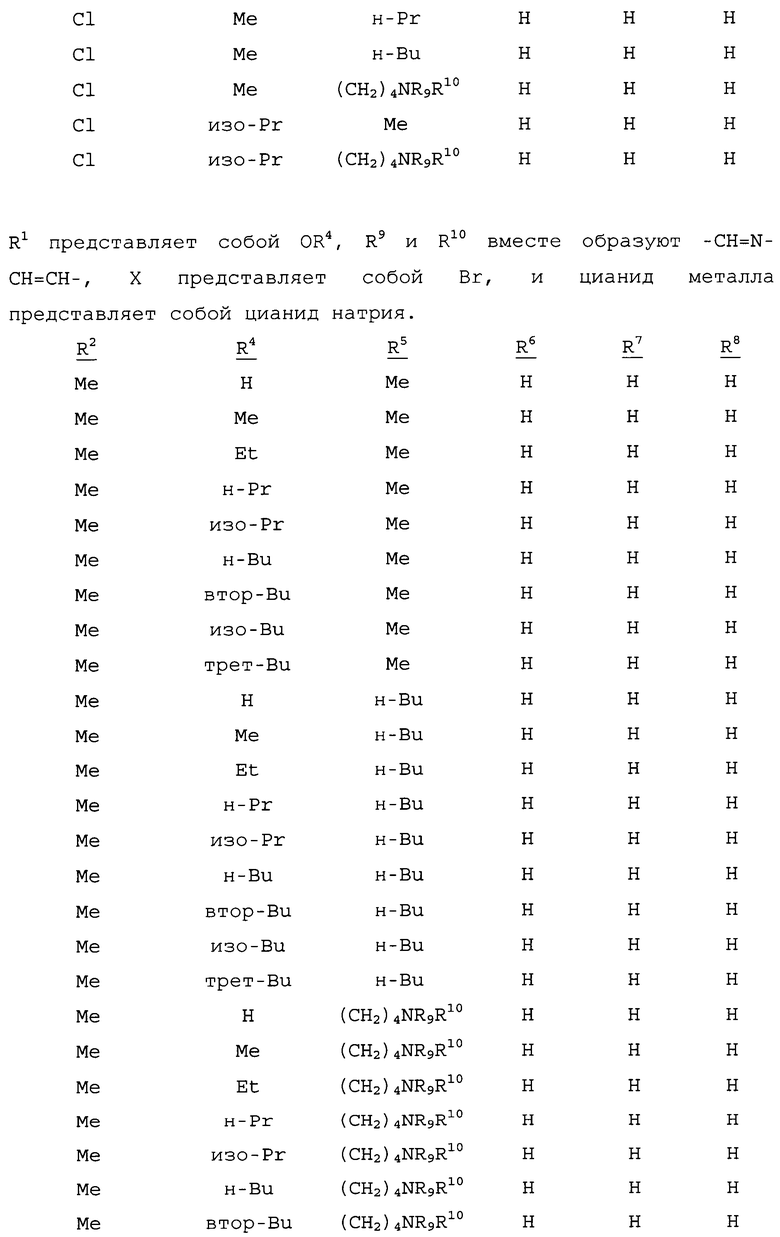
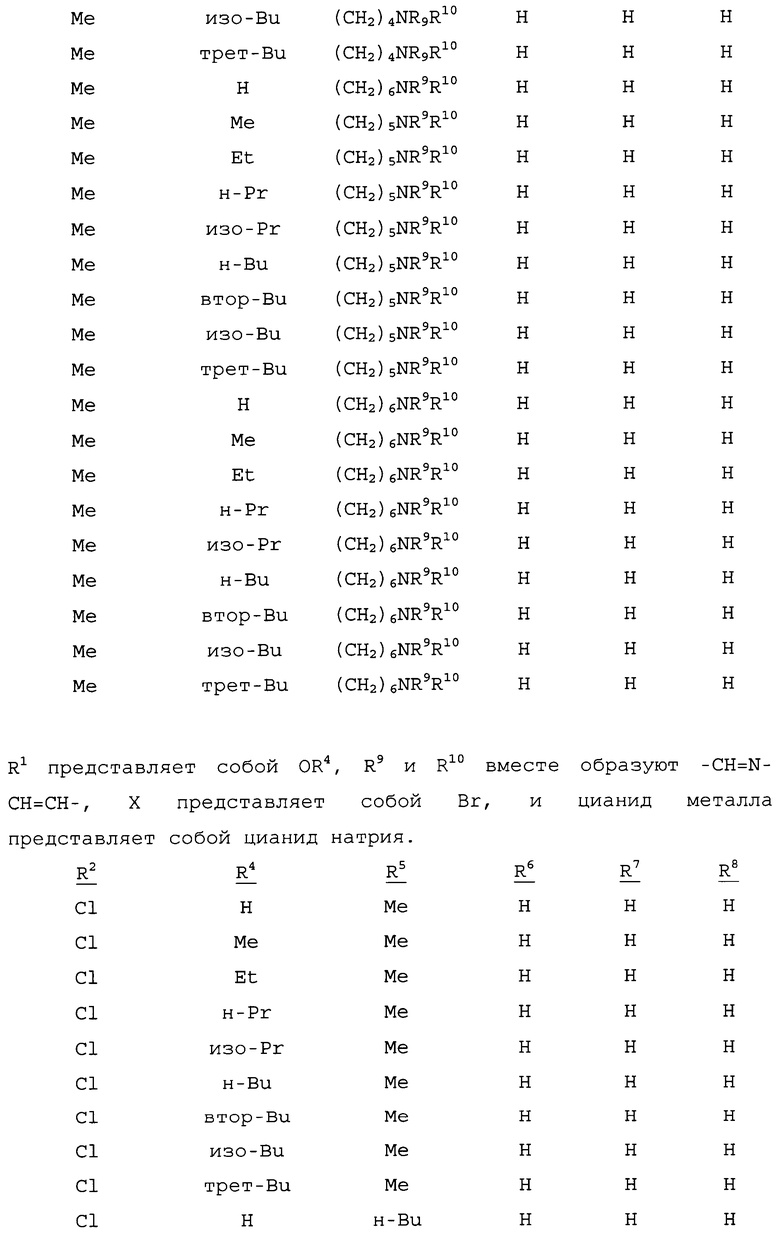
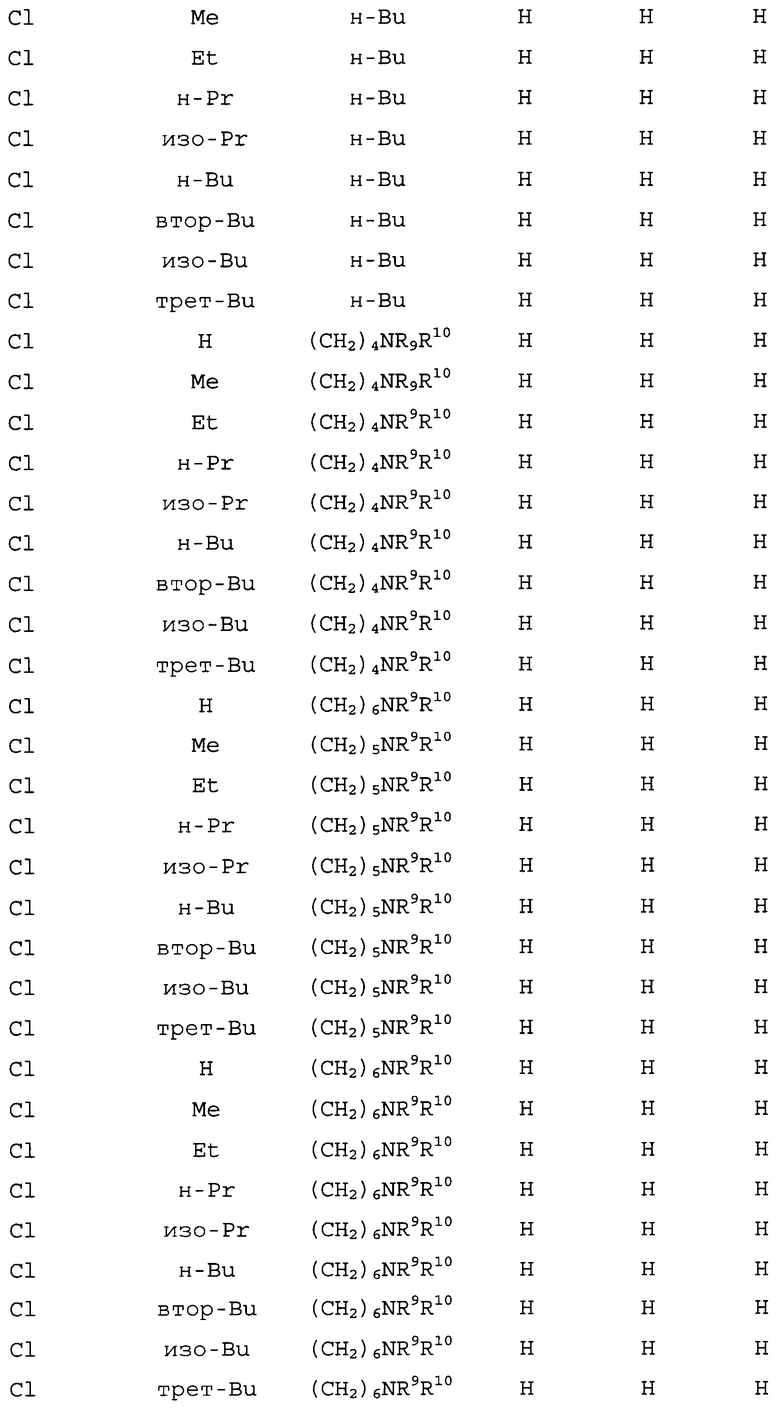
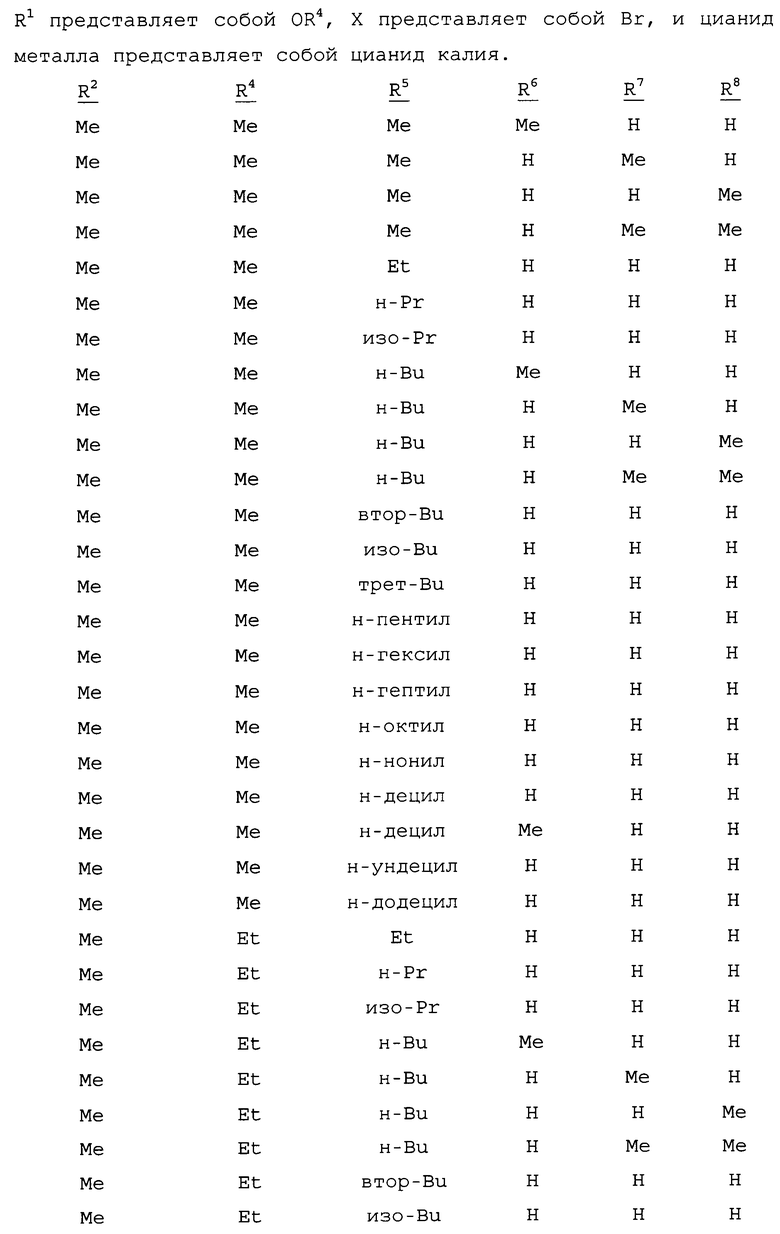

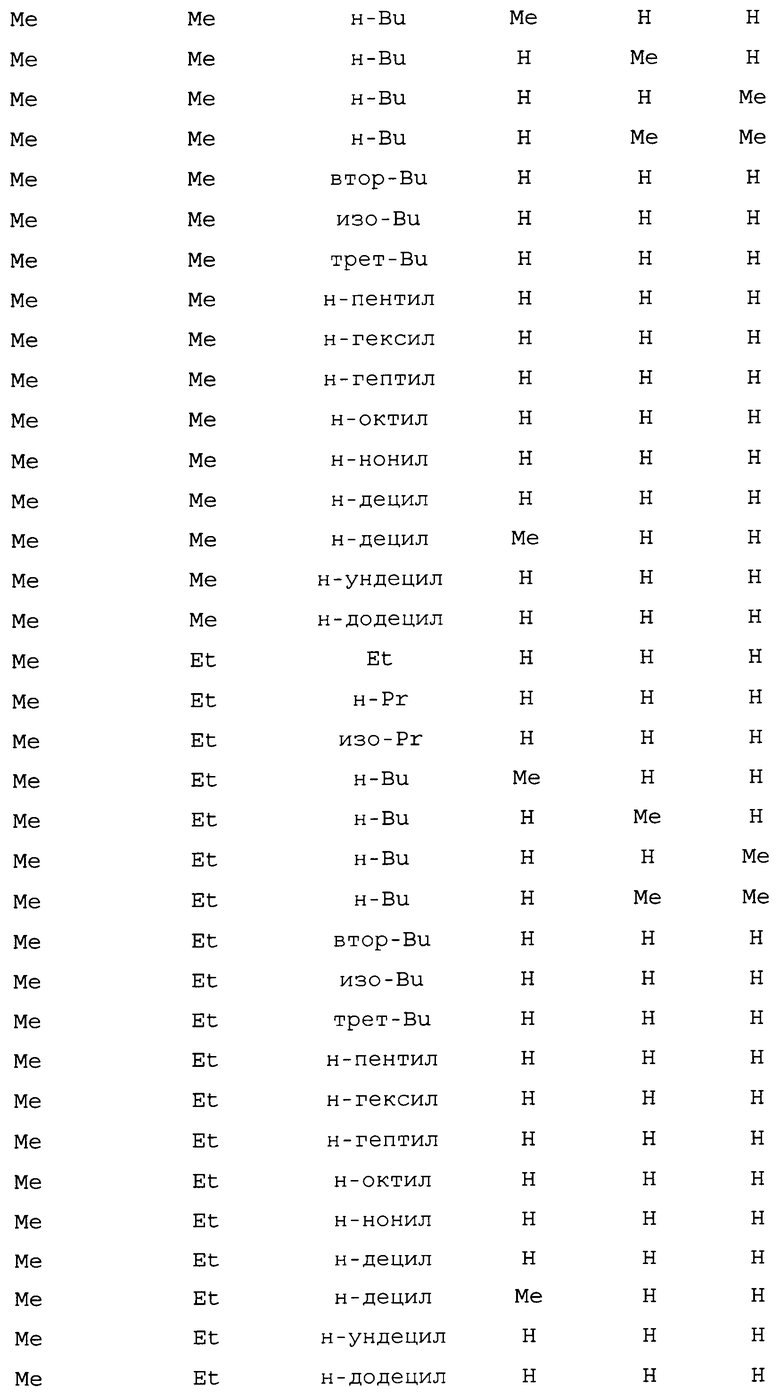

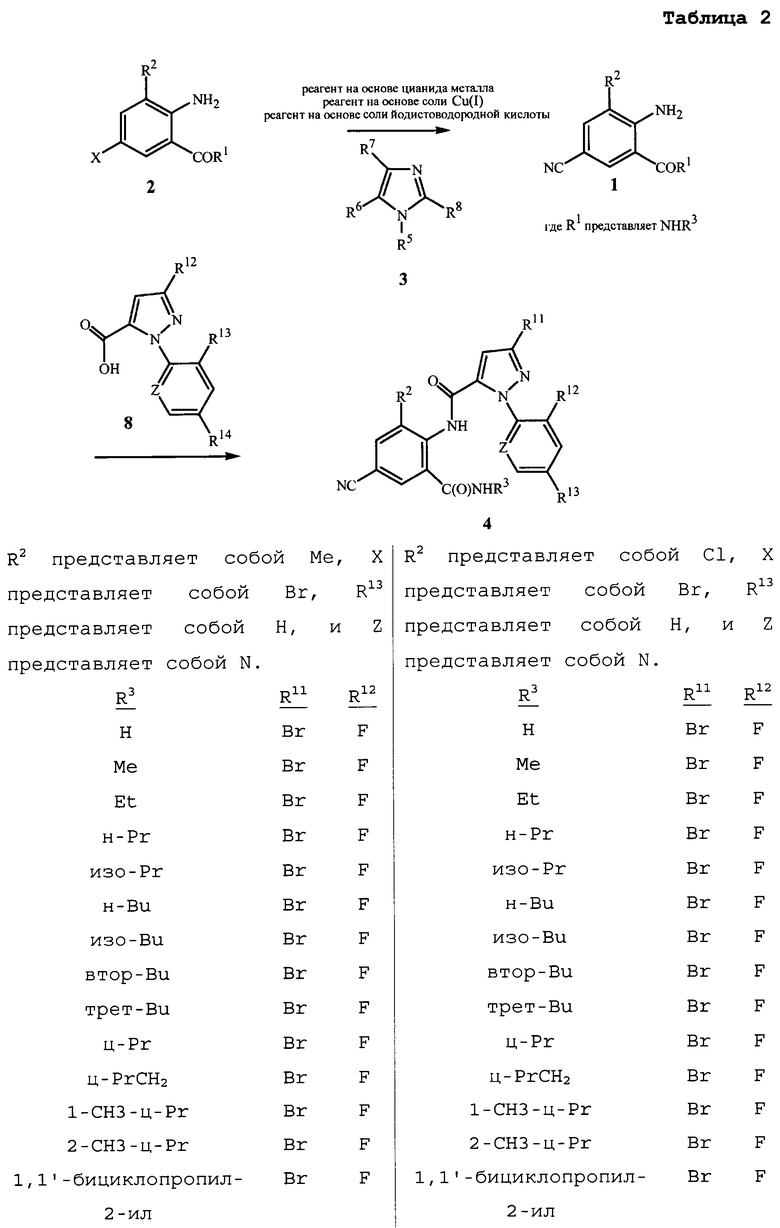




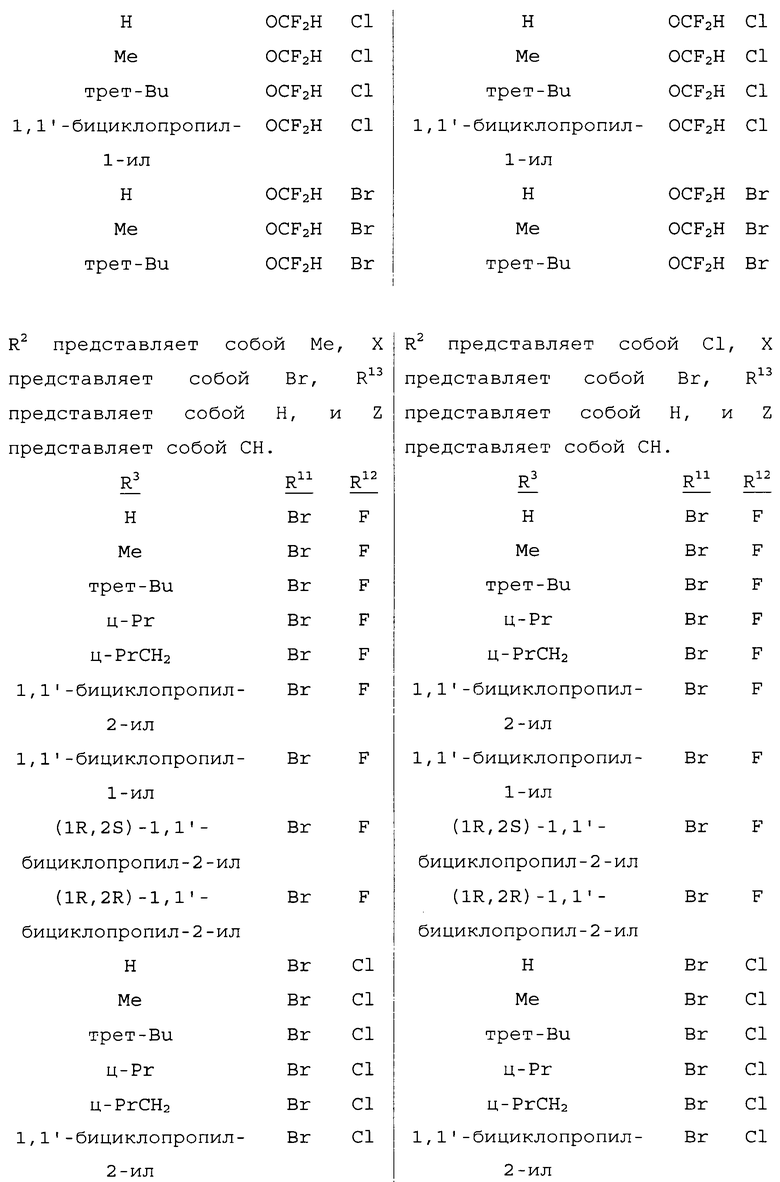










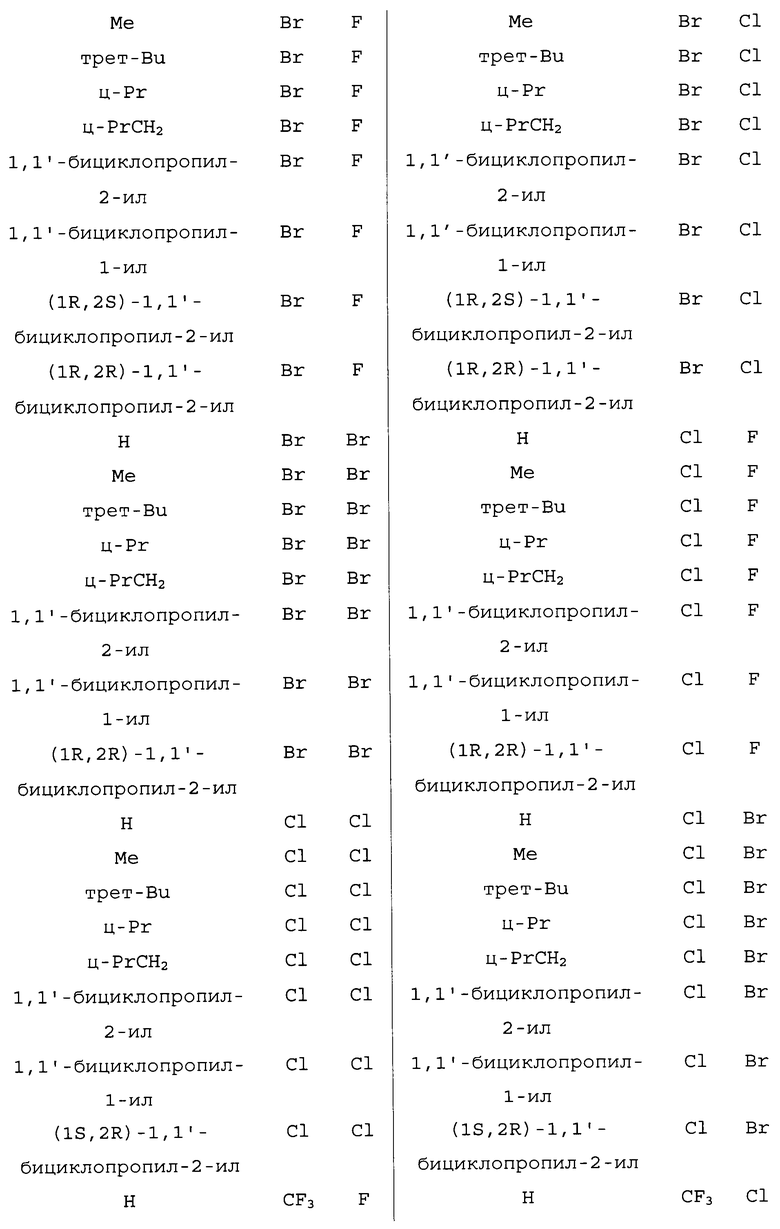




| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-ФЕНИЛПИРАЗОЛ-1-КАРБОКСАМИДОВ | 2005 |
|
RU2397165C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ЗАМЕЩЕННЫХ 2-АМИНО-5-ГАЛОГЕНБЕНЗАМИДОВ | 2007 |
|
RU2443679C2 |
| СИНТЕЗ ЗАМЕЩЕННЫХ КОНДЕНСИРОВАННЫХ С ГЕТЕРОЦИКЛОМ ГАММАКАРБОЛИНОВ | 2019 |
|
RU2795581C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2009 |
|
RU2604066C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АЛКОКСИ-3-ГИДРОКСИПИКОЛИНОВЫХ КИСЛОТ | 2017 |
|
RU2744834C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-БРОМ-4,5-ДИАЛКОКСИБЕНЗОЙНОЙ КИСЛОТЫ | 2011 |
|
RU2576520C2 |
| ПРОИЗВОДНОЕ 1-ЗАМЕЩЕННОГО 4-НИТРОИМИДАЗОЛА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2324682C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 3-(4-(2,4-ДИФТОРБЕНЗИЛОКСИ)-3-БРОМ-6-МЕТИЛ-2-ОКСОПИРИДИН-1(2Н)-ИЛ)-N,4-ДИМЕТИЛБЕНЗАМИДА | 2007 |
|
RU2411236C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2013 |
|
RU2632203C2 |
| 2-(ФЕНИЛ)-1H-ФЕНАНТРО[9.10-d]ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ mPGES-1 | 2006 |
|
RU2421448C2 |
Настоящее изобретение относится к области органической химии, а именно к способу получения производных 2-амино-5-цианобензойной кислоты формулы (1), где R1 представляет собой NHR3; R2 представляет собой СН3 или Cl; и R3 представляет собой Н, С1-С4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил; включающий приведение в контакт (1) соединения формулы 2, где Х представляет собой Br или Cl; с (2) реагентом на основе цианида металла, включающим одно или более соединений, выбранных из группы, состоящей из цианида щелочного металла и цианида меди (I), (3) реагентом на основе соли меди (I), (4) реагентом на основе соли йодистоводородной кислоты и (5) по меньшей мере одним соединением формулы 3, где R5 представляет собой Н, фенил или бензил; или С1-С12алкил, необязательно замещенный NR9R10; каждый R6, R7 и R8 представляет собой независимо Н, С1-С12алкил, фенил или бензил; или R6 и R7 вместе образуют -СН=СН-СН=СН-; и R9 и R10 вместе образуют -CH=N-CH=CH-, необязательно имеющий до 3 заместителей, независимо выбранных из С1-С12алкила; при условии, что когда Х представляет собой Cl, то R2 представляет собой метил. Технический результат: разработан новый способ получения производных 2-амино-5-цианобензойной кислоты, отличающийся быстротой и экономичностью. 13 з.п. ф-лы, 2 табл.
1. Способ получения соединения формулы 1
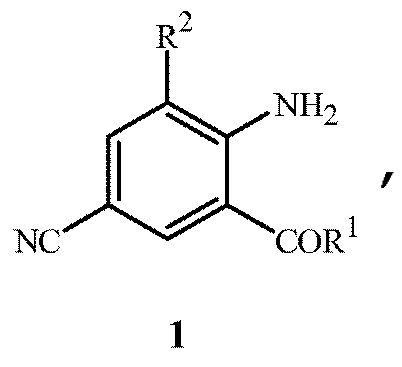
где R1 представляет собой NHR3;
R2 представляет собой СН3 или Cl; и
R3 представляет собой Н, С1-С4алкил, циклопропил, циклопропилциклопропил, циклопропилметил или метилциклопропил;
включающий приведение в контакт (1) соединения формулы 2
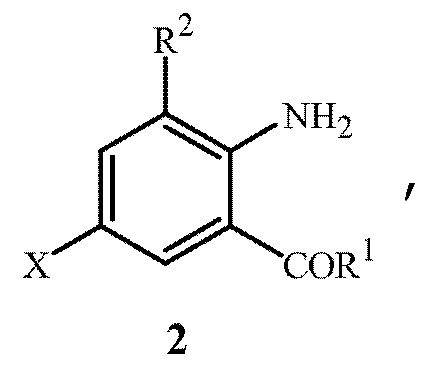
где Х представляет собой Br или Cl;
с (2) реагентом на основе цианида металла, включающим один или более соединений, выбранных из группы, состоящей из цианида щелочного металла и цианида меди (I), (3) реагентом на основе соли меди (I), (4) реагентом на основе соли йодистоводородной кислоты и (5) по меньшей мере одним соединением формулы 3
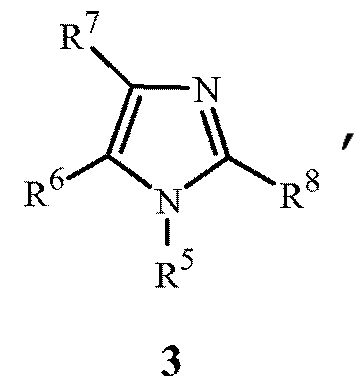
где R5 представляет собой Н, фенил или бензил; или С1-С12алкил, необязательно замещенный NR9R10;
каждый R6, R7 и R8 представляет собой независимо Н, С1-С12алкил, фенил или бензил; или
R6 и R7 вместе образуют -СН=СН-СН=СН-; и
R9 и R10 вместе образуют -CH=N-CH=CH-, необязательно имеющий до 3 заместителей, независимо выбранных из С1-С12алкила;
при условии, что когда Х представляет собой Сl, то R2 представляет собой метил.
2. Способ по п.1, где реагент на основе соли меди (I) и реагент на основе соли йодистоводородной кислоты содержат йодид меди (I).
3. Способ по п.1, где по меньшей мере одно соединение формулы 3 содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-этил-1H-имидазол, 1-пропил-1H-имидазол, 1-бутил-1H-имидазол, 1-пентил-1H-имидазол, 1-гексил-1Н-имидазол, 4-метилимидазол, 1,1'-(1,4-бутандиил)бис-1H-имидазол, 1,1'-(1,5-пентандиил)бис-1Н-имидазол и 1,1'-(1,6-гександиил)бис-1Н-имидазол.
4. Способ по п.3, где по меньшей мере одно соединение формулы 3 содержит одно или более соединений, выбранных из группы, содержащей 1-метил-1H-имидазол, 1-бутил-1H-имидазол, 4-метилимидазол и 1,1'-(1,6-гександиил)бис-1Н-имидазол.
5. Способ по п.4, где по меньшей мере одно соединение формулы 3 содержит 1-метил-1H-имидазол или 1-бутил-1H-имидазол.
6. Способ по п.1, где соединение формулы 2 приводят в контакт с подходящим органическим растворителем с образованием смеси, и затем реагент на основе цианида металла, реагент на основе соли меди (I), реагент на основе соли йодистоводородной кислоты и по меньшей мере одно соединение формулы 3 последовательно добавляют к смеси.
7. Способ по п.6, где подходящий органический растворитель включает один или более растворителей, выбранных из группы, содержащей ксилолы, толуол, хлорбензол, метоксибензол, 1,2,4-триметилбензол, 1,3,5-триметилбензол, этилбензол, (1-метилэтил)бензол, C1-С3алкил-замещенные нафталины, ShellSol А100 и ShellSol A150.
8. Способ по п.7, где подходящий органический растворитель включает один или более растворителей, выбранных из группы, содержащей ксилолы, толуол, 1,2,4-триметилбензол, 1,3,5-триметилбензол и 1-метилнафталин.
9. Способ по п.1, где реагент на основе цианида металла содержит одно или более соединений, выбранных из группы, состоящей из цианида натрия и цианида калия.
10. Способ по п.9, где реагент на основе цианида металла содержит цианид натрия.
11. Способ по п.1, где Х представляет собой Br, и соединение формулы 1 получают в виде твердого вещества, включающий приведение в контакт соединения формулы 2 с подходящим органическим растворителем с образованием смеси и затем последовательное добавление реагента на основе цианида металла, реагента на основе соли меди (I), реагента на основе соли йодистоводородной кислоты и соединения или соединений формулы 3, поддержание температуры смеси примерно от 145 до 180°С в течение примерно от 5 до примерно 8 ч, охлаждение смеси до примерно 0-50°С, добавление воды к смеси, необязательно перемешивание в течение примерно от 1 до примерно 24 ч и затем выделение соединения формулы 1 в виде твердого вещества из смеси.
12. Способ по п.11, где соединение формулы 1 представляет собой 2-амино-5-циано-N,3-диметилбензамид.
13. Способ по п.1, где Х представляет собой Cl, и соединение формулы 1 получают в виде твердого вещества, включающий приведение в контакт соединения формулы 2 с подходящим органическим растворителем с образованием смеси и затем последовательное добавление реагента на основе цианида металла, реагента на основе соли меди (I), реагента на основе соли йодистоводородной кислоты и соединения или соединений формулы 3, поддержание температуры смеси примерно от 150 до 200°С в течение примерно от 5 до примерно 24 ч, охлаждение смеси до примерно 0-50°С, добавление воды к смеси, необязательно перемешивание в течение примерно от 1 до примерно 24 ч и затем выделение соединения формулы 1 в виде твердого вещества из смеси.
14. Способ по п.13, где соединение формулы 1 представляет собой 2-амино-5-циано-N,3-диметилбензамид.
| Schareina Т | |||
| et al: "A state-of-the-art cyanation of aryl bromides: a novel and versatile cooper catalyst system inspired by nature", Chem | |||
| Eur | |||
| J | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| СПОСОБ БОРЬБЫ С КОНКРЕТНЫМИ НАСЕКОМЫМИ-ВРЕДИТЕЛЯМИ ПУТЕМ НАНЕСЕНИЯ АНТРАНИЛАМИДНЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2262231C1 |

