Область изобретения
Данное изобретение относится к быстро образующей гель твердой пероральной фармацевтической лекарственной форме, а также получаемым из нее водным суспензиям, содержащим кислоточувствительный ингибитор протонного насоса в качестве активного ингредиента, распределенный в массе гранул, покрытых энтеросолюбильной оболочкой, и к грануляту, модифицирующему суспензию. Кроме того, изобретение относится к улучшенному способу ее получения и к применению такой комопзиции в лечении желудочно-кишечных расстройств у людей, включая их предупреждение.
Предпосылки изобретения и предшествующий уровень техники
Соединения-ингибиторы протонного насоса (также обозначенные далее как «PPI»), обладающие эффектом ингибиторов Н+К+-АТФазы, представляют собой, например, соединения, известные под непатентованными наименованиями омепразол, лансопразол, пантопразол, рабепразол, тенатопразол и эзомепразол.
Эти активные вещества полезны для ингибирования секреции желудочной кислоты у млекопитающих и человека. В более общем смысле, их можно применять для предупреждения и лечения заболеваний, связанных с желудочной кислотой, у млекопитающих и человека, включая, например, рефлюкс-эзофагит, гастрит, дуоденит, язву желудка и язву двенадцатиперстной кишки. Кроме того, их можно применять для лечения других желудочно-кишечных расстройств, при которых желательным является эффект ингибирования желудочной кислоты, например, у пациентов, получающих терапию NSAID (нестероидными противовоспалительными средствами), у пациентов с неязвенной диспепсией, у пациентов с симптоматической гастроэзофагеальной рефлюксной болезнью и у пациентов с гастриномами. Также их можно применять у пациентов в ситуациях, требующих интенсивной терапии, у пациентов с острым кровотечением из верхних отделов желудочно-кишечного тракта, в до- и послеоперационном периоде для предупреждения кислотной аспирации желудочной кислоты и для предупреждения и лечения стрессового изъязвления. Кроме того, их можно применять для предупреждения и лечения синдрома раздраженной толстой кишки (IBS), воспаления кишечника (IBD), язвенного колита, болезни Крона, астмы, ларингита, синдрома Баррета, апноэ во сне, нарушения сна, псориаза, а также они полезны для предупреждения и лечения инфекционных заболеваний, вызванных Helicobacter, и заболеваний, родственных указанным выше.
Эти активные соединения, однако, подвержены разрушению/трансформации в кислой и нейтральной среде. Разрушение катализируется кислыми соединениями и стабилизируется в смесях с щелочными соединениями. На стабильность активных веществ также влияют влажность, нагревание, содержание органических растворителей и, до некоторой степени, освещение.
Пероральные лекарственные формы остаются существенной проблемой для многих пациентов, поскольку многие не могут или не хотят глотать твердую лекарственную форму. Эта проблема имеет место главным образом среди детей и пожилых людей. Она влияет на соблюдение пациентом режима лечения и, следовательно, является проблемой терапевтического лечения.
Потребность в форме для перорального введения, которая позволяет избежать трудностей при глотании, связанных с традиционными таблетками, была признана много лет назад. Были разработаны сиропы, эликсиры, микрокапсулы, содержащие взвеси, и другие новые таблетированные или инкапсулированные лекарственные формы. Среди альтернативных форм для перорального введения фармакологически активных веществ предложено применение раствора или суспензии активного ингредиента в водной среде.
Несмотря на то что готовые к употреблению суспензии (или растворы) имеют недостатки, связанные с большей хранимой емкостью и часто с ограниченным сроком годности или необходимостью хранения в холодильнике, особой проблемой, иногда возникающей для водных суспензий, является то, что некоторые твердые частицы имеют выраженную тенденцию к оседанию на дно сосуда, используемого для введения. Это может привести к тому, что часть дозы останется в сосуде, и не вся доза поступит при пероральном пути введения. Другая иногда встречаемая проблема возникает при применении суспензии частиц в жидкой среде для введения через назогастральный зонд, когда частицы могут иметь тенденцию к агрегации или образованию агломератов, что делает невозможным их прохождение через используемый зонд. Еще одна проблема возникает, когда жидкая среда имеет слишком высокую вязкость/вязкоупругость, что делает невозможным ее введение через назогастральный зонд при практически осуществимом давлении.
Очень желательно, особенно при введении кислотно-лабильных соединений, таких как ингибиторы протонного насоса, например омепразол, эзомепразол, пантопразол и лансопразол, получить легко и быстро получаемую, легко проглатываемую гомогенную суспензию, содержащую ингибитор протонного насоса в форме, защищающей его от контакта с кислотными факторами окружающей среды (например, с кислыми желудочными жидкостями). К тому же желательно, чтобы суспензия обладала вязкоупругими свойствами и вязкостью, подходящими для обеспечения ее введения через желудочный зонд или проглатывания. Также для стабильности во времени жидкого суспензионного препарата требуется определенная вязкость.
Другие задачи/проблемы встают для лекарственного препарата, который необходимо хранить в виде сухой порошковой смеси, содержащей нерастворимые в воде компоненты, и который следует вводить в качестве приготовленной для немедленного приема гомогенной суспензии.
Для некоторых композиций из уровня техники проблемой является то, что максимальный уровень вязкости достигается только после длительного периода времени, то есть вязкость не является постоянной в течение коротких временных рамок с момента получения суспензию до момента, когда ее обычно вводят пациенту. Также могут быть проблемы с колебаниями между партиями в отношении времени, требующегося для получения стабильного максимального уровня вязкости в суспензии, полученной из сухой порошковой смеси.
Распространенной проблемой является непереносимость лактозосодержащих пищевых продуктов. Таким образом, лекарственные средства, содержащие лактозу, могут представлять собой проблему для таких людей.
В данной области техники есть предложения, относящиеся к композициям, содержащим ингибитор протонного насоса, и есть другие предложения, относящиеся к способам получения быстро диспергируемых и/или растворимых препаратов.
В US 5731002 описана стабильная пероральная фармацевтическая композиция, содержащая ингибитор протонного насоса в пастообразном геле, предназначенная для лечения заболеваний, связанных с желудочной кислотой, у животных.
В US 5840737 раскрыт способ лечения расстройств, связанных с желудочной кислотой, с помощью композиций, содержащих омепразол или лансопразол вместе с гидрокарбонатами.
Проблемы, связанные с введением бикарбонатов, таких как бикарбонат натрия или калия, i.a. людям, включают то, что в результате нейтрализации карбоната в желудке может возникнуть отрыжка. Пациенты с гастроэзофагеальным рефлюксом могут обострить или ухудшить свое заболевание, поскольку отрыжка может вызвать восходящее движение желудочной кислоты (Brunton, Agents for the control of gastric acidity and treatment of peptic ulcers. B: Goodman A.G., et al. The pharmacologic basis of therapeutics, p.907. (New York, 1990.)). Более того, существует возможность того, что введение гидрокарбоната натрия может вызвать метаболический алкалоз.
Кроме того, существуют другие опубликованные патентные заявки того же патентного семейства, например US 2002/0045646 А1, которые раскрывают композиции твердых покрытых неэнтеросолюбильной оболочкой лекарственных форм, содержащие ингибитор протонного насоса и буфер. Другие заявки данного семейства, US 2003/118669, US 2003/144306, US 2003/191159, US 2003/215527, US 2004/048896 и US 2004/171646, раскрывают, например, жидкие пероральные фармацевтические композиции, содержащие ингибитор протонного насоса и буферный агент, и способ усиления абсорбции ингибитора протонного насоса.
В US 2004/0005362 А1 (Taneja) и US 2004/0082618 (Taneja) описан фармацевтический препарат, содержащий кислотолабильное лекарственное средство с энтеросолюбильной оболочкой и жидкий носитель с рН менее 6,0. Другие опубликованные заявки того же автора описывают, например, жидкий носитель с вязкостью, подходящей для суспендирования микрогранул, содержащих PPI (US 2004/0081700 A1), или (US 2004/0006109 A1 и US 2004/0081671 A1) в основном тот же состав с рН жидкого носителя более 6,5.
В WO 2004/004690 A1 (Taneja) раскрыта жидкая лекарственная форма, имеющая микрогранулы с энтеросолюбильной оболочкой, содержащие кислотолабильное лекарственное средство, и жидкая суспензия, имеющая рН менее 6,0, и вязкость, достаточную для суспендирования микрогранул. В этих лекарственных формах можно использовать карбонаты или бикарбонаты.
В US 2004/0022854 A1 раскрыта форма для перорального введения кислотолабильных активных соединений, где вспомогательные вещества не пригодны для образования энтеросолюбильных слоев (энтеросолюбильной оболочки). Полученные единицы активного соединения могут быть приготовлены в виде препарата в саше, например, вместе с лактозой, или приготовлены вместе с эксципиентами, содержащими карбонаты, в форме шипучей композиции.
В ЕР 1232746 описана быстросуспендируемая композиция сухой порошковой смеси, содержащая гелеобразующий агент или загуститель, содержащий по меньшей мере одну ксантановую камедь с определенным распределением частиц по размеру, наполнитель, увлажнитель или поверхностно-активное вещество и фармакологически активное вещество.
В US 4886669 описана водно-диспергируемая таблетка, содержащая фармацевтически активный агент, по меньшей мере, один разрыхлитель и набухающее вещество. Утверждается, что данная таблетка быстро распадается в воде с образованием гомогенной суспензии с высокой вязкостью, которую можно легко проглотить.
US 5008117 относится к способу получения быстродиспергируемого и растворимого препарата из загущающих или суспендирующих агентов и других эксципиентов, в котором легко диспергируются микрокапсулы лекарственного средства. Ингибиторы протонного насоса не упомянуты.
В ЕР 0491910 В1 раскрыта твердая фармацевтическая композиция для добавления к воде с получением суспензии лекарственного средства. Композиция содержит загущающий и суспендирующий агент, кислоту и карбонат или гидрокарбонат.
В US 6261602 описана гранулированная композиция, применимая в качестве фармацевтического носителя, которую можно использовать для получения фармацевтических композиций, способных к быстрому образованию суспензии в воде или водной среде. Композиция может быть получена способом, включающим в себя влажное гранулирование смеси загущающего агента и разрыхляющего агента с водной средой в качестве увлажнителя или сухое гранулирование с получением гранулированного продукта.
Краткое описание изобретения
Настоящее изобретение лишено недостатков композиций, известных из уровня техники, которые обсуждались выше, и представляет собой решение ранее указанных проблем. Кроме того, оно предоставляет средство изготовления носителя лекарственного средства, подходящего для введения через желудочный зонд благодаря хорошей вязкости и вязкоупругим свойствам полученного носителя (суспензии). Например, в том смысле, что он, например, достаточно устойчив для обеспечения приблизительно той же вязкости, даже если количество применяемой воды варьируется от 50% до 150% от предписанного количества.
Настоящее изобретение относится к быстро образующей гель твердой пероральной фармацевтической лекарственной форме, содержащей кислоточувствительное соединение-ингибитор протонного насоса в качестве активного ингредиента, распределенное в массе гранул с энтеросолюбильной оболочкой, и гранулят, модифицирующий суспензию.
Кроме того, неожиданно обнаружено, что полезно использовать гранулят особого состава для смешивания с большим количеством гранул, покрытых энтеросолюбильной оболочкой, содержащих ингибитор протонного насоса, причем этот гранулят при суспендировании в воде быстро и воспроизводимым образом будет образовывать водный носитель с нужным рН, нужным стабильным уровнем вязкости и удовлетворительной вязкоупругостью. Далее этот гранулят также упоминается как «гранулят, модифицирующий суспензию». Кроме того, этот гранулят не должен содержать бикарбонатных и карбонатных солей. Согласно одному воплощению данного изобретения можно изготовить этот гранулят без лактозы, то есть приемлемым для людей с непереносимостью лактозы.
Лекарственные формы по настоящему изобретению обеспечивают возможность быстрого образования вязкой стабильной суспензии. Перед введением твердый сухой гранулят, модифицирующий суспензию, и гранулы с энтеросолюбильной оболочкой растворяют/суспендируют в водной жидкости, такой как водопроводная вода, с получением вязкой жидкой композиции для перорального введения. При введении лекарственной формы по данному изобретению пациенту важно, чтобы препарат была растворен/суспендирован настолько быстро, насколько это возможно, и в то же время обеспечивал получение гомогенной суспензии с учетом распределения твердых частиц, содержащих фармакологически активный ингредиент. Следовательно, конечная жидкая композиция должна гарантировать доставку практически всей дозы, даже если эта доза содержится в суспендированной, корпускулярной форме, в ротовую полость, то есть в начало перорального пути введения, безопасным, надежным и воспроизводимым образом.
При включении активного ингредиента в состав гранул с энтеросолюбильной оболочкой необходимо, чтобы суспензионная среда имела значение рН, не вызывающее преждевременного растворения слоя энтеросолюбильной оболочки гранул, содержащих активный ингредиент. Введение через назогастральный зонд также предъявляет требования к конечной жидкой композиции в отношении таких свойств, как подходящая и стабильная вязкость, вязкоупругие свойства и отсутствие тенденции к агломерации суспендированных частиц.
Следующей особенностью является то, что суспензия пригодна для введения с помощью тонких зондов, предназначенных для педиатрического применения. Термин "желудочный зонд" включает назогастральные зонды, а также другие зонды или шприцы, предназначенные для подачи суспензии или дисперсии в желудок пациента.
Вязкоупругие и вязкие свойства становятся особенно важными, поскольку зонды, применяемые в педиатрическом лечении, могут иметь узкий внутренний диаметр и, следовательно, быть чувствительными к жидкостям с неподходящими свойствами, что приводит к высоким противодавления при введении. Одним таким примером зонда с узким внутренним диаметром является «Детский зонд для кормления, FT 1606/105 (CH/FG 6-2,0 мм наружный диаметр, 1,4 мм внутренний диаметр), Pennine Healthcare».
Лекарственные формы по данному изобретению быстрее образуют гель в воде при комнатной температуре, чем препараты из уровня техники, с получением гомогенной стабильной дисперсии. Таким образом, они обеспечивают стабильную вязкость за более короткое время, чем известный уровень техники, и, кроме того, они являются устойчивыми в отношении полученных вязких свойств.
Описывая кратко, лекарственные формы по данному изобретению содержат два основных компонента: гранулят, модифицирующий суспензию, и массу покрытых энтеросолюбильной оболочкой гранул, содержащих активный ингредиент.
Гранулят, модифицирующий суспензию, содержит:
- быстрорастворимый разбавитель,
- гелеобразующий агент,
- кислый рН-регулирующий агент,
- связующее вещество, и
- возможно разрыхлитель, и
кроме того, гранулят не содержит бикарбонатных солей и/или карбонатных солей.
Согласно одному аспекту, описанный выше гранулят, модифицирующий суспензию, не содержит лактозы. Это дополнительное преимущество делает его пригодным для людей, страдающих от непереносимости лактозы, и их можно лечить с помощью воплощений данного изобретения.
Одним аспектом данного изобретения является то, что быстрорастворимый разбавитель приводят в близкий/тесный контакт с гелеобразующим агентом. Это не только обеспечивает очень короткое время гелеобразования по сравнению с гелеобразующим агентом в отдельности, но также очень быстрое образование стабильного геля. Выбор правильного разбавителя, который также может выполнять функцию подсластителя, представляет собой одно воплощение данного изобретения.
Согласно одному аспекту данного изобретения быстрая распадаемость и быстрое гелеобразование до стабильного и воспроизводимого уровня вязкости при суспендировании в воде гранулята, модифицирующего суспензию, обеспечиваются особым способом получения. Согласно данному аспекту, способ включает смешивание и гранулирование гелеобразующего агента вместе с разбавителем/подсластителем и последующую сушку с получением низкого содержания влаги и/или растворителя.
Получение гранул с энтеросолюбильной оболочкой описано в разделе «Подробное описание изобретения», но в общем случае они могут быть получены согласно указаниям в WO 9601624 А1 с учетом особых требований к размеру. Кроме того, нет необходимости в каком-либо «дополнительном покрытии» гранул с энтеросолюбильной оболочкой.
Настоящее изобретение обеспечивает безопасные и надежные лекарственные формы для введения гранул с энтеросолюбильной оболочкой, содержащих кислотолабильные ингибиторы протонного насоса, такие как омепразол, эзомепразол, пантопразол и лансопразол, диспергированные в водной жидкой среде, а также посредством желудочного зонда. Это особенно пригодно и полезно в лечении гериатрических или педиатрических больных.
Композиции по настоящему изобретению также дают возможность включения в состав широкого интервала уровней дозировок и добавочных агентов, таких как агенты, маскирующие/улучшающие вкус, и тонических агентов.
Краткое описание графических материалов
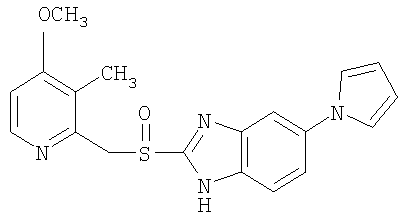
На Фиг.1 показан график зависимости вязкости от времени для воплощения по данному изобретению (5 образцов).
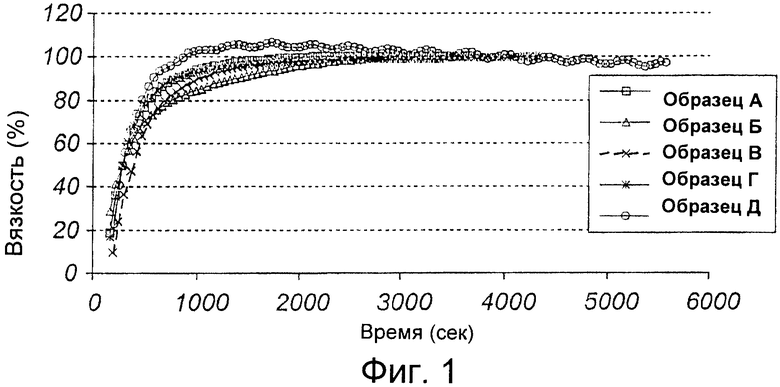
На Фиг.2 показан график зависимости вязкости от времени для воплощения из уровня техники (Lanzoтм, 4 образца).
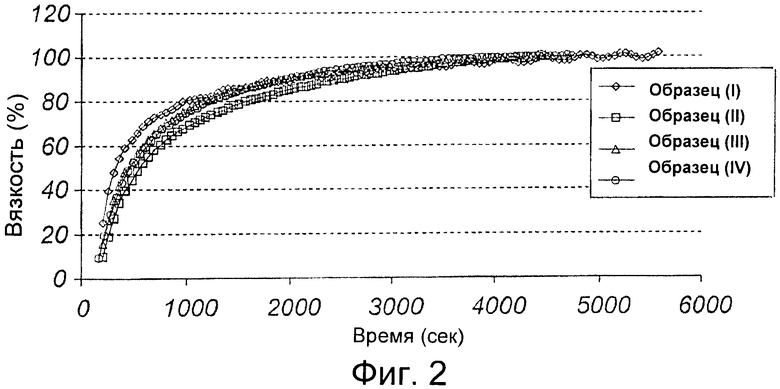
На Фиг.3 показаны средние профили концентрация-время эзомепразола в плазме после введения однократной дозы 40 мг в виде пакетированной формы, капсулы или таблетки. Через 8 часов уровни в плазме были ниже порога количественного обнаружения для каждой формы, и подсчет средних значений был невозможен.
Подробное описание изобретения
Одним аспектом настоящего изобретения является лекарственная форма, представляющая собой смесь первого компонента (I), представляющего собой массу гранул с энтеросолюбильной оболочкой, и еще одного компонента (II), представляющего собой гранулят, модифицирующий суспензию, причем эту смесь распределяют в контейнеры, например саше. Данная смесь быстро распадается и образует гель при суспендировании в водной среде, такой как водопроводная вода, образуя, таким образом, гомогенную стабильную и устойчивую суспензию с воспроизводимой и стабильной вязкостью, суспензию, которую пациент может легко проглотить или которую можно ввести пациенту через, например, назогастральный зонд. Жидкая, готовая к употреблению композиция представляет собой другой аспект настоящего изобретения, то есть она содержит три компонента: два компонента, упомянутых выше, (I) и (II), и, кроме того, жидкую среду (III).
Быстрое гелеобразование, то есть короткое время получения геля, по настоящему изобретению можно, среди прочего, рассматривать как влияние на время, необходимое до того, как по существу все гранулы с энтеросолюбильной оболочкой в полученной суспензии остаются суспендированными в жидкой среде и не опускаются на дно сосуда (склянки, мензурки), применяемого для ее получения. Время гелеобразования, требуемое для воплощений данного изобретения, обычно составляет менее 3 минут, и предпочтительно менее 2 минут при тестировании, описанном в Примере 5.
Лекарственная форма не содержит бикарбонатных солей и/или карбонатных солей. Одно воплощение данного изобретения, кроме того, не содержит лактозы. «Не содержит» означает, что такое соединение не добавляют в данную композицию. Следовые количества, присутствующие в другом сырье и сопровождающие другое сырье, применяемое в данной композиции, не принимаются во внимание данным термином.
Гранулы с энтеросолюбильной оболочкой
Гранулы с энтеросолюбильной оболочкой, содержащие активный ингредиент, изготавливают так, что наиболее удаленный от центра слой представляет собой слой энтеросолюбильной оболочки. Такие гранулы могут быть получены в соответствии со способами, известным в данной области, например описанным в WO 9601624 А1, с учетом особых требований к размеру гранул. Кроме того, нет необходимости в каком-либо «дополнительном покрытии» полученных гранул с энтеросолюбильной оболочкой.
Согласно одному аспекту изобретения средний диаметр гранул с энтеросолюбильной оболочкой составляет 0,2-1,8 мм в диаметре, предпочтительно 0,4-1,0 мм в диаметре и более предпочтительно 0,5-0,8 мм в диаметре.
В другом аспекте данного изобретения размер гранулы с энтеросолюбильной оболочкой находится в интервале 1,0-1,4 мм в диаметре.
Гранулы с энтеросолюбильной оболочкой состоят из следующих структурных компонентов:
- вещество ядра, содержащее активный ингредиент,
- возможный разделяющий или подоболочечный слой, и
- слой энтеросолюбильной оболочки,
но без дополнительного покрывающего слоя на слое энтеросолюбильной оболочки.
Вещество ядра
Вещество ядра получают способами, известными в данной области техники, такими как выдавливание - сферонизация, методы наслаивания, такие как наслаивание порошка или раствора/суспензии, распылительная сушка, комкование, методы замораживания или методы замораживания спрея. Вещество ядра содержит активный ингредиент и также может содержать затравки, связующие вещества, поверхностно-активные вещества, наполнители, разрыхлители, щелочные добавки или другие фармацевтически приемлемые ингредиенты, по отдельности или в смеси.
Активный ингредиент
Фармацевтические композиции по данному изобретению содержат в качестве активного ингредиента кислоточувствительный ингибитор протонного насоса или его щелочную соль, или индивидуальный энантиомер или щелочную соль этого энантиомера. Индивидуальный энантиомер, рацемические смеси (50% каждого энантиомера) и неравные смеси двух энантиомеров пригодны для фармацевтической композиции по настоящему изобретению.
Активный ингредиент возможно входит в состав небольших гранул/шариков с энтеросолюбильной оболочкой вместе с эксципиентами.
Соединения/активные ингредиенты, представляющие интерес для новых фармацевтических композиций по настоящему изобретению, представляют собой соединения общей формулы I, их щелочную соль, один из их индивидуальных энантиомеров или соль одного из этих энантиомеров
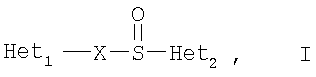
где Het1 представляет собой
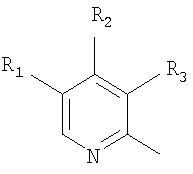 или
или 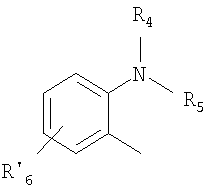
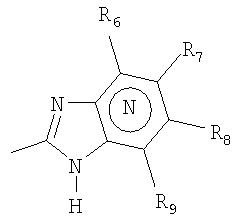
Het2 представляет собой
 или
или 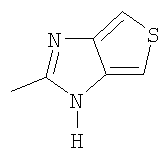
Х=
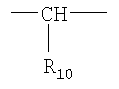 или
или 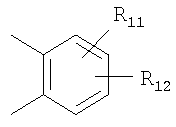 ,
,
где N в бензимидазольной части означает, что один из кольцевых атомов углерода, замещенных R6-R9, возможно может быть заменен атомом азота без каких-либо заместителей;
R1, R2 и R3 являются одинаковыми или разными и выбраны из водорода, алкила, алкокси, возможно замещенных фтором, алкилтио, алкоксиалкокси, диалкиламино, пиперидино, морфолино, галогеном, фенилом и фенилалкокси;
R4 и R5 являются одинаковыми или разными и выбраны из водорода, алкила и арилалкила;
R'6 представляет собой водород, галоген, трифторметил, алкил или алкокси;
R6-R9 являются одинаковыми или разными и выбраны из водорода, алкила, алкокси, галогена, галогеноалкокси, алкилкарбонила, алкоксикарбонила, оксазолинила, пирролила и трифторалкила, или соседние группы R6-R9 образуют кольцевые структуры, которые могут быть дополнительно замещены;
R10 представляет собой водород или вместе с R3 образует алкиленовую цепь, и
R11 и R12 являются одинаковыми или разными и выбраны из водорода, галогена и алкила.
В вышеприведенных определениях алкильные группы, алкоксигруппы и их части могут представлять собой разветвленные или прямые С1-С9-цепи или содержать циклические алкильные группы, например циклоалкилалкил.
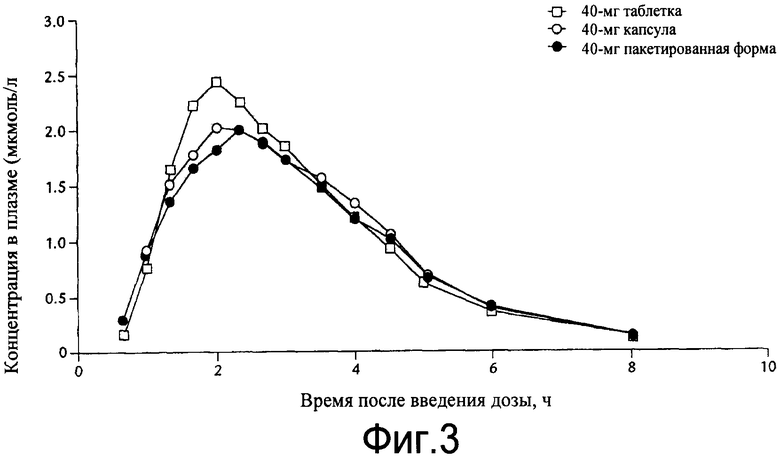
Примерами особенно интересных соединений формулы I являются
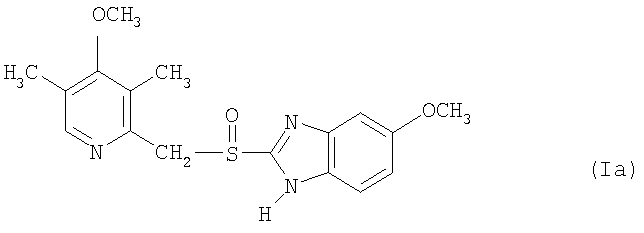
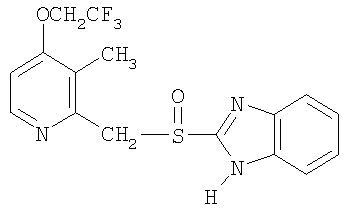
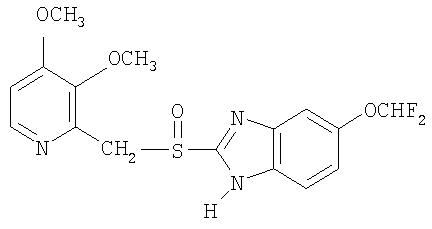
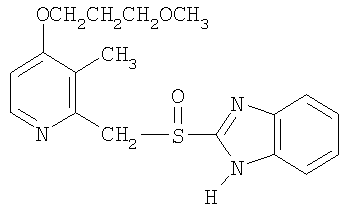
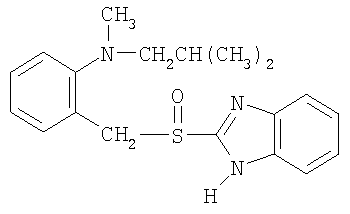
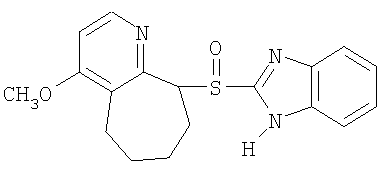
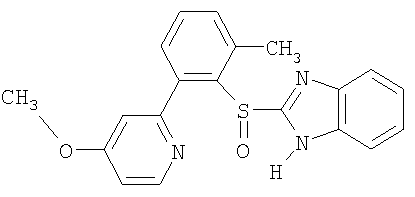
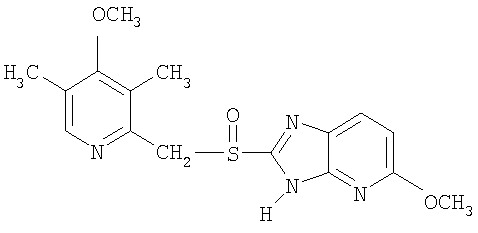
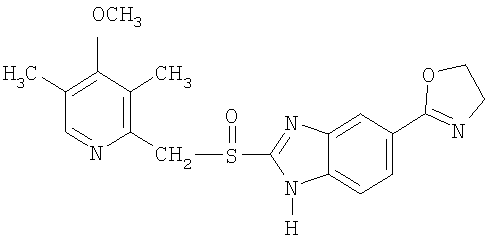
включая их таутомерные формы.
Предпочтительными соединениями для перорального фармацевтического препарата по настоящему изобретению являются омепразол, магниевая соль омепразола или магниевая соль (-)-энантиомера омепразола. Последняя имеет непатентованное название эзомепразол. Согласно одному воплощению, активный ингредиент представляет собой эзомепразола магния тригидрат. В другом воплощении данного изобретения активным лекарственным средством является тенатопразол или его фармацевтически приемлемая соль, или индивидуальный энантиомер любого их них.
Согласно другому аспекту изобретения соединение/активный ингредиент представляет собой гидратированную форму любого из вышеупомянутых соединений/активных ингредиентов.
В одном аспекте данного изобретения количество активного ингредиента в препарате находится в интервале 1 мг-100 мг, 2 мг-80 мг или 5 мг-50 мг.
Затравки
Затравки, которые следует наслаивать с активным веществом, могут представлять собой нерастворимые в воде затравки, содержащие различные оксиды, целлюлозы, органические полимеры и другие вещества по отдельности или в смесях, или растворимые в воде затравки, содержащие различные неорганические соли, сахара (исключая лактозу), нонпарель и другие вещества, по отдельности или в смесях. Кроме того, затравки могут содержать активное вещество в форме агломератов, компактов и т.п.
Связующие вещества
Связующие вещества представляют собой, например, целлюлозы, такие как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза и натрий-карбоксиметилцеллюлоза, поливинилпирролидон, полиэтиленгликоли, поливиниловые спирты, сахара (исключая лактозу), крахмалы и другие фармацевтически приемлемые вещества с когезионными свойствами.
Поверхностно-активные вещества
В лекарственных формах можно применять поверхностно-активные вещества. Подходящие поверхностно-активные вещества найдены в группах фармацевтически приемлемых неионных поверхностно-активных веществ, таких как, например, Полисорбат 80, или ионных поверхностно-активных веществ, таких как, например, лаурилсульфат натрия.
Наполнители
В лекарственной форме можно применять наполнители. Примеры наполнителей включают в себя, например, маннитол и двухзамещенный фосфат кальция.
Разрыхлители
В лекарственных формах можно применять разрыхлители. Примерами разрыхлителей, которые можно использовать, являются, например, сшитый поливинилпирролидон, клейстеризованный крахмал, микрокристаллическая целлюлоза и сшитая натрий-карбоксиметилцеллюлоза.
Щелочные добавки
Согласно одному воплощению изобретения, активное вещество также можно смешать с щелочным фармацевтически приемлемым веществом (или веществами). Такие вещества, после исключения бикарбонатных солей или карбонатных солей, могут быть выбраны из таких веществ, как натриевые, калиевые, кальциевые, магниевые и алюминиевые соли фосфорной кислоты, лимонной кислоты или других подходящих слабых неорганических или органических кислот; веществ, обычно используемых в антацидных препаратах, таких как гидроксиды алюминия, кальция и магния; оксида магния; органических рН-буферных веществ, таких как тригидроксиметиламинометан, основные амины или аминокислоты и их соли или другие похожие фармацевтически приемлемые рН-буферные вещества, но не ограничены ими.
Разделяющий или подоболочечный слой
Разделяющий(е) или подоболочечный(е) слой(и) может(гут) быть нанесен(ы) на вещество ядра посредством процессов нанесения оболочки или наслаивания в подходящем оборудовании, таком как котел для нанесения оболочки, гранулятор для нанесения оболочки или в аппарате с псевдоожиженным слоем с применением воды и/или органических растворителей для процесса нанесения оболочки. В качестве альтернативы разделяющий(е) слой(и) может(гут) быть нанесен(ы) на вещество ядра с применением метода порошкового покрытия. Вещества для разделяющих слоев представляют собой фармацевтически приемлемые соединения, такие как, например, сахар, полиэтиленгликоль, поливинилпирролидон, поливиниловый спирт, поливинилацетат, гидроксипропилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие, применяемые по отдельности или в смесях. В разделяющий(е) слой(и) также могут быть включены добавки, такие как пластификаторы, красители, пигменты, наполнители, антиадгезивные агенты и антистатики, такие как, например, стеарат магния, диоксид титана, белая сажа, тальк и другие добавки.
Разделяющий(е) слой(и) может(гут) служить диффузионным барьером и может функционировать как рН-буферная зона. рН-буферные свойства разделяющего(их) слоя(слоев) могут быть дополнительно усилены введением в слой(слои) веществ, после исключения бикарбонатных солей или карбонатных солей, выбранных из группы соединений, обычно используемых в антацидных препаратах, таких как, например, оксид, гидроксид магния, гидроксид или силикат алюминия или кальция; сложных соединений алюминия/магния, таких как, например, MgO·Al2O3·2SiO2·nH2O или других фармацевтически приемлемых рН-буферных соединений, таких как, например, натриевые, калиевые, кальциевые, магниевые и алюминиевые соли фосфорной, лимонной или других подходящих слабых неорганических или органических кислот; или подходящих органических оснований, включая основные аминокислоты или амины и их соли. Для увеличения толщины слоя(ев) и, следовательно, усиления диффузионного барьера могут быть добавлены тальк или другие соединения.
Слой энтеросолюбильной оболочки
Один или более чем один слой энтеросолюбильной оболочки наносят на вещество ядра, или на вещество ядра, покрытое разделяющим(и) слоем(слоями), применяя подходящую методику нанесения оболочки. Вещество слоя энтеросолюбильной оболочки может быть диспергировано или растворено либо в воде, либо в подходящих органических растворителях. В качестве полимеров для слоя энтеросолюбильной оболочки можно использовать один или более чем один из приведенных ниже полимеров, отдельно или в комбинации: например, растворы или дисперсии сополимеров метакриловой кислоты, ацетатфталата целлюлозы, фталата гидроксипропилметилцеллюлозы, ацетатсукцината гидроксипропилметилцеллюлозы, поливинилацетатфталата, ацетаттримеллитата целлюлозы, карбоксиметилцеллюлозы, шеллака или другого подходящего для слоя энтеросолюбильной оболочки полимера(ов).
Слои энтеросолюбильной оболочки содержат фармацевтически приемлемые пластификаторы для получения нужных механических свойств, таких как гибкость и твердость слоев энтеросолюбильной оболочки. Такие пластификаторы представляют собой, например, триацетин, эфиры лимонной кислоты, эфиры фталевой кислоты, дибутилсебацинат, цетиловый спирт, полиэтиленгликоль, полисорбаты или другие пластификаторы, но не ограничены ими.
Гранулят, модифицирующий суспензию
Гранулят, модифицирующий суспензию, содержит:
- быстрорастворимый разбавитель,
- гелеобразующий агент,
- кислый рН-регулирующий агент,
- связующее вещество и
- возможно разрыхлитель
и, кроме того, он не содержит бикарбонатных солей и/или карбонатных солей, особенно компонентов, которые могут привести к образованию газа.
Согласно одному воплощению гранулят, модифицирующий суспензию, изготавливают способом, согласно которому быстрорастворимый разбавитель и гелеобразующий агент смешивают и гранулируют вместе, а затем сушат.
Конечная влажность гранулята, модифицирующего суспензию, измеряемая как потери при сушке, составляет менее 3% (мас./мас.), предпочтительно менее 1% (мас./мас.). Конечное содержание этанола составляет менее 0,2% (мас./мас.), предпочтительно менее 0,12% (мас./мас.).
При суспендировании гранулята, модифицирующего суспензию, в водопроводной воде за короткое время получают стабильную и близкую к максимальной вязкость. Кроме того, полученная суспензия не содержит комков и является устойчивой в том смысле, что ее вязкие свойства являются приблизительно такими же, даже если пациент добавляет слишком мало или слишком много воды при приготовлении суспензии из гранулята. Таким образом, возможно добавить одну дозу активного ингредиента и гранулята, модифицирующего суспензию, к 50%-150% предписанного количества воды и тем не менее получить желаемые свойства композиции.
Гель, образующийся при добавлении гранулята, модифицирующего суспензию, к водной среде, такой как вода, имеет вязкость 3,0-6,0 log (мПа·с) = 103-106 мПа·с, предпочтительно 3,6-4,7 log (мПа·с) = 103,6-104,7 мПа·с.
Эту вязкость оценивают при 20°С, отсекая отрезок по координатной оси вязкости для линии графика зависимости log (вязкости) от log (частоты вращения (об/мин)). Эту линию получают линейным аппроксимированием с применением линейной регрессии наименьших квадратов и определяют отрезок аппроксимированной линии. Применяют подходящее для определения вязкости оборудование, такое как вискозиметр Physica DV-1 Р, измерительная конфигурация представляет собой стержень №2, диаметром 18,7 мм, длиной 6,9 мм, вращаемый с частотой вращения 3,0, 6,0, 30 и 100 об/мин, и измерения производят до получения стабильного значения (приблизительно 1 минуту).
Быструю распадаемость и быстрое гелеобразование до стабильного и воспроизводимого уровня вязкости при суспендировании в воде гранулята, модифицирующего суспензию, достигают, согласно одному аспекту данного изобретения, при применении специального способа получения для изготовления гранулята, модифицирующего суспензию.
Этот способ получения включает нижеприведенные стадии в нижеприведенном порядке, не исключая альтернативного варианта, когда можно изменить порядок стадий I и II:
I) смешивание гелеобразующего агента с рН-регулирующим агентом, быстрорастворимым разбавителем и возможно разрыхлителем;
II) растворение связующего вещества в этаноле;
III) увлажнение смеси, полученной на стадии I (альтернативно на стадии II, если изменен порядок), раствором, полученным на стадии II (альтернативно на стадии I, если изменен порядок);
IV) перемешивание влажной смеси, полученной на стадии III, для того, чтобы практически каждая частица гелеобразующего агента оказалась в близком/тесном контакте с вышеупомянутым быстрорастворимым разбавителем;
V) сушка перемешанной влажной смеси со стадии IV до тех пор, пока конечное влагосодержание гранулята, модифицирующего суспензию, измеренное как потери при сушке, не будет составлять менее 3% (мас./мас.), предпочтительно менее 1% (мас./мас.);
VI) растирание или размалывание сухих гранул, полученных на стадии V, до тех пор, пока более 95% (мас./мас.) гранул не будут проходить сквозь сито с отверстиями 1,0 мм.
Одним аспектом данного изобретения является приведение быстрорастворимого разбавителя в близкий/тесный контакт с гелеобразующим агентом, тем самым обеспечивается не только очень быстрое время гелеобразования по сравнению с гелеобразующим агентом самим по себе, но также очень быстрое образование стабильного геля. Одно воплощение данного изобретения представляет собой выбор подходящего быстрораспадающегося разбавителя, который также может выполнять функцию подсластителя.
В общем случае быстрое образование геля сухого гранулята, модифицирующего суспензию, при добавлении к воде, такой как водопроводная вода, выражается в том, что гелеобразование обычно достаточно, то есть достигает 75% от максимально получаемого уровня, уже в пределах приблизительно 10 минут. 90% или более от максимальной вязкости обычно достигают в пределах 15 минут. Для сравнения смотри таблицу в примере 2.
В частности, при суспендировании в воде одного гранулята, модифицирующего суспензию, по данному изобретению и легком перемешивании, он обеспечивает образование суспензии, достигающей более 75% от максимально получаемой вязкости в пределах 13 минут, предпочтительно более 75% в пределах 10 минут, при испытании с 1 г гранулята, модифицирующего суспензию, добавленного к 5 мл воды. Более 90% от максимально получаемой вязкости достигается в пределах 30 минут, предпочтительно более 90% в пределах 25 минут, при испытании с 1 г гранулята, модифицирующего суспензию, добавленного к 5 мл воды.
Согласно одному воплощению данного изобретения гранулят, модифицирующий суспензию (и PPI-содержащие гранулы с энтеросолюбильной оболочкой), не содержат лактозу.
Гелеобразующий агент
Гелеобразующий агент обеспечивает образование геля, пригодного для введения через желудочный зонд/назогастральный зонд, то есть выбран так, чтобы гель, полученный при диспергировании в водной среде, такой как вода, обладал подходящей вязкоупругостью, а также подходящей вязкостью. Такой способ введения является желательным в педиатрической и гериатрической терапии.
Время растворения также будет влиять на выбор гелеобразующих агентов.
Подходящие гелеобразующие агенты по данному изобретению представляют собой различные сорта ксантановых камедей.
Могут быть рассмотрены также и другие гелеобразующие агенты, но в случае, например, некоторых крахмальных продуктов, подходящий интервал концентраций очень ограничен, примером является Thick-Itтм регулярный, содержащий модифицированный кукурузный крахмал и мальтодекстрин. Этот продукт следует использовать лишь в узком интервале от приблизительно 6 до 8% от конечной суспензии, соответствующем содержанию гелеобразующего агента в грануляте, модифицирующем суспензию, от 34 до 48%, что является неприемлемо большой частью композиции.
Другим примером является кукурузный крахмал, который во многих случаях обеспечивает быстрое проглатывание, но имеет нежелательные вязкоупругие свойства.
Гелеобразующие агенты типа Na-карбоксиметилцеллюлозы (CMC) и каррагинана не могут использоваться в настоящем изобретении из-за отсутствия подходящих вязкоупругих свойств или из-за неудовлетворительных для введения полученной суспензии через желудочный зонд свойств.
Таким образом, гелеобразующие агенты в данном изобретении выбирают из ксантановых камедей.
Концентрация гелеобразующего агента составляет от 0,6 до 12% мас./мас. гранулята, модифицирующего суспензию. В предпочтительном воплощении концентрация гелеобразующего агента составляет от 1,8 до 4,8% мас./мас. Исходя из практических соображений, для пациента целесообразно иметь такой широкий интервал концентрации гелеобразующего агента при сохранении приемлемых свойств вязкоупругого геля.
В одном воплощении данного изобретения получение проводят с гелеообразующим агентом, имеющим средний размер частиц более 150 микрон.
Быстрорастворимый разбавитель
Разбавитель выполняет функцию разбавления, но он также может выполнять функцию подсластителя.
Разбавитель выбирают из группы, состоящей из моно- и дисахаридов и гидратов любого из них. Согласно одному аспекту изобретения, предпочтительными разбавителями являются глюкоза или сахароза, или гидраты любого из них. Согласно настоящему изобретению под быстрым растворением подразумевают, что время растворения разбавителя составляет менее 2 минут при растворении 2 г вещества в 10 мл воды при медленном продолжительном перемешивании при 14°С. Одним из разбавителей, особенно не удовлетворяющих этому требованию, является маннитол.
Как следствие способа получения, гранулят, модифицирующий суспензию, по изобретению содержит быстрорастворимый разбавитель, случайным образом распределенный в полученных отдельных гранулированных частицах и на них.
Кислый рН-регулирующий агент
Гранулят, модифицирующий суспензию, при суспендировании в воде образует суспензию с рН в интервале от 3,0 до 6,0, предпочтительно от 3,0 до 5,0, и более предпочтительно в интервале от 3,5 до 4,5.
Этого можно достичь добавлением подходящего кислого рН-регулирующего агента. Этот агент может состоять из одного кислого химического соединения или из смеси соединений, выбранных из кислотных и щелочных соединений, за исключением любых карбонатных солей. Любую смесь таких влияющих на рН соединений выбирают таким образом, чтобы при растворении/суспендировании данной смеси в воде она обеспечила рН в рамках нужного (кислого) интервала согласно вышесказанному.
Неограничивающими примерами подходящих кислых соединений являются: лимонная кислота, винная кислота и яблочная кислота. Не ограничивающим примером смеси соединений, выбранных из кислых и щелочных соединений, является однозамещенный фосфат натрий и двухзамещенный фосфат натрий (в соотношении, подходящем для получения рН в рамках нужного интервала).
Разрыхлитель
Возможный разрыхлитель, применяемый в сухом грануляте, модифицирующем суспензию, может представлять собой один разрыхлитель или смесь разрыхлителей.
Неограничивающий пример подходящих разрыхлителей включает:
сшитый поливинилпирролидон, сшитую натрий-карбоксиметилцеллюлозу (Ас-Di-Sol®) и клейстеризованный крахмал (Sta-Rx® 1500).
Связующее вещество
Подходящим связующим веществом, применяемым согласно настоящему изобретению, является полимер, растворимый в воде и в этаноле. Подходящими связующими веществами являются выбранные марки гидроксипропилцеллюлозы. При выборе в качестве связующего вещества гидроксипропилцеллюлозы (ниже также упоминаемой как НРС), она имеет содержание гидроксипропила в интервале 50-90%, или более предпочтительно в интервале 60-81%, и вязкость менее 450 мПа·с (сПз), определенную при 5%-ной концентрации. Такими полимерами являются, например, Klucel®JF и Klucel® LF oт Aqualon.
Гидроксипропилцеллюлоза, предполагаемая для применения в данном аспекте изобретения в качестве связующего вещества, не включает низкозамещенной гидроксипропилцеллюлозы, также упоминаемой как L-HPC.
Соотношение между связующим веществом и гелеобразующим агентом в грануляте, модифицирующем суспензию, по данному изобретению предпочтительно составляет от 1:2 до 1:3 мас./мас.
Активность лекарственной формы
Различных активностей продукта достигают, фасуя определенные количества гранул ингибитора протонного насоса с энтеросолюбильной оболочкой и гранулята, модифицирующего суспензию, по данному изобретению в саше, содержащие стандартную дозу. Согласно одному воплощению данного изобретения гранулы с энтеросолюбильной оболочкой, содержащие эзомепразола магния тригидрат, фасуют вместе с гранулятом, модифицирующим суспензию, в саше, содержащие стандартную дозу.
Соотношение масс между двумя компонентами смеси, то есть между гранулами с энтеросолюбильной оболочкой, содержащими ингибитор протонного насоса, с одной стороны, и (сухим) гранулятом, модифицирующим суспензию, с другой стороны, может варьироваться от 1:1000 до 100:1000, предпочтительно от 4:1000 до 80:1000 и наиболее предпочтительно от 8:1000 до 60:1000.
Количество гранул с энтеросолюбильной оболочкой в одном саше
Содержание лекарственного средства в PPI-содержащих гранулах с энтеросолюбильной оболочкой составляет от 5% мас./мас. гранул с энтеросолюбильной оболочкой до 40% мас./мас. гранул с энтеросолюбильной оболочкой. Это означает, что наибольшее теоретическое количество гранул на одну дозу может быть рассчитано с учетом ситуации, когда рассматривается наименьшая концентрация лекарственного средства в гранулах для наибольшей дозы лекарственного средства (100 мг согласно изобретению), с получением общего количества (100/0,05=) 2000 мг гранул.
Наименьшее возможное количество гранул может быть, в соответствии с аналогичным рассуждением, рассчитано для противоположной ситуации, исходя из наибольшей концентрации и наименьшей дозы (1 мг согласно изобретению) с получением минимального количества гранул (1/0,4=) 2,5 мг гранул.
В предпочтительном воплощении изобретения содержание лекарственного средства в гранулах с энтеросолюбильной оболочкой составляет 8-30% мас./мас.
Количество гранул с энтеросолюбильной оболочкой в одном саше согласно данному изобретению находится в интервале 2,5-2000 мг, и в предпочтительном воплощении данного изобретения количество гранул с энтеросолюбильной оболочкой в одном саше находится в интервале 3-1250 мг.
В альтернативном воплощении данного изобретения содержание лекарственного средства в гранулах с энтеросолюбильной оболочкой адаптировано к дозе лекарственного средства, предполагаемой в одном саше, согласно нижеследующей таблице:
Таким образом, в одном воплощении данного изобретения доза в одном саше составляет 1-40 мг, и содержание лекарственного средства в гранулах с энтеросолюбильной оболочкой составляет 8-12% (мас./мас.).
В другом воплощении данного изобретения доза в одном саше составляет более 40 мг-70 мг, и содержание лекарственного средства в гранулах с энтеросолюбильной оболочкой составляет 15-25% (мас./мас.).
В следующем воплощении данного изобретения доза в одном саше составляет более 70 мг-100 мг, и содержание лекарственного средства в гранулах с энтеросолюбильной оболочкой составляет 25-40% (мас./мас.).
Готовая к применению жидкая композиция
Перед применением содержимое саше высыпают в заданный объем водной жидкости. После перемешивания образуется вязкая суспензия. Эта жидкая суспензия представляет собой еще один аспект изобретения и содержит три главных компонента, то есть гранулы с энтеросолюбильной оболочкой, содержащие ингибитор протонного насоса, (сухой) гранулят, модифицирующий суспензию, и водную жидкость.
Предполагается, что количество водной жидкости будет в 5 раз больше количества гранулята, модифицирующего суспензию, но пациенту будет предоставлена возможность менять это количество жидкости от 50% до 150% от заданного количества. Это означает, что количество водной жидкости в готовой к употреблению жидкой смеси будет в 2,5-7,5 больше количества гранулята, модифицирующего суспензию.
В одном аспекте данного изобретения водная жидкость представляет собой воду.
Концентрация гелеобразующего агента должна составлять от 0,1 до 2% мас./мас.(двадцатикратный интервал концентрации) суспензии, предпочтительно от 0,3 до 0,8% мас./мас. Исходя из практических соображений, для пациента полезно иметь такой широкий интервал концентрации гелеобразующего агента при сохранении важных свойств вязкоупругого геля.
Примеры
Пример 1а
Получение гранулята. модифицирующего суспензию, по изобретению
Гидроксипропилцеллюлозу растворяют в этаноле. Раствор добавляют к сухой смеси оставшихся эксципиентов с получением влажной массы, которую подвергают гранулированию в процессе добавления раствора. Влажную массу сушат и размалывают (максимум 5% гранул более 1 мм).
3 г данного гранулята, модифицирующего суспензию, растворяли в 15 мл воды, и жидкую композицию перемешивали в течение 60 секунд. рН измеряли с помощью стеклянного электрода, используя калиброванный рН-метр и определили его как 4,0.
(Сравнительный) Пример 1б
Гранулят, модифицирующий суспензию, из уровня техники
В качестве сравнения использовали коммерческий продукт «Lanzoтм 30 мг, гранулят» от Wyeth Lederle (партия ЗЕТ032, годен до июля 2006, и 3ЕТ010, годен до марта 2006).
Композиция гранулята для суспензии (за исключением гранул с энтеросолюбильной оболочкой), применяемая для этого продукта, получена согласно SWEDIS:
Пример 2. Измерение вязкости
Условия эксперимента
Воплощение согласно данному изобретению: 3 г гранулята, модифицирующего суспензию, полученного согласно Примеру 1а, растворяли в 15 мл воды, и жидкую композицию перемешивали в течение 60 секунд.
Образец из уровня техники (Lanzoтм 30 мг, гранулят): гранулы, содержащие лансопразол, удалили из общей массы твердых частиц (5,7 г) продукта, описанного в Примере 1б, и к оставшемуся порошку/грануляту (5,4 г) добавляли 30 мл воды, после чего жидкую композицию перемешивали в течение 60 секунд.
Для обоих образцов измерения вязкости начали через еще 1 минуту.
Инструмент: Reologica Stresstech
Принцип измерения: колебания пластинки/пластинка Р 30 со щелью 2 мм.
Параметры измерения: частота 0,1 Гц; нагрузка 0,07146 Па.
Обсуждение
Вариант с лансопразолом (Пример 1б), где быстрорастворимый разбавитель (сахароза) также добавлен к композиции гранулята для суспензии, но эта композиция не образует стабильного геля в рамках желаемого более короткого временного периода, смотри Фиг.2 следует сравнить с результатом, полученным по настоящему изобретению (Пример 1а), смотри Фиг.1 и результаты приведены в таблице выше.
Результатом использования медленно растворимого разбавителя будет композиция с более медленным гелеобразованием и непрерывным увеличением вязкости в рамках приемлемого и адекватного временного периода. Таким образом, настоящее изобретение решило несколько проблем в получении не содержащей лактозу и не содержащей бикарбонатов/карбонатов композиции с коротким временем гелеобразования, с вязкостью/вязкоупругостью, приемлемыми для проглатывания или введения через зонд, такими как постоянная во времени вязкость и отсутствие комков в конечной суспензии для введения.
Пример 3. Получение гранул с энтеросолюбильной оболочкой, содержащих эзомепразол-Mg тригидрат
Вещество ядра
Подоболочечный слой
Слой энтеросолюбильной оболочки
Суспензионное наслаивание осуществляли в аппарате с псевдоожиженным слоем с применением методики нижнего распыления. Эзомепразол распыляли на сферические крупинки сахара из водной суспензии, содержащей растворенные связующее вещество и поверхностно-активное вещество. Размер сферических крупинок сахара попадал в интервал от 0,25 до 0,35 мм.
Полученное вещество ядра покрывали подоболочечным слоем в аппарате с псевдоожиженным слоем, используя раствор гидроксипропилцеллюлозы, содержащий тальк и стеарат магния. Слой энтеросолюбильной оболочки распыляли в виде водной дисперсии на гранулы, покрытые разделяющим слоем в аппарате с псевдоожиженным слоем.
Пример 4. Примеры соотношений компонентов для получения конечной жидкой композиции с разной величиной доз
Пример 5. Иллюстрация короткого времени гелеобразования по настоящему изобретению
Содержимое саше, содержащего конечную композицию с величиной дозы 40 мг согласно Примеру 4, высыпали в мензурку, содержащую номинально заданное количество воды, 15 мл.
Затем образец перемешивали в течение 15 секунд и затем оставляли в покое до 55 секунды от начала. После этого его снова перемешивали в течение 5 секунд для равномерного распределения гранул действующего лекарственного средства в суспензии.
Затем суспензию исследовали в течение 30 секунд для определения того, распределены ли по существу все гранулы с энтеросолюбильной оболочкой в суспензии или собрались на дне мензурки.
Если гранулы не распределены в жидкой среде, а собраны на дне мензурки, процесс повторяли, что означает ожидание в течение еще 25 секунд и перемешивание в течение 5 секунд, то есть до 2 минут, после чего следовало исследование в течение 30 секунд, пока по существу все гранулы не останутся распределенными в жидкой среде. Записывали время, необходимое для того, чтобы гранулы оставались в жидкой среде.
Образцы в таблице ниже оценивали описанным способом со следующими результатами:
Пример 6: Исследования биоэквивалентности
Одноцентровое, рандомизированное, открытое, тройное перекрестное исследование проводили на здоровых взрослых добровольцах для сравнения фармакокинетических свойств пакетированной формы эзомепразола (40 мг) и имеющихся в продаже таблетированной и капсулированной форм эзомепразола такой же дозировки (товарный знак Nexium, AstraZeneca).
В каждый из трех дней исследования, которые разделялись периодами вымывания препарата по меньшей мере в 6 дней, добровольцы получали 40 мг эзомепразола в виде пакетированной композиции, таблетки или капсулы. Добровольцам не разрешалось принимать пищу с 10 часов вечера и пить менее чем за час до приема лекарства. Эзомепразол в пакетированной форме суспендировали в 15 мл воды в пластиковом стакане и вводили перорально. Добавляли еще 15 мл воды, и содержимое стакана проглатывали с тем, чтобы ввести оставшиеся в стакане частички эзомепразола. Для стандартизации приема воды все добровольцы затем выпивали еще 170 мл воды. Таблетки и капсулы глотали целиком и запивали 200 мл воды.
Главная задача исследования заключалась в том, чтобы удостовериться в биоэквивалентности пакетированной формы эзомепразола имеющимся в продаже таблетированной и капсулированной формам. Эту биоэквивалентность оценивали на основании полной площади под кривой (AUC) и максимальной концентрации Cmax. Другими фармакокинетическими параметрами, интересующими исследователей, были площадь под кривой до последней поддающейся количественному измерению концентрации AUCt, Tmax и t1/2. Для определения концентрации эзомепразола в плазме образцы крови 5 мл собирали через катетер из вены предплечья перед введением дозы и через 0,33, 0,66, 1, 1,33, 1,66, 2, 2,33, 2,66, 3, 3,5, 4, 4,5, 5, 6, 7, 8, 9 и 10 часов после введения.
Образцы крови анализировали с помощью жидкостного нормально-фазового хроматографа с УФ-детекцией при 302 нм. Использовали колонку Superspher SI-Genesis Silica (размер частиц 4 мкм, 150 мм × 4,6 мм, Jones Chromatography Ltd), подвижная фаза состояла из смеси 1,4 мл 25% раствора аммиака, 2 мл метанола и 98 мл 2-пропанола, разбавленной до 1000 мл дихлорметаном (рН 6,5-7,0). Количественный анализ осуществляли против внутреннего стандарта. Предел определения эзомепразола составлял 25 нмоль/л.
Результаты. Профили концентрация-время в плазме эзомепразола были похожими для пакетированной, таблетированной и капсулированной форм (см. Фиг.3).
Рассчитанные средние геометрические и 95% Cl (доверительный интервал) для AUC, Cmax и AUCt эзомепразола 40 мг, введенного в виде 3 форм, и рассчитанные соотношения средних геометрических и 90% Cl представлены в таблице. В общем, 90% Cl для соотношений геометрических средних AUC и Cmax находились в пределах интервала 0,80-1,25. Для всех трех форм среднее t1/2 составляло приблизительно 1,1 ч и среднее Tmax составляло 2 ч.
Заключение
Из представленных данных видно, что доза 40 мг эзомепразола в пакетированной форме является биоэквивалентной дозе 40 мг эзомепразола имеющихся в продаже капсулированной и таблетированной форм.
Изобретение относится к области фармацевтики, более конкретно к пероральной лекарственной форме, представляющей собой быстро образующую гель твердую гранулированную смесь, пригодную для получения суспензии. Гранулированная смесь содержит в качестве активного ингредиента кислоточувствительный ингибитор протонного насоса, распределенный в массе гранул с энтеросолюбильной оболочкой, и гранулят, модифицирующий суспензию, содержащий быстрорастворимый разбавитель, гелеобразующий агент, кислотный рН-регулирующий агент, и связующее вещество, причем гранулят не содержит бикарбонатных и карбонатных солей и соотношение между связующим веществом и гелеобразующим агентом в грануляте, модифицирующем суспензию, составляет от 1:2 до 1:3 мас./мас. Изобретение позволяет быстро получать гомогенную, вязкую, стабильную суспензию, содержащую ингибитор протонного насоса в форме, защищающей его от контакта с кислотными факторами окружающей среды. 6 н. и 25 з.п. ф-лы, 5 табл., 3 ил.
1. Пероральная фармацевтическая лекарственная форма, представляющая собой быстро образующую гель твердую гранулированную смесь, пригодную для получения суспензии, содержащая I) в качестве активного ингредиента кислоточувствительный ингибитор протонного насоса, распределенный в массе гранул с энтеросолюбильной оболочкой, и II) гранулят, отличающаяся тем, что этот гранулят представляет собой гранулят, модифицирующий суспензию, содержащий быстрорастворимый разбавитель, выбранный из моно- и дисахаридов и их гидратов, случайным образом распределенных в отдельных гранулированных частицах и на них, гелеобразующий агент, выбранный из ксантановых камедей, кислотный рН-регулирующий агент, связующее вещество и, возможно, разрыхляющий агент, и этот гранулят не содержит бикарбонатных солей и карбонатных солей, и где соотношение между связующим веществом и гелеобразующим агентом в грануляте, модифицирующем суспензию, составляет от 1:2 до 1:3 мас./мас.
2. Лекарственная форма по п.1, которая не содержит лактозы.
3. Лекарственная форма по п.1, где быстрорастворимый разбавитель и гелеобразующий агент смешивают и гранулируют вместе таким образом, что быстрорастворимый разбавитель случайным образом распределен в полученных гранулированных частицах и на них.
4. Лекарственная форма по п.1, где концентрация гелеобразующего агента составляет от 0,6 до 12% мас./мас. гранулята, модифицирующего суспензию.
5. Лекарственная форма по п.1, где концентрация гелеобразующего агента составляет от 1,8 до 4,8% мас./мас. гранулята, модифицирующего суспензию.
6. Лекарственная форма по п.1, где гранулят, модифицирующий суспензию, при суспендировании в воде образует суспензию, имеющую рН в интервале от 3,0 до 6,0.
7. Лекарственная форма по п.1, где гранулят, модифицирующий суспензию, при суспендировании в воде образует суспензию, имеющую рН в интервале от 3,0 до 5,0.
8. Лекарственная форма по п.1, где быстрорастворимый разбавитель в грануляте, модифицирующем суспензию, выбран из глюкозы, сахарозы и гидратов любого из них.
9. Лекарственная форма по п.1, где связующее вещество представляет собой полимерное связующее вещество.
10. Лекарственная форма по п.1, где связующее вещество представляет собой полимерное связующее вещество, растворимое в воде и в этаноле.
11. Лекарственная форма по п.1, где кислоточувствительный ингибитор протонного насоса представляет собой омепразол или магниевую соль омепразола.
12. Лекарственная форма по п.1, где кислоточувствительный ингибитор протонного насоса представляет собой эзомепразол, его щелочную соль или гидратированную форму любого из них.
13. Лекарственная форма по п.1, где кислоточувствительный ингибитор протонного насоса представляет собой тенатопразол, его фармацевтически приемлемую соль или индивидуальный энантиомер любого из них.
14. Лекарственная форма по п.1, где гранулы с энтеросолюбильной оболочкой состоят из следующих структурных компонентов: вещество ядра, содержащее активный ингредиент, подоболочечный слой, слой энтеросолюбильной оболочки; и отсутствуют дополнительный покрывающий слой на энтеросолюбильной оболочке.
15. Лекарственная форма по любому из пп.1-14, где гранулы с энтеросолюбильной оболочкой имеют средний диаметр 0,2-1,8 мм в диаметре.
16. Лекарственная форма по любому из пп.1-14, где гранулы с энтеросолюбильной оболочкой имеют средний диаметр 0,4-1,0 мм в диаметре.
17. Саше, содержащее лекарственную форму по любому из пп.1-16.
18. Саше по п.17, где количество активного ингредиента составляет 1 - 100 мг.
19. Саше по п.17, где количество активного ингредиента составляет 1 - 40 мг.
20. Готовая к употреблению жидкая композиция, содержащая водную жидкость и лекарственную форму по любому из пп.1-19.
21. Жидкая композиция по п.20, где количество водной жидкости в 2,5-7,5 раз больше количества гранулята, модифицирующего суспензию.
22. Жидкая композиция по п.20, где гранулят, модифицирующий суспензию, при суспендировании и перемешивании в водной жидкости образует суспензию, достигающую более 75% от максимально получаемой вязкости в течение 13 мин.
23. Жидкая композиция по п.20, где гранулят, модифицирующий суспензию, при суспендировании и перемешивании в водной жидкости образует суспензию, достигающую более 75% от максимально получаемой вязкости в течение 10 мин.
24. Жидкая композиция по п.20, где гранулят, модифицирующий суспензию, при суспендировании и перемешивании в водной жидкости образует суспензию, достигающую более 90% от максимально получаемой вязкости в течение 30 мин.
25. Жидкая композиция по п.20, где гранулят, модифицирующий суспензию, при суспендировании и перемешивании в водной жидкости образует суспензию, достигающую более 90% от максимально получаемой вязкости в течение 25 мин.
26. Жидкая композиция по любому из пп.20-25, где водная жидкость представляет собой воду.
27. Способ получения гранулята, модифицирующего суспензию, применяемого в лекарственной форме по пп.1-16, включающий нижеприведенные стадии в следующем порядке, но не исключающий альтернативный вариант, когда может быть изменен порядок стадий I и II:
I) смешивание гелеобразующего агента с рН-регулирующим агентом, быстрорастворимым разбавителем и, возможно, разрыхлителем;
II) растворение связующего вещества в этаноле;
III) увлажнение смеси, полученной на стадии I (альтернативно на стадии II, если изменен порядок), раствором, полученным на стадии II (альтернативно на стадии I, если изменен порядок);
IV) перемешивание влажной смеси, полученной на стадии III, для того, чтобы практически все частицы гелеобразующего агента оказались в близком/тесном контакте с вышеупомянутым быстрорастворимым разбавителем;
V) сушка перемешанной влажной смеси со стадии IV до тех пор, пока конечное влагосодержание гранулята, модифицирующего суспензию, измеренное как потери при сушке, не будет составлять менее 3% (мас./мас.), предпочтительно менее 1% (мас./мас.);
VI) растирание или размалывание сухих гранул, полученных на стадии V до тех пор, пока более 95% (мас./мас.) гранул не будут проходить сквозь сито с отверстиями 1,0 мм.
28. Способ по п.27, где стадии I) и II) осуществляют в обратном порядке.
29. Способ лечения заболеваний человека, связанных с желудочной кислотой, включающий введение пациенту, нуждающемуся в лечении, пероральной фармацевтической лекарственной формы, как она определена в любом из пп.1-26.
30. Способ по п.29, где пациенты, нуждающиеся в лечении, представляют собой детей или пожилых людей.
31. Применение фармацевтической лекарственной формы по любому из пп.1-26 в лечении желудочно-кишечных заболеваний.
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| Найдено в Интернет <URL:http://www.lakemedelsverket.se/Sok-efter-lakemedel-och-mediciner-i-Lakemedelsfakta/Produktinformation-lakemedel/?NPLId=19991007000057> | |||
| US 6261602 В1, 17.07.2001 | |||
| ФАРМАЦЕВТИЧЕСКИЕ ЛЕКАРСТВЕННЫЕ ФОРМЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИЕМА, СОДЕРЖАЩИЕ ИНГИБИТОР ПРОТОННОГО НАСОСА И СРЕДСТВО НЕСТЕРОИДНОЙ ПРОТИВОВОСПАЛИТЕЛЬНОЙ ТЕРАПИИ | 1996 |
|
RU2158138C2 |

