Уровень техники
Множество расстройств у людей и других млекопитающих включают в себя аномальную резорбцию костей или ассоциируются с ней. Такие расстройства включают, но, не ограничиваясь этим, остеопороз, остеопороз, вызываемый глюкокортикоидами, болезнь Педжета, аномально увеличенный метаболизм костной ткани, периодонтальное заболевание, потерю зубов, переломы костей, ревматоидный артрит, остеоартрит, перипротезный остеолиз, несовершенный остеогенез, злокачественную гиперкальцемию или множественную миелому. Среди них, одно из наиболее распространенных расстройств представляет собой остеопороз, который встречается в своем наиболее частом проявлении у женщин в период после менопаузы. Остеопороз представляет собой системное заболевание скелета, отличающееся низкой массой костей и микроархитектурным ухудшением костной ткани, как следствие, вызывает увеличение ломкости костей и предрасположенность к переломам. Остеопоротические переломы являются главной причиной заболеваемости и смертности у населения старшего возраста. До 50% женщин и треть мужчин получают остеопоротический перелом. Большая доля населения старшего возраста уже имеет низкую плотность костей и высокий риск переломов. Имеется значительная необходимость, как в предотвращении, так и в лечении остеопороза и других состояний, ассоциируемых с резорбцией костей. Поскольку остеопороз, как и другие расстройства, связанные с потерей костей, представляют собой, как правило, хронические состояния, считается, что и соответствующая терапия будет, как правило, требовать хронического лечения.
Катепсины принадлежат к папаиновому суперсемейству цистеинпротеаз. Эти протеазы функционируют при нормальной физиологической, а также паталогической деградации соединительной ткани. Катепсины играют главную роль во внутриклеточной деградации и метаболизме и обновлении белков. К настоящему времени, идентифицирован и секвенирован ряд катепсинов из ряда источников. Эти катепсины встречаются в природе в разнообразных тканях. Например, клонированы катепсины B, C, F, H, L, K, O, S, V, W и Z. Катепсин L участвует в нормальном лизосомальном протеолизе, а также в нескольких болезненных состояниях, включая, но, не ограничиваясь этим, метастазирование меланом. Катепсин S участвует в болезни Альцгеймера, атеросклерозе, хроническом обструктивном легочном заболевании и в определенным аутоиммунных расстройствах, включая, но, не ограничиваясь этим, ювенильный диабет, множественный склероз, вульгарную пузырчатку, болезнь Грейвса, миастения гравис, системную красную волчанку, ревматоидный артрит и тиреоидит Хашимото; аллергические расстройства, включая, но, не ограничиваясь этим, астму; и аллогенные иммунные реакции, включая, но, не ограничиваясь этим, отторжение пересаженных органов или тканевых трансплантатов. Повышенные уровни катепсина B и перераспределение фермента обнаруживаются в опухолях, это говорит о его роли в инвазии и метастазировании опухолей. В дополнение к этому, аберрантная активность катепсина B участвует в таких болезненных состояниях как ревматоидный артрит, остеоартрит, пневмоцистная пневмония, острый панкреатит, заболевание дыхательных путей и расстройства костей и суставов.
Катепсины млекопитающих связаны с папаиноподобными цистеинпротеазами, экспрессируемыми болезнетворными паразитами, включая паразиты семейств протозоа, платигельминтов, нематод и членистоногих. Эти цистеинпротеазы играют главную роль в жизненном цикле этих организмов.
Коллаген тип I человека, главный коллаген в костях является хорошим субстратом для катепсина K. Смотри Kafienah, W., et al., 1998, Biochem J 331:727-732, которая тем самым включается в качестве ссылки во всей своей полноте. Эксперименты in vitro с использованием антисмысловых олигонуклеотидов к катепсину K, показали уменьшение резорбции костей in vitro, которая вероятно вызывается уменьшением трансляции mRNA катепсина K. Смотри Inui, T., et al., 1997, J Biol Chem 272:8109-8112, которая тем самым включается в качестве ссылки во всей своей полноте. Разрешена кристаллическая структура катепсина K. Смотри McGrath, M. E., et al., 1997, Nat Struct Biol 4:105-109; Zhao, B., et al., 1997, Nat Struct Biol 4: 109-11, которые тем самым включаются в качестве ссылок во всей их полноте. Также, разработаны селективные ингибиторы катепсина K на основе пептида. Смотри Bromme, D., et al., 1996, Biochem J 315:85-89; Thompson, S. K., et al., 1997, Proc Natl Acad Sci USA 94:14249-14254, которые тем самым включаются в качестве ссылок во всей их полноте. Соответственно, ингибиторы катепсина K могут уменьшить резорбцию костей. Такие ингибиторы были бы пригодными для использования при лечении расстройств, включающих резорбцию костей, таких как остеопороз.
То, что необходимо в данной области - это терапевтические агенты для лечения заболеваний, ассоциируемых с активностью катепсина K, включая остеопороз, остеопороз, вызываемый глюкокортикоидами, болезнь Педжета, аномально увеличенный метаболизм костной ткани, потерю зубов, переломы костей, ревматоидный артрит, остеоартрит, перипротезный остеолиз, несовершенный остеогенез, атеросклероз, тучность, глаукому, хроническое обструктивное легочное заболевание и рак, включая метастатическое заболевание костей, злокачественную гиперкальцемию и множественную миелому.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые способны лечить и/или предотвращать катепсин-зависимые состояния или болезненные состояния у млекопитающего, нуждающегося в этом. Один из вариантов осуществления настоящего изобретения иллюстрируется соединением формулы I и его фармацевтически приемлемыми солями, стереоизомерами и их N-оксидными производными:
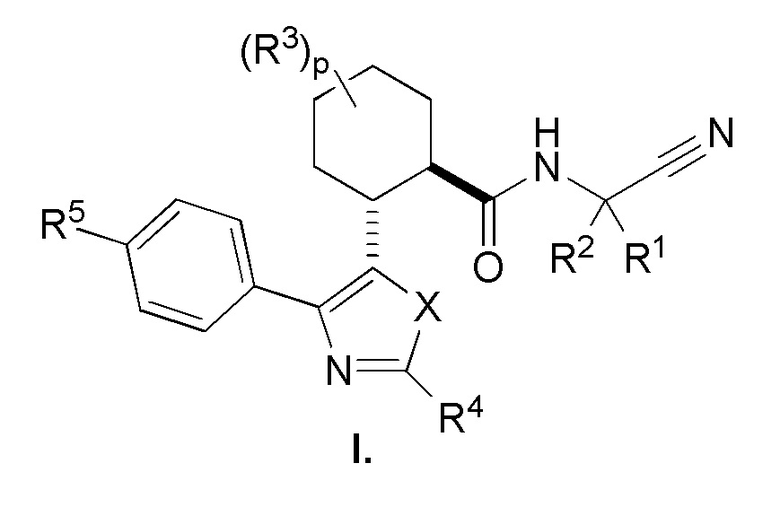
Соединения формулы I являются ингибиторами катепсина K. Как результат, соединения формулы I могут быть применимы в способах лечения, ингибирования или облегчения одного или нескольких болезненных состояний, которые могли бы получить улучшение от ингибирования катепсина K, включая остеопороз. Соединения по настоящему изобретению могут дополнительно использоваться в комбинации с другими терапевтическими эффективными агентами, включая, но, не ограничиваясь этим, другие лекарственные средства, пригодные для использования при лечении остеопороза, остеопороза, вызываемого глюкокортикоидами, болезни Педжета, аномально увеличенного метаболизма костной ткани, периодонтального заболевания, потери зубов, перелома костей, ревматоидного артрита, остеоартрита, перипротезного остеолиза, несовершенного остеогенеза, атеросклероза, тучности, глаукомы, хронического обструктивного легочного заболевания, метастатического заболевания костей, злокачественной гиперкальцемии или множественной миеломы. Кроме того, настоящее изобретение относится к способам получения соединений формулы I и к фармацевтическим композициям, которые содержат соединения формулы I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям следующей химической формулы:
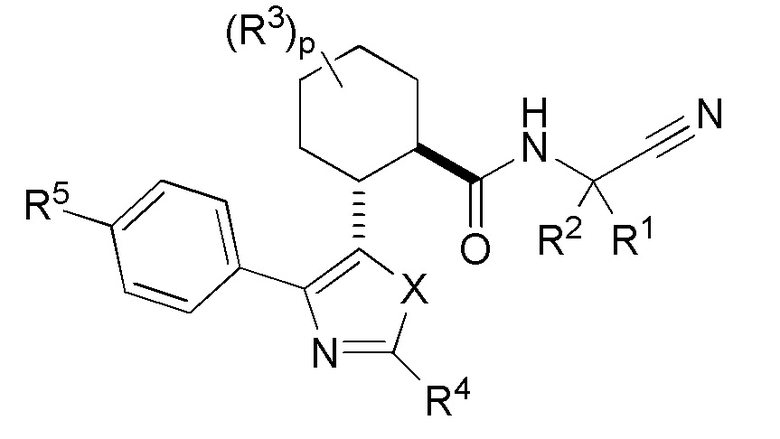
где X представляет собой атом кислорода, серы или азота;
R1 представляет собой атом водорода, C1-6 алкил, C2-6 алкенил, C3-8 циклоалкил или гетероциклил, где указанные алкильные и алкенильные группы являются необязательно замещенными C3-6 циклоалкилом, одним-шестью атомами галогена, гидрокси или R8; и где указанные циклоалкильные и гетероциклильные группы являются необязательно замещенными одним или двумя заместителями, выбранными из группы, состоящей из C1-6 алкила, атома галогена, OR6 и кето;
R2 представляет собой атом водорода, C1-6 алкил или C2-6 алкенил, где указанные алкильные и алкенильные группы являются необязательно замещенными C3-6 циклоалкилом, одним-шестью атомами галогена или R8;
или R1 и R2 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-8 циклоалкильного или гетероциклильного кольца, где указанные циклоалкильные и гетероциклильные кольца являются необязательно замещенными одним или двумя заместителями, независимо выбранными из группы, состоящей из R6, C1-6 галогеналкила и галогена;
Каждый R3 независимо выбирается из группы, состоящей из атома водорода, галогена и C1-2 алкила, где указанная алкильная группа является необязательно замещенной одним-тремя атомами галогена; или
две группы R3 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-4 циклоалкильного кольца, где указанное кольцо является необязательно замещенным одним-тремя атомами галогена;
R4 представляет собой атом водорода, C1-6 алкил, C2-6 алкенил, C2-3 алкинил, NR6R7, OR6, OR8 или R8, где указанные алкильные группы являются необязательно замещенными одним-шестью заместителями независимо выбранными из группы, состоящей из OR7, атома галогена, гидрокси, циано и R8;
R5 представляет собой атом водорода, C1-6 алкил, C2-6 алкенил, арил, гетероарил, C3-8 циклоалкил, гетероциклил, C(O)NR6R8, C(O)R8, NR6C(O)OR7 или SOmR6, где указанные арильные, гетероарильные, C3-8 циклоалкильные и гетероциклильные группы являются необязательно замещенными одним-пятью заместителями, независимо выбранными из группы, состоящей из C1-6 алкила, атома галогена, оксо, циано, C1-6 галогеналкила и SOmR6;
R6 представляет собой атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из атома галогена, гидрокси, циано и O(C1-6 алкил);
R7 представляет собой атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из атома галогена, гидрокси, циано и O(C1-6 алкил);
R8 представляет собой C3-8 циклоалкил, арил, гетероарил или гетероциклил, где указанные циклоалкильные, арильные, гетероарильные и гетероциклильные группы являются необязательно замещенным одним-четырьмя заместителями, независимо выбранными из группы, состоящей из атома галогена, циано, оксо, C1-6 галогеналкила, R6, OR6, C3-6 циклоалкила, арила, гетероарила, гетероциклила, SOmR6 и SF5;
m представляет собой целое число от нуля до двух;
p представляет собой целое число от нуля до двух.
В одном из вариантов осуществления настоящего изобретения, X представляет собой атом кислорода. В другом варианте осуществления настоящего изобретения, X представляет собой атом серы. В другом варианте осуществления настоящего изобретения, X представляет собой атом азота.
В одном из вариантов осуществления настоящего изобретения, R1 представляет собой атом водорода, C1-3 алкил, C3-8 циклоалкил или гетероциклил, где указанная алкильная группа является необязательно замещенной одним-шестью атомами галогена или гидрокси. В одном из классов настоящего изобретения, R1 представляет собой атом водорода, C1-3 алкил, C3-8 циклоалкил или гетероциклил, где указанная алкильная группа является необязательно замещенной одним-шестью атомами галогена. В одном из классов настоящего изобретения, R1 представляет собой атом водорода. В другом классе настоящего изобретения, R1 представляет собой метил. В другом классе настоящего изобретения, R1 представляет собой этил. В другом классе настоящего изобретения, R1 представляет собой изопропил. В другом классе настоящего изобретения, R1 представляет собой гетероциклил. В другом классе настоящего изобретения R1 представляет собой тетрагидропиранил.
В одном из вариантов осуществления настоящего изобретения, R2 представляет собой атом водорода или C1-3 алкил, где указанная алкильная группа является необязательно замещенной одним-шестью атомами галогена. В одном из классов настоящего изобретения, R2 представляет собой атом водорода. В другом классе настоящего изобретения, R2 представляет собой метил.
В одном из вариантов осуществления настоящего изобретения, R1 и R2 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-6 циклоалкильного кольца, которое является необязательно замещенным одним или двумя R6. В одном из классов настоящего изобретения, R1 и R2 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием циклопропильного кольца. В другом классе настоящего изобретения, R1 и R2 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием циклопропильного кольца, которое является замещенным двумя метильными группами.
В одном из вариантов осуществления настоящего изобретения, каждый R3 независимо выбирается из группы, состоящей из атома водорода или галогена, или две группы R3 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-4 циклоалкильного кольца, где указанное кольцо является необязательно замещенным одним-тремя атомами галогена. В одном из классов настоящего изобретения, R3 представляет собой атом водорода. В одном из классов настоящего изобретения, R3 представляет собой атом галогена. В подкласс по настоящему изобретению, R3 представляет собой атом фтора. В одном из классов настоящего изобретения, две группы R3 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием циклопропильного кольца.
В одном из вариантов осуществления настоящего изобретения, R4 представляет собой атом водорода, C1-6 алкил, OR6 или R8, где указанные алкильные группы являются необязательно замещенными одним-шестью заместителями, независимо выбранными из группы, состоящей из OR7, атома галогена, гидрокси, циано и R8. В одном из классов настоящего изобретения, R4 представляет собой атом водорода. В одном из классов настоящего изобретения, R4 представляет собой C1-6 алкил. В одном из классов настоящего изобретения, R4 представляет собой OR6. В одном из классов настоящего изобретения, R4 представляет собой R8. В одном из подклассов настоящего изобретения, R4 представляет собой арил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из атома галогена, метила, этила, циано или SO2CH3. В дополнительном подклассе настоящего изобретения, R4 представляет собой фенил, который является необязательно замещенным атомом галогена.
В одном из вариантов осуществления настоящего изобретения, R5 представляет собой гетероарил, гетероциклил, C(O)NR6R8, C(O)R8 или NR6C(O)OR7, где указанные гетероарильные и гетероциклильные группы являются необязательно замещенными одним-пятью заместителями, независимо выбранными из группы, состоящей из C1-6 алкила, атома галогена, оксо, циано, C1-6 галогеналкила и SOmR6. В одном из классов настоящего изобретения, R5 представляет собой гетероциклил или C(O)R8. В одном из классов настоящего изобретения, R5 представляет собой гетероциклил, который является необязательно замещенным одним-пятью заместителями, независимо выбранными из группы, состоящей из C1-6 алкила, атома галогена, оксо, циано, C1-6 галогеналкила и SOmR6. В одном из подклассов настоящего изобретения, R5 представляет собой 1,1-диоксидотиоморфолинил.
В одном из вариантов осуществления настоящего изобретения, R6 представляет собой атом водорода. В другом варианте осуществления настоящего изобретения, R6 представляет собой C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из атома галогена, гидрокси, циано и O(C1-6 алкил). В одном из классов настоящего изобретения, R6 представляет собой метил. В другом классе настоящего изобретения, R6 представляет собой трифторметил.
В одном из вариантов осуществления настоящего изобретения, m равно двум. В другом варианте осуществления настоящего изобретения, m равно единице.
В одном из вариантов осуществления настоящего изобретения, p равно нулю. В другом варианте осуществления настоящего изобретения, p равно единице. В другом варианте осуществления настоящего изобретения, p равно двум.
Упоминание предпочтительных классов и подклассов, приведенных выше, как подразумевается, включает все сочетания конкретных и предпочтительных групп, если не утверждается иного.
Конкретные варианты осуществления настоящего изобретения включают, но, не ограничиваясь этим, соединения, идентифицируемые в настоящем документе как Примеры 1-159 или их фармацевтически приемлемые соли.
Также включенной в рамки настоящего изобретения является фармацевтическая композиция, которая состоит из соединения формулы I, как описано выше, и фармацевтически приемлемого носителя. Настоящее изобретение, как предполагается, также охватывает фармацевтическую композицию, которая состоит из фармацевтически приемлемого носителя и любого из соединений, конкретно описанных в настоящей заявке. Эти и другие аспекты настоящего изобретения будут очевидны из концепций, содержащихся в настоящем документе.
Применения
Соединения по настоящему изобретению являются ингибиторами катепсинов и могут быть пригодными для использования при лечении или предотвращении катепсин-зависимых заболеваний или состояний у млекопитающих, предпочтительно, у людей. Конкретно, соединения по настоящему изобретению являются ингибиторами катепсина K и могут быть пригодными для использования при лечении или предотвращении катепсин K - зависимых заболеваний или состояний у млекопитающих, предпочтительно, у людей.
Соединения по настоящему изобретению имеют преимущества по сравнению со структурно сходными соединениями, известными в данной области, в том, что они имеют значительно улучшенные профили селективности по отношению к родственным катепсинам, в особенности, к катепсинам F.
“Катепсин-зависимые заболевания или состояния” относится к патологическим состояниям, которые зависят от активности одного или нескольких катепсинов. “Катепсин K - зависимые заболевания или состояния” относится к патологическим состояниям, которые зависят от активности катепсина K. Заболевания, ассоциируемые с активностями Катепсина K, включают остеопороз, остеопороз, вызываемый глюкокортикоидами, болезнь Педжета, аномально увеличенный метаболизм костной ткани, потерю зубов, переломы костей, ревматоидный артрит, остеоартрит, перипротезный остеолиз, несовершенный остеогенез, атеросклероз, тучность, глаукому, хроническое обструктивное легочное заболевание и рак, включая метастатическое костное заболевание, злокачественную гиперкальцемию и множественную миелому. При лечении таких состояний с помощью соединений, заявляемых в настоящем документе, необходимое терапевтическое количество будет изменяться в соответствии с конкретным заболеванием и может легко быть получено специалистом в данной области. Хотя в рамках настоящего изобретения предполагаются как лечение, так и предотвращение, лечение этих состояний представляет собой предпочтительное использование.
Один из вариантов осуществления настоящего изобретения представляет собой способ ингибирования активности катепсина у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше.
Один из классов вариантов осуществления представляет собой способ, где активность катепсина представляет собой активность катепсина K.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения или предотвращения катепсин-зависимых состояний у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше.
Один из классов вариантов осуществления представляет собой способ, где активность катепсина представляет собой активность катепсина K.
Другой вариант осуществления настоящего изобретения представляет собой способ ингибирования потери костей у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Другой вариант осуществления настоящего изобретения представляет собой способ уменьшения потери костей у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Применение ингибиторов катепсина K при ингибировании резорбции кости, которая включает аномально повышенный метаболизм костной ткани, переломы костей, болезнь Педжета, несовершенный остеогенез и перипротезный остеолиз, является известным в литературе, смотри Stroup, G.B., Lark, M.W., Veber, DF., Bhattacharrya, A., Blake, S., Dare, L.C., Erhard, K.F., Hoffman, S.J., James, I.E., Marquis, R.w., Ru, Y., Vasko-Moser, J.A., Smith, B.R., Tomaszek, T. and Gowen, M. Potent and selective inhibition of human cathepsin K leads to inhibition of bone resorption in vivo in a nonhuman primate. J. Bone Miner. Res., 16:1739-1746; 2001; и Votta, B.J., Levy, M.A., Badger, A., Dodds, R.A., James, I.E., Thompson, S., Bossard, M.J., Carr, T., Connor, J.R., Tomaszek, T.A., Szewczuk, L., Drake, F.H., Veber, D. and Gowen, M. Peptide aldehyde inhibitors of cathepsin K inhibit bone resorption both in vivo and in vitro. J. Bone Miner. Res. 12:1396-1406; 1997.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения или предотвращение остеопороза, включая остеопороз, вызываемый глюкокортикоидами, у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из указанных выше фармацевтических композиций, описанных выше. Применение ингибиторов катепсина K при лечении или предотвращение остеопороза является известным из литературы, смотри Saftig, P., Hunziker, E., Wehmeyer, O., Jones, S., Boyde, A., Rommerskirch, W., Moritz, J.D., Schu, P., and Vonfigura, K. Impaired osteoclast bone resorption leads to osteoporosis in cathepsin K-deficient mice. Proc. Natl. Acad. Sci. USA 95:13453-13458; 1998.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения или предотвращения периодонтального заболевания, включая потерю зубов, у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из указанных выше фармацевтических композиций описанных выше. Применение ингибиторов катепсина K при лечении или предотвращении периодонтального заболевания и потери зубов является известным из литературы, смотри Tsuji Y, et al., Expression of cathepsin K mRNA and protein in odontoclasts after experimental tooth movement in the mouse maxilla by in situ hybridization and immunoelectron microscopy. Cell Tissue Res. 2001 Mar; 303(3):359-69.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения или предотвращения состояния ревматоидного артрита у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что прогрессирующее разрушение околосуставной костной ткани является главной причиной дисфункции суставов и инвалидности у пациентов с ревматоидным артритом (RA), смотри Goldring SR, “Pathogenesis of bone erosions and rheumatoid arthritis”. Curr. Opin. Rheumatol. 2002; 14: 406-10. Анализ тканей суставов у пациентов с RA дает доказательство того, что катепсин K - положительные остеокласты представляют собой типы клеток, которые медиируют резорбцию фокальных костей, ассоциируемую с ревматоидным синовиальным повреждением, смотри Hou, W-S, Li, W, Keyszer, G, Weber, E, Levy, R, Klein, MJ, Gravallese, EM, Goldring, SR, Bromme, D, “Comparison Cathepsin K and S expression within the Rheumatoid and Osteoarthritic Synovium”, Arthritis Rheumatism 2002; 46: 663-74. В дополнение к этому, генерализованная потеря костей является главной причиной заболеваемости, ассоциируемой с острым RA. Частота переломов тазобедренного сустава и позвоночника значительно увеличивается у пациентов с хроническим RA, смотри Gould A, Sambrook, P, Devlin J et al, “Osteoclastic activation is principal mechanism leading to secondary osteoporosis in rheumatoid arthritis”. J. Rheumatol. 1998; 25: 1282-9. Применение ингибирования катепсина K при лечении или предотвращении резорбции околосуставной костной ткани и генерализованной потери костей представляет собой рациональный подход фармакологического вмешательства при развитии ревматоидного артрита.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения или предотвращения развития остеоартрита у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что остеоартрит (OA) сопровождается четко выраженными изменениями в суставах, включая изъязвление поверхности суставного хряща, околосуставную эндохондральную оссификацию/остеофитоз, и субхондральный костный и образование кист, смотри Oettmeier R, Abendroth, K, “Osteoarthritis and bone: osteologic types of osteoarthritis of the hip”, Skeletal Radiol. 1989; 18:165-74. В последнее время предполагается потенциальный вклад склероза субхондральных костей в инициирование и развитие OA. Повышение жесткости субхондральных костей, когда сустав реагирует на повторяющуюся импульсную нагрузку, уменьшает способность к ослаблению и распределению усилий по суставу, подвергая его воздействию более высоких механических напряжений на поверхности суставного хряща. Это, в свою очередь, ускоряет износ хряща и образование волокон, смотри Radin, EL and Rose RM, “Role of subchondral bone in initiation and progression of cartilage damage”, Clin. Orthop. 1986; 213: 34-40. Ингибирование избыточной резорбции околосуставных костей с помощью антирезорпционного агента, такого как ингибитор катепсина K, будет приводить к ингибированию метаболизма субхондральных костей, и таким образом, он может оказывать благоприятное воздействие на развития OA.
В дополнение к приведенной выше гипотезе, экспрессия белка катепсина K недавно идентифицирована в синовиальных фибробластах, макрофагоподобных клетках и хондроцитах из образцов синовиальных мембран и суставных хрящей, полученных от пациентов с OA, смотри Hou, W-S, Li, W, Keyszer, G, Weber, E, Levy, R, Klein, MJ, Gravallese, EM, Goldring, SR, Bromme, D, “Comparison of Cathepsin K and S expression within the Rheumatoid and Osteoarthritic Synovium”, Arthritis Rheumatism 2002; 46: 663-74; и Dodd, RA, Connor, JR, Drake, FH, Gowen, M, “Expression of Cathepsin K messenger RNA in giant cells and their prodrugs in human osteoarthritic synovial tissues”. Arthritis Rheumatism 1999; 42: 1588-93; и Konttinen, YT, Mandelin, J, Li, T-F, Salo, J, Lassus, J et al. “Acidic cysteine endoproteinase cathepsin K in the degeneration of the superficial articular hyaline cartilage in osteoarthritis”, Arthritis Rheumatism 2002; 46: 953-60. Таким образом, эти недавние исследования говорят о роли катепсина K в разрушении коллагена типа II в суставном хряще, ассоциируемом с развитием остеоартрита. Таким образом, применение ингибиторов катепсина K при лечении или предотвращении остеоартрита, как описано в настоящем изобретении, включает два различных механизма, один из них действует на ингибирование приводимого в действие остеокластами метаболизма субхондральных костей, а другой представляет собой непосредственное ингибирование дегенерации коллагена типа II в синовиальных мембранах и в хрящах пациентов с OA.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения рака у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций описанных выше. Из литературы известно, что катепсин K экспрессируется при карциноме грудной железы человека, раке простаты и хордоме и имеет способность деградировать матрикс, смотри Littlewood-Evans AJ, Bilbe G, Bowler WB, Farley D, Wlodarski B, Kokubo T, Inaoka T, Sloane J, Evans DB, Gallagher JA, “The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma.” Cancer Res 1997 Dec 1; 57(23):5386-90, Brubaker KD, Vessella RL, True LD, Thomas R, Corey E “Cathepsin K mRNA and protein expression in prostate cancer progression.” J Bone Miner Res 2003 18, 222-30, Haeckel C, Krueger S, Kuester D, Ostertag H, Samii M, Buehling F, Broemme D, Czerniak B, Roessner A. “Expression of cathepsin K in chordoma.” Hum Pathol 2000 Jul; 31(7):834-40.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения атеросклероза у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что катепсин K экспрессируется в атеросклеротических бляшках человека и имеет значительную эластазную активность, смотри Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. “Expression of elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells.” J Clin Invest 1998 Aug 102, 576-83. Также известно, что Cat K - нокаутированные мыши, когда скрещиваются c ApoE - нокаутированными мышами, показывают уменьшение площади атеросклеротических бляшек и увеличение стойкости к разрушению бляшек, смотри E. Lutgens, S.P.M. Lutgens, B.C.G. Faber, S. Heeneman, M.M.J. Gijbels, M.P.J. de Winther, P. Frederik, I. van der Made, D. Black, M.J.A.P. Daemen, K.B.J.M. Cleutjens “Disruption of the Cathepsin K Gene Reduces Atherosclerosis Progression and Induces Plaque Fibrosis but Accelerates Macrophage Foam Cell Formation.” Circulation 2006 113:98-107. Увеличение стабильности бляшек должно приводить к уменьшению вероятности сердечного приступа и инсульта у пациента, которому вводят терапевтически эффективные количества любого из соединений или любой из фармацевтических композиций, описанных выше.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения тучности у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что уровень mRNA катепсина K увеличивается в адипозной ткани на нескольких моделях тучности у мышей, а также в адипозной ткани тучных мужчин, смотри Chiellini C, Costa M, Novelli SE, Amri EZ, Benzi L, Bertacca A, Cohen P, Del Prato S, Friedman JM, Maffei M. “Identification of cathepsine K as novel marker of adiposity in white adipose tissue”. J Cell Physiol 2003, 195, 309-21.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения глаукомы у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Катепсин K сильно экспрессируется в радужной оболочке глаза, в силиарном теле и в эпителии пигментного слоя сетчатки и как таковой может быть пригодным для использования при лечении глаукомы, смотри Ortega, J., et al., “Gene Expression of Proteases and Proteases Inhibitors in the Human Ciliary Epithelium and ODM-2 cells,” Exp. Eye Res (1997) 65, 289-299; International Publication WO 2004/058238 (Alcon, Inc.).
Другой вариант осуществления настоящего изобретения представляет собой способ лечения хронического обструктивного легочного заболевания у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что катепсин K играет роль при фиброзе легких, смотри Buhling, F., et al., “Pivotal role of cathepsin K in lung fibrosis”, Am J Pathol. 2004 Jun; 164(6):2203-16.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения паразитарных инфекций у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что катепсины млекопитающих являются родственными папаиноподобным цистеинпротеазам, которые играют важную роль в жизненном цикле этих паразитов. Такие паразиты участвуют в заболеваниях малярии, американского трипаносомоза, африканского трипаносомоза, лейшманиоза, лямблиоза, трихомониаза, амебиаза, шистосомоза, фасциолеза, парагонимоза и кишечных круглых глистов, смотри Lecaille F, Kaleta J, Bromme D., Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem Rev 2002 102, 4459-88.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения тяжелого острого респираторного синдрома (SARS) у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения метастатического костного заболевания у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что остеокласты ответственны за резорбцию костей, и что разрушение костей и гиперкальцемия, вызываемая метастатическими опухолями, осуществляются с помощью остеокластов. Соответственно, ингибирование остеокластов может предотвратить разрушение костей и распространение метастаз в костях, смотри Miyamoto, T. и Suda, T., “Differentiation and function of osteoclasts”, Keio J Med 2003 Mar; 52(1):1-7.
Другой вариант осуществления настоящего изобретения представляет собой способ предотвращения метастатического костного заболевания у млекопитающего с первичной опухолью, которая имеет риск распространения метастаз в костях, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. В литературе описано, что соединения, которые ингибируют функцию остеокластов, могут предотвращать адгезию опухолевых клеток на костях, смотри S. Boissier, M. Ferreras, O. Peyruchaud, S. Magnetto, F. H. Ebetino, M. Colombel, P. Delmas, J.-M. Delaissé and P. Clézardin “bisphosphonates Inhibit Breast and Prostate Carcinoma Cell Invasion, an Early Event in the Formation of Bone Metastases” Cancer Research 60, 2949-2954, 2000.
Другой вариант осуществления настоящего изобретения представляет собой способ лечения злокачественной гиперкальцемии или множественной миеломы у млекопитающего, нуждающегося в этом, включающий введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что катепсин K играет роль при злокачественной гиперкальцемии и множественной миеломе, смотри Faust, J. et al., Multiple myeloma and cells of the human osteoclast lineage share morphological and cell surface markers. J Cell Biochem. 1998 Dec 15; 71(4):559-68; A. Lipton, New therapeutic agents for the treatment of bone diseases. Expert Opin Biol Ther. 2005 Jun;5(6):817-32.
Другой вариант осуществления настоящего изобретения представляет собой введение млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше, для лечения заболеваний млекопитающих, ассоциируемых с катепсином S, включая болезнь Альцгеймера, атеросклероз, хроническое обструктивное легочное заболевание, рак и определенные аутоиммунные расстройства, включая, но, не ограничиваясь этим, ювенильный диабет, множественный склероз, вульгарную пузырчатку, болезнь Грейвса, миастению гравис, системную красную волчанку, ревматоидный артрит и тиреоидит Хашимото; аллергические расстройства, включая, но, не ограничиваясь этим, астму; и аллогенную иммунную реакцию, включая, но, не ограничиваясь этим, отторжение пересаженных органов или тканевых трансплантатов. Из литературы известно, что активность катепсина S ассоциируется с указанными выше болезненными состояниями, смотри Munger JS, Haass C, Lemere CA, Shi GP, Wong WS, Teplow DB, Selkoe DJ, Chapman HA. Lysosomal processing of amyloid precursor protein to A beta peptides: a distinct role for cathepsin S. Biochem J 1995 311, 299-305, Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, Sakara Y, Chin MT, Libby P, Shi GP. Deficiency of cathepsin S reduces atherosclerosis in LDL receptor-deficient mice. J Clin Invest 2003 111, 897-906, Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr, Chapman HA Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 2000 106,1081-93, Shi GP, Sukhova GK, Kuzuya M, Ye Q, Du J, Zhang Y, Pan JH, Lu ML, Cheng XW, Iguchi A, Perrey S, Lee AM, Chapman HA, Libby P. Deficiency of the cysteine protease cathepsin S impairs microvessel growth. Circ Res 2003 92, 493-500, Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, McNeish JD, Eastman SE, Howard ED, Clarke SR, Rosloniec EF, Elliott EA, Rudensky AY. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity 1999 10, 207-17.
Иллюстрация настоящего изобретения представляет собой использования любого из соединений, описанных выше, при приготовлении лекарственного средства, пригодного для лечения и/или предотвращения остеопороза у млекопитающего, нуждающегося в этом. Кроме того, иллюстрация настоящего изобретения представляет собой использование любого из соединений, описанных выше, при приготовлении лекарственного средства пригодного для лечения и/или предотвращения: потери костей, резорбции костей, переломов костей, метастатического костного заболевания и/или расстройств, связанных с функционированием катепсина.
Соединения по настоящему изобретению могут вводиться млекопитающим, предпочтительно, людям, либо по отдельности, либо, предпочтительно, в комбинации с фармацевтически приемлемыми носителями или разбавителями, необязательно, вместе с известными вспомогательными веществами, такими как алюминиевые квасцы, в фармацевтической композиции, в соответствии со стандартной фармацевтической практикой. Соединения могут вводиться перорально или парентерально, включая внутривенный, внутримышечный, внутрибрюшинный, подкожный, ректальный и местный способы введения.
В случае таблеток для перорального применения, носители, которые повсеместно используются, включают лактозу и кукурузный крахмал, и обычно добавляются смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсул, пригодные для использования разбавители включают лактозу и высушенный кукурузный крахмал. Для перорального применения терапевтического соединения в соответствии с настоящим изобретением, выбранное соединение может вводиться, например, в форме таблеток или капсул или в виде водного раствора или суспензии. Для перорального введения в форме таблетки или капсулы, активный компонент лекарственного средства может объединяться с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния, дикальций фосфат, сульфат кальция, маннитол, сорбитол, и тому подобное; для перорального введения в жидкой форме, компоненты перорального лекарственного средстваа могут объединяться с любым пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерол, вода, и тому подобное. Кроме того, при желании или при необходимости, пригодные для использования связующие вещества, смазывающие вещества, разрыхляющие агенты и окрашивающие агенты также могут включаться в смесь. Пригодные для использования связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, подсластители из кукурузы, природные и синтетические смолы, такие как смола акации, трагаканта или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски, и тому подобное. Смазывающие вещества, используемые в этих дозированных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия, и тому подобное. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую смолу, и тому подобное. Когда необходимы водные суспензии для перорального использования, активный ингредиент объединяется с эмульгирующими и суспендирующими агентами. При желании, могут добавляться определенные подслащивающие и/или ароматизирующие агенты. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного применения, обычно приготавливают стерильные растворы активного ингредиента, и значение pH растворов должно соответствующим образом регулироваться и поддерживаться с помощью буфера. Для внутривенного применения, общая концентрация растворенного вещества должна контролироваться, чтобы сделать средство изотоническим.
Соединения по настоящему изобретению могут также вводиться в форме липосомных систем доставки, таких как малые однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут формироваться из разнообразных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Соединения по настоящему изобретению могут также доставляться с использованием моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Соединения по настоящему изобретению могут также связываться с растворимыми полимерами в качестве нацеливаемых носителей лекарственного средства. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксид-полилизин, замещенный пальмитоильными остатками. Кроме того, соединения по настоящему изобретению могут связываться с классом биодеградируемых полимеров, пригодных для использования при достижении контролируемого высвобождения лекарственного средства, например, с полимолочной кислотой, полигликолевой кислотой, с сополимерами полимолочной и полигликолевой кислот, с полиэпсилон-капролактоном, полигидроксимасляной кислотой, со сложными полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и с поперечно сшитыми или амфипатическими блок-сополимерами гидрогелей.
Настоящие соединения могут также быть пригодными для использования в комбинации с известными агентами, пригодными для использования при лечении или предотвращении остеопороза, остеопороза, вызываемого глюкокортикоидами, болезни Педжета, аномально увеличенного метаболизма костной ткани, периодонтального заболевания, потери зубов, переломов костей, ревматоидного артрита, остеоартрита, перипротезного остеолиза, несовершенного остеогенеза, метастатического костного заболевания, злокачественной гиперкальцемии и множественной миеломы. Комбинации соединений, описанных в настоящем документе, с другими агентами, пригодными для использования при лечении или предотвращении остеопороза или других расстройств костей, находятся в рамках настоящего изобретения. Специалист в данной области должен быть способен различить, какие именно комбинации агентов должны быть пригодными для использования, на основе конкретных характеристик лекарственных средств и рассматриваемого заболевания. Такие агенты включают следующее: органический бисфосфонат; селективный модулятор рецептора эстрогенов; модулятор рецепторов андрогенов; ингибитор протонной АТФазы остеокластов; ингибитор HMG-CoA-редуктазы; антагонист рецептора интегрина; анаболический агент остеобластов, такой как PTH; Витамин D; синтетический аналог витамина D; нестероидное противовоспалительное лекарственное средство; селективный ингибитор циклооксигеназы-2; ингибитор интерлейкина-1 бета; ингибитор LOX/COX и их фармацевтически приемлемые соли и смеси. Предпочтительная комбинация представляет собой соединение по настоящему изобретению и органический бисфосфонат. Другая предпочтительная комбинация представляет собой соединение по настоящему изобретению и селективный модулятор рецепторов эстрогенов. Другая предпочтительная комбинация представляет собой соединение по настоящему изобретению и модулятор рецепторов андрогенов. Другая предпочтительная комбинация представляет собой соединение по настоящему изобретению и анаболический агент остеобластов.
“Органический бисфосфонат” включает, но, не ограничиваясь этим, соединения химической формулы
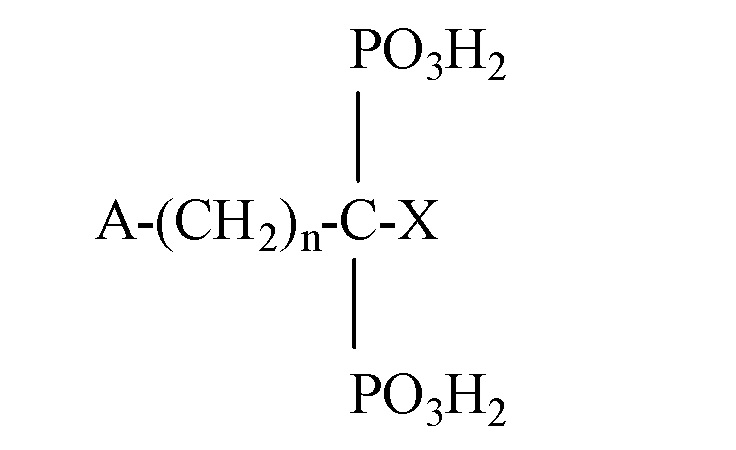
где n представляет собой целое число от 0 до 7, и где A и X независимо выбираются из группы, состоящей из H, OH, атома галогена, NH2, SH, фенила, C1-C30 алкила, C3-C30 разветвленного алкила или циклоалкила, бициклической кольцевой структуры, содержащей два или три атома N, C1-C30 замещенного алкила, C1-C10 алкила, замещенного NH2, C3-C10 разветвленного алкила или циклоалкила, замещенного NH2, C1-C10 диалкила, замещенного NH2, C1-C10 алкокси, C1-C10 алкила, замещенного тио, тиофенилом, галогенфенилтио, C1-C10 алкилзамещенного фенила, пиридила, фуранила, пирролидинила, имидазолила, имидазопиридинила и бензила, так что, как A, так и X не выбираются из H или OH, когда n равно 0; или A и X берутся вместе с атомом или атомами, углерода, к которым они присоединены, с образованием C3-C10 кольца.
В рассмотренной выше химической формуле, алкильные группы могут быть прямыми, разветвленными или циклическими при условии, что выбирается достаточное количество атомов для химической формулы. C1-C30 замещенный алкил может включать большое разнообразие заместителей, неограничивающие примеры которых включают заместители, выбранные из группы, состоящей из фенила, пиридила, фуранила, пирролидинила, имидазонила, NH2, C1-C10 алкила или диалкила, замещенного NH2, OH, SH, и C1-C10 алкокси.
Рассмотренная выше химическая формула, как предполагается, также охватывает сложные карбоциклические, ароматические и гетероатомные структуры для заместителей A и/или X, неограничивающие примеры которых включают нафтил, хинолил, изохинолил, адамантил и хлорфенилтио.
Фармацевтически приемлемые соли и производные бисфосфонатов также являются пригодными для использования в настоящем документе. Неограничивающие примеры солей включают соли, выбранные из группы, состоящей из солей щелочного металла, щелочноземельного металла, аммония и моно-, ди-, три- или тетра- C1-C10-алкилзамещенного аммония. Предпочтительные соли представляют собой соли, выбранные из группы, состоящей из солей натрия, калия, кальция, магния и аммония. Наиболее предпочтительными являются соли натрия. Неограничивающие примеры производных включают соединения, выбранные из группы, состоящей из сложных эфиров, гидратов и амидов.
Необходимо отметить, что термины "бисфосфонат" и "бисфосфонаты", как используется в настоящем документе, при упоминании терапевтических агентов по настоящему изобретению, как предполагается, охватывают также дифосфонаты, бифосфоновые кислоты и дифосфоновые кислоты, а также соли и производные этих веществ. Применение конкретной номенклатуры при упоминании бисфосфоната или бисфосфонатов, как подразумевается, не ограничивает рамки настоящего изобретения, если не указано конкретно иного. Из-за смешанной номенклатуры, используемой в настоящее время специалистами в данной области, упоминание конкретной массы или процента бисфосфонатного соединения по настоящему изобретению, относится к активной массе кислоты, если в настоящем документе не указано иного. Например, фраза "примерно 5 мг бисфосфоната, ингибирующего резорбцию костей, выбранного из группы, состоящей из алендроната, его фармацевтически приемлемых солей и его смесей, по отношению к активной массе алендроновой кислоты" означает, что количество выбранного соединения бисфосфоната вычисляется по отношению к 5 мг алендроновой кислоты.
Неограничивающие примеры бисфосфонатов, пригодных для использования в настоящем документе, включают следующие:
Алендронат, который известен также как алендроновая кислота, 4-амино-1-гидроксибутилиден-1,1-бисфосфоновая кислота, натрий алендронат или мононатрий алендронат тригидрат, мононатрий тригидрат 4-амино-1-гидроксибутилиден-1,1-бисфосфоновой кислоты.
Алендронат описан в патентах США №№ 4922007, Kieczykowski et al., опубликован 1 мая 1990 года; 5019651, Kieczykowski et al., опубликован 28 мая 1991 года; 5510517, Dauer et al., опубликован 23 апреля 1996 года; 5648491, Dauer et al., опубликован 15 июля 1997 года, все они тем самым включаются в настоящий документ в качестве ссылок во всей их полноте.
Циклогептиламинометилен-1,1-бисфосфоновая кислота, YM 175, Yamanouchi (инкадронат, ранее известный как цимадронат), как описано в патенте США № 4970335, Isomura et al., опубликованном 13 ноября 1990 года, который включается в настоящий документ в качестве ссылки во всей своей полноте.
1,1-дихлорметилен-1,1-дифосфоновая кислота (клодроновая кислота) и ее динатриевая соль (клодронат, Procter and Gamble), описаны в патенте Бельгии 672,205 (1966) и в J. Org. Chem 32, 4111 (1967), оба они включаются в настоящий документ в качестве ссылок во всей их полноте.
1-гидрокси-3-(1-пирролидинил)пропилиден-1,1-бисфосфоновая кислота (EB-1053).
1-гидроксиэтан-1,1-дифосфоновая кислота (этидроновая кислота).
1-гидрокси-3-(N-метил-N-пентиламино)пропилиден-1,1-бисфосфоновая кислота, также известная как BM-210955, Boehringer-Mannheim (ибандронат), описанная в патенте США № 4927814, опубликованном 22 мая 1990 года, который включается в настоящий документ в качестве ссылки во всей своей полноте.
1-гидрокси-2-имидазо-(1,2-a)пиридин-3-илэтилиден (минодронат).
6-амино-1-гидроксигексилиден-1,1-бисфосфоновая кислота (неридронат).
3-(диметиламино)-1-гидроксипропилиден-1,1-бисфосфоновая кислота (олпадронат).
3-амино-1-гидроксипропилиден-1,1-бисфосфоновая кислота (памидронат).
[2-(2-пиридинил)этилиден]-1,1-бисфосфоновая кислота (пиридронат), описанная в патенте США № 4761406, который включается в качестве ссылки во всей своей полноте.
1-гидрокси-2-(3-пиридинил)этилиден-1,1-бисфосфоновая кислота (ризендронат).
(4-хлорфенил)тиометан-1,1-дифосфоновая кислота (тилудронат) как описано в патенте США № 4876248, Breliere et al., 24 октября 1989 года, который включается в настоящий документ в качестве ссылки во всей своей полноте.
1-гидрокси-2-(1H-имидазол-1-ил)этилиден-1,1-бисфосфоновая кислота (золедронат).
Неограничивающие примеры бисфосфонатов включают алендронат, цимадронат, клодронат, этидронат, ибандронат, инкадронат, минодронат, неридронат, олпадронат, памидронат, пиридронат, ризендронат, тилудронат, и золендронат, и их фармацевтически приемлемы соли и сложные эфиры. Особенно предпочтительный бисфосфонат представляет собой алендронат, в особенности натриевую, калиевую, кальциевую, магниевую или аммониевую соль алендроновой кислоты. Иллюстрация предпочтительного бисфосфоната представляет собой натриевую соль алендроновой кислоты, в особенности, гидратированную натриевую соль алендроновой кислоты. Соль может гидратироваться целым числом молей воды или дробным числом молей воды. Другая иллюстрация предпочтительного бисфосфоната представляет собой гидратированную натриевую соль алендроновой кислоты, в особенности, когда гидратированная соль представляет собой алендронат мононатрий тригидрат.
Наблюдается, что можно использовать смеси двух или более бисфосфонатных активных веществ.
Точная дозировка органического бисфосфоната будет изменяться вместе с временным графиком дозирования, конкретным выбранным бисфосфонатом, возрастом, размерами, полом и состоянием млекопитающего или человека, природой и тяжестью расстройства, которое должно лечиться, и другими релевантными медицинскими и физическими факторами. Таким образом, точное фармацевтически эффективное количество не может быть указано заранее и может легко определяться медицинским работником или лечащим врачом. Соответствующие количества могут определяться с помощью рутинных экспериментов при клинических исследованиях на моделях животных и людях. Как правило, выбирается соответствующее количество бисфосфоната для получения эффекта ингибирования резорбции костей, то есть, вводится количество бисфосфоната, ингибирующего резорбцию костей. Для людей, эффективная пероральная доза бисфосфоната, как правило, составляет примерно от 1,5 примерно до 6000 мкг/кг массы тела, а предпочтительно, примерно от 10 примерно до 2000 мкг/кг массы тела. Для мононатрия алендроната тригидрата, обычные дозы для людей, которые вводятся, как правило, находятся в пределах примерно от 2 мг/день примерно до 40 мг/день, предпочтительно, примерно от 5 мг/день примерно до 40 мг/день. В США, одобренные в настоящее время дозировки мононатрия алендроната тригидрата составляют 5 мг/день для предотвращения остеопороза, 10 мг/день при лечении остеопороза и 40 мг/день, при лечении болезни Педжета.
При альтернативных режимах дозирования, бисфосфонат может вводиться при интервалах, иных, чем ежедневные, например, при дозировании раз в неделю, при дозировании два раза в неделю, при дозировании раз в две недели и при дозировании дважды в месяц. В режиме дозирования раз в неделю, мононатрий алендронат тригидрат должен вводиться при дозировках 35 мг/неделя или 70 мг/неделя.
“Селективные модуляторы рецепторов эстрогенов” относится к соединениям, которые изменяют или ингибируют связывание эстрогена с рецептором, независимо от механизма. Примеры модуляторов рецепторов эстрогенов включают, но, не ограничиваясь этим, эстроген, прогестоген, эстрадиол, дролоксифен, ралоксифен, лазофоксифен, TSE-424, тамоксифен, идоксифен, LY353381, LY117081, торемифен, фулвестран, 4-[7-(2,2-диметил-1-оксопропокси-4-метил-2-[4-[2-(1-пиперидинил)этокси]фенил]-2H-1-бензопиран-3-ил]-фенил-2,2-диметилпропаноат, 4,4’-дигидроксибензофенон-2,4-динитрофенилгидразон и SH646.
“Модулятор рецептора бета эстрогенов” представляет собой соединение, которое селективно агонизирует или антагонизирует рецептор бета эстрогенов (агонизирование ERβ повышает транскрипцию гена триптофангидроксилазы (TPH представляет собой ключевой фермент при синтезе серотонина) посредством события, медиируемого ERβ. Примеры агонистов рецепторов бета эстрогенов можно найти в Международной публикации PCT WO 01/82923, которая опубликована 8 ноября 2001 года, и в WO 02/41835, который опубликован 20 мая 2002 года, обе они тем самым включаются в качестве ссылок во всей их полноте.
“Модуляторы рецепторов андрогенов” относятся к соединениям, которые изменяют или ингибируют связывание андрогенов с рецептором, независимо от механизма. Примеры модуляторов рецептора андрогенов включают финастерид и другие ингибиторы 5α-редуктазы, нилутамид, флутамид, бикалутамид, лиарозол и абиратерон ацетат.
“Ингибитор протонной АТФазы остеокластов” относится к ингибитору протонной АТФазы, которая находится на апикальной мембране остеокласта и, как сообщается, играет значительную роль в процессе резорбции костей. Этот протонный насос представляет собой привлекательную мишень для конструирования ингибиторов резорбции кости, которые являются потенциально пригодными для использования при лечении и предотвращении остеопороза и родственных метаболических заболеваний. Смотри C. Farina et al., “Selective inhibitors of the osteoclast vacuolar proton ATPase as novel bone antiresorptive agents”, DDT, 4: 163-172 (1999)), которая тем самым включается в качестве ссылки во всей своей полноте.
“Ингибиторы HMG-CoA-редуктазы” относятся к ингибиторам 3-гидрокси-3-метилглютарил-CoA редуктазы. Соединения, которые имеют ингибиторную активность по отношению к HMG-CoA-редуктазе, можно легко идентифицировать с использованием анализов, хорошо известных в данной области. Например, смотри анализы, описанные или цитируемые в патенте США № 4231938, в столбце 6, и в WO 84/02131, pp. 30-33. Термины “ингибитор HMG-CoA-редуктазы” и “ингибитор HMG-CoA-редуктазы” имеют одинаковое значение, когда используются в настоящем документе.
Примеры ингибиторов HMG-CoA-редуктазы, которые можно использовать, включают но, не ограничиваясь этим ловастатин (MEVACOR®; смотри патенты США №№ 4231938, 4294926 и 4319039), симвастатин (ZOCOR®; смотри патенты США №№ 4444784, 4820850 и 4916239), розувастатин (в частности, кальциевую соль, продаваемую в CRESTOR®), правастатин (PRAVACHOL®; смотри патенты США №№ 4346227, 4537859, 4410629, 5030447 и 5180589), флуфастатин (LESCOL®; смотри патенты США №№ 5354772, 4911165, 4929437, 5189,164, 5118853, 5290946 и 5356896), аторвастатин (LIPITOR®; смотри патенты США №№ 5273995, 4681893, 5489691 и 5342952) и церивастатин (также известный как ривастатин и BAYCHOL®; cмотри патент США № 5177080). Структурные формулы этих и дополнительных ингибиторов HMG-CoA-редуктазы, которые могут использоваться в способах по настоящему изобретению, описаны на странице 87, M. Yalpani, "Cholesterol Lowering Drugs", Chemistry & Industry, pp. 85-89 (5 February 1996) и в патентах США №№ 4782084 и 4885314. Термин ингибитор HMG-CoA-редуктазы, как используется в настоящем документе, включает все фармацевтически приемлемые формы лактонов и кислот с открытым кольцом (то есть, такие, где лактоновое кольцо раскрывается с образованием свободной кислоты), а также солевые и сложноэфирные формы соединений, которые имеют ингибиторную активность по отношению к HMG-CoA-редуктазе, и по этой причине, использование таких солей, сложных эфиров, форм кислот с открытым кольцом и лактонов, включается в рамки настоящего изобретения. Иллюстрация лактоновой части и соответствующей ей формы кислоты с открытым кольцом показана ниже как структуры I и II.
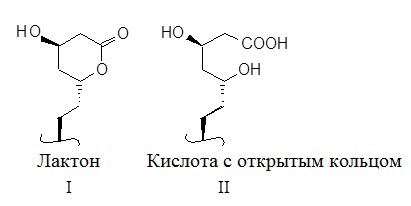
В ингибиторах HMG-CoA-редуктазы, где может существовать форма кислоты с открытым кольцом, солевые и сложноэфирные формы могут предпочтительно формироваться из формы кислоты с открытым кольцом, и все такие формы включаются в значение термина “ингибитор HMG-CoA-редуктазы”, как используется в настоящем документе. Предпочтительно, ингибитор HMG-CoA-редуктазы выбирается из ловастатина и симвастатина, а наиболее предпочтительно, из симвастатина. В настоящем документе, термин "фармацевтически приемлемые соли" по отношению к ингибитору HMG-CoA-редуктазы должен обозначать нетоксичные соли соединений, используемые в настоящем изобретении, которые, как правило, приготавливают посредством взаимодействия свободной кислоты с соответствующим органическим или неорганическим основанием, в частности, соли, которые образуются из катионов, таких как натрий, калий, алюминий, кальций, литий, магний, цинк и тетраметиламмоний, а также соли, сформированные из аминов, таких как аммиак, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N’-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, 1-п-хлорбензил-2-пирролидин-1’-ил-метилбензимидазол, диэтиламин, пиперазин и трис(гидроксиметил)аминометан. Дополнительные примеры солевых форм ингибиторов HMG-CoA-редуктазы могут включать, но, не ограничиваясь этим, ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камсилат, карбонат, хлорид, клавулант, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эсилат, фумарат, глуцептат, глюконат, глютамат, гликолилларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памоат, пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сабацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат.
Сложноэфирные производные описанных соединений ингибиторов HMG-CoA-редуктазы могут действовать в качестве пролекарств, которые, когда поглощаются в кровотоке теплокровного животного, могут расщепляться таким образом, чтобы высвобождать лекарственную форму и давать возможность лекарственному средству демонстрировать улучшенную терапевтическую эффективность.
Как используется выше, “антагонисты рецепторов интегрина” относятся к соединениям, которые селективно антагонизируют, ингибируют связывание или противодействуют связыванию физиологического лиганда с интегрином ανβ3, к соединениям, которые селективно антагонизируют, ингибируют связывание или противодействуют связыванию физиологического лиганда с интегрином ανβ5, к соединениям, которые антагонизируют, ингибируют связывание или противодействуют связыванию физиологического лиганда как с интегрином ανβ3, так и с интегрином ανβ5, и к соединениям, которые антагонизируют, ингибируют активность или противодействуют активности конкретного интегрина (интегринов), экспрессируемого в капиллярных эндотелиальных клетках. Термин также относится к антагонистам интегринов ανβ6, ανβ8, α1β1, α2β1, α5β, α6β1 и α6β4. Термин также относится к антагонистам любого сочетания интегринов ανβ3, ανβ5, ανβ6, ανβ8, α1β1, α2β1, α5β1, α6β1 и α6β4. H.N. Lode и сотрудники в PNAS USA 96: 1591-1596 (1999) наблюдали синергические эффекты между антагонистом антиангиогенного интегрина α ν и опухоле-специфичным белком слияния антитело-цитокин (интерлейкин-2) при уничтожении спонтанных метастаз опухоли. Их результаты говорят об этом сочетании как об имеющем потенциал лечения роста раковой и метастатической опухоли. Антагонисты рецептора интегрина ανβ3 ингибируют резорбцию костей с помощью нового механизма, отличного от механизма всех доступных в настоящее время лекарственных средств. Интегрины представляют собой гетеродимерные трансмембранные рецепторы адгезии, которые медиируют взаимодействия клетка-клетка и клетка-матрикс. α и β субъединицы интегрина взаимодействуют не-ковалентно и связывают лиганды внеклеточного матрикса способом, зависимым от двухвалентных катионов. Наиболее распространенный интегрин на остеокластах представляет собой ανβ3 (>107/остеокласт), который, видимо, играет роль ограничения скорости при цитоскелетальной организации, важной для миграции и поляризации клеток. Антагонизирующее воздействие ανβ3 выбирается из ингибирования резорбции кости, ингибирования рестеноза, ингибирования дистрофии желтого пятна, ингибирования артрита и ингибирования рака и роста метастаз.
“Анаболический агент остеобластов” относится к агентам, которые формируют кость, таким как PTH. Периодическое введение паратиреоидного гормона (PTH) или его амино-конечных фрагментов и аналогов, как показано, предотвращает, приостанавливает, частично обращает потерю костей и стимулирует формирование костей у животных и людей. Относительно дискуссии, сошлемся на D.W. Dempster et al., “Anabolic actions of parathyroid hormone on bone,” Endocr Rev 14: 690-709 (1993). Исследования продемонстрировали клинические преимущества паратиреоидного гормона при стимулировании формирования костей и при этом увеличение массы и прочности костей. Результаты сообщаются RM Neer et al., в New Eng J Med 344 1434-1441 (2001).
В дополнение к этому, фрагменты или аналоги белков, родственные паратиреоидному гормону, такие как PTHrP-(1-36), демонстрируют сильные антикальциурические воздействия [смотри M.A. Syed et al., “Parathyroid hormone-related protein-(1-36) stimulates renal tubular calcium reabsorption in normal human volunteers: implications for pathogenesis of humoral hypercalcemia of malignancy” JCEM 86: 1525-1531 (2001)] и могут также иметь потенциал в качестве анаболических агентов при лечении остеопороза.
“Витамин D” включает, но, не ограничиваясь этим, витамин D3 (холекальциферол) и витамин D2 (эргокальциферол), которые представляют собой встречающиеся в природе биологически неактивные предшественники гидроксилированных биологически активных метаболитов витамина D: 1α-гидроксивитамина D; 25-гидроксивитамина D, и 1α,25-дигидроксивитамина D. Витамин D2 и витамин D3 имеют одинаковую биологическую эффективность для людей. Когда витамин, либо D2, либо D3, попадает в кровоток, он гидроксилируется посредством цитохрома P450-витамин D-25-гидроксилазы с получением 25-гидроксивитамина D. Метаболит 25-гидроксивитамин D является биологически инертным и дополнительно гидроксилируется в почках посредством цитохрома P450-монооксигеназы, 25(OH) D-1α-гидроксилазы с получением 1,25-дигидроксивитамина D. Когда уровень кальция в сыворотке уменьшается, происходит увеличение продуцирования паратиреоидного гормона (PTH), который регулирует гомеостаз кальция и увеличивает уровни кальция в плазме посредством увеличения преобразования 25-гидроксивитамина D в 1,25-дигидроксивитамин D.
1,25-дигидроксивитамин D, как считается, является ответственным за воздействие витамина D на метаболизм кальция и костей. Метаболит 1,25-дигидрокси представляет собой активный гормон, необходимый для поддержания поглощения кальция и целостности скелета. Гомеостаз кальция поддерживается посредством 1,25-дигидроксивитамина D посредством индуцирования дифференциации стволовых клеток моноцитов в остеокласты и посредством поддержания кальция в нормальном диапазоне, что дает в результате минерализацию костей посредством осаждения гидроксиапатита кальция на поверхности костей, смотри Holick, MF, Vitamin D photobiology, metabolism, and clinical applications, In: DeGroot L, Besser H, Burger HG, et al.,eds. Endocrinology, 3rd ed., 990-1013 (1995). Однако повышенные уровни 1α,25-дигидроксивитамина D3 могут давать в результате увеличение концентрации кальция в крови и аномальный контроль концентрации кальция посредством метаболизма костей, дающий в результате гиперкальцемию. 1α,25-дигидроксивитамин D3 также опосредованно регулирует активность остеокластов при метаболизме костей, и его повышенные уровни могут, как ожидается, вызывать избыточную резорбцию костей при остеопорозе.
“Синтетические аналоги витамина D” включают не встречающиеся в природе соединения, которые действуют подобно витамину D.
“Нестероидные противовоспалительные лекарственные средства” или NSAID, ингибируют метаболизм арахидоновой кислоты до провоспалительных простагландинов посредством циклооксигеназы (COX)-1 и COX-2. Неограничивающие примеры NSAID включают: аспирин, ибупрофен, напроксен, диклофенак, этодолак, фенопрофен, флубипрофен, индометацин, кетопрофен, кеторолак, мелоксикам, набуметон, оксапрозин, пироксикам, сулиндак, толметин, дифлунизал, меклофенамат и фенилбутазон.
“Селективный ингибитор циклооксигеназы-2” или ингибитор COX-2 относится к типу нестероидных противовоспалительных лекарственных средств (NSAID), которые ингибируют кофермент COX-2, который вносит вклад в боль и воспаление в организме. Неограничивающие примеры ингибиторов COX-2 включают: целекоксиб, эторикоксиб, парекоксиб, рофекоксиб, вальдекоксиб и лумиракоксиб.
“Ингибитор интерлейкина-1 бета” или IL-1β относится к ингибиторам IL-1, которые представляет собой растворимый фактор, производимый моноцитами, макрофагами и другими клетками, который активирует T-лимфоциты и усиливает их реакцию на митогены или антигены. Неограничивающие примеры ингибиторов IL-1β включают диацерин и реин.
“Ингибитор LOX/COX” относится к ингибитору всех трех главных ферментов, участвующих в пути арахидоновой кислоты, а именно, 5-LOX, COX-1 и COX-2. Неограничивающие пример ингибитора LOX/COX представляет собой ликофелон.
При получении в виде единичной фиксированной дозы, такие комбинированные продукты содержат соединения по настоящему изобретению в диапазоне доз, описанном ниже, и другие фармацевтически активные агенты в одобренном диапазоне их дозировок. Альтернативно, соединения по настоящему изобретению могут использоваться последовательно с известным фармацевтически приемлемым агентом (агентами), когда комбинированный состав является непригодным для использования.
Термин "введение" и его варианты (например, "введение соединения") при упоминании соединения по настоящему изобретению означает введение соединения или пролекарства соединения в систему животного, нуждающегося в лечении. Когда соединение по настоящему изобретению или его пролекарство получают в комбинации с одним или несколькими другими активными агентами (например, с цитотоксическим агентом, и тому подобное), "введение" и его варианты, каждый, понимаются как включение в рассмотрение одновременного и последовательного введения соединения или его пролекарства и других агентов. Настоящее изобретение включает в свои рамки пролекарства соединений по настоящему изобретению. Как правило, такие пролекарства будут представлять собой функциональные производные соединений по настоящему изобретению, которые легко преобразуются in vivo в необходимое соединение. Таким образом, в способах лечения по настоящему изобретению, термин "введение" должен охватывать лечение различных описанных состояний с помощью конкретно описанного соединения или с помощью соединения, которое может и не быть конкретно описанным, но которое преобразуется в указанное соединение in vivo после введения пациенту. Обычные процедуры для выбора и приготовления пригодных для использования производных пролекарств описаны, например, в "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985, которая включается в настоящий документ в качестве ссылки во всей своей полноте. Метаболиты этих соединений включают активные частицы, полученные при введении соединений по настоящему изобретению в биологическую среду.
Как используется в настоящем документе, термин "композиция" предназначен для охвата продукта, содержащего указанные ингредиенты в указанных количествах, а также любого продукта, который получается в результате, непосредственно или опосредованно, объединения указанных ингредиентов в указанных количествах.
Термин "терапевтически эффективное количество", как используется в настоящем документе, означает, что количество активного соединения или фармацевтического агента, который проявляет биологическую или лекарственную реакцию в ткани, системе, животном или человеке, которая рассматривается исследователем, ветеринаром, врачом или другим клиницистом.
Термины “лечить” или “лечение” заболевания, как используется в настоящем документе, включают: ингибирование заболевания, то есть приостановку или замедление развития заболевания или его клинических симптомов; или облегчение заболевания, то есть регрессию заболевания или его клинических симптомов.
Термины “предотвращать” или “предотвращение” заболевания, как используется в настоящем документе, включают: прекращение развития клинических симптомов заболевания у млекопитающего, которое может экспонироваться или быть предрасположено к заболеванию, но еще не испытывает или не проявляет симптомов заболевания.
Термин “резорбция костей”, как используется в настоящем документе, относится к способу, с помощью которого остеокласты деградируют кость.
Настоящее изобретение также охватывает фармацевтическую композицию, пригодную для использования при лечении остеопороза или других расстройств костей, включающем введение терапевтически эффективного количества соединения формулы I по настоящему изобретению вместе с фармацевтически приемлемыми носителями или разбавителями или без них. Соответствующие композиции по настоящему изобретению включают водные растворы, содержащие соединения по настоящему изобретению и фармакологически приемлемые носители, например, солевой раствор, при уровне pH, например, 7,4. Растворы могут вводиться в кровоток пациента посредством местной инъекции болюса.
Когда соединение в соответствии с настоящим изобретением вводится субъекту человеку, ежедневная доза будет обычно определяться предписывающим лечащим врачом, при этом доза, как правило, изменяется в соответствии с возрастом, массой тела и реакцией отдельного пациента, а также с тяжестью симптомов у пациента.
В одном из иллюстративных применений, соответствующее количество соединения вводится млекопитающему, подвергающемуся воздействию лечения катепсин-зависимого состояния. Пероральные дозы по настоящему изобретению, когда оно используется для указанных воздействий, будут находиться в пределах примерно между 0,01 мг на кг массы тела в день (мг/кг/день) и примерно 100 мг/кг/день, предпочтительно, от 0,01 до 10 мг/кг/день, и наиболее предпочтительно, от 0,1 до 5,0 мг/кг/день. Для перорального введения, композиции предпочтительно приготавливаются в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100 и 500 миллиграмм активного ингредиента для симптоматического установления дозы для пациента, который должен лечиться. Лекарственное средство, как правило, содержит примерно от 0,01 мг примерно до 500 мг активного ингредиента, предпочтительно, примерно от 1 мг примерно до 100 мг активного ингредиента. При внутривенном введении, наиболее предпочтительные дозы будут находиться в пределах примерно от 0,1 примерно до 10 мг/кг/минута в течение вливания с постоянной скоростью. Преимущественно, соединения по настоящему изобретению могут вводиться в виде одной ежедневной дозе, или общая ежедневная доза может вводиться в разделенных дозах два, три или четыре раза в день. Кроме того, предпочтительные соединения по настоящему изобретению могут вводиться в интраназальной форме посредством местного применения соответствующих интраназальных носителей или посредством трансдермальных способов, используя формы трансдермальных кожных пластырей, хорошо известных специалистам в данной области. Для введения в форме трансдермальной системы доставки, введение дозы будет, разумеется, непрерывным, а не периодическим, в течение всего режима дозирования.
Соединения по настоящему изобретению можно использовать в комбинации с другими агентами, пригодными для использования при лечении состояний, медиируемых катепсином. Индивидуальные компоненты таких комбинаций могут вводиться по отдельности в различные моменты времени в ходе терапии или одновременно в разделенных или единых комбинированных формах. Следовательно, настоящее изобретение должно пониматься как охватывающее все такие режимы одновременного или поочередного лечения и термин "введение" должен интерпретироваться соответствующим образом. Необходимо понимать, что рамки комбинаций соединений по настоящему изобретению с другими агентами, пригодными для использования при лечении состояний, медиируемых катепсином, включают в принципе любое сочетание с любой фармацевтической композицией, пригодной для использования при лечении расстройств, связанных с функционированием эстрогенов.
Следовательно, рамки настоящего изобретения охватывают использование соединения формулы I в комбинации со вторым агентом, выбранным из: органического бисфосфоната; селективного модулятора рецепторов эстрогенов; модулятора рецепторов андрогенов; ингибитора протонной АТФазы остеокластов; ингибитора HMG-CoA-редуктазы; антагониста рецепторов интегрина; анаболического агента остеобластов, такого как PTH; витамина D; синтетического аналога витамина D; нестероидного противовоспалительного лекарственного средства; селективного ингибитора циклооксигеназы-2; ингибитора интерлейкина-1 бета; ингибитора LOX/COX и их фармацевтически приемлемых солей и смесей.
Эти и другие аспекты настоящего изобретения будут понятны из концепций, содержащихся в настоящем документе.
Определения
Соединения по настоящему изобретению могут содержать один или несколько асимметричных центров, и таким образом, они могут существовать как рацематы и рацемические смеси, индивидуальные энантиомеры, диастереомерные смеси и индивидуальные диастереомеры. Дополнительные асимметричные центры могут присутствовать в зависимости от природы различных заместителей на молекуле. Каждый такой асимметричный центр будет независимо давать два оптических изомера, и предполагается, что все возможные оптические изомеры и диастереомеры в смесях и в виде чистых или частично очищенных соединений включаются в предмет настоящего изобретения. Если только не указана конкретная стереохимия, настоящее изобретение, как считается, охватывает все такие изомерные формы этих соединений.
Независимый синтез этих диастереомеров или их хроматографическое разделение может осуществляться, как известно в данной области, с помощью соответствующей модификации методологии, описанной в настоящем документе. Их абсолютная стереохимия может определяться с помощью рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизируются, при необходимости, с помощью реагента, содержащего асимметричный центр известной абсолютной конфигурации.
Если это желательно, рацемические смеси соединений могут разделяться таким образом, что выделяются индивидуальные энантиомеры. Разделение может осуществляться с помощью способов, хорошо известных в данной области, таких как связывание рацемической смеси соединений с энантиомерно чистым соединением, с образованием диастереомерной смеси, с последующим разделением индивидуальных диастереомеров с помощью стандартных способов, таких как фракционная кристаллизация или хроматография. Реакция связывания часто представляет собой образование солей с использованием энантиомерно чистой кислоты или основания. Диастереомерные производные могут затем преобразовываться в чистые энантиомеры посредством отщепления добавленного хирального остатка. Рацемическая смесь соединений может также разделяться непосредственно с помощью хроматографических методов, с использованием хиральных стационарных фаз, эти методы хорошо известны в данной области.
Альтернативно, любой энантиомер соединения может быть получен с помощью стереоселективного синтеза с использованием оптически чистых исходных материалов или реагентов известной конфигурации с помощью способов, хорошо известных в данной области.
В соединениях формулы I, атомы могут демонстрировать их природный изотопный состав или один или несколько видов атомов могут искусственно обогащаться конкретным изотопом, имеющим такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, преобладающего в природе. Настоящее изобретение, как подразумевается, включает все пригодные для использования изотопные варианты соединений общей Формулы I. Например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H). Протий представляет собой преобладающий изотоп водорода, находящийся в природе. Обогащение дейтерием может придавать определенные терапевтические преимущества, такие как увеличение половинного времени жизни in vivo или уменьшение требований к дозировке, или может давать соединение, пригодное для использования в качестве стандарта для характеризации биологических образцов. Изотопно-обогащенные соединения в пределах общей Формулы I могут быть получены без излишних экспериментов с помощью обычных методик, хорошо известных специалистам в данной области, или с помощью способов, аналогичных тем, которые описаны на Схемах и в Примерах в настоящем документе, с использованием соответствующих изотопно-обогащенных реагентов и/или промежуточных соединений.
Таутомеры соединений, определенных в Формуле I, также включаются в рамки настоящего изобретения. Например, соединения, содержащие карбонильные группы -CH2C(O)- (кето формы), могут подвергаться воздействию таутомерии с образованием гидроксильных групп -CH=C(OH)- (энольные формы). Как кето, так и энольные формы включаются в рамки настоящего изобретения.
Когда любое переменное (например, R3, и тому подобное) встречается больше одного раза в любом составляющем, его определение в каждом случае не зависит от любого другого случая. Также, сочетания заместителей и переменных являются допустимыми, только если такие сочетания дают в результате стабильные соединения. Линии, нарисованные в кольцевых системах от заместителей, представляют то, что указанная связь может присоединяться к любому из замещаемых кольцевых атомов. Если кольцевая система является бициклической, предполагается, что связь присоединяется к любому из соответствующих атомов на любом кольце бициклического остатка.
Понятно, что один или несколько атомов кремния (Si) могут включаться в соединения по настоящему изобретению вместо одного или нескольких атомов углерода с помощью специалиста в данной области, с получением соединений, которые являются химически стабильными и которые можно легко синтезировать с помощью методик, известных в данной области, из легкодоступных исходных материалов. Один из примеров представляет собой силинан. Углерод и кремний отличаются по их ковалентным радиусам, что приводит к различиям в длине химической связи и по стерическому расположению, при сравнении аналогичных связей с элементарным C и элементарным Si. Эти различия приводят к небольшим изменениям в размере и форме кремнийсодержащих соединений, по сравнению с углеродом. Специалист в данной области должен понять, что различия в размерах и форме может приводить к небольшим или большим изменениям в сильнодействии, растворимости, отсутствии активности вне мишени, в свойствах при упаковке, и так далее. (Diass, J. O. et al. Organometallics (2006) 5:1188-1198; Showell, G.A. et al. Bioorganic & Medicinal Chemistry Letters (2006) 16:2555-2558).
Понятно, что заместители и структуры замещения на соединениях по настоящему изобретению могут выбираться специалистом в данной области для получения соединений, которые являются химически стабильными и которые можно легко синтезировать с помощью методик, известных в данной области, а также способов, приведенных ниже, из легкодоступных исходных материалов. Если заместитель сам замещен несколькими группами, понятно, что это множество групп может находиться на одном и том же атоме углерода или на различных атомах углерода, постольку, поскольку в результате получается стабильная структура. Фраза “необязательно замещенный одним или несколькими заместителями” должна пониматься как обозначающая то, что рассматриваемая группа либо является незамещенной, либо может замещаться одним или несколькими заместителями.
Диатомит “Celite®” (Fluka) представляет собой диатомовую землю и может упоминаться как “целит”.
Как используется в настоящем документе, "алкил", как предполагается, включает как разветвленные, так и прямоцепные насыщенные алифатические углеводородные группы, имеющие от одного до десяти атомов углерода, если не указано иного. Например, C1-C10, как в “C1-C10 алкиле” как определено, включают группы, имеющие 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода в линейном или разветвленном расположении. Например, “C1-C10 алкил” конкретно включает метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил, и так далее.
Термин “галогеналкил" обозначает алкильный радикал, как определено выше, если не указано иного, который является замещенным одним-пятью, предпочтительно, одним-тремя атомами галогена. Репрезентативные примеры включают, но, не ограничиваясь этим, дифторметил, трифторметил, трифторэтил, перфторэтил, дихлорэтил, и тому подобное.
Термин “циклоалкил” означает моноциклическую или бициклическую насыщенную алифатическую углеводородную группу, имеющую указанное количество атомов углерода. Например, “циклоалкил” включает циклопропил, метилциклопропил, 2,2-диметилциклобутил, 2-этилциклопентил, циклогексил, и так далее. Термин “циклоалкил” включает мостиковые системы, включая бицикло[1,1,1]пентанил, бицикло[3,1,0]гексанил, азабицикло[3,2,1]гептанил и бицикло[3,2,1]октанил.
Если количество атомов углерода не указано, термин “алкенил” относится к неароматическому углеводородному радикалу, прямому или разветвленному, содержащему от 2 до 10 атомов углерода и, по меньшей мере, 1 двойную связь одного атома углерода с другим атомом углерода. Предпочтительно 1 двойная связь одного атома углерода с другим атомом углерода присутствует, и могут присутствовать до 4 неароматическим двойных связей углерод-углерод. Таким образом, “C2-C6 алкенил” означает алкенильный радикал, имеющий от 2 до 6 атомов углерода. Алкенильные группы включают этенил, пропенил, бутенил и циклогексенил. Как описано выше по отношению к алкилу, прямая или разветвленная часть алкенильной группы может содержать двойные связи, и она может быть замещенной, если указана замещенная алкенильная группа.
Если количество атомов углерода не указано, термин “алкинил” относится к неароматическому углеводородному радикалу, прямому или разветвленному, содержащему от 2 до 10 атомов углерода и, по меньшей мере, 1 тройную связь одного атома углерода с другим атомом углерода. Предпочтительно, присутствует 1 тройная связь одного атома углерода с другим атомом углерода, и может присутствовать до 4 неароматическим тройных связей углерод-углерод. Таким образом, “C2-C6 алкинил” означает алкинильный радикал, имеющий от 2 до 6 атомов углерода. Алкинильные группы включают этинил, пропинил, бутинил и циклогексинил. Как описано выше по отношению к алкилу, прямая или разветвленная часть алкинильной группы может содержать тройные связи, и она может быть замещенной, если указана замещенная алкинильная группа.
Как используется в настоящем документе, “арил”, как предполагается, означает любое стабильное моноциклическое или бициклическое углеродное кольцо, содержащее до 12 атомов в каждом кольце, где, по меньшей мере, одно кольцо является ароматическим. Примеры таких арильных элементов включают фенил, нафтил, тетрагидронафтил, инданил, бифенил, пенантрил, антрил или аценафтил.
Термин “гетероарил”, как используется в настоящем документе, представляет стабильное моноциклическое, бициклическое или трициклическое кольцо, содержащее до 10 атомов в каждом кольце, где, по меньшей мере, одно кольцо является ароматическим и содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N и S. Термин “гетероарил” включает бициклические кольца, где одно кольцо является ароматическим, а другое нет; в этих случаях, гетероатом (гетероатомы) может находиться либо в ароматическом, либо в неароматическом кольце, например, в индолиниле. Гетероарильные группы в рамках этого определения включают но, не ограничиваясь этим: бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазоил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тетразолил, тетразолопиридил, тиадиазолил, тиазолил, тиенил, триазолил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидроиндолил, дигидрохинолинил, метилендиоксибензол, бензотиазолил, бензотиенил, хинолинил, изохинолинил, оксазолил и тетрагидрохинолин. Если гетероарил содержит атомы азота, понятно, что соответствующие их N-оксиды также охватываются этим определением.
Термин "гетероцикл" или “гетероциклил”, как используется в настоящем документе, как предполагается, обозначает 5- - 10-членное неароматическое кольцо, если не указано иного, содержащее от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, N, S, SO или SO2, и включает бициклические группы. Следовательно “гетероциклил” включает, но, не ограничиваясь этим, следующее: пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, диоксидоморфолинил, иминооксидотиоморфолинил, тетрагидропиранил, дигидропиперидинил, тетрагидротиофенил, и тому подобное. Если гетероцикл содержит азот, понятно, что его соответствующие N-оксиды также охватываются этим определением.
Как очевидно специалистам в данной области, “гало” или “галоген”, как используется в настоящем документе, как предполагается, включает хлор, фтор, бром и йод.
В определенных случаях, заместители могут определяться с некоторым диапазоном количества атомов углерода, который включает ноль, например, (C0-C6)алкиленарил. Если арил берется как фенил, это определение должно включать сам фенил, а также -CH2Ph, -CH2CH2Ph, CH(CH3) CH2CH(CH3)Ph, и так далее.
Настоящее изобретение также включает N-оксидные производные и защищенные производные соединений формулы I. Например, когда соединения формулы I содержат окисляемый атом азота, этот атом азота может преобразоваться в N-оксид с помощью способов, хорошо известных в данной области. Также, когда соединения формулы I содержат такие группы, как гидрокси, карбокси, тиол или любая группа, содержащая атом (атомы) азота, эти группы могут защищаться с помощью соответствующих защитных групп. Полный список пригодных для использования защитных групп можно найти в T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, Inc. 1981, описание которой включается в настоящий документ в качестве ссылки во всей своей полноте. Защищенные производные соединений формулы I могут быть получены с помощью способов, хорошо известных в данной области.
Как используется в настоящем документе, "фармацевтически приемлемые соли" относятся к производным, где исходные соединение модифицируются посредством получения его кислотных или основных солей. Соли в твердой форме могут существовать в виде нескольких кристаллических структур, и они могут также находиться в форме гидратов. Примеры фармацевтически приемлемых солей включают, но, не ограничиваясь этим, соли минеральных или органических кислот с основными остатками, такими как амины; щелочные или органические соли с кислотными остатками, такими как карбоновые кислоты; и тому подобное. Фармацевтически приемлемые соли включают обычные нетоксичные соли или соли четвертичного аммония и исходного соединения, образующиеся, например, из нетоксичных неорганических или органических кислот. Например, такие обычные нетоксичные соли включают соли, полученные из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная, и тому подобное; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая, и тому подобное. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, соли марганца (III), марганца (II), калия, натрия, цинка, и тому подобное.
Когда соединение по настоящему изобретению является основным, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту, и тому подобное. В одном из аспектов настоящего изобретения соли представляют собой соли лимонной, бромистоводородной, хлористоводородной, малеиновой, фосфорной, серной, фумаровой и винной кислоты. Будет понятно, что, как используется в настоящем документе, упоминание соединений формулы I, как предполагается, также включают фармацевтически приемлемые соли.
Для целей настоящего описания, следующие далее сокращения имеют указанные обозначения:
Соединения, описанные в настоящей заявке, демонстрируют активность в следующих анализах. В дополнение к этому, соединения, описанные в настоящей заявке, имеют улучшенный фармакологический профиль по отношению к ранее описанным соединениям.
АНАЛИЗ С КАТЕПСИНОМ K (HRBCAT K)
Приготавливают последовательные разбавления (1/3) исследуемых соединений от 500 мкМ вплоть до 0,025 мкМ в диметилсульфоксиде (DMSO). Затем 0,5 мкл DMSO от каждого разбавления добавляют к 18,75 мкл буфера для анализа (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% DMSO), содержащего катепсин K человека (0,4 нМ) в растворе в буфере для анализов. Растворы для анализов перемешивают в течение 5-10 секунд на планшетном шейкере и инкубируют в течение 15 минут при комнатной температуре. К растворам для анализов добавляют Z-Leu-Arg-AMC (8 мкМ) в 6,25 мкл буфера для анализов. После гидролиза кумариновой уходящей группы (AMC) следует спектрофлуорометрия (Exλ (длина волны возбуждения)=360 нм; Emλ (длина волны испускания)=460 нм) в течение 30 минут. Процент ингибирования вычисляют посредством подгонки экспериментальных значений с помощью стандартной математической модели для кривой доза - реакция.
Анализ с катепсином L
Приготавливают последовательные разбавления (1/3) исследуемых соединений от 500 мкМ до 0,025 мкМ в диметилсульфоксиде (DMSO). Затем 0,5 мкл DMSO от каждого разбавления добавляют к 18,75 мкл буфера для анализов (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% DMSO), содержащего катепсин L человека (0,027 нМ) в растворе в буфере для анализов. Растворы для анализов перемешивают в течение 5-10 секунд на планшетном шейкере и инкубируют в течение 15 минут при комнатной температуре. К растворам для анализов добавляют Z-Leu-Arg-AMC (8 мкМ) в 6,25 мкл буфера для анализов. После гидролиза кумариновой уходящей группы (AMC) следует спектрофлуорометрия (Exλ=360 нм; Emλ=460 нм) в течение 30 минут. Процент ингибирования вычисляют посредством подгонки экспериментальных значений с помощью стандартной математической модели для кривой доза - реакция.
Анализ с катепсином B
Приготавливают последовательные разбавления (1/3) исследуемых соединений от 500 мкМ до 0,025 мкМ в диметилсульфоксиде (DMSO). Затем 0,5 мкл DMSO от каждого разбавления добавляют к 18,75 мкл буфера для анализов (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% DMSO), содержащего катепсин B человека (0,1 нМ) в растворе в буфере для анализов. Растворы для анализов перемешивают в течение 5-10 секунд на планшетном шейкере и инкубируют в течение 15 минут при комнатной температуре. К растворам для анализов добавляют Boc-Leu-Lys-Arg-AMC (8 мкМ) в 6,25 мкл буфера для анализов. После гидролиза кумариновой уходящей группы (AMC) следует спектрофлуорометрия (Exλ=360 нм; Emλ=460 нм) в течение 30 минут. Процент ингибирования вычисляют посредством подгонки экспериментальных значений с помощью стандартной математической модели для кривой доза - реакция.
Анализ с катепсином S
Приготавливают последовательные разбавления (1/3) исследуемых соединений от 500 мкМ до 0,025 мкМ в диметилсульфоксиде (DMSO). Затем 0,5 мкл DMSO от каждого разбавления добавляют к 18,75 мкл буфера для анализов (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% DMSO), содержащего катепсин S человека (0,66 нМ) в растворе в буфере для анализов. Растворы для анализов перемешивают в течение 5-10 секунд на планшетном шейкере и инкубируют в течение 15 минут при комнатной температуре. К растворам для анализов добавляют Z-Val-Val-Arg-AMC (8 мкМ) в 6,25 мкл буфера для анализов. После гидролиза кумариновой уходящей группы (AMC) следует спектрофлуорометрия (Exλ=360 нм; Emλ=460 нм) в течение 20 минут. Процент ингибирования вычисляют посредством подгонки экспериментальных значений с помощью стандартной математической модели для кривой доза - реакция.
Анализ с катепсином F (hCat F)
Приготавливают последовательные разбавления (1/3) исследуемых соединений от 500 мкМ до 0,025 мкМ в диметилсульфоксиде (DMSO). Затем 0,5 мкл DMSO от каждого разбавления добавляют к 18,75 мкл буфера для анализов (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% DMSO), содержащего катепсин F человека (10 нМ) в растворе в буфере для анализов. Растворы для анализов перемешивают в течение 5-10 секунд на планшетном шейкере и инкубируют в течение 15 минут при комнатной температуре. К растворам для анализов добавляют Z-Phe-Arg-AMC (8 мкМ) в 6,25 мкл буфера для анализов. После гидролиза кумариновой уходящей группы (AMC) следует спектрофлуорометрия (Exλ=360 нм; Emλ=460 нм) в течение 20 минут. Процент ингибирования вычисляют посредством подгонки экспериментальных значений с помощью стандартной математической модели для кривой доза - реакция.
Фармакокинетика на крысах
Фармакокинетика Per Os (PO) на крысах
ПРОЦЕДУРА:
Животных содержат в клетках, кормят и за ними ухаживают в соответствии с Guidelines of Canadian Council on Animal Care.
Самцы крыс Sprague Dawley (250-400 г) выдерживают без кормления в течение ночи перед каждым исследованием уровня PO в крови.
Крыс помещают в клетку для иммобилизации в одно и то же время, и коробку жестко закрепляют. Нулевой образец крови получают посредством отрезания малого (1 мм или меньше) кусочка кончика хвоста. Затем хвост поглаживают твердым, но осторожным движением сверху донизу для сцеживания крови. Приблизительно 0,5 мл крови собирают в гепаринизированую пробирку Vacutainer.
Соединения приготавливают по потребности, при стандартном объеме дозирования 10 мл/кг, и вводят перорально посредством введения зонда для принудительного кормления калибром 16 длиной 3 дюйма в пищевод.
Последующие сборы крови осуществляют таким же образом в качестве нулевого образца крови, за исключением того, что нет необходимости подрезать хвост снова. Хвост очищают с помощью куска марли и поглаживают/сцеживают, как описано выше, в соответствующим образом меченные пробирки.
Непосредственно после отбора образца, кровь центрифугируют, разделяют, плазму помещают в четко помеченные флаконы и хранят в морозилке до анализа.
Типичные моменты времени для определения уровней в крови крыс после дозирования PO представляют собой:
0, 15 мин, 30 мин, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 24 часа
После момента времени отбора крови 4 час, крыс кормят ad libitum. Вода подается все время в течение исследования.
Носители:
Следующие носители (при соответствующих объемах доз) можно использовать при определениях уровня PO в крови крыс:
Соединения для определения уровней PO в крови могут находиться в форме суспензии. Для улучшения гомогенности, суспензия может помещаться в устройство для обработки ультразвуком приблизительно на 5 минут.
Для анализа, аликвоты разбавляют 1,2-1,5 объемами ацетонитрила, необязательно содержащего внутренний стандарт, и центрифугируют для удаления преципитата белка. Супернатант непосредственно инжектируют в колонку для ВЭЖХ с C-18 с детектированием с помощью масс-спектрометрии (MS) или поглощения ультрафиолетового излучения (УФ) или флуоресценции (Fluo). Количественное определение осуществляют по отношению к стандартной кривой, полученной с использованием чистых образцов крови, в которые импульсно вводят известные количества лекарственного средства в ацетонитриле, необязательно содержащем внутренний стандарт. Дополнительный ацетонитрил, необязательно содержащий внутренний стандарт, добавляют до количества 1,2-1,5 объемов начального количества крови, чтобы соответствовать тому, что делается в случае образцов. Биологическую доступность (F) оценивают посредством сравнения площади под кривой (AUC) для внутривенного введения по сравнению с пероральным
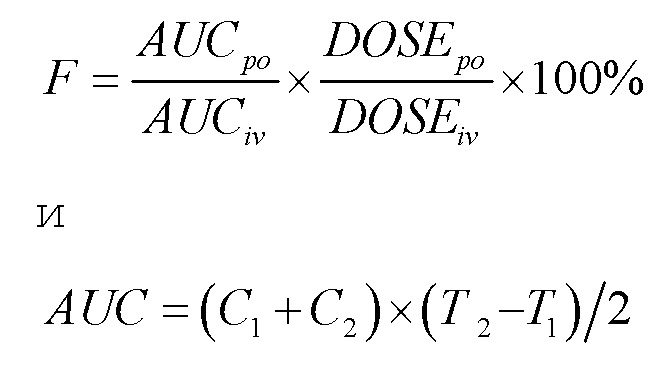
где C представляет собой концентрацию, измеренную с помощью MS или УФ или Fluo в данный момент T
Внутривенная фармакокинетика для крыс
ПРОЦЕДУРА:
Животных помещают в клетки, кормят, и за ними ухаживают в соответствии с Guidelines of Canadian Council on Animal Care.
В этих исследованиях используют некормленых самцов крыс Sprague Dawley (325-375 г).
Соединение приготавливают по потребности, при стандартном объеме дозирования 1 мл/кг.
Дозирование здоровых крыс для внутривенного введения осуществляют через яремную вену с использованием иглы 25 калибра. Это составляет нулевой момент времени.
Осуществляют 5-минутное кровопускание посредством отрезания малого (1-2 мм) кусочка кончика хвоста. Затем хвост поглаживают твердым, но осторожным движением сверху донизу для сцеживания крови из хвоста. Приблизительно 0,5 мл крови собирают в гепаринизированный флакон для сбора. Последующие кровопускания осуществляют таким же образом, за исключением того, что нет необходимости подрезать хвост снова. Хвост очищают с помощью куска марли, и собирают кровь, как описано выше, в соответствующие меченые пробирки.
Типичные моменты времени для определения уровней в крови крыс после внутривенного дозирования представляют собой либо:
0, 5 мин, 15 мин, 30 мин, 1 час, 2 часа, 4 часа, 6 часов
либо
0, 5 мин, 30 мин, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 24 часа
Носители:
При определении уровней в крови крыс при внутривенном дозировании можно использовать следующие носители:
Вместе с декстрозой можно добавлять бикарбонат натрия, если раствор мутный.
Для анализа, аликвоты разбавляют 1,2-1,5 объемами ацетонитрила, необязательно содержащего внутренний стандарт, и центрифугируют для удаления преципитата белка. Супернатант непосредственно инжектируют в колонку для ВЭЖХ с C-18 с детектированием с помощью масс-спектрометрии (MS) или поглощения ультрафиолетового излучения (УФ) или флуоресценции (Fluo). Количественное определение осуществляют по отношению к стандартной кривой, полученной с использованием чистых образцов крови, в которые импульсно вводят известные количества лекарственного средства в ацетонитриле, необязательно содержащем внутренний стандарт. Дополнительный ацетонитрил, необязательно содержащий внутренний стандарт, добавляют до количества 1,2-1,5 объема начального количества крови, чтобы соответствовать тому, что делается в случае образцов. Биологическую доступность (F) оценивают посредством сравнения площади под кривой (AUC) для внутривенного введения по сравнению с пероральным
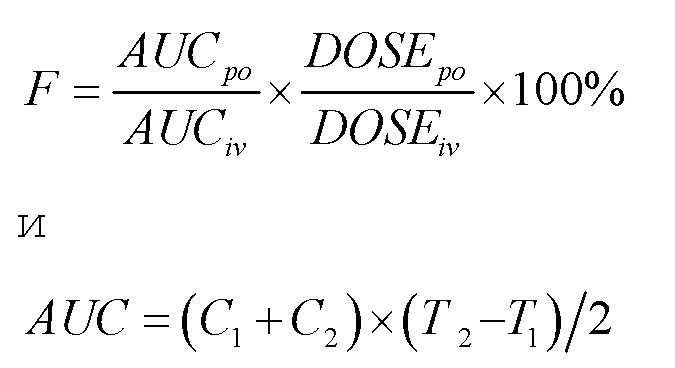
где C представляет собой концентрацию, измеренную с помощью MS или УФ или Fluo в данный момент T
Инкубирование гепатоцитов
Для инкубирования гепатоцитов крыс, 1×106 клеток, разбавленных в 0,5 мл буфера Кребса-Хензеляйта, приготавливают сначала при 37°C в течение 20 мин в атмосфере 95%:5% O2:CO2 (BOC gases: Montreal, Canada) в 48-луночном планшете, и 5 мкл 10 мМ раствора соединения, растворенного в ацетонитриле, добавляют в каждую лунку до конечной концентрации 50 мкМ. После 2 часов инкубирования при 37ºC в атмосфере 95%:5% O2:CO2, добавляют один объем ацетонитрила в каждую лунку. Погашенное инкубирование, в которое импульсно вводят исходное соединение, и пустой препарат также приготавливают в качестве контроля. После переноса, образцы центрифугируют в течение 10 мин при 14000 об/мин с использованием центрифуги Eppendorf 5415C (Hamburg, Germany), и супернатант используют для анализа LC/УФ/MS.
СХЕМА 1
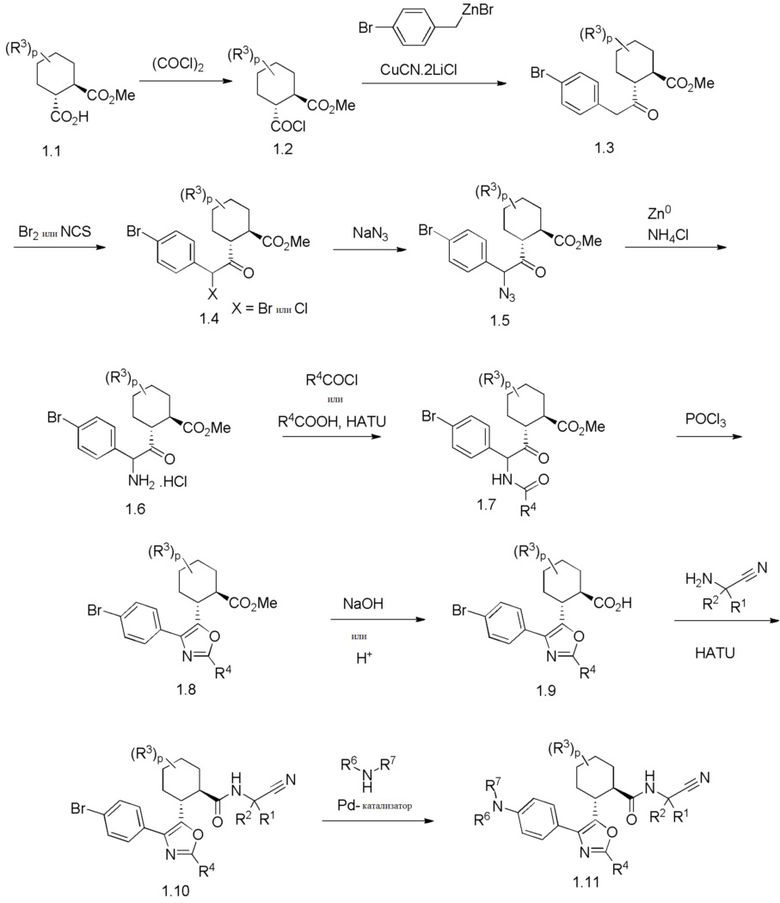
Соединения по настоящему изобретению могут быть получены в соответствии со Схемой 1. Таким образом, соединение 1.1 может быть преобразовано в хлорангидрид 1.2 посредством обработки оксалилхлоридом. Образовавшийся хлорангидрид 1.2 может обрабатываться 4-бромбензилцинком бромидом в присутствии комплекса цианида меди (I) и ди(лития хлорида), что приводит к получению соединения 1.3. Кетон 1.3 может быть преобразован в соединение 1.4 либо посредством галогенирования бромом, либо с помощью NCS. После обработки соединения 1.4 азидом натрия, соединение 1.5 может быть восстановлено до соответствующего амина посредством взаимодействия с Zn и хлоридом аммония. Гидрохлоридная соль амина 1.6 может быть преобразована в амид 1.7 посредством реакции с хлорангидридом или соответствующей кислотой в присутствии реагента для амидного связывания, такого как, но, не ограничиваясь этим, HATU. Замещенные оксазолы 1.8 могут формироваться посредством внутримолекулярной конденсации 1.7 в присутствии дегидриратирующего реагента, такого как, но, не ограничиваясь этим, POCl3. Гидролиз сложного эфира 1.8 с помощью водного раствора либо основания, либо кислоты приводит к образованию соединений 1.9, которые могут связываться с соответствующими аминами в присутствии реагентов для амидного связывания с получением желаемого амида 1.10. Катализируемое палладием аминирование соединения 1.10 соответствующими аминами дает соединение 1.11 по настоящему изобретению.
СХЕМА 2
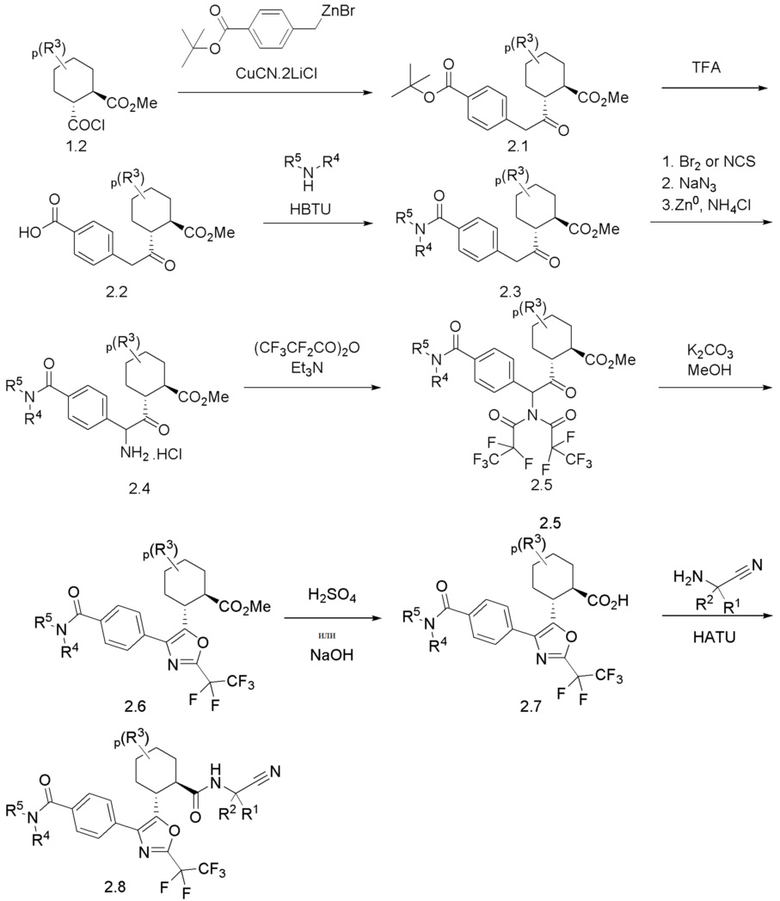
Соединения по настоящему изобретению могут также быть получены в соответствии со Схемой 2. Взаимодействие хлорангидрида 1.2 с (4-(трет-бутоксикарбонил)бензил)цинком (II) бромидом в присутствии комплекса цианида меди (I) и ди(лития хлорида) приводит к получению соединения 2.1. Гидролиз сложного трет-бутилового эфира с помощью TFA дает производное 2.2 бензойной кислоты, которое преобразуется в амид 2.3 посредством взаимодействия с соответствующими аминами в присутствии реагента для амидного связывания? такого как, но, не ограничиваясь этим, HBTU. Амид 2.3 преобразуется в соединение 2.4 либо посредством галогенирования бензильного положения с помощью брома, либо с помощью NCS с последующим замещением азидом натрия, а затем восстановлением азида до соответствующего амина с помощью Zn и хлорида аммония. Амин 2.4 преобразуется в бисамид 2.5 посредством взаимодействия с пентафторпропановым ангидридом. Частичное снятие защиты посредством карбоната калия в метаноле приводит к получению замещенного оксазола 2.6. Гидролиз функциональной группы сложного эфира с помощью водного раствора либо основания, либо кислоты приводит к получению кислоты 2.7, которая может связываться с аминами в присутствии реагента для амидного связывания, такого как, но, не ограничиваясь этим, HATU, с получением желаемого соединения 2.8 по настоящему изобретению.
СХЕМА 3
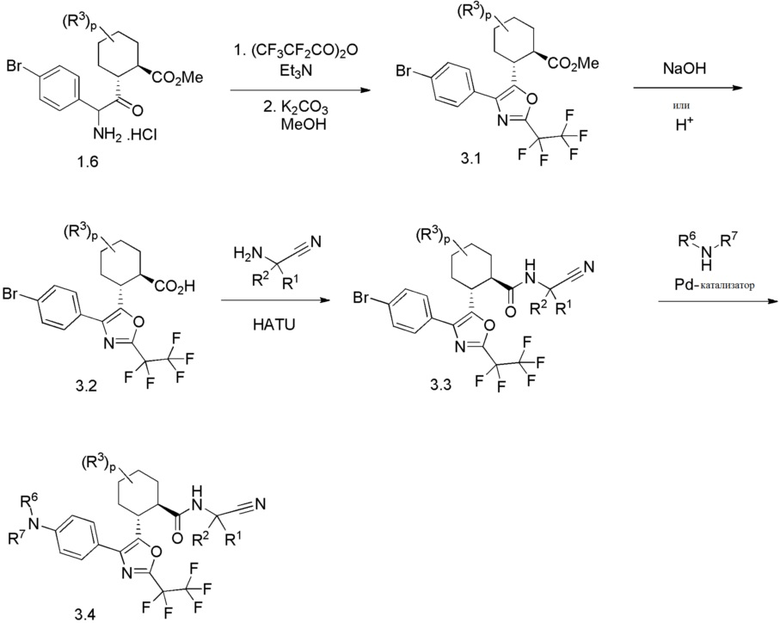
Соединения по настоящему изобретению могут также быть получены в соответствии со Схемой 3. Гидрохлоридная соль амина 1.6 преобразуется в биспентафторпропиламид посредством взаимодействия с пентафторпропановым ангидридом с последующим частичным снятием защиты с помощью карбоната калия в метаноле с получением замещенного оксазола 3.1. Гидролиз функциональной группы сложного эфира с помощью водного раствора либо основания, либо кислоты приводит к получению соединения 3.2, которое может связываться с аминами в присутствии реагента для амидного связывания, такого как, но, не ограничиваясь этим, HATU, с получением амида 3.3. Катализируемое палладием аминирование бромида 3.3 с помощью соответствующих аминов дает соединение 3.4 по настоящему изобретению.
СХЕМА 4
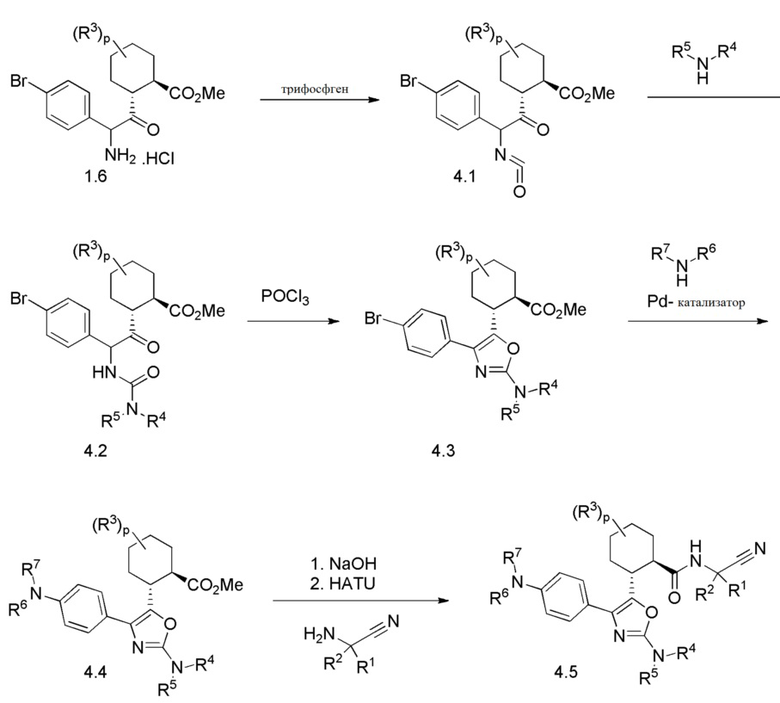
Соединения по настоящему изобретению могут также быть получены в соответствии со Схемой 4. Амин 1.6 преобразуется в изоцианат 4.1 посредством взаимодействия с трифосгеном, а затем обрабатывается с помощью соответствующих аминов с получением замещенной мочевины 4.2, внутримолекулярная конденсация 4.2 в присутствии дегидрирующего реагента такого как, но, не ограничиваясь этим, POCl3, дает замещенный оксазол 4.3. Катализируемое палладием аминирование бромида 4.3 с помощью соответствующих аминов приводит к получению 4.4. Гидролиз сложного метилового эфира с помощью водного раствора либо основания, либо кислоты с последующим связыванием с аминами в присутствии реагента для амидного связывания, такого как, но, не ограничиваясь этим, HATU, дает соединение 4.5 по настоящему изобретению.
СХЕМА 5
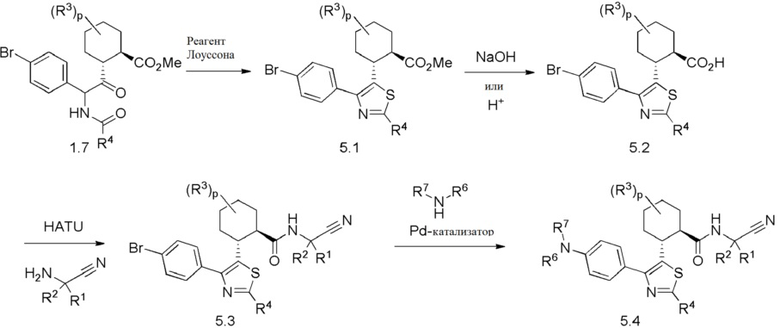
Соединения по настоящему изобретению могут также быть получены в соответствии со Схемой 5. Соединение 1.7 может обрабатываться реагентом Лоуссона с получением желаемого замещенного тиазола 5.1. Гидролиз сложного метилового эфира 5.1 с помощью водного раствора либо основания, либо кислоты приводит к получению карбоновой кислоты 5.2, которая может связываться с соответствующими аминами в присутствии реагента для амидного связывания, такого как, но, не ограничиваясь этим, HATU. Катализируемое палладием аминирование бромида 5.3 с помощью соответствующих аминов дает соединение 5.4 по настоящему изобретению.
Следующие далее примеры описывают синтез выбранных соединений по настоящему изобретению:
СИНТЕЗ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1
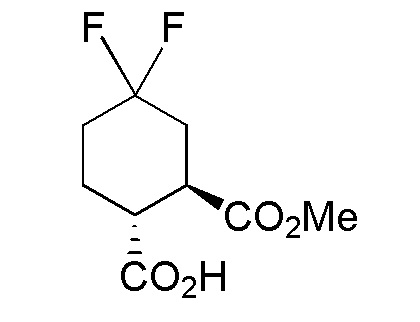
(1 R ,2 R )-Метил-2-(хлоркарбонил)циклогексанкарбоксилат
Стадия A: (1 R ,2 R )-2-(метоксикарбонил)циклогексанкарбоновая кислота
Это соединение может быть получено из (1R,2S)-диметилциклогекс-4-ен-1,2-дикарбоксилата, следуя процедуре, сходной с той, которая описана в J. Am. Chem. SOC. 1995, 117, 10905-10913 и в HELVETICA CHIMICA ACTA-Vol. 70 (1987), p.142.
Стадия B: (1 R ,2 R )-Метил-2-(хлоркарбонил)-циклогексанкарбоксилат
К раствору (1R,2R)-2-(метоксикарбонил)-циклогексанкарбоновой кислоты (1,99 г, 10,69 ммоль) в толуоле (5 мл) добавляют оксалилхлорид (1,424 г, 11,22 ммоль) и перемешивают при rt в течение 18 часов. Барботирование газообразного азота через реакционную смесь в течение 20 мин, а затем концентрирование дает бесцветную жидкость, которую используют без дополнительной очистки.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 2
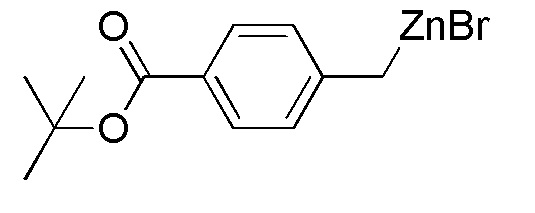
(4 -(трет -Бутоксикарбонил)бензил)цинк(II) бромид
Это соединение приготавливают, следуя процедуре, сходной с той, которая описана в Org. Lett., 2008, 6, 1107.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 3
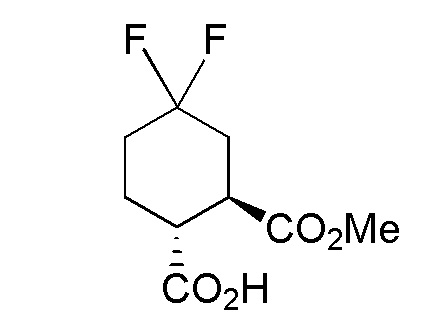
(1R,2R)-4,4-дифтор-2-(метоксикарбонил)циклогексанкарбоновая кислота
Стадия A: (1R,2R)-диметил-4-оксоциклогексан-1,2-дикарбоксилат
Рацемический диметил-4-оксоциклогексан-1,2-дикарбоксилат (660 г) загружают в 11 л 50 мМ фосфатного буфера, pH=6,8. К раствору добавляют эстеразу K310-903 (25 мл), и полученную в результате смесь перемешивают при 30°C в течение ночи. SFC показывает, что реакция завершается. Добавляют 1 н NaOH до получения pH=7, и раствор экстрагируют MTBE (4×8 л). Объединенную органическую фракцию промывают насыщенным раствором соли, сушат (Na2SO4) и фильтруют. Удаление растворителя с помощью выпаривания дает 225 г (выход 34%, энантиомерный избыток 98%) (1R,2R)-диметил-4-оксоциклогексан-1,2-дикарбоксилата в виде масла.
Стадия B: (1R,2R)-диметил-4,4-дифторциклогексан-1,2-дикарбоксилат
К раствору Et3N.3HF (224 г, 1,38 моль) в DCE (2,8 л) добавляют DAST.BF3 (630 г, 2,74 моль). Полученную в результате суспензию перемешивают в течение 10 мин, а затем добавляют (1R,2R)-диметил-4-оксоциклогексан-1,2-дикарбоксилат (280 г, 1,31 моль). Смесь нагревают при 55°C в течение ночи. Реакционную смесь выливают в насыщенный водный раствор NaHCO3 (5,6 л), а затем экстрагируют DCM (2 л ×3). Органический слой сушат (Na2SO4), концентрируют и очищают с помощью флэш-хроматографии (простой петролейный эфир/EtOAc=15:1) с получением 260 г бледно-желтого масла.
Желтое масло растворяют в 4 л DCM. Добавляют 3 л 5% водного раствора KMnO4. Смесь перемешивают при 23°C в течение ночи. Органический слой отделяют, промывают 3 л 5% KMnO4 в течение еще 3 часов. Органический слой отделяют, промывают насыщенным раствором соли, сушат (Na2SO4) и концентрируют досуха с получением (1R,2R)-диметил 4,4-дифторциклогексан-1,2-дикарбоксилата в виде белого твердого продукта (230 г, 74,5%). 1H ЯМР 400 МГц (CDCl3): δ 8,19-8,22 (м, 2H), 7,83 (д, J=8,1 Гц, 1H), 7,65-7,69 (м, 1H), 7,52-7,55 (м, 2H), 7,37 (с, 1H), 7,22 (дд, J=8,1 Гц, J=2,0 Гц, 1H), 3,17-3,20 (м, 2H), 2,73-2,76 (м, 2H).
Стадия C: (1R,2R)-4,4-дифтор-2-(метоксикарбонил)-циклогексанкарбоновая кислота
Фермент Cal B (26,5 мл) растворяют в 0,1M фосфатном буфере при pH 7,0 (6,8 л). К реакционной смеси добавляют (1R,2R)-диметил 4,4-дифторциклогексан-1,2-дикарбоксилат (100 г) в DMSO (300 мл) и перемешивают ее при 25°C в течение 6 часов. Раствор промывают насыщенным раствором соли (1 л) и экстрагируют MTBE (3 л ×4). Объединенный органический слой промывают насыщенным раствором соли, сушат (Na2SO4), фильтруют и концентрируют досуха с получением (1R,2R)-4,4-дифтор-2-(метоксикарбонил)-циклогексанкарбоновой кислоты в виде белого твердого продукта (90 г, 94%). 1H ЯМР 400 МГц CDCl3 δ 3,65 (S, 3H), 2,93 (м, 1H), 2,68 (м, 1H), 2,39 (м, 1H), 2,12 (м, 2H), 1,74 (м, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 4
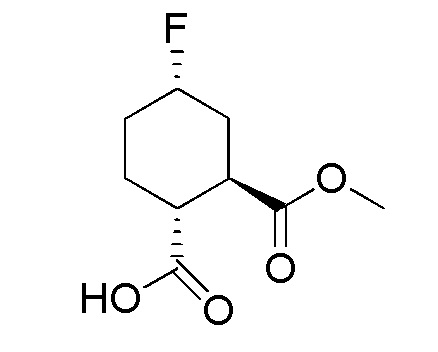
(1R,2R,4S)-4-фтор-2-(метоксикарбонил)циклогексанкарбоновая кислота
Стадия A: (7R,8R)-диметил-1,4-диоксаспиро[4,5]декан-7,8-дикарбоксилат
К раствору (1R,2R)-диметил-4-оксоциклогексан-1,2-дикарбоксилата (300 г, 1,4 моль) в толуоле (6 л) добавляют этиленгликоль (117 мл, 2,1 моль) и TsOH (5,29 г, 0,028 моль). Полученный в результате раствор нагревают с обратным холодильником в течение 6 часов. Реакционную смесь охлаждают до 23°C и промывают насыщенным раствором NaHCO3 (2×5 л) и один раз водой. Органическую фазу концентрируют с получением (7R,8R)-диметил 1,4-диоксаспиро[4,5]декан-7,8-дикарбоксилата (330 г, 91,2%) в виде желтого масла.
Стадия B: (7R,8R)-7-(метоксикарбонил)-1,4-диоксаспиро[4,5]декан-8-карбоновая кислота
В 50-л круглодонной колбе, снабженной подвесной мешалкой и связанной с pH Stat, растворяют фермент NZL-102 (60 г (5 г/л)) в 24 л фосфатного буфера при pH 7. Добавляют по каплям (7R,8R)-диметил 1,4-диоксаспиро[4,5]декан-7,8-дикарбоксилат (300 г, 1,16 моль) в 300 мл раствора DMSO и состаривают при 23°C в течение ночи. Реакционные смеси растворяют в смеси 3:1 EtOAc:IPA, экстрагируют, и органический слой промывают насыщенным раствором соли, сушат (Na2SO4), фильтруют и концентрируют с получением (7R,8R)-7-(метоксикарбонил)-1,4-диоксаспиро[4,5]декан-8-карбоновой кислоты (220 г, 77,5%) в виде желтого твердого продукта.
Стадия C: (7R,8R)-8-бензил-7-метил-1,4-диоксаспиро[4,5]декан-7,8-дикарбоксилат
К раствору (7R,8R)-7-(метоксикарбонил)-1,4-диоксаспиро[4,5]декан-8-карбоновой кислоты (2,9 г, 11,87 ммоль) в DMF (20 мл) добавляют K2CO3 (2,461 г, 17,81 ммоль), а затем (бромметил)бензол (3,05 г, 17,81 ммоль) и KI (394 мг, 2,375 ммоль). Полученную в результате суспензию перемешивают при RT в течение ночи. Реакционную смесь экстрагируют EtOAc и очищают с помощью флэш-хроматографии на силикагеле с получением 2,95 г (74%) (7R,8R)-8-бензил-7-метил-1,4-диоксаспиро[4,5]декан-7,8-дикарбоксилата в виде масла.
Стадия D: (1R,2R)-1-бензил-2-метил-4-оксоциклогексан-1,2-дикарбоксилат
К раствору (7R,8R)-8-бензил-7-метил-1,4-диоксаспиро[4,5]декан-7,8-дикарбоксилата (2,95 г, 8,82 ммоль) в ацетоне (40 мл) добавляют 1 н HCl (40 мл), и полученную в результате смесь нагревают при 60°C в течение 1 часа. Реакционную смесь разбавляют смесью EtOAc/вода, органическую фазу сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают с помощью флэш-хроматографии на силикагеле с получением 2,56 г (100%) (1R,2R)-1-бензил-2-метил-4-оксоциклогексан-1,2-дикарбоксилата в виде прозрачного масла.
Стадия E: (1R,2R,4R)-1-бензил-2-метил-4-гидроксициклогексан-1,2-дикарбоксилат
Раствор (1R,2R)-1-бензил-2-метил-4-оксоциклогексан-1,2-дикарбоксилата (2,6 г, 8,96 ммоль) в THF (45 мл) охлаждают на бане со льдосоляной смесью (~ -5°C), и добавляют NaBH4 (0,678 г, 17,91 ммоль). Полученный в результате раствор перемешивают при 0°C в течение 2 часов. Реакционную смесь осторожно гасят водой и EtOAc. Органическую фазу сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают с помощью флэш-хроматографии на силикагеле с получением 2,05 г (78%) желаемого спирта (1R,2R,4R)-1-бензил-2-метил-4-гидроксициклогексан-1,2-дикарбоксилат.
Стадия F: (1R,2R,4S)-1-бензил-2-метил-4-фторциклогексан-1,2-дикарбоксилат
К раствору морфолинодифторсульфиния тетрафторбората (1,704 г, 7,01 ммоль) в DCM (4 мл) добавляют триэтиламин тригидрофторид (2,261 г, 14,03 ммоль). Полученную в результате суспензию перемешивают в течение 10 мин при RT и охлаждают до -78°C. Раствор (1R,2R,4R)-1-бензил-2-метил-4-гидроксициклогексан-1,2-дикарбоксилата (2,05 г, 7,01 ммоль) в DCM (10 мл) добавляют при поддержании внутренней температуры < -40°C. Смесь перемешивают при -40°C в течение ~30 мин. Реакцию гасят при низкой температуре с помощью воды. Выделенную органическую фазу промывают водой, насыщенным раствором соли, сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают с помощью флэш-хроматографии на силикагеле (колонка ISCO, 12 г силикагеля, 0-20% EtOAc/гептан) с получением 525 мг (25,4%) (1R,2R,4S)-1-бензил-2-метил-4-фторциклогексан-1,2-дикарбоксилата в виде бесцветного масла.
Стадия G: (1R,2R,4S)-4-фтор-2-(метоксикарбонил)-циклогексанкарбоновая кислота
Суспензию Pd/C (10%) (122 мг, 0,115 ммоль) в EtOAc (1 мл) добавляют к (1R,2R,4S)-1-бензил-2-метил-4-фторциклогексан-1,2-дикарбоксилату (675 мг, 2,293 ммоль). Полученную в результате суспензию перемешивают при давлении H2 1 атм из раздуваемой камеры в течение 5 часов. Реакционную смесь фильтруют и концентрируют с получением 770 мг (100%) желаемой (1R,2R,4S)-4-фтор-2-(метоксикарбонил)циклогексанкарбоновой кислоты в виде бесцветного масла.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 5
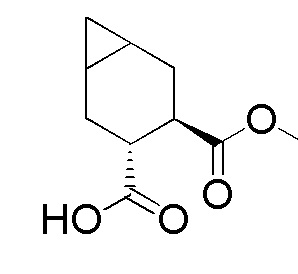
(3 R ,4 R )-4-(метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновая кислота
Стадия A: (1R,2S)-1-Бензил-2-метилциклогекс-4-ен-1,2-дикарбоксилат
В 50-мл круглодонную колбу помещают раствор (1R,6S)-6-(метоксикарбонил)циклогекс-3-енкарбоновой кислоты (2,40 г, 13,0 ммоль, 1,00 эквив.) в N,N-диметилформамиде (30 мл). К этому раствору добавляют карбонат калия (3,59 г, 26,0 ммоль, 2,00 эквив.). За этим следует добавление бензилбромида (3,34 г, 19,5 ммоль, 1,50 эквив.) по каплям при перемешивании при температуре окружающей среды. Реакционную смесь перемешивают в течение 16 часов при температуре окружающей среды. Полученную в результате смесь разбавляют насыщенным раствором соли (50 мл) и экстрагируют этилацетатом (3×50 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток используют для колоночной хроматографии на силикагеле с помощью смеси этилацетат/простой петролейный эфир (1:30-1:15). Это дает в результате 3,20 г (90%) (1R,2S)-1-бензил-2-метилциклогекс-4-ен-1,2-дикарбоксилата в виде бесцветного масла: 1H ЯМР (300 МГц, CDCl3) δ м.д. 7,43-7,30 (м, 5H), 5,69 (с, 2H), 5,14 (с, 2H), 3,58 (с, 3H), 3,14-3,05 (м, 2H), 2,64-2,53 (м, 2H), 2,42-2,34 (м, 2H); MS (ES, m/z): 275,1 (M+1).
Стадия B: (3R,4S)-3-Бензил-4-метилбицикло[4.1.0]гептан-3,4-дикарбоксилат
В 500-мл 3-горлую круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, помещают раствор диэтилцинка (1 M в гексане, 46,8 мл, 4,00 эквив.) в безводном дихлорметане (150 мл). За этим следует добавление трифторуксусной кислоты (5,34 г, 46,8 ммоль, 4,00 эквив.) по каплям при перемешивании при 0°C. К этому раствору добавляют по каплям дийодометан (12,5 г, 46,8 ммоль, 4,00 эквив.) при перемешивании при 0°C, и смесь перемешивают в течение 15 мин при 0°C. Затем добавляют по каплям раствор (1R,2S)-1-бензил-2-метилциклогекс-4-ен-1,2-дикарбоксилата (3,20 г, 11,7 ммоль, 1,00 эквив.) в безводном дихлорметане (50 мл) при перемешивании при 0°C. Реакционный раствор перемешивают и медленно нагревают до температуры окружающей среды в течение 16 часов. Реакционный раствор гасят посредством добавления воды (300 мл) и экстрагируют дихлорметаном (3×200 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток используют для колоночной хроматографии на силикагеле с помощью смеси этилацетат/простой петролейный эфир (1:20-1:10). Это дает в результате 3,10 г (92%) (3R,4S)-3-бензил-4-метилбицикло[4.1.0]гептан-3,4-дикарбоксилата в виде светло-желтого масла: MS (ES, m/z): 289,0 (M+1).
Стадия C: (3R,4S)-4-(Метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновая кислота
В 250-мл круглодонную колбу помещают раствор (3R,4S)-3-бензил-4-метилбицикло[4.1.0]гептан-3,4-дикарбоксилата (3,10 г, 10,8 ммоль, 1,00 эквив.) в метаноле (100 мл), с последующим добавлением палладия 10% на угле (0,300 г, смачивают примерно 55% воды). Реакционную смесь дегазируют водородом 3 раза и перемешивают в атмосфере водорода из раздуваемой камеры в течение 16 часов при температуре окружающей среды. Твердый продукт отфильтровывают. Фильтрат концентрируют в вакууме. Это дает в результате 2,10 г (89%) сырой (3R,4S)-4-(метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновой кислоты в виде бесцветного масла, которое используют непосредственно на следующей стадии без дополнительной очистки: MS (ES, m/z): 199,2 (M+1).
Стадия D: (3R,4R)-4-(Метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновая кислота
В 250-мл 3-горлую круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, помещают безводный метанол (130 мл). За этим следует добавление натрия (2,00 г, 87,0 ммоль, 8,21 эквив.) порциями при 0°C. Полученную в результате смесь перемешивают в течение 30 мин при температуре окружающей среды до исчезновения натрия. К этому раствору добавляют раствор (3R,4S)-4-(метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновой кислоты (2,10 г, 10,6 ммоль, 1,00 эквив.) в метаноле (20 мл). Полученный в результате раствор нагревают с обратным холодильником в течение 16 часов. Реакционную смесь охлаждают до 0°C. Значение pH доводят до 4 с помощью водного раствора хлористоводородной кислоты (1 M). Полученную в результате смесь экстрагируют этилацетатом (3×200 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток используют для колоночной хроматографии на силикагеле с помощью смеси этилацетат/простой петролейный эфир (1:5-1:3). Это дает в результате 1,40 г (60%) (3R,4R)-4-(метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновой кислоты в виде светло-желтого масла: 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 12,30-12,10 (ушир., 1H), 3,55 (с, 3H), 2,36-2,25 (м, 3H), 2,12-2,04 (м, 1H), 1,79-1,74 (м, 1H), 1,36-1,34 (м, 1H), 0,99-0,95 (м, 2H), 0,63-0,60 (м, 1H), 0,10-0,08 (м, 1H); MS (ES, m/z): 199,2 (M+1).
ПРИМЕР 1
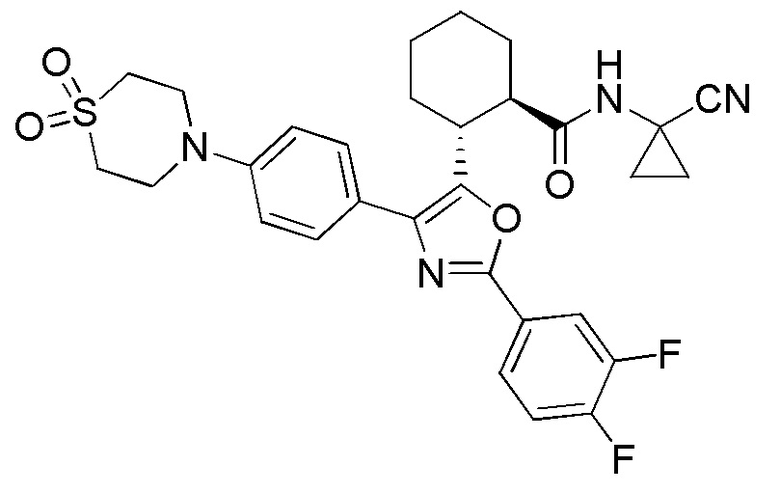
(1R,2R)- N -(1-Цианоциклопропил)-2-(2-(3,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоксамид (Соединение 1)
Стадия A: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)ацетил)циклогексанкарбоксилат
Приготовление 1 M CuCN.2LiCl: смесь CuCN (1,792 г, 20 ммоль) и LiCl (1,696 г, 40 ммоль) сушат в вакууме при 140°C в течение 5 часов. После охлаждения смеси до rt, добавляют 20 мл THF и перемешивают ее при rt в течение ночи с получением светло-желтого раствора.
К раствору CuCN.2LiCl (1 M, 12,82 мл) в THF при -25°C добавляют по каплям раствор 4-бромбензилцинка бромида (0,5 M, 25,6 мл) в THF и перемешивают в течение 15 мин. Раствор (1R,2R)-метил 2-(хлоркарбонил)циклогексанкарбоксилата (2,187 г, 10,69 ммоль) в THF (7 мл) добавляют по каплям, и полученную в результате смесь перемешивают в течение 5 мин при -25°C, в течение 15 мин при 0°C и 4 часа при rt. Реакционную смесь распределяют между EtOAc и смесью водный раствор NH4OH - водный насыщенный раствор NH4Cl (1:2 объем/объем, 200 мл). Водный слой экстрагируют EtOAc (3×). Объединенные органические растворы сушат над Na2SO4, фильтруют и концентрируют. Очистка с помощью флэш-хроматографии (40 г SiO2) с использованием линейного градиента от 3 до 15% EtOAc в гексане дает белый твердый продукт (2,45 г, 67%).
Стадия B: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-хлорацетил)циклогексанкарбоксилат
Раствор (1R,2R)-метил-2-(2-(4-бромфенил)ацетил)-циклогексанкарбоксилата (5,9 г, 17,39 ммоль) и N-хлорсукцинимида (2,79 г, 20,87 ммоль) в сухом DMF (25 мл) нагревают при 65°C в течение 1 часа, а затем охлаждают до комнатной температуры. Его распределяют между насыщенным водным раствором Na2S2O3 и EtOAc. Водный слой экстрагируют EtOAc (3×). Объединенные органические растворы промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Очистка с помощью флэш-хроматографии (120 г SiO2) с использованием линейного градиента 3-15% EtOAc в гексане дает белый твердый продукт (5,83 г, 90%).
Стадия C: (1 R ,2 R )-Метил-2-(2-азидо-2-(4-бромфенил)ацетил)-циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-(4-бромфенил)-2-хлорацетил)циклогексанкарбоксилата (4,794 г, 12,83 ммоль) в DMSO (30 мл) добавляют азид натрия (1,001 г, 15,4 ммоль). Полученную в результате смесь перемешивают при rt в течение 15 мин, разбавляют водой и экстрагируют EtOAc (3×). Объединенные органические растворы промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением светло-желтой вязкой жидкости, которую используют без дополнительной очистки.
Стадия D: (1 R ,2 R )-Метил-2-(2-амино-2-(4-бромфенил)ацетил)-циклогексанкарбоксилат гидрохлорид
К смеси (1R,2R)-метил-2-(2-азидо-2-(4-бромфенил)ацетил)-циклогексанкарбоксилата (4,794 г, 12,61 ммоль) и NH4Cl (1,686 г, 31,5 ммоль) в EtOH (29 мл) добавляют H2O (10 мл), а затем цинковую пыль (1,154 г, 17,65 ммоль). Смесь концентрируют после 1 час перемешивания при rt. Остаток перетирают с THF и фильтруют. Затем фильтрат концентрируют с получением светло-желтой пены, которую используют без дополнительной очистки.
Стадия E: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-(3,4-дифторбензамидо)ацетил)циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-амино-2-(4-бромфенил)ацетил)циклогексанкарбоксилата гидрохлорида (157 мг, 0,402 ммоль) в дихлорметане (3 мл) при 0°C добавляют 3,4-дифторбензоилхлорид (284 мг, 1,607 ммоль), а затем основание Хунига (312 мг, 2,411 ммоль). Раствор перемешивают при 0°C в течение 15 мин, а затем концентрируют. Остаток растворяют в DMSO и очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 40-95% MeCN в воде. 0,1% Модификатор TFA для MeCN и воды) с получением желаемого продукта в виде бесцветной вязкой жидкости (77 мг, 39%).
Стадия F: (1 R ,2 R )-Метил-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)циклогексанкарбоксилат
Раствор (1R,2R)-метил-2-(2-(4-бромфенил)-2-(3,4-дифторбензамидо)ацетил)циклогексанкарбоксилата (77 мг, 0,156 ммоль) в POCl3 (3 мл) нагревают при 100°C в течение 10 часов и при 80°C в течение 60 часов. Раствор концентрируют, и полученный в результате остаток растворяют в дихлорметане и охлаждают до 0°C. Насыщенный водный раствор NaHCO3 добавляют к раствору дихлорметана до тех пор, пока водный слой не станет основным. Слои разделяют. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4, фильтруют и концентрируют с получением белого твердого продукта, который используют без дополнительной очистки.
Стадия G: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-(3,4-дифторфенил)-оксазол-5-ил)циклогексанкарбоновая кислота
Суспензию (1R,2R)-метил-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)циклогексанкарбоксилата (74 мг, 0,155 ммоль) в MeOH-THF (1:2, 1,8 мл) обрабатывают 1 н водным раствором NaOH (0,621 мл) и перемешивают при 60°C в течение 4 часов. После охлаждения до rt, добавляют 0,621 мл 1 н водного раствора HCl. Полученный в результате раствор концентрируют до белого твердого продукта, который используют без дополнительной очистки.
Стадия H: (1 R ,2 R )-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)- N -(1-цианоциклопропил)-циклогексанкарбоксамид
К раствору (1R,2R)-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)циклогексанкарбоновой кислоты (72 мг, 0,156 ммоль) и HATU (89 мг, 0,389 ммоль) в DMF (1 мл) добавляют 1-цианоциклопропанаминийхлорид (46,2 мг, 0,389 ммоль) с последующим добавлением основания Хунига (101 мг, 0,779 ммоль) и перемешивают при 40°C в течение ночи. Полученный в результате раствор очищают непосредственно с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 40-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде белого твердого продукта (32 мг, 39%).
Стадия I: (1 R ,2 R )- N -(1-Цианоциклопропил)-2-(2-(3,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид (Соединение 1)
К смеси (1R,2R)-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)-N-(1-цианоциклопропил)-циклогексанкарбоксамида (20 мг, 0,038 ммоль), тиоморфолин 1,1-диоксида (15,4 мг, 0,114 ммоль), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил)]-палладия(II) (2,81 мг, 0,004 ммоль) и K3PO4.H2O (10,5 мг, 0,046 ммоль) во флаконе в инертной атмосфере добавляют THF (0,7 мл). Флакон герметизируют и нагревают при 100°C в течение 5 часов. Его охлаждают до комнатной температуры и концентрируют. Остаток извлекают в DMSO, фильтруют, и фильтрат подвергают воздействию очистки с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 30-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды), которая не дает чистого продукта. Затем этот продукт с примесями очищают с помощью флэш-хроматографии (4 г SiO2) с использованием линейного градиента 3-90% EtOAc в гексане, с получением белого твердого продукта (5 мг, 23%). MS [M+H]+ 581,1, 1H ЯМР (400 МГц, CDCl3) δ 7,88-7,78 (м, 2H), 7,66 (д, J=8,8 Гц, 2H), 7,31-7,27 (м, 1H), 6,99 (д, J=8,8 Гц, 2H), 5,67 (с, 1H), 3,95-3,92 (м, 4H), 3,35-3,24 (м, 1H), 3,14-3,12 (м, 4H), 2,48-2,44 (м, 1H), 2,25-1,64 (м, 6H), 1,48-1,26 (м, 4H), 0,79-0,62 (м, 2H).
ПРИМЕР 2
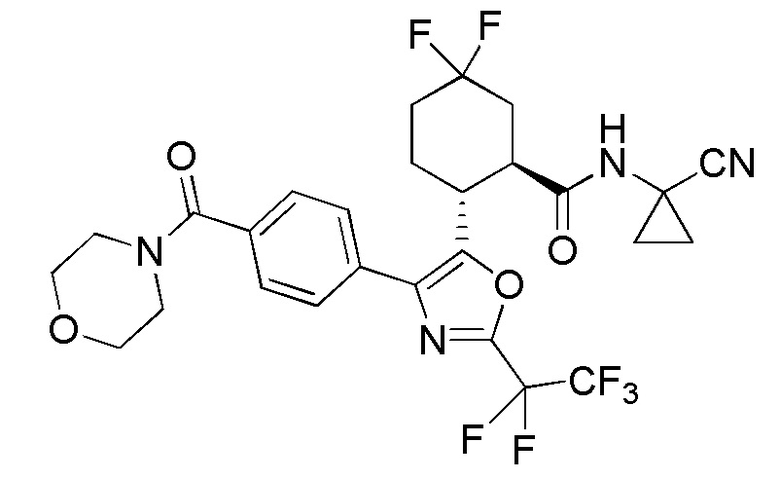
(1 R ,2 R )-N-(1-Цианоциклопропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)-циклогексанкарбоксамид (Соединение 2)
Стадия A: трет -Бутил-4-(2-((1 R ,2 R )-4,4-дифтор-2-(метоксикарбонил)циклогексил)-2-оксоэтил)бензоат
Это соединение приготавливают из (1R,2R)-4,4-дифтор-2-(метоксикарбонил)циклогексанкарбоновой кислоты (промежуточное соединение 3) с помощью ее хлорангидрида и (4-(трет-бутоксикарбонил)бензил)цинка(II) бромида (промежуточное соединение 2) с использованием процедуры подобной той, которая описана на Стадии A Примера 1.
Стадия B: 4-(2-((1 R ,2 R )-4,4-Дифтор-2-(метоксикарбонил)-циклогексил)-2-оксоэтил)бензойная кислота
К раствору трет-бутил-4-(2-((1R,2R)-4,4-дифтор-2-(метоксикарбонил)циклогексил)-2-оксоэтил)бензоата (845 мг, 2,132 ммоль) в дихлорметане (4 мл) добавляют TFA (5 мл). После перемешивания при 23°C в течение 30 мин, реакционную смесь разбавляют EtOAc, промывают водой (2×) и насыщенным раствором соли (1×), сушат над Na2SO4 и концентрируют с получением белого твердого продукта, который используют без дополнительной очистки.
Стадия C: (1 R ,2 R )-Метил-5,5-дифтор-2-(2-(4-(морфолин-4-карбонил)фенил)ацетил)циклогексанкарбоксилат
К раствору 4-(2-((1R,2R)-4,4-дифтор-2-(метоксикарбонил)циклогексил)-2-оксоэтил)бензойной кислоты (725 мг, 2,13 ммоль) и HBTU (1,212 г, 3,20 ммоль) в DMF (3 мл) добавляют морфолин (1,3 г, 14,91 ммоль). Полученную в результате смесь перемешивают при 23°C в течение 1,5 часа, а затем очищают непосредственно с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 5-85% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде бесцветной вязкой жидкости (434 мг, 50%).
Стадия D: (1 R ,2 R )-Метил-2-(2-бром-2-(4-(морфолин-4-карбонил)фенил)ацетил)-5,5-дифторциклогексанкарбоксилат
К раствору (1R,2R)-метил-5,5-дифтор-2-(2-(4-(морфолин-4-карбонил)фенил)ацетил)циклогексанкарбоксилата (434 мг, 1,06 ммоль) в дихлорметане (24 мл) добавляют раствор Br2 (169 мг, 1,06 ммоль) в дихлорметане (1 мл). Реакционную смесь перемешивают при 60°C в течение 6 часов, а затем охлаждают до комнатной температуры и распределяют между насыщенным водным раствором Na2S2O3 и дихлорметаном. Водный слой экстрагируют дихлорметаном (4×). Объединенные органические растворы сушат над Na2SO4 и концентрируют с получением белой пены, которую используют без дополнительной очистки.
Стадия E: (1 R ,2 R )-Метил-2-(2-азидо-2-(4-(морфолин-4-карбонил)фенил)ацетил)-5,5-дифторциклогексанкарбоксилат
Твердый азид натрия (48,5 мг, 0,746 ммоль) добавляют к раствору (1R,2R)-метил-2-(2-бром-2-(4-(морфолин-4-карбонил)фенил)ацетил)-5,5-дифторциклогексанкарбоксилата (331 мг, 0,678 ммоль) в MeCN (5 мл). После перемешивания реакционной смеси при 60°C в течение 30 мин, добавляют еще 50 мг азида натрия и продолжают перемешивание при 60°C в течение 1 часа. Его охлаждают до 23°C и распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc (2×). Объединенные органические растворы сушат над Na2SO4 и концентрируют с получением коричневого твердого продукта, который используют без дополнительной очистки.
Стадия F: (1 R ,2 R )-Метил-2-(2-амино-2-(4-(морфолин-4-карбонил)фенил)ацетил)-5,5-дифторциклогексанкарбоксилат гидрохлорид
Это соединение приготавливают из (1R,2R)-метил-2-(2-азидо-2-(4-(морфолин-4-карбонил)фенил)ацетил)-5,5-дифторциклогексанкарбоксилата с использованием процедуры сходной с той, которая описана для (1R,2R)-метил-2-(2-амино-2-(4-бромфенил)ацетил)циклогексанкарбоксилата гидрохлорида на Стадии D Примера 1.
Стадия G: (1 R ,2 R )-Метил-5,5-дифтор-2-(2-(4-(морфолин-4-карбонил)фенил)-2-(2,2,3,3,3-пентафтор- N -(2,2,3,3,3-пентафторпропаноил)пропанамидо)ацетил)циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-амино-2-(4-(морфолин-4-карбонил)фенил)ацетил)-5,5-дифторциклогексанкарбоксилата гидрохлорида (100 мг, 0,217 ммоль) и Et3N (88 мг, 0,868 ммоль) в дихлорметане (3 мл) добавляют по каплям раствор 2,2,3,3,3-пентафторпропанового ангидрида (135 мг, 0,434 ммоль) в дихлорметане (3 мл) и перемешивают в течение 30 мин. Затем реакционную смесь концентрируют и очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 5-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде белого твердого продукта (44 мг, 28%).
Стадия H: (1 R ,2 R )-Метил-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил-)циклогексанкарбоксилат
Раствор (1R,2R)-метил 5,5-дифтор-2-(2-(4-(морфолин-4-карбонил)фенил)-2-(2,2,3,3,3-пентафтор-N-(2,2,3,3,3-пентафторпропаноил)пропанамидо)ацетил)циклогексанкарбоксилата (44 мг, 0,061 ммоль) в MeOH (2 мл) обрабатывают твердым K2CO3 (212 мг, 1,535 ммоль) и перемешивают при 23°C в течение 1 часа. Затем реакционную смесь распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc (3×). Объединенные органические растворы сушат над Na2SO4 и концентрируют с получением коричневого твердого продукта, который используют без дополнительной очистки.
Стадия I: (1 R ,2 R )-5,5-Дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоновая кислота
Концентрированную серную кислоту (0,5 мл) добавляют по каплям к раствору (1R,2R)-метил-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксилата (34 мг, 0,062 ммоль) в смеси THF-вода (1:1, 1 мл) и перемешивают при 60°C в течение 2 часов. Затем реакционную смесь распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc (3×). Объединенные органические растворы промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют с получением коричневого твердого продукта, который используют без дополнительной очистки.
Стадия J: (1 R ,2 R )- N -(1-Цианоциклопропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид (Соединение 2)
Это соединение приготавливают из (1R,2R)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоновой кислоты, как описано для (1R,2R)-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамида на Стадии H Примера 1, MS [M+H]+ 603,0, 1H ЯМР (400 МГц, CDCl3) δ 7,77 (д, J=8,3 Гц, 2H), 7,52 (д, J=8,3 Гц, 2H), 6,17 (с, 1H), 3,90-3,42 (м, 10H), 2,88-2,82 (м, 1H), 2,35-1,80 (м, 5H), 1,35-1,43 (м, 2H), 0,68-0,98 (м, 2H).
ПРИМЕР 3
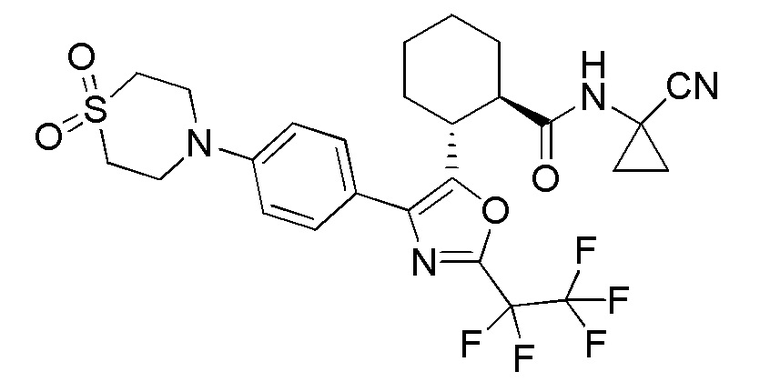
(1 R ,2 R )- N -(1-Цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(перфторэтил)оксазол-5-ил)-циклогексанкарбоксамид (Соединение 3)
Стадия A: (1 R ,2 R )-Метил-2-(4-(4-бромфенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-амино-2-(4-бромфенил)ацетил)циклогексанкарбоксилата гидрохлорида со Стадии D Примера 1 (250 мг, 0,64 ммоль) и Et3N (259 мг, 2,56 ммоль) в дихлорметане (3 мл) добавляют по каплям раствор 2,2,3,3,3-пентафторпропанового ангидрида (397 мг, 1,28 ммоль) в дихлорметане (3 мл), и перемешивают его в течение 15 мин. Реакционную смесь распределяют между дихлорметаном и насыщенным водным раствором NaHCO3. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4 и концентрируют, и очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 30-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде вязкой желтой жидкости (60 мг, 20%).
Стадия B: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-(перфторэтил)-оксазол-5-ил)циклогексанкарбонов кислота
Это соединение приготавливают из (1R,2R)-метил-2-(4-(4-бромфенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксилата, как описано для (1R,2R)-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)циклогексанкарбоновой кислоты на Стадии G Примера 1.
Стадия C: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-(перфторэтил)-оксазол-5-ил)- N -(1-цианоциклопропил)циклогексанкарбоксамид
Это соединение приготавливают из (1R,2R)-2-(4-(4-бромфенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоновой кислоты, как описано для (1R,2R)-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)-N-(1-цианоциклопропил)-циклогексанкарбоксамида на Стадии H Примера 1.
Стадия D: (1 R ,2 R )- N -(1-Цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(перфторэтил)оксазол-5-ил)-циклогексанкарбоксамид (Соединение 3)
Это соединение приготавливают из (1R,2R)-2-(4-(4-бромфенил)-2-(перфторэтил)оксазол-5-ил)-N-(1-цианоциклопропил)-циклогексанкарбоксамида, как описано для (1R,2R)-N-(1-цианоциклопропил)-2-(2-(3,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамида на Стадии I Примера 1, MS [M+H]+ 587,5, 1H ЯМР (400 МГц, CDCl3) δ 7,64 (д, J=8,8 Гц, 2H), 6,98 (д, J=8,9 Гц, 2H), 5,82 (с, 1H), 3,95-3,92 (м, 4H), 3,41-3,34 (м, 1H), 3,14-3,11 (м, 4H), 2,50-2,44 (м, 1H), 2,01-1,08 (м, 10H), 0,89-0,73 (м, 2H).
ПРИМЕР 4
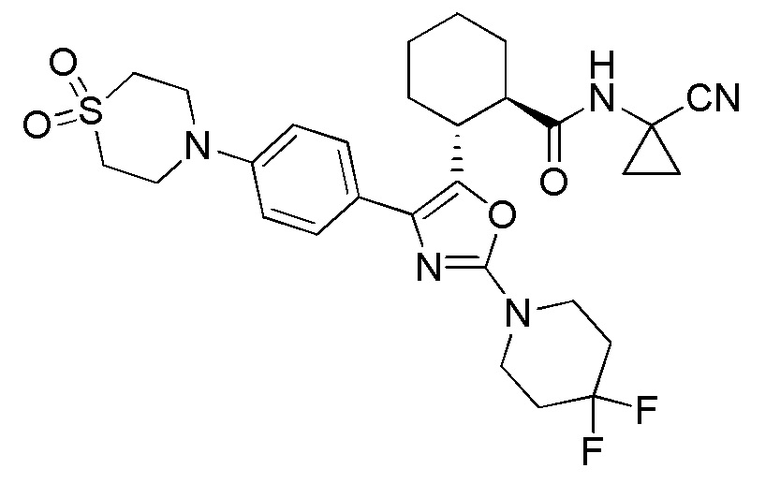
(1 R ,2 R )- N -(1-Цианоциклопропил)-2-(2-(4,4-дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоксамид (Соединение 4)
Стадия A: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-изоцианатоацетил)циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-амино-2-(4-бромфенил)ацетил)циклогексанкарбоксилата гидрохлорида со Стадии D Примера 1 (100 мг, 0,256 ммоль) в дихлорметане (3 мл) при 0°C добавляют водный насыщенный раствор NaHCO3 (5 мл). Трифосген (53 мг, 0,179 ммоль) добавляют к этой двухфазной смеси при быстром перемешивании. После 15 мин при 0°C, реакционную смесь перемешивают при 23°C в течение 2 часов, а затем разбавляют дихлорметаном. Слои разделяют. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4 и концентрируют с получением бесцветной вязкой жидкости, которую используют без дополнительной очистки.
Стадия B: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-(4,4-дифторпиперидинe-1-карбоксамидо)ацетил)циклогексанкарбоксилат
К суспензии 4,4-дифторпиперидин-1-ия хлорида (121 мг, 0,765 ммоль) в дихлорметане (0,5 мл) добавляют основание Хунига (99 мг, 0,765 ммоль). Эту смесь обрабатывают ультразвуком до тех пор, пока все твердые продукты не растворятся. К этому раствору добавляют раствор (1R,2R)-метил-2-(2-(4-бромфенил)-2-изоцианатоацетил)циклогексанкарбоксилата (97 мг, 0,255 ммоль) в дихлорметане (1,5 мл), и перемешивают их при rt в течение ночи. Затем реакционную смесь концентрируют и очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 10-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде бесцветной вязкой жидкости (48 мг, 38%).
Стадия C: (1 R ,2 R )-Метил-2-(4-(4-бромфенил)-2-(4,4-дифторпиперидин-1-ил)оксазол-5-ил)циклогексанкарбоксилат
Раствор (1R,2R)-метил-2-(2-(4-бромфенил)-2-(4,4-дифторпиперидинe-1-карбоксамидо)ацетил)циклогексанкарбоксилата (48 мг, 0,096 ммоль) в POCl3 (1 мл) перемешивают при 50°C в течение 4 часов. Раствор концентрируют и полученный в результате остаток растворяют в дихлорметане и охлаждают до 0°C. Насыщенный водный раствор NaHCO3 добавляют к раствору дихлорметана до тех пор, пока водный слой не станет основным. Слои разделяют. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4, фильтруют и концентрируют. Их очищают с помощью флэш-хроматографии (4 г SiO2) с использованием линейного градиента 3-30% EtOAc в гексане с получением белого твердого продукта (22 мг, 48%).
Стадия D: (1 R ,2 R )-Метил-2-(2-(4,4-дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоксилат
Это соединение приготавливают из (1R,2R)-метил-2-(4-(4-бромфенил)-2-(4,4-дифторпиперидин-1-ил)оксазол-5-ил)циклогексанкарбоксилата, как описано для (1R,2R)-N-(1-цианоциклопропил)-2-(2-(3,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамида на Стадии I Примера 1.
Стадия E: (1 R ,2 R )-2-(2-(4,4-Дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоновая кислота
К раствору (1R,2R)-метил-2-(2-(4,4-дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоксилата (23 мг, 0,043 ммоль) в MeOH-THF (1:3, 0,8 мл) добавляют водный раствор NaOH (1 н, 0,193 мл) и перемешивают при 40°C в течение ночи. Затем реакционную смесь распределяют между водным насыщенным раствором NH4Cl и дихлорметаном. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4 и концентрируют с получением светло-желтого липкого твердого продукта, который используют без дополнительной очистки.
Стадия F: (1 R ,2 R )- N -(1-Цианоциклопропил)-2-(2-(4,4-дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)-оксазол-5-ил)циклогексанкарбоксамид (Соединение 4)
Это соединение приготавливают из (1R,2R)-2-(2-(4,4-дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)-оксазол-5-ил)циклогексанкарбоновой кислоты, как описано для (1R,2R)-2-(4-(4-бромфенил)-2-(3,4-дифторфенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамида на Стадии H Примера 1. MS [M+H]+ 588,2, 1H ЯМР (400 МГц, CDCl3) δ 7,53 (д, J=8,6 Гц, 2H), 6,94 (д, J=8,8 Гц, 2H), 5,61 (с, 1H), 3,91-3,88 (м, 4H), 3,70-3,66 (м, 4H), 3,12-3,06 (м, 5H), 2,37-2,99 (м, 1H), 2,12-1,59 (м, 9H), 1,40-1,21 (м, 5H), 0,70-0,76 (м, 2H).
ПРИМЕР 5
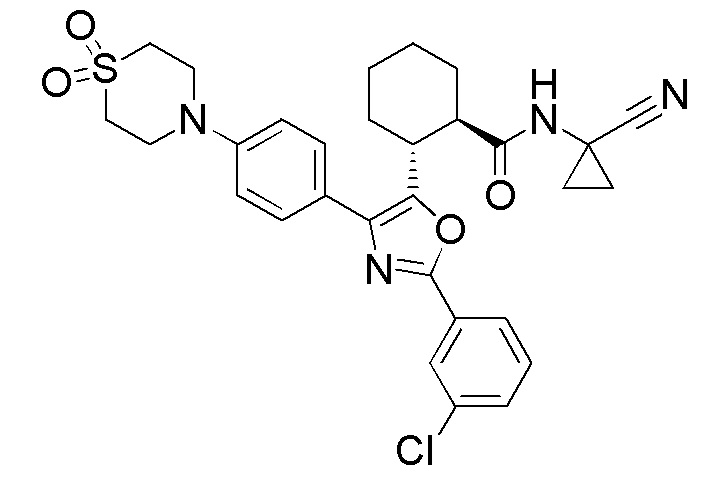
(1 R ,2 R )-2-(2-(3-Хлорфенил)-4-(4-тиоморфолино-1,1-диоксид-фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид (Соединение 5)
Стадия A: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-(3-хлорбензамидо)ацетил)циклогексанкарбоксилат
В 25-мл 3-горлую круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, добавляют (1R,2R)-метил-2-[2-амино-2-(4-бромфенил)ацетил]циклогексан-1-карбоксилат гидрохлорид со Стадии D Примера 1 (0,15 г, 0,38 ммоль, 1,00 эквив.) и дихлорметан (5 мл). К этому раствору добавляют раствор 3-хлорбензоилхлорида (0,27 г, 1,54 ммоль, 4,00 эквив.) в дихлорметане (5 мл) и N,N-диизопропилэтиламин (0,30 г, 2,31 ммоль, 6,00 эквив.) при 0°C. Реакционный раствор перемешивают в течение 30 мин при 0°C. Реакционный раствор гасят посредством добавления насыщенного водного раствора бикарбоната натрия (10 мл). Смесь экстрагируют дихлорметаном (2×10 мл). Объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над безводным раствором сульфата натрия и фильтруют. Фильтрат концентрируют в вакууме. Это дает в результате 0,20 г (1R,2R)-метил-2-(2-(4-бромфенил)-2-(3-хлорбензамидо)ацетил)-циклогексанкарбоксилата виде светло-желтого масла: MS (ES, m/z): 492,1 (M+1), 494,1 (M+1).
Стадия B: (1 R ,2 R )-Метил-2-(4-(4-бромфенил)-2-(3-хлорфенил)оксазол-5-ил)циклогексанкарбоксилат
В 25-мл круглодонную колбу помещают (1R,2R)-метил-2-[2-(4-бромфенил)-2-[(3-хлорфенил)формамидо]ацетил]циклогексан-1-карбоксилат (0,200 г, 0,410 ммоль, 1,00 эквив.), толуол (4 мл) и фосфороилтрихлорид (6 мл). Реакционный раствор перемешивают в течение 16 часов при 80°C на масляной бане. Растворитель удаляют при пониженном давлении. Остаток разбавляют смесью лед/вода (10 мл). Водный слой экстрагируют этилацетатом (3×8 мл). Объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток используют для колоночной хроматографии на силикагеле с помощью смеси этилацетат/простой петролейный эфир (5%-40%). Это дает в результате 0,13 г (67%) (1R,2R)-метил-2-[4-(4-бромфенил)-2-(3-хлорфенил)оксазол-5-ил)циклогексанкарбоксилата в виде светло-желтого твердого продукта: MS (ES, m/z): 473,9 (M+1), 475,9 (M+1)
Стадия C: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-(3-хлорфенил)-оксазол-5-ил)циклогексанкарбоновая кислота
В 25-мл круглодонную колбу помещают (1R,2R)-метил-2-[4-(4-бромфенил)-2-(3-хлорфенил)-1,3-оксазол-5-ил]циклогексан-1-карбоксилат (0,22 г, 0,46 ммоль, 1,00 эквив.), водный раствор гидроксида натрия (1 M, 1,90 мл, 4,00 эквив.), метанол (3 мл) и тетрагидрофуран (5 мл). Реакционную смесь перемешивают в течение 3 часов при 60°C на масляной бане. Значение pH смеси доводят до 2 с помощью водного раствора хлористоводородной кислоты (1 M). Смесь экстрагируют этилацетатом (3×10 мл). Объединенные органические слои промывают насыщенным раствором соли (10 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Это дает в результате 0,21 г (98%) (1R,2R)-2-(4-(4-бромфенил)-2-(3-хлорфенил)оксазол-5-ил)-циклогексанкарбоновой кислоты в виде желтого твердого продукта: MS (ES, m/z): 460,1 (M+1), 462,1 (M+1).
Стадия D: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-(3-хлорфенил)оксазол-5-ил)- N -(1-цианоциклопропил)-циклогексанкарбоксамид
В 10-мл круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, помещают раствор (1R,2R)-2-[4-(4-бромфенил)-2-(3-хлорфенил)оксазол-5-ил)циклогексанкарбоновой кислоты (0,21 г, 0,46 ммоль, 1,00 эквив.) в N,N-диметилформамиде (5 мл), 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат (0,26 г, 0,68 ммоль, 1,50 эквив.), 1-аминоциклопропан-1-карбонитрил гидрохлорид (0,14 г, 1,14 ммоль, 2,50 эквив.) и N,N-диизопропилэтиламин (0,29 г, 2,28 ммоль, 5,00 эквив.). Реакционный раствор перемешивают в течение 16 часов при 40°C на масляной бане и гасят посредством добавления смеси вода/лед (10 мл). Смесь экстрагируют этилацетатом (2×10 мл). Объединенные органические слои промывают насыщенным раствором соли (2×6 мл), сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии с помощью смеси этилацетат/простой петролейный эфир (1:1). Это дает в результате 0,17 г (71%) (1R,2R)-2-(4-(4-Бромфенил)-2-(3-хлорфенил)оксазол-5-ил)-N-(1-цианоциклопропил)-циклогексанкарбоксамида в виде светло-желтого твердого продукта: MS (ES, m/z): 524,2 (M+1), 526,2 (M+1).
Стадия E: (1 R ,2 R )-2-(2-(3-Хлорфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)- N -(1-цианоциклопропил)-циклогексанкарбоксамид (Соединение 5).
В 10-мл герметичную пробирку, продуваемую и поддерживаемую в инертной атмосфере азота, помещают (1R,2R)-2-[4-(4-бромфенил)-2-(3-хлорфенил)оксазол-5-ил)-N-(1-цианоциклопропил)-циклогексанкарбоксамид (60,0 мг, 0,11 ммоль, 1,00 эквив.), тиоморфолин-1,1-дион гидрохлорид (59,0 мг, 0,34 ммоль, 3,00 эквив.), фосфат калия (0,12 г, 0,57 ммоль, 5,00 эквив.), хлор{[2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил][2-(2-аминоэтилфенил]палладий(II)]} (8,40 мг, 0,01 ммоль, 0,10 эквив.) и тетрагидрофуран (3 мл). Смесь слегка дегазируют с помощью трех повторяющихся циклов вакуумирования (1-2 сек) и повторного заполнения азотом. Реакционную смесь перемешивают в течение 6 часов при 120°C на масляной бане. Полученную в результате смесь фильтруют, и фильтрат концентрируют в вакууме. Сырой продукт очищают с помощью препаративной ВЭЖХ при следующих условиях: колонка Xbridge C18, 5 мкм, 25×150 мм; подвижная фаза: вода (0,05% бикарбоната аммония) и ацетонитрил (от 47% до 60% ацетонитрила через 10 мин, выдерживают 100% в течение 3 мин, понижают до 47% через 1 мин); детектор, УФ 220 и 254 нм. Это дает в результате 16,0 мг (24%) (1R,2R)-2-(2-(3-хлорфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1- цианоциклопропил)циклогексанкарбоксамида (Соединение 5) в виде бесцветного твердого продукта: 1H ЯМР (300 МГц, CD3OD) δ м.д. 8,10 (с, 1H), 8,06-7,95 (м, 1H), 7,67 (д, J=8,4 Гц, 2H), 7,56-7,50 (м, 2H), 7,16 (д, J=8,7 Гц, 2H), 4,10-3,93 (м, 4H), 3,33-3,27 (м, 1H), 3,21-3,17 (м, 4H), 2,74-2,66 (м, 1H), 2,06-1,65 (м, 6H), 1,62-1,49 (м, 2H), 1,35-1,30 (м, 2H), 0,79-0,64 (м, 2H); MS (ES, m/z): 579,3 (M+1), 581,3 (M+1).
ПРИМЕР 6
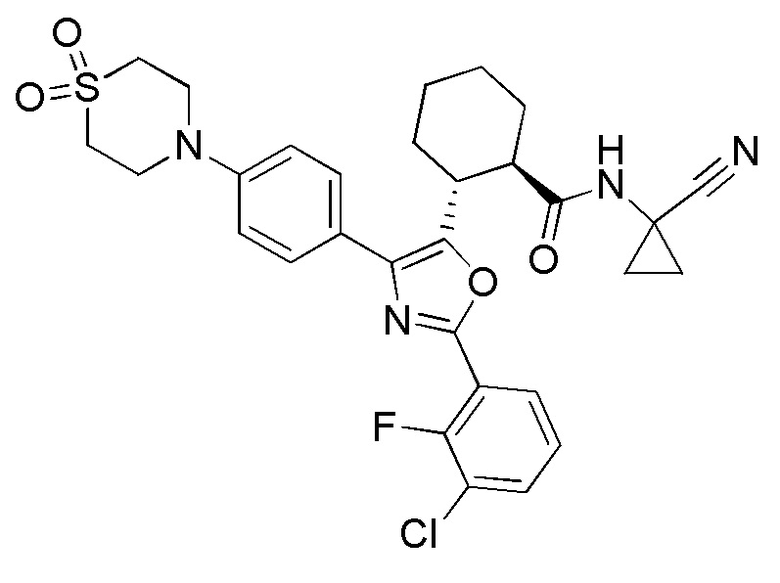
(1 R ,2 R )-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)- N -(1-цианоциклопропил)-циклогексанкарбоксамид (Соединение 6)
Стадия A: 3-Хлор-2-фторбензоилхлорид
В 50-мл круглодонную колбу помещают раствор 3-хлор-2-фторбензойной кислоты (0,30 г, 1,72 ммоль, 1,00 эквив.) в дихлорметане (10 мл). К этому раствору добавляют по каплям оксалилхлорид (0,2 мл) и одну каплю N,N-диметилформамида. Реакционный раствор перемешивают в течение 15 часов при температуре окружающей среды. Полученный в результате раствор концентрируют в вакууме. Это дает в результате 0,33 г (98%) 3-хлор-2-фторбензоилхлорида в виде желтого масла.
Стадия B: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-(3-хлор-2-фторбензамидо)ацетил)циклогексанкарбоксилат
В 50-мл круглодонную колбу помещают раствор (1R,2R)-метил-2-[2-амино-2-(4-бромфенил)ацетил]циклогексан-1-карбоксилата гидрохлорида (0,25 г, 0,64 ммоль, 1,00 эквив.) в дихлорметане (10 мл). За этим следует добавление триэтиламина (0,5 мл). Реакционный раствор перемешивают в течение 10 мин при 0°C. К нему добавляют 3-хлор-2-фторбензоилхлорид (0,308 г, 1,60 ммоль, 2,50 эквив.). Реакционный раствор перемешивают в течение 12 часов, при этом медленно нагревая до температуры окружающей среды. Полученную в результате смесь концентрируют в вакууме и разбавляют водой (10 мл). Полученную в результате смесь экстрагируют дихлорметаном (3×30 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Это дает в результате 0,41 г сырого (1R,2R)-метил-2-[2-(4-бромфенил)-2-(3-хлор-2-фторбензамидо)-ацетил)циклогексанкарбоксилата в виде желтого масла: MS (ES, m/z): 532,2 (M+23), 534,2 (M+23).
Стадия C: (1 R ,2 R )-Метил-2-(4-(4-бромфенил)-2-(3-хлор-2-фторфенил)оксазол-5-ил)циклогексанкарбоксилат
В 25-мл круглодонную колбу помещают раствор (1R,2R)-Метил-2-(2-(4-бромфенил)-2-(3-хлор-2-фторбензамидо)ацетил)циклогексанкарбоксилата (0,41 г, 0,80 ммоль, 1,00 эквив.) в толуоле (0,5 мл). К этому раствору добавляют фосфорилтрихлорид (4 мл). Реакционный раствор перемешивают в течение 5 часов при 80°C на масляной бане. Полученный в результате раствор концентрируют в вакууме и гасят посредством добавления смеси вода/лед (10 мл). Значение pH смеси доводят до 10 с помощью насыщенного водного раствора бикарбоната натрия. Полученную в результате смесь экстрагируют этилацетатом (2×30 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток используют для колоночной хроматографии на силикагеле с помощью смеси этилацетат/простой петролейный эфир (1:9). Это дает в результате 0,12 г (30%) (1R,2R)-метил 2-(4-(4-бромфенил)-2-(3-хлор-2-фторфенил)оксазол-5-ил)циклогексанкарбоксилата в виде желтого масла: MS (ES, m/z): 492,0 (M+1), 494,0 (M+1).
Стадия D: (1 R ,2 R )-Метил-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоксилат
В 10-мл круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, помещают раствор (1R,2R)-метил-2-(4-(4-бромфенил)-2-(3-хлор-2-фторфенил)оксазол-5-ил)-циклогексанкарбоксилата (0,12 г, 0,24 ммоль, 1,00 эквив.) в тетрагидрофуране (2 мл). К этому раствору добавляют тиоморфолин-1,1-диоксид гидрохлорид (0,13 г, 0,73 ммоль, 3,00 эквив.), фосфат калия (0,26 г, 1,22 ммоль, 5,00 эквив.), хлор{[2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил][2-(2-аминоэтилфенил]палладий(II)]} (18,0 мг, 0,020 ммоль, 0,100 эквив.). Реакционную смесь слегка дегазируют с помощью трех повторяющихся циклов вакуумирования (1-2 сек) и повторного заполнения азотом. Реакционную смесь перемешивают в течение 48 часов при 100°C на масляной бане. Полученную в результате смесь разбавляют водой (10 мл) и экстрагируют этилацетатом (2×30 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток используют для колоночной хроматографии на силикагеле с помощью смеси этилацетат/простой петролейный эфир (1:1). Это дает в результате 60,0 мг (45%) (1R,2R)-метил-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксилата в виде желтого масла: MS (ES, m/z): 569,1 (M+23), 571,1 (M+23).
Стадия E: (1 R ,2 R )-2-(2-(3-Хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоновая кислота
В 25-мл круглодонную колбу помещают (1R,2R)-метил-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксилата (60,0 мг, 0,11 ммоль, 1,00 эквив.). К нему добавляют 1 M водный раствор гидроксида натрия (2 мл, 18,8 эквив.), метанол (2 мл) и тетрагидрофуран (2 мл). Реакционную смесь перемешивают в течение 1 часа при 60°C на масляной бане. Полученную в результате смесь концентрируют в вакууме и разбавляют водой (5 мл). Значение pH раствора доводят до 4 с помощью водного раствора хлористоводородной кислоты (10%). Полученную в результате смесь экстрагируют этилацетатом (2×15 мл). Объединенные органические слои сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Это дает в результате 60,0 мг (92%) (1R,2R)-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоновой кислоты в виде желтого масла: MS (ES, m/z): 533,1 (M+1), 535,1 (M+1).
Стадия F: (1 R ,2 R )-2-(2-(3-Хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)- N -(1-цианоциклопропил)-циклогексанкарбоксамид (Соединение 6)
В 10-мл круглодонную колбу, продуваемую и поддерживаемую в инертной атмосфере азота, помещают раствор (1R,2R)-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоновой кислоты (60,0 мг, 0,11 ммоль, 1,00 эквив.) в N,N-диметилформамиде (5,0 мл). К этому раствору добавляют 1-аминоциклопропан-1-карбонитрил гидрохлорид (66,7 мг, 0,56 ммоль, 5,00 эквив.), 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфатметанаминий (68,5 мг, 0,18 ммоль, 1,60 эквив.) и N,N-диизопропилэтиламин (0,12 г, 0,90 ммоль, 8,00 эквив.). Реакционный раствор перемешивают в течение 24 часов при температуре окружающей среде. Полученный в результате раствор разбавляют этилацетатом (30 мл) и промывают водой (3×10 мл). Органический слой сушат над безводным сульфатом натрия и фильтруют. Фильтрат концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии с помощью смеси этилацетат/простой петролейный эфир (1:1). Это дает в результате 18,5 мг (28%) (1R,2R)-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамида (Соединение 6) в виде бесцветного твердого продукта: 1H-ЯМР (300 МГц, DMSO-d6) δ м.д. 8,80 (с, 1H), 8,07-7,97 (м, 1H), 7,85-7,73 (м, 1H), 7,63 (д, J=8,4 Гц, 2H), 7,47-7,35 (м, 1H), 7,14 (д, J=8,1 Гц, 2H), 4,00-3,80 (м, 4H), 3,33-3,22 (м, 1H), 3,21-3,10 (м, 4H), 2,67-2,55 (м, 1H), 2,00-1,72 (м, 4H), 1,70-1,23 (м, 6H), 0,72-0,60 (м, 1H), 0,50-0,40 (м, 1H); MS (ES, m/z): 597,3 (M+1), 599,3 (M+1).
ПРИМЕР 7
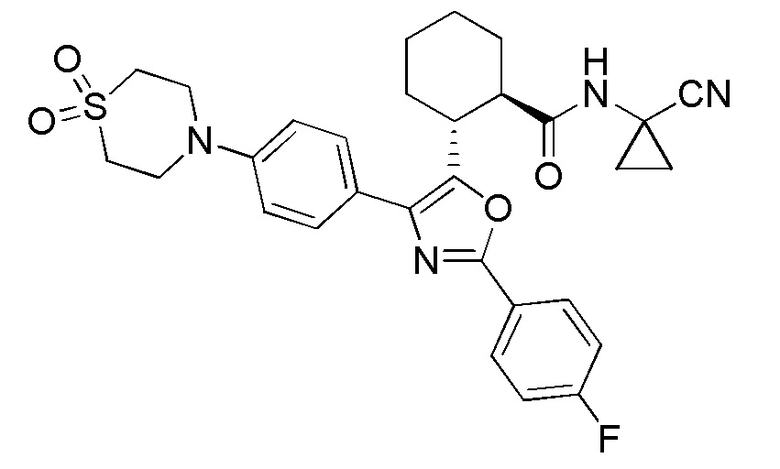
(1 R ,2 R )-N-(1-Цианоциклопропил)-2-{4-[4-(1,1-диоксо-1λ6-тиоморфолин-4-ил)фенил]-2-(4-фторфенил)-1,3-оксазол-5-ил}-циклогексанкарбоксамид (Соединение 7)
Стадия-A: (1 R ,2 R )-метил-2-(2-(4-бромфенил)-2-(4-фторбензамидо)ацетил)циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-амино-2-(4-бромфенил)ацетил)циклогексанкарбоксилата гидрохлорида (250 мг, 0,64 ммоль) в дихлорметане при 0°C добавляют 4-фторбензоилхлорид (406 мг, 2,56 ммоль), а затем основание Хунига (496 мг, 3,84 ммоль); раствор перемешивают при 0°C в течение 15 мин, а затем концентрируют. Остаток растворяют в DMSO и очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 20-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде белого твердого продукта (108 мг, 35%).
Стадия-B: (1R,2R)-метил-2-(4-(4-бромфенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксилат
Раствор (1R,2R)-метил-2-(2-(4-бромфенил)-2-(4-фторбензамидо)ацетил)циклогексанкарбоксилата (122 мг, 0,256 ммоль) в POCl3 (2 мл) нагревают при 80°C в течение 5,5 часа и при 40°C в течение 14 часов. Раствор концентрируют, и полученный в результате остаток растворяют в дихлорметане и охлаждают до 0°C. Насыщенный водный раствор NaHCO3 добавляют к раствору дихлорметана до тех пор, пока водный слой не станет основным. Слои разделяют. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4, фильтруют и концентрируют с получением белого твердого продукта, который используют без дополнительной очистки.
Стадия-C: (1 R ,2 R )-2-(4-(4-бромфенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоновая кислота
Раствор (1R,2R)-метил-2-(4-(4-бромфенил)-2-(4-фторфенил)-оксазол-5-ил)циклогексанкарбоксилата (117 мг, 0,255 ммоль) в MeOH-THF (1:1, 2 мл) обрабатывают с помощью 1 н водного раствора NaOH (1,021 мл) и перемешивают при 40°C в течение 14 часов. После охлаждения до rt, добавляют 1,021 мл 1 н водного раствора HCl. Полученный в результате раствор концентрируют до белого твердого продукта, который используют без дополнительной очистки.
Стадия-D: (1 R ,2 R )-2-(4-(4-бромфенил)-2-(4-фторфенил)оксазол-5-ил)- N -(1-цианоциклопропил)-циклогексанкарбоксамид
К раствору (1R,2R)-2-(4-(4-бромфенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоновой кислоты (113 мг, 0,254 ммоль) и HATU (75 мг, 0,636 ммоль) в DMF (2 мл) добавляют основание Хунига (164 мг, 1,272 ммоль), и раствор перемешивают при 40°C в течение 47 часов. Полученный в результате раствор очищают непосредственно с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 30-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде белого твердого продукта (85 мг, 66%).
Стадия-E: (1 R ,2 R )-N-(1-Цианоциклопропил)-2-{4-[4-(1,1-диоксо-1λ6-тиоморфолин-4-ил)фенил]-2-(4-фторфенил)-1,3-оксазол-5-ил}циклогексанкарбоксамид (Соединение 7)
К смеси (1R,2R)-2-(4-(4-бромфенил)-2-(4-фторфенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамида (116 мг, 0,228 ммоль), тиоморфолин 1,1-диоксида (93 мг, 0,685 ммоль), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил)]палладия(II) (8,43 мг, 0,011 ммоль) и K3PO4,H2O (63,1 мг, 0,274 ммоль) во флаконе в инертной атмосфере добавляют THF (1,5 мл). Флакон герметизируют и нагревают при 100°C в течение 2 часов, охлаждают до комнатной температуры, и смесь концентрируют. Остаток извлекают в DMSO, фильтруют, и фильтрат подвергают воздействию очистки с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 10-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды). Желаемые фракции распределяют между насыщенным водным раствором бикарбоната натрия и дихлорметаном. Слои разделяют. Водный слой экстрагируют дихлорметаном (3×). Объединенные органические растворы сушат над Na2SO4 и концентрируют с получением желаемого продукта в виде белого твердого продукта (120 мг, 93%). MS [M+H]+ 563,1, 1H ЯМР (400 МГц, CDCl3) δ 8,05-8,02 (м, 2H), 7,68-7,63 (м, 2H), 7,21-7,14 (м, 2H), 7,20-6,97 (м, 2H), 5,66 (с, 1H), 3,94-3,91 (м, 1H), 3,30-3,22 (м, 1H), 3,14-3,12 (м, 4H), 2,51-2,44 (м, 1H), 2,1-1,3 (м, 10H), 0,78-0,58 (м, 2H).
ПРИМЕРЫ 8 и 9
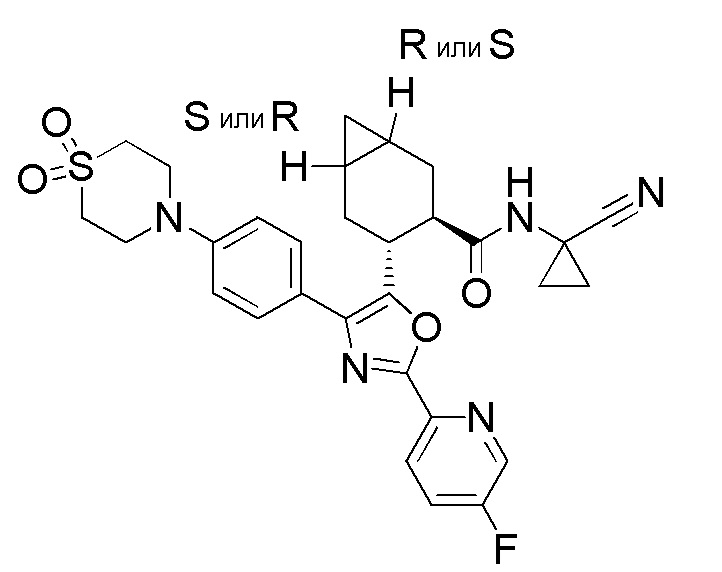
Соединение 8 или Соединение 9
(1 S или 1 R ,3 R ,4 R , 6R или 6 S )-N-(1-цианоциклопропил)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)-оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксамид (Соединение 8 и Соединение 9).
Стадия A: (3 R ,4 R )-Метил-4-(хлоркарбонил)-бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(хлоркарбонил)бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как для синтеза (1R,2R)-метил-2-(хлоркарбонил)циклогексанкарбоксилата с использованием (3R,4R)-4-(метоксикарбонил)бицикло[4.1.0]гептан-3-карбоновой кислоты (1,40 г, 7,06 ммоль, 1,00 эквив.) и оксалилхлорида (1,80 г, 14,2 ммоль, 2,00 эквив.) в толуоле (30 мл). Это дает в результате 1,5 г (98%) сырого (3R,4R)-метил-4-(хлоркарбонил)бицикло[4.1.0]гептан-3-карбоксилата в виде желтого масла, которое используют непосредственно на следующей стадии без дополнительной очистки.
Стадия B: (3 R ,4 R )-Метил-4-(2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(2-(4-бромфенил)ацетил)-бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как на Стадии A Примера 1, с использованием (3R,4R)-метил 4-(хлоркарбонил)бицикло[4.1.0]гептан-3-карбоксилата (0,62 г, 2,86 ммоль, 1,00 эквив.), комплекса цианида меди(I)и бис(хлорида лития) (1 M в тетрагидрофуране, 4,29 мл, 4,29 ммоль, 1,50 эквив.) и бром[(4-бромфенил)метил]цинка (1 M в тетрагидрофуране, 4,29 мл, 4,29 ммоль, 1,50 эквив.) в тетрагидрофуране (10 мл). Это дает в результате 0,51 г (50%) (3R,4R)-метил-4-(2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилата в виде бесцветного твердого продукта: 1H ЯМР (300 МГц, CDCl3) δ м.д. 7,44 (д, J=8,4 Гц, 2H), 7,05 (д, J=8,4 Гц, 2H), 3,76 (с, 2H), 3,65 (с, 3H), 2,73-2,08 (м, 4H), 1,80-1,65 (м, 1H), 1,46-1,24 (м, 1H), 1,04-0,94 (м, 2H), 0,71-0,64 (м, 1H), 0,04-0,00 (м, 1H); MS (ES, m/z): 351,2 (M+1), 353,2 (M+1).
Стадия C: (3 R ,4 R )-Метил-4-(2-(4-бромфенил)-2-хлорацетил)бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(2-(4-бромфенил)-2-хлорацетил)-бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как на Стадии B Примера 1, с использованием (3R,4R)-метил-4-(2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилата (0,51 г, 1,45 ммоль, 1,00 эквив.) и N-хлорсукцинимида (0,23 г, 1,74 ммоль, 1,20 эквив.) в N,N-диметилформамиде (10 мл). Это дает в результате 0,47 г (84%) (3R,4R)-метил-4-(2-(4-бромфенил)-2-хлорацетил)-бицикло[4.1.0]гептан-3-карбоксилата в виде желтого масла: MS (ES, m/z): 384,8 (M+1), 386,8 (M+1).
Стадия D: (3 R ,4 R )-Метил 4-(2-азидо-2-(4-бромфенил)-ацетил)бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(2-азидо-2-(4-бромфенил)ацетил)-бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как на Стадии C Примера 1, с использованием (3R,4R)-метил-4-(2-(4-бромфенил)-2-хлорацетил)бицикло[4.1.0]гептан-3-карбоксилата (0,47 г, 1,22 ммоль, 1,00 эквив.) и азида натрия (94,9 мг, 1,46 ммоль, 1,20 эквив.) в диметилсульфоксиде (10 мл). Это дает в результате 0,47 г (3R,4R)-метил-4-(2-азидо-2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилата в виде светло-желтого масла, которое используют непосредственно на следующей стадии без дополнительной очистки.
Стадия E: (3 R ,4 R )-Метил-4-(2-амино-2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилат гидрохлорид
(3R,4R)-Метил-4-(2-амино-2-(4-бромфенил)ацетил)-бицикло[4.1.0]гептан-3-карбоксилат гидрохлорид синтезируют, следуя такой же процедуре, как на Стадии D Примера 1, с использованием (3R,4R)-метил-4-(2-азидо-2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилата (0,47 г, 1,20 ммоль, 1,00 эквив.), порошка цинка (0,11 г, 1,68 ммоль, 1,40 эквив.) и хлорида аммония (0,16 г, 3,00 ммоль, 2,50 эквив.) в этаноле и воде (3:1 (объем/объем), 12 мл). Это дает в результате 0,43 г (89%) (3R,4R)-метил-4-(2-амино-2-(4-бромфенил)ацетил)бицикло[4.1.0]гептан-3-карбоксилата гидрохлорида в виде желтого твердого продукта: MS (ES, m/z): 366,2 (M+1), 368,2 (M+1).
Стадия F: (3 R ,4 R )-Метил-4-(2-(4-бромфенил)-2-(5-фторпиколинамидо)ацетил)бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(2-(4-бромфенил)-2-(5-фторпиколинамидо)-ацетил)бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как на Стадии H Примера 1, с использованием (3R,4R)-метил-4-(2-амино-2-(4-бромфенил)ацетил)-бицикло[4.1.0]гептан-3-карбоксилата гидрохлорида (0,43 г, 1,07 ммоль, 1,00 эквив.), 5-фторпиридин-2-карбоновой кислоты (0,38 г, 2,68 ммоль, 2,50 эквив.), HATU (1,02 г, 2,68 ммоль, 2,50 эквив.) и N,N-диизопропилэтиламина (0,69 г, 5,35 ммоль, 5,00 эквив.) в N,N-диметилформамиде (15 мл). Это дает в результате 0,33 г сырого (3R,4R)-метил-4-(2-(4-бромфенил)-2-(5-фторпиколинамидо)ацетил)бицикло[4.1.0]гептан-3-карбоксилата в виде желтого масла: MS (ES, m/z): 489,3 (M+1), 491,3 (M+1).
Стадия G: (3 R ,4 R )-Метил-4-(4-(4-бромфенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(4-(4-бромфенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как на Стадии F Примера 1, с использованием (3R,4R)-метил-4-(2-(4-бромфенил)-2-(5-фторпиколинамидо)ацетил)бицикло[4.1.0]гептан-3-карбоксилата (0,33 г, 0,67 ммоль, 1,00 эквив.) и фосфороилтрихлорида (10 мл) в толуоле (4 мл). Это дает в результате 0,17 г (3R,4R)-метил 4-(4-(4-бромфенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилата в виде светло-желтого твердого продукта: MS (ES, m/z): 471,1 (M+1), 473,1 (M+1).
Стадия H: (3 R ,4 R )-Метил-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилат
(3R,4R)-Метил-4-(2-(5-фторпиридин-2-ил)-4-(4-тиоморфолино-1,1-диоксид-фенил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилат синтезируют, следуя такой же процедуре, как на Стадии I Примера 1, с использованием (3R,4R)-метил-4-(4-(4-бромфенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилата (0,10 г, 0,21 ммоль, 1,00 эквив.), тиоморфолин-1,1-диоксида гидрохлорида (0,11 г, 0,64 ммоль, 3,00 эквив.), трис(дибензилиденацетон)дипалладия (0)-хлороформа (40 мг, 0,0386 ммоль, 0,182 эквив.), дициклогексилфосфино-2',4',6'-триизопропилбифенила (30 мг, 0,0629 ммоль, 0,297 эквив.) и карбоната цезия (0,33 г, 1,01 ммоль, 5,00 эквив.) в толуоле (5 мл). Это дает в результате 65,0 мг (3R,4R)-метил-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилата в виде желтого твердого продукта: MS (ES, m/z): 526,3 (M+1).
Стадия I: (3 R ,4 R )-4-(4-(4-(1,1-Диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоновая кислота
(3R,4R)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоновую кислоту синтезируют, следуя такой же процедуре, как на Стадии G Примера 1, с использованием (3R,4R)-метил 4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксилата (55,0 мг, 0,11 ммоль, 1,00 эквив.) в водном растворе гидроксида натрия (1 M, 2 мл) и тетрагидрофурана (2 мл). Это дает в результате 50,0 мг (93%) желаемого продукта карбоновой кислоты в виде желтого масла: MS (ES, m/z): 512,1 (M+1).
Стадия J: (1 S или 1 R ,3 R ,4 R ,6 R или 6 S )-N-(1-Цианоциклопропил)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксамид (Соединение 8 и Соединение 9)
Соединение 8 и соединение 9 синтезируют, следуя такой же процедуре, как на Стадии H Примера 1, с использованием (3R,4R)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоновой кислоты (50,0 мг, 0,0977 ммоль, 1,00 эквив.), 1-аминоциклопропан-1-карбонитрила гидрохлорида (28,9 мг, 0,24 ммоль, 2,50 эквив.), HATU (55,9 мг, 0,15 ммоль, 1,50 эквив.) и N,N-диизопропилэтиламина (63,2 мг, 0,49 ммоль, 5,00 эквив.) в N,N-диметилформамиде (5 мл). Это дает в результате 10,0 мг (14%) (3R,4R)-N-(1-цианоциклопропил)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксамид в виде бесцветного твердого продукта: MS (ES, m/z): 576,2 (M+1). Рацемический продукт (10 мг) разделяют с помощью хиральной препаративной ВЭЖХ с при следующих условиях: колонка, Chiralpak IB, 0,46×25 см, 5-мкм Chiral-A(IB) 001 IB00CE-LA026; подвижная фаза, Hex:EtOH=60:40; детектор, УФ 254 нм и 220 нм. Это дает в результате 3,6 мг соединения 8 при 16,1 мин в виде серого твердого продукта: 1H ЯМР (300 МГц, CDCl3) δ м.д. 8,68 (с, 1H), 8,44-8,39 (м, 1H), 7,87-7,84 (м, 1H), 7,75 (д, J=8,7 Гц, 2H), 7,68-7,65 (м, 1H), 7,01 (д, J=8,7 Гц, 2H), 3,96-3,92 (м, 4H), 3,16-3,13 (м, 4H), 3,09-3,03 (м, 1H), 2,81-2,70 (м, 1H), 2,37-2,31 (м, 1H), 2,26-2,16 (м, 2H), 1,94-1,86 (м, 1H), 1,35-1,20 (м, 2H), 1,09-1,04 (м, 2H), 0,90-0,85 (м, 2H), 0,78-0,72 (м, 1H), 0,20-0,16 (м, 1H); MS (ES, m/z): 576,0 (M+1). Это также дает в результате 2,1 мг Соединения 9 при 13,4 мин в виде беловатого твердого продукта: 1H ЯМР (300 МГц, CDCl3) δ м.д. 8,59 (с, 1H), 8,32-8,28 (м, 1H), 7,70-7,69 (м, 1H), 7,69 (д, J=8,7 Гц, 2H), 7,02 (д, J=8,7 Гц, 2H), 6,98 (ушир., 1H), 3,95-3,92 (м, 4H), 3,15-3,12 (м, 4H), 3,09-3,04 (м, 1H), 2,64-2,62 (м, 1H), 2,38-2,30 (м, 1H), 2,25-2,15 (м, 2H), 1,98-1,94 (м, 1H), 1,35-1,25 (м, 2H), 1,10-1,03 (м, 2H), 0,95-0,88 (м, 1H), 0,78-0,68 (м, 2H), 0,17-0,16 (м, 1H); MS (ES, m/z): 576,0 (M+1).
ПРИМЕР 10
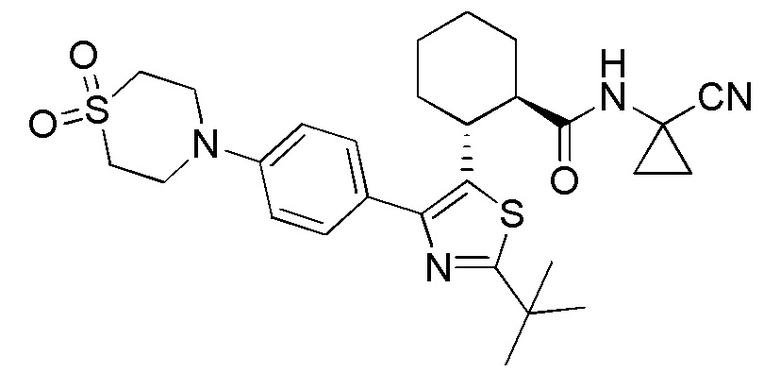
(1 R ,2 R )-2-(2-(трет-бутил)-4-(4-(1,1-диоксидотиоморфолино)-фенил)тиазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид (Соединение 10).
Стадия A: (1 R ,2 R )-Метил-2-(2-(4-бромфенил)-2-пиваламидоацетил)циклогексанкарбоксилат
К раствору сырого 1-(4-бромфенил)-2-((1R,2R)-2-(метоксикарбонил)циклогексил)-2-оксоэтанаминия хлорида (287 мг, 0,735 ммоль) в дихлорметане (5 мл) при 0°C добавляют пивалоилхлорид (0,362 мл, 2,94 ммоль, 4 эквив.) с последующим добавлением по каплям основания Хунига (0,77 мл, 4,41 ммоль). Полученный в результате раствор перемешивают при 0°C в течение 15 мин, а затем концентрируют при пониженном давлении. Остаток растворяют в DMSO и очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 15-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде белого твердого продукта (149 мг, 46%).
Стадия B: (1 R ,2 R )-Метил-2-(4-(4-бромфенил)-2-( трет -бутил)тиазол-5-ил)циклогексанкарбоксилат
К раствору (1R,2R)-метил-2-(2-(4-бромфенил)-2 пиваламидоацетил)циклогексанкарбоксилата (74 мг, 0,169 ммоль) в толуоле (2 мл) добавляют реагент Лоуссона (54,6 мг, 0,135 ммоль, 0,8 эквив.). Полученную в результате смесь перемешивают при 80°C в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры и гасят с помощью насыщенного раствора NaHCO3. Полученную в результате смесь экстрагируют EtOAc (3×5 мл). Объединенные органические фазы сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают с помощью флэш-хроматографии (4 г, SiO2): 3-15% EtOAc в гексане с получением 52 мг (71%) желаемого продукта в виде белого твердого продукта.
Стадия C: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-( трет -бутил)тиазол-5-ил)циклогексанкарбоновая кислота
К раствору (1R,2R)-метил-2-(4-(4-бромфенил)-2-(трет-бутил)тиазол-5-ил)циклогексанкарбоксилата (52 мг, 0,119 ммоль) в MeOH/THF=1/1 (2 мл) добавляют 1 н раствор NaOH. Полученный в результате раствор перемешивают при 60°C в течение 4,5 часа и при 40°C в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры с последующим добавлением 1 н HCl. Полученную в результате смесь концентрируют с получением желаемого продукта в виде белого твердого продукта. Сырой продукт используют без дополнительной очистки.
Стадия D: (1 R ,2 R )-2-(4-(4-Бромфенил)-2-( трет -бутил)тиазол-5-ил)- N -(1-цианоциклопропил)циклогексанкарбоксамид
К раствору (1R,2R)-2-(4-(4-бромфенил)-2-(трет-бутил)тиазол-5-ил)циклогексанкарбоновой кислоты (50 мг, 0,188 ммоль) и HATU (67,5 мг, 0,178 ммоль) в DMF (2 мл) добавляют 1-цианоциклопропанаминийхлорид (35 мг, 0,296 ммоль) с последующим добавлением DIEA (0,103 мл, 0,592 ммоль). Полученную в результате смесь перемешивают при 40°C в течение 18 часов. Полученный в результате раствор фильтруют и непосредственно очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 30-90% MeCN в воде, 0,1% модификатор TFA для MeCN и воды) с получением желаемого продукта в виде белого твердого продукта (49 мг, 85%).
Стадия E: (1 R ,2 R )-2-(2-( трет -Бутил)-4-(4-(1,1-диоксидотиоморфолино)фенил)тиазол-5-ил)- N -(1-цианоциклопропил)циклогексанкарбоксамид (Соединение 10)
К смеси (1R,2R)-2-(4-(4-бромфенил)-2-(трет-бутил)тиазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамида (25 мг, 0,051 ммоль), тиоморфолин 1,1-диоксида (20,84 мг, 0,154 ммоль, 3 эквив.), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил)]палладия(II) (3,8 мг, 5,14 мкмоль, 0,1 эквив.) и K3PO4,H2O (14,2 мг, 0,062 ммоль) во флаконе в атмосфере N2 добавляют THF (0,7 мл). Флакон герметизируют, и полученную в результате смесь нагревают при 100°C в течение 4 часов, а затем оставляют при комнатной температуре в течение 18 часов. Реакционную смесь концентрируют при пониженном давлении, остаток растворяют в DMSO, и раствор очищают с помощью ВЭЖХ с обращенной фазой (колонка Sunfire C18 30×150 мм; 5-95% MeCN в воде. 0,1% модификатор TFA для MeCN и воды) с получением 24,5 мг (73%) желаемого продукта (Соединение 9) в виде белого твердого продукта. MS (ES, m/z): 541,1 (M+1); 1H ЯМР (400 МГц, CDCl3) 7,48 (д, J=8,6 Гц, 2H), 6,97(д, J=8,6, 2H), 6,16 (с, 1H), 3,95-3,92 (м, 4H), 3,30-3,25 (м, 1H), 3,12-3,09 (м, 4H), 2,24-2,17 (м, 1H), 2,01-1,79 (м, 4H), 1,51-1,56 (м, 1H).
Следующие соединения приготавливают с использованием способов, аналогичных тем, которые описаны в предыдущих примерах:
m/z (M+H)
IP (нМ)
IP (нМ)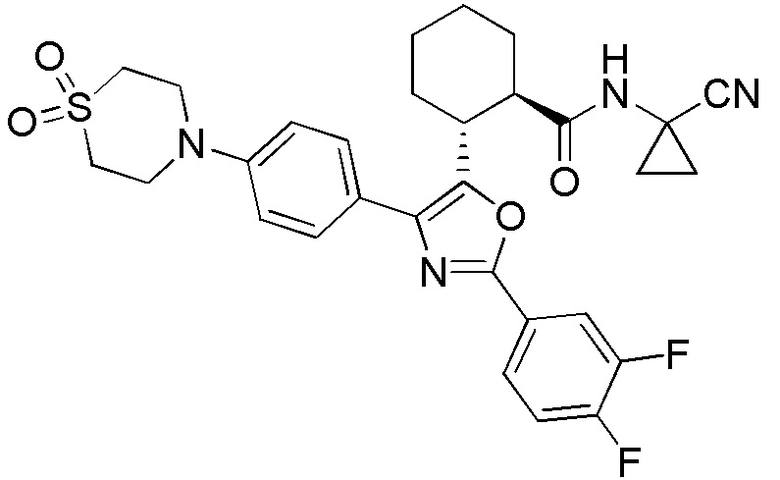
фенил)оксазол-5-ил)циклогексанкарбоксамид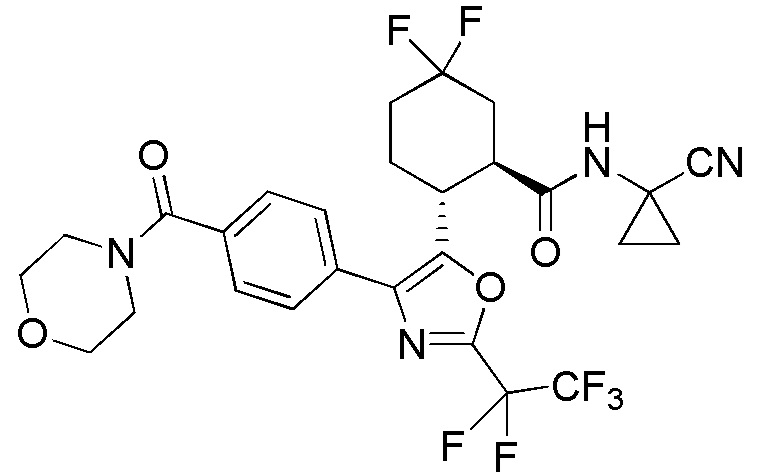
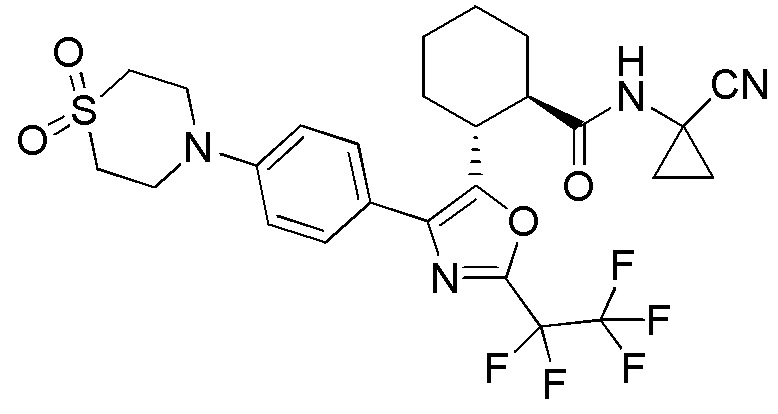
фенил)-2-(перфторэтил)-оксазол-5-ил)-циклогексанкарбоксамид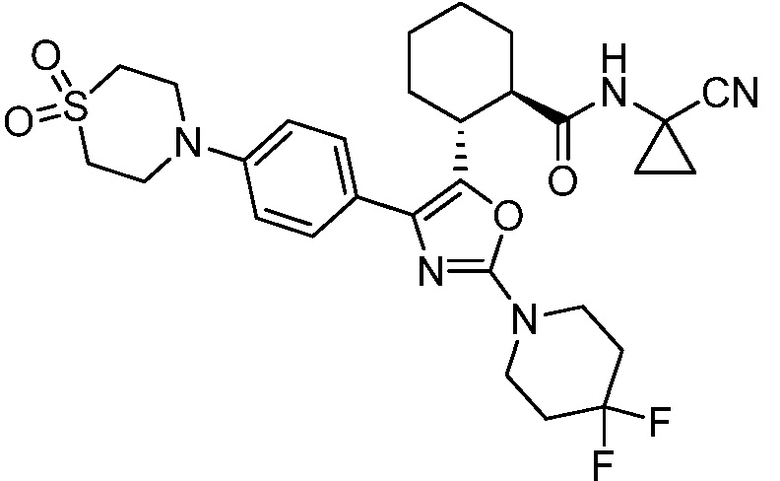
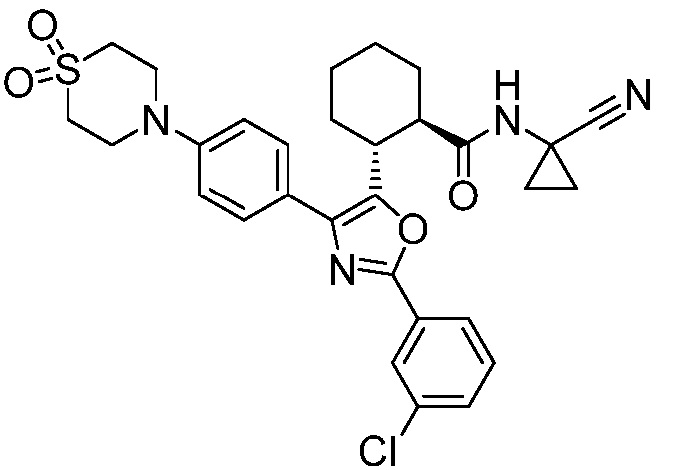
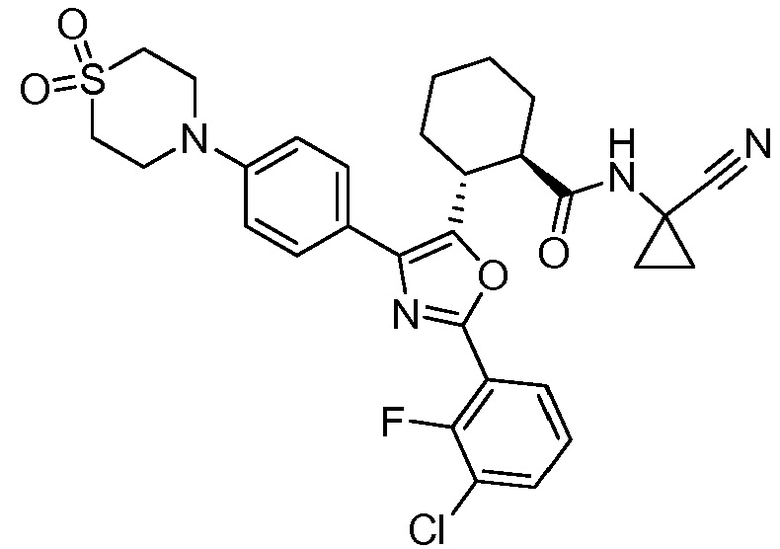
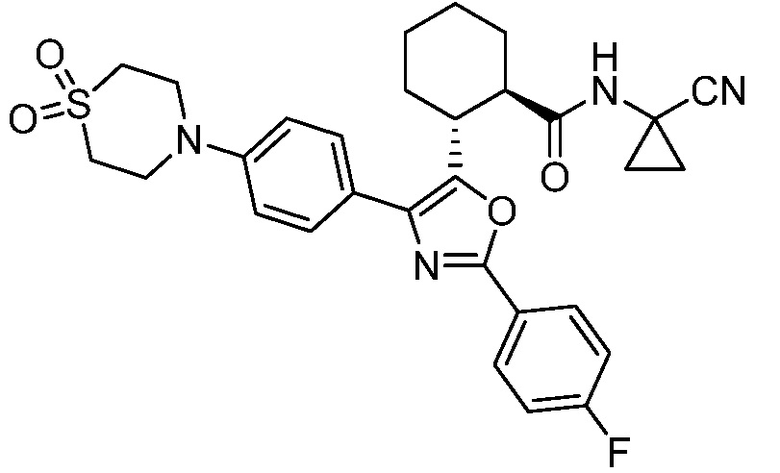
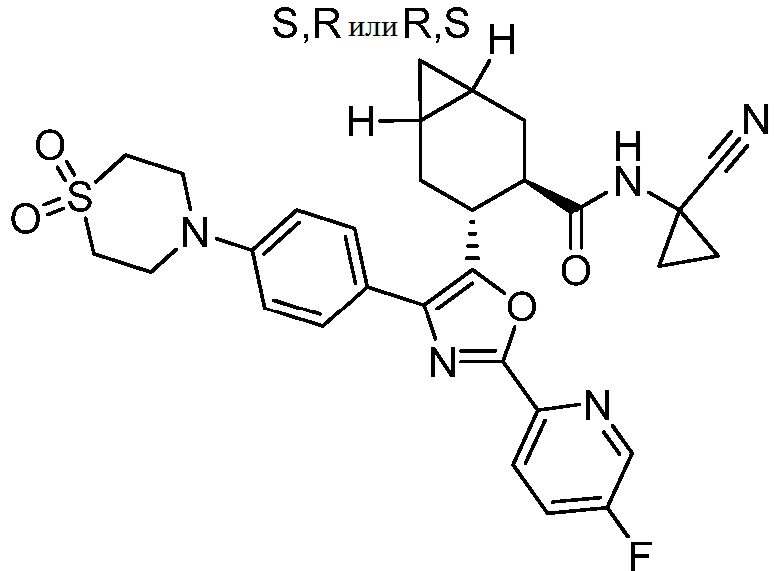
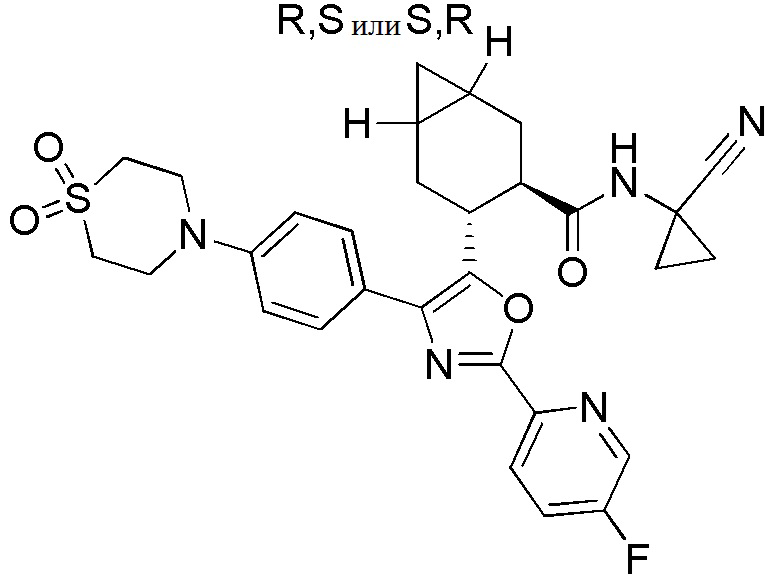
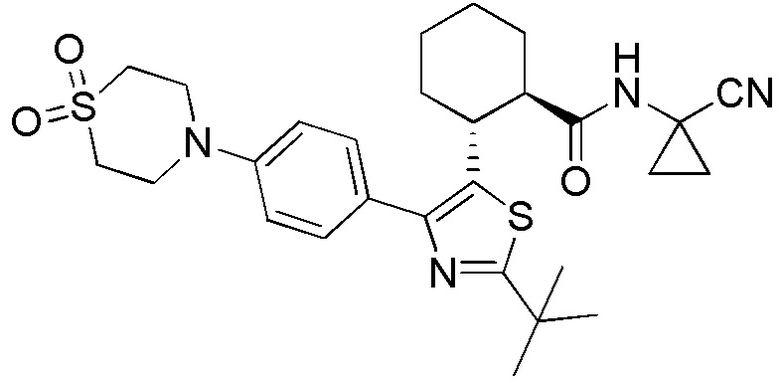
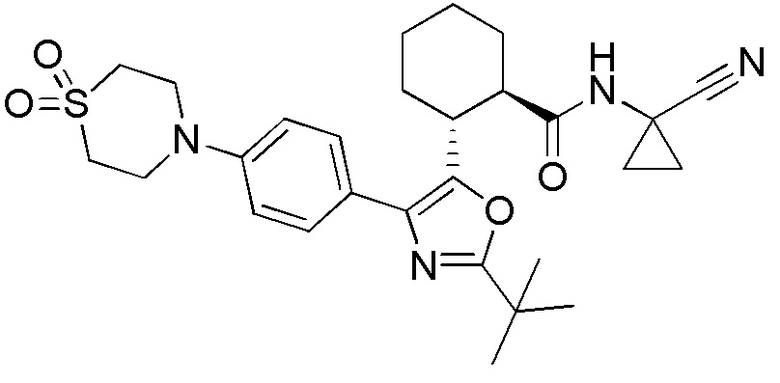
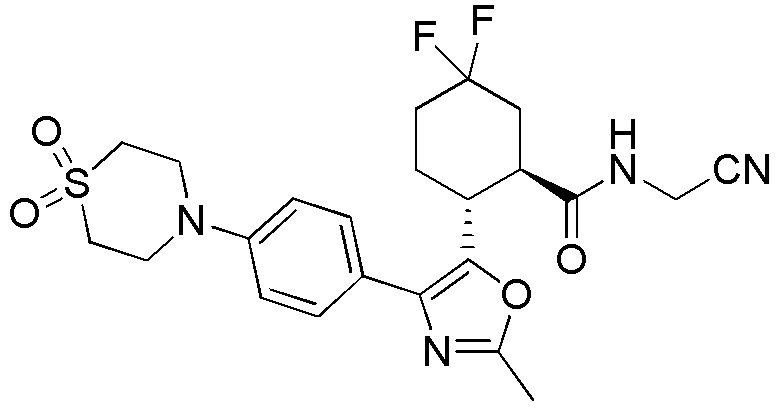
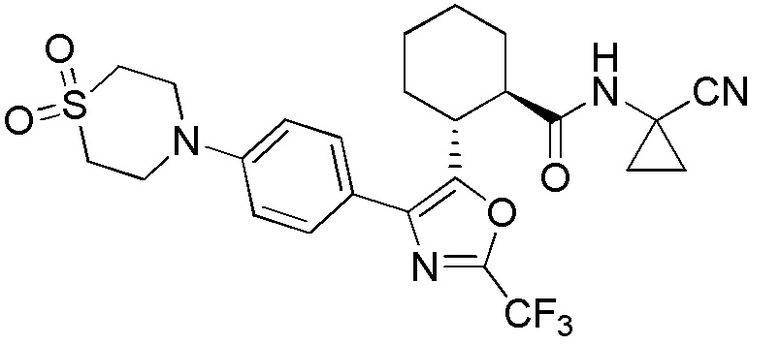
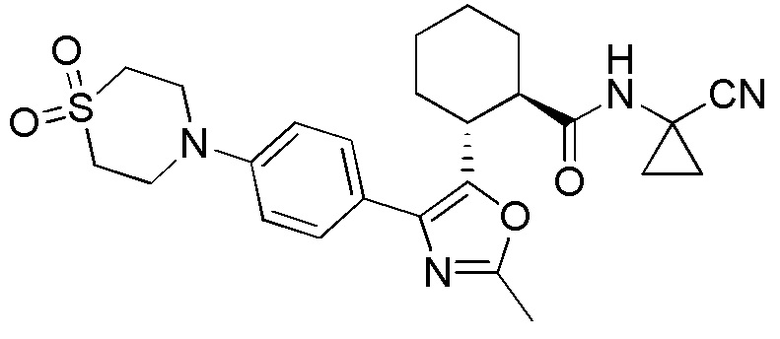
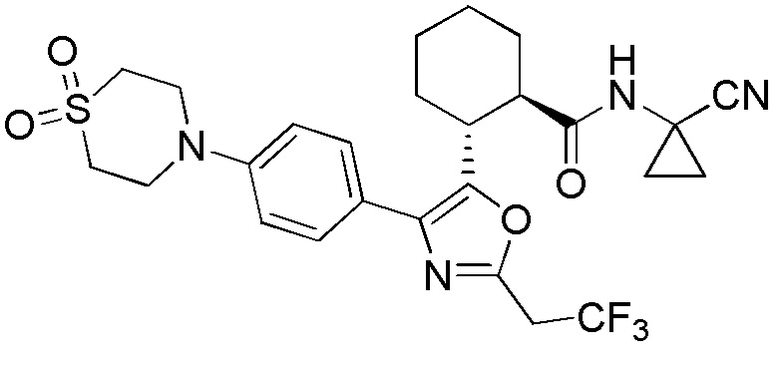
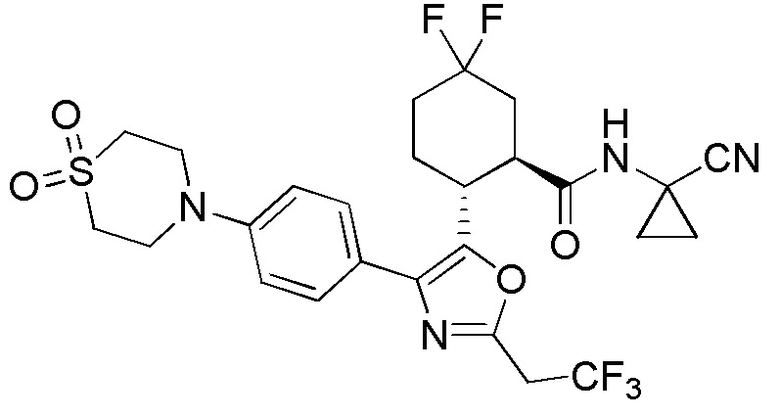
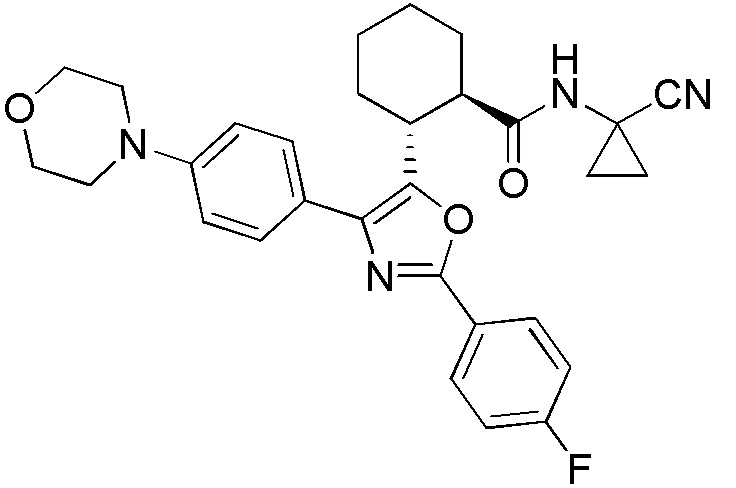
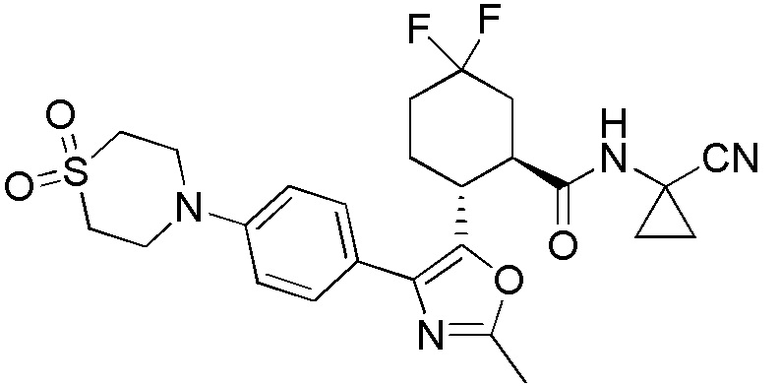
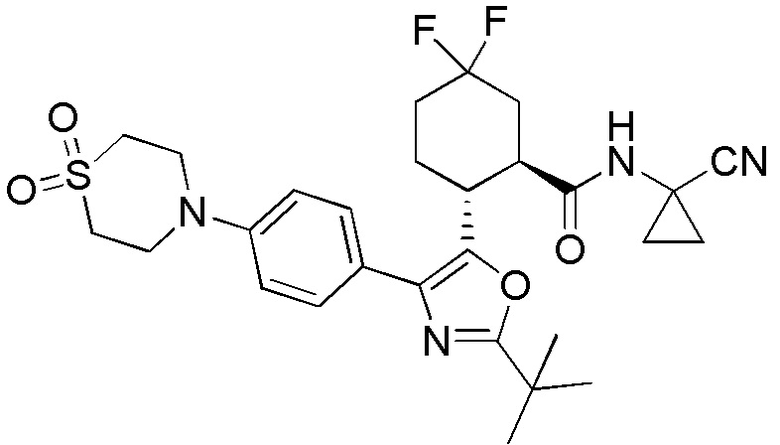
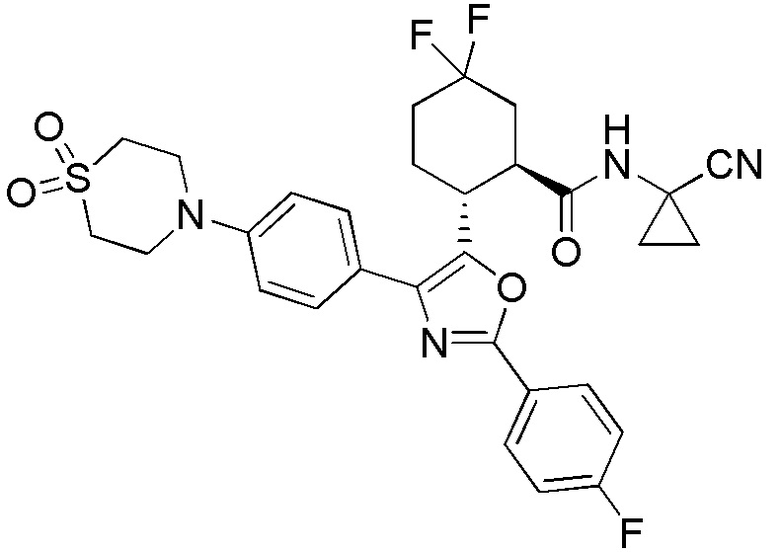
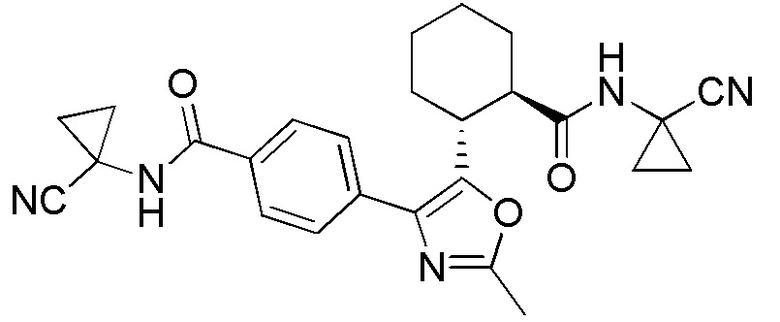
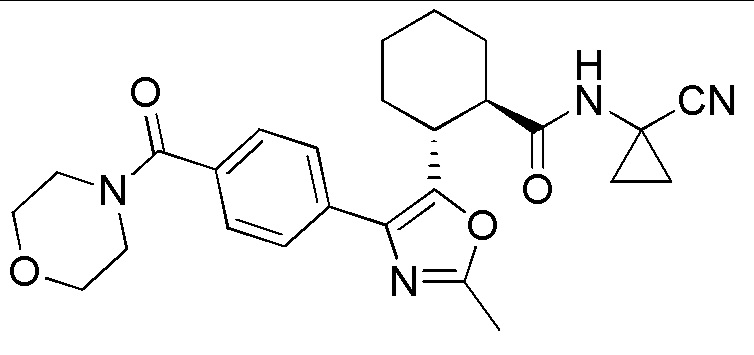
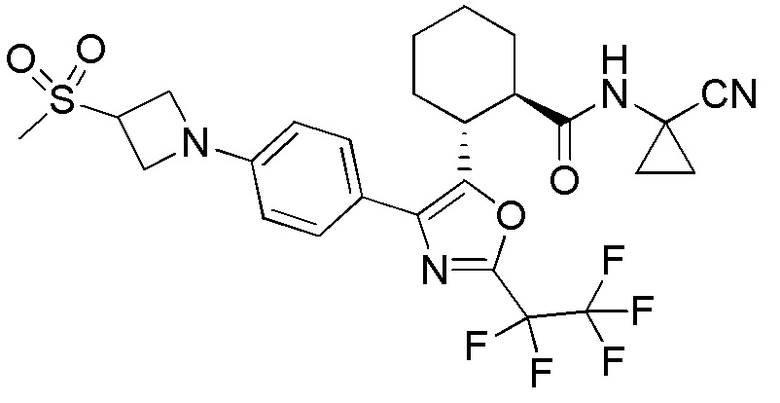
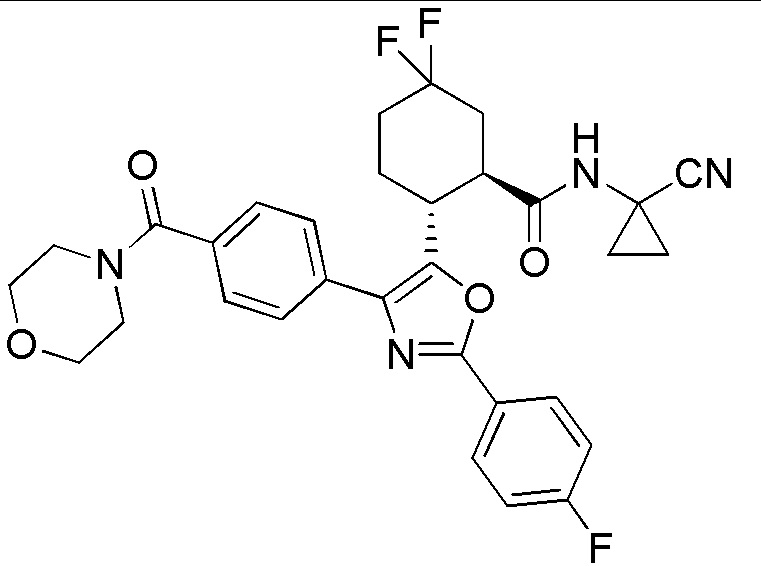
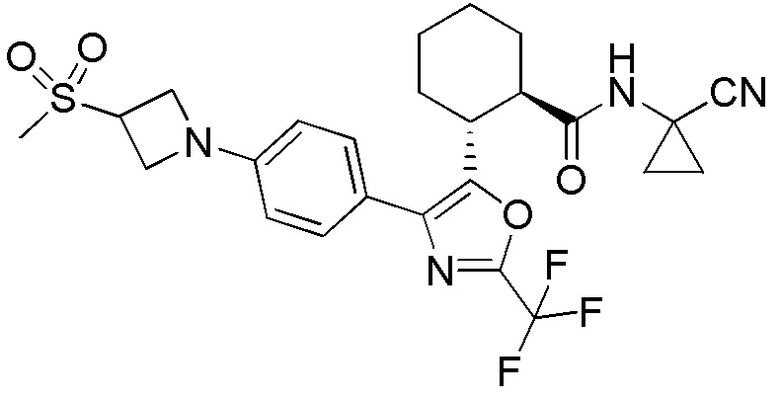
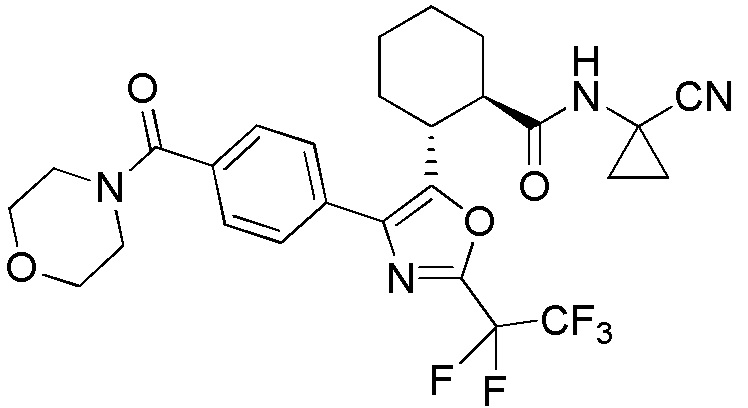
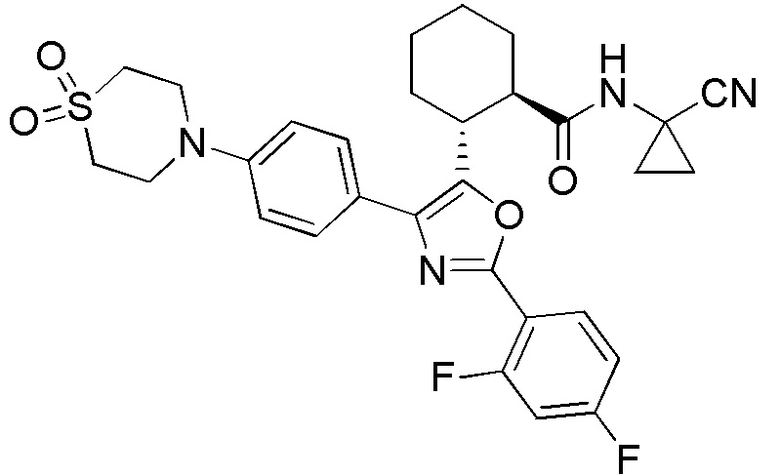
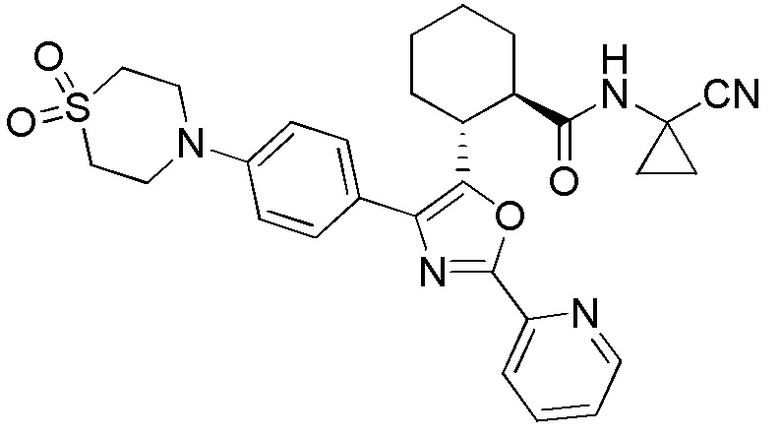
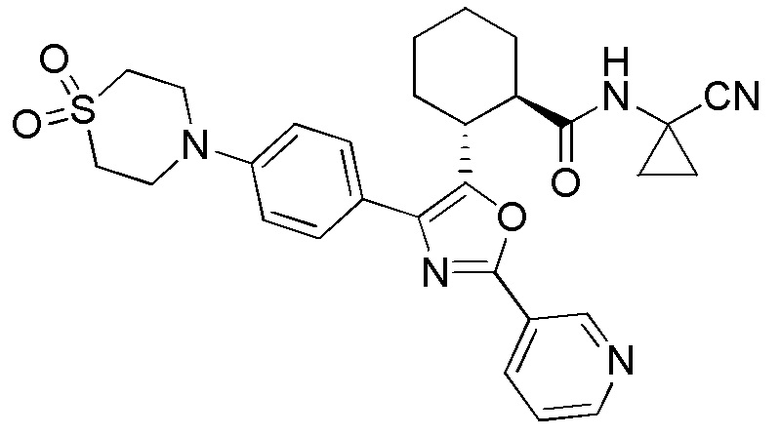
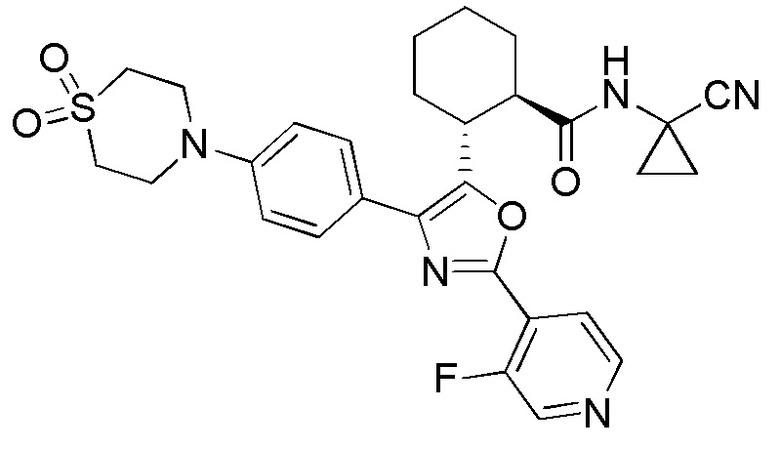
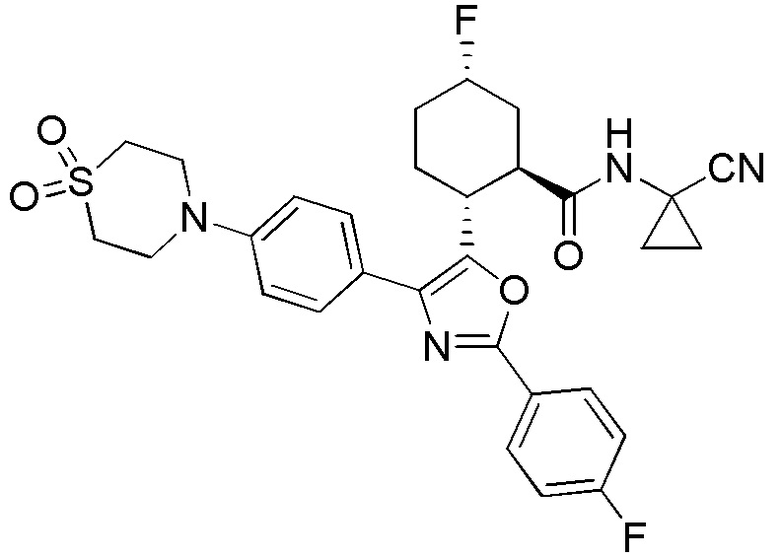
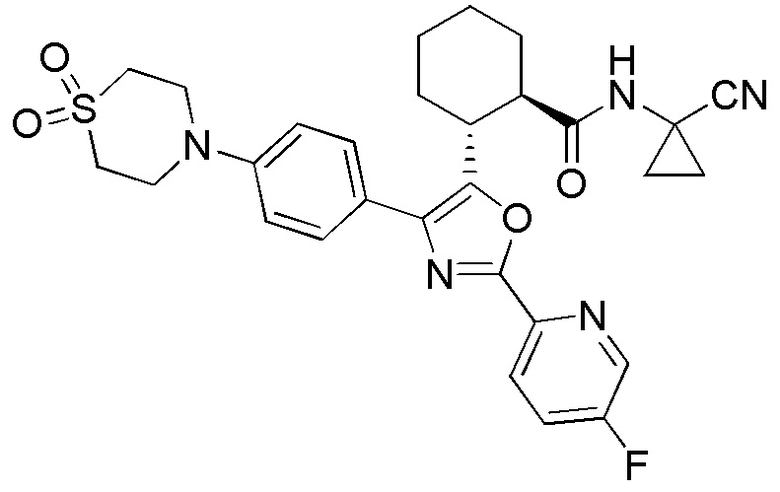
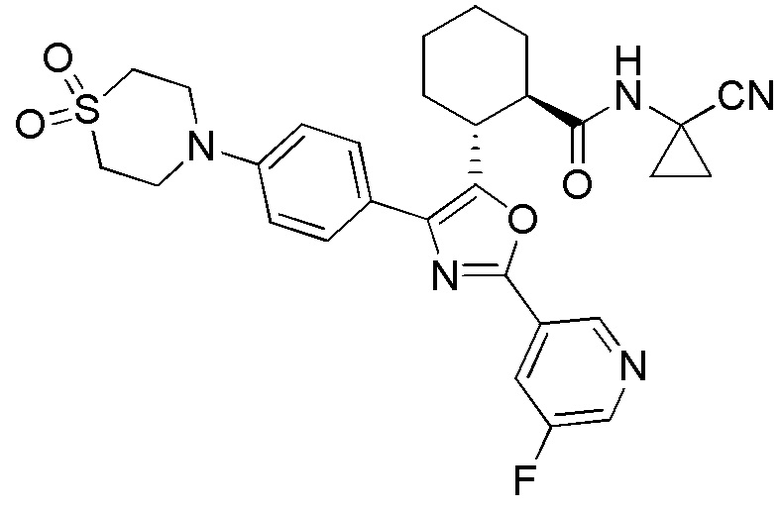
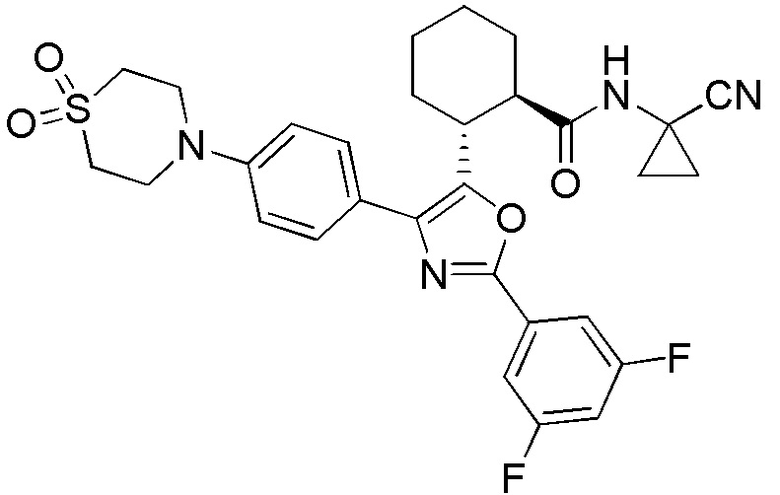
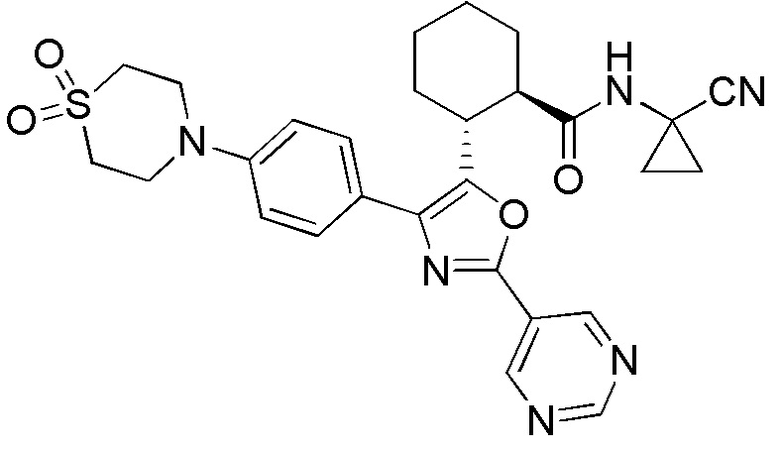
(4-(1,1-диоксидотиоморфолино)-фенил)-2-(пиримидин-5-ил)-оксазол-5-ил)-циклогексанкарбоксамид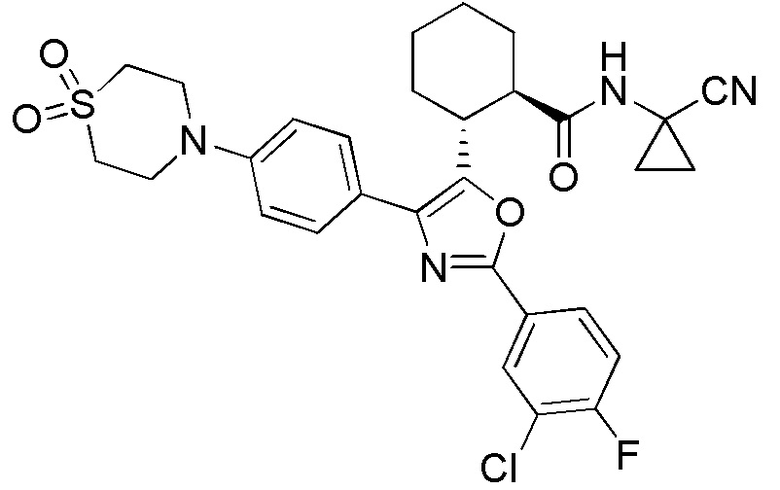
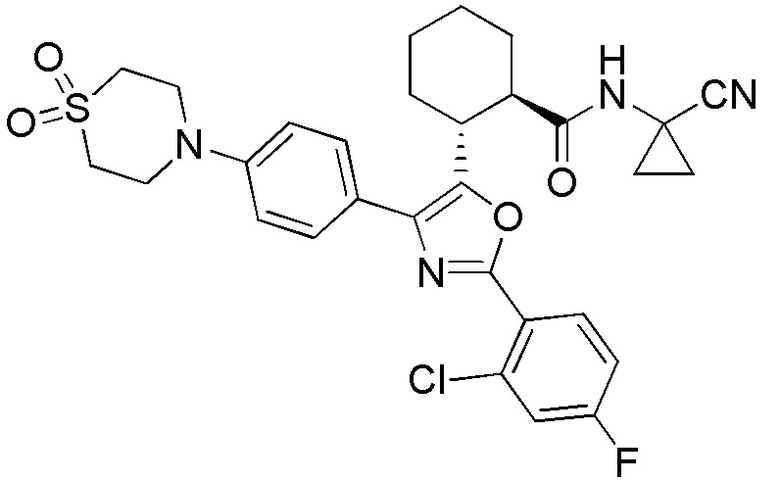
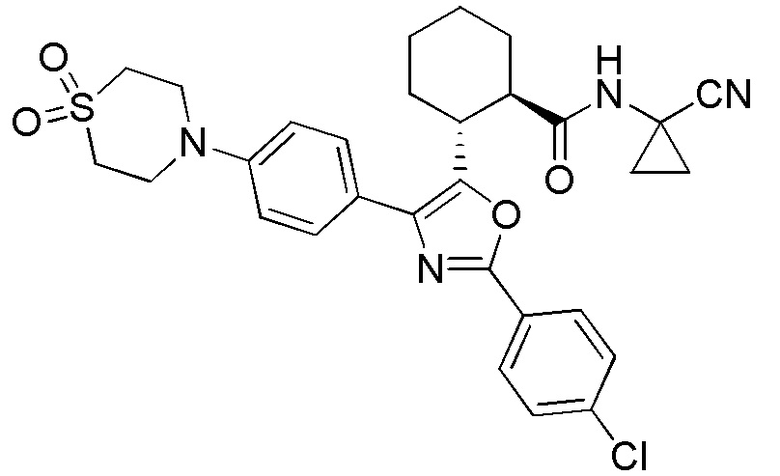
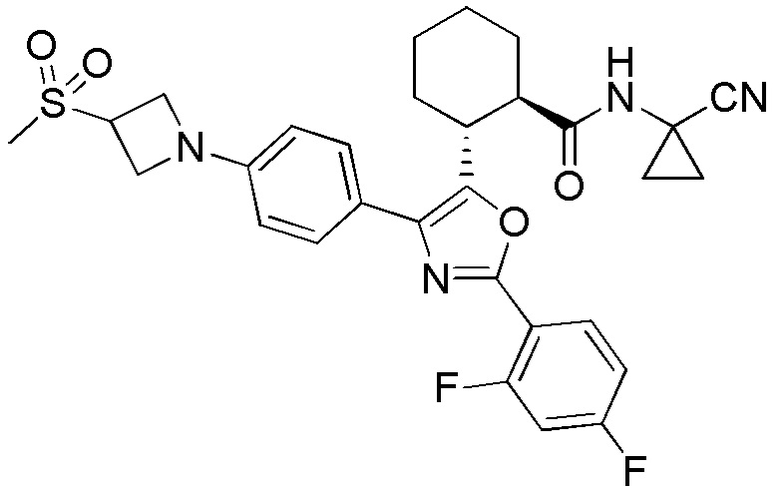
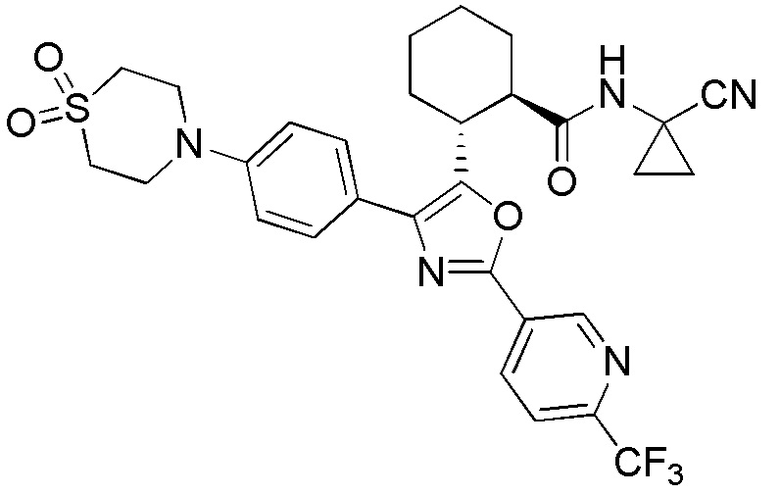
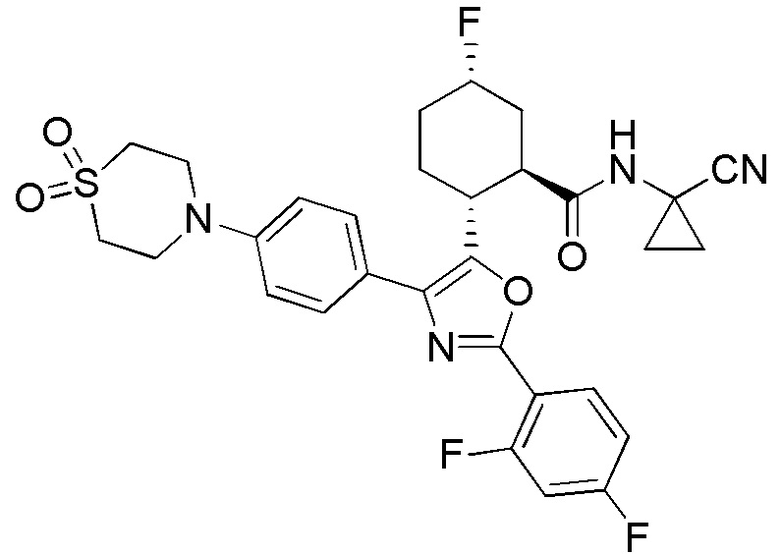
4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)-фенил)оксазол-5-ил)-5-фторциклогексан-карбоксамид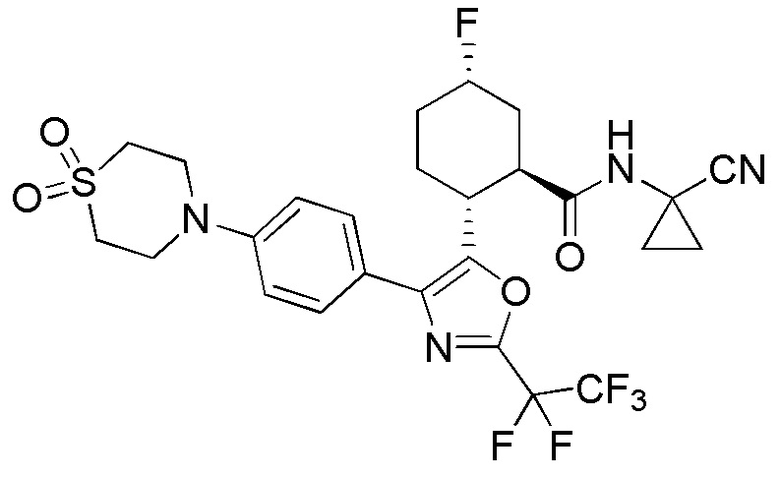
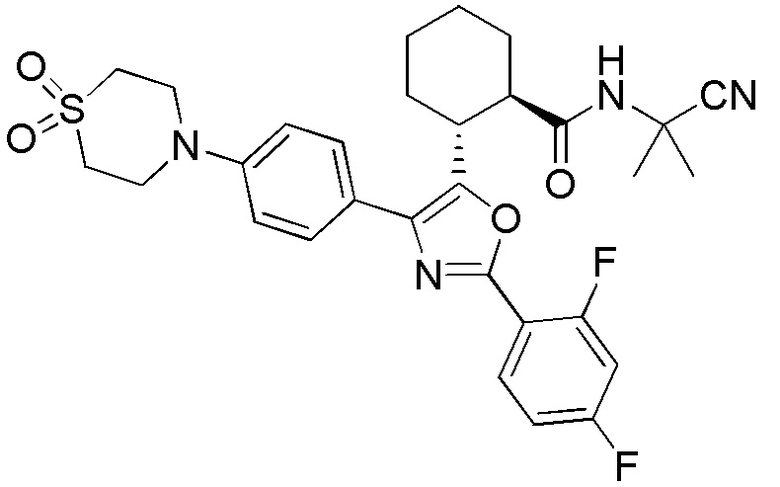
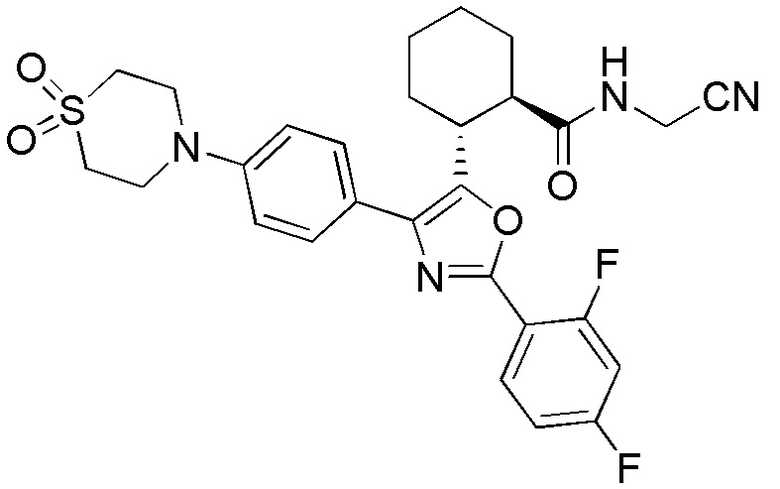
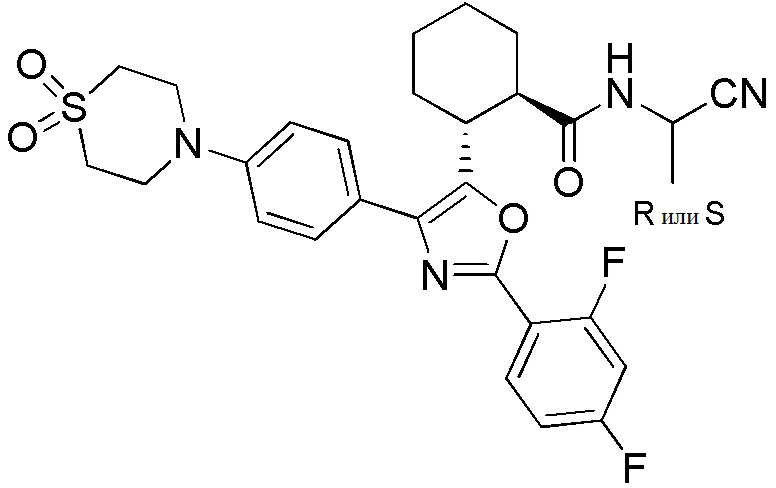
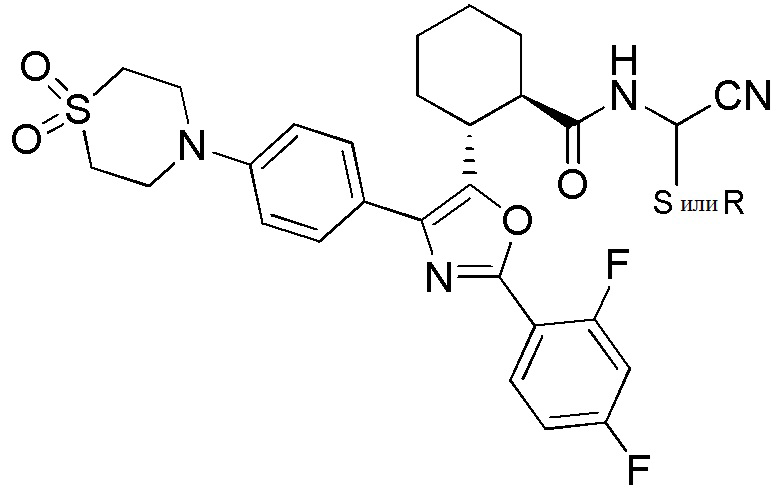
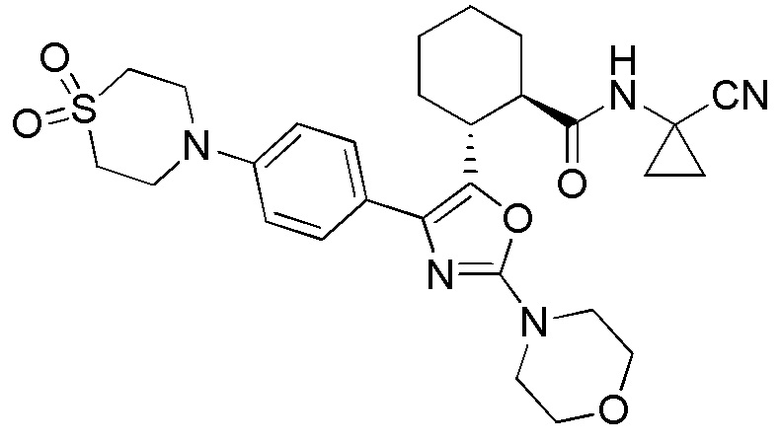
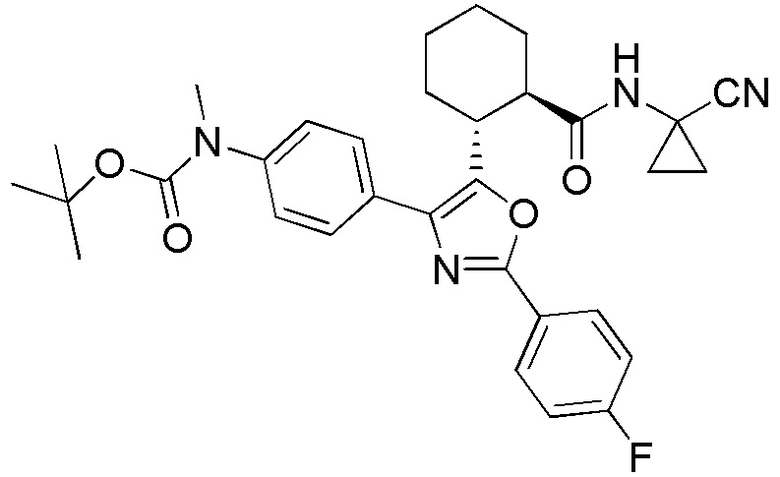
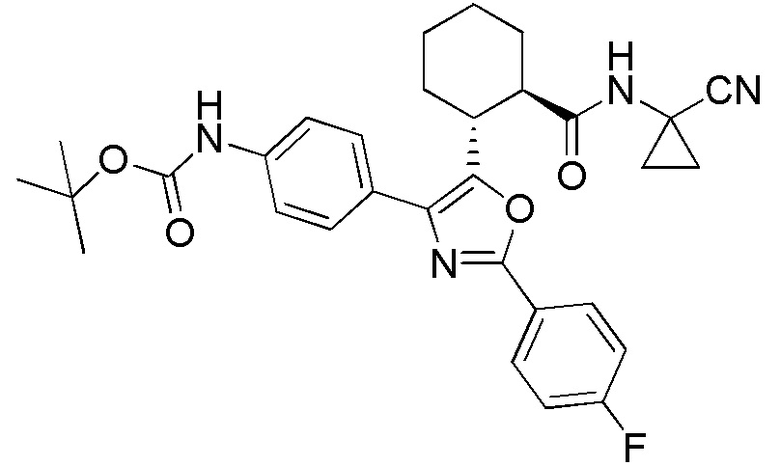
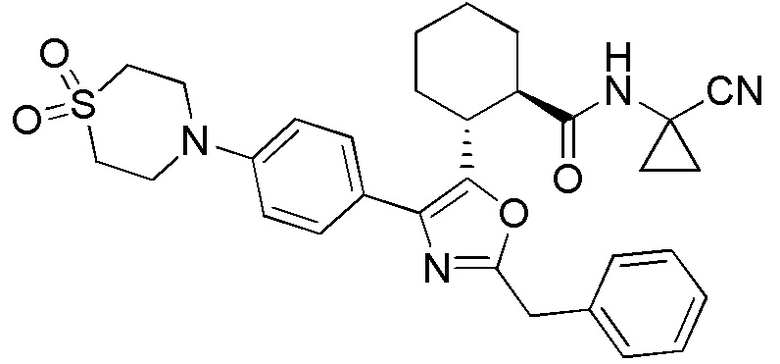
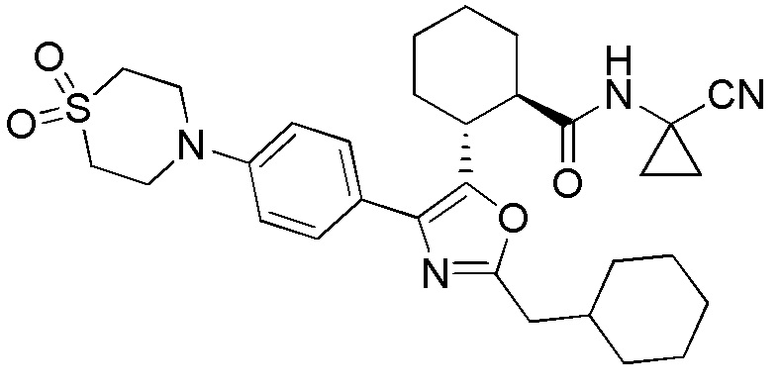
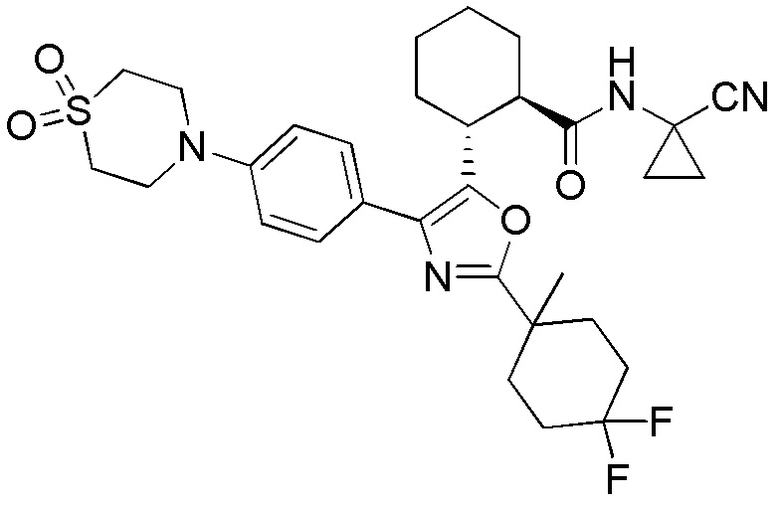
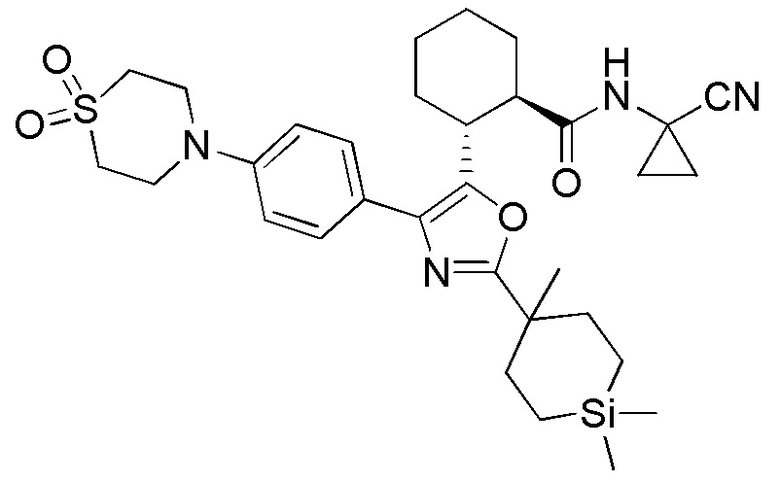
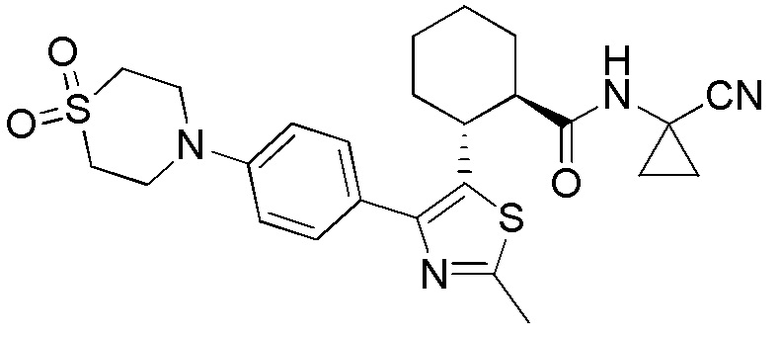
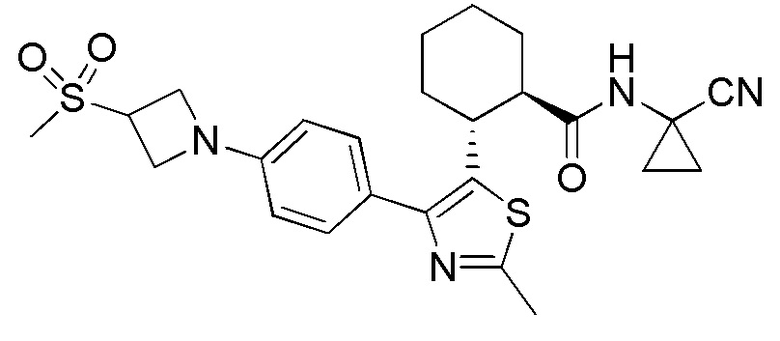
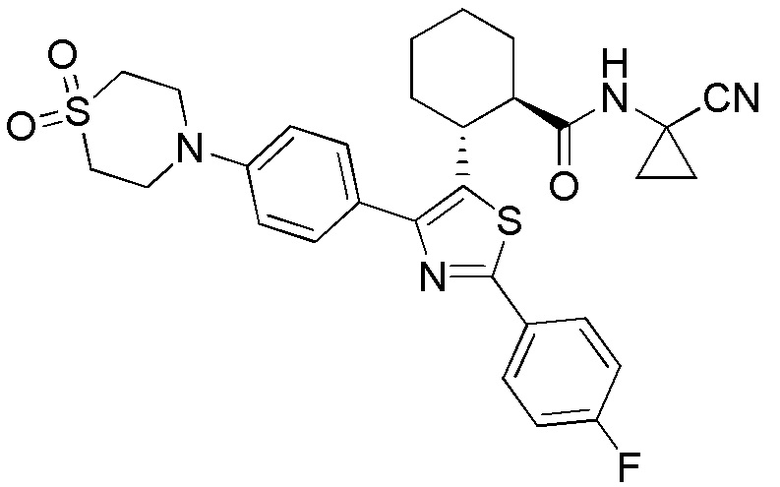
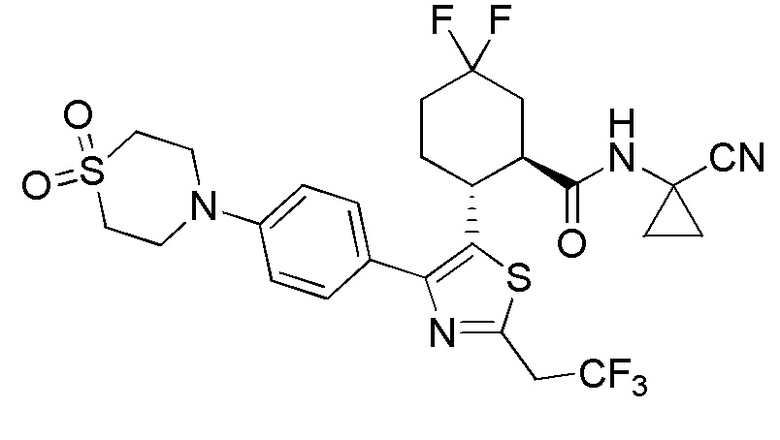
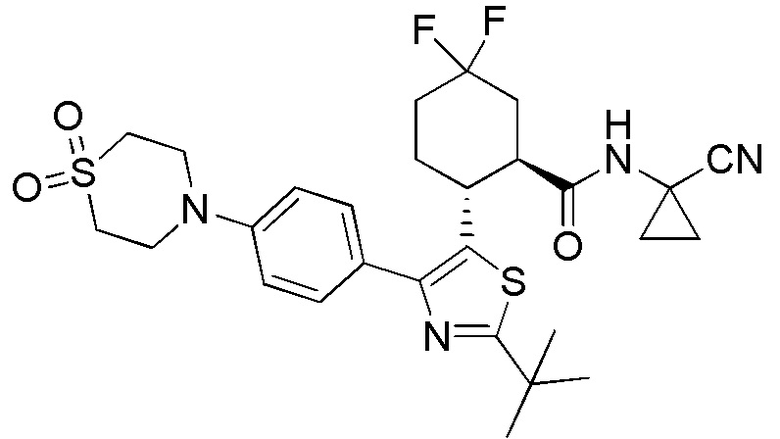
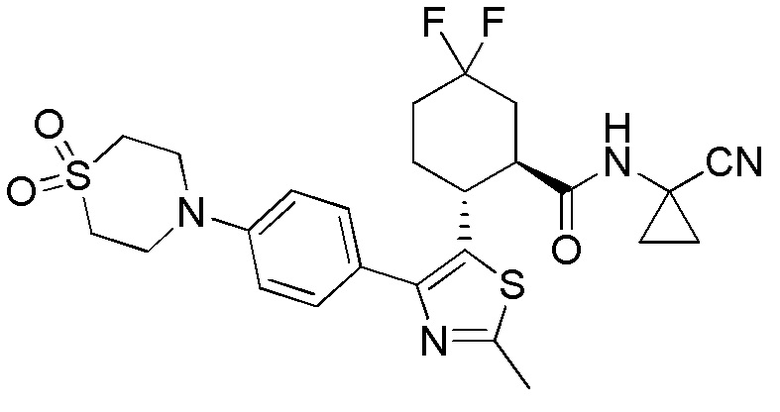
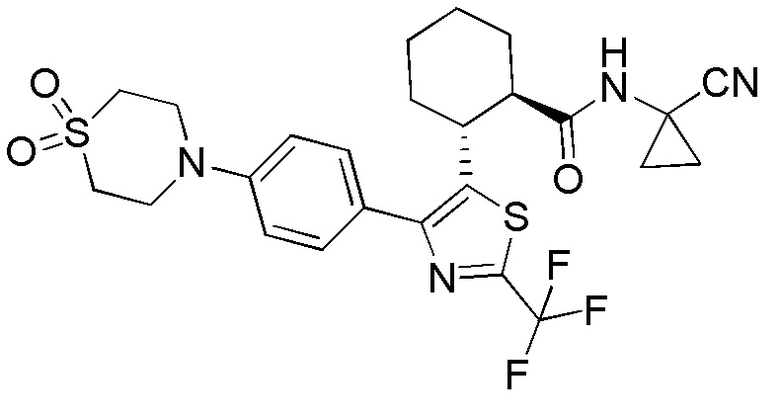
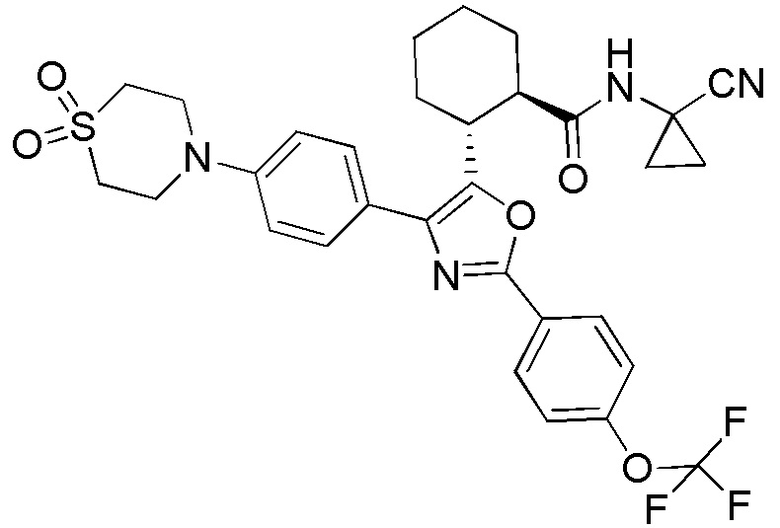
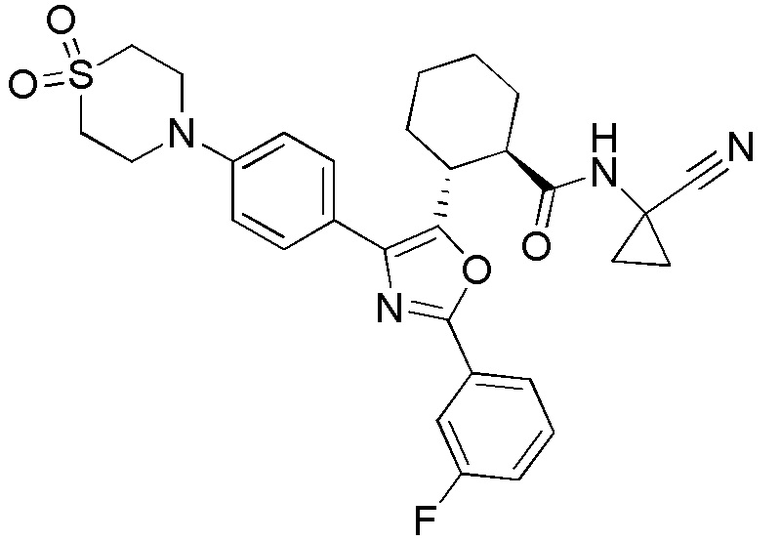
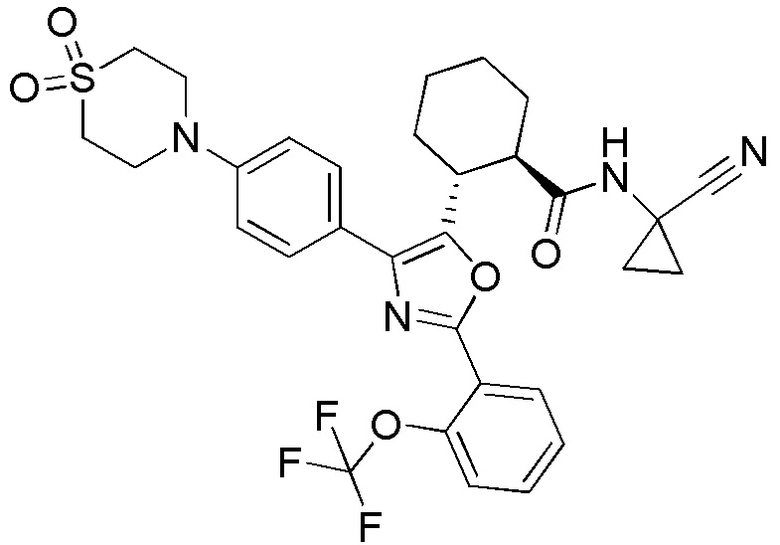
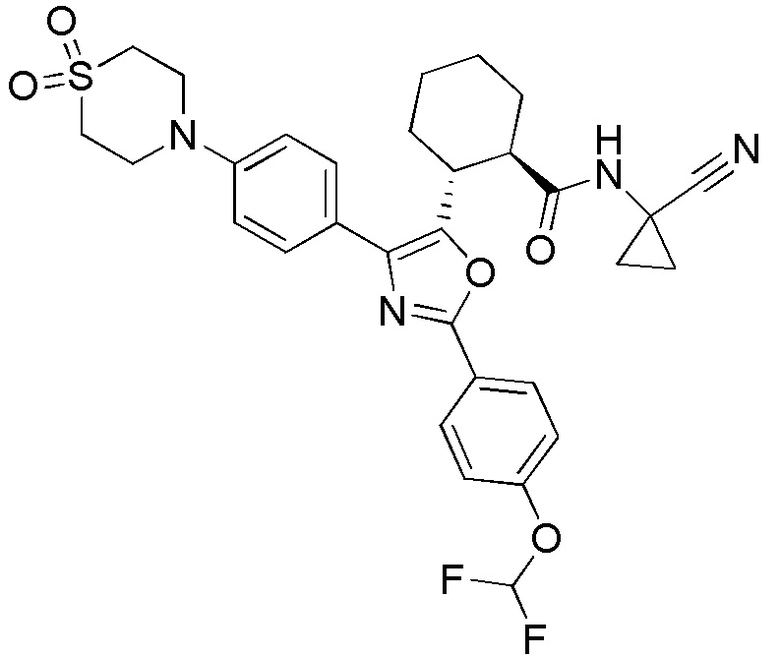
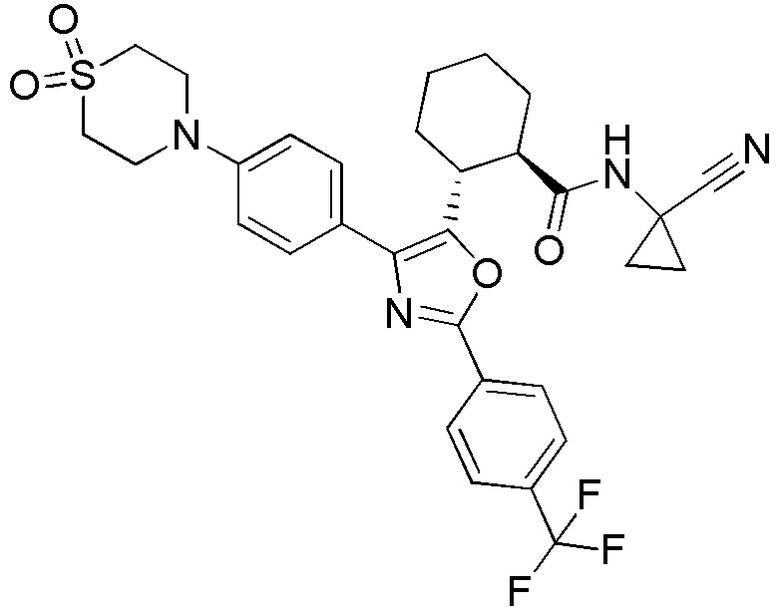
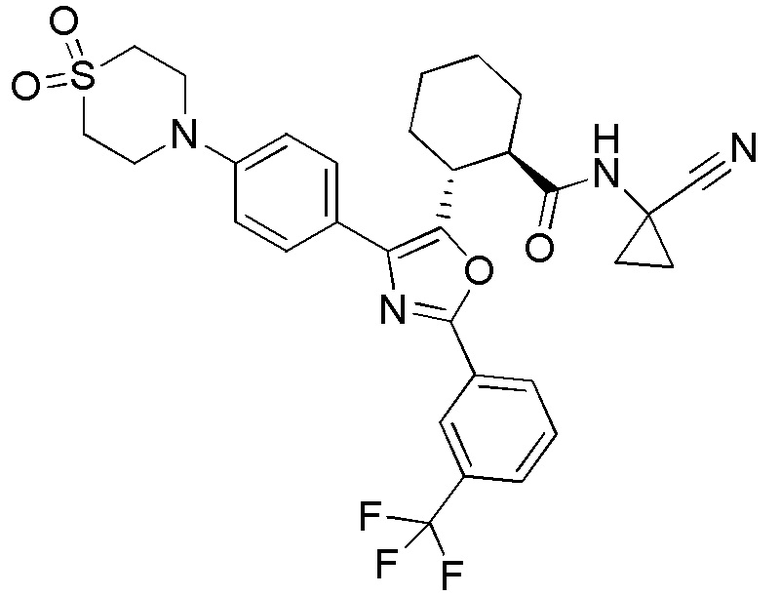
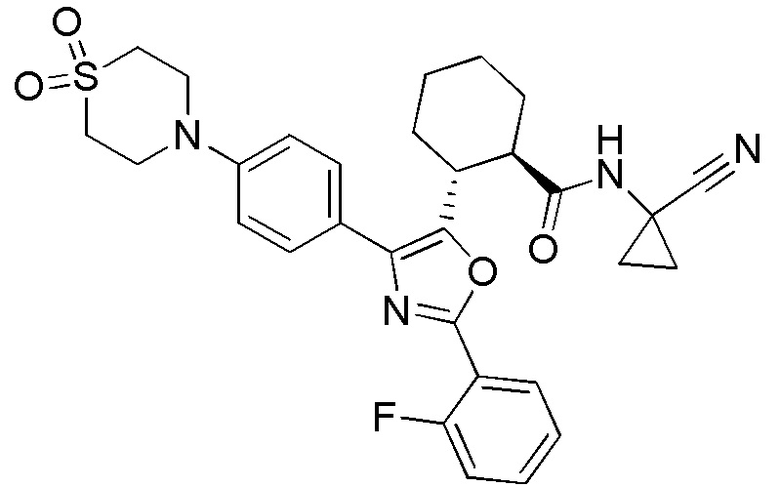
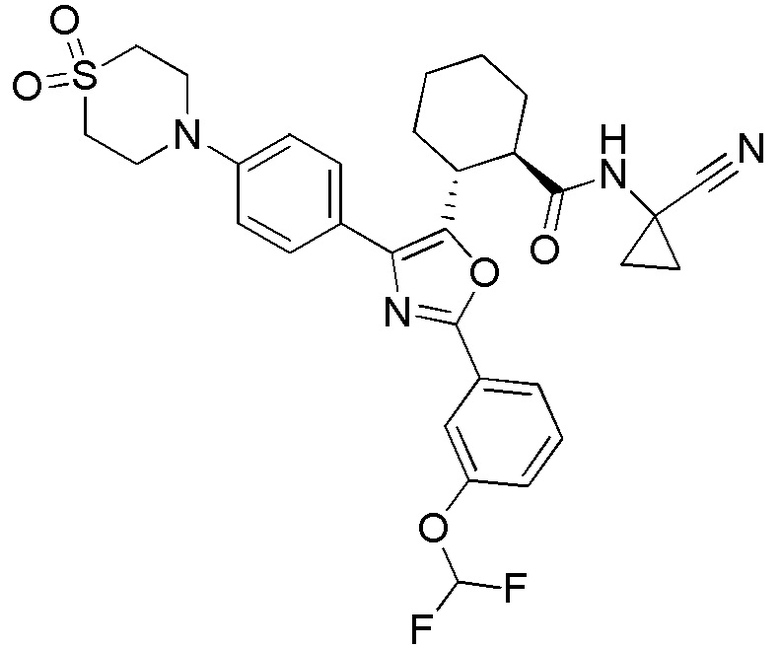
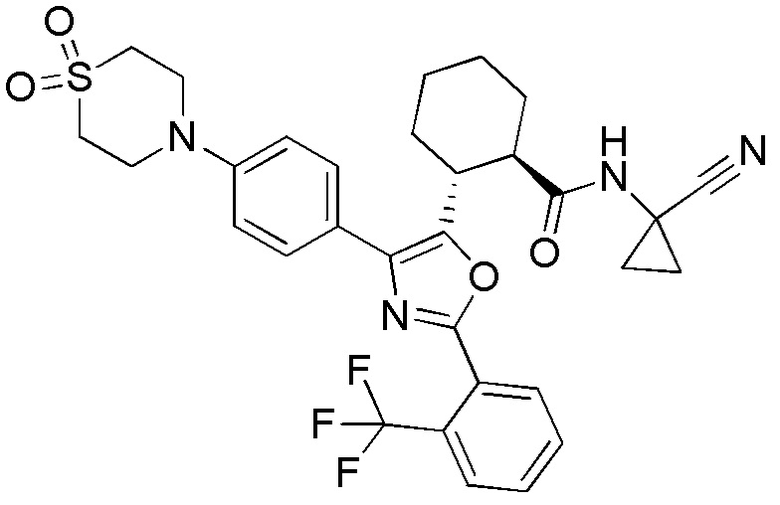
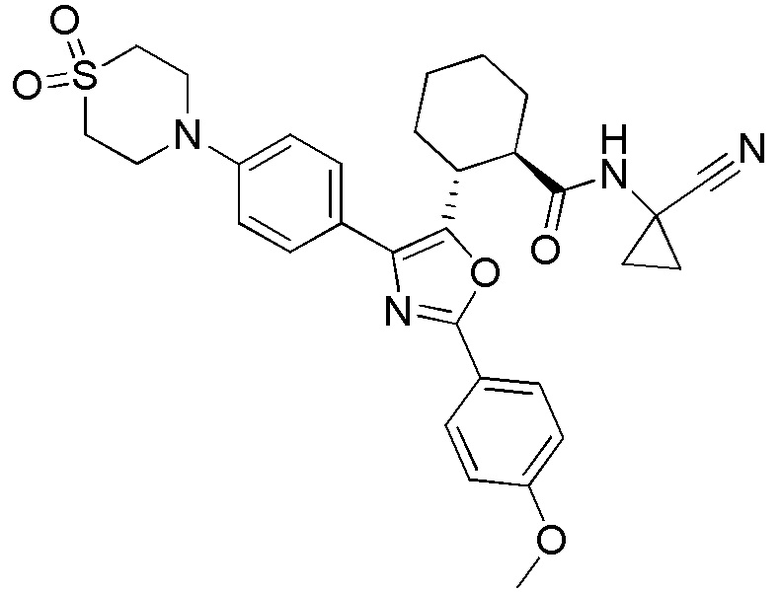
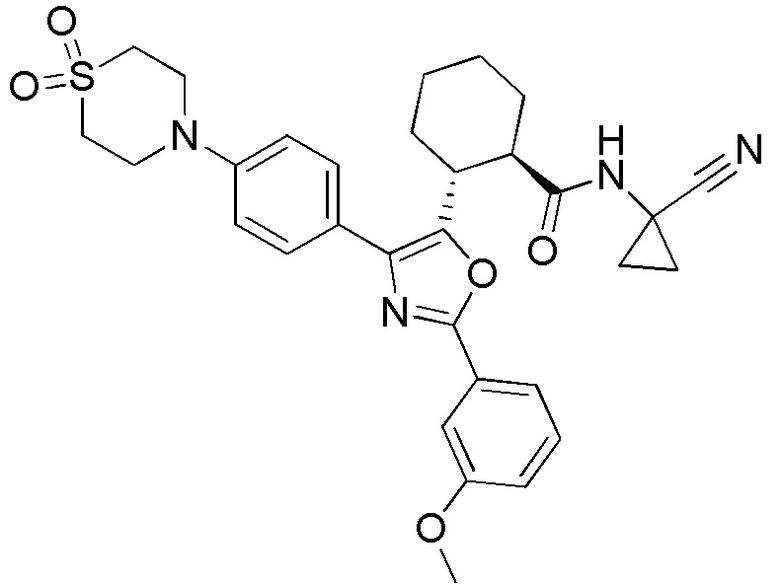
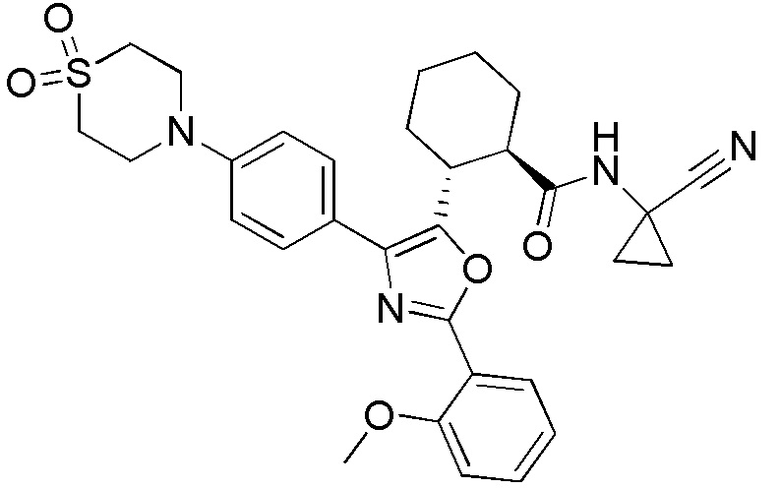
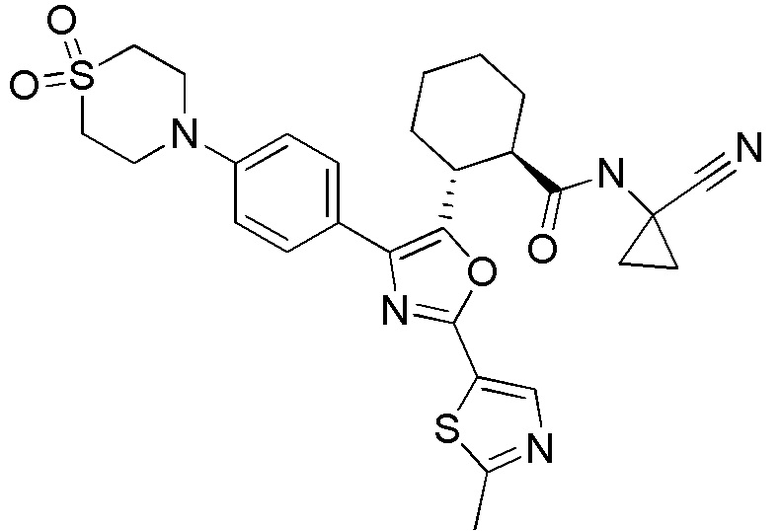
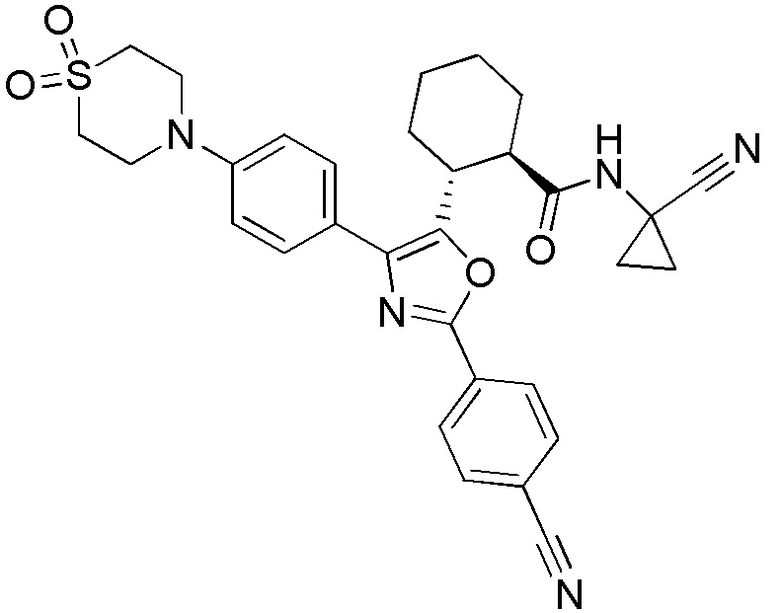
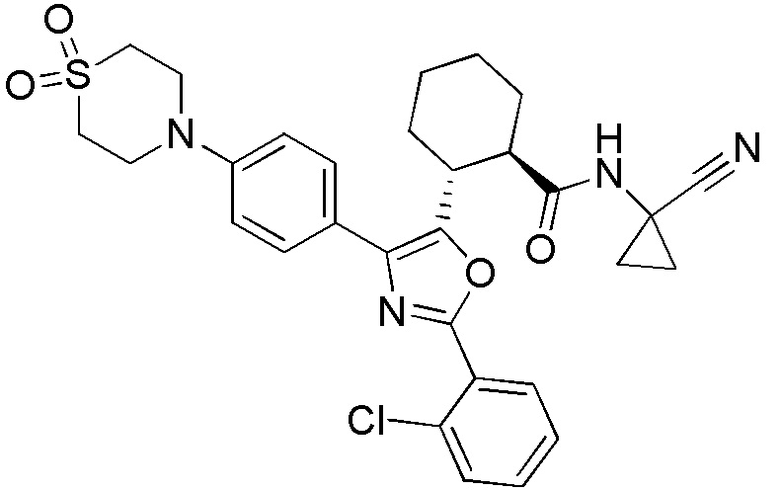
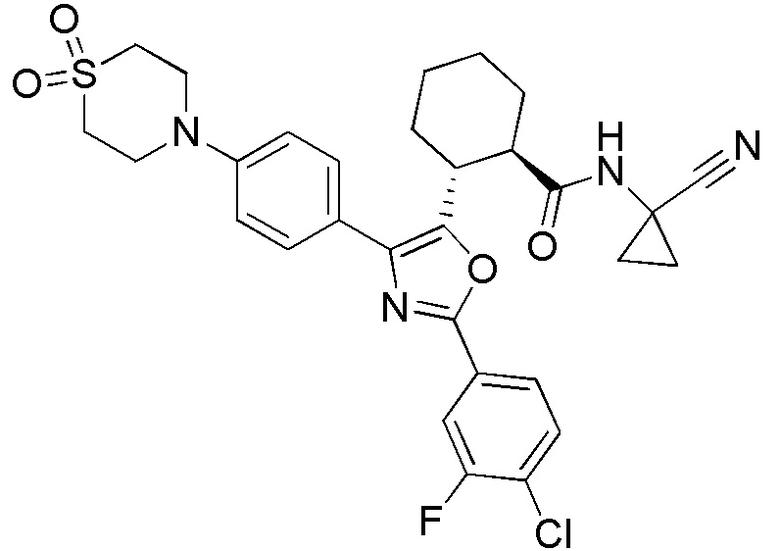
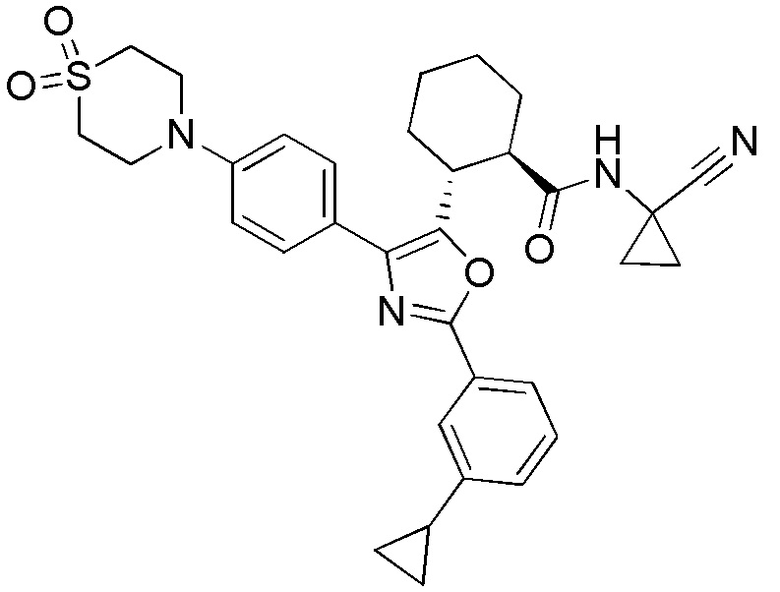
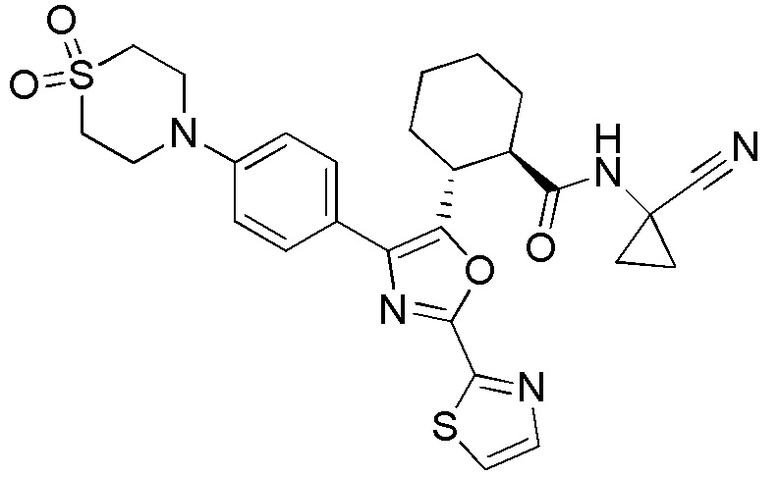
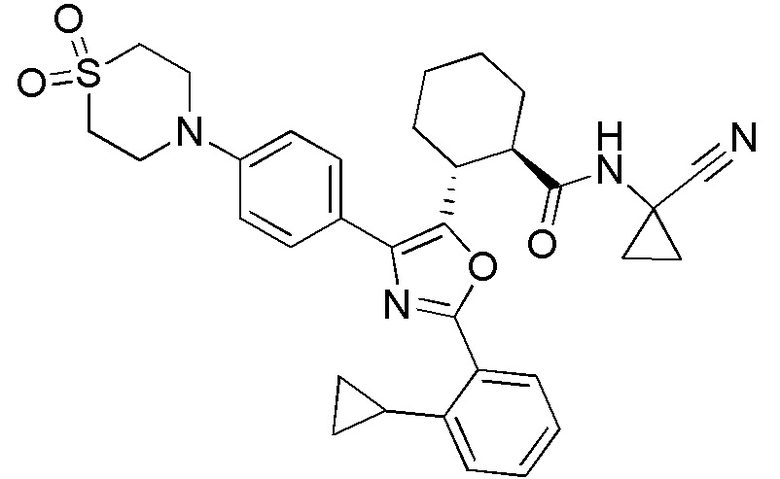
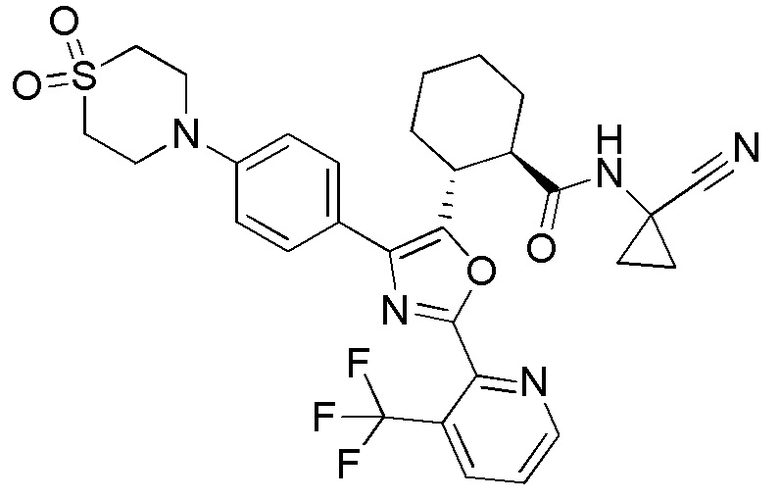
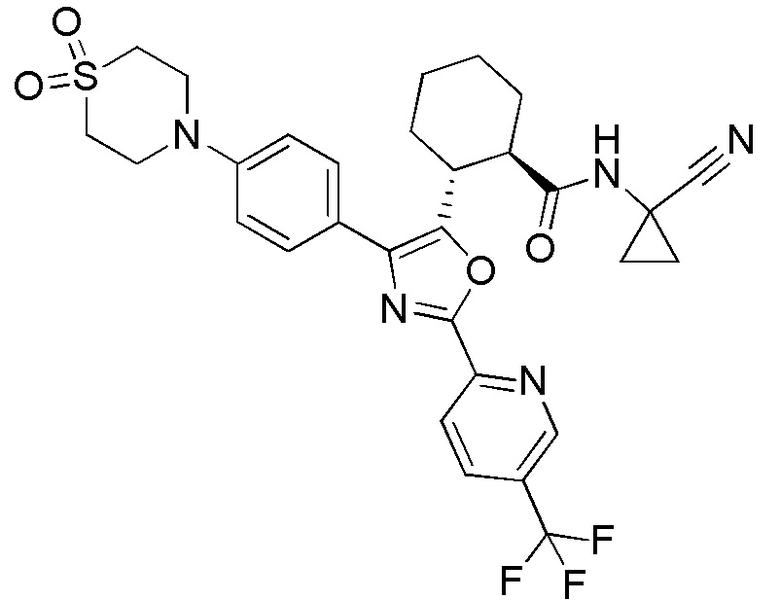
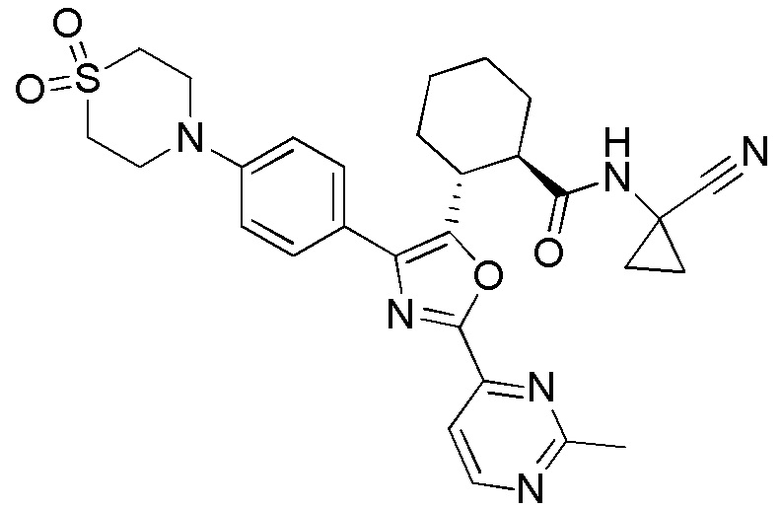
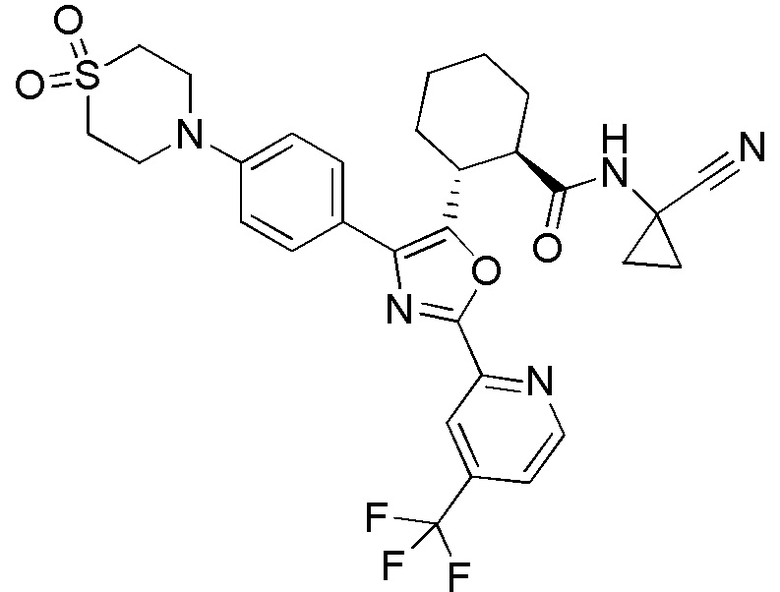
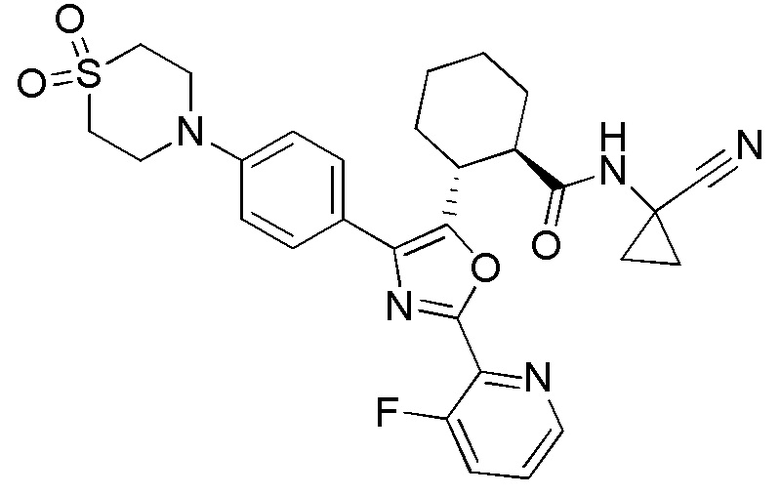
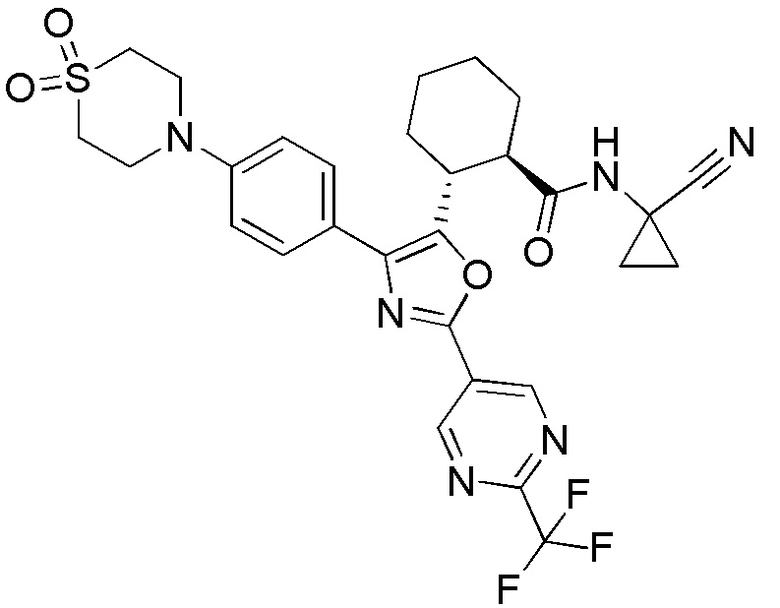
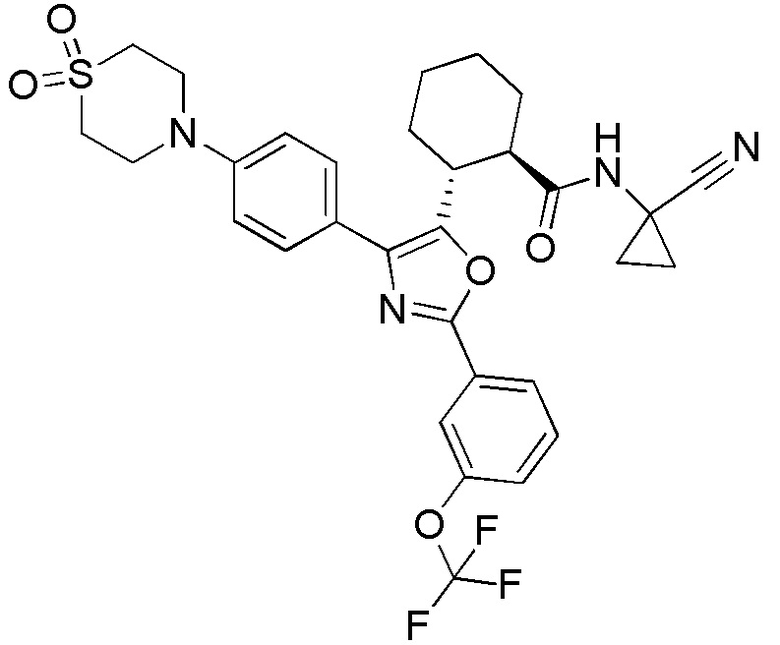
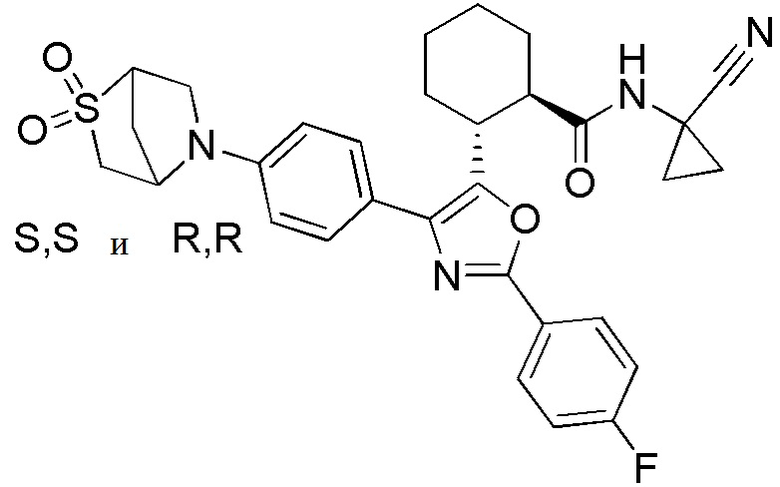
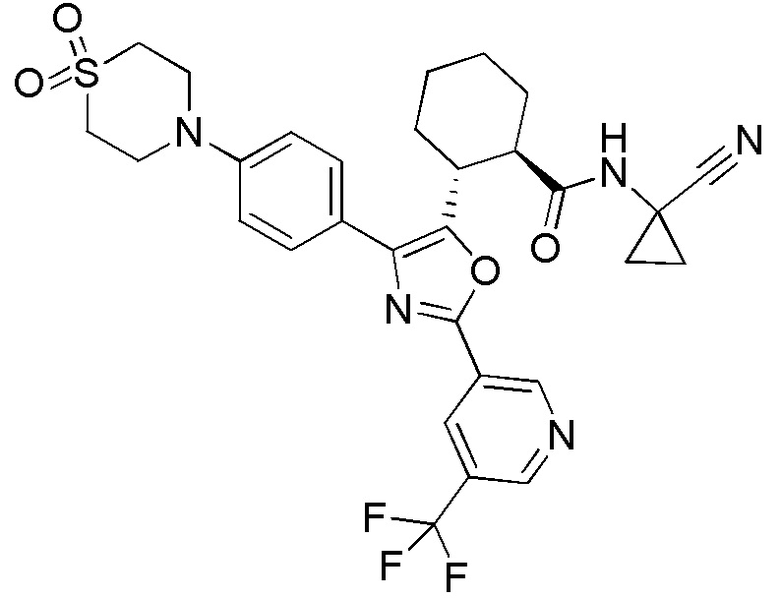
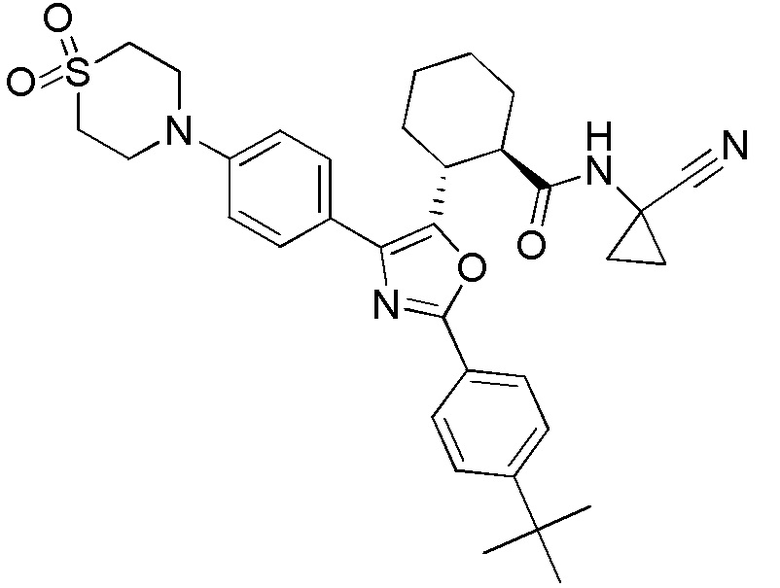
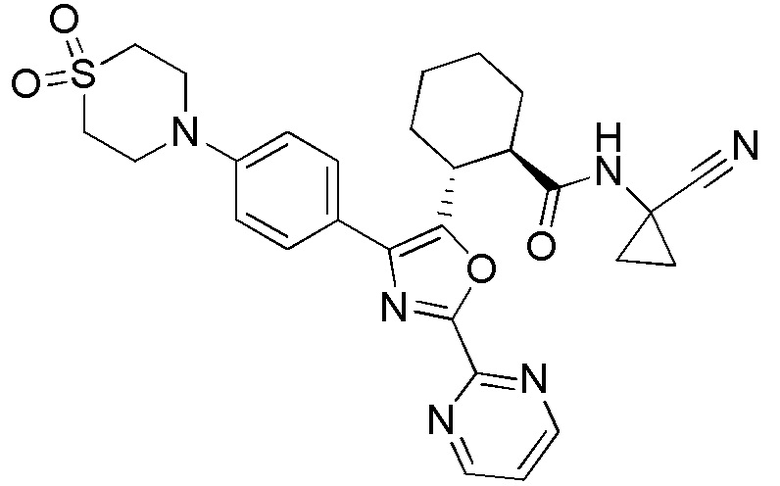
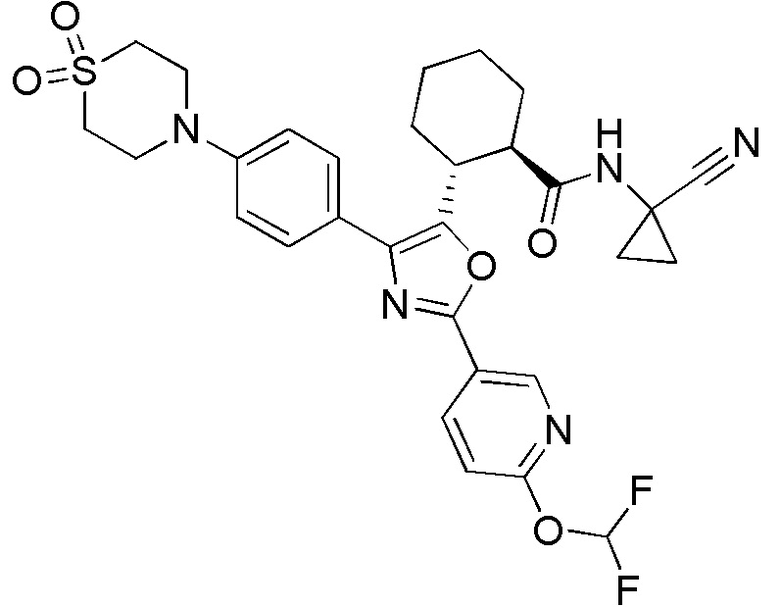
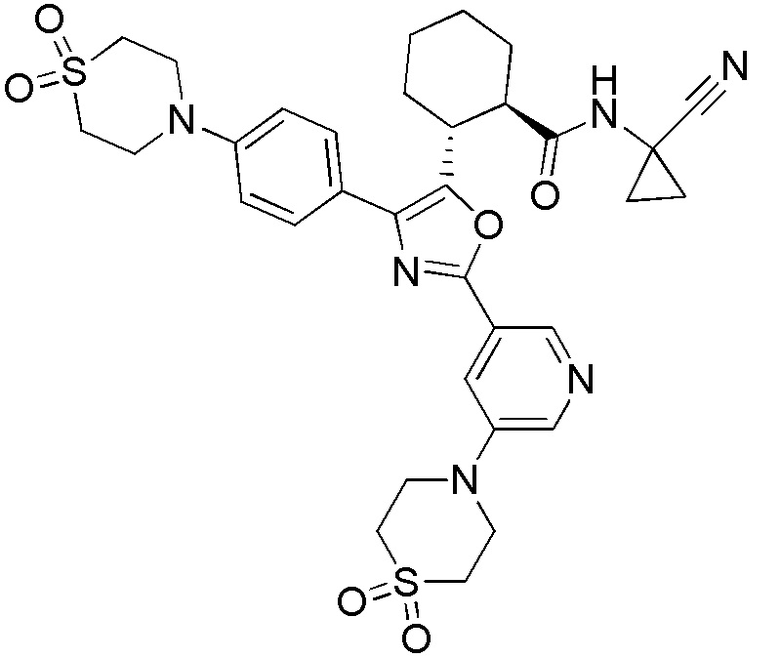
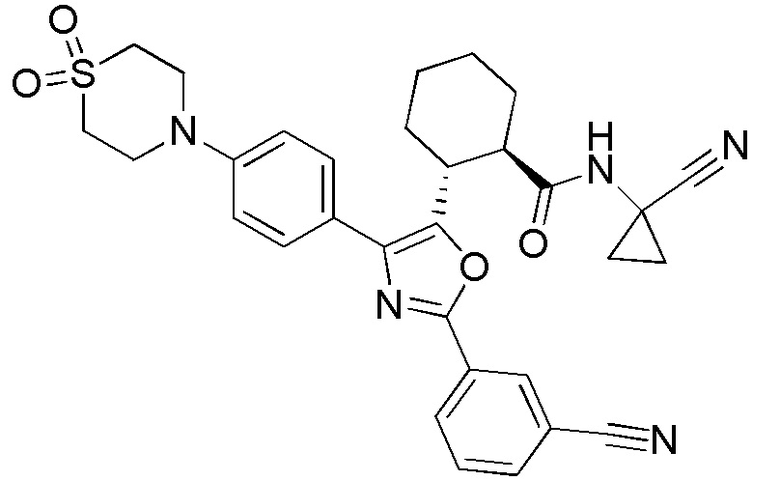
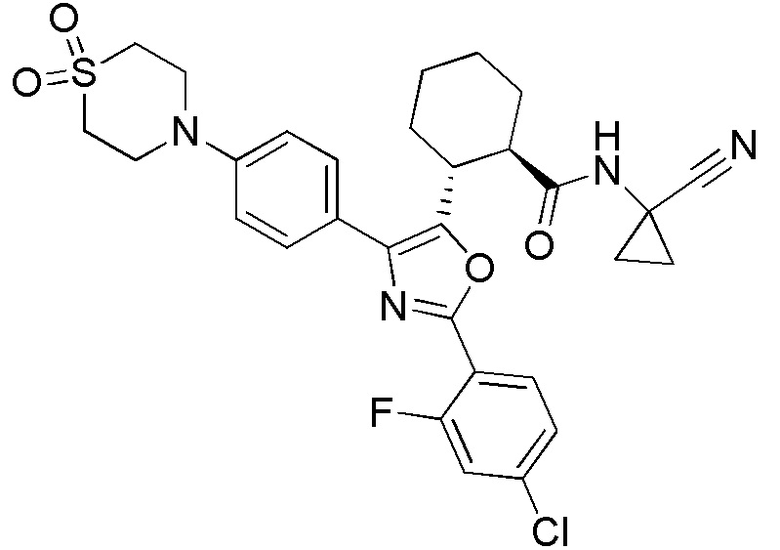
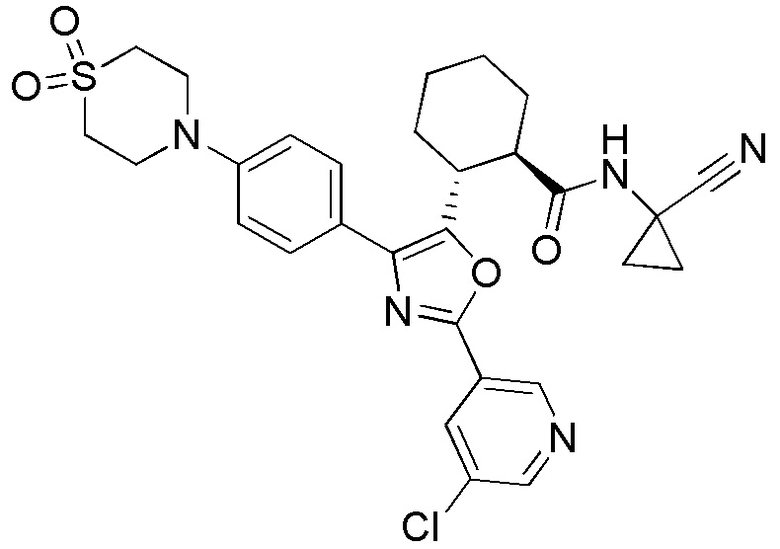
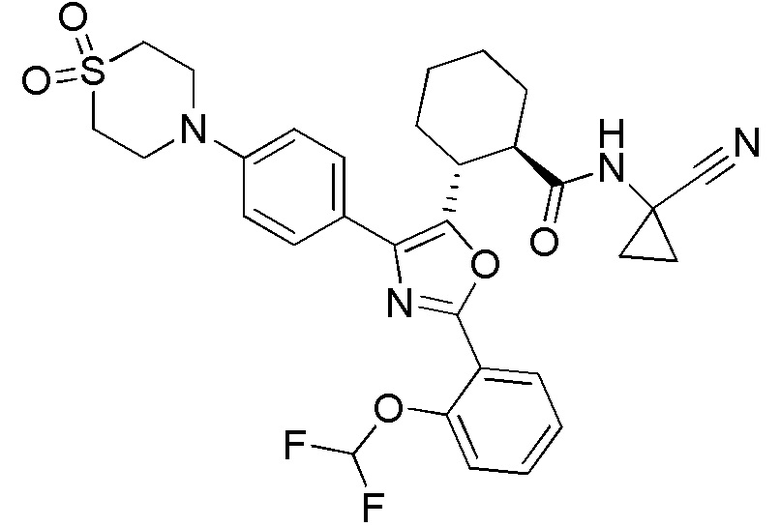
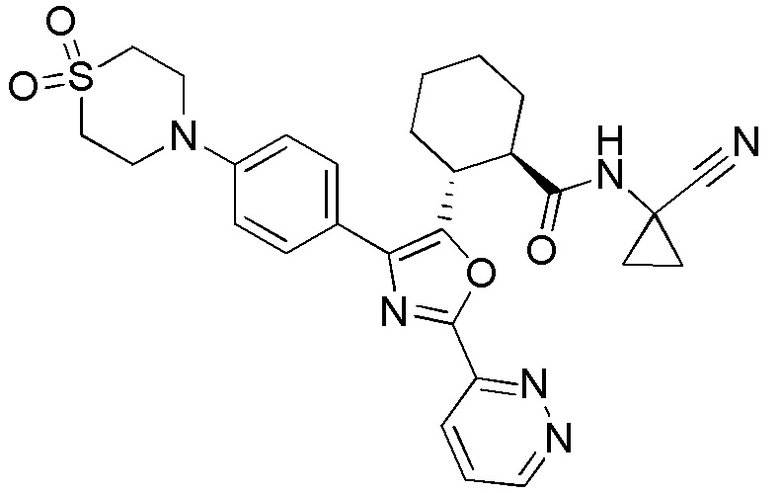
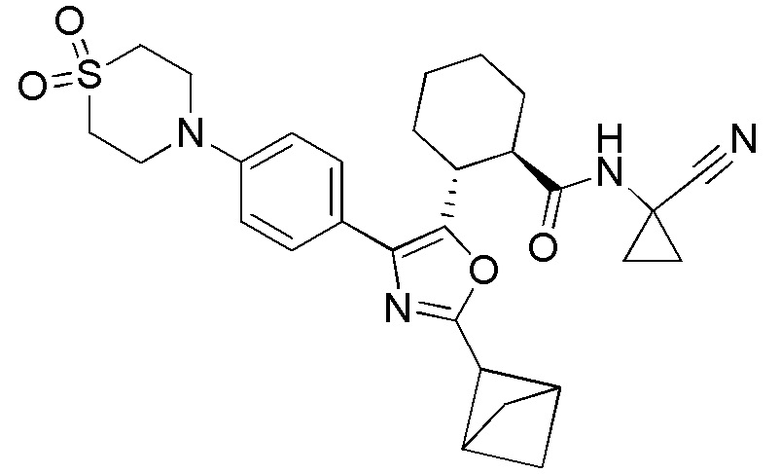
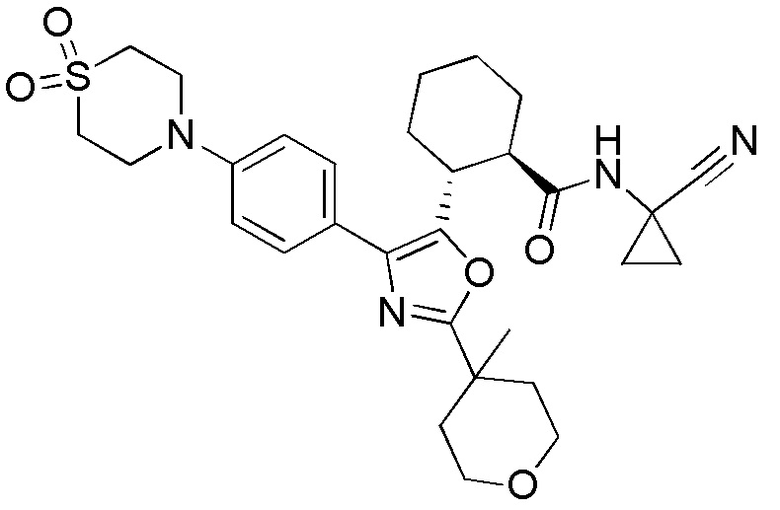
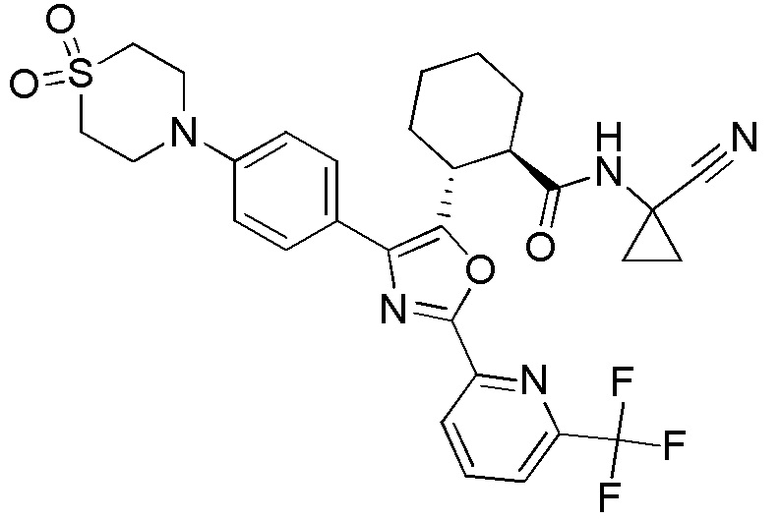
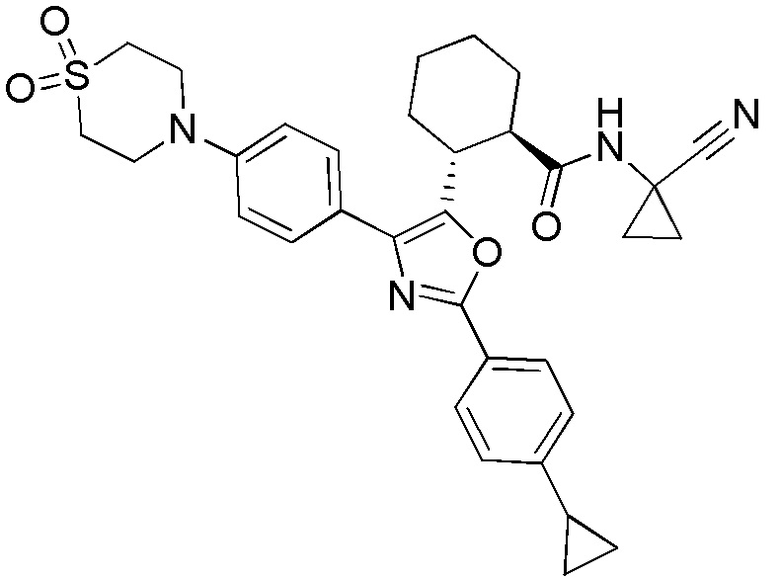
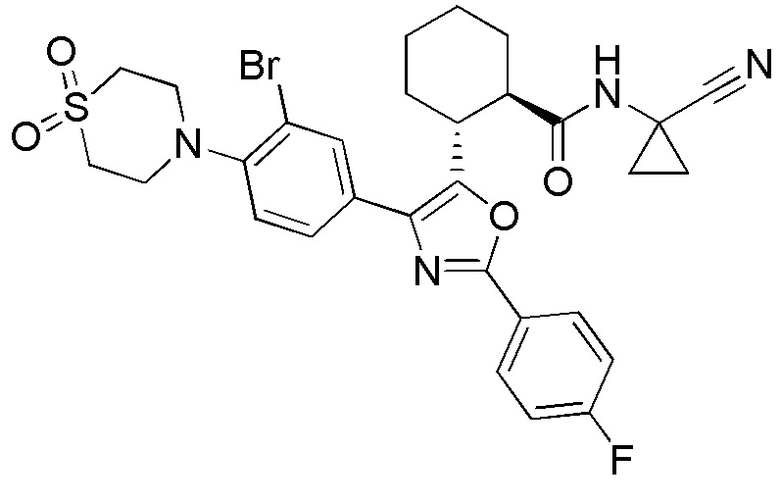
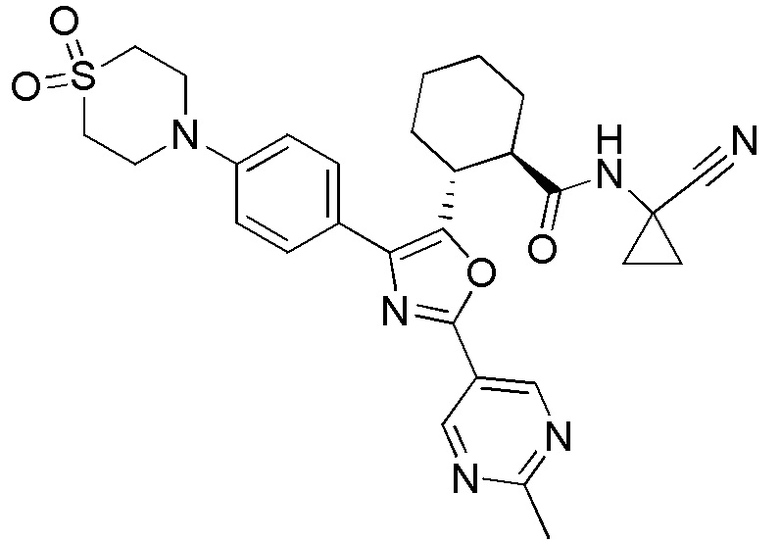
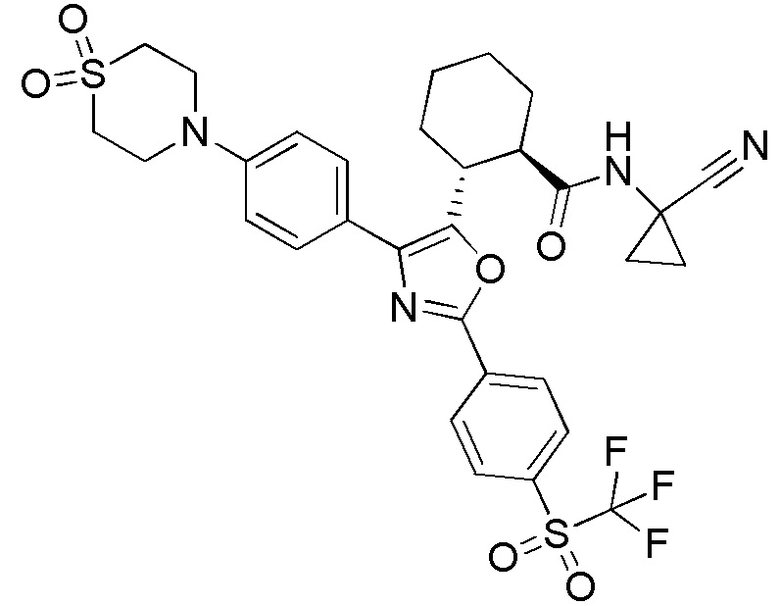
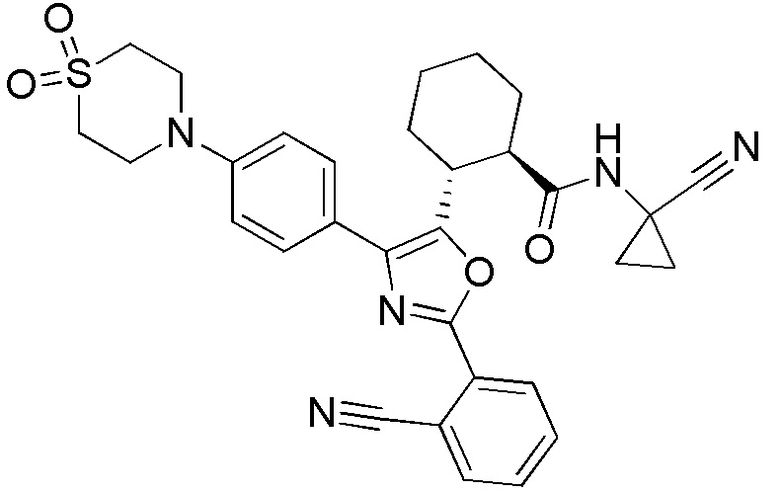
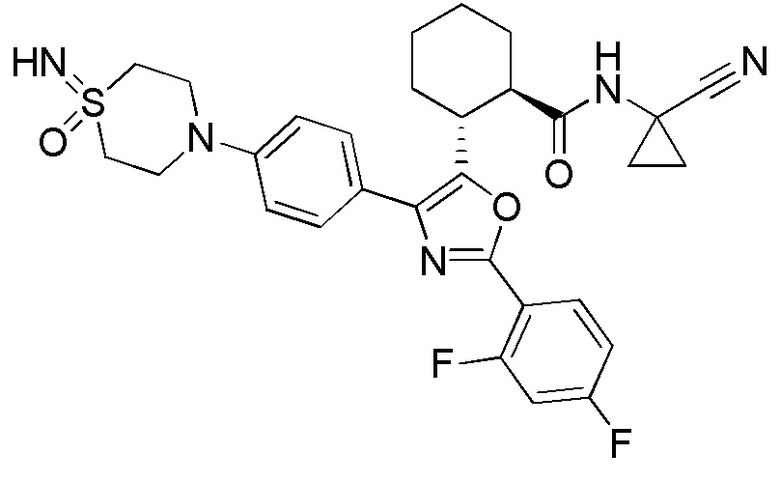
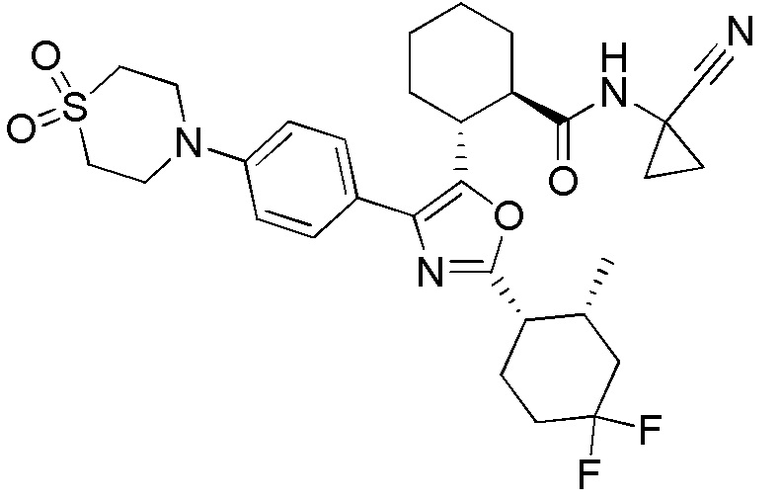
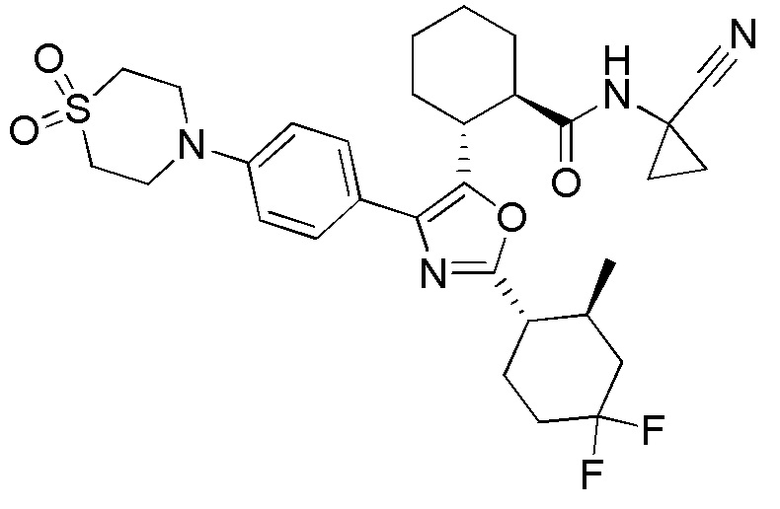
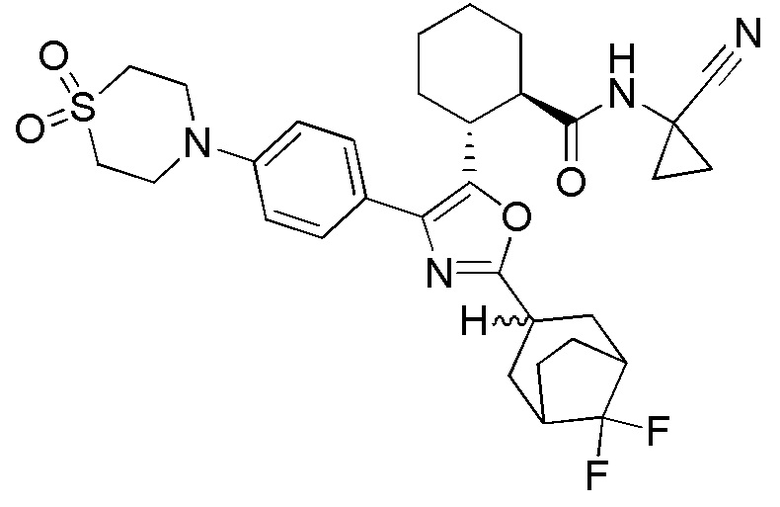
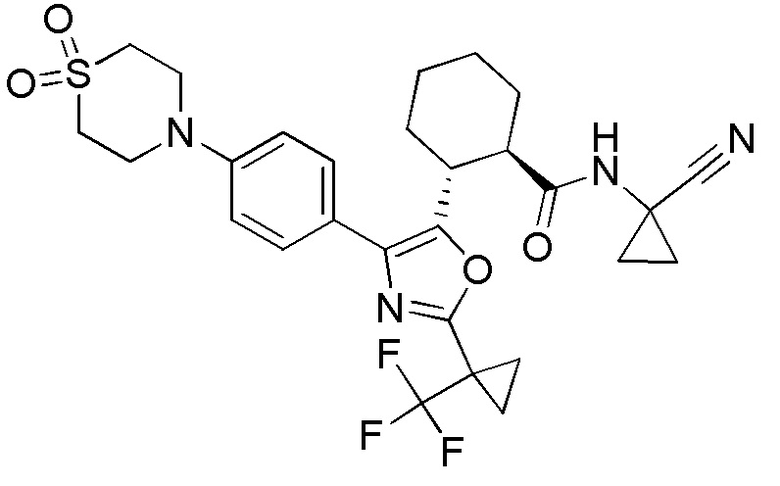
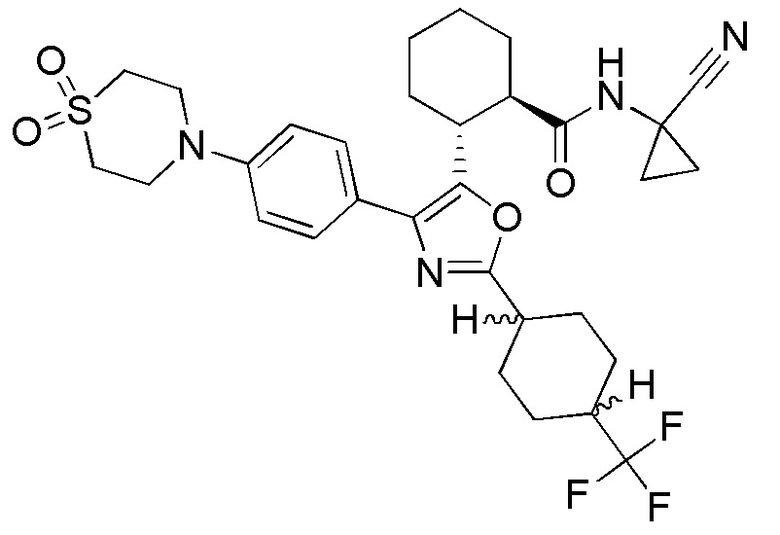
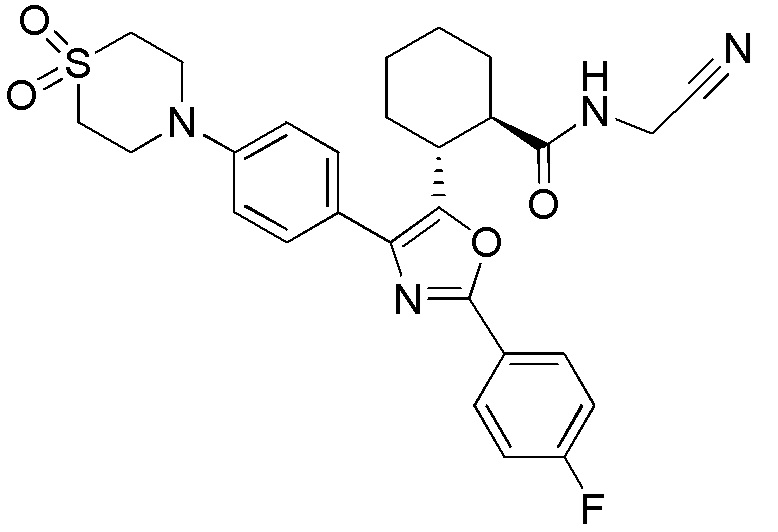
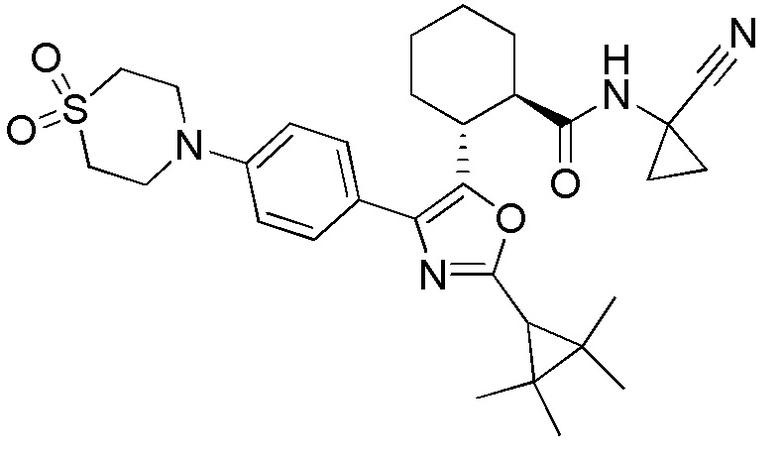
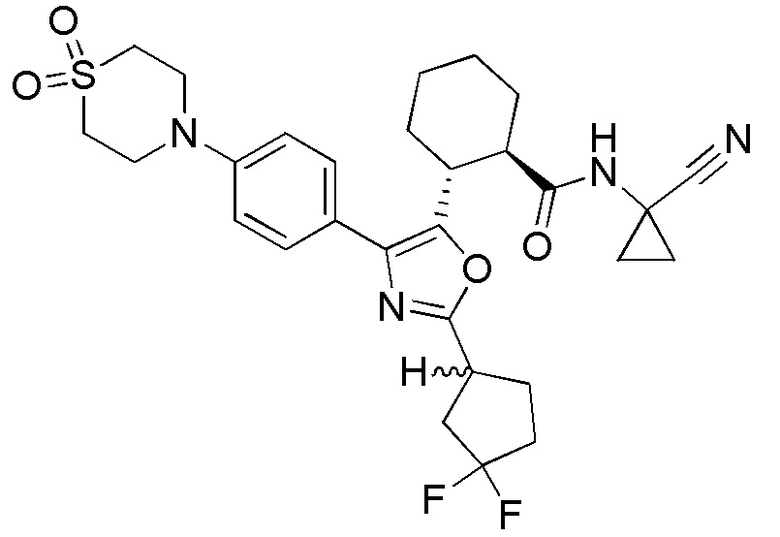
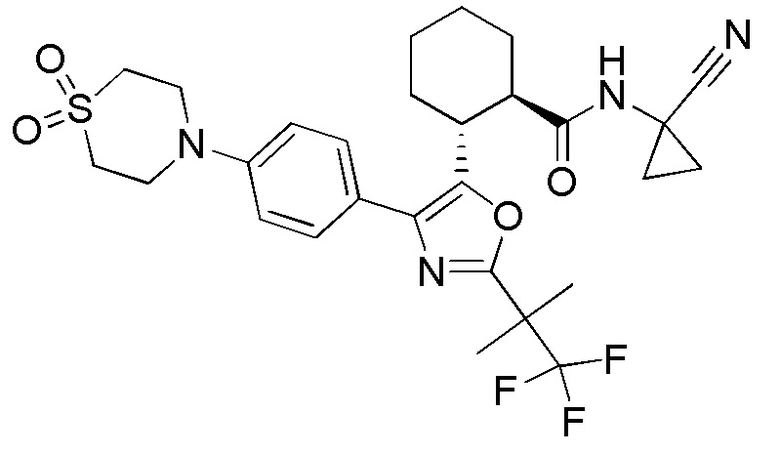
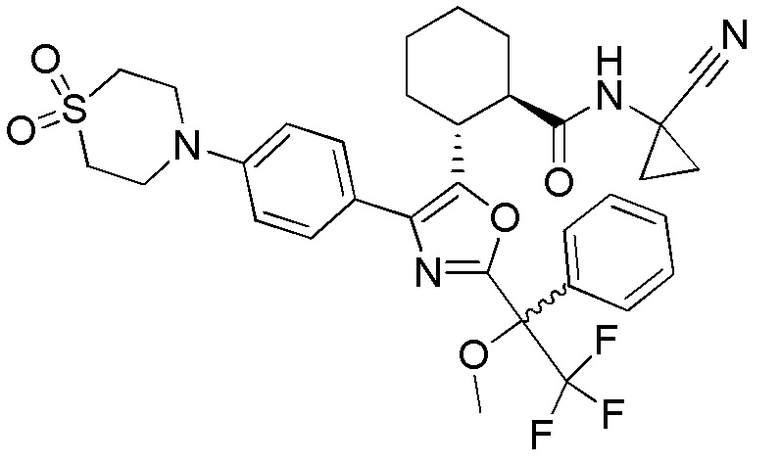
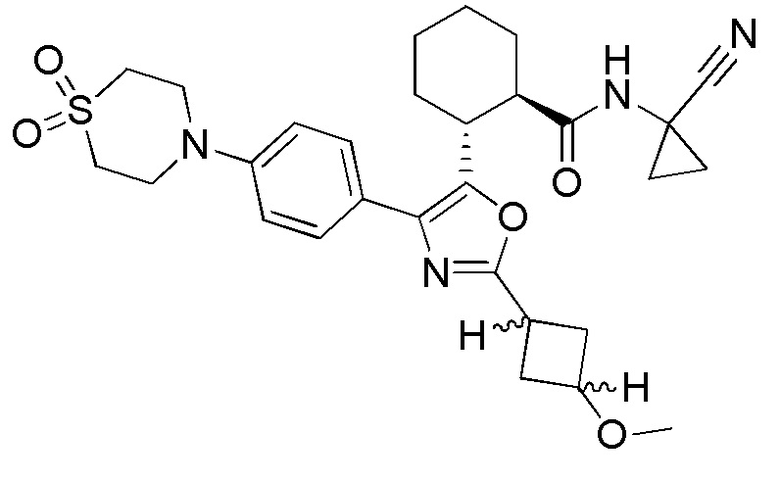
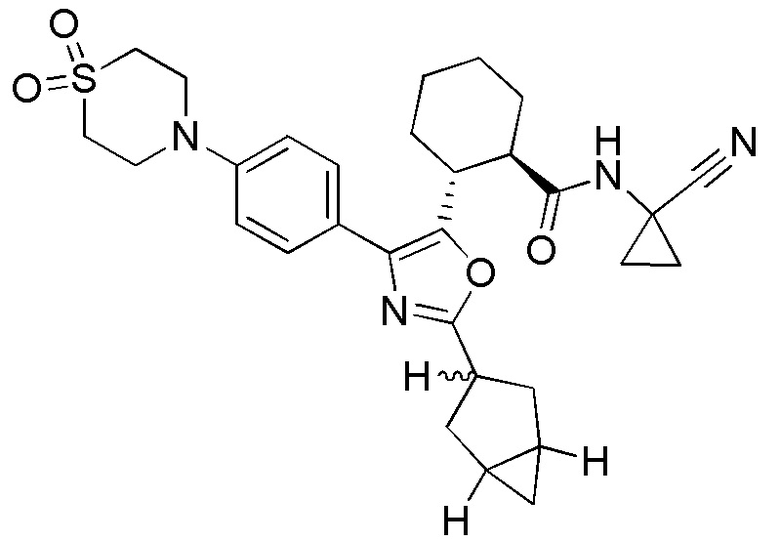
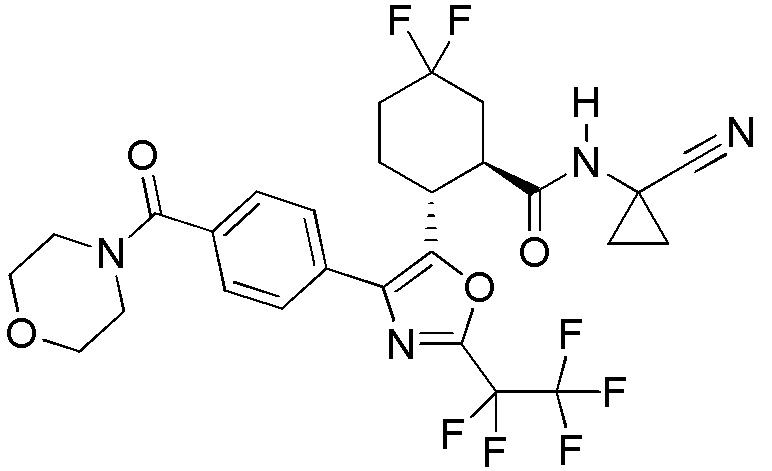
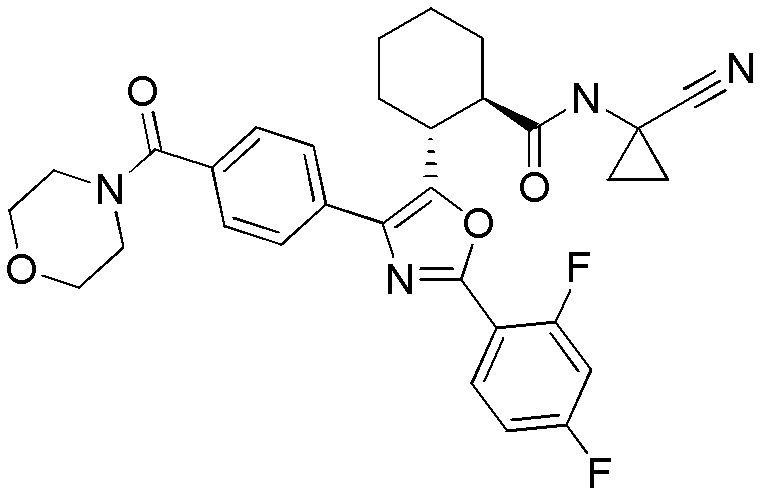
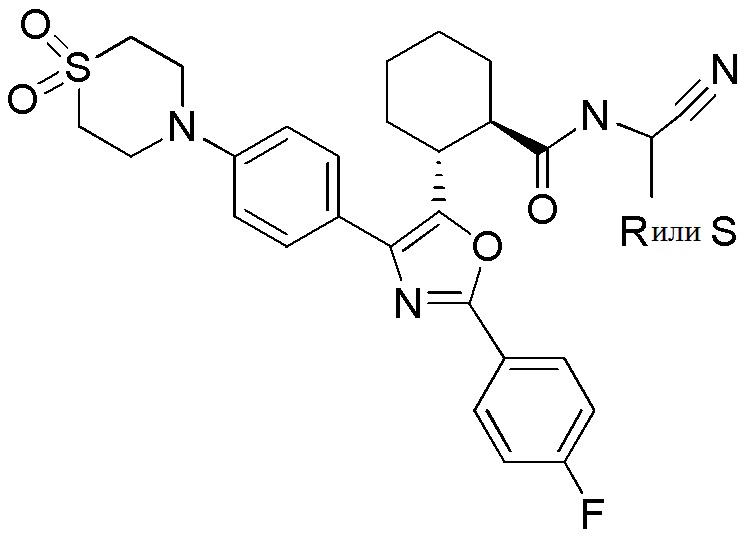
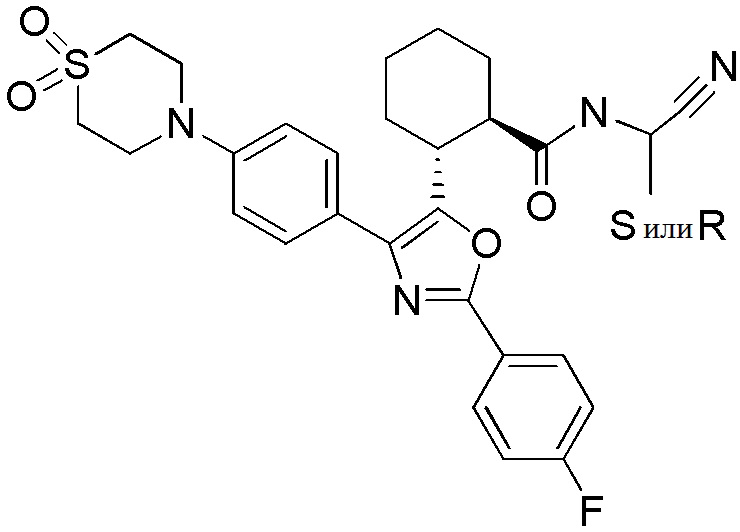
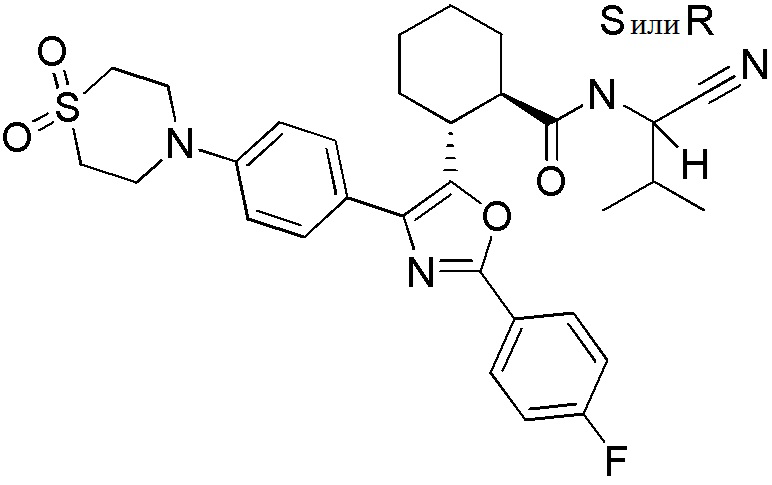
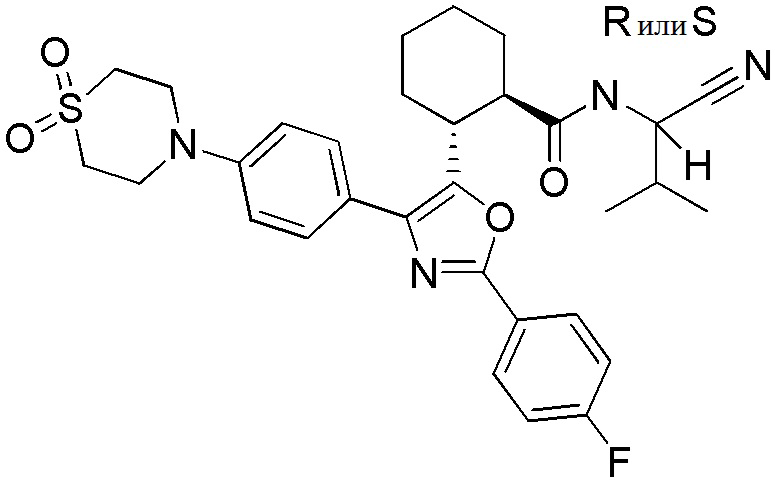
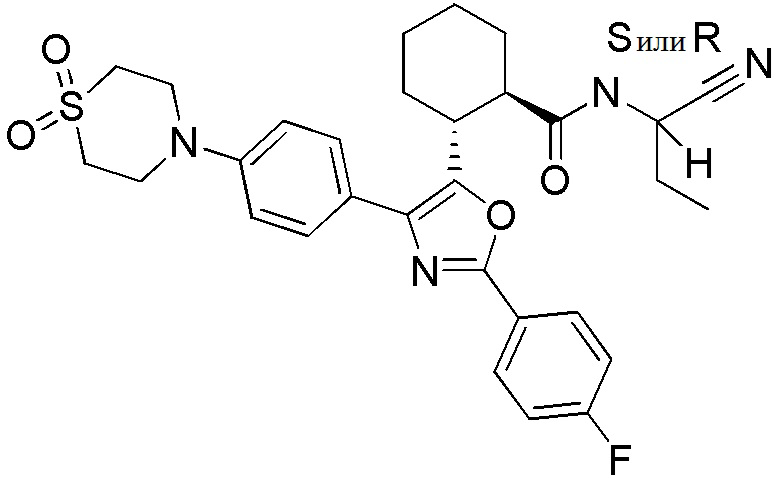
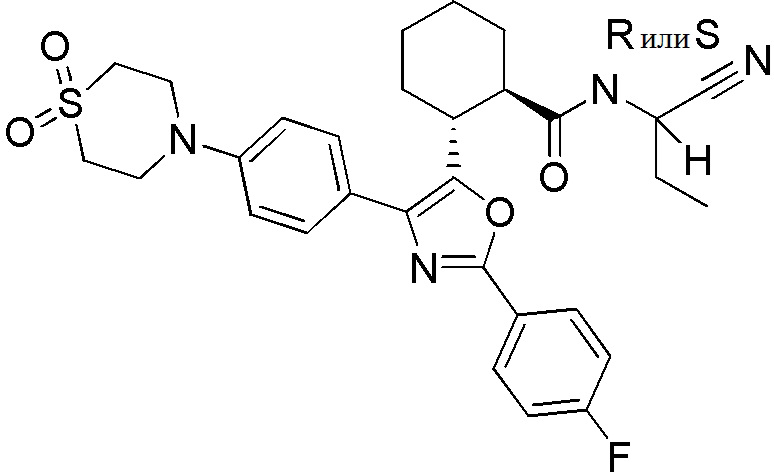
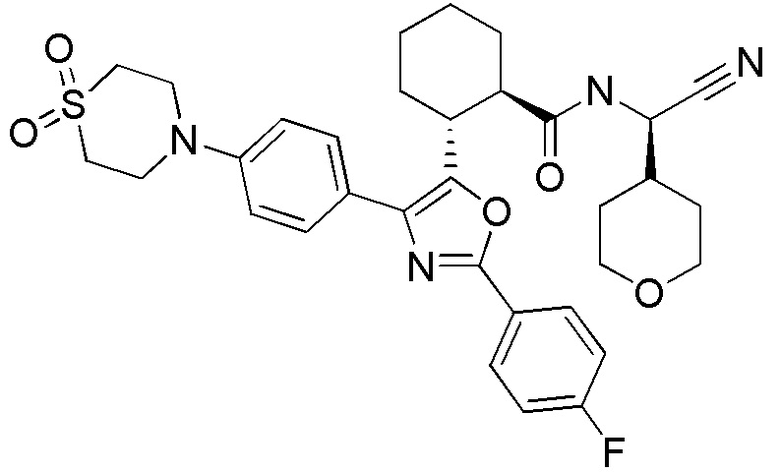
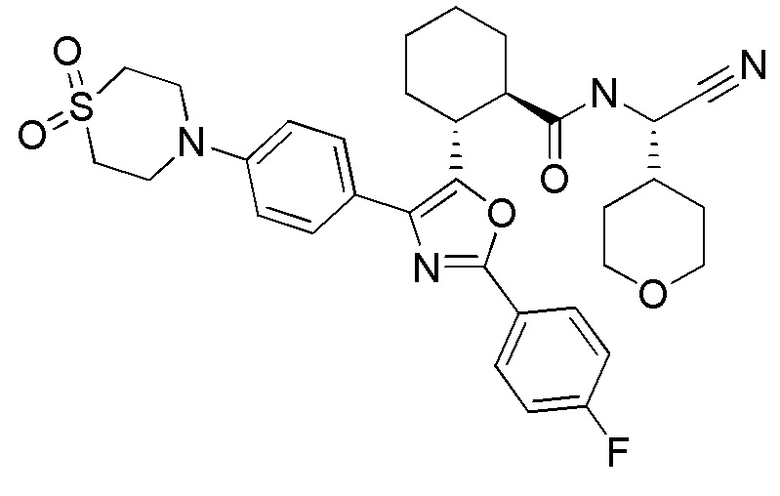
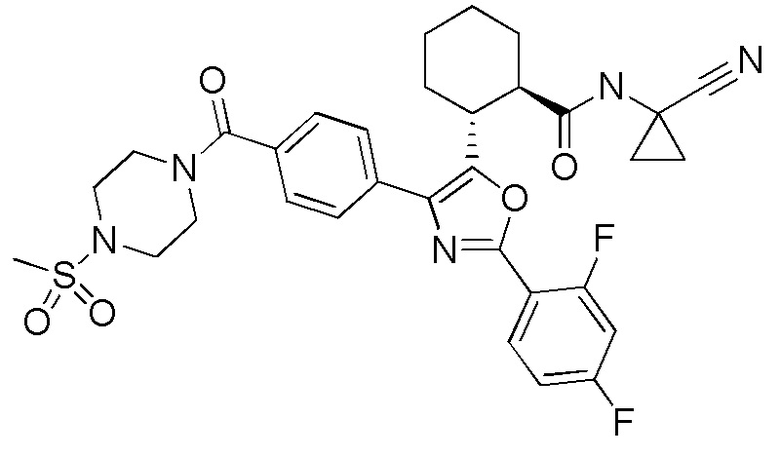
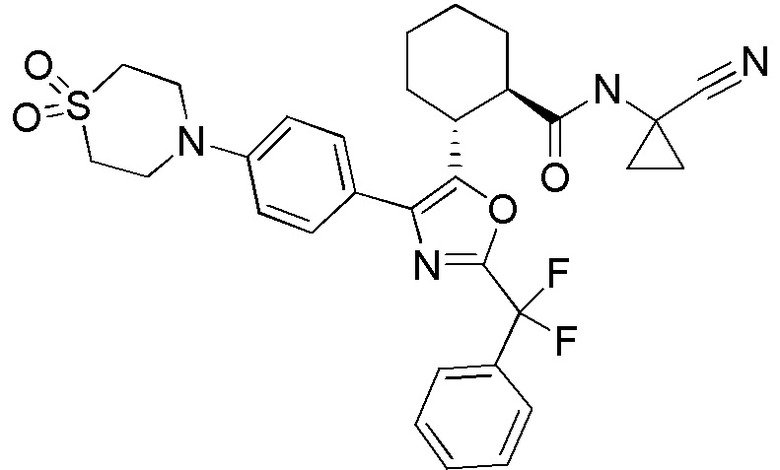
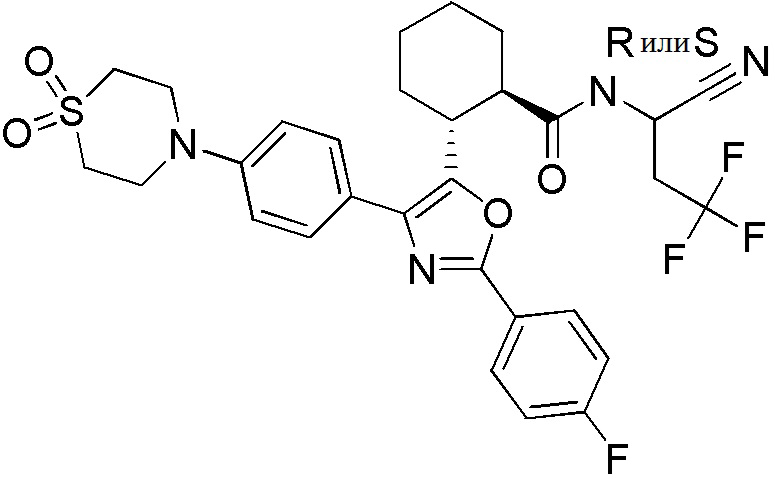
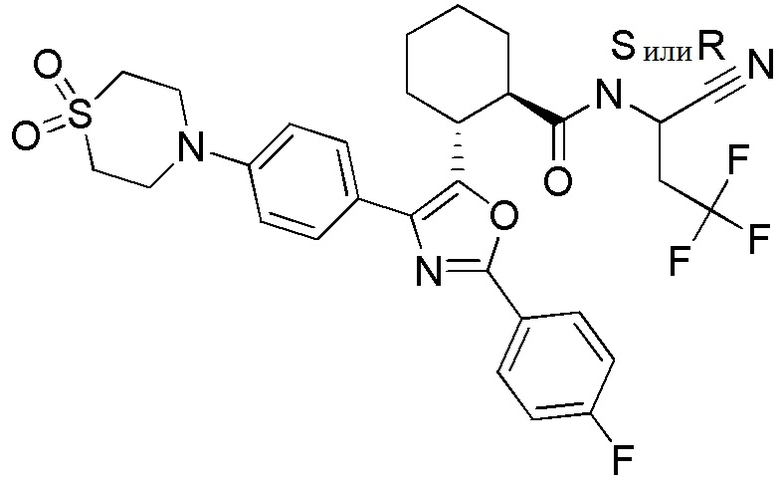
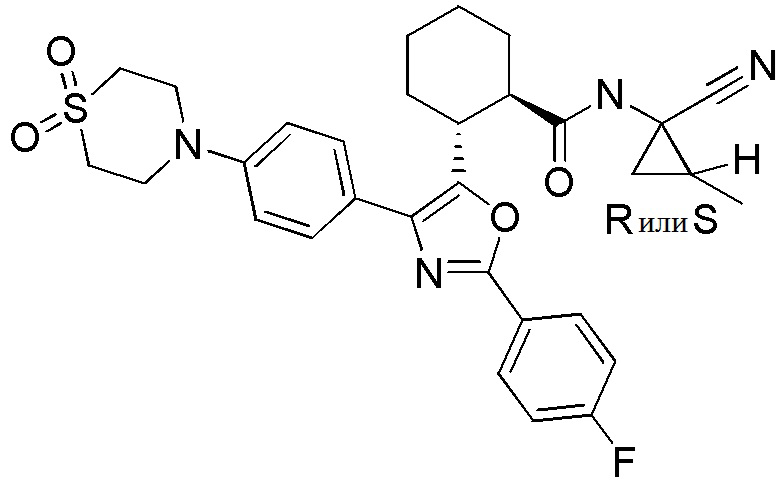
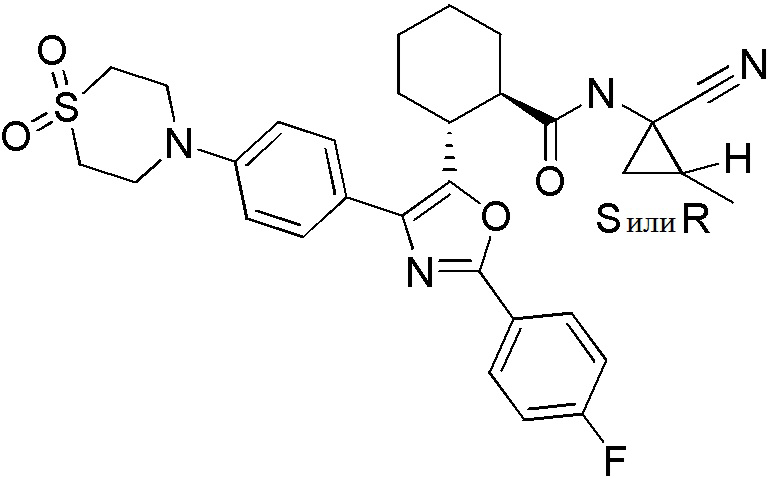
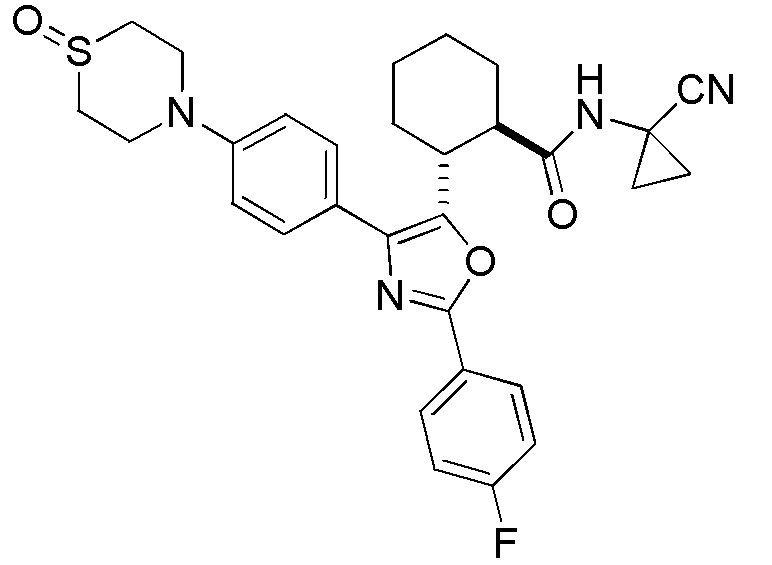
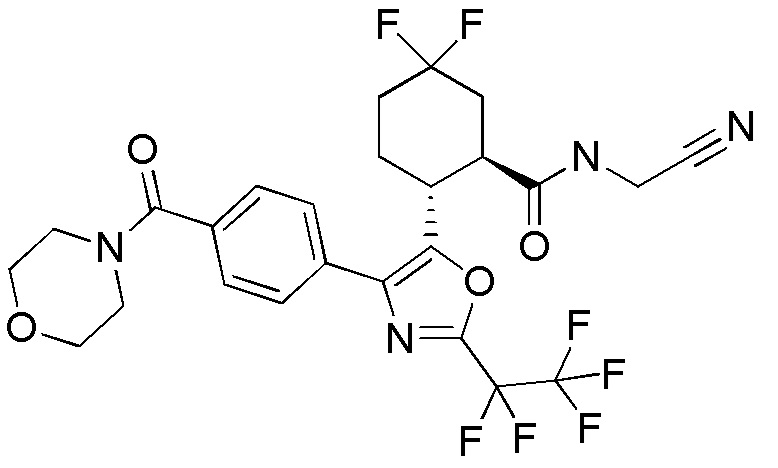
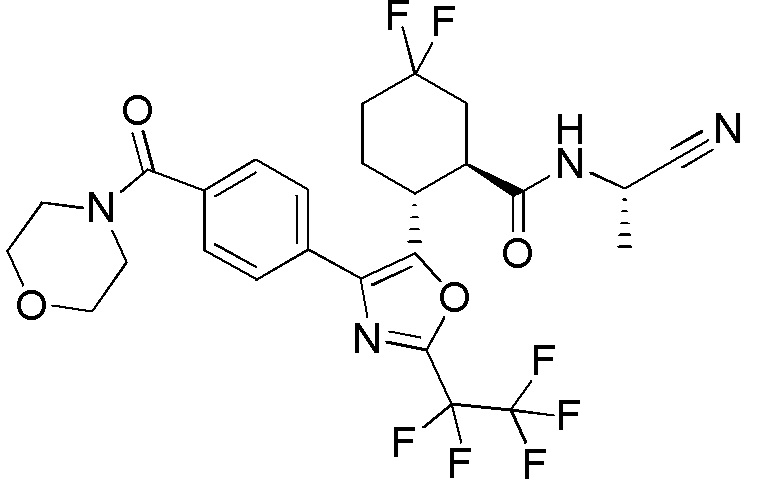
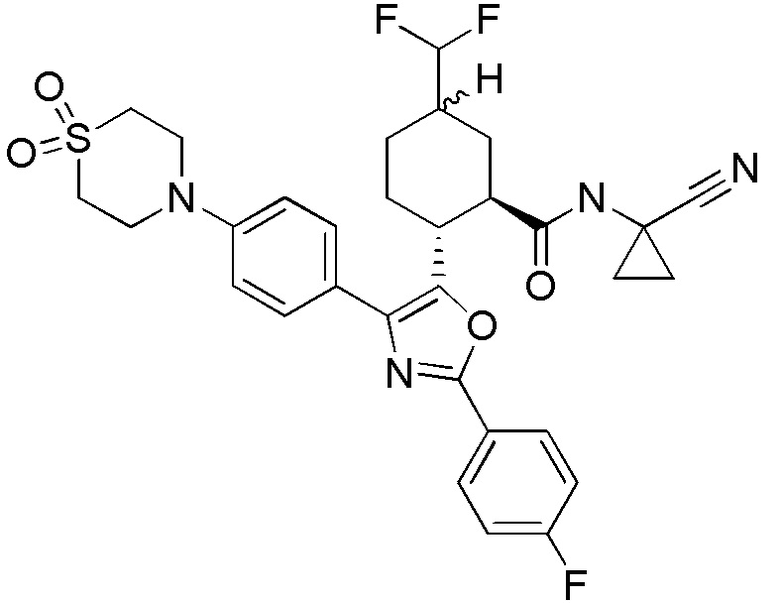
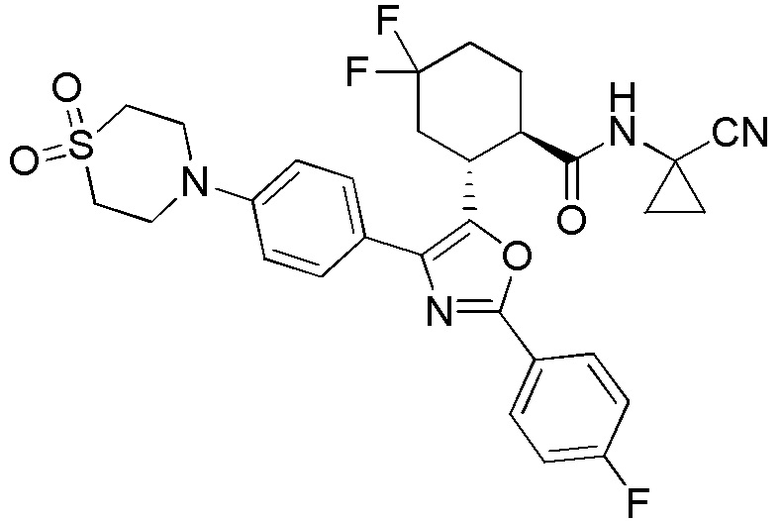
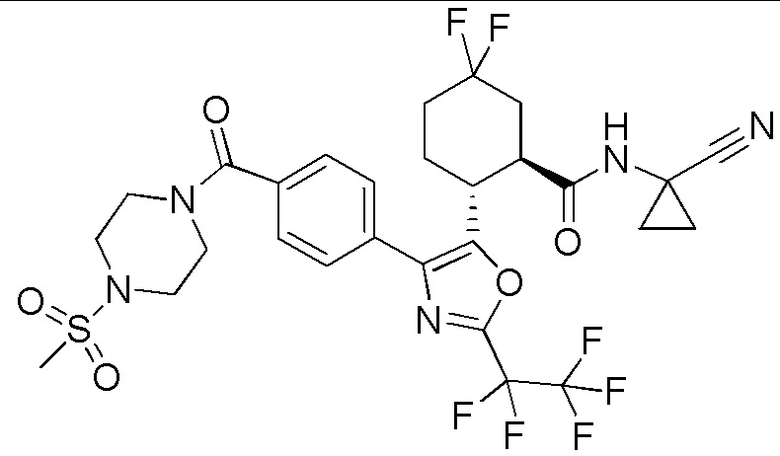
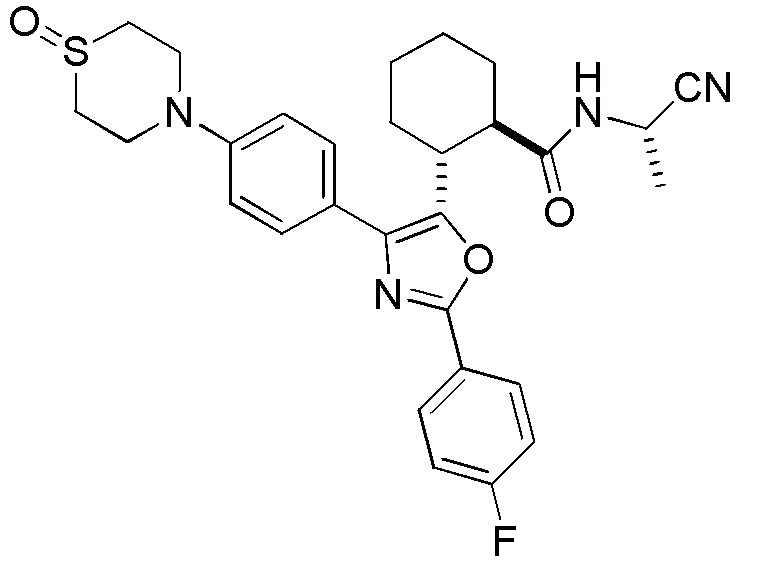
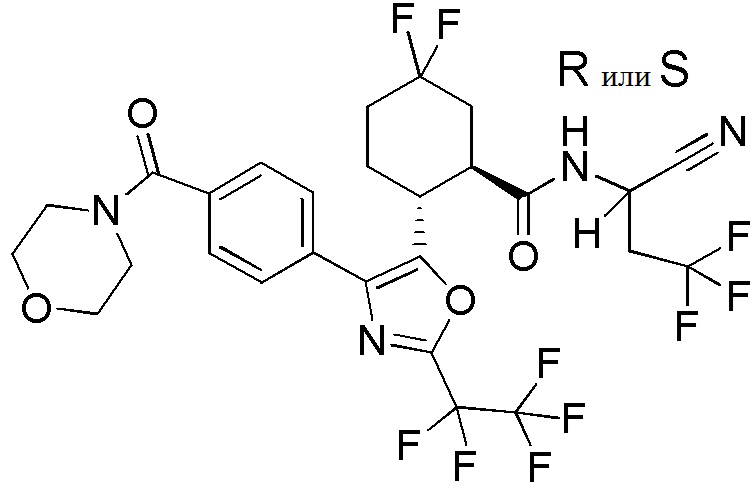
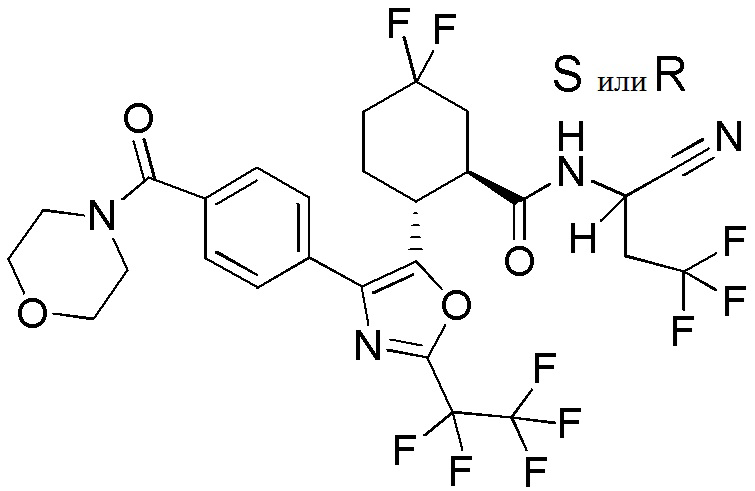
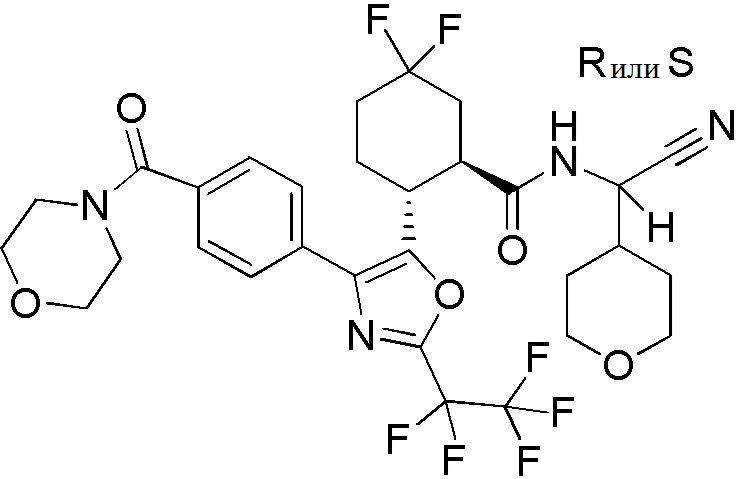
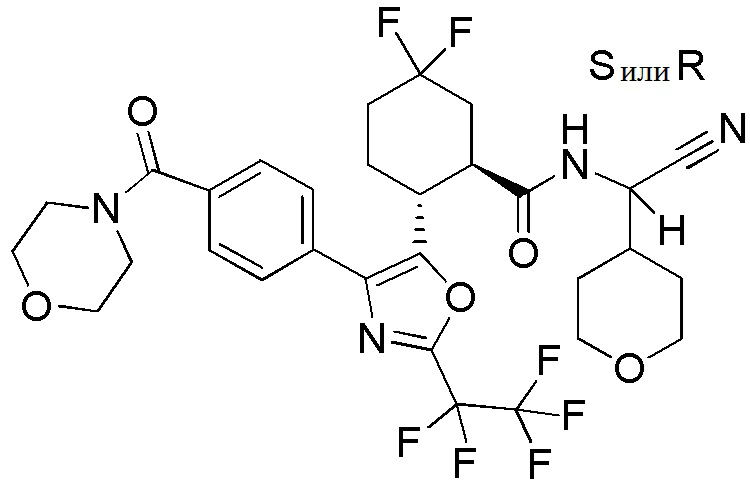
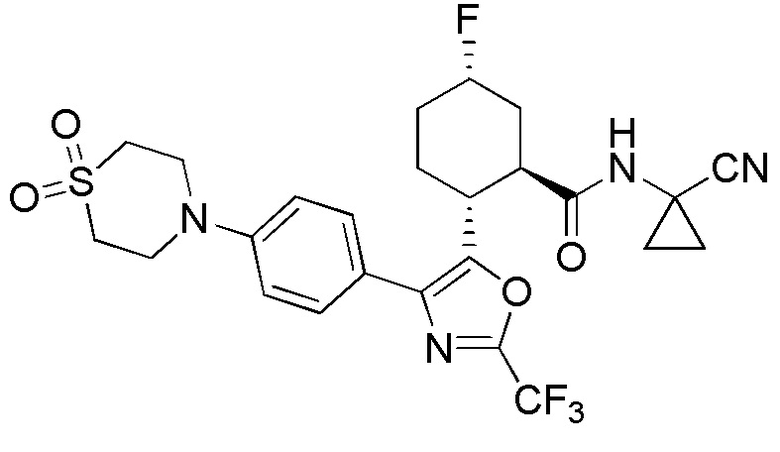
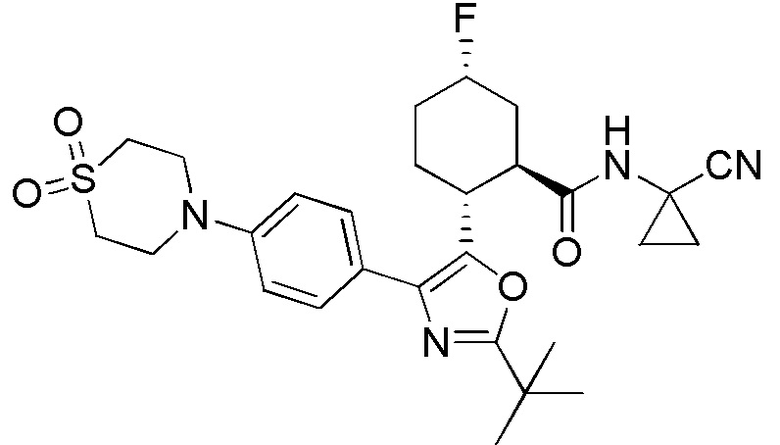
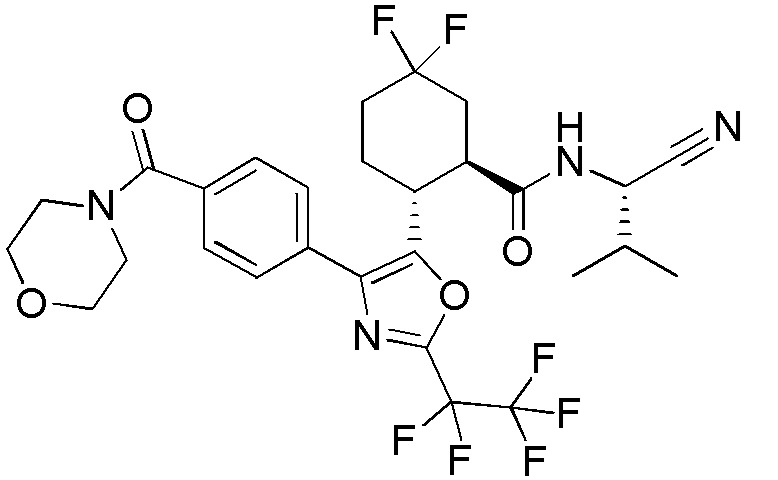
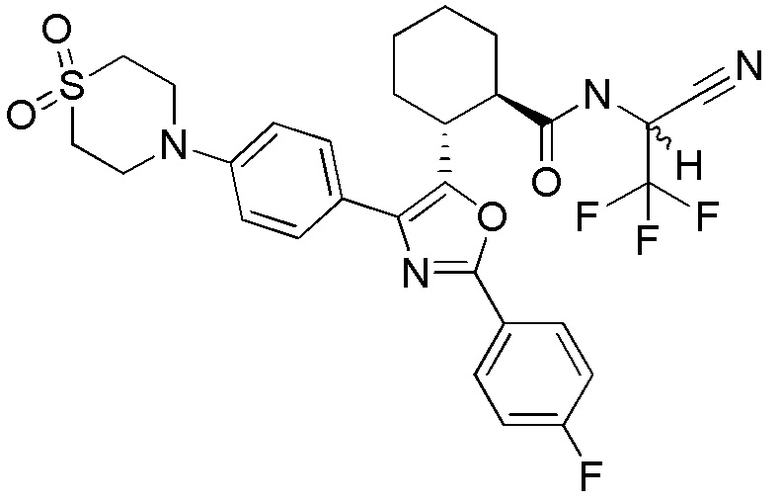
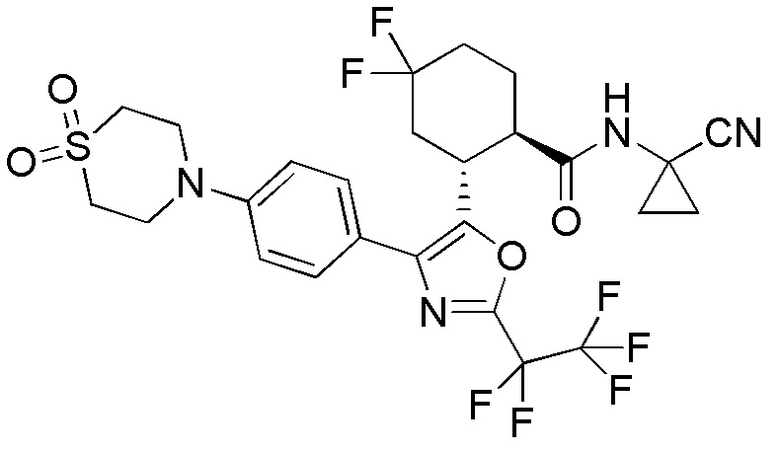
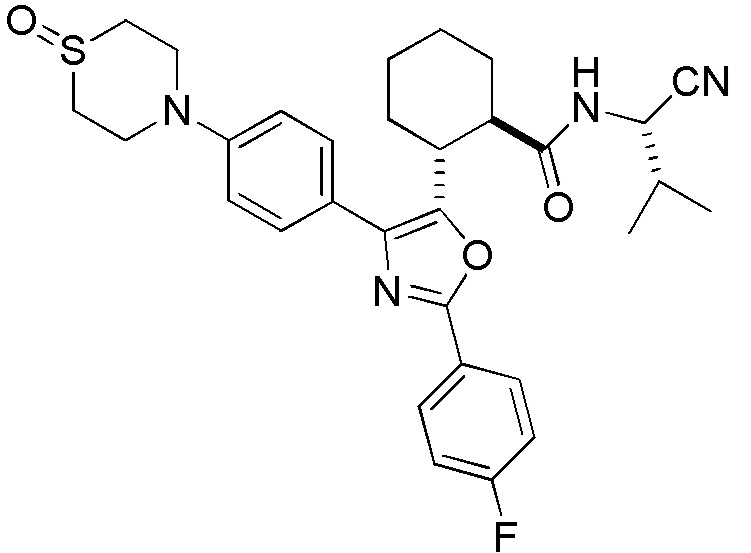
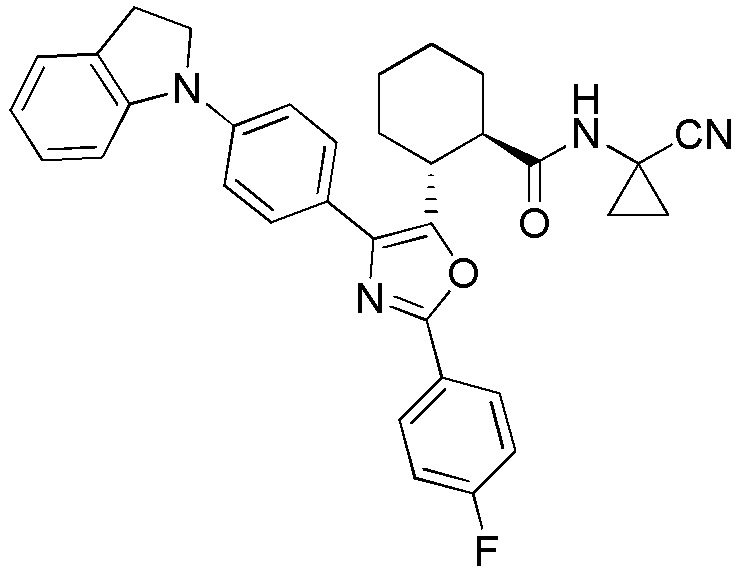
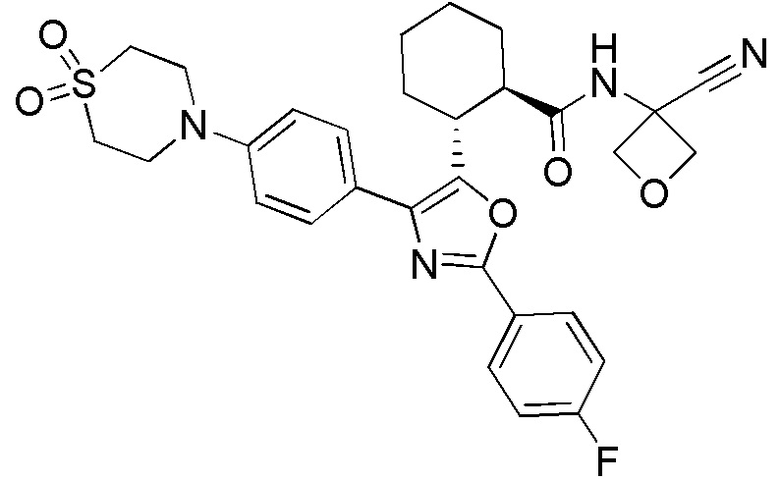
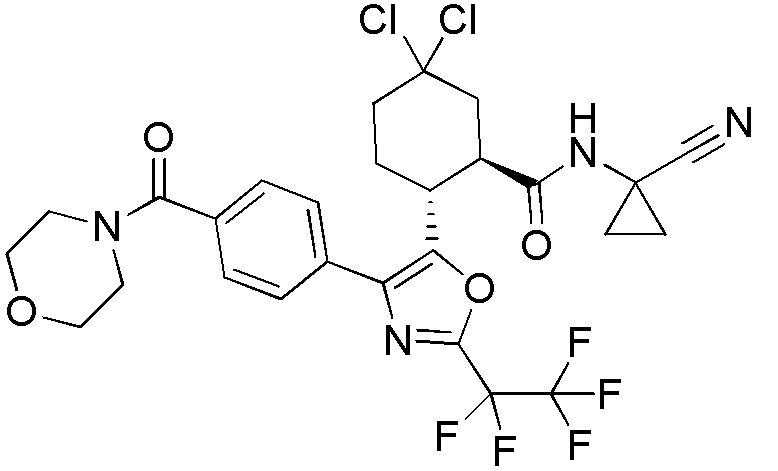
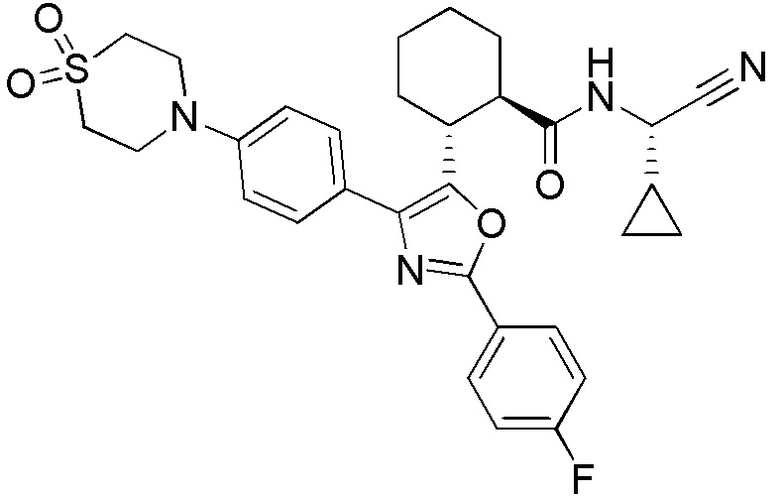
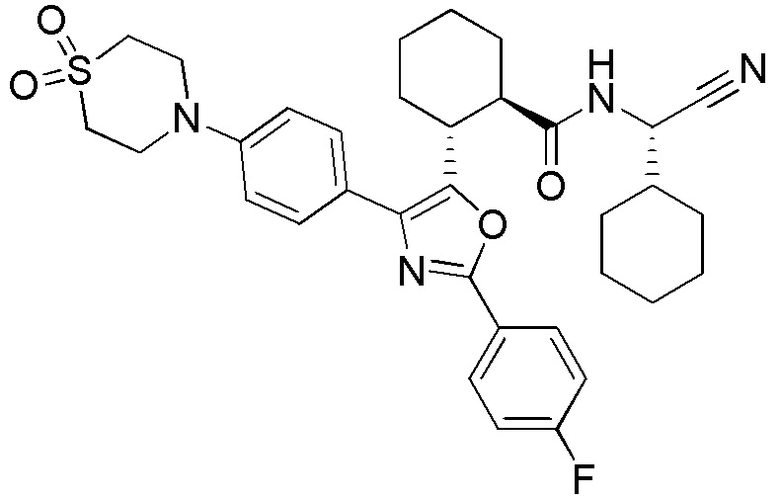
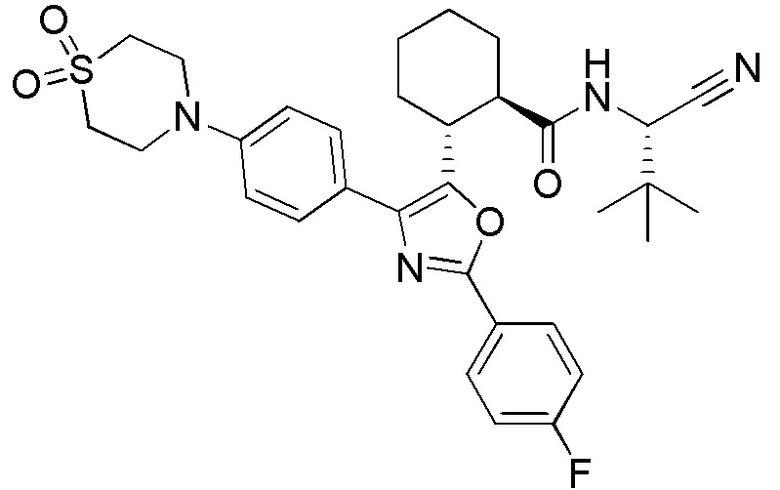

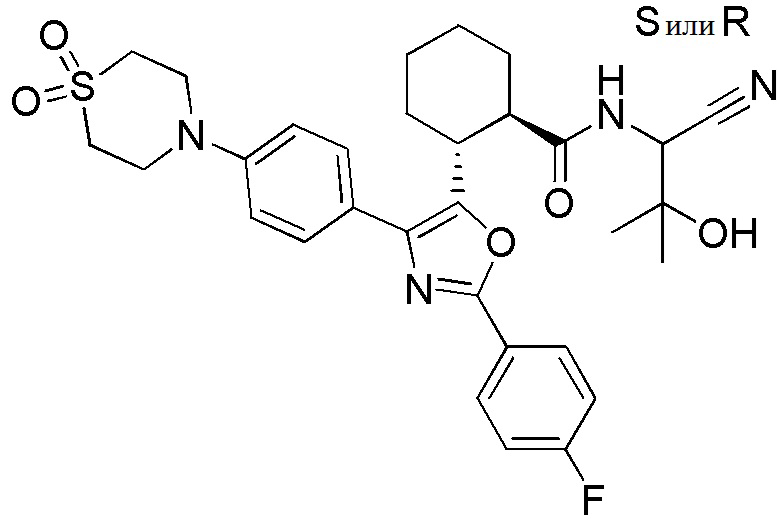
Фармацевтическая композиция
В качестве конкретного варианта осуществления настоящего изобретения, 100 мг соединения Примера 1 приготавливают вместе с достаточно мелкодисперсно измельченной лактозой с получением общего количества 580-590 мг для заполнения твердой желатиновой капсулы размером 0.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТАНА | 2010 |
|
RU2572555C2 |
| 1-ЦИАНОЦИКЛОПРОПИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА К | 2008 |
|
RU2470023C2 |
| ИНГИБИТОРЫ ЦИСТЕИНОВОЙ ПРОТЕАЗЫ КАТЕПСИНА | 2005 |
|
RU2399613C2 |
| ИНДАНИЛЬНЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2474572C1 |
| ЗАМЕЩЕННЫЕ ДИКЕТОПИПЕРАЗИНЫ КАК АНТАГОНИСТЫ ОКСИТОЦИНА | 2002 |
|
RU2303032C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛО [3.1.0]ГЕКСАНА, ПРИМЕНИМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДОПАМИНОВЫХ РЕЦЕПТОРОВ D3 | 2005 |
|
RU2434011C2 |
| ПРОИЗВОДНЫЕ ПРОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2010 |
|
RU2535479C2 |
Изобретение относится к конкретным соединениям, приведенным в формуле изобретения. Соединения по изобретению предназначены для ингибирования активности катепсина K. Соединения предназначены для приготовления лекарственного средства, пригодного для лечения остеопороза, остеопороза, вызываемого глюкокортикоидами, болезни Педжета, аномально увеличенного метаболизма костной ткани, периодонтального заболевания, потери зубов, переломов костей, ревматоидного артрита, остеоартрита, перипротезного остеолиза, несовершенного остеогенеза, атеросклероза, тучности, глаукомы, хронического обструктивного легочного заболевания, метастатического костного заболевания, злокачественной гиперкальцемии или множественной миеломы. 4 н. и 2 з.п. ф-лы, 1 табл., 159 пр.
1. Соединение, которое представляет собой:
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(3,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4,4-дифторпиперидин-1-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(3-хлорфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(3-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-Цианоциклопропил)-2-{4-[4-(1,1-диоксидо-1λ6-тиоморфолин-4-ил)фенил]-2-(4-фторфенил)-1,3-оксазол-5-ил}циклогексанкарбоксамид;
(1S,3R,4R,6R или 1R,3R,4R,6S)-N-(1-Цианоциклопропил)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксамид;
(1R,3R,4R,6S или 1S,3R,4R,6R)-N-(1-цианоциклопропил)-4-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)бицикло[4.1.0]гептан-3-карбоксамид;
(1R,2R)-2-(2-(трет-бутил)-4-(4-(1,1-диоксидотиоморфолино)фенил)тиазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(трет-бутил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(цианометил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-метилоксазол-5-ил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(трифторметил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-метилоксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2,2,2-трифторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2,2,2-трифторэтил)оксазол-5-ил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4-фторфенил)-4-(4-морфолинофенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-метилоксазол-5-ил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-2-(2-(трет-бутил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)-5,5-дифторциклогексанкарбоксамид;
N-(1-цианоциклопропил)-4-(5-((1R,2R)-2-((1-цианоциклопропил)карбамоил)циклогексил)-2-метилоксазол-4-ил)бензамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-метил-4-(4-(морфолин-4-карбонил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(3-(метилсульфонил)азетидин-1-ил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-5,5-дифтор-2-(2-(4-фторфенил)-4-(4-(морфолин-4-карбонил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(3-(метилсульфонил)азетидин-1-ил)фенил)-2-(трифторметил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(пиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(пиридин-3-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-фторпиридин-4-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R,5S)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)-5-фторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-фторпиридин-3-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(3,5-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(пиримидин-5-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(3-хлор-4-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(2-хлор-4-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(4-хлорфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2,4-дифторфенил)-4-(4-(3-(метилсульфонил)азетидин-1-ил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(6-(трифторметил)пиридин-3-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R,5S)-N-(1-цианоциклопропил)-2-(2-(2,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-5-фторциклогексанкарбоксамид;
(1R,2R,5S)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(перфторэтил)оксазол-5-ил)-5-фторциклогексанкарбоксамид;
(1R,2R)-N-(2-цианопропан-2-ил)-2-(2-(2,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(цианометил)-2-(2-(2,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-цианоэтил)-2-(2-(2,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-1-цианоэтил)-2-(2-(2,4-дифторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-морфолинооксазол-5-ил)циклогексанкарбоксамид;
трет-бутил(4-(5-((1R,2R)-2-((1-цианоциклопропил)-карбамоил)циклогексил)-2-(4-фторфенил)оксазол-4-ил)фенил)(метил)карбамат;
трет-бутил(4-(5-((1R,2R)-2-((1-цианоциклопропил)-карбамоил)-циклогексил)-2-(4-фторфенил)оксазол-4-ил)фенил)карбамат;
(1R,2R)-2-(2-бензил-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(циклогексилметил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4,4-дифтор-1-метилциклогексил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(1,1,4-триметилсилилан-4-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-метилтиазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-метил-4-(4-(3-(метилсульфонил)азетидин-1-ил)фенил)тиазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)тиазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2,2,2-трифторэтил)тиазол-5-ил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-2-(2-(трет-бутил)-4-(4-(1,1-диоксидотиоморфолино)фенил)тиазол-5-ил)-N-(1-цианоциклопропил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-метилтиазол-5-ил)-5,5-дифторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(трифторметил)тиазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-(трифторметокси)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-(трифторметокси)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4-(дифторметокси)фенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)-оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-(трифторметил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-(трифторметил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(3-(дифторметокси)фенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-(трифторметил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-метоксифенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-метоксифенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-метоксифенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-метилтиазол-5-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4-цианофенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(2-хлорфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(4-хлор-3-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(3-циклопропилфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(тиазол-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2-циклопропилфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-(трифторметил)пиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-(трифторметил)пиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-метилпиримидин-4-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-(трифторметил)пиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-фторпиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-(трифторметил)пиримидин-5-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(3-(трифторметокси)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-((1R,4R и 1S,4S)-2,2-диоксидо-2-тиа-5-азабицикло[2,2,1]гептан-5-ил)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-(трифторметил)пиридин-3-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(4-(трет-бутил)фенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(пиримидин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(6-(дифторметокси)пиридин-3-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(5-(1,1-диоксидотиоморфолино)пиридин-3-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(3-цианофенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(4-хлор-2-фторфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(5-хлорпиридин-3-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2-(дифторметокси)фенил)-4-(4-(1,1-диоксидотиоморфолино)-фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(пиридазин-3-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-(бицикло[1,1,1]пентан-2-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-метилтетрагидро-2H-пиран-4-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(6-(трифторметил)пиридин-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4-циклопропилфенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(4-(3-бром-4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)-N-(1-цианоциклопропил)-циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2-метилпиримидин-5-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-((трифторметил)сульфонил)-фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2-цианофенил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2,4-дифторфенил)-4-(4-(1-имино-1-оксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-((1S,2R)-4,4-дифтор-2-метилциклогексил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-((1S,2S)-4,4-дифтор-2-метилциклогексил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-((1R,3R,5S и 1R,3S,5S)-8,8-дифторбицикло[3,2,1]октан-3-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(1-(трифторметил)циклопропил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-((1R,4R и 1S,4S)-4-(трифторметил)циклогексил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(цианометил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(2,2,3,3-тетраметилциклопропил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-((R и S)-3,3-дифторциклопентил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(1,1,1-трифтор-2-метилпропан-2-ил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-((R и S)-2,2,2-трифтор-1-метокси-1-фенилэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-((1R,3R и 1S,3S)-3-метоксициклобутил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-2-(2-((1R,3R,5S и 1R,3S,5S)-бицикло[3,1,0]гексан-3-ил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2,4-дифторфенил)-4-(4-(морфолин-4-карбонил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-цианоэтил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-1-цианоэтил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-1-циано-2-метилпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-циано-2-метилпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-1-цианопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-цианопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R)-циано(тетрагидро-2H-пиран-4-ил)метил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S)-циано(тетрагидро-2H-пиран-4-ил)метил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(2,4-дифторфенил)-4-(4-(4-(метилсульфонил)пиперазин-1-карбонил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(дифтор(фенил)метил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-циано-3,3,3-трифторпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-1-циано-3,3,3-трифторпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((2R или 2S)-1-циано-2-метилциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((2S или 2R)-1-циано-2-метилциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4-фторфенил)-4-(4-(1-оксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(цианометил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S)-1-цианоэтил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R,5R и 1R,2R,5S)-N-(1-цианоциклопропил)-5-(дифторметил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)-4,4-дифторциклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-5,5-дифтор-2-(4-(4-(4-(метилсульфонил)пиперазин-1-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S)-1-цианоэтил)-2-(2-(4-фторфенил)-4-(4-(1-оксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-циано-3,3,3-трифторпропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-1-циано-3,3,3-трифторпропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-циано(тетрагидро-2H-пиран-4-ил)метил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S или R)-циано(тетрагидро-2H-пиран-4-ил)метил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R,5S)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(трифторметил)оксазол-5-ил)-5-фторциклогексанкарбоксамид;
(1R,2R,5S)-2-(2-(трет-бутил)-4-(4-(1,1-диоксидотиоморфолино)фенил)оксазол-5-ил)-N-(1-цианоциклопропил)-5-фторциклогексанкарбоксамид;
(1R,2R)-N-((S)-1-циано-2-метилпропил)-5,5-дифтор-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S и R)-1-циано-2,2,2-трифторэтил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(перфторэтил)оксазол-5-ил)-4,4-дифторциклогексанкарбоксамид;
(1R,2R)-N-((S)-1-циано-2-метилпропил)-2-(2-(4-фторфенил)-4-(4-(1-оксидотиоморфолино)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(1-цианоциклопропил)-2-(2-(4-фторфенил)-4-(4-(индолин-1-ил)фенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-(3-цианооксетан-3-ил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-5,5-дихлор-N-(1-цианоциклопропил)-2-(4-(4-(морфолин-4-карбонил)фенил)-2-(перфторэтил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S)-циано(циклопропил)метил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S)-циано(циклогексил)метил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((S)-1-циано-2,2-диметилпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
(1R,2R)-N-((R или S)-1-циано-2-гидрокси-2-метилпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид или
(1R,2R)-N-((S или R)-1-циано-2-гидрокси-2-метилпропил)-2-(4-(4-(1,1-диоксидотиоморфолино)фенил)-2-(4-фторфенил)оксазол-5-ил)циклогексанкарбоксамид;
или его фармацевтически приемлемая соль.
2. Соединение формулы:
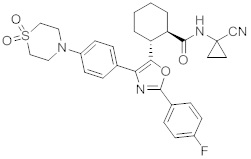 .
.
3. Фармацевтическая композиция, обладающая способностью ингибировать активность катепсина K, содержащая терапевтически эффективное количество соединения по п.1 или 2 и фармацевтически приемлемый носитель.
4. Применение соединения по п.1 или 2 при приготовлении лекарственного средства, пригодного для лечения: остеопороза, остеопороза, вызываемого глюкокортикоидами, болезни Педжета, аномально увеличенного метаболизма костной ткани, периодонтального заболевания, потери зубов, переломов костей, ревматоидного артрита, остеоартрита, перипротезного остеолиза, несовершенного остеогенеза, атеросклероза, тучности, глаукомы, хронического обструктивного легочного заболевания, метастатического костного заболевания, злокачественной гиперкальцемии или множественной миеломы.
5. Соединение по п.1 или 2 для применения в качестве ингибитора катепсина K.
6. Соединение по п.1 или 2 для применения при лечении остеопороза.
| Robichaud J | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |

