ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение касается способа получения ароматических соединений и, более конкретно, способа промышленного получения ароматических соединений каталитической реакцией низших углеводородов, таких как метан. Кроме того, настоящее изобретение касается способа получения гидрированных ароматических соединений гидрированием ароматических соединений, полученных по настоящему изобретению.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Традиционно были предложены различные способы улучшения катализаторов, применяемых для получения ароматических соединений каталитической реакцией низших углеводородов, таких как метан (японские выложенные патентные заявки (КОКАI) №№ 10-272366, 11-60514, 2001-334151, 2001-334152, 2002-336704, 2004-97891 и 2004-269398).
Однако не было предложений по улучшению самих способов получения ароматических соединений в достаточной степени. Согласно исследованию настоящих изобретателей обнаружено, что традиционные способы получения имеют следующие проблемы.
А именно, при промышленном получении ароматических соединений каталитической реакцией низших углеводородов непрореагировавший газ, содержащий низшие углеводороды, остающиеся после каталитической реакции, необходимо возвращать и повторно использовать. Однако водород является побочным продуктом реакции ароматизации низших углеводородов, так что газ, получаемый на стадии синтеза ароматических соединений, неизбежно содержит водород. Следовательно, если водородсодержащий газ, который получается после отделения ароматического соединения от газа продуктов реакции, непосредственно возвращается на стадию синтеза ароматических соединений, низшие углеводороды имеют тенденцию нежелательно разбавляться водородом, что мешает достижению удовлетворительных результатов реакции с промышленной точки зрения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ПРОБЛЕМА, РЕШАЕМАЯ ИЗОБРЕТЕНИЕМ
Настоящее изобретение осуществляли в точки зрения вышеуказанных проблем. Целью настоящего изобретения является обеспечить промышленно приемлемый способ получения ароматических соединений каталитической реакцией низших углеводородов, таких как метан.
СРЕДСТВО ДЛЯ РЕШЕНИЯ ПРОБЛЕМЫ
В результате исследования настоящими изобретателями решения вышеуказанных проблем обнаружено, что они могут быть решены подходящим объединением стадии метанирования и стадии синтеза ароматического соединения. Настоящее изобретение достигнуто на основе вышеуказанных результатов. А именно, настоящее изобретение включает в себя следующие объекты от (1) до (12).
(1) Способ получения ароматических соединений, включающий:
стадию метанирования при контакте водородсодержащего газа с моноксидом углерода и/или диоксидом углерода в присутствии катализатора с реакцией водорода, содержащегося в газе, с моноксидом углерода и/или диоксидом углерода и превращением этих компонентов в метан и воду; и
стадия синтеза ароматического соединения с реакцией низшего углеводорода с метаном, полученным на стадии метанирования, в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения и водород.
(2) Способ согласно вышеуказанному объекту (1), где ароматические соединения отделяются от газа продуктов реакции, получаемого на стадии синтеза ароматического соединения, и затем оставшийся водородсодержащий газ подается на стадию метанирования.
(3) Способ согласно вышеуказанному объекту (2), где ароматические соединения отделяются от газа продуктов реакции, получаемого на стадии синтеза ароматического соединения, затем водород отделяется от оставшегося водородсодержащего газа, полученного отделением ароматического соединения от газа продуктов реакции, отделенный таким образом водород подается на стадию метанирования и оставшийся газ, полученный отделением водорода от водородсодержащего газа, подается на стадию синтеза ароматического соединения.
(4) Способ получения ароматических соединений, включающий следующие стадии от (i) до (iii):
(i) стадия синтеза ароматического соединения с реакцией низшего углеводорода в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения, низший углеводород и водород;
(ii) стадия отделения ароматических соединений с отделением и возвратом ароматических соединений и газа, содержащего низший углеводород и водород, от газа продуктов реакции, полученного на стадии (i) синтеза ароматических соединений;
(iii) стадия метанирования с контактом газа, содержащего низший углеводород и водород, который отделили от ароматических соединений на стадии (ii) отделения ароматических соединений, с моноксидом углерода и/или диоксидом углерода в присутствии катализатора с реакцией водорода, содержащегося в газе, с моноксидом углерода и/или диоксидом углерода и превращением этих компонентов в метан и воду, причем упомянутый способ обеспечивается средством для возврата газа, получаемого на стадии метанирования, на стадию синтеза ароматического соединения.
(5) Способ согласно вышеуказанному объекту (4), где водород отделяется от водородсодержащего газа, полученного на стадии отделения ароматического соединения, и подается на стадию метанирования, а оставшийся газ, полученный отделением водорода от водородсодержащего газа, подается на стадию синтеза ароматических соединений.
(6) Способ согласно любому из вышеуказанных объектов от (1) до (5), где низший углеводород, содержащий моноксид углерода и/или диоксид углерода, подается на стадию метанирования.
(7) Способ согласно любому из вышеуказанных объектов от (1) до (5), где низший углеводород, по существу не содержащий моноксида углерода и/или диоксида углерода, подается на стадию синтеза ароматических соединений.
(8) Способ согласно любому из вышеуказанных объектов от (1) до (7), где моноксид углерода и/или диоксид углерода, подаваемые на стадию метанирования, представляют собой моноксид углерода и/или диоксид углерода, возвращаемые извне реакционной системы.
(9) Способ согласно любому из вышеуказанных объектов от (1) до (8), где подача диоксида углерода на стадию метанирования осуществляется делением оставшегося газа, полученного отделением ароматических соединений от газа продуктов реакции на стадии отделения ароматических соединений, на первую и вторую фракции, сжиганием второй фракции, возвратом диоксида углерода из полученного отходящего газа сжигания и подачей возвращенного таким образом диоксида углерода вместе с первой фракцией на стадию метанирования.
(10) Способ получения гидрированных ароматических соединений, содержащий гидрирование ароматических соединений, полученных посредством способа, определенного в любом из вышеприведенных объектов от (1) до (9), в присутствии катализатора.
(11) Способ согласно вышеприведенному объекту (10), где гидрированные ароматические соединения отделяются от газа продуктов реакции, получаемого на стадии гидрирования, и оставшийся газ, получаемый отделением гидрированных ароматических соединений от газа продуктов реакции, возвращается на стадию метанирования.
(12) Способ согласно любому из вышеуказанных объектов от (10) до (11), где газ продуктов реакции, содержащий ароматические соединения и водород, который получается на стадии синтеза ароматических соединений, подается на стадию гидрирования.
РЕЗУЛЬТАТ ИЗОБРЕТЕНИЯ
В способе получения ароматического соединения согласно настоящему изобретению количество водорода, подаваемого на стадию синтеза ароматических соединений, может быть уменьшено, и диоксид углерода, который считается основной причиной, вызывающей явление глобального потепления, может расходоваться в качестве исходного материала для низших углеводородов (таких, как метан), тем самым делая возможным промышленное получение ароматических соединений.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой блок-схему, показывающую пример способа получения ароматических соединений согласно настоящему изобретению.
Фиг.2 представляет собой блок-схему, показывающую другой пример способа получения ароматических соединений по настоящему изобретению.
ОБЪЯСНЕНИЕ ССЫЛОЧНЫХ ОБОЗНАЧЕНИЙ
А: Стадия синтеза ароматического соединения
В: Стадия отделения ароматического соединения
С: Стадия метанирования
D: Стадия удаления диоксида углерода
Е: Стадия реакции сдвига
F: Стадия гидрирования
G: Стадия отделения гидрированного ароматического соединения
ПРЕДПОЧТИТЕЛЬНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение подробно описано ниже. Последующее описание настоящего изобретения является только иллюстративным и не претендует на ограничение объема изобретения.
Способ получения ароматических соединений согласно настоящему изобретению содержит в качестве основных стадий "стадию метанирования с контактом водородсодержащего газа с моноксидом углерода и/или диоксидом углерода в присутствии катализатора с реакцией водорода, содержащегося в газе, с моноксидом углерода и/или диоксидом углерода и превращением этих компонентов в метан и воду" и "стадию синтеза ароматического соединения с реакцией низшего углеводорода с метаном, получаемым на стадии метанирования, в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения и водород".
В предпочтительном варианте осуществления настоящего изобретения данный способ получения содержит в качестве основных стадий стадию синтеза ароматического соединения, стадию отделения ароматического соединения и стадию метанирования, и дополнительно в данном способе получения обеспечивается средство для возврата газа, получаемого на стадии метанирования, на стадию синтеза ароматического соединения.
В способе настоящего изобретения при получении ароматических соединений могут быть использованы различные исходные газы, такие как содержащие низшие углеводороды газы, по существу не содержащие моноксида углерода и/или диоксида углерода, и содержащие низшие углеводороды газы, содержащие моноксид углерода и/или диоксид углерода. Выражение "по существу не содержащие моноксида углерода и/или диоксида углерода" означает, что исходные газы могут содержать моноксид углерода и/или диоксид углерода в таком количестве, которое не оказывает вредного воздействия на каталитическую реакцию на стадии синтеза ароматического соединения. Кроме того, следующие уравнения реакций соответственно показывают пример реакций, происходящих на нижеуказанных стадиях, где уравнения (1) и (1') представляют реакции, происходящие на стадии синтеза ароматического соединения, а уравнения (2) и (3) представляют реакции, происходящие на стадии метанирования.
<Исходные газы>
Термин "низшие углеводороды", применяемый в настоящем изобретении, означает углеводородное соединение, имеющее от 1 до 4 атомов углерода. Конкретные примеры низших углеводородов могут включать в себя метан, этан, пропан, н-бутан, изобутан и производные от них ненасыщенные соединения. Среди этих углеводородных соединений предпочтительными являются метан и ненасыщенные углеводородные соединения, имеющие от 2 до 4 атомов углерода, и более предпочтительным является метан.
Типичные примеры содержащих низшие углеводороды газов, по существу не содержащих моноксид углерода и/или диоксид углерода, могут включать в себя природные газы, такие как LNG и NG, LPG, метан-гидраты и отходящие газы установок нефтехимии или установок нефтепереработки. Примеры содержащих низшие углеводороды газов, содержащих моноксид углерода и/или диоксид углерода, могут включать в себя коксовые газы, газы газификации угля, газы газификации асфальта, газы газификации остатка тяжелой нефти, газы газификации нефтяного кокса, газы печи реформинга, оксогазы, биогазы, газы газификации биомассы и газы газификации отходов. Коксовые газы содержат не только низшие углеводороды, но также моноксид углерода и/или диоксид углерода, и следовательно, могут подходяще использоваться в качестве исходного газа для получения метана. Вышеуказанные содержащие низшие углеводороды газы содержат водород в таком количестве, как определено согласно их типов. Также в качестве источника водорода может использоваться получаемый или побочно получаемый водород.
Низший углеводород или содержащий низший углеводород газ подается на следующую стадию синтеза ароматического соединения. Вышеуказанный исходный газ для получения метана предпочтительно подается на указанную ниже стадию метанирования. Содержащий низший углеводород газ, по существу не содержащий моноксида углерода и/или диоксида углерода, может подаваться на стадию синтеза ароматического соединения или может подаваться вместе с моноксидом углерода и/или диоксидом углерода на стадию метанирования. Также различные содержащие низшие углеводороды газы, содержащие моноксид углерода и/или диоксид углерода, могут подаваться на стадию метанирования. Моноксид углерода и/или диоксид углерода, содержащиеся в различных содержащих низшие углеводороды газах, содержащих моноксид углерода и/или диоксид углерода, превращаются в метан, как показано в вышеприведенных уравнениях (2) и (3), и затем полученный таким образом метан подается на стадию синтеза ароматического соединения. Таким образом, подачей различных содержащих низшие углеводороды газов, содержащих моноксид углерода и/или диоксид углерода, на стадию метанирования может быть устранена проблема, касающаяся разбавления низшего углеводорода моноксидом углерода и/или диоксидом углерода, которая будет возникать при непосредственной подаче этих газов на стадию синтеза ароматического соединения.
<(i) Стадия синтеза ароматического соединения>
На стадии синтеза ароматического соединения низший углеводород реагирует в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения, низший углеводород и водород.
Примеры подходящего катализатора могут включать в себя катализаторы, описанные в японской выложенной патентной заявке (КОКАI) №2001-334151, т.е. катализаторы, состоящие из каталитического материала, содержащего, по меньшей мере, один необходимый компонент, выбранный из группы, состоящей из Rе и его соединений, если требуется вместе с, по меньшей мере, одним компонентом, выбранным из группы, состоящей из Zn, Ga, Co, Fe и их соединений, по меньшей мере, одним компонентом, выбранным из группы, состоящей из Cr, W, Mo и их соединений, и, по меньшей мере, одним компонентом, выбранным из группы, состоящей из редкоземельных металлов и их соединений; и металлосиликат.
Металлосиликат, используемый в качестве носителя, предпочтительно присутствует в форме пористого вещества, имеющего некоторое количество пор. Например, в качестве алюмосиликата могут применяться пористые носители, имеющие различные композиции, составленные из оксида кремния и оксида алюминия, такие как молекулярное сито 5А (UТА), фожазит (NаY и NаХ), ZSM-5, ZSM-11, ZSM-22, ZSM-48, морденит, МСМ-22 и др. Примеры носителей, содержащих фосфорную кислоту в качестве основного компонента, могут включать в себя пористые носители, имеющие микропоры или каналы, имеющие размер от 4 до 8 Å, обычно такие, как SАРО-5, SАРО-34 и VРI-5.
Кроме того, в качестве носителя также могут применяться модифицированные мезопористые материалы, полученные регулированием диаметра мезопор носителей, содержащих оксид кремния в качестве основного компонента вместе с оксидом алюминия в качестве дополнительного компонента, которые отличаются цилиндрическими порами (каналами) мезопор (от 10 до 100 Å), такие как FSМ-16 и МСМ-41, от 4 до 8 Å с помощью СVD способа, используя алкоксид кремния, и др.
В качестве металлосиликата в добавление к алюмосиликатам, состоящим из оксида кремния и оксида алюминия, также могут применяться такие пористые носители, как титаносиликат, состоящим из оксида кремния и оксида титана, который содержит, по меньшей мере, один компонент, выбранный из группы, состоящей из Fe, Ti, Mn, Cr, In, Ga, Mo, W, Co, V, Zn, и имеет диаметр пор от 4 до 8 Å.
Среди этих носителей предпочтительными являются носители, имеющие диаметр микро- и мезопор от 4 до 8 Å, более предпочтительны металлосиликаты, имеющие диаметр пор 5,5±1 Å, и еще более предпочтительны металлосиликаты, имеющие площадь поверхности от 200 до 1000 м2/г. Кроме того, например, в качестве алюмосиликатов могут использоваться обычно доступные пористые носители, имеющие отношение оксида кремния к оксиду алюминия от 1 до 8000. Чтобы достичь практически эффективной величины конверсии и селективности, отношение оксида кремния к оксиду алюминия предпочтительно составляет от 10 до 100.
При нанесении на металлосиликат каталитический материал, такой как Rе, может быть приготовлен в форме его предшественника. Примеры предшественников могут включать в себя галогениды, такие как хлориды и бромиды, соли минеральных кислот, такие как нитраты, сульфаты и фосфаты, карбонаты, соли карбоновых кислот, такие как ацетаты и оксалаты, и металлоорганические соли, такие как металлкарбонильные комплексы и циклопентадиенильные комплексы. В частности, примеры рениевого предшественника могут включать в себя ренийкарбонильные соединения, такие как Rе2(СО)10, Rе6(СО), (С2Н5)Rе(СО)2, и СН3RеО3, а также галогениды Rе, такие как хлориды и бромиды, соли Rе минеральных кислот, такие как нитраты, сульфаты и фосфаты, карбонаты Rе и соли Rе карбоновых кислот, такие как ацетаты и оксалаты. В качестве предшественника также могут использоваться сложные комплексы и сложные оксиды.
Количество каждого каталитического материала, нанесенного на металлосиликат, не ограничивается конкретно и обычно составляет от 0,001 до 50% по массе, предпочтительно от 0,01 до 40% по массе относительно всей массы катализатора. Когда используется множество каталитических материалов, полное количество каталитических материалов, нанесенных на носитель, обычно составляет от 0,002 до 50% по массе, предпочтительно от 0,02 до 40% по массе относительно всей массы катализатора. Между тем, при использовании предшественника в качестве каталитического материала количество каталитического материала подразумевает количество нанесенного предшественника.
Каталитический материал может наноситься на металлосиликат с помощью (i) способа пропитки металлосиликата водным раствором вышеуказанного металлического предшественника или его раствором в органическом растворителе, таком как спирты, или (ii) способа нанесения каталитического материала на металлосиликат с помощью способа ионного обмена и затем тепловой обработки полученного нанесенного катализатора в инертном газе или кислородсодержащем газе. Пример вышеуказанных способов более конкретно описывается ниже. Сначала металлосиликат пропитывают, например, водным раствором нитрата рения, чтобы поместить соль рения на металлосиликат, сушат для удаления из него соответствующего количества растворителя и затем нагревают до температуры обычно от 250 до 800°С, предпочтительно от 350 до 600°С в азотсодержащей атмосфере или в потоке чистого кислородсодержащего газа, получая рений-нанесенный металлосиликатный катализатор. Кроме того, при использовании сложных оксидов или сложных комплексов в качестве каталитического материала катализаторы, образованные из сложных оксидов или сложных комплексов, также можно получать, используя такой же способ нанесения или такой же способ тепловой обработки.
Катализатор, содержащий рений и/или соединение рения (здесь и далее называемые просто "первый компонент") и возможно содержащий, по меньшей мере, один материал, выбранный из группы, состоящей из цинка, галлия, железа, кобальта и их соединений (здесь и далее называемые просто "второй компонент"), по меньшей мере, один материал, выбранный из группы, состоящей из хрома, вольфрама, молибдена и их соединений (здесь и далее называемые просто "третий компонент"), по меньшей мере, один материал, выбранный из группы, состоящей из редкоземельных металлов и их соединений, и носитель, может быть получен нанесением первого компонента на металлосиликат и затем последовательного нанесения необязательного выбранного второго и последующих компонентов на металлосиликат; нанесением первого компонента и необязательно выбранного второго и последующих компонентов на металлосиликат в подходящем необязательном порядке; или нанесением соответствующих компонентов на металлосиликат одновременно. Среди этих способов предпочтительны способы с первоначальным нанесением первого компонента на металлосиликат. После нанесения первого компонента множество других компонентов можно наносить последовательно или одновременно.
Катализатор может быть использован в форме частиц, таблеток или любой другой форме. Кроме того, чтобы уменьшить период индукции, требуемый для получения ароматического соединения, катализатор может быть подвергнут активации, включая предварительные обработки водородсодержащим газом, гидразином или такими металловодородными соединениями, как ВН3, NаН и АlН3.
В качестве низшего углеводорода, в качестве исходного реакционного материала могут применяться различные газы, содержащие низший углеводород в преобладающем количестве. Конкретные примеры содержащих низшие углеводороды газов могут включать в себя LРG, содержащий метан в количестве обычно не меньше чем 50% по массе, предпочтительно не меньше чем 70% по массе и т.д.
Реакция может осуществляться периодически или непрерывно. Среди этих способов предпочтительным является непрерывный способ реакции с использованием неподвижного, подвижного или псевдоожиженного слоев. Температура реакции обычно составляет от 300 до 800°С, предпочтительно от 450 до 775°С; реакционное давление обычно составляет от 0,1 до 10 кг/см2 (избыточное давление; это определение также применимо к последующим описаниям), предпочтительно от 1 до 7 кг/см2; и массовая часовая объемная скорость (МЧОС) обычно составляет от 0,1 до 10, предпочтительно от 0,5 до 5,0.
В указанной реакции могут быть получены ароматические соединения, содержащие ароматические углеводороды, такие как бензол и толуол, в качестве основного компонента, и водород в качестве подобного продукта.
<(ii) Стадия отделения ароматического соединения>
На стадии отделения ароматического соединения газ продуктов реакции, полученный на вышеуказанной стадии синтеза ароматического соединения, разделяется на "ароматические соединения" и "газ, содержащий непрореагировавший низший углеводород, получаемый низший углеводород и водород", для возврата соответствующих компонентов. Газ, содержащий "низший углеводород (включая непрореагировавший низший углеводород и получаемый низший углеводород) и водород", который получается отделением ароматического соединения от газа продуктов реакции, подается на последующую стадию метанирования. Между тем, примеры компонентов, содержащихся в ароматических соединениях настоящего изобретения, могут включать в себя бензол, толуол, ксилол, нафталин, триметилбензол, метилнафталин и диметилнафталин. Среди этих компонентов предпочтительными являются бензол, толуол и нафталин. Также получаемый низший углеводород включает в себя этан, этилен и т.д., которые являются побочными продуктами маршрута получения ароматических соединений из низшего углеводорода.
Способ отделения ароматического соединения от газа продуктов реакции не ограничивается конкретно. Может использоваться способ охлаждения газа с помощью теплообменника, чтобы конденсировать высококипящие соединения, содержащиеся в газе, и затем отделением конденсированного продукта с использованием сепаратора газ-жижкость, оборудованного каплеотбойником. В данном случае, чтобы увеличить количество конденсированных жидких компонентов, охлаждающая температура может быть снижена с использованием хладагента. Когда ароматическое соединение представляет собой бензол, например, реакционное давление увеличивают до давления, используемого на следующей стадии (стадия метанирования), а температуру снижают до 6°С, чтобы отделить ароматические соединения от газа продуктов реакции. Использование более высокого реакционного давления является предпочтительным. Однако, когда реакционное давление поднимается до слишком высокого уровня, имеет место тенденция к потере рабочей мощности. Также применение более низкой температуры реакции является предпочтительным. Однако, когда температура реакции уменьшается до менее чем 6°С, ароматическое соединение (бензол) имеет тенденцию к затвердеванию, то есть может быть трудно его отделить. Кроме того, когда температура реакции уменьшается до менее чем 1°С, требуется отделение и удаление воды, а также увеличение размера охлаждающего оборудования, что приводит к увеличению затрат на оборудование. Примеры другого способа отделения могут включать в себя способ отделения, использующий поглощающий раствор.
Ароматические соединения отделяются в форме жидкого компонента. С другой стороны, газ, содержащий непрореагировавший низший углеводород и водород, который отделяется от ароматических соединений, может содержать в добавление к этану и водороду моноксид углерода, диоксид углерода, углеводороды, имеющие от 2 до 5 атомов углерода, хотя они меняются в зависимости от состава исходных газов и, следовательно, не ограничиваются конкретно.
Между тем, хотя водородсодержащий газ, полученный на стадии отделения ароматического соединения, может непосредственно направляться на стадию метанирования, с точки зрения промышленности и эффективности предпочтительно, когда водород отделяется от водородсодержащего газа и подается на стадию метанирования, а оставшийся газ, полученный отделением водорода от водородсодержащего газа, который содержит непрореагировавший низший углеводород в качестве основного компонента, подается на стадию синтеза ароматического соединения.
Водород может отделяться от водородсодержащего газа способом, использующим мембрану отделения водорода, способом поглощения с изменением давления (ПИД) и т.д.
<(iii) Стадия метанирования>
На стадии метанирования водород, содержащийся в подаваемом газе, реагирует с моноксидом углерода и/или диоксидом углерода, превращая эти компоненты в метан и воду.
В качестве источника водорода может использоваться водородсодержащий газ, обычно применяемый в промышленности, водород, выделяющийся на вышеописанной стадии (i) синтеза ароматического соединения, водород, содержащийся в Н2-содержащем газе, используемом в качестве исходного газа на стадии (i) синтеза ароматического соединения и т.д. Примеры источника водорода могут включать в себя коксовые газы, газы газификации угля, газы газификации асфальта, газы газификации остатка тяжелой нефти, газы газификации нефтяного кокса, газы печи реформинга, оксогазы, биогазы, газы газификации биомассы и газы газификации отходов.
Кроме того, получаемый или побочно получаемый водород может использоваться в качестве источника водорода. Примеры получаемого или побочно получаемого водорода могут включать в себя водород, отделяемый от вышеуказанных исходных газов или отходящих газов нефтехимических или нефтеперерабатывающих процессов, водород, получаемый реформингом таких углеводородов, как нафта, LNG и LРG, при использовании пара, кислорода, диоксида углерода и т.д., водород, получаемый прямым термическим разложением метана с использованием плазмы и т.д., водород, побочно получаемый на содовых заводах, и водород, получаемый электролизом воды с помощью электрической энергии, которая генерируется электростанциями, использующими гидравлическую энергию, энергию горения, энергию ветра или атомную энергию. Для электролиза воды может применяться способ электролиза щелочной воды, способ электролиза воды высокого давления/высокой температуры, способ электролиза воды с твердым полимерным электролитом, способ электролиза высокотемпературного пара и т.д. Кроме того, также может быть использован водород, получаемый при генерации электроэнергии с помощью солнечных элементов, а также водород, получаемый разложением воды с применением катализатора фоторазложения, такого как диоксид титана. Кроме того, в качестве водорода, получаемого способом, иным чем электролиз, также может применяться водород, получаемый термохимическим способом, в котором термическое разложение воды разделяется на несколько химических реакций, чтобы разделить воду на водород и кислород только с помощью тепла, генерируемого при меньшей температуре, чем тепло, требуемое для прямого термического разложения. В этом случае в качестве источника тепла для термического разложения также могут использоваться отходящие газы, выпускаемые из высокотемпературной газовой печи, использующей ядерное топливо. Кроме того, разложение воды может осуществляться с использованием излучения, такого как γ-лучи или лучи близкого ультрафиолета, в качестве источника энергии. Более того, когда в будущем установится общество, способное использовать водород в качестве постоянного источника энергии, водород также можно будет использовать в качестве источника энергии для термического разложения.
Между тем, так как моноксид углерода и/или диоксид углерода расходуются на стадии метанирования, настоящее изобретение может рассматриваться как эффективная промышленная технология переработки диоксида углерода.
Стадия метанирования выражается вышеприведенными формулами (2) и (3). Более конкретно, например, стадия метанирования может осуществляться, как показано в следующих процедурах от (А) до (Е).
(А) "Газ, содержащий низший углеводород (включая непрореагировавший низший углеводород и получаемый низший углеводород) и водород", который отделяется от ароматических соединений на вышеупомянутом этапе отделения ароматического соединения, контактирует с моноксидом углерода и/или диоксидом углерода в присутствии катализатора с реакцией водорода, содержащегося в данном газе, с моноксидом углерода и/или диоксидом углерода, превращая эти компоненты в метан и воду. Между тем, когда водород находится в недостатке относительно большого количества подаваемого моноксида углерода и/или диоксида углерода, дополнительное количество водородсодержащего газа может подаваться извне.
(В) Когда "содержащий низший углеводород газ, содержащий моноксид углерода и/или диоксид углерода" подается на стадию метанирования, моноксид углерода и/или диоксид углерода, содержащиеся в данном газе, реагируют с водородом, содержащимся в данном газе, или "водородом, содержащимся в газе, содержащем низший углеводород (включая непрореагировавший низший углеводород и получаемый низший углеводород) и водород", который отделяется от ароматических соединений на стадии отделения ароматического соединения, в присутствии катализатора, превращая эти компоненты в метан. Между тем, когда количество моноксида углерода и/или диоксида углерода, содержащихся в содержащем низший углеводород газе, находится в недостатке по сравнению с количеством содержащегося в нем водорода, дополнительное количество моноксида углерода и/или диоксида углерода может подаваться извне. Кроме того, дополнительное количество водородсодержащего газа также может подаваться извне.
(С) Когда "содержащий низший углеводород газ, по существу не содержащий моноксида углерода и/или диоксида углерода", подается на стадию метанирования, моноксид углерода и/или диоксид углерода подаются и реагируют с водородом, содержащимся в данном газе, или "водородом, содержащимся в газе, содержащем низший углеводород (включая непрореагировавший низший углеводород и получаемый низший углеводород) и водород", который отделяется от ароматических соединений на стадии отделения ароматического соединения, в присутствии катализатора, превращая эти компоненты в метан. Между тем, в вышеописанном случае, когда количество подаваемого моноксида углерода и/или диоксида углерода слишком велико и, следовательно, количество водорода, содержащегося в газе, находится в относительном дефиците, дополнительное количество водородсодержащего газа может подаваться извне.
(D) В соответствующих способах, описанных в вышеприведенных процедурах от (А) до (С), реакция осуществляется в присутствии катализатора при дополнительной подаче водородсодержащего газа в качестве исходного материала извне, превращая эти компоненты в метан.
(Е) Водородсодержащий газ контактирует с моноксидом углерода и/или диоксидом углерода в присутствии катализатора, вызывая реакцию водорода, содержащегося в данном газе, с моноксидом углерода и/или диоксидом углерода и превращая эти компоненты в метан и воду.
Газ, получаемый на вышеописанной стадии метанирования, возвращается на стадию синтеза ароматического соединения. В данном случае, так как реакция на стадии синтеза ароматического соединения (реакция, выражаемая вышеприведенной формулой (1)) является равновесной реакцией, состав исходного газа на стадии синтеза ароматического соединения может выгодным образом регулироваться так, что содержание в нем углеводорода является настолько низким, насколько возможно для увеличения скорости его реакции. Следовательно, в вышеописанных процедурах (А) и (В), когда количество водорода, содержащегося в газе, относительно больше, чем количество соответствующего моноксида углерода и/или диоксида углерода, дополнительное количество моноксида углерода и/или диоксида углерода предпочтительно подается на стадию метанирования, тем самым регулируя содержание водорода на данном этапе до величины, подходящей для превращения исходных материалов в метан и воду.
В качестве моноксида углерода и/или диоксида углерода, подаваемых на стадию метанирования, могут использоваться моноксид углерода и/или диоксид углерода, возвращаемые из внешней реакционной системы способа получения согласно настоящему изобретению. Более конкретно, в качестве диоксида углерода может использоваться диоксид углерода, извлекаемый из различных отходящих газов сгорания. Примеры отходящих газов сгорания, из которых может быть извлечен диоксид углерода, могут включать в себя газы сгорания, выходящие из турбин или бойлеров электростанций, и отходящие газы, выпускаемые из различных нагревательных печей химических заводов и различных печей сжигания.
Между тем, в настоящем изобретении, так как моноксид углерода и/или диоксид углерода удерживаются стадией метанирования, способ настоящего изобретения является выгодным с точки зрения снижения количества выпускаемого диоксида углерода.
В качестве катализатора любые катализаторы, известные как катализаторы реакции метанирования, могут быть использованы без особых ограничений. Обычными катализаторами, применимыми в настоящем изобретении, являются никелевые катализаторы. Температура реакции метанирования составляет обычно от 200 до 500°С. Так как реакция метанирования является сильно изотермической реакцией, когда концентрация моноксида углерода и/или диоксида углерода во входящем газе высока, требуется применять многостадийные, например 2- или 3-стадийные, реакторы и помещать между ними хладоагент, или повторно использовать реакционный газ, тем самым регулируя температуру реакции. Моноксид углерода и/или диоксид углерода превращается в метан до достижения по существу равновесного состава реакции. Между тем, когда исходный газ содержит углеводородные соединения, имеющие большое число атомов углерода, пар может добавляться на стадию метанирования для преобразования этих углеводородов. Подходящее количество пара, которое добавляется на стадию метанирования, составляет от 0,8 до 4,5 от массы углерода, подаваемого на этап метанирования.
В предпочтительном варианте осуществления настоящего изобретения рекомендуется использовать способ деления газа, отделяемого от ароматических соединений на вышеописанной стадии отделения, на первую и вторую фракции; сжигания второй фракции; извлечения диоксида углерода из полученного газа сгорания; и подачи этого извлеченного диоксида углерода вместе с первой фракцией на стадию метанирования. Согласно вышеуказанному предпочтительному варианту осуществления настоящего изобретения тепло, генерируемое при сжигании второй фракции, может быть использовано в качестве источника тепла для поддержания температуры реакции на стадии синтеза ароматического соединения, и дополнительный диоксид углерода, выходящий из реакционной системы, извлекается подходящим образом, что создает привлекательный способ с точки зрения защиты окружающей среды, а также предотвращения накопления несжимаемых газов, таких как азот, и примесей в реакционной системе. Между тем, отношение деления первой и второй фракций может быть определено при рассмотрении температуры реакции на этапе синтеза ароматического соединения.
Извлечение диоксида углерода из отходящего газа сгорания может подходящим способом осуществляться, например, с помощью способа, описанного в выложенной японской патентной заявке (КОКАI) №5-184865 (1993), более конкретно, с помощью способа взаимодействия отходящего газа сгорания с водным раствором моноэтаноламина (МЭА) при нормальном давлении для удаления и извлечения диоксида углерода, содержащегося в отходящем газе сгорания. В данном случае водный раствор МЭА предпочтительно представляет собой водный раствор, содержащий МЭА в количестве не менее чем 35% по массе.
Способ, описанный в выложенной японской патентной заявке (КОКАI) №5-184865 (1993), излагается более подробно ниже. То есть, в данном способе существует главным образом применяемое оборудование, включающее в себя охладитель для отходящего газа сгорания, башню извлечения СО2 и башню регенерации водного раствора МЭА.
Охладитель для отходящего газа сгорания имеет башенную структуру, снабженную в его верней части разбрызгивающими соплами и в его центральной части наполняющей секцией и оборудованную насосом циркуляции увлажняющей охлаждающей воды. Отходящий газ сгорания, имеющий температуру обычно от 100 до 150°С, вводится в охладитель отходящего газа сгорания из его верхней части, выпускается из его нижней части и затем подается в нижнюю часть башни извлечения СО2. В охладителе отходящий газ сгорания контактирует с увлажняющей охлаждающей водой, разбрызгиваемой из разбрызгивающих сопел, и охлаждается до температуры обычно от 50 до 150°С.
Башня извлечения СО2 снабжена в ее верхней части разбрызгивающими соплами для водного раствора МЭА и в ее центральной части заполняющей секцией и оборудована насосом для выпуска водного раствора МЭА с поглощенным СО2. Отходящий газ сгорания, введенный в нижнюю часть башни извлечения СО2, контактирует с водным раствором МЭА в противотоке, так что СО2, содержащийся в отходящем газе сгорания, поглощается в водном растворе МЭА и удаляется. Отходящий газ сгорания, из которого удален СО2, выпускается из реакционной системы из верхней части башни извлечения СО2.
Башня регенерации водного раствора МЭА обеспечена в ее верхней части разбрызгивающими соплами для использованного водного раствора МЭА и в ее центральной части заполняющей секцией и оборудована нагревателем регенерации (ребойлером). Водный раствор МЭА с поглощенным СО2 охлаждается теплообменником и затем подается в башню регенерации водного раствора МЭА, где использованный водный раствор МЭА регенерируется ребойлером. Диоксид углерода, освобожденный из водного раствора МЭА, выпускается из реакционной системы из верхней части башни регенерации водного раствора МЭА.
В настоящем изобретении с помощью использования водорода, содержащегося в исходном газе, в качестве источника метана получаемые выше ароматические соединения могут легко превращаться в гидрированные ароматические соединения посредством последующего этапа.
<(iv) Стадия синтеза гидрированного ароматического соединения>
На стадии синтеза гидрированного ароматического соединения ароматические соединения, извлекаемые на вышеописанной стадии отделения ароматического соединения, могут гидрироваться в присутствии катализатора с получением гидрированных ароматических соединений.
Реакция гидрирования ароматических соединений представляет собой обычную технологию, известную в течение долгого времени. В настоящем изобретении любые известные обычные технологии могут быть использованы для реакции гидрирования. Примеры катализатора, применимого в реакции гидрирования, могут включать в себя нанесенные металлические катализаторы, содержащие в качестве активного металла, по меньшей мере, один металл, выбранный из группы, состоящей из родия, иридия, платины, рутения, рения, палладия, молибдена, никеля, вольфрама, ванадия, осмия, кобальта, хрома, железа и их оксидов и сульфидов. Температура реакции гидрирования обычно составляет от 150 до 300°С, предпочтительно от 180 до 270°С, а реакционное давление обычно составляет от 4 до 80 кг/см2, предпочтительно от 9 до 70 кг/см2.
В настоящем изобретении в качестве источника водорода предпочтительно используется, например, водородсодержащий газ, полученный действием на вышеуказанный коксовый газ реакции сдвига, чтобы уменьшить количество моноксида углерода, содержащегося в газе. Реакция сдвига выражается уравнением СО + Н2О → СО2 + Н2, и хорошо известна специалистам в данной области техники. Реакция сдвига осуществляется в присутствии такого катализатора, как железохромовый катализатор или медноцинковый катализатор. Температура реакции сдвига обычно составляет от 180 до 480°С, и реакционное давление обычно составляет от 1 до 34 кг/см2. Между тем, в качестве способа снижения количества моноксида углерода, содержащегося в газе, также может быть использована вышеописанная реакция метанирования. Кроме того, в качестве источника водорода может быть использован, например, водородсодержащий газ, полученный снижением количества моноксида углерода, содержащегося в коксовом газе и т.д., поглощением с изменением давления (ПИД) или способом, использующим мембрану отделения водорода.
<(v) Стадия отделения гидрированного ароматического соединения>
На стадии отделения гидрированного ароматического соединения газ продуктов реакции, полученных на вышеописанном этапе синтеза гидрированного ароматического соединения, разделяется на гидрированные ароматические соединения и водородсодержащий газ для возврата соответствующих компонентов. Компонент гидрированных ароматических соединений может включать в себя вышеуказанный гидрид бензола (С6Н12), и т.д., и данный газовый компонент может включать в себя в добавление к низшему углеводороду и диоксиду углерода водород и т.д.
Способ отделения гидрированных ароматических соединений от газа продуктов реакции не ограничивается конкретно. Гидрированные ароматические соединения могут подходящим образом отделяться от газа продуктов реакции охлаждением данного газа теплообменником с конденсацией содержащихся в нем высококипящих соединений и воздействия на этот конденсированный продукт разделения газ/жидкость с использованием сепаратора, оборудованного каплеотбойником. В данном способе при поддержании реакционного давления гидрирования газ продуктов реакции охлаждается до 6°С для выделения из него гидрированных ароматических соединений.
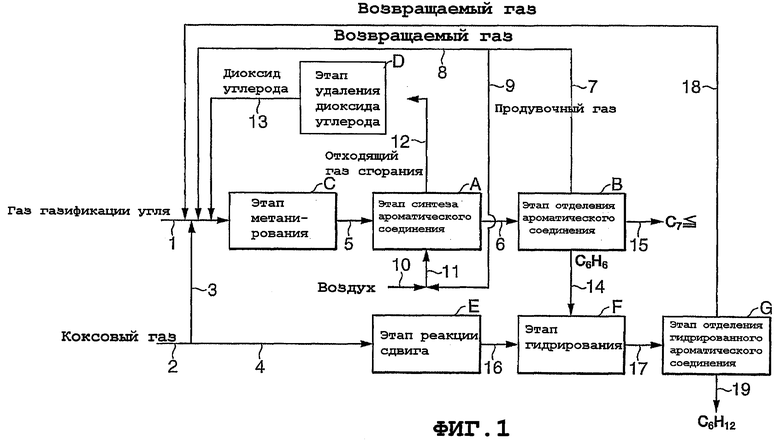
Фиг.1 представляет собой блок-схему, показывающую пример способа получения ароматических соединений согласно настоящему изобретению. Способ получения, показанный на фиг.1, использует в качестве исходного газа смешанный газ, состоящий из газа газификации угля (содержащего Н2, СО, СО2 и N2) и коксового газа (содержащего Н2, СН4, СО, СО2 и N2), и включает в себя в добавление к стадии (А) синтеза ароматического соединения, этапу (В) отделения ароматического соединения и стадии (С) метанирования стадию (D) удаления диоксида углерода согласно предпочтительному варианту осуществления настоящего изобретения. Кроме того, данный способ получения может дополнительно включать в себя стадию (Е) реакции сдвига, стадию (F) гидрирования и стадию (G) отделения гидрированного ароматического соединения, чтобы реформировать часть коксового газа и получать гидрированные ароматические соединения в добавление к ароматическим соединениям. Поток газа через соответствующие стадии протекает следующим образом.
Газ газификации угля подается по линии (1) на стадию (С) метанирования. Коксовый газ вводится по линии (2) и делится на две текущие фракции, одна из которых подается по линии (3), соединяясь с подачей газа газификации угля по линии (1) и затем подаваясь в виде смешанного газа на стадию (С) метанирования, а другая подается по линии (4) на стадию (Е) реакции сдвига.
Газ (метансодержащий газ), полученный на стадии (С) метанирования, подается по линии (5) на стадию (А) синтеза ароматического соединения, а газ продуктов реакции, полученный на стадии (А), подается по линии (6) на стадию (В) отделения ароматического соединения.
Газ, содержащий непрореагировавший метан и водород, который отделяется от ароматических соединений на стадии (В) отделения ароматического соединения, выпускается по линии (7) и делится на две текущих фракции, одна из которых подается в качестве возвращаемого газа по линии (8) на стадию (С) метанирования, а другая подается по линии (9) на стадию (А) синтеза ароматического соединения и используется в качестве топлива для подвода туда теплоты реакции.
Более конкретно, газ, подаваемый по линии (9), смешивается с воздухом, подаваемым по линии (10), и полученный смешанный газ подается по линии (11) и используется в качестве топлива на стадии (А) синтеза ароматического соединения. Отходящий газ сгорания подается по линии (12) на стадию (D) удаления диоксида углерода, а диоксид углерода, извлеченный на стадии (D), подается по линии (13) вместе с возвращаемым газом, подаваемым по линии (8), на стадию (С) метанирования.
Ароматические соединения, отделяемые на стадии (В) отделения ароматического соединения, могут быть разделены, если требуется, на фракцию бензола (С6Н6) и другие фракции (высококипящие компоненты, такие как типичные компоненты, имеющие 7 или больше атомов углерода). Такое разделение может быть легко выполнено, например, с помощью соответствующей дистилляционной колонны.
Между тем, в варианте осуществления, показанном на фиг.1, дистилляционная колонна пропущена, и схематически показано, что свыше двух видов фракций выпускается со стадии (В) отделения ароматического соединения по линиям (14) и (15) соответственно.
Бензол, выпускаемый по линии (14), подается на стадию (F) гидрирования и гидрируется там. Водород, требуемый для гидрирующей обработки, подается со стадии (Е) реакции сдвига по линии (16). Газ, полученный на стадии (F) гидрирования, подается по линии (17) на этап (G) отделения гидрированного ароматического соединения, а газ, от которого отделяется гидрированное ароматическое соединение, подается в качестве возвращаемого газа по линии (18) на стадию (С) метанирования. Извлеченное гидрированное ароматическое соединение забирается по линии (19).
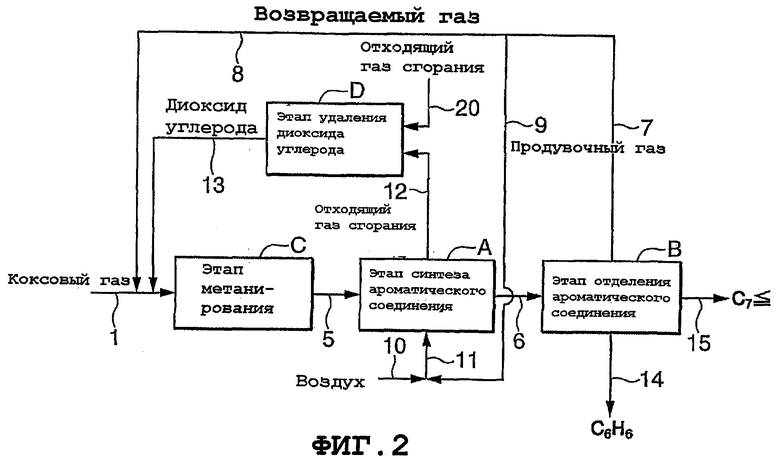
Фиг.2 представляет собой блок-схему, показывающую другой пример способа получения ароматических соединений согласно настоящему изобретению. Способ получения, показанный на фиг.2, использует в качестве исходного газа коксовый газ (содержащий Н2, СН4, СО, СО2 и N2) и отходящий газ сгорания, возвращаемый извне, и включает в себя в добавление к стадии (А) синтеза ароматического соединения, стадии (В) отделения ароматического соединения и стадии (С) метанирования стадию (D) удаления диоксида углерода согласно предпочтительному варианту осуществления настоящего изобретения. Поток газа через соответствующие стадии протекает следующим образом.
Коксовый газ подается по линии (1) на стадию (С) метанирования. Отходящий газ сгорания, возвращаемый извне, подается по линии (20) на стадию (D) удаления диоксида углерода.
Газ (метансодержащий газ), полученный на стадии (С) метанирования, подается по линии (5) на стадию (А) синтеза ароматического соединения, а газ продуктов реакции, полученный на стадии (А), подается по линии (6) на стадию (В) отделения ароматического соединения.
Газ, содержащий непрореагировавший низший углеводород и водород, который отделяется от ароматических соединений на стадии (В) отделения ароматического соединения, выпускается по линии (7) и делится на две текущих фракции, одна из которых подается в качестве возвращаемого газа по линии (8) на стадию (С) метанирования, а другая подается по линии (9) на стадию (А) синтеза ароматического соединения и используется в качестве топлива для подвода туда теплоты реакции.
Более конкретно, газ, подаваемый по линии (9), смешивается с воздухом, подаваемым по линии (10), и полученный смешанный газ подается по линии (11) и используется в качестве топлива на этапе (А) синтеза ароматического соединения. Отходящий газ сгорания подается по линии (12) на стадию (D) удаления диоксида углерода, тогда как отходящий газ сгорания, возвращаемый извне, подается по линии (20) на стадию (D) удаления диоксида углерода. Диоксид углерода, извлеченный на стадии (D) удаления диоксида углерода, подается по линии (13) на стадии (С) метанирования.
Ароматические соединения, отделяемые на этапе (В) отделения ароматического соединения, могут быть разделены, если требуется, на фракцию бензола (С6Н6) и другие фракции (высококипящие компоненты, такие как типичные компоненты, имеющие 7 или больше атомов углерода). Такое разделение может быть легко выполнено, например, с помощью соответствующей дистилляционной колонны.
Между тем, в варианте осуществления, показанном на фиг.2, дистилляционная колонна пропущена, и схематически показано, что свыше двух видов фракций выпускается со стадии (В) отделения ароматического соединения по линиям (14) и (15) соответственно.
Между тем, ароматические соединения, полученные с помощью вышеописанного способа настоящего изобретения, могут быть использованы в качестве исходного материала для получения не только гидрированных ароматических соединений, но также всех обычно получаемых производных ароматических соединений. В случае, когда ароматическое соединение представляет собой бензол, примеры соединений, которые могут быть получены из бензола, могут включать в себя этилбензол, получаемый алкилированием бензола этиленом (исходный материал для стирола и полистироловых смол), кумол, получаемый алкилированием бензола пропиленом (исходный материал для фенола, бифенола А и поликабонатных смол), и высшие алкилбензолы, получаемые алкилированием бензола высшими олефинами (исходные материалы для алкилбензолсульфонововых кислот), а также такие алкилбензолы, как толуол и ксилол, которые могут быть получены алкилированием бензола метанолом и т.д. Кроме того, например, подвергая п-ксилол реакции окисления, может быть получена терефталевая кислота, и далее терефталевая кислота может реагировать с этиленгликолем, давая полиэтилентерефталат.
В случае, когда гидрированное ароматическое соединение представляет собой циклогексан, примеры соединений, которые могут быть получены из циклогексана, могут включать в себя циклогексанон, циклогексанол и капролактам. Капролактам может быть подвергнут реакции раскрытия цикла с получением 6-нейлона. Также циклогексанон может быть подвергнут дегидрированию с получением циклогексена, и далее циклогексен может быть использован в качестве исходного материала для получения адипиновой кислоты. Адипиновая кислота может реагировать с гексаметилендиамином с получением 6,6-найлона.
Ароматическое соединение, в частности бензол, может быть сначала подвергнуто реакции селективного окисления с получением малеинового ангидрида, а затем малеиновый ангидрид может быть подвергнут реакции каталитического гидрирования с получением γ-бутиролактона, тетрагидрофрана, 1,4-бутандиола и т.д. Кроме того, γ-бутиролактон может реагировать с алкиламином или аммиаком с получением N-алкил-2-пирролидона. Также 1,4-бутандиол может быть подвергнут реакции дегидратирования с селективным получением тетрагидрофурана, и тетрагидрофуран может далее реагировать в присутствии кислотного катализатора и т.д. с получением политетраметиленгликолевого эфира в качестве низкосортного полимерного продукта.
Когда 1,4-бутандиол и терефталевая кислота подвергаются реакции конденсации, может быть получен полибутилентерефталат. Также нафталины могут быть окислены с получением фталевой кислоты и ее производных. Кроме того, ароматическое соединение или гидрированное ароматическое соединение могут каталитически разлагаться с получением низших олефинов, таких как этилен, пропилен и бутилен. Примеры производных низших олефинов, производимых из этих низших олефинов, могут включать в себя производные этилена, например окисленные продукты, такие как этиленоксид, этиленгликоль, этаноламин и гликолевые эфиры; и хлорированные продукты, такие как винилхлоридный мономер, 1,1,1-трихлорэтан, поливинилхлоридные смолы и винилиденхлорид. Кроме того, этилен может полимеризоваться с получением α-олефинов, полиэтиленов низкой или высокой плотности и т.д. Далее α-олефины могут быть использованы в качестве исходных материалов для получения высших спиртов путем последовательного действия на α-олефины оксореакции и затем реакции гидрирования. Этилен может также реагировать с уксусной кислотой с получением винилацетата, и может также подвергаться реакции Вакера с получением ацетальдегида и этилацетата в качестве производного. Примеры производных пропилена могут включать в себя продукты аммоксидирования, такие как акрилонитрил; продукты селективного окисления, такие как акролеин, акриловая кислота и акриловые сложные эфиры; продукты оксореакции, такие как н-бутиральдегид и оксоспирты, такие как 2-этилгексанол; пропиленовые полимеры, такие как полипропилен; продукты селективного окисления пропилена, такие как пропиленоксид и пропиленгликоль; и продукты гидратирования пропилена, такие как изопропиловый спирт. Кроме того, ацетон может получаться по реакции Вакера. Затем ацетон может быть использован в качестве исходного материала для получения метилизопропилкетона или ацетонцианогидрина. Ацетонцианогидрин также может быть использован в качестве исходного материала для получения метилметакрилата. Далее бутен может подвергаться реакции окислительного гидрирования с получением бутадиена. Бутадиен также может быть последовательно подвергнут ацетоксилированию, гидрированию и гидролизу с получением 1,4-бутандиола. 1,4-бутандиол может быть использован в качестве исходного материала для получения γ-бутиролактона и пирролидонов, таких как N-метилпирролидон, и также может быть подвергнут реакции дегидратации с получением тетрагидрофурана, политетраметиленгликоля и т.д. Также бутадиен может быть использован в качестве исходного материала для получения различных синтетических каучуков.
ПРИМЕРЫ
Настоящее изобретение более подробно описывается примерами. Однако примеры являются только иллюстративными и не предназначены для ограничения объема настоящего изобретения.
Пример 1
Используя смешанный газ, состоящий из газа газификации угля (содержащего Н2, СО, СО2 и N2) и коксового газа (содержащего Н2, СН4, СО, СО2 и N2), в качестве исходного газа, подаваемого извне, ароматическое соединение непрерывно получали согласно блок-схеме, показанной на фиг.1. Коксовый газ перед использованием подвергали предварительным обработкам, таким как обессеривание и удаление смолы и пыли, с помощью обычных способов.
Между тем, катализатор для синтеза ароматического соединения получали по способу, описанному в примере 2 выложенной японской патентной заявки (КОКАI) №2005-255605. Более конкретно, в качестве исходного материала катализатора смесь, состоящую из неорганического компонента, органического связующего и воды при массовом отношении 65,4:13,6:21,0, где неорганический компонент состоял из цеолита ZSM-5 (цеолит МFI-типа, SiO/Al2O3=40 (молярное отношение)), глины и стекловолокна при массовом отношении 82,5: 10,5: 7,0, замешивали, получая формованное изделие. Формованное изделие сушили при температуре 100°С в течение 5 часов и затем прокаливали при температуре 750°С. Затем прокаленное изделие погружали в раствор молибдата аммония, пропитывая 6% Мо по массе, и затем прокаливали при температуре 550°С в течение 10 часов, получая предшественник Мо-нанесенного катализатора. Полученный таким образом предшественник Мо-нанесенного катализатора подвергали зауглероживающей обработке при температуре 350°С в течение 24 часов в атмосфере (С4Н10 и Н2) смешанного газа, получая целевой катализатор.
<Стадия (С) метанирования: использование никелевого катализатора>
На стадии (С) метанирования соответственно подавали газ газификации угля по линии (1), коксовый газ по линии (3), возвращаемый газ (метан и водородсодержащий газ) с нижеуказанной стадии (В) отделения ароматического соединения по линии (8), диоксид углерода по линии (13) и возвращаемый газ (метан и водородсодержащий газ) из нижеуказанной стадии (G) отделения гидрированного ароматического соединения по линии (18). Этап (С) метанирования осуществляли при следующих условиях: давление: 10 кг/см2; температура (вход): 350°С; ЧОСГ: 300 ч-1. Составы газов, подаваемых на этап метанирования, а также состав газа продуктов реакции (метансодержащий газ), получаемого на этапе метанирования (состав газа, полученного охлаждением газа продуктов реакции до 40°С с помощью хладоагента и затем от него конденсированной воды), показаны в таблице 1.
<Стадия (А) синтеза ароматического соединения: использование Мо/цеолитного катализатора>
Давление газа (метансодержащий газ), получаемого на стадии (С) метанирования, понижали до 3 кг/см2 и затем этот газ подавали по линии (5) на стадию (А) синтеза ароматического соединения, где газ подвергали каталитической реакции метана. Стадия (А) синтеза ароматического соединения осуществляли при следующих условиях: давление: 3 кг/см2; температура: 750°С; ЧОСГ: 1000 ч-1. Состав газа продуктов реакции, получаемого на стадии (А) синтеза ароматического соединения, показан в таблице 2.
(скорость потока: 44 кНм3/ч)
Температуру реакции на стадии (А) синтеза ароматического соединения поддерживали путем использования теплоты сжигания смешанного газа, состоящего из непрореагировавшего метана и водородсодержащего газа (его состав показан в таблице 3), подаваемого по линии (9), и воздуха, подаваемого по линии (10), используя теплообменник.
Отходящий газ сгорания, получаемый с помощью вышеуказанного сжигания, подавали по линии (12) на стадию (D) удаления диоксида углерода, из которого извлекали 11 кНм3/ч диоксида углерода. Давление извлекаемого таким образом диоксида углерода повышали до 10 кг/см2 и затем диоксид углерода подавали по линии (13) на стадию (С) метанирования. Этап (D) удаления диоксида углерода осуществляли согласно способу, описанному в выложенной японской патентной заявке (КОКАI) №5-184865 (1993), используя 40 мас.% водный раствор МЭА в качестве поглощающего раствора для диоксида углерода.
<Стадия (В) отделения ароматического соединения>
Газ продуктов реакции, получаемый на стадии (А) синтеза ароматического соединения, подавали по линии (6) на стадию (В) отделения ароматического соединения и там обрабатывали. То есть, давление газа продуктов реакции повышали до 10 кг/см2, используя компрессор, и затем газ продуктов реакции охлаждали до температуры газа 6°С с помощью хладагента. После этого газ продуктов реакции разделяли на конденсированную жидкость и газ с помощью сепаратора, оборудованного каплеотбойником.
Отделенный таким образом газ (непрореагировавший метан и водородсодержащий газ) выпускали по линии (7) и делили на две фракции, одну из которых подавали в качестве возвращаемого газа по линии (8) на стадию (С) метанирования, а другую подавали по линии (9) на стадию (А) синтеза ароматического соединения и использовали там в качестве топлива для реакции. С другой стороны, конденсированную жидкость, отделенную от газа, обрабатывали дистилляцией при давлении 10 кг/см2 и коэффициенте опрокидывания потока 0,5, и разделяли на бензол и другие фракции. Скорость выпуска бензола была 22 т/ч.
<Стадия (Е) реакции сдвига: использование железохромового катализатора>
Коксовый газ подавали по линии (4) на стадию (Е) реакции сдвига и обрабатывали там. Стадию (Е) реакции сдвига осуществляли при следующих условиях: давление: 20 кг/см2; температура: 250°С; ЧОСГ: 300 ч-1.
<Стадия (F) гидрирования: использование никелевого катализатора>
Вышеуказанный бензол подавали со скоростью 22 т/ч по линии (14), и водородсодержащий газ, получаемый на вышеуказанном стадии (Е) реакции сдвига, подавали по линии (16) на стадию (F) гидрирования, поддерживаемый при давлении 20 кг/см2 и температуре 200°С. Соответствующие газы подавали при ЧОСГ 500 ч-1. Составы исходных газов, подаваемых по соответствующим линиям, перед гидрирующей обработкой и состав газа продуктов реакции, получаемого после гидрирующей обработки, показаны в таблице 4.
(скорость потока: 38 кНм3/ч)
(скорость потока: 19 кНм3/ч)
<Стадия (G) отделения гидрированного ароматического соединения>
Газ продуктов реакции, получаемый на этапе (F) гидрирования, подавали по линии (17) на стадию (G) отделения гидрированного ароматического соединения и обрабатывали там. То есть, газ продуктов реакции охлаждали до температуры газа 1°С с помощью хладоагента и затем разделяли на конденсированную жидкость и газ с помощью сепаратора, оборудованного каплеотбойником. Количество извлекаемой конденсированной жидкости (С6Н12) составляло 23 т/ч. Извлеченный таким образом газ подавали в качестве возвращаемого газа (его состав показан в таблице 5) по линии (18) на стадию (С) метанирования.
(скорость потока: 25 кНм3/ч)
Пример 2:
Используя коксовый газа (содержащий Н2, СН4, СО, СО2 и N2) в качестве исходного газа, подаваемого извне, ароматическое соединение непрерывно получали согласно блок-схеме, показанной на фиг.2. Коксовый газ перед использованием подвергали предварительным обработкам, таким как обессеривание и удаление смолы и пыли, с помощью обычных способов.
<Стадия (С) метанирования: использование никелевого катализатора>
На стадии (С) метанирования соответственно подавали коксовый газ по линии (1), возвращаемый газ (метан и водородсодержащий газ) из нижеуказанной стадии (В) отделения ароматического соединения по линии (8) и диоксид углерода по линии (13). Стадию (С) метанирования осуществляли при следующих условиях: давление: 10 кг/см2; температура (вход): 280°С; ЧОСГ: 300 ч-1. Составы газов, подаваемых на стадию метанирования, а также состав газа продуктов реакции (метансодержащий газ), получаемого на стадии метанирования (состав газа, полученного охлаждением газа продуктов реакции до 40°С с помощью охладителя и затем отделения от него конденсированной воды), показаны в таблице 6.
<Стадия (А) синтеза ароматического соединения: использование Мо/цеолитного катализатора>
Давление газа (метансодержащий газ), получаемого на стадии (С) метанирования, понижали до 3 кг/см2 и затем этот газ подавали по линии (5) на стадию (А) синтеза ароматического соединения, где газ подвергали каталитической реакции метана. Стадию (А) синтеза ароматического соединения осуществляли при следующих условиях: давление: 3 кг/см2; температура: 750°С; ЧОСГ: 1000 ч-1. Состав газа продуктов реакции, получаемого на стадии (А) синтеза ароматического соединения, показан в таблице 7.
(скорость потока: 615 кНм3/ч)
Температуру реакции на стадии (А) синтеза ароматического соединения поддерживали за счет использования теплоты сжигания топлива, подаваемого по линии (11), а именно смешанного газа, состоящего из непрореагировавшего метана и водородсодержащего газа (его состав показан в таблице 8), подаваемого по линии (9), и воздуха, подаваемого по линии (10), используя теплообменник.
(скорость потока: 15 кНм3/ч)
Отходящий газ сгорания, получаемый с помощью вышеуказанного сжигания, подавали по линии (12) на стадию (D) удаления диоксида углерода, и отходящий газ сгорания, возвращаемый извне, подавали по линии (20) на стадию (D) удаления диоксида углерода. Диоксид углерода, извлекаемый на стадии (D) удаления диоксида углерода, составлял 24 кНм3/ч. Давление извлекаемого таким образом диоксида углерода повышали до 10 кг/см2 и затем диоксид углерода подавали по линии (13) на стадию (С) метанирования. Стадию (D) удаления диоксида углерода осуществляли согласно способу, описанному в выложенной японской патентной заявке (КОКАI) №5-184865 (1993), используя 40 мас.% водный раствор МЭА в качестве поглощающего раствора для диоксида углерода.
<Стадия (В) отделения ароматического соединения>
Газ продуктов реакции, получаемый на стадии (А) синтеза ароматического соединения, подавали по линии (6) на стадию (В) отделения ароматического соединения и там обрабатывали. То есть, давление газа продуктов реакции повышали до 10 кг/см2, используя компрессор, и затем газ продуктов реакции охлаждали до температуры газа 6°С с помощью охладителя. После этого газ продуктов реакции разделяли на конденсированную жидкость и газ с помощью сепаратора, оборудованного каплеотбойником.
Отделенный таким образом газ (непрореагировавший метан и водородсодержащий газ) выпускали по линии (7) и делили на две фракции, одну из которых подавали в качестве возвращаемого газа по линии (8) на стадию (С) метанирования, а другую подавали по линии (9) на стадию (А) синтеза ароматического соединения и использовали там в качестве топлива для реакции. С другой стороны, конденсированную жидкость, отделенную от газа, обрабатывали дистилляцией при давлении 10 кг/см2 и коэффициенте опрокидывания потока 0,5 и разделяли на бензол и другие фракции. Скорость выпуска бензола была 29 т/ч.
Пример 3:
Используя смешанный газ, состоящий из коксового газа (содержащего Н2, СН4, СО, СО2 и N2) и диоксида углерода, извлекаемого из отходящего газа сгорания в качестве исходного газа, подаваемого извне, ароматическое соединение непрерывно получали согласно блок-схеме, показанной на фиг.2. Коксовый газ перед использованием подвергали предварительным обработкам, таким как обессеривание и удаление смолы и пыли, с помощью обычных способов.
<Стадия (С) метанирования: использование никелевого катализатора>
На cтадии (С) метанирования соответственно подавали коксовый газ по линии (3), возвращаемый газ (низший углеводород и водородсодержащий газ) из нижеуказанной стадии (В) отделения ароматического соединения по линии (8), диоксид углерода по линии (13) и возвращаемый газ (низший углеводород и водородсодержащий газ) из нижеуказанной стадии (G) отделения гидрированного ароматического соединения по линии (18). Все из вышеуказанных газов подавали по линии (1) на стадию (С) метанирования. Стадию (С) метанирования осуществляли при следующих условиях: давление: 10 кг/см2; температура (вход): 350°С; ЧОСГ: 300 ч-1. Составы газов, подаваемых на стадии метанирования, а также состав газа продуктов реакции (метансодержащий газ), получаемого на этапе метанирования (состав газа, полученного охлаждением газа продуктов реакции до 40°С с помощью хладоагента и затем отделения от него конденсированной воды), показаны в таблице 9.
<Стадия (А) синтеза ароматического соединения: использование Мо/цеолитного катализатора>
Давление газа (метансодержащий газ), получаемого на стадии (С) метанирования, понижали до 3 кг/см2 и затем этот газ подавали по линии (5) на стадии (А) синтеза ароматического соединения, где газ подвергали каталитической реакции метана. Стадию (А) синтеза ароматического соединения осуществляли при следующих условиях: давление: 3 кг/см2; температура: 750°С; ЧОСГ: 1000 ч-1. Состав газа продуктов реакции, получаемого на стадии (А) синтеза ароматического соединения, показан в таблице 10.
(скорость потока: 437 кНм3/ч)
Температуру реакции на стадии (А) синтеза ароматического соединения поддерживали за счет использования теплоты сжигания топлива, подаваемого по линии (11), а именно смешанного газа, состоящего из непрореагировавшего низшего углеводорода и водородсодержащего газа (его состав показан в таблице 11), подаваемого по линии (9), и воздуха, подаваемого по линии (10), используя теплообменник.
(скорость потока: 13 кНм3/ч)
Отходящий газ сгорания, получаемый с помощью вышеуказанного сжигания, подавали по линии (12) на стадию (D) удаления диоксида углерода, и отходящий газ сгорания, возвращаемый извне, подавали по линии (20) на стадию (D) удаления диоксида углерода. Количество диоксида углерода, извлекаемого на стадии (D) удаления диоксида углерода, составляло 21 кНм3/ч. Давление извлекаемого таким образом диоксида углерода повышали до 10 кг/см2 и затем диоксид углерода подавали по линии (13) на стадию (С) метанирования. Стадию (D) удаления диоксида углерода осуществляли согласно способу, описанному в выложенной японской патентной заявке (КОКАI) №5-184865 (1993), используя 40 мас.% водный раствор МЭА в качестве поглощающего раствора для диоксида углерода.
<Стадия (В) отделения ароматического соединения>
Газ продуктов реакции, получаемый на стадии (А) синтеза ароматического соединения, подавали по линии (6) на стадию (В) отделения ароматического соединения и там обрабатывали. То есть, давление газа продуктов реакции повышали до 10 кг/см2, используя компрессор, и затем газ продуктов реакции охлаждали до температуры газа 6°С с помощью охладителя. После этого газ продуктов реакции разделяли на конденсированную жидкость и газ с помощью сепаратора, оборудованного каплеотбойником.
Отделенный таким образом газ (непрореагировавший низший углеводород и водородсодержащий газ) выпускали по линии (7) и делили на две фракции, одну из которых подавали в качестве возвращаемого газа по линии (8) и затем по линии (1) на стадию (С) метанирования, а другую подавали по линии (9) на стадию (А) синтеза ароматического соединения и использовали там в качестве топлива для реакции. С другой стороны, конденсированную жидкость, отделенную от газа, обрабатывали дистилляцией при давлении 10 кг/см2 и коэффициенте возврата потока 0,5 и разделяли на бензол и другие фракции. Скорость выпуска бензола была 20 т/ч.
<Стадия (Е) реакции сдвига: использование железохромового катализатора>
Коксовый газ подавали по линии (4) на стадию (Е) реакции сдвига и обрабатывали там. Стадию (Е) реакции сдвига осуществляли при следующих условиях: давление: 20 кг/см2; температура: 250°С; ЧОСГ: 300 ч-1.
<Стадия (F) гидрирования: использование никелевого катализатора>
Вышеуказанный бензол подавали со скоростью 20 т/ч по линии (14), и водородсодержащий газ, получаемый на вышеуказанной стадии (Е) реакции сдвига, подавали по линии (16) на стадию (F) гидрирования, поддерживаемый при давлении 20 кг/см2 и температуре 200°С. Соответствующие газы подавали при ЧОСГ 500 ч-1. Составы исходных газов, подаваемых по соответствующим линиям, перед гидрирующей обработкой и состав газа продуктов реакции, получаемого после гидрирующей обработки, показаны в таблице 12.
<Стадия (G) отделения гидрированного ароматического соединения>
Газ продуктов реакции, получаемый на стадии (F) гидрирования, подавали по линии (17) на стадию (G) отделения гидрированного ароматического соединения и обрабатывали там. То есть, газ продуктов реакции охлаждали до температуры газа 1°С с помощью хладоагента и затем разделяли на конденсированную жидкость и газ с помощью сепаратора, оборудованного каплеотбойником. Количество извлекаемой конденсированной жидкости (С6Н12) составляло 21 т/ч. Извлеченный таким образом газ подавали в качестве возвращаемого газа (его состав показан в таблице 13) по линии (18) и затем по линии (1) на этап (С) метанирования.
(скорость потока: 12 кНм3/ч)
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ АЛИФАТИЧЕСКИХ | 2008 |
|
RU2461537C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2418780C2 |
| ПОЛУЧЕНИЕ АЛКИЛИРОВАННЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2417974C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2454390C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2459789C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2462444C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2008 |
|
RU2460581C2 |
| СИСТЕМА ДЛЯ ПРОИЗВОДСТВА АРОМАТИЧЕСКОГО СОЕДИНЕНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2015 |
|
RU2650513C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2008 |
|
RU2491120C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2006 |
|
RU2408565C2 |
Изобретение относится к двум вариантам способа получения ароматических соединений, один из которых включает: стадию метанирования с контактом водородсодержащего газа с моноксидом углерода и/или диоксидом углерода в присутствии катализатора, вызывающего реакцию водорода, содержащегося в газе, с моноксидом углерода и/или диоксидом углерода и превращение этих компонентов в метан и воду; и стадию синтеза ароматического соединения с реакцией низшего углеводорода с метаном, получаемым на стадии метанирования, в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения и водород, причем ароматические соединения отделяют от газа продуктов реакции, получаемого на стадии синтеза ароматического соединения, и затем остающийся полученный водородсодержащий газ подают на стадию метанирования. Также изобретение относится к способу получения гидрированных ароматических соединений, которые получены вышеотмеченными способами. Применение настоящего способа позволяет получать ароматические соединения каталитической реакцией низших углеводородов, таких как метан. 3 н. и 12 з.п. ф-лы, 13 табл., 2 ил.
1. Способ получения ароматических соединений, включающий:
стадию метанирования с контактом водородсодержащего газа с моноксидом углерода и/или диоксидом углерода в присутствии катализатора, вызывающего реакцию водорода, содержащегося в газе, с моноксидом углерода и/или диоксидом углерода и превращение этих компонентов в метан и воду; и
стадию синтеза ароматического соединения с реакцией низшего углеводорода с метаном, получаемым на стадии метанирования, в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения и водород, причем
ароматические соединения отделяют от газа продуктов реакции, получаемого на стадии синтеза ароматического соединения, и затем остающийся полученный водородсодержащий газ подают на стадию метанирования.
2. Способ по п.1, в котором ароматические соединения отделяют от газа продуктов реакции, получаемого на стадии синтеза ароматического соединения, затем водород отделяют от остающегося водородсодержащего газа, получаемого отделением ароматических соединений от газа продуктов реакции, отделенный таким образом водород подают на стадию метанирования, а остающийся газ, получаемый отделением водорода от водородсодержащего газа, подают на стадию синтеза ароматического соединения.
3. Способ по любому из пп.1 и 2, в котором низший углеводород, содержащий моноксид углерода и/или диоксид углерода, подают на стадию метанирования.
4. Способ по любому из пп.1 и 2, в котором низший углеводород, по существу не содержащий моноксида углерода и/или диоксида углерода, подают на стадию синтеза ароматического соединения.
5. Способ по любому из пп.1 и 2, в котором моноксид углерода и/или диоксид углерода, подаваемые на стадию метанирования, представляют собой газы, возвращаемые извне реакционной системы.
6. Способ по любому из пп.1 и 2, в котором подачу диоксида углерода на стадию метанирования осуществляют путем деления остающегося газа, получаемого отделением ароматических соединений от газа продуктов реакции на стадии отделения ароматического соединения, на первую и вторую фракции, сжигания второй фракции, извлечения диоксида углерода из полученного отходящего газа сгорания и подачи извлеченного таким образом диоксида углерода вместе с первой фракцией на стадию метанирования.
7. Способ получения ароматического соединения, включающий следующие стадии от (i) до (iii):
(i) стадия синтеза ароматического соединения с реакцией низшего углеводорода в присутствии катализатора с получением газа продуктов реакции, содержащего ароматические соединения, низший углеводород и водород;
(ii) стадия отделения ароматического соединения с отделением и извлечением ароматических соединений и газа, содержащего низший углеводород и водород, из газа продуктов реакции, получаемого на стадии (i) синтеза ароматического соединения; и
(iii) стадия метанирования с контактом газа, содержащего низший углеводород и водород, который отделяют от ароматических соединений на стадии (ii) отделения ароматического соединения, с моноксидом углерода и/или диоксидом углерода в присутствии катализатора, вызывающего реакцию водорода, содержащегося в газе, с моноксидом углерода и/или диоксидом углерода и превращение этих компонентов в метан и воду,
причем упомянутый способ обеспечивают средством для возврата газа, получаемого на стадии метанирования, на стадию синтеза ароматического соединения.
8. Способ по п.7, в котором водород отделяют от водородсодержащего газа, получаемого на стадии отделения ароматического соединения, и подают на стадию метанирования, а остающийся газ, получаемый отделением водорода от водородсодержащего газа, подают на стадию синтеза ароматического соединения.
9. Способ по п.7, в котором низший углеводород, содержащий моноксид углерода и/или диоксид углерода, подают на стадию метанирования.
10. Способ по п.7 или 8, в котором низший углеводород, по существу не содержащий моноксида углерода и/или диоксида углерода, подают на стадию синтеза ароматического соединения.
11. Способ по п.7 или 8, в котором моноксид углерода и/или диоксид углерода, подаваемые на стадию метанирования, представляют собой газы, возвращаемые извне реакционной системы.
12. Способ по п.7 или 8, в котором подачу диоксида углерода на стадию метанирования осуществляют путем деления остающегося газа, получаемого отделением ароматических соединений от газа продуктов реакции на стадии отделения ароматического соединения, на первую и вторую фракции, сжигания второй фракции, извлечения диоксида углерода из полученного отходящего газа сгорания и подачи извлеченного таким образом диоксида углерода вместе с первой фракцией на стадию метанирования.
13. Способ получения гидрированных ароматических соединений, включающий гидрирование ароматических соединений, полученных с помощью способа, определенного в п.1 или 7, в присутствии катализатора.
14. Способ по п.13, в котором гидрированные ароматические соединения отделяют от газа продуктов реакции, получаемого на стадии гидрирования, и остающийся газ, получаемый отделением гидрированных ароматических соединений от газа продуктов реакции, возвращают на стадию метанирования.
15. Способ по п.13 или 14, в котором газ продуктов реакции, содержащий ароматические соединения и водород, который получают на стадии синтеза ароматического соединения, подают на стадию гидрирования.
| JP 8127544 A, 21.05.1996 | |||
| JP 8127545 A, 21.05.1996 | |||
| JP 11179204 A, 06.07.1999 | |||
| JP 2001334151 A, 04.12.2001 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| JP 2004269398 A, 30.09.2004 | |||
| СПОСОБ ГИДРИРОВАНИЯ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 0 |
|
SU254496A1 |
| RU 97101490 A, 20.02.1999. | |||

