Настоящее изобретение относится к способу получения винилароматических (со)полимеров, привитых на эластомер.
Более конкретно, настоящее изобретение относится к способу получения композиций, включающих жесткую матрицу, сформированную винилароматическими полимерами или сополимерами, и эластомерную или каучукоподобную фазу, диспергированную в матрице в виде частиц со строго бимодальным распределением.
Еще более конкретно, настоящее изобретение относится к способу получения ударопрочного полистирола (УППС), содержащего эластомерную фазу, диспергированную в матрице в форме частиц со строго бимодальным распределением.
Термин «строго бимодальное распределение», используемый в описании и формуле изобретения, подразумевает набор частиц эластомера, диспергированных случайным образом в жесткой полимерной матрице, где указанные частицы подразделяются на первый класс частиц (преобладающий тип) со среднеобъемным размером от 0,15 до 0,50 мкм и второй класс частиц (менее распространенный тип) со среднеобъемным размером от 1 до 8 мкм при полном отсутствии частиц с промежуточными размерами между двумя упомянутыми классами. Среднеобъемный диаметр (Dv) или [D(4,3)] частиц представляет собой подходящую для использования среднюю величину диаметра при необходимости учитывать вклад, который дает каждая частица во фракционный объем дисперсной фазы (фазы геля). Он равен отношению между статистическими моментами, соответственно 4-ой степени [ΣNi(Di)4] и 3-ей степени [ΣNi(Di)3] в статистическом распределении диаметров частиц и выражается следующей общей формулой:
Dv=D(4,3)=ΣNi(Di)4/ΣNi(Di)3,
где Ni и Di представляют число Ni частиц, имеющих диаметр Di (R.D.Cadle "Particle Size Analysis" New York (1965), pages 27-50), и его можно легко определить экспериментально:
- посредством электронной микроскопии (просвечивающий электронный микроскоп, ПЭМ) на тонких слоях полимера, например УППС, с тетроксидом осмия в качестве контрастного средства, с последующим обнаружением видимых диаметров частиц, их уточнением и применением стереологической поправки на толщину тонкого слоя;
посредством лазерной гранулометрии (ЛГ) с применением метилэтилкетона в качестве суспензионной среды, на образцах в виде гранул после повторного нагревания их при 280°С в азоте в течение времени, достаточного для образования обширной сетки из каучукоподобных частиц, сводя к минимуму явление набухания при приготовлении суспензии в метилэтилкетоне.
Для более простого использования этого метода в способе согласно настоящему изобретению интервалы, определяемые для размеров частиц, и экспериментальные измерения относят к методу лазерной гранулометрии (ЛГ), полагая, что:
D(4,3)[ПЭМ]=k·D(4,3)[ЛГ]+h,
где k и h являются параметрами, которые можно получить путем калибровки с некоторым количеством образцов, и в данном случае они имеют значения, соответственно, 0,76 и 0 по отношению к прибору MS MASTERSIZER MICROPLUS компании MALVERN.
Известно, что физико-химические характеристики и механические свойства винилароматических полимеров, упрочненных каучуком, в частности ударопрочного полистирола, зависят от множества факторов, среди которых присутствуют размеры образующих сетку частиц каучука, привитых на полимерную матрицу.
Также известно, что на некоторые свойства, такие как ударная вязкость и поверхностный глянец, в частности у УППС, средний размер и распределение диаметров частиц каучука для данной концентрации каучука, оказывают влияние в двух противоположных направлениях. А именно, «крупные» частицы увеличивают ударную вязкость материала за счет глянца, в то время как «мелкие» частицы снижают прочность на разрыв, но способствуют получению материала с очень хорошей глянцевой поверхностью.
В литературе предложены различные способы получения упрочненных каучуком винилароматических (со)полимеров, например упрочненных каучуком полистиролов, имеющих хорошую глянцевую поверхность в сочетании с хорошей стойкостью к ударным нагрузкам. Например, один из упомянутых способов включает добавление в полимерную матрицу ограниченного числа «крупных» частиц к преобладающему количеству уже присутствующих «мелких» частиц. Полученные таким образом продукты обычно определяют как ударопрочные винилароматические полимеры с бимодальным распределением размеров частиц каучука.
В случае УППС указанное сочетание приводит к получению продукта с положительным синергизмом ударной прочности с отличным поверхностным глянцем.
Например, в патенте США 4153645 описан УППС с улучшенным соотношением свойств, полученный механическим перемешиванием (смешивание в расплаве) 50-80% масс. ударопрочного полистирола, содержащего мелкие частицы каучука (со средним диаметром приблизительно 0,2-0,9 мкм), с 15-50 мас.% ударопрочного полистирола, содержащего частицы каучука большего размера (средний диаметр приблизительно 2-5 мкм). Согласно указанному патенту конечный продукт, получаемый смешиванием двух УППС, показывает значения стойкости к ударным нагрузкам или сопротивление изгибу выше, чем величины, ожидаемые при применении правила смесей, без проявления какого-либо ухудшения других физических свойств.
В патенте США 4493922 описан полученный аналогичным способом (смешивание в расплаве) УППС с бимодальной морфологией, образованной 60-95 мас.% «капсюльных» частиц, имеющих диаметр от 0,2 до 0,6 мкм, и 40-5 мас.% частиц с морфологией «ячейки» и/или «клубка», имеющих диаметр от 2 до 8 мкм.
В патентах США 4221883, 4344039, 4254236; EP(D)M 15752 и 96447 и в международных патентных заявках WO 98/52985 и WO/09080 описан так называемый способ «полимеризации с раздельной подачей» для получения УППС с бимодальной морфологией, позволяющей улучшить соотношение ударной прочности и глянца. Согласно указанному типу процесса в двух третях реактора предварительной полимеризации получают преобладающий вид с меньшими размерами частиц путем подачи раствора полибутадиенового каучука с низкой вязкостью в стироле или раствора блок-сополимера соответствующего состава в стироле. В оставшуюся треть реактора подают раствор полибутадиенового каучука с высокой вязкостью в стироле. Полибутадиен с высокой вязкостью в контакте с ранее образованным форполимером подвергается быстрому обращению фаз, образуя большие частицы, почти не привитые и неспособные легко изменяться в размерах.
В патенте США 5240993 описан способ «параллельной полимеризации» для получения ударопрочных винилароматических полимеров, характеризующихся бимодальным распределением каучуковой фазы, согласно непрерывному способу в массе, где используют два реактора идеального вытеснения. В одном из двух реакторов получают первый форполимер, содержащий каучуковую фазу с мелкими частицами, в то время как в другом реакторе получают второй форполимер с крупными частицами. На выходе из двух реакторов полимерные потоки смешивают, и полимеризацию завершают в третьем реакторе, всегда представляющем собой реактор идеального вытеснения и называемым конечным реактором.
Упрощенный вариант способа «параллельной полимеризации» описан в международной патентной заявке WO 97/39040, согласно которому крупные частицы получают в первой половине реактора предварительной полимеризации, подавая подходящий раствор каучука с высокой вязкостью в винилароматическом мономере при таких условиях, чтобы гарантировать хорошую эффективность прививки и точное регулирование размеров. Во второй половине указанного реактора форполимер с крупными частицами смешивают в подходящих пропорциях со вторым форполимером с мелкими частицами, предварительно полученным в реакторе, установленном последовательно с первым реактором.
Недостатками обоих вышеописанных способов является главным образом следующее:
- в случае «смешивания в расплаве» использование стадии смешивания, что приводит к увеличению производственных затрат или к получению компонентов УППС, которые как таковые трудно продать;
- в случае «параллельной полимеризации» или «полимеризации с раздельной подачей» разработка и инфрастуктура промышленных установок, скомпонованных значительно сложнее (реакторы предварительной полимеризации, расположенные параллельно, отложенные подачи каучуковых растворов, реактор с разделяющей перегородкой) и снабженных гораздо более сложными системами регулирования, чем традиционные установки с последовательно соединенными реакторами полимеризации, используемыми для производства традиционного УППС;
- в обоих случаях получение каучуковой фазы с «мелкими» частицами требует обязательного использования стирол-бутадиеновых блок-сополимеров и/или полибутадиенов радиальной структуры (низкой вязкости), высокая стоимость которых хорошо известна.
Кроме систем, которые предусматривают получение винилароматических сополимеров с бимодальным распределением упрочняющих частиц каучука посредством смешивания предварительно полученных продуктов, были предложены различные «химические» способы, которые позволяют получать указанные особые морфологии путем оперирования составами реакционной смеси и использовать обычные производственные мощности, используемые для традиционных УППС.
Например, в европейском патенте ЕР 418042 описан способ получения упрочненных каучуком винилароматических полимеров, в которых частицы имеют «в общем бимодальное» распределение или расширенное распределение и включают, помимо преобладающего вида мелких частиц (0,1-0,8 мкм) и менее распространенного вида крупных частиц (2-6 мкм), также третий класс частиц с промежуточными размерами (0,8-2,0 мкм). Указанное распределение получают с использованием промежуточного цис-полибутадиена, имеющего бимодальное молекулярно-массовое распределение и продаваемого под наименованием ASAPRENE 760 А.
Аналогично, в европейском патенте ЕР 731016 описан способ получения УППС, имеющего бимодальную морфологию, с использованием традиционной компоновки реакторов, с эластомерной фазой (растворенной в полистироле), образованной полибутадиеном со средним содержанием цис-звеньев и с низкой вязкостью, и полибутадиеном с высоким содержанием цис-звеньев и с высокой вязкостью.
В европейском патенте 726280 описан способ получения УППС, имеющего бимодальную морфологию, путем введения особых концентраций стабильных нитроксильных радикалов при полимеризации с получением УППС с традиционной компоновкой реакторов. Аналогично, в патентной заявке WO 03/033559 описан УППС, имеющий псевдо-бимодальную морфологию, полученный введением особых концентраций функционализованных нанокомпозитных материалов при полимеризации с получением УППС с традиционной компоновкой реакторов. Функцией нанокомпозитного материала является превращение части крупных частиц каучука в мелкие частицы каучука.
Однако во всех указанных патентах предлагаемые способы имеют по меньшей мере тот недостаток, что они не обеспечивают какого-либо «строго бимодального» распределения частиц каучука. Они обеспечивают только «главным образом бимодальные», или просто «широкие» распределения, а также не позволяют регулировать соотношение между «преобладающим» и «менее распространенным» видом.
Наконец, в европейском патенте ЕР 620236 предложен способ получения УППС с «бимодальным» распределением. Согласно указанному способу небольшое количество УППС с крупными частицами растворяют в стироле вместе с полибутадиеновым каучуком или со стирол-бутадиеновым блок-сополимером, необходимым для получения преобладающего вида мелких частиц. Полученный таким образом раствор затем полимеризуют с традиционной компоновкой реакторов. Во время всей полимеризации образующие сетку частицы каучука в предварительно полученном УППС не претерпевают обратной инверсии, но сохраняют неизменными свою структуру и размеры, а полибутадиеновый каучук или стирол-бутадиеновый блок-сополимер дает мелкие частицы соответствующей структуры и размеров. Существенным ограничением предложенного технического решения указанного патента является максимальное процентное содержание предварительно полученного УППС, которое можно растворить в стироле вместе с каучуком (менее 5%).
Более того, следует отметить, что согласно большинству способов известного уровня техники, в которых осуществляют полимеризацию винилароматического мономера и смеси, образованной полибутадиеном и каучуком с полистирольными и полибутадиеновыми блоками, с традиционной компоновкой реакторов структура соответствующего ударопрочного (со)полимера не является строго бимодальной, не только увеличивая отношение η[ПБ]/η[С-Б] от 6 до 25, где η представляет собой вязкость в 5% растворе в стирольном мономере при 25°С.
Заявитель обнаружил способ получения регулируемым образом винилароматических (со)полимеров, привитых на эластомер, с помощью каталитической системы, которая включает стабильный инициатор свободнорадикальной полимеризации, позволяющий получить конечный продукт, в котором распределение диаметров частиц дисперсной эластомерной фазы является «строго бимодальным» и не обязательно зависит от типа используемого эластомера, который может также быть полибутадиеном. Более того, если на стадии полимеризации используют растворитель, конечный непрореагировавший продукт (растворитель + мономер), извлекаемый после стадий удаления летучих продуктов или отпаривания, не требует разделения на его отдельные составляющие, но его можно использовать и возвращать в цикл как таковой, самое большее предусматривая удаление возможного полярного мономера (если он присутствует), такого как производные акриловой или метакриловой кислоты, чтобы улучшить растворение эластомера.
Соответственно, задачей настоящего изобретения является способ получения ударопрочных винилароматических (со)полимеров, привитых на эластомер, содержащий жесткую матрицу, образованную винилароматическими полимерами или сополимерами, и эластомерную фазу, диспергированную в матрице в виде частиц со строго бимодальным распределением диаметров, который включает:
1) функционализацию первой части (X1) эластомера с каталитической системой функционализации/полимеризации, образованной инициатором (G) свободнорадикальной полимеризации, с количеством F функциональных групп, способным отрывать протон от полимерной цепи эластомера, и стабильным инициатором (I) свободнорадикальной полимеризации, включающим группу = N-O и группу = N-O-R', при молярных соотношениях (I)/G·F от 1 до 3, предпочтительно от 1 до 2, причем F равно числу функциональных групп на молекулу инициатора, которая путем разложения дает два свободных радикала, R' является C1-C6 (изо)алкильным радикалом или С7-С20 арилалкильным радикалом, возможно содержащим гетероатомы, такие как N, О, S; R' предпочтительно является 2-фенилэтилом или 2-метил-2-цианопропилом;
2) смешивание функционализованного эластомера со второй частью (Х2) эластомера в течение времени, достаточного для получения однородной композиции;
3) добавление эластомерной однородной композиции в жидкую фазу, по существу состоящую из смеси винилароматического мономера и растворителя полимеризации с массовым соотношением от 60/40 до 90/10, массовым соотношением (Х1+Х2)/винилароматический мономер больше или равным 8/92, обычно от 8/92 до 22/78;
4) полимеризацию винилароматического мономера, возможно в присутствии одного или более сомономеров, при температуре больше или равной 120°С;
5) извлечение полученного винилароматического полимера по окончании полимеризации и
6) возможное возвращение смеси растворителя и мономера, поступающей со стадии извлечения, на стадию (1) после отделения возможных полярных сомономеров.
В способе данного изобретения можно использовать любой способ функционализации эластомера. Обычно предпочтительно функционализировать эластомер в расплавленном состоянии или в растворителе. В первом случае эластомер и каталитическую систему загружают в экструдер или аналогичное устройство типа Брабендера, доводят до температуры и поддерживают в указанных условиях в течение времени, достаточного для функционализации эластомера. После охлаждения и отвердевания функционализованный эластомер можно также хранить в течение нескольких дней перед обработкой согласно указанному изобретению.
Во втором случае эластомер и каталитическую систему растворяют в специально предназначенном растворителе, например таком же растворителе, как и используемый во время полимеризации, и функционализация протекает при температуре ниже 110°С, предпочтительно от 80 до 110°С. После этого полученный таким образом функционализованный раствор переносят на последующие стадии полимеризации.
Растворитель, когда он отличается от используемого в полимеризации, является инертным для компонентов каталитической системы и имеет температуру кипения выше, чем температура функционализации.
По окончании функционализации функционализованную часть смешивают с оставшейся нефункционализованной частью. Указанная операция смешивания предпочтительно происходит в растворителе при поддержании двух частей в контакте в течение времени, по существу необходимого для получения однородной композиции. В конце предпочтительно подавать полученную таким образом однородную композицию непосредственно на стадию полимеризации без промежуточного хранения.
Смешивание частей происходит в растворителе. Последний может быть таким же растворителем, как и используемый на стадии функционализации и/или полимеризации.
В случае функционализации и смешивания в присутствии такого же растворителя, как и используемый в полимеризации, способ настоящего изобретения возможно осуществлять согласно альтернативному приему.
Соответственно, задачей настоящего изобретения также является способ получения ударопрочных винилароматических (со)полимеров, привитых на эластомер, содержащих жесткую матрицу, образованную винилароматическими полимерами или сополимерами, и эластомерную фазу, диспергированную в матрице в виде частиц со строго бимодальным распределением диаметров, который включает:
а) растворение первой части (X1) эластомера в жидкой фазе, состоящей из смеси винилароматического мономера и растворителя полимеризации с массовым соотношением от 60/40 до 100/0, предпочтительно от 60/40 до 90/10, при соотношении эластомер/винилароматический мономер больше или равном 8/92, обычно от 8/92 до 22/78;
б) добавление к раствору каталитической системы функционализации/ полимеризации, образованной инициатором (G) свободнорадикальной полимеризации, с количеством F функциональных групп, способным отрывать протон от полимерной цепи эластомера, и стабильным инициатором (I) свободнорадикальной полимеризации, включающим группу = N-O и группу = N-O-R' с молярными соотношениями (I)/G-F от 1 до 3, предпочтительно от 1 до 2, причем F равно числу функциональных групп на молекулу инициатора, которая путем разложения дает два свободных радикала, причем R' является С1-С6 (изо)алкильным радикалом или С7-С20 арилалкильным радикалом, возможно содержащим гетероатомы, такие как N, О, S; R', предпочтительно является 2-фенилэтилом или 2-метил-2-цианопропилом;
в) нагревание при перемешивании смеси, полученной на стадии (б), при температуре от 80 до 110°С в течение времени, достаточного для функционализации эластомера;
г) подачу в смесь, содержащую функционализованный эластомер в растворе, второй части (X2) эластомера и возможно растворителя и/или винилароматического мономера и/или сомономеров, гомогенизацию получаемой смеси и полимеризацию при температуре выше или равной 120°С, предпочтительно от 120 до 200°С;
д) извлечение полученного винилароматического полимера по окончании полимеризации и
е) возможное возвращение смеси растворителя и мономера, поступающей со стадии извлечения, на стадию (а) после отделения возможных полярных сомономеров.
Согласно способу настоящего изобретения возможно получать винилароматический (со)полимер, упрочненный эластомером со строго бимодальным распределением, при этом возможно регулировать соотношение между мелкими и крупными частицами просто путем регулирования массового отношения между первой частью Х1 и второй частью Х2 эластомера до и после стадии функционализации/растворения эластомера. Предпочтительные массовые соотношения X1/X2 составляют от 99/1 до 40/60.
Согласно настоящему изобретению способ получения винилароматического полимера можно осуществлять в массе, в присутствии растворителя, по периодическому, полупериодическому или непрерывному типу способа. Предпочтительным способом согласно настоящему изобретению является непрерывный способ, описанный, например, в европейском патенте ЕР 400479.
Альтернативно, способ настоящего изобретения можно осуществлять полностью эквивалентным путем при периодическом способе в массе-суспензии, используя снабженные мешалкой автоклавы периодического действия. В таком примере после растворения и гомогенизации эластомерных фракций X1 и Х2 массу нагревают, полимеризуют до обращения фаз и затем ее переносят в автоклав, содержащий воду, где полимеризацию продолжают «в суспензии» согласно традиционным способам.
Термин «винилароматический (со)полимер», используемый в настоящем описании и в пунктах формулы изобретения, по существу означает (со)полимер, получаемый при (со)полимеризации по меньшей мере мономера, соответствующего следующей общей формуле (II):

где R - водород или метильная группа, n равно нулю или целому числу от 1 до 5, и Y - галоген, такой как хлор или бром, или алкильный, или алкоксильный радикал, имеющий от 1 до 4 атомов углерода.
Некоторыми примерами винилароматических мономеров, имеющих вышеуказанную общую формулу, являются стирол, α-метилстирол, β-метилстирол, этилстирол, бутилстирол, диметилстирол, моно-, ди-, три-, тетра-и пентахлорстирол, бромстирол, метоксистирол, ацетоксистирол и т.п. Предпочтительными винилароматическими мономерами являются стирол и/или α-метилстирол.
Винилароматические мономеры общей формулы (I) можно использовать сами по себе или в смеси, содержащей до 50 мас.% других сополимеризуемых мономеров. Некоторыми примерами указанных мономеров являются (мет)акриловая кислота, C1-C4алкиловые эфиры (мет)акриловой кислоты, такие как метилакрилат, метилметакрилат, этилакрилат, этилметакрилат, изопропилакрилат, бутилакрилат, амиды, и нитрилы (мет)акриловой кислоты, такие как акриламид, метакриламид, акрилонитрил, метакрилонитрил, бутадиен, этилен, дивинилбензол, малеиновый ангидрид и т.п. Предпочтительными сополимеризуемыми мономерами являются акрилонитрил и метилметакрилат.
Любой эластомер, пригодный к использованию в качестве упрочняющего продукта в винилароматическом (со)полимере, можно использовать в способе данного изобретения. Однако предпочтительным продуктом из экономических соображений является полибутадиеновый гомополимер со среднечисленной молекулярной массой (Mn) от 50000 до 350000 и среднемассовой молекулярной массой (Mw) от 100000 до 500000. Альтернативно, также можно использовать этилен-пропиленовые эластомеры (ЭПК) или этилен-пропилен-диеновые эластомеры (ЭПДК).
Другие эластомеры, которые можно использовать в смеси с полибутадиеном, можно выбрать среди гомополимеров и сополимеров 1,3-алкадиенов, содержащих 40-100 мас.% мономера 1,2-алкадиена, например бутадиена, изопрена или пентадиена, и 0-60 мас.% одного или более мономера с одной ненасыщенной двойной связью, выбираемого из стирола, акрилонитрила, α-метилстирола, метилакрилата и этилакрилата.
Некоторыми примерами 1,3-алкадиенов являются блок-сополимеры стирола и бутадиена, такие как линейные эластомеры с двумя блоками типа С-Б, где С представляет полистирольный блок со среднемассовой молекулярной массой Mw от 500 до 80000, а Б представляет полибутадиеновый блок со среднемассовой молекулярной массой Mw от 2000 до 250000. В указанных эластомерах количество блока С находится в интервале от 10 до 50 мас.% относительно общего количества сополимера С-Б. Предпочтительным продуктом является блок-сополимер стирола и бутадиена, имеющий содержание стирола 40 мас.% и вязкость, измеренную при 23°С в 5 мас.% растворе в стироле, от 35 до 50 сП.
Другими примерами эластомеров, которые можно использовать в способе настоящего изобретения, являются эластомеры, упомянутые в европейском патенте 606931. Вне зависимости от вида эластомера его используют с конечной концентрацией от 5 до 16% относительно суммарного количества эластомер + винилароматический полимер + растворитель.
Описанные выше эластомеры можно растворять в жидкой фазе, включающей винилароматический мономер и растворитель полимеризации. Предпочтительным растворителем, согласно настоящему изобретению, является этилбензол, но можно использовать другие ароматические растворители, такие как толуол или ксилолы, или алифатические растворители, такие как гексан или циклогексан.
Каталитическую систему функционализации и полимеризации используют в количествах от 0,1 до 2,5 мас.% относительно общего количества эластомера. Каталитическая система состоит из инициатора свободнорадикальной полимеризации и стабильного инициатора свободно-радикальной полимеризации при указанных выше соотношениях. Оказалось, что в случае функционализации эластомера, как описано в стадии (в), не происходит какого-либо существенного образования полимера, и даже если он образуется, его количество не превышает 2 мас.%, и не отмечалось образование сетки эластомера.
Инициаторы свободнорадикальной полимеризации, способные отрывать протон от полимерной цепи эластомера, выбирают из азо-производных, таких как 4,4'-бис(4-цианопентановая кислота), 2,2'-азобис(2-амидинопропан)дигидрохлорид и т.п., или из пероксидов, гидропероксидов, перкарбонатов, перэфиров и солей перкислот, например персульфатов, таких как персульфат натрия или аммония. В общем, предпочтительными инициаторами свободнорадикальной полимеризации являются пероксиды, выбираемые из трет-бутил-изопропил-монопероксикарбоната, трет-бутил-2-этил-монопероксикарбоната, дикумилпероксида, ди-трет-бутилпероксида, 1,1-ди(трет-бутилперокси)циклогексана, 1,1-ди(трет-бутилперокси)-3,3,5-триметилциклогексана, трет-бутилпероксиацетата, кумил-трет-бутилпероксида, трет-бутилпероксибензоата и трет-бутилперокси-2-этилгексаноата.
Стабильный инициатор свободнорадикальной полимеризации, характеризующийся группой = N-O и группой = N-O-R', выбирают из соединений общей формулы (III):

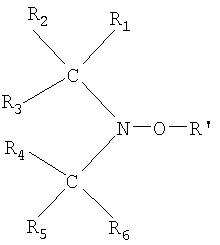
где группы R1, R2, R5 и R6, одинаковые или отличные друг от друга, являются линейными или разветвленными алкильными радикалами, замещенными или незамещенными, содержащими от 1 до 20 атомов углерода, или алкилароматическими радикалами, в которых алкильная группа содержит от 1 до 4 атомов углерода, а группы R3 и R4, одинаковые или отличные друг от друга, являются такими же как R1, R2, R5 и R6, или R3-CNC-R4 является частью циклической структуры, например с 4 или 5 атомами углерода, возможно конденсированной с ароматическим кольцом или насыщенным кольцом, содержащим от 3 до 20 атомов углерода, где по меньшей мере атом водорода циклической структуры может быть замещен гидроксильной группой.
Общая формула (III) инициаторов и их получение описаны в патенте США 4581429.
Некоторыми особенно предпочтительными инициаторами формулы (III), которые можно использовать в способе настоящего изобретения, являются 2,2,5,5-тетраметил-1-пирролидинилокси, известный под торговой маркой PROXYL, 2,2,6,6-тетраметил-1-пиперидинилокси, известный под торговой маркой TEMPO, 4-гидрокси-2,2,6,6-тетраметил-1-пиперидинилокси, известный под торговой маркой 4OH-ТЕМРО, и 1,1,3,3-тетраэтилизоиндолин-2-хилоксил (hiloxyl) (TEDIO). Другие примеры стабильных инициаторов, которые можно использовать в способе настоящего изобретения и которые относятся к общей формуле (III), описаны в вышеупомянутом патенте США 4581429.
Полимеризация протекает в присутствии возможных добавок, например агентов переноса цепи, антиоксидантов, суспендирующих агентов, стабилизирующих агентов и других известных специалистам в этой области. Подробную информацию можно найти в (1) С.В.Bucknall. "Toughened Plastics", Applied Science Publishers Ltd. London, (1977), pages 66-105; (2) A. Echte, "Rubber Toughened Plastics", Advances in Chemistry Series №222, (1989), pages 15-64 (Riew K.S. Ed.); (3) A.E.Platt, "Comprehensive Polymer in Science", Pergamon Press, (1989), vol. 6, pages 437-450; (4) J.L.Hanfeld, B.D.Dalke, "Encyclopedia of Polymer Science and Engineering" 2nd edition, Wiley Interscience, (1989), vol.16, pages 1-71.
По окончании полимеризации полимер отделяют от возможного растворителя и от непрореагировавших мономеров, которые могут находиться в таких соотношениях, что будут пригодными для использования без отделения друг от друга, за исключением возможных полярных сополимеров.
Для лучшего понимания настоящего изобретения и его практического применения ниже приведены некоторые иллюстративные и не ограничивающие изобретение примеры.
ПРИМЕР 1 (Сравнительный)
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, вводят при комнатной температуре 2,63 кг этилбензола, 5,06 кг стирольного мономера (СМ) и 2,17 кг полибутадиена INTENE 50 (вязкость 5% раствора в стирольном мономере составляет 150 сП). Число оборотов мешалки доводят до 80 об/мин. Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение последующих четырех часов. После этого добавляют 28,7 г бензоилпероксида (БПО) и 20 г 4OH-ТЕМРО. Температуру поднимают до 105°С в течение трех часов и поддерживают постоянной в течение последующих двух часов. В реакционную смесь добавляют 24,1 кг стирольного мономера, и смесь затем нагревают до 125°С за 30 минут. Температуру поддерживают при этом значении в течение 6 часов. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler и содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000, 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса полимеризационного раствора добавляют 30 г ди-трет-бутилпероксида, и обороты мешалки увеличивают до 270 об/мин. Температуру смеси повышают до 120°С в течение 45 минут и поддерживают постоянной в течение часа, затем повышают до 140°С в течение 30 минут и поддерживают постоянной в течение 2 часов, и наконец, повышают до 155°С в течение 45 минут и поддерживают постоянной в течение 3 часов. По прошествии трех часов проводят охлаждение до 115°С в течение 40 минут, и смесь воды и органического материала отпаривают от легких фракций (5 литров/час, добавляя 5 литров деминерализованной воды) нагреванием в автоклаве до 145°С в течение трех часов. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого проводят охлаждение до 40°С и автоклав разгружают. Полученный таким образом полимер фильтруют и сушат при 80°С в течение 5 часов.
Анализ полимера с помощью лазерной гранулометрии показывает мономодальную эластомерную фазу с частицами 0,77 мкм (фиг.1).
ПРИМЕР 2
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, вводят при комнатной температуре 2,63 кг этилбензола, 6,06 кг стирольного мономера и 1,52 кг полибутадиена INTENE 50 (вязкость 5% раствора в стирольном мономере составляет 150 сП). Число оборотов мешалки доводят до 80 об/мин. Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение последующих четырех часов. После этого добавляют 20,1 г бензоилпероксида (БПО) и 14 г 4OH-ТЕМРО. Температуру поднимают до 105°С в течение трех часов и поддерживают постоянной в течение последующих двух часов. Затем реакционную смесь охлаждают до 40°С за один час и к реакционной смеси добавляют 24,1 кг стирольного мономера и 0,65 кг INTENE 50. Температуру реакционной ванны снова увеличивают до 80°С за 30 минут и поддерживают 80°С в течение трех часов. Когда заканчивается растворение второй части эластомера, температуру поднимают до 125°С за тридцать минут. Температуру поддерживают при указанном значении в течение пяти с половиной часов. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler, содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000, 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса реакционной смеси добавляют 30 г ди-трет-бутилпероксида и вращение мешалки увеличивают до 270 об/мин. Температуру смеси поднимают до 120°С за 45 минут и поддерживают постоянной в течение часа, после чего ее поднимают до 140°С за 30 минут и поддерживают постоянной в течение двух часов, и наконец, ее поднимают до 155°С за 45 минут и поддерживают постоянной в течение трех часов. По прошествии трех часов проводят охлаждение до 115°С за 40 минут и смесь воды и органического материала отпаривают (5 литров/час, добавляя 5 литров деминерализованной воды) нагреванием в автоклаве до 145°С за 3 часа. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого смесь охлаждают до 40°С и автоклав разгружают. Полученный таким образом полимер фильтруют и сушат при 80°С в течение 5 часов.
Анализ полимера с помощью лазерной гранулометрии показывает бимодальную эластомерную фазу с 62% крупных частиц размером 2,3 мкм и 38% мелких частиц размерами 0,37 мкм (фиг.2).
ПРИМЕР 3
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, вводят при комнатной температуре 2,63 кг этилбензола, 6,02 кг стирольного мономера и 2,13 кг полибутадиена INTENE 40 (вязкость 5% раствора в стирольном мономере составляет 95 сП). Число оборотов мешалки доводят до 80 об/мин. Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение последующих четырех часов. После этого добавляют 28,8 г бензоилпероксида (БПО) и 22,6 г 4OH-ТЕМРО. Температуру поднимают до 105°С за три часа и поддерживают постоянной в течение последующих двух часов. Затем реакционную смесь охлаждают до 40°С за один час и к реакционной смеси добавляют 23,6 кг стирольного мономера и 0,64 кг INTENE 40. Температуру реакционной ванны снова увеличивают до 80°С за 30 минут и поддерживают 80°С в течение трех часов. Когда заканчивается растворение второй части эластомера, температуру поднимают до 125°С за тридцать минут. Температуру поддерживают при указанном значении в течение пяти с половиной часов. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler, содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000; 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса реакционной смеси добавляют 30 г ди-трет-бутилпероксида и вращение мешалки увеличивают до 270 об/мин. Температуру смеси поднимают до 120°С за 45 минут и поддерживают постоянной в течение часа, после чего ее поднимают до 140°С за 30 минут и поддерживают постоянной в течение 2 часов, и наконец, ее поднимают до 155°С и поддерживают постоянной в течение трех часов. По прошествии трех часов проводят охлаждение до 115°С за 40 минут и смесь воды и органического материала отпаривают (5 литров/час, добавляя 5 литров деминерализованной воды) нагреванием в автоклаве до 145°С за три часа. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого смесь охлаждают до 40°С и автоклав разгружают. Полученный таким образом полимер фильтруют и сушат при 80°С в течение 5 часов.
Анализ полимера с помощью лазерной гранулометрии показывает бимодальную эластомерную фазу с 20% крупных частиц размером 2,1 мкм и 80% мелких частиц размерами 0,3 мкм (фиг.3).
ПРИМЕР 4
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, вводят при комнатной температуре 2,61 кг этилбензола, 6,09 кг стирольного мономера и 2,15 кг полибутадиена INTENE 40 (вязкость 5% раствора в стирольном мономере составляет 95 сП). Число оборотов мешалки доводят до 80 об/мин. Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение последующих четырех часов. После этого добавляют 28,5 г бензоилпероксида (БПО) и 22,8 г 4OH-ТЕМРО. Температуру поднимают до 105°С в течение трех часов и поддерживают постоянной в течение последующих двух часов. Затем реакционную смесь охлаждают до 40°С за один час и к реакционной смеси добавляют 23,9 кг стирольного мономера и 0,21 кг INTENE 60 (вязкость 5% раствора в стирольном мономере составляет 250 сП). Температуру реакционной ванны снова увеличивают до 80°С за 30 минут и поддерживают 80°С в течение трех часов. Когда заканчивается растворение второй части эластомера, температуру поднимают до 125°С за тридцать минут. Температуру поддерживают при указанном значении в течение пяти с половиной часов. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler, содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000; 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса реакционной смеси добавляют 30 г ди-трет-бутилпероксида и вращение мешалки увеличивают до 270 об/мин. Температуру смеси поднимают до 120°С за 45 минут и поддерживают постоянной в течение часа, после чего ее поднимают до 140°С за 30 минут и поддерживают постоянной в течение 2 часов, и наконец, ее поднимают до 155°С за 45 минут и поддерживают постоянной в течение трех часов. По прошествии трех часов проводят охлаждение до 115°С за 40 минут и смесь воды и органического материала отпаривают (5 литров/час, добавляя 5 литров деминерализованной воды) нагреванием в автоклаве до 145°С за 3 часа. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого смесь охлаждают до 40°С и автоклав разгружают. Полученный таким образом полимер фильтруют и сушат при 80°С в течение 5 часов.
Анализ полимера с помощью лазерной гранулометрии показывает бимодальную эластомерную фазу с 20% крупных частиц размером 2,1 мкм и 80% мелких частиц размерами 0,3 мкм (фиг.4).
ПРИМЕР 4А
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, при комнатной температуре вводят 2,61 кг этилбензола, 27 кг стирольного мономера и 2,15 кг полибутадиена INTENE 40 (вязкость 5% раствора в стироле при 20°С равна 95 сП), предварительно функционализированного 0,3 масс.ч 4OH-ТЕМРО при 150°С в атмосфере воздуха с использованием двухшнекового экструдера (время пребывания 2-3 мин). Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение четырех часов. После этого реакционную смесь охлаждают до 40°С в течение одного часа и добавляют к ней 0,21 кг INTENE 60 (вязкость по Муни М(1+4)100°С=60, вязкость 5% раствора в СМ составляет 260 сП). Температуру реакционной смеси снова увеличивают до 80°С за 30 мин и поддерживают постоянной в течение одного часа. После завершения растворения второй части эластомера температуру системы поднимают до 125°С за 30 мин. Температуру поддерживают постоянной в течение 5 ч 30 мин. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler, содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000, 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса реакционной смеси добавляют 30 г ди-трет-бутилпероксида и вращение мешалки увеличивают до 270 об/мин. Температуру смеси поднимают до 120°С за 45 мин и поддерживают постоянной в течение часа, после чего ее поднимают до 140°С за 30 минут и поддерживают постоянной в течение 2 часов, и наконец, ее поднимают до 155°С за 45 мин и поддерживают постоянной в течение 3 часов. В завершение реакционную смесь охлаждают до 115°С за 40 мин, и отпаривают смесь воды и органических веществ (5 л/час, добавляя 5 л деминерализованной воды) нагреванием в автоклаве до 145°С за 3 часа. Указанную температуру поддерживают в течение 8 часов, продолжая непрерывное отпаривание, как описано выше. После этого смесь охлаждают до 40°С и гранулы полимера выгружают на фильтр, промывают водой и сушат воздухом при 80°С в течение 5 часов.
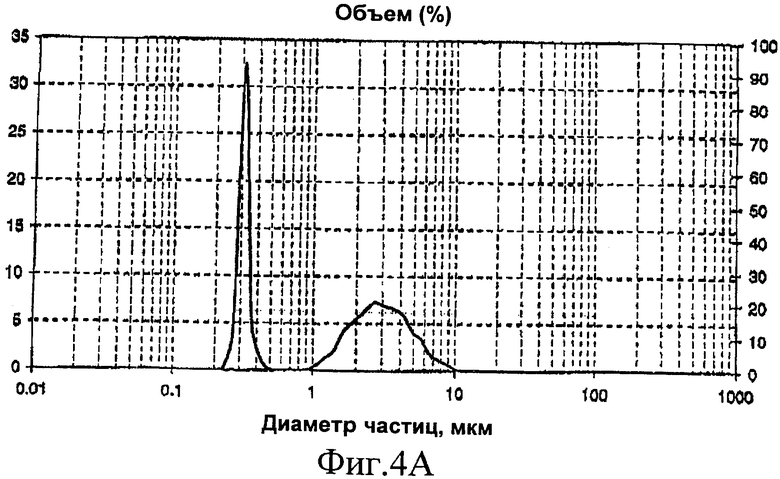
Анализ полимера с помощью лазерной гранулометрии показывает бимодальную эластомерную фазу с 18% крупных частиц размером 2,3 мкм и 82% мелких частиц размером 0,28 мкм (фиг.4А).
ПРИМЕР 5
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, вводят при комнатной температуре 2,63 кг этилбензола, 6,02 кг стирольного мономера и 2,13 кг полибутадиена INTENE 40 (вязкость 5% раствора в СМ составляет 95 сП). Число оборотов мешалки доводят до 80 об/мин. Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение последующих четырех часов. После этого добавляют 28,8 г бензоилпероксида (БПО) и 22,6 г 4OH-ТЕМРО. Температуру поднимают до 105°С в течение трех часов и поддерживают постоянной в течение последующих двух часов. Затем реакционную смесь охлаждают до 40°С за один час и к реакционной смеси добавляют 23,6 кг стирольного мономера и 0,64 кг INTENE 40. Температуру реакционной ванны снова увеличивают до 80°С за 30 минут и поддерживают 80°С в течение трех часов. Когда заканчивается растворение второй части эластомера, температуру поднимают до 125°С за тридцать минут. Температуру поддерживают при указанном значении в течение пяти с половиной часов. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler, содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000; 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса реакционной смеси добавляют 30 г ди-трет-бутилпероксида и вращение мешалки увеличивают до 270 об/мин. Температуру смеси поднимают до 120°С за 45 минут и поддерживают постоянной в течение часа, после чего ее поднимают до 140°С за 30 минут и поддерживают постоянной в течение 2 часов, и наконец, ее поднимают до 155°С и поддерживают постоянной в течение трех часов. По прошествии трех часов проводят охлаждение до 115°С за 40 минут и смесь воды и органического материала отпаривают (5 литров/час, добавляя 5 литров деминерализованной воды) нагреванием в автоклаве до 145°С за 3 часа. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого смесь охлаждают до 40°С и автоклав разгружают. Полученный таким образом полимер фильтруют и сушат при 80°С в течение 5 часов.
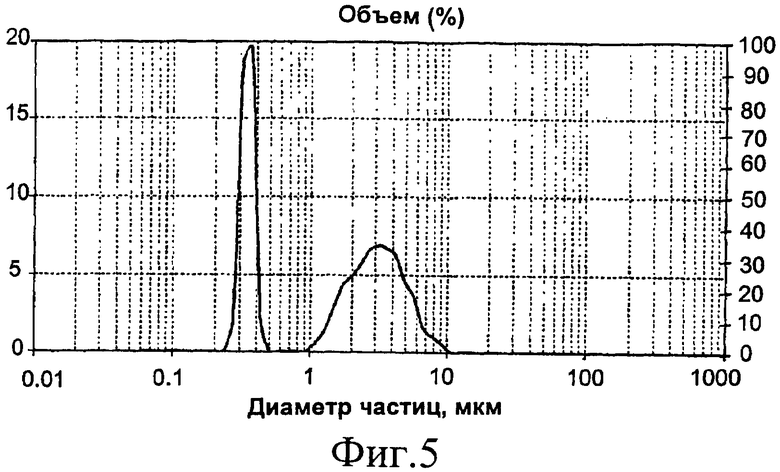
Анализ полимера с помощью лазерной гранулометрии показывает бимодальную эластомерную фазу с 57% крупных частиц размером 3,1 мкм и 43% мелких частиц размером 0,36 мкм (фиг.5).
ПРИМЕР 6
В автоклав периодического действия емкостью 60 л, снабженный регулятором температуры и системой с якорной мешалкой, вводят при комнатной температуре 2,61 кг этилбензола, 6,09 кг стирольного мономера и 2,15 кг полибутадиена INTENE 40 (вязкость 5% раствора в СМ составляет 95 сП). Число оборотов мешалки доводят до 80 об/мин. Температуру системы поднимают до 80°С в течение часа и поддерживают постоянной в течение последующих четырех часов. После этого добавляют 28,5 г бензоилпероксида (БПО) и 22,8 г 4OH-ТЕМРО. Температуру поднимают до 105°С в течение трех часов и поддерживают постоянной в течение последующих двух часов. Затем реакционную смесь охлаждают до 40°С за один час и к реакционной смеси добавляют 23,9 кг стирольного мономера и 0,21 кг INTENE 60 (вязкость 5% раствора в СМ составляет 250 сП). Температуру реакционной ванны снова увеличивают до 80°С за 30 минут и поддерживают 80°С в течение трех часов. Когда заканчивается растворение второй части эластомера, температуру поднимают до 125°С за тридцать минут. Температуру поддерживают при указанном значении в течение пяти с половиной часов. После этого реакционную смесь переносят в автоклав емкостью 100 л, снабженный мешалкой Pfaudler, содержащий 31,5 кг деминерализованной воды (при температуре 103°С), 40,5 г ETHAPOL 1000; 93 г натрийсульфонатного нафталина и 33 г хлорида натрия. После завершения переноса реакционной смеси добавляют 30 г ди-трет-бутилпероксида и вращение мешалки увеличивают до 270 об/мин. Температуру смеси поднимают до 120°С за 45 минут и поддерживают постоянной в течение часа, после чего ее поднимают до 140°С за 30 минут и поддерживают постоянной в течение 2 часов, и наконец ее поднимают до 155°С за 45 минут и поддерживают постоянной в течение трех часов. По прошествии трех часов проводят охлаждение до 115°С за 40 минут и смесь воды и органического материала отпаривают (5 литров/час, добавляя 5 литров деминерализованной воды), нагревая автоклав до 145°С за три часа. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого смесь охлаждают до 40°С и автоклав разгружают. Полученный таким образом полимер фильтруют и сушат при 80°С в течение 5 часов.
Анализ полимера с помощью лазерной гранулометрии показывает бимодальную эластомерную фазу с 38% крупных частиц размером 3,6 мкм и 62% мелких частиц размером 0,36 мкм (фиг.6).
Далее приведены некоторые сравнительные испытания, которые подтверждают, что использование смесей на основе полибутадиена и стирол-бутадиенового блок-сополимера не приводит к получению эластомерных частиц со строгим бимодальным распределением диаметров.
Испытание 1
В автоклаве периодического действия емкостью 60 л, снабженном регулятором температуры и системой с якорной мешалкой, готовят раствор, используя 4,2 кг стирол-бутадиенового сополимера 40/60 BUNA BL 6533 TC (Bayer) (вязкость 5% раствора в СМ составляет 40 сП), 0,90 кг вазелинового масла PRIMOL 352 (ESSO) и 30 г антиоксиданта ANOX РР 18 в 24,9 (кг) стирольного мономера, перемешивая в течение 5 часов при 85°С. Затем добавляют 24 г агента переноса цепи (TDM) и выполняют предварительную полимеризацию с прививкой и обращением фаз, нагревая и перемешивая полученный таким образом раствор в течение 5 часов и 30 минут при 120°С. Во время предварительной полимеризации добавляют две дозы по 3 г TDM через 3 часа и через 5 часов после начала нагревания до 120°С. В конце форполимер переносят во второй автоклав емкостью 100 л, снабженный мешалкой Pfaudler, и его суспендируют в водной фазе (отношение вода/органический материал = 1/1), содержащей NaCl (0,11 мас.%), нафталинсульфонат натрия (0,31 мас.%) и ETHAPOL 1000 (0,13 мас.%). Добавляют 30 г ди-трет-бутилпероксида и выполняют полимеризацию до полного превращения мономера и полного образования сетки из эластомерной фазы, нагревая при перемешивании в течение часа при 120°С, в течение 2 часов при 140°С и в течение 3 часов при 155°С. По прошествии трех часов проводят охлаждение до 115°С за 40 минут и смесь воды и органического материала отпаривают (5 литров/час, добавляя 5 литров деминерализованной воды), нагревая автоклав до 145°С за три часа. Указанную температуру поддерживают в течение восьми часов, продолжая отпаривать 5 литров смеси воды и органического материала каждый час (добавляя 5 литров деминерализованной воды). После этого смесь охлаждают до 40°С и автоклав разгружают. Полимер в форме шариков отмывают и сушат при 80°С в течение 5 часов, а затем гранулируют в экструдере.
Анализ полимера с помощью лазерной гранулометрии показывает мономодальную эластомерную фазу с D(4,3)=0,38 мкм (фиг.7).
Испытание 2
Повторяют испытание 1 с разницей в том, что вместо использования только сополимера BUNA 6533 ТС используют смесь, состоящую из 3,6 кг сополимера BUNA BL 6533 ТС и 0,6 кг полибутадиена INTENE 60 AF (вязкость 5% раствора в СМ составляет 250 сП).
Анализ полимера с помощью лазерной гранулометрии показывает мономодальную эластомерную фазу с D(4,3)=0,43 мкм (фиг.8).
Испытание 3
Повторяют испытание 1 с разницей в том, что вместо использования только сополимера BUNA BL 6533 ТС используют смесь, состоящую из 2,9 кг сополимера BUNA BL 6533 ТС и 1,3 кг полибутадиена INTENE 60 AF (вязкость 5% раствора в CM составляет 250 сП).
Анализ полимера с помощью лазерной гранулометрии показывает мономодальную эластомерную фазу с D(4,3)=0,61 мкм (фиг.9).
Испытание 4
Повторяют испытание 2 с разницей в том, что вместо сополимера BUNA BL 6533 ТС используют сополимер такого же состава (NS 318 S Nippon Zeon), но с еще более низкой вязкостью в растворе (вязкость 5% раствора в СМ составляет 10 сП).
Анализ полимера с помощью лазерной гранулометрии показывает мономодальную эластомерную фазу с частицами, имеющими D(4,3)=0,43 мкм (фиг.10).
Испытание 5
Повторяют испытание 3 с разницей в том, что вместо сополимера BUNA BL 6533 ТС используют сополимер такого же состава (NS 318 S Nippon Zeon), но с еще более низкой вязкостью в растворе (вязкость 5% раствора в СМ составляет 10 сП).
Анализ полимера с помощью лазерной гранулометрии показывает мономодальную эластомерную фазу с частицами, имеющими D(4,3)=0,65 мкм (фиг.11).
Характеристики используемых веществ
ETHAPOL 1000 - сополимер акриловой кислоты и 2-этилгексилакрилата.
Порошок, суспендирующий агент. Применение: полистирол (ПС) общего назначения, вспениваемый полистирол, сополимерный полистирол. Номер CAS: 25134-51-4.
INTENE 40 - полибутадиен литиевый со средним содержанием цис-звеньев (38%). Mw=200 кДа, Mw/Mn=2,2, вязкость 5% (мас.) раствора в стироле = 100 сП.
INTENE 50 - полибутадиен литиевый со средним содержанием цис-звеньев (38%). Вязкость по Муни = 48, Mw=230 кДа, Mw/Mn=2,2, вязкость 5% (мас.) раствора в стироле = 170 сП.
INTENE 60 - полибутадиен литиевый со средним содержанием цис-звеньев (38%). Вязкость по Муни=60, Mw=290 кДа, Mw/Mn=2,2, вязкость 5% (мас.) раствора в стироле = 250 сП.
Полимеры INTENE соответствуют техническим требованиям фирмы-производителя «Polimeri Europa S.p.A.».









Настоящее изобретение относится к способу получения винилароматических (со)полимеров, привитых на эластомер. Описан способ получения ударопрочных винилароматических (со)полимеров, привитых на эластомер, включающих жесткую матрицу, образованную винилароматическими полимерами или сополимерами, и эластомерную фазу, диспергированную в матрице в форме частиц со строго бимодальным распределением по диаметру, включающий: 1) функционализацию первой части (X1) эластомера с помощью каталитической системы функционализации/полимеризации, образованной инициатором (G) свободно-радикальной полимеризации, с количеством F функциональных групп, способным отрывать протон от полимерной цепи эластомера, и стабильным инициатором (I) свободно-радикальной полимеризации, включающим группу=N-O. и группу = N-O-R', при молярных соотношениях (I)/G·F от 1 до 3, причем F равно числу функциональных групп на молекулу инициатора, которые путем разложения дают два свободных радикала, R' является C1-С6 (изо)алкильным радикалом или C7-C20 арилалкильным радикалом, возможно содержащим гетероатомы; 2) смешивание функционализованного эластомера со второй частью (Х2) эластомера в течение времени, достаточного для получения однородной композиции; 3) добавление однородной эластомерной композиции в жидкую фазу, по существу образованную смесью винилароматического мономера и растворителя полимеризации с массовым соотношением от 60/40 до 100/0 и соотношением (Х1+Х2)/винилароматический мономер больше или равным 8/92; 4) полимеризацию винилароматического мономера, возможно в присутствии одного или более сомономеров, при температуре больше или равной 120°С; 5) извлечение полученного винилароматического полимера по окончании полимеризации и 6) возможное возвращение смеси растворителя и мономера, поступающей с операции извлечения, на стадию (1), после отделения возможных полярных сомономеров. Также описан способ получения ударопрочных винилароматических (со)полимеров, привитых на эластомер, включающих жесткую матрицу, образованную винилароматическими полимерами или сополимерами, и эластомерную фазу, диспергированную в матрице в форме частиц со строго бимодальным распределением по диаметру (вариант). Технический результат - получение конечного продукта, в котором распределение диаметров частиц дисперсной эластомерной фазы является "строго бимодальным". 2 н. и 12 з.п. ф-лы, 11 ил.
1. Способ получения ударопрочных винилароматических (со)полимеров, привитых на эластомер, включающих жесткую матрицу, образованную винилароматическими полимерами или сополимерами, и эластомерную фазу, диспергированную в матрице в форме частиц со строго бимодальным распределением по диаметру, включающий:
1) функционализацию первой части (X1) эластомера с помощью каталитической системы функционализации/полимеризации, образованной инициатором (G) свободно-радикальной полимеризации, с количеством F функциональных групп, способным отрывать протон от полимерной цепи эластомера, и стабильным инициатором (I) свободно-радикальной полимеризации, включающим группу = N-O. и группу = N-O-R', при молярных соотношениях (I)/G·F от 1 до 3, причем F равно числу функциональных групп на молекулу инициатора, которые путем разложения дают два свободных радикала, R' является C1-С6 (изо)алкильным радикалом или С7-С20 арилалкильным радикалом, возможно содержащим гетероатомы;
2) смешивание функционализованного эластомера со второй частью (Х2) эластомера в течение времени, достаточного для получения однородной композиции;
3) добавление однородной эластомерной композиции в жидкую фазу, по существу образованную смесью винилароматического мономера и растворителя полимеризации с массовым соотношением от 60/40 до 100/0 и соотношением (Х1+Х2)/винилароматический мономер больше или равным 8/92;
4) полимеризацию винилароматического мономера, возможно в присутствии одного или более сомономеров, при температуре больше или равной 120°С;
5) извлечение полученного винилароматического полимера по окончании полимеризации; и
6) возможное возвращение смеси растворителя и мономера, поступающей с операции извлечения, на стадию (1), после отделения возможных полярных сомономеров.
2. Способ по п.1, в котором функционализацию эластомера осуществляют в расплавленном состоянии.
3. Способ по п.1, в котором функционализацию эластомера осуществляют в растворителе.
4. Способ по п.1 или 3, в котором используют тот же растворитель, что и при полимеризации, и функционализацию осуществляют при температуре ниже 110°С.
5. Способ по п.1, в котором смешивание функционализованного эластомера X1 и нефункционализованного эластомера Х2 осуществляют в растворителе.
6. Способ по п.1 или 5, в котором используют тот же растворитель, что и при полимеризации.
7. Способ получения ударопрочных винилароматических (со)полимеров, привитых на эластомер, включающих жесткую матрицу, образованную винилароматическими полимерами или сополимерами, и эластомерную фазу, диспергированную в матрице в форме частиц со строго бимодальным распределением по диаметру, включающий:
а) растворение первой части (X1) эластомера в жидкой фазе, состоящей из смеси винилароматического мономера и растворителя полимеризации с массовым соотношением от 60/40 до 100/0, при соотношении эластомер/винилароматический мономер больше или равном 8/92;
б) добавление к раствору каталитической системы функционализации и полимеризации, образованной инициатором (G) свободно-радикальной полимеризации с количеством F функциональных групп, способным отрывать протон от полимерной цепи эластомера, и стабильным инициатором (I) свободно-радикальной полимеризации, включающим группу = N-O. и группу = N-O-R', при молярных соотношениях (I)/G·F от 1 до 3, причем F равно числу функциональных групп на молекулу инициатора, которые путем разложения дают два свободных радикала, R' является С1-С6 (изо)алкильным радикалом или С7-С20 арилалкильным радикалом, возможно содержащим гетероатомы;
в) нагревание при перемешивании смеси, полученной на стадии (б), при температуре от 80 до 110°С в течение времени, достаточного для полной функционализации эластомера;
г) подачу в смесь, содержащую функционализованный эластомер в растворе, второй части (X2) эластомера и, возможно, растворителя и/или винилароматического мономера, гомогенизацию полученной смеси и полимеризацию при температуре выше или равной 120°С, предпочтительно от 120 до 200°С;
д) извлечение полученного винилароматического полимера по окончании полимеризации; и
е) возможное возвращение смеси растворителя и мономера, поступающей с операции извлечения, на стадию (а) после отделения возможных полярных (со)мономеров в случае полимеризации в растворителе.
8. Способ по п.1 или 7, в котором массовые соотношения между первой частью X1 и второй частью Х2 эластомера составляют от 99/1 до 40/60.
9. Способ по п.1 или 7, в котором эластомером является полибутадиеновый гомополимер со среднечисленной молекулярной массой (Mn) от 50000 до 350000 и среднемассовой молекулярной массой (Mw) от 100000 до 500000.
10. Способ по п.1 или 7, в котором эластомер выбирают из этилен-пропиленового каучука (ЭПК) или тройного этилен-пропилен-диенового каучука (ЭПДК).
11. Способ по п.1 или 7, в котором каталитическую систему полимеризации добавляют в количествах от 0,1 до 2,5 мас.% по отношению к общему количеству эластомера.
12. Способ по п.1 или 7, в котором инициатор свободно-радикальной полимеризации, способный отрывать протон от полимерной цепи эластомера, выбирают из азопроизводных, пероксидов, гидропероксидов, перкарбонатов, перэфиров и солей перкислот.
13. Способ по п.1 или 7, в котором стабильный инициатор свободно-радикальной полимеризации выбирают из соединений общей формулы (III)
где группы R1, R2, R5 и R6, одинаковые или отличные друг от друга, являются свободными линейными или разветвленными алкильными радикалами, замещенными или незамещенными, содержащими от 1 до 20 атомов углерода, или алкилароматическими радикалами, в которых алкильная группа содержит от 1 до 4 атомов углерода, а группы R3 и R4, одинаковые или отличные друг от друга, являются такими же, как R1, R2, R5 и R6, или R3-CNC-R4 является частью циклической структуры, возможно конденсированной с ароматическим кольцом или насыщенным кольцом, содержащим от 3 до 20 атомов углерода, где по меньшей мере атом водорода циклической структуры может быть замещен гидроксильной группой.
14. Способ по п.13, в котором стабильный инициатор свободно-радикальной полимеризации выбирают из 2,2,5,5-тетраметил-1-пирролидинилокси, 4-гидрокси-2,2,6,6-тетраметил-1-пиперидинилокси и 1,1,3,3-тетраэтилизоиндолин-2-хилоксила.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 6255402 B1, 03.07.2001 | |||
| US 6262179 B1, 17.07.2001 | |||
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛАРОМАТИЧЕСКИХ ПОЛИМЕРОВ, УСИЛЕННЫХ КАУЧУКОМ | 1997 |
|
RU2142475C1 |
| WO 00/77078 A1, 21.12.2000. | |||

