Изобретение относится к биодеградируемым полимерным носителям для доставки противоопухолевых лекарственных средств, в частности, соединений класса таксанов, таких как паклитаксел, доцетаксел, антрациклиновых антибиотиков, таких как доксорубицин, эпирубицин и других.
Современная медицина имеет в своем распоряжении целый ряд эффективных противоопухолевых лекарственных средств, однако, их широкое применение в клинической практике может быть ограничено некоторыми негативными факторами, в частности, такими, как высокая токсичность, низкая растворимость в воде.
Одним из возможных путей решения указанной проблемы является связывание противоопухолевых лекарственных средств с полимерными носителями. Как установлено исследованиями, полимеры, имеющие высокую молекулярную массу, не способны легко диффундировать через нормальные капилляры и гломерулярный эндотелий, что предотвращает интоксикацию нормальной ткани. С другой стороны установлено, что злокачественные опухоли часто имеют нарушенный капиллярный эндотелий и более высокую проницаемость, чем сосудистая сеть нормальной ткани. Благодаря указанным факторам конъюгат полимер-лекарственное средство может селективно просачиваться из кровеносных сосудов в опухоли, приводя к накоплению в опухоли активного терапевтического средства. Кроме того, полимеры могут обеспечивать солюбилизацию коньюгированных с ними нерастворимых лекарственных средств.
В настоящее время разработаны различные синтетические и природные полимеры, способные выполнять функцию носителей противоопухолевых лекарственных средств с обеспечением их опухоль - специфической доставки.
Так, в частности, известны полимерные носители для противоопухолевых лекарственных средств класса таксанов, представляющие собой полиэтиленгликоль или его производные [RU 2002109594], гиалуроновую кислоту или ее производные [RU 2384593].
Указанные полимерные носители не являются биологически деградируемыми соединениями, вследствие чего могут накапливаться в организме, вызывая тем самым нежелательные последствия.
Известны полимерные носители на основе природных аминокислот, достоинством которых является биосовместимость и биодеградируемость.
Так, в частности, известен полимерный носитель для противоопухолевых лекарственных средств [ЕА 002400], представляющий собой полиаспарагиновую или полиглутаминовую кислоту или сополимер указанных аминокислот. Данные полимерные носители имеют молекулярный вес от 5000 до 100000 Да, хорошо растворимы в воде, легко разрушаются лизосомными ферментами, стабильны в плазме, содержат достаточное количество функциональных групп для присоединения лекарственного средства.
В рассматриваемом изобретении связь полимерного носителя с противоопухолевым лекарственным средством осуществляется с участием карбоксильных групп полимера и гидроксильных групп лекарственного средства, при этом образуются трудно разрушаемые сложноэфирные связи, расщепление которых происходит в условиях высокой щелочности среды. Это затрудняет высвобождение лекарственного средства в физиологических средах организма человека, рН которых лежат в области значений, близких к нейтральным или слабокислым.
Задачей заявляемого изобретения является создание нового биодеградируемого полимерного носителя для доставки противоопухолевого лекарственного средства.
По первому варианту изобретения биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства представляет собой полимер, структурными звеньями макромолекулы которого являются фрагменты L-лизина формулы
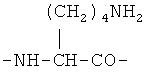
и фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты общей формулы
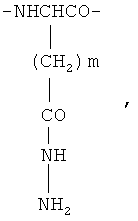
где m=1 или 2,
фрагменты L-лизина образуют полимерную цепь линейной конфигурации, фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты образуют полимерные цепи линейной конфигурации, при этом последние привиты к έ-аминогруппам L-лизина.
По второму варианту изобретения биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства представляет собой полимер, структурными звеньями макромолекулы которого являются фрагменты L-лизина формулы
и фрагменты β-гидразида аспарагиновой кислоты и/или γ- гидразида глутаминовой кислоты общей формулы
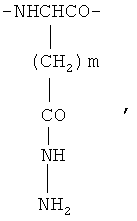
где m=1 или 2,
при этом макромолекула представляет собой гиперразветвленный гетерополи-L-лизин, содержащий вне точек ветвления полимерные цепи, образованные фрагментами β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты.
По третьему варианту изобретения биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства представляет собой полимер, структурными звеньями макромолекулы которого являются фрагменты L-лизина формулы
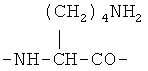
и фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты общей формулы
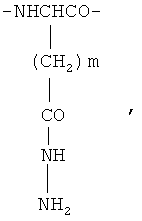
где m=1 или 2,
при этом фрагменты L-лизина образуют полимерную цепь в виде дендримера, а фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты привиты к концевым аминогруппам фрагментов L-лизина, расположенных во внешней сфере дендримера.
Предлагаемый носитель для противоопухолевого лекарственного средства по первому, второму и третьему вариантам изобретения представляет собой полимер на основе природных аминокислот: L-лизина и аспарагиновой и/или глутаминовой кислоты, что обуславливает его биодеградируемость и биосовместимость.
Характерной особенностью полимерного носителя по первому, второму и третьему вариантам изобретения является наличие ацил-гидразидных группировок CONHNH2 в боковых цепях фрагментов β-гидразида аспарагиновой или γ-гидразида глутаминовой кислоты, образующих полимерные цепи, привитые к поли-L-лизину или встроенные в его полимерную цепь.
За счет наличия указанных ацил-гидразидных группировок обеспечивается химическое или физическое связывание с лекарственным средством заявляемого полимерного носителя по первому, второму и третьему вариантам изобретения.
Химическое связывание полимерного носителя по первому, второму и третьему вариантам изобретения может осуществляться с лекарственным средством, в составе молекул которого имеются альдегидные или кетонные группы, в частности, с соединениями класса таксанов или антрациклиновыми антибиотиками. При этом ацил-гидразидные группировки полимерного носителя и альдегидные или кетонные группы лекарственного средства образуют хемодеградируемые альдиминные или кетиминные связи. Распад указанных альдиминных или кетиминных связей происходит при значениях pH среды около 7, что облегчает высвобождение лекарственного средства из полимерного носителя в физиологических средах организма человека, характеризующихся значением pH, близким к нейтральному или слабокислому.
Физическое связывание полимерного носителя по первому, второму и третьему вариантам изобретения может осуществляться с лекарственным средством после его включения в гидрофобное ядро полимерного носителя и формирования полимерных сеток в результате внутримолекулярных подшивок ацил-гидразидных группировок полимерных цепей.
Таким образом, техническим результатом, достигаемым при реализации заявляемого изобретения по первому и второму его вариантам, является создание нового полимерного биодеградируемого носителя, способного к химическому или физическому связыванию с противоопухолевым лекарственным средством с образованием легко разрушаемых связей в физиологических средах организма человека.
В первом варианте изобретения фрагменты L-лизина образуют полимерную цепь линейной конфигурации, фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты также образуют полимерные цепи линейной конфигурации, при этом последние привиты к έ-аминогруппам L-лизина (графт-сополимер типа "щетки").
Молекулярная масса (ММ) указанного полимерного носителя составляет от 30000 до 500000 Да, количество звеньев L-лизина в линейной цепи его макромолекулы составляет от 300 до 5000, отношение фрагментов β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты к фрагментам L-лизина составляет от 3 до 30.
Во втором варианте изобретения фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты образуют олигомерные или полимерные цепи, расположенные вне точек ветвления гиперразветвленного поли-L-лизина.
ММ указанного полимерного носителя составляет от 30000 до 600000 Да, отношение фрагментов β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты к фрагментам L-лизина составляет от 3 до 30, степень ветвления указанного полимера составляет от 2 до 30.
В третьем варианте изобретения моно- или олигомерные цепи, образованные фрагментами β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты, располагаются во внешней сфере лизинового дендримера ("звездообразный" коньюгат лизинового дендримера).
ММ указанного полимерного носителя составляет от 1500 до 1000000 Да, число генераций лизинового дендримера составляет от 1 до 6, отношение фрагментов β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты к фрагментам L-лизина составляет от 30 до 300.
Полимерный носитель по первому варианту изобретения получают, в частности, по следующей схеме:
1. Получение исходных мономеров: N-карбоксиангидридов Nε-карбобензокси-L-лизина, N-карбоксиангидридов сложных эфиров аспарагиновой или глутаминовой кислоты: γ-бензил-глутамата или β-бензил-аспартата, γ-метил-глутамата или β-метил-аспартата по известной методике [см., например, W. Daly, D. Poche // Tetrahedron Lett., 1988, V.29, №46, P5859];
2. Синтез линейного поли-L-лизина, к έ-аминогруппам звеньев которого привиты остатки аспарагиновой и/или глутаминовой кислоты, имеющие на конце боковой цепи сложноэфирные защитные группировки (-O-бензил- или -O-метил-), который осуществляют, в частности, следующим образом:
Осуществляют полимеризацию Nε-карбобензокси-L-лизина с использованием изопропиламина в качестве инициатора. Полученный полимер деблокируют при действии системы трифторметансульфокислота/трифторуксусная кислота. Полученную трифторацетатную соль поли-L-лизина высаживают в безводный диэтиловый эфир, а затем очищают с помощью ГПХ (носитель - сефадекс G-25) в системе 5% уксусной кислоты и лиофилизуют.
Проводят полимеризацию с N-карбоксиангидрида сложного эфира аспарагиновой кислоты и/или глутаминовой кислоты с использованием трифторацетатной соли поли-L-лизина как инициатора полимеризации. Время реакции составляет от 3 до 10 суток.
Полученный графт-сополимер осаждают в диэтиловый эфир, а затем промывают и сушат.
3. Обработка полученного полимера, в ходе которой сложноэфирные защитные группы (-O-бензил- или -O-метил-) превращаются в ацил-гидразидные группировки. В частности, указанную обработку осуществляют путем проведения реакции 50-100-кратного избытка гидразин-гидрата или безводного гидразина с раствором полученного полимера в метаноле, этаноле или другом инертном растворителе при температуре от 35 до 50°С. Продолжительность указанной реакции составляет от 1 до 15 суток. По окончании реакции полимер высаживают в диэтиловый эфир, а затем промывают и сушат.
Полимерный носитель по второму варианту изобретения получают, в частности, по следующей схеме:
1. Получение исходных мономеров: N-карбоксиангидридов Nε-карбобензокси-L-лизина, N-карбоксиангидридов сложных эфиров аспарагиновой или глутаминовой кислоты: γ-бензил-глутамата или β-бензил-аспартата, γ-метил-глутамата или β-метил-аспартата по известной методике [см., например, W. Daly, D. Poche. Tetrahedron Lett., 1988, V.29, №46, P.5859];
2. Проведение реакции каталитического гидрирования в реакционной среде, содержащей N-карбоксиангидрид Nε-карбобензокси-L-лизина и N-карбоксиангидрид одного из вышеперечисленных сложных эфиров, в ходе которого осуществляется восстановительное удаление Nε-карбобензоксизащитной группировки с N-карбоксиангидрида Nε-карбобензокси-L-лизина при пропускании водорода через раствор указанного N-карбоксиангидрида в диоксане или ТГФ в присутствии активированного палладия на угле. При этом s-аминогруппа N-карбоксиангидрида L-лизина, освободившаяся в ходе каталитического удаления, сразу же выступает в роли инициатора полимеризации N-карбоксиангидрида L-лизина. В процессе каталитического гидрирования продолжается удаление карбобензоксизащитных группы с Nε-аминогрупп N-карбоксиангидрида лизина (это ведет к появлению новых инициирующих центров полимеризации) и с Nε-аминогрупп образовавшихся олигомерных/полимерных цепочек (это дает разветвление полимера). Наличие в реакционной среде наряду с N-карбоксиангидридом Nε-карбобензокси-L-лизина одного из вышеуказанных N-карбоксиангидридов сложных эфиров аспарагиновой и/или глутаминовой кислоты приводит к образованию гиперразветвленного гетерополи-L-лизина, содержащего вне точек ветвления фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты [Власов Г.П. и др. // Высокомолекулярные соединения 2005, Сер. А, Т.47, №5, С.731-739];
3. Обработка полученного полимера, в ходе которой сложноэфирные защитные группы (-O-бензил- или -O-метил-) превращаются в ацил-гидразидные группировки. В частности, указанную обработку осуществляют путем проведения реакции 50-100-кратного избытка гидразин-гидрата или безводного гидразина с раствором полученного полимера в метаноле, этаноле или другом инертном растворителе при температуре от 35 до 50°С. Продолжительность указанной реакции составляет от 1 до 15 суток. По окончании реакции полимер высаживают в диэтиловый эфир, а затем промывают и сушат.
Полимерный носитель по третьему варианту изобретения получают, в частности, по следующей схеме:
1. Получение исходных мономеров: N-карбоксиангидридов Nε-карбобензокси-L-лизина, N-карбоксиангидридов сложных эфиров аспарагиновой или глутаминовой кислоты: γ-бензил-глутамата или β-бензил-аспартата, γ-метил-глутамата или β-метил-аспартата по известной методике [см., например, W. Daly, D. Poche. Tetrahedron Lett., 1988, V.29, №46, P.5859];
2. Синтез лизинового дендримера по известной методике Г.П.Власов и др. // Биоорганическая химия, 2004, Т.30. №1, С.1],
3. Полимеризация N-карбоксиангидрида сложного эфира аспарагиновой и/или глутаминовой кислоты (γ - бензил-глутамата, β - бензил-аспартата, γ - метил-глутамата, β - метил-аспартата) на концевых аминогруппах L-лизина, расположенных во внешней сфере дендримера. Указанную полимеризацию осуществляют, в частности, следующим образом. К депротонированному дендримеру добавляют 10-кратный избыток N-карбоксиангидрида сложного эфира аспарагиновой и/или глутаминовой кислоты. Реакционную смесь оставляют на 3-7 суток при температуре от 35 до 50°С. После окончания реакции полимеризации полученный полимер высаживают в диэтиловый эфир, промывают и сушат [Г.П.Власов и др. // Высокомолекулярные соединения, Сер. А, 2009, том 51, №12, с.2191-2192];
4. Обработка полученного полимера, в ходе которой сложноэфирные защитные группы (-O-бензил- или -O-метил-) превращаются в ацил-гидразидные группировки. В частности, указанную обработку осуществляют путем проведения реакции 50-100 кратного избытка гидразин-гидрата или безводного гидразина с раствором полученного полимера в метаноле, этаноле или другом инертном растворителе при температуре от 35 до 50°С. Продолжительность указанной реакции составляет от 1 до 15 суток. По окончании реакции полимер высаживают в диэтиловый эфир, а затем промывают и сушат.
Возможность реализации первого, второго и третьего вариантов заявляемого изобретения показана в примерах конкретного выполнения.
Пример 1 (первый вариант изобретения).
Получали полимерный носитель, представляющий собой линейный поли-L-лизин, к έ-аминогруппам которого привиты полимерные цепи, образованные фрагментами β-гидразида аспарагиновой кислоты.
Проводили синтез N-карбоксиангидрида β-метилового эфира аспарагиновой кислоты:
К суспензии, содержащей 4,5 г (0,0245 моль) β-метилового эфира аспарагиновой кислоты в 100 мл диоксана, нагретого до 50°С, прибавляли 2,43 г (1/3 эквивалент) трифосгена. Периодически реакционную смесь продували азотом для удаления остатков HCl. Через 4 ч по окончания реакции смесь фильтровали от непрореагировавшего хлоргидрата β-метиласпартата и раствор концентрировали на вакуумном испарителе. Полученную массу растворили в этилацетате и затем высаживали в петролейный эфир. Перекристаллизацию проводили несколько раз, чтобы избавиться от примесей хлористого водорода. Чистоту N-карбоксиангидрида β-метил-аспартата оценивали с помощью ТСХ анализа, в системе бензол; ацетон 1:1.
Выход N-карбоксиангидрида β-метил-аспартата 1,5 г. Тпл. составила 59-61°С, что является близким к теоретическому значению Тпл.
Проводили синтез N-карбоксиангидрида Nε-карбобензокси-L-лизина следующим образом.
К суспензии, содержащей Nε-карбобензокси-L-лизин в количестве 2,8 г (0,01 моль) и 50 мл диоксана, нагретого до 50°С, прибавляли 1,0 г (0,0033 моль) трифосгена. Реакционную смесь непрерывно продували азотом для удаления остатков HCl. Контроль процесса осуществляли при помощи ТСХ, используя систему бензол/ацетон в соотношении 1:1. По окончании реакции диоксан отогоняли на вакуумном испарителе. Перекристаллизацию проводили в системе растворителей этилацетат/петролейный эфир. Перекристаллизацию проводили несколько раз. Чистоту N-карбоксиангидрида Nε-карбобензокси-L-лизина оценивали с помощью ТСХ анализа.
Выход N- карбоксиангидрида Nε-карбобензокси-L-лизина составил 1,5 г. Тпл. составила 100-101°С, что является близким к теоретическому значению Тпл.
Получали линейный поли-L-лизин путем полимеризации N-карбоксиангидрида Nε-карбобензокси-L-лизина при использовании изопропиламина в качестве инициатора. Соотношение мономера и инициатора составляло 100:1.
Полученный полимер был деблокирован при действии системы трифторметансульфокислота/трифторуксусная кислота. После высаживания деблокированного полимера в безводный диэтиловый эфир полимер был очищен с помощью ГПХ (носитель - сефадекс G-25) в системе 5% уксусной кислоты и лиофилизован. В результате получили трифторацетатную соль поли-L-лизина.
К раствору 43 мг трифторацетатной соли поли-L-лизина в 30 мл диметилформамида добавляли 1,5 г (0,008 моль) N-карбоксиангидрида β-метилового эфира аспарагиновой кислоты. Полимеризацию проводили в течение 14 суток. За ходом полимеризации следили с помощью ТСХ по исчезновению N-карбоксиангидрида β-метилового эфира аспарагиновой кислоты. Полученный графт-сополимер осаждали в диэтиловый эфир, промывали диэтиловым эфиром и сушили. Выход графт-сополимера составил 130 мг.
1,0 г графт-сополимера растворяли в 30 мл диметилфорамида, к раствору прибавляли 50-кратный избыток гидразин-гидрата. Раствор выдерживали в течение 10 суток при температуре 45°С. Полученный полимер высаживали в диэтиловый эфир, промывали и сушили. Выход полимера составил 0,7 г.
По данным ИК спектров гидразинолиз прошел полностью.
ММ полученного полимера, определенная с использованием ГПХ, составила 400000 Да. Отношение фрагментов β-гидразида аспарагиновой кислоты к L-лизину составило 4,2.
Пример 2 (второй вариант изобретения).
Получали полимерный носитель, представляющий собой гиперразветвленный гетерополи-L-лизин, содержащий вне точек ветвления структурные звенья β-гидразида аспарагиновой кислоты.
Проводили синтез N-карбоксиангидрида β-метил-аспартата и синтез N-карбоксиангидрида Nε-карбобензокси-L-лизина как описано в примере 1.
Осуществляли сополимеризацию N-карбоксиангидридов β-метил-аспартата и Nε-карбобензокси-L-лизина следующим образом.
К раствору, содержащему 1,3 г (0,0075 моль) N-карбоксиангидрида β-метил-аспартата и 1,15 г (0,00375 моль) N-карбоксиангидрида Nε-карбобснзокси-L-лизина и 61 мл диоксана, добавляли 115 мг палладиевой черни (Pd/C) и 0,212 мл безводной муравьиной кислоты. Смесь ангидридов была оставлена на 7 суток для полноты протекания реакции. За процессом превращения N-карбоксиангидридов в полимер следили по исчезновению N-карбоксиангидридов с помощью тонкослойной хроматографии в системе бензол/ацетон в соотношении 1:1. После завершения реакции полимерный раствор отфильтровали от палладиевой черни, концентрировали под вакуумом и осаждали в диэтиловый эфир. Готовый полимер промывали диэтиловым эфиром и сушили в эксикаторе. Выход полимера составил 1,64 г.
0,8 г сополимера, растворяли в 15 мл диметилфорамида, прибавляли 50-кратный избыток гидразин-гидрата. Раствор выдерживали в течение 4-х дней при температуре 45°С. Полученный сополимер высаживали в диэтиловый эфир, промывали и сушили. Выход составил - 0,7 г.
По данным ИК спектров гидразинолиз прошел полностью.
По данным аминокислотного анализа отношение фрагментов β-гидразида аспарагиновой кислоты к L-лизину составило 6,0.
MM полученного полимера, определенная с использованием ГПХ, составила 400000 Да.
Пример 3 (третий вариант изобретения).
Получали полимерный носитель, представляющий собой лизиновый дендример третьей генерации, к концевым аминогруппам структурных звеньев которого, расположенным во внешней сфере дендримера, привиты структурные звенья γ-гидразида глутаминовой кислоты.
Получали N-карбоксиангидрид γ-метилового эфира глутаминовой кислоты следующим образом.
К суспензии, содержащей 8 г γ-метилового эфира глутаминовой кислоты в 200 мл диоксана, нагретого до 50°С, прибавляли 3,3 г (1/3 эквивалент) трифосгена. Периодически реакционную смесь продували азотом для удаления HCl. Через 3 ч диоксан отгоняли на вакуумном роторе, полученную массу растворяли в этилацетате, а затем высаживали в петролейный эфир. Перекристаллизацию проводили из смеси этилацетата с петролейным эфиром несколько раз, чтобы избавиться от примесей хлористого водорода. Чистоту N-карбоксиангидрида γ-метил глутамата оценивали с помощью тонкослойной хроматографии (ТСХ) в системе бензол - ацетон, взятых в отношении 1:1.
Выход N-карбоксиангидрида γ-метил-глутамата составил 5,3 г. Тпл. составила 95-97°С, что является близким к теоретическому значению Тпл.
Получали лизиновый дендример третьей генерации по методике, описанной в [Г.П. Власов и др. Биоорганическая химия, 2004, Т.30. №1, С.1].
Осуществляли полимеризацию N-карбоксиангидрида γ-метил-глутамата на концевых аминогруппах L-лизина, расположенных во внешней сфере дендримера следующим образом.
К 100 мг (0,0498 ммоль) депротонированного дендримера, растворенного в 30 мл диметилформамида, добавляли 10-кратный избыток N-карбоксиангидрида γ-метил глутамата - 1,5 г (0,079 моль). Реакционную смесь оставляли на 7 суток при 30°С. За исчезновением карбоксиангидрида следили по ТСХ в системе бензол/ацетон в соотношении 1:1. После окончания реакции полимеризации полученный сополимер высаживали в диэтиловый эфир, промывали и сушили на воздухе. Выход составил 0,9 г.
Осуществляли обработку полученного сополимера гидразин - гидратом следующим образом.
К 0,8 г сополимера, растворенного в 15 мл диметилфорамида, прибавляли 50-кратный избыток гидразин-гидрата. Раствор выдерживали в течение 4-х суток при температуре 45°С. Полученный сополимер высаживали в диэтиловый эфир, промывали и сушили. Выход составил - 0,7 г.
По данным ИК спектров гидразинолиз прошел полностью.
ММ сополимера, определенная с помощью ГПХ, составила 400000 Да.
По данным аминокислотного анализа отношение фрагментов гидразида глутаминовой кислоты к L-лизину составило 8,0.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОДЕГРАДИРУЕМЫЙ ПОЛИМЕРНЫЙ НОСИТЕЛЬ ДЛЯ ДОСТАВКИ ПРОТИВООПУХОЛЕВОГО ЛЕКАРСТВЕННОГО СРЕДСТВА (ВАРИАНТЫ) | 2012 |
|
RU2493848C1 |
| ПЕПТИДНЫЙ СОПОЛИМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2009 |
|
RU2402576C1 |
| Конъюгат дексаметазона с синтетическим статистическим полипептидом | 2020 |
|
RU2792146C2 |
| КОНЪЮГАТЫ ПОЛИГЛУТАМАТ-АМИНОКИСЛОТА И СПОСОБЫ | 2006 |
|
RU2472812C2 |
| НОВЫЕ АНАЛОГИ ГЛЮКАГОН-ПОДОБНОГО ПЕПТИДА, КОМПОЗИЦИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2010 |
|
RU2557301C2 |
| Способ получения высокоочищенного тетрадекапептида | 2020 |
|
RU2759377C1 |
| СОЛИ КОНЪЮГАТОВ ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2747528C2 |
| ГЛИКОКОНЪЮГАТЫ 20(S)-КАМПТОТЕЦИНА | 1998 |
|
RU2184122C2 |
| АМПЛИФИКАЦИЯ СИСТЕМЫ ПОГЛОЩЕНИЯ ВИТАМИНА B ПРИ ПОМОЩИ ПОЛИМЕРОВ | 1994 |
|
RU2139732C1 |
| СОПОЛИМЕР-1, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И АНАЛИТИЧЕСКИЕ МЕТОДЫ | 2012 |
|
RU2604521C2 |
Изобретение относится к медицине и описывает биодеградируемые полимерные носители для доставки противоопухолевых лекарственных средств, в частности соединений класса таксанов и антрациклиновых антибиотиков. Биодеградируемый полимерный носитель представляет собой полимер, структурными звеньями макромолекулы которого являются фрагменты L-лизина и фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты. Фрагменты L-лизина образуют полимерную цепь линейной конфигурации, фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты образуют полимерные цепи линейной конфигурации, при этом последние привиты к έ-аминогруппам L-лизина. Биодеградируемый полимерный носитель способен к связыванию с противоопухолевым лекарственным средством с образованием легко разрушаемых связей в физиологических средах организма человека. 3 пр.
Биодеградируемый полимерный носитель для доставки противоопухолевого лекарственного средства, структурными звеньями макромолекулы которого являются фрагменты L-лизина формулы
и фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты общей формулы
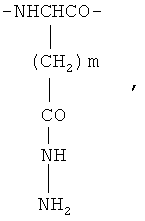
где m=1 или 2,
фрагменты L-лизина образуют полимерную цепь линейной конфигурации, фрагменты β-гидразида аспарагиновой кислоты и/или γ-гидразида глутаминовой кислоты образуют полимерные цепи линейной конфигурации, при этом последние привиты к έ-аминогруппам L-лизина.
| US 20090232762 A1, 17.09.2009 | |||
| WO 2006115293 A1, 02.11.2006 | |||
| ВЛАСОВ Г.П | |||
| и др | |||
| Гиперразветвленный поли-L-лизин, содержащий между точками «ветвления» дополнительные аминокислоты или их олигомеры: синтез и структура | |||
| - Высокомол | |||
| соединения | |||
| Серия А, 2005, т.47 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ВЛАСОВ Г.П | |||
| и др | |||
| Изучение синтеза полиглутаминовых | |||

