Родственная заявка
Данная заявка связана со следующими заявками США: серийный номер 60/605020, поданная 27 августа 2004 года, серийный номер 60/617170, поданная 8 октября 2004 года, серийный номер 60/625676, поданная 5 ноября 2004 года, и серийный номер 60/683169, поданная 19 мая 2005 года, каждая из которых включена сюда в качестве ссылки в их полноте.
Уровень техники
Hedgehog сигнальный путь является существенной частью многочисленных процессов в составе эмбрионального развития. Члены секретируемых белков семейства Hedgehog управляют пролиферацией клетки, дифференциацией и образованием ткани. Путь был вначале расшифрован в организме плодовой мухи Drosophila, но с тех пор показано, что он в большой степени сохраняется в беспозвоночных и позвоночных, включая людей. Полная активность сигнального пути Hedgehog снижается после эмбриогенеза в большинстве клеток, но путь остается активным в некоторых видах взрослых клеток. Недавно показано, что нерегулируемая активация сигнального пути Hedgehog приводит к определенным видам рака, как детально описано ниже.
Hedgehog полипептид представляет собой секретируемый белок, который функционирует как сигнальный лиганд в Hedgehog пути. Типичные гены и белки Hedgehog описаны в публикациях РСТ WO 95/18856 и WO 96/17924. Три различные формы белка Hedgehog найдены у людей; Sonic Hedgehog (Shh), Desert Hedgehog (Dhh) и Indian Hedgehog (Ihh). Sonic Hedgehog является самым распространенным представителем Hedgehog у млекопитающих и также наиболее хорошо описанным лигандом семейства Hedgehog. Перед секрецией Shh подвергается внутримолекулярному расщеплению и реакции модификации липида. Липид-модифицированный пептид отвечает за всю активность передачи сигналов.
Два трансмембранных белка участвуют в трансдукции сигнала по Hedgehog пути; двенадцать-трансмембранный Patched рецептор (Ptc) и семь-трансмембранный Smoothened белок (Smo).
Исследования в технике предполагают, что Hedgehog действует посредством связывания Ptc, таким образом деблокируя ингибирующее действие Ptc на Smo. Так как Ptc и Smo, оба, представляют собой трансмембранные белки, предложенным сценарием является то, что они физически связываются, формируя рецепторный комплекс, хотя непрямые механизмы действия также вероятны. Депрессия Smo от ингибирования Ptc наиболее вероятно включает конформационное изменение в Smo. Ptc, однако не является необходимым для активности Smo, так как Smo становится конститутивно активированным при полном отсутствии Pathed белка (Alcedo et al., выше; Quirk et al. (1997) Cold Spring Harbor Symp. Quant. Biol. 62: 217-226). Как только Smo инициируется, он быстро и высоко фосфорилируется и преобразовывает сигнал, который активизирует транскрипцию через факторы транскрипции Gli (гомолог белка Ci Drosophila) (Alexandre et al. (1996) Genes Dev. 10: 2003-13 ). Фактор транскрипции Gli1 повышающе регулирует многие гены, включаемые в рост и развитие (Alexandre et al., выше). Hedgehog передача сигналов является необходимой частью многих стадий развития, особенно в формировании лево-правосторонней симметрии. Потеря или сокращение Hedgehog передачи сигналов приводит к развитию множества дефицитов и уродств, из которых наиболее серьезным является циклопия (Belloni et al. (1996) Nature Genetics, 14: 353-6).
Недавно сообщалось, что активация мутаций формирования Hedgehog пути происходит при спорадической базально-клеточной карциноме (Xie et al. (1998) Nature 391: 90-2) и первичных нейроэктодермальных опухолях центральной нервной системы (Reifenberger et al. (1998) Cancer Res. 58: 1798-803). Нерегулируемая активация Hedgehog пути также выявлена при многочисленных типах рака, таких как рак желудочно-кишечного тракта, включая рак поджелудочной железы, рак пищевода, рак желудка (Berman et al. (2003) Nature 425: 846-51, Thayer et al. (2003) Nature 425: 851-56), рак легкого (Watkins et al. (2003) Nature 422: 313-317, рак простаты (Karhadkar et al. (2004) Nature 431: 707-12, Sheng et al. (2004) Molecular Cancer 3: 29-42, Fan et al. (2004) Endocrinology 145: 3961-70), рак молочной железы (Kubo et al. (2004) Cancer Research 64: 6071-74, Lewis et al. (2004) Journal of Mammary Gland Biology and Neoplasia 2: 165-181) и гепатоцеллюлярный рак (Sicklick et al. (2005) ASCO conference, Mohini et al. (2005) AACR conference).
Показано, что ингибирование низкомолекулярными соединениями активности Hedgehog пути приводит к смерти клеток в ряду различных типов рака, имеющих нерегулируемую активацию Hedgehog пути (см. например, Berman et al., 2003 Nature 425: 846-51).
Антагонисты Hedgehog пути в настоящее время исследуются в большом количестве клинических условий, где терапевтический эффект для состояния или расстройства может быть получен посредством ингибирования одного или более аспектов активности Hedgehog пути. Хотя основное внимание сосредоточено на раке, исследователи нашли, что ингибирование низкомолекулярными соединениями Hedgehog пути показало улучшение симптомов псориаза (Tas, et al., 2004 Dermatology 209: 126-131, published US patent application 20040072913 (включенный здесь посредством ссылки)). Псориаз представляет собой очень распространенное хроническое расстройство кожи, обычно характеризующееся поражением кожи, включающим эритематозные папулы и пятна с серебристой чешуйкой, хотя имеются изменения и на коже, и в других частях тела. Псориаз, как в настоящее время полагают, является аутоиммунным заболеванием, но его этиология все еще плохо понята.
Ингибитором Hedgehog пути, который привлек значительный интерес, является природный продукт циклопамин. Циклопамин был вначале выделен из лилии Veratrum californicum в 1966 году после того, как было найдено, что потомство овец, которых содержали на подножном корму, родилось с тяжелыми врожденными деформациями. Чтобы идентифицировать средство(а), вызывающее данные врожденные деформации, FDA исследовал возможные источники тератогенов и идентифицировал иервиновое семейство стероидных алкалоидов, включающее соединение циклопамин, как тератогены, ответственные за врожденные деформации.
Намного позже найдено, что механизмом действия циклопамина было прямое ингибирование активности Hedgehog пути (Cooper et al. (1998) Science 280: 1603-7, Chen et al. (2002) Genes and Development 16: 2743-8). Показано, что циклопамин и родственные соединения имеют противораковую активность через действие на Hedgehog путь. Несмотря на первоначальную перспективу, никакие члены данного семейства соединений или их аналогов не привели к успешной разработке антиракового средства. Настоящее изобретение выполняет данную потребность и имеет другие соответствующие преимущества.
Сущность изобретения
Настоящее изобретение предоставляет аналоги стероидных алкалоидов семейства циклопамина, которые являются полезными для ингибирования пролиферации клеток и/или поддержания апоптоза в клетке, в том числе для лечения пролиферативных расстройств, таких как рак. Антагонисты Hedgehog пути настоящего изобретения могут использоваться для подавления пролиферации (или других биологических последствий) клеток или тканей, таких как у пациентов, характеризующихся как имеющие фенотип сниженной функции Ptc, фенотип повышенной функции Smo или фенотип повышенной функции Hedgehog.
В определенных областях настоящие способы используются, чтобы противодействовать фенотипическим эффектам нежелательной активации Hedgehog пути, таким как происходящее в результате мутаций увеличение функции Hedgehog, снижение функции Ptc или увеличение функции Smo. Например, настоящие способы могут включать приведение клетки в контакт (in vitro или in vivo) с антагонистом Hedgehog пути настоящего изобретения (как определено ниже) в количестве, достаточном, чтобы оказывать антагонистическое воздействие на Smo-зависимую активацию пути. Такой антагонизм остановит или замедлит нежелательную пролиферацию клетки и может привести к смерти клетки.
В некоторых вариантах осуществления способы и соединения настоящего изобретения могут использоваться для регуляции пролиферации клеток и/или гибели клетки in vitro и/или in vivo, таких как лечение онкологических заболеваний головы, шеи, полости носа, параназальных пазух, носоглотки, полости рта, ротоглотки, гортани, гортаноглотки, слюнных желез, параганглиом, поджелудочной железы, желудка, кожи, пищевода, печени и желчного дерева, кости, тонкой кишки, толстой кишки, прямой кишки, яичников, простаты, легкого, молочной железы, лимфатической системы, крови, центральной нервной системы, костного мозга или головного мозга.
В некоторых вариантах осуществления способы и соединения настоящего изобретения могут использоваться для лечения симптомов псориаза у пациента. Соединения настоящего изобретения могут использоваться для лечения псориаза как единственное средство или в комбинации с одним или более средствами для лечения псориаза. В предпочтительных воплощениях соединения настоящего изобретения применяются местно у пациента, нуждающегося в этом.
Соединения настоящего изобретения могут быть далее включены в состав как фармацевтической композиции, включающей фармацевтически приемлемый инертный наполнитель, для введения пациенту как средство лечения рака. Антагонисты Hedgehog пути настоящего изобретения и/или композиции, включающие их, могут вводиться пациенту для лечения состояний, включающих нежелательную пролиферацию клетки, например, рак и/или опухоли головы, шеи, полости носа, параназальных пазух, носоглотки, полости рта, ротоглотки, гортани, гортаноглотки, слюнных желез, параганглиом, поджелудочной железы, желудка, кожи, пищевода, печени и желчного дерева, кости, тонкой кишки, толстой кишки, прямой кишки, яичников, простаты, легкого, груди, лимфатической системы, крови, центральной нервной системы, костного мозга или головного мозга. В некоторых вариантах осуществления такие соединения или композиции применяются системно, например парентерально и/или локально, например местно.
Подробное описание изобретения
Определения
Определения терминов, используемых здесь, предназначаются, чтобы включить настоящие современные определения, признанные для каждого термина в химических и фармацевтических областях. Где необходимо, приводятся примеры. Определения применяются к терминам, поскольку они используются везде по этой спецификации, если не ограничено иначе в специфических случаях или индивидуально, или как часть большей группы.
Термин "гетероатом" известен в данной области техники и относится к атому любого элемента, кроме атомов углерода или водорода. Иллюстративные гетероатомы включают бор, азот, кислород, фосфор, серу и селен.
Термин "алкил" известен в данной области техники и включает насыщенные алифатические группы, включая алкильные группы с неразветвленной цепью, алкильные группы с разветвленной цепью, циклоалкильные (полициклические) группы, алкилзамещенные циклоалкильные группы и циклоалкилзамещенные алкильные группы. В некоторых вариантах осуществления алкил с неразветвленной цепью или разветвленной цепью имеет приблизительно 30 или меньше атомов углерода в основной цепи (например, C1-С30 для неразветвленной цепи, C3-C30 для разветвленной цепи), и альтернативно, приблизительно 20 или меньше. Аналогично, циклоалкилы имеют от приблизительно 3 до приблизительно 10 атомов углерода в их кольцевой структуре, и альтернативно приблизительно 5, 6 или 7 атомов углерода в кольцевой структуре. Алкильные группы, если не определено иначе, могут необязательно замещаться подходящими заместителями. Количество заместителей обычно ограничено количеством доступных валентностей на алкильной группе; таким образом, алкильная группа может быть замещена одним или большим количеством атомов водорода, которые могли бы присутствовать на незамещенной группе. Подходящие заместители для алкильных групп включают галоген, =О, =N-CN, =N-OR', =NR', OR', NR'2, SR', SO2R', SO2NR'2, NR'SO2R', NR'CONR'2, NR'COOR', NR'COR', CN, COOR', CONR'2, OOCR', COR' и NO2, где каждый R' является независимым Н, C1-C6 алкилом, C2-C6 гетероалкилом, C1-C6, ацилом, C2-C6 гетероацилом, C6-C10 арилом, С6-C10 гетероарилом, C7-C12 арилалкилом или C6-C12 гетероарилалкилом, каждый из которых необязательно замещен одной или более группой, выбранных из галогена, C1-C4 алкила, C1-C4 гетероалкила, C1-C6 арила, C1-C6 гетероацила, гидрокси, амино и =О; и где два R' в составе одного заместителя или на смежных атомах, могут быть связаны в форме 3-7-членных колец, необязательно содержащих до трех гетероатомов, выбранных из N, O и S.
Если количество атомов углерода не определено иначе, низший алкил относится к алкильной группе, как определено выше, но имеющей от одного до приблизительно десяти атомов углерода, альтернативно от одного до приблизительно шести атомов углерода в структуре основной цепи. Аналогично, низший алкенил и низший алкинил имеют подобные длины цепи.
Термин "аралкил" известен в данной области техники и относится к алкильной группе, замещенной группой арила (например, ароматической или гетероароматической группой).
Термины "алкенил" и "алкинил" известны в данной области техники и относятся к ненасыщенным алифатическим группам, аналогичным по длине и возможному замещению алкилам, описанным выше, но они содержат по меньшей мере одну двойную или тройную связь соответственно и могут содержать смесь и двойных, и тройных связей. Алкенильные и алкинильные группы также необязательно замещены, если не определено иначе, теми же самыми заместителями, описанными выше для алкильных групп.
Гетероалкил, гетероалкенил и гетероалкинил и подобные определяются аналогично соответственным гидрокарбильным (алкильным, алкенильным и алкинильным) группам, но термины «гетеро» относятся к группам, которые содержат 1-3 О, S или N гетероатомы или их комбинации в пределах остатка основной цепи; таким образом, по меньшей мере, один атом углерода соответственно алкильной, алкенильной или алкинильной групп заменен одним из указанных гетероатомов, формируя гетероалкильную, гетероалкенильную или гетероалкинильную группу. Типичные и предпочтительные размеры для гетероформ алкильных, алкенильных и алкинильных групп являются обычно теми же самыми, что и для соответственных гидрокарбильных групп, и заместители, которые могут присутствовать на гетерогруппах, являются теми же, что и описанные выше для гидрокарбильных групп. Также понимается, что если не определено иначе, по причинам химической стабильности такие группы не включают больше, чем два смежных гетероатома, исключая те, где оксогруппа присутствует на N или S, как в сульфонильной группе.
Термин "арил" известен в данной области техники и относится к 5-, 6- и 7-членным моноциклическим ароматическим радикалам, которые могут включать от нуля до четырех гетероатомов, например бензол, нафталин, антрацен, пирен, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин и пиримидин и подобные. Арильные группы, имеющие гетероатомы в кольцевой структуре, могут также упоминаться как арильные гетероциклы или гетероароматические. Ароматическое кольцо может замещаться в одном или более положениях кольца с такими заместителями, как описано выше, например, галогеном, азидом, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, гидроксилом, алкоксильной группой, амино, нитро, сульфгидрильной, имино, амидо, фосфонатом, фосфинатом, карбонилом, карбоксильной группой, силилом, простым эфиром, алкилтио, сульфонилом, сульфонамидо, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматическими или гетероароматическими остатками, -CF3, -CN или подобными. Термин "арил" также включает полициклические системы, имеющие два или больше циклических кольца, в которых два или больше атомов углерода являются общими для двух смежных колец (кольца представляют собой "конденсированные кольца"), где, по меньшей мере, одно из колец является ароматическим, например, другие циклические кольца могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами и/или гетероциклами.
Термины орто, мета и пара известны в данной области техники и относятся к 1,2-, 1,3- и 1,4- дважды замещенным бензолам соответственно. Например, названия "1,2-диметилбензол" и "орто-диметилбензол" синонимичны.
Термины "гетероциклил", "гетероарил" или "гетероциклическая группа" являются известными в данной области техники и относятся к от 3- до приблизительно 10-членным кольцевым структурам, альтернативно от 3- до приблизительно 7-членным кольцам, кольцевые структуры которых включают 1-4 гетероатома. Гетероциклы могут также быть полициклами. Гетероциклические группы включают, например, тиофен, тиантрен, фуран, пиран, изобензофуран, хромен, ксантен, феноксантен, пиррол, имидазол, пиразол, изотиазол, изоксазол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, индол, индазол, пурин, хинолизин, изохинолин, хинолин, фталазин, нафтиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карболин, фенантридин, акридин, пиримидин, фенантролин, феназин, фенарсазин, фентиазин, фуразан, феноксазин, пирролидин, оксолан, тиолан, оксазол, пиперидин, пиперазин, морфолин, лактоны, лактамы, такие как азетидиноны и пирролидиноны, сультамы, сультоны и т.п. Гетероциклическое ядро может быть замещено в одном или более положениях такими заместителями, как описано выше, например, галоген, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксильная группа, силил, простой эфир, алкилтио, сульфонил, кетон, альдегид, сложный эфир, гетероциклил, ароматический или гетероароматический остатки, -CF3, -CN или подобные.
Термины "полициклил" или "полициклическая группа" являются известными в данной области техники и относятся к двум или более кольцам (например, циклоалкилы, циклоалкенилы, циклоалкинилы, арилы и/или гетероциклилы), в которых два или более атомов углерода являются общими для двух присоединенных колец, например кольца представляют собой конденсированные кольца. Кольца, соединенные через несмежные атомы, называются мостиковыми кольцами. Каждое из колец полицикла может быть замещено такими заместителями, как описано выше, например, галоген, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксильная группа, силил, эфир, алкилтио, сульфонил, кетон, альдегид, сложный эфир, гетероциклил, ароматический или гетероароматический остаток, -CF3, -CN или подобными.
Термин "карбоцикл" известен в данной области техники и относится к ароматическому или неароматическому кольцу, в котором каждый атом кольца является углеродом.
Термин "нитро" известен в данной области техники и относится к -NO2, термин "галоген" известен в данной области техники и относится к -F, -Cl, -Br или -I; термин "сульфгидрильный" известен в данной области техники и относится к -SH, термин "гидроксил" означает -ОН; и термин "сульфонил" известен в данной области техники и относится к -SO2. Галогенид определяет соответствующий анион галогенов, и "псевдогалоидному соединению" сформулировали определение на стр.560 из "Advanced Inorganic Chemistry" by Cotton and Wilkinson.
Термины "амин", "амино" и "аммоний" известны в данной области техники и относятся к незамещенным и к замещенным аминам, например остатку, который может быть представлен общими формулами:
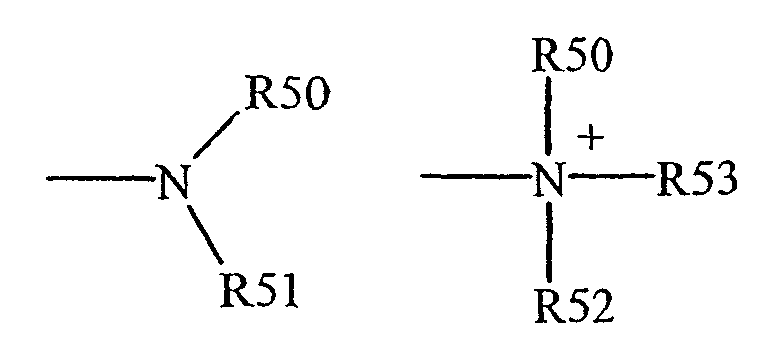
где R50, R51 и R52, каждый, независимо представляет собой водород, алкил, алкенил, -(CH2)m-R61 или R50 и R51, взятые вместе с атомом N, к которому они присоединены, образуют гетероцикл от 4 до 8 атомов в кольцевой структуре; R61 представляет арил, циклоалкил, циклоалкенил, гетероцикл или полицикл; и m представляет собой нуль или целое число в диапазоне 1-8. В других воплощениях R50 и R51 (и, необязательно, R52), каждый, независимо представляют водород, алкил, алкенил, или -(CH2)m-R61. Таким образом, термин "алкиламин" включает группу амина, как определено выше, имеющую присоединенный замещенный или незамещенный алкил, т.е., по меньшей мере, один из R50 и R51 представляет собой алкильную группу.
Термин "ациламино" известен в данной области техники и относится к остатку, который может быть представлен общей формулой:
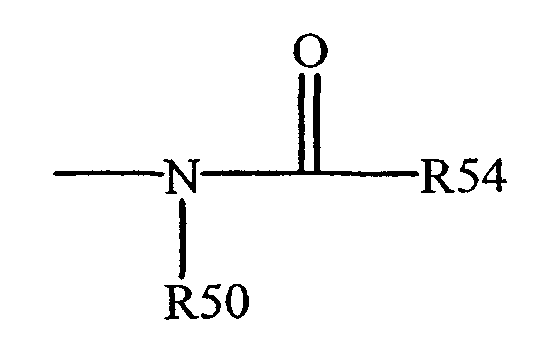
где R50, как определено выше, и R54 представляют водород, алкил, алкенил или (CH2)m-R61, где m и R61 определены выше.
Термин "амидо" известен в данной области техники как аминозамещенный карбонил и включает остаток, который может быть представлен общей формулой:
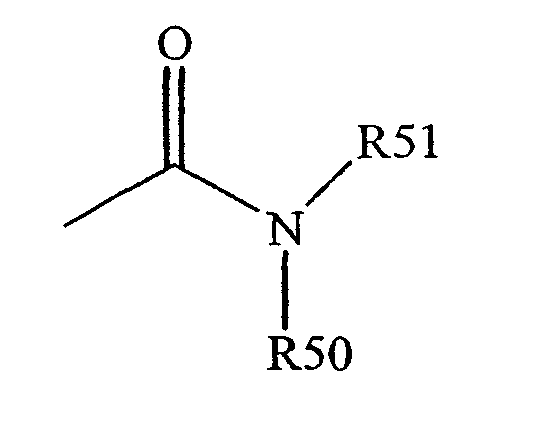
где R50 и R51, как определены выше. Определенные воплощения амида в существующем изобретении не будут включать имиды, которые могут быть нестабильными.
Термин "алкилтио" относится к алкильной группе, как определено выше, присоединенной через радикал серы. В некоторых вариантах осуществления остаток алкилтио представлен одним из -S-алкил, -S-алкенил, -S-алкинил и -S-(CH2)m-R61, где m и R61 определены выше. Типичные алкилтиогруппы включают метилтио, этилтио и подобные.
Термин "карбоксильная группа" известен в данной области техники и включает такие остатки, которые могут быть представлены общими формулами:
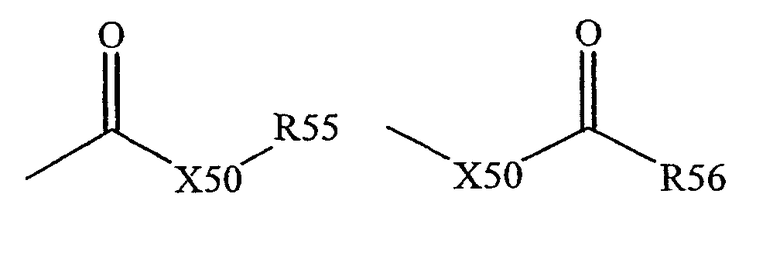
где X50 представляет собой связь или кислород, или серу, и R55 и R56 представляют собой водород, алкил, алкенил, -(CH2)m-R61 или фармацевтически приемлемую соль, R56 представляет собой водород, алкил, алкенил или -(CH2)m-R61, где m и R61 определены выше. В тех случаях, когда X50 представляет собой кислород и R55 или R56 не являются водородом, формула представляет собой сложный эфир. В тех случаях, когда X50 представляет собой кислород и R55, как определено выше, остаток упоминается здесь как карбоксильная группа, и, предпочтительно, когда R55 представляет собой водород, формула представляет собой карбоновую кислоту. В тех случаях, когда X50 представляет собой кислород и R56 представляет собой водород, формула представляет собой формиат. Обычно в тех случаях, когда атом кислорода вышеупомянутой формулы замещен серой, формула представляет собой тиокарбонильную группу. В тех случаях, когда X50 представляет собой серу и R55 или R56 не представляет собой водород, формула представляет собой сложный тиоэфир. В тех случаях, когда X50 представляет собой серу и R55 представляет собой водород, формула представляет собой тиокарбоновую кислоту. В тех случаях, когда X50 представляет собой серу и R56 представляет собой водород, формула представляет собой тиоформиат. С другой стороны, в тех случаях, когда X50 представляет собой связь и R55 не является водородом, вышеупомянутая формула представляет собой кетоновую группу. В тех случаях, когда X50 представляет собой связь и R55 представляет собой водород, вышеупомянутая формула представляет собой альдегидную группу.
Термин "карбамоил" относится к -O(C=O)NRR', где R и R' представляют собой независимо Н, алифатические группы, арильные группы или гетероарильные группы.
Термин "оксо" относится к кислороду карбонила (=О).
Термины "алкоксил" или "алкокси" являются известными в данной области техники и относятся к алкильной группе, как определено выше, присоединенной через радикал кислорода. Типичные алкоксильные группы включают метокси, этокси, пропилокси, трет-бутокси и подобные. Простой эфир представляет собой два углеводорода, соединенные ковалентной связью посредством кислорода. Соответственно, заместитель алкила, который образует алкиловый эфир или входит в состав алкоксильной группы, которая может быть представлена одним из -О-алкил, -O-алкенил, -О-алкинил, -О-(CH2)m-R61, где m и R61 описаны выше.
Термин "сульфонат" известен в данной области техники и относится к остатку, который может быть представлен общей формулой:
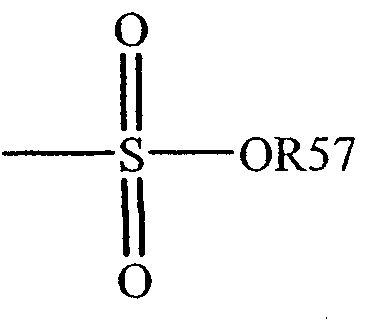
в которой R57 является электронной парой, водородом, алкилом, циклоалкилом или арилом.
Термин "сульфат" является известным в данной области техники и включает остаток, который может быть представлен общей формулой:
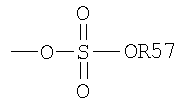
в которой R57, как определено выше.
Термин "сульфонамидо" известен в данной области техники и
включает остаток, который может быть представлен общей формулой:
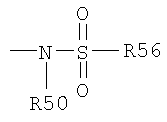
в которой R50 и R56, как определено выше.
Термин "сульфамоил" известен в данной области техники и относится к остатку, который может быть представлен общей формулой:
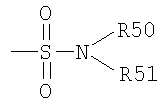
в которой R50 и R51, как определено выше.
Термин "сульфонил" известен в данной области техники и относится к остатку, который может быть представлен общей формулой:
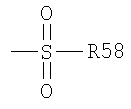
в которой R58 является одним из следующих: водород, алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил или гетероарил.
Термин "сульфоксидо" известен в данной области техники и относится к остатку, который может быть представлен общей формулой:

в котором R58 определен выше.
Термин "фосфорил" известен в данной области техники и может быть представлен общей формулой:
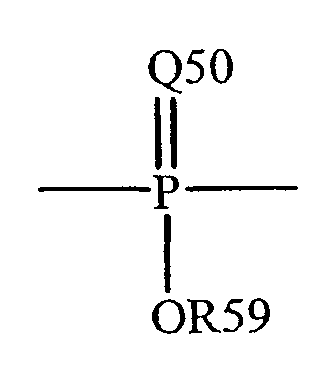
где Q50 представляет собой S или O, и R59 представляет собой водород, низший алкил или арил. Когда имеет место замещение, например, алкила, фосфорильная группа фосфорилалкила может быть представлена общими формулами:
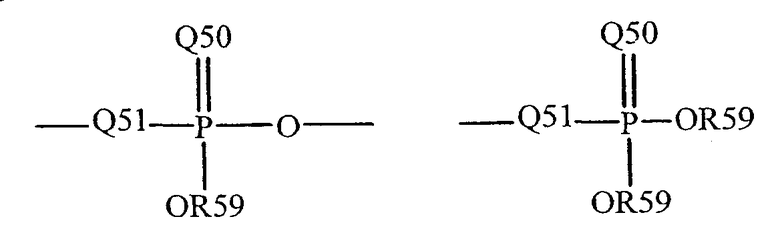
где Q50 и R59, каждый независимо, определены выше, и Q51 представляет собой О, S или N. Когда Q50 представляет собой S, фосфорильный остаток представляет собой "фосфортиоат".
Термин "фосфорамидит" известен в данной области техники и может быть представлен общими формулами:
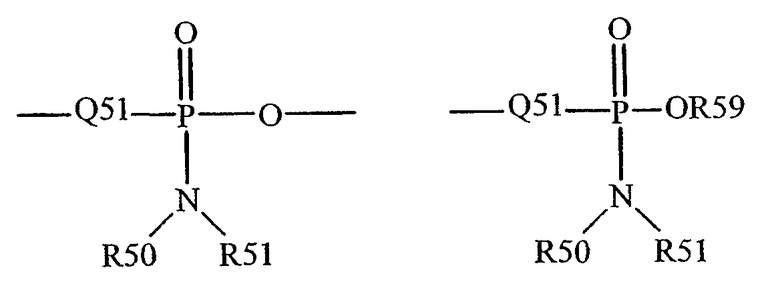
где Q51, R50, R51 и R59, как определено выше.
Термин "фосфонамидит" известен в данной области техники и может быть представлен общими формулами:
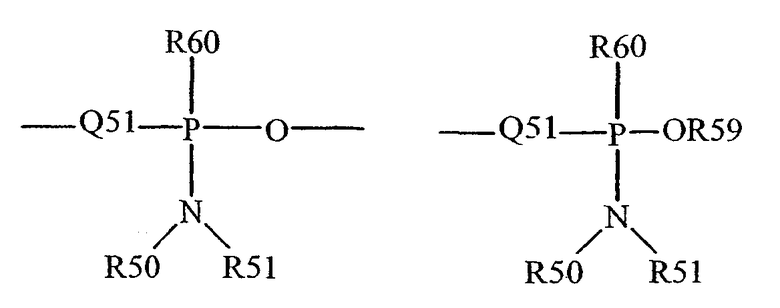
где Q51, R50, R51 и R59, как определено выше, и R60 представляет собой низший алкил или арил.
Может производиться замещение алкенильных и алкинильных групп аналогичными заместителями с получением, например, аминоалкенилов, аминоалкинилов, амидоалкенилов, амидоалкинилов, иминоалкенилов, иминоалкинилов, тиоалкенилов, тиоалкинилов, карбонилзамещенных алкенилов или алкинилов.
Определение каждого выражения, например, "алкил", "m", "n" и подобных предназначено для того, чтобы быть независимым от определения в другом месте той же самой структуры, когда она неоднократно встречается в этой структуре.
Термин "селеноалкил" известен в данной области техники и относится к алкильной группе, присоединенной через селеногруппу. Типичный "селеноэфир" может быть заместителем алкила, который выбирают из -Se-алкила, -Se-алкенила, -Se-алкинила и -Se-(CH2)m-R61, m и R61, определенных выше.
Термины "трифлил", "тозил", "метилсульфонил" и "нонафлил" известны в данной области техники и относятся к трифторметансульфонильным, p-толуолсульфонильным, метансульфонильным и нонафторбутансульфонильным группам соответственно. Термины "трифлат", "тозилат", "мезилат" и "нонафлат" известны в данной области техники и относятся к трифторметансульфонату, p-толуолсульфонату, метансульфонату и нонафторбутансульфонату, функциональные группы и молекулы которых содержат названные группы.
Сокращения Ме, Et, Ph, Tf, Nf, Ts и Ms означают метил, этил, фенил, трифторметансульфонил, нонафторбутансульфонил, p-толуолсульфонил и метансульфонил соответственно. Более полный список сокращений, используемых в данной области техники химиками-органиками обычной квалификации, появляется в первом выпуске каждого тома Journal of Organic Chemistry; данный список обычно представлен в таблице, озаглавленной Standard List of Abbreviations.
Некоторые соединения, включенные в композиции настоящего изобретения, могут существовать в предпочтительных геометрических или стереоизомерных формах. Настоящее изобретение рассматривает все такие соединения, включая цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, попадающие в диапазон изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителе, таком как алкильная группа. Все такие изомеры, так же как их смеси, предназначены для включения в данное изобретение.
Если, например, желателен предпочтительный энантиомер соединений настоящего изобретения, он может быть получен посредством асимметричного синтеза или посредством модификации с хиральным вспомогательным соединением, где получающаяся диастереомерная смесь разделяется и вспомогательная группа отщепляется, чтобы обеспечить химически чистые желательные энантиомеры. Альтернативно, в случаях, когда молекула содержит основную функциональную группу, такую как амино, или кислую функциональную группу, такую как карбоксильная группа, диастереомерные соли формируются с подходящей оптически активной кислотой или основанием, с последующим разделением диастереомеров, которое осуществляют фракционной кристаллизацией или хроматографическим способом, известным в технике, и последующим выделением чистых энантиомеров. Точно так же предпочтительный энантиомер может быть выделен из рацемической смеси его энантиомеров с помощью хиральных хроматографических методов, известных в данной области техники.
Подразумевается, что заместитель или замещенный подразумевает, что такое замещение находится в соответствии с допустимой валентностью замещенного атома и заместителя и что замещение приводит к стабильному соединению, например, которое не подвергается спонтанной трансформации, такой как перегруппировка, циклизация, элиминирование или другая реакция.
Термин "замещенный" также рассматривается как включающий все допустимые заместители органических соединений. В широком аспекте допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Иллюстративные заместители включают, например, описанные здесь выше. Допустимые заместители могут быть одним или более и теми же самыми или отличными для подходящих органических соединений. В целях данного изобретения, гетероатомы, такие как азот, могут иметь водородные заместители и/или любые допустимые заместители органических соединений, описанных здесь, которые удовлетворяют валентности гетероатомов. Не подразумевают, что данное изобретение ограничивается любыми допустимыми заместителями органических соединений.
Как используется здесь, фраза "защитная группа" означает временные заместители, которые защищают потенциально реактивную функциональную группу от нежелательных химических превращений. Примеры таких защитных групп включают сложные эфиры карбоновых кислот, силиловые эфиры спиртов и ацетали и кетали альдегидов и кетонов соответственно. Область химии защитных групп была рассмотрена (Green, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991). Защищенные формы соединений изобретения включены в рамки этого изобретения.
В целях данного изобретения химические элементы идентифицированы в соответствии с Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 67th Ed., 1986-87, на обложке.
Фраза "аберрантная модификация или мутация" гена относится к таким генетическим поражениям, как, например, делеция, замена или вставка нуклеотидов в гене, так же как к существенным хромосомным перегруппировкам гена и/или патологическому метилированию гена. Аналогично, неправильная экспрессия гена относится к аберрантным уровням транскрипции гена относительно таких уровней в нормальной клетке в подобных условиях, а также сплайсингу нестабильного типа мРНК, транскрибированной с гена.
Базально-клеточные карциномы существуют в различных клинических и гистологических формах, таких как узловато-язвенная, поверхностная, пигментированная, кольцевидная, фиброэпителиома и невоидный синдром. Базально-клеточные виды рака представляют собой самые распространенные кожные опухоли, найденные у людей. Большинство новых случаев рака кожи, не относящихся к меланоме, относятся к этой категории.
Термин "карцинома" относится к злокачественному новообразованию, составленному из эпителиальных клеток, имеющих тенденцию инфильтрироваться в окружающие ткани и давать начало метастазам. Типичные карциномы включают базально-клеточный рак, который является эпителиальной опухолью кожи, которая, редко метастазируя, имеет потенциал для местной инвазии и деструкции; плоскоклеточную карциному, которая относится к карциномам, исходящим из сквамозного эпителия и имеющим кубические клетки; карциносаркому, которая включает злокачественные опухоли, составленные из карциноматозных и саркоматозных тканей; аденокистозную карциному, карциному, характеризующуюся цилиндрами или полосами или муцинозной стромой, отделенными или окруженными гнездами или шнурами малых эпителиальных клеток, встречающихся в грудных и слюнных железах и слизистых железах дыхательных путей; эпидермоидную карциному, которая относится к злокачественным клеткам, которые имеют тенденцию дифференцироваться таким же образом, как им подобные из эпидермиса; т.е. они имеют тенденцию к формированию шиповидных клеток и переносят ороговение; назофарингеальную карциному, которая относится к злокачественной опухоли, возникающей в эпителиальном покрове пространства, находящегося позади носа; и почечно-клеточный рак, который относится к раку почечной паренхимы, составленной из канальцевых клеток в изменившемся местоположении.
Другими карциноматозными эпителиальными разрастаниями являются папилломы, которые относятся к доброкачественным опухолям, исходящим из эпителия, и имеют возбудитель - вирус папилломы; и эпидермоидомы, которые относятся к опухоли мозга или мозговой оболочки, формирующей включение эктодермальных элементов во время закладки желобка нервной трубки.
Термин "ED50" означает дозу лекарственного средства, которое производит 50% его максимальной реакции или эффекта.
Эффективное количество рассматриваемого соединения, относительно существующих методов лечения, относится к количеству антагониста в составе композиции, которое, будучи примененным как часть желательного режима дозировки, вызывает, например, изменение уровня пролиферации клетки и/или уровня выживания клетки согласно клинически приемлемым стандартам для заболеваний, к которым будет применено лечение.
Термины "эпителиальные" и "эпителий" относятся к клеточному покрытию внутренних и внешних поверхностей тела (кожа, слизистые и серозные оболочки), включая железы и другие структуры, исходящие из них, например роговичному, пищеводному, эпидермальному и волосяным эпителиальным клеткам фолликула. Другая типичная эпителиальная ткань включает обонятельный эпителий, который представляет собой псевдомногослойный эпителий, выстилающий обонятельную область полости носа и содержащий обонятельные рецепторы; железистый эпителий, который относится к эпителию, состоящему из секретирующих клеток; сквамозный эпителий, который относится к эпителию, составленному из сглаженных пластинчатых клеток. Термин "эпителий" может также относиться к переходному эпителию, который типично находится в выстилке полых органов, которые подвергаются большому механическому изменению, такому как сокращение и растяжение, например, ткань, которая представляет собой переход между многослойным сквамозным и столбчатым эпителием.
Состояние роста клетки относится к скорости пролиферации клетки и/или состоянию дифференциации клетки. Измененное состояние роста является состоянием роста, характеризующимся патологическим уровнем пролиферации, например клетка, показывающая увеличенный или уменьшенный уровень пролиферации относительно нормальной клетки.
Термин "антагонист Hedgehog пути" относится к средству, которое замедляет функцию Hedgehog пути, например, подавляет транскрипцию генов-мишеней (Gli1 и гены Ptc), которая в нормальных клетках индуцируется посредством контакта клетки с Hedgehog. В дополнение к изменению Smoothened-зависимого пути в некоторых вариантах осуществления антагонисты Hedgehog пути настоящего изобретения могут использоваться, чтобы преодолеть снижение функции Ptc, Smoothened повышение функции и/или повышение функции Hedgehog. Термины "снижение функции" и "увеличение функции" соответственно относятся к аберрантной модификации или мутации, например, гена Ptc, Hedgehog гена или Smoothened гена, или уменьшению или увеличению уровня экспрессии такого гена, который приводит к фенотипу, который, например, напоминает контакт клетки с Hedgehog белком, такой как аберрантная активация Hedgehog пути или напоминает снижение функции Smo. Мутация может включать снижение способности продукции генов Ptc или Smo для регуляции уровня активности белков Gli/Ci, например Gli1, Gli2 и Gli3.
Как используется здесь, иммортализованные клетки относятся к клеткам, которые были химически изменены, и/или рекомбинантным клеткам, и означают, что такие клетки имеют способность расти через неограниченное количество делений в культуре.
Термин "LD50" означает дозу лекарственного средства, которая является летальной для 50% тестируемых пациентов.
"Пациент" или "субъект" означает организм, подвергающийся лечению способом настоящего изобретения, и может подразумевать или человека, или животное.
Фраза "фармацевтически приемлемый" используется здесь по отношению к тем соединениям, материалам, композициям и/или дозированным формам, которые в пределах исследованного медицинского суждения являются подходящими для использования в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерного с приемлемым соотношением выгода/риск.
Фраза "фармацевтически приемлемый носитель", как используется здесь, означает фармацевтически приемлемый материал, композицию или связующее средство, такое как жидкий или твердый наполнитель, растворитель, инертный наполнитель, вспомогательное вещество (например, лубрикант, порошок магнезии, кальций или стеарат цинка или стеариновая кислота) или растворимый инкапсулирующий материал, включенный в перенос или транспортировку рассматриваемого соединения от одного органа или части тела к другому органу или части тела. Каждый носитель должен быть приемлемым в том смысле, чтобы быть совместимым с другими ингредиентами композиции и не вредным для пациента. Некоторые примеры материалов, которые могут служить фармацевтически приемлемыми носителями, включают (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза и ее производные, такие как натрий-карбоксиметилцеллюлозы, этилцеллюлоза и ацетат целлюлозы; (4) измельченный в порошок трагакант; (5) солод; (6) желатин; (7) тальк; (8) инертные наполнители, такие как масло какао и воск для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннитол и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар-агар; (14) буферные средства, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) рН-буферизованные растворы; (21) полиэфиры, поликарбонаты и/или полиангидриды и (22) другие нетоксичные совместимые вещества, используемые в фармацевтических составах.
Термин "пролекарство" предназначается, чтобы охватить соединения, которые при физиологических условиях преобразуются в терапевтически активные средства настоящего изобретения. Распространенный способ производства пролекарств должен включать отобранные остатки, которые гидролизуются при физиологических условиях, образуя желательную молекулу. В других воплощениях пролекарство преобразуется ферментативной активностью (или другой физиологической активностью) животного-хозяина.
Как используется здесь, "пролиферирующие" и "пролиферация" относится к клеткам в состоянии митоза.
На всем протяжении данной заявки термин "пролиферативное расстройство кожи" относится к любому заболеванию/расстройству кожи, отмеченному нежелательной или аберрантной пролиферацией кожной ткани. Эти состояния обычно характеризуются пролиферацией эпидермальной клетки или неполной дифференциацией клетки и включают, например, X-сцепленный ихтиоз, псориаз, атопический дерматит, аллергический контактный дерматит, эпидермолитический гиперкератоз и себорейный дерматит. Например, эпидермодисплазия представляет собой форму дефектного развития эпидермиса. Другим примером является эпидермолиз, который относится к ослабленному состоянию эпидермиса с формированием булл или спонтанно, или на участке травмы.
Термин "терапевтический индекс" относится к терапевтическому индексу лекарственного средства, определенного как LD50/ED50.
Термин "трансформированные клетки" относится к клеткам, которые спонтанно преобразовались в состояние неограниченного роста, т.е. они приобрели способность расти через неопределенное количество делений в культуре. Трансформированные клетки могут характеризоваться такими терминами, как "неопластические", "анапластические" и/или "гиперпластические", по отношению к их снижению контроля роста.
Термин "субъект", как используется здесь, относится к животному, обычно млекопитающему, или человеку, который был целью лечения, наблюдения и/или эксперимента. Когда этот термин используется в сочетании с введением соединения или лекарственного средства, субъекта подвергают лечению, наблюдению и/или введению соединения или лекарственного средства. Фраза "терапевтически эффективное количество", как используется здесь, означает количество соединения, материала или композиции, включающей соединение настоящего изобретения, которое является эффективным для того, чтобы произвести некоторый желательный терапевтический эффект в, по меньшей мере, субпопуляции клеток животного в приемлемом соотношении выгоды/риска, применимом к любому лечению.
Фразы "парентеральное введение" и "применяемый парентерально", как используется здесь, означают способы введения, иные, чем энтеральное и местное введение, обычно посредством инъекции, и включают, без ограничения, внутривенную, внутримышечную, внутриартериальную, интратекальную, интракапсулярную, внутриорбитальную, внутрисердечную, интраперитонеальную, транстрахеальную, подкожную, субкутикулярную, внутрисуставную, подкапсулярную, субарахноидальную, спинальную и внутригрудинную инъекцию и инфузию.
Фразы "системное воздействие", "применяемый системно", "периферическое воздействие" и "применяемый периферический", как используется здесь, означают введение соединения, лекарственного средства или другого материала, иное, чем непосредственно к остатку Hedgehog пути, связанному с расстройством, такое, что поступает в систему пациента и, таким образом, подчинено метаболизму и другим подобным процессам, например, подкожное введение.
Термин «сахар», как используется здесь, относится к природному или неприродному моносахариду, дисахариду или олигосахариду, включающему одно или более колец пиранозы или фуранозы. Сахар может быть ковалентно связан со стероидным алкалоидом настоящего изобретения через простую эфирную связь или через алкильную связь. В определенных вариантах осуществления сахаридный остаток может быть ковалентно связан со стероидным алкалоидом настоящего изобретения в аномерном центре сахаридного кольца.
Термин «бирадикал», как используется в данном описании, относится к любому ряду двухвалентных групп, например, алкильные, алкенильные, алкинильные, арильные, циклоалкильные, гетероциклоалкильные, аралкильные, гетероарильные и гетероаралкильные группы. Например, 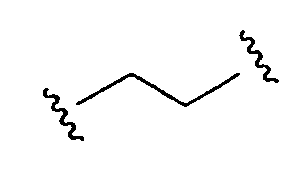 является алкильным двухвалентным радикалом;
является алкильным двухвалентным радикалом; 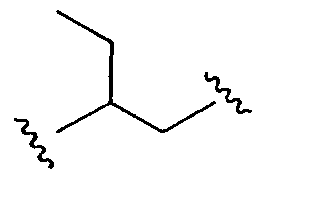 является также алкильным двухвалентным радикалом;
является также алкильным двухвалентным радикалом; 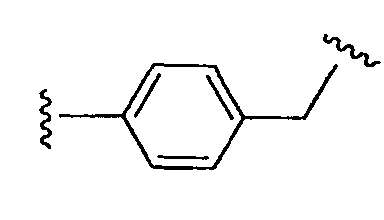 является аралкильным двухвалентным радикалом; и
является аралкильным двухвалентным радикалом; и 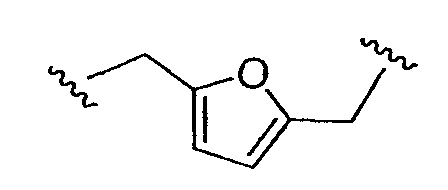 является (алкил)гетероалкильным двухвалентным радикалом. Типичные примеры включают алкилены общей структуры (СН2)х, где Х равно 1-6 и соответствующие алкениленовые и алкиниленовые линкеры, имеющие 2-6 углеродных атомов и одну или более двойных или тройных связей; циклоалкиленовые группы, имеющие 3-8 кольцевых элементов; и аралкильные группы, где одна открытая валентность находится на кольце арила, и каждый из алкильных фрагментов, таких как
является (алкил)гетероалкильным двухвалентным радикалом. Типичные примеры включают алкилены общей структуры (СН2)х, где Х равно 1-6 и соответствующие алкениленовые и алкиниленовые линкеры, имеющие 2-6 углеродных атомов и одну или более двойных или тройных связей; циклоалкиленовые группы, имеющие 3-8 кольцевых элементов; и аралкильные группы, где одна открытая валентность находится на кольце арила, и каждый из алкильных фрагментов, таких как 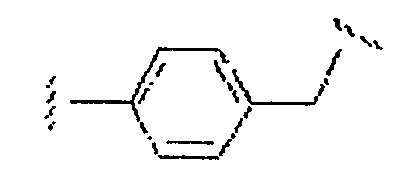 , и его изомеры.
, и его изомеры.
Соединения изобретения
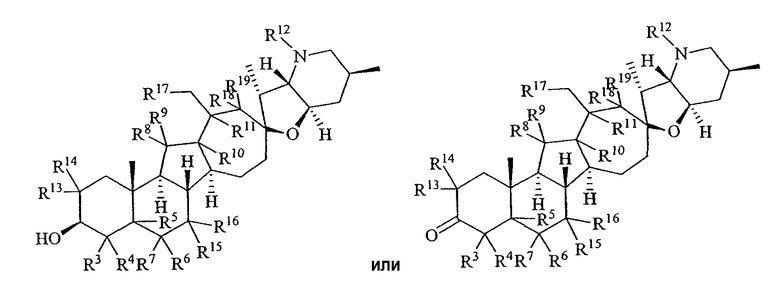
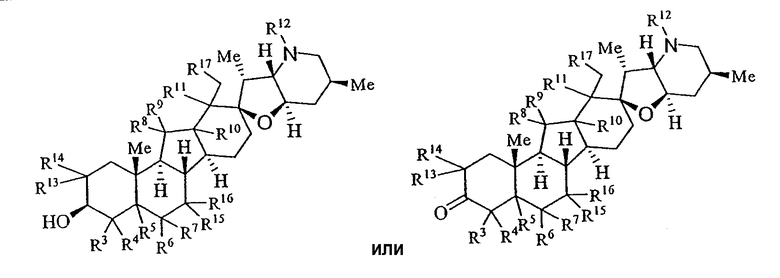
Настоящее изобретение предоставляет аналоги циклопамина, а также их выделенные и очищенные формы, включая синтетические и полусинтетические аналоги, а также фармацевтические композиции, содержащие такие аналоги. В одном варианте осуществления настоящее изобретение предоставляет соединения, представленные соединением формулы 1:
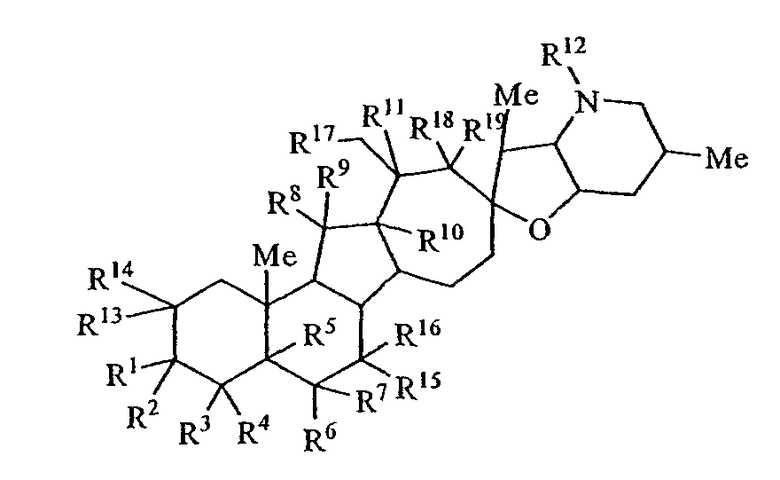 (1)
(1)
или их фармацевтически приемлемую соль;
в котором каждый R1 и R8 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогенид, сульфгидрил, алкилтио, арилтио, аралкилтио, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, сульфонамид, карбоксил, нитрил, сульфат, -OP(L)(OR20)2, -X-C(L)-R21 или -X-C(L)-Х-R21;
где Rl может также представлять собой сахар;
X представляет собой O или NR, где R представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил или аралкил;
L представляет собой O или S;
R2 и R9 независимо представляют собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, алкоксил, арилокси, ацилокси, галогенид, сульфгидрил, алкилтио, арилтио, аралкилтио, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино, гетероарил или гетероаралкил;
каждый R5 и R11 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, алкоксил, арилокси, ацилокси, галогенид, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино, алкилселено, аралкилселено, арилселено, алкилтио, аралкилтио, арилтио, гетероарил или гетероаралкил;
каждый R3, R4, R6, R7, R13 и R14 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, алкоксил, арилокси, ацилокси, галогенид, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино, гетероарил или гетероаралкил;
или R1 и R2 и/или R8 и R9, взятые вместе с углеродом, с которым они связаны, образуют -(C=O)-, -(C=S)-, -(C=N(OR20))-, -(C=N(R20))-, -(C=N(N(R20)(R20))) или образуют необязательно замещенный 3-8-членный цикл; или
R4 и R5, взятые вместе, и/или R5 и R6, взятые вместе, и/или R10 и R11, взятые вместе, образуют двойную связь или образуют группу, представленную 1b
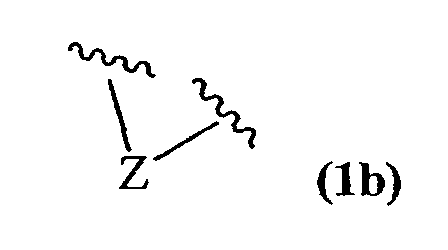
где Z представляет NR21, O или C(R23)(R23);
R12 представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, гидроксил, аралкил, гетероарил, гетероаралкил, галогеналкил, алкоксил, -C(O)R21, -CO2R21, -SO2R21, -C(O)N(R21)(R21), -[C(R21)2]q-R21, -[(W)-N(R21)C(O)]qR21, -[(W)-C(O)]qR21, -[(W)-C(O)O]qR21, -[(W)-(O)C(O)]qR21, -[(W)-SO2]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-N(R21)]qR21 или -[(W)-S]qR21;
где каждый W представляет собой бирадикал, а q равен 1, 2, 3, 4, 5 или 6;
R15, R16 и R17 независимо представляют собой Н, алкоксил, арилокси, ацилокси, галогенид, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино; или R15 и R16, взятые вместе с углеродом, с которым они связаны, образуют -C(O)- или -C(S)-;
R18 и R19 независимо представляют собой Н, алкил, аралкил, галогенид, амидо или сложный эфир;
R20 представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил или гетероаралкил; или любые два R20 в составе одного заместителя могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R21 представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[C(R20)2]р-R25, где p равно 0-6; или любые два R21 в составе одного заместителя могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R23 представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогенид, алкоксил, арилокси, ацилокси, силилокси, нитрил, -C(O)R21, -CO2R21, -SO2R21 и -C(O)N(R21)2;
R25 представляет собой гидроксил, ациламино, -N(R20)COR20, -N(R20)C(O)OR20, -N(R20)SO2(R20), -COR20N(R20)2, -ОС(O)R20N(R20)(R20), -SO2N(R20)(R20), -N(R20)(R20), -COOR20, -C(O)N(ОН)(R21), -OS(O)2OR20, -S(О)2OR20, -OP(L)(OR20)(OR20), -NP(O)(OR20)(OR20) или -P(O)(OR20)(OR20).
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R13, R14, R15, R16 и R17 представляют собой водород.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 представляет собой гидроксил, сахар, -OP(L)(OR20)2, -X-C(L)-R21 или -X-C(L)-Х-R21; или R1 и R2, взятые вместе с углеродом, c которым они связаны, образуют -С(O)-.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R4 и R5, взятые вместе, образуют двойную связь.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -C(O)-.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 представляет собой гидроксил, а R2 представляет собой Н.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 представляет собой гидроксил, R2 представляет собой Н; и R5 и R6, взятые вместе, образуют двойную связь; или R5 и R6, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R10 и R11, взятые вместе, образуют двойную связь; или R10 и Rll, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R5 и R6, взятые вместе, образуют двойную связь и R10 и Rll, взятые вместе, образуют двойную связь.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -С(О)-; R4 и R5, взятые вместе, образуют двойную связь; и R10 и R11, взятые вместе, образуют двойную связь; или R10 и R11, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой С(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 представляет собой гидроксил, а R2 представляет собой Н; R10 и Rl1, взятые вместе, образуют двойную связь; или R10 и R11, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R8 и R9 представляют собой водород; или R8 и R9, взятые вместе с углеродом, с которым они связаны, являются -C(O)-.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-C(O)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R13, R14, R15, R16 и R17 представляют собой водород; и R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-C(O)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R4 и R5, взятые вместе, образуют двойную связь; R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -С(О)-; и R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-C(O)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены 1 и сопутствующими определениями, где R1 представляет собой гидроксил и R2 представляет собой Н; и R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-С(О)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-C(O)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены соединением формулы:
где
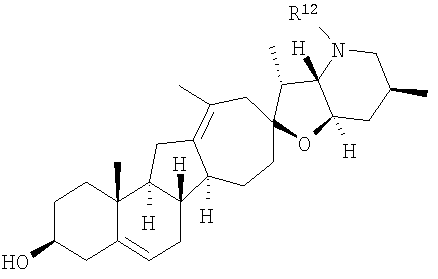 или
или 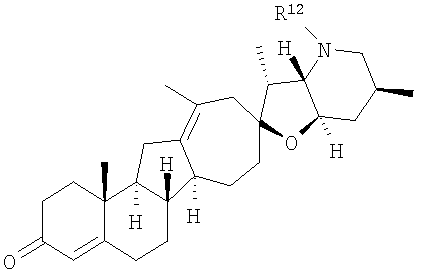
R12 представляет собой Н, алкил, арил, циклоалкил, гетероциклоалкил, гидроксил, аралкил, гетероарил, гетероаралкил, галогеналкил, алкоксил, -C(O)R21, -CO2R21, -SO2R21, -C(O)N(R21)(R21), -[C(R21)2]p-R21, -[(W)-N(R21)C(O)]qR21, -[(W)-C(O)]qR21, -[(W)-C(O)O]qR21, -[(W)-OC(O)]qR21, -[(W)-SO2]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-N(R21)]qR21 или -[(W)-S]qR21; где каждый W независимо представляет собой бирадикал;
q равен 1, 2, 3, 4, 5 или 6;
R20 представляют собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил или гетероаралкил; или любые два R20 в составе одного заместителя могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R21 представляет собой Н, алкил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[C(R20)2]р-R25; или любые два R21 могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R25 представляет собой гидроксил, ациламино, -N(R20)COR20, -N(R20)C(O)OR20, -N(R20)SO2(R20), -COR20N(R20)2, -ОС(O)R20N(R20)(R20), -SO2N(R20)(R20), -N(R20)(R20), -COOR20, -C(O)N(ОН)(R21), -OS(O)2OR19, -S(O)2OR20, -OP(L)(OR20)(OR20), -NP(O)(OR20)(OR20) или -P(O)(OR20)(OR20).
Настоящее изобретение конкретно предоставляет соединения, представленные группой, состоящей из
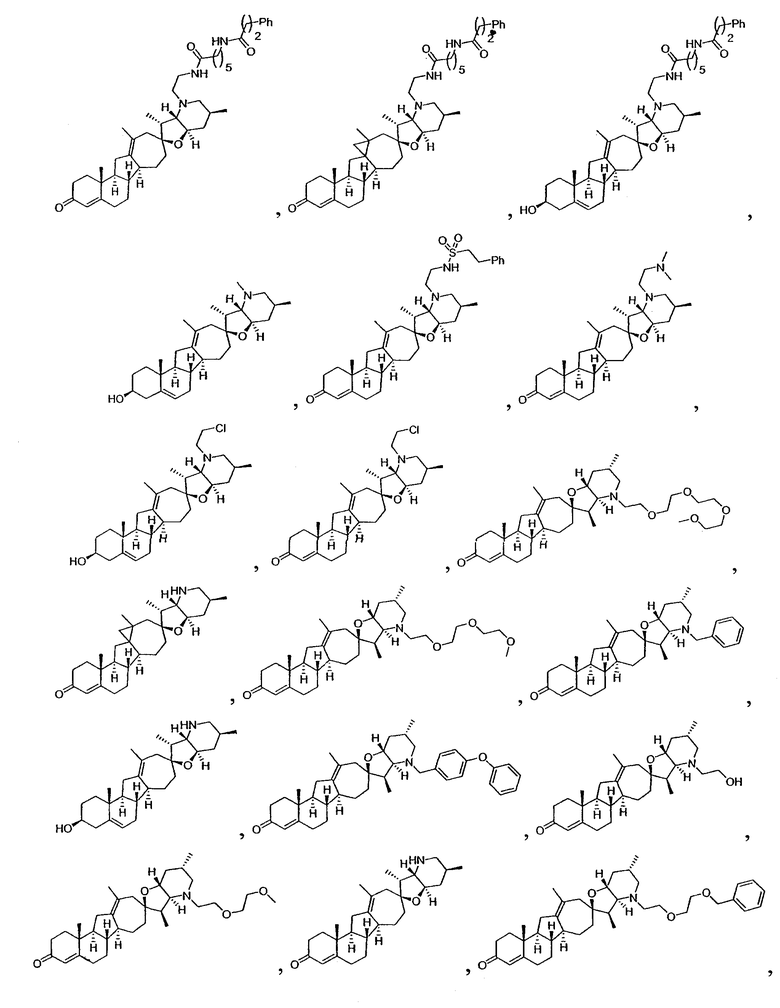
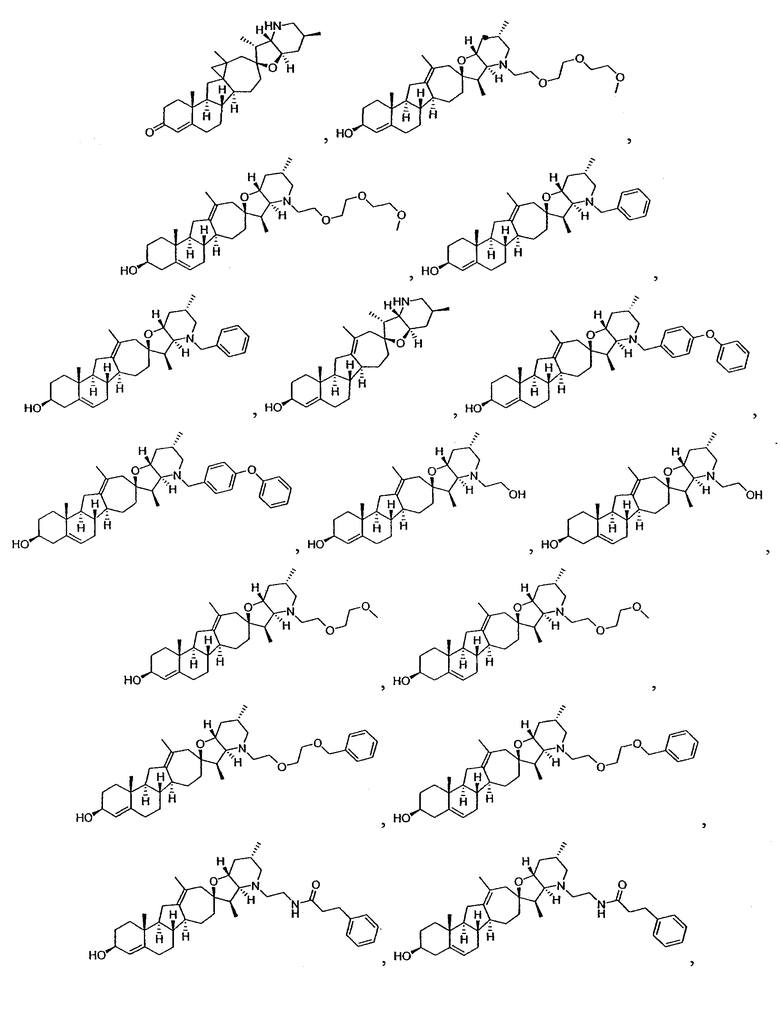

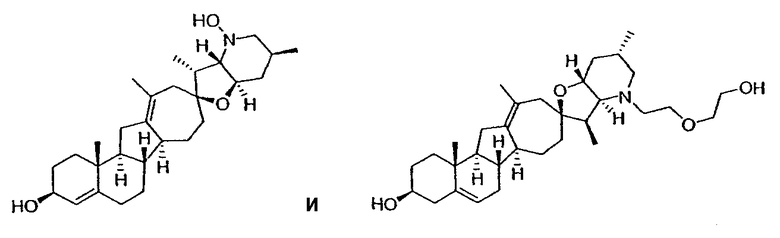
В некоторых вариантах осуществления соединения настоящего изобретения представлены любым из вышеупомянутых соединением и сопутствующими определениями, где соединение представлено формулой:
В одном варианте осуществления настоящее изобретение предоставляет соединения, представленные соединением формулы 2:
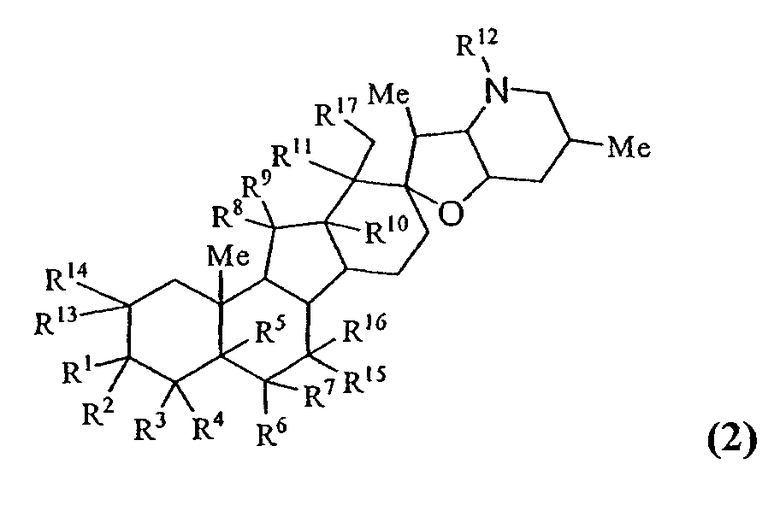
или его фармацевтически приемлемой солью;
в котором каждый R1 и R8 независимо представляют собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогенид, сульфгидрил, алкилтио, арилтио, аралкилтио, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, сульфонамид, карбоксил, нитрил, сульфат, -OP(L)(OR20)2, -X-C(L)-R21 или -X-C(L)-Х-R21;
где Rl может также представлять собой сахар;
X представляет собой O или NR, где R представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил или аралкил;
L представляет собой O или S;
R2 и R9 независимо представляют собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, алкоксил, арилокси, ацилокси, карбоксил, галогенид, сульфгидрил, алкилтио, арилтио, аралкилтио, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино, гетероарил или гетероаралкил;
каждый R5 и R11 независимо представляют собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, алкоксил, арилокси, ацилокси, галогенид, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино, алкилселено, аралкилселено, арилселено, алкилтио, аралкилтио, арилтио, гетероарил или гетероаралкил;
каждый R3, R4, R6, R7, R13 и R14 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, алкоксил, арилокси, ацилокси, галогенид, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино, гетероарил или гетероаралкил;
где R1 и R2 и/или R8 и R9, взятые вместе с углеродом, с которым они связаны, образуют -(C=O)-, -(C=S)-, -(C=N(OR20))-, -(C=N(R20))-, -(C=N(N(R20)(R20))) или образуют необязательно замещенный 3-8-членный цикл; или
R4 и R5, взятые вместе, и/или R5 и R6, взятые вместе, и/или R10 и R11, взятые вместе, образуют двойную связь или образуют группу, представленную 1b
где Z представляет NR21, O или C(R23)(R23);
R12 представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, гидроксил, аралкил, гетероарил, гетероаралкил, галогеналкил, алкоксил, -C(O)R21, -CO2R21, -SO2R21, -C(O)N(R21)(R21), -[C(R21)2]q-R21, -[(W)-N(R21)C(O)]qR21, -[(W)-C(O)]qR21, -[(W)-C(O)O]qR21, -[(W)-OC(O)]qR21, -[(W)-SO2]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-N(R21)]qR21 или -[(W)-S]qR21;
где W представляет собой бирадикал, а q равен 1, 2, 3, 4, 5 или 6;
R15, R16 и R17 независимо представляют собой Н, алкоксил, арилокси, ацилокси, галогенид, гидроксил, амино, алкиламино, ариламино, ациламино, аралкиламино; или R15 и R16, взятые вместе с углеродом, с которым они связаны, образуют -C(O)- или -C(S)-;
R18 и R19 независимо представляют собой Н, алкил, аралкил, галогенид, амидо или сложный эфир;
R20 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил или гетероаралкил; или любые два R20 в составе одного заместителя могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R21 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[C(R20)2]р-R25, где p равно 0-6; или любые два R21 в составе одного заместителя могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R23 независимо представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогенид, алкоксил, арилокси, ацилокси, силилокси, нитрил, -C(O)R21, -CO2R21, -SO2R21 и -C(O)N(R21)2; и
R25 представляет собой гидроксил, ациламино, -N(R20)COR20, -N(R20)C(O)R20, -N(R20)SO2(R20), -COR20N(R20)2, -ОС(О)R20N(R20)(R20), -SO2N(R20)(R20), -N(R20)(R20), -COOR20, -C(O)N(ОН)(R21), -OS(O)2OR20, -S(O)2OR20, -OP(L)(OR20)(OR20), -NP(O)(OR20)(OR20) или -P(O)(OR20)(OR20),
при условии, что существует, по меньшей мере, одна группа, представленная формулой 1b, на указанном соединении формулы 2.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где в данном описании R13, R14, R15, R16 и R17 представляют собой водород.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 представляет собой гидроксил, сахар, OP(L)(OR20)2, -X-C(L)-R21 или -X-C(L)-X-R21; или R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -С(О)-.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R4 и R5, взятые вместе, образуют двойную связь.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -С(О)-.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 представляет собой гидроксил, a R2 представляет собой Н.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 представляет собой гидроксил, а R2 представляет собой Н; R5 и R6, взятые вместе, образуют двойную связь; или R5 и R6, взятые вместе, образуют группу, представленную 1b;
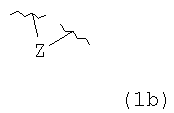
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R10 и R11, взятые вместе, образуют двойную связь; или R10 и R11, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R5 и R6, взятые вместе, образуют двойную связь, и R10 и R11, взятые вместе, образуют двойную связь.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -С(О)-; R4 и R5, взятые вместе, образуют двойную связь; и R10 и R11, взятые вместе, образуют двойную связь; или R10 и R11, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 представляет собой гидроксил и R2 представляет собой Н; R10 и R11, взятые вместе, образуют двойную связь; или R10 и R11, взятые вместе, образуют группу, представленную 1b;
где
Z представляет собой C(R23)(R23).
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R8 и R9 представляют собой водород; или R8 и R9, взятые вместе d углеродом, с которым они связаны, являются -С(O)-.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-С(О)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R13, R14, R15, R16 и R17 представляют собой водород, а R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]q 21, -[(W)-N(R21)SO2]qR21, -[(W)-С(O)N(R21)]qR21, -[(W)-О]qR21, -[(W)-С(О)]qR21 или - [(W)-С(О)О]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R4 и R5, взятые вместе, образуют двойную связь; R1 и R2, взятые вместе с углеродом, с которым они связаны, образуют -С(О)-; а R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-С(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-C(O)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены формулой 2 и сопутствующими определениями, где R1 представляет собой гидроксил и R2 представляет собой Н; а R12 представляет собой Н, алкил, циклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, гидроксил, алкоксил, -[(W)-N(R21)С(О)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-C(O)]qR21 или -[(W)-C(O)O]qR21.
В некоторых вариантах осуществления соединения настоящего изобретения представлены соединением формулы:
или 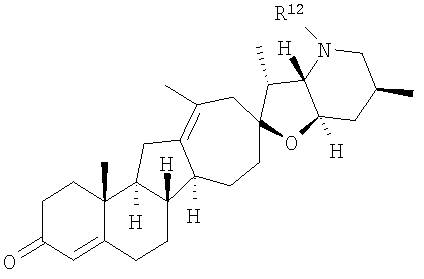
где
R12 представляет собой Н, алкил, арил, циклоалкил, гетероциклоалкил, гидроксил, аралкил, гетероарил, гетероаралкил, галогеналкил, алкоксил, -C(O)R21, -C(O)N(R21)(R21), -[C(R21)2]p-R21, -[(W)-N(R21)C(O)]qR21, -[(W)-C(O)]qR21, -[(W)-C(O)O]qR21, -[(W)-OC(O)]qR21, -[(W)-SO2]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-C(O)N(R21)]qR21, -[(W)-O]qR21, -[(W)-N(R21)]qR21 или -[(W)-S]qR21;
q равен 1, 2, 3, 4, 5 или 6;
R20 представляет собой Н, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, или гетероаралкил; или любые два R20 в составе одного заместителя могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R21 представляет собой Н, алкил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[С(R20)2]p-R25; или любые два R21 могут быть взяты вместе для образования 4-8-членного необязательно замещенного цикла;
R25 представляет собой гидроксил, ациламино, -N(R20)COR20, -N(R20)C(O)OR20, -N(R20)SO2(R20), -COR20N(R20)2, -ОС(О)R20N(R20)(R20), -SO2N(R20)(R20), -N(R20)(R20), -COOR20, -С(О)N(ОН)(R21), -OS(O)2OR19, -S(O)2OR20, -OP(L)(OR20)(OR20), -NP(O)(OR20)(OR20) или -P(O)(OR20)(OR20).
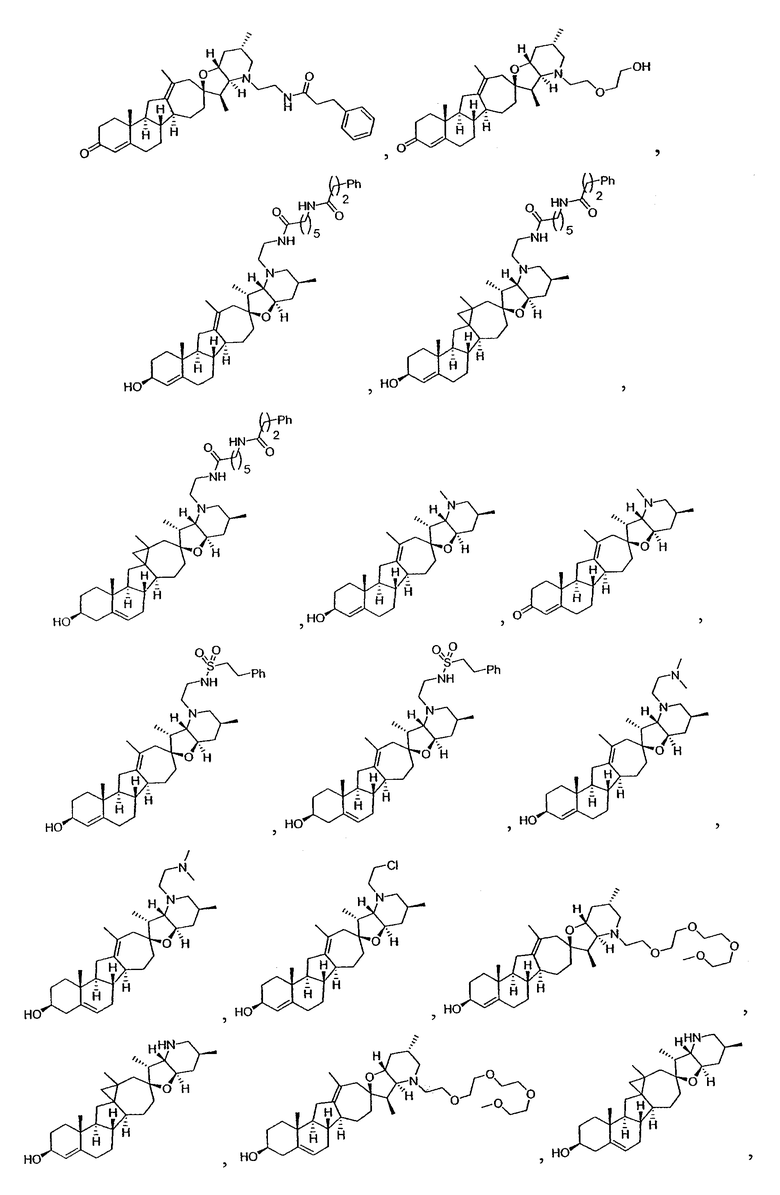
Настоящее изобретение конкретно предоставляет соединения, представленные группой, состоящей из;
 ,
,  ,
, 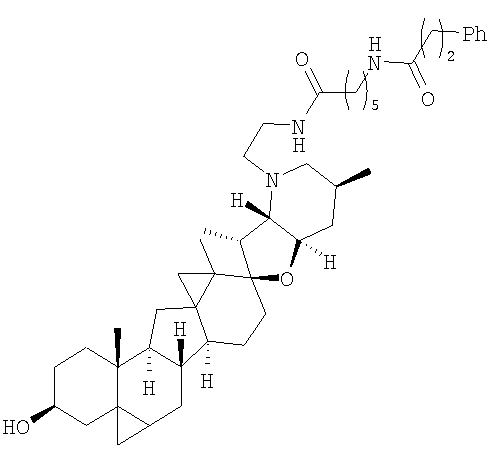
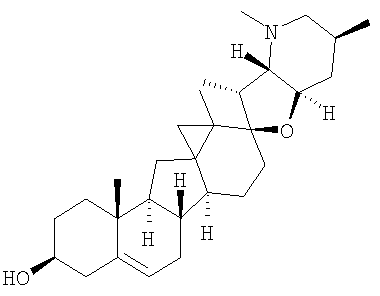 ,
, 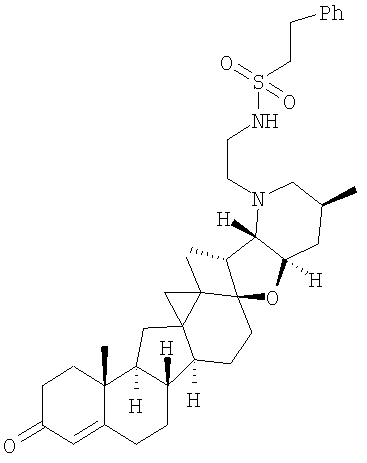 ,
, 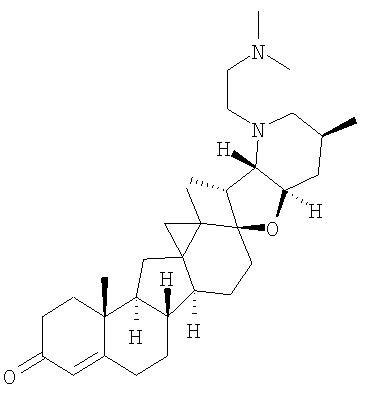 ,
, 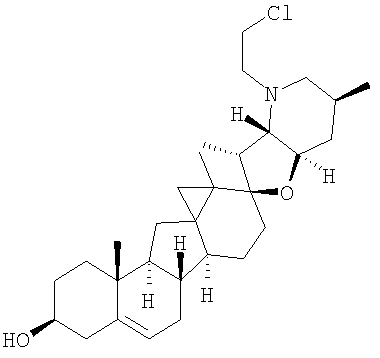 ,
,
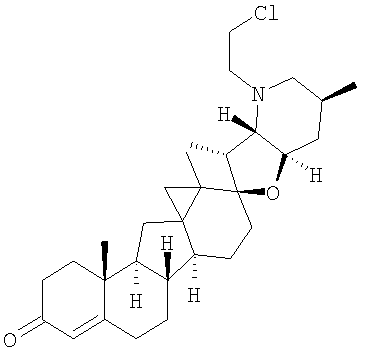 ,
, 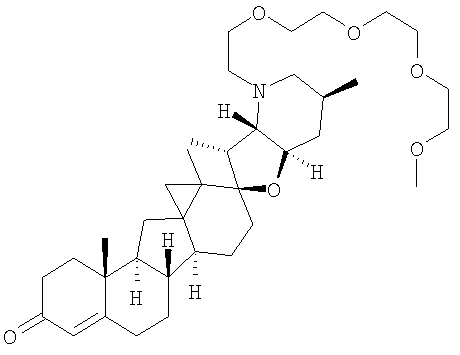 ,
, 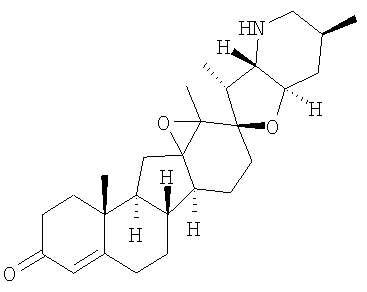 ,
, 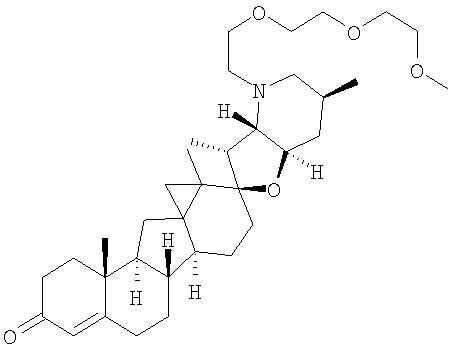
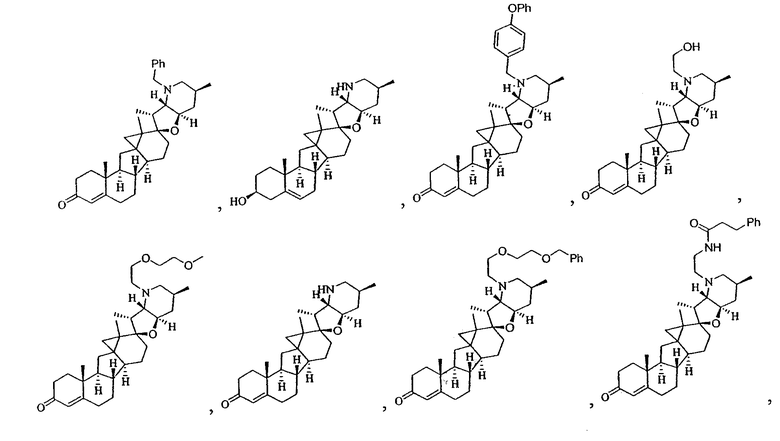

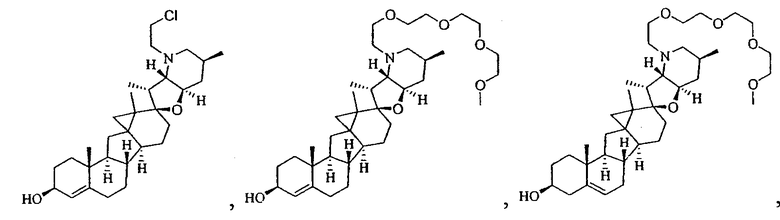
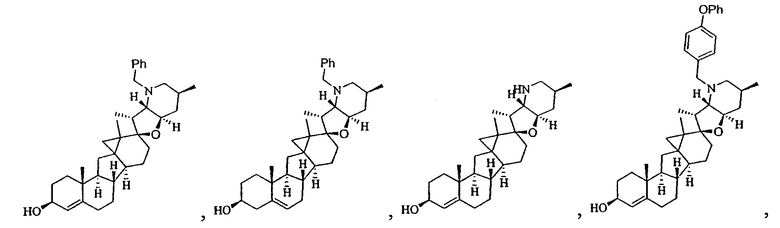

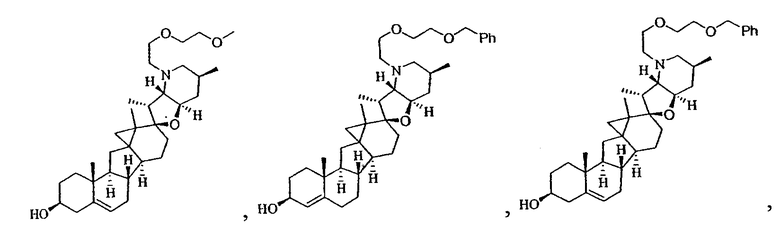
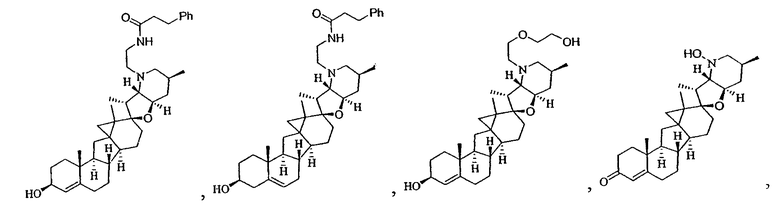
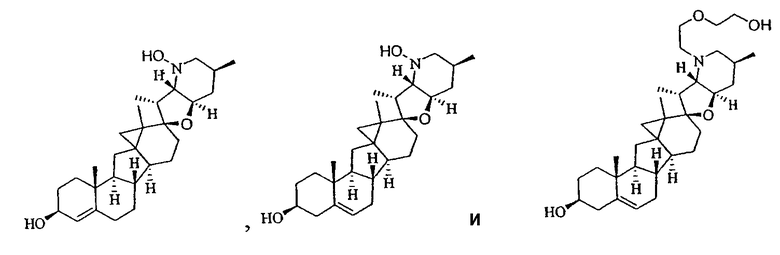
В некоторых вариантах осуществления соединения настоящего изобретения представлены любым из вышеупомянутых соединений и сопутствующими определениями, где соединение представлено формулой:
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, содержащей любые одно или несколько из вышеупомянутых соединений; и фармацевтически приемлемый наполнитель.
Синтез стероидных алкалоидных соединений
Циклопропильные стероидные алкалоидные производные настоящего изобретения могут быть получены непосредственно из стероидного алкалоида, выделенного в виде природного продукта (или синтезированного) или N-защищенных форм этих соединений. Подходящие защитные группы для азота включают, но не ограничены, Fmoc, Alloc, Boc, Troc, трифторацетатом, тозилом, Cbz, этилцианидом и Bn.
Для циклопропанирования стероидного алкалоида могут применяться разнообразные циклопропанирующие средства. 1,1-Галогеналкилметаллические комплексы, включая реакционно-способные вещества, известные как карбеноиды, обычно применяемые для циклопропанирования олефинов. Эти реагенты типовым образом получают, применяя дийодалкан или диазоалкан и металлические или металлоорганические соединения, такие как Et2Zn, iBu3Al, самарий, медь, родий или палладий. В некоторых вариантах осуществления Et2Zn и дийодметан используют для воздействия на циклопропанирование. Могут также применяться другие известные способы циклопропанирования, такие как способы с применением илидов серы для взаимодействия с олефином, сопряженным с карбонилом для присоединения групп CH2 или СН-алкил или СН-арил, и катализируемое металлом разложение диазоалкильных и α-диазокарбонильных соединений, таких как диазометан и этилдиазоацетат: эти способы легко предоставляют циклопропаны, имеющие алкильные, арильные, алкоксикарбонил(-COOR) или ацильные заместители. Например, присоединение этилдиазопропионата (EtO2C-C(N2)-Me) к олефиновому соединению в органическом растворителе, содержащем металлический катализатор, такой как медь или палладий, приводит к образованию циклопропана, содержащего группу, представленную формулой 1b, где Z представляет С(R23)2, в которой один R23 представляет собой Me и другой R23 представляет собой COOEt.
Посредством тщательного выбора циклопропанирующего средства, селективность по положению может достигаться при циклопропанировании стероидных алкалоидов более чем одним олефином. Например, если дийодметан и Et2Zn применяют для циклопропанирования иервина в определенных условиях, только олефин, более богатый электронами, будет взаимодействовать.
Диастереоселективность циклопропанирования может контролироваться разными способами. Например, снижение температуры реакции циклопропанирования может приводить к более высокой диастереоселективности. Альтернативно, может применяться хиральное циклопропанирующее средство, которое может различать каждую диастереолицевую сторону стероидного алкалоида. Энантиоселективность при циклопропанировании может также достигаться использованием субстрат-направленных реакций (т.е. циклопропанированием аллильных спиртов с применением Et2Zn/CH2I2 реагентов).
Реакции циклопропанирования могут проводиться в апротонном растворителе, предпочтительно таком, в котором ингредиенты реакции по существу являются растворимыми. Подходящие растворители включают простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, диглим, трет-бутилметиловый эфир, тетрагидрофуран и т.п.; галогенированные растворители, такие как хлороформ, дихлорметан, дихлорэтан и т.п.; алифатические или ароматические углеводородные растворители, такие как бензол, ксилол, толуол, гексан, пентан и т.п.; сложные эфиры и кетоны, такие как этилацетат, ацетон и 2-бутанон; полярные апротонные растворители, такие как ацетонитрил, диметилсульфоксид, диметилформамид и т.п.; или сочетания двух или более растворителей. В предпочтительном варианте осуществления дихлорметан является растворителем, используемым для циклопропанирования, когда применяют диалкилцинк и дийодметан.
Вслед за синтезом циклопропанированной стероидной алкалоидной центральной части соединение может быть дериватизировано с использованием разнообразных реакций функционализации, известных в данной области. Представительные примеры включают реакции сочетания с палладием до алкенгалогенидов или арилгалогенидов, окисления, восстановления, реакции с нуклеофилами, реакции с электрофилами, реакции перициклизации, введение защитных групп, удаление защитных групп и т.п.
В присутствии кислот Льюиса или Бренстеда циклопропилциклопаминовые аналоги настоящего изобретения претерпевают неизвестную ранее перегруппировку и расширение цикла, что позволяет получить новые циклопаминовые аналоги, в которых кольцо D увеличивается на один атом углерода. Циклопропильное кольцо может быть замещенным или незамещенным. В тех случаях, где циклопропильное кольцо является замещенным, группы, присоединенные к метилену циклопропана, будут встраиваться в кольцо D после перегруппировки и расширения кольца. Подходящие кислоты включают, но не ограничены, ZnI2, BF3, метансульфоновую кислоту, диарилоксифосфорные кислоты и HCl. В предпочтительном варианте осуществления изобретения используемая кислота Льюиса представляет собой BF3. Эти гомологенизированные аналоги могут быть далее функционализированы с использованием разнообразных реакций функционализации, известных в данной области. Представительные примеры включают реакции сочетания с палладием до алкенгалогенидов или арилгалогенидов, окисления, восстановления, реакции с нуклеофилами, реакции с электрофилами, реакции перициклизации, введение защитных групп, удаление защитных групп и т.п.
Способы изобретения
Настоящее изобретение далее предоставляет способы лечения, улучшения одного или более симптомов и уменьшения тяжести гиперпролиферативных расстройств, например рака, также как других расстройств и состояний, опосредованных через Hedgehog путь.
В настоящее время антагонисты Hedgehog пути исследуются в большом количестве клинических условий, где терапевтический эффект может быть получен для состояния или расстройства, замедляя один или более аспектов активности Hedgehog пути. Хотя основное внимание исследований было направлено на рак, исследователи нашли, что ингибирование пути Hedgehog низкомолекулярными соединениями улучшает симптомы псориаза (Tas, et al., 2004 Dermatology 209: 126-131, опубликовано как US 20040072913 (включенный сюда в качестве ссылки)). Псориаз представляет собой очень распространенное, хроническое кожное расстройство, обычно характеризующееся поражениями кожи, обычно включающее эритематозные папулы и пятна с серебристой чешуйкой, хотя имеются изменения и на коже, и в других частях тела. В настоящее время полагают, что псориаз представляет собой аутоиммунное заболевание, но его этиология все еще плохо изучена. В первой стадии местное нанесение циклопамина на псориатические поражения приводило к полной или частичной регрессии поражения с уменьшением воспалительных клеток (Tas и др., выше).
Антагонисты Hedgehog пути настоящего изобретения могут использоваться для лечения или профилактики псориаза, когда они применяются как единственное средство или когда применяются в комбинации с одним или более другими противопсориатическими средствами, включая, но не ограничиваясь данными, кортикостероиды, деготь, кальципотриен, тазаротен, ингибиторы кальциневрина, облучение ультрафиолетовыми лучами, метотрексат, ретиноиды, циклоспорин, иммуномодулирующие препараты, этанерсепт, алефасепт, эфализумаб и инфликсимаб.
Показано, что многие опухоли и пролиферативные состояния зависят от пути Hedgehog. На рост и выживание таких клеток можно повлиять лечением соединениями настоящего изобретения. Например, показано ингибирование Hedgehog пути низкомолекулярными соединениями для замедления роста базально-клеточного рака (Williams, и др., 2003 PNAS 100: 4616-21), медуллобластомы (Berman et al., 2002 Science 297: 1559-61), рака поджелудочной железы (Berman et al., 2003 Nature 425: 846-51), рака желудка (Berman et al., 2003 Nature 425: 846-51, опубликованная заявка PCT WO 05/013800), рака пищевода (Berman et al., 2003 Nature 425: 846-51), рака легкого (Watkins et al., 2003. Nature 422: 313-7) и рака простаты (Karhadkar et al., 2004. Nature 431: 707-12).
Кроме того, было показано, что многие типы рака имеют неконтролируемую активацию Hedgehog пути, например рак молочной железы (Kubo et al., 2004. Cancer Research 64: 6071-4), гепатоцеллюлярный рак (Patil et al., 2005. 96 th Annual AACR conference, abstract #2942 Sicklick et al., 2005. ASCO annual meeting, abstract #9610), гематологические злокачественные заболевания (Watkins and Matsui, неопубликованные результаты), базально-клеточная карцинома (Bale & Yu, 2001. Human Molec. Genet. 10: 757-762 Xie et al., 1998 Nature 391: 90-92), медуллобластома (Pietsch et al., 1997. Cancer Res. 57: 2085-88) и рак желудка (Ma et al., 2005 Carcinogenesis May 19, 2005 (Epub)).
Рак или неопластические заболевания и относящиеся к ним расстройства, которые можно лечить введением соединений и композиций настоящего изобретения, включают, но не ограничиваются данными, рак коры надпочечников, рак анального канала, апластическая анемия, рак желчного протока, рак мочевого пузыря, рак кости, опухоли мозга/ЦНС, рак молочной железы, рак шейки матки, не-Ходжкинская лимфома, рак толстой кишки, рак прямой кишки, рак эндометрия, рак пищевода, опухоли семейства Юинга, рак глаза, рак желчного пузыря, желудочно-кишечные карциноидные опухоли, желудочно-кишечные стромальные опухоли, гестационная трофобластическая болезнь, болезнь Ходжкина, саркома Капоши, рак почки, рак гортани и подглоточный рак, острый лимфоцитарный лейкоз, острая миелоидная лейкемия, детская лейкемия, хронический лимфоцитарный лейкоз, хроническая миелоидная лейкемия, рак печени, рак легкого, карциноидная опухоль легкого, не-Ходжскинская лимфома, мужской рак молочной железы, злокачественная мезотелиома, множественная миелома, миелодиспластический синдром, рак полости носа и параназальной области, рак носоглотки, нейробластома, рак полости рта, рак ротоглотки, остеосаркома, рак яичника, рак поджелудочной железы, рак пениса, опухоль гипофиза, рак простаты, ретинобластома, рабдомиосаркома, рак слюнной железы, саркома, меланома, рак кожи, не относящийся к меланомам, рак желудка, рак тестикул, рак тимуса, рак щитовидной железы, саркома матки, рак влагалища, рак вульвы, макроглобулинемия Вальденстрема и опухоль Вилмса.
Способы и композиции настоящего изобретения могут использоваться при лечении рака у человека, например базально-клеточных раков и других опухолей эпителиальных тканей, таких как кожа. Дополнительно, соединения настоящего изобретения могут использоваться как часть лечения базально-клеточной карциномы, рака поджелудочной железы, рака простаты, саркомы, лимфом, лейкоза, рака желудка, рака желудочно-кишечного тракта, множественной миеломы, мелкоклеточного рака легкого, глиомы, рака молочной железы, гепатоцеллюлярного рака или медуллобластомы посредством применения терапевтически эффективного количества по меньшей мере одного из соединений настоящего изобретения как единственного средства или в комбинации с другим противораковым средством.
Способы и композиции настоящего изобретения могут использоваться при лечении неопластических или гиперпластических трансформаций, таких как те, которые могут встречаться в центральной нервной системе. Например, соединения настоящего изобретения могут использоваться, чтобы заставить такие трансформированные клетки становиться или постмитотическими, или апоптотическими. Предоставляемый способ может поэтому использоваться как часть лечения, например, злокачественных глиом, менингиом, медуллобластом, нейроэктодермальных опухолей и эпендимомы.
В одном варианте осуществления существующий метод может использоваться как часть режима лечения злокачественной медуллобластомы и других первичных злокачественных нейроэктодермальных опухолей ЦНС.
В некоторых вариантах осуществления настоящее изобретение относится к способу лечения рака, включающему введение пациенту, если он нуждается в этом, терапевтически эффективного количества одного или более какого-либо из вышеупомянутых соединений.
В некоторых вариантах осуществления настоящее изобретение относится к способу лечения рака, включающему введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения какого-либо одного или более вышеупомянутых соединений, в тех случаях, когда рак локализуется в голове пациентов, шее, полости носа, параназальных пазухах, носоглотке, полости рта, ротоглотке, гортани, гортаноглотке, слюнных железах, параганглионарно, в поджелудочной железе, желудке, коже, пищеводе, печени и желчном дереве, кости, тонкой кишке, толстой кишке, прямой кишке, яичниках, простате, легком, молочной железе, лимфатической системе, крови, центральной нервной системе, костном мозге или головном мозге.
В некоторых вариантах осуществления настоящее изобретение относится к способу лечения рака, включающему введение нуждающемуся в этом пациенту терапевтически эффективного количества соединения какого-либо одного или более вышеупомянутых соединений, в тех случаях, когда рак представляет собой базально-клеточный рак, рак поджелудочной железы, рак простаты, саркому, лимфому, лейкоз, рак желудка, рак пищевода, рак желчных протоков, рак толстой кишки, множественную миелому, мелкоклеточный рак легкого, глиому, рак молочной железы, гепатоцеллюлярный рак или медуллобластому.
В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу, где соединение применяется в комбинации с лучевой терапией или другим противораковым химиотерапевтическим средством.
В некоторых вариантах осуществления настоящее изобретение относится к любому вышеупомянутому способу, где соединение применяется локально к опухоли или системно к организму пациента.
В некоторых вариантах осуществления настоящее изобретение относится к любому вышеупомянутому способу, где способ введения указанного соединения представляет собой ингаляцию, пероральный, внутривенный, сублингвальный, глазной, чрескожный, ректальный, влагалищный, местный, внутримышечный, внутриартериальный, интратекальный, подкожный, щечный или интраназальный.
В некоторых вариантах осуществления настоящее изобретение относится к любому вышеупомянутому способу, где способ введения является пероральным, внутривенным или местным.
В некоторых вариантах осуществления настоящее изобретение относится к способу антагонистического воздействия на Hedgehog путь в клетке, включающему приведение в контакт клетки, экспрессирующей Smoothened, с эффективным количеством какого-либо одного или более вышеупомянутых соединений.
В некоторых вариантах осуществления настоящее изобретение относится к способу антагонистического воздействия на Hedgehog путь в клетке, включающему приведение в контакт клетки экспрессирующей Smoothened, с эффективным количеством какого-либо одного или более вышеупомянутых соединений, где указанная клетка, экспрессирующая Smoothened, контактирует с указанным соединением in vitro.
В некоторых вариантах осуществления настоящее изобретение относится к способу антагонистического воздействия на Hedgehog путь в клетке, включающему приведение в контакт клетки, экспрессирующей Smoothened, с эффективным количеством какого-либо одного или более вышеупомянутых соединений, где указанная клетка, экспрессирующая Smoothened, контактирует с указанным соединением in vivo.
В некоторых вариантах осуществления настоящее изобретение относится к способу антагонистического воздействия на Hedgehog путь в клетке, включающему приведение в контакт клетки, экспрессирующей Smoothened, с эффективным количеством какого-либо одного или более вышеупомянутых соединений, где указанная клетка, экспрессирующая Smo, контактирует с указанным соединением в пределах тела пациента.
В некоторых вариантах осуществления настоящее изобретение относится к способу лечения или профилактики псориаза у пациента, включая введение нуждающемуся в этом пациенту терапевтически эффективного количества какого-либо одного или более вышеупомянутых соединений.
В некоторых вариантах осуществления настоящее изобретение относится к вышеупомянутому способу лечения или профилактики псориаза, где способ введения указанного состава является местным.
В некоторых вариантах осуществления одно или более соединений настоящего изобретения применяются для лечения и профилактики псориаза в комбинации с одним или более противопсориатическими средствами, включая, но не ограничиваясь данными, кортикостероиды, деготь, кальципотриен, тазаротен, ингибиторы кальциневрина, облучение ультрафиолетовыми лучами, метотрексат, ретиноиды, циклоспорин, иммуномодулирующие лекарственные средства, этанерсепт, алефасепт, эфализумаб и инфликсимаб.
Лечение рака в комбинации с химиотерапией или лучевой терапией
В некоторых вариантах осуществления одно или более соединений настоящего изобретения применяются для лечения или профилактики рака или неопластического заболевания в комбинации с одним или более противораковыми химиотерапевтическими средствами, включая, но не ограничиваясь данными, гемцитабин, метотрексат, таксол, меркаптопурин, тиогуанин, гидроксимочевина, цитарабин, циклофосфамид, ифосфамид, нитрозомочевины, цисплатин, карбоплатин, митомицин, дакарбазин, прокарбизин, этопозид, преднизолон, дексаметазон, цитарбин, кампатекин, блеомицин, доксорубицин, идарубицин, даунорубицин, дактиномицин, пликамицин, митоксантрон, аспарагиназу, винбластин, винкристин, винорелбин, паклитаксел и доцетаксел. В предпочтительном воплощении одно или более соединений настоящего изобретения применяются для лечения и профилактики рака или неопластического заболевания в комбинации с одним или более химиотерапевтическими или другими противораковыми средствами, включая, но не ограничиваясь данными, излучение (например, гамма-излучение), азотистый иприт (например, циклофосфамид, ифосфамид, трофосфамид, хлорамбуцил, эстрамустин и мелфалан), нитрозомочевины (например, кармустин (BCNU) и ломустин (CCNU)), алкилсульфонаты (например, бусульфан и треосульфан), триазены (например, дакарбазин и темозоломид), соединения платины, (например, цисплатин, карбоплатин и оксалиплатин), алкалоиды барвинка (например, винкристин, винбластин, виндезин и винорелбин), таксоиды (например, паклитаксел и доцетаксол), эпинодофиллины (например, этопозид, тенипозид, топотекан 9-аминокамптотецин, камптоиринотекан, криснатол, митомицин С и митомицин С), антиметаболиты, ингибиторы DHFR (например, метотрексат и триметрексат), ингибиторы IMP дегидрогеназы (например, mycophenolic, тиазофурин, рибавирин и EICAR), ингибиторы рибонуклеотидредуктазы (например, гидроксимочевина и дефероксамин), аналоги урацила (например, фторурацил, флоксуридин, доксифлуридин, ратитрексед и капецитабин), аналоги цитозина (например, цитарабин (ара С), арабинозид цитозина и флударабин), аналоги пурина (например, меркаптопурин и тиогуанин), антиэстрогены (например, тамоксифен, ралоксифен и мегестрол), агонисты LHRH (например, госерелин и ацетат лейпролида), антиандрогены (например, флутамид и бикалутамид), аналоги витамина D3 (например, EB 1089, CB 1093 и KH 1060), фотодинамическая терапия (например, вертопорфирин (BPD-MA), фталоцианин, фотосенсибилизатор Pc4 и деметоксигипокреллин А (2BA-2-DMHA)), цитокины (например, α-интерферон, γ-интерферон и фактор некроза опухоли), ингибиторы изопренилирования (например, ловастатин), допаминергические нейротоксины (например, ион 1-метил-4-фенилпиридина), ингибиторы клеточного цикла (например, стауроспорин), актиномицины (например, актиномицин D и дактиномицин), блеомицины (например, блеомицин A2, блеомицин B2 и пепломицин), антрациклины (например, даунорубицин, доксорубицин (адриамицин), идарубицин, эпирубицин, пирарубицин, зорубицин и митоксантрон), ингибиторы MDR (например, верапамил), ингибиторы Ca2+-АТФ-азы (например, тапсигаргин), антитела (например, авастин, эрбитукс, ритуксан и бекссар), кортикостероиды (например, преднилон, предизон и т.д.), иматиниб, талидомид, леналидомид, бортезомиб, гемцитабин, эрлотиниб, гефитиниб, сорафениб и сутиниб.
Химиотерапевтическое средство и/или лучевая терапия могут вводиться согласно терапевтическим протоколам, известным в данной области техники. Специалистам в данной области техники будет очевидно, что введение химиотерапевтического средства и/или лучевой терапии может варьироваться различно в зависимости от заболевания, против которого применяется лечение, и известных эффектов химиотерапевтического средства и/или лучевой терапии на данное заболевание. Кроме того, в соответствии со знаниями квалифицированного врача-клинициста, терапевтические протоколы (например, дозировки и время введения) могут быть различны ввиду наблюдаемых у пациента эффектов от применяемых терапевтических средств (т.е. антинеопластического средства или излучения) и ввиду наблюдаемых ответных реакций заболевания на вводимые терапевтические средства и наблюдаемых неблагоприятных эффектов.
Кроме того, в общем, соединения настоящего изобретения и химиотерапевтическое средство не должны вводиться в одной и той же фармацевтической композиции, и возможно, так как физические и химические характеристики различны, должны вводиться посредством различных путей. Например, соединения настоящего изобретения могут вводиться внутривенно для создания и поддержания хороших уровней в крови, в то время как химиотерапевтическое средство может вводиться перорально. Определение способа введения и целесообразности введения при возможности в одной и той же фармацевтической композиции находится в пределах знаний квалифицированного врача-клинициста. Первоначальное введение может производиться согласно установленным протоколам, известным в данной области техники, и затем квалифицированный врач-клиницист может изменять дозировку, способы введения и время введения, основываясь на наблюдаемых эффектах.
Предпочтительный выбор химиотерапевтического средства или вида излучения будет зависеть от диагноза врачей и их суждения о состоянии пациента и подходящем протоколе введения.
Соединение настоящего изобретения и химиотерапевтическое средство и/или излучение могут вводиться одновременно (например, одновременно, по существу одновременно или в пределах одного и того же протокола лечения) или последовательно, в зависимости от характера пролиферативного заболевания; состояния пациента и фактического выбора химиотерапевтического средства и/или излучения, которые вводятся совместно (т.е. в пределах единственного протокола введения) с соединением настоящего изобретения.
Если соединение настоящего изобретения и химиотерапевтическое средство и/или излучение не вводятся одновременно или по существу одновременно, то оптимальный порядок введения соединения настоящего изобретения и химиотерапевтического средства и/или излучения может быть различным для различных опухолей. Таким образом, в определенных ситуациях соединение настоящего изобретения может вводиться вначале, сопровождаемое назначением химиотерапевтического средства и/или излучения; и в других ситуациях химиотерапевтическое средство и/или излучение могут вводиться вначале, сопровождаемые назначением соединения настоящего изобретения. Данное дополнительное назначение может быть повторено в течение одного протокола лечения. Определение порядка введения и количества повторений введения каждого терапевтического средства в течение протокола лечения полностью находится в пределах знаний квалифицированного врача после оценки болезни, подвергаемой лечению, и состояния пациента. Например, химиотерапевтическое средство и/или излучение могут вводиться вначале, особенно если это цитотоксическое средство и затем лечение продолжаются с введением соединения настоящего изобретения, сопровождаясь, при уставленной полезности, введением химиотерапевтического средства и/или излучения, и так далее, пока протокол лечения не закончится.
Таким образом, в соответствии с опытом и знаниями практикующий врач может в продолжение лечения изменять каждый протокол введения компонента (терапевтическое средство, например, соединение настоящего изобретения, химиотерапевтическое средство или излучение) для лечения согласно потребностям индивидуального пациента.
Фармацевтические композиции
В еще одном варианте осуществления настоящее изобретение предоставляет фармацевтически приемлемые композиции, которые включают терапевтически эффективное количество одного или более соединений, описанных выше (формула 1 и 2), сформированные вместе с одним или более фармацевтически приемлемыми носителями и/или растворителями. Фармацевтические композиции настоящего изобретения могут быть специально сформированы для введения в твердой или жидкой форме, включая адаптированные для следующего: (1) пероральное введение, например, микстуры (водные или неводные растворы или суспензии), таблетки, например, предназначенные для буккального, сублингвального и системного употребления, пилюли, порошки, гранулы, пасты для приклеивания к языку; (2) парентеральное введение, например, посредством подкожной, внутримышечной, внутривенной или эпидуральной инъекции, как, например, стерильный раствор или суспензия, или состав для непрерывного введения; (3) местное применение, например, как крем, мазь или пластырь с контролируемым высвобождением или распыляемый раствор, применяемые к коже; (4) внутривлагалищно или ректально, например, как маточное кольцо, крем или пена; (5) сублингвально; (6) через глаза; (7) чрескожно; (8) через легкие или (9) через нос. Как изложено выше, определенные воплощения существующих соединений могут содержать основную функциональную группу, такую как амино или алкиламино, и, таким образом, способны к формированию фармацевтически приемлемых солей с фармацевтически приемлемыми кислотами. Термин "фармацевтически приемлемые соли" в этом отношении относится к относительно нетоксичным солям соединений настоящего изобретения с неорганической и органической кислотами. Данные соли могут приготовляться in situ в связующем веществе для введения или в виде дозированных форм в производственном процессе, или посредством раздельного реагирования очищенного соединения изобретения в его свободной основной форме с подходящей органической или неорганической кислотой, и выделения соли, которая формируется в течение последующей очистки. Типичные соли включают бромгидрат, хлоргидрат, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонат, и подобные (см., например, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19).
Фармацевтически приемлемые соли соединений настоящего изобретения включают общепринятые нетоксичные соли или четвертичные аммониевые соли соединений, например, нетоксичных органических или неорганических кислот. Например, такие общепринятые нетоксичные соли включают соли, являющиеся производными неорганических кислот, таких как хлористоводородной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной и т.п.; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая и т.п.
В других случаях соединения настоящего изобретения могут содержать одну или более кислотных функциональных групп и, таким образом, обладать способностью к образованию фармацевтически приемлемых солей с фармацевтически приемлемыми основаниями. Термин “фармацевтически приемлемые соли” в данных случаях относится к относительно нетоксичным, неорганическим и органическим основно-аддитивным солям соединений настоящего изобретения. Данные соли могут, вероятно, быть получены in situ в среде для введения или способе производства дозированной формы или посредством отдельного взаимодействия очищенного соединения в его свободной кислотной форме с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым ораническим первичным, вторичным или третичным амином. Представительные щелочные или щелочноземельные соли включают соли лития, натрия, калия, кальция, магния и алюминия и т.п. Представительные органические амины, применимые для образования основно-аддитивных солей, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и т.п. (см., например, Berge et al., supra).
В композициях могут также присутствовать увлажняющие средства, эмульгаторы и смазки, такие как лаурилсульфат натрия и стеарат магния, а также красители, средства высвобождения, покрывающие средства, подсластители, отдушки и ароматизаторы, консерванты и антиоксиданты.
Примеры фармацевтически приемлемых антиоксидантов включают (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п.; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (БГА), бутилированный гидрокситолуол (БГТ), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) металлхелатирующие средства, такие как лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТА), сорбит, винная кислота, фосфорная кислота и т.п.
Способы получения данных готовых форм или композиций включают стадию приведения в ассоциацию соединения настоящего изобретения с носителем и, необязательно, одним или более вспомогательных ингредиентов. В целом, готовые формы получают посредством однородного и непосредственного приведения в ассоциацию соединения настоящего изобретения с жидкими носителями или мелкоизмельченными твердыми носителями или обоими и далее, если необходимо, формования продукта.
Фармацевтические композиции настоящего изобретения, пригодные для парентерального введения, содержат одно или более соединений изобретения в сочетании с одним или более фармацевтически приемлемыми стерильными изотоническими водными или неводными растворами, дисперсиями, суспензиями или эмульсиями, или стерильными порошками, которые могут быть внедрены в стерильные инъецируемые растворы или дисперсии непосредственно перед применением, которые могут содержать сахара, спирты, антиоксиданты, буферы, бактериостатики, растворимые вещества, которые придают готовой форме изотонические свойства по отношению к крови предназначенного реципиента или суспендирующие, или сгущающие средства.
Примеры подходящих водных и неводных носителей, которые могут использоваться в фармацевтических композициях изобретения, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.) и их подходящие смеси, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Нужная текучесть может устанавливаться, например, посредством применения покрывающих материалов, таких как лецитин, посредством поддержания требуемого размера частиц в случае дисперсий и посредством использования поверхностно-активных частиц.
Данные композиции могут также содержать адъюванты, такие как консерванты, увлажнители, эмульгирующие и диспергирующие средства. Предотвращение действия микроорганизмов на соединения настоящего изобретения может быть обеспечено посредством включения различных антибактериальных и противогрибковых средств, например, парабена, хлорбутанола, фенолсорбиновой кислоты и т.п. Также может быть желательным включать в композиции изотонические средства, такие как сахара, хлорид натрия и т.п. Дополнительно, пролонгированное всасывание инъецируемой фармацевтической формы может быть придано посредством включения средств, которые замедляют всасывание, таких как моностеарат алюминия и желатин.
В некоторых случаях для пролонгирования эффекта лекарственного средства желательно замедлить всасывание лекарственного средства в результате подкожной или внутримышечной инъекции. Это может достигаться посредством применения жидкой суспензии кристаллического или аморфного вещества, имеющего плохую растворимость в воде. Степень всасывания лекарственного средства тогда зависит от его степени растворения, которая в свою очередь может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленное всасывание лекарственной формы для парентерального введения достигается посредством растворения или суспендирования лекарственного средства в масляной среде.
Инъецируемые депонируемые формы получают посредством образования микроинкапсулированных матриц соединений настоящего изобретения в биодеградируемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и природы конкретного используемого полимера, скорость высвобождения лекарственного средства может контролироваться. Примеры других биодеградируемых полимеров включают поли(ортосложные эфиры) и поли(ангидриды). Депонируемые инъецируемые готовые формы также получают внедрением лекарства в липосомы или микроэмульсии, которые являются совместимыми с тканями организма.
Готовые композиции изобретения, пригодные для перорального введения, могут находиться в виде капсул, каше, пилюль, таблеток, пастилок (с использованием ароматизированной основы, обычно сахарозы и акации или трагаканта), порошков, гранул или в виде раствора или суспензии в водной или неводной жидкости, или в виде жидкой эмульсии масло-в-воде или вода-в-масле, или в виде эликсира или сиропа, или в виде пастил (с использованием инертной основы, такой как желатин и глицерин или сахарозы и акации), и/или в виде полосканий для рта и т.п., причем каждая из них содержит предварительно определенное количество соединения настоящего изобретения в виде активного ингредиента. Соединение настоящего изобретения может также вводиться в виде болюса, электуария или пасты.
Когда соединения настоящего изобретения вводят в виде фармацевтических средств людям и животным, их могут вводить как таковые или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99% (более предпочтительно, от 10 до 30%) активного ингредиента в сочетании с фармацевтически приемлемым носителем.
Эти соединения могут вводиться людям или другим животным для терапии посредством любого пригодного пути введения, включая пероральный, назальный, или посредством, например, спрея, ректально, интравагинально, парентерально, интрацистернально или местно, или посредством порошков, мазей или капель, включая буккальный и подъязычный пути.
Независимо от выбранного пути введения соединения настоящего изобретения, которые могут применяться в подходящей гидратированной форме, и/или фармацевтические композиции настоящего изобретения получают в виде фармацевтически приемлемых дозированных форм общепринятыми способами, известными специалистам в данной области.
Действительные уровни дозировки активных ингредиентов в фармацевтических композициях настоящего изобретения могут изменяться таким образом, чтобы получить количество активного ингредиента, которое является эффективным для достижения требуемого терапевтического эффекта для конкретного пациента, композиции и способа введения, без токсичности для пациента.
Выбранный уровень дозировки будет зависеть от множества факторов, включая активность используемого конкретного соединения настоящего изобретения или его сложного эфира, соли или амида, пути введения, времени введения, скорости экскреции или метаболизма используемого конкретного соединения, скорости и степени всасывания, продолжительности лечения, других лекарств, соединений и/или веществ, применяемых в сочетании с конкретным используемым соединением, возраста, пола, массы, состояния, общего состояния здоровья и предшествующей истории болезни пациента, подлежащего лечению, и подобных факторов, хорошо известных в области медицины.
Терапевт или ветеринар, являющиеся рядовыми специалистами в данной области, могут легко определить и предписать требуемое эффективное количество фармацевтической композиции. Например, терапевт или ветеринар могут начать с доз соединений изобретения, используемых в фармацевтической композиции на уровнях, меньших, чем требуется, для достижения желательного терапевтического эффекта и постепенно увеличивать дозировку, пока не будет достигнут желательный эффект.
В целом, подходящая суточная доза соединения изобретения будет составлять такое количество соединения, которое является наименьшей дозой, эффективной для получения терапевтического эффекта. Такая эффективная доза будет, в целом, зависеть от факторов, описанных выше. В общем случае, пероральные, внутривенные, интрацеребровентрикулярные и подкожные дозы соединений настоящего изобретения для пациента при использовании для указанных анальгетических эффектов будут находиться в интервале приблизительно от 0,0001 до 100 мг на килограмм массы тела в день.
По желанию, эффективная суточная доза активного соединения может вводиться в виде двух, трех, четырех, пяти, шести или более субдоз, вводимых раздельно в соответствующие интервалы в течение дня, необязательно, в разовых дозированных формах. Предпочтительной дозировкой является одно введение в день.
В то время как возможно введение только одного соединения настоящего изобретения, предпочтительно вводить соединения в виде фармацевтической готовой формы (композиции).
Пациент, получающий данное лечение, представляет собой любое животное, нуждающееся в этом лечении, включая приматов, особенно людей, и других млекопитающих, таких как лошади, крупный рогатый скот, свиньи и овцы; в общем случае, домашнюю птицу и домашних животных.
Соединения изобретения могут вводиться как таковые или в смесях с фармацевтически приемлемыми носителями и могут также вводиться в соединении с противомикробными средствами, такими как пенициллины, цефалоспорины, аминогликозиды и гликопептиды. Сочетанная терапия, таким образом, включает последовательное, одновременное и раздельное введение активного соединения таким образом, что терапевтические эффекты первого вводимого соединения не исчезают полностью, когда вводят последующее.
Иллюстрирование примерами
Теперь, когда изобретение в целом описано, оно будет более легко понятно со ссылкой на следующие примеры, которые включены просто с целью иллюстрации некоторых аспектов настоящего изобретения и подразумевают, что они не ограничивают изобретение.
Пример 1
Получение производного циклопамина
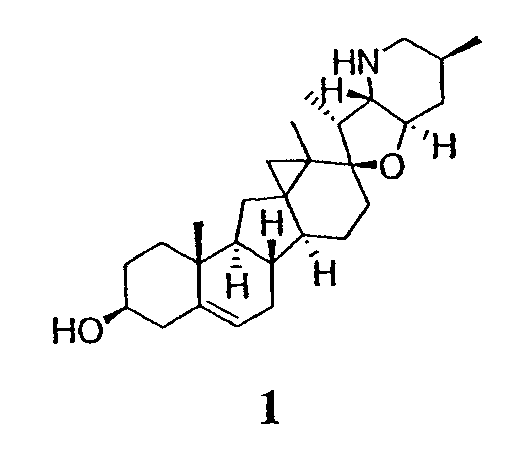
Стадия А
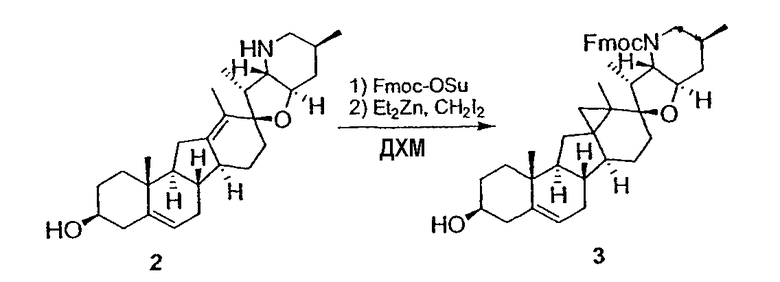
К раствору циклопамина 2 (250 мг, 0,6 ммоль, 1 экв.) в ДХМ при комнатной температуре добавляют Fmoc-OSu (205 мг, 0,6 ммоль, 1 экв.) и полученную в результате смесь перемешивают при комнатной температуре в течение ночи. Полученный в результате раствор неочищенного Fmoc-циклопамина далее охлаждают до 0°С и обрабатывают 15% диэтилцинком в толуоле (0,5 мл, 0,6 ммоль, 1 экв.) и перемешивают в течение 30 мин (колба А).
Дийодметан (0,4 мл, 6 ммоль, 10 экв.) в ДХМ (20 мл) при 0°С обрабатывают 15% диэтилцинком в толуоле (3 мл, 3 ммоль, 5 экв.) и полученный в результате раствор перемешивают в течение 5 мин (колба В).
Содержимое колбы В переносят в колбу А через канюлю и полученную в результате суспензию перемешивают в течение 5 ч при комнатной температуре. Реакцию гасят HCl (1 М), перемешивают в течение 10 мин (до тех пор, пока не растворится все белое вещество) и экстрагируют ДХМ (5×). Органические экстракты сушат (MgSO4), фильтруют через целит и концентрируют в вакууме. Остаток очищают флэш-хроматографией (1:1 гексан/AcOEt). Целевой 11,12-моноциклопропан получают в виде 9:1 смеси диастереоизомеров, наряду с 20% диастереомерными бис-циклопропанированными продуктами (80% общего извлечения). Данную смесь разделяют, используя препаративную SFC хроматографию.
Стадия В

Fmoc-моноциклопропилциклопамин 3 (100 мг, 0,15 ммоль, 1 экв.) в ДХМ (2 мл) при комнатной температуре обрабатывают диэтиламином (0,5 мл, 4,8 ммоль, 32 экв.) в течение ночи, полученный в результате раствор концентрируют в вакууме и остаток абсорбируют на силикагеле и очищают флэш-хроматографией (2:1→1:1 гексан/AcOEt и далее 95:5→90:10→20:80 ДХМ/МеОН). Требуемый продукт получают в виде белого твердого вещества (95% выход). МС (ESI(+)) m/e 426,31 (M+H)+.
Пример 2
Получение производного циклопамина
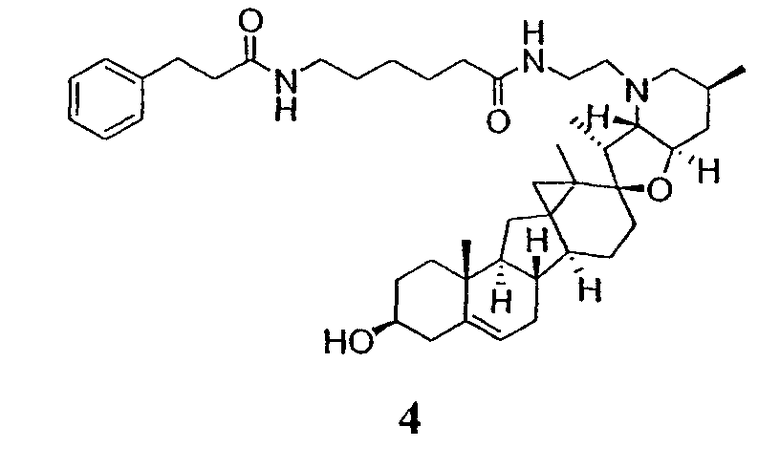
Стадия А
К раствору гидрокоричной кислоты 5 (3,01 г, 20 ммоль, 1 экв.) в безводном хлороформе (30 мл) при 75°С добавляют тионилхлорид (1,75 мл, 24,1 ммоль, 1,2 экв.) по каплям в течение периода 3 мин. Смесь кипятят с обратным холодильником в течение 3,5 ч. Растворитель отгоняют с получением неочищенного хлорангидрида кислоты в виде светло-желтой вязкой жидкости. Неочищенное вещество используют без дальнейшей очистки.
Стадия В
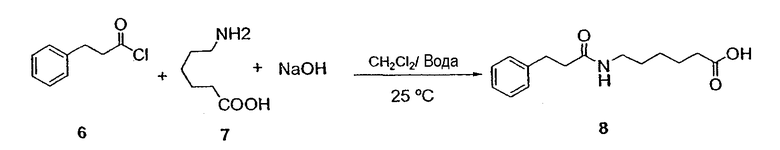
К бифазной смеси 7 (3,16 г, 24,1 ммоль, 1,2 экв.) в ДХМ (30 мл) и водного раствора NaOH (2,0 М, 30 мл, 3 экв.) при 25°С добавляют раствор хлорангидрида кислоты 6 (3,38 г, 20 ммоль, 1 экв.) в ДХМ (10 мл) и полученную в результате смесь перемешивают при 25°С в течение 3 ч. Смесь далее нейтрализуют водным раствором HCl (2 M, 30 мл). Органический слой далее отделяют и водный слой экстрагируют ДХМ (3×50 мл). Объединенные органические слои промывают HCl (2,0 М, 25 мл), водой (3×50 мл), насыщенным солевым раствором (50 мл), сушат над сульфатом магния и раствор упаривают при пониженном давлении. Неочищенное вещество хроматографируют на силикагеле, используя 5% МеОН:ДХМ в качестве элюента и колонку далее элюируют 10% МеОН:ДХМ с получением 1,141 г соединения 8.
Стадия С
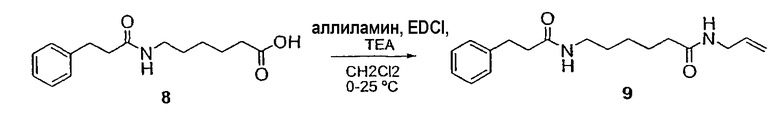
К смеси кислоты 8 (264 мг, 1 ммоль, 1 экв.), EDCI (231 мг, 1,2 ммоль, 1,2 экв.) и триэтиламина (168 мкл, 1,2 ммоль, 1,2 экв.) в ДХМ (2 мл) при 0°С добавляют аллиламин (90,3 мкл, 1,2 ммоль, 1,2 экв.), полученную в результате смесь перемешивают при 0°С и дают ей нагреться до 25°С в течение периода, равного 2 ч. Реакционную смесь добавляют к воде (50 мл), экстрагируют ДХМ (4×25 мл), объединенные органические слои промывают 1М HCl (2×25 мл), водой (3×25 мл), насыщенным солевым раствором (25 мл), сушат над сульфатом магния и растворитель упаривают при пониженном давлении с получением 287,5 мг требуемого продукта. Данное вещество используют без дальнейшей очистки.
Стадия D
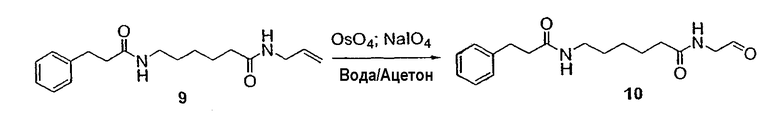
К раствору аллиламида 9 (70 мг, 0,23 ммоль, 1 экв.) в ацетоне (1 мл) и воде (0,3 мл) добавляют раствор тетроксида осмия (0,35 мл, 0,035 ммоль, 0,15 экв., 2,5 мас./мас. в трет-бутаноле). Реакционную смесь немедленно охлаждают на льду вскоре после добавления раствора OsO4 и полученный в результате темно-коричневый раствор перемешивают при 0°С в течение 15 мин. Перйодат натрия (110 мг, 0,51 ммоль, 2,2 экв.) добавляют 5 порциями к вышеуказанной смеси и перемешивание продолжают в течение 1 ч при 0°С, и смеси дают нагреться до 25°С в течение периода, равного 2 ч. Реакционную смесь разбавляют ДХМ (3 мл) фильтруют через небольшой слой сульфата магния и осадок на фильтре промывают ДХМ (4×3 мл). Фильтрат концентрируют и остаток (67,9 мг) фильтруют через небольшой слой RP силикагеля, используя 5% МеОН:ДХМ с получением 38,9 мг требуемого продукта.
Стадия Е
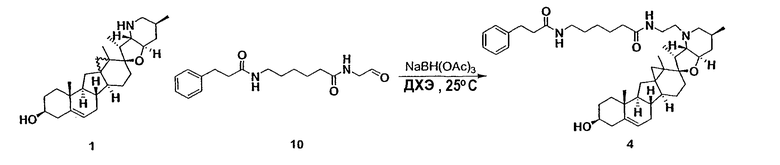
К суспензии 1 (10 мг, 0,023 ммоль, 1 экв.) в ацетонитриле (2 мл) добавляют раствор альдегида 10 (17 мг, 0,056 ммоль, 2,4 экв.) в ацетонитриле (0,3 мл) с последующим добавлением триацетоксиборгидрида натрия (6,5 мг, 0,031 ммоль, 1,3 экв.) и реакционную смесь перемешивают при 25°С в течение 16 ч. Растворитель далее упаривают при пониженном давлении и остаток хроматографируют на силикагеле (7 см × 10 мм), используя 3% метанол:ДХМ с получением 24,6 мг неочищенного вещества. Это вещество повторно подвергают колоночной хроматографии на силикагеле, используя 2% метанол:ДХМ и извлекают 18,2 мг неочищенного продукта, который далее очищают препаративной ТСХ, используя 3% метанол:ДХМ в качестве проявляющего растворителя (2 прохода) с получением 6,3 мг требуемого продукта. МС (ESI(+)) m/e 714,4 (M+H)+.
Пример 3
Получение производного циклопамина
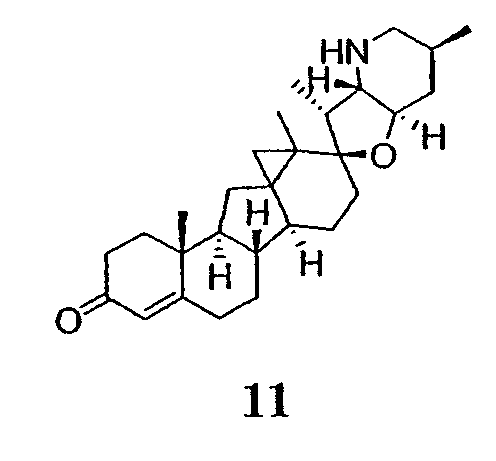
Стадия А
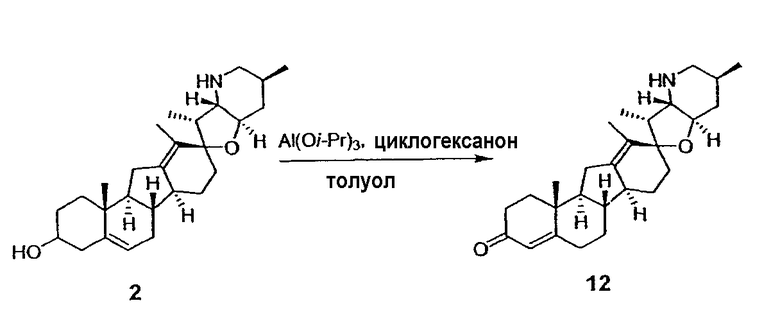
Циклопамин 2 (20 мг, 0,049 ммоль, 1 экв.) суспендируют в сухом толуоле (0,6 мл) и добавляют циклогексанон (150 мкл, 1,47 ммоль, 30 экв.) с последующим добавлением изопропоксида алюминия (79 мг, 0,392 ммоль, 8 экв.). Полученную в результате смесь нагревают при кипячении с обратным холодильником в течение 2 ч, охлаждают до комнатной температуры, разбавляют AcOEt и гасят солевым раствором Рочеля. Бифазную смесь перемешивают в течение ночи, слои разделяют, водный слой экстрагируют AcOEt и объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (ДХМ, ДХМ/МеОН 98:2 и 95:5). Целевое вещество получают в виде белого кристаллического твердого вещества (70% выход).
Стадия В
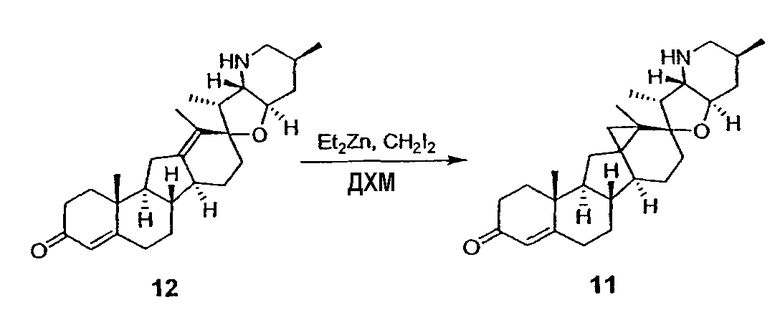
Дийодметан (40 мкл, 0,5 ммоль, 25 экв.) в ДХМ (0,52 мл) при 0°С обрабатывают 15% диэтилцинком в толуоле (0,2 мл, 0,2 ммоль, 10 экв.) и полученный в результате раствор перемешивают в течение 5 мин. Добавляют соединение 12 (10 мг, 0,02 ммоль, 1 экв.) в ДХМ (0,35 мл) и полученную в результате смесь перемешивают при комнатной температуре (при удалении бани со льдом) в течение 3 ч, гасят 2 н. NaOH и перемешивают в течение 10 мин, слои разделяют и водный слой экстрагируют ДХМ (2×). Органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (ДХМ/МеОН 92:8). Циклопропанированное вещество получают в виде белого твердого вещества. МС (ESI(+)) m/e 424,5 (M+H)+.
Пример 4
Получение производного циклопамина
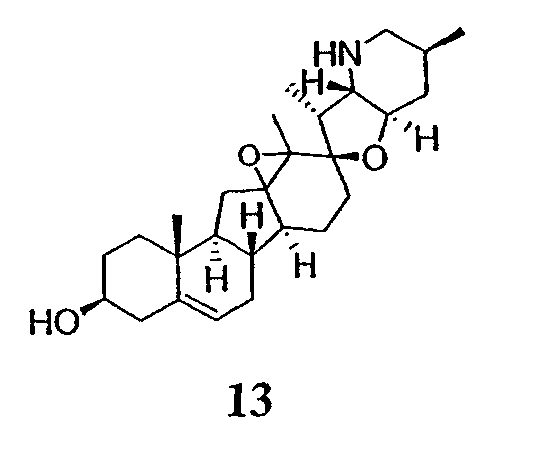
Стадия А
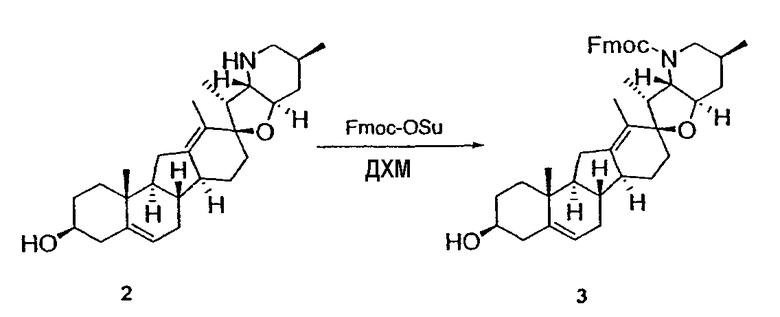
К раствору циклопамина 2 (250 мг, 0,6 ммоль, 1 экв.) в ДХМ (10 мл) при комнатной температуре добавляют Fmoc-OSu (205 мг, 0,6 ммоль, 1 экв.), полученную в результате смесь перемешивают при комнатной температуре в течение ночи и концентрируют в вакууме. Неочищенный Fmoc-циклопамин получают в виде не совсем белой пены.
Стадия В
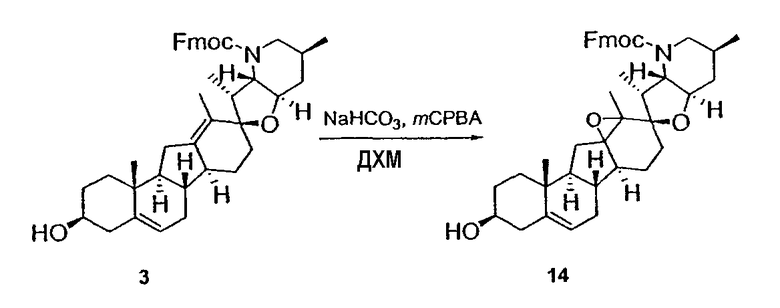
Раствор неочищенного Fmoc-циклопамина 3 (15 мг, 0,024 ммоль, 1 экв.) в ДХМ (0,5 мл) охлаждают до -78°С и обрабатывают гидрокарбонатом натрия (4 мг, 0,047 ммоль, 1,96 экв.) с последующим добавлением мСРВА (4 мг, 0,024 ммоль, 1 экв.). Реакционную смесь перемешивают при -78°С в течение 1 ч, разбавляют H2O и экстрагируют ДХМ (3×). Органические экстракты промывают 10% NaHCO3 и насыщенным солевым раствором, сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенное вещество очищают препаративной ТСХ (гексан/AcOEt 1:2) с получением эпоксида в виде белой пены (70% выход).
Стадия С
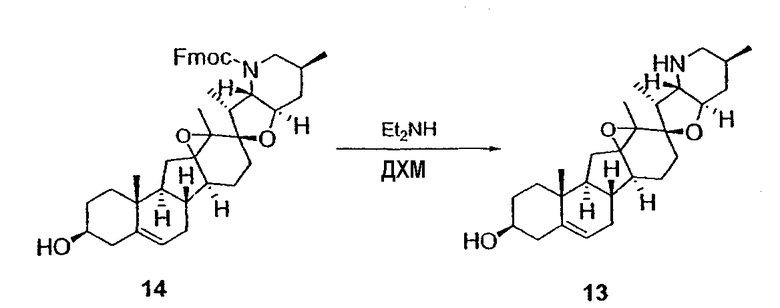
Раствор соединения 14 (11 мг, 0,017 ммоль, 1 экв.) в ДХМ (0,5 мл) обрабатывают при комнатной температуре Et2NH (0,5 мл, 4,8 ммоль, 282 экв.), полученный в результате раствор перемешивают при комнатной температуре в течение ночи и концентрируют в вакууме. Остаток очищают препаративной ТСХ (ДХМ/МеОН 9:1). Соединение 13 получают в виде белого твердого вещества (90% выход). МС (ESI(+)) m/e 428,5 (M+H)+.
Пример 5
Получение производного циклопамина
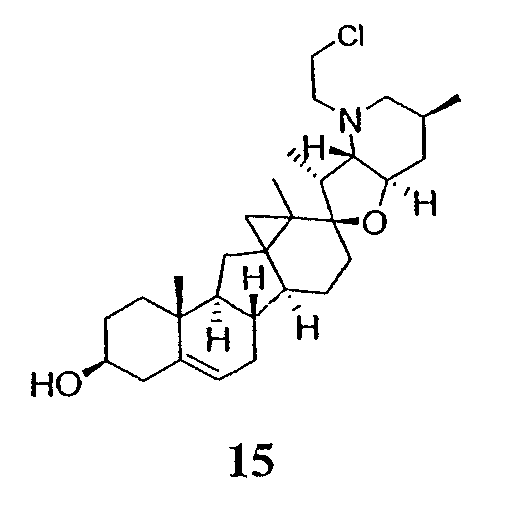
Стадия А
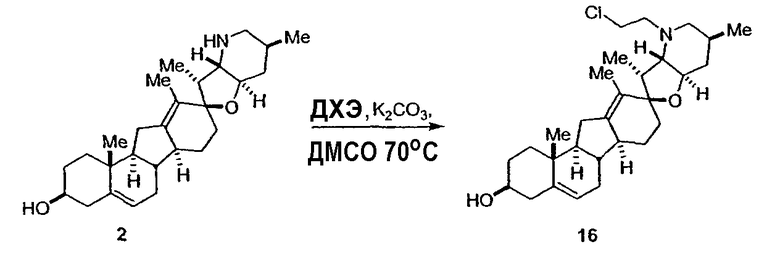
Соединение 2 (1,30 г, 3,2 ммоль, 1 экв.) взвешивают и загружают в реакционную емкость. Карбонат калия (0,91 г, 6,6 ммоль, 2,1 экв.) взвешивают и загружают в реакционную емкость с последующим добавлением дихлорэтана (6,0 мл, 76 ммоль, 23,8 экв.) и безводного ДМСО (5 мл). Реакционную смесь нагревают до 70°С в течение 36 час в атмосфере азота. Реакционную смесь охлаждают до комнатной температуры, разбавляют ДХМ (15 мл) и промывают дважды водой (2×15 мл). Органический слой сушат над сульфатом натрия, фильтруют (промывка ДХМ, как необходимо), и концентрируют досуха с получением бледно-желтого твердого вещества. Флэш-хроматографией (ДХМ/EtOAc) получают целевое вещество в виде белого кристаллического твердого вещества.
Стадия В
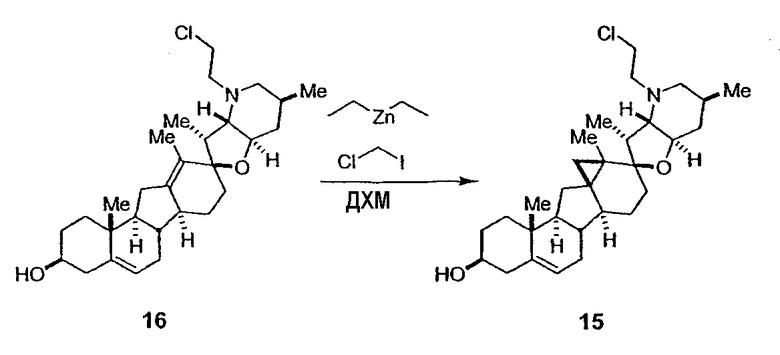
Соединение 16 (0,111 г, 0,233 ммоль, 1 экв.) переносят в реакционную колбу, помещают в атмосферу азота и растворяют в безводном ДХМ (2 мл). Добавляют хлорйодметан (0,238 мл, 3,27 ммоль, 14 экв.). Раствор охлаждают до -15°С. Диэтилцинк (1М в гептане, 1,63 мл, 1,63 ммоль, 6,5 экв.) добавляют по каплям в течение 30 мин, осторожно контролируя экзотермический эффект. Реакционную смесь выдерживают между -10°С и -14°С в течение нескольких часов до тех пор, пока ТСХ не указывает на расход исходного вещества. Реакцию далее гасят осторожным добавлением ТГФ (6 мл) и затем водного цитратного буфера (рН 4,5, 10 мл). Слоям дают нагреться до комнатной температуры. Добавляют насыщенный раствор сульфата натрия. Слои хорошо смешивают, переносят в соединительную воронку с избытком ДХМ и собирают органический слой. Органический слой промывают водным гидроксидом натрия (1 н., 10 мл) и насыщенным сульфатом натрия (10 мл), сушат над сульфатом натрия перед концентрированием досуха. Неочищенное вещество очищают флэш-хроматографией с получением требуемого продукта с 55% выходом.
Пример 6
Получение производного циклопамина
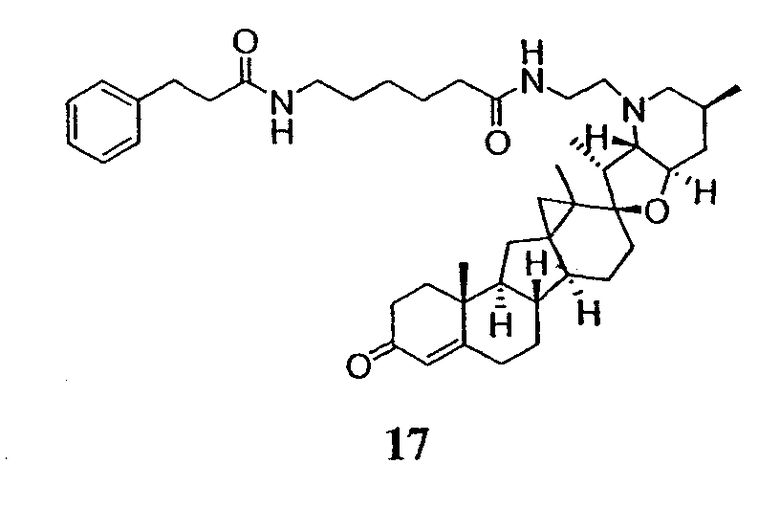
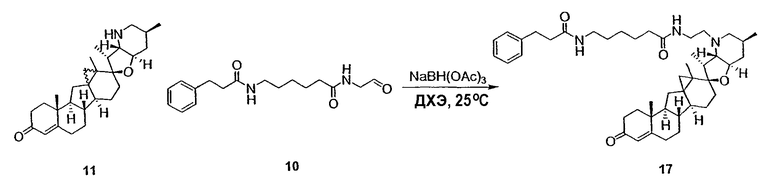
К раствору соединения 11 (5 мг, 0,01 ммоль, 1 экв.) и соединения 10 (10 мг, 0,04 ммоль, 3 экв.) в безводном ДХМ (5 мл) добавляют твердый триацетоксиборгидрид (8 мг, 0,004 ммоль, 3 экв.) и полученную в результате суспензию перемешивают при 25°С в течение 2 ч. Реакционную смесь гасят бикарбонатом натрия, экстрагируют ДХМ (4×10 мл), органический слой собирают и промывают насыщенным солевым раствором (1×20 мл), сушат над сульфатом магния и концентрируют при пониженном давлении. Неочищенное вещество очищают ПТСХ (ДХМ/МеОН 95:5) с получением 8 мг требуемого продукта.
Пример 7
Получение производного циклопамина

Стадия А
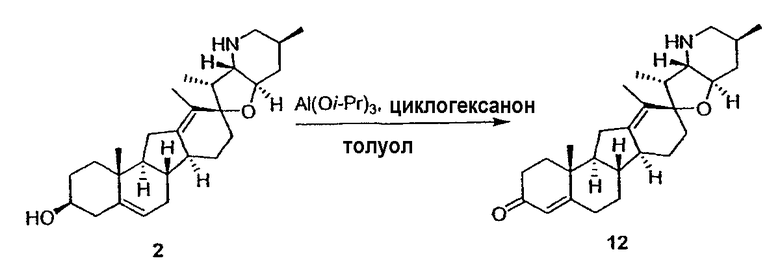
Циклопамин 2 (20 мг, 0,049 ммоль) суспендируют в сухом толуоле (0,6 мл) и к нему добавляют циклогексанон (150 мкл, 1,47 ммоль, 30 экв.) с последующим добавлением изопропоксида алюминия (79 мг, 0,392 ммоль, 8 экв.). Полученную в результате смесь нагревают при кипячении с обратным холодильником в течение 2 ч, охлаждают до комнатной температуры, разбавляют этилацетатом и гасят солевым раствором Рочеля. Бифазную смесь перемешивают в течение ночи, слои соединяют, водный слой экстрагируют этилацетатом и объединенные органические экстракты сушат (над MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (ДХМ, ДХМ/метанол 98:2 и 95:5). Целевое вещество 12 получают в виде белого кристаллического твердого вещества (70% выход).
Стадия В
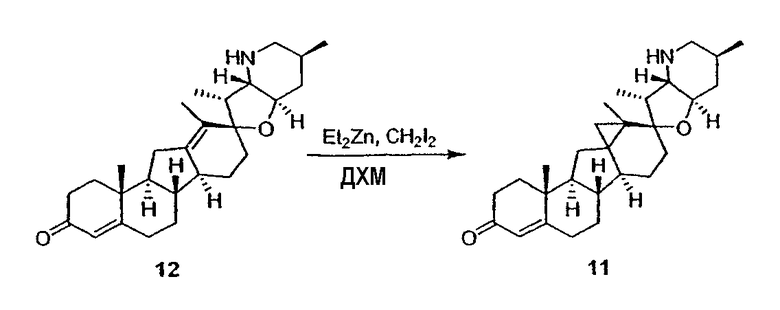
Дийодметан (40 мкл, 0,5 ммоль, 2,5 экв.) в ДХМ (0,52 мл) при 0°С обрабатывают 15% диэтилцинком в толуоле (0,2 мл, 0,2 ммоль, 1 экв.) и полученный в результате раствор перемешивают в течение 5 мин (где наблюдают белый осадок). Добавляют енон 12 (10 мг, 0,02 ммоль, 1 экв.) в ДХМ (0,35 мл) и полученную в результате смесь перемешивают при комнатной температуре (при удалении бани со льдом) в течение 3 ч, гасят NaOH (2N) и перемешивают в течение 10 мин, слои разделяют и водный слой экстрагируют ДХМ (2×). Органические экстракты сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией (ДХМ/МеОН 92:8). Циклопропанированное вещество 11 получают в виде белого твердого вещества.
Стадия С

К раствору циклопропиленона 11 (10 мг, 24 мкмоль, 1 экв.) в ДХМ (0,5 мл) при 0°С в атмосфере аргона добавляют BF3·Et2O (30 мкл, 0,24 ммоль, 10 экв.) и полученный в результате раствор перемешивают при 0°С в течение 1,5 ч, разбавляют ДХМ и гасят насыщенным раствором бикарбоната натрия. Органическую фазу промывают насыщенным бикарбонатом натрия и объединенные водные слои экстрагируют ДХМ. Объединенные органические слои промывают насыщенным солевым раствором, сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают препаративной ТСХ (ДХМ/МеОН 9:1). Целевое соединение 18 получают в виде белого твердого вещества (90% выход). МС (ESI(+)) m/e 424,62 (M+H)+.
Пример 8
Получение производного циклопамина
Стадия А
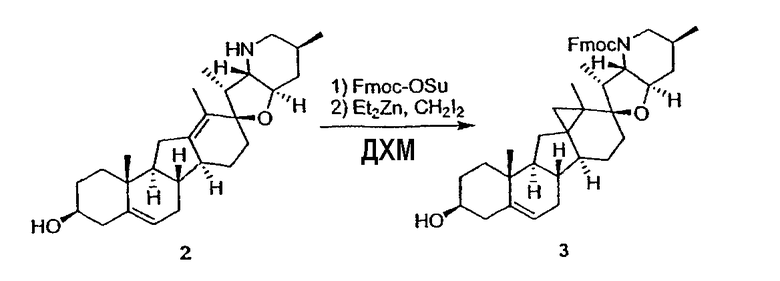
К раствору циклопамина 2 (250 мг, 0,6 ммоль, 1 экв.) в ДХМ (10 мл) при комнатной температуре добавляют Fmoc-OSu (205 мг, 0,6 ммоль, 1 экв.) и полученную в результате смесь перемешивают при комнатной температуре в течение ночи. Полученный в результате раствор неочищенного Fmoc-циклопамина далее охлаждают до 0°С и обрабатывают 15% диэтилцинком в толуоле (0,5 мл, 0,6 ммоль, 1 экв.) и перемешивают в течение 30 мин (колба А, желтоватый раствор).
Дийодметан (0,4 мл, 6 ммоль, 10 экв.) в ДХМ (20 мл) при 0°С обрабатывают 15% диэтилцинком в толуоле (3 мл, 3 ммоль, 5 экв.) и полученный в результате раствор перемешивают в течение 5 мин (колба В, белый осадок).
Содержимое колбы В переносят в колбу А через канюлю и полученную в результате суспензию перемешивают в течение 5 ч при комнатной температуре. Реакцию гасят 1 н. хлористоводородной кислотой, перемешивают в течение 10 мин (до тех пор, пока не растворится все белое вещество) и экстрагируют ДХМ (5×). Органические экстракты сушат над MgSO4, фильтруют через целит и концентрируют в вакууме. Остаток очищают флэш-хроматографией (гексан/этилацетат 1:1). Целевой 11,12-моноциклопропан 5 получают в виде 9:1 смеси диастереоизомеров, наряду с 20% диастереомерными бис-циклопропанированными продуктами (80% общего извлечения). Данную смесь разделяют, используя препаративную SFC хроматографию.
Стадия В
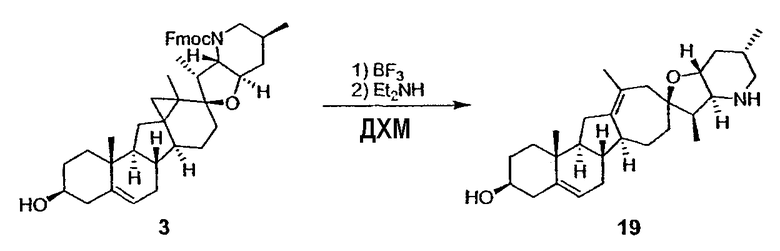
Fmoc-циклопропилциклопамин 3 (14 мг, 22 мкмоль, 1 экв.) растворяют в ДХМ (0,5 мл), охлаждают до 0°С и обрабатывают BF3 .Et2O (27 мкл, 0,22 ммоль, 10 экв.) в течение 1 ч, гасят насыщенным раствором бикарбоната натрия, слои разделяют и водный слой экстрагируют ДХМ. Объединенные органические экстракты сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают препаративной тонкослойной хроматографией (гексан/этилацетат 2:1). Целевое вещество, Fmoc-защищенный циклопамин получают в виде прозрачного масла.
Раствор неочищенного Fmoc-защищенного циклопамина (20 мг, 31 мкмоль, 1 экв.) в ДХМ (0,5 мл) обрабатывают Et2NH (0,5 мл, 4,8 ммоль, 154 экв.) в течение ночи, концентрируют в вакууме и остаток очищают флэш-хроматографией (ДХМ, ДХМ/метанол 98:2 и 95:5). Требуемое соединение получают в виде масла, которое кристаллизуется при стоянии. МС (ESI(+)) m/e 426,29 (M+H)+.
Пример 9
Получение производного циклопамина
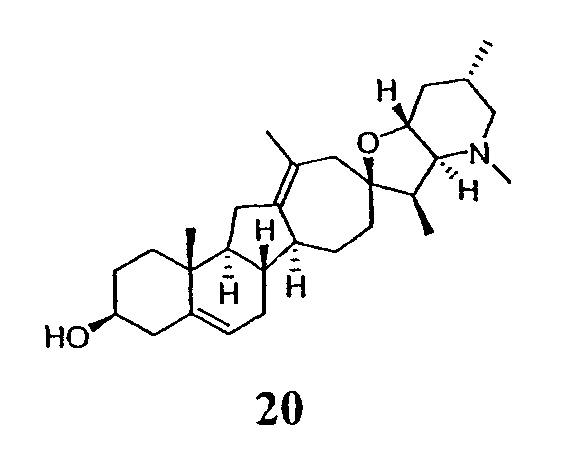
Стадия А
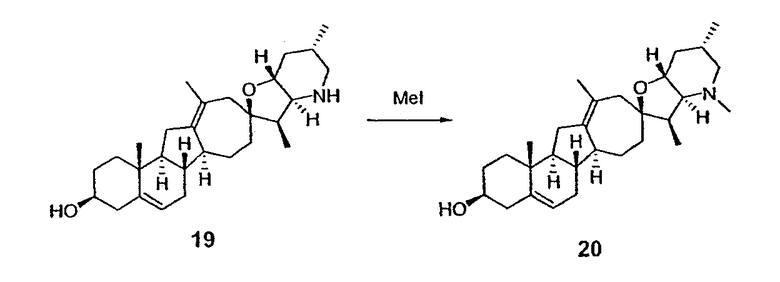
Соединение 6 (23 мг, 54 мкмоль, 1 экв.) растворяют в ДХМ (1 мл) и к нему добавляют метилйодид (0,17 мл, 0,54 ммоль, 10 экв.). Реакционной смеси дают перемешаться при комнатной температуре в атмосфере азота в течение ночи. На следующее утро ТСХ/ЖХ-МС указывает на то, что остается еще немного ИВ (исходного вещества). Добавляют шпатель Na2CO3 и смесь перемешивают в течение еще одного часа. Неочищенное вещество загружают в систему Biotage 25 Si+M и элюируют ДХМ/EtOAc/MeOH (82,5/10/7,5). Получают аморфное вещество: 16 мг. МС (ESI(+)) m/e 440,32 (M+H)+.
Пример 10
Получение производного циклопамина

Стадия А

Гидроциннамоилхлорид 22 (1,13 г, 6,7 ммоль, 1 экв.) и аллиламин (0,77 мл, 10 ммоль, 1,5 экв.) солюбилизируют в ТГФ (20 мл) и реакционную смесь перемешивают при комнатной температуре в течение 24 час. Образуется белый осадок. Реакционную смесь фильтруют. Фильтрат сушат над MgSO4, фильтруют и концентрируют в вакууме. Выделяют бесцветное масло, которое превращается в воскоподобное твердое вещество (1,1 г).
К раствору аллиламида (0,81 мг, 0,27 ммоль, 1 экв.) в смеси ацетон:вода (9 мл; 3:1) при 0°С добавляют раствор OsO4 (0,55 мл, 2,5 мас./мас. в трет-BuOH) и полученную в результате коричневатую смесь перемешивают в течение 10 мин. Твердый перйодат натрия (0,13 г, 0,59 ммоль, 2,2 экв.) добавляют тремя порциями и смесь перемешивают при 0°С и дают нагреться до 25°С в течение периода, равного 2 ч. Светлую грязно-белую кремоподобную смесь разбавляют ДХМ (25 мл), сушат над сульфатом магния, твердые вещества отфильтровывают через слой целита, фильтрат концентрируют при пониженном давлении. Неочищенное вещество медленно приобретает желто-черный цвет. Неочищенное вещество загружают в систему Biotage 25+S и очищают, элюируя смесью гексан/EtOAc (1:1 до 1:2) с получением бесцветного масла, которое отверждается при сушке (250 мг).
Стадия В
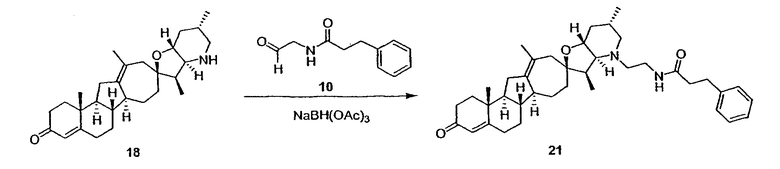
К раствору 18 (108 мг, 0,25 ммоль, 1 экв.) и альдегида 21 (100 мг, 0,52 ммоль, 2,1 экв.) в ДХМ (5 мл) добавляют триацетоксиборгидрид натрия (100 мг, 0,47 ммоль, 1,9 экв.) одной порцией и взвесь перемешивают при комнатной температуре в течение 7 ч. Реакцию гасят добавлением МеОН и фильтруют через целит. Упариванием досуха получают 230 мг масла. Вещество очищают хроматографией (SiO2, колонка 3 см × 4 см), элюируя смесью гексан/EtOAc (4:6 до 2:8) с получением 38 мг требуемого продукта. МС (ESI(+)) m/e 599,74 (M+H)+.
Пример 11
Получение производного циклопамина
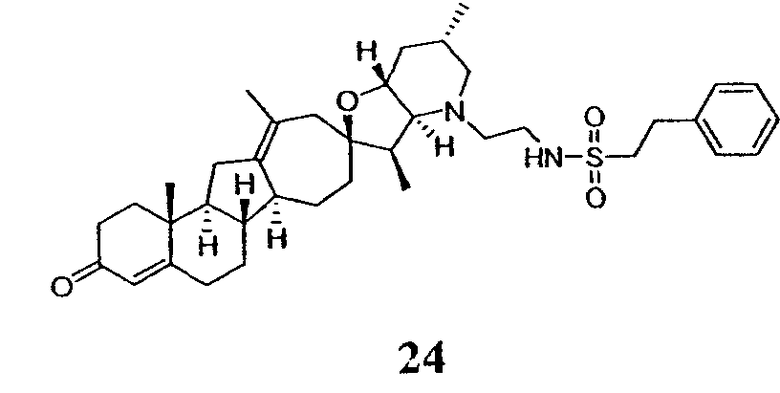
Стадия А
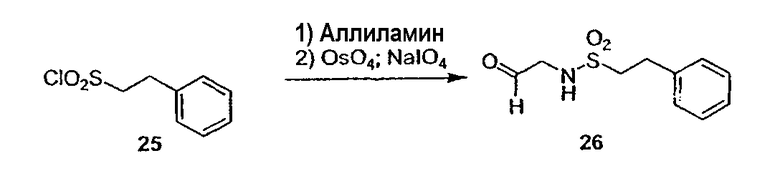
2-Фенилэтансульфонилхлорид 25 (1,13 г, 5,5 ммоль, 1 экв. и аллиламин (0,56 мл, 7,3 ммоль, 1,3 экв.) солюбилизируют в ТГФ (20 мл) и дают взаимодействовать при комнатной температуре в течение 24 ч. Образуется белый осадок. Реакционную смесь фильтруют. Фильтрат сушат над MgSO4, фильтруют и концентрируют в вакууме. Выделяют слегка желтое масло (1,1 г). Неочищенное вещество используют непосредственно в следующей стадии.
К раствору аллилсульфонамида (0,15 г, 0,66 ммоль, 1 экв.) в смеси ацетон:вода (4 мл; 3:1) при 0°С добавляют раствор OsO4 (0,13 мл, 2,5 мас./мас. в трет-BuOH) и полученную в результате коричневатую смесь перемешивают в течение 10 мин. Твердый перйодат натрия (0,31 г, 1,46 ммоль, 2,2 экв.) добавляют тремя порциями и смесь перемешивают при 0°С и ей дают нагреться до 25°С в течение периода, равного 2 ч. Светлую грязно-белую кремоподобную смесь разбавляют ДХМ (25 мл), сушат над сульфатом магния, твердые вещества отфильтровывают через слой целита, фильтрат концентрируют при пониженном давлении. Неочищенное вещество очищают на SiO2 (колонка 2 см × 12 см), элюируя смесью гексан/EtOAc (7:3) с получением требуемого вещества (16 мг).
Стадия В
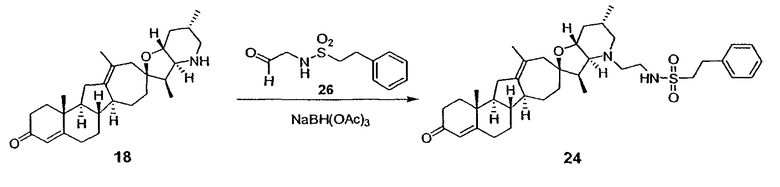
К раствору 18 (15 мг, 35,4 мкмоль, 1 экв.) и альдегида 26 (16 мг, 70 мкмоль, 2 экв.) в ДХМ (3 мл) добавляют триацетоксиборгидрид натрия (20 мг, 94 мкмоль, 2,6 экв.) одной порцией при комнатной температуре. Через 24 ч реакцию гасят добавлением нескольких капель МеОН и фильтруют через целит. Неочищенное вещество очищают препаративной ТСХ 1 мм (первое элюирование: толуол/ацетон (9:1), второе элюирование: толуол/ацетон (4:1)) с получением 4 мг бесцветного масла. МС (ESI(+)) m/e 635,43 (M+H)+.
Пример 12
Получение производного циклопамина
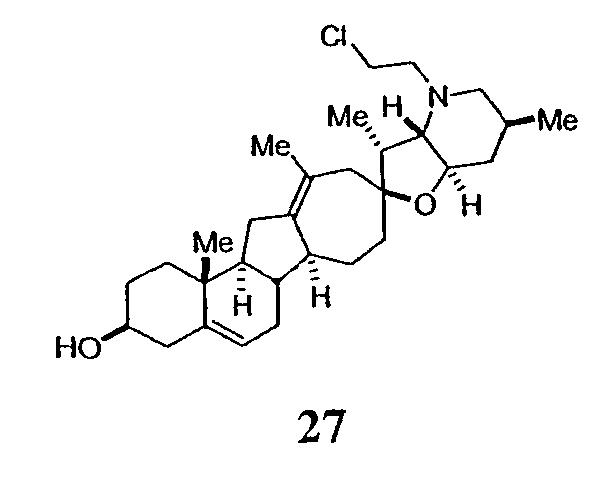
Стадия А
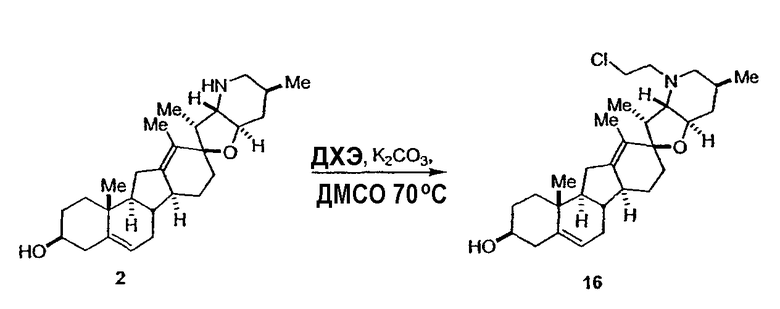
Соединение 2 (1,30 г, 3,2 ммоль, 1 экв.) взвешивают и загружают в реакционную емкость. Карбонат калия (0,91 г, 6,6 ммоль, 2,1 экв.) взвешивают и загружают в реакционную емкость с последующим добавлением дихлорэтана (6,0 мл, 76 ммоль, 23,8 экв.) и безводного ДМСО (5 мл). Реакционную смесь нагревают до 70°С в течение 36 ч в атмосфере азота. Реакционную смесь охлаждают до комнатной температуры, разбавляют ДХМ (15 мл) и промывают дважды водой (2×15 мл). Органический слой сушат над сульфатом натрия, фильтруют (промывка ДХМ, как необходимо) и концентрируют досуха с получением бледно-желтого твердого вещества. Флэш-хроматографией (ДХМ/EtOAc) получают целевое вещество в виде белого кристаллического твердого вещества.
Стадия В
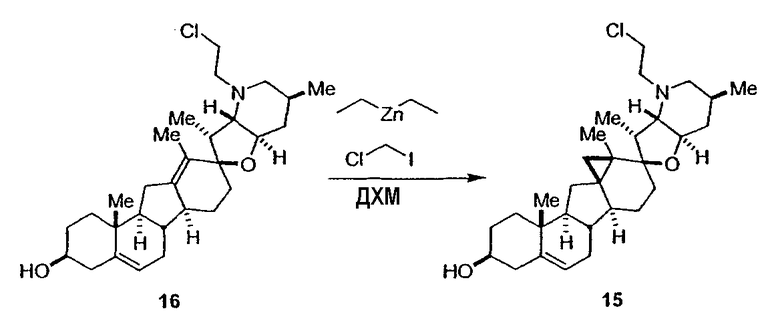
Соединение 16 (0,111 г, 0,233 ммоль, 1 экв.) переносят в реакционную колбу, помещают в атмосферу азота и растворяют в безводном ДХМ (2 мл). Добавляют хлорйодметан (0,238 мл, 3,27 ммоль, 14 экв.). Раствор охлаждают до -15°С. Диэтилцинк (1М в гептане, 1,63 мл, 1,63 ммоль, 6,5 экв.) добавляют по каплям в течение 30 минут, осторожно контролируя экзотермический эффект. Реакционную смесь выдерживают между -10°С и -14°С в течение нескольких часов до тех пор, пока ТСХ не указывает на расход исходного вещества. Реакцию далее гасят осторожным добавлением ТГФ (6 мл) и затем водного цитратного буфера (рН 4,5, 10 мл). Слоям дают нагреться до комнатной температуры. Добавляют насыщенный раствор сульфата натрия (10 мл). Слои хорошо смешивают, переносят в разделительную воронку с избытком ДХМ и собирают органический слой. Органический слой промывают водным гидроксидом натрия (1М, 10 мл) и насыщенным раствором сульфата натрия (10 мл), сушат над сульфатом натрия перед концентрированием досуха. Неочищенное вещество очищают флэш-хроматографией с получением требуемого продукта с 55% выходом.
Стадия С
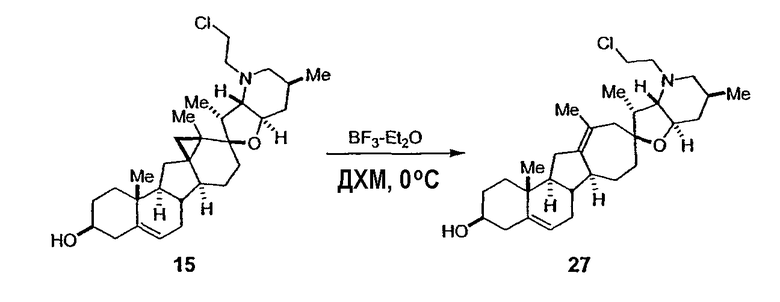
Соединение 15 (1,25 г, 2,56 ммоль, 1 экв.) растворяют в ДХМ (22 мл) в атмосфере азота и раствор охлаждают до внутренней температуры, равной 0,9°С. Неразбавленный BF3-OEt2 (1,6 мл, 12,8 ммоль, 5 экв.) добавляют порциями в течение нескольких часов, в то же время проводя мониторинг реакции с помощью ЖХМС. Реакционной смеси дают медленно нагреться до 10°С до завершения реакции. Реакцию гасят МеОН (5 мл) при 0°С, разбавляют КОН (2 М, 30 мл) и перемешивают при комнатной температуре в течение 2 часов. Слои разделяют и органический слой промывают водой, сушат над Na2SO4, фильтруют и концентрируют досуха. Хроматографией с ДХМ/EtOAc получают требуемый продукт.
Пример 13
Получение производного циклопамина
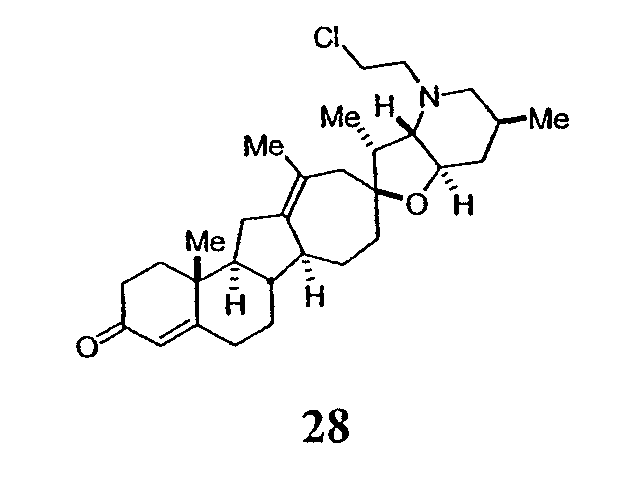
Стадия А
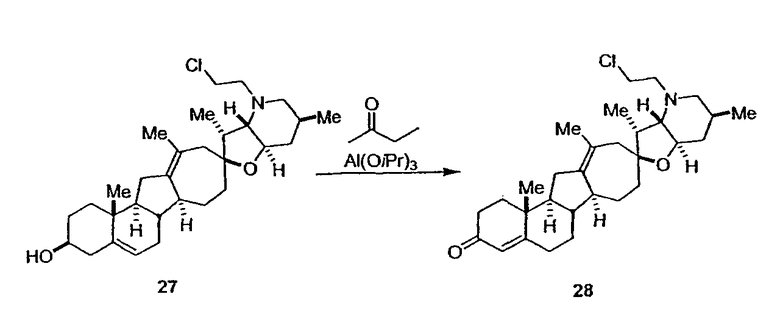
Соединение 27 (29 мг, 60 мкмоль, 1 экв.) помещают в 5-мл круглодонную колбу. Добавляют бутанон (2 мл) и Al(OiPr)3 (12,3 мг, 60 мкмоль, 1 экв.). Содержимое круглодонной колбы нагревают при кипячении с обратным холодильником в атмосфере аргона в течение 7 ч. Реакционную смесь далее гасят раствором (2 мл), образованным смешением лимонной кислоты (500 г), NaOH (15,7 г) и воды (500 мл). Полученную в результате смесь перемешивают с высокой скоростью до рассеивания эмульсии. Смесь далее экстрагируют EtOAc (3×10 мл). Органические слои собирают, сушат над Na2SO4 и концентрируют. Неочищенное вещество очищают колоночной хроматографией. МС (ESI(+)) m/e 486,26 (M+H)+.
Пример 14
Получение производного циклопамина
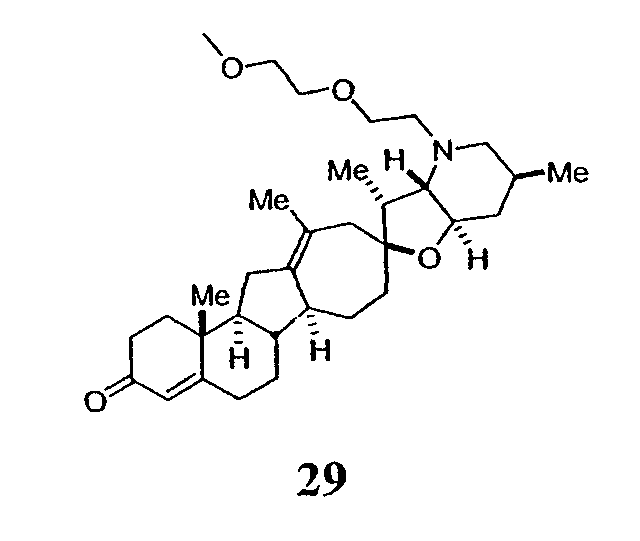
Стадия А
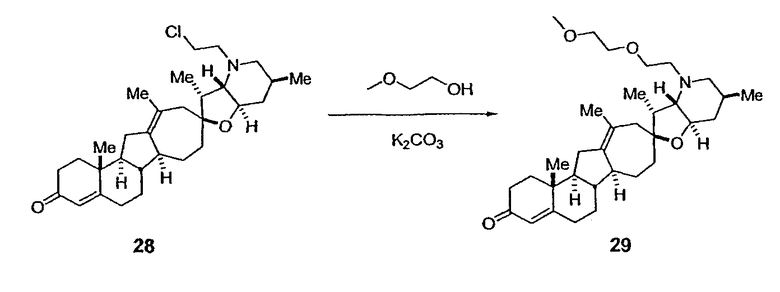
Соединение 28 (25 мг, 0,051 ммоль, 1 экв.) растворяют в безводном 2-метоксиэтаноле (1 мл, 12,7 ммоль, 234 экв.). Добавляют карбонат калия (7,1 мг, 0,051 ммоль, 1 экв.) и реакционную смесь нагревают до 120°С. Мониторинг реакции проводят с помощью ТСХ. Когда ТСХ указывает на остановку реакции, реакционную смесь охлаждают до комнатной температуры. Реакционную смесь далее разбавляют этилацетатом и промывают водой. Органический слой сушат над сульфатом натрия и концентрируют досуха. Хроматографией с ДХМ/EtOAc получают требуемый продукт. МС (ESI(+)) m/e 526,66 (M+H)+.
Пример 15
Получение производного циклопамина
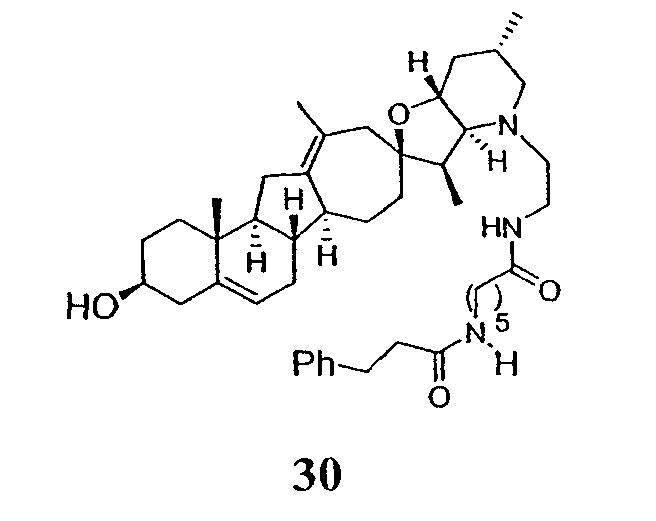
Стадия А
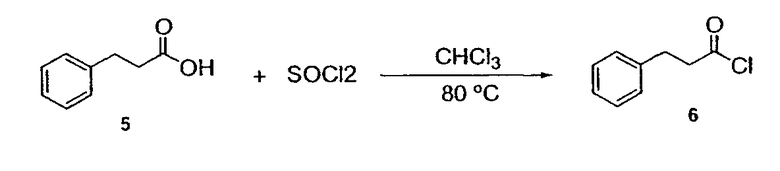
К раствору гидрокоричной кислоты 5 (3,01 г, 20 ммоль, 1 экв.) в 30 мл безводного хлороформа при 75°С добавляют тионилхлорид (1,75 мл, 24,1 ммоль, 1,2 экв.) по каплям в течение периода 3 мин. Смесь кипятят с обратным холодильником в течение 3,5 ч. Растворитель отгоняют с получением неочищенного хлорангидрида кислоты в виде светло-желтой вязкой жидкости. Неочищенное вещество используют без дальнейшей очистки.
Стадия В
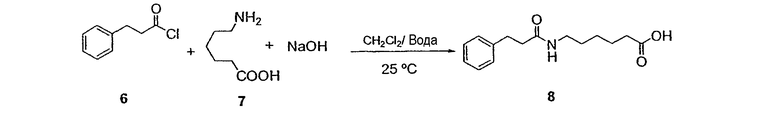
К бифазной смеси 7 (3,16 г, 24,1 ммоль, 1,2 экв.) в ДХМ (30 мл) и водного раствора NaOH (2,0 М, 30 мл, 3 экв.) при 25°С добавляют раствор хлорангидрида кислоты 6 (3,38 г, 20 ммоль, 1 экв.) в ДХМ (10 мл) и полученную в результате смесь перемешивают при 25°С в течение 3 ч. Смесь далее нейтрализуют водным раствором HCl (2 M, 30 мл). Органический слой далее отделяют и водный слой экстрагируют ДХМ (3×50 мл). Объединенные органические слои промывают HCl (2,0 М, 25 мл), водой (3×50 мл), насыщенным солевым раствором (50 мл), сушат над сульфатом магния и раствор упаривают при пониженном давлении. Неочищенное вещество хроматографируют на силикагеле, используя 5% МеОН:ДХМ в качестве элюента и колонку далее элюируют 10% МеОН:ДХМ с получением 1,141 г соединения 8.
Стадия С
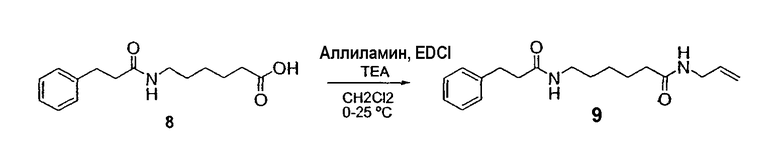
К смеси кислоты 8 (264 мг, 1 ммоль, 1 экв.), EDCI (231 мг, 1,2 ммоль, 1,2 экв.) и триэтиламина (168 мкл, 1,2 ммоль, 1,2 экв.) в ДХМ (2 мл) при 0°С добавляют аллиламин (90,3 мкл, 1,2 ммоль, 1,2 экв.) и полученную в результате смесь перемешивают при 0°С и дают ей нагреться до 25°С в течение периода, равного 2 ч. Реакционную смесь добавляют к воде (50 мл), экстрагируют ДХМ (4×25 мл), объединенные органические слои промывают 1М HCl (2×25 мл), водой (3×25 мл), насыщенным солевым раствором (25 мл), сушат над сульфатом магния и растворитель упаривают при пониженном давлении с получением 287,5 мг требуемого продукта. Данное вещество используют без дальнейшей очистки.
Стадия D
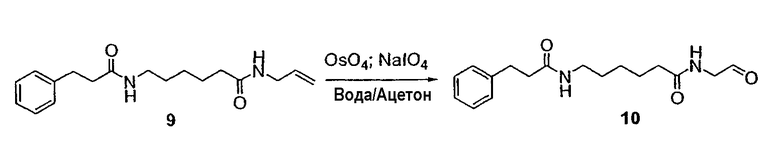
К раствору аллиламида 9 (70 мг, 0,23 ммоль, 1 экв.) в ацетоне (1 мл) и воде (0,3 мл) добавляют раствор тетроксида осмия (0,35 мл, 0,035 ммоль, 2,5 мас./мас. в трет-бутаноле) и реакционную смесь немедленно охлаждают на бане со льдом после добавления раствора OsO4. Полученный в результате темно-коричневый раствор перемешивают при 0°С в течение 15 мин. Перйодат натрия (110 мг, 0,51 ммоль, 2,2 экв.) добавляют 5 порциями к вышеуказанной смеси и перемешивание продолжают в течение 1 ч при 0°С, и смеси дают нагреться до 25°С в течение периода, равного 2 ч. Реакционную смесь разбавляют ДХМ (3 мл) фильтруют через небольшой слой сульфата магния и осадок на фильтре промывают ДХМ (несколько раз). Фильтрат концентрируют и остаток (67,9 мг) фильтруют через небольшой слой RP силикагеля, используя 5% МеОН:ДХМ с получением 38,9 мг требуемого продукта.
Стадия Е
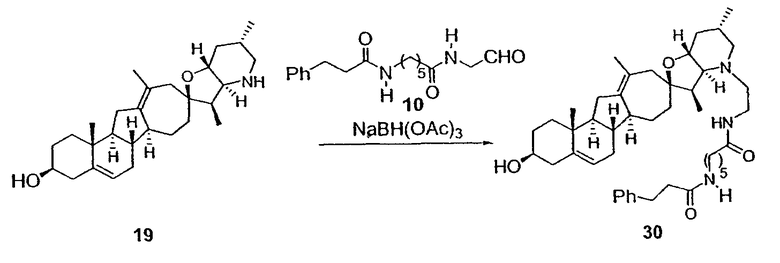
К раствору 19 (0,0242 г, 0,0569 ммоль, 1 экв.) и альдегида 10 (0,0346 г, 0,114 ммоль) в 3,0 мл ДХМ при 23°С добавляют триацетоксиборгидрид натрия (24,1 мг, 0,114 ммоль, 2 экв.) одной порцией и полученную в результате смесь перемешивают в течение 16 час. После полной конверсии исходного вещества в требуемый продукт, что является очевидным по данным ЖХМС и ТСХ, смесь отбирают в 2,5 мл метанола и очищают обращенно-фазовой препаративной хроматографией (ацетонитрил-буфер 20 мМ карбоната аммония, основной способ). Фракции концентрируют и отбирают в минимальный объем ацетонитрила, раствор замораживают и лиофилизируют с получением 0,007 г (0,0098 ммоль, 17%) в виде белого твердого вещества. МС (ESI(+)) m/e 714,6 (M+H)+.
Пример 16
Получение производного циклопамина
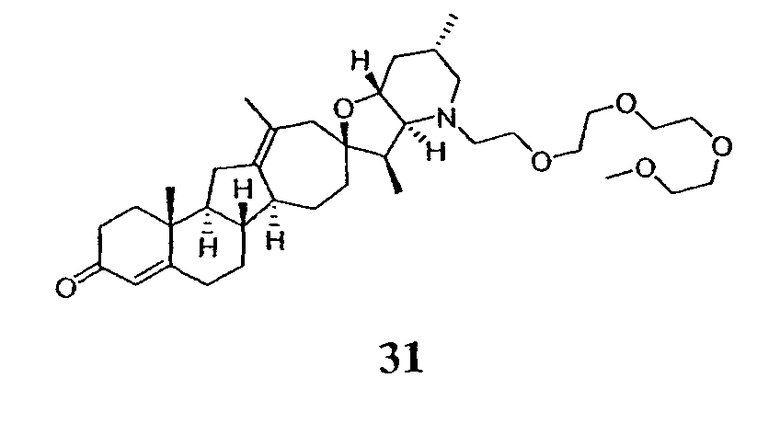
Стадия А
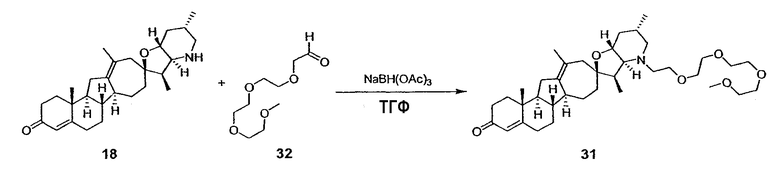
К раствору 18 (35 мг, 0,08 ммоль, 1 экв.) и альдегида 32 (34 мг, 0,17 ммоль, 2,0 экв.) в ТГФ (2,0 мл) добавляют триацетоксиборгидрид натрия (35 мг, 0,17 ммоль, 2,0 экв.) одной порцией. Раствору дают перемешаться при 23°С в течение 12 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 1:1 гексаны:этилацетат с последующими 1:2, 1:4 и непосредственно этилацетат. Некоторое вещество еще элюируется, поэтому колонку далее промывают в флэш-режиме смесью 9:1 этилацетат:метанол. Требуемый продукт совместно элюируется с некоторым количеством альдегида, поэтому вещество далее очищают препаративной ВЭЖХ. (Основной метод 50_100). Требуемые фракции замораживают и лиофилизируют с получением маслянистого остатка (12 мг, 0,02 ммоль, 24% выход). МС (ESI(+)) m/e 614,44 (M+H)+.
Пример 17
Получение производного циклопамина

Стадия А
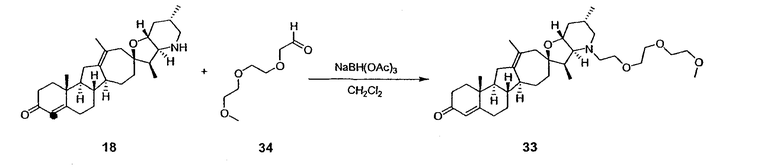
К раствору 18 (8,0 мг, 0,02 ммоль, 1 экв.) и альдегида 34 (6,0 мг, 0,04 ммоль, 2,0 экв.) в CH2Cl2 (1,0 мл) добавляют триацетоксиборгидрид натрия (8,0 мг, 0,17 ммоль, 2,0 экв.) одной порцией. Раствору дают перемешаться при 23°С в течение 12 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 1:1 гексаны:этилацетат с последующими 1:2, 1:4 для выделения требуемого продукта, соэлюируемого с некоторым количеством альдегида. Данное вещество далее очищают препаративной ВЭЖХ. Требуемые фракции замораживают и лиофилизируют с получением белого порошка (4,9 мг, 0,009 ммоль, 46% выход). МС (ESI(+)) m/e 570,41 (M+H)+.
Пример 18
Получение производного циклопамина
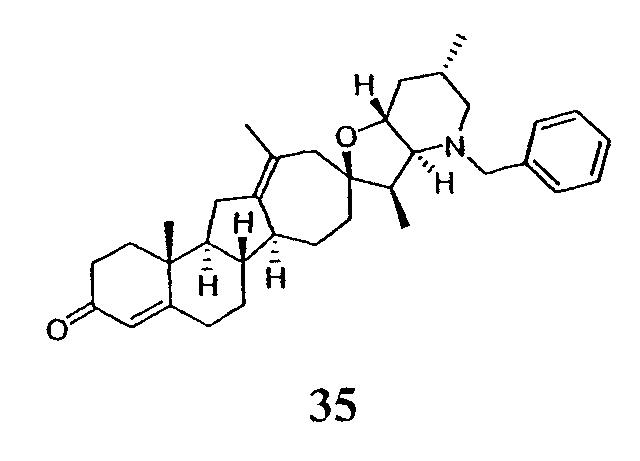
Стадия А
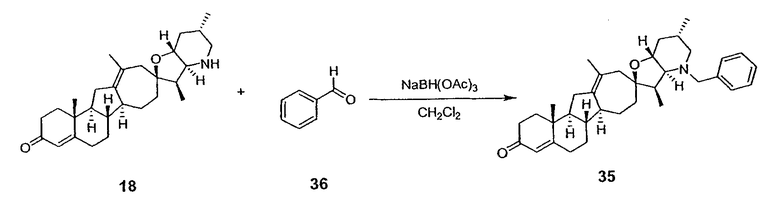
К раствору 18 (6,0 мг, 0,01 ммоль, 1 экв.) и бензальдегида 36 (3,0 мг, 0,02 ммоль, 2,0 экв.) в CH2Cl2 (0,5 мл) добавляют триацетоксиборгидрид натрия (6,0 мг, 0,02 ммоль, 2,0 экв.) одной порцией. Раствору дают перемешаться при 23°С в течение 12 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 4:1 гексаны:этилацетат с последующим 1:1 для выделения требуемого продукта, соэлюируемого с некоторым количеством альдегида. Данное вещество далее очищают препаративной ВЭЖХ. Требуемые фракции замораживают и лиофилизируют с получением белого порошка (0,6 мг, 0,001 ммоль, 8% выход).
Пример 19
Получение производного циклопамина
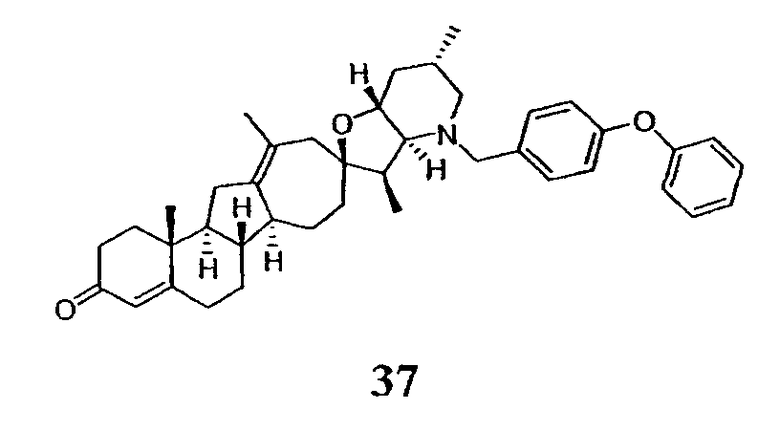
Стадия А
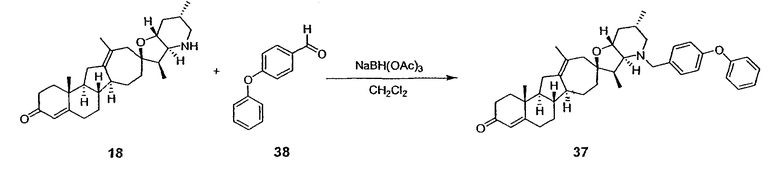
К раствору 18 (6,0 мг, 0,01 ммоль, 1 экв.) и 4-феноксибензальдегида 38 (6,0 мг, 0,02 ммоль, 2,0 экв.) в CH2Cl2 (0,5 мл) добавляют триацетоксиборгидрид натрия (6,0 мг, 0,02 ммоль, 2,0 экв.) одной порцией. Раствору дают перемешаться при 23°С в течение 12 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 4:1 гексаны:этилацетат с последующим 1:1 для выделения требуемого продукта, соэлюируемого с некоторым количеством альдегида. Данное вещество далее очищают препаративной ВЭЖХ. Требуемые фракции замораживают и лиофилизируют с получением белого порошка (1,8 мг, 0,003 ммоль, 21% выход). МС (ESI(+)) m/e 606,4 (M+H)+.
Пример 20
Получение производного циклопамина

Стадия А
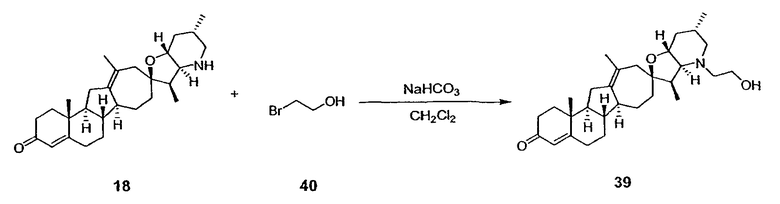
К смеси 18 (40 мг, 0,09 ммоль, 1 экв.) и бикарбоната натрия (15 мг, 0,18 ммоль, 2,0 экв.) в CH2Cl2 (0,5 мл) добавляют бромэтанол 40 (33 мкл, 0,47 ммоль, 5,0 экв.). Раствор нагревают при кипячении с обратным холодильником в течение 4 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент ДХМ, с последующим 38:1:1 дихлорметан:этилацетат:метанол, далее 36:3:1, далее 17:2:1 для выделения требуемого продукта в виде масла (12 мг, 0,026 ммоль, 27% выход). МС (ESI(+)) m/e 468,24 (M+H)+.
Пример 21
Получение производного циклопамина
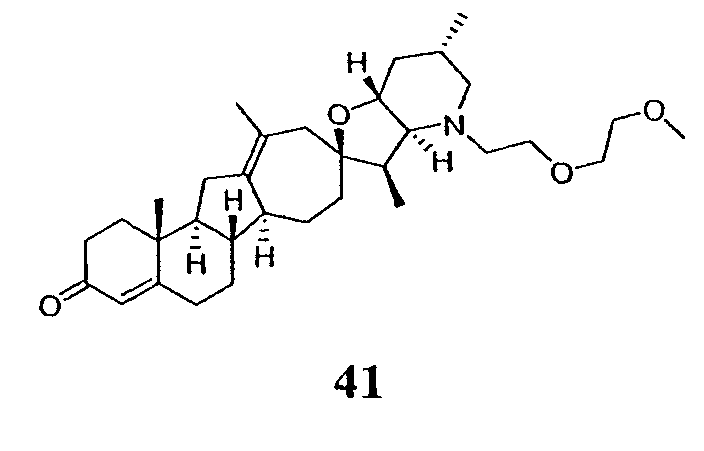
Стадия А
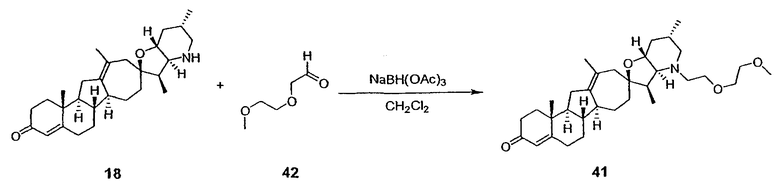
К раствору 18 (100 мг, 0,24 ммоль, 1 экв.) и альдегида 42 (42 мг, 0,35 ммоль, 1,5 экв.) в CH2Cl2 (2,5 мл) добавляют триацетоксиборгидрид натрия (100 мг, 0,47 ммоль, 2,0 экв.) одной порцией. Раствору дают перемешаться при 23°С в течение 12 ч, и ЖХМС показывает только 50% конверсии. К смеси добавляют дополнительный эквивалент альдегида 36 (26 мг, 0,24 ммоль, 1,0 экв.) и триацетоксиборгидрид натрия (48 мг, 0,24 ммоль, 1,0 экв.) и дают перемешаться в течение 2 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 1:1 гексаны:этилацетат с последующими 1:2 и 1:4 для выделения требуемого продукта, соэлюируемого с некоторым количеством альдегида. Данное вещество далее очищают препаративной ВЭЖХ. Требуемые фракции замораживают и лиофилизируют с получением белого порошка (53 мг, 0,10 ммоль, 43% выход). МС (ESI(+)) m/e 526,66 (M+H)+.
Пример 22
Получение производного циклопамина

Стадия А
К раствору 18 (15 мг, 0,04 ммоль, 1 экв.) и альдегида 44 (6,9 мг, 0,04 ммоль, 1,0 экв.) в CH2Cl2 (0,6 мл) добавляют триацетоксиборгидрид натрия (15 мг, 0,07 ммоль, 2,0 экв.) одной порцией. Раствору дают перемешаться при 23°С в течение 12 ч, и ЖХМС показывает только 50% конверсии. К смеси добавляют дополнительный эквивалент альдегида 44 (6,9 мг, 0,04 ммоль, 1,0 экв.) и триацетоксиборгидрид натрия (7,5 мг, 0,04 ммоль, 1,0 экв.) и дают перемешаться в течение 12 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 1:1 гексаны:этилацетат с последующими 1:2 и 1:4 для выделения требуемого продукта в виде масла (12 мг, 0,19 ммоль, 54% выход). МС (ESI(+)) m/e 635,43 (M+H)+.
Пример 23
Получение производного циклопамина
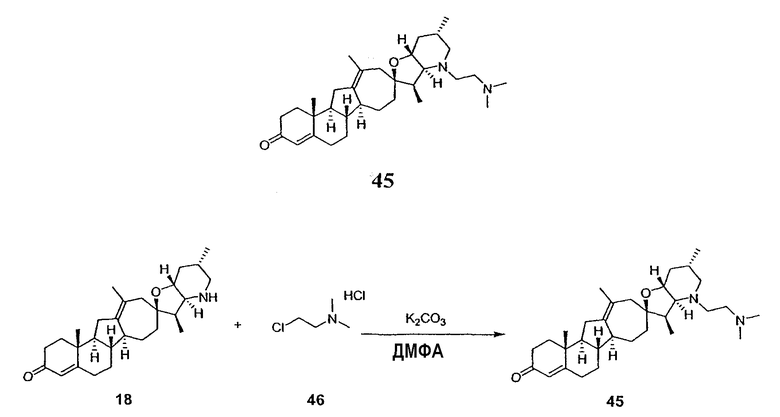
Стадия А
К смеси 18 (12 мг, 0,03 ммоль, 1 экв.) и карбоната калия (40 мг, 0,28 ммоль, 10 экв.) в ДМФА (0,5 мл) добавляют гидрохлорид 2-(диметиламино)этилхлорида 46 (20 мг, 0,14 ммоль, 5,0 экв.). Раствор перемешивают в течение 2 ч при 23°С и реакции не происходит. Раствор далее нагревают при 65°С в течение 12 ч, гасят водой (2 мл) и далее экстрагируют диэтиловым эфиром (2×10 мл). Объединенные органические экстракты промывают насыщенным солевым раствором и сушат MgSO4. Смесь далее концентрируют и очищают препаративной ВЭЖХ. Требуемые фракции замораживают и лиофилизируют с получением белого порошка (17, 1,8 мг, 0,004 ммоль, 13% выход). МС (ESI(+)) m/e 495,71 (M+H)+.
Пример 24
Получение производного циклопамина
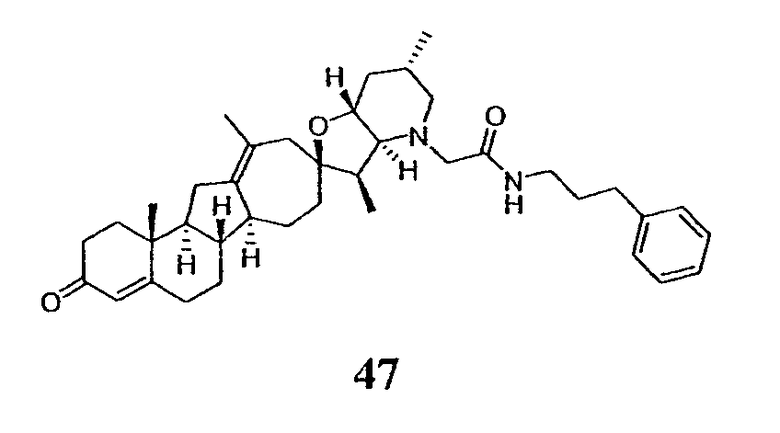
Стадия А
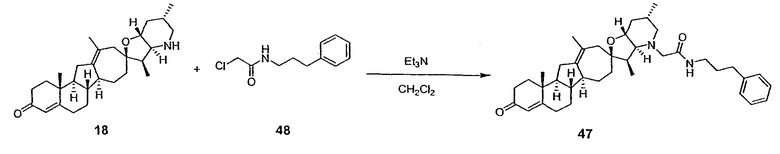
К раствору 18 (100 мг, 0,24 ммоль, 1 экв.) и хлорацетамида 48 (250 мг, 1,2 ммоль, 5,0 экв.) в CH2Cl2 (1,0 мл) добавляют триэтиламин (160 мкл, 1,2 ммоль, 5,0 экв.). Раствор нагревают при кипячении с обратным холодильником и перемешивают в течение 72 ч. Смесь далее концентрируют и очищают хроматографией на силикагеле, используя градиент 4:1 гексаны:этилацетат с последующими 2:1, 1:1 и 1:2 для выделения требуемого продукта в виде смеси двух пятен. Те же условия колонки повторяют и требуемый продукт выделяют в виде масла (17 мг, 0,14 ммоль, 12% выход).
Пример 25
Получение производного циклопамина
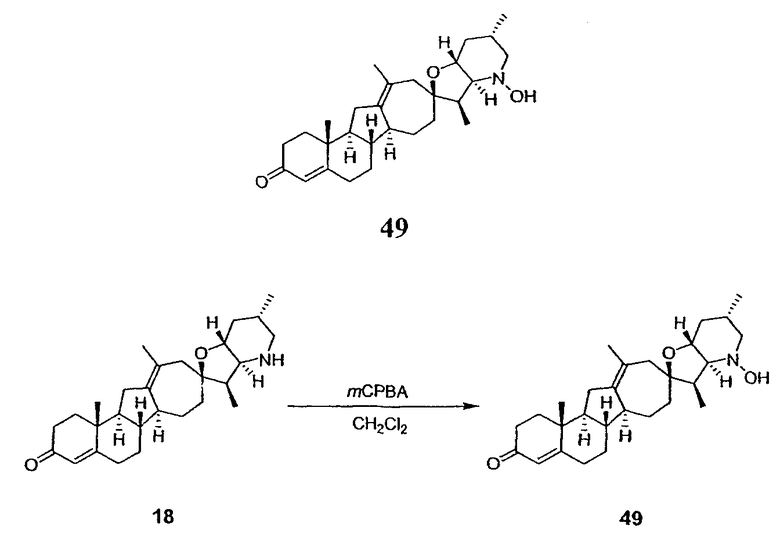
Соединение 18 (103 мг, 0,24 ммоль, 1 экв.) растворяют в ДХМ (3,0 мл) и охлаждают до -78°С. К данному раствору добавляют мСРВА (77 мас.%, 54 мг, 0,24 ммоль, 1,0 экв.) и далее раствору дают нагреться до 22°С в течение 12 ч. Реакция проходит с 50% конверсией (ЖХМС). Раствор гасят бикарбонатом натрия и экстрагируют ДХМ. Объединенные органические фазы сушат сульфатом магния, фильтруют и концентрируют. Вещество далее очищают хроматографией на силикагеле, используя градиент ДХМ:EtOAc:MeOH, равный 95:2,5:2,5, далее 92,5:5,0:2,5, далее 85:10:5, для выделения требуемого продукта, соэлюируемого с небольшим количеством примеси. Объединенные фракции концентрируют и очищают препаративной ВЭЖХ с получением 3,4 мг требуемого продукта. МС (ESI(+)) m/e 440,63 (M+H)+.
Пример 26
Ингибирование Hedgehog пути в культуре клеток, используя аналоги циклопамина
Эффекты по уничтожению специфических раковых клеток Hedgehog пути могут быть выявлены с использованием следующего анализа. Клетки С3Н10Т1/2 дифференцируют в остеобласты при контакте с озвученным Hedgehog пептидом (Shh-N). При дифференциации данные остеобласты продуцируют высокие уровни щелочной фосфатазы (АР), которые могут быть измерены в ферментативном анализе (Nakamura et al., 1997 BBRC 237: 465). Соединения, которые блокируют дифференциацию С3Н10Т1/2 в остеобласты (Shh зависимое событие), могут, следовательно, быть идентифицированы посредством восстановления при выработке АР (van der Horst et al., 2003 Bone 33: 899). Детали анализа описаны ниже. Результаты, аппроксимирующие (ЕС50 для ингибирования) анализ дифференциации, показаны ниже в таблице.
Протокол анализа
Культура клеток
С3Н10Т1/2 клетки фибробластов мезодермы мышиных эмбрионов (получены от АТСС) культивируют в базальной МЕМ среде (Gibco/Invitrogen), дополненной 10% инактивированной теплом FBS (Hyclone), 50 ед./мл пенициллина и 50 мкг/мл стрептомицина (Gibco/Invitrogen) при 37°С с 5% СО2 в атмосфере воздуха.
Анализ щелочной фосфатазы
С3Н10Т1/2 клетки помещают в 96-луночные планшеты при плотности 8×103 клеток/лунку. Клетки выращивают до слияния (72 часа). После добавления озвученного Hedgehog (250 нг/мл), (R&D Systems) и/или обработки соединением клетки лизируют в 110 мкл лизисного буфера (50 мМ Трис рН 7,4, 0,1% TritonX100), плашки озвучивают и лизаты продавливают через плашки 0,2 мкм PVDF (Corning). 40 мкл лизатов анализируют на предмет активности АР в растворе щелочного буфера (Sigma), содержащего 1 мг/мл п-нитрофенилфосфата. После инкубации в течение 30 мин при 37°С плашки считывают с помощью считывающего устройства для плашек Envision при 405 нм. Общий белок количественно оценивают с помощью набора для анализа белка ВСА от Pierce в соответствии с инструкциями производителя. Активность АР нормализуют по отношению к общему белку. Отмечают, что «А» указывает на то, что IC50 составляет менее 200 нМ, «В» указывает, что IC50 составляет 200-500 нМ, «С» указывает на то, что IC50>500 нМ.
для ингибирования
Пример 27
Способ препаративной сверхкритической жидкостной хроматографической очистки (SFC)
Описан способ препаративной сверхкритической жидкостной хроматографии для очистки соединений настоящего изобретения.
Используемое программное обеспечение:
SFC: Berger PrepSFC System
Ультрафиолетовый детектор: Knauer Model K-2501
Колонка: Berger 5 микрон диоксид кремния, 20 мм - 250 нм
Условия SFC:
Подвижные фазы: СО2 - 95%; метанол - 5%
Скорость потока: 50,00 мл/мин
Температура колонки: 35°С
Изократика в течение 40 мин при 5% метаноле в сверхкритическом СО2
Объем ввода: 1000 мкл
Концентрации образца обычно проходят при 5,0 мг/мл
Получение образца: образцы растворяют в 20% ДХМ/80% метанола
Продукты элюируют между 25 и 40 мин
Параметры ультрафиолетового детектора
Длина волны = 210 нм; и разрешение = 1,0 нм
Пример 28
Способ жидкостной хроматографии - масс-спектрометрии (ЖХМС)
Описан способ жидкостной хроматографии - масс-спектрометрии для соединений настоящего изобретения.[A] Отчет о способе ввода
Подвижная фаза Waters Alliance 2795 LC
Временная таблица внешних событий Waters Alliance 2795 LC
Входы временной таблицы внешних событий насоса отсутствуют
Параметры ввода Waters Alliance 2795
Параметры автосэмплера Waters Alliance 2795
Waters996 PDA
[B] Экспериментальный отчет
Функция 1: МС сканирования, время 0,00 до 3,50, масс 200,00 до 1000,00 ES+
Включение посредством ссылки
Все из патентов США и опубликованных патентных заявок США, цитируемые в данном описании, включены в него в качестве ссылки.
Эквиваленты
Специалисты в данной области смогут распознать или будут в состоянии выявить, используя не более чем рутинные эксперименты, множество эквивалентов конкретных вариантов осуществления изобретения, описанных в данном документе. Подразумевают, что такие эквиваленты охватываются следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И СПОСОБЫ ИНГИБИРОВАНИЯ ВЗАИМОДЕЙСТВИЯ БЕЛКОВ BCL С КОМПОНЕНТАМИ СВЯЗЫВАНИЯ | 2006 |
|
RU2424230C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ АНАЛОГОВ ЦИКЛОПАМИНА | 2007 |
|
RU2492859C2 |
| ИНГИБИТОР CD73, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2787428C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ ДЛЯ ИНГИБИРОВАНИЯ ВЗАИМОДЕЙСТВИЯ BCL БЕЛКОВ С КОМПОНЕНТАМИ ПО СВЯЗЫВАНИЮ | 2005 |
|
RU2416606C2 |
| АНАЛОГИ ЦИКЛОПАМИНА | 2007 |
|
RU2486194C2 |
| АМИНОПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2010 |
|
RU2636589C2 |
| НОВЫЕ ГИДРОКСИКИСЛОТНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2745430C1 |
| ИНГИБИТОРЫ Axl ДЛЯ ПРИМЕНЕНИЯ В КОМБИНИРОВАННОЙ ТЕРАПИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ, УСТРАНЕНИЯ ИЛИ ЛЕЧЕНИЯ МЕТАСТAЗИРУЮЩЕГО РАКА | 2010 |
|
RU2555326C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| ТРИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ ТРОМБИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2329264C9 |
Настоящее изобретение относится к новым соединениям формулы 1:
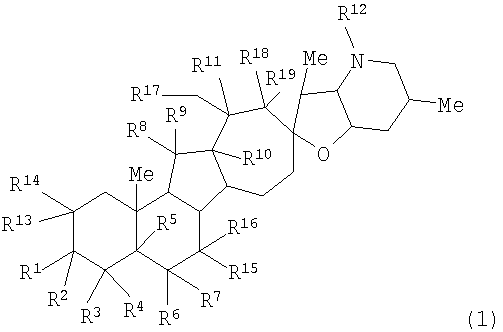
или его фармацевтически приемлемой соли, в котором каждый R1 и R8 независимо представляют собой Н или гидроксил; каждый R2 и R9 независимо представляют собой Н или гидроксил; R5 представляет собой Н; каждый R3, R4, R6, R7, R13 и R14 независимо представляют собой Н; или R1 и R2, взятые вместе, образуют =O; или R4 и R5, взятые вместе, образуют двойную связь; или R5 и R6, взятые вместе, образуют двойную связь; R10 и R11, взятые вместе, образуют двойную связь; R12 представляет собой Н, алкил, гидроксил, аралкил, галогеналкил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-O]qR21 или -[(W)-N(R21)]qR21; где каждый W независимо представляет собой двухвалентный алкильный или аралкильный радикал, a q равен 1, 2, 3 или 4; каждый R15, R16 и R17 независимо представляет собой Н; каждый R18 и R19 независимо представляет собой Н; и каждый R21 независимо представляет собой Н, алкил, арил или аралкил. Изобретение также относится к фармацевтической композиции. Настоящее изобретение предоставляет аналоги циклопамина, которые могут применяться для противодействия фенотипическим эффектам нежелательной активации Hedgehog пути, таким как приобретение функции от Hedgehog, Ptc потеря функции или смягченные приобретенные от функции мутации. Соединения настоящего изобретения являются особенно применимыми при лечении рака. 3 н. и 16 з.п. ф-лы, 1 табл.
1. Соединение формулы 1
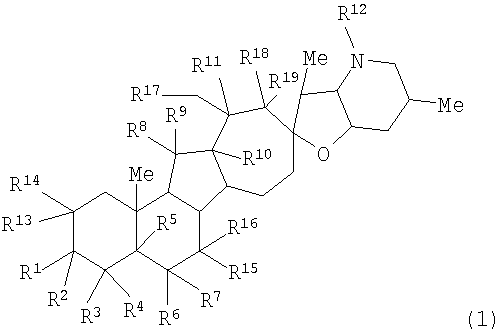
или его фармацевтически приемлемая соль,
в котором каждый R1 и R8 независимо представляет собой Н или гидроксил;
каждый R2 и R9 независимо представляет собой Н или гидроксил;
R5 представляет собой Н;
каждый R3, R4, R6, R7, R13 и R14 независимо представляет собой Н;
или R1 и R2, взятые вместе, образуют =O; или
R4 и R5, взятые вместе, образуют двойную связь; или
R5 и R6, взятые вместе, образуют двойную связь;
R10 и R11, взятые вместе, образуют двойную связь;
R12 представляет собой Н, алкил, гидроксил, аралкил,
галогеналкил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, [(W)-O]qR21 или -[(W)-N(R21)]q 21;
где каждый W независимо представляет собой двухвалентный алкильный или аралкильный радикал, a q равен 1, 2, 3 или 4;
каждый R15, R16 и R17 независимо представляет собой Н;
каждый R18 и R19 независимо представляет собой Н и
каждый R21 независимо представляет собой Н, алкил, арил или аралкил.
2. Соединение по п.1, которое имеет формулу
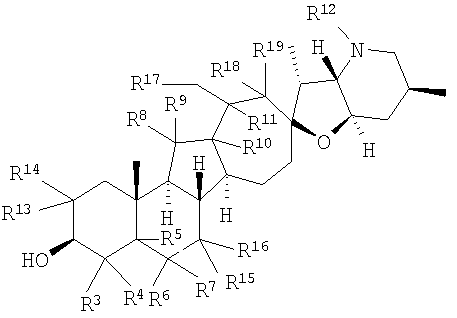 или
или 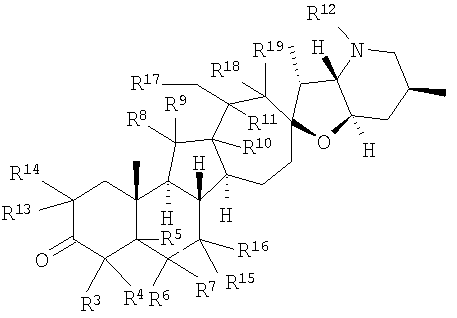
где R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18 и R19 такие, как определено в п.1, или его фармацевтически приемлемая соль.
3. Соединение по п.1, где
R1 представляет собой гидроксил;
или R1 и R2, взятые вместе, образуют =O, и/или
R4 и R5, взятые вместе, образуют двойную связь.
4. Соединение по п.1, где R1 и R2, взятые вместе, образуют =O или R1 представляет собой гидроксил, а R2 представляет собой Н.
5. Соединение по п.1, где R5 и R6, взятые вместе, образуют двойную связь.
6. Соединение по п.1, где R8 и R9 представляют собой водород.
7. Соединение по п.1, где R12 представляет собой Н, алкил, аралкил, галогеналкил, гидроксил или алкоксил.
8. Соединение по п.1 формулы
или 
или его фармацевтически приемлемая соль,
где R12 представляет собой Н, алкил, гидроксил, аралкил, галогеналкил, алкоксил, -[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-O]qR21 или
-[(W)-N(R)]qR21, где каждый W независимо представляет собой двухвалентный алкильный или аралкильный радикал;
q равен 1, 2, 3 или 4;
каждый R21 независимо представляет собой Н, алкил, арил или аралкил.
9. Соединение по п.1, выбранное из группы, состоящей из
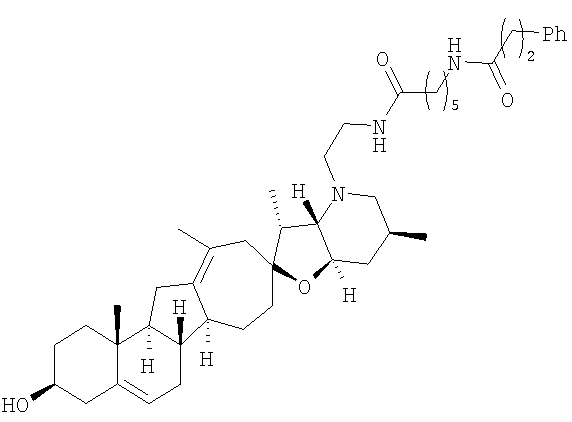
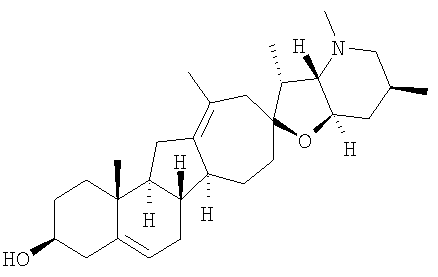
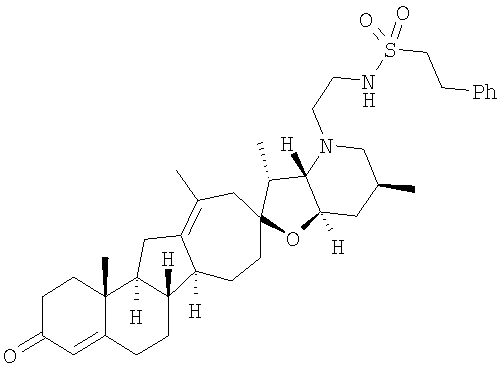
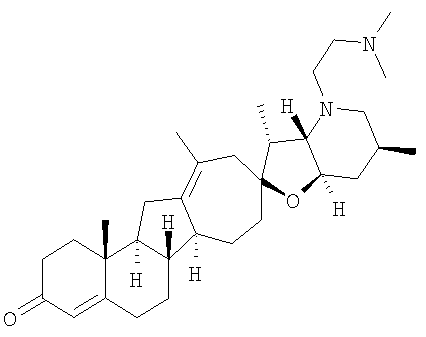

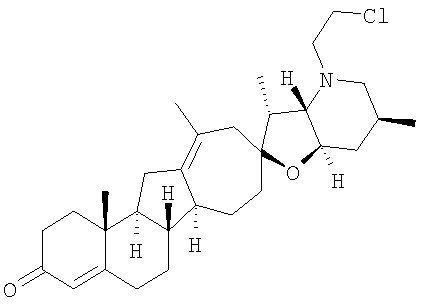

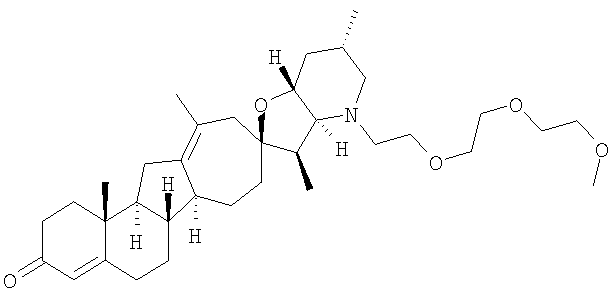
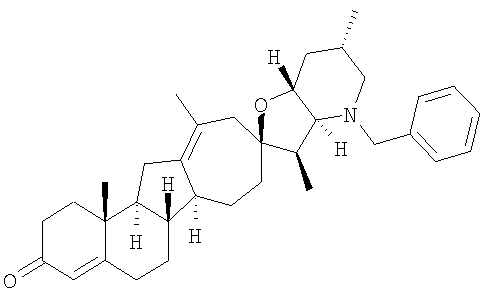
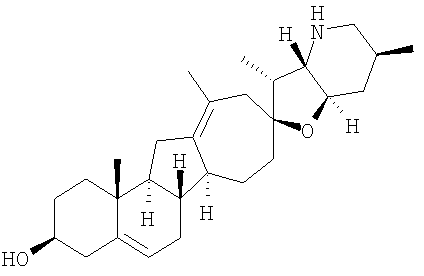
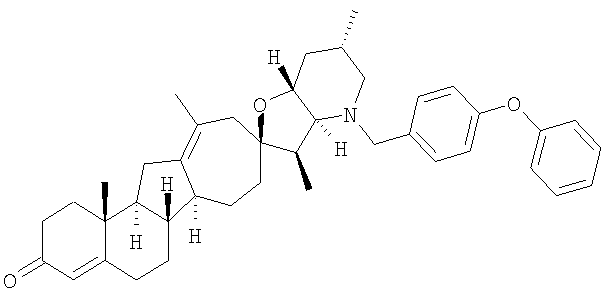
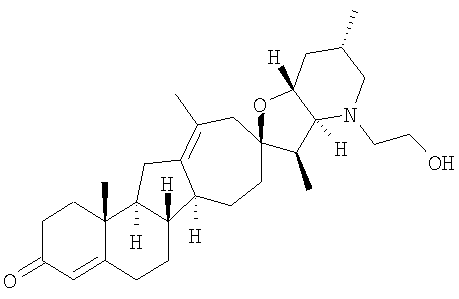
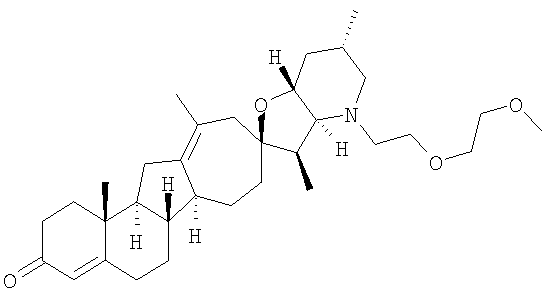
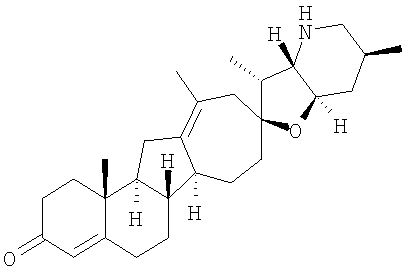
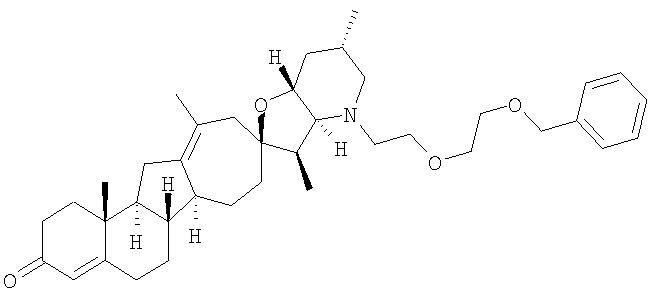
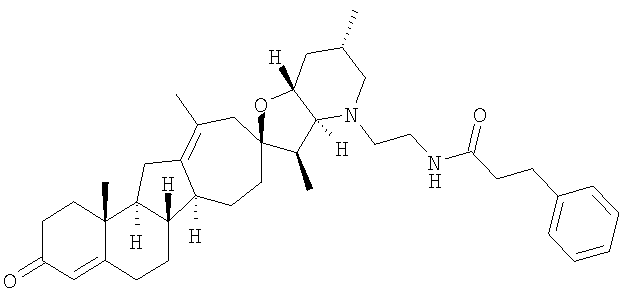
 и
и
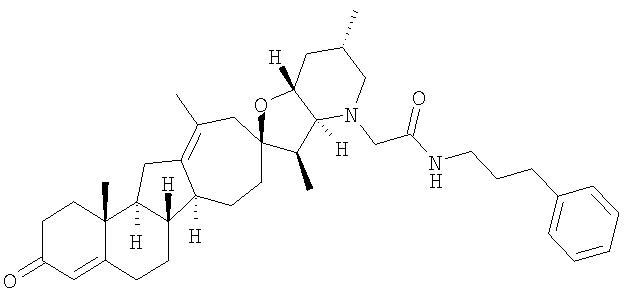
и их фармацевтически приемлемых солей.
10. Соединение формулы 2

или его фармацевтически приемлемая соль,
в котором каждый R1 и R8 независимо представляют собой Н или гидроксил;
каждый R2 и R9 независимо представляет собой Н или гидроксил;
R5 представляет собой Н;
каждый R3, R4, R6, R7, R13 и R14 независимо представляет собой Н;
или R1 и R2, взятые вместе, образуют =O; или
R4 и R5, взятые вместе, образуют двойную связь; или
R5 и R6, взятые вместе, образуют двойную связь;
R10 и R11, взятые вместе, образуют группу, представленную 1b
где Z представляет О или C(R23)(R23);
R12 представляет собой Н, алкил, гидроксил, аралкил, галогеналкил, алкоксил,
-[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-O]qR21 или -[(W)-N(R21)]qR21;
где каждый W независимо представляет собой двухвалентный алкильный или аралкильный радикал, a q равен 1, 2, 3 или 4;
каждый R15, R16 и R17 независимо представляет собой Н;
каждый R21 независимо представляет собой Н, алкил, арил или аралкил и
каждый R23 независимо представляет собой Н.
11. Соединение по п.10, которое имеет формулу
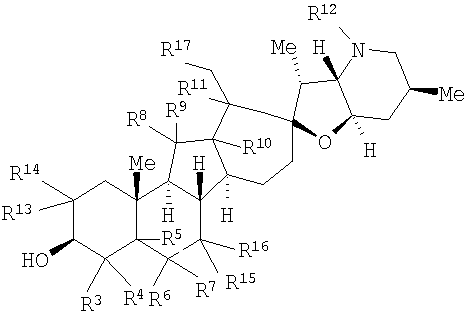
или

где R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16 и R17 такие, как определено в п.10, или его фармацевтически приемлемая соль.
12. Соединение по п.10, где
R1 представляет собой гидроксил; или R1 и R2, взятые вместе, образуют =O; и/или
R4 и R5, взятые вместе, образуют двойную связь.
13. Соединение по п.10, где R5 и R6, взятые вместе, образуют двойную связь.
14. Соединение по п.10, где R8 и R9 представляют собой водород.
15. Соединение по п.10, где R12 представляет собой Н, алкил, аралкил или галогеналкил.
16. Соединение по п.10 формулы
 или
или 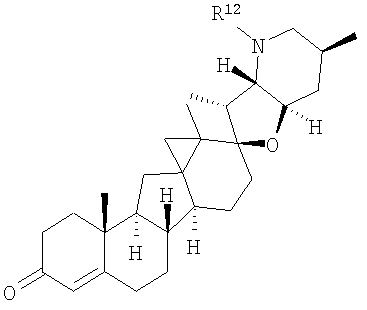
или его фармацевтически приемлемая соль,
где R12 представляет собой Н, алкил, гидроксил, аралкил, галогеналкил, алкоксил,
-[(W)-N(R21)C(O)]qR21, -[(W)-N(R21)SO2]qR21, -[(W)-O]qR21 или -[(W)-N(R21)]qR, где каждый W независимо представляет собой двухвалентный алкильный или аралкильный радикал; и
q равен 1, 2, 3 или 4; и
каждый R21 независимо представляет собой Н, алкил, арил или аралкил.
17. Соединение по п.10, выбранное из группы, состоящей из
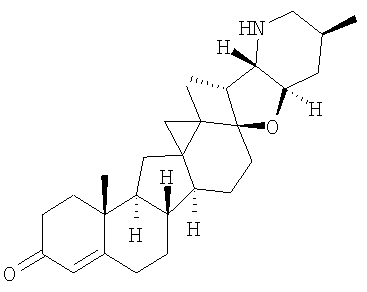
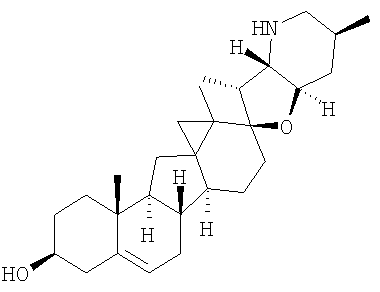 и
и 
и их фармацевтически приемлемых солей.
18. Фармацевтическая композиция, обладающая противораковой активностью, содержащая соединение по любому из пп.1-17; и фармацевтически приемлемый наполнитель.
19. Соединение по любому из пп.1-17 для применения в качестве лекарственного средства для лечения рака.
| US 6238876 B1, 29.05.2001 | |||
| WO 9518856 A1, 13.07.1995 | |||
| RU 99121846 C2, 20.07.2001. |

