Родственные заявки
Данная заявка испрашивает приоритет заявки США порядкового номера 60/878018, поданной 28 декабря 2006, и заявки США порядкового номера 60/941596, поданной 1 июня 2007; причем обе заявки во всей полноте включены посредством ссылки в данную заявку.
Правительственное фондирование
Некоторая часть описанной здесь работы была выполнена при поддержке правительства в виде гранта № К23 СА107040, присужденного NIH/NCI (национальным институтом здравоохранения/национальным онкологическим институтом). Правительство обладает определенными правами на изобретение.
Предпосылки создания изобретения
Настоящее изобретение в целом относится к способам антагонизации сигнального пути hedgehog и способам лечения различных состояний с использованием аналогов циклопамина.
Было показано, что ингибирование сигнального пути hedgehog при некоторых видах рака приводит к ингибированию роста опухоли. Например, было показано, что анти-hedgehog антитела антагонизируют функцию сигнального пути hedgehog и ингибируют рост опухоли. Также было показано, что ингибирование активности сигнального пути hedgehog малыми молекулами приводит к гибели клеток некоторых видов рака.
Исследование в этой области было главным образом сфокусировано на выяснении биологии сигнального пути hedgehog и на обнаружении новых ингибиторов сигнального пути hedgehog. Хотя ингибиторы сигнального пути hedgehog были установлены, все еще существует потребность в более сильных ингибиторах сигнального пути hedgehog.
В PCT публикации WO 2006/026430, опубликованной 9 марта 2006 года и переуступленной тому же правопреемнику, что и настоящая заявка, описывается большое разнообразие аналогов циклопамина, фокусируя внимание на соединениях с ненасыщенностью в кольцах А и В. В настоящей заявке удивительно сильные аналоги содержат полностью насыщенные кольца А и В.
Сущность изобретения
Настоящее изобретение относится к способам лечения гиперпролиферативных нарушений и состояний, опосредуемых сигнальным путем hedgehog.
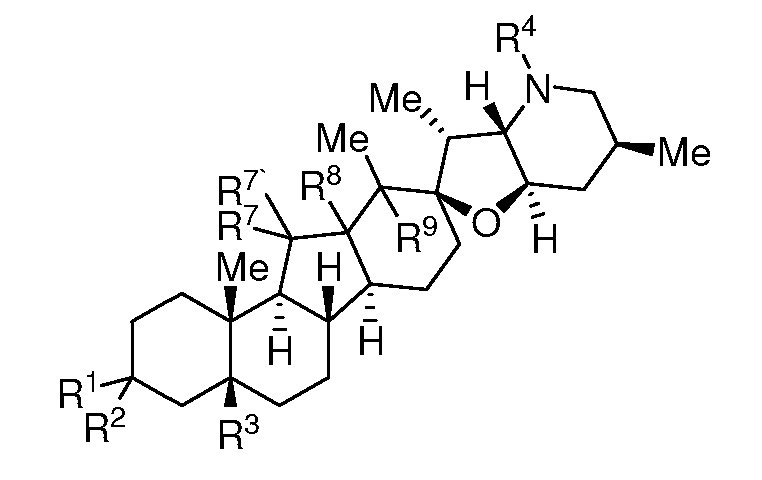
Согласно одному аспекту, изобретение относится к способу лечения гиперпролиферативного нарушения. Способ включает введение пациенту эффективного количества соединения, имеющего нижеследующую формулу:
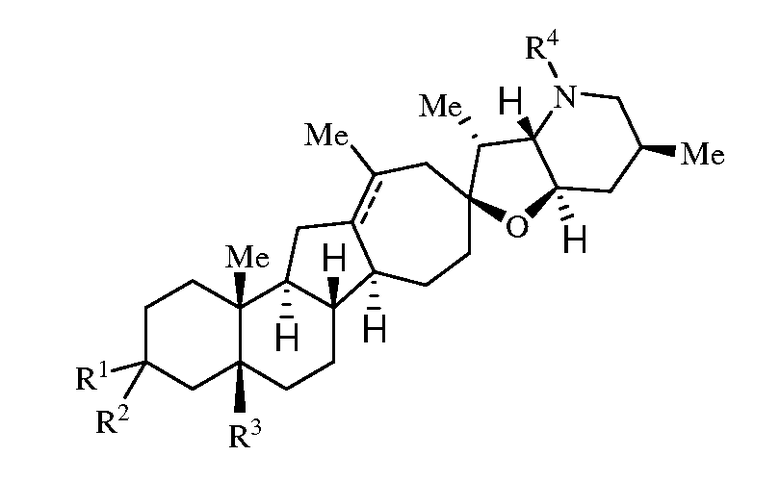
или его фармацевтически приемлемой соли;
где R1 представляет собой H, алкил, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, -N(R5)C(O)R5 или сахар;
R2 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, нитрил или гетероциклоалкил;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R), =N(NR2), =C(R)2;
R3 является H, алкилом, алкенилом или алкинилом;
R4 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, -OR5, -C(O)R5, -CO2R5, -SO2R5, -C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5 -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5 -[(W)-SO2]qR5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5, -W-NR5 3 +X- или -[(W)-S]qR5; где каждый символ W в каждом случае независимо представляет собой бирадикал;
каждый символ q в каждом случае независимо принимает значения 1, 2, 3, 4, 5 или 6;
X- является галоидом;
каждый R, независимо, представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил или аралкил;
каждый R5 в каждом случае независимо представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[C(R)2]P-R6;
где p принимает значения 0-6; или
где любые два R5 у одного и того же заместителя могут вместе образовывать необязательно замещенное 4-8 членное кольцо, которое содержит 0-3 гетероатома, выбираемых из атомов N, O, S и P;
каждый R6 независимо является гидроксилом, -N(R)COR, -N(R)C(O)OR, -N(R)SO2(R), -C(O)N(R)2, -OC(O)N(R)(R), -SO2N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(О)2OR, -S(О)2OR, -OP(O)(OR)(OR), -NP(O)(OR)(OR) или -P(O)(OR)(OR).
В некоторых вариантах осуществления, когда R2, R3 и R4 представляют собой атом H; R1 не является гидроксилом или сахаром.
В некоторых вариантах осуществления, когда R4 является гидроксилом, тогда R1 не является сахаром или гидроксилом, а R1 и R2 вместе не являются С=О.
В некоторых вариантах осуществления, R1 является сульфонамидо.
Состояние может быть выбрано из группы, состоящей из раков кожи, раков центральной нервной системы, раков желудочно-кишечного тракта, раков легочной системы, мочеполовых раков, рака молочной железы, гепатоцеллюлярного рака, рака мозга и раков кроветворной системы.
Согласно другому аспекту, изобретение относится к способу лечения состояния, опосредуемого сигнальным путем hedgehog. Способ включает введение пациенту эффективного количества соединения, имеющего формулу:
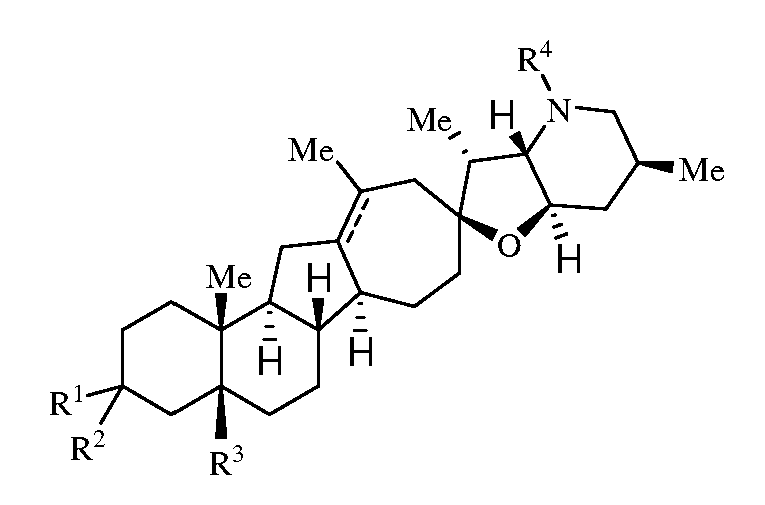
или его фармацевтически приемлемой соли;
где R1 представляет собой H, алкил, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, - N(R5)C(O)R5 или сахар;
R2 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, нитрил или гетероциклоалкил;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R), =N(NR2), =C(R)2;
R3 является H, алкилом, алкенилом или алкинилом;
R4 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, -OR5, -C(O)R5 -CO2R5, -SO2R5, -C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5, -[(W)-SO2]4R5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5, -W-NR5 3 +X- или -[(W)-S]qR5;
где каждый символ W в каждом случае независимо является бирадикалом; каждый символ q независимо для каждого случая принимает значения 1, 2, 3, 4, 5 или 6;
X- представляет собой галоид;
каждый R независимо является атомом H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом или аралкилом;
каждый R5 в каждом случае независимо является атомом H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом или -[C(R)2]P-R6;
где p принимает значения 0-6; или
где любые два R5 у одного и того же заместителя могут вместе образовывать необязательно замещенное 4-8 членное кольцо, которое содержит 0-3 гетероатома, выбираемых из атомов N, O, S и P;
каждый R6 независимо представляет собой гидроксил, -N(R)COR, -N(R)C(O)OR, -N(R)SO2(R), -C(O)N(R)2, -OC(O)N(R)(R), -SO2N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(О)2OR, -S(О)2OR, -OP(O)(OR)(OR), -NP(O)(OR)(OR) или -P(O)(OR)(OR).
В некоторых вариантах осуществления, когда R2, R3 и R4 представляют собой H; R1 не является гидроксилом или сахаром.
В некоторых вариантах осуществления, когда R4 является гидроксилом, тогда R1 не является сахаром или гидроксилом, и R1 вместе с R2 не представляют C=O.
В некоторых вариантах осуществления, R1 является сульфонамидо.
Состояние может быть выбрано из группы, состоящей из раков кожи, раков центральной нервной системы, раков желудочно-кишечного тракта, раков легочной системы, мочеполовых раков, рака молочной железы, гепатоцеллюлярного рака, рака мозга и раков кроветворной системы. Конкретные примеры включают мелкоклеточный рак легких, рак поджелудочной железы, медуллобластому, множественную миелому, лейкоз, миелодиспластический синдром, неходжкинскую лимфому и болезнь Ходжкина. Соединения могут быть введены перорально, внутривенно или местно.
Согласно еще одному аспекту, изобретение относится к способу антагонизации сигнального пути hedgehog у пациента. Способ включает введение пациенту эффективного количества соединения имеющего формулу:

или его фармацевтически приемлемой соли;
где R1 представляет собой H, алкил, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, -N(R5)C(O)R5 или сахар;
R2 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, нитрил или гетероциклоалкил;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R), =N(NR2), =C(R)2;
R3 является H, алкилом, алкенилом или алкинилом;
R4 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, -OR5, -C(O)R5, -CO2R5, -SO2R5, -C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5 -[(W)-SO2]qR5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, [(W)-N(R)]qR5, -W-NR5 3 +X- или -[(W)-S]qR5; где каждый символ W независимо в каждом случае представляет собой бирадикал; каждый символ q независимо в каждом случае принимает значения 1, 2, 3, 4, 5 или 6;
X- является галоидом;
каждый R независимо является атомом H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом или аралкилом;
каждый R5 независимо в каждом случае представляет собой атом H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[C(R)2]P-R6;
где p принимает значения от 0 до 6; или
где любые два R5 у одного и того же заместителя вместе могут образовывать 4-8 членное необязательно замещенное кольцо, которое содержит 0-3 гетероатома, выбираемых из атомов N, O, S и P;
каждый R6 независимо представляет собой гидроксил, -N(R)COR, -N(R)C(O)OR, -N(R)SO2(R), -C(O)N(R)2, -OC(O)N(R)(R), -SO2N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(О)2OR, -S(О)2OR, -OP(O)(OR)(OR), -NP(O)(OR)(OR) или -P(O)(OR)(OR).
В некоторых вариантах осуществления, когда R2, R3 и R4 представляют собой H; R1 не является гидроксилом или сахаром.
В некоторых вариантах осуществления, когда R4 является гидроксилом, тогда R1 не является сахаром или гидроксилом, и R1 и R2 вместе не являются C=O.
В описанных выше вариантах осуществления соединение может быть выбрано из группы, состоящей из:


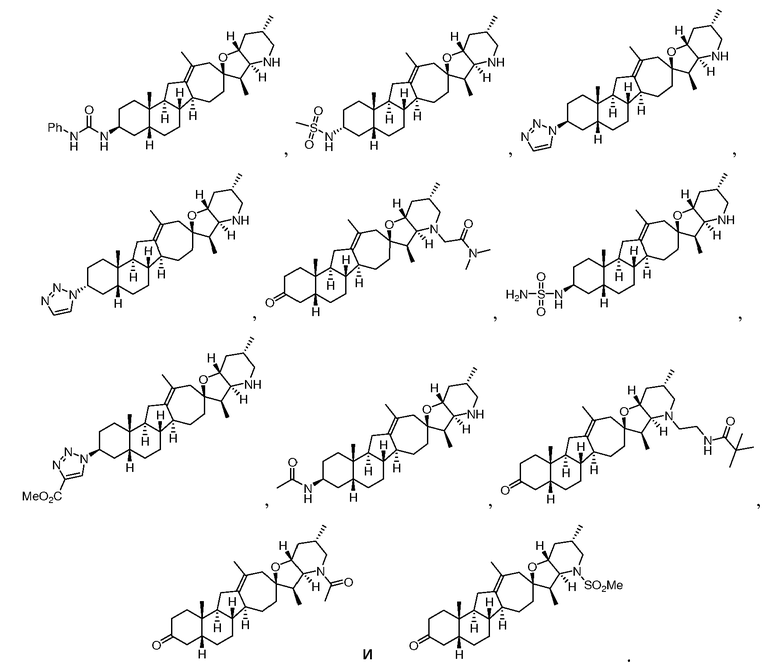
или его фармацевтически приемлемой соли.
Согласно еще одному аспекту изобретение относится к способу антагонизации у пациента сигнального пути hedgehog. Способ включает введение пациенту эффективного количества соединения, имеющего нижеследующую структуру:
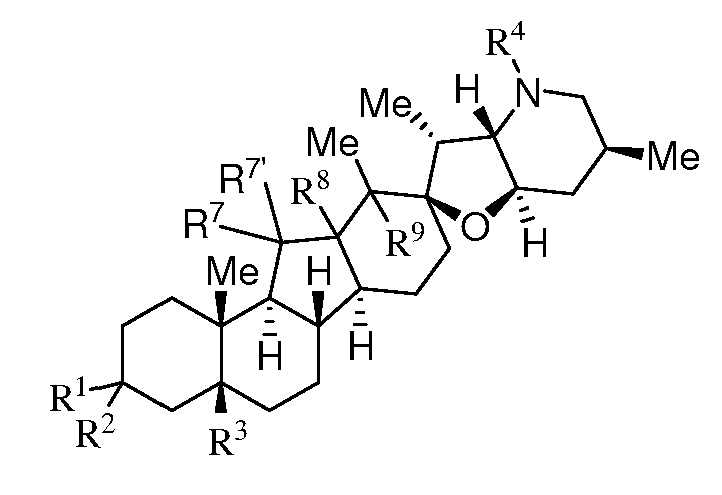
или его фармацевтически приемлемой соли;
где R1 представляет собой H, алкил, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, N(R5)C(O)R5 или сахар;
R2 представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, нитрил или гетероциклоалкил;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R)-, =N(NR2), =C(R)2;
R3 является атомом H, алкилом, алкенилом или алкинилом;
R4 представляет собой атом H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галогеналкил, -OR5, -C(O)R5, -CO2R5, -SO2R5, -C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5, -[(W)-SO2]qR5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5, -W-NR5 3 +X- или [(W)-S]qR5;
где каждый символ W независимо является бирадикалом;
каждый символ q независимо принимает значения 1, 2, 3, 4, 5 или 6;
X- является галоидом;
каждый R независимо представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил или аралкил;
каждый R5 независимо представляет собой H, алкил, алкенил, алкинил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -[C(R)2]P-R6;
где p принимает значения от 0 до 6; или
любые два R5 у одного и того же заместителя могут вместе образовывать 4-8 членное необязательно замещенное кольцо, которое содержит 0-3 гетероатома, выбираемых из атомов N, O, S и P;
каждый R6 независимо представляет собой гидроксил, -N(R)COR, -N(R)C(O)OR, -N(R)SO2(R), -C(O)N(R)2, -OC(O)N(R)(R), -SO2N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(О)2OR, -S(О)2OR, -OP(O)(OR)(OR), -NP(O)(OR)(OR) или -P(O)(OR)(OR) где каждый R независимо является атомом H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом или аралкилом;
каждый из R7 и R7' является атомом H; или
R7 и R7' вместе образуют =O;
каждый R8 и R9 является атомом H, или R8 и R9 вместе образуют связь; и
при условии что, когда R3, R4, R8, R9 представляет собой атом H; и R7 и R7' вместе образуют =O; R1 не может быть гидроксилом, а R2 не может быть H;
при условии что, когда R3, R4, R8, R9 являются атомом H, и R7 и R7' вместе образуют =O; R1 не может быть ацетатом, а R2 не может быть атомом H;
при условии что, когда R3, R4, R8, R9 являются атомом H; и R7 и R8 являются атомом H; R1 и R2 вместе не могут представлять =О, и
при условии что, когда R3, R4, R8, R9 являются атомом H; и R7 и R7' представляют собой атом H; R1 и R2 не могут быть атомом H.
В некоторых вариантах осуществления, R1 представляет собой сульфонамид.
Подробное описание изобретения
Определения
Определения терминов, используемых здесь, предназначены для введения современных определений, общепризнанных для каждого термина в химической и фармацевтической области. Там, где это уместно, приведены иллюстрации на примерах. Определения применимы к терминам, которые использованы по всему настоящему описанию изобретения, пока их не ограничивают конкретными примерами, или индивидуально или как части большей группы.
Используемое здесь определение для каждого выражения, например, алкил, m, n, и так далее, при его появлении в любой структуре более одного раза, задают как независимое от его определения где-либо в той же самой структуре.
Термин "ациламино" обозначает группу, которая может быть представлена общей формулой:
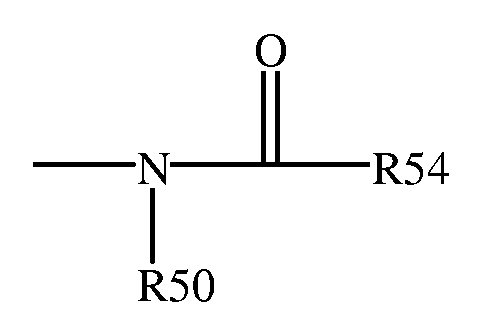
в которой R50 и R54 представляют собой водород, алкил, алкенил или -(CH2)m-R61, где R61 представляет арил, циклоалкил, циклоалкенил, гетероцикл или полицикл; а m является нулем или целым числом в интервале от 1 до 8.
Термины "алкенил" и "алкинил" относятся к ненасыщенным алифатическим группам, аналогичным по длине и возможному замещению алкилам, описанным ниже, но которые содержат, по крайней мере, одну двойную или тройную связь, соответственно.
Термины "алкоксил" или "алкокси" относятся к алкильной группе, определенной ниже, имеющей присоединенный к ней кислородный радикал. Характерные алкоксильные группа включают метокси-, этокси-, пропилокси-, трет-бутоксигруппы и тому подобное.
Термин "алкил" обозначает радикал насыщенных алифатических групп, включающий неразветвленные алкильные группы, алкильные группы с разветвленной цепью, циклоалкильные (алициклические) группы, алкилзамещенные циклоалкильные группы и циклоалкилзамещенные алкильные группы. В некоторых вариантах осуществления, алкил с неразветвленной или с разветвленной цепью имеет 30 или менее углеродных атомов в основной цепи (например, C1-C30 для неразветвленной цепи, C3-C30 для разветвленной цепи), 20 или менее углеродных атомов.
Аналогично, некоторые циклоалкилы имеют 3-10 углеродных атомов в их циклической структуре, а другие имеют 5, 6 или 7 атомов углерода в циклической структуре.
Термин "алкилтио" относится к алкильной группе определенной выше, имеющей присоединенный к ней серосодержащий радикал. В некоторых вариантах осуществления, "алкилтио" фрагмент представляют одним из радикалов -S-алкил, -S-алкенил, -S-алкинил, и -S-(CH2)m-R61, где m и R61 определены выше. Представитель алкилтиогрупп включает метилтио, этилтио и тому подобное.
Термин "амидо", определяемый в данной области техники как аминозамещенный карбонил и включает фрагмент, который может быть представлен общей формулой:

где R50 и R51, каждый независимо, представляют собой водород, алкил, алкенил, -(CH2)m-R61; или R50 и R51, ваместе с атомом N, к которому они присоединены, завершают гетероцикл, имеющий в циклической структуре от 4 до 8 атомов;
R61 представляет собой арил, циклоалкил, циклоалкенил, гетероцикл или полицикл; и «m» представляют собой нуль или целое число, находящееся в интервале от 1 до 8. Некоторые варианты осуществления амида по настоящему изобретению не будут включать имиды, которые могут оказаться неустойчивыми.
Термина "амин" и "амино" идентифицируют в данной области техники и обозначают как незамещенные, так и замещенные амины, а именно, как фрагмент, который может быть представлен общими формулами:
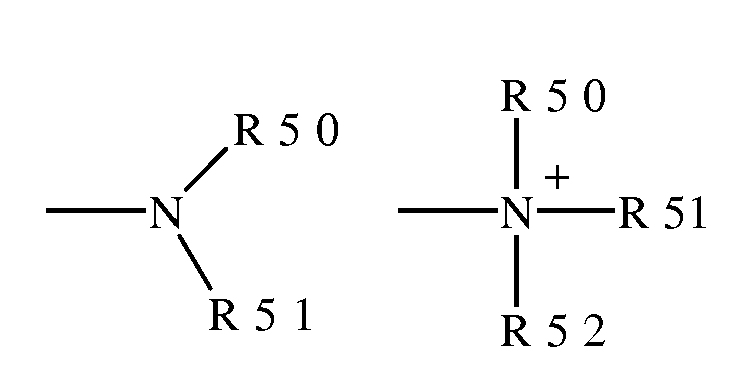
где R50, R51 и R52, каждый независимо, представляет собой водород, алкил, алкенил, -(CH2)m-R61; или R50 и R51, вместе с атомом N, к которому они присоединены, завершают гетероцикл, имеющий от 4 до 8 атомов в циклической структуре; R61 представляет собой арил, циклоалкил, циклоалкенил, гетероцикл или полицикл; а m является нулем или целым числом, находящемся в интервале от 1 до 8. Таким образом, термин "алкиламин" включает аминогруппу, определенную выше, имеющую присоединенный к ней замещенный или незамещенный алкил, то есть, по крайней мере, один из R50 и R51 представляет собой алкильную группу.
Термин "аралкил", используемый здесь, обозначает алкильную группу, замещенную арильной группой (например, ароматической или гетероароматической группой).
Термин "арил" используемый здесь включает 5-, 6- и 7-членную моноциклическую ароматическую группу, которая может содержать от нуля до четырех гетероатомов, например, такую как бензол, антрацен, нафталин, пирен, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин и пиримидин и тому подобное. Эти арильные группы, содержащие гетероатомы в циклической структуре, также можно обозначать как "арильные гетероциклы" или "гетероароматические системы". Ароматическое кольцо может быть замещено у одного или нескольких положений кольца такими заместителями, как описанные выше, например, галогеном, азидом, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, гидроксилом, алкоксилом, амино, нитро, сульфгидрилом, имино, амидо, фосфонатом, фосфинатом, карбонилом, карбоксилом, силилом, простым эфиром, алкилтио, сульфонилом, сульфонамидо, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматической или гетероароматической группой, -CF3, -CN или тому подобным. Термин "арил" также включает полициклические системы, имеющие два или более колец в циклической системе, в которой два или более атомов углерода являются общими для двух соседних колец (кольца представляют собой "конденсированные кольца"), где, по крайней мере, одно из колец является ароматическим, другие кольца циклической системы могут, например, представлять собой циклоалкилы, циклоалкенилы, циклоалкинилы, арилы и/или гетероциклилы.
Термин "кислота Бренстеда обозначает любое вещество, которое может функционировать как донор водородного иона (протона).
Термин "карбоксил" включает такие фрагменты, которые могут быть представлены общими формулами:
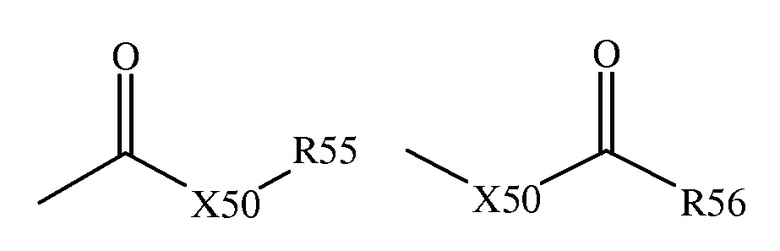
в которых X50 является связью или представляет атом кислорода или серы; а каждый из R55 и R56 независимо представляет атом водорода, алкил, алкенил, -(CH2)m-R61 или фармацевтически приемлемую соль, где m и R61 определены выше.
Термин "бирадикал" относится к любой из ряда двухвалентных групп из алкильной, алкенильной, алкинильной, арильной, циклоалкильной, гетероциклоалкильной, аралкильной, гетероарильной и гетероаралкильной групп. Например:  является алкильным бирадикалом;
является алкильным бирадикалом; 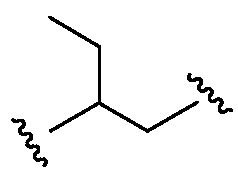 также является алкильным бирадикалом;
также является алкильным бирадикалом;  является аралкильным бирадикалом; и
является аралкильным бирадикалом; и  является (алкил)гетероаралкильным бирадикалом. Характерные примеры включают алкилены общей структуры (CH2)X, где X принимает значения 1-6, и соответствующие алкениленовый и алкиниленовый линкеры, имеющие 2-6 углеродных атомов и одну или несколько двойных или тройных связей; циклоалкиленовые группы, содержащие 3-8 членов кольца и аралкильные группы, в которых одна открытая валентность находится у арильного кольца, а одна - у алкильной части, такие как показано ниже:
является (алкил)гетероаралкильным бирадикалом. Характерные примеры включают алкилены общей структуры (CH2)X, где X принимает значения 1-6, и соответствующие алкениленовый и алкиниленовый линкеры, имеющие 2-6 углеродных атомов и одну или несколько двойных или тройных связей; циклоалкиленовые группы, содержащие 3-8 членов кольца и аралкильные группы, в которых одна открытая валентность находится у арильного кольца, а одна - у алкильной части, такие как показано ниже: 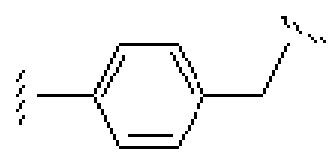 и их изомеры.
и их изомеры.
Термин "эффективное количество" относится к количеству соединения, которое при введении, как часть требуемой схемы введения, вызывает желаемый эффект, например, изменение в скорости пролиферации клеток и/или уровня выживания клетки, в соответствии с клинически приемлемыми нормами для нарушения, подвергаемого лечению.
Термин "галогеналкил", используемый здесь, относится к алкильной группе, где заменены на галоид от 1 до всех атомов водорода. Термин "пергалогеналкил" означает, что все водороды заменены на галоид.
Термин "гетероатом", используемый здесь, означает атом любого элемента, оличного от атомов углерода или водорода. Примеры гетероатомов включают атомы бора, азота, кислорода, фосфора, серы и селена.
Термины "гетероциклил" или "гетероциклическая группа" обозначают 3-10-членные циклические структуры, в некоторых примерах 3-7-членные кольца; эти циклические структуры содержат от одного до четырех гетероатомов. Гетероциклы также могут представлять собой полициклы. Гетероциклильные группы включают, например, тиофен, тиантрен, фуран, пиран, изобензофуран, хромен, ксантен, феноксатиин, пиррол, имидазол, пиразол, изотиазол, изоксазол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, индол, индазол, пурин, хинолизин, изохинолин, хинолин, фталазин, нафтиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карболин, фенантридин, акридин, пиримидин, фенантролин, феназин, фенарсазин, фенотиазин, фуразан, феноксазин, пирролидин, оксолан, тиолан, оксазол, пиперидин, пиперазин, морфолин, лактоны, лактамы, такие как азетидиноны и пирролидиноны, султамы, сультоны и тому подобное. Гетероцикл может быть замещен у одного или нескольких положений такими заместителями, как описанные выше, как например, галогеном, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, гидроксилом, амино, нитро, сульфгидрилом, имино, амидо, фосфонатом, фосфинатом, карбонилом, карбоксилом, силилом, простым эфиром, алкилтио, сульфонилом, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматической или гетероароматической группой, -CF3, -CN или тому подобным.
Термин "выделенный" в связи с соединением по настоящему изобретению, означает соединение, не находящиеся в клетке или организме, и соединение, отделенное от некоторых или всех компонентов, которые обычно сопровождают его в природе.
Термин "кислота Льюиса" относится к любому веществу, которое может функционировать как акцептор электронной пары.
До тех пор пока число атомов углерода не задано иначе, термин "низший алкил", используемый здесь, означает алкильную группу, определенную выше, но имеющую от одного до десяти атомов углерода, в некоторых вариантах осуществления от одного до шести атомов углерода в структуре ее основной цепи. Аналогично, "низший алкенил" и "низший алкинил" имеют такую же длину цепи. Некоторые алкильные группы являются низшими алкилами. в некоторых вариантах осуществления, заместитель, обозначенный здесь как алкил, является низшим алкилом.
При использовании здесь, термин "нитро" означает -NO2; термин "галоген" обозначает -F, -CI, -Br или -I; термин "сульфгидрил" означает -SH; термин "гидроксил" означает -OH; и термин "сульфонил" означает -SO2-.
Термин "оксо" обозначает карбонильный кислород (=O).
Термины "полициклил" или "полициклическая группа" обозначают два или более колец (например, циклоалкилы, циклоалкенилы, циклоалкинилы, арилы и/или гетероциклилы), в которых два или более атомов углерода являются общими для двух соседних колец, например, кольца являющиеся "конденсированными кольцами". Кольца, которые связаны через несоседние атомы, называются "мостиковыми" кольцами. Каждое из колец полицикла может быть замещено такими заместителями, как описанные выше, как например, галоген, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, кетон, альдегид, сложный эфир, гетероциклил, ароматическая или гетероароматическая часть, -CF3, -CN или тому подобное.
Термин "эпимерно чистый", относящийся к соединению по настоящему изобретению, означает, что соединение, по существу, не содержат своих стереоизомеров c обращенной конфигурацией стереогенного центра, с которым связан R3. Например, эпимерно чистое соединение представляет нижеследующая формула:
где R1, R2, R3, R4, R7, R7', R8 и R9 являются такими же, как определено ниже и, по существу, не содержит соединений, представляемых нижеследующей формулой:
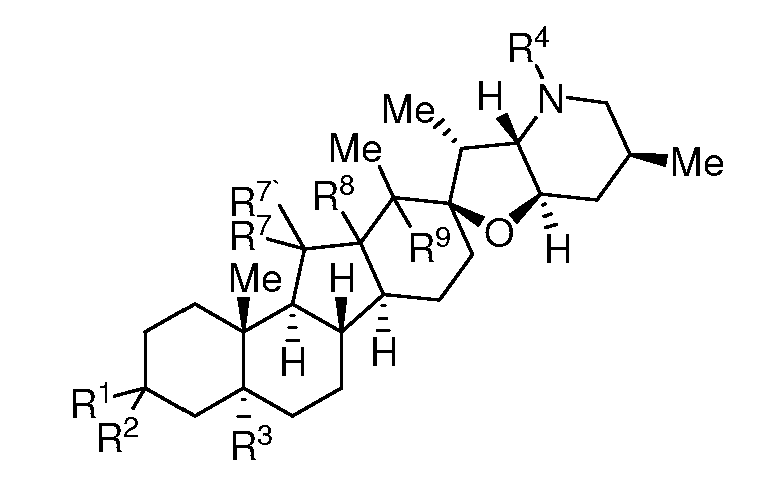
где R1, R2, R3, R4, R7, R7', R8 и R9 являются такими же как определено ниже. Эпимерно чистые соединения содержат менее, чем приблизительно 20% масс., менее, чем, приблизительно, 15% масс., менее, чем, приблизительно, 10% масс., менее, чем, приблизительно, 5% масс., или менее, чем, приблизительно, 3% масс. стереоизомерных соединений, у которых конфигурация стереогенного центра, с которым связан R3, является обращенной по отношению к данному соединению.
Фраза "защитная группа", используемая здесь, обозначает временные заместители, которые защищают потенциально реакционноспособную функциональную группу от нежелательных химических превращений. Примеры таких защитных групп включают сложные эфиры карбоновых кислот, силиловые простые эфиры спиртов и ацетали и кетали альдегидов и кетонов, соответственно. Имеются обзорные работы по химии защитных групп (Greene, T. W.; Wuts, P.G.M., Protective Groups in Organic Синтез, 2nd ed,; Wiley: New York, 1991). В некоторых случаях защищаемую функциональную группу и защитную группу обозначают вместе как один фрагмент. Например, фрагмент, показанный ниже, иногда обозначают как бензилкарбонат; то есть защищаемый (подчеркнуто) «О» (кислород) составляет часть карбоната.

Анолагично, фрагмент, показанный ниже, в котором защищаемый N (азот) составляет часть карбамата, обозначен как бензилкарбамат.
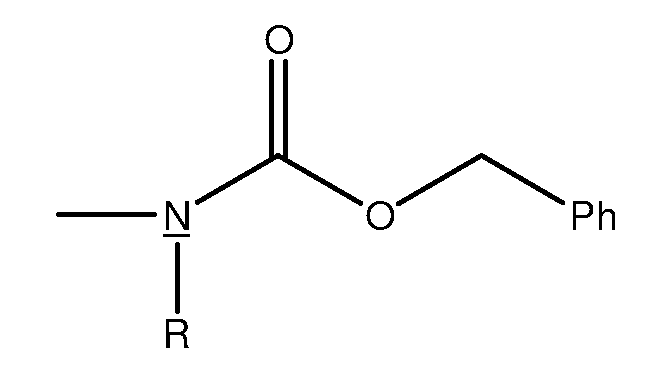
Термин "сахар" используемый здесь, относится к природному или неприродному моносахариду, дисахариду или олигосахариду, содержащему один или несколько пиранозных или фуранозных колец. Сахар может быть ковалентно связан со стероидным алкалоидом по настоящему изобретению через простую эфирную связь или через алкильную связь. В некоторых вариантах осуществления сахаридный фрагмент может быть ковалентно связан со стероидным алкалоидом по настоящему изобретению у аномерного центра сахаридного кольца. Сахара могут включать, среди прочих, рибозу, арабинозу, ксилозу, ликсозу, аллозу, альтрозу, маннозу, гулозу, идозу, галактозу, талозу, глюкозу и трегалозу.
Термин "сульфонамидо" или "сульфонамид" используемый здесь, включает фрагмент, имеющий любую из нижеследующих формул:

где R50 определен выше.
Термины "трифлил", "тозил", "мезил" и "нонафлил" обозначают трифторметансульфонильную, п-толуолсульфонильную, метансульфонильную, и нонафторбутансульфонильную группы, соответственно. Термины "трифлат", "тозилат", "мезилат" и "нонафлат" обозначают сложноэфирные функциональные группы трифторметансульфоната, п-толуолсульфоната, метансульфоната и нонафторбутансульфоната и, соответственно, молекулы, которые содержат эти группы.
Термин "тиоксо" обозначает карбонильную серу (=S).
Будет понятно, что термины "замещение" или "замещенный" включают неявное условие, что такое замещение осуществляется согласно допустимой валентности замещенного атома и заместителя, и что замещение приводит к устойчивому соединению, которое, например, не испытывает самопроизвольного превращения, такого как через перегруппировку, циклизацию, элиминирование и так далее.
Некоторые соединения по настоящему изобретению могут находиться в определенных геометрических или стереоизомерных формах. В настоящем изобретении рассматривают все такие соединения, включая цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, как входящие в объем изобретения. В таком заместителе, как алкильная группа, могут присутствовать дополнительные асимметрические атомы углерода. Все такие изомеры, так же как их смеси, подразумевают включенными в данное изобретение.
Как изложено выше, некоторые варианты осуществления настоящих соединений могут содержать основную функциональную группу, такую как амино или алкиламино, и, таким образом, способны к образованию фармацевтически приемлемых солей с фармацевтически приемлемыми кислотами. Термин "фармацевтически приемлемые соли" в этом отношении обозначает относительно нетоксичные, неорганические и органические кислотно-аддитивные соли соединений по настоящему изобретению. Эти соли могут быть получены in situ во введенном носителе или при производственном процессе получения дозированных форм, или посредством отдельного взаимодействия очищенного соединения по изобретению в форме свободного основания с пригодной органической или неорганической кислотой и выделения во время последующей очистки образованной таким образом соли. Характерные соли включают такие соли, как гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонат, и тому подобное (Смотри, например, Berge, et al. "pharmaceutical salts", J. Pharm. Sci. (1977) 66:1-19).
Фармацевтически приемлемые соли соединений по настоящему изобретению включают общепринятые нетоксичные соли или четвертичные аммонийные соли соединений, например, из нетоксичных органических или неорганических кислот. Например, такие общепринятые нетоксичные соли включают соли, полученные из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, ортофосфорная, азотная и тому подобное; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфокислота, метансульфоновая, этандисульфоновая, щавелевая, изотионовая и тому подобное.
В других случаях, соединения по настоящему изобретению могут содержать одну или несколько кислотных функциональных групп и, таким образом, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин "фармацевтически приемлемый соли" в этих примерах обозначает относительно нетоксичные, неорганические и органические основно-аддитивные соли соединений по настоящему изобретению. Аналогично, эти соли могут быть получены in situ во вводимом носителе или при производственном процессе получения дозированных форм, или посредством отдельного взаимодействия очищенного соединения по изобретению в форме его свободной кислоты с соответствующим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла; с аммиаком, или с фармацевтически приемлемым первичным, вторичным или третичным органическим амином. Характерные соли щелочных или щелочноземельных металлов включают соли лития, натрия, калия, кальция, магния и алюминия, и тому подобное. Характерные органические амины, применимые для образования основно-аддитивных солей, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и тому подобное (Смотри, например, Berge, et al., supra).
Синтез соединений стероидных алкалоидов
Производные стероидных алкалоидов с расширенным циклом, описанные выше, могут быть получены непосредственно из природных стероидных алкалоидов или их синтетических аналогов. В отдельных примерах исходными веществами, являющимися стероидными алкалоидами, могут быть циклопамин или иервин. Эти стероидные алкалоиды можно приобрести коммерческим путем или экстрагировать из Veratrum Californicum. Вкратце, способ по настоящему изобретению включает стадии циклопропанирования соответствующих исходных производных стероидных алкалоидов, с последующей перегруппировкой с расширением цикла производных циклопропила. В некоторых примерах может оказаться желательным перед циклопропанированием соответствующим образом защитить или иначе преобразовать реакционноспособные функциональные группы, присутствующие в молекуле. Например, спиртовая группа, находящаяся у R1 и вторичный азот, находящийся на конденсированном фуранопиперидиновом кольце, оба могут быть защищены перед циклопропанированием. В некоторых вариантах осуществления, защитные группы, которые эффективно присоединяют и удаляют из алкалоида, в синтетическом способе дают промежуточные соединения с улучшенными свойствами для переработки, что предполагает эффективную очистку образованных синтетических промежуточных соединений, и могут оказаться предпочтительными.
Примеры защитных групп кислорода включают, среди прочих, формиат, ацетат, хлорацетат, дихлорацетат, трихлорацетат, пивалоат, бензоаты, алкилкарбонат, алкенилкарбонат, арилкарбонаты, аралкилкарбонат (например, бензилкарбонат), 2,2,2-трихлорэтилкарбонат, простой алкоксиметиловый эфир, простой аралкоксиметиловый эфир, простой алкилтиометиловый эфир, простой аралкилтиоэфир, простой арилтиоэфир, простой триалкилсилиловый эфир, простой алкиларилсилиловый эфир, простой бензиловый эфир, простой арилметиловый эфир, и простой аллиловый эфир.
Примеры защитных групп азота включают, среди прочих, формил, хлорацетил, трихлорацетил, трифторацетил, фенилацетил, бензоилы, бензамиды, алкилкарбаматы, аралкилкарбаматы (например, бензилкарбаматы), арилкарбаматы, аллил, аралкил, алкоксиметил, аралкоксиметил, N-2-цианоэтил, диарилфосфинамиды, диалкилфосфинамидаты, диарилфосфинамидаты и триалкилсилил.
Дополнительные защитные группы, которые могут быть использованы в способах по настоящему изобретению, описаны в Green, T. W.; Wuts, P. G., Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. 1999.
Множество циклопропанирующих агентов могут быть использованы для циклопропанирования стероидного алкалоида. 1,1-Галогеналкилметаллические комплексы и реакционноспособные группы, обозначаемые как карбеноиды, обычно используют для циклопропанирования олефинов. Эти реагенты обычно получают, используя дииодалкан или диазоалкан и металлы или органические вещества, такие как Et2Zn, i-Bu3Al, самарий, медь, родий или палладий. В некоторых вариантах осуществления, Et2Zn и дииодметан используют для образования 1,1-галогеналкилметаллов.
Химическая активность и легкость переработки 1,1-галогеналкилцинковых комплексов могут быть модифицированы добавлением некоторых реагентов, таких как кислоты. Полагают, что при добавлении кислоты к 1,1-галогеналкилцинковым соединениям образуется алкилцинковая смешанная соль. В примерах, описанных ниже, диарилортофосфорную кислоту смешивают с дииодметаном и диэтилцинком с образованием предполагаемого циклопропанирующего агента, галогеналкилцинкфосфата. Множество фосфорных кислот может быть использовано для образования предполагаемого галогеналкилцинкфосфата.
Также могут быть использованы другие известные способы циклопропанирования, такие как способы, использующие выходы серы для взаимодействия с олефином, сопряженным с карбонилом, с присоединением CH2- или CH-алкильной или CH-арильной группы; и катализируемое металлом разложение диазоалкильных и α-диазокарбонильных соединений, таких как диазометан и этилдиазоацетат. Этими способами легко получить циклопропаны, имеющие алкильные, арильные, алкоксикарбонильные (-COOR) или ацильные заместители. Дополнительные агенты циклопропанирования описаны в работах Masalov, et al, Organic Letters (2004) 6:2365-2368; и Hansen, et al., Chem. Comm. (2006) 4838-4840.
Циклопропильное кольцо может быть замещенным или незамещенным. В случаях, когда циклопропильное кольцо замещено, группы, присоединенные к метилену циклопропана после перегруппировки и расширения цикла, будут расположены у D кольца.
Реакциии циклопропанирования могут проходить в апротонном растворителе. Подходящие растворители включают простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, диглим, трет-бутилметиловый эфир, тетрагидрофуран и тому подобное; галогенированные растворители, такие как хлороформ, дихлорметан, дихлорэтан и тому подобное; алифатические или ароматические углеводородные растворители, такие как бензол, ксилол, толуол, гексан, пентан и тому подобное; сложные эфиры и кетоны, такие как этилацетат, ацетон, и 2-бутанон; полярные апротонные растворители, такие как ацетонитрил, диметилсульфоксид, диметилформамид, и тому подобное; или комбинации двух или более растворителей. В некоторых вариантах осуществления, дихлорметан является растворителем, применяемым для циклопропанирования, когда используют диалкилцинк и дииодметан.
В примерах, описанных ниже, раствор, содержащий циклопропанирующий агент, получают, сначала добавляя раствор ортофосфорной кислоты к раствору диэтилцинка, с последующим добавлением к реакционному раствору дииодметана. Затем к этому раствору добавляют субстрат для циклопропанирования. Альтернативно, циклопропанирующий агент может быть получен в присутствии субстрата циклопропанирования, изменяя порядок добавления реагентов. В некоторых вариантах осуществления реакцию циклопропанирования проводят, добавляя сначала ортофосфорную кислоту к раствору диалкилцинка, с последующим добавлением субстрата циклопропанирования, и, в заключение, добавляют дигалогеналкан. Используя этот способ, циклопропанирующий агент получают в контролируемых условиях, и он немедленно взаимодействует с субстратом циклопропанирования.
После синтеза центрального каркаса циклопропанированного стероидного алкалоида соединение может быть преобразовано, используя множество реакций изменения функциональности, известных в данной области техники. Характерные примеры включают реакции сочетания палладия с алкенилгалогенидами или арилгалогенидами, реакции окисления, реакции восстановления, реакции с нуклеофилами, реакции с электрофилами, перициклические реакции, радикальные реакции, введение защитных групп, удаление защитных групп, и тому подобное.
В присутствии кислот Льюиса или Бренстеда аналоги циклопропила претерпевают перегруппировку и расширение кольца с получением аналогов стероидных алкалоидов, у которых D кольцо расширено на один углеродный атом.
Циклопропанирование и расширение кольца могут проходить как двухстадийный процесс, протекающий в одном реакционном сосуде или как двухстадийный процесс, протекающий в двух реакционных сосудах. Когда циклопропанирование и расширение цикла проводят в одном и том же реакционном сосуде, то после завершения реакции циклопропанирования добавляют кислоту, чтобы инициировать перегруппировку с расширением кольца. При определенных условиях, соли цинка, образующиеся в ходе циклопропанирования стероидного алкалоида, сами могут действовать как кислоты Льюиса, катализирующие перегруппировку с расширением кольца. Химическую активность солей цинка, образовавшихся после циклопропанирования, можно менять, добавляя кислоты, чтобы генерировать более активные кислоты Льюиса.
Как описано ниже в разделе примеров, после завершения реакции циклопропанирования в реакционный сосуд для циклопропанирования добавляют метансульфоновую кислоту. Дополнительные примеры подходящих кислот включают, среди прочих, соли цинка, соединения бора, соли магния, соли титана, соли индия, соли алюминия, соли олова, соли лантана, трифторметансульфоновую кислоту, диарилоксифосфорную кислоту, уксусную кислоту, и HCl. В некоторых вариантах осуществления изобретения, используемой кислотой Льюиса является соль цинка или BF3.
У этих аналогов с расширенным циклом можно далее менять функциональность, используя множество известных в данной области реакций изменения функциональности. Характерные примеры включают реакции сочетания палладия с алкенилгалогенидами или арилгалогенидами, реакции окисления, реакции восстановления, реакции с нуклеофилами, реакции с электрофилами, перициклические реакции, радикальные реакции, введение защитных групп, удаление защитных групп и тому подобное.
Фармацевтические композиции
Соединения, описанные здесь, могут быть сформированы в композицию, пригодную для введения, используя один или несколько фармацевтически приемлемых носителей (добавок) и/или разбавителей. Фармацевтические композиции могут быть специально сформированы для введения в твердой или жидкой форме, включая композиции, приспособленные для нижеследующих путей введения: (1) перорального введения, например, препараты для орошения (водными или неводными растворами или суспензиями), таблетки, например, таблетки, предназначенные для буккального, сублингвального введения и системной абсорбции; капсулы, шарики, порошки, гранулы, пасты для нанесения на язык; (2) парентерального введения, например, подкожного, внутримышечного, внутривенного или эпидруального инъецирования, например, стерильного раствора или суспензии, или препараты замедленного высвобождения; (3) местного нанесения, например, в виде крема, мази или медицинского пластыря с контролируемым высвобождением или аэрозоля, наносимого на кожу; (4) внутривагинального или внутриректального введения, например, в виде пессария, крема или пены; (5) сублингвального введения; (6) введения через глаз; (7) трансдермального введения; (8) пульмонального введения или (9) назального введения.
Примеры пригодных водных и неводных носителей, которые могут быть использованы в фармацевтической композиции, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и тому подобное), и их пригодные смеси, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Надлежащая текучесть может быть сохранена, например, посредством использования покрывающих веществ, таких как лецитин, путем сохранения необходимого размера частицы в случае дисперсионных систем, и посредством использования поверхностно-активных веществ.
Эти композиции могут также содержать такие адъюванты, как консерванты, увлажняющие агенты, эмульгаторы, диспергирующие агенты, лубриканты, и/или антиоксиданты. Защита от действия микроорганизмов соединений, описанных здесь, может быть гарантирована добавлением разнообразных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенолсорбиновой кислоты, и тому подобного. Может также оказаться желательным включение в композиции изотонических агентов, таких как сахара, хлорид натрия и тому подобное. Кроме того, пролонгированное поглощение инъецируемой фармацевтической формы может быть осуществлено путем включения агентов, которые замедляют поглощение, таких как моностеарат алюминия и желатин.
Способы получения этих препаратов или композиций включают стадию внесения в ассоциацию соединения с носителем и, необязательно, одного или нескольких дополнительных ингредиентов. В целом, препараты получают, непрерывно и равномерно внося в ассоциацию соединение с жидкими носителями, или мелкодисперсными твердыми носителями, или и теми и другими, и затем, при необходимости, формуя продукт.
Когда соединения, описанные здесь, вводят в форме фармацевтических средств людям и животным, их могут давать в чистом виде или в виде фармацевтической композиции, содержащей, например, приблизительно 0,1-99% или приблизительно 10-50%, или приблизительно 10-40%, или приблизительно 10-30%, или приблизительно 10-20%, или приблизительно 10-15% активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Фактические дозировочные уровни активных ингредиентов в фармацевтических композициях можно варьировать, таким образом, чтобы получить количество активного ингредиента, которое, не будучи токсичным для пациента, является эффективным для достижения желаемой ответной трапевтической реакции для конкретных пациента, композиции и способа введения.
Выбираемый уровень дозирования будет зависеть от множества факторов, включая активность конкретного используемого соединения или его сложного эфира, соли или амида, пути введения, времени введения, скорости экскреции или метаболизма конкретно используемого соединения, скорости и степени абсорбции, продолжительности лечения, других лекарственных средств, соединений и/или веществ, используемых в комбинации с конкретно используемым соединением, возраста, пола, массы тела, состояния болезни, общего состояния здоровья и предшествующей истории болезни больного, подвергаемого лечению, и тому подобных факторов, хорошо известных в медицине.
Вообще, пригодной ежедневной дозой соединения, описанного здесь, будет такое количество соединения, которое является самой низкой дозой, эффективной для продуцирования терапевтического эффекта. Такая эффективная доза будет, как правило, зависеть от факторов, описанных выше. Как правило, дозы соединений для перорального, внутривенного и подкожного введений пациенту, применяемые для получения указанных эффектов, будут находиться в интервале от, приблизительно, 0,0001 до, приблизительно, 200 мг, или от, приблизительно, 0,001 до, приблизительно, 100 мг, или от, приблизительно, 0,01 до, приблизительно, 100 мг, или от, приблизительно, 0,1 до, приблизительно, 100 мг, или от, приблизительно, 1 до, приблизительно, 50 мг на килограмм массы тела в день.
Соединения могут быть введены ежедневно, через день, три раза в неделю, дважды в неделю, еженедельно, или раз в две недели. Режим дозирования может включать "отдых от лекарств", то есть лекарственное средство могут вводить в течение двух недель с перерывом в одну неделю, или вводить три недели с перерывом в одну неделю, или вводить четыре недели с перерывом в одну неделю и так далее, или вводить непрерывно без «отдыха от лекарств». Соединения могут быть введены перорально, внутривенно, интраперитонально, местно, трансдермально, внутримышечно, подкожно, интраназально, сублингвально или любым другим путем.
Пациентом, принимающим такое лечение, является любое животное, в нем нуждающееся, включая приматов, в частности, человека, и других млекопитающих, таких как лошади, крупный рогатый скот, свиньи и овцы; и домашнюю птицу и в целом комнатных домашних животных.
Способы лечения
Сигнальный путь Hedghog является существенным на многих стадиях развития, особенно при формировании лево-правой симметрии. Потеря или уменьшение сигнального пути hedghog ведет к множественной недостаточности развития и врожденным порокам, одним из самых поразительных из которых является циклопия.
Было показано, что многие опухоли и пролиферативные состояния зависят от сигнального пути hedgehog. Лечение соединениями, описанными здесь, может воздействовать на рост таких клеток и выживаемость. В последнее время сообщали, что мутации, активирующие сигнальный путь hedgehog, происходят при спорадическом базально-клеточном раке (Xie, et al, Nature (1998) 391:90-92) и незрелых нейроэктодермальных опухолях центральной нервной системы (Reifenberger, et al., Рак Res (1998) 58:1798-1803). На многочисленных видах рака, таких как раки желудочно-кишечного тракта, включая рак поджелудочной железы, рак пищевода и рак желудка (Berman, et al, Nature (2003) 425:846-851; Thayer, et al, Nature (2003) 425:851-856); рак легких (Watkins, et al, Nature (2003) 422:313-317); рак простаты (Karhadkar, et al, Nature (2004) 431:707-712; Sheng, et al, Molecular Cancer (2004) 3:29-42; Fan, et al., Endocrinology (2004) 145:3961-3970); рак молочной железы (Kubo et al Cancer Research (2004) 64:6071-6074, Lewis, et al, Journal of Mammary Gland Biology and Neoplasia (2004) 2:165-181); и гепатоцеллюлярный рак (Sicklick, et al, ASCO conference (2005), Mohini, et al, AACR conference (2005)), была также показана неуправляемая активация сигнального пути hedgehog.
Например, было показано, что ингибирование малыми молекулами сигнального пути hedgehog ингибирует рост базально-клеточного рака (Williams, et al, PNAS (2003) 100:4616-4621), медуллобластомы (Berman, etal, Science (2002) 297:1559-1561), рака поджелудочной железы (Berman, et al, Nature (2003) 425:846-851), желудочно-кишечных раков (Berman, et al, Nature (2003) 425:846-851, published PCT application WO 05/013800), рака пищевода (Berman, et al, Nature (2003) 425:846-851), рака легких (Watkins, et al, Nature (2003) 422:313-317), и рака простаты (Karhadkar, et al, Nature (2004) 431:707-712).
Было дополнительно показано, что при многих видах рака имеется неуправляемая активация сигнального пути hedgehog, например, для таких видов рака, как рак молочной железы (Kubo, et al, Cancer Research (2004) 64:6071-6074); гепатоцеллюлярный рак (Patil, et al, 96th Annual AACR conference, abstract #2942 (2005); Sicklick, et al, ASCO annua] meeting, abstract #9610 (2005)); гемобластозы (Watkins and Matsui, unpublished results); базально-клеточный рак (Bale & Yu, Human Molec. Genet. (2001) 10:757-762; Xie, et al, Nature (1998) 391:90-92), медуллобластома (Pietsch, et al, Cancer Res. (1997) 57:2085-2088) и рак желудка (Ma, et al, Carcinogenesis, May 19, 2005 (Epub) (2005)). Кроме того, исследователи обнаружили, что ингибирование малыми молекулами сигнального пути hedgehog дает улучшение симптомов псориаза (Tas, et ai, Dermatology (2004) 209:126-131). Как показано на примерах, соединения, описанные здесь, демонстрировали модуляцию сигнального пути hedgehog, и выбранные соединения демонстрировали ингибирование роста опухоли. Поэтому полагают, что эти соединения могут оказаться применимыми для лечения множества гиперпролиферативных нарушений, таких как разнообразные виды раков.
Пролиферативные нарушения, которые можно лечить, используя описанные здесь способы, включают: рак легких (включая мелкоклеточный рак легких и немелкоклеточный рак легких), другие виды рака легочной системы, медуллобластому и другие раки мозга, рак поджелудочной железы, базально-клеточный рак, рак молочной железы, рак простаты и другие мочеполовые раки, опухоль гастроинтестинальной стромы (GIST) и другие виды рака желудочно-кишечного тракта, рак толстой кишки, рак кишечника, рак яичников, раки кроветворной системы (включая множественную миелому, острый лимфолейкоз, острый миелоидный лейкоз, хронический миелоидный лейкоз, хронический лимфолейкоз, Ходжкинскую лимфому, и неходжкинскую лимфому, и миелодиспластический синдром), полицитиемию Vera, макроглобулинемию Вальденстрема, заболевание тяжелой цепи, саркомы мягких тканей, такие как фибросаркому, миксосаркому, липосаркому, хондросаркому, остеогенную саркому, хордому, ангиосаркому, эндотелиосаркому, лимфоангиосаркому, лимфоангиоэндотелиальную саркому, синовиому, мезотелиому, опухоль Юнга, лейомиосаркому, рабдомиосаркому, плоскоклеточный рак, базально-клеточный рак, меланому и другие кожные раки, аденокарциному, рак потовых желез, рак сальных желез, папиллокарциному, папиллярные аденокарциномы, стаденокарциному, медуллярный рак, бронхогенный рак, рак почки, гепатому, рак желчных протоков, хориокарциному, семиному, эмбриональный рак, опухоль Вильямса, цервикальный рак, рак матки, тестикулярный рак, рак мочевого пузыря, и другие мочеполовые виды рака, эпителиальный рак, глиому, астроцитому, медуллобластому, карниофарингиому, эпендиому, пинеалому, гемангиобластому, невриному слухового нерва, олигодендроглиому, менингиому, нейробластому, ретинобластому, рак эндометрия, фолликулярную лимфому, диффузную крупно-В-клеточную лимфому, лимфому клеток тканей, одевающих спорангий, гепатоцеллюлярный рак, рак щитовидной железы, рак желудка, рак пищевода, рак шеи и головы, мелкоклеточные виды рака, эссенциальную тромбоцитемию, идиопатическую миелоидную метаплазию, синдром гиперэозинофилии, системный мастоцитоз, семейную гиперэозинофилию, хронический эозинофильный лейкоз, рак щитовидной железы, нейроэндокринные раки и карциноидные опухоли. Дополнительные нарушения включают синдром Горлина-Гольца и псориаз.
Пациент, получающий это лечение, является любым животным, в нем нуждающимся, включая приматов, в частности, человека и других млекопитающих, таких как лошади, крупный рогатый скот, свиньи и овцы; и домашнюю птицу и комнатных домашних животных в целом.
Ингибиторы сигнального пути hedgehog, описанные здесь, можно комбинировать с другими видами лечения рака. Например, их можно совмещать с хирургическими видами лечения; облучением; биотерапией (а именно, интерферонами, цитокинами, например, интерферон α, интерферон γ, и фактор некроза опухолей, гемопоэтические факторы роста, моноклональная серотерапия, вакцины и иммуностимуляторы;) лечением антителами (например, препаратами авастин, эрбитукс, ритуксан, и бексар); эндокринной терапией (включающей пептидные гормоны, кортикостероиды, эстрогены, андрогены и ингибиторы ароматазы); лечением анти-эстрогенами (например, препаратами тамоксифен, ралоксифен, и мегестрол); лечением агонистами LHRH (релизинг-фактора лютеинизирующего гормона) (например, препаратами госкрклин и леупролидацетат); лечением анти-андрогенами (например, препаратами флутамид и бикалутамид); генной терапией; пересадкой костного мозга; фотодинамическими видами терапии (например, используя препараты вертопрофин (BPD-MA), фталоцианин, фотосенсибилизатор Pc4, и деметоксигипокреллин A (2BA-2-DMHA)) и химиотерапией.
Примеры химиотерапии включают гемцитабин, метотрексат, таксол, меркаптопурин, тиогуанин, гидроксимочевину, цитрабин, циклофосфамид, ифосфамид, нитрозомочевины, цисплатин, карбоплатин, ммитомицин, дакарбазин, прокарбизин, этопозиды, преднизалон, дексаметазон, цитарабин, кампатецины, блеомицин, докорубицин, идарубицин, даунорубицин, дактиномицин, пликамицин, митоксантрон, аспарагиназа, винбластин, винкристин и винорелбин. Дополнительные агенты включают азотистый иприт (например, циклофосфамид, ифосфамид, трофосфамид, хлорамбуцил, эстрамустин и мелфалан); нитрозомочевины (например, кармустин (BCNU) и ломустин (CCNU)); алкилсульфонаты (например, бусулфан и треосулфан); триазены (например, дакарбазин и темозоломид); соединения, содержащие платину, (например, цис-платин, карбоплатин, и оксалиплатин); алкалоиды барвинка (например, винкристин, винбластин, виндезин и винорелбин); таксоиды (например, паклитаксел и доцетаксол), эпиподофилины (например, этопозид, тенипозид, топотецин, 9-аминокамптотецин, камптоиринотекан, криснатол, митомицин C и митомицин C), антиметаболиты, ингибиторы DHFR (дегидрофолат-редуктазы) (например, метотрексат и триметрексат); ингибиторы IMP дегидрогеназы (например, микофенольная кислота, тиазофурин, рибварин, и EICAR), ингибиторы рибонуклеотидредуктазы (например, гидроксимочевина и деферохамин); аналоги урацила (например, Фторурацил, флоксиридин, доксифлуридин, ратитрексед и капецитабин); аналоги цитозина (например, цитарабин (araC) цитозин арабинозид, и флударабин); аналоги пурина (например, меркаптопурин и Тиогуанин), аналоги Витамина D3 (например, EB 1089, CB 1093, и KH 1060), ингибиторы изопренилирования (например, ловастатин), дофаминергические нейротоксины (например, ион 1-метил-4-фенилпиридиния); ингибиторы клеточного цикла (например, стауроспорин); актиномицины (например, актиномицин D и дактиномицин); блеомицины (например, блеомицин A2, блеомицин B2, и пепломицин); антрациклины (например, даунорубицин, докорубицин (адриамицин), идарубицин, эпирубицин, пирарубицин, зорубицин и митоксантрон); ингибиторы MDR (мультирезистентность) (например, верапамил); ингибиторы Ca2+ АТФ-азы (например, тапсигарджин, иматиниб, талидомид, леналидомид, эрлотиниб, гефитиниб, сорафениб и санитиниб), и ингибиторы протеасомы, включающие бортезомиб.
Когда ингибиторы hedgehog, описанные здесь, вводят в комбинации с другими видами лечения, таким как дополнительные виды терапевтического лечения, или совместно с облучением или хирургическим лечением, дозы каждого агента или вида терапии будут в большинстве случаев ниже, чем соответствующая доза в терапии, использующей один агент. Также, вообще говоря, ингибиторы hedgehog, описанные здесь, и второй терапевтический агент не обязаны быть введены в одной и той же фармацевтической композиции, и могут, вследствие различных физических и химических характеристик, быть введены разными путями. Например, одно соединение может быть введено перорально, тогда как второе терапевтическое средство вводят внутривенно. Определение путей введения и целесообразности введения, где это возможно, в одной и той же фармацевтической композиции, находится в полной компетенции квалифицированного практикующего врача. Первоначальное введение может быть выполнено, в соответствии с известными учрежденными протоколами в данной области техники, и затем, на основе наблюдаемых эффектов, дозировка, пути введения и количество введений могут быть откорректированы квалифицированным практикующим врачом.
В зависимости от природы пролиферативного заболевания, состояния больного и фактического выбора для введения пациенту второго терапевтического агента и/или облучения, ингибитор hedgehog и второй терапевтический агент, и/или облучение могут быть введены параллельно (например, одновременно; по существу, одновременно; или согласно одному и тому же протоколу лечения) или последовательно (то есть, одно следом за другим, с необязательным временным интервалом между ними).
Если ингибитор сигнального пути hedgehog, и второй терапевтический агент и/или облучение не вводят одновременно или, по существу, одновременно, тогда для различных состояний оптимальный порядок введения может быть различным. Таким образом, в некоторых ситуациях ингибитор сигнального пути hedgehog может быть введен первым, с последующим введением второго терапевтического агента и/или облучения; а в других ситуациях второй терапевтический агент и/или облучение могут быть введены первыми, с последующим введением ингибитора сигнального пути hedgehog. Такое альтернативное введение может быть повторено в процессе единого протокола лечения. Определение порядка введения, и числа повторных введений каждого терапевтического агента, в процессе протокола лечения, находится полностью в компетенции квалифицированного практикующего врача после определения им заболевания, подвергаемого лечению, и оценки состояния больного. Например, второй терапевтический агент и/или облучение могут быть введены первыми, в особенности, если это цитотоксичный агент; и затем лечение продолжают введением ингибитора hedgehog и, при установлении благоприятного эффекта, с последующим введением второго терапевтического агента и/или облучения, и так далее до завершения протокола лечения.
Иллюстративные примеры
Описанное в целом изобретение будет более понятным с помощью ссылок на нижеследующие примеры, которые включены единственно с целью иллюстрации некоторых сторон и вариантов осуществления настоящего изобретения, и не предназначены для ограничения самого изобретения.
Пример 1
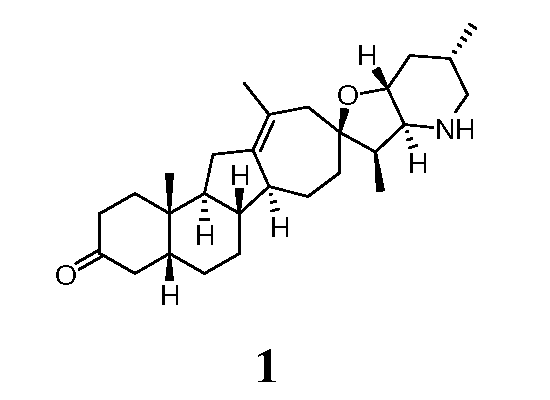
Стадия A

Перекристаллизованный циклопамин 2 (14,1 г, 34,0 ммоль, 1 экв.) растворяют в безводном DCM (дихлорметан) (70 мл) и безводном MeOH (29 мл). Прозрачный раствор охлаждают и добавляют триэтиламин (10,4 г, 102,7 ммоль, 3 экв.) с последующим добавлением бензилхлорформиата (6,20 г, 36,3 ммоль, 1,1 экв.) После того как добавление завершено, раствор 30 мин перемешивают на ледяной бане. На протяжении 3 часов добавляют три порции бензилхлорформиата (3×0,35 г, 3,46 ммоль, 0,03 экв.). Реакцию медленно гасят водой (71 мл), при этом поддерживая температуру ниже 20oC. До отстаивания и разделения слоев, смесь 15 мин перемешивают. Органический слой сушат над сульфатом натрия и фильтруют. Объединенный фильтрат буферизуют безводным пиридином (30 мл), концентрируют, и растворитель заменяют дополнительным количеством безводного пиридина (43 мл) и концентрируют.
Раствор соединения в пиридине (43 мл) далее разбавляют дополнительным количеством безводного пиридина (85 мл). К реакционной смеси медленно добавляют триметилацетилхлорид (8,3 г, 68,7 ммоль, 2 экв.), и реакционную смесь нагревают до 45°C. Реакционную смесь 30 мин перемешивают при 45°C. Реакционную смесь охлаждают, и реакцию гасят, добавляя безводный MeOH (4,5 мл). Загашенную реакционную смесь 40 мин перемешивают при комнатной температуре, затем разбавляют толуолом (97 мл), и последовательно обрабатывают водой (35 мл) и 10% масс. водным раствором карбоната натрия (100 мл). После интенсивного перемешивания слои разделяют, и органический слой дважды промывают водой (2×100 мл), сушат над сульфатом натрия, и фильтруют. Осадок на фильтре промывают толуолом (49 мл) и отбрасывают. Объединенные фильтраты концентрируют, и растворитель обменивают с концентрированием на толуол (145 мл), концентрируя далее до сухого состояния. Продукт перекристаллизовывают из толуола и гептана. Кристаллический продукт отделяют фильтрованием с отсосом, промывают холодным гептаном и сушат до постоянной массы, чтобы получить 15,1 г требуемого продукта.
Стадия В

Бис(2,6-диметилфенил)фосфат (10,65 г, 34,8 ммоль, 3,1 экв.) сушат концентрированием из безводного DCM (42 мл) и выдерживают в атмосфере азота. Затем фосфат повторно растворяют в безводном DCM (110 мл). В отдельной колбе получают раствор беспримесного диэтилцинка (4,17 г, 33,8 ммоль, 3,0 экв.) в безводном DCM (35 мл) и охлаждают до -25°C. Раствор фосфата на протяжении 1 часа медленно перемещают в сосуд, содержащий раствор диэтилцинка, сохраняя температуру -10°C или ниже. Прозрачный раствор этилцинкфосфата нагревают до 0°C и перемешивают 15 мин. Дииодметан (9,25 г, 34,5 ммоль, 3,0 экв.) медленно добавляют к раствору этилцинкфосфата, поддерживая температуру реакции между 0 и 5°C. После завершения добавления, раствор цинккарбеноида дополнительно перемешивают в течение 20 мин.
В отдельной колбе, соединение 3 (7,20 г, 11,4 ммоль, 1 экв.) растворяют в безводном DCM (36 мл) и перемещают в реакционную колбу. После того как добавление завершено, удаляют ледяную баню, и реакционную смесь оставляют нагреваться до комнатной температуры. Через 6 часов содержимое колбы охлаждают до -53°C. Добавляют раствор метансульфоновой кислоты (3,38 г, 35,2 ммоль, 3,1 экв.) в безводном DCM (3 мл), поддерживая реакционную температуру ниже -45°C. Через 10 мин к реакционной смеси добавляют морфолин (20 г, 230 ммоль, 20 экв.), поддерживая температуру реакции ниже - 40°C. Реакцию оставляют нагреваться до комнатной температуры на протяжении ночи. Соли морфолина удаляют фильтрованием, и осадок на фильтре промывают DCM (22 мл). Объединенные фильтраты промывают 2н. водным раствором хлористоводородной кислоты (2×140 мл), 5% водным бикарбонатом натрия (140 мл), 5% водным бикарбонатом натрия (70 мл) и 5% водным бисульфитом натрия (70 мл), и насыщенным солевым раствором (144 мл). Органический слой сушат над сульфатом магния и фильтруют. Раствор DCM концентрируют, не доходя досуха, и растворитель обменивают на метанол (280 мл). Суспензию охлаждают на ледяной бане и 40 минут перемешивают. Твердые частицы отделяют фильтрованием, дважды промывают холодным метанолом (2×25 мл), и сушат до постоянной массы, чтобы получить 5,94 г требуемого продукта.
Стадия С

Соединение 4 (11,67 г, 18,1 ммоль, 1 экв.) и 20% гидроксид палладия на влажном угле (2,40 г, 1,71 ммоль, 0,09 экв.) в круглодонной колбе помещают в атмосферу азота и разбавляют EtOAc (115 мл) и толуолом (60 мл). Раствор дегазируют азотом (3 раза), используя кольца вакуумирование/продувка, и процесс повторяют с водородом. Суспензию 1,5 часа интенсивно перемешивают при комнатной температуре. Водородную атмосферу заменяют на азотную. К реакции добавляют этилендиамин (0,57 г, 9,5 ммоль, 0,52 экв.), и полученную смесь перемешивают 20 мин. Раствор фильтруют в атмосфере азота, и фильтрат промывают 2% (масс./масс.) водным раствором этилендиамина (125 мл), затем водой (130 мл), и затем сушат над сульфатом натрия. Осушитель удаляют фильтрованием, и фильтрат концентрируют досуха в вакууме. Остающиеся твердые частицы отгоняют с толуолом (2×55 мл) на роторном испарителе, и полученное вещество используют без дополнительной очистки на следующей стадии.
Вещество предшествующей стадии растворяют в безводном DCM (26 мл). Получающийся прозрачный раствор добавляют к 1 M раствору DIBAL в DCM (65 мл, 65 ммоль, 3,6 экв.), при этом поддерживая реакционную температуру между -10 и -25°C. Через 30 мин. реакцию гасят ацетоном (13 мл), поддерживая реакционную температуру около или ниже 0°C. После перемешивания загашенной реакционной смеси в течение 17 мин, ее порциями добавляют в колбу, содержащую холодный, перемешанный раствор 20% (масс./масс.) водной сегнетовой соли (200 мл). Получающуюся гелеобразную суспензию 15 часов перемешивают при комнатной температуре. После перемешивания, прозрачные слои разделяют, и водный слой обратно экстрагируют DCM (30 мл). Объединенные органические слои промывают водой (60 мл) и сушат над сульфатом натрия. Осушитель удаляют фильтрованием и отбрасывают. Фильтрат концентрируют в вакууме, и растворитель обменивают на толуол (225 мл, добавляемые порциями). Получающийся раствор дополнительно концентрируют до суспензии (50 мл) и разбавляют гептаном (115 мл). Получающуюся смесь нагревают до превращения ее в гомогенную смесь (92°C). Раствор на протяжении 12 часов медленно охлаждают до 15°C, и затем дополнительно выдерживают в течение 16 часов. Кристаллический продукт отделяют фильтрованием с отсосом, промывают гептаном (2×75 мл) и сушат до постоянной массы, чтобы получить 7,70 г требуемого продукта.
В круглодонную колбу последовательно загружают гомоаллиловый спирт (7,50 г, 17,6 ммоль, 1 экв.), трет-бутилат алюминия (6,10 г, 24,8 ммоль, 1,4 экв.), безводный толуол (115 мл) и 2-бутанон (90 г, 1,24 моль, 7 экв.). В атмосфере азота суспензию 16 часов нагревают до 75°C. Затем, реакцию оставляют охлаждаться до температуры реакции 49°C. Водный 20% (w/w) раствор тартрата калия натрия (226 г) добавляют к перемешанной суспензии. Суспензию 3,5 часа перемешивают при комнатной температуре. Слои разделяют. Органический слой промывают водной 20% сегнетовой солью (2×250 мл) и водой (225 мл), затем сушат над сульфатом натрия и фильтруют. Остаток промывают толуолом (30 мл) и отбрасывают. Объединенные органические вещества концентрируют досуха. Остаточные реакционные растворители удаляют из вещества концентрированием из 2-пропанола (250 мл, добавляемых порциями), доводя до конечной массы раствора в 44 г. Растворитель 2-пропанол заменяют гептаном (275 мл, добавляемых порциями) до конечной массы 41 г раствора полностью осаждаемого требуемого продукта. Суспензию дополнительно разбавляют гептаном (40 мл), 1 час перемешивают при комнатной температуре и фильтруют. Продукт промывают н-гептаном (17 мл) и сушат, чтобы получить 5,4 г требуемого продукта.
Стадия D
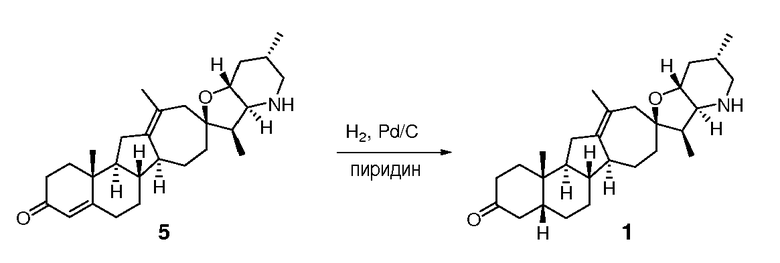
В круглодонную колбу загружают исходное вещество (110 мг, 0,26 ммоль, 1 экв.) и 10% палладий на угле (106 мг). Твердые частицы суспендируют в пиридине (4 мл). Суспензию помещают в атмосферу водорода (1 атм.), и смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют через Celite®, и фильтрат концентрируют в вакууме. Неочищенное вещество очищают, используя флэш хроматографию на силикагеле (смесью MeOH/DCM 5:95), чтобы получить требуемое соединение. ([M+H] = 426,6 m/z).
Пример 2
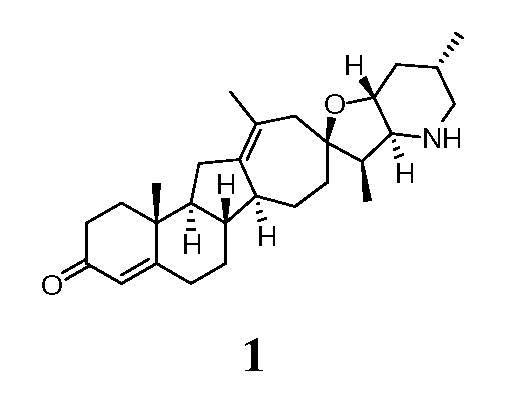
Стадия A
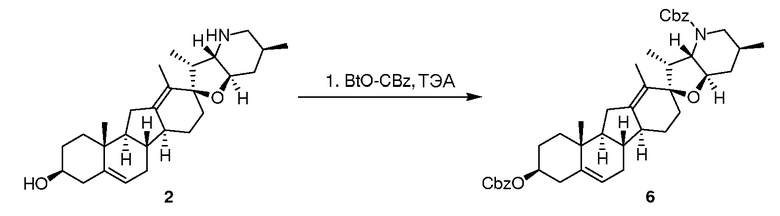
Циклопамин 2 (5,02 г, 12,2 ммоль, 1,0 экв.) растворяют в безводном пиридине (25 мл). Добавляют DMAP (300 мг, 2.44 ммоль, 0,2 экв.) и триэтиламин (5,5 мл, 39,1 ммоль, 3,2 экв.), с последующим добавлением BtO-Cbz (10,5 г, 39,1 ммоль, 3,2 экв.) и греют 2 часа при 40°C. Смесь охлаждают до комнатной температуры, обрабатывают 30 мл воды, нагревают до получения гомогенного раствора и оставляют охлаждаться до комнатной температуры. Образующийся белый осадок собирают фильтрованием, осадок на фильтре промывают порциями воды (3×50 мл), и сушат на воздухе, чтобы получить 9,53 г неочищенного вещества, которое кристаллизуют из смеси толуол/гептаны (1:9, 70 мл) с получением 6,75 г требуемого продукта.
Стадия В
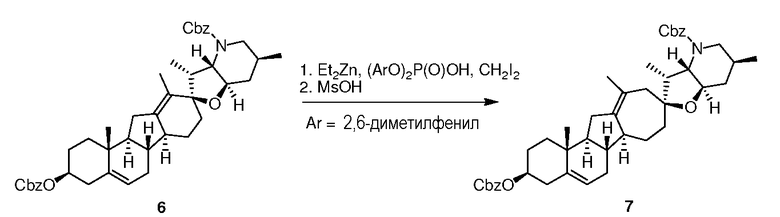
К раствору диэтилцинка (572 мг, 482 мкл, 4,63 ммоль, 3,00 экв.) в 5,0 мл DCM добавляют при -20°C раствор бис-(2,6-диметилфенил)фосфорной кислоты (1,42 г, 4,63 ммоль, 3,00 экв.) в DCM (15 мл), поддерживая температуру реакции ниже -8°C. Раствор оставляют стареть при 0°C в течение 15 мин., добавляют беспримесный дииодметан (1,24 г, 374 мкл, 3,00 экв.), оставляют стареть при 0°C в течение 15 мин перед добавлением раствора (бисCBz)циклопамина (1,05 г, 1,54 ммоль, 1,0 экв.) в DCM (10 мл). Охлаждающую баню заменяют водяной баней при комнатной температуре и сохраняют 4,5 часа при комнатной температуре. Смесь охлаждают до -76oC на бане из смеси сухой лед/ацетон и обрабатывают, добавляя по каплям раствор метансульфоновой кислоты в DCM (0,6 мл 50% об./об. раствор, 4,63 ммоль, 3,0 экв.), поддерживая температуру реакции ниже -74°C. Смесь оставляют стареть в течение 15-20 мин, и по каплям гасят морфолином (2,69 г, 2,70 мл, 20 экв.), поддерживая температуру реакции ниже -65°C. Охлаждающую баню удаляют, реакционную смесь перемешивают в течение 16-18 часов. Белый осадок отфильтровывают, и фильтрат последовательно промывают 2,0 M HCl (2×20 мл), насыщ. бикарбонатом натрия (2×20 мл), водой (2×20 мл) и насыщенным солевым раствором (20 мл). Сушат над сульфатом магния, концентрируют в вакууме досуха, и неочищенный продукт очищают флэш хроматографией на силикагеле (смесью гексаны/EtAc, 17:3 → 4:1), чтобы получить 924 мг (1,33 ммоль, 86%) требуемого продукта.
Стадия С

К раствору соединения 7 (4,05 г, 5,83 ммоль, 1 экв.) в растворе смеси EtOAc:толуол (2:1, 60 мл) добавляют 20% гидроксид палладия на угле (823 мг, 0,583 ммоль, 0,1 экв.). Колбу вакуумируют и заполняют водородом три раза. Смесь перемешивают в атмосфере водорода 1 час. Добавляют беспримесный этилендиамин (0,38 мл), перемешивают 1 час, и катализатор отфильтровывают. Осадок на фильтре дважды промывают смесью EtOAc:толуол (2:1, 12 мл). Объединенные фильтраты промывают 2% водным раствором этилендиамина (3×20 мл), сушат над сульфатом натрия и концентрируют в вакууме, чтобы получить 2,46 г продукта в виде белого кристаллический твердого вещества.
Стадия D
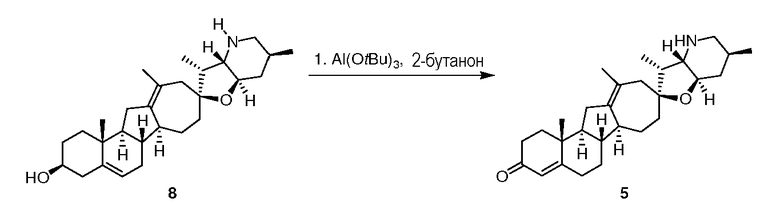
В круглодонную колбу последовательно загружают гомоаллиловый спирт 8 (7,50 г, 17,6 ммоль, 1 экв.), трет-бутилат алюминия (6,10 г, 24,8 ммоль, 1,4 экв.), безводный толуол (115 мл) и 2-бутанон (90 г, 1,24 моль, 7 экв.). Суспензию 16 часов нагревают в атмосфере азота до 75°C. Затем реакцию оставляют охлаждаться до температуры 49°C. Водный 20% (масс./масс.) раствор тартрата калия натрия (226 г) добавляют к перемешанной суспензии. Суспензию перемешивают при комнатной температуре 3,5 часа. Слои разделяют. Органический слой промывают водной 20% сегнетовой солью (2×250 мл) и водой (225 мл), затем сушат над сульфатом натрия и фильтруют. Остаток промывают толуолом (30 мл) и отбрасывают. Объединенные органические вещества концентрируют досуха. Остаточные растворители реакции удаляют из вещества концентрированием из 2-пропанола (250 мл, добавляемых порциями), доводя до конечной массы раствора в 44 г. Растворитель 2-пропанол заменяют гептаном (275 мл, добавляемых порциями) до конечной массы 41 г раствора полностью осаждаемого требуемого продукта. Суспензию дополнительно разбавляет н-гептаном (40 мл), 1 час перемешивают при комнатной температуре и фильтруют. Продукт промывают н-гептаном (17 мл) и сушат, чтобы получить 5,4 г требуемого продукта.
Стадия E
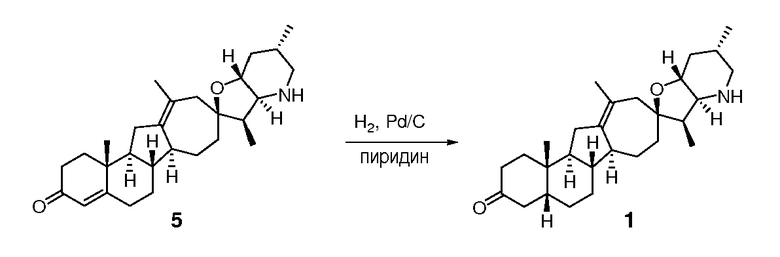
В круглодонную колбу загружают исходное вещество (110 мг, 0,26 ммоль, 1 экв.) и 10% палладий на угле (106 мг). Твердые частицы суспендируют в пиридине (4 мл). Суспензию помещают в атмосферу водорода (1 атом), и смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют через Celite®, и фильтрат концентрируют в вакууме. Неочищенное вещество очищают, используя флэш хроматографию на силикагеле (смесь MeOH/DCM 5:95), чтобы получить 93 мг требуемого соединения.
([M+H] = 426,6 m/z).
Пример 3

В трубку для запаивания загружали кетон 6 (85 мг, 0,199 ммоль, 1 экв.) и добавляли триэтиленгликоль (2 мл), с последующим добавлением моногидрата гидразина (500 мг, 10 ммоль, 50 экв.) и карбоната калия (138 мг, 1 ммоль, 5 экв.). Трубку запаивали, и реакционную смесь нагревали 16 часов при 150°C. Реакционную смесь охлаждали до комнатной температуры и добавляли воду. Остаток экстрагировали хлороформом (3X). Объединенные органические слои промывали водой, сушили над Na2SO4, и концентрировали досуха. Бесцветное масло очищали, используя флэш хроматографию на силикагеле (DCM/MeOH 96:4). Очищенные фракции объединяли и концентрировали досуха. Получающееся масло растворяли в MTBE (трет-бутилметиловый эфир) и промывали водой (2X), 2н. NaOH и затем насыщенным солевым раствором. Объединенные органические слои сушили над Na2SO4, фильтровали и выпаривали, чтобы получить 64 мг требуемого вещества в виде белой пены. ([M+H] = 412,7 m/z).
Пример 4
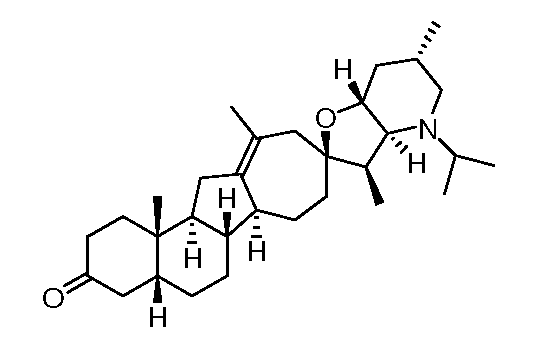
10

В трубку для запаивания загружали соединение 5 (223 мг, 0,52 ммоль, 1 экв.) и добавляли DMF (ДМФА) (lмл). Добавляли 2-бромпропан (1,3 г, 10,5 ммоль, 20 экв.) и Na2CO3 (73 мг, 0,68 ммоль, 1,3 экв.) и колбу запаивали и нагревали до 50°C. Смесь перемешивали 16 часов, когда наблюдали ~ 70% превращение. Дополнительно добавляли 2-бромропан (0,26 г, 2,12 ммоль, 4 экв.). Реакционную смесь перемешивали 2 часа и дополнительно добавляли 2-бромпропан (0,13 г, 1,1 ммоль, 2 экв.). Реакционную смесь перемешивали еще l час. Реакционную смесь охлаждали до комнатной температуры, и добавляли воду. Осадок экстрагировали MTBE (3X). Органические слои объединяли, промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали досуха. Белую пену очищали, используя флэш хроматографию на силикагеле (смесью DCM/MeOH 99:1), чтобы получить 206 мг N-изопропильного производного в виде белой пены.
N-изопропильное производное (205 мг, 0,44 ммоль, 1 экв.) растворяли в 4-метоксипиридине (1,5 мл). Колбу помещали в инертную атмосферу и добавляли Pd/C 10% (влажный, Aldrich Degussa тип E101, 40 мг). Колбу запаивали, три раза продували водородом и оставляли на 16 часов в атмосфере водорода (1 атм). К реакционной смеси добавляли Celite®. Смесь фильтровали через небольшой рыхлый слой Celite® и промывали EtOAc. Органический слой промывали 1 н. водной HCl (2x), затем водой. Органический слой сушили над Na2SO4, фильтровали через хлопчатобумажную ткань и упаривали, чтобы получить 34 мг неочищенного продукта. Водный слой нейтрализовали 2 н. KOH и экстрагировали DCM (3X). Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали через хлопчатобумажную ткань и объединяли с первоначальными 34 мг неочищенного продукта. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью гексан/EtOAc (6:4)), чтобы получить 80 мг требуемого продукта. ([M+H] = 468,7 m/z).
Пример 5
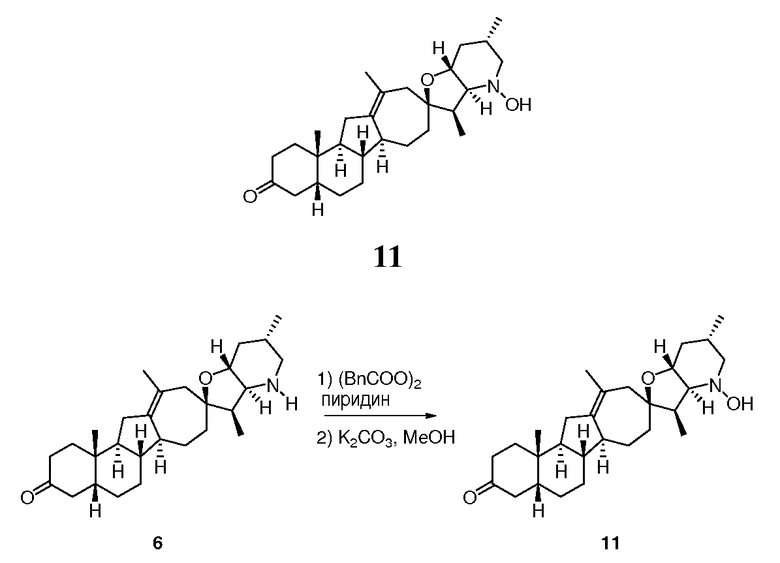
В круглодонной колбе соединение 6 (88 мг, 0,21 ммоль, 1 экв.) растворяли в безводном ТГФ (THF) (1 мл). Смесь охлаждали до 0°C. Последовательно добавляли пиридин (84 мкл, 1 ммоль, 5 экв.) и пероксид бензоила (150 мг, 0,62 ммоль, 3 экв.). Гомогенную смесь на протяжении 2 часов постепенно нагревали до комнатной температуры и при комнатной температуре перемешивали в течение ночи. Реакцию гасили, добавляя насыщенный раствор NaHCO3. Осадок экстрагировали MTBE. Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью гексан/EtOAc (от 9:1 до 4:1)), чтобы получить продукт (N-О) производного (60 мг, 0,11 ммоль) в виде белой пены. Эту пену растворяли в 2 мл MeOH, с последующим добавление 2 н. водного раствора KOH (0,4 мл). Реакционную смесь перемешивали l час. Большую часть MeOH выпаривали в токе азота и добавляли 1 н. HCl (500 мкл). Вещество экстрагировали DCM (3X). Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью гексаны/EtOAc (88:12 → 1:1)), чтобы получить 11 мг требуемого продукта. ([M+H] = 442,5 m/z).
Пример 6
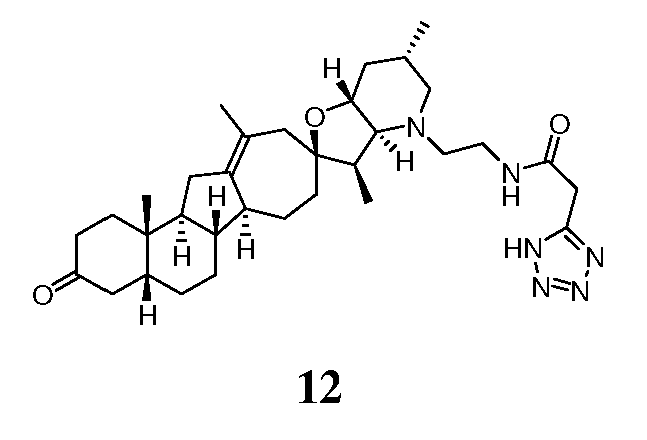
Стадия A

В круглодонной колбе растворяли соединение 6 (89 мг, 0,209 ммоль, 1 экв.) и N-(бензилоксикарбонил)аминоацетальдегид (148 мг, 0,85 ммоль, 4 экв.) в DCM (2 мл). Добавляли триацетоксиборогидрид натрия (177 мг, 0,85 ммоль, 4 экв.), и реакционную смесь перемешивали 3 часа при комнатной температуре. Смесь вливали в насыщенный водный раствор NaHCO3, и осадок экстрагировали DCM (3x). Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали через хлопчатобумажную ткань и упаривали, чтобы получить вспененное твердое вещество (247 мг). Неочищенное вещество растворяли в EtOAc (2 мл) и обрабатывали 4М HCl (156 мкл). Через 30 мин медленно образовывался белый осадок. Получающуюся взвесь перемешивали 15 мин. Фильтрование дало 120 мг белого твердого вещества. Вещество растворяли в EtOAc и обрабатывали насыщенным водным раствором NaHCO3. Органический слой собирали, а водный слой экстрагировали EtOAc (2X). Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4. Фильтрование и упаривание дало требуемое промежуточное соединение. Это вещество использовали без очистки на следующей стадии.
Стадия В

Все вещество, полученное на стадии A, растворяли в EtOAc (3 мл) и обрабатывали Pd/C 10% (30 мг, влажный, Aldrich Degussa тип E101). Колбу запаивали, три раза продували водородом, и на протяжении ночи оставляли в атмосфере водорода (1 атм). Через 16 часов смесь фильтровали через небольшой рыхлый слой Celite® и промывали EtOAc, чтобы получить 52 мг амина в виде белой пены.
Стадия С

В круглодонную колбу, содержащую амин 14 (52 мг, 0,11 ммоль, 1 экв.), загружали 1H-тетразол-5-уксусную кислоту (21 мг, 0,166 ммоль, 1,5 экв.), DCM (2 мл), EDCl (дихлорэтан) (42 мг, 0,22 ммоль, 2 экв.) и N,N-диизопропилэтиламин (57 мг, 0,44 ммоль, 4 экв.). Получающийся желтый раствор 4 часа перемешивали при комнатной температуре. Реакцию гасили, добавляя насыщенный водный раствором NaHCO3, и осадок экстрагировали DCM (3X). Объединенные органические слои сушили над Na2SO4, фильтровали через хлопчатобумажную ткань и упаривали, чтобы получить 62 мг не совсем белого твердого вещества. Это вещество очищали, используя флэш хроматографию на силикагеле (смесью MeOH/DCM 5:95 → 10:90), чтобы получить 31 мг требуемого продукта. ([M+H] = 579,7 m/z).
Пример 7
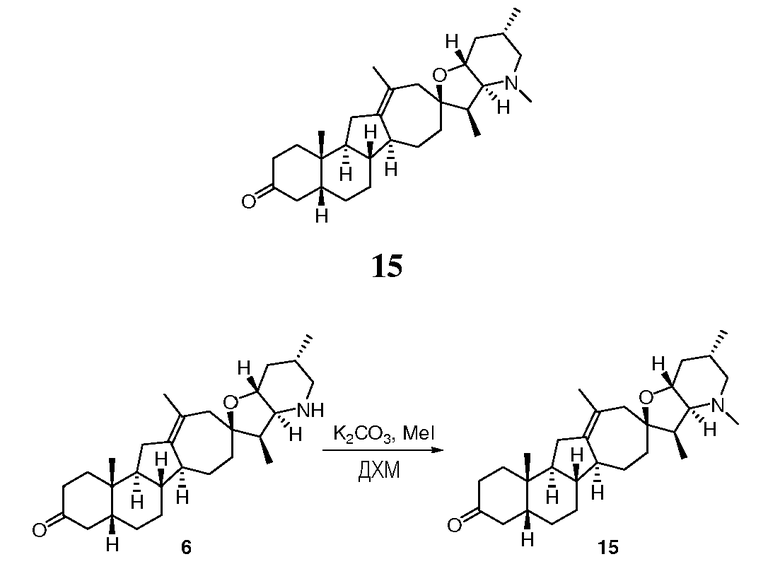
В круглодонную колбу загружали исходное вещество (47 мг, 0,110 ммоль, 1 экв.) и карбонат калия (150 мг, 1,09 ммоль, 10 экв.). Твердые частицы суспендировали в 2 мл DCM. Добавляли иодметан (14 мкл, 0,22 ммоль, 2 экв.), и смесь перемешивали 2 часа при комнатной температуре. Анализ ТСХ (TLC) (тонкослойная хроматография) (смесь DCM/MeOH 95:5) показывает более чем 90% завершение взаимодействия. К реакционной смеси добавляли иодметан (14 мкл, 0,22 ммоль, 2 экв.), перемешивали 2 часа. К реакционной смеси добавляли воду. Фазы разделяли, и органические вещества сушили и концентрировали досуха. Остаток очищали, используя флэш хроматографию на силикагеле (смесью DCM/MeOH 100:0->98:2), чтобы получить 34 мг требуемого продукта. ([M+H] = 440,5 m/z).
Пример 8


В круглодонную колбу загружали исходное вещество (59 мг, 0,126 ммоль, 1 экв.) и карбонат калия (350 мг, 2,5 ммоль, 20 экв.). Твердые частицы суспендировали в 3 мл DCM. В реакционную смесь загружали иодметан (80 мкл, 1,29 ммоль, 10 экв.), и смесь перемешивали в течение ночи при комнатной температуре. В реакционную смесь загружали воду. Органическую фазу отделяли, и водный слой обратно экстрагировали DCM. Объединенные органические слои сушили и концентрировали досуха. Остаток очищали, используя флэш хроматографию на силикагеле (смесью DCM/MeOH (95:5 → 90:10)), чтобы получить 52 мг требуемого продукта. ([M+H] = 639,5 m/z).
Пример 9

Стадия A

В круглодонной колбе растворяли соединение 5 (50 мг, 0,12 ммоль, 1 экв.) и N-{трет-бутоксикарбонил)аминоацетальдегид (6 мг, 0,38 ммоль, 3,1 экв.) в DCM (2 мл). Добавляли триацетоксиборгидрид натрия (8 мг, 0,38 ммоль, 3,1 экв.), и реакционную смесь перемешивали 2 часа при комнатной температуре. Смесь вливали в насыщенный водный раствор NaHCO3, и осадок экстрагировали DCM (3x). Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали через хлопчатобумажную ткань и упаривали, чтобы получить вспененное твердое вещество (95 мг). Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью EtOAc/Гексаны 1:1), чтобы получить 55 мг соединения 18.
Стадия В
В круглодонную колбу загружали исходное вещество 18 (800 мг, 1,4 ммоль, 1 экв.). Твердое вещество растворяли в растворе DCM и TFA (трифторуксусная кислота) (10 мл, 1:1). Раствор перемешивали 45 мин при комнатной температуре. Реакционную смесь распределяли между 10% раствором карбоната натрия и DCM. Органическую часть отделяли и промывали 10% карбонатом натрия. Органическую фазу концентрировали досуха. Остаток использовали без дополнительной очистки на следующей стадии.
Стадия С
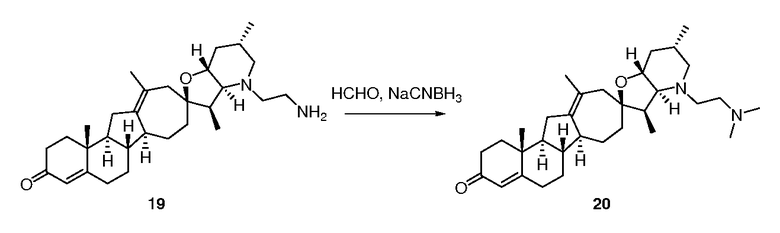
В круглодонную колбу загружали исходное вещество (300 мг, 0,64 ммоль, 1 экв.), растворяли в смеси ТГФ/ACN (ацетонитрил) (1:1,4 мл). В реакционную смесь загружали 37% формальдегид в воде (240 мкл, 3,22 ммоль, 5 экв.) и цианоборгидрид натрия (64 мг, 1 ммоль, 1,6 экв.). Смесь перемешивали 30 мин при комнатной температуре. Затем, реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия и DCM. Органическое вещество отделяли, сушили и концентрировали досуха. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смеью MeOH/DCM (5:95 → 10:90)), чтобы получить требуемое вещество.
Стадия D
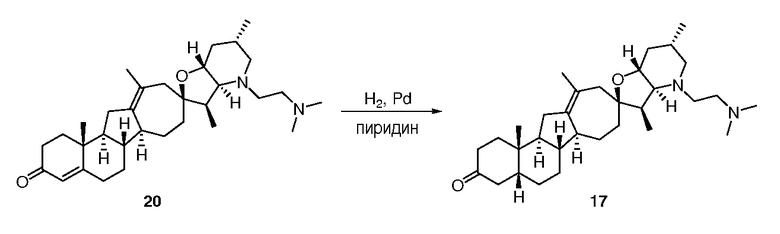
В круглодонную колбу загружали исходное вещество 20 (30 мг, 0,06 ммоль, 1 экв.) и 10% палладий на угле (30 мг). Твердые частицы суспендировали в пиридине (2 мл). Суспензию помещали в атмосферу водорода, и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь фильтровали на Celite®, фильтрат концентрировали досуха. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью MeOH/DCM (5:95 → 10:90)), чтобы получить требуемое вещество. ([M+H] = 497,7 m/z).
Пример 10
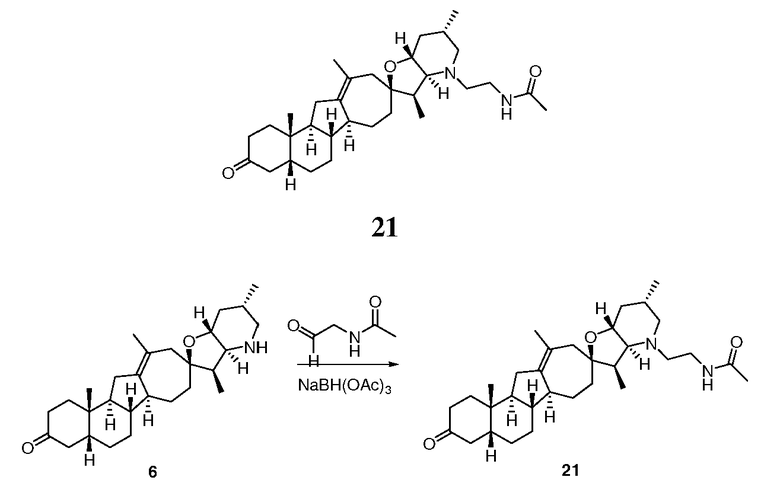
В круглодонную колбу загружали исходное вещество (85 мг, 0,20 ммоль, 1 экв.), растворяли в DCM (4 мл). В реакционную смесь загружали N-(2-оксоэтил)ацетамид (80 мг, 0,70 ммоль, 3,5 экв.) и триацетоксиборгидрид натрия (170 мг, 0,80, 4 экв.). Смесь перемешивали 1 час при комнатной температуре. Реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия и DCM. Органическое вещество отделяли, сушили и концентрировали досуха. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью MeOH/DCM 5:95), чтобы получить требуемое вещество. ([M+H] = 511,7 m/z).
Пример 11
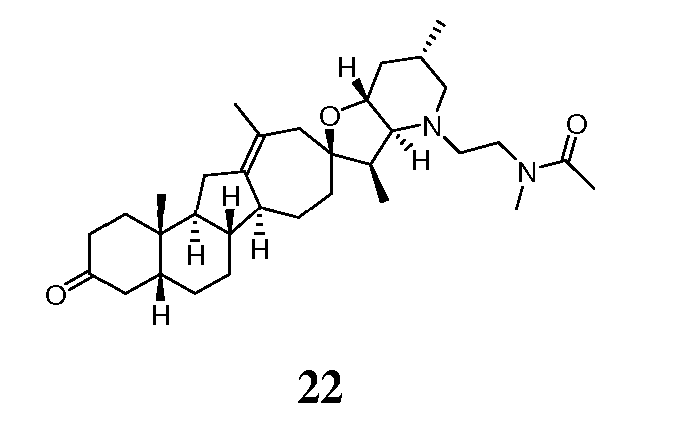
Соединения 22 синтезировали, по методике, описанной в примере 9, используя N-метил-N-(2-оксоэтил)ацетамид вместо N-(2-оксоэтил)ацетамида. ([M+H] = 525,7 m/z).
Пример 12

Соединение 23 синтезировали, по методике, описанной в примере 10, используя N-(2-оксоэтил)-3-фенилпропанамид вместо N- (2-оксоэтил)ацетамида. ([M+H] = 601,8 m/z).
Пример 13

Соединение 23 синтезировали, по методике, описанной в примере 10, используя N-метил-N-(2-оксоэтил)-3-фенилпропанамид вместо N-(2-оксоэтил)ацетамида. ([M+H] 615,9 m/z).
Пример 14
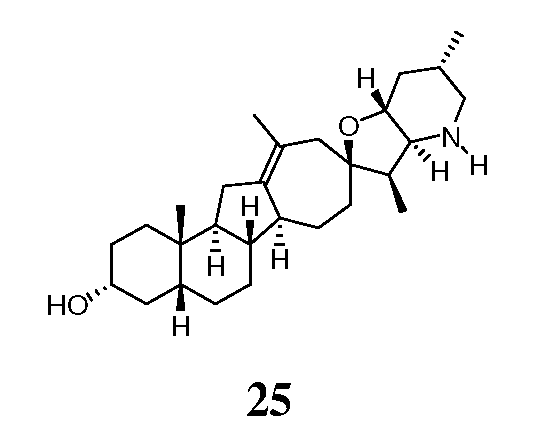
Стадия A

В круглодонную колбу загружали соединение 6 (4,23 г, 9,94 ммоль, 1 экв.) и ТГФ (60 мл). Добавляли триэтиламин (6,92 мл, 49,7 ммоль, 5,0 экв.) и бензилхлорформиат (1,54 мл, 10,93 ммоль, 1,1 экв.), и смесь перемешивали 1 час при комнатной температуре. Реакционную смесь распределяли между насыщенным водным бикарбонатом (100 мл) и EtOAc (100 мл). Фазы разделяли, и органические вещества сушили (Na2SO4) и концентрировали досуха. Неочищенное вещество очищали, используя флэш хроматографию на силикагеле (смесью EtOAc/Гексаны 2:98 → 14:86) чтобы получить 3,75 г вещества.
Стадия В

Раствор гептагидрата трихлорида церия (260 мг, 0,69 ммоль, 1,3 экв.) в MeOH (10 мл) обрабатывали при 0°C боргидридом натрия (24 мг, 0,65 ммоль, 1.2 экв.), перемешивали 15 мин и затем охлаждали до -78°C. Добавляли раствор (10 мл) кетона 26 (300 мг, 0,54 ммоль, 1 экв.) в ТГФ, и смесь перемешивали 1 час и затем нагревали до комнатной температуры. Добавляли воду (50 мл) и EtOAc (50 мл), смешивали, и слои разделяли. Органический слой собирали, промывали насыщенным солевым раствором (30 мл), сушили над сульфатом натрия, и концентрировали до белого осадка. Неочищенный продукт очищали используя флэш хроматографию на силикагеле (смесью простой эфир/гексаны 2:3 → 1:1), чтобы получить 235 мг 3-бета-спирта 27.
Стадия С

Соединение 27 (235 мг, 0,42 ммоль, 1 экв.) растворяли в EtOAc (7 мл) в колбе, снабженной мешалкой и каучуковой перегородкой. Азот барботировали через раствор, и добавляли Pd/C 10% (влажный, Aldrich Degussa тип E101, 50 мг). Азот барботировали через эту смесь и затем газообразный водород, и 3 часа перемешивали при комнатной температуре. Затем, азот барботировали через смесь, фильтровали через полиэтиленовую мембрану 0,45 мкм и концентрировали до прозрачного масла. Масло очищали флэш хроматографией на силикагеле (смесью NH4OH(вод.)/MeOH/DCM 0,5:2:97,5→ 0,5:6:93,5), чтобы получить 130 мг соединения 25 в виде белого порошка. ([M+H] = 427,4 m/z).
Пример 15
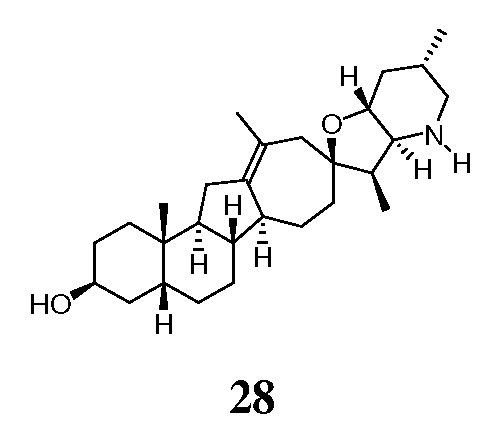
Стадия A
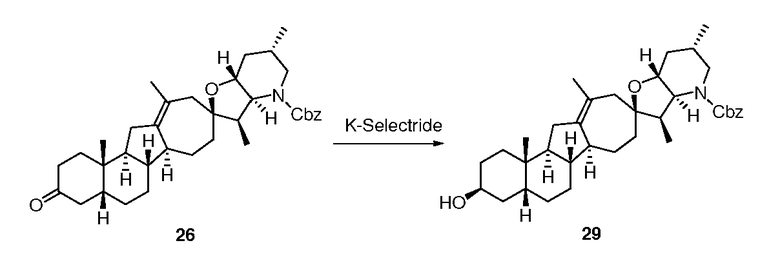
ТГФ раствор (10 мл) кетона 26 (300 мг, 0,54 ммоль, 1 экв.) при -78°C обрабатывали составом K-Selectride® (три-втор-бутилборгидрид калия) (0,58 мл, 0,58 ммоль, 1,1 экв.) и перемешивали 60 мин. Добавляли метанол (1 мл), и раствор нагревали до комнатной температуры. Добавляли воду (50 мл) и EtOAc (50 мл), смешивали, и слои разделяли. Органический слой промывали насыщенным солевым раствором (30 мл), сушили над сульфатом натрия, и концентрировали до белого остатка. Неочищенный продукт очищали флэш хроматографиией на силикагеле (смесью простой эфир/гексаны 2:3 → 1:14), чтобы получить 170 мг чистого 3-альфа-спирта 29.
Стадия В
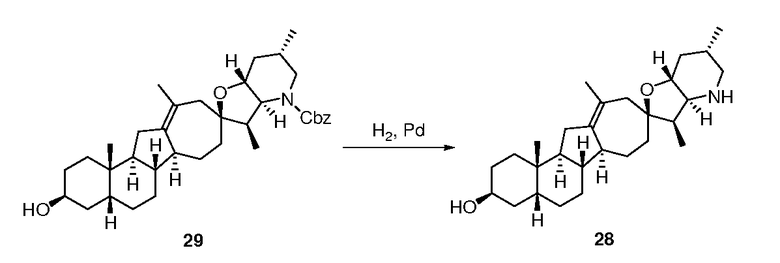
Соединение 29 (170 мг, 0,30 ммоль, 1 экв.) растворяли в EtOAc (5 мл) в колбе, снабженной мешалкой и каучуковой перегородкой. Азот барботировали через раствор, и добавляли Pd/C 10% (влажный, Aldrich Degussa тип E101, 35 мг). Азот барботировали через эту смесь и затем газообразный водород, и 3 часа перемешивали при комнатной температуре. Затем, азот барботировали через смесь, фильтровали через полиэтиленовую мембрану 0,45 мкм и концентрировали до прозрачного масла. Масло очищали флэш хроматографиией на силикагеле (смесью NH4OH(водн)/MeOH/DCM 0,5:2:97,5 → 0,5:6:93,5), чтобы получить 76 мг соединения 28 в виде белого порошка. ([M+H] = 427,4 m/z).
Пример 16
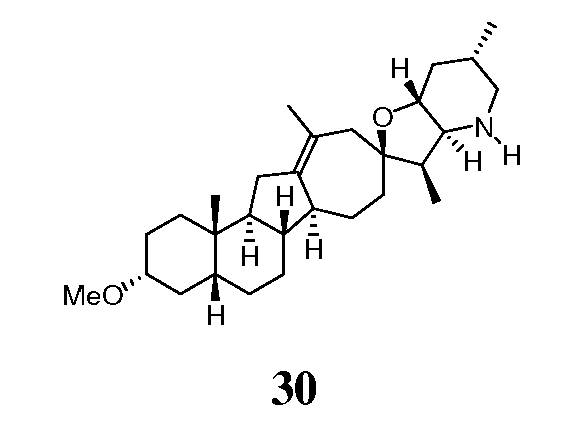
Стадия A

Соединение 27 (100 мг, 0,18 ммоль, 1 экв.) и бензилтриэтиламмонийхлорид (8 мг, 0,36 ммоль, 0,2 экв.) растворяли в DCM (5 мл) и 18 часов интенсивно взбалтывали при комнатной температуре с диметилсульфатом (130 мкл 1,43 ммоль, 8 экв.) и 50% водным гидроксидом калия (0,5 мл). Смесь распределяли между водой (30 мл) и EtOAc (30 мл). Затем органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали до прозрачного масла. Неочищенный простой эфир очищали флэш хроматографиией на силикагеле (смесью простой эфир/гексаны 3:7 → 9:113), чтобы получить 75 мг простого метилового эфира в виде прозрачного масла.
Стадия В
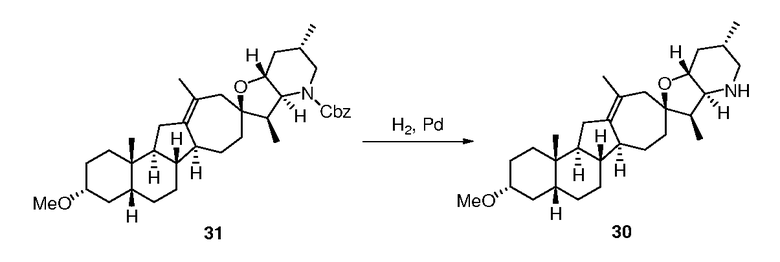
Соединение 31 (66 мг, 0,115 ммоль, 1 экв.) растворяли в EtOAc (5 мл) в колбе, снабженной мешалкой и каучуковой перегородкой. Азот барботировали через раствор, и добавляли 10% Pd/C (влажный, Aldrich Degussa тип E101, 20 мг). Азот барботировали через эту смесь и затем газообразный водород, и 3 часа перемешивали при комнатной температуре. Затем через эту смесь барботировали азот, фильтровали через полиэтиленовую мембрану 0,45 мкм, и концентрировали до прозрачного масла. Масло очищали флэш хроматографиией на силикагеле (смесью NH4OH(водн)/MeOH/DCM 0,5:2:97,5 → 0,5:6:93,5), чтобы получить 22 мг соединения 30 в виде белого порошка. ( [M+H] = 441,4 m/z).
Пример 17
Стадия A
Соединение 27 (100 мг, 0,18 ммоль, 1 экв.) растворяли в DCM (5 мл) и добавляли 4-диметиламинопиридин (4 мг, 0,35 ммоль, 0,2 экв.), N,N-диизопропилэтиламин (0,15 мл, 0,9 ммоль, 5 экв.) и уксусный ангидрид (0,070 мл, 0,2 ммоль, 4 экв.). После перемешивания в течение 12 часов при комнатной температуре раствор распределяли между EtOAc (30 мл) и 5% водным бикарбонатом натрия (15 мл). Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия, и концентрировали до прозрачного масла. Неочищенный сложный эфир очищали флэш хроматографиией на силикагеле (смесью простой эфир/гексаны 3:7 → 9:l13), чтобы получить 100 мг сложного эфира в виде прозрачного масла.
Стадия В

Соединение 33 (100 мг, 0,18 ммоль, 1 экв.) растворяли в EtOAc (5 мл) в колбе, снабженной мешалкой и каучуковой перегородкой. Азот барботировали через раствор, и добавляли 10% Pd/C (влажный, Aldrich Degussa тип E101, 20 мг). Азот барботировали через эту смесь и затем газообразный водород, и 3 часа перемешивали при комнатной температуре. Затем через эту смесь барботировали азот, фильтровали через полиэтиленовую мембрану 0,45 мкм и концентрировали до прозрачного масла. Масло очищали флэш хроматографиией на силикагеле (смесью NH4OH(водн)/MeOH/DCM 0,5:2:97,5 → 0,5:6:93,5), чтобы получить 45 мг соединения 32 в виде белого порошка ([M+H] = 469,4 m/z).
Пример 18

Соединение 34 синтезировали по методике, описанной в примере 16, используя соединение 29 вместо соединения 27. ([M+H] = 441,4 m/z).
Пример 19
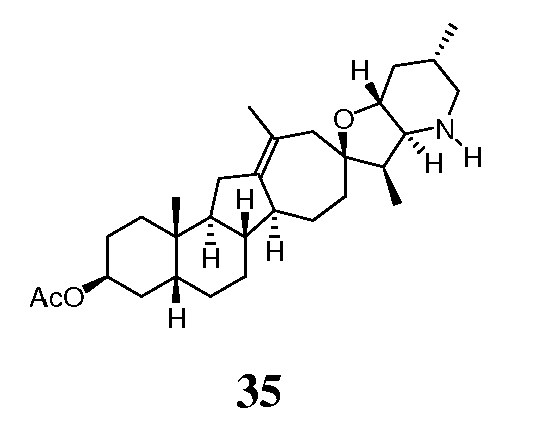
Соединение 34 синтезировали по методике, описанной в примере 17, используя соединение 29 вместо соединения 27. МС([M+H] = 469,4 m/z).
Пример 20

Стадия A
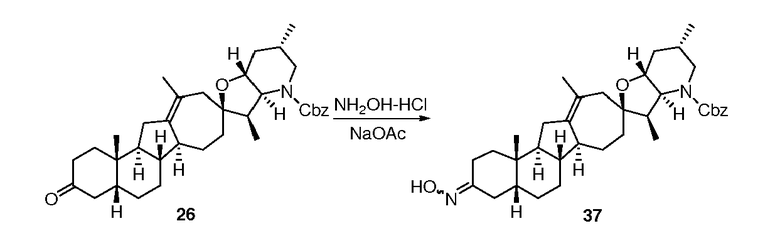
Раствор в этаноле (5 мл) соединения 26 (185 мг, 0,3 ммоль, 1 экв.) обрабатывали гидрохлоридом гидроксиламина (140 мг, 2 ммоль, 6 экв.), ацетатом натрия (160 мг, 2 ммоль, 6 экв.) и водой (0,5 мл), и смесь перемешивали при комнатной температуре 1 час. Смесь разделяли между EtOAc и водой (50 мл каждого вещества). Органический слой промывали насыщенным солевым раствором (30 мл), сушили над сульфатом натрия и концентрировали до белого остатка. Неочищенный продукт очищали хроматографией на силикагеле (смесью простой эфир/гексаны 2:3 → 1:1), чтобы получить 193 мг оксима 37.
Стадия В

Соединение 37 (65 мг, 0,113 ммоль) растворяли в EtOAc (7 мл) в колбе, снабженной мешалкой и каучуковой перегородкой. Азот барботировали через раствор и добавляли Pd/C 10% (влажный, Aldrich Degussa тип E101, 20 мг). Азот барботировали через эту смесь и затем газообразный водород, и перемешивали при комнатной температуре 3 часа. Затем через эту смесь барботировали азот, фильтровали через полиэтиленовую мембрану 0,45 мкм и концентрировали до прозрачного масла. Масло очищали флэш хроматографией на силикагеле (смесью NH4OH(водн)/MeOH/DCM 0,5:2:97,5 → 0,5:6:93,5), чтобы получить 15 мг соединения 36 в виде белого порошка, представляющего собой смесь цис- и транс- изомеров оксима. ([M+H] = 440,3 m/z).
Пример 21

Стадия A

Соединение 27 (42 мг, 0,075 ммоль, 1 экв.) растворяли в DCM (5 мл) и добавляли 4-диметиламинопиридин (2 мг, 0,02 ммоль, 0,2 экв.), (N-Cbz)глицин (23 мг, 0110 ммоль, 1,5 экв.), и диизопропилкарбодиимид (0,023 мл, 0,150 ммоль, 2 экв.). После перемешивания в течение 12 часов при комнатной температуре раствор распределяли между EtOAc (30 мл) и 5% водным бикарбонатом натрия (15 мл). Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия, и концентрировали до прозрачного масла. Неочищенный сложный эфир очищали флэш хроматографией на силикагеле (смесью простой эфир/гексаны 2:3 → 1:1) чтобы получить в виде прозрачного масла 35 мг сложного эфира.
Стадия В

Соединение 39 (235 мг, 0,42 ммоль, 1 экв.) растворяли в EtOAc (7 мл) в колбе, снабженной мешалкой и каучуковой перегородкой. Азот барботировали через раствор, и добавляли Pd/C 10% (влажный, Aldrich Degussa тип E101, 50 мг). Азот барботировали через эту смесь и затем газообразный водород, и перемешивали при комнатной температуре 3 часа. Затем через эту смесь барботировали азот, фильтровали через полиэтиленовую мембрану 0,45 мкм и концентрировали до прозрачного масла. Масло очищали флэш хроматографией на силикагеле (смесью NH4OH(водн)/MeOH/DCM 0,5:2:97,5 → 0,5:6:93,5), чтобы получить 17 мг требуемого продукта в виде белого порошка ([M+H] = 452,4 m/z).
Пример 22
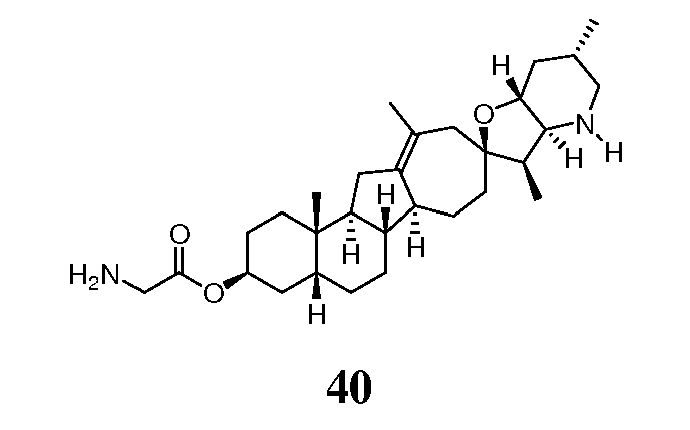
Соединение 40 синтезировали по методике, описанной в примере 21, используя соединения 29 вместо соединения 27. ([M+H] = 452,4 m/z).
Пример 23
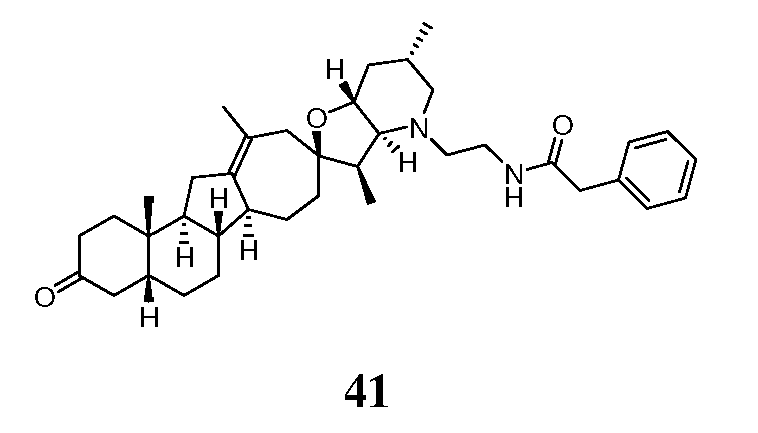
Соединение 41 синтезировали по методике, описанной в примере 10, используя N-(2-оксоэтил)-2-фенилацетамид вместо N -(2-оксоэтил)ацетамида. ([M+H] = 587,7 m/z).
Пример 24

42
Стадия A
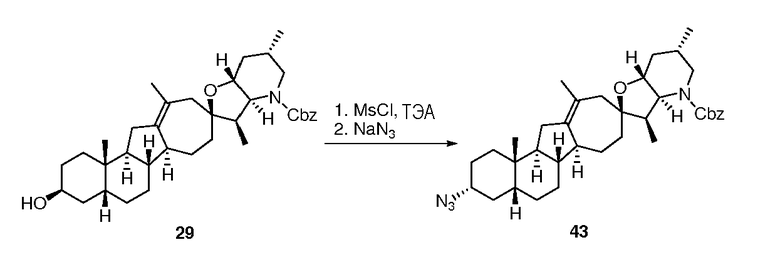
В круглодонную колбу загружали спирт 29 (7,60 г, 13,53 ммоль, 1 экв.) и растворяли в DCM (115 мл). В реакционную смесь загружали триэтиламин (8,21 г, 81 ммоль, 6,0 экв.). Смесь охлаждали до 0°C, и загружали метансульфонилхлорид (1,86 г, 16,2 ммоль, 1,2 экв.). Через 30 мин реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия и EtOAc. Органический слой отделяли, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали, используя флэш хроматографию на силикагеле (смесью EtOAc/гексаны 10 → 25%), получая требуемое вещество, мезилат.
В круглодонную колбу загружали мезилат (9,1 г, 14,22 ммоль, 1 экв.) и растворяли в 50 мл DMPU. В реакционную смесь загружали азид натрия (4,62 г, 71,1 ммоль, 5,0 экв.) и нагревали до 60°C. Смесь перемешивали 17 часов. Затем реакционную смесь охлаждали до комнатной температуры и загружали воду. Смесь перемешивали 30 мин. Смесь фильтровали в вакууме, промывали водой и сушили на открытом воздухе и сразу использовали на следующей стадии без очистки.
Стадия В

В круглодонную колбу загружали азид 43 (8,35 г, 14,23 ммоль, 1 экв.) и добавляли ТГФ (120 мл). Затем в реакционную смесь загружали трифенилфосфин (11,2 г, 42,7 ммоль, 3,0 экв.). Смесь нагревали до 50°C и перемешивали 20 часов. Затем реакционную смесь охлаждали до комнатной температуры, и растворитель удаляли в вакууме. Остаток очищали флэш хроматографией на силикагеле (смесью MeOH/DCM 10% → 20%), чтобы получить амин.
В круглодонную колбу загружали амин (5,10 г, 9,09 ммоль, 1 экв.) и растворяли в DCM (60 мл). В реакционную смесь загружали N,N-диизопропилэтиламин (5,88 г, 45,5 ммоль, 5,0 экв.). Смесь охлаждали до 0°C и загружали метансульфонилхлорид (2,08 г, 18,2 ммоль, 2,0 экв.). Через 30 минут реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия и EtOAc, Органический слой собирали, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали флэш хроматографией на силикагеле (смесью EtOAc/гексаны 10 → 30%), чтобы получить Cbz -защищенный метансульфонамид.
Стадия С

В круглодонную колбу загружали метансульфонамид (5,37 г, 8,41 ммоль, 1 экв.), защищенный Cbz (бензилоксикарбонилом), и 10% палладий на угле (1,0 г). Твердые частицы суспендировали в 2-пропаноле (50 мл). Суспензию помещали в атмосферу водорода, и смесь перемешивали 4 часа при 25°C. Затем реакционную смесь фильтровали через Celite®, и фильтрат концентрировали досуха. Затем остаток очищали флэш хроматографией на силикагеле (смесью DCM/MeOH 0 → 5%), чтобы получить требуемый продукт. [M+H] = 505,6 m/z.
Альтернативный синтез соединения 42
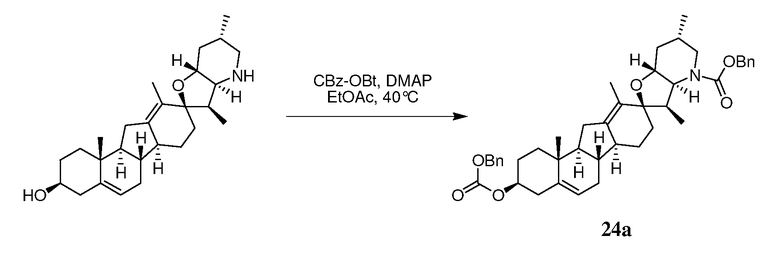
Перекристаллизованный циклопамин (2,07 г) загружают в реакционный сосуд соответствующего размера и помещают в инертную атмосферу. Последовательно добавляют EtOAc (7,6 г), триэтиламин (1,53 Г), и DMAP (307 мг). Суспензию нагревают до 40°C. На протяжении 90 минут тремя порциями добавляют Cbz-OBt, поддерживая внутреннюю температуру ниже 45°C. Реакционную смесь 90 минут перемешивают при 40°C. Сохраняют температуру, пока метанол (26,4 г) медленно добавляют к реакционной смеси. Получающуюся суспензию охлаждают до комнатной температуры и перемешивают в течение, по крайней мере, 15 часов. Неочищенный продукт собирают фильтрованием и промывают метанолом (5 г). Белое твердое вещество сушат в вакууме до постоянной массы и перекристаллизовывают из гептана (30,3 г) и толуола (3,2 г), чтобы получить соединения 24a (3,0 г).
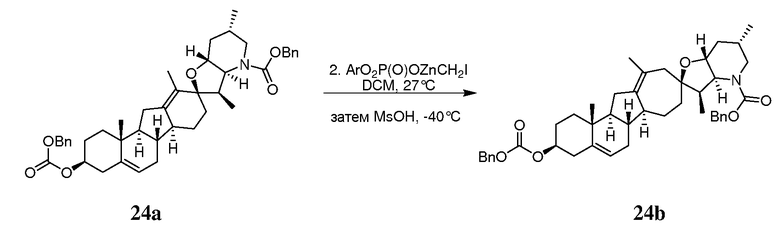
Твердый бис(2,6-диметилфенил)гидрофосфат и соединение 24a предварительно сушат и помещают в атмосферу азота. Беспримесный диэтилцинк (722 мг) загружают в реакционный сосуд соответствующего размера, содержащий DCM (9,0 г). Последовательно добавляют при температуре 25ºC или ниже растворы фосфата (1,83 г в 17,9 г) и IPI-332690 (1,34 г в 3,6 г) в DCM. Загружают дииодметан (1,58 г), и реакционную смесь 4-6 часов перемешивают при 28°C. Реакционную смесь охлаждают до -45°C, и загружают раствор метансульфоновой кислоты в DCM (566 мг в 1,5 г). Через 15 минут добавляют морфолин (1,711 г), и смесь оставляют нагреваться до комнатной температуры в течение ночи. Органический слой дважды промывают 2 н. HCl (2×13,6 г), затем последовательно промывают 4,8% масс. карбонатом натрия (водн.), 4,8% масс. сульфитом натрия (водн.), и 4,8% масс. насыщенным солевым раствором (13,6 г каждый). Органический слой сушат, фильтруют, концентрируют до 4 г и разбавляют изопропанолом (4 г). Продукт кристаллизуют из раствора, медленно добавляя метанол (9,3 г). Фильтруют с промыванием метанолом (2,6 г) и сушат, чтобы получить 1,09 г соединения 24b (с 79% выходом выделенного продукта).
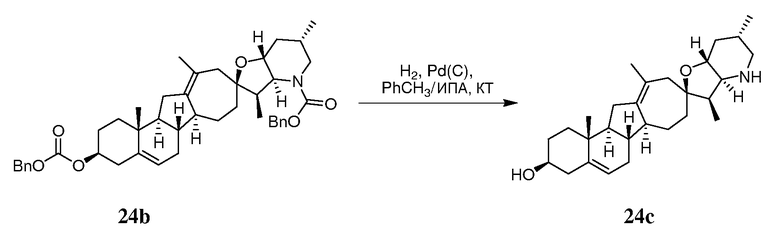
Johnson Matthey Pd/C катализатор A-305038-5 (890 мг) загружают в реакционный сосуд соответствующего размера, добавляя затем соединение 24b (2,24 г). Реакционный сосуд продувают N2 и последовательно добавляют толуол (21,8 г) и 2-пропанол (6,7 г). Систему дегазируют и помещают в атмосферу азота, и процесс повторяют с водородом. Систему интенсивно перемешивают и в течение 4-5 часов сохраняют водородный защитный слой с давлением в одну атмосферу. Реакцию контролируют с помощью или ТСХ-анализа, или ВЭЖХ-анализа (HPLC - жидкостная хроматография высокого разрешения). Если, не дойдя до конца, реакция становится неактивной, то дополнительно загружают катализатор (145 мг), и возвращают атмосферу водорода еще на один час. Загружают этилендиамин (12,9 мг), и смесь перемешивают 15 минут. Катализатор удаляют фильтрованием, с промыванием смесью толуол/IPA (изофталевая кислота) (3:1). Фильтрат и промывные жидкости концентрируют, и растворитель заменяют на толуол. Продукт кристаллизуют из толуола (19,0 г) и гептана (18,0 г), чтобы получить соединение 24c в виде белого кристаллического твердого вещества (1,34 г, 98% выход).
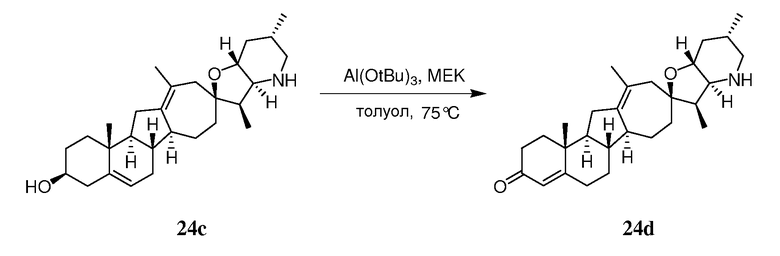
Соединение 24c (644 мг) загружают в реакционный сосуд соответствующего размера, затем добавляют трет-бутилат алюминия (525 мг), толуол (8,34 г, 15 об.), и 2-бутанон (7,83 г, 15 об.). Для удаления кислорода содержимое колбы дегазируют кольцами вакуумирование/продувка азотом. Реакционную смесь нагревают при 75°C с интенсивным перемешиванием в течение 16-18 часов. Реакцию гасят, добавляя водную сегнетовую соль (2,6 г в 10,3 г воды), и смесь интенсивно перемешивают один час при 45°C. Водный и органический слои разделяют. Водный слой обратно экстрагируют смесью толуола (2,9 г) и EtOAc (2,9 г). Органические слои объединяют и промывают раствором свежей сегнетовой соли (2,6 г в 10,3 г воды) и затем - водой (12,9 г). Получающийся органический слой сушат над сульфатом натрия (1,97 г), фильтруют, и концентрируют в вакууме. Продукт кристаллизуют посредством загрузки и обмена растворителя с концентрированием сначала на IPA (6,5 г) и затем на гептан (7,7 г). Крупнодисперсную взвесь в гептане (~2,7 г) перемешивают в течение ночи, и твердые частицы собирают фильтрованием. Вакуумная сушка позволяет получить соединение 24d (550 мг) с выходом 85%.
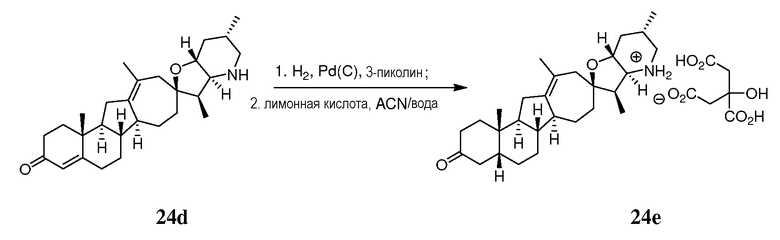
Енон 24d (459 мг) и Johnson-Matthey 5% палладий на угле (A503023-5, 101 мг) загружают в многогорловый реакционный сосуд соответствующего размера. Сосуд продувают азотом, и в качестве растворителя загружают 3-пиколин (2,2 г). Начинают перемешивание, и сосуд сначала дегазируют азотом, и затем 8 часов перемешивают в атмосфере водорода. В конце реакции катализатор удаляют фильтрованием через фильтрующий материал толщиной в 0,2 микрона, промывая ACN (1,4 мл). В атмосфере азота в чистом реакционном сосуде, снабженном механической мешалкой и внутренней термопарой, объединяют фильтрат и промывную жидкость. Раствор лимонной кислоты (3,7 г) в воде (9,2 мл) загружают при температуре 30°C или ниже в реакционный сосуд и оставляют IPI-335589 медленно выкристаллизовываться из раствора в виде цитратной соли при 20, и затем при 0°C. Кристаллический продукт извлекают фильтрованием с отсосом и промывают водой (3,7 мл). После сушки, цитратную соль, 24e, выделяют в виде гидрата (3-5% масс. воды) с выходом 89,5% (622 мг), при отношении β-формы к α-форме, достигающем 90:1.
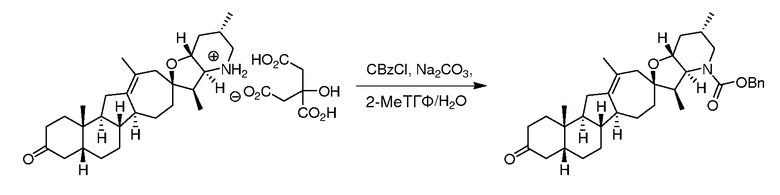
Соединение 24e (1,50 г) совместно с 2-метилтетрагидрофураном (7,7 г) и 1 М карбонатом натрия (9,0 мл) загружают в реактор соответствующего размера. Раствор бензилхлорформиата (454 мг) в 2-метилтетрагидрофуране (300 мг) добавляют через капельную воронку; и в течение 1-2 часов реакция идет при температуре окружающей среды. Когда реакция полностью завершена, перемешивание прекращают, слои разделяют, и органический слой дважды промывают водой (2×6г). Органический слой сушат над сульфатом натрия (3 г), фильтруют и концентрируют. Остаточную воду далее снижают концентрированием из свежеприготовленного 2-метилтетрагидрофурана (6,5 г) и вещество в виде раствора в безводном 2-метилтетрагидрофуране используют в следующей реакции.

1 M Раствор коммерческого реагента «K-Selectride» в ТГФ (1,20 г) в атмосфере азота загружают в сухой реакционный сосуд, разбавляют безводным 2-метилтетрагидрофураном (2,10 г) и охлаждают до -65°C. Затем, чтобы отрегулировать внутреннюю температуру при -65±5°C, в реакционный сосуд медленно добавляют раствор соединения 24f (0,41 г) в 2-метилтетрагидрофуране (1,5 г). Реакционную смесь перемешивают 2 часа и в течение приблизительно 1 часа нагревают до -20°C, и дополнительно перемешивают еще 1 час. Реакционную смесь контролируют по ВЭЖХ и те реакции, которые не закончены, доводят до завершения с помощью дополнительного количества реагента «K-selectride». Реакцию гасят при низкой температуре сначала MeOH (0,33 г), затем 3M NaOH (2,4 г) при -20°C и 15% раствором пероксида водорода в воде (1,04 г) при температуре 5°C или ниже, перемешивая затем в течение ночи при температурах окружающей среды. Слои разделяют, и органический слой промывают последовательно 1M водным раствором NaOH (2 мл), 0,5 M водным раствором Na2SO3 (2 мл), и водой (2 мл), отрегулированной до pH = 3 с помощью HCl. Органический слой сушат над сульфатом натрия (0,82 г), фильтруют и концентрируют. Продукт 24g (0,457 г) повторно концентрируют из DCM (0,9 г) и используют в следующей реакции.
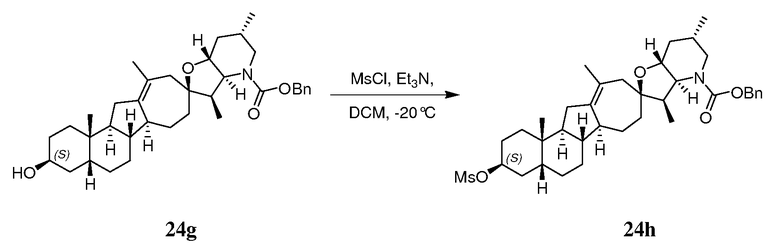
Соединение 24g (1,36 г) загружают вместе с безводным DCM (18,1 г) в реакционный сосуд соответствующего размера, помещают в инертную атмосферу и охлаждают до -20°C. Загружают триэтиламин (0,61 мг) с последующим медленным добавлением метансульфонилхлорида (373 мг) в безводном DCM (300 мг). Реакционную смесь перемешивают 1 час при -20°C. Реакцию контролируют по ВЭЖХ. Незаконченные реакции доводят до конца с помощью дополнительного количества метансульфонилхлорида. При завершении, реакцию гасят водой (13,6 г) и оставляют нагреваться. Слои разделяют, и органический слой промывают 2,5% масс. бикарбонатом натрия (13,8 г) и затем - водой (10,9 г). Органический слой сушат над сульфатом натрия (4 г), фильтруют и концентрируют. Растворитель в растворе продукта заменяют, сначала загружая и концентрируя трет-бутилметиловый эфир (10,9 мл), а затем l,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон (DMPU, 4,7 мл). Раствор DMPU сразу используют в следующей реакции.

Азид натрия (0,74 г) загружают в реакционный сосуд соответствующего размера. Раствор соединения 24h (1,46 г) в DMPU (5,9 г) загружают в реакционный сосуд, промывая дополнительным количеством DMPU (1,9 г). Суспензию 15 часов нагревают до 60°C, сохраняя для полноты реакции продувание азотом. Реакцию охлаждают до температуры окружающей среды и разбавляют MTBE (11,7 г). Органический раствор 3 раза промывают 2% солевым раствором (3×8 г), сушат над сульфатом натрия (4,4 г), фильтруют и концентрируют. Продукт концентрируют из ТГФ (6,4 г) и сразу используют в следующей реакции.
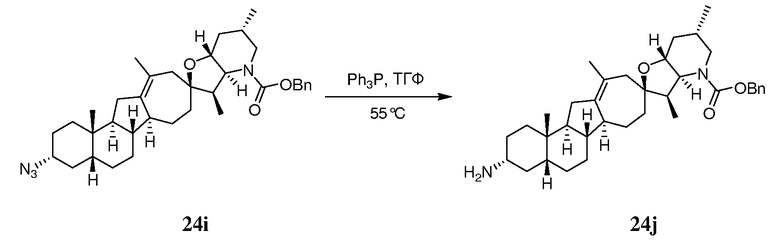
Неочищенное соединение 24i (1,34 г) растворяют и перемещают в реакционный сосуд соответствующего размера с ТГФ (12,6 г). Загружают трифенилфосфин (0,70 г) и воду (0,44 г), и реакционную смесь 15-24 часа нагревают до 55°C. По завершении, реакцию охлаждают до температуры окружающей среды, сушат сульфатом магния (4 г), фильтруют и концентрируют. Твердые частицы растворяют и концентрируют из трех порций DCM (3×9 г) очищают хроматографией на силикагеле, используя градиентное элюирование смесью DCM/MeOH/Et3N для удаления примесей, привносимых реагентами. Объединенные фракции концентрируют досуха, растворяют в DCM (6,8 г) и снова концентрируют досуха, чтобы получить аморфное твердое вещество (1,12 г), которое используют в следующей реакции.
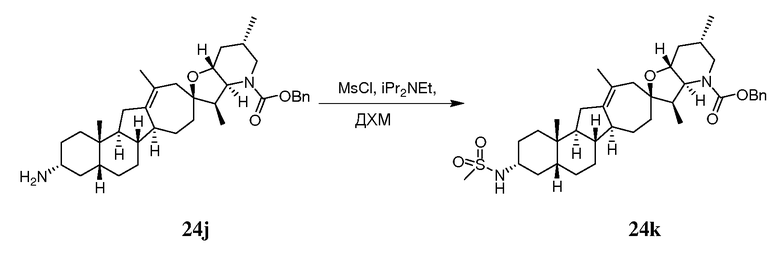
Соединение 24j (1,09 г) растворяют и перемещают в реакционный сосуд соответствующего размера, содержащий безводный DCM (15,8 г), и помещают в атмосферу азота. Раствор охлаждают до 0°C. Последовательно загружают N,N-диизопропилэтиламин (357 мг) и беспримесный метансульфонилхлорид (0,165 мл), поддерживая при этом температуру ниже 5°C. Реакцию контролируют по ВЭЖХ. Незавершенные реакции доводят до конца, добавляя дополнительное количество метансульфонилхлорида. Реакцию гасят 0,4 M водного бикарбоната натрия (11,4 г) и нагревают до температуры окружающей среды. Слои разделяют, и водную фазу обратно экстрагируют DCM (5,8 г). Объединенные органические слои сушат над сульфатом магния (0,55 г), фильтруют и концентрируют. Продукт 24k растворяют и отгоняют из 2-пропанола (4,0 г), чтобы удалить остаточный DCM, и сразу используют в следующей реакции.

В реакционный сосуд соответствующего размера загружают Pd/C 10% (249 мг) (Aldrich Degussa тип El01 NE/W) и помещают в атмосферу азота. В реакционный сосуд загружают раствор соединения 24k (1,24 г) в 2-пропаноле (9,8 г). Систему дегазируют и помещают в атмосферу азота; и процесс повторяют с водородом. Реакционную смесь 8 часов перемешивают в атмосфере водорода (1 атм.) при температуре окружающей среды. Сосуд возвращают в инертную атмосферу, и в реакционную смесь осуществляют вторую загрузку катализатора (125 мг), суспендированного в 2-пропаноле (0,5 г). Реакционную смесь дегазируют и помещают в атмосферу азота, и процесс повторяют с водородом. Реакционную смесь перемешивают еще 15 часов при температуре окружающей среды в атмосфере водорода (1 атм.). Реакцию контролируют по анализу ВЭЖХ. Незавершенные реакции обрабатывают дополнительным количеством катализатора и водорода. По завершении, реакционную смесь фильтруют, обрабатывают углем (200 мг), активированным водяным паром, и снова фильтруют. Раствор сушат частичным концентрированием, перемещают в реакционный сосуд и разбавляют безводным 2-пропанолом до концентрации 0,09 М, основываясь на теоретическом выходе. На протяжении 20 минут загружают 1,25 М раствор HCl в 2-пропаноле (1,64 г). Гидрохлорид соли медленно кристаллизуется при осторожном перемешивании, и его отделяют фильтрованием. Кристаллы промывают 2-пропанолом (2,5 г) и сушат в вакууме, чтобы получить соединение 42 (916 мг, 80% выход) в виде 1:1 сольвата IPA.
Пример 25
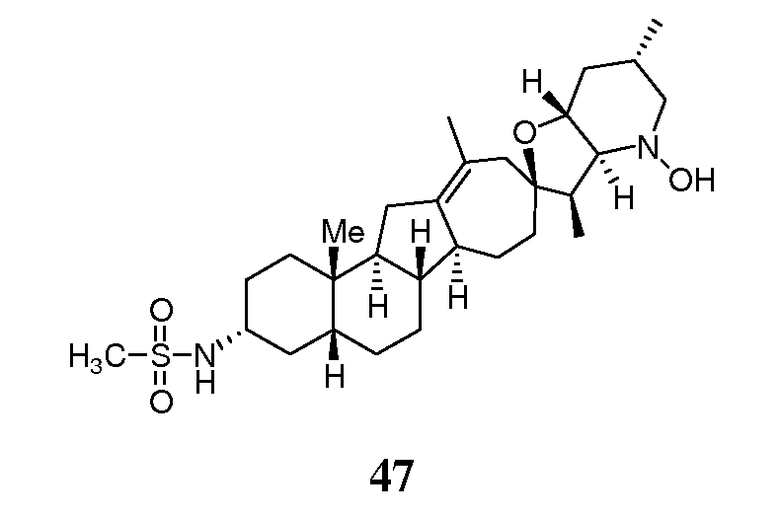
Стадия A
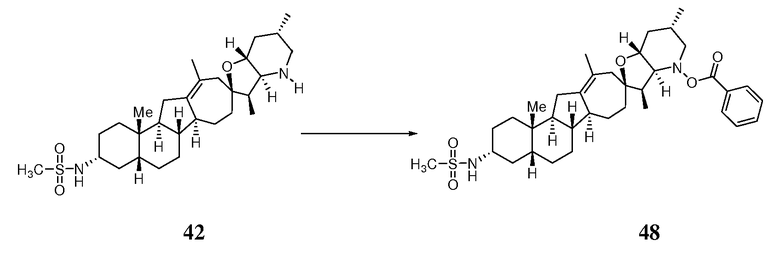
В круглодонную колбу загружали амин 42 (1,1 г, 2,1 ммоль, 1 экв.), сухой тетрагидрофуран (10 мл) и пиридин (880 мкл), 10,9 ммоль, 5 экв.). Охлажденную (0°C) смесь обрабатывали пероксидом бензоила (1,6 г, 6,5 ммоль, 3 экв.). Смесь перемешивали 2 часа при 0oC, затем в течение ночи при 25°C. Реакционную смесь разбавляли MTBE и промывали смесью насыщенного водного раствора NaHCO3 и 1 н. раствора NaOH до разделения слоев. Органический слой собирали, а водный слой один раз повторно экстрагировали MTBE. Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенное масло растворяли в 5 мл CH2Cl2, загружали в SiO2 колонку (40 г) и элюировали смесью гексаны/EtOAc (10% до 50%), чтобы получить бензоильное производное 48 (380 мг) ([M+H] = 625,4 m/z).
Стадия В

В круглодонную колбу загружали соединение 48 (374 мг, 0,6 ммоль, 1 экв.) и MeOH (5 мл). Раствор обрабатывали при 25°C в присутствии 2 н. KOH (0,3 мл, 0,6 ммоль, 1 экв.). Смесь перемешивали 3 часа. Растворитель удаляли в вакууме. Добавляли MTBE к остатку, который нейтрализовали 1н. раствором HCl. Слои разделяли, и водный слой экстрагировали двумя порциями CH2Cl2. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенное вещество (380 мг) растворяли CH2Cl2, загружали в SiO2 колонку (12 г) и элюировали смесью гексаны/EtOAc (от 0% до 100%), чтобы получить гидроксиламин 47. Вещество лиофилизовали из смеси т-BuOH/7% H20, чтобы получить 213 мг соединения 47 в виде белого порошка ([M+H] = 521,4 m/z).
Пример 26

Стадия A

51
В колбу, высушенную с использованием струйной воздушной сушилки, загружали сухой CH2Cl2 (5 мл) и бензиловый спирт (785 мкл, 7,58 ммоль, 1,3 экв.). Охлажденный (0°C) раствор обрабатывали хлорсульфонилизоцианатом (506 мкл), 5,83 ммоль, 1 экв.). Затем добавляли DMAP (1,4 г, 11,6 ммоль, 2 экв.), и смесь перемешивали 1 час при 25°C. После полного растворения DMAP, реакционная смесь незначительное время оставалась прозрачной. Затем образовывался белый рыхлый осадок. Смесь разбавляли CH2Cl2 (30 мл) и промывали тремя порциями (30 мл, каждая) воды. Органический слой сушили над Na2SO4, фильтровали и упаривали досуха. Требуемое белое твердое соединение 51 без очистки использовали на следующей стадии.
Стадия В

В круглодонную колбу загружали соединение 52 (30 мг, 0,053 ммоль, 1 экв.) и соединение 51 (18 мг, 0,053 ммоль, 1 экв.). Оба реагента растворяли в CH2Cl2 (2 мл), и раствор перемешивали при 25°C. Неочищенное вещество загружали в SiO2 колонку (4 г) и элюировали смесью гексаны/EtOAc (от 0% до 50%), чтобы получить 16 мг сульфамоильного производного 53. ([M+Na] = 796,4 m/z).
Стадия С
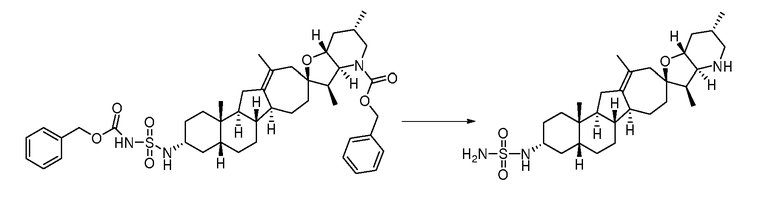

В круглодонную колбу загружали соединение 53 (16 мг, 0,021 ммоль, 1 экв.) и 11 мг 10% Pd/C (влажный, Aldrich Degussa тип El0l). Вещество суспендировали в 2-пропаноле (3 мл). Колбу герметизировали и три раза продували водородом, и оставляли в течение ночи в атмосфере водорода (1 атм). Взвесь фильтровали через 0,2 микронный фильтр Acrodisc, промывали 2-пропанолом, и растворитель удаляли в вакууме. Остаток очищали на SiO2 колонке (1 г), элюируя смесью CH2Cl2/MeOH (от 5% до 10%). Основной продукт лиофилизовали из смеси т-BuOH/7% H20, чтобы получить 9 мг сульфамида 50. ([M+H] = 506,4 m/z).
Пример 27
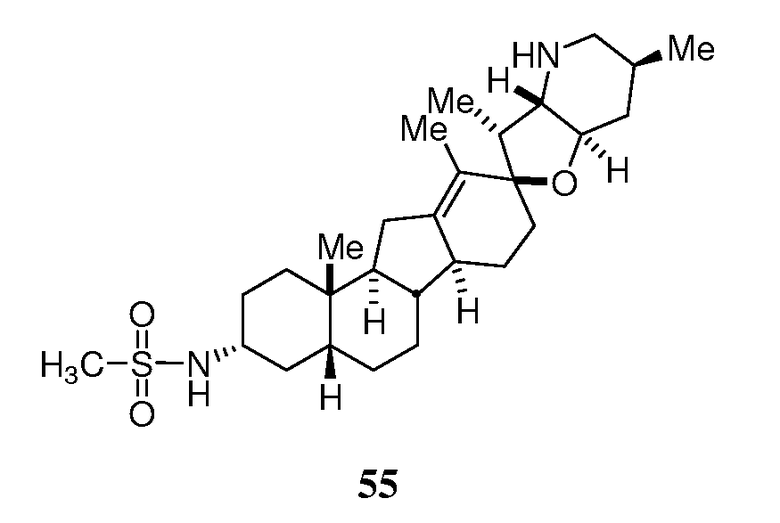
Стадия A
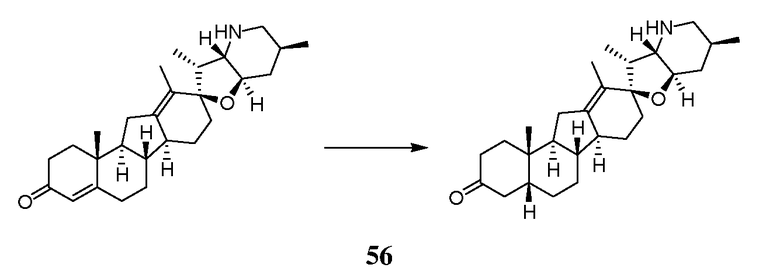
В круглодонную колбу загружали циклопамин-4-ен-3-он (3,5 г, 8,5 ммоль, 1 экв.) и пиридин (70 мл). В реактор загружали Pd/C (10% Pd, 500 мг). Реакционную смесь помещали в атмосферу водорода (1 атм). Через 3,5 часа, анализ LCMS (жидкостная хроматомасс-спектрометрия) показал полный расход исходного вещества. Катализатор отфильтровывали на 0,2 микронном фильтре Acrodisk и промывали толуолом. Растворитель удаляли в виде азеотропной смеси с толуолом (2×10 мл). Требуемое вещество 56, (3,5 г) ([M+H] = 412,5 m/z) использовали таким, как получили, на следующий стадии.
Стадия В
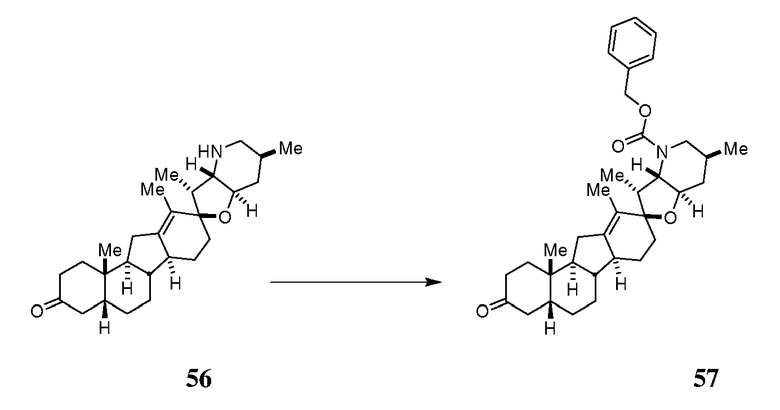
В круглодонную колбу загружали вещество 56 (1,2 г, 2,8 ммоль, 1 экв.), CH2Cl2 (10 мл) и триэтиламин (1,9 мл, 14,2 ммоль, 5 экв.). Охлажденный (0°C) раствор обрабатывали CBz-Cl (440 мкл, 2,8 ммоль, 1 экв.). Через 1 час, анализ LCMS показал полный расход исходного вещества. Смесь разбавляли водой. Слои разделяли, и органический слой дважды промывали водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали досуха. Продукт очищали колоночной хроматографией (SiO2, 40 г), элюируя смесью гексан/EtOAc (от 0 до 20%), чтобы получить соединение 57. (891 мг) ([M+Na] - 468,4 m/z).
Стадия С
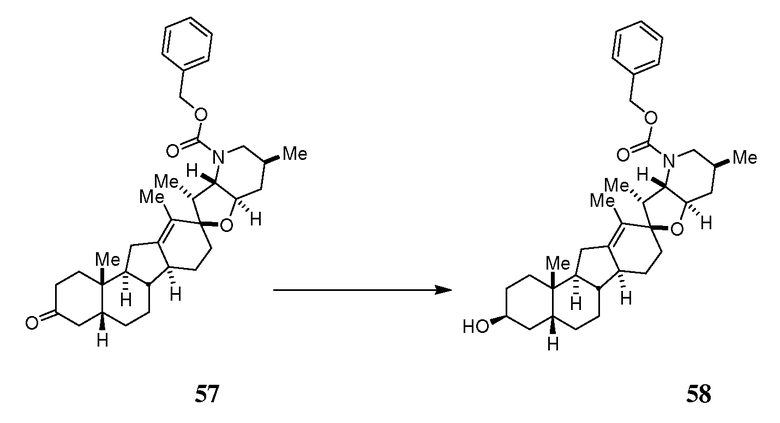
В круглодонной колбе, кетон 57 отгоняли два раза в виде азеотропной смеси с CH2Cl2 и сушили 1 час в вакууме. В атмосфере азота, кетон 2 (693 мг, 1,27 ммоль, 1 экв.) растворяли в безводном ТГФ (20 мл), и раствор охлаждали до -78C. По каплям добавляли 1М раствор реагента K-selectride в ТГФ (1,9 мл, 1,9 ммоль, 1,5 экв.). Через 1 час, согласно ТСХ, реакция была завершена. Реакцию гасили, добавляя 2,6 мл 5 н. NaOH, с последующим медленным добавлением 2,6 мл 30% масс. H2O2. Получающуюся смесь оставляли перемешиваться в течение ночи. Смесь распределяли между водой и EtOAc. Водный слой экстрагировали обратно EtOAc. Объединенные органические вещества промывали сначала водой (буферизованной небольшой порцией хлорида аммония), затем насыщенным солевым раствором. Органическое вещество сушили, фильтровали и концентрировали до неочищенной пены (840 мг). Неочищенное вещество растворяли в CH2Cl2, загружали в SiO2 колонку (40г) и элюировали смесью гексаны/EtOAc (от 0 до 50%), чтобы получить соединение 58 (565 мг).
Стадия D

В круглодонной колбе в атмосфере азота растворяли спирт 58 (530 мг, 0,98 ммоль, 1 экв.) в 5 мл безводного CH2Cl2 и триэтиламина (800 мкл), 5,81 ммоль, 6 экв.). Реакционную смесь охлаждали до 0°C и по каплям добавляли Ms-Cl (112 мкл), 1,45 ммоль, 1,5 экв.). Смесь перемешивали 30 мин при 0°C. ТСХ (смесью гексан:EtOAC, 7:3) показала ~ 70% превращение. В реакционный сосуд загружали 70 мкл триэтиламина (70 мкл), 0,5 экв.) и Ms-Cl (10 мкл), 0,1 экв.). Через 90 мин загружали раствор насыщенного бикарбоната, и остаток экстрагировали CH2Cl2. Органический слой промывали водой, сушили и концентрировали до не совсем белой пены. Вещество растворяли в CH2Cl2 и очищали на колонке SiO2 (40 г), элюируя смесью гексаны/EtOAc (от 0% до 50%), чтобы получить соединение 59 (430 мг).
Стадия E

В круглодонной колбе, растворяли мезилат 59 (420 мг, 0,67 ммоль, 1 экв.) в 2 мл DMPU. Раствор при 60°C обрабатывали азидом натрия (218 мг, 3,4 ммоль, 5 экв.) в течение 5 часов. Смесь охлаждали до 25ºC, затем вливали в ледяную воду с образованием белого твердого вещества. Соединение экстрагировали MTBE (3 раза). Объединенные органические слои промывали водой (2X), затем насыщенным солевым раствором. Органические слои сушили над Na2SO4, фильтровали и концентрировали до белой пены (342 мг). Требуемое вещество 60 использовали таким, как получили, на следующей стадии.
Стадия F
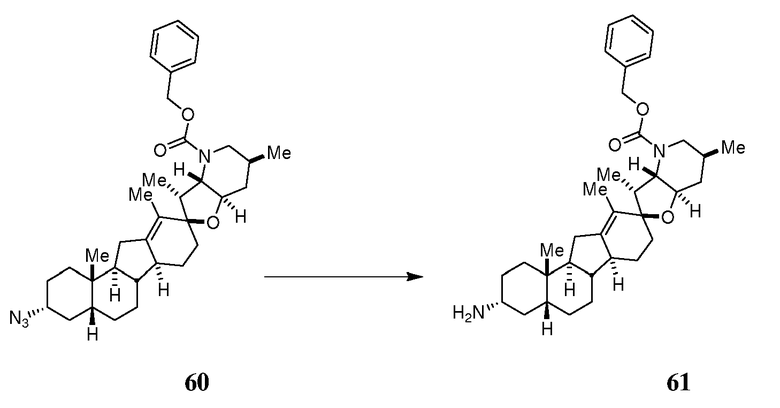
В круглодонной колбе, снабженной холодильником, растворяли азид 60 (336 мг, 0,58 ммоль, 1 экв.) в 7 мл ТГФ и 140 мкл воды и обрабатывали трифенилфосфином (462 мг, 1,76 ммоль, 3 экв.). Смесь в течение ночи нагревали до 70°C. Анализ ТСХ (смесью гексан/EtOAc, 7:3) подтвердил, что реакция завершена. Реакционную смесь концентрировали досуха. Неочищенное вещество растворяли в CH2Cl2, загружали в SiO2 колонку (12 г) и элюировали смесью CH2Cl2/MeOH (от 0 до 20%), чтобы получить амин 61 (254 мг).
Стадия G

В круглодонной колбе в атмосфере азота, растворяли амин 61 (248 мг, 0,45 ммоль, 1 экв.) в 7 мл безводного CH2Cl2 и N,N-диизопропилэтиламина (237 мкл), 0,91 ммоль, 2 экв.). Реакционную смесь охлаждали до 0°C и по каплям добавляли Ms-Cl (70 мкл), 1,45 ммоль, 1,5 экв.). Смесь перемешивали 2 часа при 0°C. Анализ ТСХ (смесью гексан/EtOAc, 7:3) показал незначительное количество амина. В смесь загружали 10 мкл Ms-Cl (0,2 экв.), и нагревали l час до 25°C. Реакционную смесь разбавляли CH2Cl2, затем насыщенным раствором NaHCO3. Слои разделяли. Водный слой экстрагировали одной порцией CH2Cl2. Объединенные органические слои промывали водой, сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенное вещество (326 мг) загружали в SiO2 колонку (12 г) и элюировали смесью гексаны/EtOAc (0 до 50%), чтобы получить сульфонамид 62 (256 мг).
Стадия H

В круглодонную колбу загружали сульфонамид 62 (250 мг, 0,4 ммоль, 1 экв.) и 50 мг 10% Pd/C (влажный, Aldrich Degussa тип E101 партия 08331KC). Вещество суспендировали в EtOAc (5 мл). Колбу герметизировали и три раза продували водородом, и перемешивали в атмосфере водорода (1 атм). Через 3 часа наблюдали некоторое превращение, но оставалось много исходного вещества. Взвесь фильтровали через 0,2 микронный фильтр Acrodisc, промывали 2-пропанолом. В раствор фильтрата добавляли 54 мг катализатора, повторно создавая условия протекания реакции. Через 3 часа реакция была завершена. Взвесь фильтровали через 0,2 микронный фильтр Acrodisc, промывали 2-пропанолом, и растворитель концентрировали досуха. Неочищенное вещество (200 мг) загружали в SiO2 колонку (12 г), и соединение подвергали градиентному элюированию смесью CH2Cl2/MeOH (от 0 до 10%), чтобы получить свободный амин. Вещество лиофилизовали из смеси т-BuOH/7% H2O, чтобы получить 175 мг соединения 55 в виде белого порошка ([M+H] = 491,3 m/z).
Пример 28
Ингибирование сигнального пути hedgehog в клеточной культуре
При использовании нижеследующего теста могут быть выявлены эффекты гибели специфических раковых клеток, обусловленные сигнальным путем Hedgehog. Клетки C3H10T1/2 дифференцируются в остеобласты, при контакте с обработанным ультразвуком пептидом hedgehog (Shh-N). После дифферецировки эти остеобласты продуцируют высокие уровни щелочной фосфатазы (AP), которые могут быть измерены при ферментативном анализе (Nakamura, et at., BBRC (1997) 237:465). Соединения, которые блокируют дифференцировку клеток C3H10T1/2 в остеобласты (Shh-зависимое событие), можно, поэтому, идентифицировать по ослаблению продуцирования AP (van der Horst, et al., Bone (2003) 33:899). Подробности анализа описаны ниже. Апрксимация результатов теста на дифференцировку (EC50 для ингибирования) приведена ниже в таблице 1.
Протокол опыта
Клеточная культура
Клетки C3H10T1/2 мышиных эмбриональных мезодермальных фибробластов (получаемых в ATCC (американская коллекция типовых культур)) культивировали при 37°C в основной среде MEM (минимальной незаменимой среде) (Gibco/Invitrogen) с 10% инактивированной нагреванием FBS (телячьей эмбриональной сывороткой) (Hyclone), 50 ед./мл пенициллина и 50 мкг/мл стрептомицина (Gibco/Invitrogen), в воздушной атмосфере с 5% CO2.
Анализ щелочной фосфатазы
Клетки C3H10T1/2 высевали в 96 луночные планшеты с плотностью 8x103 клеток/лунка. Клетки выращивали до слияния (72 часа). После обработки обработанным ультразвуком, Hedgehog (250 нг/мл) и/или соединением, клетки лизировали в 110 мкл лизисного буфера (50 мМ Tris pH 7,4, 0,1% TritonXlOO). Планшеты обрабатывали ультразвуком, и лизаты центрифугировали с применением фильтровальных плашек с PVDF мембраной с диаметром 0,2 мкм (Corning). Лизаты (40 мкл) анализировали на AP активность в щелочном буферном растворе (Sigma), содержащем l мг/мл п-нитрофенил фосфата. После инкубирования при 37°C в течение 30 мин, планшеты считывали на Envision планшет-ридере на 405 нм. Общий белок определяли, используя набор для белкового анализа BCA (BCA protein assay kit from Pierce), согласно инструкции производителя. AP активность нормировали по общему белку. Заметим, что знак "A" указывает на то, что IC50 составляет менее 20 нМ (nM), знак "B" указывает, что IC50 составляет 20-100 нМ, знак "C" указывает, что IC50 составляет > 100 нМ.
Приблизительные EC50 для анализа ингибирования дифференцировки
Пример 29
Модель рака поджелудочной железы
Активность соединения 42 далее тестировали на модели поджелудочной железы человека: Мышам подкожно в боковую часть нижней правой конечности имплантировали клетки BXPC-3. На 42 день после имплантирования опухоли, мышей разделяли методом случайного отбора на две группы, для получения либо носителя (30% HPBCD), либо соединения 42. Соединения 42 вводили перорально в дозе 40 мг/кг/день. После введения 25-и ежедневных доз, соединение 42 статистически снижало объемный рост опухоли на 40%, по сравнению с контрольной группой, которой вводили носитель (p=0,0309). В конце исследования, через 4 часа после последней дозы, осуществляли забор опухолевых клеток, чтобы оценить ответную реакцию мишени по q-ОТ-ПЦР (q-RT PCR) анализу генов сигнального пути HH. Анализ Gli-1 человека показал отсутствие модуляции. Анализ мышиных Gli-1 мРНК уровней показал устойчивую даун-регуляцию в группе, которую лечили соединением, по сравнению с группой, которую лечили носителем. Ингибирование сигнального пути hedgehog в мышиных клетках, но не в клетках опухоли человека, показывает, что единственным действием ингибитора hedgehog является воздействие на взаимодействие опухоли и стромы.
Пример 30
Модель Медуллобластомы
Активность соединения 42 оценивали также на модели медуллобластомы трансгенных мышей. Мыши, которые являются гетерозиготными, вследствие мутаций с потерей функций в опухолевых суппрессорах Patchedl (Ptchl), и гиперметилированными при раковом (Hicl) развитии спонтанной медуллобластомы. Аналогично человеческой медуллобластоме, эти опухоли демонстрируют полное гиперметилирование промотора остающегося Hicl аллеля, так же как потерю экспрессии Ptchl аллеля дикого типа. При пассаже (пересеве) в виде подкожных аллотрансплантантов эти опухоли растут агрессивно и зависимо от сигнального пути hedgehog. Эту модель использовали, чтобы оценить эффективность перорально вводимого соединения и откорректировать активность с экспозицией лекарственного средства в плазме и опухоли. Пероральное введение (PO) единичной дозы соединения 42 вело к дозозависимой даун-регуляции сигнального пути HH в подкожно имплантированных опухолях, что измеряли по сниженной Gli-1 мРНК экспрессии через 8 часов после введения дозы.
Ежедневное (QD) введение соединения PO приводило к, дозозависимому ингибированию роста опухоли, с явной регрессией опухоли, наблюдаемой при повышенных дозах. Эффективная ежедневная приблизительная пероральная доза для 50% ингибирования роста опухоли (ED50) составляет 4 мг/кг. Когда животных обрабатывали QD в течение 21 дня, после прекращения лечения наблюдали длительное продолжение выживаемости (>60 дней), c незначительным возобновлением или отсутствием роста опухоли. Это показывает, что ингибитор hedgehog соединение 42 ингибирует и сигнальный путь hedgehog, и рост опухоли в опухоли, зависимой, вследствие генетической мутации, от сигнального пути hedgehog.
Пример 31
Модель рака легких
Чтобы тестировать активность соединения 42 на модели человеческой SCLC опухоли, клетки LX22 подкожно имплантировали в боковую часть правой нижней конечности. LX22 представляет собой первичную модель ксенотрансплантата SCLC, получаемого от больных, не подвергавшихся химиотерапии, который сохраняли при серийном пассаже от мыши к мыши. Реакция опухоли на химиотерапию этопозидом/карбоплатином очень напоминает патологический процесс. В процессе химиотерапевтического лечения LX22 регрессирует, проходит через период ремиссии и затем начинает рецидивировать. На модели LX22 тестировали активность отдельного представителя соединения и его способность понижать частоту хемоустойчивого (хеморезистентного) рецидива. На 32 день после имплантирования опухоли мышей методом случайного отбора разделяли на три дозировочные группы, чтобы принимать носитель (30% HBPCD), соединение или химиотерапевтическую комбинацию этопозида и карбоплатина (E/P). Соединение 42 вводили перорально в дозе 40 мг/кг/день. После введения 16 последовательных доз отсутствовало измеряемое различие между группами, в которых мышей подвергали лечению, и группой, в которой мышам вводили носитель. Этопозид вводили (i.v) внутривенно при дозе 12 мг/кг на 34, 35, 36 и 48 день, тогда как карбоплатин вводили i.v. в дозе 60 мг/кг на 34, 41 и 48 день после имплантирования опухоли. На 50 день, мыши, которых лечили E/P, были разделены методом случайного отбора, чтобы для завершения лечения получать или носитель (30% HPBCD), или соединение. Соединение вводили перорально в мульти-дозе (MTD), составляющей 40 мг/кг/день. После 35 последовательных доз, в группе, подвергаемой лечению, наблюдали значительную задержку рецидива опухоли, по сравнению с группой, принимающей носитель (p=0,0101).
Пример 32
Множественная миелома
Способность соединения 42 ингибировать рост клеток множественной миеломы (ММ) тестировали in vitro, используя клеточные линии (NCI-H929 и KMS12) множественной миеломы человека и клинические мазки крови костного мозга, получаемые от больных MM. Клетки 96 часов обрабатывали соединением, промывали, затем высевали в метилцеллюлозу. Через 10-21 день подсчитывали количество колоний опухолей как индикатора способности клеточного роста после обработки. Обработка клеточных линий или образцов, получаемых от первичных больных, привела к уменьшенному клеточному росту, по сравнению с необработанным контролем. Когда необработанный контроль показывал 100% рост клеток, каждая из обработанных клеточных линий, как и клинические образцы, показывали менее 25% роста.
Пример 33
Острый миелоидный лейкоз и миелодиспластический синдром
Исследовали способность соединения 42 ингибировать in vitro рост клеточных линий человека, получаемых от больных острым миелоидным лейкозом (ОМЛ (AML), клеточная линия U937) и миелодиспластическим синдромом (МДС (MDS), клеточная линия KG1 и KG1a). Каждую из клеточных линий обрабатывали 72 часа соединением 42 (1,0 мкМ), с последующим посевом в метилцеллюлозу. Рост этих клеточных линий ингибировался соединением 42, что суммировано в таблице, приведенной ниже.
Ингибирование клеточного роста при ОМЛ и МДС
Пример 34
Неходжкинская лимфома (НХЛ) и болезнь Ходжкина (БХ)
Исследовали способность соединения 42 ингибировать in vitro рост клеточных линий человека, получаемых от больных неходжкинской лимфомой (клеточные линии RL и Jeko-1) и болезнью Ходжкина (клеточная линия L428). Каждую из клеточных линий обрабатывали 72 часа соединением 42 (1,0 мкм) с последующим посевом в метилцеллюлозу. Рост этих клеточных линий ингибировался соединением 42, что суммировано в таблице, приведенной ниже.
Ингибирование клеточного роста при БХ и НХЛ
Пример 35
Острый лимфолейкоз пре-B клеток
Изучали активность соединения 42 (1 мкМ) против трех клеточных линий (REH, RS4-11 и Nalm-6) острого лимфолейкоза пре-B клеток, применяя анализ с транзиентной трансфекцией, в котором Gli-респонсивный люциферазный репортер транзиентно трансфицировали в клетки. Лечение соединением 42 подавляло активность люциферазы, по сравнению с контролем, обработанным носителем (Таблица 4). Это показывает, что соединение 42 является эффективным антагонистом сигнального пути hedgehog.
Подавление активности люциферазы
Изучали также действие соединения 42 на рост двух из этих клеточных линий, обрабатываемых 72 часа in vitro. После обработки клетки промывали и высевали в метилцеллюлозу. Наблюдали слабое ингибирование образования колонии, но следующий повторный посев колоний демонстрировал значительное ингибирование клеточного роста (таблица 5).
Ингибирование клеточного роста в целом
Включение путем ссылки
Все из цитированных здесь патентов США и опубликованных заявок США на патент настоящим включены в данный документ путем ссылки.
Эквиваленты
Специалисты в данной области техники поймут или будут способны обнаружить, применяя только обычное экспериментирование, много эквивалентов для конкретных вариантов осуществления изобретения, описанных здесь. Такие эквиваленты предназначены для включения в нижеследующую формулу изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ ЦИКЛОПАМИНА | 2007 |
|
RU2486194C2 |
| АНАЛОГИ ЦИКЛОПАМИНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2403256C2 |
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| ПИРИДИЛПИПЕРИДИНОВЫЕ АНТАГОНИСТЫ РЕЦЕПТОРА ОРЕКСИНОВ | 2008 |
|
RU2470021C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| СПИРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2018 |
|
RU2781639C2 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ИНГИБИТОРЫ IAP | 2005 |
|
RU2401840C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
Изобретение относится к области фармацевтики и медицины и касается способов лечения рака и антагонизации сигнального пути hedgehog в клетке с помощью производного циклопамина общей формулы (1). 3 н. и 31 з.п. ф-лы, 5 табл., 35 пр.

1. Способ лечения рака у пациента, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения, имеющего структуру:
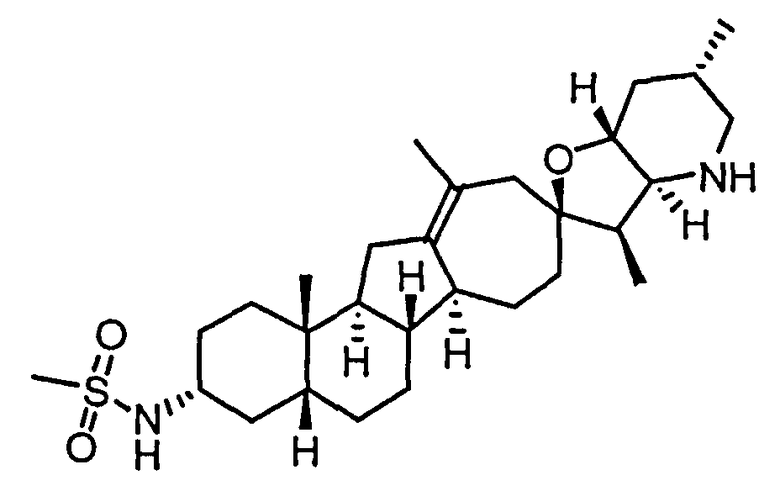
или его фармацевтически приемлемой соли;
где указанный рак выбирают из группы, включающей острый лимфолейкоз, базально-клеточный рак, рак желчных протоков, глиому, рак молочной железы, хондросаркому, хронический лимфолейкоз, хронический миелоидный лейкоз, рак толстой кишки, рак пищевода, рак желудка, опухоль гастроинтестинальной стромы, гепатоцеллюлярный рак, рак почки, рак легких, медуллобластому, меланому, множественную миелому, нейроэндокринные опухоли, неходжкинскую лимфому, остеосаркому, рак яичников, рак поджелудочной железы, рак простаты, тестикулярный рак и саркому.
2. Способ по п.1, где рак представляет собой базально-клеточный рак, рак поджелудочной железы, рак простаты, остеосаркому, хондросаркому, неходжкинскую лимфому, острый лимфолейкоз, хронический лимфолейкоз, хронический миелоидный лейкоз, рак желудка, рак пищевода, рак желчных протоков, множественную миелому, рак легких, глиому, рак молочной железы, гепатоцеллюлярный рак, рак яичников, рак толстой кишки или медуллобластому.
3. Способ по п.1, где рак представляет собой рак легких.
4. Способ по п.3, где рак легких представляет собой мелкоклеточный рак легких.
5. Способ по п.1, где рак представляет собой рак поджелудочной железы.
6. Способ по п.1, где рак представляет собой базально-клеточный рак.
7. Способ по п.1, где рак представляет собой медуллобластому.
8. Способ по п.1, где рак представляет собой острый лимфолейкоз.
9. Способ по п.1, где рак представляет собой хронический лимфолейкоз.
10. Способ по п.1, где рак представляет собой рак яичников.
11. Способ по п.1, где рак представляет собой хондросаркому.
12. Способ по п.1, где рак представляет собой остеосаркому.
13. Способ по п.1, где рак представляет собой хронический миелоидный лейкоз.
14. Способ по п.1, где соединение применяется в комбинации с одним или несколькими химиотерапевтическими или другими противораковыми средствами.
15. Способ по п.14, где другое противораковое средство представляет собой облучение.
16. Способ по п.1, где соединение вводится локально в опухоль.
17. Способ по п.1, где соединение вводится системно.
18. Способ по п.1, где путь введения указанного соединения представляет собой ингаляционный, пероральный, внутривенный, сублингвальный, глазной, трансдермальный, ректальный, вагинальный, местный, внутримышечный, внутриартериальный, интратекальный, подкожный, буккальный или назальный.
19. Способ по п.18, где путь введения представляет собой пероральный, внутривенный или местный.
20. Способ антагонизации сигнального пути hedgehog в клетке, включающий контактирование клетки, экспрессирующей Smoothened белок, с эффективным количеством соединения, имеющего следующую формулу:
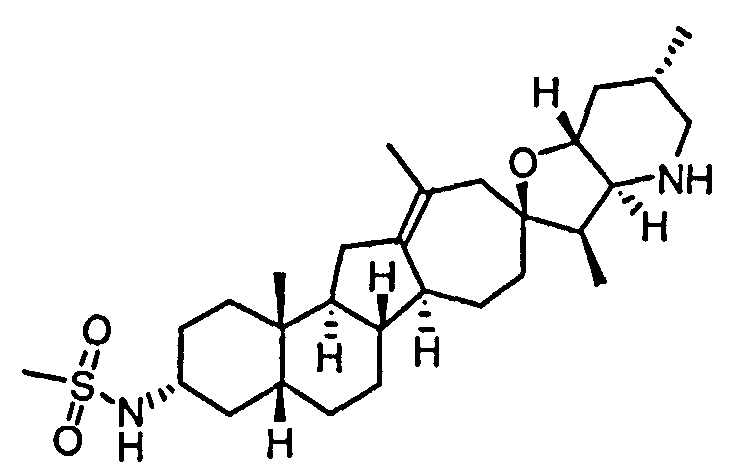
или его фармацевтически приемлемой солью.
21. Способ по п.20, где указанное контактирование осуществляется in vitro.
22. Способ по п.20, где указанное контактирование осуществляется in vivo.
23. Способ по п.20, где указанная клетка, экспрессирующая Smoothened белок, находится внутри тела организма.
24. Способ лечения рака у пациента, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения, имеющего формулу:
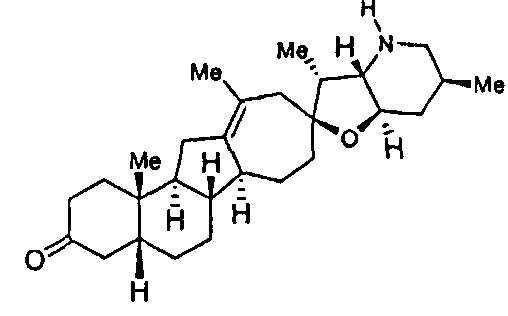 ,
,  ,
,
 ,
,  ,
,
 ,
,  ,
,
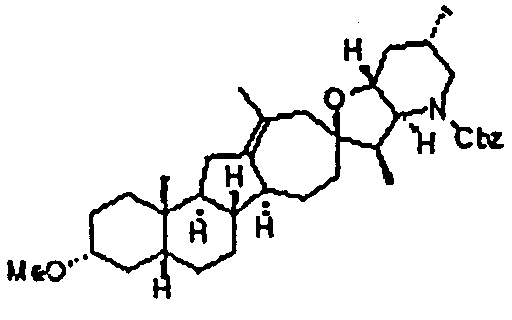 ,
,  ,
,
 ,
, 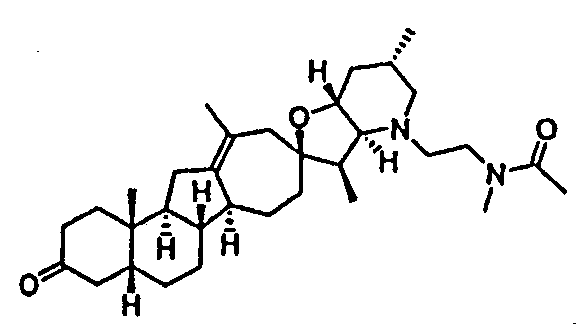 ,
,
 ,
,  ,
,
 ,
, 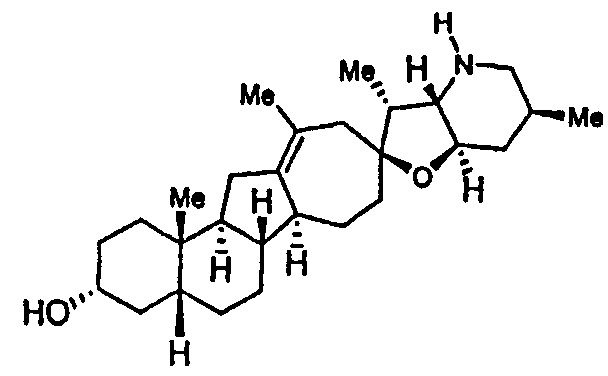 ,
,
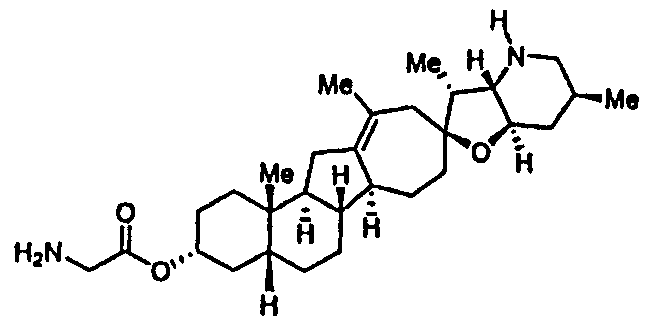 ,
, 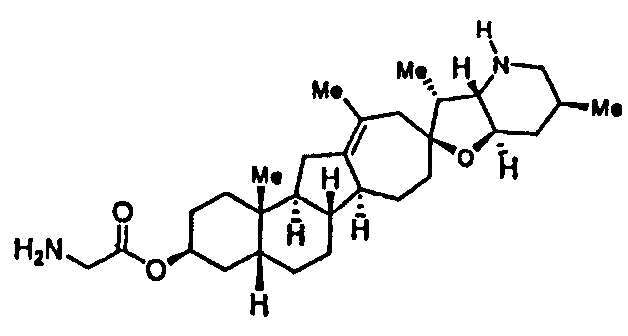 ,
,
 и
и 
или его фармацевтически приемлемой соли,
где указанный рак выбирают из группы, включающей острый лимфолейкоз, базально-клеточный рак, рак желчных протоков, глиому, рак молочной железы, хондросаркому, хронический лимфолейкоз, хронический миелоидный лейкоз, рак толстой кишки, рак пищевода, рак желудка, опухоль гастроинтестинальной стромы, гепатоцеллюлярный рак, рак почки, рак легких, медуллобластому, меланому, множественную миелому, нейроэндокринные опухоли, неходжкинскую лимфому, остеосаркому, рак яичников, рак поджелудочной железы, рак простаты, тестикулярный рак и саркому.
25. Способ по п.24, где раком является множественная миелома.
26. Способ по п.24, где лейкозом является острый лимфолейкоз, хронический миелоцитарный лейкоз или хронический лимфолейкоз.
27. Способ по п.24, где раком является неходжкинская лимфома.
28. Способ по п.24, где соединение применяется в комбинации с одним или несколькими другими химиотерапевтическими средствами или противораковыми средствами.
29. Способ по п.28, где другим противораковым средством является облучение или хирургическое лечение.
30. Способ по п.24, где соединение вводится локально в опухоль.
31. Способ по п.24, где соединение вводится системно.
32. Способ по п.24, где путь введения указанного соединения представляет собой ингаляционный, пероральный, внутривенный, сублингвальный, глазной, трансдермальный, ректальный, вагинальный, местный, внутримышечный, внутриартериальный, интратекальный, подкожный, буккальный или назальный.
33. Способ по п.32, где путь введения представляет собой пероральный, внутривенный или местный.
34. Способ по п.24, где фармацевтически приемлемой солью является гидрохлоридная соль.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 2006142245 A1, 29.06.2006 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 2004103072 A, 27.03.2005. | |||

