Родственные заявки
В настоящей заявке испрашивается приоритет в связи с предварительными заявками на выдачу патента США серийный номер 60/580616, поданной 17 июня 2004 г., и №60/659301, поданной 7 марта 2005 г., которые полностью включены в объем настоящей заявки в качестве ссылок.
Область техники
Настоящее изобретение относится к лекарственным средствам для лечения рака. Прежде всего, настоящее изобретение относится к средствам для лечения рака, которые вызывают апоптоз опухолевых клеток с использованием аналогов изоксазолидина. Соединения изоксазолидина по настоящему изобретению связываются с белками Bcl и блокируют антиапоптозную функцию белков Bcl в опухолевых клетках и тканях, экспрессирующих белки Bcl. Соединения и фармацевтические композиции, включающие такие соединения, можно использовать для лечения рака в отдельности или в комбинации с химиотерапевтическими агентами или другими лекарственными средствами.
Предпосылки создания настоящего изобретения
Апоптоз или запрограммированная гибель клетки является важной стадией нормального эмбрионального/анатомического развития, защиты организма и подавления онкогенеза. Нарушенная регуляция апоптоза наблюдается при развитии рака и многих других заболеваний человека, которые возникают в результате дисбаланса между процессами деления клеток и гибели клеток. Впервые Bcl-2 был идентифицирован при изучении хромосомального точечного разрыва t(14;18)-содержащих В-клеток лимфомы и относится к все возрастающему семейству белков, регулирующих апоптоз (Gross A., McDonnell J.M., Korsmeyer S.J., BCL-2 family members and the mitochondria in apoptosis, Genes & Development, т.13, cc.1899-1911 (1999), Cory S., Huang D.C.S., Adams J.M., The Bcl-2 family: roles in cell survival and oncogenesis, Oncogene, т.22, cc.8590-8607 (2003), Danial N.N., Korsmeyer S.J., Cell death: Critical control points, Cell, т.116, cc.205-218 (2004), Chao D.Т., Korsmeyer S.J., Вс1-2 family: regulators of cell death, Annu. Rev. Immunol., т.16, cc.395-419 (1998), Apoptosis, Christopher Potten, James Wilson, Cambridge University Press, (2004)). Семейство белков Bcl-2 включает как антиапоптозные молекулы, такие как Bcl-2 и Bcl-XL, так и проапоптозные молекулы, такие как Вах, Bak, Bid и Bad. Bcl-2 вносит вклад в прогрессирование раковых клеток за счет предотвращения нормального обновления клеток, которое регулируется механизмами физиологической гибели клеток. Избыточная экспрессия Bcl-2 наблюдается в 70% случаев рака молочной железы и многих других форм рака (Buolaniwini J.К., Novel anticancer drug discovery., Curr. Opin. Chem. Biol., т.3, cc.500-509 (1999)). Уровни экспрессии белков Bcl-2 коррелируют также с устойчивостью к множеству химиотерапевтических лекарственных средств и к лечению лучевой терапией (Reed J.С., Miyashita Т., Takayama S., Wang H.-G., Sato Т., Krajewski S., Aime-Sempe C., Bodrug S., Kitada S., Hanada M., Bcl-2 family proteins: Regulators of cell-death involved in the pathogenesis of cancer and resistance to therapy, J. Cell. Biochem. т.60, cc.23-32 (1996), Reed J.С., Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer, Advances in Pharmocology, т.41, cc.501-553 (1997), Strasser A., Huang D.C.S., Vaux D.L., The role of the Bcl-2/ced-9 gene family in cancer and general implications of defects in cell death control for tumorigenesis and resistance to chemotherapy, Biochem. Biophys. Acta, т.1333, cc. F151-F189 (1997), DiPaola R.S., Aisner J., Overcoming Bcl-2- and p53-mediated resistance in prostate cancer, Semin. Oncol., т.26, cc.112-116 (1999)).
Члены семейства белков Bcl-2 являются основными регуляторами апоптоза и проявляют проапоптозные свойства (например, Вах, Bak, Bid, Bim, Noxa, Puma) и антиапоптозные свойства (например, Bcl-2, Bcl-xL, Mcl-1). Избирательная и конкурентная димеризация про- и антиапоптозных членов семейства определяет судьбу клетки после проапоптозной стимуляции. Несмотря на то, что точная роль, которую играют белки Bcl-2 и Bcl-xL в развитии рака, в настоящее время еще не известна, получены некоторые данные о том, что Bcl-2 и Bcl-xL не только вносят вклад в прогрессирование рака за счет предотвращения нормального обновления клеток, но и принимают участие в устойчивости опухолевых клеток к современным противоопухолевым лекарственным средствам. Экспериментальная сверхэкспрессия Bcl-2 (Bcl-xL) придает опухолевым клеткам устойчивость к множеству химиотерапевтических агентов и облучению (Reed, J.С., Bcl-2 family proteins: Regulators of cell-death involved in the pathogenesis of cancer and resistance to therapy. J. Cell. Biochem., т.60, cc.23-32 (1996)). Сверхэкспрессия белков Bcl-2 и/или Bcl-xL наблюдается в более 50% всех опухолей, как показано ниже (см. Wang S., Yang D., Lippman M.E., Targeting Bcl-2 and Bcl-xL with nonpeptidic small-molecule antagonists, Seminars in Oncology, т.5, cc.133-142 (2003)).
Было установлено, что биологические подходы к модуляции функции Bcl-2 при использовании антисмысловых олигонуклеотидов или одноцепочечных антител повышают чувствительность опухолевых клеток к химиотерапевтическим агентам (Ziegler A., Luedke G.H., Fabbro D., Altmann К.Н., Stahel R.A., Zangemeister-Wittke U., Induction ofapoptosis in small-cell lung cancer cells by an antisense oligodeoxynucleotide targeting the Bcl-2 coding sequence, J. Natl. Cancer. Inst., т.89, cc.1027-1036 (1997), Webb A., Cunningham D., Cotter F., Clarke P.A., Di Stefano F., Ross P., Corpo M., Dziewanowska Z., Bcl-2 antisense therapy in patients with non-hodgkin lymphoma, Lancet, т.349, cc.1137-1141 (1997), Cotter F.E., Phase I clinical and pharmacokinetic study of Bcl-2 antisense oligonucleotide therapy in patients with non-hodgkin's lymphoma, J. Clin. Oncol., т.18, cc.1812-1823 (2000), Piche A., Grim J., Rancourt C., Gomez-Navarro J., Reed J.C., Curiel D.Т., Modulation of Bcl-2 protein levels by an intracellular anti-Bcl-2 single-chain antibody increases drug-induced cytotoxicity in the breast cancer cell line MCF-7, Cancer Res., т.58, cc.2134-2140 (1998)).
Было также установлено, что антисмысловой олигонуклеотид (G3139, Raynaud F.I., Orr R.M., Goddard P.M., Lacey H.A., Lancashire H., Judson I.R., Beck Т., Bryan В., Cotter F.E., Pharmacokinetics of G3139, a phosphorothioate oligodeoxynucleotide antisense to Bcl-2, after intravenous administration or continuous subcutaneous infusion to mice, J. Pharmacol. Exp.Ther., т.281, cc.420-427 (1997)), предназначенный для гибридизации с последовательностью мРНК Bcl-2, ингибирует экспрессию Bcl-2, индуцирует апоптоз и подавляет рост клеток и опухолевой ткани при раке молочной железы человека, харакеризующихся сверхэкспрессией Bcl-2 (Chen Н.X., Marchall J.L., Trocky N., Baidas S., Rizvi N., Ling Y., Bhagava P., Lippman M.E., Yang D. и Hayes D.F., A Phase I study of Bcl-2 antisense G3139 (Genta) and weekly docetaxel in patients with advanced breast cancer and other solid tumors. Proceedings of American Society of Clinical Oncology (2000)). Следует отметить, что при совместном введении G3139 и доцетакселя наблюдается синергетическое действие и полная регрессия опухоли in vivo. Таким образом, белок Bcl-2 представляет собой перспективную мишень при разработке нового способа лечения многих видов рака.
Ограничения, связанные с использованием в качестве лекарственных агентов больших молекул, таких как олигонуклеотиды, белки и полипептиды, связаны с низкой биодоступностью при пероральном введении, низкой стабильностью in vivo и высокой стоимостью. Более предпочтительные лекарственные препараты не должны содержать пептидов, должны представлять собой низкомолекулярные соединения, способные проникать через клеточную мембрану, связываться с белками Bcl-2, блокировать антиапоптозную функцию при раке и вызывать гибель опухолевых клеток.
Было установлено, что различные низкомолекулярные соединения подавляют функцию белков Bcl-2. По данным биохимических исследований и анализа in vitro было установлено, например, что ацилсульфамиды подавляют функции Bcl-2 и Bcl-xL в (Nature, т.435, cc.677-681 (2005)). Тем не менее, в настоящее время существует необходимость в разработке низкомолекулярных органических соединений, способных связываться с белком Bcl-2 и блокировать его антиапоптозную функцию при раке и вызывать гибель опухолевых клеток. Настоящее изобретение удовлетворяет этим условиям и характеризуется другими преимуществами.
Краткое описание сущности настоящего изобретения
Один объект настоящего изобретения относится к соединениям изоксазолидина. В некоторых случаях атом азота в изоксазолидиновом цикле связан с замещенной аралкильной группой. В некоторых вариантах замещенная аралкильная группа является замещенной бензильной группой. В других вариантах изоксазолидиновый цикл содержит в качестве заместителей гидроксиметильную или гидроксиэтильную группу. В некоторых вариантах изоксазолидиновый цикл содержит в качестве заместителей гидроксиметильную или гидроксиэтильную группу. В некоторых вариантах изоксазолидиновый цикл содержит в качестве заместителей амидную группу. В настоящем изобретении предлагаются также фармацевтически активные соли указанных выше изоксазолидиновых соединений. Другой объект настоящего изобретения относится к фармацевтическим композициям, включающим изоксазолидиновое соединение по настоящему изобретению. Еще один объект настоящего изобретения относится к способу применения описанных выше соединений или их фармацевтически активных солей, в отдельности или в комбинации с другими агентами для лечения рака. Более подробно, в настоящем изобретении предлагается способ лечения, включающий лечение состояния, характеризующегося патологической пролиферацией клеток у млекопитающих, таких как опухолевые клетки (например, при раке молочной железы или миелоидном лейкозе), при введении млекопитающему или человеку, страдающему от такого состояния, эффективного количества соединения по настоящему изобретению. В некоторых вариантах соединение по настоящему изобретению вводят в комбинации с фармацевтически приемлемым носителем.
Краткое описание чертежей
На фиг.1 показаны результаты изучения эффективности соединения 221 на модели ксенотрансплантанта опухоли RL у мышей SCID/NOD.
Подробное описание вариантов осуществления настоящего изобретения
Настоящее изобретение в основном относится к соединениям изоксазолидина, предназначенным для лечения рака. Соединения изоксазолидина по настоящему изобретению связываются с одним или более белками Bcl и блокируют антиапоптозную функцию белков Bcl в опухолевых клетках и тканях, экспрессирующих белок Bcl. В некоторых вариантах определенные соединения по настоящему изобретению селективно ингибируют антиапоптозную активность только одного члена подсемейства антиапоптозных белков Bcl-2. Соединения изоксазолидина по настоящему изобретению можно использовать для лечения пациентов, страдающих от заболевания, связанного с Bcl. В некоторых вариантах соединения изоксазолидина по настоящему изобретению можно использовать для лечения пациентов, страдающих от рака. Соединения изоксазолидина по настоящему изобретению вводят пациенту в виде фармацевтической композиции. Фармацевтическая копозиция включает соединение изоксазолидина по настоящему изобретению и один или более фармацевтически приемлемых эксципиентов. В некоторых вариантах фармацевтическая композиция включает соединение изоксазолидина по настоящему изобретению, химиотерапевтический агент и один или более фармацевтически приемлемых эксципиентов. В некоторых вариантах химиотерапевтическим агентом является доцетаксель, паклитаксель, цисплатин, 5-FU, доксрубинцин, эпиподофилотоксин, камптотецин, 17-AAG или циклофосфамид.
Синтез соединений изоксазолидина
Соединения изоксазолидина по настоящему изобретению получают по реакции циклоприсоединения [3+2] нитрона и алкена. Нитроновый субстрат и алкен содержат функциональные группы, пригодные для химической модификации с последующим синтезом изоксазолидинового фрагмента. В некоторых вариантах в реакционную смесь добавляют кислоту Льюиса. В предпочтительном варианте кислотой Льюиса является Ti(Oi-Pr)4. В некоторых вариантах реакционную смесь подвергают микроволновому облучению. В большинстве случаев реакцию проводят в жидкой реакционной среде, но иногда реакцию проводят на твердом носителе. Реакцию проводят в апротонном растворителе, предпочтительно, в котором полностью растворяются компоненты реакционной смеси. Пригодные растворители включают простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, диглим, трет-бутилметиловый эфир, тетрагидрофуран и т.п., галогенированные растворители, такие как хлороформ, дихлорметан, дихлорэтан, хлорбензол, четыреххлористый углерод и т.п., алифатические или ароматические углеводородные растворители, такие как бензол, ксилол, толуол, гексан, пентан и т.п., сложные эфиры и кетоны, такие как этилацетат, ацетон и 2-бутанон, полярные апротонные растворители, такие как ацетонитрил, диметилсульфоксид, диметилформамид, пиридин и т.п., или комбинации двух или более растворителей. Реакцию проводят при различной температуре. В большинстве случаев при проведении реакции при низкой температуре продолжительность реакции возрастает. В некоторых вариантах реакцию циклоприсоединения проводят в интервале от приблизительно 15°С до приблизительно 60°С. В других вариантах реакцию циклоприсоединения проводят в интервале от приблизительно 15°С до приблизительно 30°С. В некоторых вариантах реакцию циклоприсоединения проводят при приблизительно комнатной температуре. В других вариантах реакцию циклоприсоединения проводят в интервале от приблизительно 80°С до приблизительно 150°С. В некоторых вариантах реакцию циклоприсоединения проводят в интервале от приблизительно 90°С до приблизительно 120°С. В других вариантах реакцию циклоприсоединения проводят в интервале от приблизительно 95°С до приблизительно 105°С. В некоторых вариантах реакцию циклоприсоединения проводят на субстрате, иммобилизованном на твердом носителе. После синтеза изоксазолидинового фрагмента соединение изоксазолидина модифицируют по известным реакциям. Типичные примеры таких реакций включают реакции конденсации в присутствии палладия к алкенилгалогенидам или арилгалогенидам, реакции окисления, реакции восстановления, реакции с нуклеофильными агентами, реакции с электрофильными агентами, перициклические реакции, введение защитных групп, удаление защитных групп и т.п.
Определение биологической активности
Для определения активности и специфичности соединений по настоящему изобретению при связывании с белком Bcl-2 и ингибировании функции белка Bcl-2 в клетках используют следующие методы анализа связывания in vitro и анализа с использованием клеток.
Связывание с белком Bcl-2
Связывание с белками Bcl-2 и Bcl-xL определяют по известным методикам. Одна из таких методик является чувствительным и количественным методом анализа связывания in vitro с использованием поляризации флуоресценции (FP), описанной в работе Wang J.-L., Zhang Z-J., Choksi S., Sjam S., Lu Z., Croce C. M., Alnemri E.S., Komgold R., Huang Z., Cell permeable Bcl-2 binding peptides: a chemical approach to apoptosis induction in tumor cells, Cancer Res., т.60, cc.1498-1502 (2000).
Анализ с использованием клеток
Было установлено, что соединения изоксазолидина по настоящему изобретению способны подавлять жизнеспособность опухолевых клеток, которые характеризуются сверхэкспрессией белка Bcl-2. При обработке клеток RL соединениями изоксазолидина по настоящему изобретению наблюдается дозозависимая гибель клеток по данным анализа на цитотоксичность с использованием красителя аламарового синего, при этом величины IC50 составляют от приблизительно 100 мкМ до приблизительно 1 мкМ (см. раздел примеры). При обработке клеток Panc1 соединениями изоксазолидина по настоящему изобретению в комбинации с камптотецином наблюдается синергетическая дозозависимая гибель клеток по данным анализа на выживаемость клеток при проявлении иодидом пропидия, при этом величины IC50 составляют от приблизительно 100 мкМ до приблизительно 1 мкМ (см. раздел примеры).
Было установлено, что ингибиторы Всl-2 проявляют активность в отношении ряда линий опухолевых клеток при обработке соединениями в отдельности, включая, без ограничения перечисленным, рак молочной железы (заявки US 2003/0119894, опубликованные заявки РСТ WO 02/097053 и WO 02/13833), лимфомы (Nature, т.435, cc.677-681 (2005)), мелкоклеточный рак легких (Nature, т.435, cc.677-681 (2005)), рак головы и шеи (опубликованная заявка РСТ WO 02/097053), и лейкозы (опубликованная заявка РСТ WO 02/13833).
Установлено, что ингибиторы Bcl-2 проявляют активность в отношении ряда линий опухолевых клеток в комбинации с другими противоопухолевыми агентами и облучением, включая, без ограничения перечисленным, рак молочной железы (в комбинации с доцетакселем, опубликованная заявка РСТ WO 02/097053), рак предстательной железы (в комбинации с доцетакселем, опубликованная заявка РСТ WO 02/097053), рак головы и шеи (в комбинации с доцетакселем, опубликованная заявка РСТ WO 02/097053) и немелкоклеточный рак легких (в комбинации с паклитакселем, Nature, т.435, cc.677-681 (2005)). Кроме упомянутой выше комбинации с химиотерапевтическими агентами, низкомолекулярные ингибиторы белков Bcl-2 проявляют синергизм с другими противоопухолевыми агентами, включающими, без ограничения перечисленным, доксорубицин, цисплатин, паклитаксель и облучение (Nature, т.435, cc.677-681 (2005)).
Способы лечения
В настоящем изобретении предлагаются также способы лечения и снижения тяжести ракового заболевания, а также других опосредованных белком Bcl нарушений или состояний.
Раковые или неопластические заболевания и связанные с ними заболевания, которые можно лечить при введении соединений или композиций по настоящему изобретению, без ограничения перечисленным, перечислены в таблице 1 (обзор, в котором описаны такие заболевания, представлен в книге Fishman и др., Medicine, 2-е изд., J.В.Lippincott Co., Philadelphia (1985)).
В предпочтительном варианте соединения по настоящему изобретению используют для лечения рака, включающего, без ограничения перечисленным, лимфомы (предпочтительно фолликулярную лимфому, диффузную крупноклеточную В-клеточную лимфому, лимфому спорангических клеток или хронический лимфоцитарный лейкоз), рак предстательной железы (наиболее предпочтительно нечувствительный к гормонам), рак молочной железы (предпочтительно положительный в отношении эстрогенных рецепторов), нейробластому, колоректальный рак, рак яичников, рак легких (предпочтительно мелкоклеточный рак), гепатоцеллюлярную карциному, множественную миелому, рак головы и шеи или рак яичек (предпочтительно эмбриональный).
Лечение рака в комбинации с химиотерапевтическими агентами или с лучевой терапией
В некоторых вариантах осуществления настоящего изобретения одно или более соединений по настоящему изобретению используют для лечения или профилактики рака или неопластических заболеваний в комбинации с одним или более противоопухолевых химиотерпевтических агентов, включающих, без ограничения перечисленным, метотрексат, таксол, меркаптопурин, тиогуанин, гидроксимочевину, цитарабин, циклофосфамид, ифосфамид, нитрозомочевины, цисплатин, карбоплатин, митомицин, дакарбазин, прокарбизин, этопозиды, преднизолон, дексаметазон, цитарбин, кампатецины, блеомицин, доксорубицин, идарубицин, даунорубицин, дактиномицин, пликамицин, митоксантрон, аспарагиназу, винбластин, винкристин, винорелбин, пактитаксель и доцетаксель. В предпочтительном варианте одно или более соединений по настоящему изобретению используют для лечения или профилактики рака или неопластических заболеваний в комбинации с одним или более химиотерпевтических или других противоопухолевых агентов, которые, без ограничения перечисленным, перечислены в таблице 2.
Химиотерапевтический агент и/или лучевую терапию проводят по известным методикам. Специалистам в данной области техники представляется очевидным, что введение химиотерапевтического агента и/или тип лучевой терапии зависят от заболевания, предназначенного для лечения, и от известного действия, которые оказывают Химиотерапевтический агент и/или лучевая терапия на указанное заболевание. Курс лечения (например, дозы и время введения) зависит также от мнения врача, от наблюдаемого действия при введении терапевтических агентов (например, противоопухолевый агент или облучение) на пациента, а также от наблюдаемой чувствительности заболевания к вводимым терапевтическим агентам.
В основном соединения по настоящему изобретению и Химиотерапевтический агент не вводят в составе одной и той же фармацевтической композиции, и вследствие различных физических и химических свойств их вводят различными способами. Например, соединения по настоящему изобретению можно вводить внутривенным способом для обеспечения и поддержания высокого уровня в крови, в то время как химиотерапевтический агент вводят пероральным способом. Специалисты в данной области опеределяют способ введения и целесообразность введения, при необходимости, в составе одной фармацевтической композиции. Сначала введение проводят стандартным методом, а затем, в зависимости от наблюдаемого действия, специалисты в данной области могут изменять величину дозы, способ введения и время введения.
Выбор химиотерапевтического агента или типа облучения зависит от диагноза лечащего врача, от его заключения о состоянии пациента и соответствущей схемы лечения.
Соединение по настоящему изобретению и химиотерапевтический агент и/или облучение используют совместно (например, одновременно, в основном одновременно или в одной схеме лечения) или последовательно, в зависимости от природы пролиферативного заболевания, состояния пациента и выбора конкретного химиотерапевтического агента и/или типа облучения, которые используют в комбинации (например, в одной схеме лечения) с соединением по настоящему изобретению.
Если соединение по настоящему изобретению и химиотерапевтический агент и/или облучение не используют одновременно или в основном одновременно, то оптимальный порядок введения соединения по настоящему изобретению и химиотерапевтического агента и/или использования облучения зависит от вида опухоли. Таким образом, в некоторых случаях сначала вводят соединение по настоящему изобретению с последующим введением химиотерапевтического агента и/или использованием облучения, а в других случаях сначала вводят химиотерапевтический агент и/или используют облучение с последующим введением соединения по настоящему изобретению. Такие различные способы введения применяют в одной схеме лечения. Порядок введения и число повторных введений каждого терапевтического агента в ходе лечения опеределяется лечащим врачом после диагностики заболевания и оценки состояния пациента. Например, сначала вводят химиотерапевтический агент и/или используют облучение, прежде всего в случае цитотоксического агента, а затем лечение продолжают при введении соединения по настоящему изобретению с последующим, при необходимости, введением химиотерапевтического агента и/или использованием облучения и т.п., до завершения курса лечения.
Таким образом, в ходе лечения лечащий врач может изменять любую схему лечения (терапевтический агент, например, соединение по настоящему изобретению, химиотерапевтический агент или облучение) в зависимости от состояния конкретного пациента.
Определения
Некоторые термины, используемые в настоящем описании, примерах и формуле изобретения, приведены ниже.
Термин «совместное введение» означает как одновременное введение (введение двух или более терапевтических агентов в одно и тоже время), так и введение в различное время (введение одного или более терапевтических агентов в различное время после введения дополнительного терапевтического агента или агентов), то есть обеспечивается присутствие терапевтических агентов в организме пациента в некоторой степени в одно и тоже время.
Термин «гетероатом», используемый в настоящем описании, означает атом любого элемента, за исключением углерода или водорода. Предпочтительные гетероатомы включают бор, азот, кислород, фосфор, серу и селен.
Термин «алкил» означает насыщенную алифатическую группу, такую как алкил с прямой цепью, алкил с разветвленной цепью, циклоалкил (алициклил), алкилзамещенные циклоалкилы, и циклоалкилзамещенные алкилы. В предпочтительных вариантах осуществления настоящего изобретения алкил с прямой или разветвленной цепью содержит 30 или менее атомов углерода в цепи (например, C1-С30алкил с прямой цепью, С3-С30алкил с разветвленной цепью) и более предпочтительно 20 или менее атомов углерода. Предпочтительные циклоалкилы содержат от 3 до 10 атомов углерода в цикле и более предпочтительно 5, 6 или 7 атомов углерода в цикле.
Если количество атомов углерода не указано, то термин «(низш.)алкил», используемый в настоящем описании, означает алкил, определенный выше, но при этом содержащий от одного до десяти атомов углерода, более предпочтительно от одного до шести атомов углерода в цепи. Термины «(низш.)алкил» и «(низш.)алкинил» означают алкенил и алкинил с цепью, содержащей аналогичное количество атомов углерода. Предпочтительные алкилы включают (низш.)алкилы. В предпочтительных вариантах осуществления настоящего изобретения заместитель, определенный в настоящем описании как алкил, является (низш.)алкилом.
Термин «галогеналкил», используемый в настоящем описании, означает алкил, в котором от 1 до всех атомов водорода заменены на атомы галогена. Термин «пергалогеналкил» означает алкил, в котором все атомы водорода заменены на атомы галогена.
Термин «аралкил», используемый в настоящем описании, означает алкил, замещенный арилом (например, ароматической или гетероароматической группой).
Термины «алкенил» и «алкинил» означают ненасыщенные необязательно замещенные алифатические группы с длиной цепи, равной длине цепи алкилов, описанных выше, но при этом содержащие по крайней мере одну двойную или тройную химическую связь, соответственно.
Термин «арил», используемый в настоящем описании, означает 5-, 6- и 7-членные моноциклические ароматические группы, которые включают от 0 до 4 гетероатомов, такие как, например, бензол, антрацен, нафталин, пирен, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин, пиримидин и т.п. Термины «гетероциклические арилы» и «гетероароматические соединения» также означают арилы, включающие гетероатомы в цикле. Ароматический цикл является замещенным в одном или более положениях, как описано выше, заместителями, такими как, например, галоген, азид, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, алкоксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, сульфонамидо, кетон, альдегид, сложный эфир, гетероциклил, ароматические или гетероароматические остатки, -CF3, -CN или т.п. Термин "арил" также включает полициклические системы, содержащие два или более цикла, в которых два или более атомов углерода входят в состав двух соседних циклов (такие циклы называются "конденсированными циклами"), где по крайней мере один из циклов является, например, ароматическим, при этом остальные циклы являются циклами, такими как циклоалкилы, циклоалкенилы, циклоалкинилы, арилы и/или гетероциклилы.
Термины «орто», «мета» и «пара» означают 1,2-, 1,3- и 1,4-двухзамещенные бензолы, соответственно. Например, названия «1,2-диметилбензол» и «орто-диметилбензол» являются синонимами.
Термины "гетероциклил" и "гетероциклическая группа" означают 3-10-членные циклы, более предпочтительно 3-7-членные циклы, которые включают от одного до четырех гетероатомов. Гетероциклы являются также полициклами. Гетероциклические группы включают, например, тиофен, тиантрен, фуран, пиран, изобензофуран, хромен, ксантен, феноксатиин, пиррол, имидазол, пиразол, изотиазол, изоксазол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, индол, индазол, пурин, хинолизин, изохинолин, хинолин, фталазин, нафтиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карболин, фенантридин, акридин, пиримидин, фенантролин, феназин, фенарсазин, фенотиазин, фуразан, феноксазин, пирролидин, оксолан, тиолан, оксазол, пиперидин, пиперазин, морфолин, лактоны, лактамы, такие как азетидиноны и пирролидиноны, сультамы, сультоны и т.п. Гетероцикл является замещенным в одном или более положениях, как описано выше, заместителями, такими как, например, галоген, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, кетон, альдегид, сложный эфир, гетероциклил, ароматические или гетероароматические остатки, -CF3, -CN или т.п.
Термины "полициклил" или "полициклическая группа" означают два или более циклов (например, циклоалкилы, циклоалкенилы, циклоалкинилы, арилы и/или гетероциклилы), в которых два или более атомов углерода входят в состав двух соседних циклов, например, такие циклы являются конденсированными циклами. Термин «циклы с мостиковой связью» означает циклы, соединенные через не соседние атомы. Каждый из циклов, входящих в состав полицикла, является замещенным, как описано выше, заместителями, такими как, например, галоген, алкил, аралкил, алкенил, алкинил, циклоалкил, гидроксил, алкоксил, амино, нитро, сульфгидрил, имино, амидо, фосфонат, фосфинат, карбонил, карбоксил, силил, простой эфир, алкилтио, сульфонил, кетон, альдегид, сложный эфир, гетероциклил, ароматические или гетероароматические остатки, -CF3, -CN или т.п.
Термин «нитро», используемый в настоящем описании, означает -NO2, термин «галоген» означает -F, -Cl, -Br или -I, термин «сульфгидрил» означает SH, термин «гидроксил» означает -ОН, а термин "сульфонил" означает -SO2-.
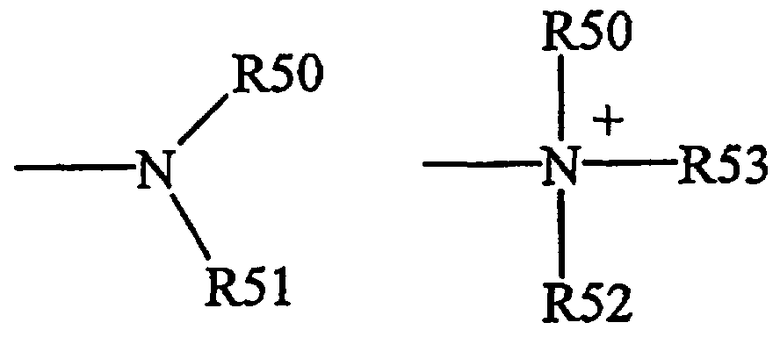
Термины "амин" и "амино" являются общепринятыми в данной области техники и означают незамещенные и замещенные амины, например остаток общих формул,
где
R50, R51 и R52 каждый независимо означает водород, алкил, алкенил, -(СН2)m-R61, или
R50 и R51 вместе с атомом N, к которому они присоединены, означают гетероцикл, содержащий от 4 до 8 атомов в цикле,
R61 означает арил, циклоалкил, циклоалкенил, гетероцикл или полицикл,
m равно нулю или целому числу от 1 до 8.
В некоторых вариантах осуществления настоящего изобретения только один из R50 и R51 означает карбонил, например, R50, R51 и азот вместе не образуют имид. В других вариантах осуществления настоящего изобретения R50 и R51 (и необязательно R52) каждый независимо означает водород, алкил, алкенил или
-(CH2)m-R61. Таким образом, термин «алкиламин» включает аминогруппу, как определено выше, содержащую замещенный или незамещенный алкил, присоединенный к атому азота, т.е. по крайней мере один из R50 и R51 означает алкил.
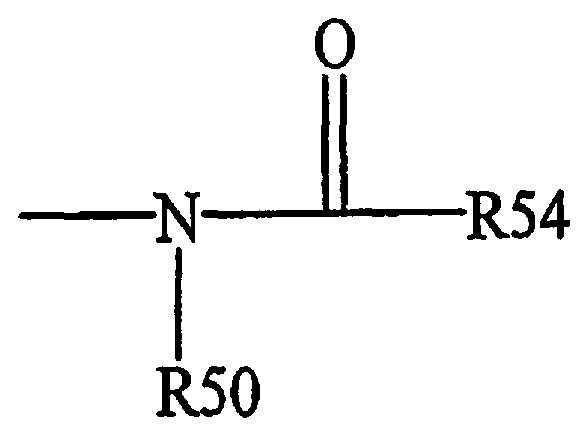
Термин «ациламино» является общепринятым в данной области техники и означает остаток общей формулы,
где
R50 имеет значения, определенные выше, и
R54 означает водород, алкил, алкенил или -(CH2)m-R61, где
m и R61 имеют значения, определенные выше.
Термин «амидо» является общепринятым в данной области техники, означает аминозамещенный карбонил и включает остаток общей формулы,
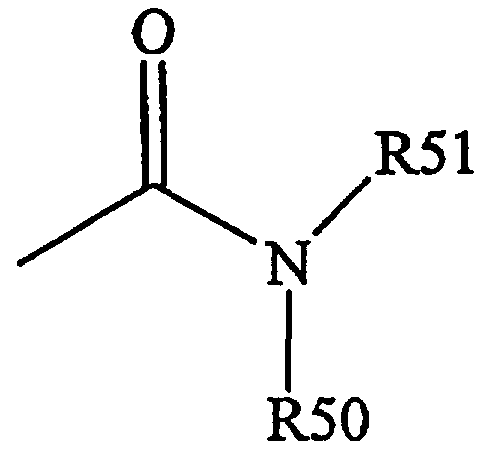
где R50 и R51 имеют значения, определенные выше.
В некоторых вариантах осуществления настоящего изобретения термин «амид» не включает имиды, которые являются неустойчивыми.
Термин «алкилтио» означает алкил, определенный выше, к которому присоединен серосодержащий радикал. В некоторых вариантах осуществления настоящего изобретения термин "алкилтио" означает один из остатков, таких как -S-алкил, -S-алкенил, -S-алкинил и S-(CH2)m-R61, где m и R61 имеют значения, определенные выше. Алкилтиогруппы включают метилтио, этилтио и т.п.
Термин «карбоксил» является общепринятым в данной области техники и включает остатки общих формул,
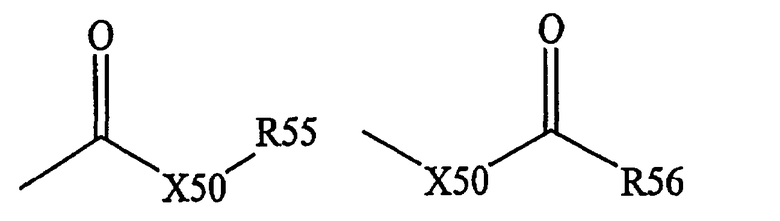
где
Х50 означает химическую связь или кислород или серу,
R55 и R56 означают водород, алкил, алкенил, -(CH2)m-R61 или фармацевтически приемлемую соль,
R56 означает водород, алкил, алкенил или -(CH2)m-R61, где
m и R61 имеют значения, определенные выше.
Если Х50 означает кислород и R55 или R56 не означает водород, то указанная формула означает сложный эфир. Если Х50 означает кислород и R55 имеет значения, определенные выше, и который обозначается термином «карбоксил», используемым в настоящем описании, и, прежде всего, если R55 означает водород, то указанная формула означает карбоновую кислоту. Если Х50 означает кислород и R56 означает водород, то указанная формула означает формиат. Как правило, остаток указанной выше формулы, где атом кислорода заменен на атом серы, означает тиокарбонил. Если Х50 означает серу и R55 или R56 не означает водород, то указанная формула означает тиоэфир. Если Х50 означает серу и R55 означает водород, то указанная формула означает тиокарбоновую кислоту. Если Х50 означает серу и R56 означает водород, то указанная формула означает тиоформиат. В другом варианте, если Х50 означает химическую связь и R55 не означает водород, то указанная формула означает кетон. Если Х50 означает химическую связь и R55 означает водород, то указанная формула означает альдегид.
Термины «алкоксил» или «алкокси», используемые в настоящем описании, означают алкил, определенный выше, к которому присоединен кислородсодержащий радикал. Алкоксилы включают метокси, этокси, пропилокси, трет-бутокси и т.п. Термин «простой эфир» означает два углеводорода, присоединенных ковалентной связью к атому кислорода. Таким образом, если заместитель в составе алкила образует простой эфир типа алкоксигруппы, то такие группы можно обозначить следующими формулами -O-алкил, -O-алкенил, -O-алкинил, -O-(CH2)m-R8, где m и R8 имеют значения, определенные выше.
Термин «сульфонат» является общепринятым в данной области техники и включает остатки общей формулы,
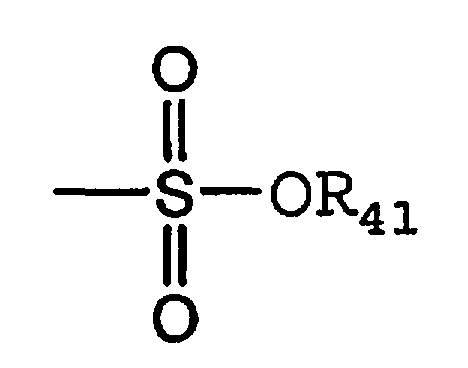
где
R41 означает электронную пару, водород, алкил, циклоалкил или арил.
Термины «трифлил», «тозил», «мезил» и «нонафлил» являются общепринятыми в данной области техники и означают трифторметансульфонил, пара-толуолсульфонил, метансульфонил и нонафторбутансульфонил, соответственно. Термины «трифлат», «тозилат», «мезилат» и «нонафлат» являются общепринятыми в данной области техники и означают функциональные группы, такие как трифторметансульфонат, пара-толуолсульфонат, метансульфонат и нонафторбутансульфонат, соответственно, и молекулы, содержащие указанные группы.
Термин «карбамоил» означает -O(C=O)NRR′ , где R и R′ независимо означают H, алифатические группы, арилы или гетероарилы.
Термин «алкиламино» означает -NHR, где R означает алкил. Термин «диалкиламино» означает -NRR′, где оба R и R′ означают алкилы. Термин «гидроксиалкил» означает -R-OH, где R означает алифатическую группу.
Термин «аминоалкил» означает -R-NH2, где R означает алифатическую группу.
Термин «алкиламиноалкил» означает -R-NH-R′ , где R и R′ означают алифатические группы.
Термин «диалкиламиноалкил» означает -R-N(R′)-R′′ , где R, R′ и R′′ означают алифатические группы.
Термин «ариламиноалкил» означает -R-NH-R′ , где R означает алифатическую группу, а R′ означает арил.
Термин «оксо» означает карбонильный кислород (=O). Термины «бирадикал» и «двухвалентный», используемые в настоящем описании, являются равнозначными и означают любые двухвалентные группы, которые образуются из следующих групп: алкил, алкенил, алкинил, алкиламино, алкоксил, циклоалкил, гетероциклоалкил, арил, аралкил, гетероциклил, гетероарил и гетероаралкил. Например, соединение формулы
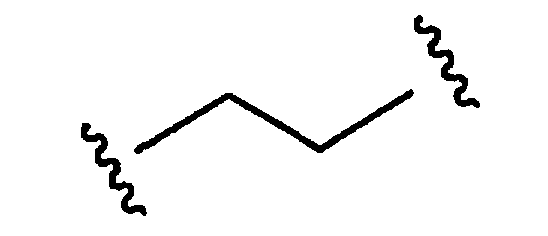
является двухвалентным алкилом или алкильным бирадикалом, соединение формулы
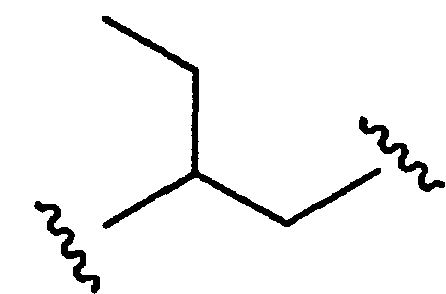
является также двухвалентным алкилом или алкильным бирадикалом, соединение формулы
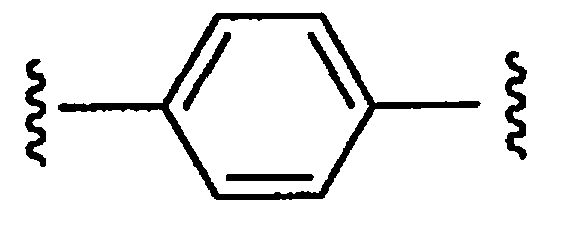
является двухвалентным арилом или арильным бирадикалом, соединение формулы
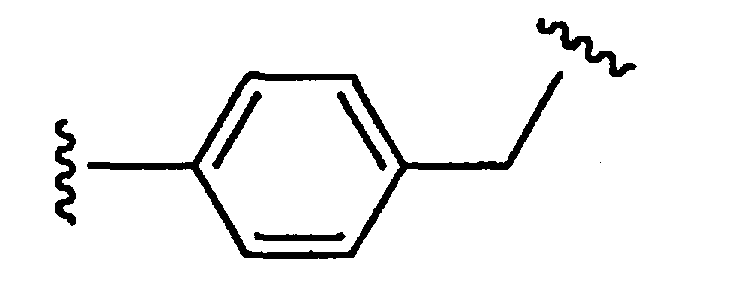
является двухвалентным аралкилом или аралкильным бирадикалом, соединение формулы
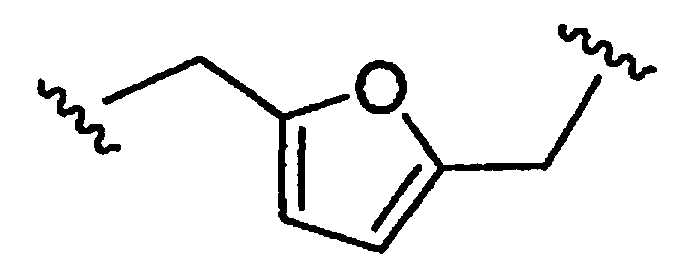
является двухвалентным (алкил)гетероаралкилом или (алкил)гетероаралкильным бирадикалом.
Сокращения Me, Et, Ph, Tf, Nf, Ts, Ms означают метил, этил, фенил, трифторметансульфонил, нонафторбутансульфонил, пара-толуолсульфонил и метансульфонил, соответственно. Более широкий список сокращений, используемых в данной области техники, представлен в первом выпуске каждого тома Journal of Organic Chemistry, указанный список, как правило, представлен в таблице, называемой список стандартных сокращений. Сокращения, входящие в указанный список, и все сокращения, используемые в данной области техники, включены в данное описание.
Термин «сульфат» является общепринятым в данной области техники и означает остаток общей формулы,
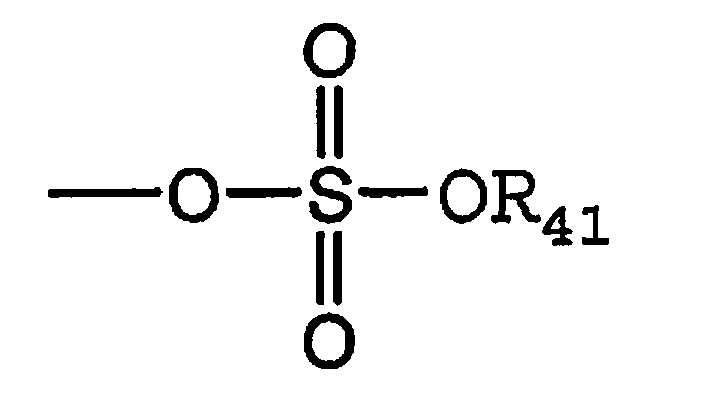
где
R41 имеет значения, определенные выше.
Термин «сульфониламино» является общепринятым в данной области техники и означает остаток общей формулы
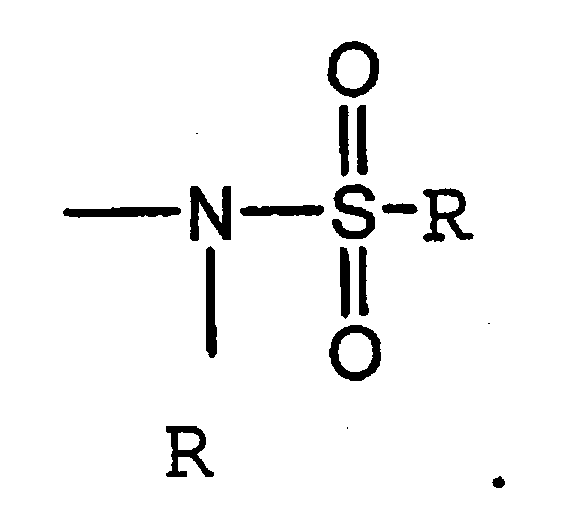
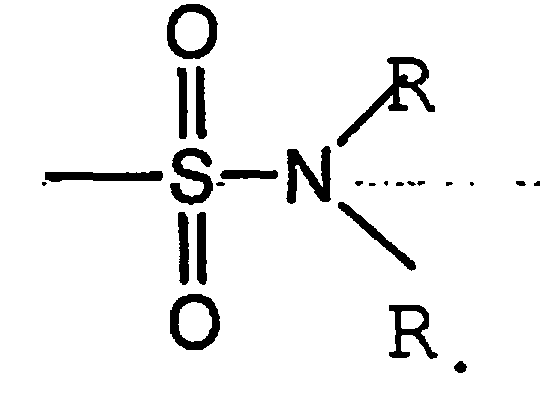
Термин «сульфамоил» является общепринятым в данной области техники и означает остаток общей формулы
Термин «сульфонил» является общепринятым в данной области техники и означает остаток общей формулы,
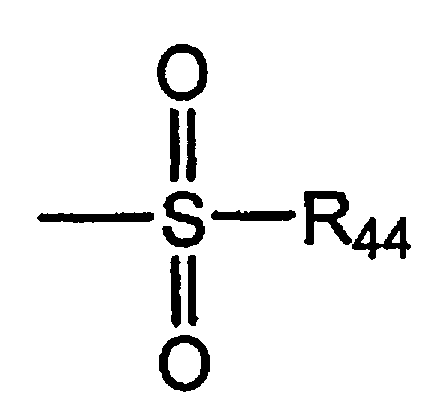
где
R44 выбирают из группы, включающей водород, алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил или гетероарил.
Термин «сульфоксидо», используемый в настоящем описании, означает остаток общей формулы,

где
R44 выбирают из группы, включающей водород, алкил, алкенил, алкинил, циклоалкил, гетероциклил, аралкил или арил.
Термин «селеноалкил» означает алкил, к которому присоединены замещенные селеногруппы. Примеры «селеноэфиров», которые являются замещенными алкилами, включают -Se-алкил, -Se-алкенил, -Se-алкинил и -Se-(CH2)m-R7, где m и R7 имеют значения, определенные выше.
При аналогичном замещении алкенилов и алкинилов образуются, например, аминоалкенилы, аминоалкинилы, амидоалкенилы, амидоалкинилы, иминоалкенилы, иминоалкинилы, тиоалкенилы, тиоалкинилы, алкенилы или алкинилы, замещенные карбонилом.
Если термин, используемый в настоящем описании, такой как, например, алкил, m, n и т.п., встречается более одного раза в любой структуре, то значение этого термина является независимым для каждого термина в данной структуре.
Следует понимать, что термин «замещение» или «замещенный» означает замещение в соответствии с разрешенной валентностью замещенного атома и заместителя, которое приводит к устойчивому соединению, которое не подвергается самопроизвольному превращению, такому как перегруппировка, циклизация, отщепление и т.п.
Термин «замещенный», используемый в настоящем описании, включает все допустимые заместители для органических соединений. Допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Примеры заместителей включают, например, заместители, описанные выше. Органическое соединение может содержать один или более заместителей, которые могут быть одинаковыми или различными. Гетероатомы по настоящему изобретению, такие как азот, замещены атомами водорода и/или любыми допустимыми заместителями для органических соединений, описанными в настоящем описании, в соответствии с валентностью гетероатомов. Допустимые заместители для органических соединений приведены только для иллюстрации и не ограничивают объем и сущность изобретения.
Термин «защитная группа», используемый в настоящем описании, означает заместители, которые временно защищают реакционноспособную функциональную группу от нежелательных химических превращений. Примеры таких защитных групп включают эфиры карбоновых кислот, силиловые эфиры спиртов и ацетали и кетали альдегидов и кетонов, соответственно. Защитные группы подробно описаны в книге Greene T.W., Wuts P.G.M. Protective Groups in Organic Synthesis, 2-е изд., Wiley, New York, (1991). Защищенные формы соединений по настоящему изобретению включены в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению находятся в определенных геометрических или стереоизомерных формах. Настоящее изобретение относится ко всем таким формам соединений, которые включают цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, включенные в объем настоящего изобретения. Заместители, такие как алкил, могут включать асимметричные атомы углерода. Настоящее изобретение относится ко всем таким изомерам и их смесям.
Если, например, существует потребность в определенном энантиомере соединения по настоящему изобретению, то его получают асимметрическим синтезом или синтезом с введением хирального вспомогательного остатка, с последующим разделением полученной смеси диастереомеров и удалением вспомогательного остатка, при этом получают требуемые индивидуальные энантиомеры. В другом варианте, если молекула содержит основную функциональную группу, такую как аминогруппу, или кислотную функциональную группу, такую как карбоксил, то сначала получают соли диастереомеров и соответствующей оптически активной кислоты или основания, затем разделяют диастереомеры фракционной кристаллизацией или хроматографическими методами, известными в данной области техники, и затем выделяют инидивидуальные энантиомеры.
Указанные аналоги соединений, описанных выше, включают соединения с близкой структурой, которые обладают аналогичными общими свойствами соединений, описанных выше (например, являются болеутоляющими средствами), при этом указанные аналоги получают простым введением различных заместителей, которые не влияют на эффективность взаимодействия соединения с сигма-рецепторами. В общем случае соединения по настоящему изобретению получают по методикам, описанным на общих схемах реакций, как, например, описано ниже, или указанными методами, модифицированными с использованием коммерческих продуктов, реагентов и стандартных методик синтеза. Указанные реакции также проводят способами, известными в данной области техники.
Химические элементы по настоящему изобретению идентифицируют в соответствии с периодической таблицой элементов, CAS version. Handbook of Chemistry and Physics, 67th Ed., на внутренней стороне обложки (1986-1987).
Термин «пациент», используемый в настоящем описании, означает животное, как правило, млекопитающее или человека, который является объектом лечения, наблюдения и/или эксперимента. Если указанный термин используется в отношении введения соединения или лекарственного средства, то пациентом является объект лечения, наблюдения и/или введения соединения или лекарственного средства.
Термин «терапевтически эффективное количество», используемый в настоящем описании, означает такое количество активного соединения или фармацевтического препарата, которое вызывает биологическую или медицинскую ответную реакцию в клеточной культуре, системе ткани, организме животного или человека, который выбран исследователем, ветеринаром, клиницистом или лечащим врачом, и которое способствует снижению интенсивности симптомов заболевания, расстройства или состояния, которые предназначены для лечения. Такое количество активного соединения по настоящему изобретению является достаточным для связывания с белком bcl-2 в клетке и по крайней мере для частичного ингибирования антиапоптозной активности белка. Такое количество активного соединения является достаточным для обеспечения терапевтической эффективности при лечении пациента или для повышения чувствительности клетки к лечению другим противоопухолевым средством.
Термин «композиция» означает продукт, включающий определенные ингредиенты в определенных количествах, и любой продукт, который получают прямым или многостадийным способом из комбинаций указанных ингредиентов в указанных количествах.
Термин «фармацевтически приемлемый носитель» означает носитель, который используют для получения лекарственной формы соединения. Фармацевтически приемлемый носитель включает один или более растворителей, разбавителей или других жидких наполнителей, диспергированных или суспендированных вспомогательных веществ, поверхностно-активный агент (ПАВ), изотонические агенты, загустители или эмульгаторы, консерванты, твердые связующие агенты, замасливатели и т.п. Различные носители, используемые для получения фармацевтических композиций, и методики, известные для получения таких композиций, описаны в книгах Remington's Pharmaceutical Sciences, 15-е изд., Е.W.Martin (Mack Publishing Co., Easton, Pa., (1975)) и Handbook of Pharmaceutical Excipients, 3-е изд., А.Н.Kibbe и др. (American Pharmaceutical Assoc. (2000)).
Термины «нарушение, опосредованное Bcl» и «нарушение, опосредованное экспрессией клетками белков Bcl» означают патологическое состояние или заболевание, связанное с белком Bcl. Такая роль белка Bcl является прямой или косвенной причиной патологического состояния и заболевания. Основной особенностью такого заболевания является снижение его симптомов при ингибировании активности и функции белков Bcl, или связывания с белками Bcl.
Термины «Bcl» и «белок Bcl» означают одно или более подсемейство антиапоптозных белков Bcl-2, Bcl-w, Mcl-1, Bcl-XL, A1, Bfl1, Bcl-B, BOO/DIVA и их гомологов.
Соединения по настоящему изобретению
В одном варианте осуществления настоящего изобретения предлагается соединение формулы 1
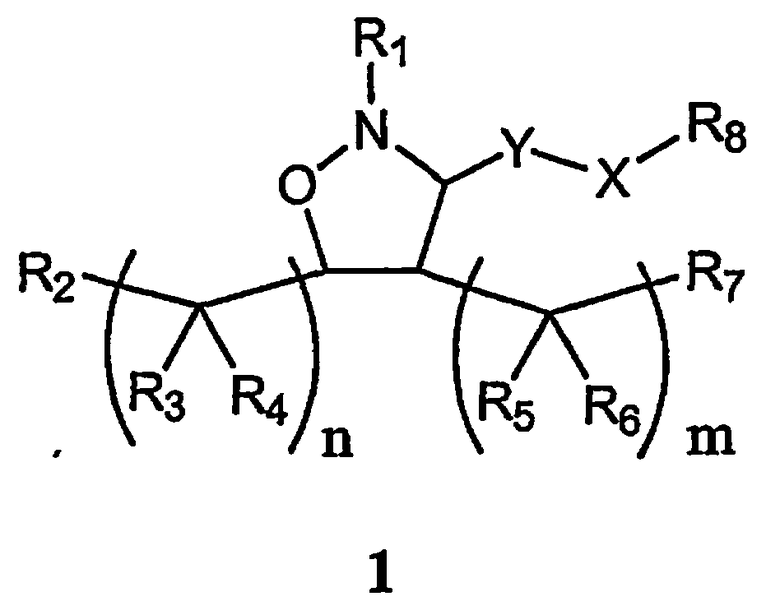
или его фармацевтически приемлемые соли, сольваты или гидраты, где
Y означает -C(R9)2-, -C(O)-, -C(S)- или -C(=NR10)-,
Х означает -N(R11)-, необязательно замещенный фенил или химическую связь,
Х каждый независимо означает О, N(R10) или S,
m равно 0, 1, 2, 3, 4, 5 или 6,
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6,
R1 означает алкил, аралкил или гетероаралкил формулы 1а,
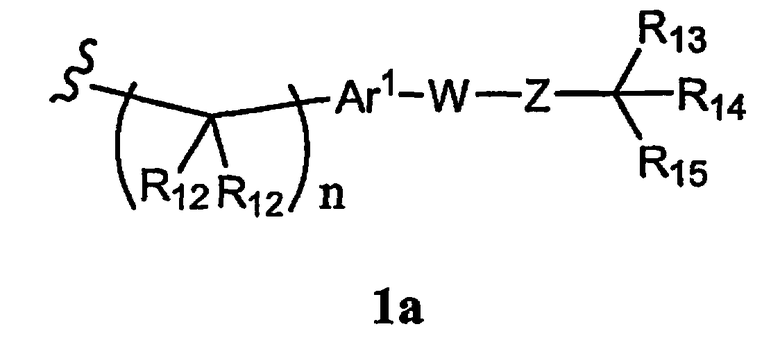
где
R12 каждый независимо означает Н, алкил, арил, гетероарил или аралкил, где любые два R12 соединены ковалентной связью,
Ar1 означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
W означает химическую связь или двухвалентный алкил, алкенил или алкинил,
Z означает химическую связь, -(C(R12)2)n- или -X′(C(R12)2)n-,
R13 и R14 независимо означают Н, алкил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3, или R13 и R14 вместе образуют моноцикл или полицикл, или R13 и R14 вместе с R15 образуют циклоалкенил, ароматический или гетероароматический цикл,
R15 означает галоген, гидроксил, алкоксил, арил, арилокси, ацилокси, -N(R10)2, ациламино, аралкил, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10,
-CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R19,
-N(R10)C(X′)N(R19)2, -N(R10)(C(R9)2)n-Al-A2-A3, -(С(R9)2)n-галоген или -CH2O-гетероциклил, или R15 вместе с R13 и R14 образуют циклоалкенил, ароматический или гетероароматический цикл, или R1 или R15 каждый независимо означает остаток формулы 1b
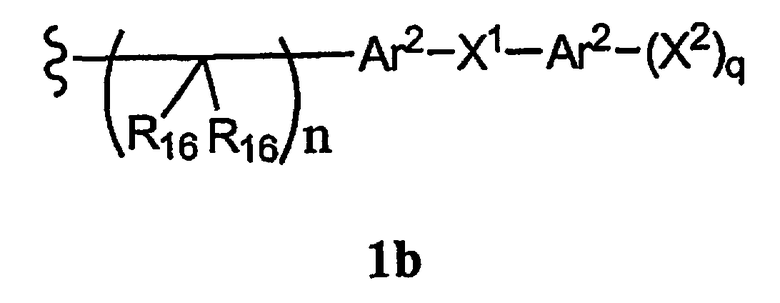
R16 каждый независимо означает Н, алкил, циклоалкил, гетероциклоалкил, арил, аралкил, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R10 или
-N(R10)C(X′)N(R10)2, где любые два R16 соединены ковалентной связью с образованием цикла,
Ar2 каждый независимо означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
Х1 каждый независимо означает химическую связь, О, S, S(O), S(O)2, S(O)3, амино, алкиламинобирадикал, алкоксилбирадикал, алкилбирадикал, алкенилбирадикал, алкинилбирадикал, амидо, карбонил, -N(R10)CO2-, -OC(O)N(R10)- или
-N(R10)C(X′ )N(R10)-,
X2 каждый независимо означает Н, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2,
-N(R10)SO2R10, -N(R10)C(X′)N(R10)2 или -СН2О-гетероциклил, и
q каждое независимо равно 1, 2, 3, 4 или 5,
R2 и R7 независимо означают Н, гидроксил, алкил, алкоксил, амино, алкиламино или ациламино, или R2 и R7 вместе образуют -ОС(O)O-связующее звено,
-N(R10)C(O)N(R10)-связующее звено или необязательно замещенное связующее звено, включающее от 1 до 6 атомов углерода и 0, 1 или 2 атома азота, кислорода или серы, образующее 5-8-членный цикл, или R7 означает химическую связь с R8,
R3 и R6 каждый независимо означает Н, гидроксил или алкил,
R4 и R5 каждый независимо означает Н или алкил,
R8 означает Н, разветвленный или неразветвленный алкил или алкенил, циклоалкил, гетероциклоалкил, бициклоалкил, химическую связь с R7, гетероциклоалкил, замещенный аралкилом, или остаток формулы 1с,
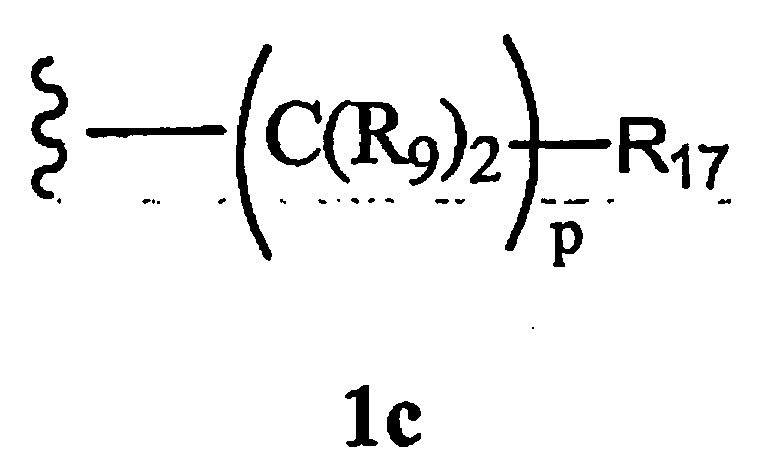
где
р равно 0, 1, 2, 3, 4, 5 или 6, и R17 означает арил, циклоалкил, циклоалкенил, гетероциклил, алкоксил, гетероарил, -OR18, -SR18, -N(R18)2, -N(R10)CO2-алкил,
-CO2R10, -С(O)N(R10)арил или полицикл, содержащий от 8 до 14 атомов углерода,
где
R18 каждый независимо означает Н, алкил, арил, аралкил, ацил, -А1-А2-А3 или
-CR9=CR9(C(R9)2)nCR9=C(R9)2, или два R18 вместе образуют цикл,
R9 каждый независимо означает Н или алкил,
R10 и R11 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил или гетероаралкил,
R19 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3,
А1 и А3 каждый независимо означает алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил или гетероаралкил,
А2 каждый независимо означает О, N(R10), S или химическую связь, при этом стереохимическая конфигурация в любом стереоцентре соединения формулы 1 означает R-, S-конфигурацию или смесь этих конфигураций.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где Y означает -С(O)-, Х означает -N(R11)-, R2 и R7 означают гидроксил, R6 означает метил, этил или пропил, а R3, R4 и R5 означают Н.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, которое является соединением формулы 1d,
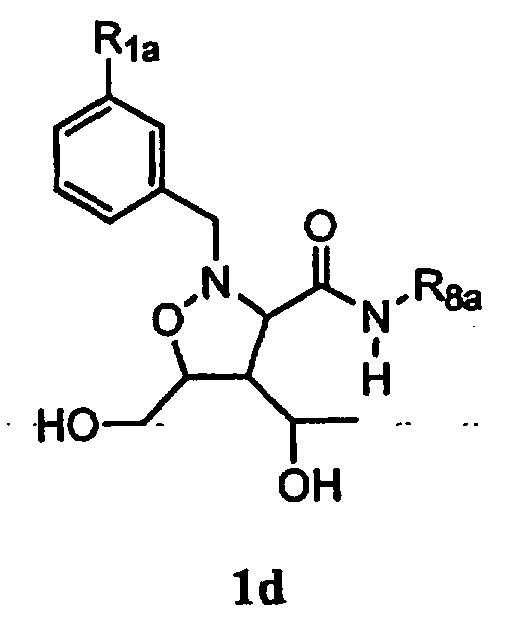
где
R1a означает соединение формулы 1е,
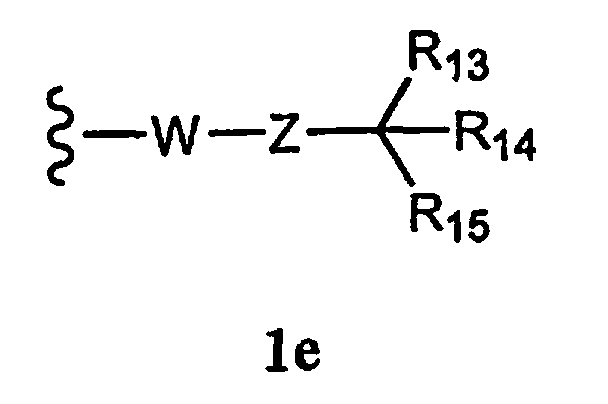
где
W означает химическую связь или двухвалентный алкил, алкенил или алкинил,
Z означает химическую связь, -(C(R12)2)n- или -O(C(R12)2)n-,
R13 и R14 независимо означают Н, алкил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3, или R13 и R14 вместе образуют моноцикл или полицикл, или R13 и R14 вместе с R15 образуют циклоалкенил, ароматический или гетероароматический цикл,
R15 означает галоген, гидроксил, алкоксил, арил, арилокси, ацилокси, -N(R10)2, ациламино, аралкил, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10,
-CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R19, -N(R10)C(O)N(R19)2,
-N(R10)(C(R9)2)n-Al-A2-A3, -(С(R9)2)n-галоген или -CH2O-гетероциклил, или R15 вместе с R13 и R14 образуют циклоалкенил, ароматический или гетероароматический цикл, или остаток формулы 1f
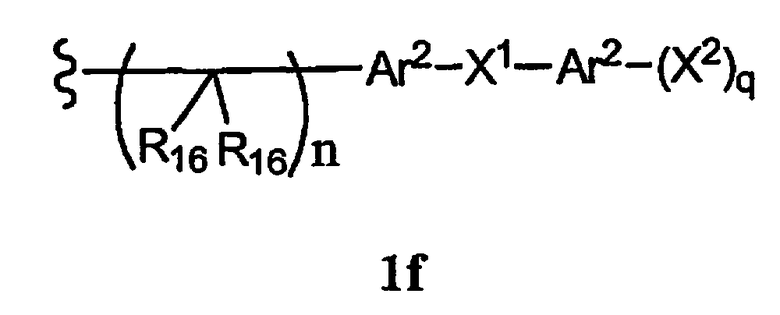
R16 каждый независимо означает Н, алкил, циклоалкил, гетероциклоалкил, арил, аралкил, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R10 или
-N(R10)C(O)N(R10)2, где любые два R16 соединены ковалентной связью с образованием цикла,
Ar2 каждый независимо означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
Х1 означает химическую связь, О, S, S(O), S(O)2, S(O)3, амино, алкиламинобирадикал, алкоксилбирадикал, алкилбирадикал, алкенилбирадикал, алкинилбирадикал, амидо, карбонил, -N(R10)CO2R10, -OC(O)N(R10)2 или
-N(R10)C(O)N(R10)2,
X2 каждый независимо означает Н, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CC2R10, -N(R10)CO2R10, -OC(O)N(R10)2,
-N(R10)SO2R10, -N(R10)C(O)N(R10)2 или -CH2O-гетероциклил, и
q равно 1, 2, 3, 4 или 5,
R8a означает Н, разветвленный или неразветвленный алкил или алкенил, циклоалкил, гетероциклоалкил, бициклоалкил, химическую связь с R7, гетероциклоалкил, замещенный аралкилом, или остаток формулы 1g,
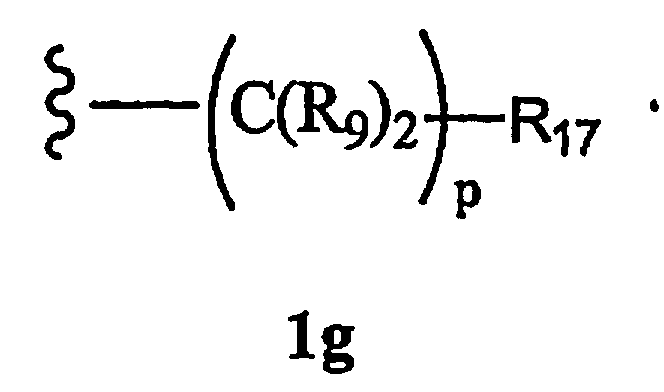
где
р равно 0, 1, 2, 3, 4, 5 или 6, и
R17 означает арил, циклоалкил, циклоалкенил, гетероциклил, алкоксил, гетероарил, -OR18, -SR18, -N(R18)2, -N(R10)CO2-алкил, -CO2R10, -С(O)N(R10)арил или полицикл, содержащий от 8 до 14 атомов углерода, где
R18 каждый независимо означает Н, алкил, арил, аралкил, ацил, -А1-А2-А3 или
-CR9=CR9(C(R9)2)nCR9=C(R9)2, или два R18 вместе образуют цикл.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R13 и R14 независимо означают Н, алкил или арил, или R13 и R14 вместе образуют моноцикл или полицикл, или R13 и R14 вместе с R15 образуют циклоалкенил или гетероароматический цикл,
R15 означает галоген, гидроксил, алкоксил, арил, арилокси, ацилокси, -N(R10)2, ациламино, аралкил, -N(R10)SO2R19 или -N(R10)C(O)N(R19)2, или R15 вместе с R13 и R14 образуют циклоалкенил или гетероароматический цикл, или остаток формулы 1f,
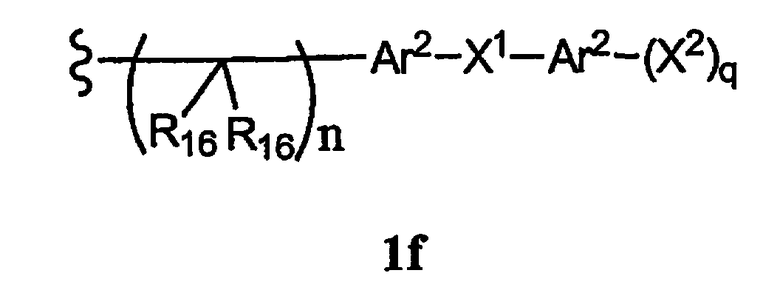
где
R16 означает Н,
Ar2 каждый независимо означает моноциклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
Х1 означает химическую связь,
Х2 каждый независимо означает Н, галоген, гидроксил или алкоксил, и
q равно 1 или 2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R8 означает бициклоалкил, гетероциклоалкил, замещенный аралкилом, или остаток формулы 1g,
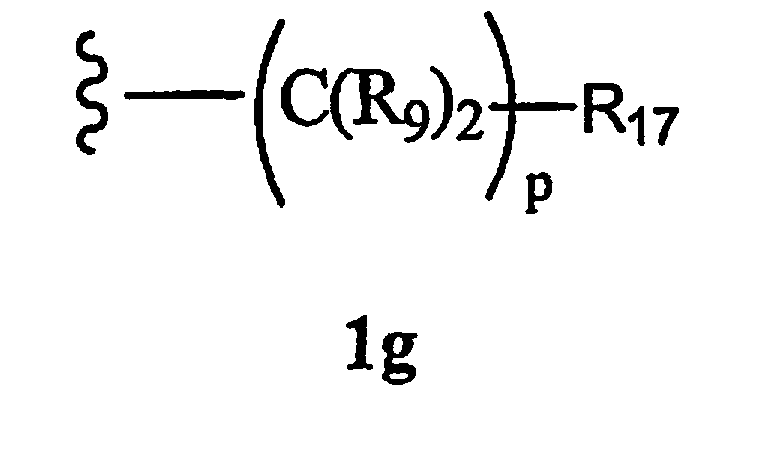
где
р равно 0, 1, 2, 3, 4, 5 или 6, и
R17 означает арил, циклоалкил, циклоалкенил, гетероциклил, алкоксил, гетероарил, -OR18, -SR18, -N(R18)2 или полицикл, содержащий от 8 до 14 атомов углерода, где
R18 каждый независимо означает Н, алкил, арил, аралкил, ацил или -А1-А2-А3.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где W означает алкинил и Z означает химическую связь.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R13 и R14 означают Н и R15 означает ациламино.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R13 и R14 вместе образуют циклогексил и R15 означает аминогруппу.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R8a означает бициклоалкил.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R8a означает остаток формулы Ig и R17 означает Н(СН3)Ph.
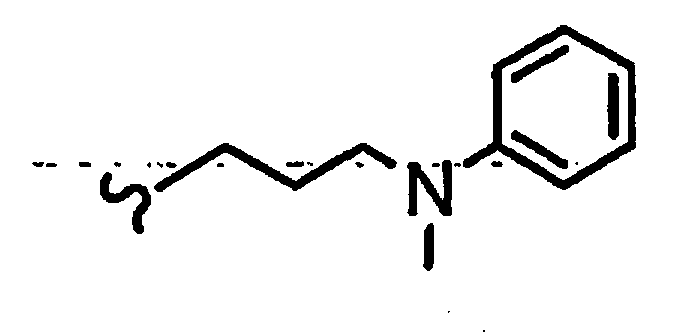
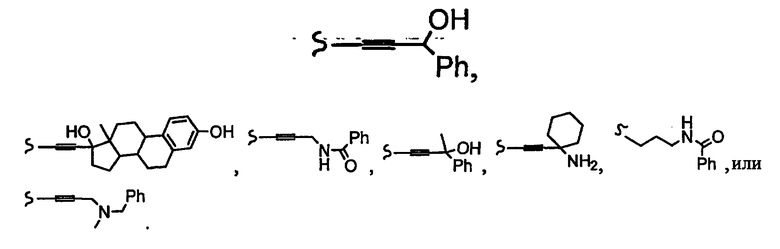
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1a означает остатки формул
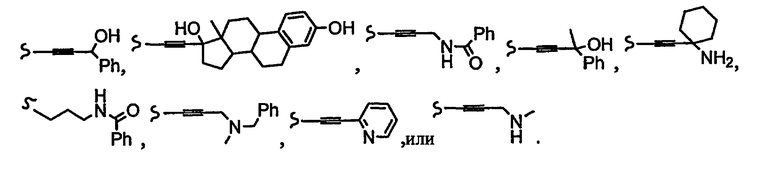
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R8a означает остатки формул
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
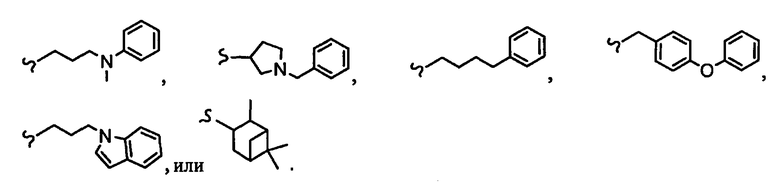
R8a означает остаток формулы
и R1a означает остатки формул
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
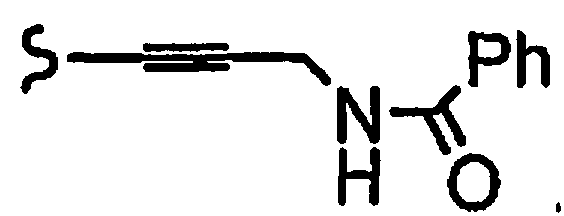
и R8a означает остатки формул
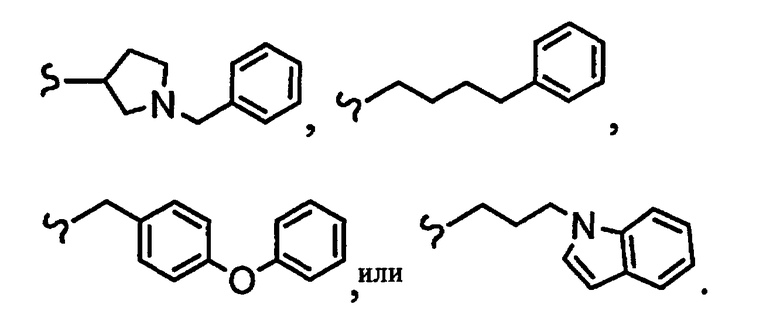
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R8a означает остаток формулы
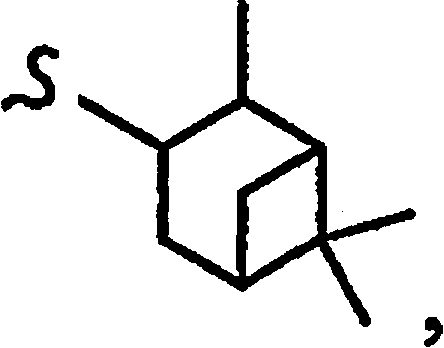
и R1a означает остатки формул
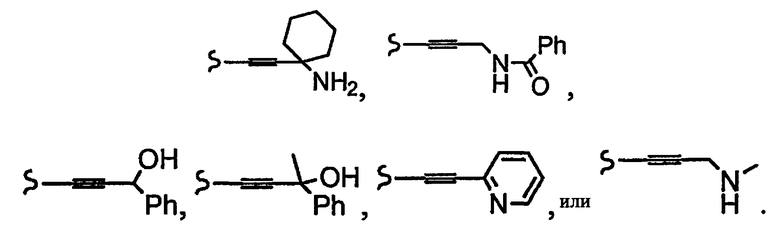
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
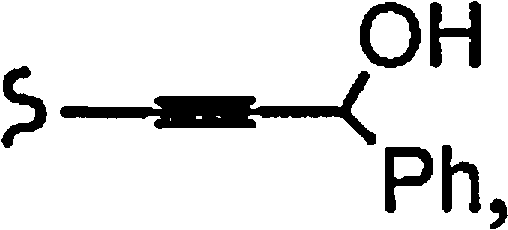
и R8a означает остаток формулы
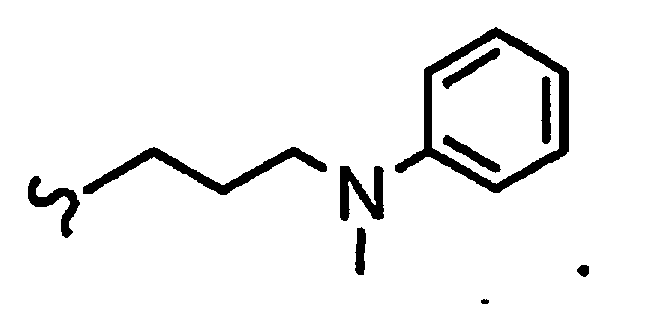
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
и R8a означает остаток формулы
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
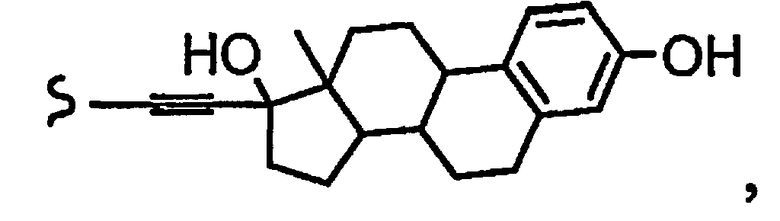
R1a означает остаток формулы
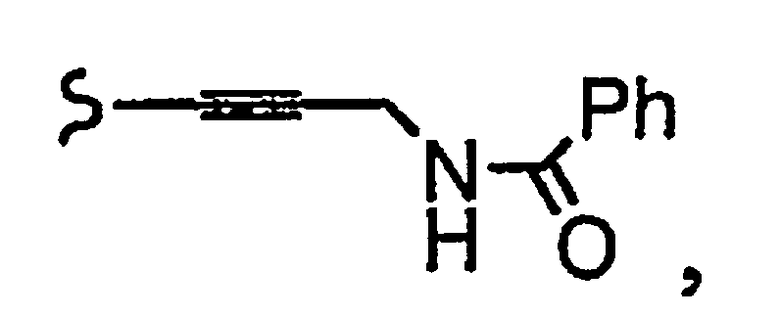
и R8a означает остаток формулы
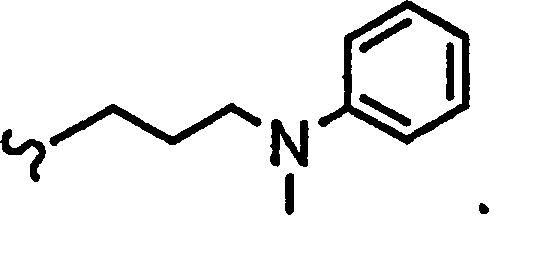
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
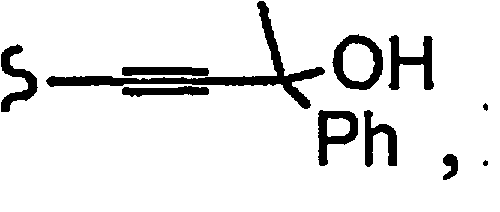
и R8a означает остаток формулы
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
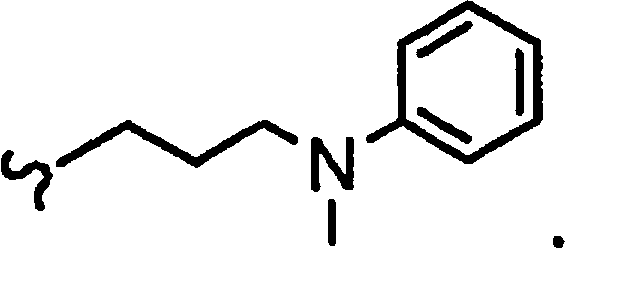
R1a означает остаток формулы
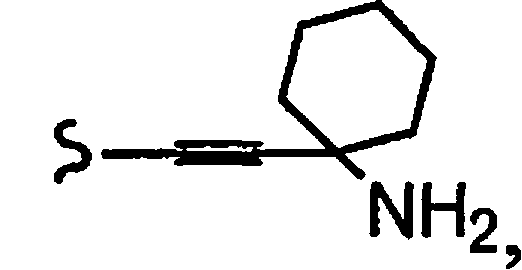
и R8a означает остаток формулы
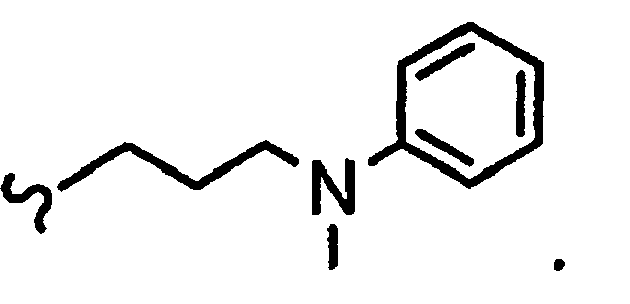
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
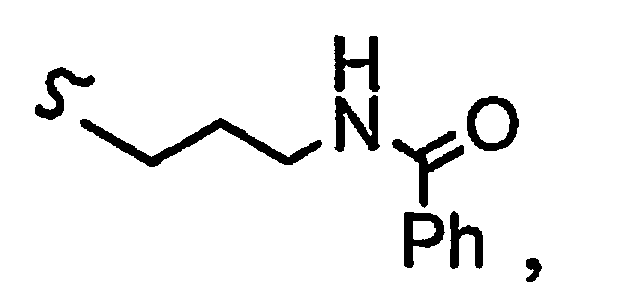
и R8a означает остаток формулы
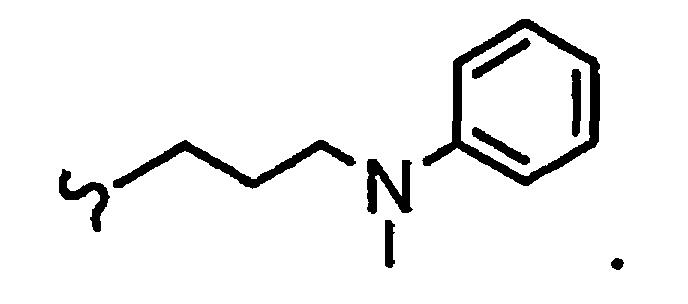
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
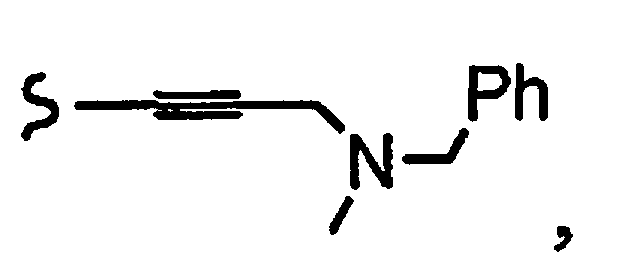
и R8a означает остаток формулы
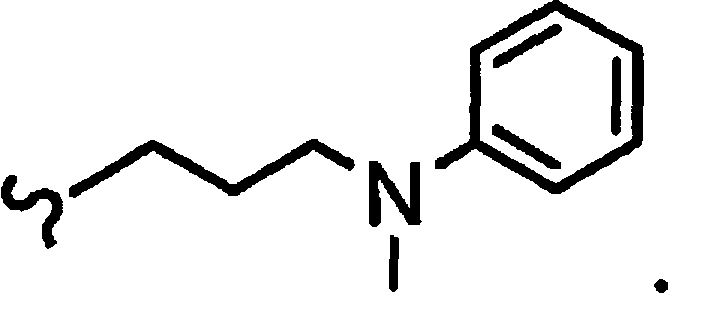
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
и R8a означает остаток формулы
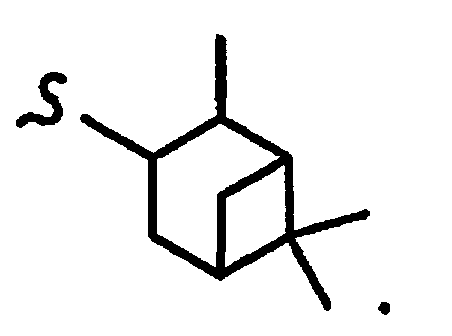
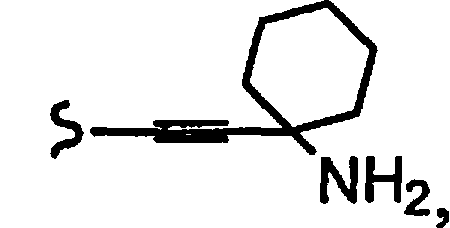
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
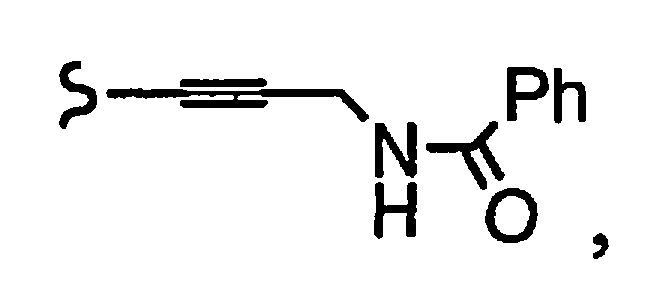
и R8a означает остаток формулы
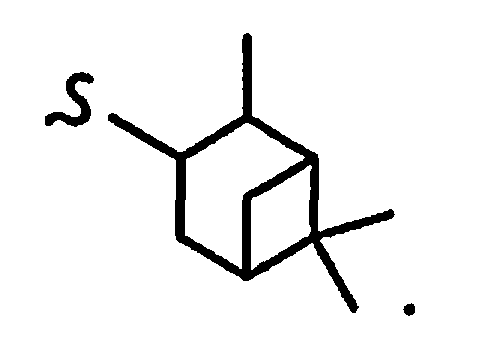
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
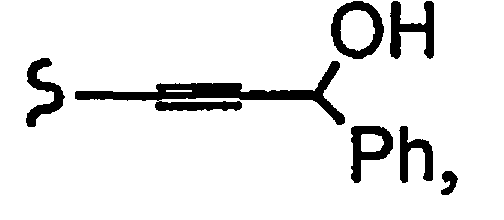
и R8a означает остаток формулы
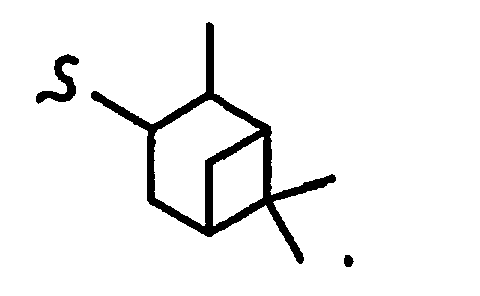
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
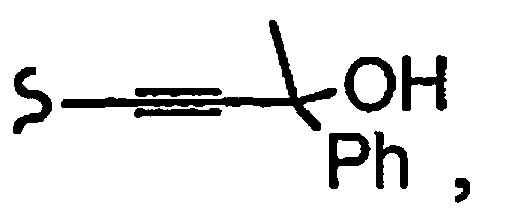
и R8a означает остаток формулы
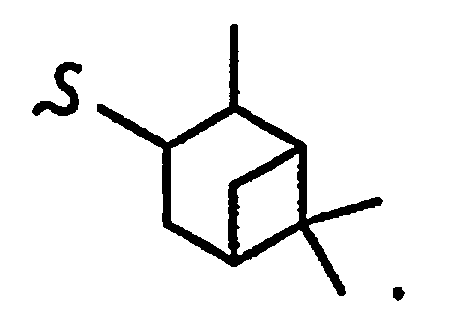
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
и R8a означает остаток формулы
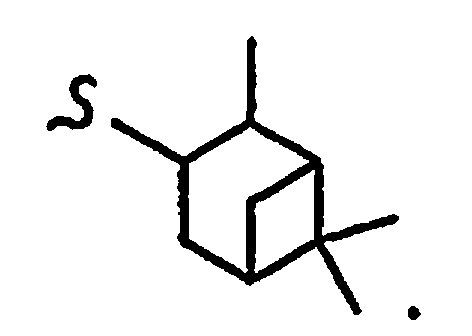
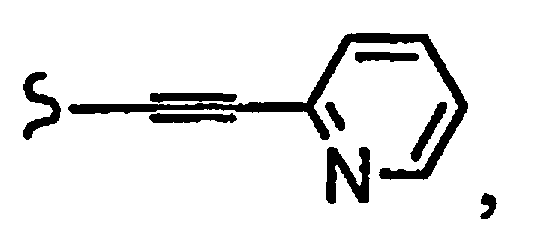
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
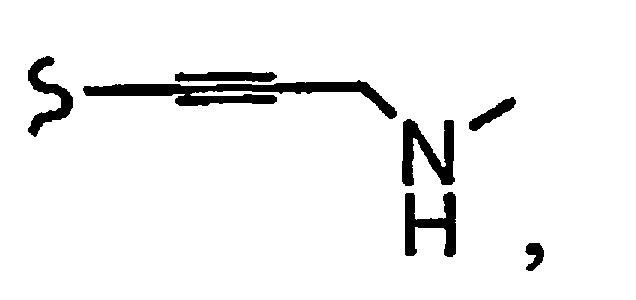
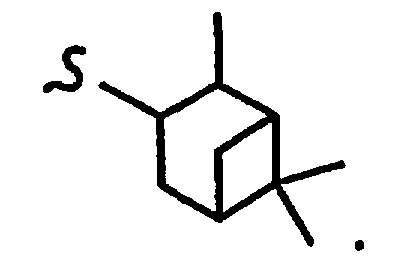
и R8a означает остаток формулы
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
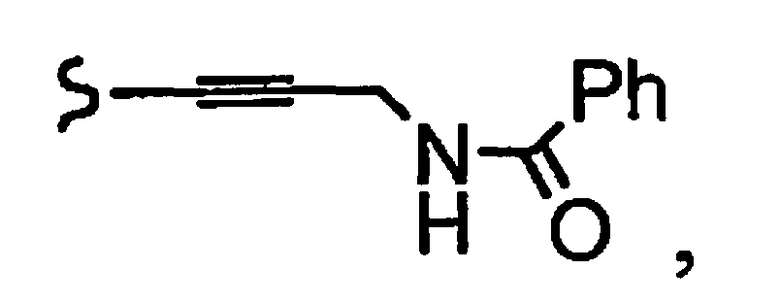
и R8a означает остаток формулы
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
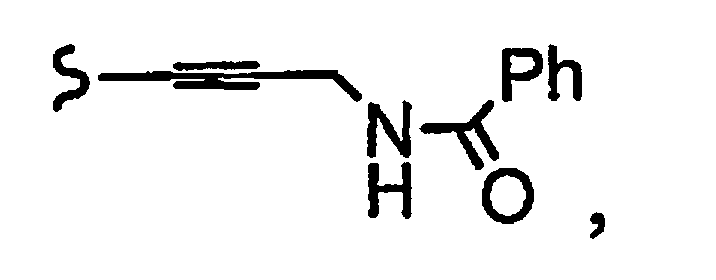
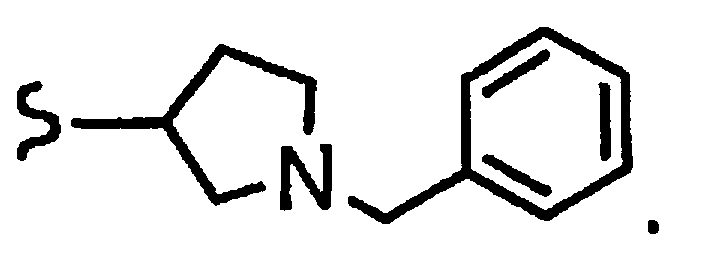
и R8a означает остаток формулы
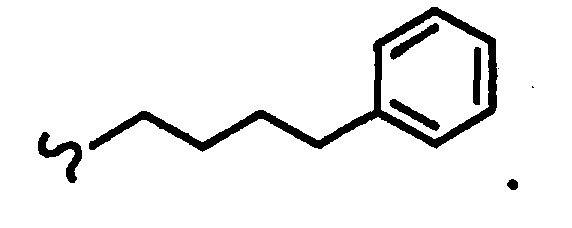
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
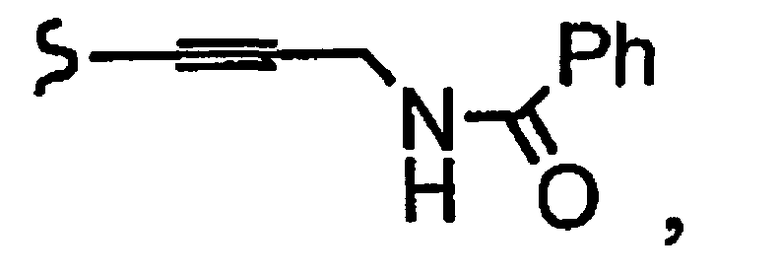
и R8a означает остаток формулы
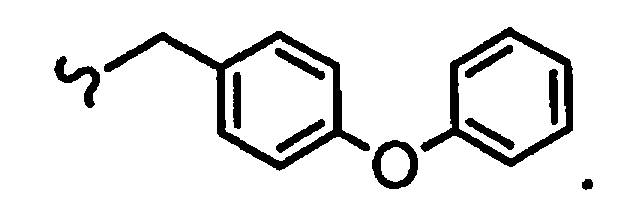
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где
R1a означает остаток формулы
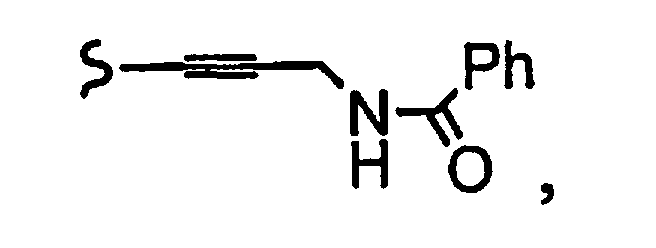
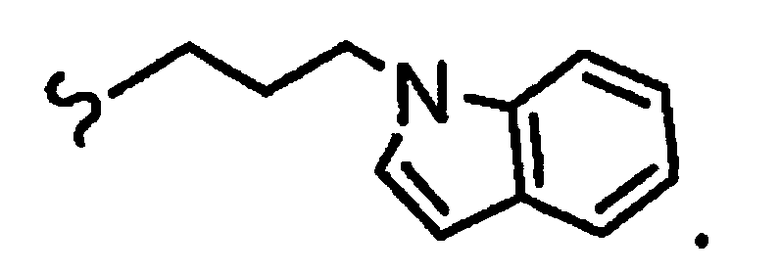
и R8a означает остаток формулы
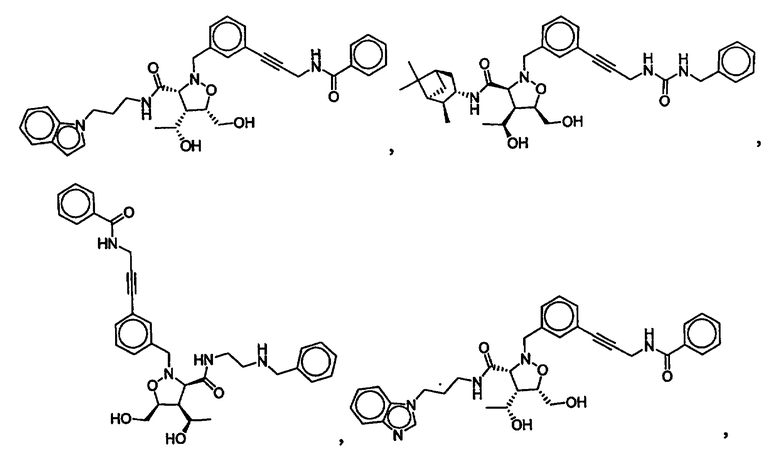
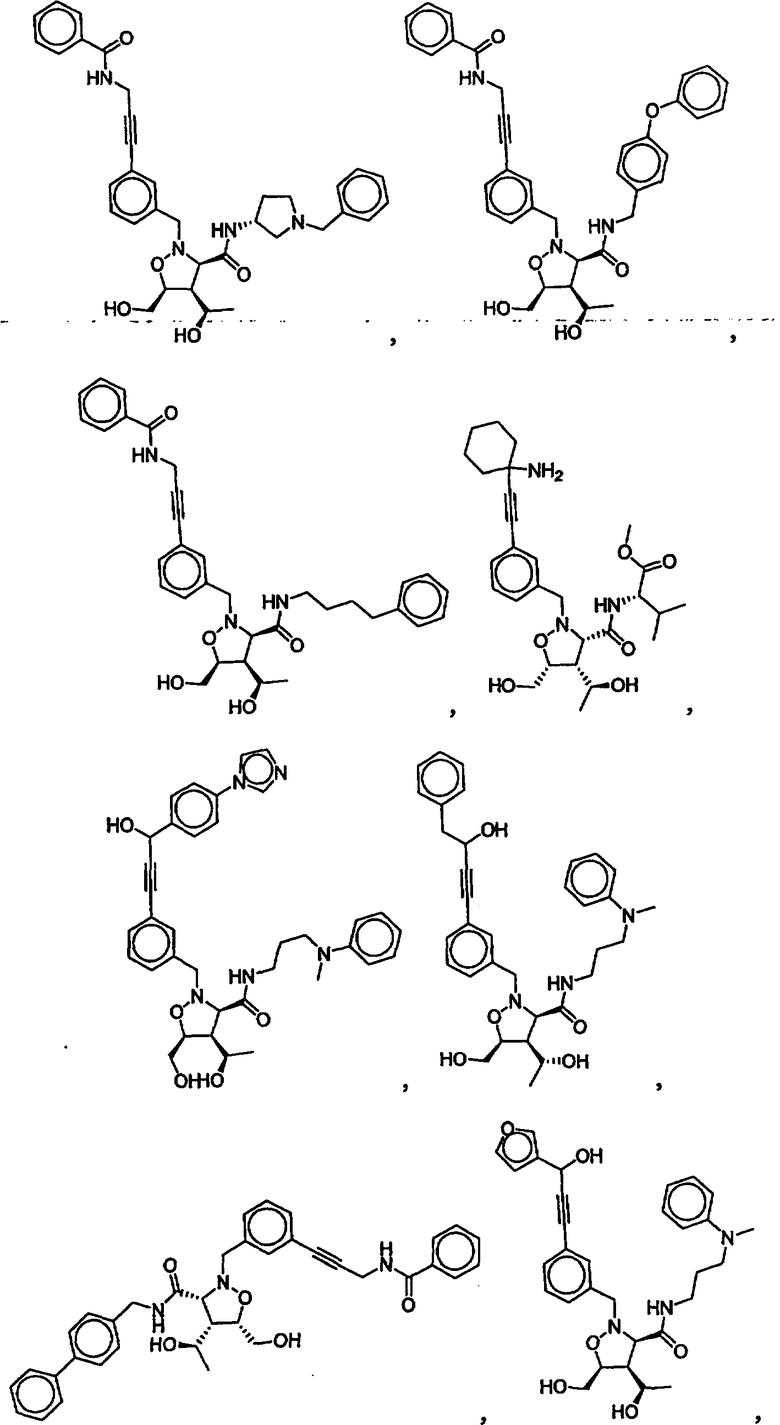
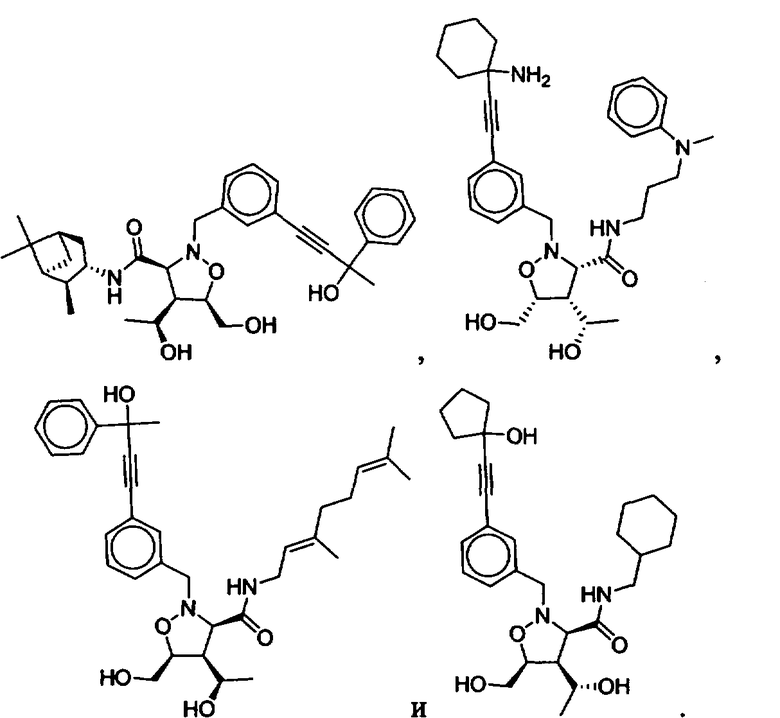
В другом варианте осуществления настоящего изобретения предлагается соединение, выбранное из группы, включающей следующие соединения
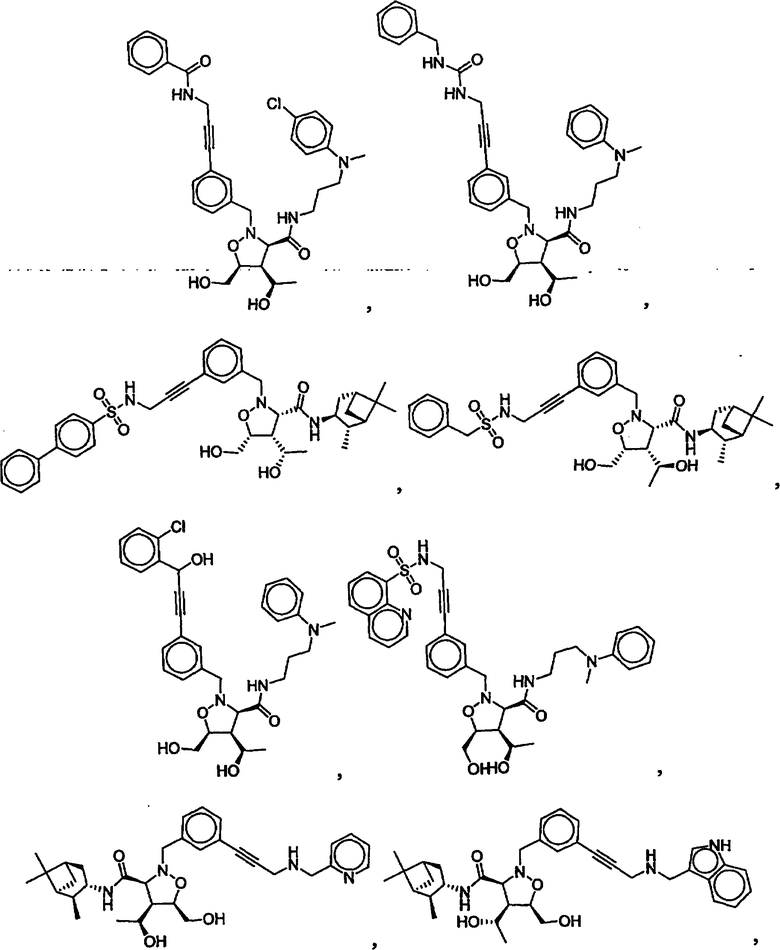
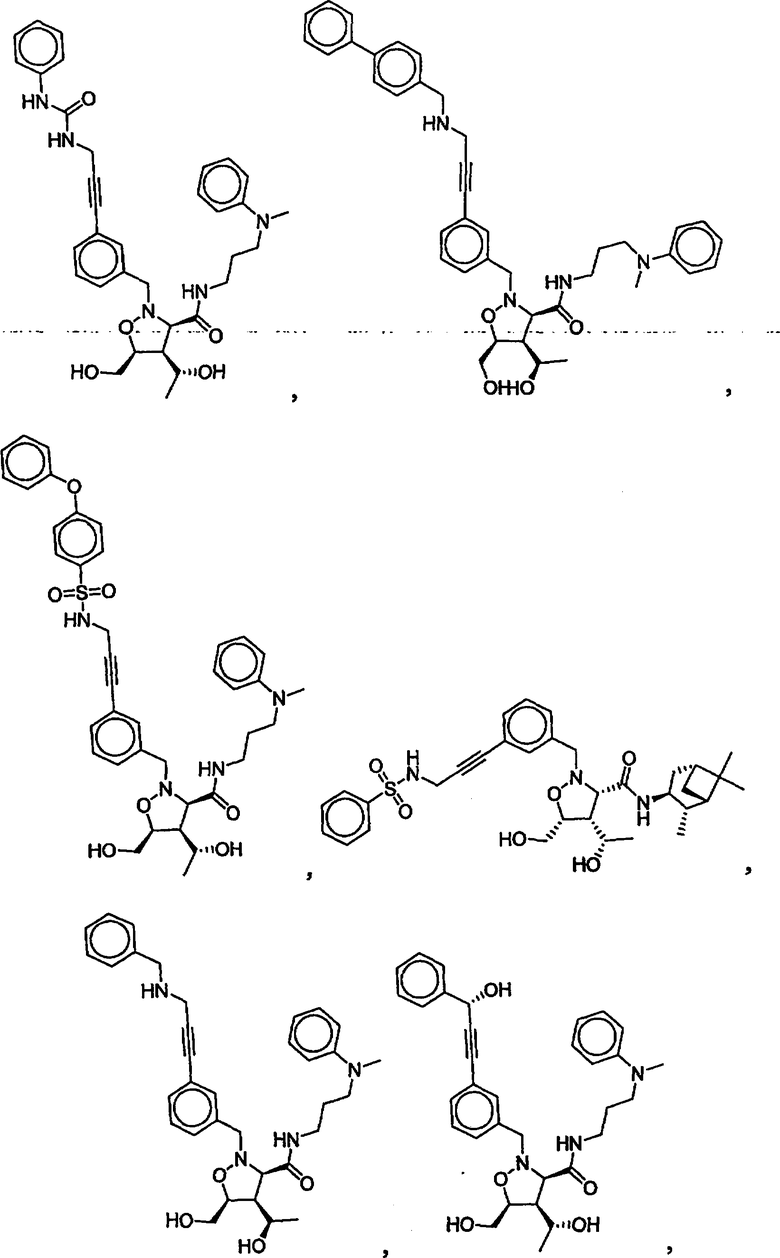
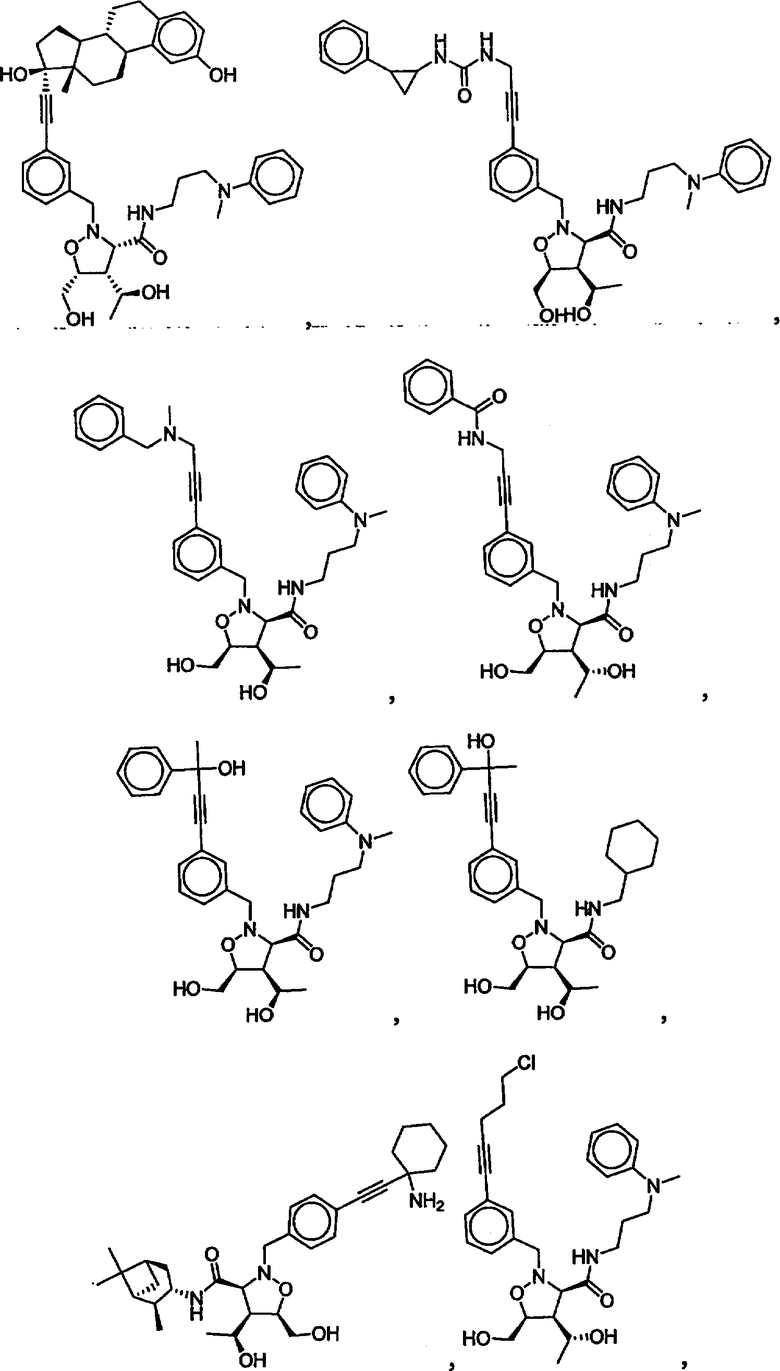
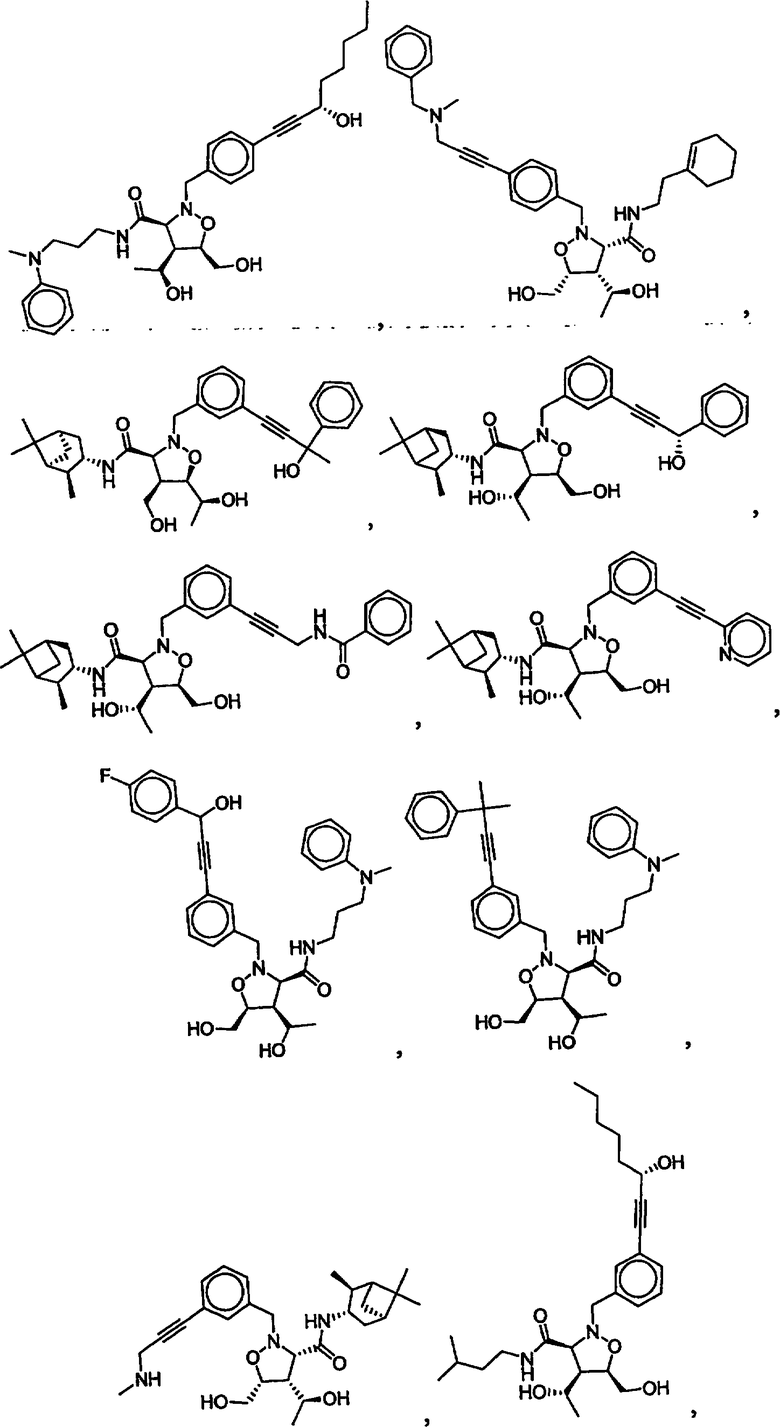
В другом варианте осуществления настоящего изобретения предлагается соединение формулы 2,
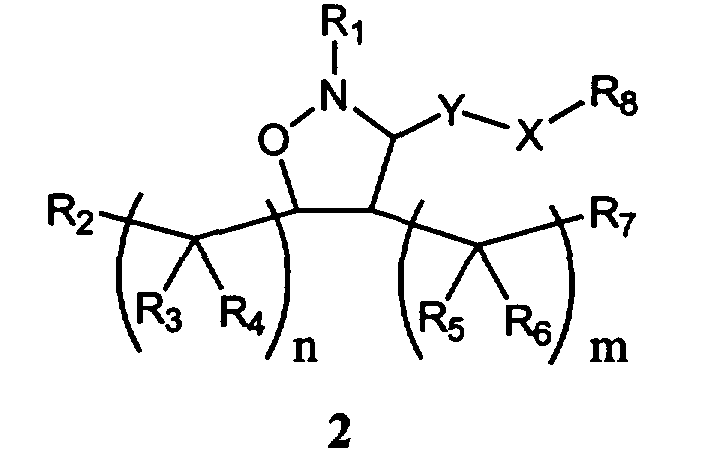
или его фармацевтически приемлемые соли, сольваты или гидраты, где
Y означает -C(R9)2-, -C(O)-, -C(S)- или -C(=NR10)-,
Х означает О, S, арил, -N(R11)- или химическую связь,
m равно 0, 1, 2, 3, 4, 5 или 6,
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6,
R1 означает остаток формулы 2а,
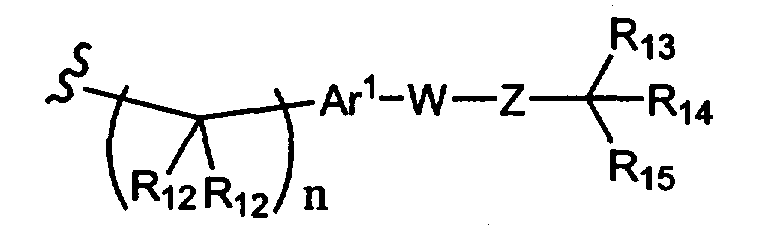
где
R12 каждый независимо означает Н или алкил, причем любые два R12 соединены ковалентной связью,
Ar1 означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
W означает химическую связь или двухвалентный алкил, алкенил или алкинил,
Z означает химическую связь, -(C(R12)2)n- или -O(C(R12)2)n-,
R13 и R14 независимо означают Н, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3, или R13 и R14 вместе образуют моноцикл или полицикл, или R13 и R14 вместе с R15 образуют циклоалкенил или гетероароматический цикл,
R15 означает галоген, гидроксил, алкоксил, арил, арилокси, ацилокси, -N(R10)2, ациламино, нитро, сульфгидрил, алкилтио, карбоксамид, карбоксил, тиоалкил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R19,
-N(R10)C(O)N(R19)2, -N(R10)(C(R9)2)n-A1-A2-A3, -(C(R9)2)n-галоген или -CH2O-гетероциклил, или R15 вместе с R13 и R14 образуют циклоалкенил или гетероароматический цикл, или остаток формулы 2b,
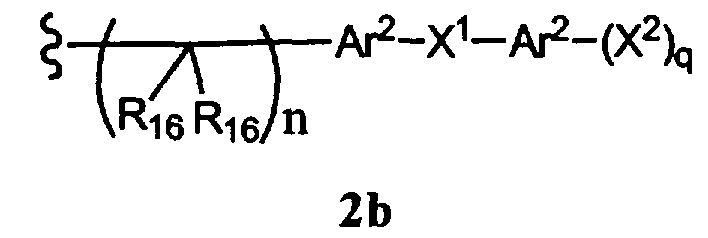
где
R16 каждый независимо означает Н или алкил,
Ar2 каждый независимо означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
Х1 означает химическую связь или О,
Х2 каждый независимо означает Н, галоген, гидроксил, алкоксил, амино, алкиламино или ариламино, и
q равно 1 или 2,
R2 и R7 независимо означают Н, гидроксил, алкил, галоген, алкоксил, арилокси, ацилокси, силилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, сульфгидрил, алкилтио, ацилтио, карбоксамид, карбоксил, фосфат, силил, тиоалкил, алкилсульфонил, арилсульфонил, алкилсульфонилокси, арилсульфонилокси, нитрил, -COR, -CO2R или -СН2О-гетероциклил, или R2 и R7 вместе образуют остаток
-ОС(O)O- или необязательно замещенный алкил, содержащий от 1 до 6 атомов углерода, или R7 означает химическую связь с R8,
R3 и R6 каждый независимо означает Н, галоген, гидроксил, амино, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил, алкоксил, арилокси, ацилокси, силилокси, алкиламино, ариламино, ациламино или аралкиламино,
R4 и R5 каждый независимо означает Н, галоген, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил, алкоксил, арилокси, ацилокси, силилокси, алкиламино, ариламино, ациламино или аралкиламино,
R8 означает Н, разветвленный или неразветвленный алкил или алкенил, циклоалкил, гетероциклоалкил, бициклоалкил, химическую связь с R7, гетероциклоалкил, замещенный аралкилом, или остаток формулы 2с,
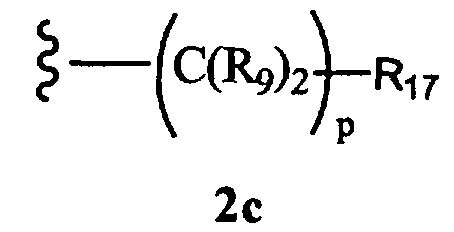
где
р равно 0, 1, 2, 3, 4, 5 или 6, и
R17 означает арил, циклоалкил, циклоалкенил, гетероциклил, алкоксил, гетероарил, -OR18, -SR18, -N(R18)2, -N(R10)CO2-алкил, -CO2R10, -С(O)N(R10)арил или полицикл, содержащий от 8 до 14 атомов углерода, где
R18 каждый независимо означает Н, алкил, арил, аралкил, ацил, -А1-А2-А3 или
-CR9=CR9(C(R9)2)nCR9=C(R9)2, или два R18 вместе образуют цикл,
R9 каждый независимо означает Н или алкил,
R10 и R11 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил или гетероаралкил,
R19 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3,
А1 и А3 каждый независимо означает алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил или гетероаралкил,
А2 каждый независимо означает О или химическую связь, при этом стереохимическая конфигурация в любом стереоцентре соединения формулы 2 означает R-, S-конфигурацию или смесь этих конфигураций.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R2 и R7 означают гидроксил.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R2 и R7 означают гидроксил, a R4, R5 и R6 означают Н.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R2 и R7 означают гидроксил, R4, R5 и R6 означают Н, a m и n равны 1.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R2 и R7 означают гидроксил, R4, R5 и R6 означают Н, m и n равны 1, а R3 означает метил.
В другом варианте осуществления настоящего изобретения предлагается соединение формулы 3,
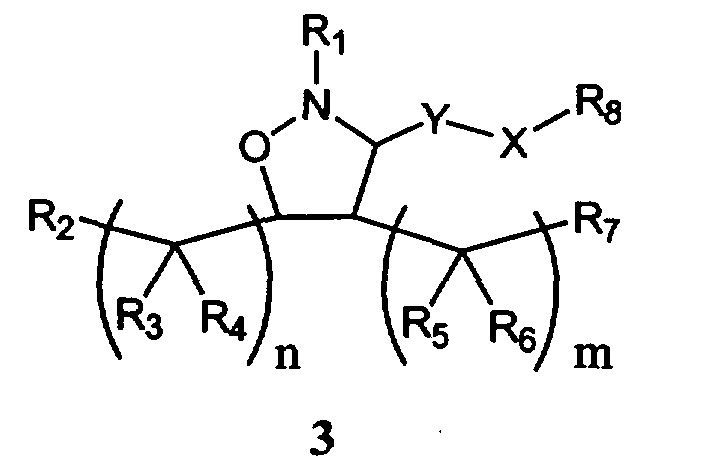
или его фармацевтически приемлемые соли, сольваты или гидраты, где
Y означает -C(R9)2-, -C(O)-, -C(S)- или -C(=NR10)-,
Х означает -N(R11)-, необязательно замещенный фенил или химическую связь,
m равно 0, 1, 2, 3, 4, 5 или 6,
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6,
R1 означает алкил, аралкил, гетероаралкил или остаток формулы 3а,
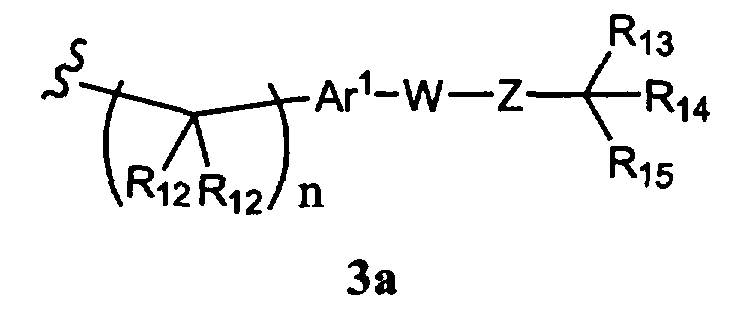
где
R12 каждый независимо означает Н или алкил, причем любые два R12 соединены ковалентной связью,
Ar1 означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
W означает химическую связь или двухвалентный алкил, алкенил или алкинил,
Z означает химическую связь, -(C(R12)2)n- или -O(C(R12)2)n-,
R13 и R14 независимо означают Н, алкил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галоген, алкенил, алкинил, аминоалкил, тиол, тиоалкил, силил, нитро, нитрил, алкоксил, ацил, ациламино, -COR10, -CO2R10 или -А1-А2-А3, или R13 и R14 вместе образуют моноцикл или полицикл, или R13 и R14 вместе с R15 образуют циклоалкенил, ароматический или гетероароматический цикл,
R15 означает галоген, гидроксил, алкоксил, арил, арилокси, ацилокси, -N(R10)2, ациламино, аралкил, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10,
-CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R19, -N(R10)C(O)N(R19)2,
-N(R10)(C(R9)2)n-Al-A2-A3, -(С(R9)2)n-галоген или -CH2O-гетероциклил, алкил, циклоалкил, алкенил, алкинил, силилокси, тиол, ацилтио, фосфат, силил, тиоалкил, алкилсульфонил, арилсульфонил, алкилсульфонилокси или арилсульфонилокси, или R15 вместе с R13 и R14 образуют циклоалкенил, ароматический или гетероароматический цикл, или R1 или R15 каждый независимо означает остаток формулы 3b,
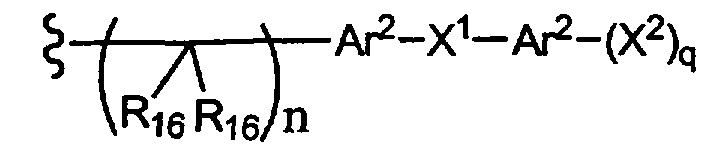
где
R16 каждый независимо означает Н, алкил, циклоалкил, гетероциклоалкил, арил, аралкил, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R10 или
-N(R10)C(O)N(R10)2, причем любые два R16 соединены ковалентной связью с образованием цикла,
Ar2 каждый независимо означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
Х1 каждый независимо означает химическую связь, О, S, S(O), S(O)2, S(O)3, амино, алкиламинобирадикал, алкоксилбирадикал, алкилбирадикал, алкенилбирадикал, алкинилбирадикал, амидо, карбонил, -N(R10)CO2R10, -OC(O)N(R10)2 или
-N(R10)C(O)N(R10)2,
X2 каждый независимо означает Н, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2,
-N(R10)SO2R10, -N(R10)C(O)N(R10)2 или -CH2O-гетероциклил, и
q каждое независимо равно 1, 2, 3, 4 или 5,
R2 и R7 независимо означают Н, гидроксил, алкил, галоген, алкоксил, арилокси, ацилокси, силилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, сульфгидрил, алкилтио, ацилтио, карбоксамид, карбоксил, фосфат, силил, тиоалкил, алкилсульфонил, арилсульфонил, алкилсульфонилокси, арилсульфонилокси, нитрил, -COR, -CO2R или -CH2O-гетероциклил, или R2 и R7 вместе образуют остаток -ОС(O)O- или необязательно замещенный алкил, содержащий от 1 до 6 атомов углерода, или R7 означает химическую связь с R8,
R3 и R6 каждый независимо означает Н, галоген, гидроксил, амино, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил, алкоксил, арилокси, ацилокси, силилокси, алкиламино, ариламино, ациламино или аралкиламино,
R4 и R5 каждый независимо означает Н, галоген, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил, алкоксил, арилокси, ацилокси, силилокси, алкиламино, ариламино, ациламино или аралкиламино,
R8 означает разветвленный или неразветвленный алкил, бициклоалкил, гетероциклоалкил, замещенный аралкилом, или остаток формулы 3с,
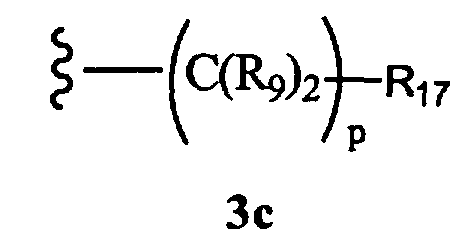
где
р равно 0, 1, 2, 3, 4, 5 или 6, и
R17 означает арил, циклоалкил, циклоалкенил, гетероциклил, гетероарил,
-N(R18)2, -OR18 или -CO2R10, где
R18 каждый независимо означает Н, алкил, арил, аралкил, -А1-А2-А3 или -CR9=CR9(C(R9)2)nCR9=C(R9)2, или два R18 вместе образуют цикл,
R9 каждый независимо означает Н или алкил,
R10 и R11 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил или гетероаралкил,
R19 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3,
А1 и А3 каждый независимо означает алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил или гетероаралкил,
А2 каждый независимо означает О или химическую связь, при этом стереохимическая конфигурация в любом стереоцентре соединения формулы 3 означает R-, S-конфигурацию или смесь этих конфигураций.
В другом варианте осуществления настоящего изобретения предлагается соединение формулы 4,
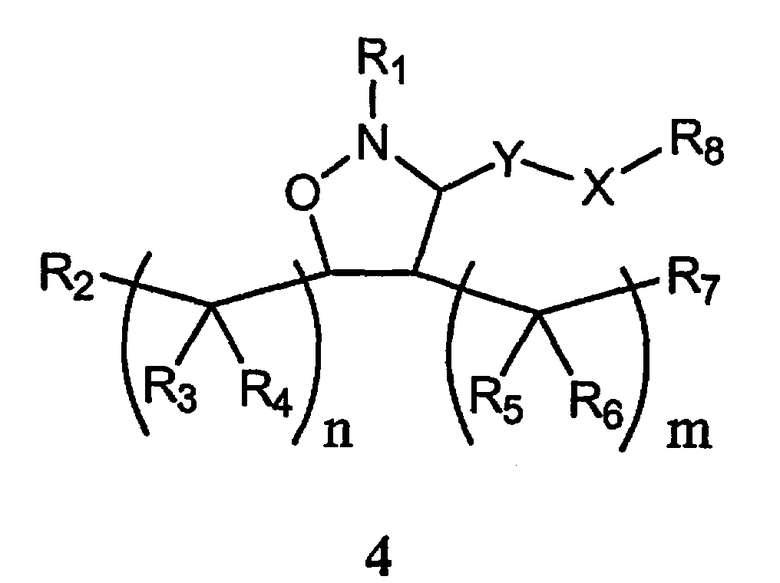
или его фармацевтически приемлемые соли, сольваты или гидраты, где
Y означает -C(R9)2-, -C(O)-, -C(S)- или -C(=NR10)-,
Х означает О, S, -N(R11)-, арил или химическую связь,
m равно 0, 1, 2, 3, 4, 5 или 6,
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6,
R1 означает алкил, аралкил, гетероаралкил или остаток формулы 4а,
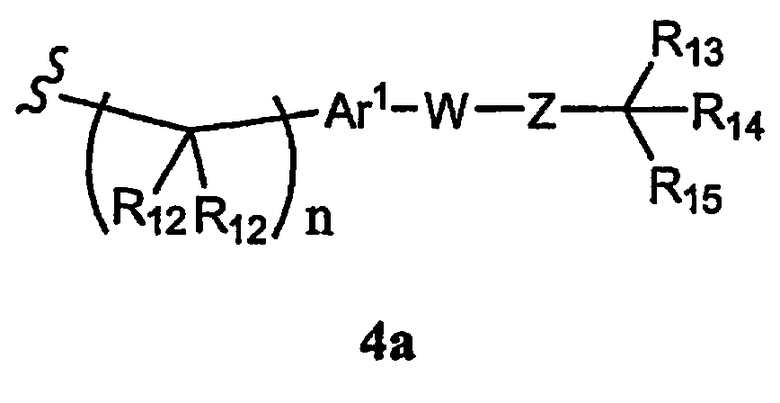
где
R12 каждый независимо означает Н или алкил, причем любые два R12 соединены ковалентной связью,
Ar1 означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
W означает химическую связь или двухвалентный алкил, алкенил или алкинил,
Z означает химическую связь, -(C(R12)2)n- или -O(C(R12)2)n-,
R13 и R14 независимо означают Н, алкил, арил, циклоалкил, гетероциклоалкил, аралкил, гетероарил, гетероаралкил, галоген, алкенил, алкинил, аминоалкил, тиол, тиоалкил, силил, нитро, нитрил, алкоксил, ацил, ациламино, -COR10, -CO2R10 или -А1-А2-А3, или R13 и R14 вместе образуют моноцикл или полицикл, или R13 и R14 вместе с R15 образуют циклоалкенил, ароматический или гетероароматический цикл,
R15 означает галоген, гидроксил, алкоксил, арил, арилокси, ацилокси, -N(R10)2, ациламино, аралкил, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10,
-CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R19, -N(R10)C(O)N(R19)2,
-N(R10)(C(R9)2)n-Al-A2-A3, -(С(R9)2)n-галоген или -CH3O-гетероциклил, алкил, циклоалкил, алкенил, алкинил, силилокси, тиол, ацилтио, фосфат, силил, тиоалкил, алкилсульфонил, арилсульфонил, алкилсульфонилокси или арилсульфонилокси, или R15 вместе с R13 и R14 образуют циклоалкенил, ароматический или гетероароматический цикл, или R1 или R15 каждый независимо означает остаток формулы 4b,
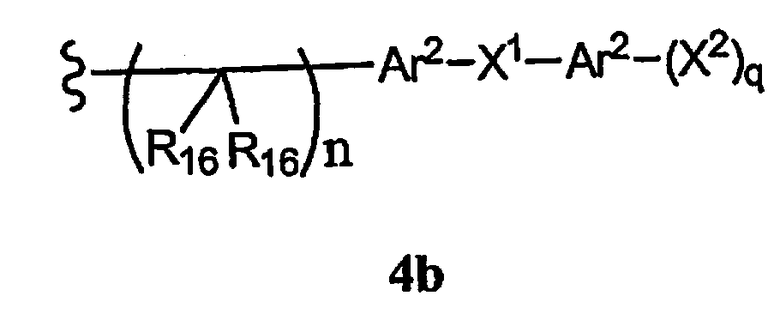
где
R16 каждый независимо означает Н, алкил, циклоалкил, гетероциклоалкил, арил, аралкил, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2, -N(R10)SO2R10 или
-N(R10)C(O)N(R10)2, причем любые два R16 соединены ковалентной связью с образованием цикла,
Ar2 каждый независимо означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N,
Х1 каждый независимо означает химическую связь, О, S, S(O), S(O)2, S(O)3, амино, алкиламинобирадикал, алкоксилбирадикал, алкилбирадикал, алкенилбирадикал, алкинилбирадикал, амидо, карбонил, -N(R10)CO2R10, -OC(O)N(R10)2 или
-N(R10)C(O)N(R10)2,
X2 каждый независимо означает Н, галоген, гидроксил, алкоксил, арилокси, ацилокси, амино, алкиламино, ариламино, ациламино, аралкиламино, нитро, ацилтио, карбоксамид, карбоксил, нитрил, -COR10, -CO2R10, -N(R10)CO2R10, -OC(O)N(R10)2,
-N(R10)SO2R10, -N(R10)C(O)N(R10)2 или -CH2O-гетероциклил, и
q каждое независимо равно 1, 2, 3, 4 или 5,
R2 и R7 независимо означают гидроксил или алкоксил,
R3, R4 и R5 означают Н,
R6 означает метил, этил или пропил,
R8 означает Н, разветвленный или неразветвленный алкил или алкенил, циклоалкил, гетероциклоалкил, бициклоалкил, химическую связь с R7, гетероциклоалкил, замещенный аралкилом, или остаток формулы 4с,
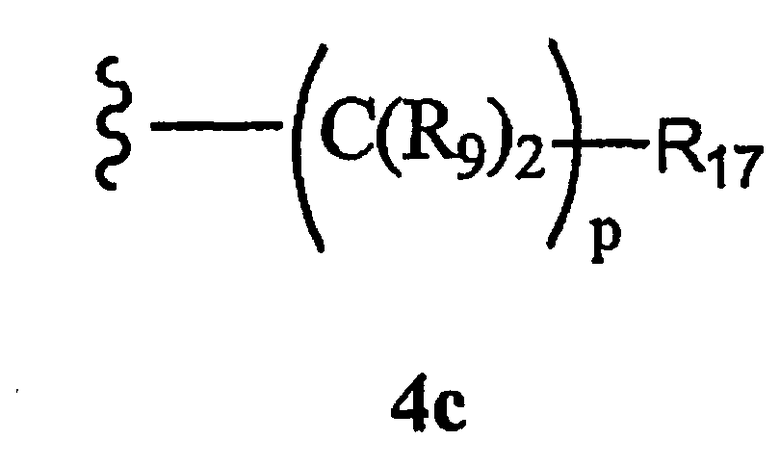
где
р равно 0, 1, 2, 3, 4, 5 или 6, и
R17 означает арил, циклоалкил, циклоалкенил, гетероциклил, алкоксил, гетероарил, -OR18, -SR18, -N(R18)2, -N(R10)CO2-алкил, -CO2R10, -С(O)N(R10)арил или полицикл, содержащий от 8 до 14 атомов углерода, где
R18 каждый независимо означает Н, алкил, арил, аралкил, ацил, -А1-А2-А3 или
-CR9=CR9(C(R9)2)nCR9=C(R9)2, или два R18 вместе образуют цикл,
R9 каждый независимо означает Н или алкил,
R10 и R11 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил или гетероаралкил,
R19 каждый независимо означает Н, алкил, арил, циклоалкил, аралкил, гетероарил, гетероаралкил или -А1-А2-А3,
А1 и А3 каждый независимо означает алкил, циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил или гетероаралкил,
А2 каждый независимо означает О или химическую связь, при этом стереохимическая конфигурация в любом стереоцентре соединения формулы 4 означает R-, S-конфигурацию или смесь этих конфигураций.
В другом варианте осуществления настоящего изобретения предлагается соединение формулы 5,
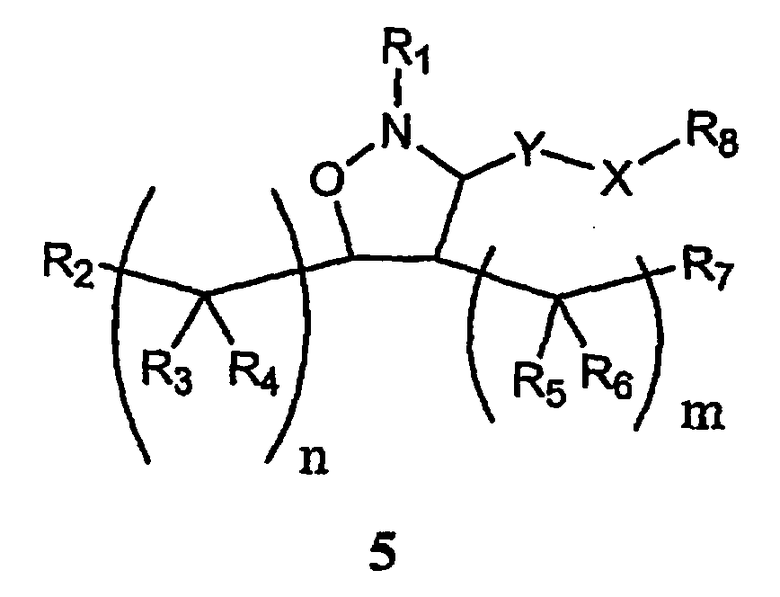
или его фармацевтически приемлемые соли, сольваты или гидраты, где
Y означает -C(R9)2-, -C(O)-, -C(S)- или -C(=NR10)-,
Х означает О, S, -N(R11)-,
m равно 0, 1, 2, 3, 4, 5 или 6,
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6,
R1 означает алкил, аралкил, гетероаралкил или остаток формул 5а или 5b,
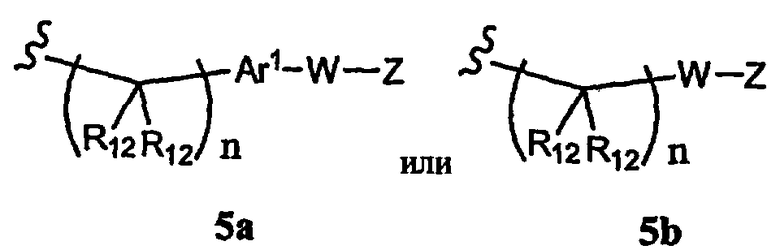
где
R12 каждый независимо означает Н или алкил, причем любые два R12 соединены ковалентной связью,
Ar1 означает моноциклический или бициклический арил, содержащий от 6 до 14 атомов в цикле, или моноциклический или бициклический гетероарил, содержащий от 5 до 14 атомов в цикле, где один, два или три атома в цикле независимо являются S, О или N, или Ar1 означает остаток формулы 5с,
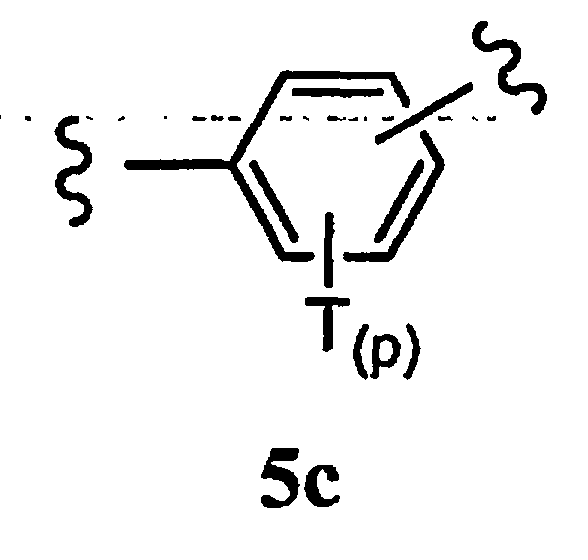
где
Т каждый независимо означает Н, галоген, разветвленный или неразветвленный алкил, алкенил, аллил, алкокси, арил, аралкил, гидроксил, амино, аминоалкил, амидо, карбоксамид, циклоалкил, циклоалкен, бициклоалкил, бициклоалкен, циклоалкилалкил, гетероароматический цикл, гетероаралкил, гетероциклил, гетероциклоалкил, галогеналкил, сложный эфир, карбоксил, бис-арил, бис-ариловый эфир, гетероцикл, замещенный арилом, или два Т вместе образуют ароматический или неароматический цикл, и
р равно 0, 1, 2, 3 или 4,
W означает химическую связь или двухвалентный алкил, арил, гетероарил или гетероциклил,
Z означает химическую связь, Н, -SR, -S(O)2R, -NRSO2R, -S(O)R, -N(R)2, -C(O)R,
-CO2R, -C(O)N(R)2, -C(S)N(R)2, -СН2С(O)гетероциклил, -NRC(O)R, -NRCO2R,
-OC(O)N(R)2, -NRC(O)(C(R9)2)nN(R)2, -NC(O)CH(R)2, -C(=NR)N(R)2, -C(=NR)R, гидроксиалкил или моно- или бициклический арил, гетероарил или гетероциклил, где
R каждый независимо означает Н, разветвленный или неразветвленный алкил, алкенил, аллил, алкокси, галогеналкил, ацил, мезилат, тозилат, сложный эфир,
-(C(R9)2)nT, -CH((C(R9)2)nT)2, или два R вместе образуют ароматический или неароматический цикл,
R2 и R7 каждый независимо означает Н, гидроксил, алкил, алкоксил, амино, алкиламино, сложный эфир или карбоксамид,
R3 и R6 каждый независимо означает Н, гидроксил или алкил,
R4 и R5 каждый независимо означает Н или алкил, и
R8 означает Н, разветвленный или неразветвленный алкил или алкенил, циклоалкил, гетероциклоалкил, бициклоалкил, разветвленный или неразветвленный аминоалкил или гетероциклоалкил, замещенный аралкилом,
R9, R10 и R12 каждый независимо означает Н, алкил, арил, аралкил, циклоалкил, циклоалкилалкил, гетероарил или гетероаралкил, если
Ar1, W и Z замещены одной или более группами, выбранными из группы, включающей галоген, амидо, алкокси, эфир, -NO2, гидроксил, -NR2 или -CN, то Ar1, W и Z связаны между собой в орто-, мета- или пара-положении, при этом стереохимическая конфигурация в любом стереоцентре соединения формулы 5 означают R-, S-конфигурацию или смесь этих конфигураций.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R2 означает ОН.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R6 означает метил или этил, a R7 означает гидроксил.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где Y означает -С(O)-, Х означает -N(R11)-, a R8 означает бициклоалкил.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5а.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5а, а R12 означает Н или метил.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5а, а Ar1 означает бензол.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5а, а W означает химическую связь, -СН2- или бензол.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5b, a R12 означает Н или метил.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5b, где n равно 4.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где R1 означает остаток формулы 5b и Z означает N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение формулы 5d или 5е
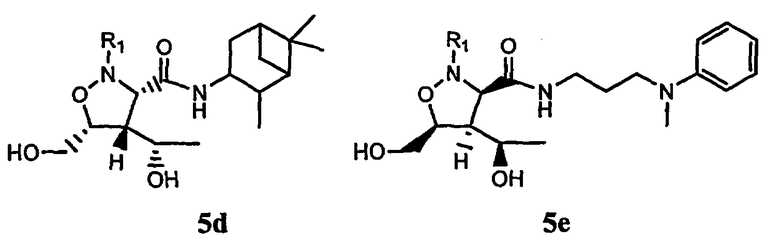
где
R1 означает остаток формулы 5f
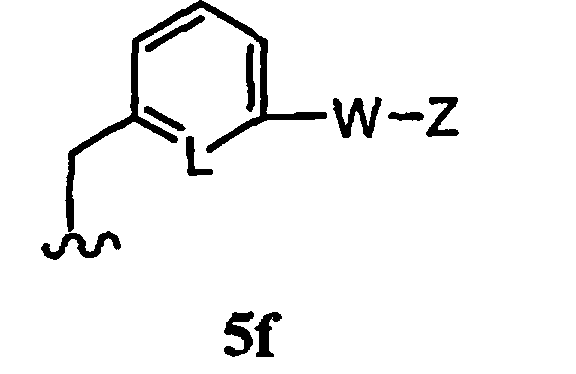
где
L означает N или CR, и
W, Z, R13, R14, R15 и
n имеют значения, определенные выше.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает бензол, а Z означает -C(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает CR, R означает алкокси, W означает бензол, a Z означает -C(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СОМе, W означает бензол, а Z означает -C(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает COEt, W означает бензол, а Z означает -C(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СОСН2 (циклопропил), W означает бензол, а Z означает -C(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает бензол, а Z означает Н.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает -СН2-, а Z означает -N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает пиперазин, а Z означает -C(S)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает пиперазин, а Z означает -C(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает химическую связь, а Z означает N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает химическую связь, а Z означает -NRCO2R или -OC(O)N(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанное выше соединение, где L означает СН, W означает химическую связь, a Z означает -NRC(O)(C(R9)2)nN(R)2.
В некоторых вариантах осуществления настоящего изобретения предлагается соединение формулы 5g
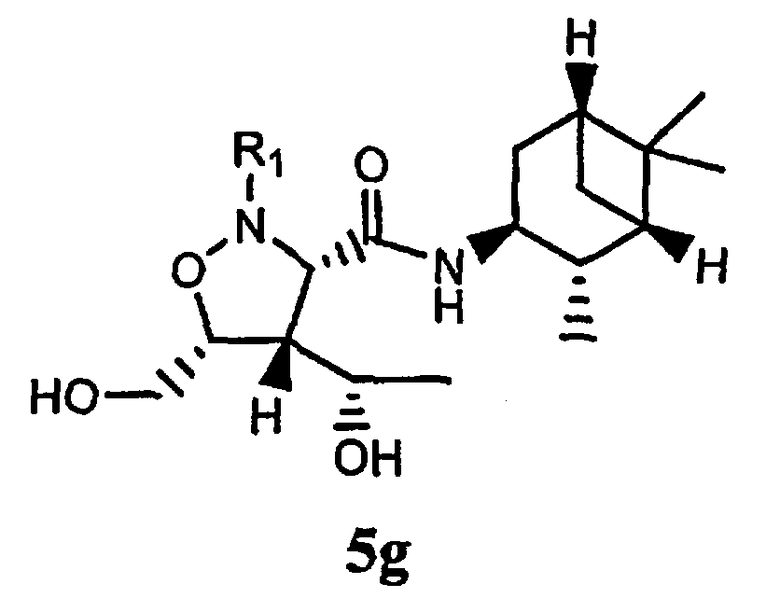
где
R1 означает
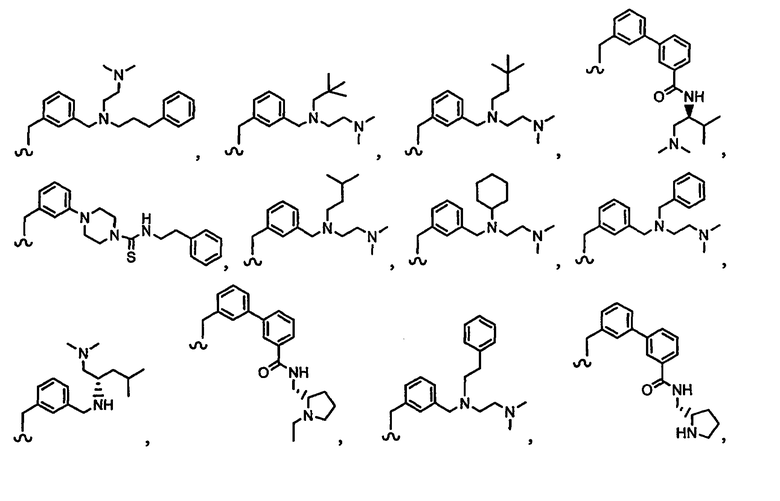
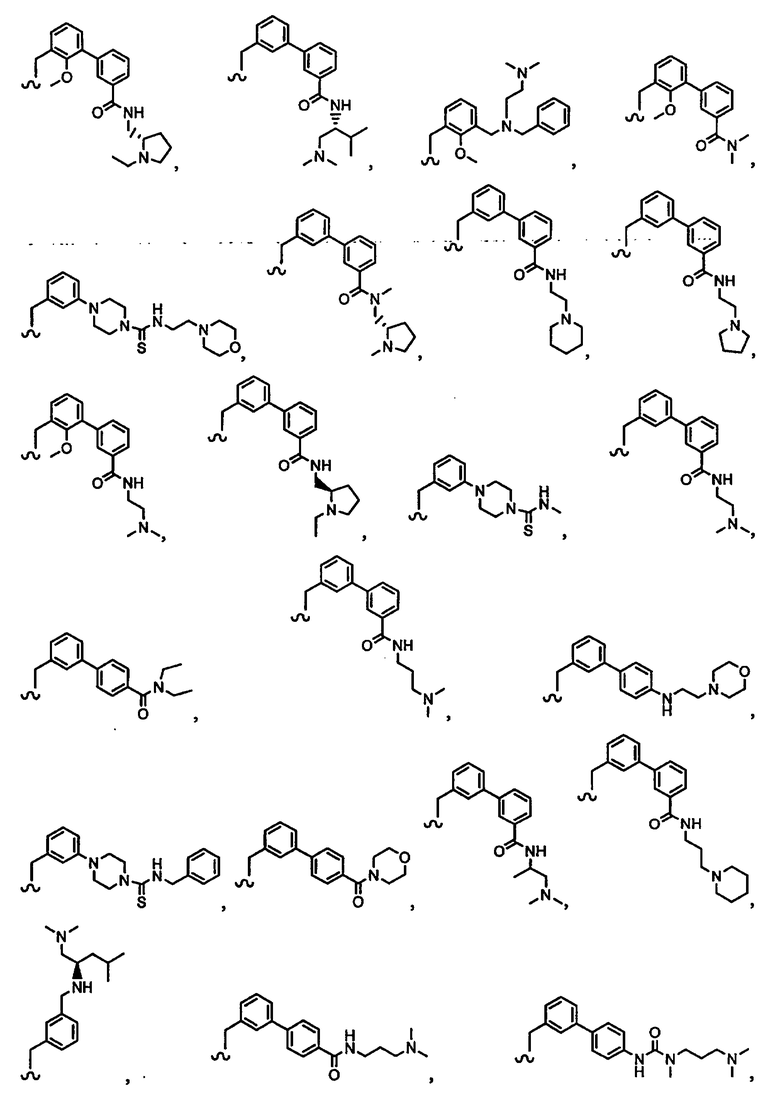
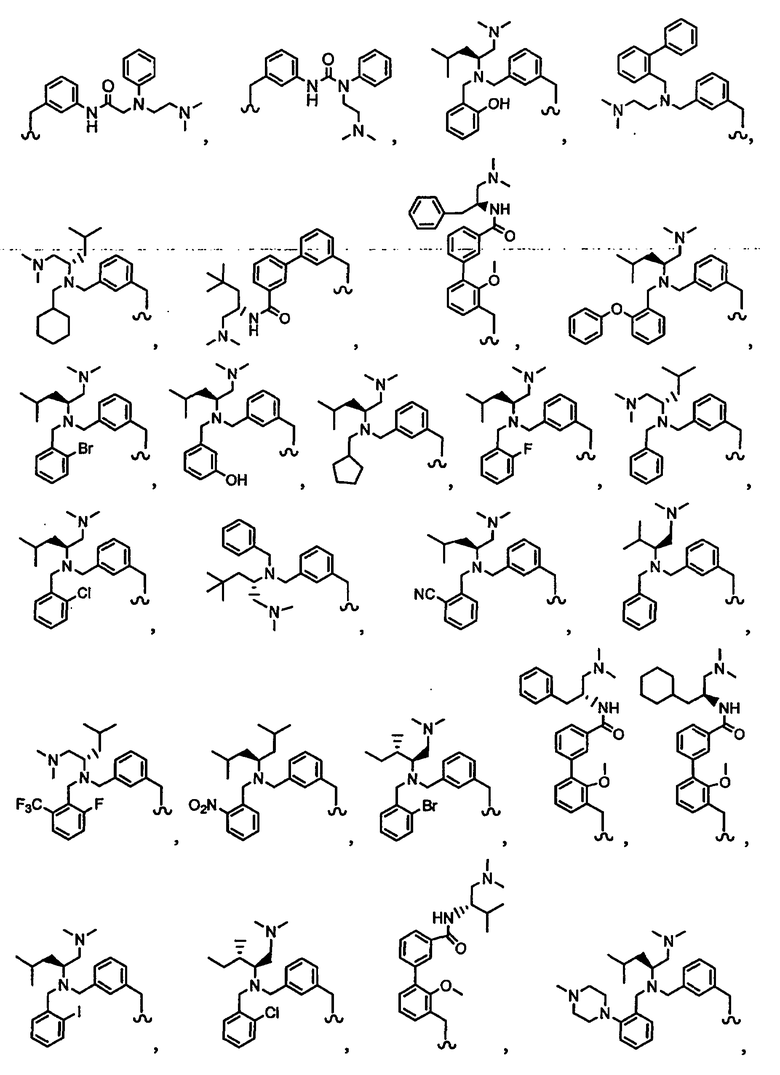
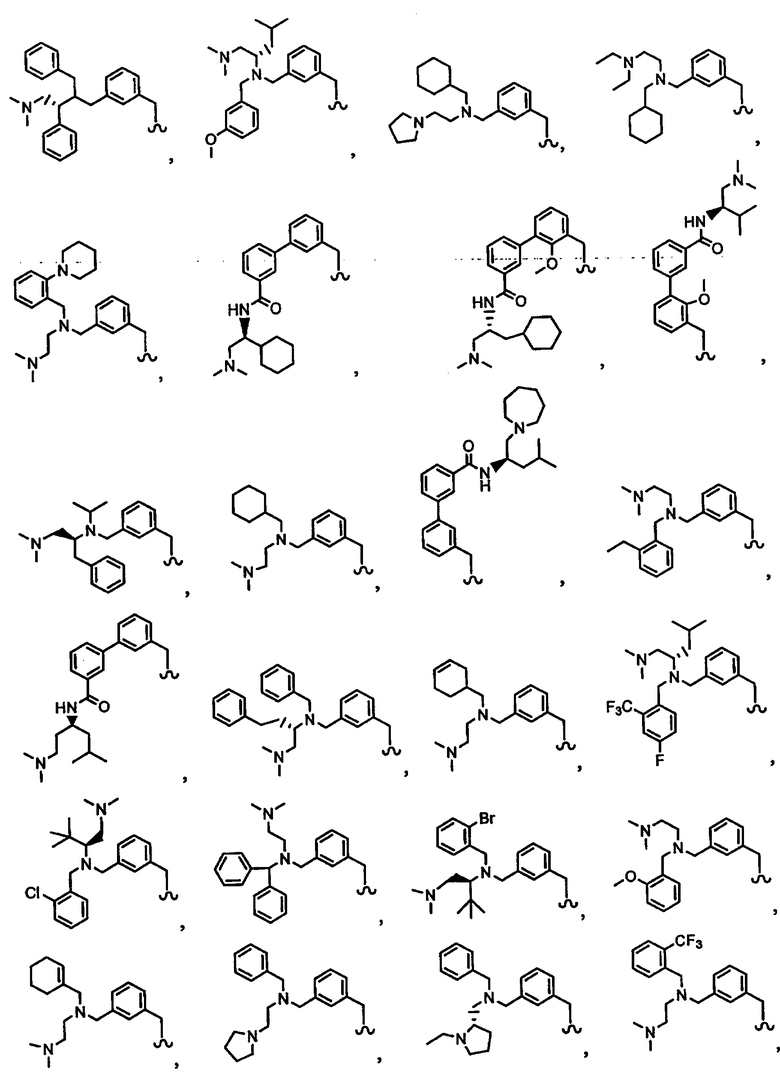
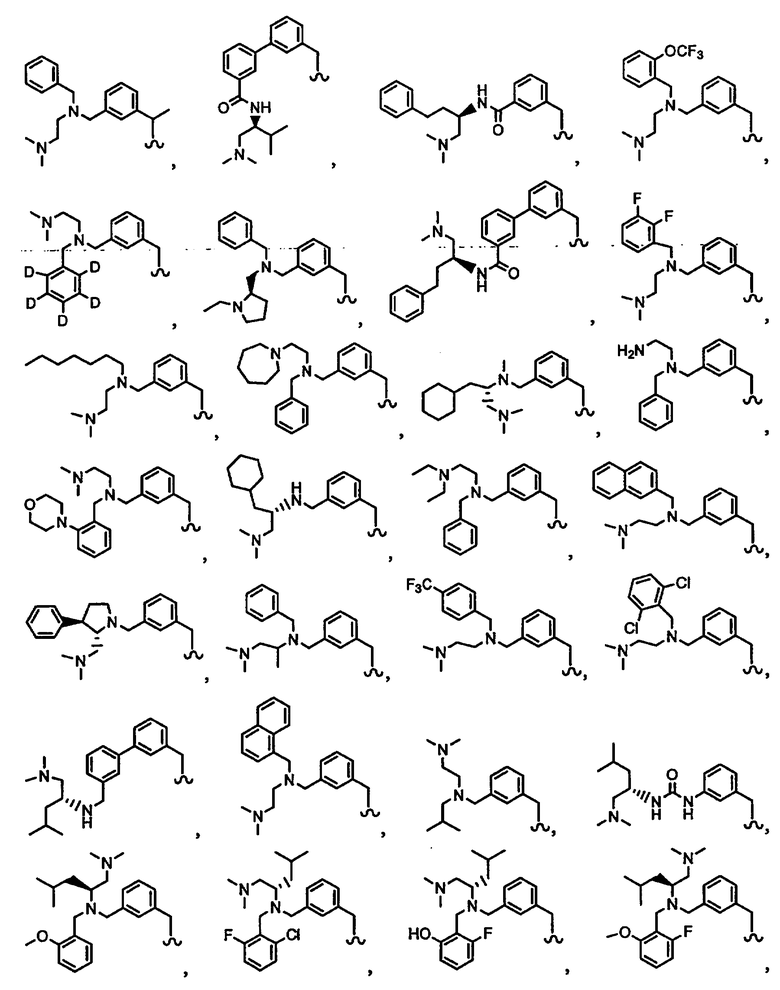
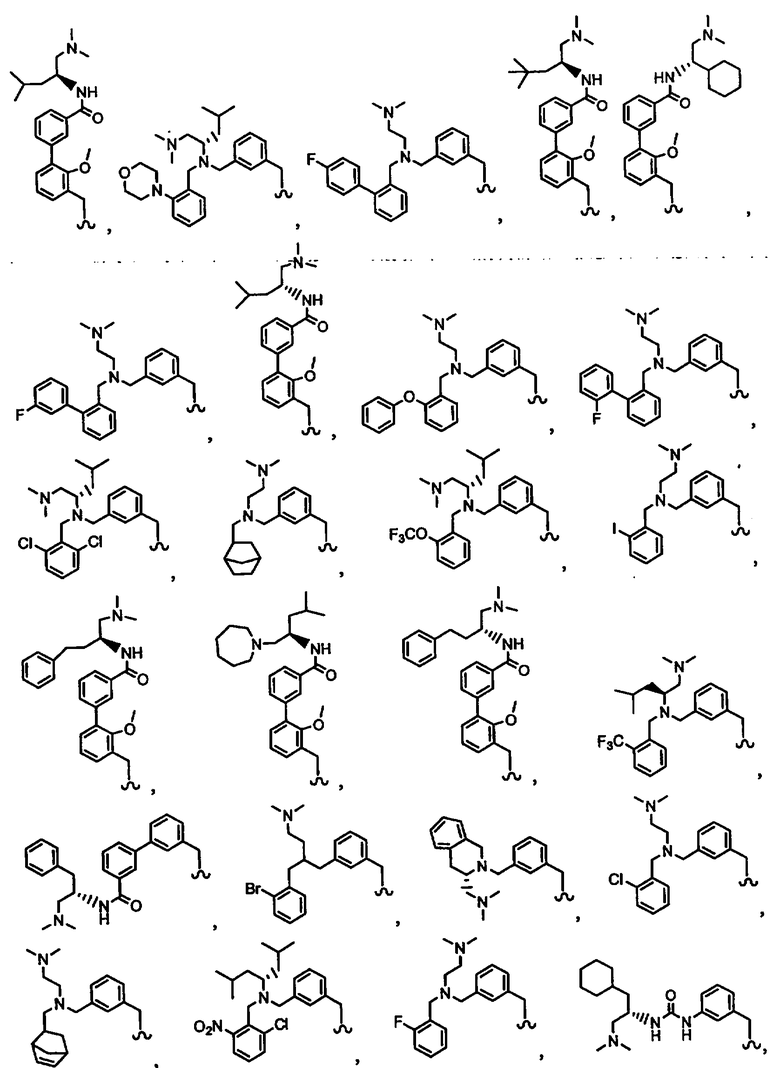
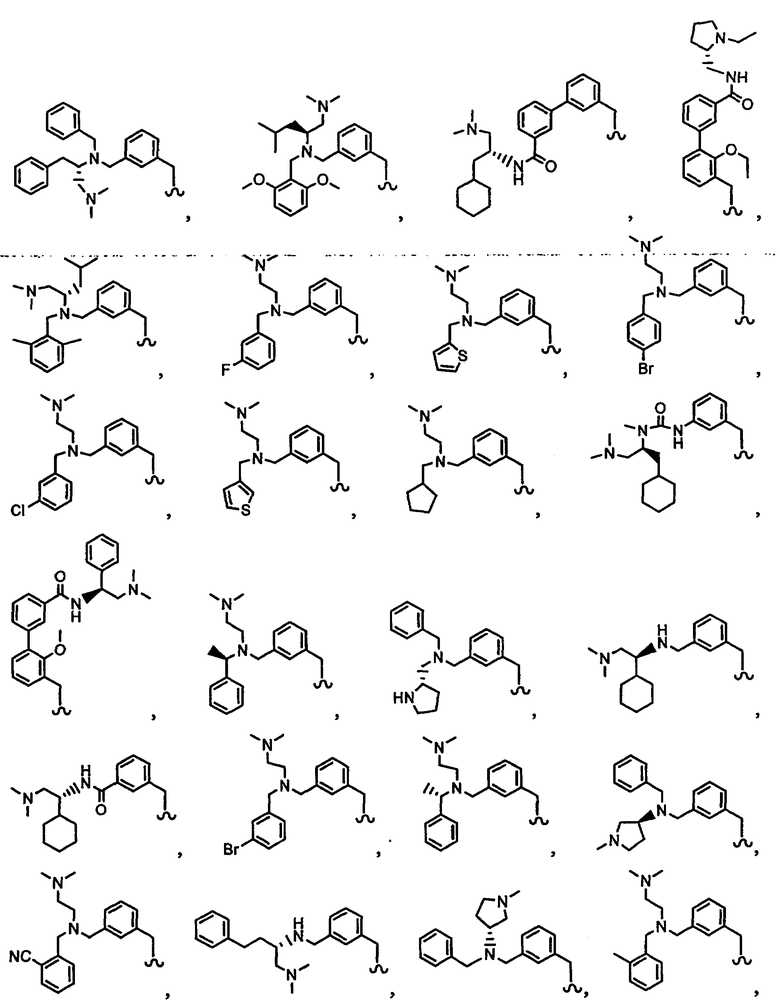
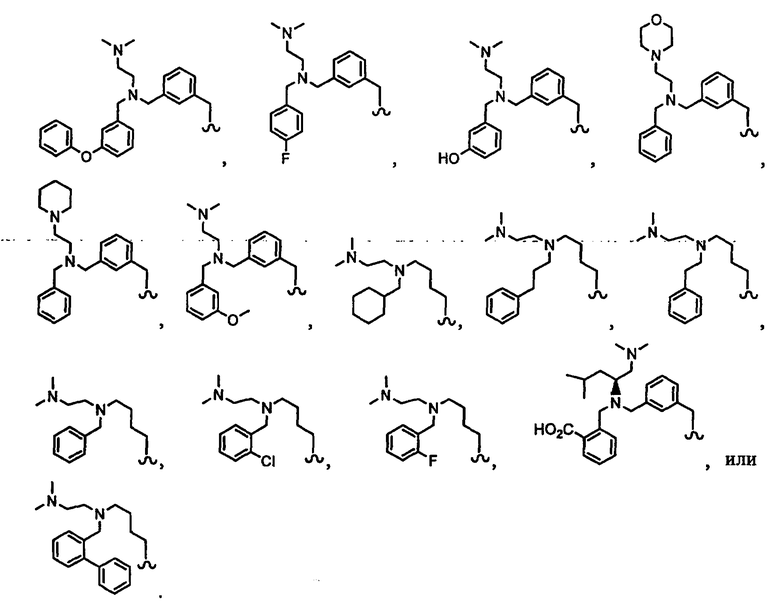
В другом варианте осуществления настоящего изобретения предлагается соединение, выбранное из группы, включающей следующие соединения
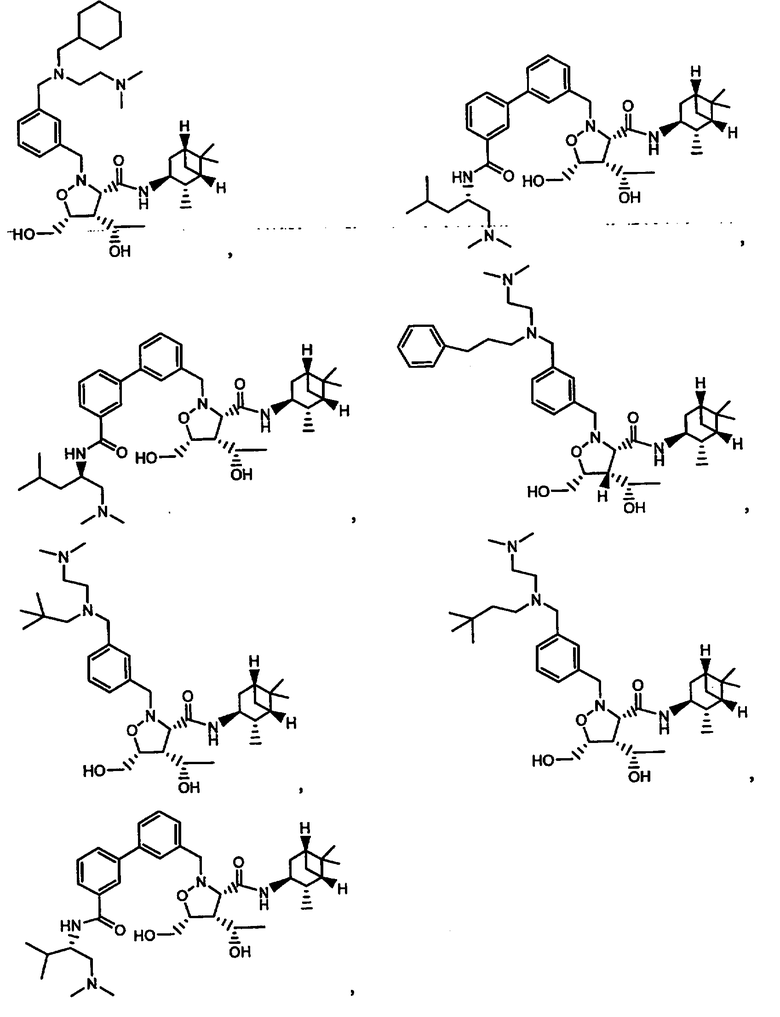
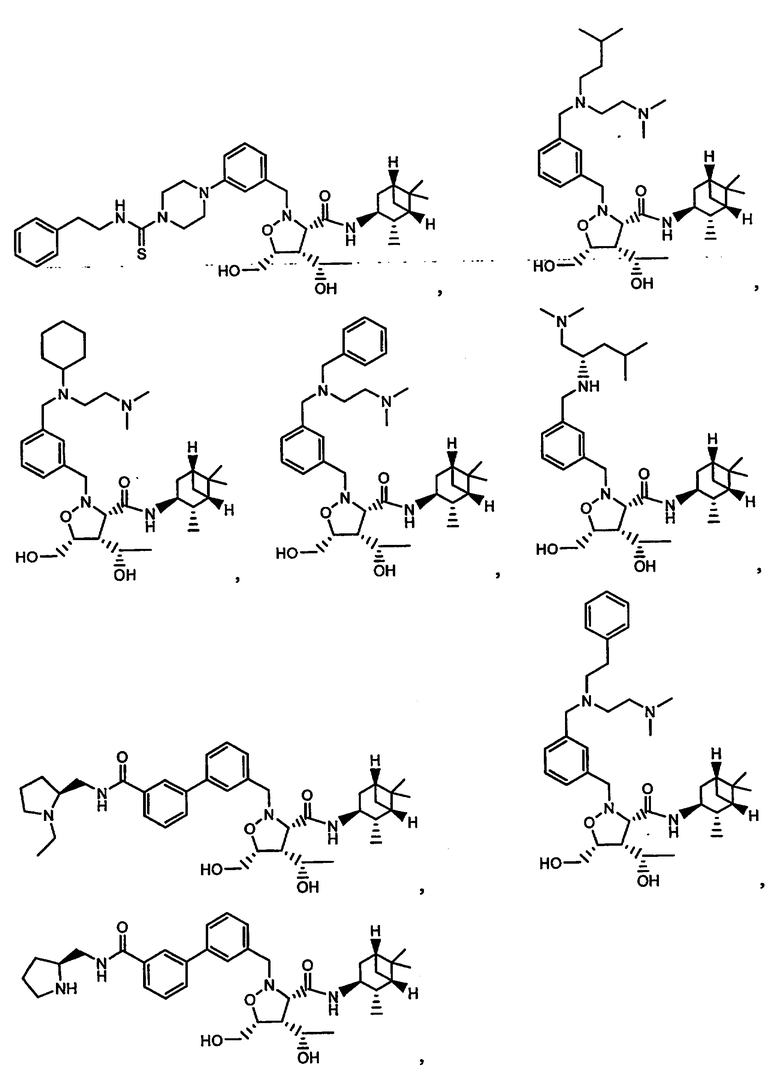
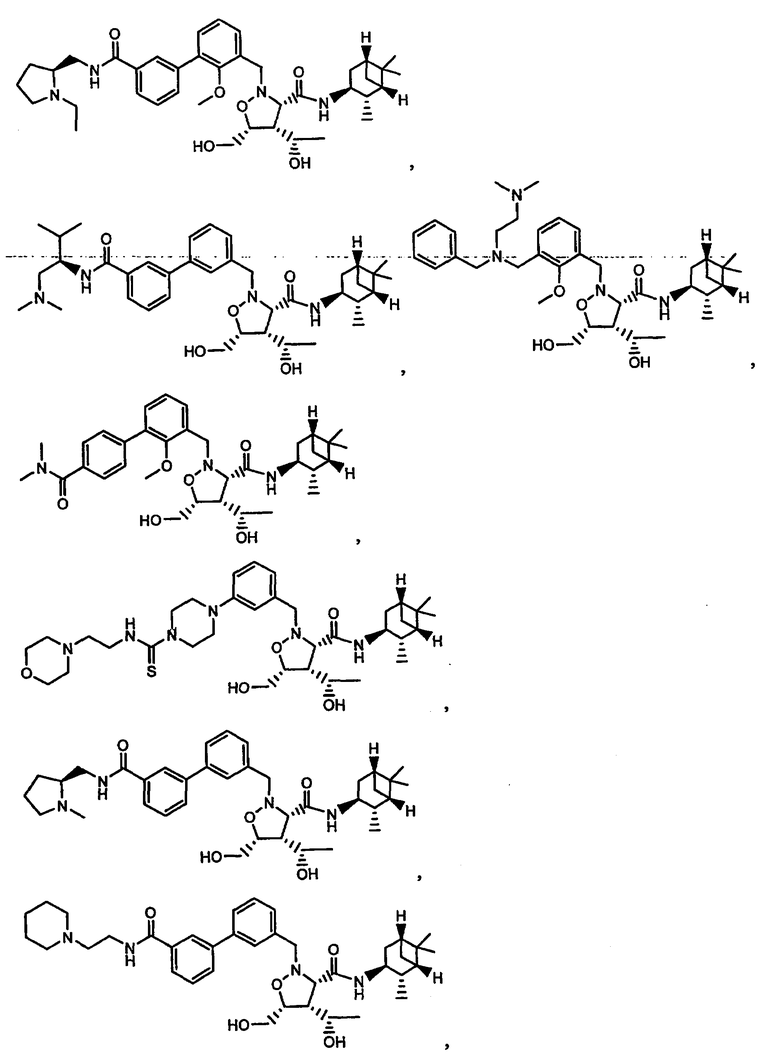
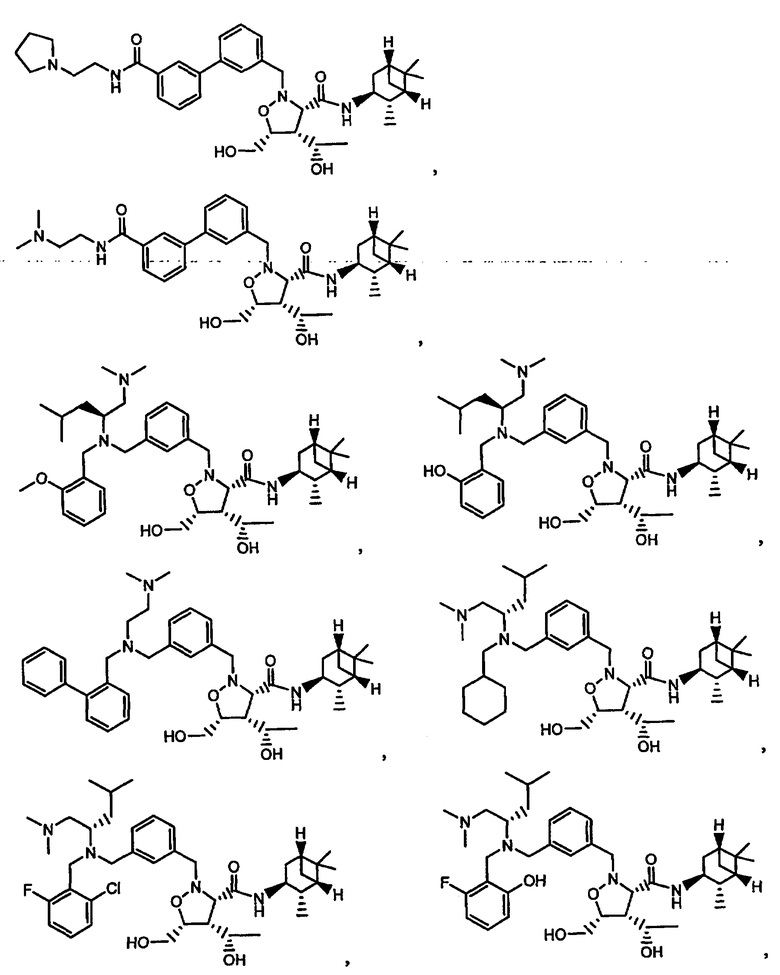
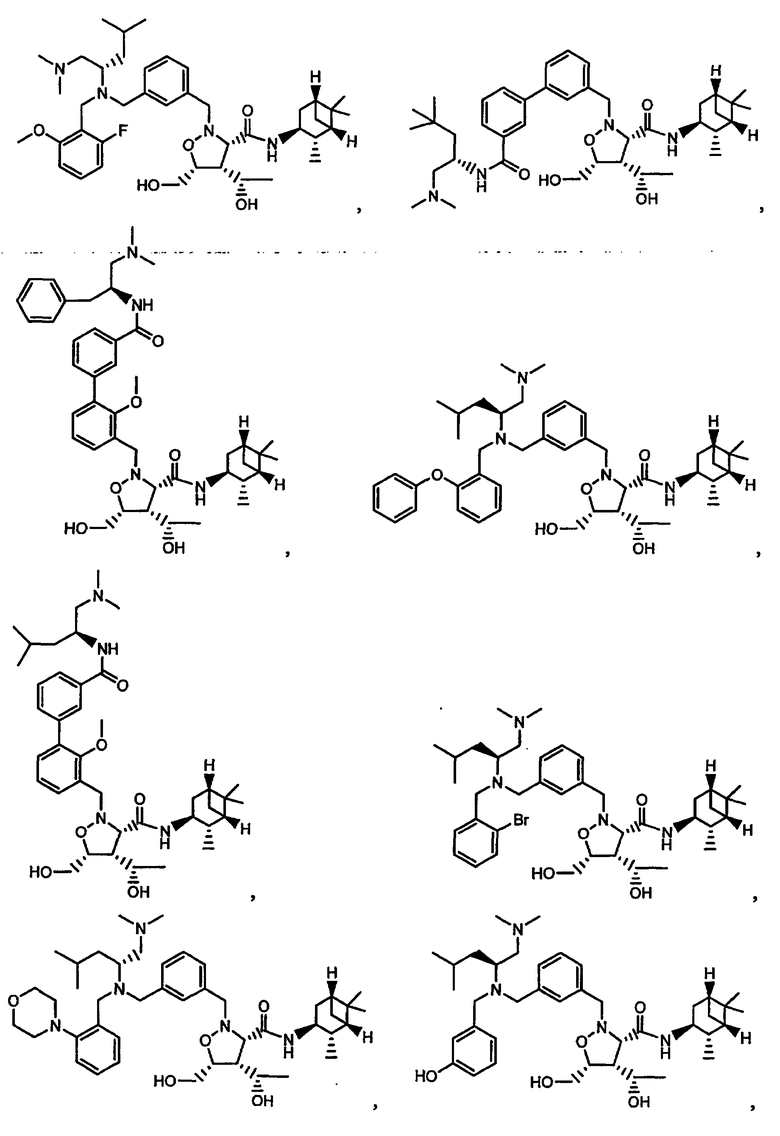
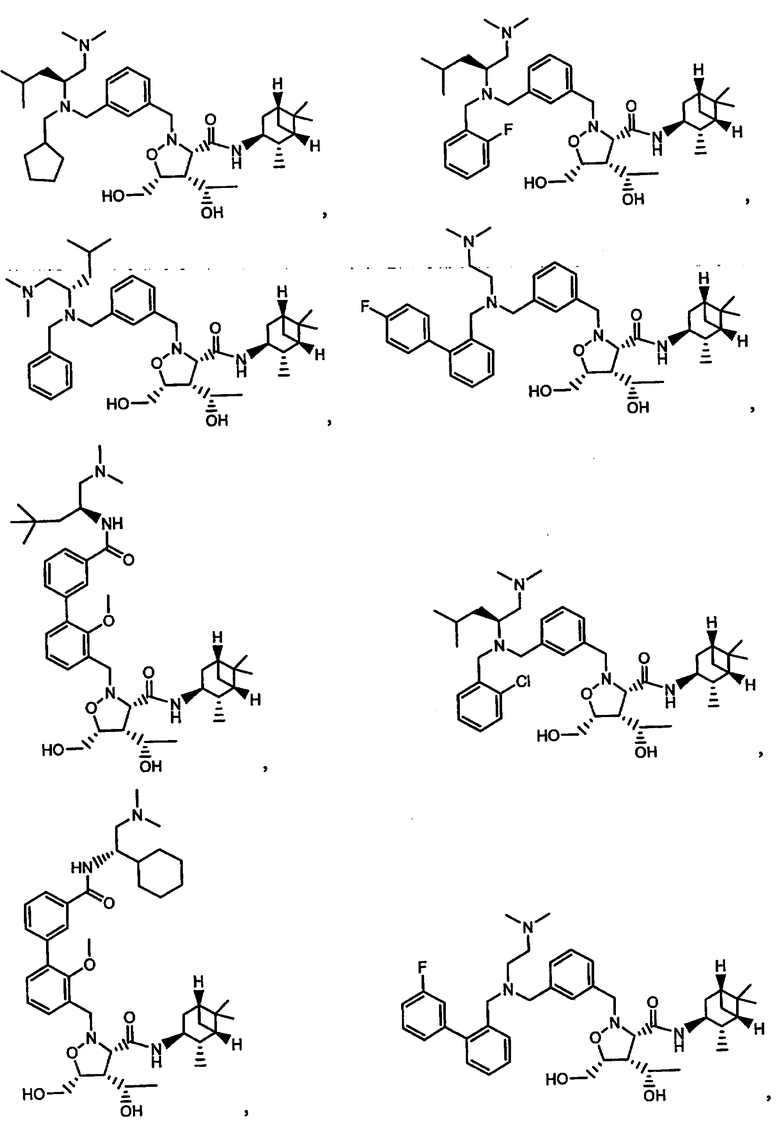
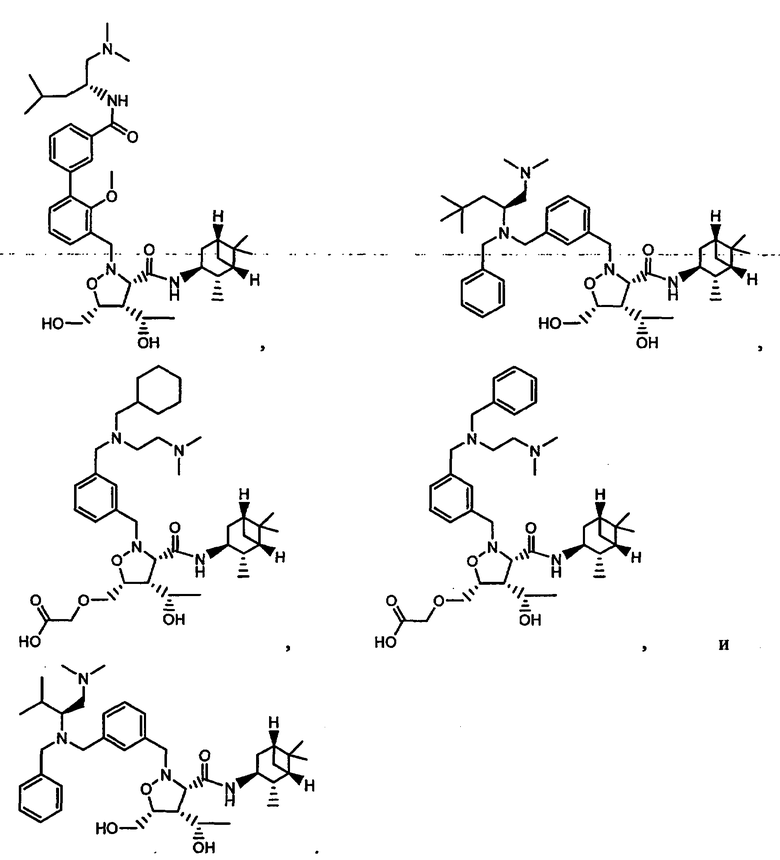
В другом объекте настоящего изобретения предлагается фармацевтическая композиция, включающая соединение формулы 1, 2, 3, 4 или 5, описанное выше, и по крайней мере один фармацевтически приемлемый экципиент.
Способы по настоящему изобретению
В одном объекте настоящего изобретения предлагается способ лечения нарушений, опосредованных Bcl, включающий стадию введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы 1, 2, 3, 4 или 5, описанного выше.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором нарушение, опосредованное Bcl, означает рак или новообразование, которые выбирают из группы, включающей острый лейкоз, острый лимфолейкоз, острый миелоизный лейкоз, миелобластный, промиелоцитарный, миеломоноцитарный, моноцитарный и эритролейкоз, хронический лейкоз, хронический миелоцитарный (гранулоцитарный) лейкоз, хронический лимфоцитарный лейкоз, истинную полицитемию, болезнь Ходжкина, болезнь не-Ходжкина, множественную миелому, болезнь Вальденстрема, болезнь тяжелых цепей, фибросаркому, миксосаркому, липосаркому, хондросаркому, остеогенную саркому, хордому, ангиосаркому, эндотелиосаркому, лимфангиосаркому, лимфангиоэндотелиальную саркому, синовиому, мезотелиому, опухоль Юинга, лейомиосаркому, рабдомиосаркому, карциному ободочной кишки, рак поджелудочной железы, рак молочной железы, рак яичников, рак предстательной железы, плоскоклеточную карциному, базально-клеточный рак, аденокарциному, карциному потовой железы, карциному сальной железы, папиллярную карциному, папиллярные аденокарциномы, стаденокарциному, медуллярный рак, карциному бронхов, рак почки, гепатому, рак желчного протока, хориокарциному, семиному, эмбриональный рак, опухоль Вильмса, рак шейки матки, рак матки, рак семенников, карциному легкого, мелкоклеточный рак легкого, тестикулярный рак, эпителиальную карциному, глиому, астроцитому, медуллобластому, краниофарингиому, эпендимому, пинеалому, гемангиобластому, акустическую неврому, олигодендроглиому, менингиому, меланому, нейробластому, ретинобластому и эндометриальный рак.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный рак выбирают из группы, включающей фолликулярную лимфому, диффузную крупноклеточную В-лимфому, лимфому коры головного мозга, хронический лимфолейкоз, рак предстательной железы, рак молочной железы, нейробластому, колорекстальный рак, рак эндометрия, яичников и легкого, злокачественную гепатому, множественную миелому, рак головы и шеи или тестикулярный рак.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный рак опосредован сверхэкспрессией белка Bcl.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором степень прогрессирования рака зависит от белков Bcl.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный белок Bcl означает белок Bcl-2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный белок Bcl означает белок Bcl-xL.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором рак связан с хромосомальной транслокацией t (14,18).
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанное соединение вводят парентеральным способом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанное соединение вводят внутримышечным, внутривенным, подкожным, пероральным, внутрилегочным, интратекальным, местным или интраназальным способом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанное соединение вводят системным способом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный пациент является млекопитающим.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный пациент является приматом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный пациент является человеком.
В другом варианте осуществления настоящего изобретения предлагается способ лечения нарушений, ассоциированных с Bcl, включающий стадию введения пациенту терапевтически эффективного количества химиотерапевтического агента в комбинации с терапевтически эффективным количеством соединения формулы 1, 2, 3, 4 или 5, описанного выше.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанные нарушения, ассоциированные с Bcl, означают рак или новоообразование.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный рак или новообразование выбирают из группы, включающей острый лейкоз, острый лимфолейкоз, острый миелоизный лейкоз, миелобластный, промиелоцитарный, миеломоноцитарный, моноцитарный и эритролейкоз, хронический лейкоз, хронический миелоцитарный (гранулоцитарный) лейкоз, хронический лимфоцитарный лейкоз, истинную полицитемию, болезнь Ходжкина, болезнь не-Ходжкина, множественную миелому, болезнь Вальденстрема, болезнь тяжелых цепей, фибросаркому, миксосаркому, липосаркому, хондросаркому, остеогенную саркому, хордому, ангиосаркому, эндотелиосаркому, лимфангиосаркому, лимфангиоэндотелиальную саркому, синовиому, мезотелиому, опухоль Юинга, лейомиосаркому, рабдомиосаркому, карциному ободочной кишки, рак поджелудочной железы, рак молочной железы, рак яичников, рак предстательной железы, плоскоклеточную карциному, базально-клеточный рак, аденокарциному, карциному потовой железы, карциному сальной железы, папиллярную карциному, папиллярные аденокарциномы, стаденокарциному, медуллярный рак, карциному бронхов, рак почки, гепатому, рак желчного протока, хориокарциному, семиному, эмбриональный рак, опухоль Вильмса, рак шейки матки, рак матки, рак семенников, карциному легкого, мелкоклеточный рак легкого, тестикулярный рак, эпителиальную карциному, глиому, астроцитому, медуллобластому, краниофарингиому, эпендимому, пинеалому, гемангиобластому, акустическую неврому, олигодендроглиому, менингиому, меланому, нейробластому, ретинобластому и рак эндометрия.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный рак опосредован сверхэкспрессией белка Bcl.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором степень прогрессирования рака зависит от белков Bcl.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный белок Bcl означает белок Bcl-2.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный белок Bcl означает белок Bcl-xL.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором рак связан с хромосомальной транслокацией t (14,18).
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором количество соединения формулы 1, 2, 3, 4 или 5 является достаточным для снижения уровня клиентных белков Bcl в клетках, а количество указанного химиотерапевтического агента является достаточным для эффективного ингибирования указанных клиентных белков Bcl.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанное соединение вводят парентеральным способом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанное соединение вводят внутримышечным, внутривенным, подкожным, пероральным, внутрилегочным, интратекальным, местным или интраназальным способом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанное соединение вводят системным способом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный пациент является млекопитающим.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный пациент является приматом.
В некоторых вариантах осуществления настоящего изобретения предлагается указанный выше способ, в котором указанный пациент является человеком.
Фармацевтические композиции
В другом объекте осуществления настоящего изобретения предлагаются фармацевтически приемлемые композиции, которые включают терапевтически эффективное количество одного или более соединений, описанных выше, в смеси с одним или более фармацевтически приемлемых носителей (добавок) и/или разбавителей. Фармацевтические композиции по настоящему изобретению, как подробно описано ниже, получают в твердых или жидких формах для введения пациенту, которые предназначены: (1) для перорального способа введения лекарственного средства, например микстуры (водные или неводные растворы или суспензии), таблетки, которые вводят, например, трансбуккальным или подъязычным и системным способами, пилюли, порошки, гранулы и пасты, которые наносят на язык, (2) для парентерального способа введения лекарственного средства, например, с использованием подкожной, внутримышечной, внутривенной или энидуральной инъекции, например стерильный раствор или суспензия, или состав для замедленного высвобождения лекарственного средства, (3) для местного применения лекарственного средства, например крем, мазь или пластырь для контролируемого высвобождения лекарственного средства или спрей для кожного применения, (4) для внутривлагалищного или внутриректального способа введения лекарственного средства, например пессарий, крем или пена, (5) для подъязычного способа введения лекарственного средства, (6) для глазного способа введения лекарственного средства, (7) для чрескожного способа введения лекарственного средства, (8) для интраназального способа введения лекарственного средства, (9) для внутрилегочного способа введения лекарственного средства или (10) для интратекального способа введения лекарственного средства.
Термин «терапевтически эффективное количество», используемый в настоящем описании, означает количество соединения, материала или композиции, включающей соединение по настоящему изобретению, которое является достаточным для обеспечения требуемого терапевтического действия по крайней мере на определенную популяцию клеток в организме животного при приемлемом соотношении польза/риск, для использования в любой области медицины.
Термин «фармацевтически приемлемый», используемый в настоящем описании, относится к соединениям, материалам, композициям и/или лекарственным формам, известным в данной области медицины, и которые являются пригодными для контактирования с тканями организма человека, не проявляют высокой токсичности и не вызывают раздражения, аллергической реакции или другого действия или осложнения при приемлемом соотношении польза/риск.
Термин «фармацевтически приемлемый носитель», используемый в настоящем описании, означает фармацевтически приемлемый материал, композицию или наполнитель, такие как жидкий или твердый наполнитель, разбавитель, экципиент, технологическое вспомогательное вещество (например, замасливатель, тальк, стеарат магния, кальция или цинка или стеариновую кислоту) или растворитель, или инкапсулирующий материал для переноса или транспортировки соединения от одного органа или части тела к другому органу или части тела. Каждый носитель является «приемлемым», т.е. совместимым с другими ингредиентами лекарственного средства и не оказывает отрицательного действия на пациента. Некоторые примеры материалов, которые являются фармацевтически приемлемыми носителями, включают: (1) сахара, такие как лактоза, глюкоза и сахароза, (2) крахмалы, такие как кукурузный и картофельный крахмал, (3) целлюлоза и ее производные, такие как натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза и ацетилцеллюлоза, (4) трагакант в виде порошка, (5) солод, (6) желатин, (7) тальк, (8) экципиенты, такие как масло какао и воски для суппозитория, (9) масла, такие как арахисовое, хлопковое, сафлоровое, кунжутное, оливковое, кукурузное и соевое масло, (10) гликоли, такие как пропиленгликоль, (11) многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль, (12) сложные эфиры, такие как этилолеат и этиллаурат, (13) агар, (14) буферные вещества, такие как гидроксид магния и алюминия, (15) альгиновую кислоту, (16) апирогенную воду, (17) изотонический раствор, (18) раствор Рингера, (19) этиловый спирт, (20) буферные растворы с определенным рН, (21) полиэфиры, поликарбонаты и/или полиангидриды, и (22) другие нетоксичные совместимые соединения, используемые в фармацевтических композициях.
В некоторых вариантах осуществления настоящего изобретения предлагаются указанные выше соединения, которые содержат основную функциональную группу, такую как амино или алкиламино, и, таким образом, способны образовывать фармацевтически приемлемые соли фармацевтически приемлемых кислот. Термин «фармацевтически приемлемые соли» означает относительно нетоксичные неорганические и органические кислотно-аддитивные соли соединений по настоящему изобретению. Указанные соли получают in situ в процессе получения лекарственной формы или при добавлении наполнителя или вспомогательного вещества, или при взаимодействии очищенного соединения по настоящему изобретению в свободной основной форме с пригодной органической или неорганической кислотой и с последующим выделением полученной соли с последующей очисткой. Примеры солей включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфонат и т.п. (см., например, статью Berge и др. "Pharmaceutical Salts", J. Pharm. Sci., 66: 1-19 (1977)).
Фармацевтически приемлемые соли соединений по настоящему изобретению включают стандартные нетоксичные соли или четвертичные аммониевые соли соединений, например нетоксичных органических или неорганических кислот. Например, такие стандартные нетоксичные соли включают соли неорганических кислот, такие как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная кислота и т.п., и соли органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, оксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изотионовая кислота и т.п.
В других случаях соединения по настоящему изобретению включают одну или более кислотных функциональных групп и, таким образом, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин «фармацевтически приемлемые соли», относящийся к таким соединениям, означает относительно нетоксичные, неорганические и органические основно-аддитивные соли соединений по настоящему изобретению. Такие соли также получают in situ в процессе получения лекарственной формы или при добавлении наполнителя или вспомогательного вещества, или при взаимодействии очищенного соединения по настоящему изобретению в свободной кислотной форме с пригодным основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Примеры солей щелочных или щелочно-земельных металлов включают соли лития, натрия, калия, кальция, магния, алюминия и т.п. Примеры органических аминов, пригодных для получения основно-аддитивных солей, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и т.п. (см. выше, например, статью Berge и др.).
Композиции также включают смачивающие агенты, эмульгаторы и замасливатели, такие как лаурилсульфат натрия и стеарат магния, и красители, антиадгезивы, покровный материал, подсластители, ароматизаторы и вкусовые добавки, консерванты и антиоксиданты.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п., (2) жирорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и т.п. и (3) агенты, образующие хелаты с металлами, такие как лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТУ), сорбит, винная кислота, фосфорная кислота и т.п.
Лекарственные средства по настоящему изобретению включают лекарственные средства, пригодные для перорального, назального, местного (включая трансбуккальный и подъязычный), ректального, вагинального и/или парентерального способа введения лекарственного средства. Лекарственные средства применяют в стандартной лекарственной форме и получают любыми методами, известными в данной области техники. Количество активного ингредиента, включенного в композицию в смеси с носителем, для получения стандартной лекарственной формы изменяется и зависит от состояния пациента, проходящего курс лечения, и способа введения лекарственного средства. Количество активного ингредиента, включенного в композицию в смеси с носителем, для получения стандартной лекарственной формы равно, как правило, количеству соединения, достаточному для обеспечения терапевтического действия. Как правило, указанное количество активного ингредиента составляет от приблизительно 0,1% до приблизительно 99%, предпочтительно от приблизительно 5% до приблизительно 70%, наиболее предпочтительно от приблизительно 10% до приблизительно 30%.
В некоторых вариантах осуществления настоящего изобретения предлагается лекарственное средство по настоящему изобретению, которое включает экципиент, выбранный из группы, включающей циклодекстрины, целлюлозы, липосомы, мицеллообразующие агенты, такие как, например, желчные кислоты и полимерные носители, такие как, например, сложные полиэфиры и полиангидриды, и соединения по настоящему изобретению. В некоторых вариантах осуществления настоящего изобретения указанный выше состав обеспечивает пероральную биодоступность соединения по настоящему изобретению.
Способы получения указанных лекарственных средств или композиций включают стадию смешивания соединения по настоящему изобретению с носителем и, необязательно, одним или более пригодными ингредиентами. Как правило, лекарственные средства получают непосредственно при смешивании соединения по настоящему изобретению с жидкими носителями или тонкоизмельченными твердыми носителями, или со смесью указанных носителей, затем при необходимости продукт перерабатывают в лекарственную форму.
Лекарственные средства по настоящему изобретению, пригодные для перорального введения, применяют в виде капсул, облаток, пилюль, таблеток, лепешек (включающих ароматизаторы, как правило, сахарозу и камедь или трагакант), порошков, гранул или раствора или суспензии в водной или неводной жидкости, или жидкой эмульсии масло-в-воде или вода-в-масле, или эликсира или сиропа, или пастилок (включающих инертный носитель, такой как желатин и глицерин, или сахароза и камедь) и/или жидкостей для полоскания ротовой полости и т.п., включающих определенное количество соединения по настоящему изобретению, которое является активным ингредиентом. Соединения по настоящему изобретению также вводят в форме болюсов, электуария или пасты.
Для получения твердых стандартных лекарственных форм по настоящему изобретению для перорального введения (таких как капсулы, таблетки, пилюли, драже, порошки, гранулы и т.п.), активный ингредиент смешивают с одним или более фармацевтически приемлемыми носителями, такими как цитрат натрия или дикальцийфосфат и/или любыми другими носителями, такими как (1) наполнители или сухие разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота, (2) связующие агенты, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или камедь, (3) увлажнители, такие как глицерин, (4) дезинтегрирующие агенты, такие как агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, (5) агенты, замедляющие растворение, такие как парафин, (6) ускорители впитывания, такие как четвертичные аммониевые основания и ПАВ, такие как полоксамер и лаурилсульфат натрия, (7) смачивающие агенты, такие как, например, цетиловый спирт, моностеарат глицерина и неионогенные ПАВ, (8) абсорбирующие агенты, такие как каолин и бентонитовая глина, (9) замасливатели, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, стеарат цинка, стеарат натрия, стеариновая кислота и их смеси, (10) красители и (11) агенты, предназначенные для контролируемого высвобождения активного ингредиента, такие как кросповидон или этилцеллюлоза. При применении капсул, таблеток и пилюль фармацевтические композиции также включают буферные вещества. Аналогичные твердые композиции также используют в качестве наполнителей в мягких и твердых желатиновых капсулах, включающих экципиенты, такие как лактоза или молочные сахара и высокомолекулярные полиэтиленгликоли и т.п.
Таблетку получают формованием или прессованием необязательно в смеси с одним или более вспомогательными веществами. Формованные таблетки получают при добавлении связующего агента (такого как, например, желатин или гидроксипропилметилцеллюлоза), замасливателя, инертного разбавителя, консерванта, дезинтегрирующего агента (такого как, например, натриевая соль крахмалгликолята или сшитая натриевая соль карбоксиметилцеллюлозы), ПАВ или диспергирующего агента. Прессованные таблетки получают прессованием в пригодном аппарате при смешивании соединения в виде порошка с увлажняющим инертным жидким разбавителем.
Таблетки и другие твердые стандартные лекарственные формы фармацевтических композиций по настоящему изобретению, такие как драже, капсулы, пилюли и гранулы, получают необязательно нанесением покрытий, таких как энтеросолюбильные покрытия и другие покрытия, известные в фармацевтике. Таблетки и другие твердые стандартные лекарственные формы также получают в формах, предназначенных для замедленного или контролируемого высвобождения активного ингредиента при добавлении, например, гидроксипропилметилцеллюлозы, в различных соотношениях, других полимерных матриц, липосом и/или микрогранул. Формы, предназначенные для быстрого высвобождения активного ингредиента, получают, например, при лиофилизации. Указанные формы также стерилизуют, например фильтрацией через фильтр, удерживающий бактерии, или при добавлении стерилизующих агентов в форме стерильных твердых композиций, которые растворяют в стерильной воде или другом стерильном растворителе непосредственно перед применением. Такие композиции также необязательно включают опалесцирующие агенты и композиции, предназначенные для высвобождения активного ингредиента(ов) предпочтительно только в определенном отделе желудочно-кишечного тракта необязательно с замедленным высвобождением активного ингредиента. Примеры используемых инкапсулированных композиций включают полимерные соединения и воски. Активный ингредиент можно получать также в виде микрокапсул, содержащих один или более экципиентов, описанных выше.
Жидкие стандартные лекарственные формы для перорального введения соединений по настоящему изобретению включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие стандартные лекарственные формы кроме активного ингредиента включают инертные разбавители, как правило, используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый и изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (прежде всего, хлопковое, арахисовое, кукурузное, из зародышей пшеницы, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбита и их смеси.
Композиции для перорального применения, кроме инертных разбавителей, также включают экципиенты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы, красители, вкусовые добавки и консерванты.
Суспензии, кроме активных соединений, включают суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар, трагакант и их смеси.
Лекарственные средства, включающие фармацевтические композиции по настоящему изобретению, для ректального или вагинального способа введения применяют в форме суппозиториев, которые получают смешиванием одного или более соединений по настоящему изобретению с одним или более пригодных экципиентов, не вызывающих раздражение, или носителями, включающими, например, масло какао, полиэтиленгликоль, воски для суппозиториев или салицилат, которые являются твердыми веществами при комнатной температуре и жидкостями при температуре тела и, таким образом, расплавляются в прямой кишке или влагалище с высвобождением активного соединения.
Лекарственные средства по настоящему изобретению, которые являются пригодными для вагинального способа введения, также включают пессарии, тампоны, кремы, гели, пасты, пены или аэрозоли, содержащие носители, известные в данной области техники.
Стандартные лекарственные формы для местного или чрескожного способа введения включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и лекарственные формы для ингаляции. Активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми требуемыми консервантами, буферными веществами или пропелентами.
Мази, пасты, кремы и гели содержат, кроме активного соединения по настоящему изобретению, экципиенты, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, кремнийорганические материалы, бентониты, кремниевая кислота, тальк, оксид цинка или их смеси.
Порошки и спреи содержат, кроме соединения по настоящему изобретению, экципиенты, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция, полиамидные порошки и их смеси. Спреи, кроме того, содержат обычные пропеленты, такие как хлорфторуглеводороды и летучие незамещенные углеводороды, такие как бутан и пропан.
Пластыри для чрескожного способа введения применяют для контролируемого способа введения соединения по настоящему изобретению в организм пациента. Такие стандартные лекарственные формы получают растворением или диспергированием соединения в пригодном носителе. Для повышения проницаемости соединения через кожу используют также усиливающие абсорбцию агенты. Скорость такой проницаемости соединения контролируют с использованием мембраны, регулирующей скорость прохождения соединения через кожу, или диспергированием соединения в полимерной матрице или геле.
Глазные лекарственные средства, глазные мази, порошки, растворы и т.п. также включены в объем настоящего изобретения.
Фармацевтические композиции по настоящему изобретению, пригодные для парентерального способа введения, включают одно или более соединений по настоящему изобретению в комбинации с одним или более фармацевтических приемлемых стерильных изотонических водных или неводных растворов, дисперсий, суспензий или эмульсий, или стерильных порошков, из которых перед применением получают стерильные инъекционные растворы или дисперсии, и которые содержат сахара, спирты, антиоксиданты, буферные вещества, антимикробные добавки, изотонические растворы, совместимые с кровью пациента, или суспендирующие агенты, или загустители.
Примеры пригодных водных и неводных носителей, включенных в фармацевтические композиции по настоящему изобретению, включают воду, этанол, многоатомные спирты (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.) и их смеси, растительные масла, такие как оливковое масло, и инъекционные органические сложные эфиры, таких как этилолеат. Требуемую текучесть фармацевтических композиций обеспечивают, например, применением покровных материалов, таких как лецитин, а также при получении частиц требуемого размера в случае дисперсий и при использовании ПАВ.
Указанные композиции также содержат адъюванты, такие как консерванты, смачивающие агенты, эмульгаторы и диспергирующие агенты. Воздействие микроорганизмов на активные соединения предотвращают при добавлении различных антибактериальных и противогрибковых агентов, например парабена, хлорбутанола, фенолсорбиновой кислоты и т.п. При необходимости в фармацевтические композиции также добавляют изотонические агенты, такие как сахара, хлорид натрия и т.п. Кроме того, замедленную абсорбцию вводимой лекарственной формы обеспечивают включением агентов для замедленной абсорбции, таких как моностеарат алюминия и желатин.
В некоторых случаях для замедленного действия лекарственного средства требуется замедлить доставку лекарственного средства в организм при подкожной или внутримышечной инъекции. Для замедленной доставки лекарственного средства в организм в фармацевтические композиции включают жидкие суспензии кристаллического или аморфного материала, малорастворимого в воде. Скорость доставки лекарственного средства в организм зависит от скорости растворения лекарственного средства, которая, в свою очередь, зависит от размера и формы кристаллов. В другом варианте замедленную доставку лекарственной формы, которую вводят парентерально, обеспечивают растворением или суспендированием лекарственного средства в масле.
Формы для инъекции веществ замедленного всасывания получают при образовании микрокапсулированных матриц активных соединений в биодеградабельных полимерах, таких как полилактид-полигликолид. Скорость доставки лекарственного средства регулируют изменением соотношения количества лекарственного средства и полимера и природы используемого полимера. Примеры биодеградабельных полимеров включают поли(ортосложные эфиры) и поли(ангидриды). Формы для инъекции веществ замедленного всасывания также получают включением лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями организма.
Если соединения по настоящему изобретению вводят в виде лекарственных средств человеку и животным, то соединения по настоящему изобретению вводят сами по себе или в виде фармацевтической композиции, содержащей, например, от 0,1% до 99% (более предпочтительно от 10% до 30%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Полученные лекарственные средства по настоящему изобретению вводят перорально, парентерально, местно или ректально. Лекарственные средства по настоящему изобретению вводят в формах, пригодных для определенного способа введения, например в форме таблеток или капсул, инъекцией, ингаляцией, глазных растворов, мазей, суппозиториев и т.п., при этом лекарственные средства вводят с использованием инъекции, вливания или ингаляции, при местном способе введения используют лосьон или мазь, а при ректальном способе введения используют суппозитории. Пероральные способы введения лекарственного средства являются предпочтительными.
Термины «парентеральный способ введения» и «вводят парентерально», используемые в настоящем описании, означают способы введения лекарственного средства, в отличие от энтерального и местного способа введения, обычно с использованием инъекции, и включают, без ограничения перечисленным, внутривенную, внутримышечную, внутриартериальную, интратекальную, интракапсулярную, внутриглазничную, внутрисердечную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, внутрикожную, внутрисуставную, внутриоболочечную, субарахноидальную, интраспинальную, интрастернальную инъекцию и вливание.
Термины «системное введение» «системный способ введения», «периферическое введение» и «периферический способ введения», используемые в настоящем описании, означают способ введения соединения, лекарственного средства или другого материала (в отличие от способа введения непосредственно в центральную нервную систему), при котором лекарственное средство вводят в общую систему организма пациента и, таким образом, лекарственное средство подвергается метаболизму и другим процессам в организме, например, при подкожном способе введения.
Указанные соединения можно вводить человеку и другим животным для лечения любыми пригодными способами введения, включающими пероральный, интраназальный, такой как, например, с помощью спрея, ректальный, внутривлагалищный, парентеральный, интрацистернальный и местный способ введения в форме порошков, мазей или капель, а также трансбуккальный и подъязычный способ.
Независимо от выбранного способа введения лекарственного средства соединения по настоящему изобретению, которые применяют в пригодной гидратированной форме, и/или фармацевтические композиции по настоящему изобретению перерабатывают в фармацевтически приемлемые стандартные лекарственные формы стандартными способами, известными в данной области техники.
Реальный уровень дозы активных ингредиентов в фармацевтических композициях по настоящему изобретению изменяется в зависимости от эффективного количества активного ингредиента, достаточного для обеспечения терапевтического действия на определенного пациента, в зависимости от композиции и способа введения, при отсутствии токсичности.
Уровень дозы лекарственного средства зависит от множества факторов, включающих активность конкретного соединения по настоящему изобретению, или его сложного эфира, соли или амида, способ введения лекарственного средства, продолжительность введения, скорость экскреции или метаболизма конкретного соединения, скорость и степень впитывания, продолжительность лечения, другие лекарственные средства, соединения и/или материалы, применяемые в комбинации с конкретным соединением, возраст, пол, массу, заболевание, состояние и предшествующую историю болезни пациента, проходящего курс лечения, и другие факторы, известные в данной области техники.
Лечащий врач или ветеринар определяет и назначает эффективное количество фармацевтической композиции, необходимое для лечения пациента. Например, лечащий врач или ветеринар начинают лечение с дозировки соединений по настоящему изобретению, включенных в фармацевтическую композицию, в более низкой дозе, требуемой для обеспечения терапевтического действия, и затем постепенно увеличивают дозу до достижения требуемого действия.
Как правило, пригодная суточная доза соединения по настоящему изобретению включает минимальное эффективное количество соединения, достаточное для обеспечения терапевтического действия. Эффективная доза, как правило, зависит от факторов, описанных выше. Как правило, дозы соединений по настоящему изобретению при пероральном, внутривенном, интрацеребро-желудочковом и подкожном введении пациенту для обеспечения анальгезирующего действия составляют от приблизительно 0,0001 мг до приблизительно 100 мг на килограмм массы тела в сутки.
При необходимости эффективную суточную дозу активного соединения разделяют на отдельные две, три, четыре, пять, шесть или более поддоз, которые вводят через определенные промежутки времени в течение суток необязательно в виде стандартных лекарственных форм. Предпочтительный способ введения составляет введение один раз в сутки.
Хотя соединения по настоящему изобретению можно вводить пациенту сами по себе, соединение по настоящему изобретению предпочтительно вводят в составе фармацевтической композиции.
Соединения по настоящему изобретению перерабатывают в любую стандартную лекарственную форму для введения пациенту любым пригодным способом, известным в медицине или ветеринарии, аналогичным для получения других фармацевтических препаратов.
В другом объекте настоящего изобретения предлагаются фармацевтически приемлемые композиции, которые включают терапевтически эффективное количество одного или более соединений по настоящему изобретению, как описано выше, в смеси с одним или более фармацевтически приемлемыми носителями (добавками) и/или разбавителями. Как подробно описано ниже, фармацевтические композиции по настоящему изобретению получают в твердой или жидкой форме, пригодной для введения пациенту, включая следующие формы: (1) для перорального введения, например, микстуры (водные или неводные растворы или суспензии), таблетки, болюсы, порошки, гранулы и пасты, которые наносят на язык, (2) для парентерального введения, например, с использованием подкожной, внутримышечной или внутривенной инъекции, например стерильный раствор или суспензия, (3) для местного введения, например крем, мазь или спрей для нанесения на кожу или для введения в легкие или на слизистую оболочку, (4) для внутривлагалищного или внутриректального введения, например пессарий, крем или пена, (5) для подъязычного или трансбуккального введения, (6) для глазного введения, (7) для чрескожного способа введения лекарственного средства, (8) для интраназального введения.
Термин «лечение» означает также профилактику, лечение и поддерживающий курс лечения.
Пациенты, которые нуждаются в таком лечении, включают любое животное, включая приматов, человека и других млекопитающих, таких как лошади, крупный рогатый скот, свиньи и овцы, а также домашняя птица и домашние животные.
Соединение по настоящему изобретению вводят пациенту само по себе или в виде смеси с фармацевтически приемлемыми носителями, а также в смеси с бактерицидными агентами, такими как пенициллины, цефалоспорины, аминогликозиды и гликопептиды. Комплексная терапия, таким образом, включает последовательное, одновременное и раздельное введение активного соединения, при котором терапевтическое действие первой дозы лекарственного средства еще продолжается перед введением следующей дозы.
Активное соединение по настоящему изобретению добавляют в корм для животных, т.е. предпочтительно предварительно получают соответствующую кормовую смесь, содержащую активное соединение в эффективном количестве, и включают эту предварительно приготовленную смесь в полный рацион для кормления животных.
В другом варианте промежуточный пищевой концентрат или кормовую добавку, содержащие активный ингредиент, предварительно смешивают с кормом. Такие кормовые смеси и полный рацион получают известными способами, описанными в справочниках, таких как «Applied Animal Nutrition», W.H.Freedman и CO., San Francisco, USA (1969) или «Livestock Feeds and Feeding» О и В книги, Corvallis, Ore., USA (1977).
Мицеллы
В настоящее время в фармацевтической промышленности используют технологию микроэмульгирования для повышения биодоступности некоторых липофильных (нерастворимых в воде) фармацевтических агентов, примеры включают триметрин (см. статью Dordunoo S.К. и др., Drug Development and Industrial Pharmacy, 17(12), 1685-1713, (1991)) и REV 5901 (см. статью Sheen P.С. и др., J Pharm Sci 80(7), 712-714, (1991)). Кроме основных преимуществ микроэмульгирование обеспечивает повышение биодоступности за счет предпочтительной и непосредственной доставки (впитывания) лекарственного средства в лимфатическую систему, а не в кровеносную систему, что позволяет исключить поступление лекарственного средства в печень, и таким образом предотвращает разложение соединений в печеночно-желчной системе.
Предпочтительные пригодные амфифильные носители в основном включают носители, включенные в список безопасных веществ Generally-Recognized-as-Safe (GRAS) и которые способны солюбилизировать соединение по настоящему изобретению и микроэмульгировать его на более поздней стадии, когда раствор контактирует с комплексной водной фазой (например, среда в желудочно-кишечном тракте человека). Как правило, величина гидрофильно-гидрофобного баланса (ГГБ) амфифильных ингредиентов, которые удовлетворяют указанным требованиям, составляет от 2 до 20 и указанные ингредиенты содержат в своей структуре алифатические С6-С20 радикалы с прямой цепью. Примеры амфифильных ингредиентов включают полиэтиленгликолизированные глицериды жирных кислот и полиэтиленгликоли (ПЭГ).
Коммерческие амфифильные носители прежде всего включают продукты Gelucire-series, лабрафил, лабразол или лаурогликоль (выпускаемые фирмой Gattefosse Corporation, Saint Priest, France), ПЭГ-моноолеат, ПЭГ-биолеат, ПЭГ-монолаурат и билаурат, лецитин, полисорбат 80 и т.п. (выпускаемые многими фирмами в США и во всем мире).
Полимеры
Гидрофильные полимеры, пригодные для применения по настоящему изобретению, являются высоко растворимыми в воде, могут ковалентно связываться с везикулообразующим липидом и характеризуются удовлетворительной преносимостью in vivo при отсутствии токсичности (т.е. являются биологически совместимыми). Пригодные полимеры включают полиэтиленгликоль (ПЭГ), полимолочную кислоту (называемую также полилактид), полигликолиевую кислоту (называемую также полигликолид), сополимер полимолочной-полигликолиевой кислот и поливиниловый спирт. Предпочтительные полимеры включают полимеры с молекулярной массой от приблизительно 100 дальтон или 120 дальтон до приблизительно 5000 или 10000 дальтон и более предпочтительно от приблизительно 300 дальтон до приблизительно 5000 дальтон. В одном предпочтительном варианте осуществления настоящего изобретения полимером является полиэтиленгликоль с молекулярной массой от приблизительно 100 дальтон до приблизительно 5000 дальтон и более предпочтительно с молекулярной массой от приблизительно 300 дальтон до приблизительно 5000 дальтон. В другом предпочтительном варианте полимером является полиэтиленгликоль с молекулярной массой 750 дальтон (ПЭГ 750). Полимеры по настоящему изобретению включают определенное число мономеров, в предпочтительном варианте предлагаются полимеры, включающие по крайней мере приблизительно три мономера, такие полимеры ПЭГ включают три мономера (с молекулярной массой приблизительно 150 дальтон).
Другие гидрофильные полимеры, пригодные для применения по настоящему изобретению, включают поливинилпирролидон, полиметоксазолин, полиэтилоксазолин, полигидроксипропилметакриламид, полиметакриламид, полидиметилакриламид и производные целлюлозы, такие как гидроксиметилцеллюлоза или гидроксиэтилцеллюлоза.
В некоторых предпочтительных вариантах композиция по настоящему изобретению включает биологически совместимый полимер, выбранный из группы, включающей полиамиды, поликарбонаты, полиалкилены, полимеры эфиров акриловой и метакриловой кислот, поливинилполимеры, полигликолиды, полисилоксаны, полиуретаны и их сополимеры, целлюлозы, полипропилен, полиэтилены, полистирол, полимеры молочной и гликолиевой кислот, полиангидриды, поли(орто)сложные эфиры, полибутановую кислоту, поливалериановую кислоту, поли(лактид-капролактон), полисахариды, белки, полигиалуроновые кислоты, полицианоакрилаты и их комбинации, смеси или сополимеры.
Циклодекстрины
Циклодекстрины являются циклическими олигосахаридами, включающими 6, 7 или 8 остатков глюкозы, и обозначаются греческими буквами альфа, бета или гамма соответственно. Циклодекстрины, включающие менее шести остатков глюкозы, как известно, не существуют. Остатки глюкозы связаны между собой альфа-1,4-глюкозидными связями. Остатки сахаров существует в конформации «кресло», при этом все вторичные гидроксильные группы (в положении С2, С3) располагаются с одной стороны цикла, а все первичные гидроксильные группы в положении С6 с другой стороны цикла. Таким образом, остатки, расположенные на внешней поверхности циклодекстрина, с внешней стороны цикла являются гидрофильными и отвечают за растворимость циклодекстринов в воде. И наоборот, остатки, расположенные внутри молекулы циклодекстрина, являются гидрофобными, так как указанные остатки являются соседними с атомами водорода в положениях С3 и C5 и эфирными атомами кислорода. Такие матрицы образуют комплексы с различными относительно гидрофобными соединениями, включающими, например, стероиды, такие как 17-бета-эстрадиол (см., например, статью van Uden и др., Plant Cell Tiss. Org. Cult., 38:1-3-113 (1994)). Такие комплексы образуются за счет ван-дер-ваальсовских взаимодействий и образования водородной связи (см., например, общий обзор по химии циклодекстринов в статье Wenz, Agnew. Chem. Jnt. Ed. Engl., 33:803-822 (1994).
Физико-химические свойства производных циклодекстрина в значительной степени зависят от природы и количества заместителей. Например, растворимость производных циклодекстрина в воде находится в диапазоне от полной нерастворимости (например, триацетил-бета-циклодекстрин) до 147% растворимости (мас./об.) (G-2-бета-циклодекстрин). Кроме того, производные циклодекстрина являются растворимыми во многих органических растворителях. Свойства циклодекстринов можно использовать для изменения растворимости различных компонентов композиции, снижая или повышая их растворимость.
Ряд циклодекстринов и методы их получения описаны в литературе, например, электронейтральные циклодекстрины описаны Parmeter (I) и др. в патенте США №3453259 и Gramera и др. в патенте США №3459731. Другие производные циклодекстрина включают циклодекстрины с катионными свойствами (см. патент США №3453257, Parmeter (II)), нерастворимые сшитые циклодекстрины (см. патент США №3420788, Solms) и циклодекстрины с анионными свойствами (см. патент США №3426011, Parmeter (III)). Производные циклодекстрина с анионными свойствами включают карбоновые, фосфористые, фосфинистые, фосфоновые, фосфорные, тиофосфоновые, тиосульфиновые и сульфоновые кислоты в составе исходного циклодекстрина (см. патент Parmeter (III) выше). Кроме того, сульфоалкиловые эфиры циклодекстрина описаны Stella и др. в патенте США №5134127.
Липосомы
Липосомы состоят по крайней мере из одного липидного бислоя, который окружает внутреннюю водную фазу. Липосомы различают по типу мембран и по размеру. Малые однослойные везикулы (SUV) включают одну мембрану и обычно их диаметр составляет от 0,02 до 0,05 мкм, диаметр больших однослойных везикул (LUV) составляет, как правило, более 0,05 мкм. Большие многослойные везикулы содержат множество, обычно концентрических, мембранных слоев и их диаметр, как правило, составляет более 0,1 мкм. Липосомы с несколькими неконцентрическими мембранами, т.е. включающие несколько везикул меньшего размера в одной большой везикуле, называются мультивезикулярными липосомами.
В одном варианте осуществления настоящего изобретения предлагается способ получения липосом, содержащих соединение по настоящему изобретению и липосомальную мембрану, состав которой повышает скорость доставки активного агента. В другом варианте соединение по настоящему изобретению содержится внутри бислоя липосомы или сорбируется в нем. Соединение по настоящему изобретению образует агрегат с липидной поверхностно-активной фазой липосомы и проникает во внутреннее пространство липосомы, в указанных случаях состав мембраны липосомы должен обеспечивать устойчивость к разрушающему действию агрегата ПАВ и активного агента.
В одном варианте осуществления настоящего изобретения липидный бислой липосомы содержит липиды, включающие полиэтиленгликоль (ПЭГ), при этом цепи ПЭГ расположены на внутренней поверхности липидного бислоя и проникают во внутреннее пространство липосомы, а также из внешнего слоя липосомы в окружающую среду.
Активные агенты по настоящему изобретению, содержащиеся внутри липосом, находятся в солюбилизированном состоянии. Агрегаты поверхностно-активного вещества (ПАВ) и активного агента (такие как эмульсии или мицеллы, содержащие активный агент) можно инкапсулировать во внутреннее пространство липосомы по настоящему изобретению. ПАВ способствуют диспергированию и солюбилизации активного агента, и их выбирают из группы, включающей любое пригодное алифатическое, циклоалифатическое или ароматическое ПАВ, такое как, без ограничения перечисленным, биологически совместимые лизофосфатидилхолины (LPC) с различной длиной цепи (например, от приблизительно С14 до приблизительно С20). Полимерные липиды, такие как ПЭГ-липиды, можно также использовать для образования мицеллы, так как такие липиды ингибируют слияние мицеллы с мембраной, а полимер в составе ПАВ снижает критическую константу мицеллообразования (ККМ) ПАВ и способствует образованию мицеллы. Предпочтительными ПАВ являются ПАВ, величина ККМ которых находится в микромолярном диапазоне, а ПАВ, ККМ которых составляет более высокие величины, можно использовать для получения мицелл, инкапсулированных во внутреннее пространство липосом по настоящему изобретению, однако мономеры в составе ПАВ мицеллы могут влиять на устойчивость бислоя липосомы, что можно использовать при создании липосом с требуемой стабильностью.
Липосомы по настоящему изобретению получают по любым методикам, известным в данной области техники (см., например, патент США №4235871, опубликованную заявку РСТ WO 96/14057 и книги New RRC, Liposomes: A practical approach, IRL Press, Oxford, стр.33-104 (1990), Lasic DD, Liposomes from physics to applications, Elsevier Science Publishers BV, Amsterdam, (1993)).
Например, липосомы по настоящему изобретению получают диффундированием липида, модифицированного гидрофильным полимером, в предварительно полученные липосомы, например, при взаимодействии предварительно полученных липосом с мицеллами, полученными из липид-привитых полимеров, при концентрации липидов, соответствующей конечной требуемой молярной концентрации модифицированного липида в липосоме. Липосомы, содержащие гидрофильный полимер, получают также по известной методике, например, такой как гомогенизация, липидная гидратация или экструзия.
В одном объекте настоящего изобретения липосомы характеризуются гомогенностью по размерам в выбранном диапазоне. Один эффективный метод получения гомогенных липосом включает экструзию водной суспензии липосом через серию поликарбонатных мембран с определенным размером пор, при этом размер пор мембраны приблизительно равен наибольшему размеру липосом, полученных экструзией через такую мембрану (см., например, патент США №4737323 (12 апреля 1988).
Модификаторы высвобождения активного агента
Характеристики высвобождения активного агента по настоящему изобретению зависят от материала оболочки, концентрации инкапсулированного лекарственного средства и присутствия модификаторов высвобождения. Например, высвобождение активного агента регулируют изменением рН, например, с использованием рН-чувствительного покрытия, через которое активный агент высвобождается только при низком рН, например в желудке, или при более высоком рН, например в кишечнике. Энтеросолюбильное покрытие используют для предотвращения высвобождения активного агента во время прохождения лекарственного средства через желудок. При получении многослойных покрытий или смесей цианамида, инкапсулированных в различные материалы, высвобождение активного агента начинается в желудке, а затем в кишечнике. Высвобождение активного агента также регулируют включением солей или порообразующих агентов, которые повышают поглощение воды или высвобождение лекарственного средства при диффузии из капсулы. Для регулирования высвобождения используют также экципиенты, которые изменяют растворимость лекарственного средства. Для регулирования высвобождения также используют агенты, которые ускоряют разрушение матрицы или высвобождение лекарственного средства из матрицы. Такие агенты добавляют к лекарственному средству в отдельности (т.е. в виде частиц) или растворяют в полимерной фазе в зависимости от природы соединения. Во всех случаях количество добавляемых агентов осоставляет от 0,1 мас.% до 30 мас.% в расчете на массу полимера. Примеры агентов, способствующих разрушению матрицы, включают неорганические соли, такие как сульфат и хлорид аммония, органические кислоты, такие как лимонная кислота, бензойная кислота и аскорбиновая кислота, неорганические основания, такие как карбонат натрия, карбонат калия, карбонат кальция, карбонат цинка и гидроксид цинка, и органические основания, такие как протаминсульфат, спермин, холин, этаноламин, диэтаноламин и триэтаноламин, и ПАВ, такие как твин® и плуроник®. Порообразующие агенты, которые формируют микроструктуру матрицы (т.е. растворимые в воде соединения, такие как неорганические соли и сахара), добавляют в виде частиц в концентрации от 1 мас.% до 30 мас.% в расчете на массу полимера.
Доставку лекарственного средства также регулируют изменением времени пребывания частиц в кишечнике, например покрытием частиц слизистым адгезивным полимером или при использовании в качестве материала оболочки такого полимера. Примеры таких полимеров включают множество полимеров со свободными карбоксильными группами, таких как хитозан, целлюлозы, и, прежде всего, полиакрилаты (термин «полиакрилаты», используемый в настоящем описании, означает полимеры, включающие акрилатные группы и модифицированные акрилатные группы, такие как цианоакрилаты и метакрилаты).
Примеры
Следующие примеры приведены только для иллюстрации и не ограничивают объем и сущность изобретения.
Пример 1
Синтез бензилгидроксиаминов
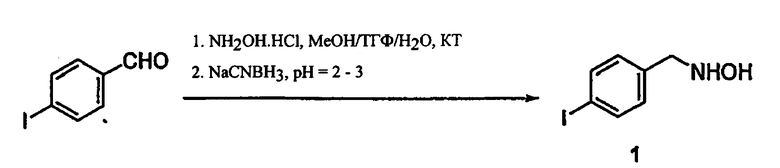
В раствор 4-иодбензальдегида (0,025 ммоля) в смеси МеОН/ТГФ (40 мл, 3:1) добавляли водный раствор NH2OH·HCl (10 мл), при этом рН реакционной смеси доводили до 9 добавлением 6 н. KOH. Реакционную смесь перемешивали при КТ в течение 2 ч и затем добавляли NaCNBH3 (1,5 г, 0,025 ммоля) и кристалл метилового оранжевого. Раствор подкисляли до рН 2, при этом ярко-красный цвет реакционной смеси поддерживали в ходе реакции добавлением 1 н. HCl. Через 2 ч добавляли вторую порцию NaCNBH3 (1,5 г, 0,025 ммоля). Смесь перемешивали в течение 14 ч, затем упаривали 2/3 объема растворителя и повышали рН реакционной смеси до 9-10 добавлением 6 н. водного раствора KOH. Полученную смесь экстрагировали CH2Cl2 (три раза по 100 мл). Органические слои объединяли, промывали водой и затем солевым раствором. Органический слой сушили над MgSO4, фильтровали и упаривали в вакууме, при этом получали соединение 1 (5,7 г, 91%) в виде твердого вещества грязно-белого цвета.
Пример 2
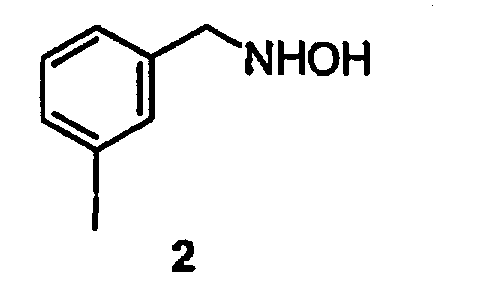
Гидроксиамин 2 синтезировали аналогично тому, как описано в примере 1, но при замене 4-иодбензальдегида на 3-иодбензальдегид, при этом получали требуемый продукт (выход 90%).
Пример 3
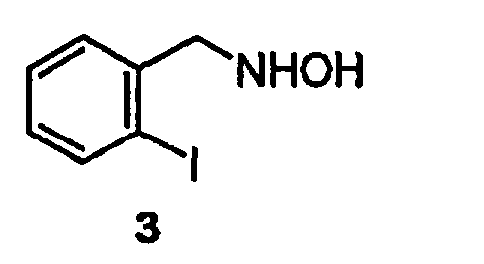
Гидроксиамин 3 синтезировали аналогично тому, как описано в примере 1, но при замене 4-иодбензальдегида на 2-иодбензальдегид, при этом получали требуемый продукт (выход 85%).
Пример 4
Синтез нитроновых кислот для проведения реакции циклоприсоединения [3+2] (см. статью Keirs D., Overton К., Heterocycles, 28, 841-848 (1989)

В суспензию N-(4-иодбензил)гидроксиламина (10 г, 42,5 ммоля) в CH2Cl2 (200 мл) добавляли моногидрат глиоксиловой кислоты (4,7 г, 51,1 ммоля) в атмосфере азота в течение 24 ч при КТ. Реакционную смесь промывали водой (2 раза по 200 мл), солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. К твердому веществу желтоватого цвета добавляли Et2O (50 мл) и суспензию титровали и фильтровали, при этом получали соединение в виде кремообразного окрашенного твердого вещества 4. Маточный раствор концентрировали в вакууме, твердое вещество промывали Et2O, затем фильтровали, при этом получали соединение 4 (11,8 г, выход 93%).
Пример 5
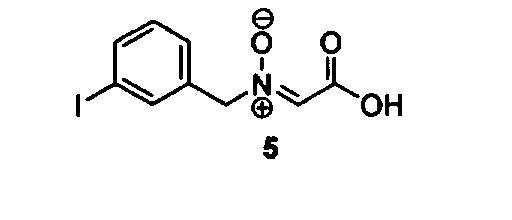
Нитроновую кислоту 5 синтезировали аналогично тому, как описано в примере 4, но при замене N-(4-иодбензил)гидроксиламина на N-(3-иодбензил)гидроксиламин, при этом получали требуемый продукт (90%).
Пример 6
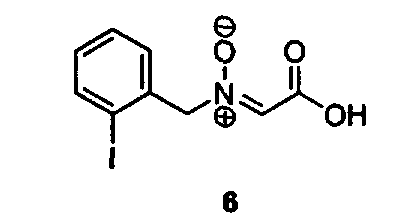
Нитроновую кислоту синтезировали аналогично тому, как описано в примере 4, но при замене N-(4-иодбензил)гидроксиламина на N-(2-иодбензил)гидроксиламин, при этом получали требуемый продукт (90%).
Пример 7
Синтез метиловых эфиров нитронкарбоновых кислот для использования в реакции циклоприсоединения [3+2]

В раствор N-(4-иодбензил)гидроксиламина (16 г, 64 ммоля) в бензоле (320 мл) добавляли метилглиоксилат (6,8 г, 80 ммолей). Смесь нагревали при 120°С в течение 3 ч с использованием ловушки Дина Старка. Раствор охлаждали до КТ и растворитель концентрировали в вакууме, при этом получали продукт 7 (19,1 г, 93%) в виде твердого вещества желтого цвета.
Пример 8
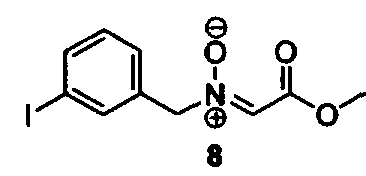
Метиловый эфир нитроновой кислоты 8 синтезировали аналогично тому, как описано в примере 7, но при замене N-(4-иодбензил)гидроксиламина на N-(3-иодбензил)гидроксиламин, при этом получали требуемый продукт (85%).
Пример 9
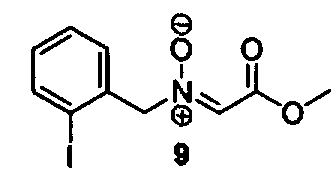
Метиловый эфир нитроновой кислоты 9 синтезировали аналогично тому, как описано в примере 7, но при замене N-(4-иодбензил) гидроксиламина на N-(2-иодбензил)гидроксиламин, при этом получали требуемый продукт (90%).
Пример 10
Синтез диполярных соединений
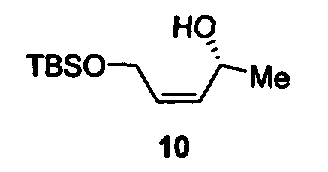
Стадия А
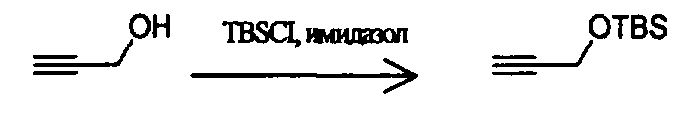
В раствор TBSC1 (215 г, 1,43 моля) в CH2Cl2 (1,2 л) при 0°С добавляли имидазол (97 г, 1,43 моля). Затем добавляли по каплям пропаргиловый спирт (83 мл, 1,43 моля), суспензию нагревали до КТ и перемешивали в течение 60 мин. Реакцию останавливали добавлением воды (500 мл). Смесь концентрировали в вакууме и остаток экстрагировали гексаном (3 раза по 500 мл). Органические экстракты объединяли, промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали перегонкой (70°С/~10 мм рт.ст.), при этом получали требуемый продукт (179 г, 74%).
Стадия В

В раствор трет-бутилдиметил(2-пропинилокси)силана (свежеперегнанный, 27 г, 0,16 моля) в ТГФ (300 мл) при -78°С добавляли н-бутиллитий (119 мл 1,6 М раствор в гексане, 0,19 моля). Через 15 мин добавляли уксусный альдегид (10 мл, 0,19 моля). Реакционную смесь перемешивали в течение 30 мин и реакцию останавливали добавлением водного раствора 5 мас./об.% NH4Cl (50 мл) и воды (100 мл). Половину объема ТГФ упаривали в вакууме и смесь выливали в воду (150 мл). Полученную смесь экстрагировали гексаном (3 раза по 200 мл) и Et2O (100 мл). Объединенные экстракты сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали перегонкой (115°С/1 мм рт.ст. (температура в бане 155°С), при этом получали продукт (30 г, 88%).
Стадия С
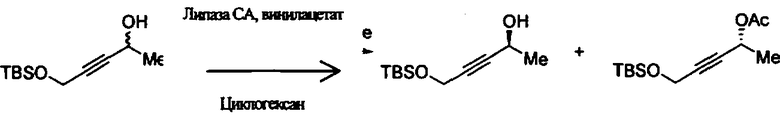
Липазу СА (Candida Antarctic, иммобилизованная на макропористой акриловой смоле, Sigma L-4777 Lot 11K127) (1 г) добавляли в смесь пропаргилового спирта (10 г, 0,47 моля) и винилацетата (129 мл, 0,14 моля) в циклогексане (380 мл). Реакционную смесь перемешивали в течение 48 ч, затем фильтровали и смолу промывали EtOAc (50 мл). Фильтрат и промывные фракции объединяли и концентрировали в вакууме. Неочищенный материал очищали колоночной хроматографией (гексан/EtOAc от 95:5 до 80:20), при этом получали 5,3 г спирта и 6,5 г ацетата (избыток энантиомера 93%).
Стадия D
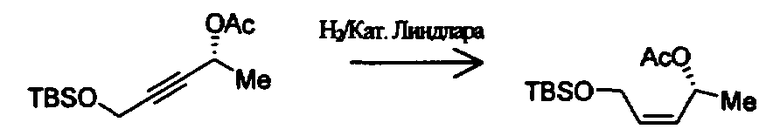
В перемешиваемый раствор пропаргилацетата (60 г, 0,23 ммоля) в EtOAc (700 мл) добавляли хинолин (30 мл) и катализатор Линдлара (6 г) в атмосфере водорода. Через 9 ч реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией (гексан/EtOAc, 95:5) при этом получали требуемый продукт (57,5 г, 97%).
Стадия Е
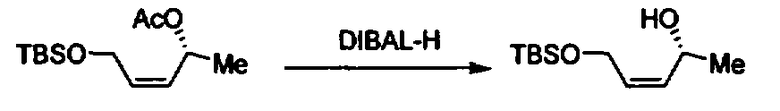
В перемешиваемый раствор аллилацетата (50 г, 0,19 моля) в CH2Cl2 (400 мл) при
-78°С добавляли DIBAL-H (426 мл, 1 М раствор в гептане, 0,43 моля). Через 15 мин реакционную смесь разбавляли Et2O (400 мл) и реакцию останавливали добавлением солевого раствора (150 мл). Реакционную смесь нагревали до КТ и перемешивали в течение еще 2 ч. Реакционную смесь сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенную смесь очищали колоночной хроматографией (гексан/EtoAc, 80:20), при этом получали 41 г требуемого продукта.
Пример 11
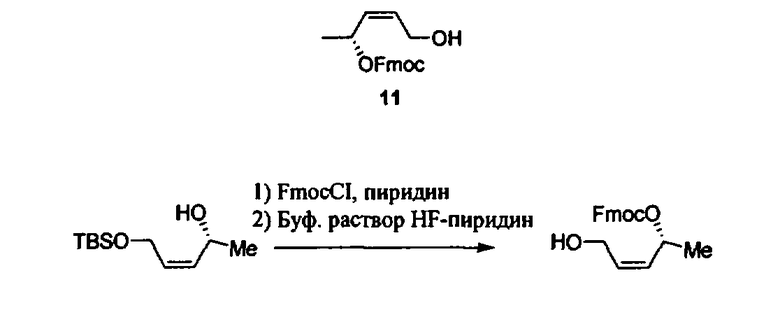
В перемешиваемый раствор аллилового спирта (40 г, 0,19 моля) в пиридине (400 мл) и CH2Cl2 (100 мл) при 0°С добавляли Fmoc-Cl (62 г). Реакционную смесь нагревали до КТ, перемешивали в течение еще 30 мин, и останавливали добавлением воды (500 мл). Слои разделяли, водную фазу экстрагировали Et2O (2 раза по 200 мл), объединенные экстракты промывали водным раствором CuSO4, солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали Fmoc-защищенный спирт. Неочищенный материал использовали на следующей стадии без предварительной очистки.
Неочищенный продукт (35 г) помещали в пластиковый сосуд и добавляли ТГФ (100 мл) с последующим добавлением раствора HF-пиридин (HF-пиридин (7,7 мл)/ пиридин (15,4 мл)/ТГФ (76,9 мл). Реакционную смесь выдерживали при КТ в течение 4 ч, обрабатывали TMSOMe в течение 60 мин и выливали в воду (500 мл). Слои разделяли, водную фазу экстрагировали Et2O (3 раза по 200 мл). Органические экстракты объединяли и промывали водным раствором NaHCO3, солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали неочищенный аллиловый спирт. Неочищенный материал очищали колоночной хроматографией, при этом получали 14,4 г требуемого продукта.
Пример 12
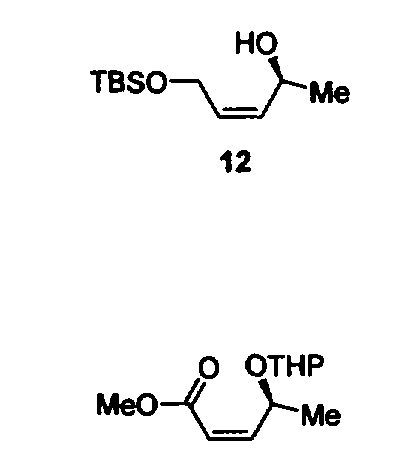
Стадия А
Раствор метилового эфира бис(2,2,2-трифторэтил)фосфоноуксусной кислоты (28 г, 0,1 ммоля) и 18-краун-6 (132 г, 0,50 ммоля) в ТГФ (2 л) охлаждали до -78°С в атмосфере азота. В охлажденный раствор добавляли 0,6 М раствор бис(триметилсилил)амида калия в толуоле (20 г, 0,1 ммоля). Затем добавляли (8)-2-(тетрагидропиранилокси)пропаналь (синтез описан в статье J. Chem. Soc., Perkin. Trans. 1, 2791 (1994), 16 г, 0,1 ммоля) и полученную смесь перемешивали в течение 30 мин при -78°С. Затем добавляли насыщенный раствор хлорида аммония и продукт экстрагировали Et2O (3 раза по 500 мл). Эфирные экстракты объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный материал очищали хроматографией на колонке с силикагелем, при этом получали 13,5 г продукта.
Стадия В
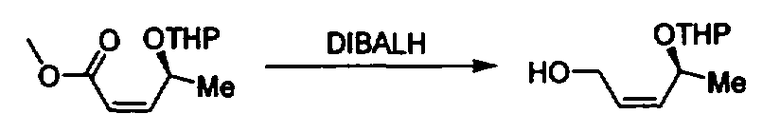
Метиловый эфир 4(S)-(тетрагидропиран-2-илокси)пент-2-еноевой кислоты (10 г, 46,7 ммоля) восстанавливали DIBAL-H по методике, описанной в статье J. Chem. Soc., Perkin. Trans., 1, 2791 (1994), при этом получали 4(S)-(тетрагидропиран-2-илокси)пент-2-ен-1-ол (выход 88%).
Стадия С
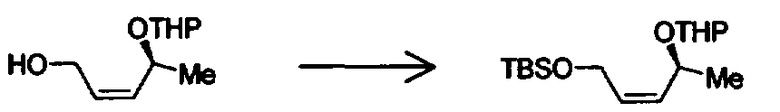
В раствор 4(S)-(тетрагидропиран-2-илокси)пент-2-ен-1-ола (4,0 г, 22 ммоля) в ТГФ (20 мл) медленно добавляли имидазол (3,66 г, 53,5 ммоля), затем TBSCl (1,2 экв., 3,89 г, 25,8 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, реакцию останавливали водой (20 мл) и смесь экстрагировали Et2O (3 раза по 10 мл). Объединенные органические экстракты промывали водой (5 раз 50 мл), солевым раствором (1 раз по 50 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Масло очищали колоночной хроматографией (гексан/EtOAc 2:1), при этом получали требуемый эфир TBDMS (5,9 г, 92%) в виде бесцветного масла.
Стадия D
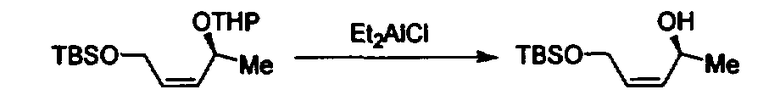
В трет-бутилдиметил[4(S)-(тетрагидропиран-2-илокси)пент-2-енилокси]силане удаляли ТНР-защитную группу (10 г, 33 ммоля) по методике, описанной в статье Tetrahedron letters, 25, 663 (1984), при этом получали продукт (выход 83%).
Пример 13
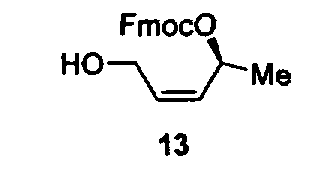
Стадия А
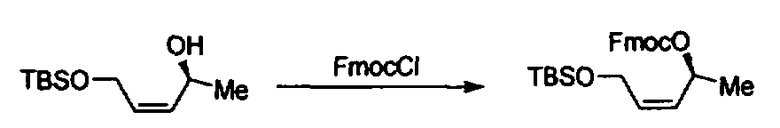
В раствор 5-(трет-бутилдиметилсиланилокси)пент-3-ен-2(S)-ола (3,95 г, 18,3 ммоля) в пиридине (20 мл) добавляли FmocCl (6,14 г, 23,7 ммоля, 1,3 экв.). Реакционную смесь перемешивали в течение ночи при КТ. Реакцию медленно останавливали водой (20 мл) и экстрагировали Et2O (3 раза по 15 мл). Объединенные органические экстракты промывали водой (3 раза по 50 мл), 5% раствором KH2PO4 (3 раза по 50 мл) и солевым раствором (1 раз по 50 мл), сушили над MgSO4 и фильтровали. После концентрирования и очистки колоночной хроматографией получали 6,98 г (87%) продукта в виде масла бледно-желтого цвета.
Стадия В
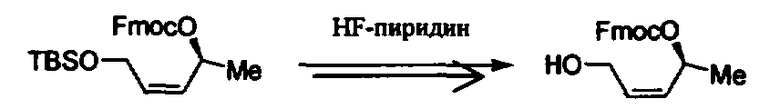
В раствор 4-(трет-бутилдиметилсиланилокси)-1-метилбутил-2(S)енилового, 9Н-флуорен-9-илметилового эфира карбоновой кислоты (700 мг, 1,60 ммоля) в Et2O (5 мл) медленно добавляли через пластиковую трубку Wharton® HF/пиридин (70% HF в пиридине, 6 мл) шестью порциями. Завершение реакции контролировали методом ТСХ. После завершения реакции смесь охлаждали до 0°С на ледяной бане, затем реакцию останавливали TMSOMe (10 мл). Реакционную смесь перемешивали в течение 30 мин при нагревании до комнатной температуры. Реакционную смесь выливали в воду (50 мл), слои разделяли, водный слой экстрагировали Et2O (3 раза по 15 мл). Объединенные органические экстракты промывали водой (3 раза по 30 мл), 5% KH2PO4 (3 раза по 30 мл), солевым раствором (1 раз 30 мл), сушили над Na2SO4 и фильтровали. После концентрирования и колоночной хроматографии получали 471 мг продукта (91%) в виде прозрачного бесцветного масла.
Пример 14
Общие методики циклоприсоединения [3+2]
Способ 1
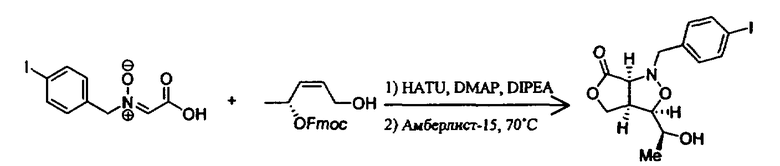
В раствор нитронкарбоновой кислоты 4 (1,4 г, 3,1 ммоля) в CH2Cl2 добавляли аллиловый спирт 11 (1,0 г, 3,1 ммоля), HATU (2,0 г, 6 ммолей) и DMAP (0,56 г, 4,6 ммоля). Раствор охлаждали на ледяной бане и перемешивали в течение 1,0 ч, затем добавляли диизопропилэтиламин (0,44 г, 0,6 мл, 4,6 ммоля) по каплям в течение 15 мин и реакционную смесь перемешивали при 0°С в течение 1 ч. Раствор разбавляли CH2Cl2-5% NaHCO3 (300 мл, 1:1) и водный слой экстрагировали CH2Cl2 (2 раза по 125 мл). Объединенные органические экстракты объединяли, промывали водой (200 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток суспендировали в ТГФ (10 мл), добавляли диизопропилэтиламин (0,12 г, 0,167 мл, 1 ммоль) и раствор нагревали с обратным холодильником в течение 1 ч, охлаждали до КТ и концентрировали в вакууме. Остаток переносили в ТГФ (8 мл), добавляли 1,33 г смолы Амберлист-15 и смесь нагревали при 70°С в течение 2 ч. Раствор снова охлаждали, смесь фильтровали и концентрировали в вакууме. Неочищенный материал очищали экспресс-хроматографией, при этом выход требуемого продукта составил 59%.
Способ 2
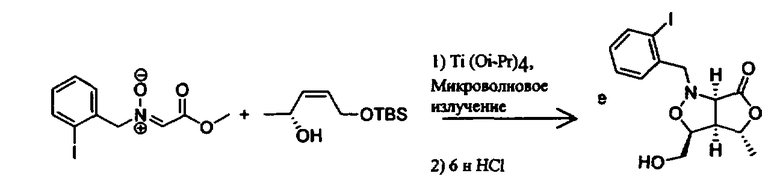
В раствор метилового эфира нитроновой кислоты 9 (8,1 г, 38 ммолей) и вторичного спирта 10 (12 г, 38 ммолей) в толуоле (40 мл) добавляли Ti(ОСН(СН3)2)4 (16 г, 17 мл, 56 ммолей). Суспензию нагревали в микроволновом реакторе при 140°С в течение 30 мин и охлаждали до КТ. Раствор разбавляли EtOAc (150 мл) и 3-(диметиламино)-1,2-пропандиолом (7 г, 7 мл, 58 ммолей), перемешивали при КТ в течение 8 ч. В раствор добавляли воду (100 мл) и органическую фазу отделяли, водную фазу промывали EtOAc (3 раза по 30 мл). Объединенные органические экстракты промывали водой (100 мл), солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный материал очищали экспресс-хроматографией (Et2O/CH2Cl2, 1:29), при этом получали лактон (13,5 г, 71%).
В раствор изоксазолидина (13,5 г, 26 ммолей) в ТГФ (120 мл) добавляли 6 н. HCl (67 мл). Раствор перемешивали при КТ в течение 1,5 ч, разбавляли водой (25 мл) и экстрагировали EtOAc (3 раза по 80 мл), органические экстракты объединяли и промывали насыщенным NaHCO3 (50 мл), солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали хроматографией на силикагеле (230-400 меш, Et2O-CH2Cl2, градиент от 1:4 до 1:2), при этом получали требуемый продукт (9,5 г, суммарный выход в расчете на 2 стадии, 64%).
Эти методики описаны также в статьях Porter, Georges и Hopkins.
Пример 15
Синтез изоксазолидиновых фрагментов
Изоксазолидин 14 синтезировали по общей методике 2, но при замене метилового эфира нитроновой кислоты 7 на метиловый эфир нитроновой кислоты 9 и аллилового спирта 10 на аллиловый спирт 12. Выход составляет 40-60%.
Пример 16
Изоксазолидин 15 синтезировали по общей методике 2, но при замене метилового эфира нитроновой кислоты 7 на метиловый эфир нитроновой кислоты 8 и аллилового спирта 10 на аллиловый спирт 12. Выход составляет: 40-60%.
Пример 17
Изоксазолидин 16 синтезировали по общей методике 2, но при использовании метилового эфира нитроновой кислоты 7 и при замене аллилового спирта 10 на аллиловый спирт 12. Выход составляет: 40-60%.
Пример 18
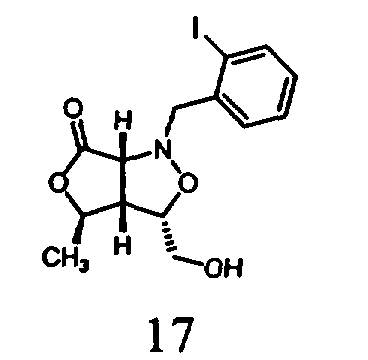
Изоксазолидин 17 синтезировали по общей методике 2, но при замене метилового эфира нитроновой кислоты 7 на метиловый эфир нитроновой кислоты 9 и с использованием аллилового спирта 10. Выход составляет: 40-60%.
Пример 19
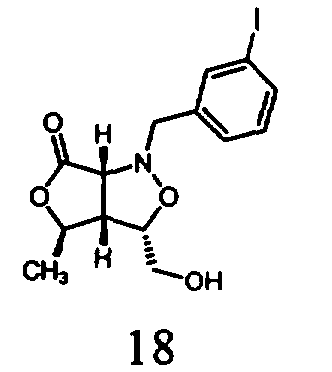
Изоксазолидин 18 синтезировали по общей методике 2, но при замене метилового эфира нитроновой кислоты 7 на метиловый эфир нитроновой кислоты 8 и при использовании аллилового спирта 10. Выход составляет: 40-60%.
Пример 20
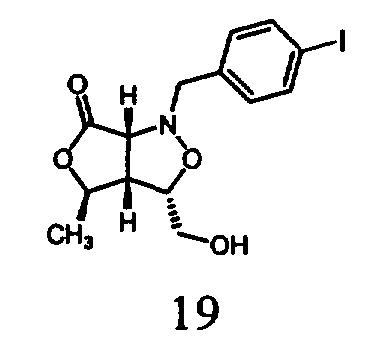
Изоксазолидин 19 синтезировали по общей методике 2, но с использованием метилового эфира нитроновой кислоты 7 и аллилового спирта 10. Выход составляет: 40-60%.
Пример 21
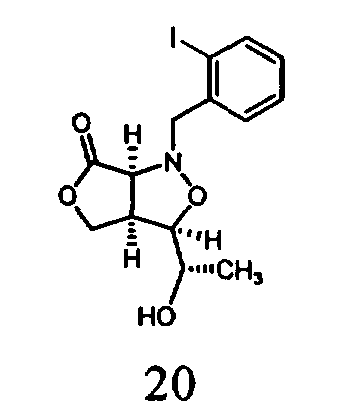
Изоксазолидин 20 синтезировали по общей методике 1, но при замене нитроновой кислоты 4 на нитроновую кислоту 6 и с использованием аллилового спирта 11. Выход составляет: 50-60%.
Пример 22
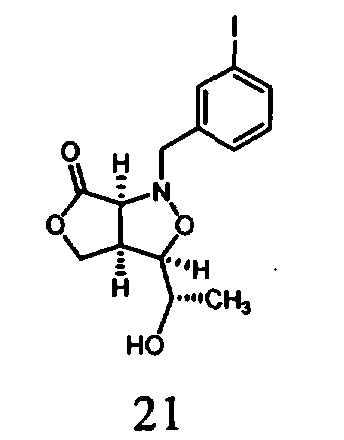
Изоксазолидин 21 синтезировали по общей методике 1, но при замене нитроновой кислоты 4 на нитроновую кислоту 5 и с использованием аллилового спирта 11. Выход составляет: 50-60%.
Пример 23
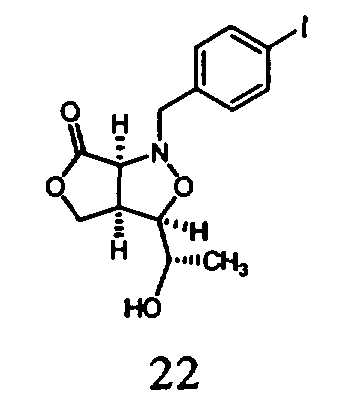
Изоксазолидин 22 синтезировали по общей методике 1, но с использованием нитроновой кислоты 4 и аллилового спирта 12. Выход составляет: 50-60%.
Пример 24
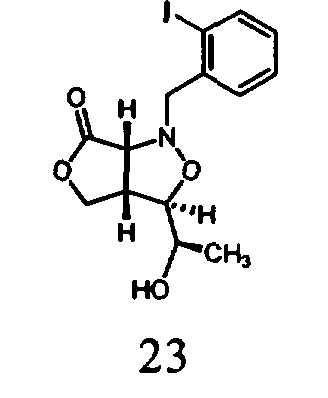
Изоксазолидин 23 синтезировали по общей методике 1, но при замене нитроновой кислоты 4 на нитроновую кислоту 6 и аллилового спирта 11 на аллиловый спирт 13. Выход составляет: 50-60%.
Пример 25
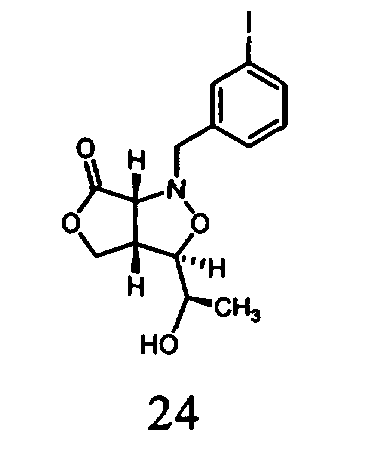
Изоксазолидин 24 синтезировали по общей методике 1, но при замене нитроновой кислоты 4 на нитроновую кислоту 5 и аллилового спирта 11 на аллиловый спирт 13. Выход составляет: 50-60%.
Пример 26
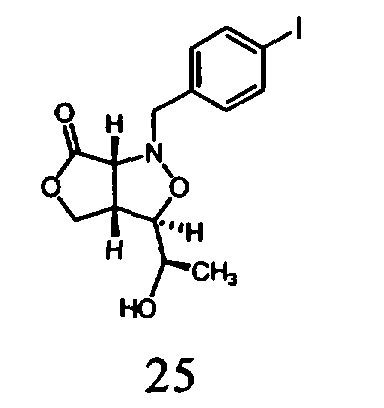
Изоксазолидин 25 синтезировали по общей методике 1, но с использованием нитроновой кислоты 4 и при замене аллилового спирта 11 на аллиловый спирт 13. Выход составляет: 50-60%.
Пример 27
Синтез аллилсилана (связующей группы)
Аллилсилан 26 синтезировали аналогично тому, как описано в статье Tallarico и др., J. Chem. Comb., 3, 312-318 (2001).
Пример 28
Модификация частиц носителя Lantern Mimotopes включением силильной связующей группы
32000 частиц носителя (2,22 моля фенильных групп, SynPhase-PS, серия L, Lantern Mimotopes, Клейтон Виктория, Австралия) добавляли в колбу реактора объемом 22 л, снабженную 5-горловой верхней разъемной насадкой и нижним выходным отверстием с тефлоновым резьбовым соединением. В верхнюю насадку устанавливали мешалку с тефлоновыми лопастями шириной 16 см с верхним приводом, шланг для подачи аргона, капельную воронку объемом 250 мл, температурный датчик и шланг для присоединения к ловушке HBr (колба объемом 1 л, содержащая 500 мл воды). Реактор продували аргоном в течение 15 мин, затем добавляли безводный ДХМ (14,8 л). Через 10 мин добавляли ацетат таллия (76 г, 0,20 моля, 0,090 экв.). Реакционный сосуд оборачивали алюминиевой фольгой и перемешивали при комнатной температуре в течение 150 мин. Затем добавляли бром (177 г, 1,11 моля, 0,50 экв.) в ДХМ (100 мл) из капельной воронки в течение 15 мин, при этом температура в реакторе поднималась от 19,3 до 27,0°С. После завершения добавления брома реакционную смесь перемешивали в течение еще 60 мин. Реакцию останавливали добавлением МеОН (1,5 л) и перемешивали при комнатной температуре в течение ночи. Реакционный раствор из реактора откачивали и отбрасывали, а частицы носителя промывали следующим образом: 12 л каждого растворителя в течение 10-20 мин: ДХМ, ТГФ/IPA 3:1, ТГФ/вода 3:1, вода и ТГФ (2 раза). Из частиц носителя удаляли растворитель при пониженном давлении. По данным элементного анализа пяти частиц носителя среднее содержание брома составляет 34,2+0,7 мкмоль/частица, содержание по теории составляет 35,0 мкмоль/частица (т.е. выход включения атомов брома составляет 98%).
К колбе реактора объемом 22 л, снабженной 5-горловой верхней насадкой и нижним выходным отверстием с тефлоновым резьбовым соединением, присоединяли шланг для подачи растворителя или аргона, тефлоновую мешалку с лопастями шириной 16 см с верхним приводом (с защитой от искр), температурный датчик и два холодильника. Реактор помещали в колбонагреватель в держателе и закрепляли к стене вытяжного шкафа. В колбу добавляли безводный ТГФ (500 мл, содержание воды по данным анализа KF составляет 40 част./млн) и интенсивно перемешивали для удаления со стенок колбы остатков воды. Растворитель удаляли через нижнее выходное отверстие в атмосфере аргона. Колбу продували аргоном в течение 10 мин, затем заливали диизопропил(4-метоксифенил)аллилсилан (353 г, 1,34 моля, 1,2 экв., Maybridre # MO01086ZZ), добавляли безводный ТГФ (11 л). Раствор интенсивно продували аргоном в течение 20 мин с использованием тефлонового шланга 1/8′′ Teflon®. Затем добавляли 9-BBN (167 г, 1,34 моля, 1,2 экв.) и раствор перемешивали при комнатной температуре в атмосфере аргона в течение 2 ч. Полноту потребления аллилсилана определяли методом ЯМР (CDCl3). Затем добавляли в атмосфере агрона бромированные частицы носителя lantern (32000, 1,11 моля Br, 1,0 экв.), Pd(PPh3)4 (65 г, 0,056 моля, 0,05 экв., Strem Chemical # 40-2150) и 2 н. NaOH (1,34 л, 2,69 моля, 2,4 экв.). Реакционную смесь нагревали до внутренней температуры 65°С при постоянной продувке аргоном при перемешивании в течение 40 ч. Реакционную смесь охлаждали, раствор отбрасывали, а частицы промывали следующим образом (10 л каждого растворителя в течение 10-20 мин): ТГФ, ТГФ/IPA 3:1, ТГФ/1 н. NaCN (водный) 3:1 (в течение 1 ч или до исчезновения черного окрашивания частиц носителя), вода (2 раза), ТГФ/вода 3:1, ТГФ (2 раза) и ДХМ. Из частиц носителя удаляли растворитель при пониженном давлении. По данным элементного анализа среднее содержание кремния составляет 22,2+2,2 мкмоль/частица. Среднее содержание остаточного брома составляет 5,5 мкмоль/частица.
Пример 29
Включение и модификация изоксазолидиновых фрагментов
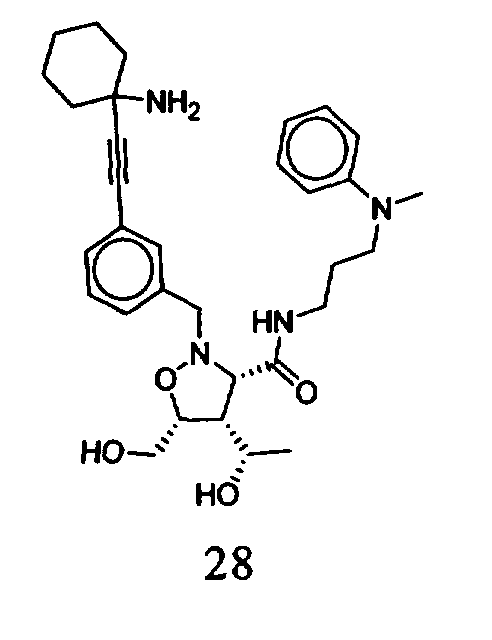
Стадия A
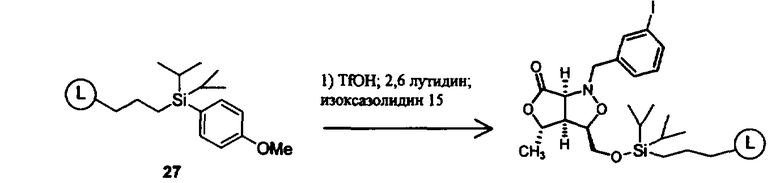
1808 частиц носителя lantern помещали в высушенную на пламени горелки колбу объемом 2 л, снабженную перемешивающим стержнем для магнитной мешалки, колбу продували азотом и закрывали каучуковой мембраной. В колбу добавляли CH2Cl2 (1,2 л) и частицы носителя выдерживали в этом растворе в течение 10 мин, затем растворитель удаляли, добавляли 3% раствор трифторметансульфоновой кислоты в безводном CH2Cl2 (1,2 л, 393 ммоля, 3 об./об.%) и частицы носителя осторожно перемешивали в течение 20 мин. Затем раствор трифторметансульфоновой кислоты удаляли через трубку и добавляли CH2Cl2 (1,2 л) и 2,6-лутидин (62 мл, 532 ммолей). Частицы носителя перемешивали в этом растворе в течение 10 мин. Затем добавляли сухой изоксазолидин 15 (10 г, 38 ммолей). Полученную смесь перемешивали в течение 18 ч, затем раствор декантировали и частицы носителя промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (1,5 л), ТГФ (1,5 л), ТГФ/IPA (3:1, 1,5 л), ТГФ/вода (3:1, 1,5 л), ТГФ/IPA (3:1, 1,5 л), и ТГФ (1,5 л). Затем частицы носителя сушили при пониженном давлении в течение ночи. Определение содержания иммобилизованного на носителе фрагмента: 5 частиц носителя lantern помещали каждую в отдельности в полипропиленовые контейнеры объемом 5 мл. В каждый контейнер добавляли ТГФ (300 мкл) и HF-пиридин (50 мкл). Частицы носителя выдерживали в этом растворе в течение 6 ч, затем добавляли TMSOMe (500 мкл) и частицы носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера затем переливали в стеклянную круглодонную колбу и концентрировали в вакууме, при этом получали изоксазолидин 15. Полученный материал взвешивали и рассчитывали содержание фрагмента в частице носителя, которое составляет 14 мкмолей/частица.
Стадия В
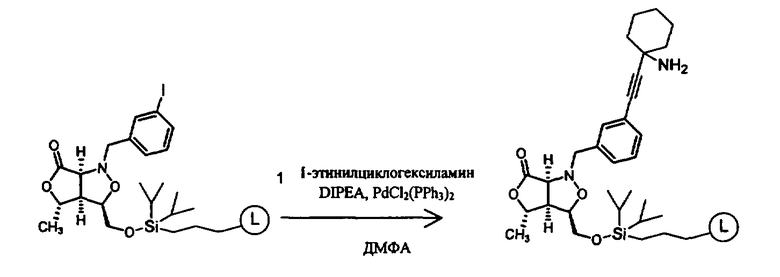
Модифицированные изоксазолидином 376 частиц носителя lantern помещали в колбу объемом 500 мл. Колбу продували азотом и закрывали. В колбу добавляли Pd(PPh3)2Cl2 (7,92 г, 11,3 ммоля) и CuI (3,22 г, 16,9 ммоля). Реакционный сосуд снова продували азотом, закрывали и добавляли безводный ДМФА (300 мл), добавляли диизопропилэтиламин (30,0 мл, 172 ммоля) с последующим добавлением с помощью шприца 1-этинилциклогексиламина (110 ммолей). Реакционный сосуд затем осторожно встряхивали в течение 2 ч. Затем раствор декантировали, а частицы носителя промывали следующим образом: (2 раза в течение 10 мин) ДМФА (300 мл), ТГФ (300 мл), ТГФ/IPA (3:1, 300 мл), ТГФ/вода (3:1, 300 мл), ТГФ/IPA (3:1, 300 мл), ТГФ (300 мл), CH2Cl2 (300 мл). Метод определения степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу выдерживали в этом растворе в течение 6 ч, затем дабавляли TMSOMe (500 мкл) и выдерживали частицу в течение еще 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и вакуумировали, при этом получали продукт реакции Соногашира.
Стадия С
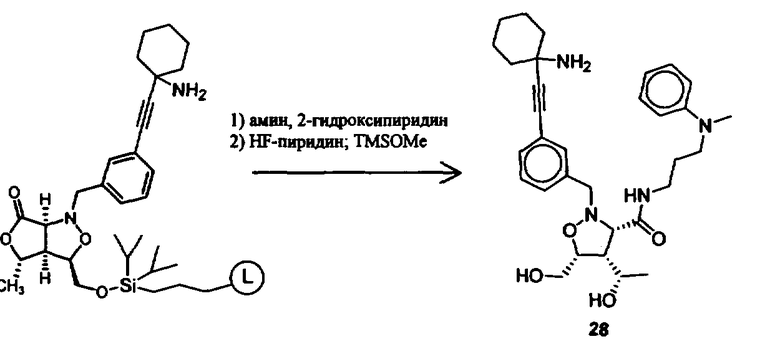
374 частиц носителя lantern помещали в круглодонную колбу объемом 500 мл, добавляли 2-гидроксипиридин (300 мл, 0,35 М раствор в ТГФ, 105 ммолей), продували N2 и колбу закрывали каучуковой мембраной, добавляли N1-метил-N1-фенилпропан-1,3-диамин (279 ммолей) и реакционную смесь нагревали при 50°С в течение 16 ч. Растворитель удаляли, частицы носителя промывали следующим образом: (2 раза 10 мин) ТГФ (300 мл), ТГФ/IPA (3:1, 300 мл), ТГФ/вода (3:1, 300 мл), ТГФ/IPA (3:1, 300 мл), ТГФ (300 мл), CH2Cl2 (300 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (160 мкл), пиридином (200 мкл) и HF-пиридином (40 мкл). Частицу выдерживали в этом растворе в течение 1 ч. Затем добавляли TMSOMe (500 мкл), затем частицу выдерживали в этом раствор в течение еще 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт.
Изоксазолидин 28 характеризовали методом ЖХ-МС. Ниже приведены общие методики и условия аналитических методов, использованных в этих и других примерах.
Условия анализа методом ЖХ-МС
Масс спектрометр: Waters ZQ
ЖХВР: Waters 2795 Alliance HT
Набор диодов: Waters 2696
Условия анализа масс-спектрометрией:
Тип ионизации масс-спектрометра: режим ионизации с переключением съемки положительных и отрицательных ионов.
Набор масс 150-1000 Дальтон
Напряжение на капилляре 3,2 кВ
Напряжение на распылителе 35 В
Напряжение на экстракторе 3 В
Радиочастотные линзы 0
Температуры источника 120°С
Температура осушающего газа 350°С
Скорость подачи газа 25 л/ч
Скорость подачи осушающего газа 550 л/ч
Условия анализа методом ЖХВР:
Подвижные фазы:
А: вода 95%, ацетонитрил 5%, муравьиная кислота 0,1%
В: вода 5%, ацетонитрил 95%, муравьиная кислота 0,1%
Скорость потока: 1,00 мл/мин
Колонка: Waters Symmetry, размер 4,6 мм на 50 мм, частицы С 18,5 мкм
Температура в колонке 50°С
Объем образца при нанесении: 5 мкл
Параметры диодной матрицы: длина волны: 220 нм-400 нм
Разрешение: 1,2 нм
Если не указано иное, оптимальная концентрация образцов составляет 0,2 мг/мл.
Условия анализа методом MC-TOF
Масс-спектрометр: Micromass LCT
ЖХВР: Waters 2795 Alliance HT
Набор диодов: Waters 2696
Условия анализа масс-спектрометрометрией:
Масс-спектрометр с ионизацией: распыление положительных частиц
Набор масс 150-1000 Дальтон
Капилляр (KB) 3,2
Сопло (В) 35
Экстрактор (V) 3 В
Линзы RF 0
Источник температуры 120°С
Температура десольватации 350°С
Скорость подачи газа 25 л/ч
Скорость подачи осушающего газа 450 л/ч
Условия анализа методом ЖХВР:
Подвижная фаза:
А: вода, содержащая муравьиную кислоту 0,1%
В: 65% метанол/35% 2-пропанол, содержащий 0,1% муравьиную кислоту
Скорость потока: 1,00 мл/мин
Колонка: Varian Polaris 2,1 мм на 50 мм; частицы С 18,5 микрон
Температура в колонке 50°С
Система из двух колонок установлена таким образом, что в то время как одна колонка уравновешивается, на другой анализируется следующий образец.
Объем образца: 5 мкл
Диодная матрица: длина волны: 220 нм-400 нм
разрешение: 1,2 нм
Если не указано иное, оптимальная концентрация образцов составляет 2,0 мг/мл.
Пример 30
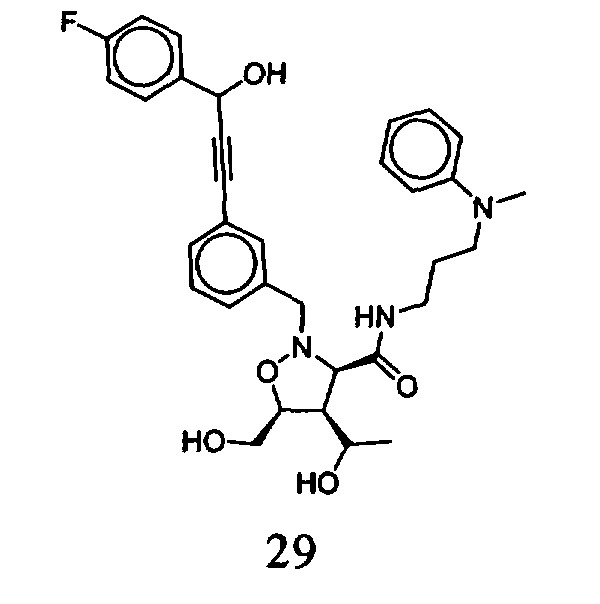
Соединение 29 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 1-(4-фторфенил)проп-2-ин-1-ол, и с использованием N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 576,4 (М+Н)+.
Пример 31
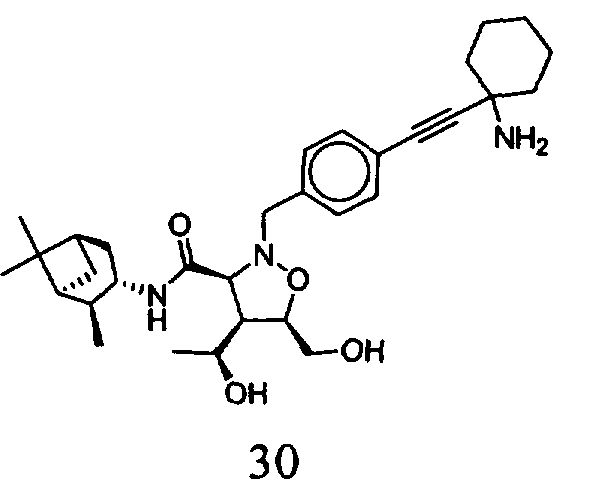
Соединение 30 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 1-(4-фторфенил)проп-2-ин-1-ол и N1-метил-N1-фенилпропан-1,3-диамин на (+)-изопинокамфеиламин. МС (ESI(+)): m/e 538,3 (М+Н)+.
Пример 32
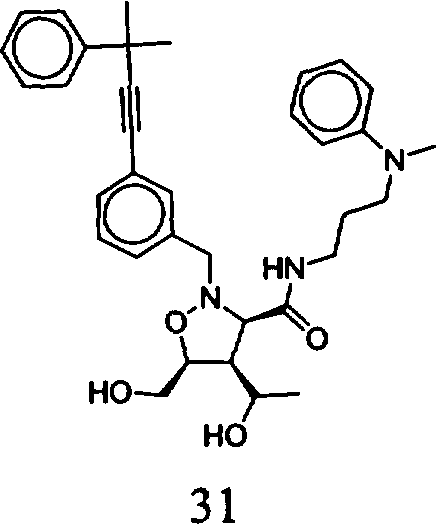
Соединение 31 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, (1,1-диметилпроп-2-инил)бензола вместо 1-этинилциклогексиламина и с использованием N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 570,4 (М+Н)+.
Пример 33
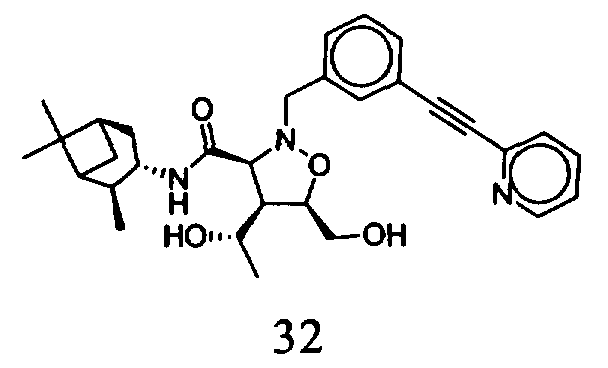
Соединение 32 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, 2-этинилпиридина вместо 1-этинилциклогексиламина и (+)-изопинокамфеиламина вместо N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 518,4 (М+Н)+.
Пример 34
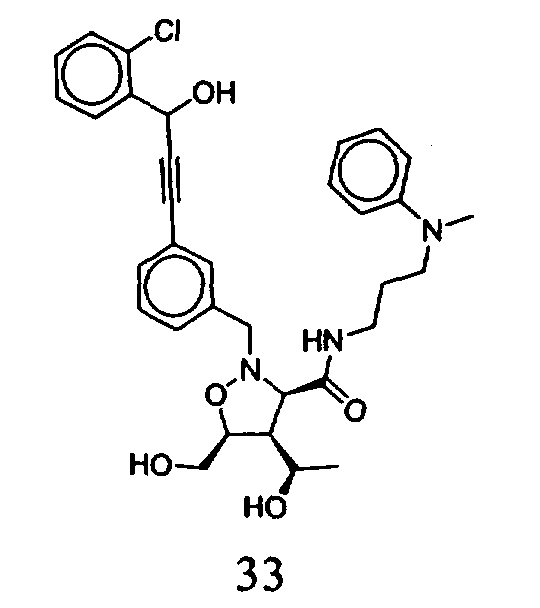
Соединение 33 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинил-циклогексиламин на 1-(2-хлорфенил)проп-2-ин-1-ол и с использованием N1-метил-N1-фенилпропан-1,3-диамина.
Пример 35
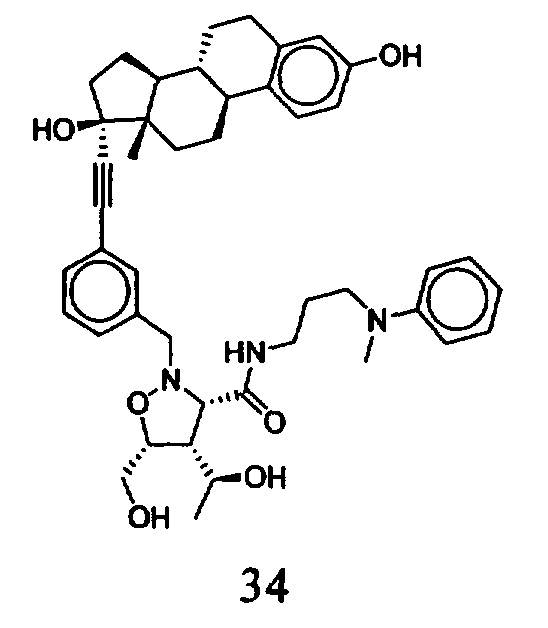
Соединение 34 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, 17-α-этинилэстрадиола вместо 1-этинилциклогексиламина и с использованием N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 722,4 (М+Н)+.
Пример 36
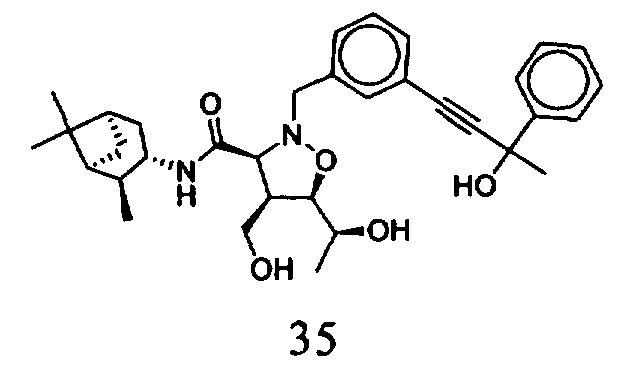
Соединение 35 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, 2-фенилбут-3-ин-2-ола вместо 1-этинилциклогексиламина и (+)-изопинокамфеиламина вместо N1-метил-N1-фенилпропан-1,3-диамина.
Пример 37
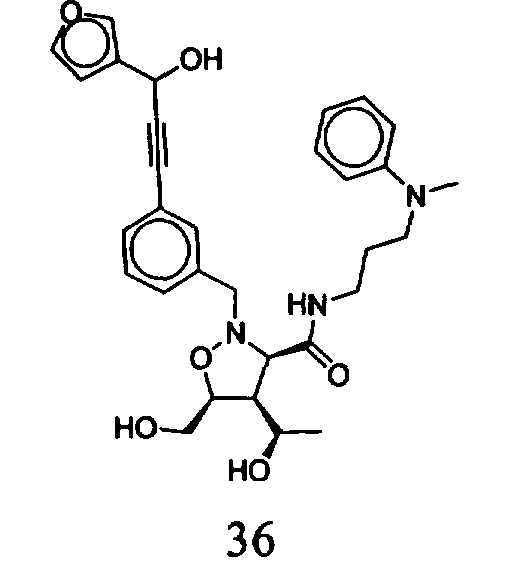
Соединение 36 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 1-фуран-3-илпроп-2-ин-1-ол и с использованием N1-метил-N1-фенилпропан-1,3-диамина.
Пример 38
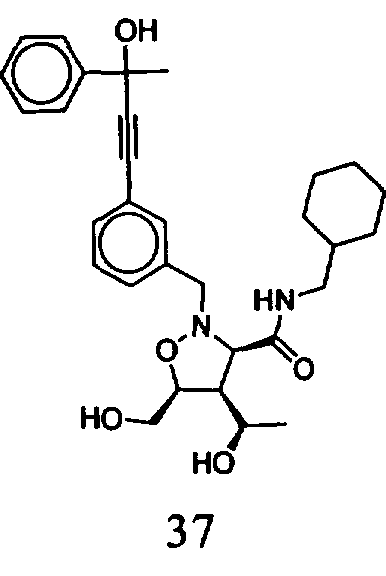
Соединение 37 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 2-фенилбут-3-ин-2-ол и N1-метил-N1-фенилпропан-1,3-диамин на циклогексилметиламин. МС (ESI(+)): m/e 521,6 (М+Н)+.
Пример 39
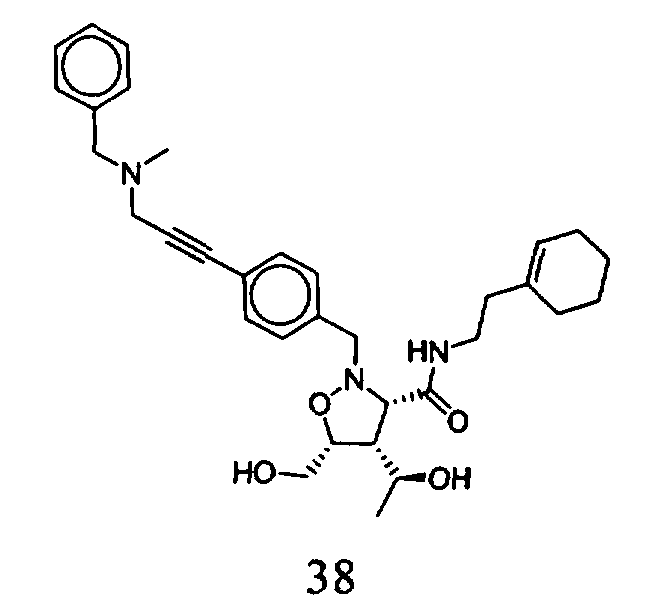
Соединение 38 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 16, 1-этинилциклогексиламина на бензилметилпроп-2-иниламин и N1-метил-N1-фенил-пропан-1,3-диамина на 2-циклогекс-1-енилэтиламин.
Пример 40
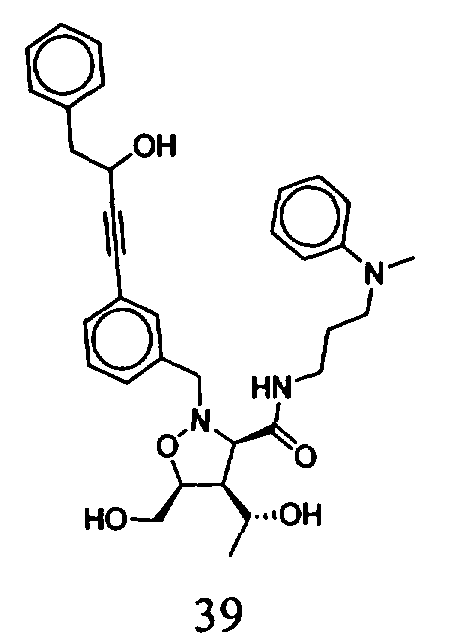
Соединение 39 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинил-циклогексиламина на 1-фенилбут-3-ин-2-ол и с использованием N1-метил-N1-фенилпропан-1,3-диамина.
Пример 41
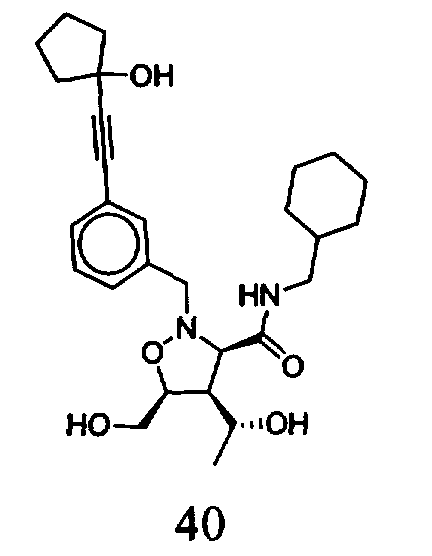
Соединение 40 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 1-этинилциклопентанол и N1-метил-N1-фенилпропан-1,3-диамина на циклогексилметиламин.
Пример 42
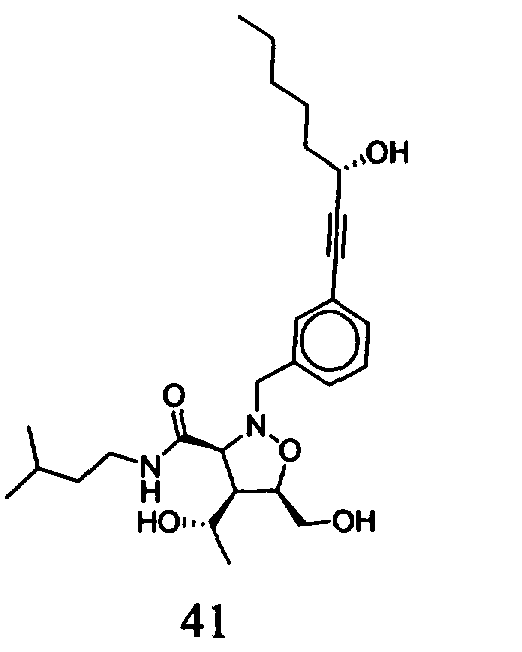
Соединение 41 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, окт-1-ин-3-ола вместо 1-этинилциклогексиламина и 3-метилбутиламина вместо N1-метил-N1-фенилпропан-1,3-диамина.
Пример 43
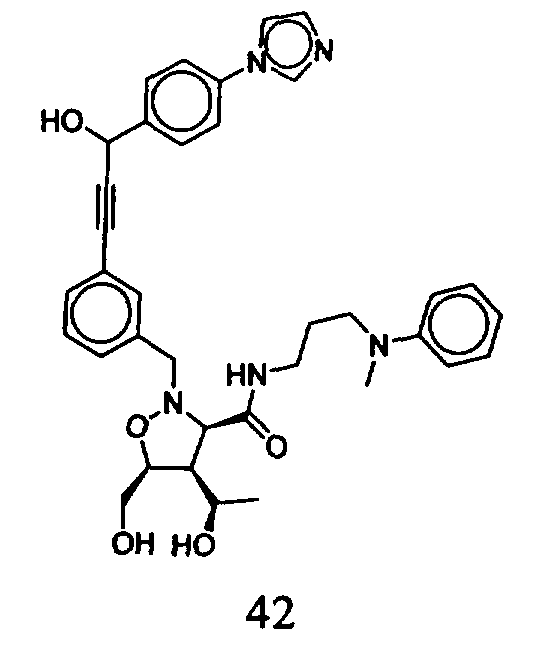
Соединение 42 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 1-(4-имидазол-1-илфенил)проп-2-ин-1-ол и с использованием N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 624,4 (М+Н)+.
Пример 44
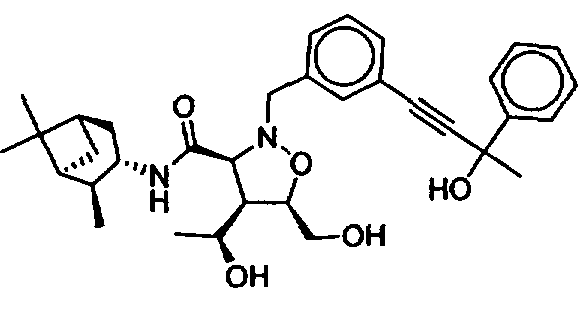
Соединение 43 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, 2-фенилбут-3-ин-2-ола вместо 1-этинилциклогексиламина и (+)-изопинокамфеиламина вместо N1-метил-N1-фенил-пропан-1,3-диамина. МС (ESI(+)): m/e 561,3 (M+H)+.
Пример 45
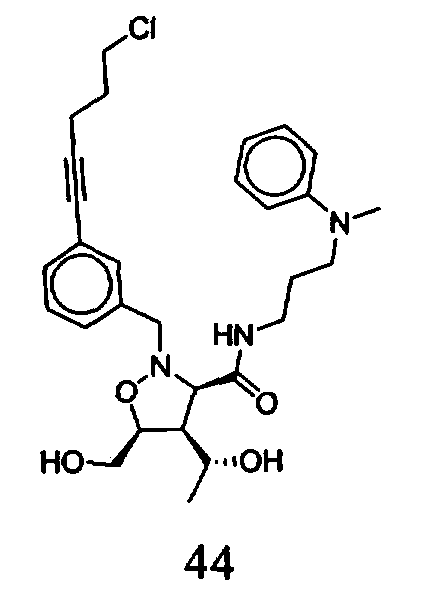
Соединение 44 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 5-хлорпент-1-ин и с использованием N1-метил-N1-фенилпропан-1,3-диамина.
Пример 46
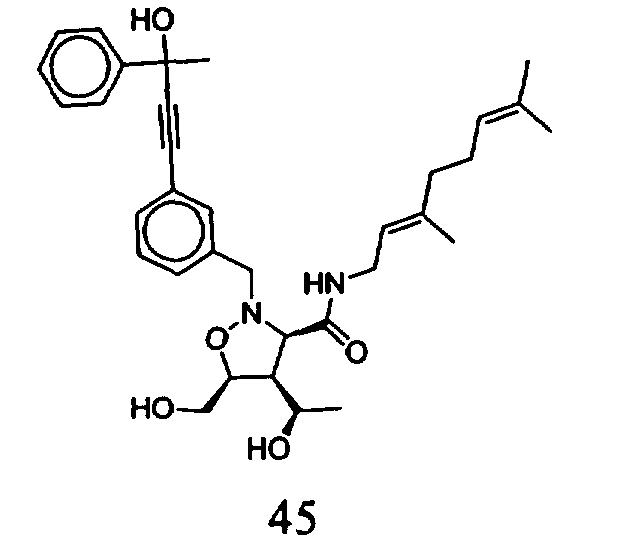
Соединение 45 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 2-фенилбут-3-ин-2-ол и N1-метил-N1-фенилпропан-1,3-диамина на гераниламин. МС (ESI(+)): m/e 561,7 (М+Н)+.
Пример 47
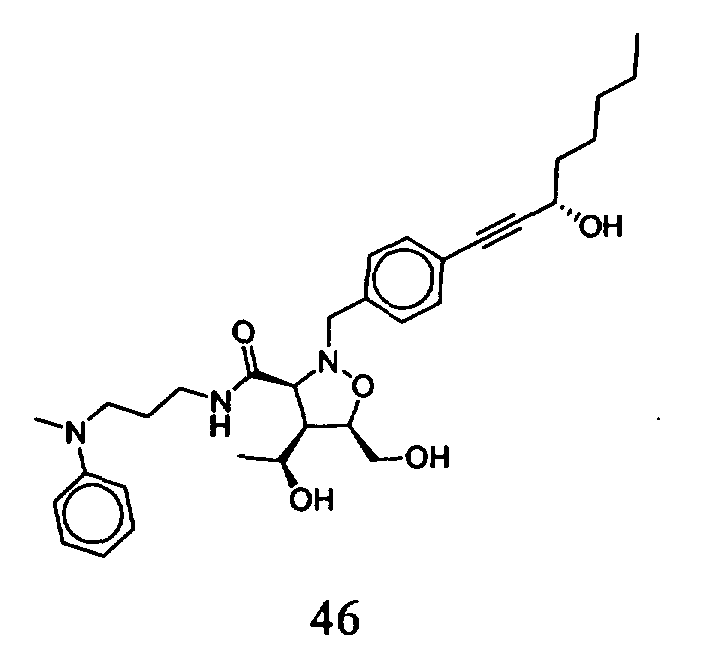
Соединение 46 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, окт-1-ин-3-ола вместо 1-этинилциклогексиламина и с использованием N1-метил-N1-фенилпропан-1,3-диамина.
Пример 48
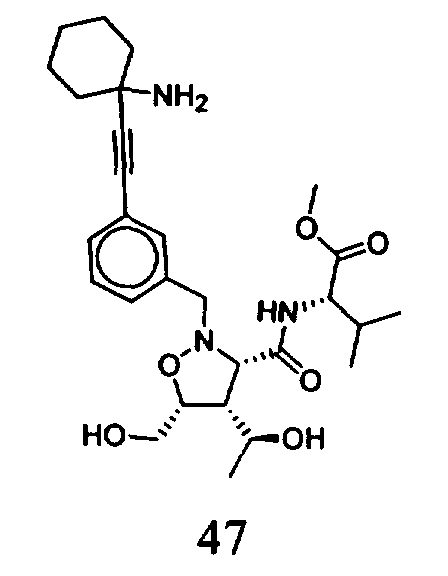
Соединение 47 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, 1-этинилциклогексиламина и при замене N1-метил-N1-фенилпропан-1,3-диамина на метиловый эфир валина.
Пример 49
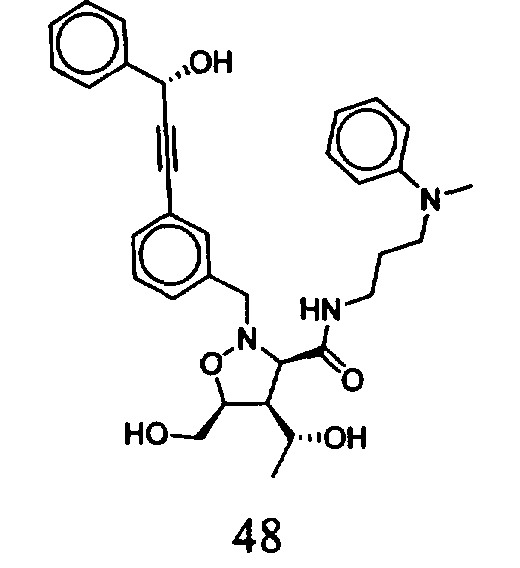
Соединение 48 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 1-фенилпроп-2-ин-1-ол и с использованием N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 558,2 (М+Н)+.
Пример 50
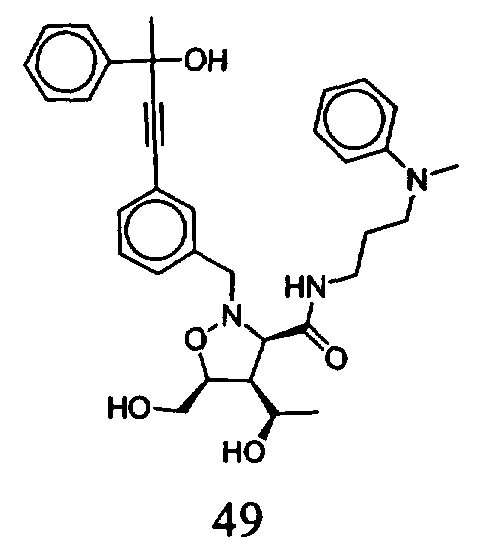
Соединение 49 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на 2-фенилбут-3-ин-2-ол и с использованием N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 572,3 (М+Н)+.
Пример 51
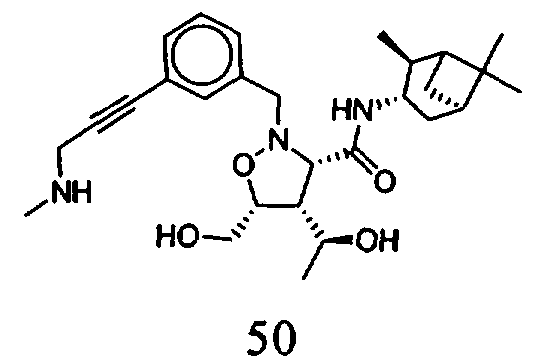
Соединение 50 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, метилпроп-2-иниламина вместо 1-этинилциклогексиламина и (+)-изопинокамфеиламина вместо N1-метил-N1-фенилпропан-1,3-диамина. МС (ESI(+)): m/e 484,2 (М+Н)+.
Пример 52
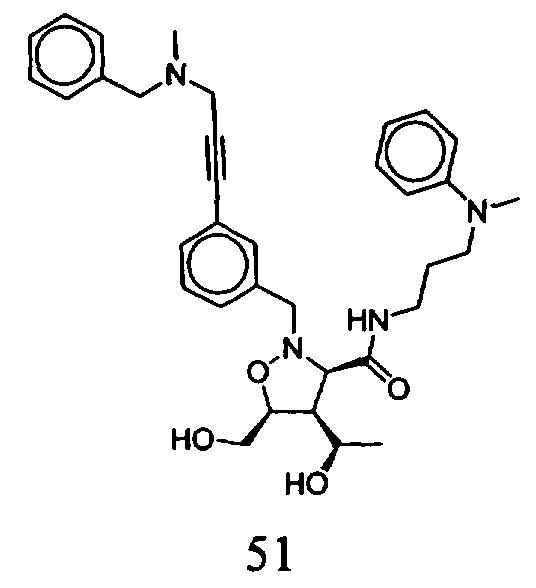
Соединение 51 синтезировали аналогично тому, как описано в примере 29, но при замене изоксазолидина 15 на изоксазолидин 18, 1-этинилциклогексиламина на бензилметилпроп-2-иниламин и с использованием N1-метил-N1-фенил-пропан-1,3-диамина. МС (ESI(+)): m/e 585,3 (M+H)+.
Пример 53
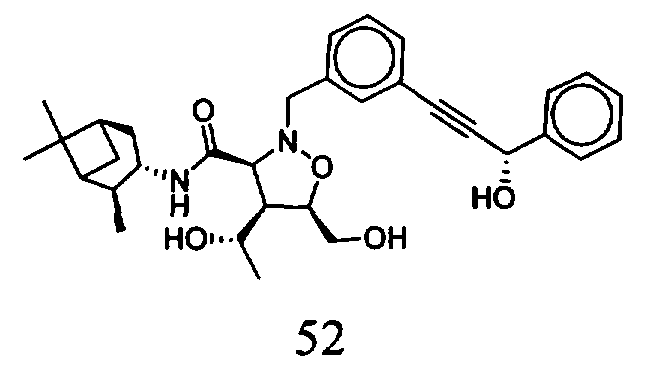
Соединение 52 синтезировали аналогично тому, как описано в примере 29, но с использованием изоксазолидина 15, 1-фенилпроп-2-ин-1-ола вместо 1-этинилциклогексиламина и (+)-изопинокамфеиламина вместо N1-метил-N1-фенил-пропан-1,3-диамина. МС (ESI(+)): m/e 547,3 (М+Н)+.
Пример 54
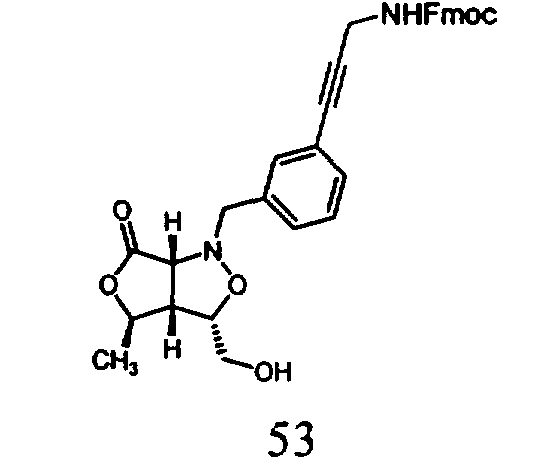
Стадия А
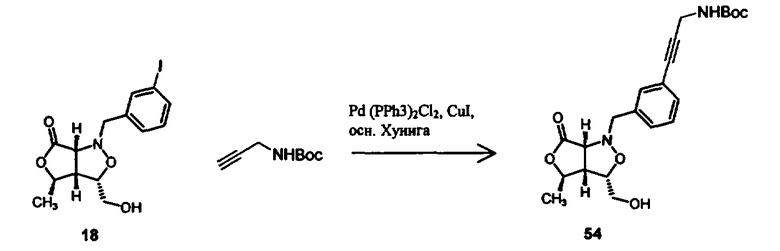
В раствор изоксазолидина 18 (0,1 г, 0,26 ммоля) добавляли N-Boc-пропаргиламин (0,08 г, 0,51 ммоля) в ДМФА (3 мл), Pd(PPh3)2Cl2 (54 мг, 0,3 ммоля), CuI (20 мг, 0,1 ммоля) и диизопропилэтиламин (0,14 мл, 0,77 ммоля). Полученную реакционную смесь выдерживали при КТ в течение 12 ч. Затем смесь распределяли между EtOAc и насыщенным водным NH4Cl (60 мл, 1:1). Водный слой экстрагировали EtOAc (2 раза по 30 мл), все органические фракции объединяли и промывали насыщенным водным раствором NH4Cl (2 раза по 50 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный материал очищали хроматографией на силикагеле (гексан/EtOAc, 1:1), при этом получали требуемый продукт (выход 90%).
Стадия В
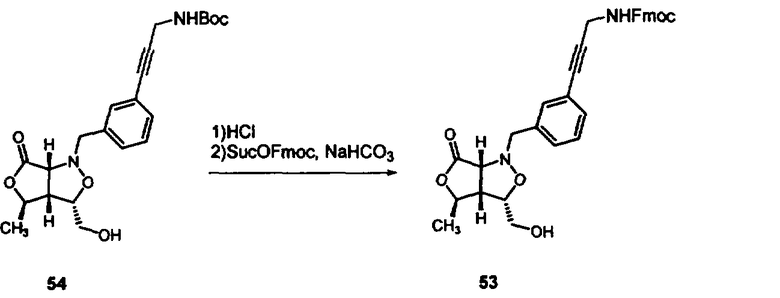
В раствор Вос-защищенного пропаргиламина 54 в EtOAc (6 мл) добавляли HCl (0,12 мл, 4 М раствор в 1,4-диоксане, 0,24 мкмоля) при 0°С. Раствор перемешивали при КТ в течение 30 мин, при этом образуется осадок. Полученную смесь перемешивали при КТ в течение 2 ч (по данным ТСХ в смеси присутствует исходный материал). В смесь добавляли дополнительное количество HCl (120 мл), смесь выдерживали при КТ в течение 24 ч. По данным ТСХ реакция полностью не завершена. Реакционную смесь концентрировали досуха, остаток растворяли в CH2Cl2 (6 мл), охлажденный до 0°С и добавляли по каплям ТФУ (1 мл). Раствор перемешивали при КТ в течение 10 мин и концентрировали в вакууме. Полученный остаток смешивали с Fmoc-сукцинамидом (0,13 г, 0,39 ммоля) и добавляли CH2Cl2/вода (1:1, 3 мл) и NaHCO3 (220 мг, 2,60 ммоля). Полученную смесь интенсивно перемешивали при КТ в течение 14 ч. Органическую фазу отделяли и водную фазу экстрагировали CH2Cl2, органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (силикагель, градиент EtOAc/гексан, от 1:1 до 5:1), при этом получали требуемый продукт (89 мг, 60%).
Пример 55
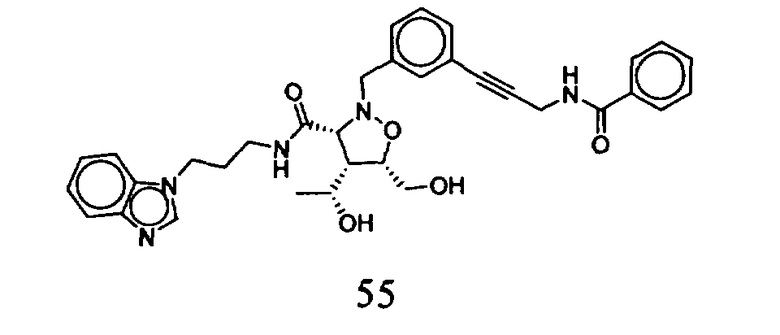
Стадия А
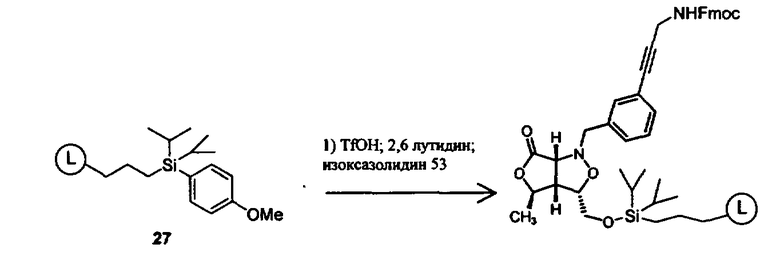
1808 частиц носителя lantern помещали в высушенную на пламени горелки колбу объемом 2 л, снабженную перемешивающим стержнем для магнитной мешалки, продували азотом и закрывали каучуковой мембраной. В колбу добавляли безводный CH2Cl2 (1,2 л), частицы носителя выдерживали в этом растворе в течение 10 мин и растворитель удаляли, добавляли 3% раствор трифторметансульфоновой кислоты в безводном CH2Cl2 (1,2 л, 393 ммолей, 3 об./об.%) и частицы носителя осторожно перемешивали в течение 20 мин. Раствор трифторметансульфоновой кислоты удаляли через трубку, добавляли безводный CH2Cl2 (1,2 л) и 2,6-лутидин (62 мл, 532 ммоля). Частицы носителя выдерживали в этом растворе при перемешивании в течение 10 мин, затем добавляли изоксазолидин 53 (10 г, 38 ммолей). Полученную смесь перемешивали в течение 18 ч, затем раствор декантировали, а частицы носителя промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (1,5 л), ТГФ (1,5 л), ТГФ/IPA (3:1, 1,5 л), ТГФ/вода (3:1, 1,5 л), ТГФ/IPA (3:1, 1,5 л) и ТГФ (1,5 л). Частицы носителя сушили в вакууме в течение ночи. Определение содержания иммобилизованного фрагмента: 5 частиц носителя каждую в отдельности помещали в полипропиленовые контейнеры объемом 5 мл. В каждый контейнер добавляли ТГФ (300 мкл) и HF-пиридин (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч. Затем добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Затем раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали изоксазолидин 53. Этот материал взвешивали и рассчитывали содержание иммобилизованного фрагмента в частице носителя, которое составляет 10 мкмолей/частица.
Стадия В
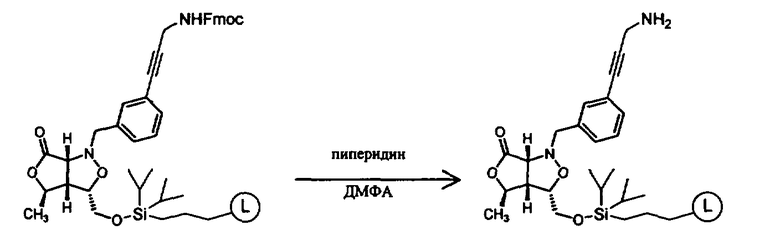
9 Частиц носителя lantern (0,09 ммоля, 1 экв.) суспендировали в 20% растворе пиперидина в ДМФА (9 мл). Реакционную смесь встряхивали в течение 1 ч, затем раствор из реактора откачивали и добавляли 20% пиперидина в ДМФА (9 мл). Реакционную смесь встряхивали в течение 1 ч. Затем частицы носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (9 мл), ТГФ (9 мл), ТГФ/IPA (3:1,9 мл), ТГФ/вода (3:1,9 мл), ТГФ/IPA (3:1, 9 мл) и ТГФ (9 мл). Определение степени превращения иммобилизованного фрагмента: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу выдерживали в этом растворе в течение 6 ч, затем добавляли TMSOMe (500 мкл) и частицу выдерживали в этом растворе еще в течение 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали свободный амин.
Стадия С
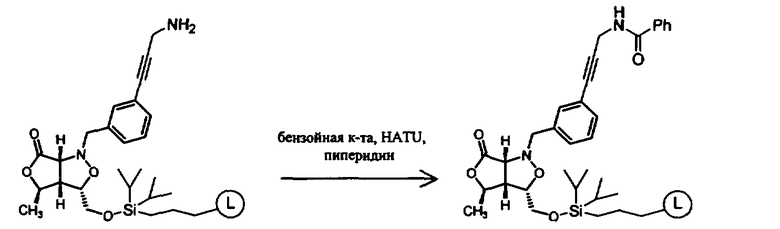
1 Частицу носителя lantern (0,01 ммоля, 1 экв.) помещали в раствор ДМФА/ТГФ (1:1, 0,8 мл). Затем добавляли бензойную кислоту (0,02 г, 0,20 ммоля, 20 экв.), HATU (0,02 г, 0,1 ммоля, 10 экв.) и пиридин (0,014 г, 0,10 ммоля, 10 экв.). Реакционную смесь встряхивали в течение 17 ч в атмосфере азота, затем частицу удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (1 мл), ТГФ (1 мл), ТГФЛРА (3:1,1 мл), ТГФ/вода (3:1,1 мл), ТГФЛРА (3:1, 1 мл) и ТГФ (1 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу выдерживали в этом растворе в течение 6 ч, затем добавляли TMSOMe (500 мкл) и частицу выдерживали в этом растворе еще в течение еще 15 мин. Раствор из контейнера переносили в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт.
Стадия D
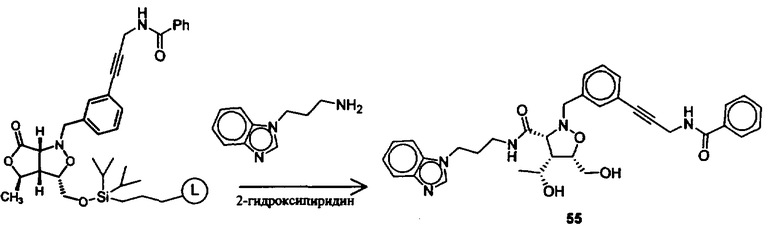
52 Частицы носителя lantern (0,52 ммоля, 1 экв.) помещали в герметичную колбу и продували азотом. В колбу добавляли 2-гидроксипиридин (30 мл, 0,35 М раствор в ТГФ, 20 экв.) и 3-бензимидазол-1-илпропиламин (6,38 г, 36,4 ммоля, 70 экв.). Реакционную смесь встряхивали в течение 27 ч при 50°С в атмосфере азота, затем частицы удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (57 мл), ТГФ (57 мл), ТГФ/1РА (3:1, 57 мл), ТГФ/вода (3:1, 57 мл), ТГФ/IPA (3:1, 57 мл) и ТГФ (57 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу выдерживали в этом растворе в течение 6 ч, добавляли TMSOMe (500 мкл) и частицу выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переносили в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт.
Пример 56
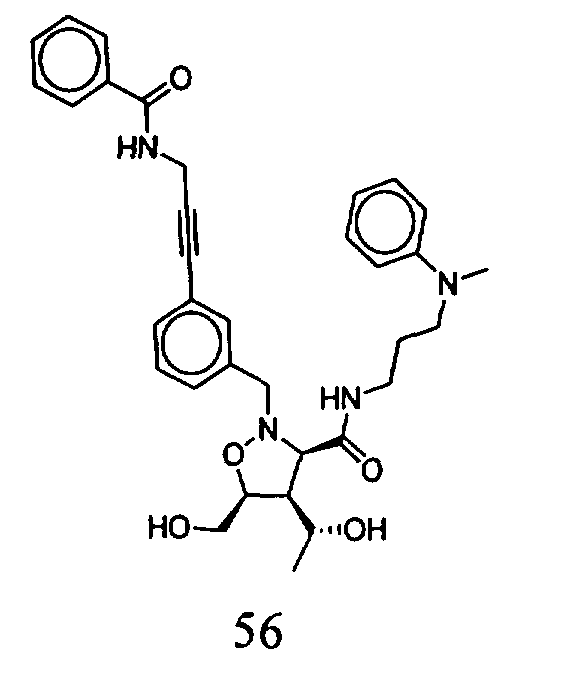
Соединение 56 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на N1-метил-N1-фенилпропан-1,3-диамин. МС (ESI(+)): m/e 585,4 (M+H)+.
Пример 57
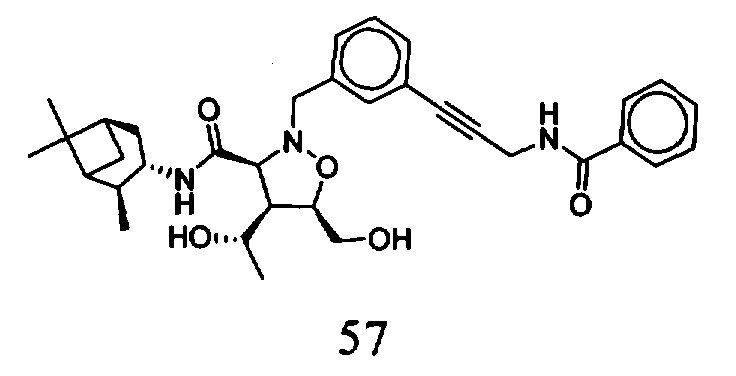
Соединение 57 синтезировали аналогично тому, как описано в примере 55, но при замене изоксазолидина 18 на изоксазолидин 15 и при замене 3-бензимидазол-1-илпропиламина на (+)-изопинокамфеиламин. МС (ESI(+)): m/e 574,4 (М+Н)+.
Пример 58
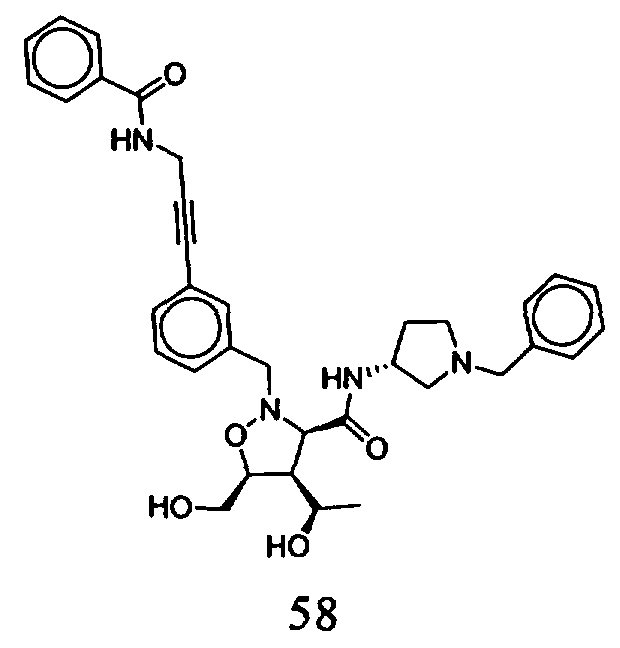
Соединение 58 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на 1-бензилпирролидин-3-иламин.
МС (ESI(+)): m/e 597,4 (М+Н)+.
Пример 59
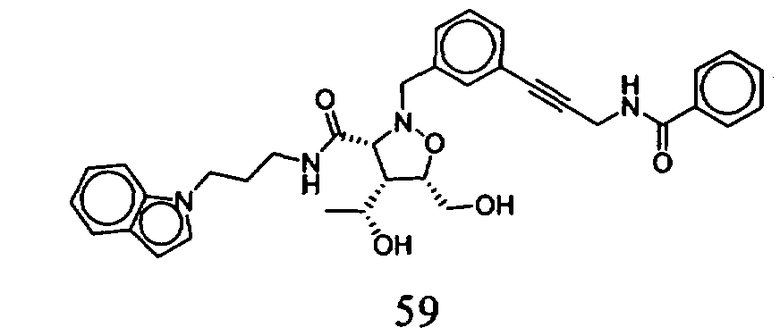
Соединение 59 синтезировали аналогично тому, как описано в примере 55, но при замене изоксазолидина 18 на изоксазолидин 15 и 3-бензимидазол-1-ил-пропиламина на 3-индол-1-илпропиламин. МС (ESI(+)): m/e 574,4 (М+Н)+.
Пример 60
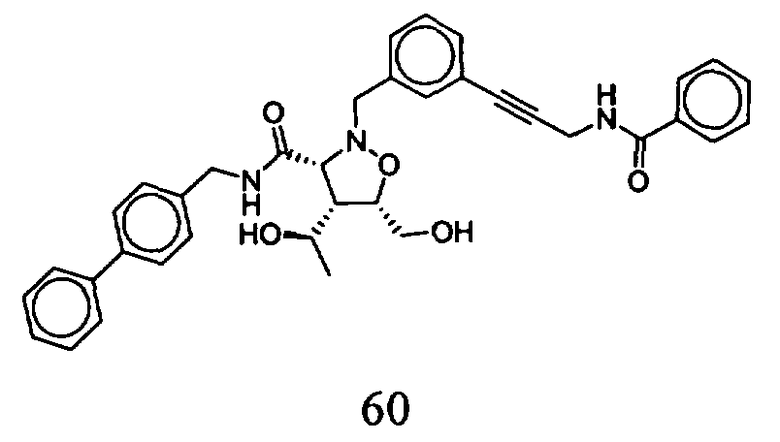
Соединение 60 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на 4-фенилбензиламин.
Пример 61
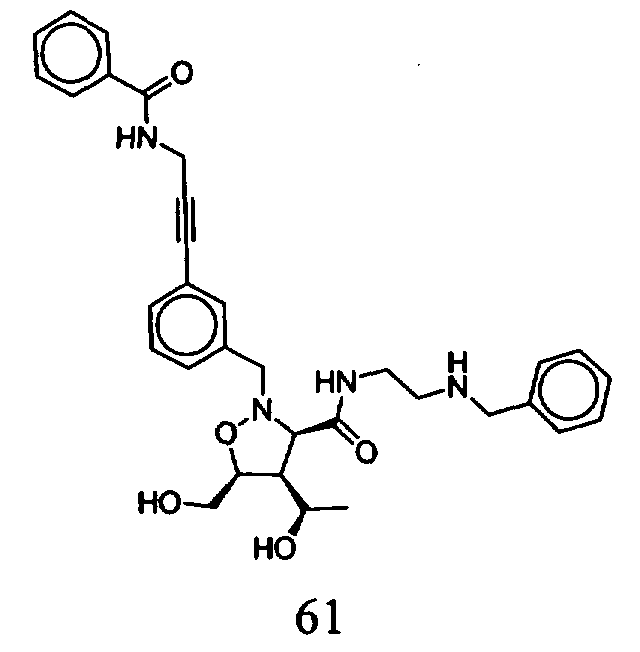
Соединение 61 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на N1-бензилэтан-1,2-диамин.
Пример 62
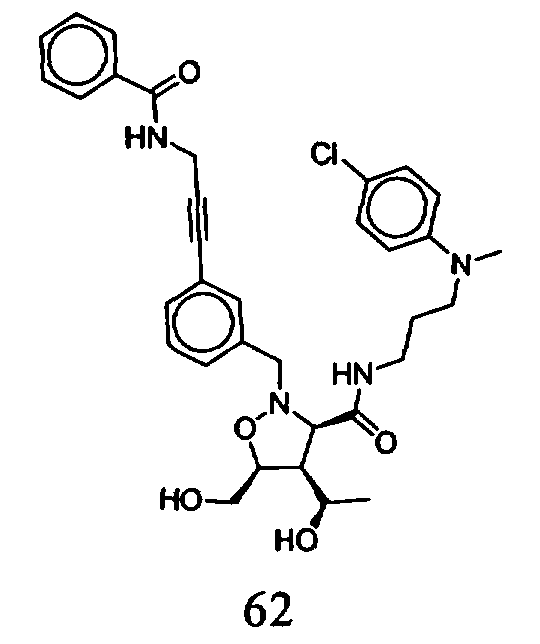
Соединение 62 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на N1-(4-хлорфенил)-N1-метилпропан-1,3-диамин.
Пример 63
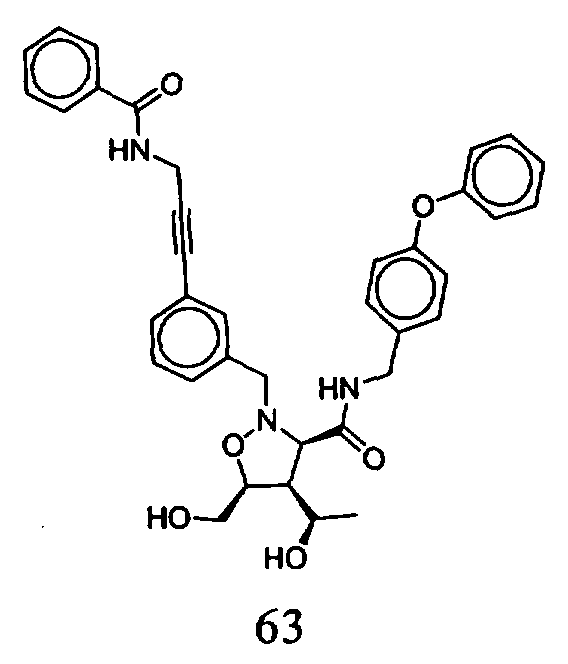
Соединение 63 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на 4-феноксибензиламин.
Пример 64
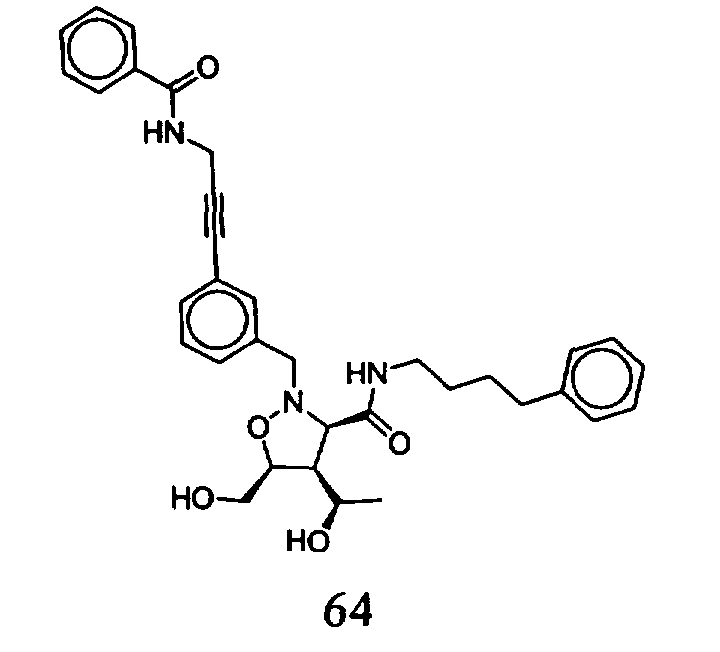
Соединение 64 синтезировали аналогично тому, как описано в примере 55, но при замене 3-бензимидазол-1-илпропиламина на 4-фенилбутиламин.
Пример 65
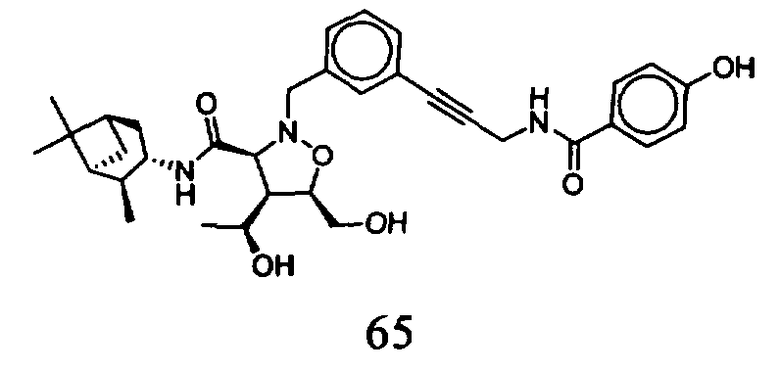
Соединение 65 синтезировали аналогично тому, как описано в примере 55, но при замене изоксазолидина 18 на изоксазолидин 15, 3-бензимидазол-1-илпропиламина на (+)-изопинокамфеиламин, и бензойной кислоты на 4-гидроксибензойную кислоту. МС (ESI(+)): m/e 590,5 (М+Н)+.
Пример 66
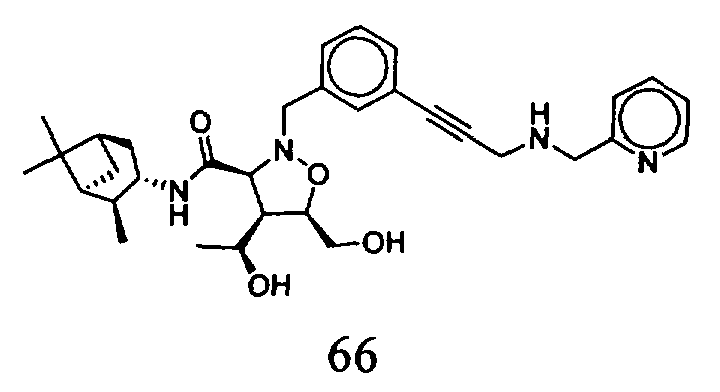
Стадия А
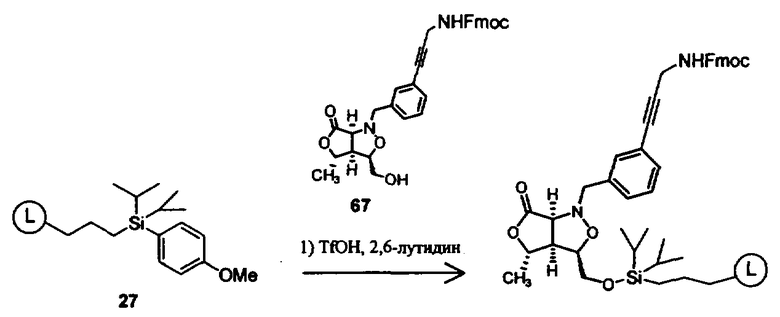
1808 Частиц носителя lantern помещали в высушенную на пламени горелки колбу объемом 2 л, снабженную перемешивающим стержнем для магнитной мешалки, колбу продували азотом и закрывали каучуковой мембраной, в колбу добавляли CH2Cl2 (1,2 л) и частицы носителя выдерживали в этом растворе в течение 10 мин, затем растворитель удаляли. В колбу добавляли раствор трифторметансульфоновой кислоты в безводном CH2Cl2 (1,2 л, 393 ммоля, 3 об./об.%) и частицы носителя осторожно перемешивали в течение 20 мин. Затем раствор трифторметансульфоновой кислоты удаляли через трубку, добавляли безводный CH2Cl2 (1,2 л) и 2,6-лутидин (62 мл, 532 ммоля). Частицы носителя перемешивали в этом растворе в течение 10 мин, добавляли изоксазолидин 67 (полученный как описано в примере 54, 10 г, 38 ммолей). Полученную смесь перемешивали в течение 18 ч, раствор из реактора откачивали, а частицы носителя промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (1,5 л), ТГФ (1,5 л), ТГФ/IPA (3:1, 1,5 л), ТГФ/вода (3:1, 1,5 л), ТГФ/IPA (3:1, 1,5 л) и ТГФ (1,5 л). Затем частицы носителя сушили при пониженном давлении. Определение содержания иммобилизованного фрагмента в частицах носителя: 5 частиц носителя помещали каждую в отдельности в полипропиленовые контейнеры объемом 5 мл. В каждый контейнер добавляли ТГФ (300 мкл) и HF-пиридин (50 мкл). Частицы носителя выдерживали в этом растворе в течение 6 ч, добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переливали в колбу и концентрировали в вакууме, при этом получали изоксазолидин 67. Полученный материал взвешивали и рассчитывали среднее содержание фрагмента на частицу носителя, которое составляет 10 мкмоля/частица.
Стадия В
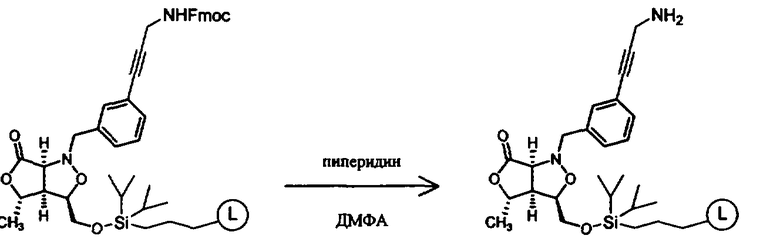
9 Частиц носителя lantern (полученных, как описано выше в примерах, 0,09 ммоля, 1 экв.) суспендировали в 20% растворе пиперидина в ДМФА (9 мл). Реакционную смесь встряхивали в течение 1 ч, раствор из реактора откачивали и добавляли дополнительную порцию пиперидина в ДМФА (20 об./об.%, 9 мл). Реакционную смесь встряхивали в течение 1 ч. Частицы носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (9 мл), ТГФ (9 мл), ТГФ/IPA (3:1, 9 мл), ТГФ/вода (3:1,9 мл), ТГФ/IPA (3:1, 9 мл) и ТГФ (9 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч, добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе еще в течение 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали в вакууме, при этом получали свободный амин.
Стадия С
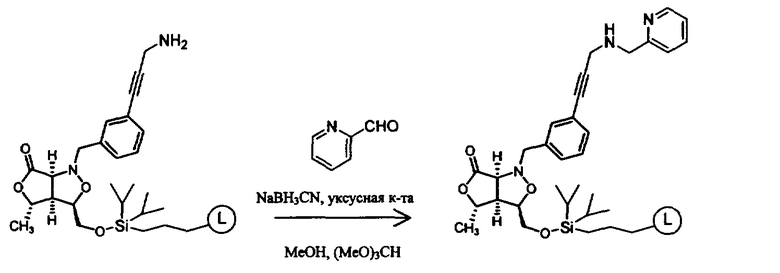
4 Частицы носителя lantern (полученных как описано выше, в примерах, 0,04 ммоля, 1 экв.) помещали в стеклянный флакон, добавляли пиридин-2-карбоксальдегид (0,08 г, 0,8 ммоля, 20 экв.), затем триметилортоформиат (4 мл). Частицы носителя выдерживали в этом растворе в течение 45 мин при осторожном встряхивании, добавляли NaBH3CN (0,13 г, 2,1 ммоля, 30 экв.), уксусную кислоту (одну каплю) и МеОН (одну каплю), реакционную смесь встряхивали в атмосфере азота в течение 36 ч. Частицы носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (4 мл), ТГФ (4 мл), ТТФ/IPA (3:1, 4 мл), ТГФ/вода (3:1,4 мл), ТТФ/IPA (3:1, 4 мл) и ТГФ (4 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч. Затем добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе еще в течение 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт.
Стадия D
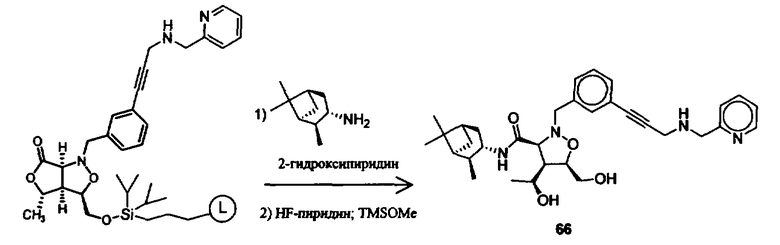
52 частицы носителя lantern (0,52 ммоля, 1 экв.) помещали в колбу высокого давления и продували азотом. В колбу добавляли 2-гидроксипиридин (30 мл, 0,35 М раствор в ТГФ, 20 экв.) и (+)-изопинокамфеиламин (5,57 г, 36,4 ммоля, 70 экв.). Реакционную смесь встряхивали в течение 27 ч при 50°С в атмосфере азота, частицы носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (57 мл), ТГФ (57 мл), ТГФ/IPA (3:1, 57 мл), ТГФ/вода (3:1, 57 мл), ТГФ/IPA (3:1, 57 мл) и ТГФ (57 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч. Затем добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт. МС (ESI(+)): m/e 561,5 (M+H)+.
Пример 67
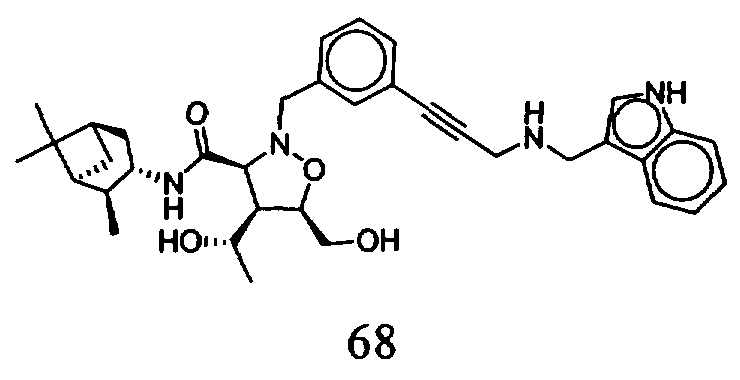
Соединение 68 синтезировали аналогично тому, как описано в примере 66, но при замене пиридин-2-карбальдегида на индол-3-карбоксальдегид. МС (ESI(+)): m/e 599,0 (М+Н)+.
Пример 68
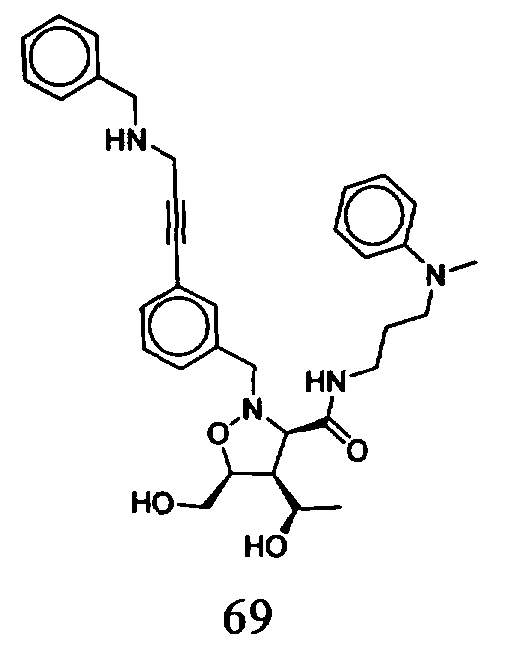
Соединение 69 синтезировали аналогично тому, как описано в примере 66, но при замене изоксазолидина 15 на изоксазолидин 18, пиридин-2-карбоксальдегида на бензальдегид и (+)-изопинокамфеиламина на N1-метил-N1-фенилпропан-1,3-диамин. МС (ESI(+)): m/e 607,0 (М+Н)+.
Пример 69
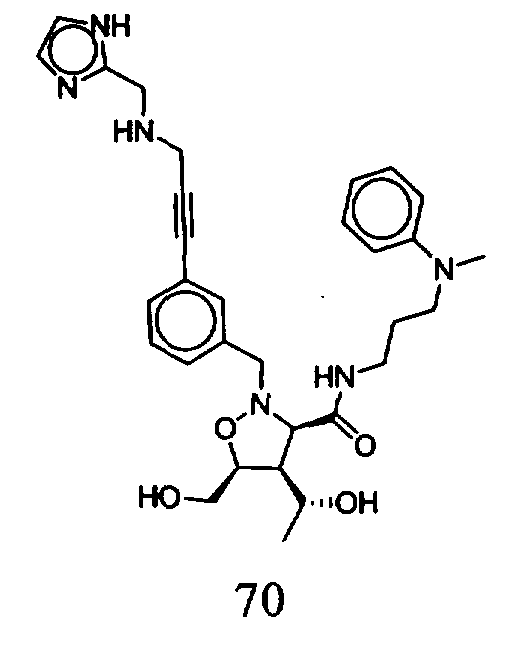
Соединение 70 синтезировали аналогично тому, как описано в примере 66, но при замене изоксазолидина 15 на изоксазолидин 18, пиридин-2-карбоксальдегида на имидазол-2-карбальдегид и (+)-изопинокамфеиламина на N1-метил-N1-фенилпропан-1,3-диамин. МС (ESI(+)): m/e 607,5 (М+Н)+.
Пример 70
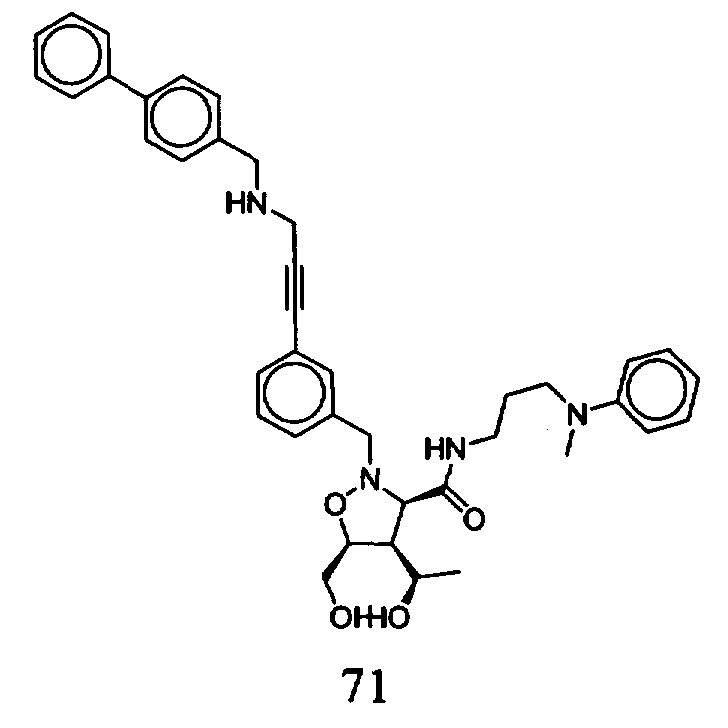
Соединение 71 синтезировали аналогично тому, как описано в примере 66, но при замене изоксазолидина 15 на изоксазолидин 18, пиридин-2-карбоксальдегида на 4-фенилбензальдегид и (+)-изопинокамфеиламина на N1-метил-N1-фенилпропан-1,3-диамин. МС (ESI(+)): m/e 647,5 (M+H)+.
Пример 71
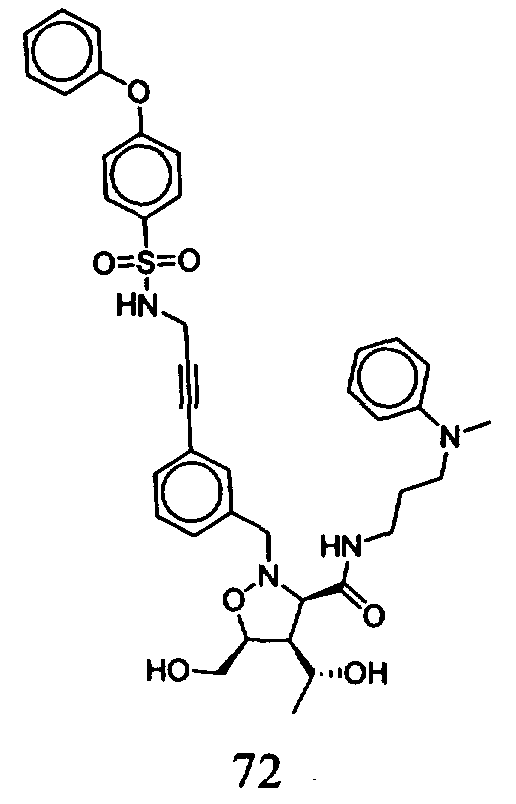
Стадия А
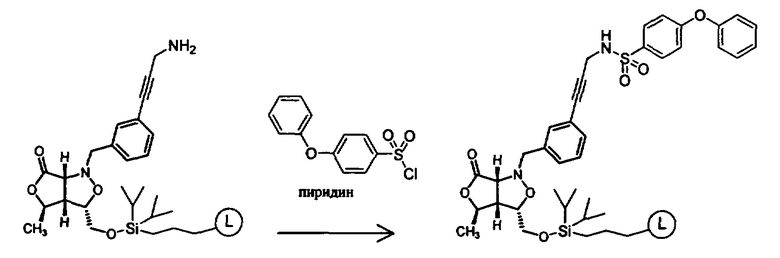
1 Частицу, полученную, как описано выше в примерах, добавляли в стеклянный флакон, затем добавляли 4-феноксибензолсульфонилхлорид (0,22 г, 20 экв.), CH2Cl2 (0,4 мл), и пиридин (18 мкл, 20 экв.). Реакционную смесь осторожно встряхивали в течение 5 сут. в атмосфере азота, частицу носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (1 мл), ТГФ (1 мл), ТГФ/IPA (3:1,1 мл), ТГФ/вода (3:1,1 мл), ТГФ/IPA (3:1, 1 мл) и ТГФ (1 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч, добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переливали и концентрировали при пониженном давлении, при этом получали требуемый продукт.
Стадия В
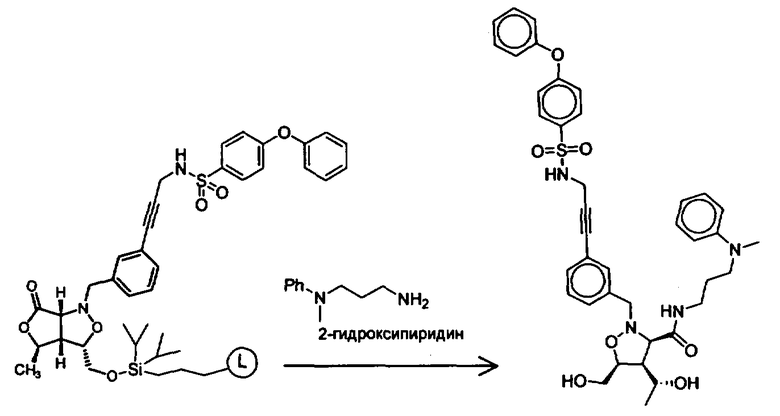
52 Частицы носителя lantern (0,52 ммоля, 1 экв.) помещали в колбу высокого давления и продували азотом. В колбу добавляли 2-гидроксипиридин (30 мл 0,35 М раствор в ТГФ, 20 экв.) и N1-метил-N1-фенил-пропан-1,3-диамин (5,97 г, 36,4 ммоля, 70 экв.). Реакционную смесь встряхивали в течение 27 ч при 50°С в атмосфере азота. Затем частицу носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (57 мл), ТГФ (57 мл), ТГФ/IPA (3:1, 57 мл), ТГФ/вода (3:1, 57 мл), ТГФ/IPA (3:1, 57 мл) и ТГФ (57 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч. Затем добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт. МС (ESI(+)): m/e 713,3 (М+Н)+.
Пример 72
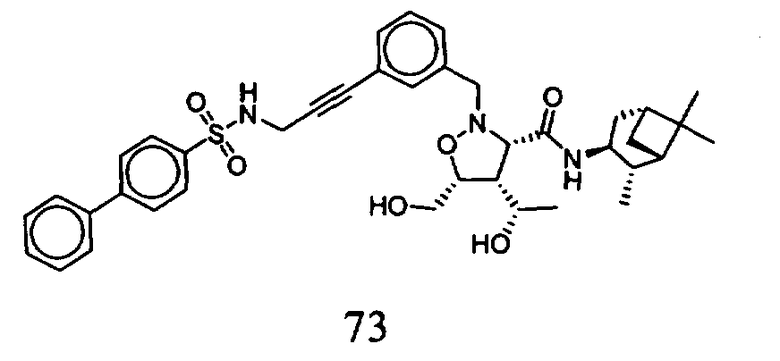
Соединение 73 синтезировали аналогично тому, как описано в примере 71, но при замене изоксазолидина 18 на изоксазолидин 15, N1-метил-N1-фенилпропан-1,3-диамина на (+)-изопинокамфеиламин, и 4-феноксибензолсульфонилхлорида на бифенил-4-сульфонилхлорид. МС (ESI(+)): m/e 686,5 (М+Н)+.
Пример 73
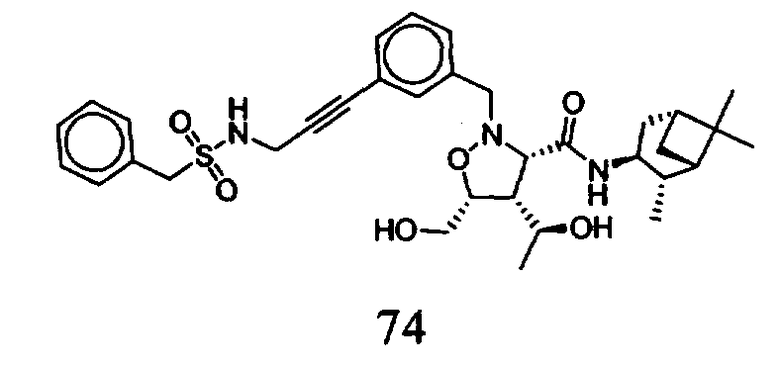
Соединение 74 синтезировали аналогично тому, как описано в примере 71, но при замене изоксазолидина 18 на изоксазолидин 15, N1-метил-N1-фенил-пропан-1,3-диамина на (+)-изопинокамфеиламин, и 4-феноксибензолсульфонилхлорида на фенилметансульфонилхлорид. МС (ESI(+)): m/e 624,5 (М+Н)+.
Пример 74
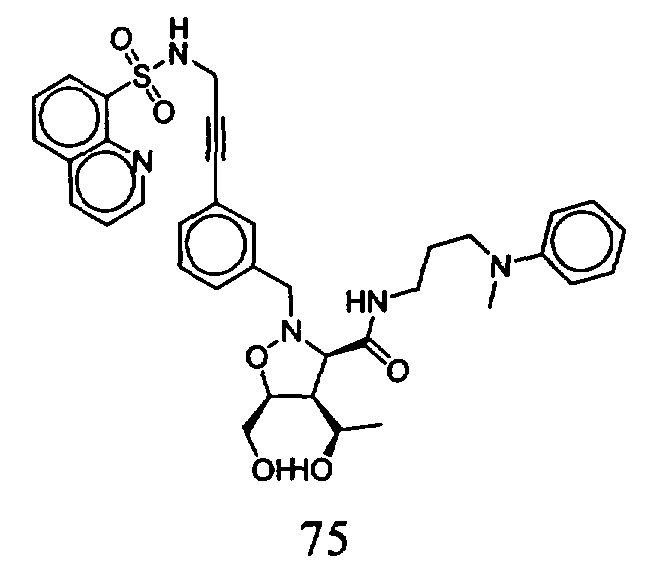
Соединение 75 синтезировали аналогично тому, как описано в примере 71, но при замене 4-феноксибензолсульфонилхлорида на хинолин-8-сульфонилхлорид. МС (ESI(+)): m/e 672,5 (М+Н)+.
Пример 75
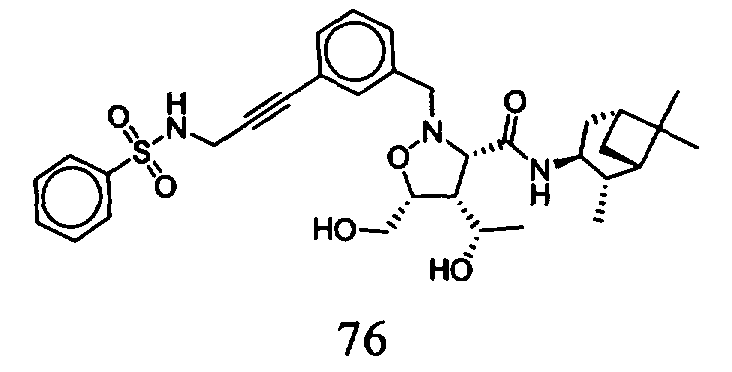
Соединение 76 синтезировали аналогично тому, как описано в примере 71, но при замене изоксазолидина 18 на изоксазолидин 15, N1-метил-N1-фенилпропан-1,3-диамина на (+)-изопинокамфеиламин, и 4-феноксибензолсульфонилхлорида на фенилсульфонилхлорид. МС (ESI(+)): m/e 610,5 (М+Н)+.
Пример 76
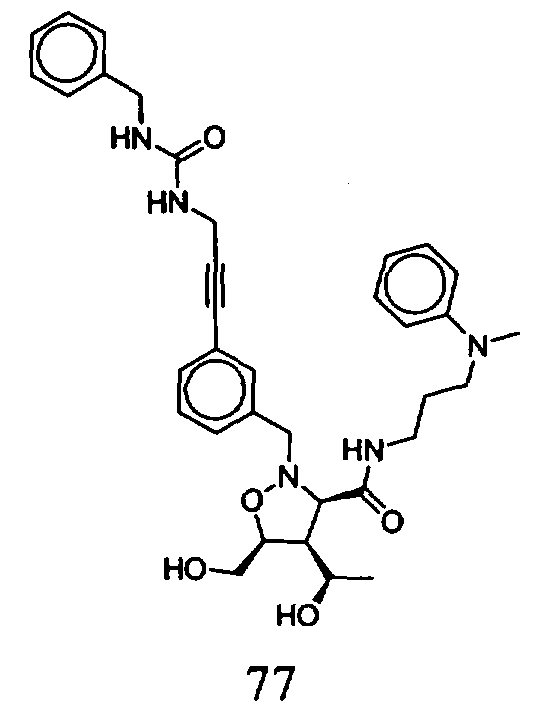
Стадия А
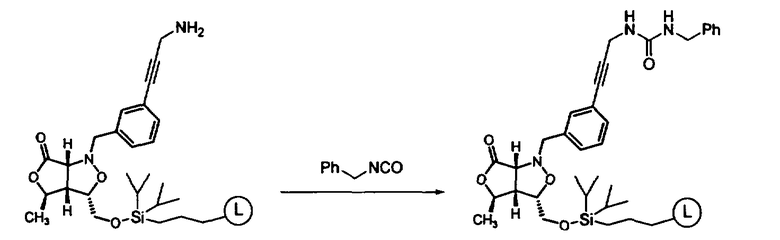
1 Частицу добавляли в стеклянный флакон, затем добавляли изоцианатометилбензол (26 мг, 10 экв.) и CH2Cl2 (0,8 мл). Реакционную смесь осторожно встряхивали в течение 24 ч, частицу удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (1 мл), ТГФ (1 мл), ТГФ/IPA (3:1,1 мл), ТГФ/вода (3:1,1 мл), ТГФ/IPA (3:1, 1 мл) и ТГФ (1 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч, добавляли TMSOMe (500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переливали в стеклянную круглодонную колбу и концентрировали при пониженном давлении, при этом получали требуемый продукт.
Стадия В
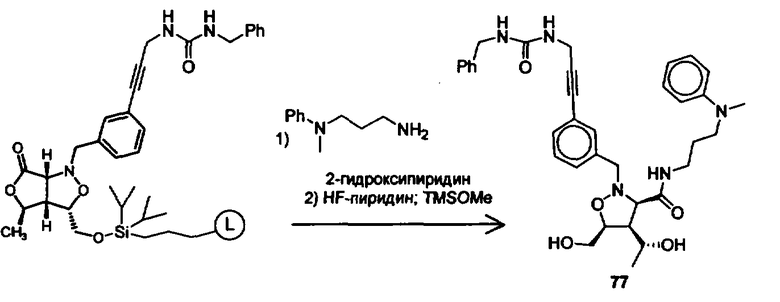
52 частицы носителя lantern (полученные как описано выше в примерах, 0,52 ммоля, 1 экв.) помещали в колбу высокого давления и продували азотом. В колбу добавляли 2-гидроксипиридин (0,35 М в ТГФ, 30 мл, 20 экв.) и N1-метил-N1-фенилпропан-1,3-диамин (5,97 г, 36,4 ммоля, 70 экв.). Реакционную смесь встряхивали в течение 27 ч при 50°С в атмосфере азота, затем частицы носителя удаляли и промывали следующим образом: (2 раза в течение 10 мин) CH2Cl2 (57 мл), ТГФ (57 мл), ТГФ/IPA (3:1, 57 мл), ТГФ/вода (3:1, 57 мл), ТГФ/IPA (3:1, 57 мл) и ТГФ (57 мл). Определение степени превращения: 1 частицу помещали в полипропиленовый контейнер объемом 5 мл и обрабатывали ТГФ (300 мкл) и HF-пиридином (50 мкл). Частицу носителя выдерживали в этом растворе в течение 6 ч, добавляли TMSOMe
(500 мкл) и частицу носителя выдерживали в этом растворе в течение еще 15 мин. Раствор из контейнера переливали и концентрировали при пониженном давлении, при этом получали требуемый продукт. МС (ESI(+)): m/e 614,5 (М+Н)+.
Пример 77
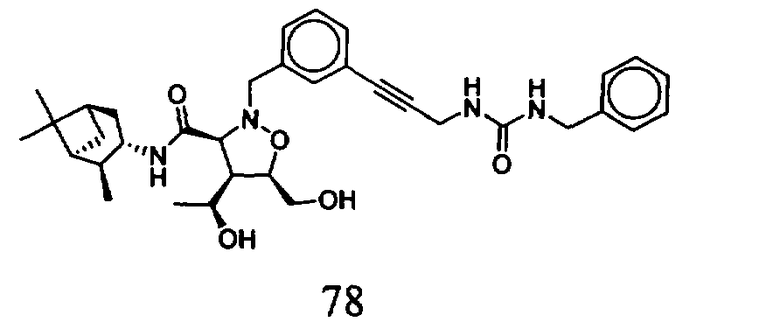
Соединение 78 синтезировали аналогично тому, как описано в примере 76, но при замене изоксазолидина 15 на изоксазолидин 18, N1-метил-N1-фенил-пропан-1,3-диамина на (+)-изопинокамфеиламин, и с использованием изоцианатометилбензола. МС (ESI(+)): m/e 603,5 (М+Н)+.
Пример 78
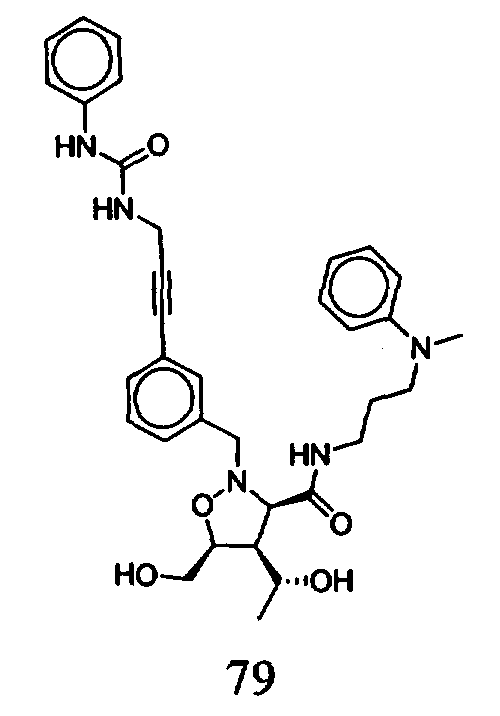
Соединение 79 синтезировали аналогично тому, как описано в примере 76, но при замене изоцианатометилбензола на фенилизоцианат. МС (ESI(+)): m/e 600,5 (М+Н)+.
Пример 79
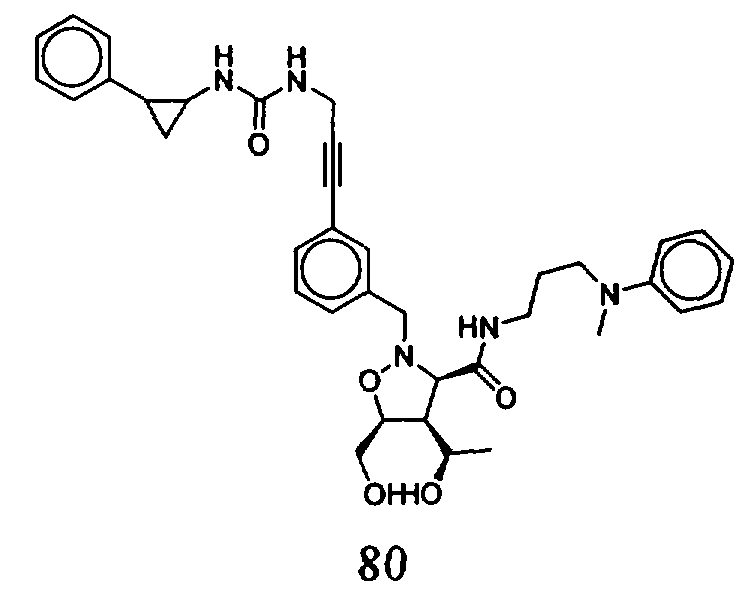
Соединение 80 синтезировали аналогично тому, как описано в примере 76, но при замене изоцианатометилбензола на 1-изоцианато-2-фенилциклопропан. МС (ESI(+)): m/e 640,5 (М+Н)+.
Пример 80
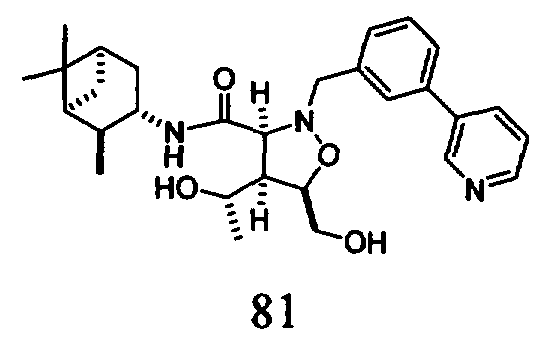
Стадия А
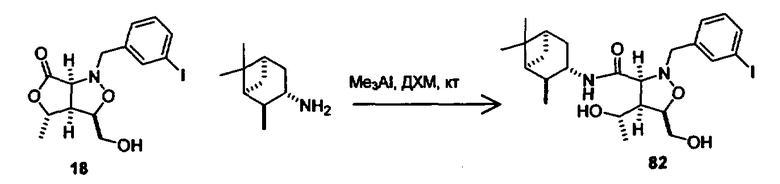
В раствор (+) изопинокамфеиламина (0,2 г, 1,3 ммоля) в CH2Cl2 (10 мл) при КТ добавляли по каплям AlMe3 (0,85 мл, 2 М раствор в толуоле, 1,7 ммоля) в течение 2,5 мин. Раствор перемешивали при КТ в течение 10 мин, затем по каплям добавляли раствор лактона 18 (0,5 г, 0,02 ммоля) в CH2Cl2 (10 мл). Реакционную смесь перемешивали в течение 1 ч, разбавляли CH2Cl2 (125 мл) и насыщенным водным раствором сегнетовой соли (125 мл). Смесь интенсивно перемешивали в течение 2 ч до образования двух фаз. Слои разделяли, органическую фазу промывали водой, солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали твердое вещество. Этот материал использовали на следующей стадии без предварительной очистки. Выход неочищенного продукта составляет 0,7 г (100%).
Стадия В
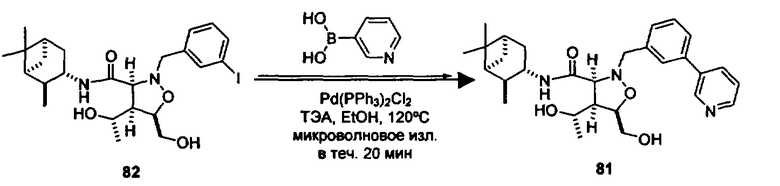
Изоксазолидин 82 (30 мг, 0,06 ммоля), 3-пиридилбороновую кислоту (7 мг, 0,06 ммоля), Pd(PPh3)2Cl2 (39 мг, 0,06 ммоля), Et3N (17 мг, 0,18 ммоля) и EtOH (0,5 мл) смешивали при КТ. Эту смесь нагревали при 120°С в микроволновом реакторе в течение 30 мин. Реакционную смесь концентрировали в вакууме и полученный остаток очищали экспресс-хроматографией, при этом получали продукт 81 (10 мг, 30%) в виде масла бледно-желтого цвета. МС (ESI(+)): m/e 494,3 (М+Н)+.
Пример 81
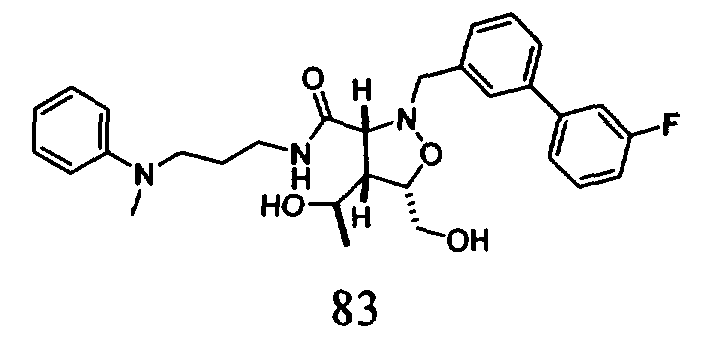
Соединение 83 синтезировали аналогично тому, как описано в примере 80, но при замене изоксазолидина 18 на изоксазолидин 15, (+)-изопинокамфеиламина на N1-метил-N1-фенилпропан-1,3-диамин, и с использованием 3-фторфенилбороновой кислоты (выход 25%). МС (ESI(+)): m/e 522,3 (М+Н)+.
Пример 82
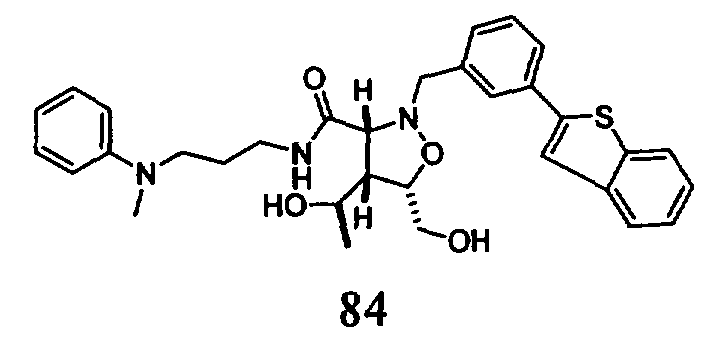
Соединение 84 синтезировали аналогично тому, как описано в примере 80, но при замене изоксазолидина 18 на изоксазолидин 15, (+)-изопинокамфеиламина на N1-метил-N1-фенилпропан-1,3-диамин, и с использованием бензотиофенил-2-бороновой кислоты (выход 17%).
Пример 83
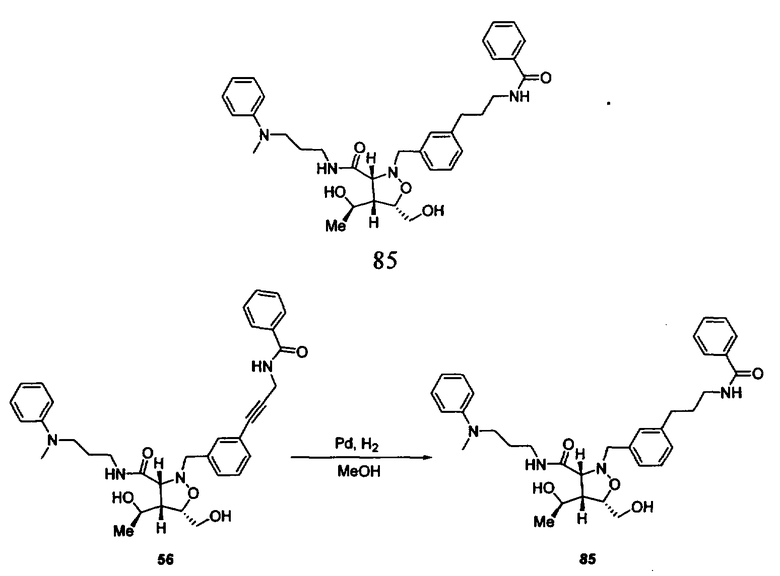
Изоксазолидин 56 (20 мг, 0,05 ммоля) растворяли в МеОН (10 мл) и обрабатывали в атмосфере водорода в присутствии 5 мас.% Pd на угле (11 мг, 0,095 ммоля) в течение 12 ч, раствор фильтровали через целит и концентрировали в вакууме, при этом получали масло, которое очищали препаративной хроматографией на пластинке (элюент: CH2Cl2/MeOH, 9:1), при этом получали продукт 85 (18 мг, 62%) в виде прозрачного масла. МС (ESI(+)): m/e 589,3 (М+Н)+.
Пример 84
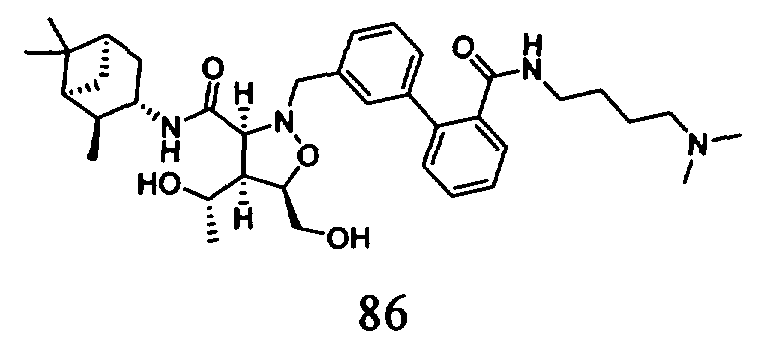
Стадия А
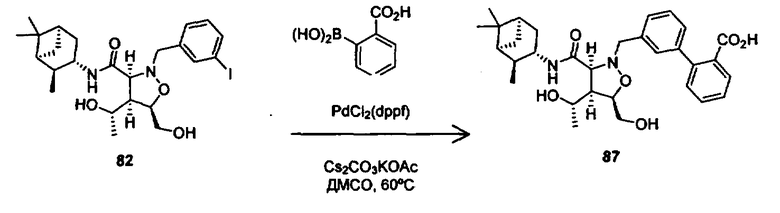
В колбу добавляли продукт 82 (0,33 г, 0,6 ммоля), 2-карбоксифенилбороновую кислоту (0,2 г, 1,2 ммоля), Cs2CO3 (0,62 г, 1,9 ммоля), КОАс (60 мг, 0,6 ммоля, 1,0 экв.) и PdCl2(dppf) (34,2 мг, 4,2 мол.%). Колбу продували аргоном, добавляли дегазированный ДМСО (продували аргоном в течение 30 мин, 10 мл) и нагревали до 60°С в атмосфере аргона. Через 5,5 ч добавляли еще одну порцию PdCl2(dppf) (34 мг, 4,1 мол.%) и смесь нагревали в течение 12 ч, завершение реакции определяли методами ЖХ-МС и ТСХ (элюент: гексан/EtOAc/АсОН 30:70:0,5, Rf0,38). Реакционную смесь разбавляли смесью CH2Cl2/вода (2:1, 45 мл) и подкисляли 6 М HCl (0,5 мл) до рН 2. Водный слой отделяли и экстрагировали CH2Cl2 (3 раза по 10 мл). Объединенные органические слои промывали водой (15 мл) и солевым раствором (15 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали хроматографией (силикагель, нанесение 75:25, элюент гексан/EtOAc/АсОН 50:50:0,5, 25:75:0,5 (2 раза), 0:100:0,5 200 мл), при этом получали продукт 87 в виде аморфного твердого вещества оранжевого цвета (265 мг, 80%).
Стадия В
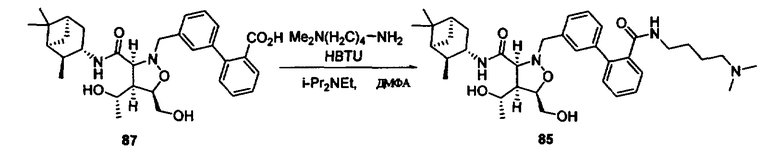
В раствор соединения 87 в ДМФА (0,7 мл) (15 мг, 0,03 ммоля) добавляли амин (13 мг, 0,12 ммоля), диизопропилэтиламин (15 мг, 20 мкл, 0,12 ммоля) и HBTU (23 мг, 0,06 ммоля). Реакционную смесь перемешивали при комнатной температуре. Через 90 мин добавляли еще одну порцию амина (6 мг, 0,03 ммоля), HBTU (11 мг, 0,03 ммоля) и диизопропилэтиламина (7 мг, 10 мкл). Через 2 ч реакционную смесь разбавляли МеОН (0,7 мл), и очищали методом ЖХВР, при этом получали продукт 85 (10 мг, 52%). МС (ESI(+)): m/e 635,4 (M+H)+.
Пример 85
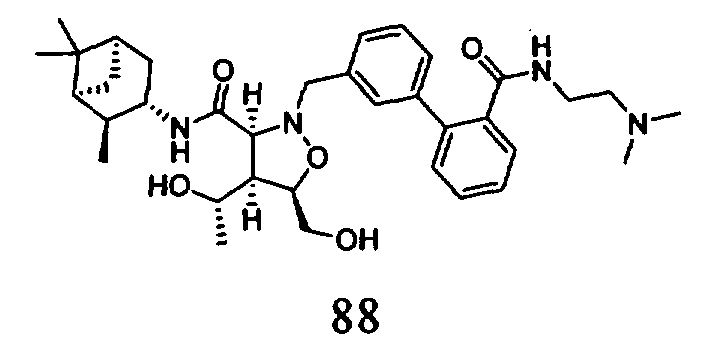
Соединение 88 синтезировали аналогично тому, как описано в примере 84, но при замене N,N-диметилбутиламина на N,N-диметилэтиламин (выход составляет 40-70%). МС (ESI(+)): m/e 635,4 (М+Н)+.
Пример 86
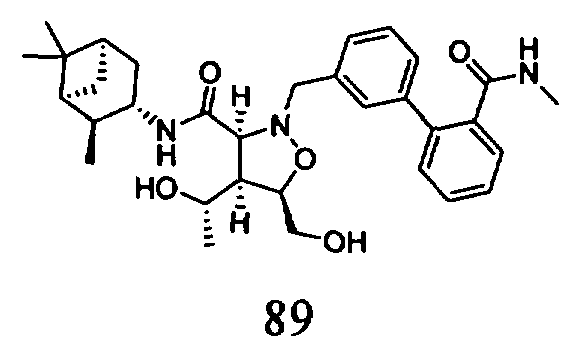
Соединение 89 синтезировали аналогично тому, как описано в примере 84, но при замене N,N-диметилбутиламина на метиламин (выход составляет 40-70%). МС (ESI(+)): m/e 550,2 (M+H)+.
Пример 87
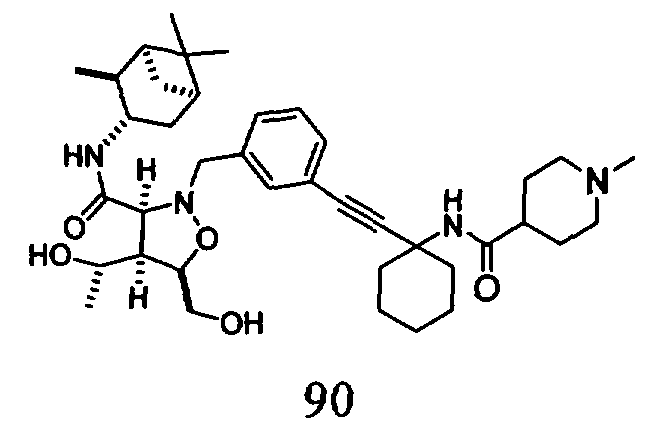
Стадия А
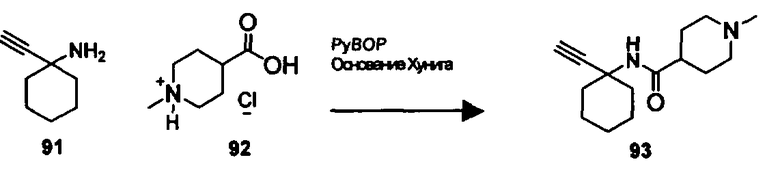
В раствор соединения 91 (0,5 г, 4,1 ммоля) в ДМФА (37 мл) добавляли диизопропилэтиламин (2,54 мл, 14,6 ммоля), соединение 92 (0,5 г, 4,1 ммоля) и PyBOP (2,53 г, 4,87 ммоля). Смесь перемешивали при КТ в течение 17 ч, по данным ТСХ (МеОН) в реакционной смеси присутствуют два основных компонента. Реакционную смесь разбавляли водой и водный слой экстрагировали EtOAc. Органические экстракты объединяли, сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали продукт в виде масла. Неочищенный продукт очищали экспресс-хроматографией, при этом получали продукт 93 (0,9 г, 90%).
Стадия В
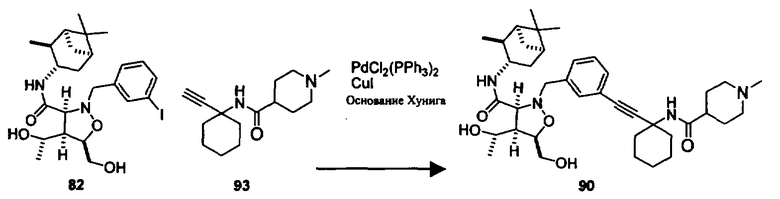
В смесь соединения 82 (70 мг, 0,1 ммоля), CuI (8 мг, 0,04 ммоля), Pd(PPh3)2Cl2 (22 мг, 0,03 ммоля) в ДМФА (3 мл) добавляли соединение 93 (50 мг, 0,21 ммоля) с последующим добавлением диизопропилэтиламина (40 мг, 54,8 мкл, 0,32 ммоля). Полученную смесь темно-коричневого цвета перемешивали при комнатной температуре в темноте в течение 2 сут. Реакционную смесь разбавляли смесью EtOAc/вода (6 мл, 1:1) и раствор нейтрализовали добавлением 1 М HCl до рН 7. Водную фазу отделяли и экстрагировали EtOAc (2 раза по 4 мл). Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, при этом получали масло. Неочищенный материал очищали колоночной хроматографией, при этом получали продукт 90 (48 мг, 70%). МС (ESI(+)): m/e 663,5 (M+H)+.
Пример 88
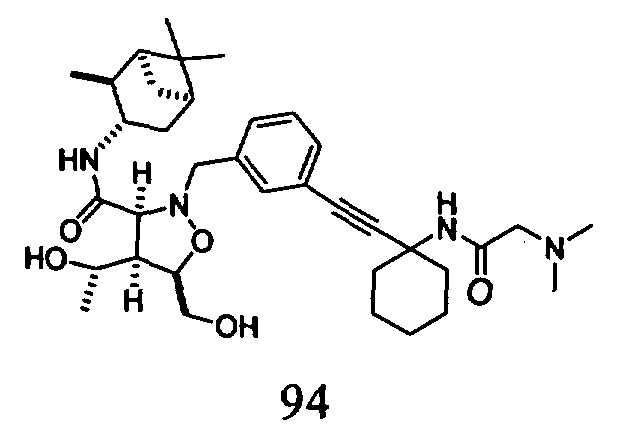
Стадия А
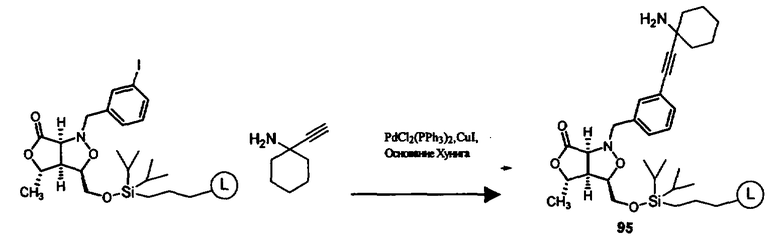
110 Частиц носителя lantern, содержащих иммобилизованный изоксазолидин (12 мкмолей/частица), помещали в реакционный сосуд и продували азотом. В реакционный сосуд добавляли ДМФА (77 мл), Pd(PPh3)2Cl2 (1,9 г, 2,6 ммоля, 2 экв.), CuI (0,75 г, 4 ммоля, 3 экв.), 1-этинилциклогексиламин
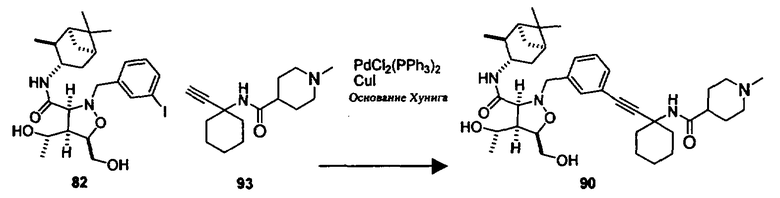
В смесь соединения 82 (70 мг, 0,1 ммоля), CuI (8 мг, 0,04 ммоля), Pd(PPh3)2Cl2 (22 мг, 0,03 ммоля) в ДМФА (3 мл) добавляли соединение 93 (50 мг, 0,21 ммоля) с последующим добавлением диизопропилэтиламина (40 мг, 54,8 мкл, 0,32 ммоля). Полученную смесь темно-коричневого цвета перемешивали при комнатной температуре в темноте в течение 2 сут. Реакционную смесь разбавляли смесью EtOAc/вода (6 мл, 1:1) и раствор нейтрализовали добавлением 1 М HCl до рН 7. Водную фазу отделяли и экстрагировали EtOAc (2 раза по 4 мл). Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, при этом получали масло. Неочищенный материал очищали колоночной хроматографией, при этом получали продукт 90 (48 мг, 70%). МС (ESI(+)): m/e 663,5 (M+H)+.
Пример 88
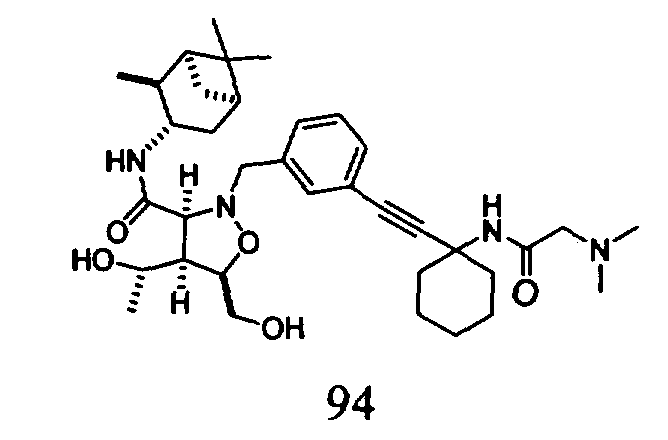
Стадия А
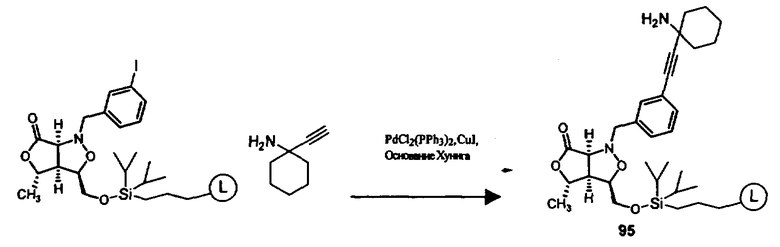
110 Частиц носителя lantern, содержащих иммобилизованный изоксазолидин (12 мкмолей/частица), помещали в реакционный сосуд и продували азотом. В реакционный сосуд добавляли ДМФА (77 мл), Pd(PPh3)2Cl2 (1,9 г, 2,6 ммоля, 2 экв.), CuI (0,75 г, 4 ммоля, 3 экв.), 1-этинилциклогексиламин (3,7 г, 3,9 мл, 29 ммолей) и диизопропилэтиламин (5 г, 6 мл, 40 ммоля, 30 экв.). Реакционный сосуд продували азотом, закрывали и осторожно встряхивали в течение 48 ч. Затем частицы носителя удаляли из реакционной смеси и промывали следующим образом: CH2Cl2 (2 раза по 100 мл), ТГФ (2 раза по 100 мл), ТГФ/IPA (2 раза по 100 мл, 3:1), ДМФА (2 раза по 100 мл), ТГФ (2 раза по 100 мл), CH2Cl2 (2 раза по 100 мл). Частицы носителя сушили при пониженном давлении.
Стадия В
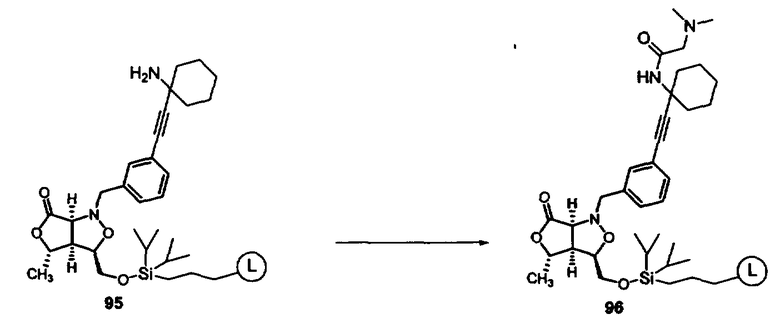
Во флакон, содержащий соединение 95 (5 частиц носителя lantern) в растворе ТГФ/ДМФА (1:1, 3 мл) добавляли HATU (76 мг, 2 ммоля), пиридин (162 мкл, 2 ммоля), N,N-диметилглицин (0,2 г, 20 ммолей) и диизопропилэтиламин (0,5 г, 0,7 мл, 4 ммоля). Смесь перемешивали при КТ в течение 3 сут. Затем частицы носителя удаляли и промывали следующим образом: CH2Cl2 (2 раза по 100 мл), ТГФ (2 раза по 100 мл), ТГФ/IPA (3:1, 2 раза по 100 мл), ДМФА (2 раза по 100 мл), ТГФ (2 раза по 100 мл), CH2Cl2 (2 раза по 100 мл). Частицы носителя сушили при пониженном давлении.
Стадия С
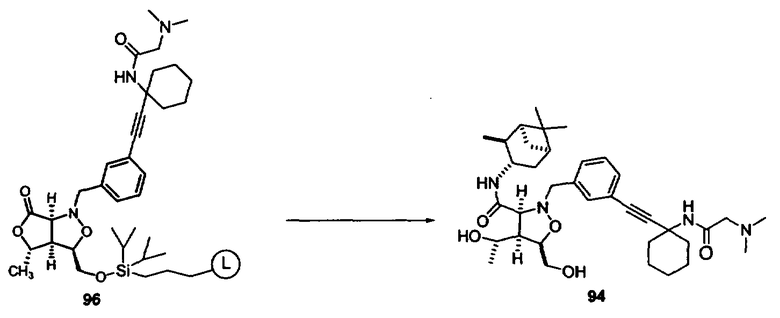
В раствор (+)-изопинокамфеиламина (1 г, 1,1 мл, 80 экв.,) в CH2Cl2 (4 мл) добавляли AlMe3 (0,4 мл, 2,0 М раствор в гексане, 0,8 ммоля, 10 экв.) и перемешивали в течение 15 мин. В колбу добавляли 4 частицы носителя lantern, содержащих иммобилизованное соединение 96, и осторожно встряхивали при КТ в течение 24 ч. Частицы носителя удаляли из реакционной смеси и промывали следующим образом: CH2Cl2 (2 раза по 100 мл), ТГФ (2 раза по 100 мл), ТГФ/IPA (3:1,2 раза по 100 мл), ДМФА (2 раза по 100 мл), ТГФ (2 раза по 100 мл), CH2Cl2 (2 раза по 100 мл). Частицы носителя сушили при пониженном давлении. Частицы носителя помещали в пластиковую пробирку и обрабатывали ТГФ (1,5 мл), добавляли HF/пиридин (0,25 мл) и частицы носителя выдерживали в этом растворе при осторожном встряхивании в течение 30 мин, затем добавляли TMSOMe (0,5 мл) и полученную смесь встряхивали в течение 30 мин. Раствор отделяли и упаривали, при этом получали продукт,
МС (ESI(+)): m/e 623,4 (М+Н)+.
Пример 89
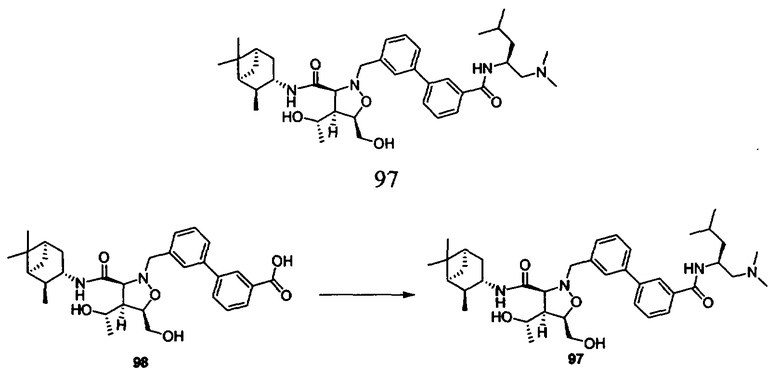
Раствор моногидрата (S)-Boc-Leu-OH (1 г, 4,1 ммоля), ДМФА (5 мл) и N,N диметиламина (4,1 мл, 2,0 М раствор в ТГФ, 8,6 ммоля) обрабатывали HBTU (2 г, 5,2 ммоля) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь добавляли в насыщенный раствор NaHCO3 (25 мл) и воду (25 мл). Слои разделяли и водный слой экстрагировали Et2O (3 раза по 40 мл). Объединенные органические экстракты в смеси с нерастворимым осадком белого цвета упаривали, обрабатывали ТФУ (10 мл) в течение 3 ч, затем ТФУ упаривали в вакууме, остаток упаривали в смеси с толуолом, остаток растворяли в ТГФ (50 мл), обрабатывали LiAlH4 (1,6 г, 42 ммоля) и суспензию нагревали с обратным холодильником в течение ночи. Реакционную смесь охлаждали в ледяной бане и обрабатывали IPA (10 мл) и 6 М NaOH (5 мл), после перемешивания в течение 2 ч суспензию фильтровали и концентрировали в вакууме. Остаток суспендировали в солевом растворе (50 мл) и экстрагировали CH2Cl2 (3 раза по 15 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали в вакууме, при этом получали (2S)-1-диметиламино-2-амино-4-метилпентан, который использовали без дополнительной очистки.
Раствор полученного неочищенного амина (25 мг, 0,17 ммоля) и неочищенного соединения 98 (25 мг, 0,05 ммоля) в ДМФА (0,7 мл) обрабатывали HBTU (40 мг, 0,1 ммоля). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (0,8 мл) и очищали ЖХВР. Соответствующие фракции концентрировали, при этом получали соединение 97 (20 мг, 25%) в виде твердого вещества белого цвета.
МС (ESI(+)): m/e 663,5 (М+Н)+.
Пример 90
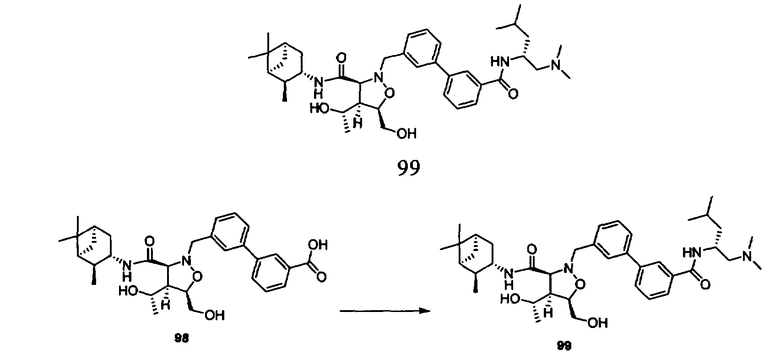
Раствор (R)-Boc-D-Leu-OH (1 г, 4,1 ммоля), ДМФА (5 мл) и диметиламина (4,3 мл, 2М раствор в ТГФ, 8,6 ммоля) обрабатывали HBTU (2 г, 5,2 ммоля) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь добавляли в насыщенный раствор NaHCO3 (25 мл) и воду (25 мл). Слои разделяли, водный слой экстрагировали Et2O (3 раза по 40 мл). Объединенные органические экстракты в смеси нерастворимым осадком белого цвета упаривали и обрабатывали ТФУ (10 мл) в течение 3 ч, затем ТФУ упаривали в вакууме. Остаток упаривали в смеси с толуолом, остаток растворяли в ТГФ (50 мл), обрабатывали LiAlH4 (1,6 г, 42 ммоля) и суспензию кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали на ледяной бане, последовательно обрабатывали IPA (10 мл) и 6 М NaOH (5 мл), после перемешивания в течение 2 ч суспензию фильтровали, концентрировали в вакууме. Остаток суспендировали в солевом растворе (50 мл) и экстрагировали CH2Cl2 (3 раза по 15 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали (2R)-1-диметиламино-2-амино-4-метилпентан, который использовали без предварительной очистки.
Раствор полученного неочищенного амина (25 мг, 0,17 ммоля) и неочищенного соединения 98 (25 мг, 0,04 ммоля) в ДМФА (0,7 мл) обрабатывали HBTU (40 мг, 0,1 ммоля). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (0,8 мл) и очищали методом ЖХВР. Соответствующие фракции концентрировали и получали соединение 99 (18 мг, 23%) в виде твердого вещества белого цвета.
МС (ESI(+)): m/e 663,5 (М+Н)+.
Пример 91
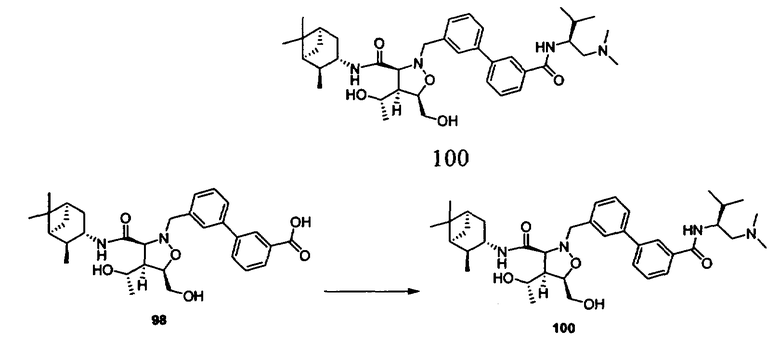
Раствор (S)-Boc-Val-OH (1 г, 4,1 ммоля), ДМФА (5 мл) и диметиламина (4,3 мл, 2 М раствор в ТГФ, 8,6 ммоля) обрабатывали HBTU (2 г, 5,3 ммоля) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь добавляли в насыщенный раствор NaHCO3 (25 мл) и воду (25 мл). Слои разделяли, водный слой экстрагировали Et2O (3 раза по 40 мл). Объединенные органические слои в смеси с белым нерастворимым осадком белого цвета упаривали и обрабатывали ТФУ (10 мл) в течение 3 ч, затем ТФУ упаривали в вакууме. Остаток упаривали в смеси с толуолом, остаток растворяли в ТГФ (50 мл), обрабатывали LiAlH4 (1,6 г, 42 ммоля), и суспензию кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали на ледяной бане и последовательно обрабатывали IPA (10 мл) и 6 М NaOH (5 мл), после перемешивания в течение 2 ч суспензию фильтровали и концентрировали в вакууме. Остаток суспендировали в солевом растворе (50 мл) и экстрагировали CH2Cl2 (3 раза по 15 мл). Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали (2S)-1-диметиламино-2-амино-3-метилбутан в виде масла желтого цвета, который использовали без предварительной очистки.
Раствор неочищенного амина (22 мг, 0,17 ммоля) и неочищенное соединение 98 (25 мг, 0,04 ммоля) в ДМФА (0,7 мл) обрабатывали HBTU (40 мг, 0,1 ммоля). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (0,8 мл) и очищали методом ЖХВР. Соответствующие фракции концентрировали и получали соединение 100 (20 мг, 26%) в виде твердого белого вещества белого цвета.
МС (ESI(+)): m/e 649,4 (М+Н)+.
Пример 94
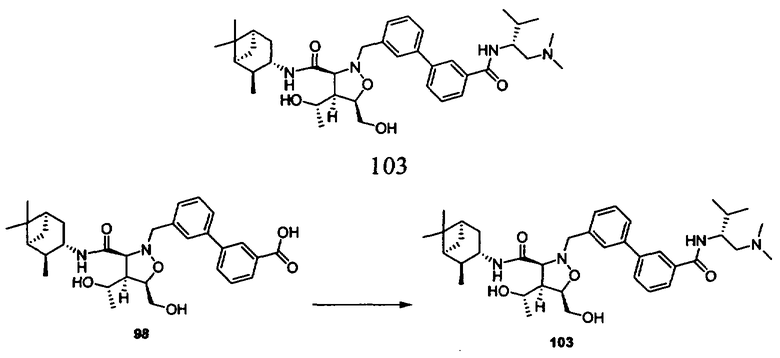
Раствор Boc-D-Val-OH (940 мг), ДМФА (5 мл) и 2,0 М диметиламин в ТГФ (4,3 мл) обрабатывали HBTU (2,0 г) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь добавляли в насыщенный раствор NaHCO3 (25 мл) и воду (25 мл), затем экстрагировали Et2O (3 раза по 40 мл). Объединенные органические слои в смеси нерастворимым осадком белого цвета упаривали и обрабатывали ТФУ (10 мл) в течение 3 ч, затем ТФУ упаривали в вакууме. Остаток упаривали в смеси с толуолом, остаток растворяли в ТГФ (50 мл), обрабатывали LiAlH4 (1,6 г), и суспензию кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали на ледяной бане и последовательно обрабатывали IPA (10 мл) и 6 М NaOH (5 мл), после перемешивания в течение 2 ч суспензию фильтровали, концентрировали и добавляли солевой раствор (50 мл). После экстракции ДХМ (3 раза по 15 мл), высушивания объединенных органических слоев над MgSO4 и концентрирования получали (2R)-1-диметиламино-2-амино-3-метилбутан в виде масла желтого цвета с неприятным запахом, которое использовали без дополнительной очистки.
Полученный раствор неочищенного амина (22 мкл) и неочищенного соединения 98 (25 мг) в ДМФА (700 мкл) обрабатывали HBTU (40 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрировали соответствующих фракций получали 20 мг соединения 103 в виде твердого вещества белого цвета. МС (ESI(+)): m/e 649,4 (М+Н)+.
Пример 95
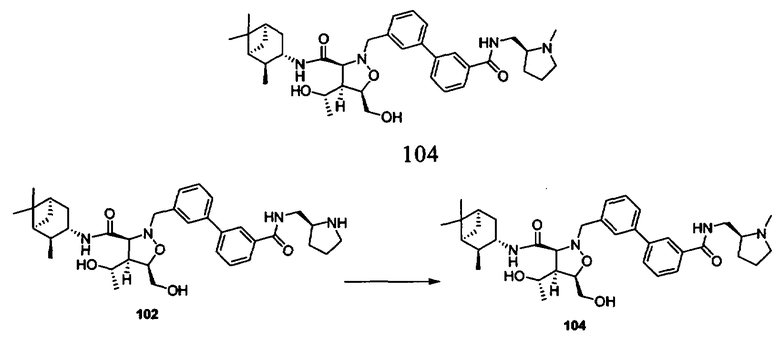
Во флакон, содержащий неочищенное соединение 102 (8 мг) в ДХМ (2 мл), добавляли 37% водный раствор формальдегида (6 мкл) и NaHB(ОАс)3 (12 мг), флакон встряхивали при комнатной температуре в течение 1,5 ч, затем реакционную смесь концентрировали, переносили в МеОН и очищали методом ЖХВР, при этом получали соединение 104 в виде твердого вещества белого цвета (8 мг). МС (ESI(+)) m/e: 633,4 (М+Н)+.
Пример 96
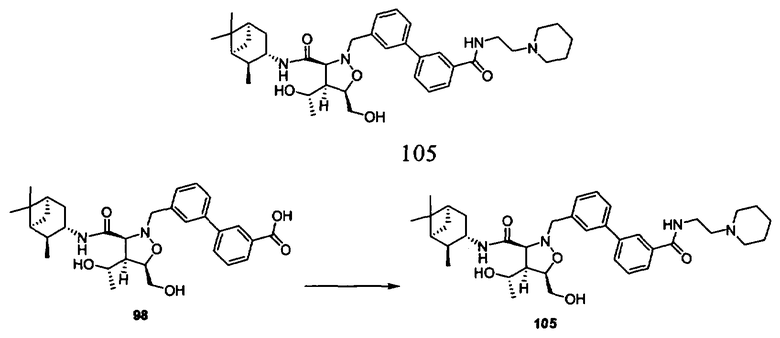
В раствор неочищенного соединения 98 (25 мг) и 1-(2-аминоэтил)пиперидина (23 мкл) в ДМФА (700 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 105 в виде твердого вещества белого цвета (20 мг). МС (ESI(+)) m/e: 647,3 (М+Н)+.
Пример 97
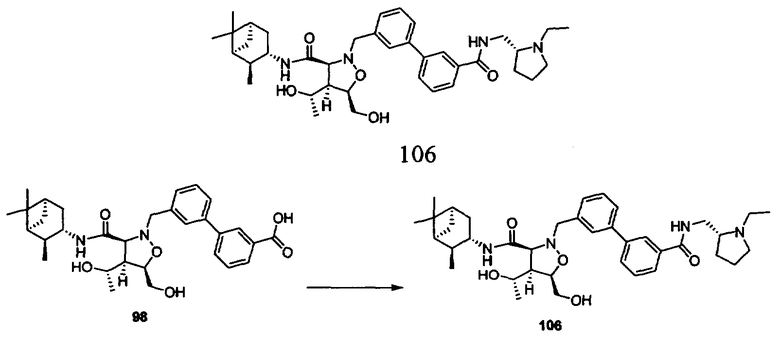
В раствор неочищенного соединения 98 (25 мг) и (2R)-1-этил-2-аминометилпирролидина (25 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 106 в виде твердого вещества белого цвета (20 мг). МС (ESI(+)) m/e: 647,3 (М+Н)+.
Пример 98
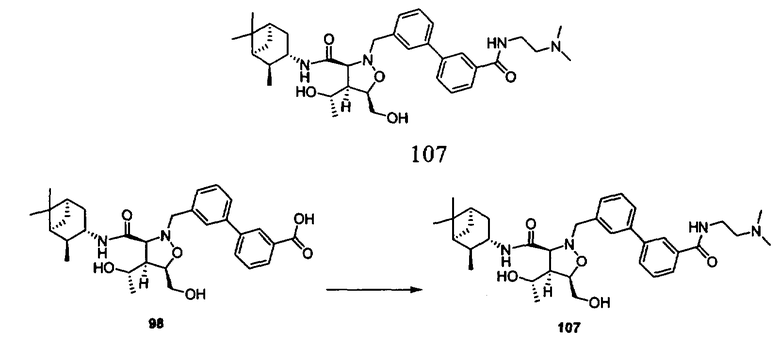
В раствор неочищенного соединения 98 (25 мг) и N,N-диметилэтилендиамина (18 мкл) в ДМФА (700 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 107 в виде твердого вещества белого цвета (23 мг). МС (ESI(+)) m/e: 607,3 (М+Н)+.
Пример 99
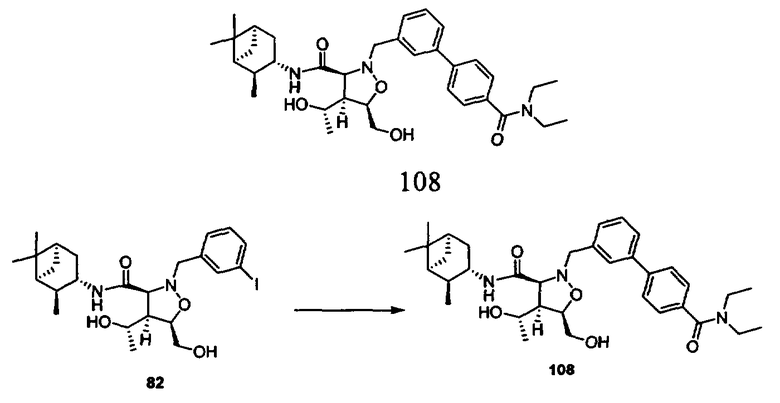
Пробирку, содержащую соединение 82 (30 мг), 4-(диэтиламинокарбонил)фенилбороновую кислоту (25 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) в EtOH (900 мкл), закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали, остаток очищали хроматографией на силикагеле (элюент: 50->100% EtOAc/гексан), при этом получали соединение 108 в виде прозрачного бесцветного масла (17 мг). МС (ESI(+)) m/e: 592,4 (М+Н)+.
Пример 100
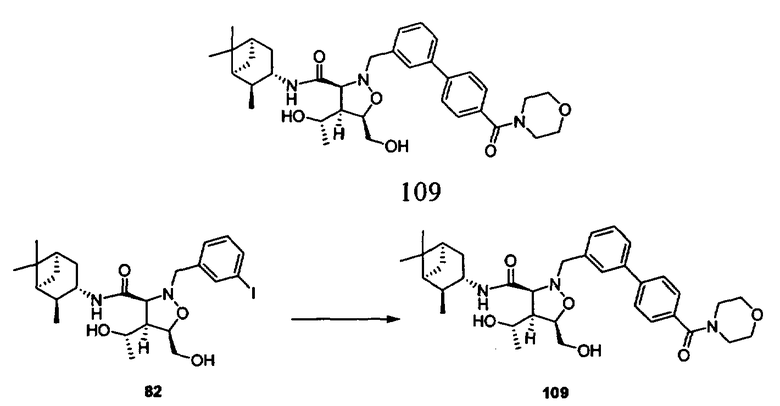
Пробирку, содержащую соединение 82 (30 мг), 4-(4-морфолинокарбонил)фенилбороновую кислоту (263 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) в EtOH (900 мкл), закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали, остаток очищали хроматографией на силикагеле (элюент: 50->100% EtOAc/гексан), при этом получали соединение 109 в виде прозрачного бесцветного масла (14 мг). МС (ESI(+)) m/e: 606,4 (М+Н)+.
Пример 101
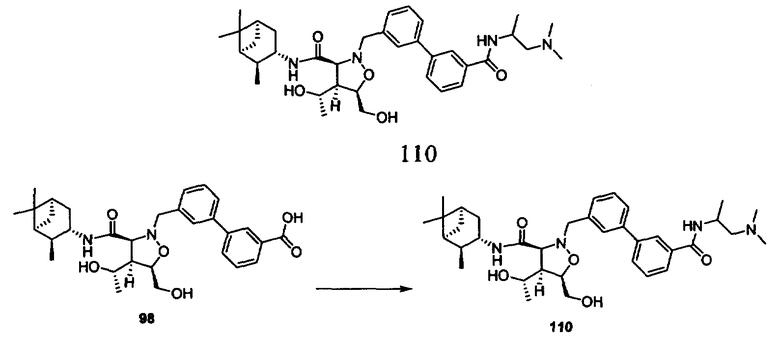
В раствор неочищенного соединения 98 (25 мг) и рацемического 1-диметиламино-2-пропиламина (19 мкл) в ДМ ФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 110 в виде твердого вещества белого цвета (21 мг). МС (ESI(+)) m/e: 621,2 (М+Н)+.
Пример 102
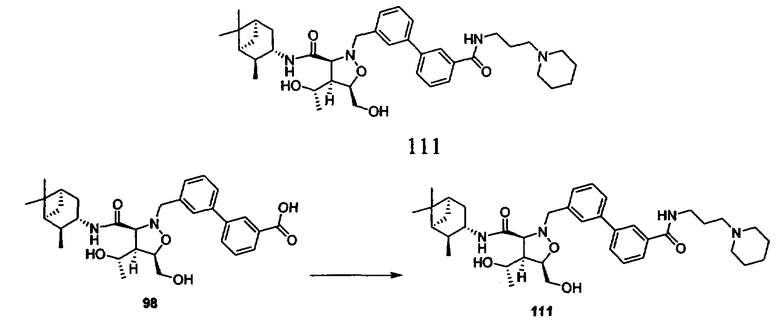
В раствор неочищенного соединения 98 (25 мг) и 1-(3-аминопропил)пиперидина (26 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 111 в виде твердого вещества белого цвета (14 мг). МС (ESI(+)) m/e: 661,4 (М+Н)+.
Пример 103
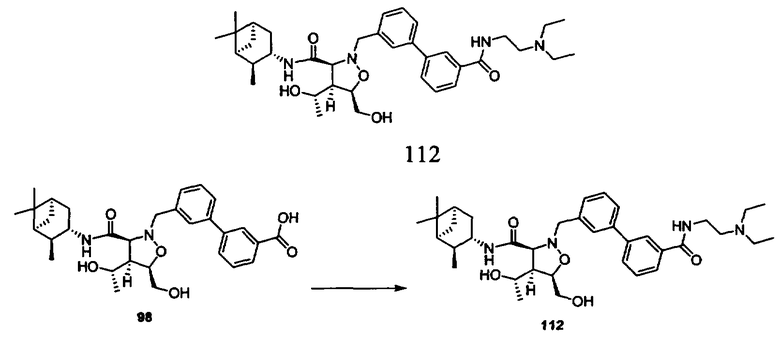
В раствор неочищенного соединения 98 (25 мг) и N,N-диэтилэтилендиамина (21 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 112 в виде твердого вещества белого цвета (22 мг). МС (ESI(+)) m/e: 635,3 (M+H)+.
Пример 104
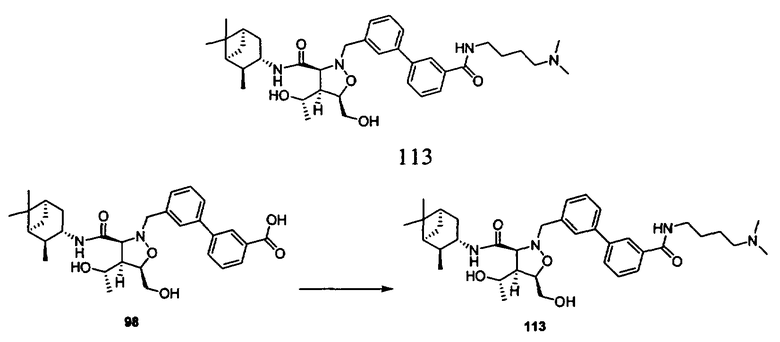
В раствор неочищенного соединения 98 (25 мг) и 4-диметиламинобутиламина (25 мкл) в ДМФА (700 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 113 в виде твердого вещества белого цвета (25 мг). МС (ESI(+)) m/e: 635,3 (М+Н)+.
Пример 105
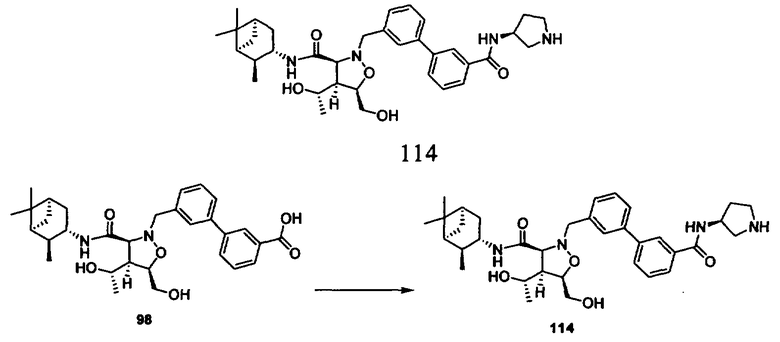
В раствор (3S)-3-трет-бутоксикарбониламинопирролидина (204 мг) в ДХМ (5 мл) добавляли DIEA (286 мкл) и бензилхлорформиат (188 мкл). Раствор перемешивали при комнатной температуре в течение ночи, затем добавляли насыщенный NaHCO3 и экстрагировали ДХМ (3×20 мл). Объединенные органические фазы сушили над MgSO4 и концентрировали, при этом получали твердое вещество белого цвета. Полученный материал растворяли в ТФУ (4 мл), перемешивали в течение 2 ч, раствор концентрировали в вакууме, при этом получали прозрачное масло.
В раствор полученного неочищенного масла (139 мг), неочищенного соединения 98 (25 мг) и DIEA (80 мкл) в ДМФА (1,4 мл) добавляли HBTU (80 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (1,5 мл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали твердое вещество белого цвета.
Половину количества полученного твердого вещества растворяли в EtOH (2 мл), добавляли НОАс (5 мкл) и влажный 20% Pd на угле (5 мг). Через смесь пропускали Н2, затем смесь перемешивали в атмосфере H2 в течение 4 ч. Полученную реакционную смесь отфильтровывали через фильтр с размером пор 0,2 мкм и очищали методом ЖХВР, при этом получали соединение 114 в виде твердого вещества белого цвета (6 мг). МС (ESI(+)) m/e: 605,2 (М+Н)+.
Пример 106
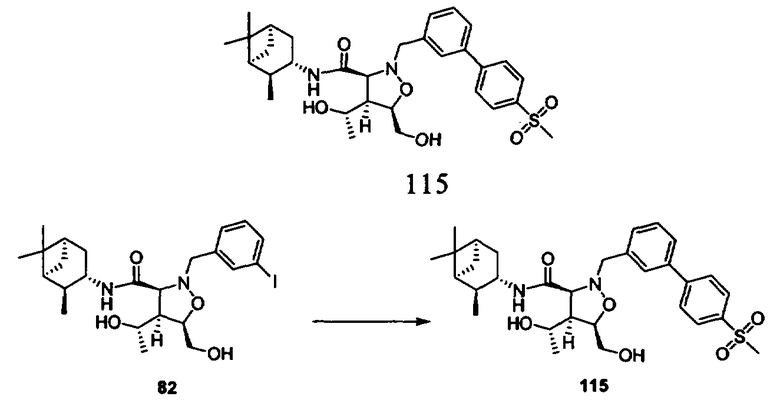
Пробирку, содержащую соединение 82 (30 мг), 4-(метансульфонил)фенилбороновую кислоту (22 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) в EtOH (1 мл), закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали. Остаток очищали хроматографией на силикагеле (элюент: 50->100% EtOAc/гексан), при этом получали соединение 115 в виде прозрачного бесцветного масла (30 мг). МС (ESI(+)) m/e: 571,3 (М+Н)+.
Пример 107
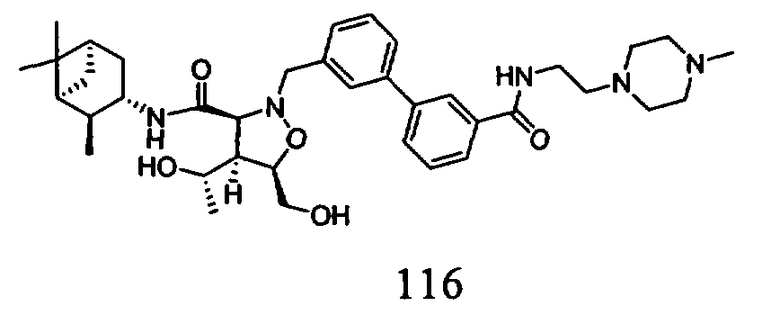
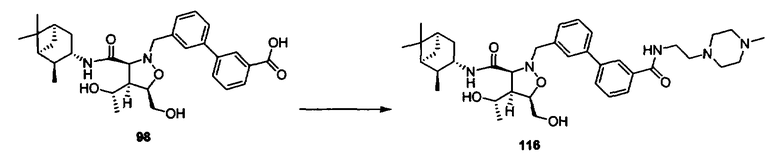
В раствор неочищенного соединения 98 (25 мг) и 1-(2-аминоэтил)-4-метилпиперазина (23 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 116 в виде твердого вещества белого цвета (26 мг). МС (ESI(+)) m/e: 662,3 (М+Н)+.
Пример 108
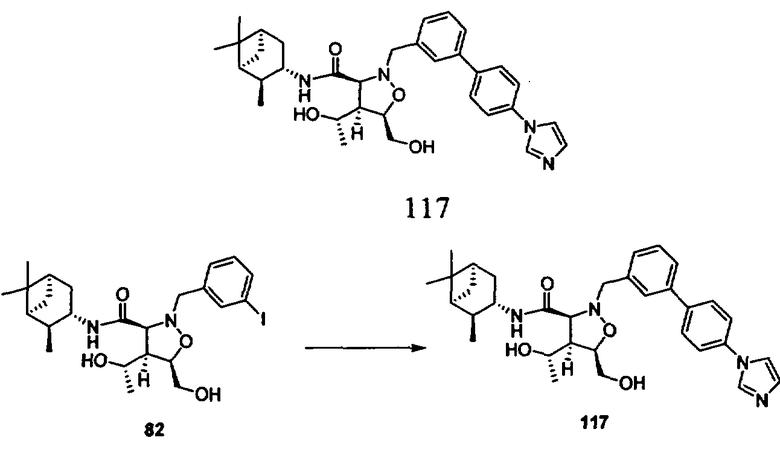
В колбу помещали 1-(4-бромфенил)имидазол (25 мг), Pd(dppf)Cl2 (4 мг), КОАс (33 мг) и бис(пинаколято)дибор (28 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 80°С в течение 1 ч, затем добавляли соединение 82 (30 мг) и Cs2CO3 (35 мг), нагревали при 60°С в течение еще 4 ч. Реакционную смесь очищали методом ЖХВР, после концентрирования соответствующих фракций получали соединение 117 в виде твердого вещества белого цвета (6,5 мг). МС (ESI(+)) m/e: 559,2 (М+Н)+.
Пример 109
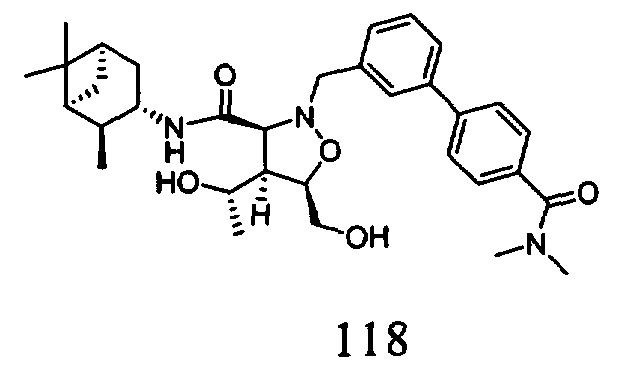
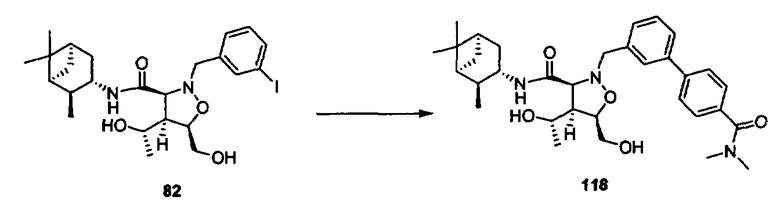
В колбу помещали соединение 82 (100 мг), 4-(диметиламинокарбонил)фенилбороновую кислоту (71 мг), Cs2CO3 (120 мг), КОАс (20 мг) и Pd(dppf)Cl2 (10 мг), колбу продували аргоном и добавляли ДМСО (6 мл). Смесь нагревали при 60°С в течение 3 ч, затем через 2,5 ч добавляли еще одну порцию Pd(dppf)Cl2 (5 мг). Реакционную смесь охлаждали и добавляли в ДХМ (25 мл) и 1% NaS2CNMe2 (75 мг), затем органическую фазу отделяли, а водную фазу экстрагировали ДХМ (3×25 мл). Объединенные органические фазы промывали водой (25 мл) и солевым раствором (25 мл), а затем сушили над Na2SO4. После концентрирования и очистки методом ЖХВР получали соединение 118 в виде твердого вещества белого цвета (64 мг). МС (ESI(+)) m/e: 564,3 (М+Н)+.
Пример 110
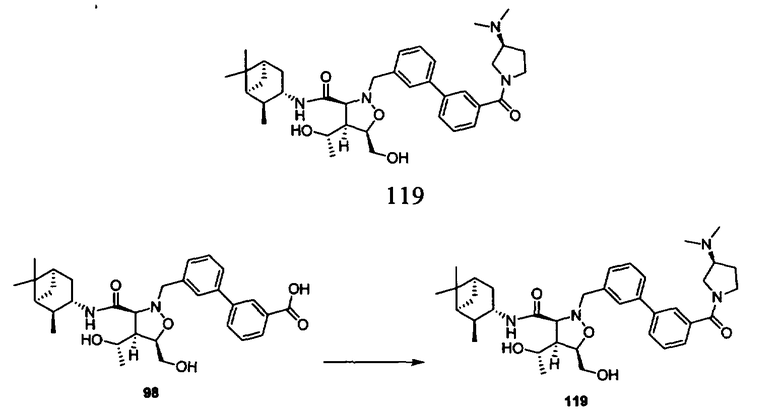
В раствор неочищенного соединения 98 (19 мг) и (3S)-3-(диметиламино)пирролидина (18 мкл) в ДМФА (525 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (1 мл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 119 в виде твердого вещества белого цвета (18 мг). МС (ESI(+)) m/e: 633,3 (М+Н)+.
Пример 111
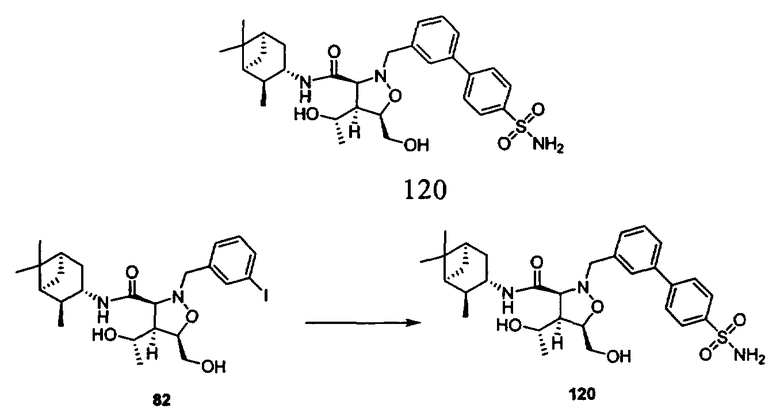
В небольшую колбу помещали соединение 82 (25 мг) 4-(сульфониламино)фенилбороновую кислоту (19 мг), Cs2CO3 (40 мг), КОАс (5 мг) и Pd(dppf)Cl2 (4 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 60°С в течение 2 ч, очищали методом ЖХВР и при этом получали соединение 120 в виде твердого вещества белого цвета (11 мг). МС (ESI(+)) m/e: 572,2 (M+H)+.
Пример 112
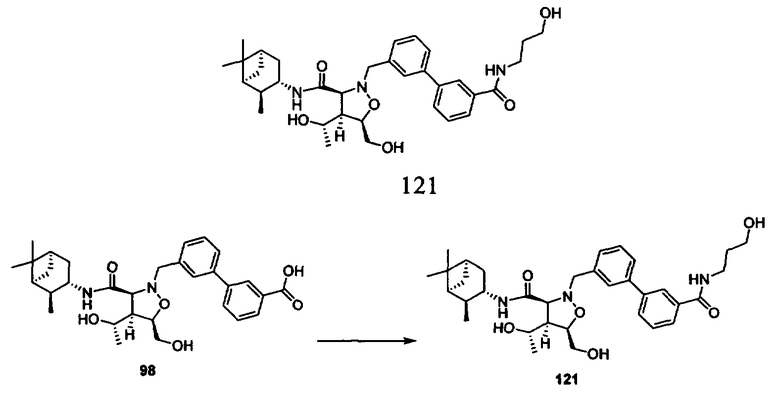
В раствор неочищенного соединения 98 (25 мг) и 3-пропаноламина (12 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 121 в виде твердого вещества белого цвета (23 мг). МС (ESI(+)) m/e: 594,3 (М+Н)+.
Пример 113
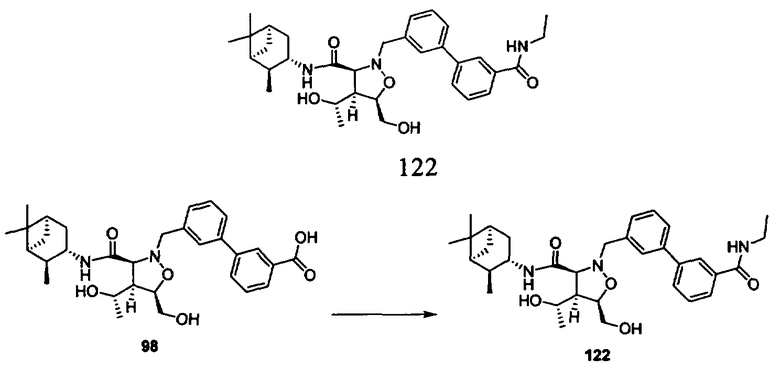
В раствор неочищенного соединения 98 (25 мг) и 2,0 М этиламина в ТГФ (74 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 122 в виде твердого вещества белого цвета (15 мг). МС (ESI(+)) m/e: 564,3 (М+Н)+.
Пример 114
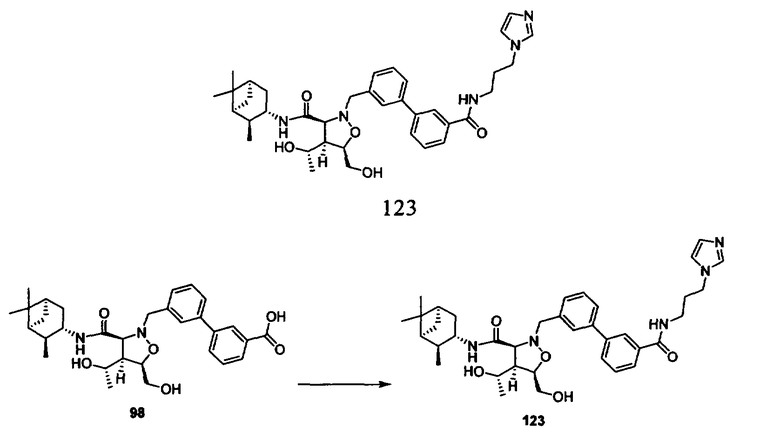
В раствор неочищенного соединения 98 (25 мг) и 1-(3-аминопропил)имидазола (19 мкл) в ДМФА (700 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 123 в виде твердого вещества белого цвета (17 мг). МС (ESI(+)) m/e: 644,3 (М+Н)+.
Пример 115
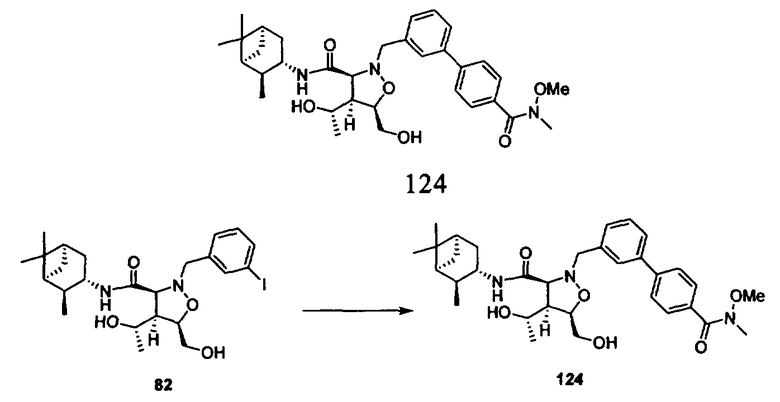
Пробирку, содержащую соединение 82 (30 мг), 4-(метокси(метил)карбамоил)фенилбороновую кислоту (23 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) в EtOH (900 мкл), закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали. Остаток очищали хроматографией на силикагеле (элюент: 50->100% EtOAc/гексан), при этом получали соединение 124 в виде прозрачного бесцветного масла (22 мг). МС (ESI(+)) m/e: 580,4 (М+Н)+.
Пример 116
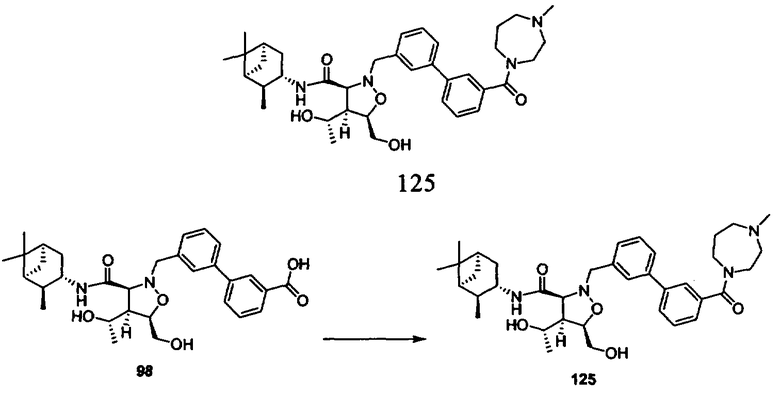
В раствор неочищенного соединения 98 (25 мг) и 1-метилгомопиперазина (20 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 125 в виде твердого вещества белого цвета (20 мг). МС (ESI(+)) m/e: 633,3 (М+Н)+.
Пример 117
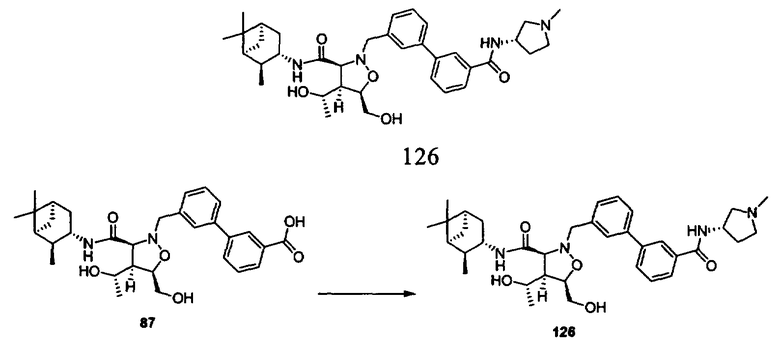
В раствор неочищенного соединения 98 (25 мг), гидрохлорида (3S)-3-амино-1-метилпирролидина (25 мг) и DIEA (50 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 126 в виде твердого вещества белого цвета (15 мг). МС (ESI(+)) m/e: 619,4 (М+Н)+.
Пример 118
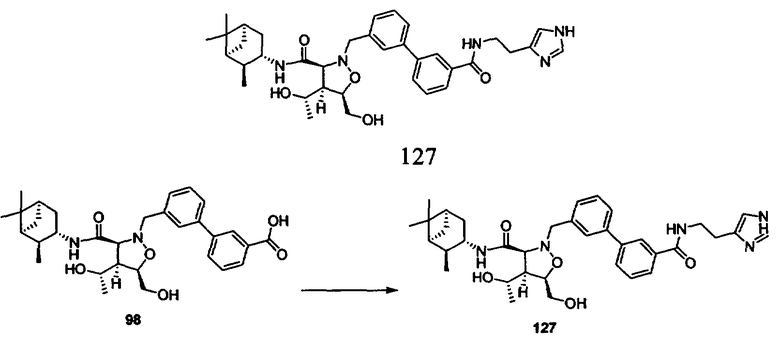
В раствор неочищенного соединения 98 (25 мг) и гистамина (18 мг) в ДМФА (700 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 127 в виде твердого вещества белого цвета (10 мг). МС (ESI(+)) m/e: 630,2 (М+Н)+.
Пример 119
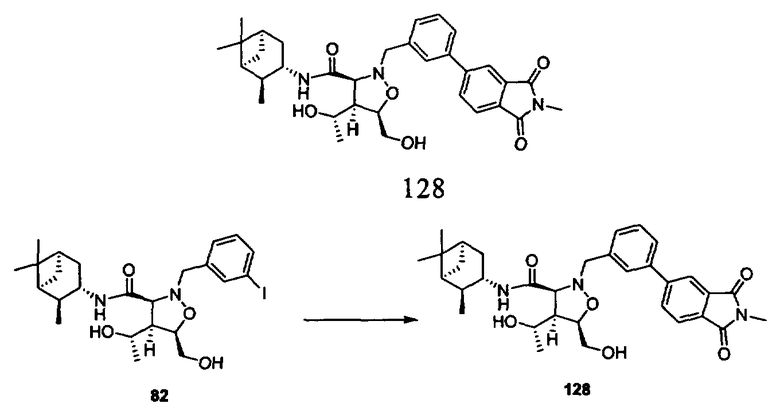
Раствор 4-бромфталевого ангидрида (2,0 г) в толуоле (20 мл) обрабатывали 33% метиламином в EtOH (1,65 мл) и кристаллическим DMAP. Смесь перемешивали при комнатной температуре в течение 1 ч, кипятили с обратным холодильником с использованием насадка Дина-Старка в течение 14 ч, затем охлаждали до -20°С, полученные кристаллы отфильтровывали и промывали гексаном, при этом получали N-метил-3-бромфталимид (1,32 г, tпл. 149-151). Маточный раствор концентрировали и перекристаллизовывали из бензола (3 мл), при этом получали вторую порцию (430 мг).
В колбу помещали N-метил-3-бромфталимид (27 мг), Pd(dppf)Cl2 (3 мг), КОАс (33 мг) и бис(пинаколято)дибор (28 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 80°С в течение 1 ч, затем добавляли соединение 82 (30 мг) и Cs2CO3 (35 мг) и нагревали при 80°С в течение еще 2,5 ч. Реакционную смесь очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 128 в виде твердого вещества белого цвета (12 мг). МС (ESI(+)) m/e: 576,3 (М+Н)+.
Пример 120
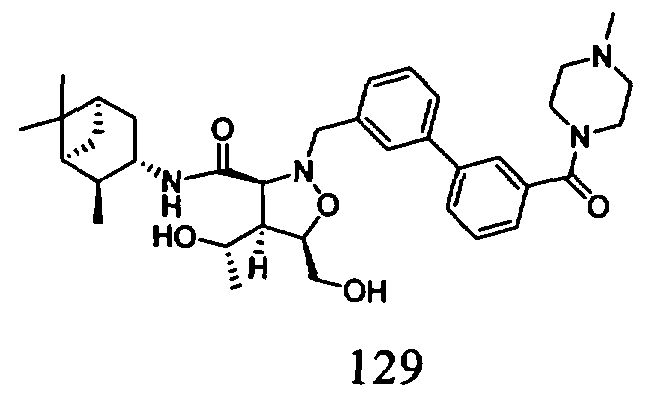
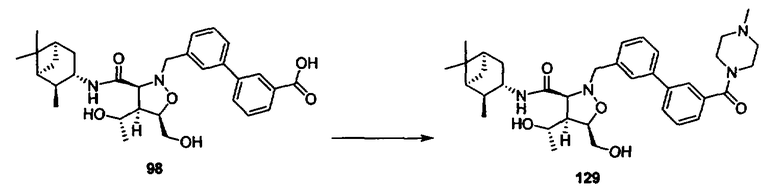
В раствор неочищенного соединения 98 (25 мг) и 1-метилпиперазина (18 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 129 в виде твердого вещества белого цвета (21 мг). МС (ESI(+)) m/e: 619,3 (М+Н)+.
Пример 121
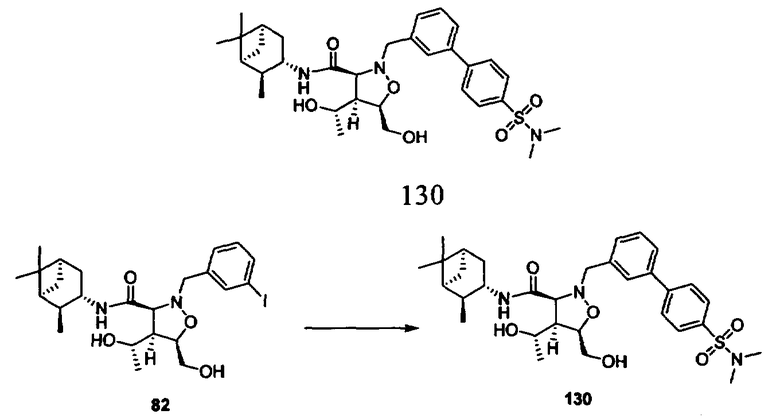
Пробирку, содержащую соединение 82 (30 мг), 4-(диметиламиносульфонил)фенилбороновую кислоту (33 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) в EtOH (900 мкл), закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали. Остаток очищали хроматографией на силикагеле (элюент: 50->75% EtOAc/гексан), при этом получали соединение 130 в виде прозрачного бесцветного масла (5 мг). МС (ESI(+)) m/e: 600,4 (М+Н)+.
Пример 122
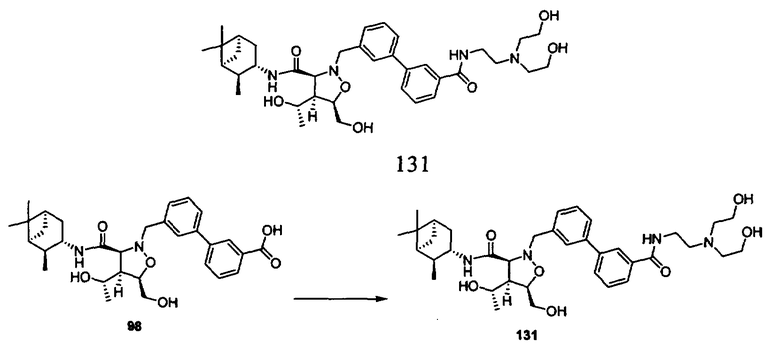
В раствор неочищенного соединения 98 (25 мг) и N,N-бис(2-гидроксиэтил)этилендиамина (20 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 131 в виде твердого вещества белого цвета (16 мг). МС (ESI(+)) m/e: 667,4 (М+Н)+.
Пример 123
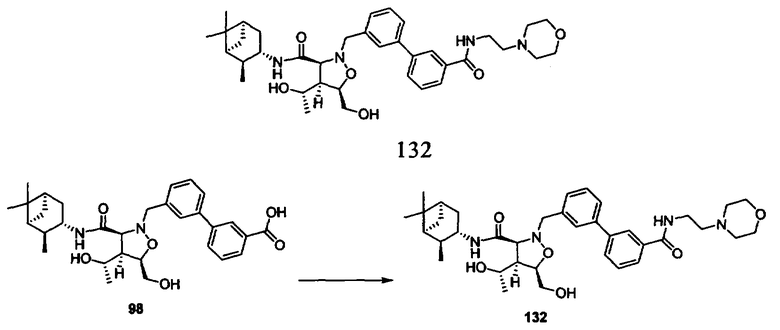
В раствор неочищенного соединения 98 (25 мг) и 4-(2-аминоэтил)морфолина (21 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 132 в виде твердого вещества белого цвета (21 мг). МС (ESI(+)) m/e: 649,3 (М+Н)+.
Пример 124
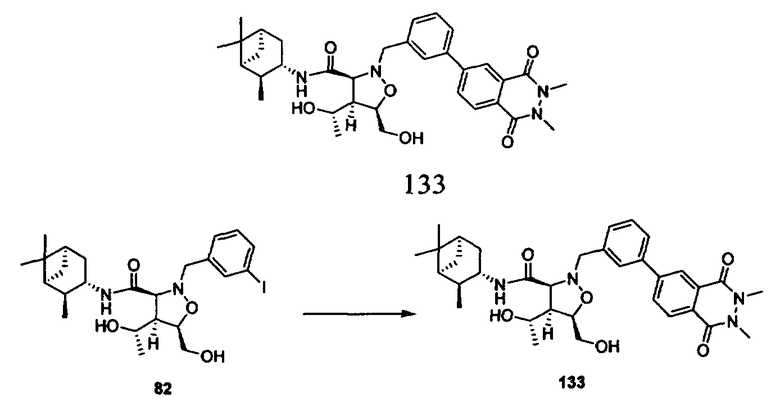
Суспензию 4-бромфталевого ангидрида (1,0 г) в EtOH (10 мл) обрабатывали дигидрохлоридом 1,2-диметилгидразина (650 мг) и NEt3 (1,35 мл), затем кипятили с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали и разбавляли 0,1 М HCl (100 мл). Остаток в виде твердого вещества белого цвета отделяли фильтрованием, промывали водой и сушили в вакууме, при этом получали 6-бром-2,3-диметил-2,3-дигидрофталазин-1,4-дион (516 мг, tпл. 207-209).
В колбу помещали полученный бромид (30 мг), Pd(dppf)Cl2 (4 мг), КОАс (33 мг) и бис(пинаколято)дибор (28 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 60°С в течение 1 ч, затем добавляли соединение 82 (30 мг) и Cs2CO3 (35 мг) и нагревали в течение еще 1 ч. Реакционную смесь очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 133 в виде твердого вещества белого цвета (16,5 мг). МС (ESI(+)) m/e: 605,2 (М+Н)+.
Пример 125
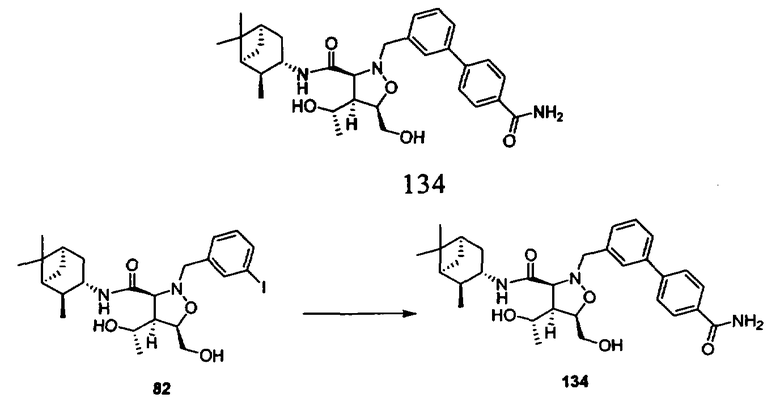
Пробирку, содержащую соединение 82 (30 мг), 4-(аминокарбонил)фенилбороновую кислоту (33 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) в EtOH (1 мл), закрывали и нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали. Остаток очищали хроматографией на силикагеле, при этом получали соединение 134 в виде твердого вещества белого цвета (8 мг). МС (ESI(+)) m/e: 536,3 (М+Н)+.
Пример 126
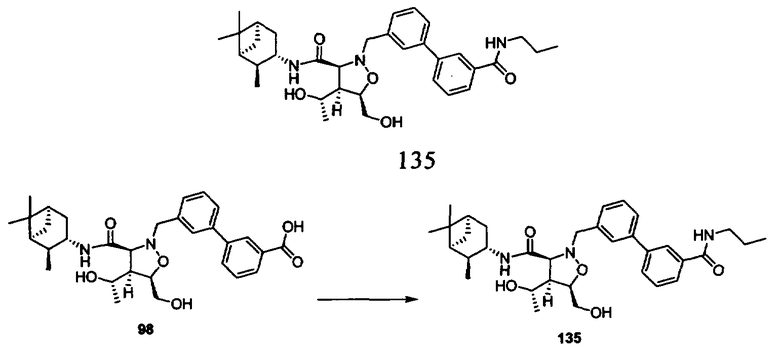
В раствор неочищенного соединения 98 (25 мг) и пропиламина (12 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 135 в виде твердого вещества белого цвета (19 мг). МС (ESI(+)) m/e: 578,3 (М+Н)+.
Пример 127
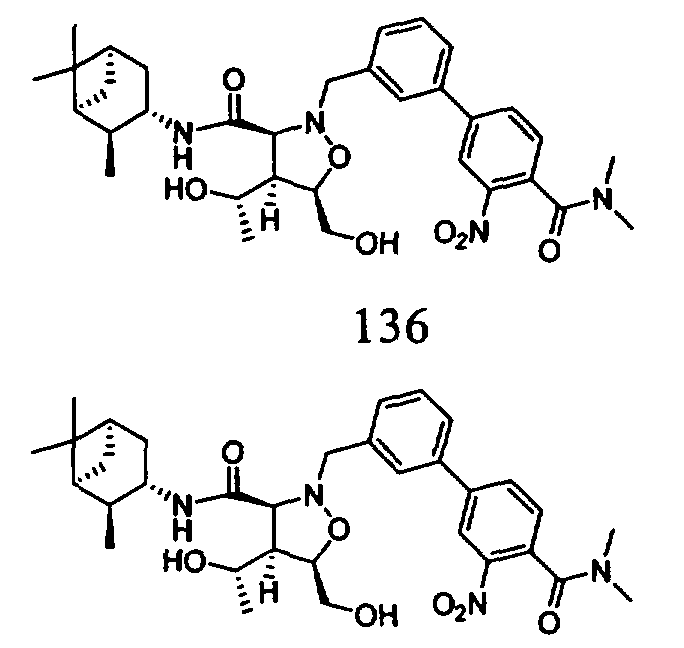
В колбу, содержащую 4-бром-2-нитробензойную кислоту (500 мг) и снабженную перемешивающим стержнем, добавляли SOCl2 (3 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Затем SOCl2 упаривали, добавляли бензол (5 мл) и снова упаривали. Остаток охлаждали на ледяной бане, растворяли в ДХМ (3 мл) и обрабатывали 2,0 М диметиламином в ТГФ (4 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, разбавляли EtOAc (80 мл), промывали 2 М HCl, 2М NaOH и водой (каждого по 20 мл). Органический слой сушили над MgSO4, фильтровали и остаток перекристаллизовывали из гексана (50 мл), при этом получали N,N-диметил-4-бром-2-нитробензамид в виде игольчатых кристаллов желтого цвета (453 мг, tпл. 104,5-105,5°С).
В колбу помещали полученный бромид (30 мг), Pd(dppf)Cl2 (4 мг), КОАс (33 мг) и бис(пинаколято)дибор (28 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 80°С в течение 1 ч, затем добавляли соединение 82 (30 мг) и Cs2CO3 (36 мг), нагревали в течение еще 3,5 ч. Реакционную смесь очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 136 в виде твердого вещества белого цвета (10 мг). МС (ESI(+)) m/e: 609,3 (М+Н)+.
Пример 128
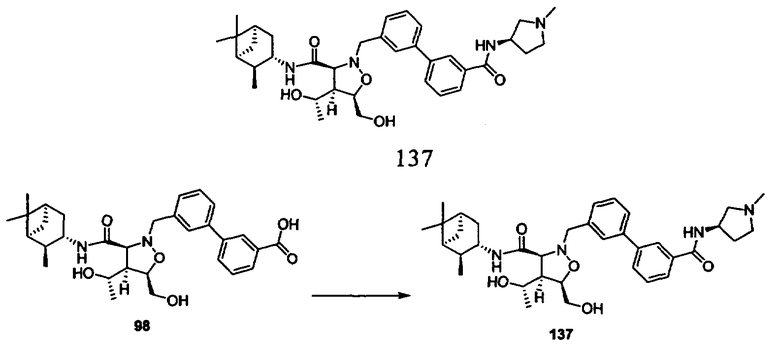
В раствор неочищенного соединения 98 (25 мг), дигидрохлорида (3R)-3-амино-1-метилпирролидина (25 мг) и DIEA (50 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 137 в виде твердого вещества белого цвета (14 мг). МС (ESI(+)) m/e: 619,4 (М+Н)+.
Пример 129
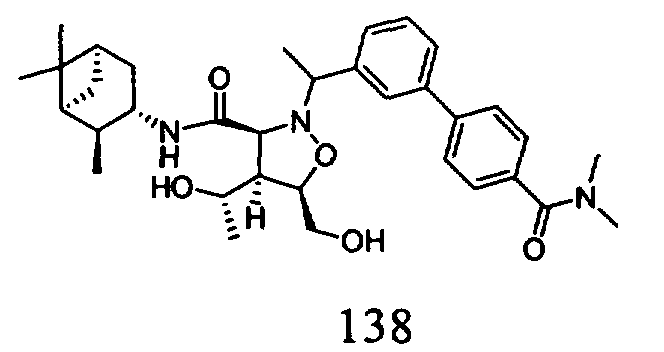
Стадия А
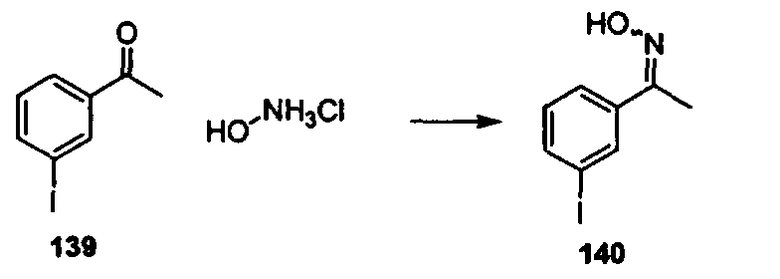
Соединение 140 (выход 94%) получали по методике, описанной в работе S.T.Pickard и Н.Е.Smith, JACS 112, 5741-5747 (1990).
Стадия В

Соединение 141 (выход 94%) получали по методике, описанной в статье М. Ueda и др. Tet. Lett. 43, 4369-4371 (2002).
Стадия С
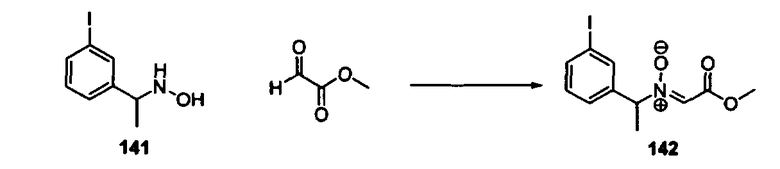
В раствор метилглиоксилата (0,29 г, 3,3 ммоля, 1,5 экв.) добавляли гидроксиламин (0,58 г, 2,2 ммоля, 1 экв.) в бензоле (15 мл), раствор кипятили с обратным холодильником в течение 72 ч в круглодонной колбе, снабженной насадкой Дина-Старка и погруженной в колбонагреватель. Полученный раствор охлаждали до КТ и концентрировали в вакууме. После концентрирования данные анализа ЯМР свидетельствовали о полном превращении в требуемый нитрон, полученный продукт выделяли в виде твердого вещества оранжевого цвета.
Стадия D
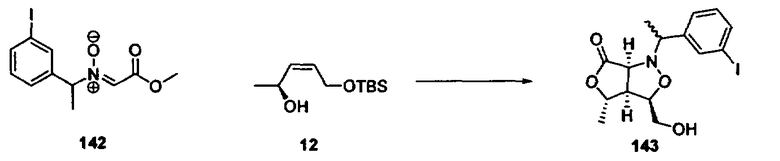
В раствор рацемического N-оксида метилового эфира 2-(1-(3-иодфенил)этилимино)уксусной кислоты 142 (429 мг) и (S,Z)-5-(трет-бутилдиметилсилилокси)пент-3-ен-2-ола 12 (275 мг) в толуоле (10 мл) добавляли Ti(Oi-Pr)4 (570 мкл). Раствор нагревали в микроволновом реакторе при 140°С в течение 15 мин, затем обрабатывали 3-(диметиламино)пропиленгликолем (1 мл) и водой (20 мл). Смесь экстрагировали EtOAc (3×30 мл), объединенные органические слои промывали водой (25 мл) и сушили над MgSO4, при этом получали остаток, который очищали колоночной хроматографией на силикагеле (элюент: 5->20% EtOAc/гексан), при этом получали два диастереомера. Масса первой фракции составляла 179 мг.
Полученный материал растворяли в ТГФ (5 мл) и добавляли 6 М HCl (1 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли EtOAc (30 мл), 1 М NaOH (5 мл), насыщенный NaHCO3 (13 мл) и воду (13 мл). Водный слой отделяли и экстрагировали EtOAc (2×30 мл), объединенные органические слои промывали солевым раствором (20 мл), сушили над MgSO4 и концентрировали, при этом получали твердое вещество желтого цвета. К полученному остатку добавляли гексан (20 мл) и декантировали, при этом получали твердое вещество белого цвета (102 мг).
Стадия Е
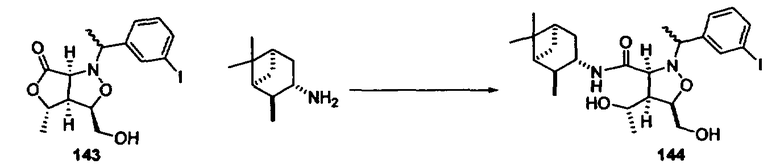
В высушенную в пламени колбу, содержащую (+)-изопинокамфеиламин (111 мг) и ДХМ (2 мл), добавляли 2,0 М AlMe3 в гексане (360 мкл), смесь перемешивали в течение 15 мин, затем добавляли соединение 143, суспендированное в ДХМ (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, затем обрабатывали насыщенным раствором сегнетовой соли (10 мл) и перемешивали в течение 1 ч. Смесь разделяли и экстрагировали ДХМ (3×10 мл), органические фазы сушили над Na2SO4, остаток очищали колоночной хроматографией, при этом получали продукт (205 мг).
Стадия F
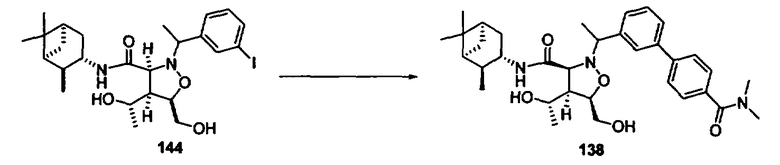
Соединение 144 (30 мг) в смеси с 4-(диметиламинокарбонил)фенилбороновой кислотой (21 мг), Pd(Ph3P)2Cl2 (2 мг) и NEt3 (31 мкл) растворяли в EtOH (900 мкл), нагревали в микроволновом реакторе при 120°С в течение 20 мин. Реакционную смесь смешивали с силикагелем (1 г) и растворитель упаривали. Остаток очищали хроматографией на силикагеле, при этом получали индивидуальный диастереомер 138 в виде твердого вещества белого цвета (30 мг). МС (ESI(+)) m/e: 578,3 (М+Н)+.
Пример 130
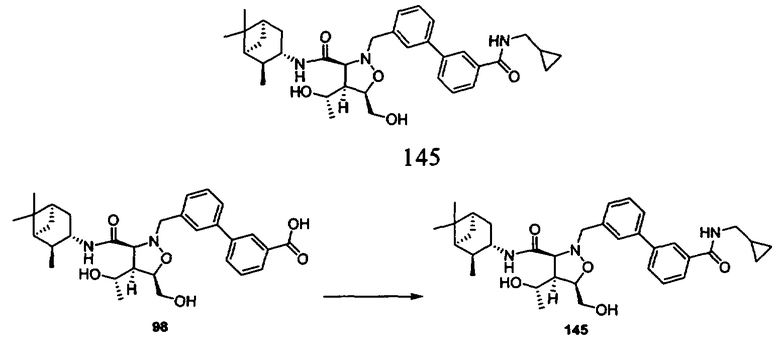
В раствор неочищенного соединения 98 (25 мг) и метиламиноциклопропана (13 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 145 в виде твердого вещества белого цвета (21 мг). МС (ESI(+)) m/e: 590,3 (М+Н)+.
Пример 131
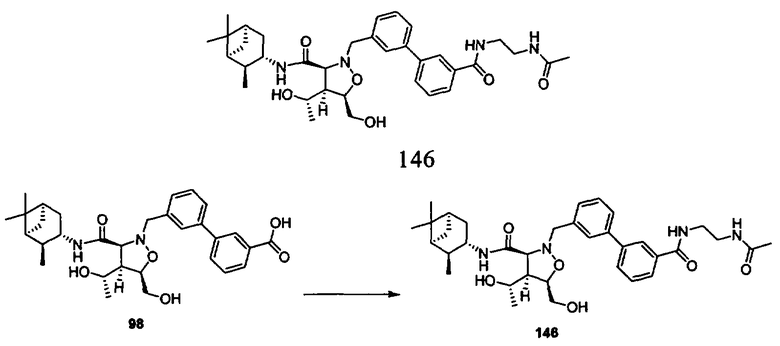
В раствор неочищенного соединения 98 (25 мг) и N-(2-аминоэтил)ацетамида (16 мкл) в ДМФА (700 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 146 в виде твердого вещества белого цвета (18 мг). МС (ESI(+)) m/e: 621,2 (М+Н)+.
Пример 132
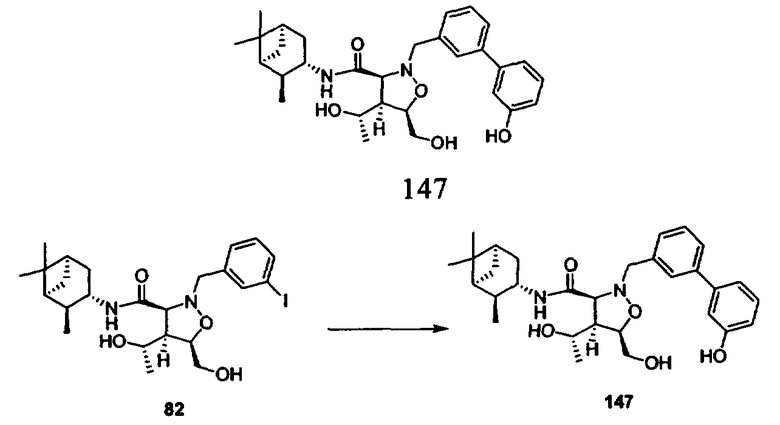
В небольшую колбу помещали соединение 82 (25 мг), 3-гидроксифенилбороновую кислоту (13 мг), Cs2CO3 (40 мг), КОАс (5 мг) и Pd(dppf)Cl2 (4 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 60°С в течение 1,5 ч, очищали методом ЖХВР и при этом получали соединение 147 в виде твердого вещества белого цвета (9 мг). МС (ESI(+)) m/e: 509,2 (М+Н)+.
Пример 133
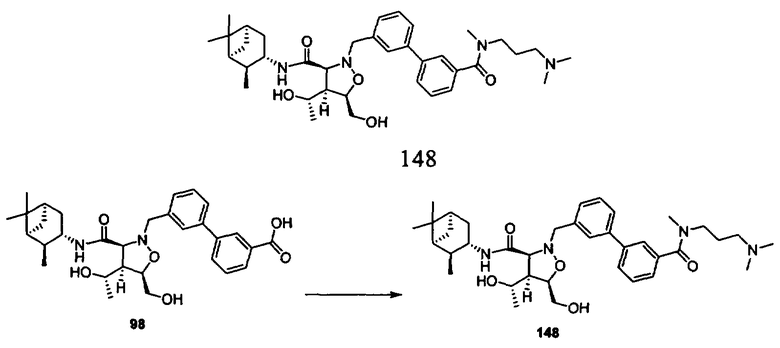
В раствор неочищенного соединения 98 (23 мг) и N,N,N′ -триметил-1,3-пропандиамина (19 мкл) в ДМФА (700 мкл) добавляли HBTU (35 мг), встряхивали в течение 4 ч, затем реакционную смесь разбавляли МеОН (1,5 мл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 148 в виде твердого вещества желтоватого цвета (6 мг). МС (ESI(+)) m/e: 635,5 (М+Н)+.
Пример 134
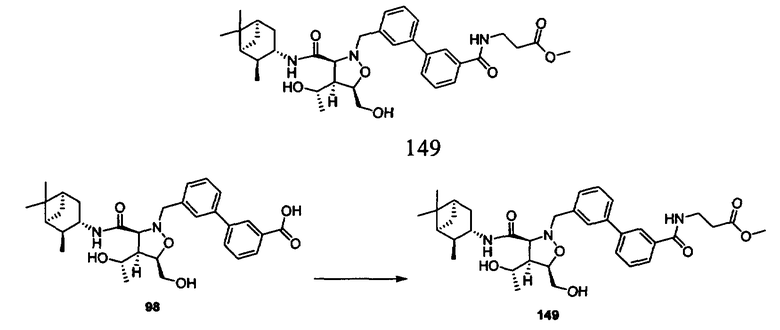
В раствор неочищенного соединения 98 (25 мг) и метилового эфира 3-аминопропионовой кислоты (20 мг) в ДМФА (700 мкл) добавляли HBTU (35 мг) и NEt3 (18 мкл), встряхивали в течение 2 ч, затем в реакционную смесь добавляли насыщенный NaHCO3 (5 мл) и воду (5 мл), экстрагировали ДХМ (3×10 мл). Органические фазы сушили над Na2SO4, частично концентрировали и смешивали с силикагелем (1 г), растворитель упаривали, остаток очищали хроматографией на силикагеле, при этом получали соединение 149 в виде твердого вещества желтоватого цвета (20 мг). МС (ESI(+)) m/e: 622,3 (М+Н)+.
Пример 135
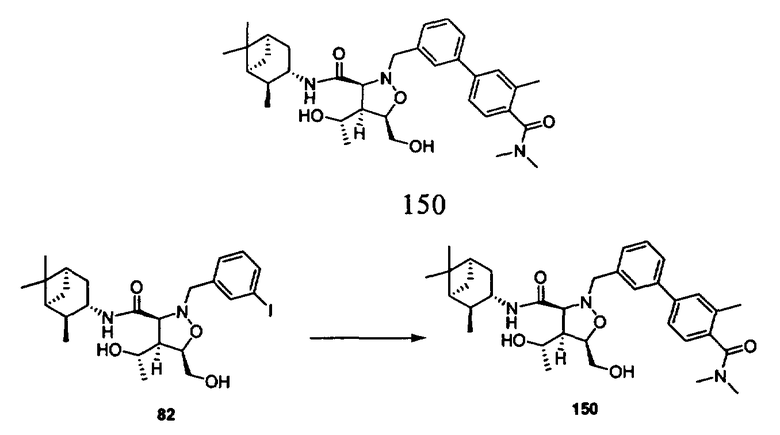
В колбу, содержащую 4-бром-2-метилбензойную кислоту (495 мг) и снабженную перемешивающим стержнем, добавляли SOCl2 (3 мл) и кипятили с обратным холодильником в течение 1 ч. Затем SOCl2 упаривали, добавляли бензол (5 мл) и снова упаривали. Остаток охлаждали на ледяной бане, растворяли в ДХМ (3 мл) и обрабатывали 2,0 М диметиламином в ТГФ (4 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, разбавляли EtOAc (80 мл), промывали 2 М HCl, 2М NaOH и водой (каждого по 20 мл). Органический слой сушили над MgSO4, остаток концентрировали, при этом получали N,N,2-триметил-4-бромбензамид в виде масла желтоватого цвета (460 мг).
В колбу помещали полученный неочищенный бромид (27 мг), Pd(dppf)Cl2 (3 мг), КОАс (38 мг) и бис(пинаколято)дибор (34 мг), колбу продували аргоном и добавляли ДМСО (3 мл). Смесь нагревали при 80°С в течение 1,5 ч, затем добавляли соединение 82 (30 мг) и Cs2CO2 (36 мг), нагревали в течение еще 2,5 ч. Реакционную смесь очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 150 в виде твердого вещества белого цвета (8 мг). МС (ESI(+)) m/e: 578,4 (М+Н)+.
Пример 136
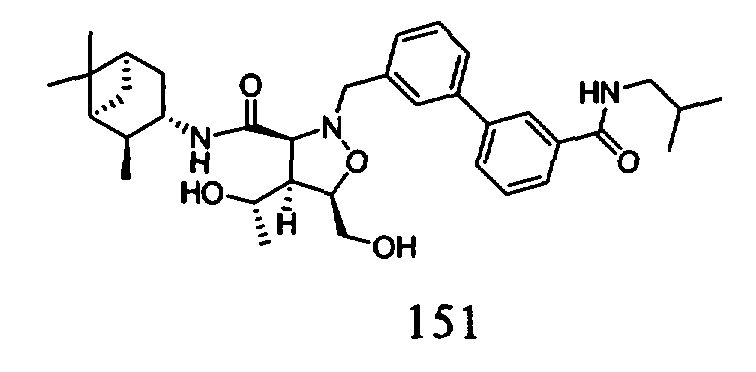
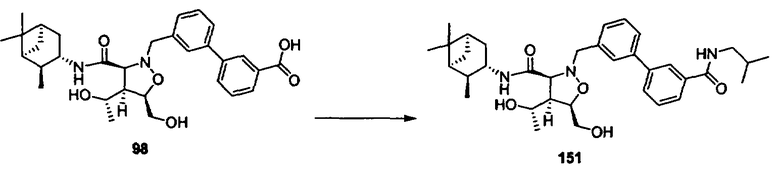
В раствор неочищенного соединения 98 (25 мг) и изобутиламина (16 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 151 в виде твердого вещества белого цвета (24 мг). МС (ESI(+)) m/e: 592,2 (М+Н)+.
Пример 137
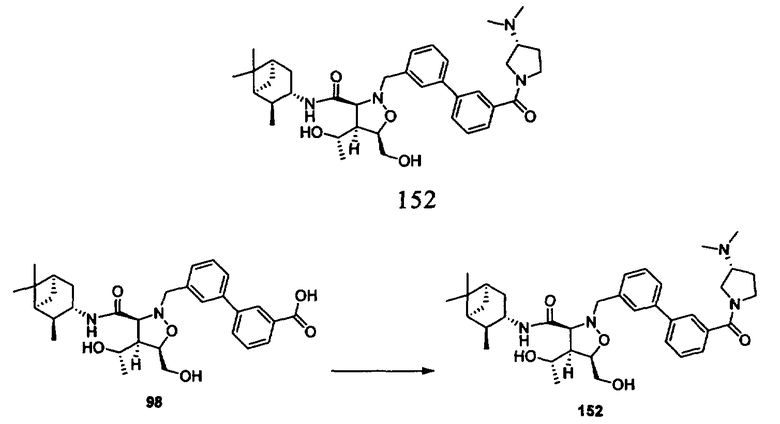
В раствор неочищенного соединения 98 (19 мг) и (3R)-3-(диметиламино)пирролидина (18 мкл) в ДМФА (525 мкл) добавляли HBTU (25 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (1 мл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 152 в виде твердого вещества белого цвета (17 мг). МС (ESI(+)) m/e: 633,3 (М+Н)+.
Пример 138
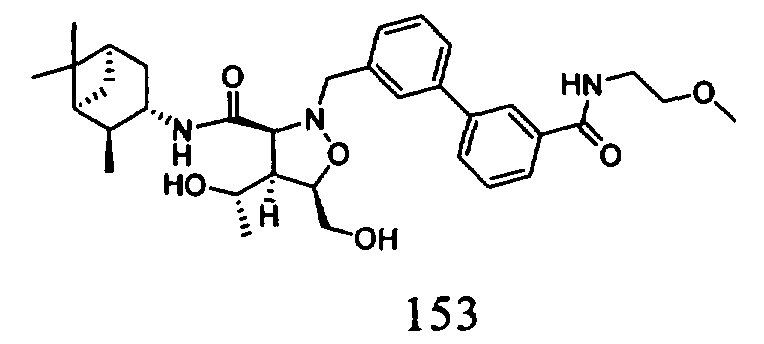
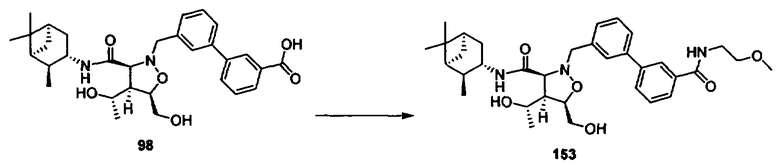
В раствор неочищенного соединения 98 (25 мг) и 2-метоксиэтиламина (13 мкл) в ДМФА (700 мкл) добавляли HBTU (40 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 153 в виде твердого вещества белого цвета (20 мг). МС (ESI(+)) m/e: 594,3 (М+Н)+.
Пример 139
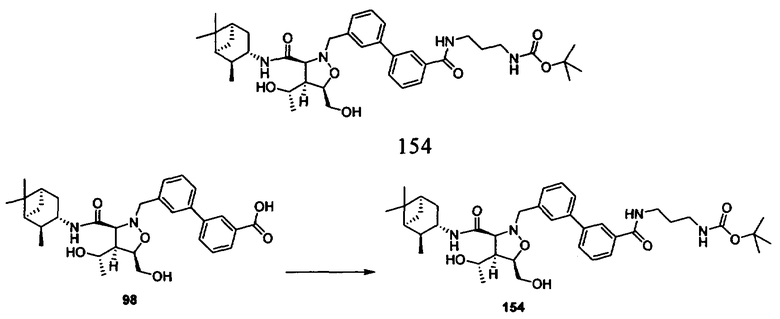
В раствор неочищенного соединения 98 (25 мг) и N-(трет-бутоксикарбонил)-1,3-диаминопропана (28 мг) в ДМФА (700 мкл) добавляли HBTU (30 мг), встряхивали в течение 1 ч, затем реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 154 в виде твердого вещества белого цвета (26 мг). МС (ESI(+)) m/e: 693,3 (М+Н)+.
Пример 140
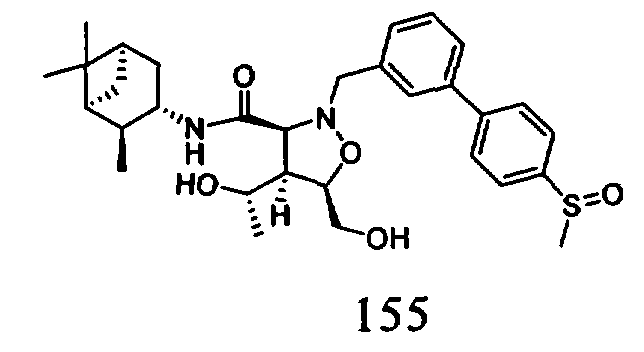
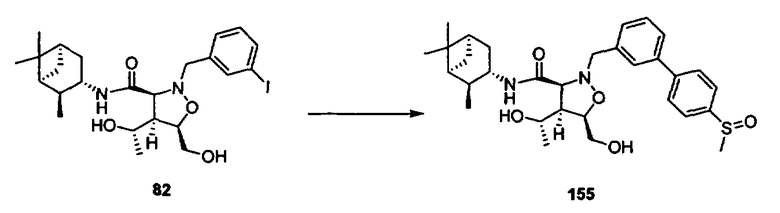
В небольшую колбу помещали соединение 82 (50 мг), 3-метилтиофенилбороновую кислоту (31 мг), Cs2CO3 (60 мг), КОАс (10 мг) и Pd(dppf)Cl2 (7 мг), колбу продували аргоном и добавляли ДМСО (5 мл). Смесь нагревали при 60°С в течение 1,5 ч, очищали методом ЖХВР и при этом получали твердое вещество белого цвета (45 мг).
Порцию полученного твердого вещества (35 мг) растворяли в МеОН (1 мл) и добавляли раствор оксона (20 мг) в воде (130 мкл). Через 30 мин реакционную смесь добавляли в 10% Na2S2O3 (500 мкл), разбавляли водой (10 мл) и экстрагировали ДХМ (3×10 мл). Объединенные органические фазы концентрировали и очищали методом ЖХВР. После концентрирования соответствующих фракций получали соединение 155 в виде твердого вещества белого цвета (28 мг). МС (ESI(+)) m/e: 555,1 (М+Н)+.
Пример 141
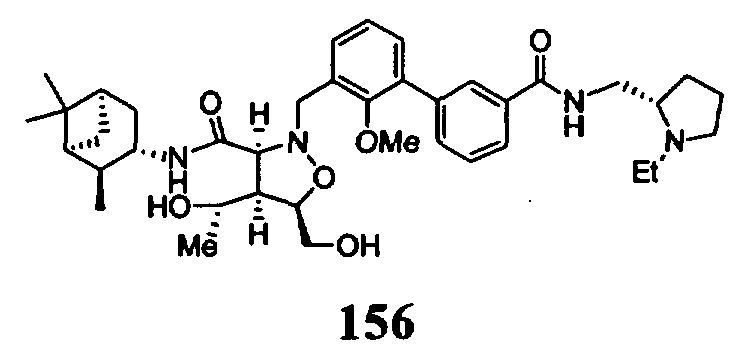
Стадия А
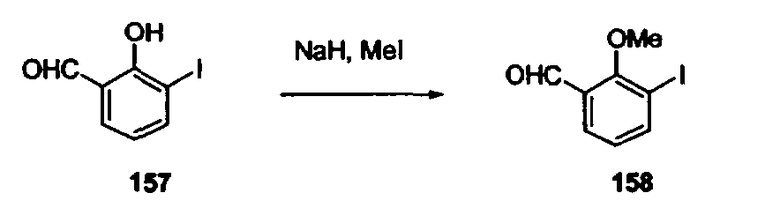
В раствор фенола 157 (750 мг, 3 ммоля) в ДМФА (5 мл) при 0°С добавляли NaH (130 мг, 3,6 ммоля), а затем Mel (280 мкл, 4,5 ммоля). Реакционную смесь перемешивали при КТ в течение 24 ч, затем реакцию останавливали добавлением воды. Смесь разбавляли EtOAc и промывали два раза водой, а затем солевым раствором. Полученный раствор сушили над MgSO4, фильтровали и концентрировали, при этом получали неочищенный продукт 157 (795 мг, 100%).
Стадия В
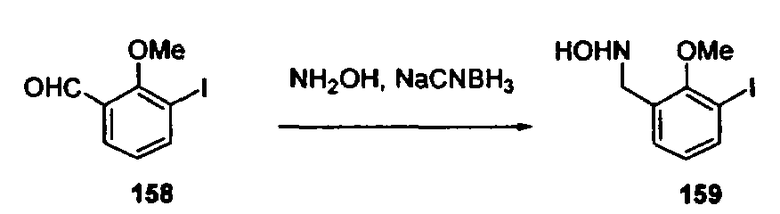
Альдегид 158 (795 мг, 3,03 ммоля) и гидрохлорид гидроксиламина (253 мг, 3,64 ммоля) растворяли в ТГФ/МеОН (10 мл, 3:2), затем добавляли воду (2 мл) и величину рН доводили до 9 добавлением 6,0 н. KOH. Реакционную смесь перемешивали при КТ в течение ночи. Через 16 ч добавляли цианоборгидрид натрия (381 мг, 6,07 ммоля), затем один кристалл метилового оранжевого. Величину рН доводили до 2, при этом раствор окрашивался в рубиново-красный цвет, который сохранялся в ходе реакции при периодическом добавлении 1н. HCl. Смесь перемешивали в течение 2 ч, затем добавляли еще одну порцию цианоборгидрида натрия (381 мг). После перемешивания в течение 16 ч величину рН доводили до 7 и добавляли ДХМ. Смесь три раза промывали водой, солевым раствором и сушили над MgSO4. Неочищенный продукт очищали экспресс-хроматографией (элюент: 50% EtOAc в гексане, затем 100% EtOAc), при этом получали гидроксиламин 159 (706 мг, 83%).
Стадия С

Раствор гидроксиламина 159 (705 мг, 2,53 ммоля) и метилглиоксилата (445 мг, 5,05 ммоля) в бензоле (15 мл) кипятили с обратным холодильником с использованием насадки Дина-Старка в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученный нитрон 160 использовали на следующей стадии без дополнительной очистки.
Стадия D
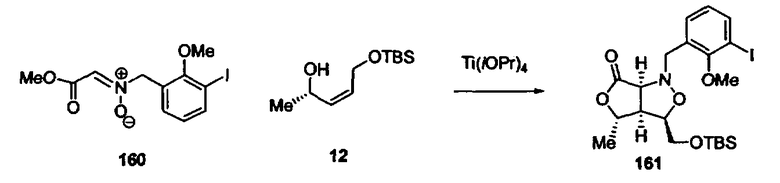
Нитрон 160 (882 мг, 2,53 ммоля), аллиловый спирт 6 (820 мг, 3,79 ммоля) и Ti(iOPr)4 (1,12 мл, 3,79 ммоля) растворяли в толуоле (5 мл) и нагревали в микроволновом реакторе при 120°С в течение 10 мин. Реакционную смесь разбавляли EtOAc (15 мл) и добавляли 3-(диметиламино)-1,2-пропандиол (500 мкл), затем перемешивали в течение 2 ч, добавляли EtOAc и смесь три раза промывали водой, солевым раствором, сушили над MgSO4, фильтровали через целит и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: гексан/EtOAc, 5:1), при этом получали лактон 161 (575 мг, 43%).
Стадия Е
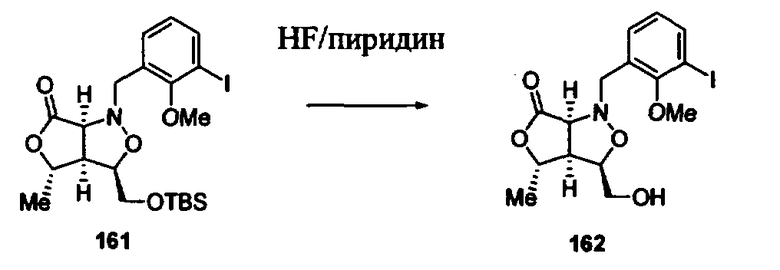
В раствор соединения 161 (225 мг, 0,042 ммоля) в ТГФ (6 мл) добавляли пиридин (2 мл) и смесь HF/пиридин (2 мл). Смесь перемешивали при КТ в течение 4 ч, затем добавляли TMSOMe (8 мл). Растворитель удаляли при пониженном давлении и неочищенный продукт очищали экспресс-хроматографией (элюент: EtOAc), при этом получали соединение 162 в виде пены белого цвета (128 мг, 72%).
Стадия F
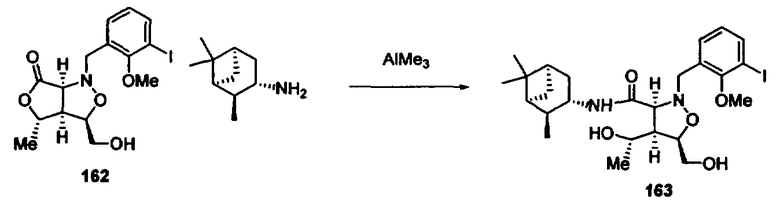
В высушенную в пламени горелки круглодонную колбу объемом 10 мл, содержащую (+)-изопинокамфеиламин (108 мкл, 0,611 ммоля) и ДХМ (2,0 мл), добавляли триметилалюминий в гексане (2,0 М, 305 мкл, 0,611 ммоля), смесь перемешивали в течение 15 мин, затем добавляли раствор лактона 162 (128 мг, 0,305 ммоля) в ДХМ (4 мл) и смесь перемешивали при КТ в течение ночи. Реакцию останавливали добавлением насыщенного водного раствора сегнетовой соли (5 мл) и смесь интенсивно перемешивали в течение 2 ч, добавляли ДХМ, смесь три раза промывали водой, солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали экспресс-хроматографией (элюент: EtOAc), при этом получали соединение 163 (160 мг, 91%).
Стадия G
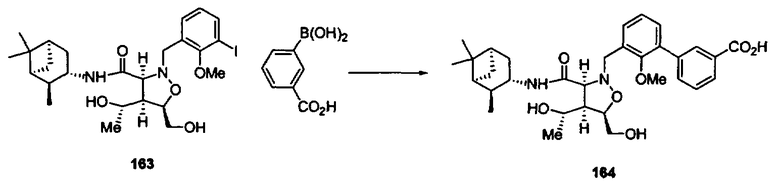
В смесь иодида 163 (45 мг, 0,079 ммоля), 3-карбоксифенилбороновой кислоты (26 мг, 0,160 ммоля), Cs2CO3 (100 мг, 0,31 ммоля), КОАс (8 мг, 0,079 ммоля) и Pd(dppf)Cl2 (6 мг, 0,008 ммоля) в атмосфере аргона добавляли ДМСО (2 мл, дегазированный). Смесь перемешивали при 60°С в течение 12 ч. По данным анализа ЖХ-МС в реакционной смеси еще оставалось некоторое количество исходного материала, поэтому добавляли еще одну порцию Pd(dppf)Cl2 (6 мг, 0,008 ммоля) и смесь перемешивали при 60°С в течение 6 ч. Смесь разбавляли ДХМ и три раза промывали водой, солевым раствором, сушили над MgSO4 и концентрировали. Неочищенный продукт 164 использовали на следующей стадии без дополнительной очистки.
Стадия Н
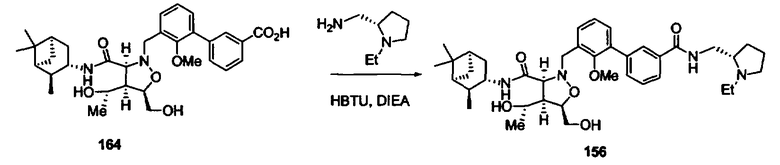
В кислоту 164 (22 мг, 0,039 ммоля), растворенную в ДМФА (700 мкл), добавляли (S)-2-аминоэтил-1-этилпирролидин (20 мг, 0,16 ммоля), DIEA (27 мкл, 0,16 ммоля) и HBTU (44 мг, 0,12 ммоля). Реакционную смесь перемешивали при КТ в течение 3 ч, затем разбавляли метанолом (800 мкл). Неочищенный продукт очищали методом ЖХВР, фракции, содержащие требуемый продукт, объединяли и лиофилизировали, при этом получали соединение 156 в виде рыхлого твердого вещества белого цвета (5 мг, 20%). МС (ESI(+)) m/e: 677,4 (М+Н)+.
Пример 142
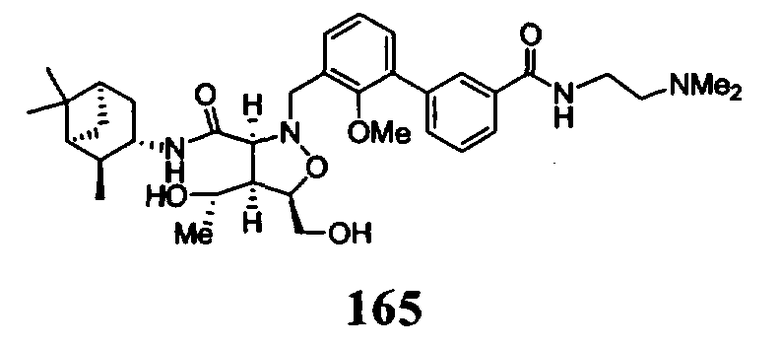
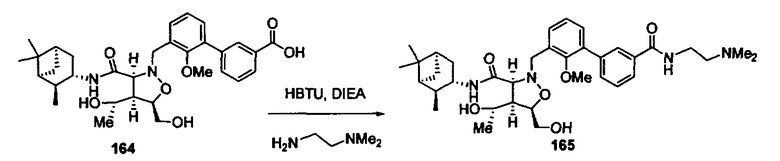
В кислоту 164 (22 мг, 0,039 ммоля), растворенную в ДМФА (700 мкл), добавляли диметилэтилендиамин (17 мкл, 0,16 ммоля), DIEA (27 мкл, 0,16 ммоля) и HBTU (44 мг, 0,12 ммоля). Реакционную смесь перемешивали при КТ в течение 3 ч, затем разбавляли метанолом (800 мкл). Неочищенный продукт очищали методом ЖХВР, фракции, содержащие требуемый продукт, объединяли и лиофилизировали, при этом получали соединение 165 в виде рыхлого твердого вещества белого цвета (5 мг, 20%).
Пример 143
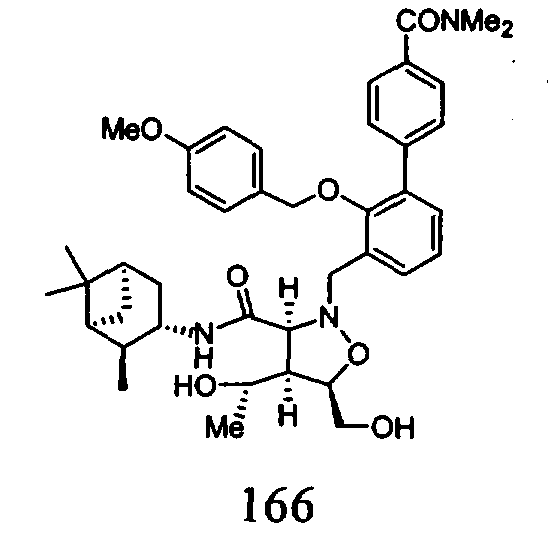
Стадия А
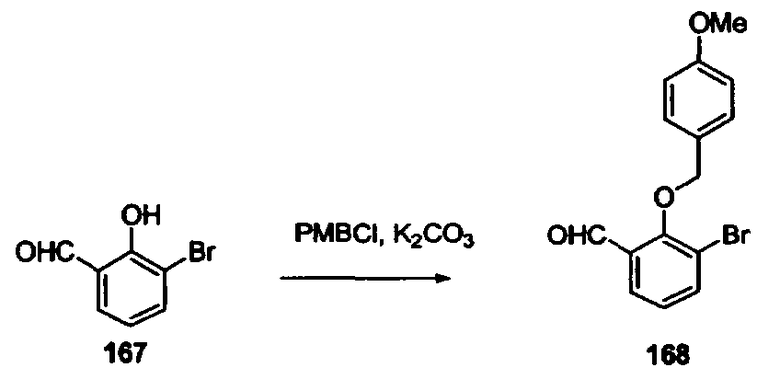
В неочищенный фенол 167 (6,1 г, 30 ммолей, полученный из 2-бромфенола по методике, описанной в J. Med. Chem. 44 (17), 2766 (2001)) добавляли ДМФА (5 мл), затем PMBCl (4,9 мл, 36 ммолей) и К2СО3 (6,3 г, 46 ммолей). Реакционную смесь перемешивали при 60°С в течение 5 мин охлаждали до КТ, смесь разбавляли водой и два раза экстрагировали EtOAc. Слой EtOAc промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Неочищенный материал (масло желтого цвета) очищали экспресс-хроматографией (элюент: гексан/EtOAc, 9:1), при этом получали соединение 168 в виде твердого вещества белого цвета (5,7 г, 58% в расчете на две стадии).
Стадия В
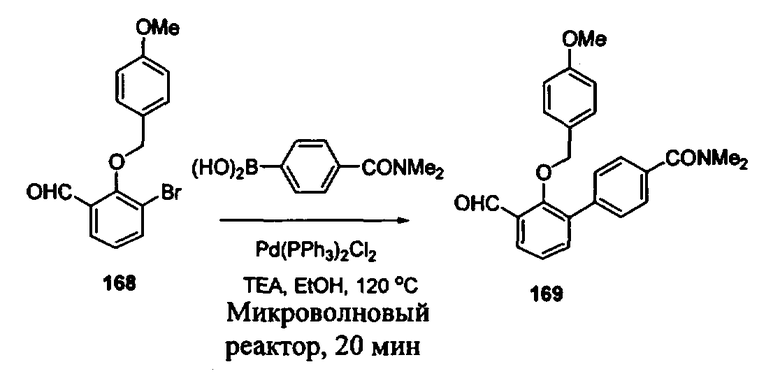
В раствор арилбромида 168 (500 мг, 2 ммоля) в EtOH (5 мл) добавляли Et3N (651 мкл, 5 ммолей), [4-(N,N-диметиламинокарбонил)фенил]бороновую кислоту (391 мг, 2 ммоля) и Pd(PPh3)2Cl2 (109 мг, 0,2 ммоля). Смесь нагревали в микроволновом реакторе при 120°С в течение 20 мин, затем концентрировали и очищали экспресс-хроматографией, при этом получали соединение 169 (320 мг, 53%).
Стадия С
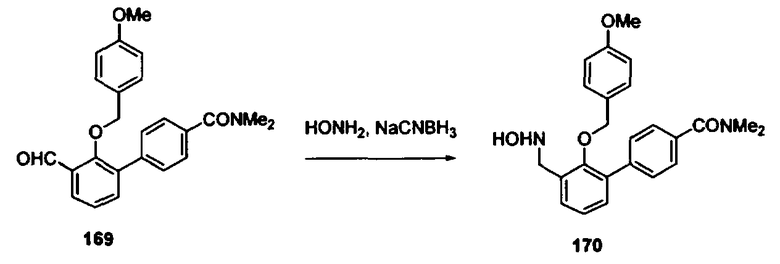
Альдегид 169 (640 мг, 1,64 ммоля) и гидрохлорид гидроксиламина (136 мг, 1,97 ммоля) растворяли в ТГФ/МеОН (10 мл, 3:2), добавляли воду (2 мл) и величину рН доводили до 9 добавлением 6,0 н. KOH. Реакционную смесь перемешивали при КТ в течение ночи. Через 16 ч добавляли цианоборгидрид натрия (207 мг, 3,29 ммоля), затем один кристалл метилового оранжевого. Величину рН доводили до 2, при этом раствор окрашивался в рубиново-красный цвет, который сохранялся в ходе реакции при периодическом добавлении 1 н. HCl. Смесь перемешивали в течение 2 ч, затем добавляли еще одну порцию цианоборгидрида натрия (207 мг). После перемешивания в течение 16 ч величину рН доводили до 7 и добавляли ДХМ. Смесь три раза промывали водой, солевым раствором и сушили над MgSO4. Неочищенный продукт очищали экспресс-хроматографией (элюент: 50% EtOAc в гексане, затем 100% EtOAc), при этом получали гидроксиламин 170 (500 мг, 75%).
Стадия D
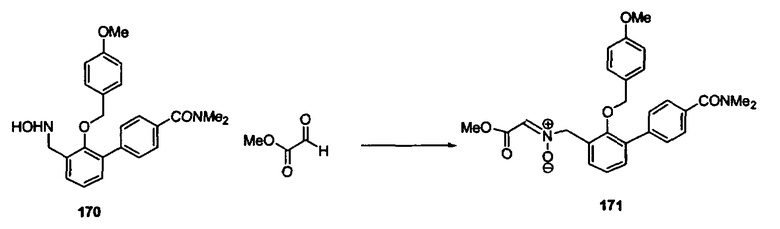
Раствор гидроксиламина 170 (500 мг, 1,23 ммоля) и метилглиоксилата (217 мг, 2,46 ммоля) в бензоле (15 мл) кипятили с обратным холодильником с использованием насадки Дина-Старка в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученный нитрон 171 использовали на следующей стадии без дополнительной очистки.
Стадия Е
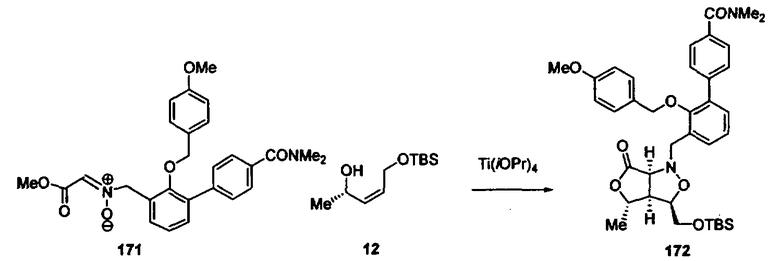
Нитрон 171 (586 мг, 1,23 ммоля), аллиловый спирт 12 (399 мг, 1,84 ммоля) и Ti(iOPr)4 (544 мкл, 1,84 ммоля) растворяли в толуоле (5 мл) и нагревали в микроволновом реакторе при 120°С в течение 10 мин. Реакционную смесь разбавляли EtOAc (10 мл) и добавляли 3-(диметиламино)-1,2-пропандиол (200 мкл), затем перемешивали в течение 2 ч, добавляли EtOAc и смесь три раза промывали водой, солевым раствором, сушили над MgSO4, фильтровали через целит и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: ДХМ, Затем 10% МеОН в ДХМ), при этом получали лактон 172 (438 мг, 54%).
Стадия F
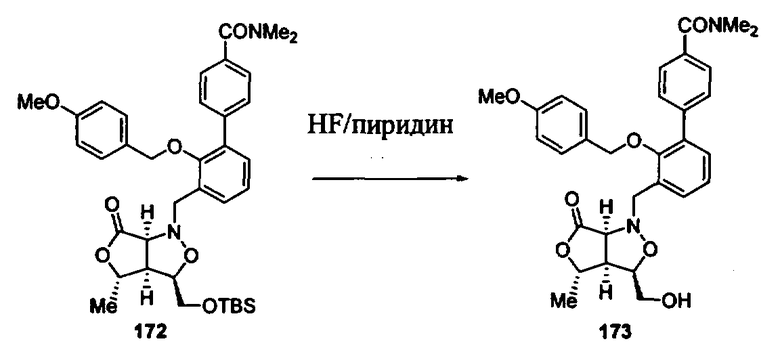
В раствор соединения 172 (25 мг, 0,038 ммоля) в ТГФ (1,5 мл) при 0°С добавляли пиридин (1,5 мл) и смесь HF/пиридин (450 мкл). Смесь перемешивали в течение 2 ч, затем добавляли TMSOMe (1 мл). Смесь концентрировали при пониженном давлении и неочищенный продукт 173 использовали на следующей стадии без дополнительной очистки.
Стадия G
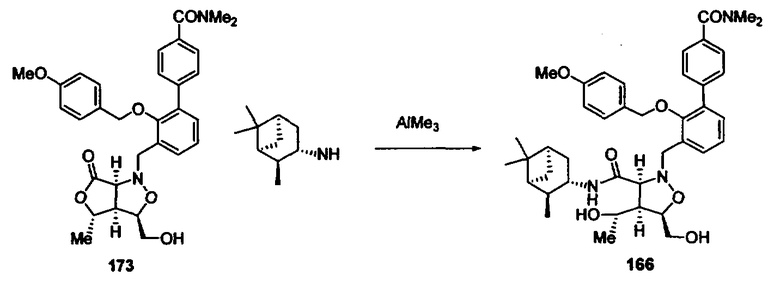
В небольшую высушенную в пламени горелки колбу, содержащую (+)-изопинокамфеиламин (16 мкл, 0,096 ммоля) и ДХМ (500 мкл) добавляли триметилалюминий в гексане (2,0 М, 38 мкл, 0,077 ммоля), смесь перемешивали в течение 15 мин, затем добавляли раствор лактона 173 (21 мг, 0,038 ммоля) в ДХМ (1 мл) и смесь перемешивали при КТ в течение ночи. Реакцию останавливали добавлением насыщенного водного раствора сегнетовой соли (2 мл) и ДХМ (3 мл), смесь интенсивно перемешивали в течение 2 ч, добавляли ДХМ, смесь три раза промывали водой, солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали экспресс-хроматографией (элюент: 2% МеОН в ДХМ), при этом получали соединение 166 (18 мг, 67%).
МС (ESI(+)) m/e: 700,3 (M+H)+.
Пример 144
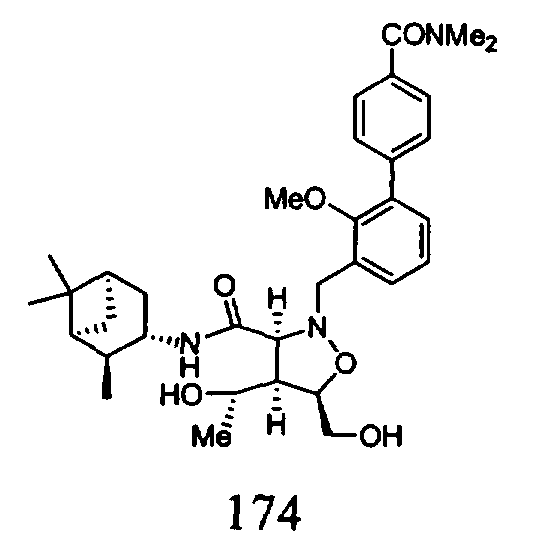
Стадия А
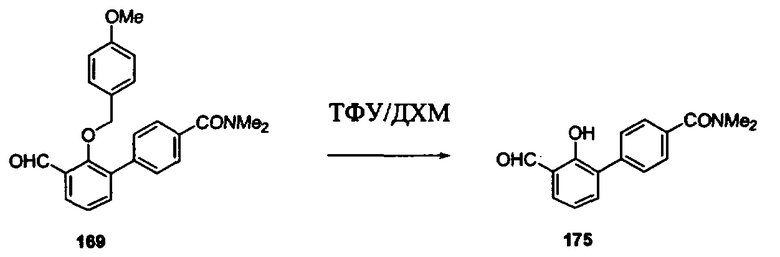
В раствор соединения 169 в ДХМ (100 мл) добавляли ТФУ (2 мл). Реакционную смесь перемешивали при КТ в течение 3 ч, а затем разбавляли ДХМ. Смесь два раза промывали насыщенным водным раствором NaHCO3 и водой, сушили над MgSO4 и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: градиент гексан/EtOAc, от 2:1 до 1:1), при этом получали соединение 175 в виде масла желтого цвета (1,8 г, 87%).
Стадия В

В раствор фенола 175 (490 мг, 1,8 ммоля) в ДМФА (8 мл) добавляли NaH (90 мг, 2,2 ммоля, 60%), затем MeI (170 мкл, 2,7 ммоля). Реакционную смесь перемешивали при КТ в течение 3 ч, а затем разбавляли водой и экстрагировали EtOAc. Слой EtOAc промывали водой, затем солевым раствором, сушили над MgSO4 и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: градиент гексан/EtOAc, от 2:1 до 1:1), при этом получали соединение 176 (453 мг, 88%).
Стадия С
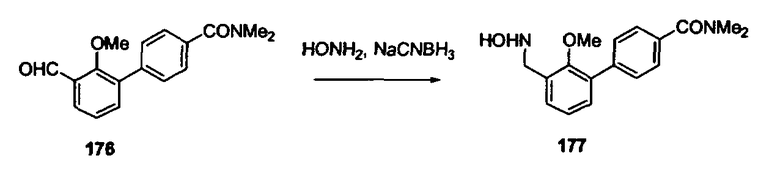
Альдегид 176 (453 мг, 1,60 ммоля) и гидрохлорид гидроксиламина (133 мг, 1,92 ммоля) растворяли в ТГФ/МеОН (10 мл, 3:2), добавляли воду (2 мл) и величину рН доводили до 9 добавлением 6,0 н. KOH. Реакционную смесь перемешивали при КТ в течение ночи. Через 16 ч добавляли цианоборгидрид натрия (201 мг, 3,20 ммоля), затем один кристалл метилового оранжевого. Величину рН доводили до 2, при этом раствор окрашивался в рубиново-красный цвет, который сохранялся в ходе реакции при периодическом добавлении 1н. HCl. Смесь перемешивали в течение 2 ч, затем добавляли еще одну порцию цианоборгидрида натрия (201 мг). После перемешивания в течение 16 ч величину рН доводили до 7 и добавляли ДХМ. Смесь три раза промывали водой, солевым раствором и сушили над MgSO4. Неочищенный продукт очищали экспресс-хроматографией (элюент: 50% EtOAc в гексане, затем 100% EtOAc), при этом получали гидроксиламин 177 (402 мг, 84%).
Стадия D

Раствор гидроксиламина 177 (402 мг, 1,34 ммоля) и метилглиоксилата (236 мг, 2,68 ммоля) в бензоле (15 мл) кипятили с обратным холодильником с использованием насадки Дина-Старка в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученный нитрон 178 использовали на следующей стадии без дополнительной очистки.
Стадия Е
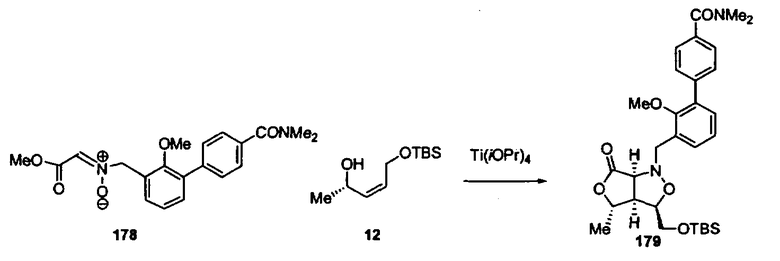
Нитрон 178 (496 мг, 2,53 ммоля), аллиловый спирт 6 (435 мг, 2,01 ммоля) и Ti(iOPr)4 (0,593 мл, 2,01 ммоля) растворяли в толуоле (5 мл) и нагревали в микроволновом реакторе при 120°С в течение 10 мин. Реакционную смесь разбавляли EtOAc (10 мл) и добавляли 3-(диметиламино)-1,2-пропандиол (200 мкл), затем перемешивали в течение 2 ч, добавляли EtOAc и смесь три раза промывали водой, солевым раствором, сушили над MgSO4, фильтровали через целит и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: ДХМ, затем 10% МеОН в ДХМ), при этом получали лактон 179 (575 мг, 77%).
Стадия F
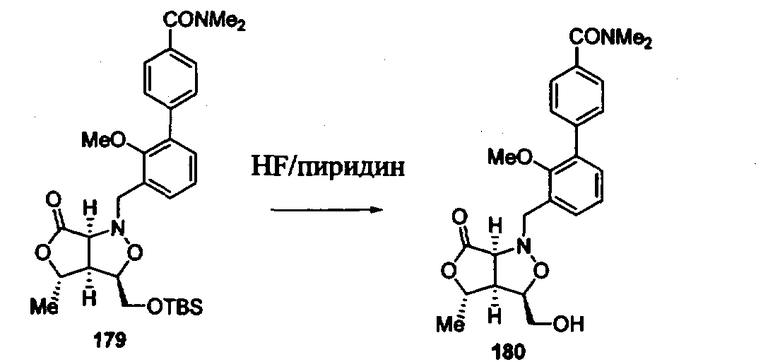
В раствор соединения 179 (510 мг, 919 ммолей) в ТГФ (10 мл) при 0°С добавляли пиридин (5 мл) и смесь HF/пиридин (2 мл). Смесь перемешивали в течение 5 ч, затем реакцию останавливали добавлением TMSOMe (15 мл) и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: 2% МеОН в ДХМ), при этом получали соединение 180 (267 мг, 66%).
Стадия G
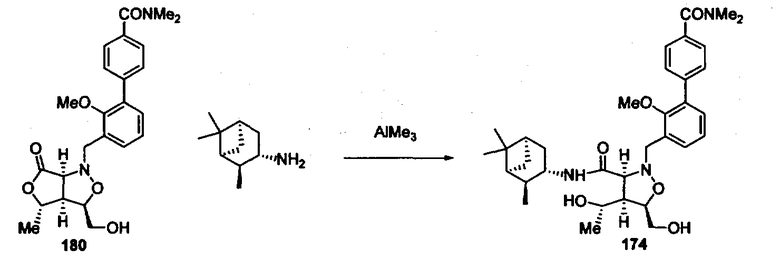
В высушенную в пламени горелки круглодонную колбу объемом 25 мл, содержащую (+)-изопинокамфеиламин (160 мкл, 0,98 ммоля) и ДХМ (2 мл), добавляли триметилалюминий в гексане (2,0 М, 490 мкл, 0,98 ммоля), смесь перемешивали в течение 20 мин, затем добавляли раствор лактона 180 (215 мг, 0,490 ммоля) в ДХМ (8 мл) и смесь перемешивали при КТ в течение ночи.
Реакцию останавливали добавлением насыщенного водного раствора сегнетовой соли (10) и ДХМ (10 мл), смесь интенсивно перемешивали в течение 2 ч, добавляли ДХМ, смесь три раза промывали водой, солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали экспресс-хроматографией (элюент: EtOAc, затем 1% МеОН в EtOAc), при этом получали соединение 174 (170 мг, 59%).
МС (ESI(+)) m/e: 594,2 (M+H)+.
Пример 145
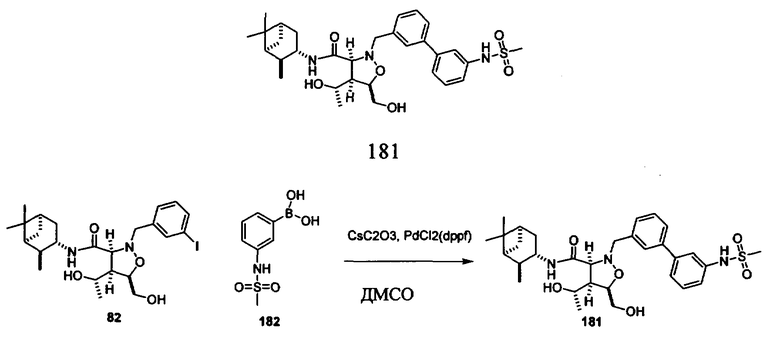
Арилиодид 82 (0,04 ммоля), бороновую кислоту 182 (0,08 ммоля), карбонат цезия (0,08 ммоля) и катализатор на основе палладия (0,008 ммоля) смешивали в реакционном сосуде и продували аргоном. Затем в реакционную смесь добавляли ДМСО (1 мл) и перемешивали в атмосфере азота при 60°С в течение 4 ч. Реакционную смесь охлаждали до КТ, разбавляли водой и экстрагировали EtOAc (3×5 мл). Органические слои отделяли, сушили над MgSO4 и концентрировали. Неочищенный материал очищали методом ЖХВР, выход 65%. МС (ESI(+)) m/e: 586,1 (М+Н)+.
Пример 146
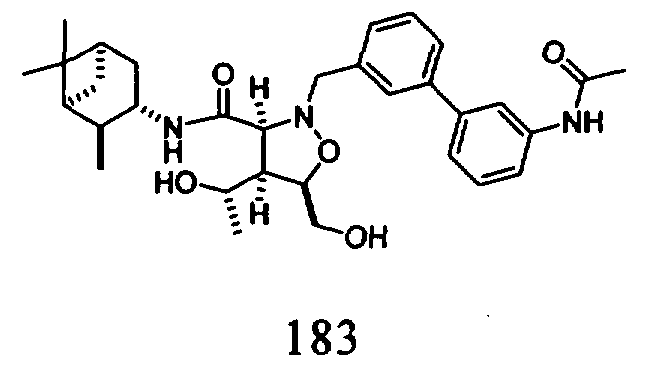
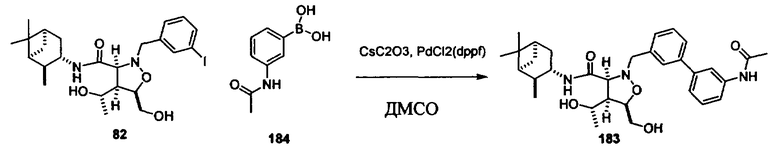
Арилиодид 82 (0,04 ммоля), бороновую кислоту 184 (0,08 ммоля), карбонат цезия (0,08 ммоля) и катализатор на основе палладия (0,008 ммоля) смешивали в реакционном сосуде и продували аргоном. Затем в реакционную смесь добавляли ДМСО (1 мл) и перемешивали в атмосфере азота при 60°С в течение 4 ч. Реакционную смесь охлаждали до КТ, разбавляли водой и экстрагировали EtOAc (3×5 мл). Органические слои отделяли, сушили над MgSO4 и концентрировали. Неочищенный материал очищали методом ЖХВР, выход 60%.
МС (ESI(+)) m/e: 550,2 (М+Н)+.
Пример 147
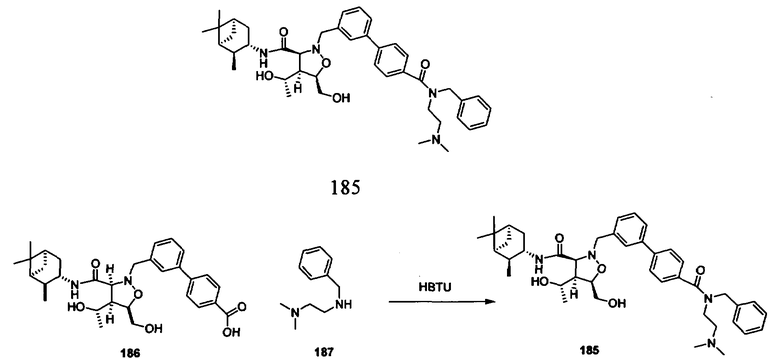
В раствор биариловой кислоты 186 (получена по методике, описанной для синтеза соединения 87, 0,03 ммоля) в ДМФА (0,6 мл) добавляли HBTU (0,06 ммоля) и амин 187 (0,10 ммоля). Затем сосуд с реакционной смесью закрывали и перемешивали в атмосфере азота при КТ в течение 1 ч. Реакционную смесь охлаждали до КТ, разбавляли водой и экстрагировали EtOAc (3×5 мл). Органические слои отделяли, сушили над MgSO4 и концентрировали. Неочищенный материал очищали методом ЖХВР, выход 85%.
МС (ESI(+)) m/e: 697,5 (М+Н)+.
Пример 148
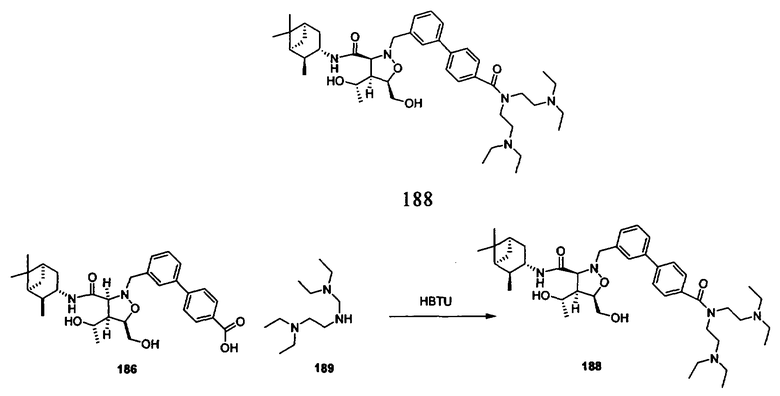
В раствор биариловой кислоты 186 (0,03 ммоля) в ДМФА (0,6 мл) добавляли HBTU (0,06 ммоля) и амин 189 (0,10 ммоля). Затем реакционный сосуд закрывали, и смесь перемешивали при КТ в течение 1 ч в атмосфере азота. Реакционную смесь разбавляли водой и экстрагировали EtOAc (3×5 мл). Органические слои объединяли, сушили над MgSO4 и концентрировали. Неочищенный материал очищали методом ЖХВР (выход 82%).
МС (ESI(+)) (m/e): 735,6 (М+Н)+.
Пример 149
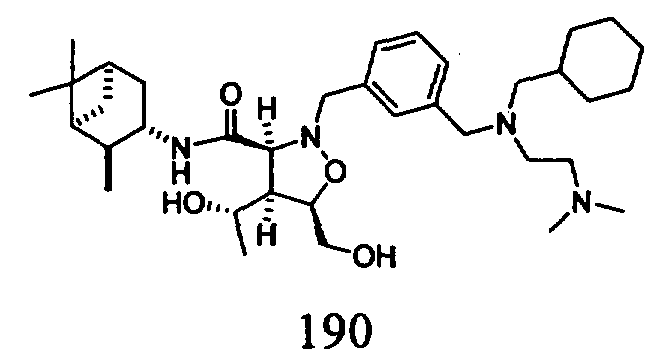
Стадия А
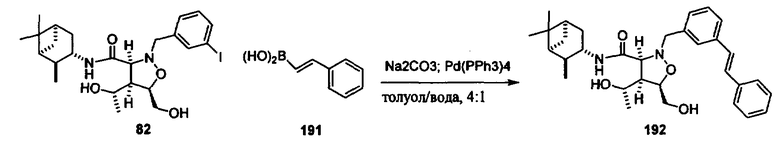
В колбу объемом 100 мл добавляли арилиодид 82 (2,65 ммоля), винилбороновую кислоту 191 (10,62 ммоля), карбонат натрия (10,62 ммоля) и тетракис палладия (0,530 ммоля). Затем колбу продували аргоном, и реагенты растворяли в смеси толуол/вода (4:1, 30 мл). Полученную смесь нагревали при 65°С в течение 3 ч. Затем органический слой отделяли. Водный слой экстрагировали EtOAc (3×20 мл). Органические слои объединяли, промывали солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный материал очищали экспресс-хроматографией (выход 85%).
Стадия В
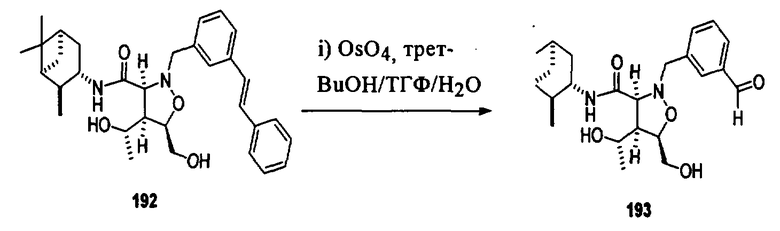
В круглодонную колбу (объем 200 мл) добавляли соединение 192 (0,110 г, 0,205 ммоля) и NMO (0,111 г, 0,820 ммоля), растворенный в трет-BuOH (16 мл), ТГФ (8 мл), Н2О (2 мл). В полученный перемешиваемый раствор по каплям добавляли раствор OsO4 в трет-BuОН (2,5% раствор, 2,9 мл, 0,210 г, 0,0205 ммоля). По данным ТСХ через 4 ч наблюдалось полное потребление исходного материала. Реакцию останавливали добавлением раствора Na2S2O3, продукт распределяли между этилацетатом и солевым раствором. Водный слой промывали, органическую фазу сушили над MgSO4, фильтровали и концентрировали. Затем полученный неочищенный продукт (0,010 г) переносили в ТГФ (0,2 мл), добавляли воду (20 мкл), периодат натрия (0,0041 г), и перемешивали в течение ночи. Реакцию останавливали добавлением Na2S2O3, продукт промывали солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный материал очищали экспресс-хроматографией (выход 85%).
Стадия С
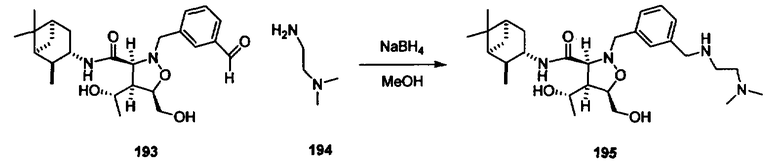
Альдегид 193 (0,274 ммоля) переносили в круглодонную колбу (объем 25 мл) и растворяли в метаноле (6 мл). Затем в полученный раствор добавляли амин 194 (0,549 ммоля) и перемешивали в течение 1 ч, при этом получали прозрачный раствор. В полученный раствор одной порцией добавляли боргидрид натрия (0,549 ммоля). Затем добавляли еще одну порцию амина (1 экв.), и реакционную смесь перемешивали в течение 20 мин, добавляли еще одну порцию боргидрида (0,5 экв.) и перемешивали. Реакцию останавливали добавлением уксусной кислоты (40 мкл). После добавления бензола (2 мл) реакционную смесь концентрировали при пониженном давлении. Неочищенный материал несколько раз перегоняли в азеотропной смеси с бензолом (3 мл), при этом получали твердое вещество белого цвета, которое сушили в высоком вакууме в течение ночи. Полученный материал использовали на следующей стадии без дополнительной очистки.
Стадия D
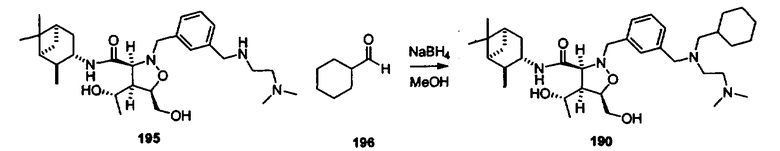
Соединение 195 (18 мг, 0,035 ммоля) переносили в флакон (1/8 унций), добавляли ДХМ (1 мл), затем добавляли альдегид 196 (0,070 ммоля). Затем одной порцией добавляли триацетоксиборгидрид натрия (0,052 ммоля), и реакционную смесь перемешивали при КТ. Неочищенный материал очищали методом ЖХВР (выход 77%).
МС (ESI(+)) (m/e): 613,5 (М+Н)+.
Пример 150
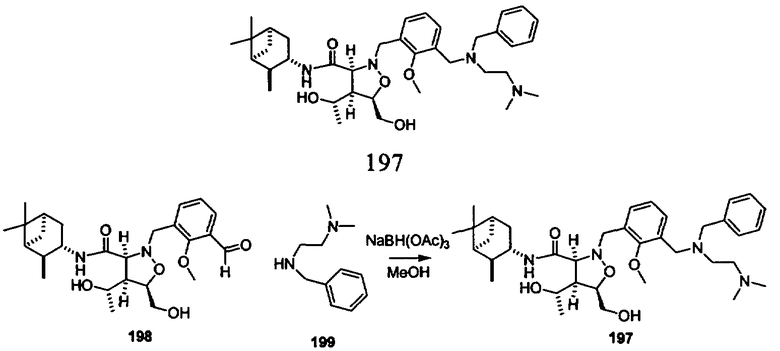
Альдегид 198 получали по методике, описанной в примере 149, но при замене арилиодида 82 на арилиодид 163 (выход 88%). Альдегид 198 (0,02 ммоля) переносили в колбу, растворяли в ДХМ (0,4 мл), и добавляли амин 199 (0,08 ммоля). Затем в полученную смесь добавляли триацетоксиборгидрид натрия (0,03 ммоля) и перемешивали в течение ночи. Неочищенную реакционную смесь очищали методом ЖХВР (выход 91%).
МС (ESI(+)) (m/e): 637,5 (М+Н)+.
Пример 151
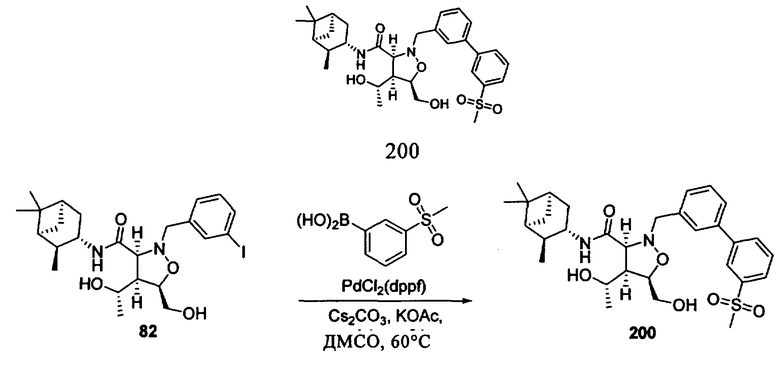
В круглодонную колбу добавляли соединение 82 (20 мг, 0,035 ммоля), 3-метилсульфон фенилбороновой кислоты (14 мг, 0,070 ммоля), карбонат цезия (34 мг, 0,11 ммоля), ацетат калия (3,5 мг, 0,035 экв.) и PdCl2(dppf) (2,9 мг, 0,0035 ммоля). Колбу продували аргоном и добавляли 1 мл ДМСО (предварительно дегазированный аргоном в течение 30 мин). Реакционную смесь в атмосфере аргона нагревали на масляной бане при 60°С в течение 3 ч, затем охлаждали и перемешивали при 23°С в течение еще 12 ч. Смесь разбавляли CH2Cl2 (5 мл), насыщенным солевым раствором (5 мл) и экстрагировали CH2Cl2 (2×25 мл). Органические слои объединяли и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное масло очищали экспресс-хроматографией (SiO2, 5 г, элюент: градиент EtOAc/гексан от 90 до 100%), при этом получали бифенилметилсульфон 200 (13 мг, 65%) в виде бесцветного масла.
МС (ESI(+)) (m/e): 571,2 (М+Н)+.
Пример 152
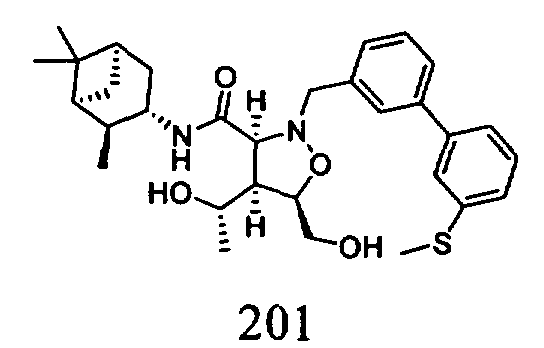
Соединение 201 получали по методике, описанной в примере 151, но при замене 3-метилсульфон фенилбороновой кислоты на 3-метилсульфид фенилбороновой кислоты. Выход 70%.
МС (ESI(+)) (m/e): 539,2 (М+Н)+.
Пример 153
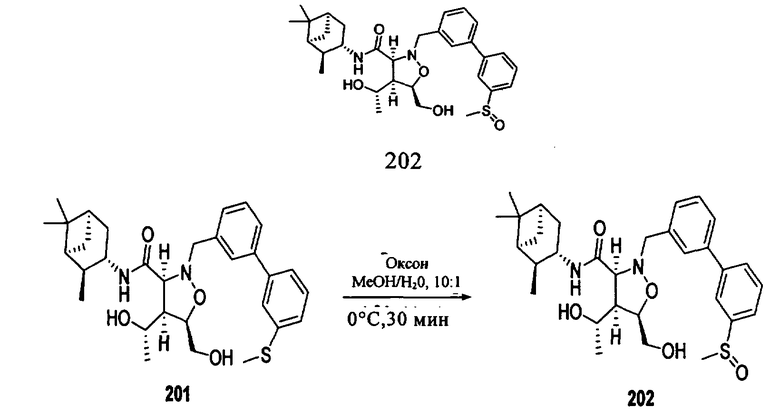
В раствор соединения 201 (22 мг, 0,041 ммоля) в МеОН/ТГФ (10:1, 2 мл), при 0°С одной порцией добавляли оксон (13 мг, 0,02 экв.). После перемешивания в течение 1 ч при 0°С реакционную смесь разбавляли CH2Cl2 (5 мл), насыщенным солевым раствором (5 мл) и экстрагировали CH2Cl2 (2×10 мл). Органические слои объединяли и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное масло очищали методом препаративной тонкослойной хроматографии (пластинка 20×20 см, толщина слоя 250 мкм, элюент: 5% МеОН в CH2Cl2), при этом получали смесь диастереомеров сульфоксидов (1:1, 18 мг, 79%) в виде пены белого цвета.
МС (ESI(+)) (m/e): 555,2 (М+Н)+.
Пример 154
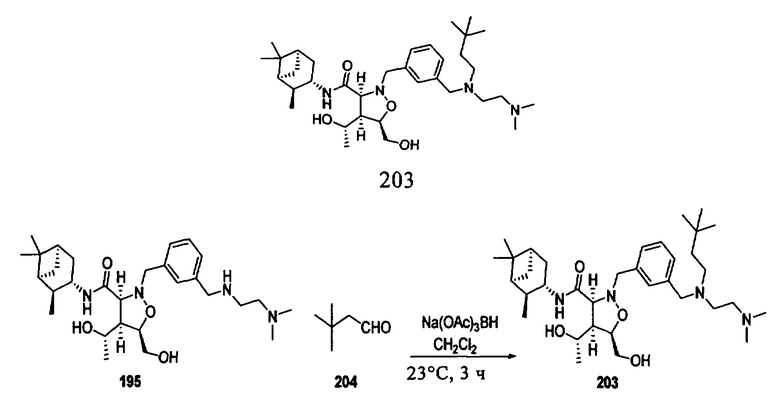
В раствор соединения 195 (20 мг, 0,04 ммоля) в CH2Cl2 (1 мл) при 23°С добавляли 3,3-диметилбутиральдегид (0,08 ммоля), а затем Na(OAc)3BH (10 мг, 0,06 экв.). После перемешивания в течение 3 ч при 23°С реакционную смесь фильтровали через слой стекловолокна и очищали методом ЖХВР, при этом получали требуемый продукт (приблизительно 5-10 мг, 20-50%).
MS (ESI(+)) (m/e): 601,5 (M+H)+.
Пример 155
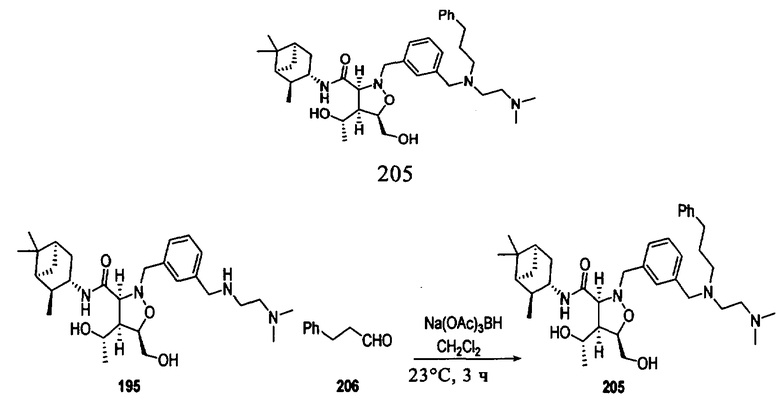
В раствор соединения 195 (20 мг, 0,04 ммоля) в CH2Cl2 (1 мл) при 23°С добавляли гидрокоричный альдегид (0,08 ммоля), а затем Na(ОАс)3ВН (10 мг, 0,06 экв.). После перемешивания при 23°С в течение 3 ч реакционную смесь фильтровали через слой стекловолокна и очищали методом ЖХВР, при этом получали требуемый продукт (приблизительно 5-10 мг, 20-50%).
МС (ESI(+)) (m/e): 635,4 (М+Н)+.
Пример 156
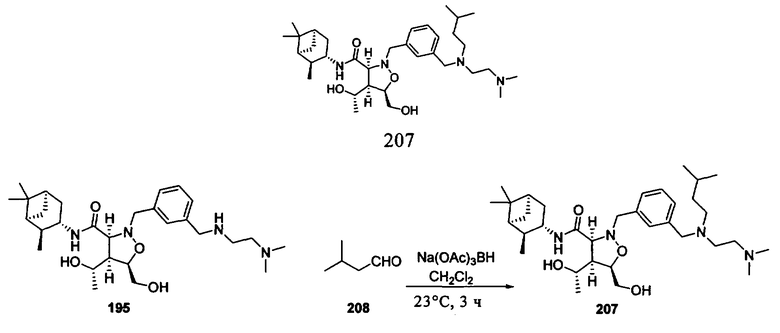
В раствор соединения 195 (20 мг, 0,04 ммоля) в CH2Cl2 (1 мл) при 23°С добавляли изомасляный альдегид (0,08 ммоля), а затем Na(ОАс)3ВН (10 мг, 0,06 экв.). После перемешивания при 23°С в течение 3 ч реакционную смесь фильтровали через слой стекловолокна и очищали методом ЖХВР, при этом получали требуемый продукт (приблизительно 5-10 мг, 20-50%).
МС (ESI(+)) (m/e): 587,5 (М+Н)+.
Пример 157
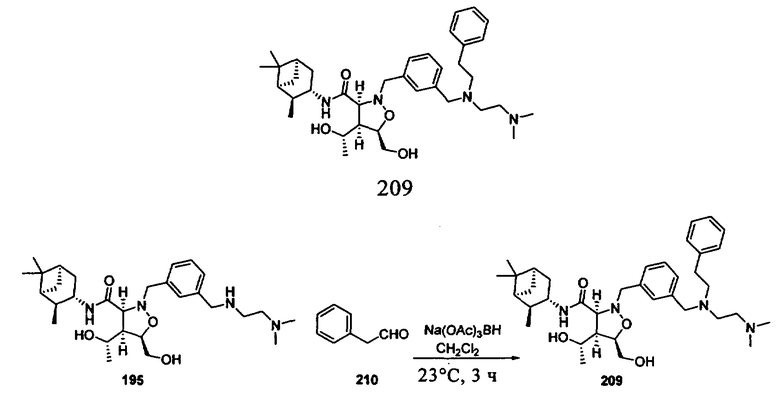
В раствор соединения 195 (20 мг, 0,04 ммоля) в CH2Cl2 (1 мл) при 23°С добавляли фенилацетальдегид (0,08 ммоля), а затем Na(OAc)3BH (10 мг, 0,06 экв.). После перемешивания при 23°С в течение 3 ч реакционную смесь фильтровали через слой стекловолокна и очищали методом ЖХВР, при этом получали требуемый продукт (приблизительно 5-10 мг, 20-50%).
МС (ESI(+)) (m/e): 621,5 (М+Н)+.
Пример 158
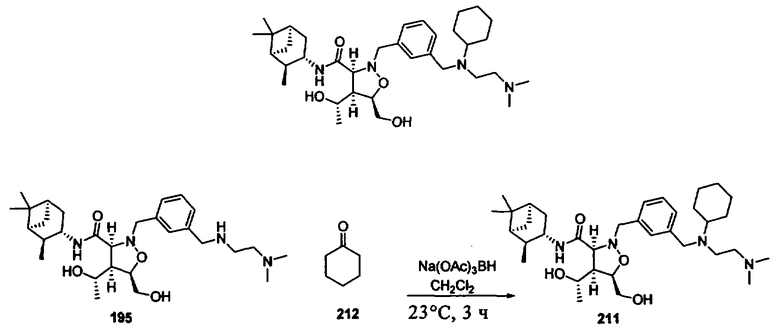
В раствор соединения 195 (20 мг, 0,04 ммоля) в CH2Cl2 (1 мл) при 23°С добавляли циклогексанон (0,08 ммоля), а затем Na(ОАс)3ВН (10 мг, 0,06 экв.). После перемешивания при 23°С в течение 3 ч реакционную смесь фильтровали через слой стекловолокна и очищали методом ЖХВР, при этом получали требуемый продукт (приблизительно 5-10 мг, 20-50%).
МС (ESI(+)) (m/e): 599,5 (М+Н)+.
Пример 159
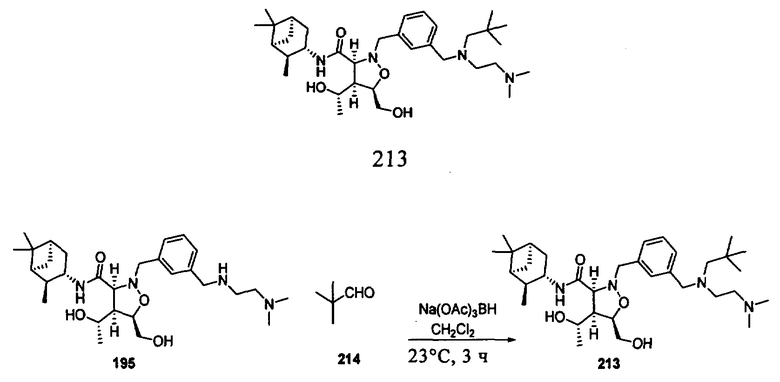
В раствор соединения 195 (20 мг, 0,04 ммоля) в CH2Cl2 (1 мл) при 23°С добавляли пивалоилальдегид (0,08 ммоля), а затем Na(ОАс)3 ВН (10 мг, 0,06 экв.). После перемешивания при 23°С в течение 3 ч реакционную смесь фильтровали через слой стекловолокна и очищали методом ЖХВР, при этом получали требуемый продукт (приблизительно 5-10 мг, 20-50%).
МС (ESI(+)) (m/e): 587,5 (М+Н)+.
Пример 160
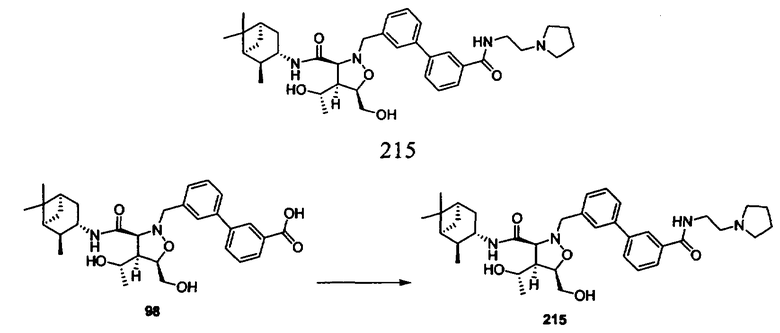
В раствор неочищенного соединения 98 (25 мг) и 1-(2-аминоэтил)пирролидина (16 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. Фракции, содержащие продукт, концентрировали, при этом получали соединение 215 (14 мг) в виде твердого вещества белого цвета.
МС (ESI(+)) (m/e): 633,3 (М+Н)+.
Пример 161
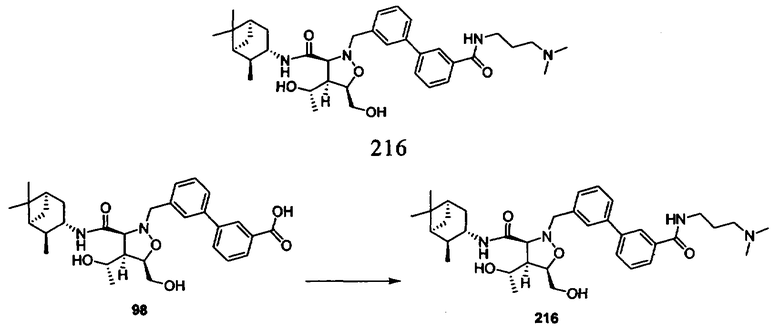
В раствор неочищенного соединения 98 (25 мг) и N,N-диметил-1,3-пропандиамина (19 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. Фракции, содержащие продукт, концентрировали, при этом получали соединение 216 (13 мг) в виде твердого вещества белого цвета.
MS (ESI(+)) (m/e): 621,3 (M+H)+.
Пример 162
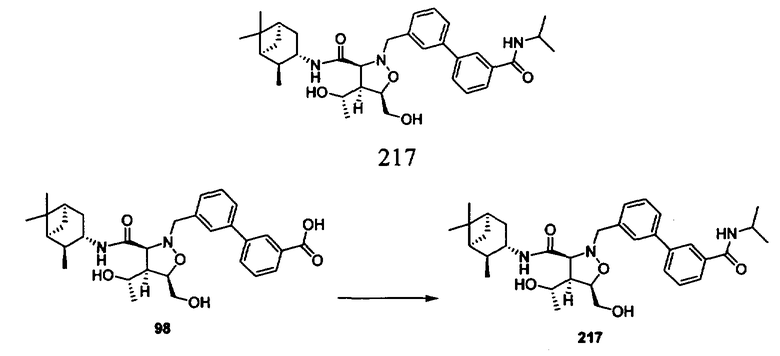
В раствор неочищенного соединения 98 (25 мг) и изопропиламина (13 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. Фракции, содержащие продукт, концентрировали, при этом получали соединение 217 (14 мг) в виде твердого вещества белого цвета.
MS (ESI(+)) (m/e): 578,3 (M+H)+.
Пример 163
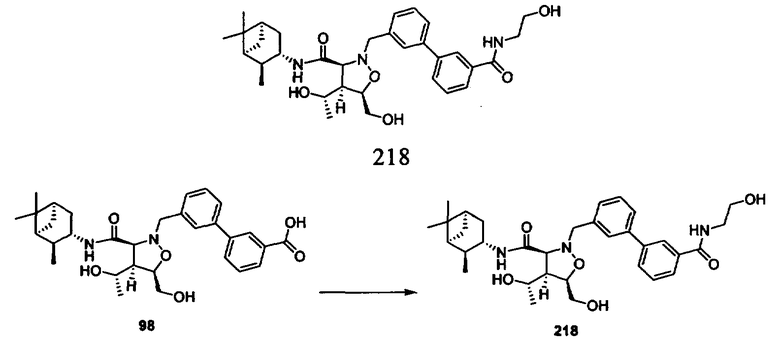
В раствор неочищенного соединения 98 (25 мг) и этаноламина (9 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. Фракции, содержащие продукт, концентрировали, при этом получали соединение 218 (14 мг) в виде твердого вещества белого цвета.
МС (ESI(+)) (m/e): 580,3 (М+Н)+.
Пример 164
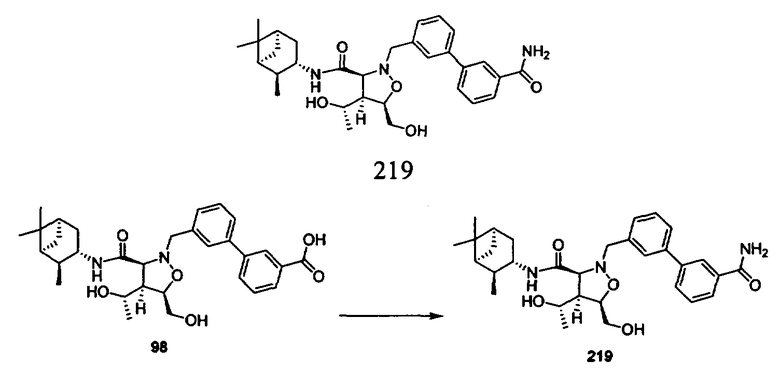
В раствор неочищенного соединения 98 (25 мг), NH4Cl (8 мг) и DIEA (40 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. Фракции, содержащие продукт, концентрировали, при этом получали соединение 219 (16 мг) в виде твердого вещества белого цвета.
МС (ESI(+)) (m/e): 536,2 (М+Н)+.
Пример 165

В раствор неочищенного соединения 98 (25 мг) и метиламина в ТГФ (2,0 М, 74 мкл) в ДМФА (700 мкл) добавляли HBTU (30 мг). После встряхивания в течение 1 ч реакционную смесь разбавляли МеОН (800 мкл) и очищали методом ЖХВР. Фракции, содержащие продукт, концентрировали, при этом получали соединение 220 (14 мг) в виде твердого вещества белого цвета.
МС (ESI(+)) (m/e): 550,2 (М+Н)+.
Пример 166
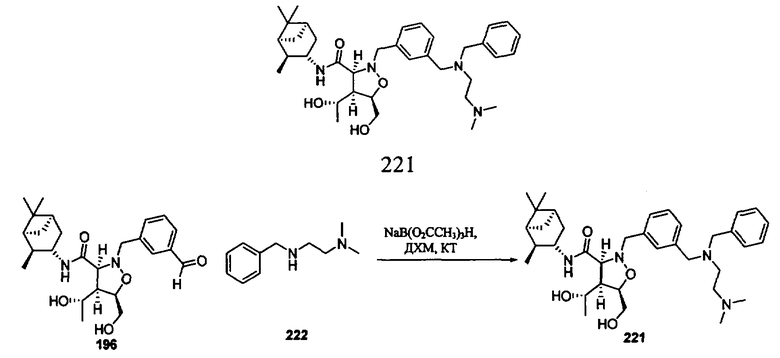
В раствор соединения 196 (0,65 ммоля) в ДХМ (17 мл) при 0°С добавляли соединение 222 (0,92 ммоля), а затем одной порцией Na(OAc)3BH (1,30 ммоля). Раствор нагревали до КТ и перемешивали в течение 2 ч, затем разбавляли водой (20 мл), органическую фазу отделяли, промывали солевым раствором (10 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали твердое вещество белого цвета. Твердое вещество растворяли в ТГФ (15 мл) и очищали методом обращенно-фазовой хроматографии (колонка С 18, элюент: 40% ацетонитрил в воде, содержащий 0,01% муравьиную кислоту), при этом получали требуемый продукт.
МС (ESI(+)) (m/e): 607,7 (М+Н)+.
Пример 167
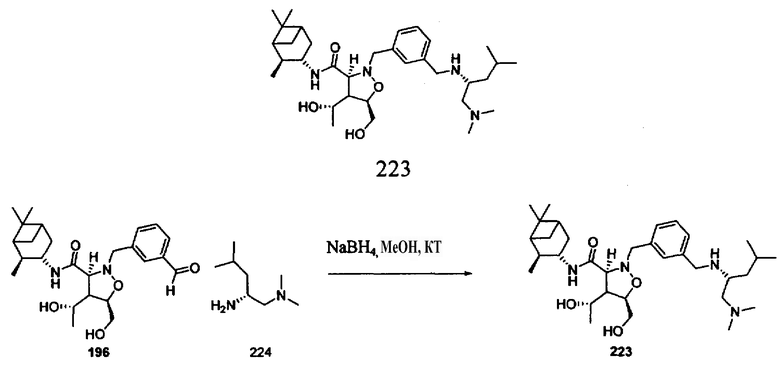
В раствор соединения 196 (0,03 ммоля) в МеОН (0,5 мл) добавляли соединение 224 (0,05 ммоля). Полученный раствор перемешивали в течение 2 ч при КТ, затем одной порцией добавляли NaBH4 (0,07 ммоля). Раствор инкубировали при КТ в течение 1 ч, разбавляли водой (3 мл) и экстрагировали EtOAc (2×2 мл). Объединенные органические экстракты промывали солевым раствором (4 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С 18, элюент: 20% ацетонитрил в воде, содержащий 0,01% муравьиную кислоту), при этом получали требуемый продукт.
МС (ESI(+)) (m/e): 573,8 (M+H)+.
Пример 168
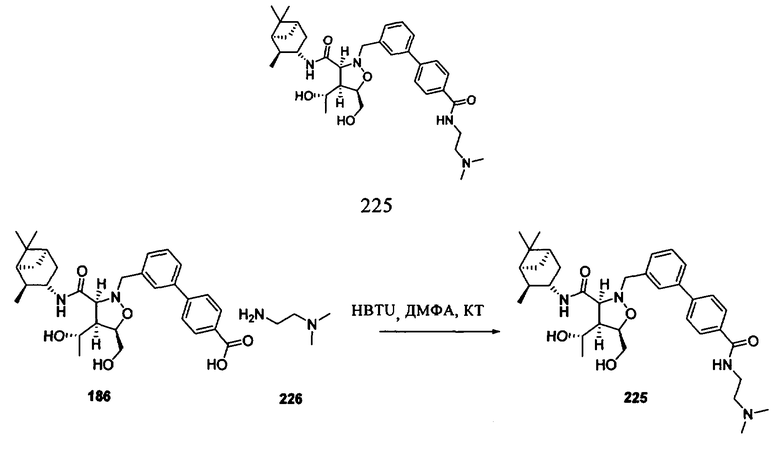
В раствор кислоты 186 (0,028 ммоля) в ДМФА (0,5 мл) при КТ добавляли N,N-диметилэтилендиамин 226 (0,11 ммоля) и HBTU (0,084 ммоля). Раствор перемешивали в течение 2 ч, разбавляли водой (1 мл) и экстрагировали EtOAc (1 мл), органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С 18, элюент: 10% ацетонитрил в воде, содержащий 0,1% бикарбонат аммония), при этом получали требуемый продукт.
Пример 169
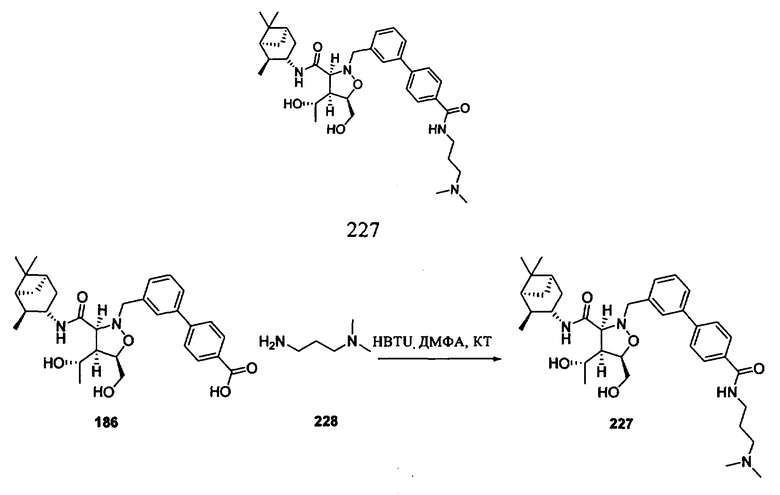
В раствор кислоты 186 (0,028 ммоля) в ДМФА (0,5 мл) при КТ добавляли N,N-диметил-1,3-пропандиамин 228 (0,11 ммоля) и HBTU (0,084 ммоля). Раствор перемешивали в течение 2 ч, разбавляли водой (1 мл) и экстрагировали EtOAc (1 мл), органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С 18, элюент: 10% ацетонитрил в воде, содержащий 0,1% бикарбонат аммония), при этом получали требуемый продукт.
Пример 170
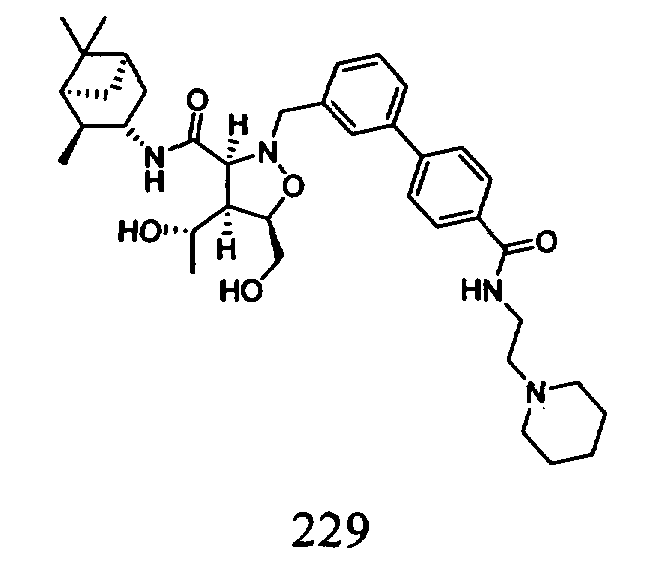
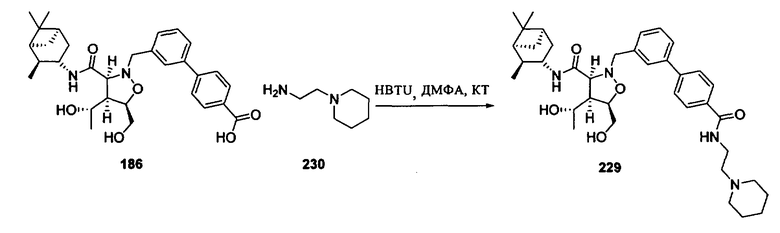
В раствор кислоты 186 (0,028 ммоля) в ДМФА (0,5 мл) при КТ добавляли 1-(2-аминоэтил)пиперидин 230 (0,11 ммоля) и HBTU (0,084 ммоля). Раствор перемешивали в течение 2 ч, разбавляли водой (1 мл) и экстрагировали EtOAc (1 мл), экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С18, элюент: 10% ацетонитрил в воде, содержащий 0,1% бикарбонат аммония), при этом получали требуемый продукт.
Пример 171
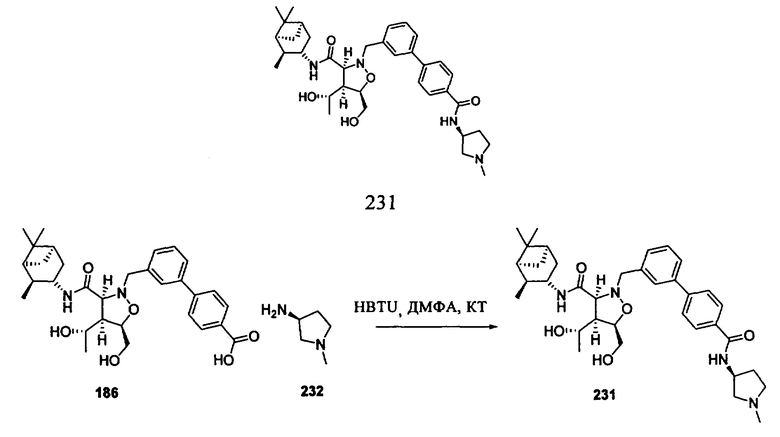
В раствор кислоты 186 (0,028 ммоля) в ДМФА (0,5 мл) при КТ добавляли (S)-N-метил-3-аминопирролидин 232 (0,11 ммоля) и HBTU (0,084 ммоля). Раствор перемешивали в течение 2 ч, разбавляли водой (1 мл) и экстрагировали EtOAc (1 мл), органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С 18, элюент: 10% ацетонитрил в воде, содержащий 0,1% бикарбонат аммония), при этом получали требуемый продукт.
МС (ESI(+)) (m/e): 619,4 (M+H)+.
Пример 172
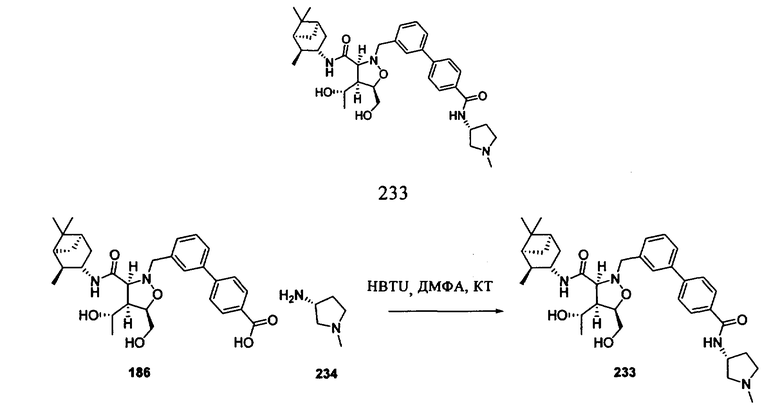
В раствор кислоты 186 (0,028 ммоля) в ДМФА (0,5 мл) при КТ добавляли (R)-N-метил-3-аминопирролидин 234 (0,11 ммоля) и HBTU (0,084 ммоля). Раствор перемешивали в течение 2 ч, разбавляли водой (1 мл) и экстрагировали EtOAc (1 мл), органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С 18, элюент: 10% ацетонитрил в воде, содержащий 0,1% бикарбонат аммония), при этом получали требуемый продукт.
Пример 173
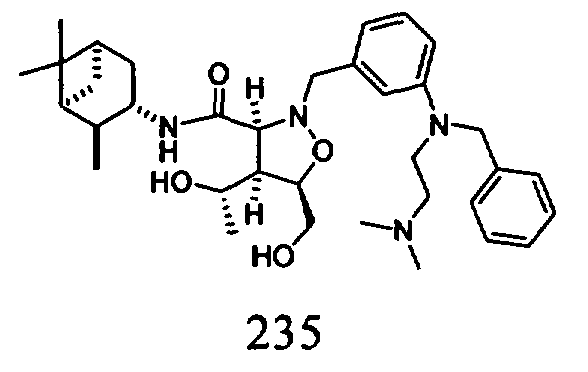
Стадия А
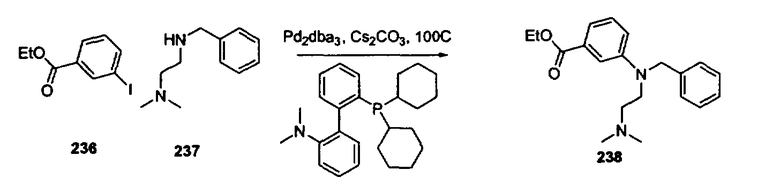
В сухой колбе в атмосфере N2 смешивали этиловый эфир 3-иодбензойной кислоты (20 г, 0,077 моля), N-бензил-N,N-диметилэтилендиамин (18 г, 1,4 экв.), Pd2DBA3 (3,3 г, 5%), Cs2CO3 (33 г, 1,4 экв.) и 2-дициклогексилфосфино-2′-(N,N-диметиламино)бифенил (2,9 г, 10%). Затем в колбу добавляли диоксан (45 мл) и триэтиламин. Через реакционную смесь пропускали аргон в течение 5 мин, затем нагревали при 100°С в течение 14 ч. Смесь охлаждали до КТ, разбавляли EtOAc (200 мл) и фильтровали через слой целита. Неочищенную смесь концентрировали, при этом получали неочищенный остаток (32 г), который по данным ЖХ/МС анализа содержал требуемый продукт.Продукт очищали (элюент: градиент гексан/EtOAc от 20% до 60%, затем этилацетат), при этом получали требуемый продукт (10 г).
Стадия В
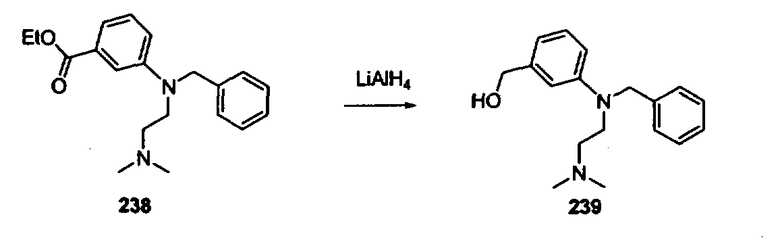
Соединение 238 растворяли в ТГФ (30 мл) и добавляли LAH (18 мл, 1 М, 0,6 экв.) при 0°С. Затем реакционную смесь нагревали при КТ. Через 5 ч реакцию останавливали добавлением воды (5 мл), затем промывали/экстрагировали этилацетатом, сушили (Na2SO4) и концентрировали, при этом получали неочищенный продукт (8 г), который очищали на силикагеле (элюент: 50% этилацетат в гексане, затем этилацетат и 5% МеОН в ДХМ), при этом получали требуемый продукт (6 г).
Стадия С
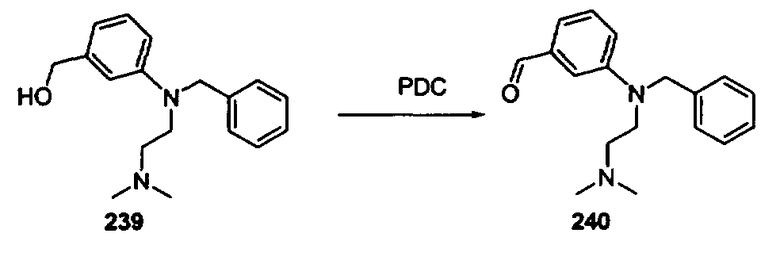
Соединение 239 растворяли в ДХМ (20 мл). В раствор добавляли PDC (16 г, 2 экв.). Реакционную смесь перемешивали при КТ в течение 14 ч. Неочищенную смесь фильтровали через слой целита и промывали этилацетатом. Органические растворы объединяли, концентрировали и очищали на силикагеле (элюент: ДХМ, 5% МеОН в ДХМ), при этом получали требуемый продукт (2,8 г), содержащий исходный спирт (3 г).
Стадия D
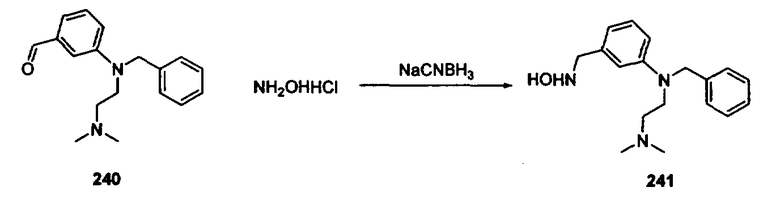
В раствор бензилальдегида 240 (2,8 г) в МеОН/ТГФ (3:1, 40 мл) добавляли одной порцией водный раствор гидроксиламина (0,9 г в 3 мл воды). Смесь подщелачивали добавлением 6 н. KOH до рН 9, и перемешивали при КТ в течение 2 ч. После полного потребления альдегида (по данным ТСХ) в раствор добавляли NaBH3CN (2 экв.). Раствор подкисляли добавлением HCl в МеОН (20 об./об.) до рН 2-3, и перемешивали в течение ночи. Затем раствор подщелачивали добавлением 2 н. KOH до рН 11 и экстрагировали CH2Cl2 (3 раза), сушили и упаривали в вакууме, при этом получали твердое вещество желтого цвета (2,8 г), которое использовали на следующей стадии без дополнительной очистки.
Стадия Е
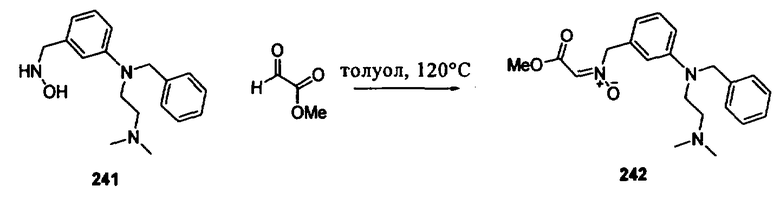
Соединение 241 и метиловый эфир глиоксиловой кислоты (1 г) растворяли в 100 мл толуола. Смесь нагревали при 120°С в течение 3 ч и концентрировали, при этом получали продукт (15 мл), который использовали на следующей стадии.
Стадия F
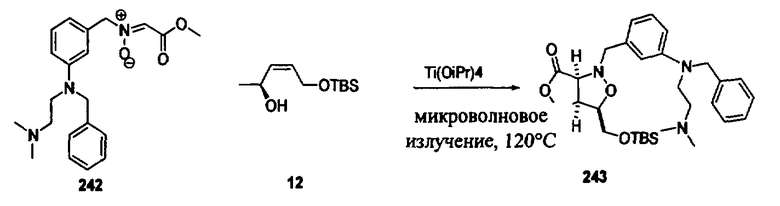
В 5 флаконов добавляли раствор соединения 242 (полученного на предыдущей стадии) в толуоле (по 3 мл в каждый флакон). В каждый флакон добавляли TBS-аллиловый спирт 12 (0,4 г) и изопропоксид титана (0,6 г). Смесь нагревали при 140°С в течение 10 мин в микроволновом реакторе. Реакционную смесь охлаждали до КТ и объединяли. К объединенным фракциям добавляли этилацетат, и органический слой промывали водой, солевым раствором, сушили над Mg2SO4, фильтровали и концентрировали. Остаток очищали экспресс-хроматографией на силикагеле (элюент: 3% МеОН в ДХМ), при этом получали частично очищенный продукт (900 мг).
Стадия G
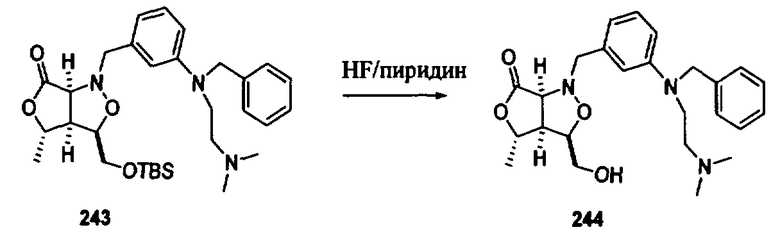
Частично очищенный продукт (соединение 243), полученный на предыдущей стадии (0,90 г) растворяли в 3 мл безводного ТГФ, затем по каплям добавляли HF/пиридин (0,5 мл). Через 1 ч смесь концентрировали в вакууме, при этом получали требуемый продукт (180 мг), содержащий основной примесный компонент (который не отделяется при хроматографии на колонке).
Стадия Н
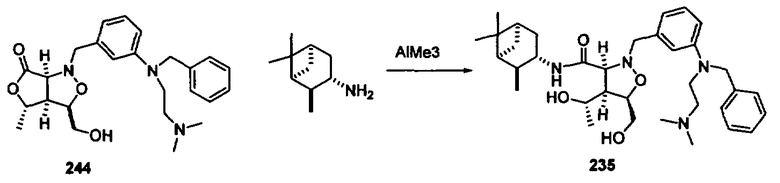
Изопинокамфеиламин (0,18 г, 0,41 ммоля) растворяли в безводном ДХМ (3 мл). В раствор по каплям добавляли Al(Ме)3 (2 М, 0,41 мл, 0,82 ммоля). Смесь перемешивали при КТ в течение 20 мин. Соединение 244 растворяли в ДХМ (1 мл), полученный раствор осторожно добавляли в раствор амина и Al(Ме)3. Реакционную смесь перемешивали при КТ в течение 12 ч, затем разбавляли ДХМ (25 мл), водный раствор сегнетовой соли (5 мл, тартрат калий-натрия), полученную двухслойную смесь перемешивали при КТ в течение 2 ч. Органический слой отделяли, промывали водой и солевым раствором, сушили (Na2SO4), фильтровали и концентрировали, при этом получали неочищенный продукт (80 мг), который очищали экспресс-хроматографией на силикагеле (элюент: ДХМ, 2% МеОН в ДХМ, 5% МеОН в ДХМ), при этом получали требуемый продукт (40 мг).
МС (ESI(+)) (m/e): 593,4 (М+Н)+.
Пример 174
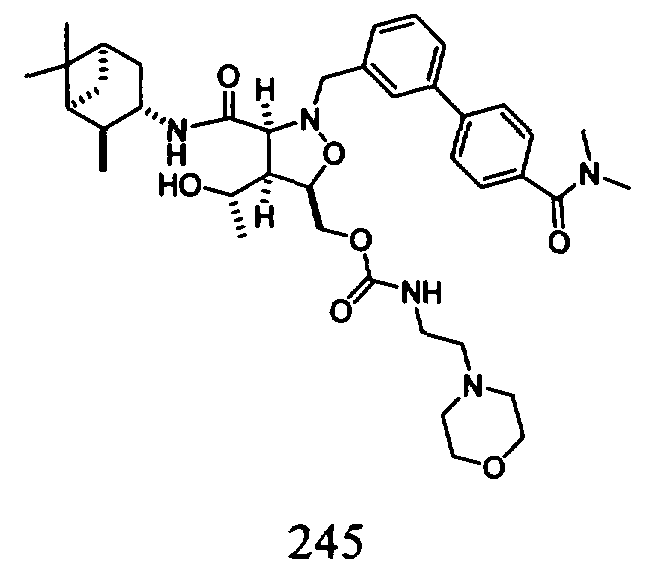
Стадия А
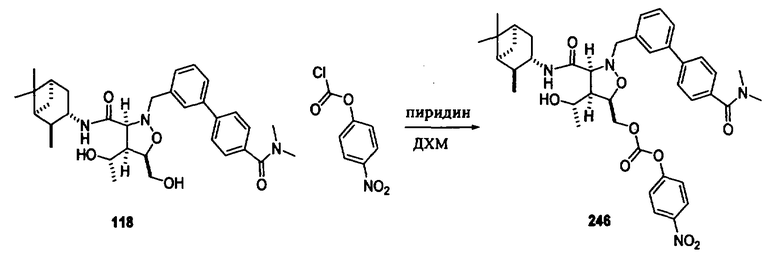
пара-нитрофенилхлорформиат (40 мг) растворяли в ДХМ (2 мл) и пиридине (20 мкл, хх ммоля, хх экв.). Полученную суспензию белого цвета охлаждали до 0°С и добавляли соединение 118 (80 мг, хх ммоля, хх экв.) в ДХМ (1 мл). Реакционную смесь перемешивали при КТ в течение 14 ч, разбавляли ДХМ (30 мл), промывали водой, солевым раствором и сушили над Na2SO4, фильтровали и концентрировали, при этом получали неочищенный продукт (77 мг), который очищали экспресс-хроматографией (элюент: градиент гексан/EtOAc от 10:1 до 1:2), при этом получали требуемый продукт (60 мг).
Стадия В
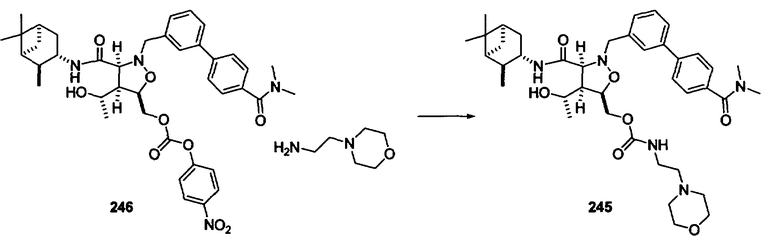
Соединение 246 (8 мг) и морфолинамин (6 мг, 4 экв.) растворяли в ДХМ (1 мл), и смесь перемешивали при КТ в течение ночи. Реакционную смесь разбавляли ДХМ (30 мл), промывали водой, солевым раствором, сушили, фильтровали и концентрировали. Неочищенный продукт очищали на силикагеле (элюент: градиент от EtOAc до EtOAc/МеОН, 10:1), при этом получали очищенный продукт (7 мг).
МС (ESI(+)) (m/e): 720,6 (М+Н)+.
Пример 175
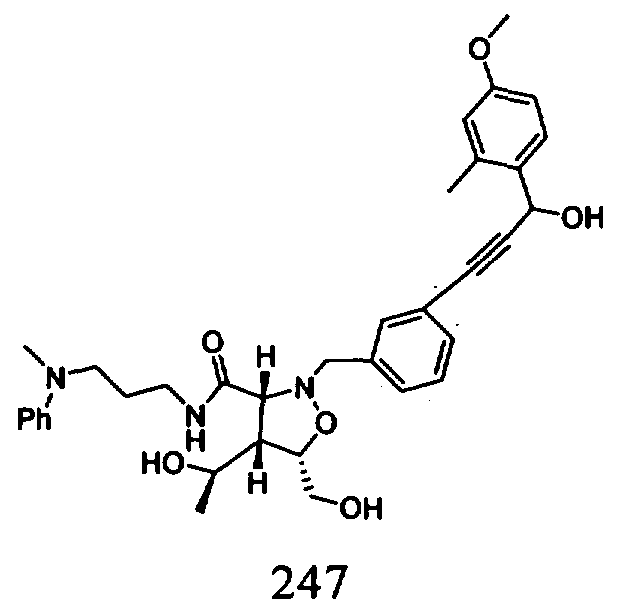
Стадия А
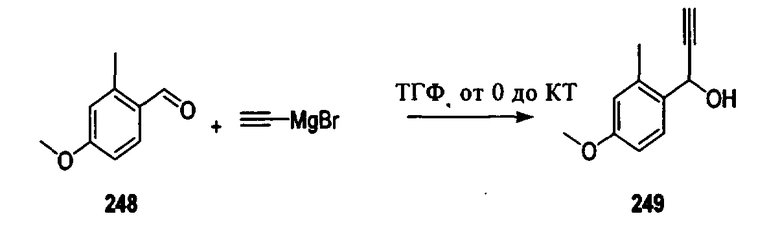
Этинилмагнийбромид (0,5 М, 8 мл) охлаждали в атмосфере N2 до 0°С. Альдегид 248 (0,4 г) растворяли в безводном ТГФ (3 мл) атмосфере N2, полученный раствор по каплям добавляли к реактиву Гриньяра. Смесь перемешивали при 0°С в течение 10 мин, затем при КТ в течение 4 ч. Реакцию останавливали добавлением насыщенного раствора NH4Cl, требуемый продукт экстрагировали EtOAc. Органические экстракты промывали солевым раствором, сушили и фильтровали, при этом получали требуемый продукт (0,41 г).
Стадия В
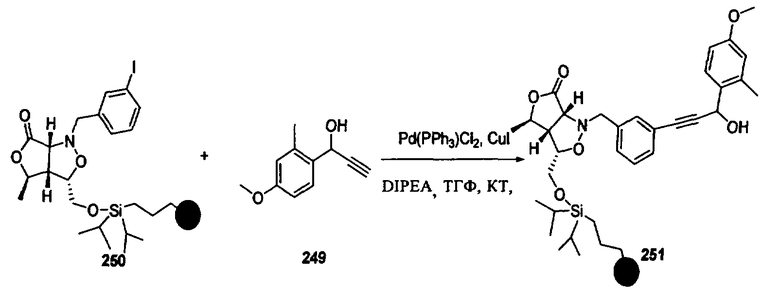
Изоксазолидин 18 иммобилизовали на частицах носителя lantern аналогично тому, как описано в примере 29, стадия А. В стеклянный флакон добавляли 3 частицы lantern 250 (содержание иммобилизованного фрагмента 18 мкмолей/частица lantern), сушили в вакууме (18 ч), затем продували азотом.
В указанный флакон последовательно добавляли ДМФА (хх мл), дихлор-бис(трифенилфосфин)палладий(II) (63 мг, 2 экв.), иодид меди (26 мг, 3 экв.) и DIPEA (0,3 мл, 40 экв.). Пропаргиловый спирт 249 (0,32 г, хх ммоля, 40 экв.) растворяли в безводном ДМФА (2 мл) и добавляли во флакон. Флакон встряхивали в атмосфере азота в течение 16 ч, непрореагировавшие реагенты удаляли декантацией. Частицы lantern промывали ДМФА, ТГФ, ТГФ/Н2О (3:1), ТГФ/IPA (3:1), ТГФ, CH2Cl2. Частицы lantern переносили в круглодонную колбу и упаривали при пониженном давлении, при этом удаляли избыток растворителя. Частицы lantern сушили в высоком вакууме в течение ночи.
Продукт отщепляли от частиц lantern обработкой HF/пиридин (20 мкл) в ТГФ (200 мкл), а затем обрабатывали TMSOMe (100 мкл), при этом получали требуемый продукт.
Стадия С
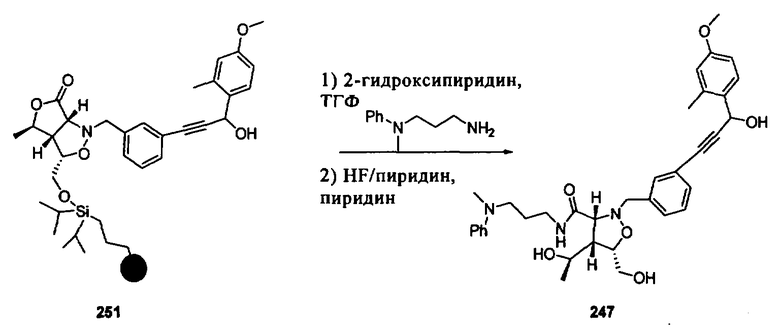
В небольшую круглодонную колбу, заполненную азотом, добавляли соединение 251 (2 частицы lantern), затем добавляли ТГФ (2 мл), 2-гидроксипиридин (40 мг, 10 экв.) и амин (660 мг, 100 экв.). Реакционный сосуд встряхивали при 50°С в течение 12 ч. Непрореагировавшие реагенты удаляли декантацией. Частицы lantern промывали ДМФА, ТГФ, ТГФ/Н2О (3:1), ТГФ/IPA (3:1), ТГФ, CH2Cl2, переносили в круглодонную колбу и упаривали при пониженном давлении в течение ночи, при этом удаляли избыток растворителя.
Продукт отщепляли от частиц lantern обработкой HF/пиридин (80 мкл) в ТГФ (400 мкл), а затем TMSOMe (1 мл), при этом получали требуемый продукт (4 мг, по данным ЖХ/МС и ЯМР анализа).
МС (ESI(+)) (m/e): 602,2 (М+Н)+.
Пример 176
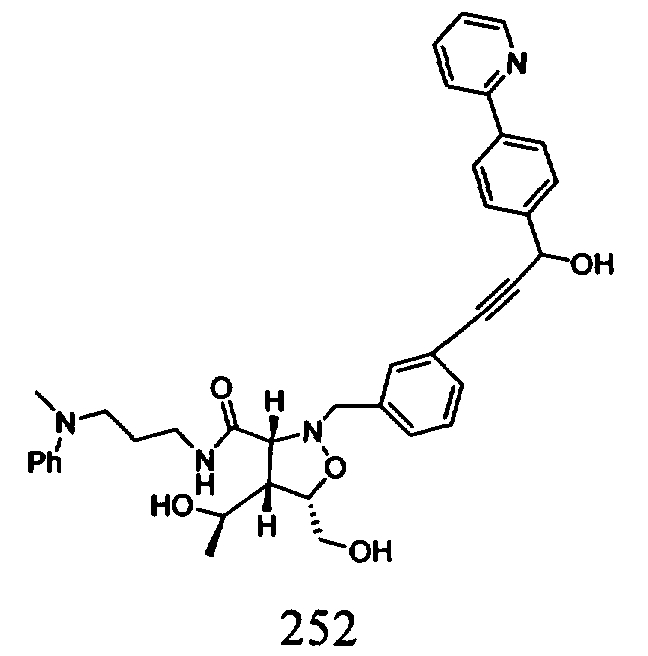
Соединение 252 получали по методике, описанной в примере 186, но при замене 4-метокси-2-метилбензальдегида на 4-(пиридин-2-ил)бензальдегид. МС (ESI(+)) (m/e): 635,4 (М+Н)+.
Пример 177
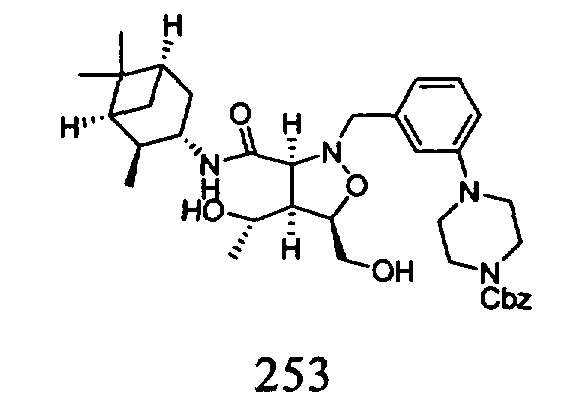
Стадия А
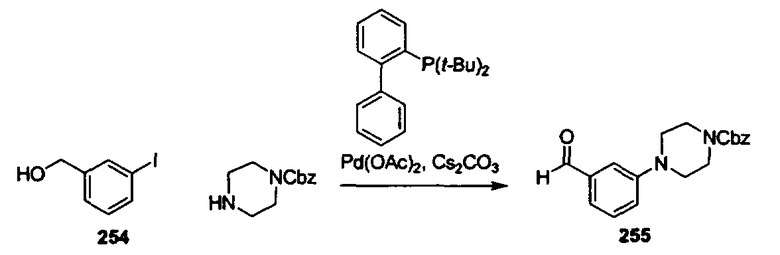
В бензол (72 мл) при комнатной температуре добавляли N-Cbz-пиперазин (8,24 г, 37,4 ммоля), 3-иодбензиловый спирт 265 (7 г, 29,9 ммоля), 2-бис(трет-бутил)фосфинобифенил (1,8 г, 6,0 ммоля), ацетат палладия (1,34 г, 6,0 ммоля) и карбонат цезия (14,6 г, 44,9 ммоля). Полученную смесь продували аргоном и нагревали при 70°С в течение 14 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc (40 мл), полученную смесь интенсивно перемешивали в течение 10 мин. Твердое вещество отделяли фильтрованием, промывали EtOAc (2×20 мл) и фильтрат концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: гексан/EtOAc, 4:1), при этом получали соединение 255 (2,29 г, 24%) в виде полутвердого вещества светло-желтого цвета.
Стадия В
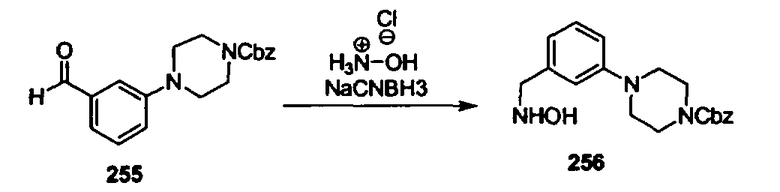
В перемешиваемый раствор альдегида 255 в МеОН (100 мл) и ТГФ (40 мл) при 15°С в течение 2 мин по каплям добавляли водный раствор гидрохлорида гидроксиламина (5 мл). Полученный раствор перемешивали при 15°С в течение 30 мин. По данным ТСХ анализа (силикагель, элюент: гексан/EtOAc, 2:1) наблюдается полное потребление исходного материала. Смесь подщелачивали добавлением 1 н. NaOH (14 мл) до рН 10 и одной порцией добавляли NaCNBH3 (1,5 г). Затем реакционную смесь при 0°С подкисляли добавлением по каплям раствора HCl в МеОН (6 М, 14 мл) до рН 2-4. После добавления HCl охлаждающую баню удаляли и реакционную смесь перемешивали при 23°С в течение 16 ч. По данным ТСХ анализа (силикагель, элюент: гексан/EtOAc, 2:1) реакция завершается через 16 ч. Смесь подщелачивали добавлением насыщенного водного раствора NaHCO3 (40 мл) до рН 8. Затем добавляли ДХМ/Н2О (1:1, 400 мл), полученные слои разделяли. Водный слой экстрагировали ДХМ (2×200 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме, при этом получали требуемый продукт (5,4 г, >100%) в виде масла бледно-желтого цвета.
Стадия С
Гидроксиламин 256 (5,1 г, 15 ммолей) и метиловый эфир глиоксиловой кислоты (1,3 г, 15 ммолей) добавляли в безводный толуол (100 мл). Полученную смесь нагревали при 100°С в течение 6 ч. Смесь охлаждали до комнатной температуры и концентрировали в вакууме, при этом получали требуемый продукт (6,2 г, 100%) в виде вязкого масла желтого цвета.
Стадия D
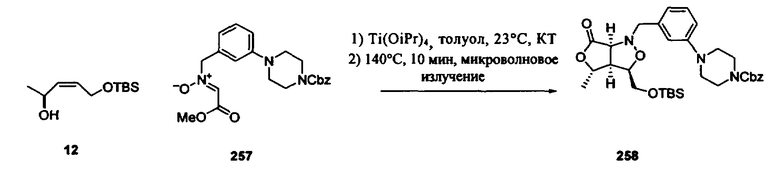
В смесь аллилового спирта 12 (3,2 г, 15 ммолей) и нитрона 257 (6,2 г, 15 ммолей) в безводном толуоле (40 мл) при комнатной температуре в течение 3 мин по каплям добавляли Ti(OiPr)4. Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Затем раствор переносили в 8 флаконов для микроволнового реактора (~6 мл/флакон). Каждый флакон в отдельности нагревали при 140°С в микроволновом реакторе в течение 10 мин. Растворы объединяли, разбавляли EtOAc (200 мл), 3-(диметиламино)-1,2-пропандиолом (3,6 г, 30,0 ммоля), и перемешивали при комнатной температуре в течение 14 ч. Неочищенный продукт очищали хроматографией на силикагеле (элюент: гексан/EtOAc, 3:1), при этом получали требуемый продукт (3,4 г, 38%) в виде масла светло-желтого цвета.
Стадия Е
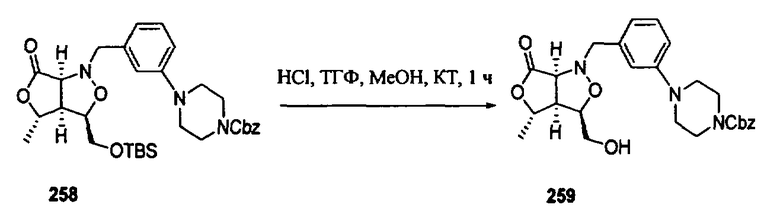
В раствор соединения 258 (3,4 г, 5,7 ммоля) в МеОН (20 мл) при комнатной температуре в течение 5 мин четырьмя порциями добавляли HCl/MeOH (1:1,8 мл). Полученный раствор перемешивали в течение 1 ч. По данным ТСХ анализа через указанное время происходит полное потребление исходного материала. Затем при интенсивном перемешивании в течение 30 мин пятью порциями добавляли насыщенный водный раствор NaHCO3 (100 мл). Полученные слои разделяли, водный слой экстрагировали ДХМ (3×250 мл). Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (элюент: ДХМ/МеОН, 25:1), при этом получали требуемый продукт (2,45 г, 89%) в виде твердого вещества желтого цвета.
Стадия F
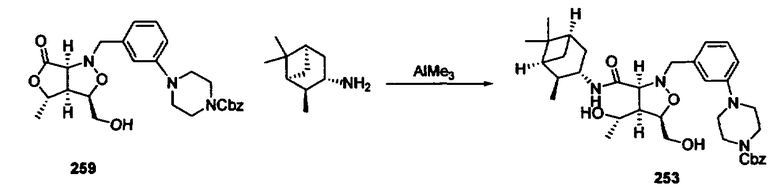
(+)-Изопинокамфеиламин (0,2 ммоля) растворяли в ДХМ (0,3 мл) при КТ, добавляли триметилалюминий (2,0 М в гексане, 0,2 ммоля), раствор перемешивали в течение 10 мин. Затем добавляли раствор соединения 259 (0,1 ммоля) в ДХМ (0,2 мл), и реакционную смесь перемешивали в течение 16 ч. Раствор разбавляли насыщенным водным раствором сегнетовой соли (15 мл). Полученную смесь перемешивали в течение 2 ч до образования двухфазной смеси. Водный слой отделяли и экстрагировали ДХМ (2×15 мл), органические экстракты объединяли, промывали водой (15 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, при этом получали требуемый продукт (60%) в виде масла бледно-желтого цвета.
МС (ESI(+)) (m/e): 635,4 (М+Н)+.
Пример 178
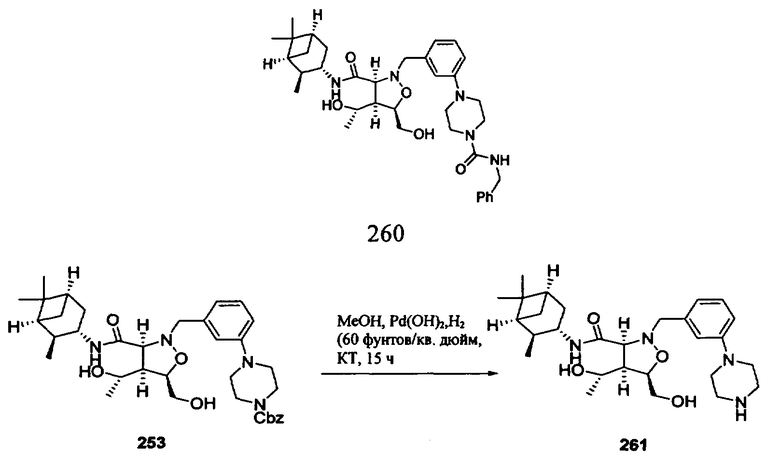
Соединение 253 (2,1 г, 3,3 ммоля) в МеОН (20 мл) добавляли в толстостенный стеклянный флакон для реактора Парра для гидрирования, затем добавляли суспензию гидроксида палладия (850 мг) в метаноле (20 мл). Флакон помещали в реактор для гидрирования. Для удаления из растворителя кислорода реакционную смесь вакуумировали и продували водородом (несколько циклов). Затем флакон встряхивали в течение 15 ч в атмосфере водорода (при давлении 60 фунтов на кв. дюйм). Катализатор отделяли фильтрованием и фильтрат концентрировали в вакууме. Полученный неочищенный продукт очищали хроматографией на силикагеле (элюент: ДХМ/МеОН, 30:1, затем 20:1, и 10:1), при этом получали требуемый продукт (1,1 г, 66%) в виде пены светло-желтого цвета.
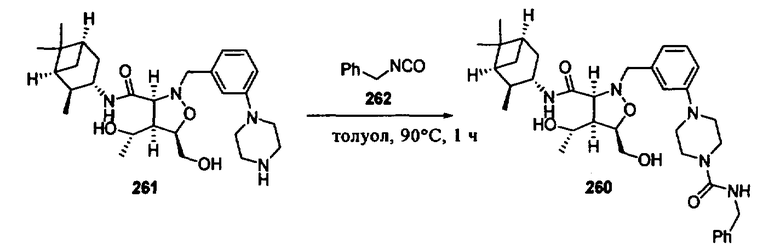
Соединение 261 (25 мг, 0,05 ммоля) и бензилизоцианат 262 (7 мг, 0,05 ммоля) добавляли в безводный толуол (0,3 мл) и нагревали при 90°С в течение 1 ч. Раствор охлаждали до комнатной температуры и концентрировали в вакууме. Неочищенный продукт очищали хроматографией на силикагеле (элюент: ДХМ/МеОН, 40:1), при этом получали требуемый продукт (10 мг, 33%) в виде пены бледно-желтого цвета.
МС (ESI(+)) (m/e): 634,3 (М+Н)+.
Пример 179
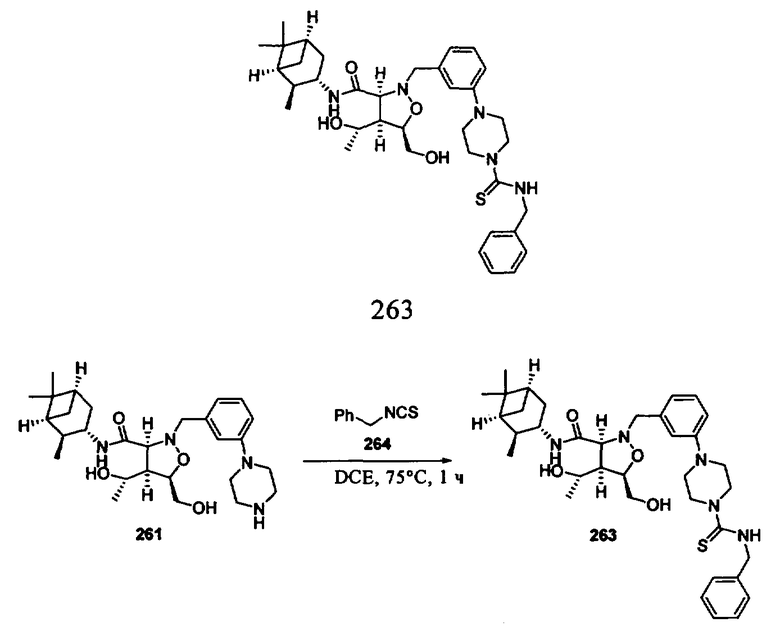
Соединение 263 получали по методике, описанной в примере 189, но при замене соединения 262 на соединение 264 (выход 36%).
МС (ESI(+)) (m/e): 650,4 (М+Н)+.
Пример 180
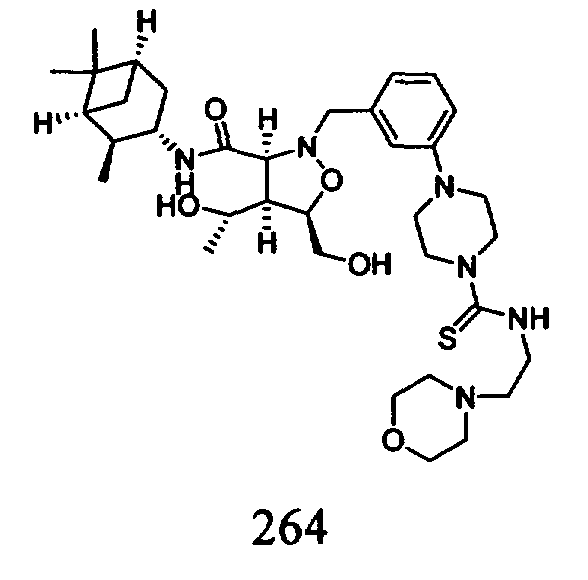
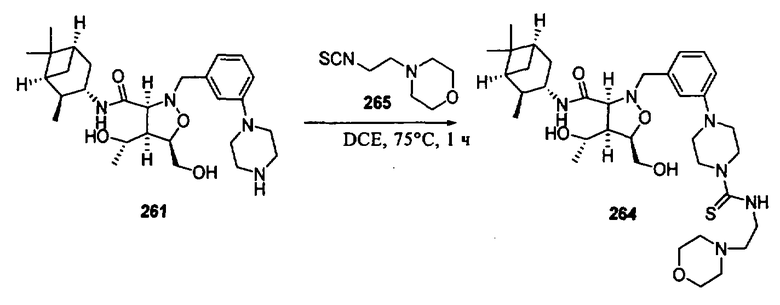
Соединение 264 получали по методике, описанной в примере 189, но при замене соединения 262 на соединение 265 (выход 34%).
МС (ESI(+)) (m/e): 673,5 (М+Н)+.
Пример 181

Соединение 266 получали по методике, описанной в примере 189, но при замене соединения 222 на соединение 267 (выход 30%).
МС (ESI(+)) (m/e): 664,5 (М+Н)+.
Пример 182
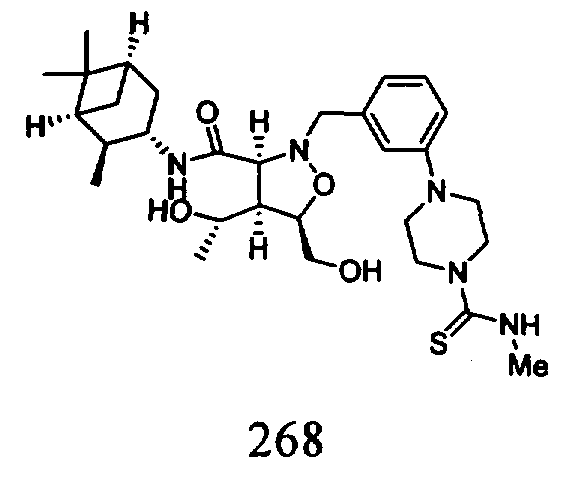
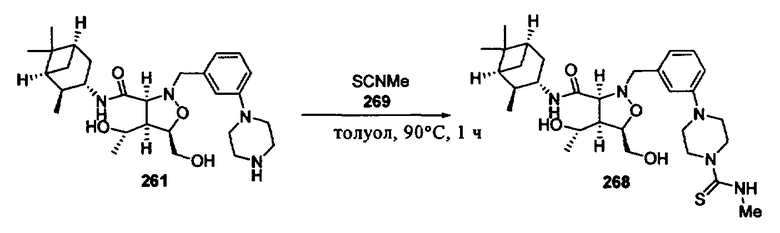
Соединение 268 получали по методике, описанной в примере 189, но при замене соединения 262 на соединение 269 (выход 35%).
МС (ESI(+)) (m/e): 574,3 (М+Н)+.
Пример 183
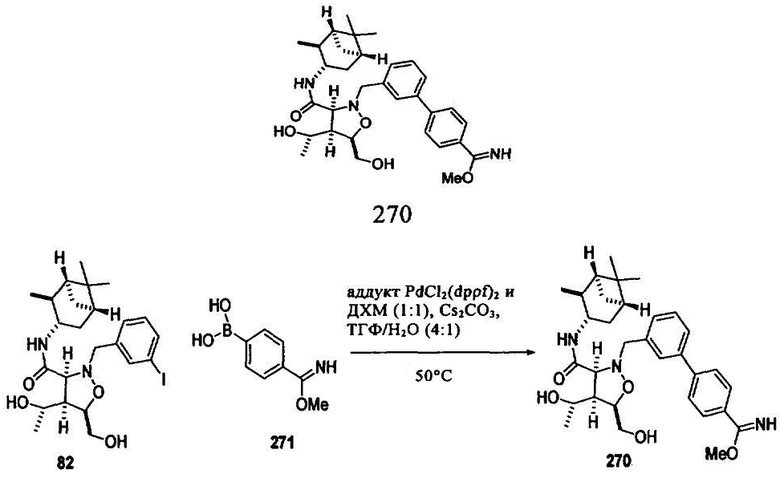
Арилиодид 82 (0,03 ммоля), бороновую кислоту 271 (0,03 ммоля), аддукт PdCl2(dppf)2 и ДХМ (1:1, 0,006 ммоля), карбонат цезия (0,03 ммоля) при комнатной температуре добавляли в ТГФ/Н2О (4:1, 0,3 мл). Затем полученную смесь дегазировали (замораживание/вакуумирование/размораживание, 3 цикла). Затем смесь нагревали при 50°С в течение 12 ч. Реакционную смесь концентрировали в вакууме при 50°С, остаток очищали хроматографией на силикагеле, при этом получали биарил (выход 75%).
МС (ESI(+)) (m/e): 550,3 (М+Н)+.
Пример 184
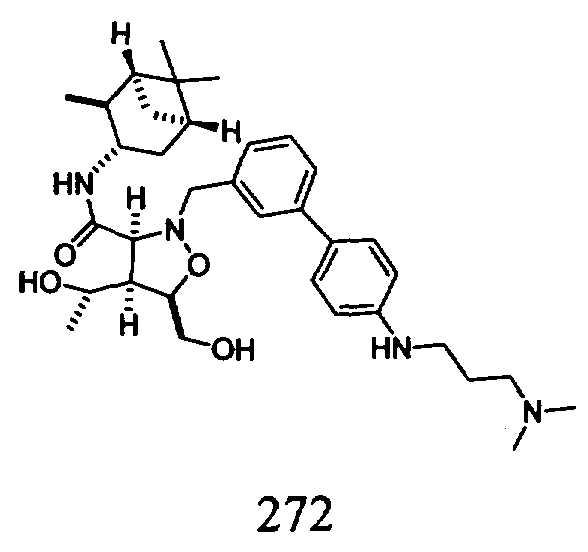
Стадия А
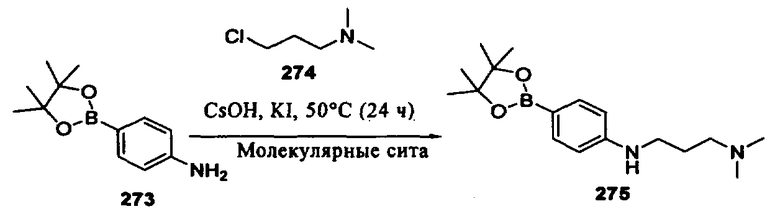
Соединение 273 (300 мг, 1,0 ммоля), алкилхлорид 274 (2 г, 14 ммоля), гидроксид цезия (200 мг, 1,0 ммоля), KI (один кристалл) и молекулярные сита (4А, 500 мг) добавляли в безводный ДМФА (1,5 мл). Полученную смесь нагревали при 50°С в течение 24 ч. Затем охлаждали до комнатной температуры и разбавляли EtOAc (100 мл). Твердое вещество отделяли фильтрованием. Остаток очищали хроматографией на силикагеле (элюент: ДХМ/МеОН, 25:1), при этом получали биарил (8,5 мг, 2%) в виде твердого вещества бледно-желтого цвета.
Стадия В
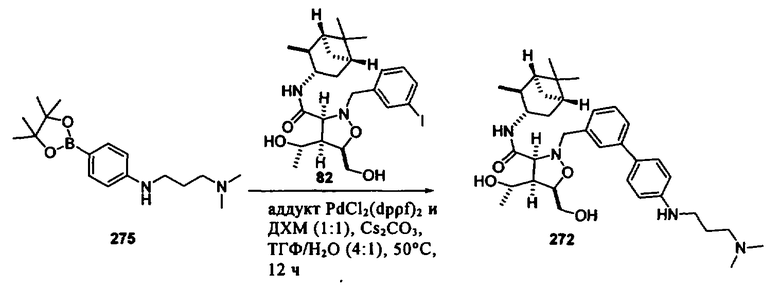
Соединение 82 (15 мг, 0,03 ммоля), соединение 275 (8 мг, 0,03 ммоля), аддукт PdCl2(dppf)2 и ДХМ (1:1, 5 мг, 0,006 ммоля), карбонат цезия (9 мг, 0,03 ммоля) при комнатной температуре добавляли в ТТФ/Н2О (4:1, 0,3 мл). Затем полученную смесь дегазировали (вакуумирование/продувка аргоном, 3 цикла) и нагревали при 50°С в течение 12 ч. Реакционную смесь концентрировали в вакууме при 50°С, остаток очищали хроматографией на силикагеле (элюент: ДХМ/МеОН, 15:1), при этом получали соединение 272 (5 мг, 31%) в виде пены бледно-желтого цвета.
МС (ESI(+)) (m/e): 593,6 (М+Н)+.
Пример 185
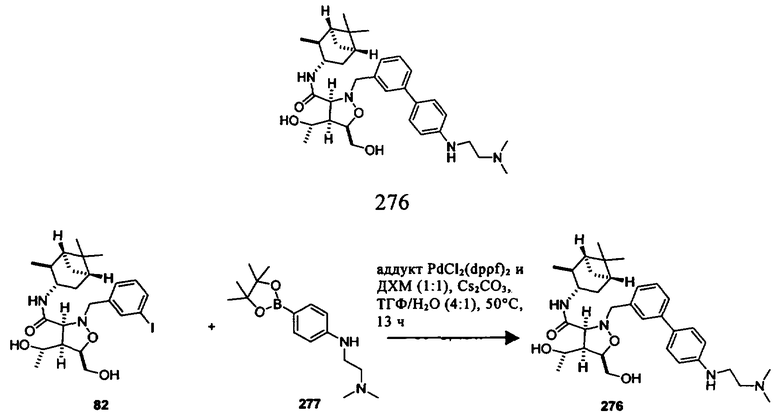
Соединение 277 получали по методике, описанной в примере 184 (стадия А), но при замене 2-хлор-N,N-диметилпропанамина на 2-хлор-N,N-диметилэтанамин.
Соединение 276 получали по методике, описанной в примере 184 (выход 38%).
МС (ESI(+)) (m/e): 579,3 (M+H)+.
Пример 186
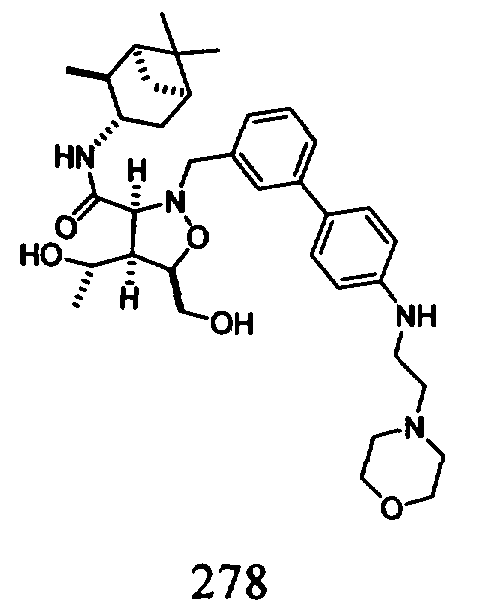
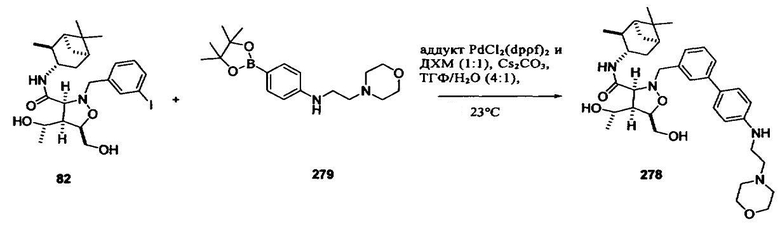
Соединение 279 получали по методике, описанной в примере 184 (стадия А), но при замене 2-хлор-N,N-диметилпропанамина на 4-(2-хлорэтил)морфолин.
Соединение 278 получали по методике, описанной в примере 184, но при замене соединения 275 на соединение 279 (выход 23%).
МС (ESI(+)) (m/e): 621,5 (М+Н)+.
Пример 187
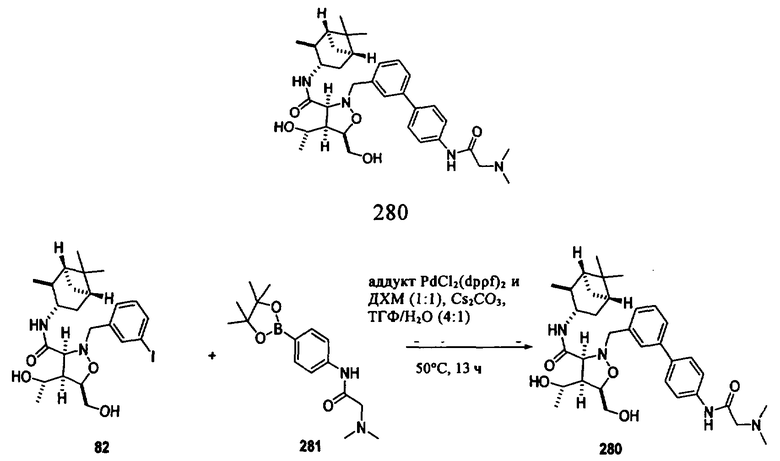
Соединение 290 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на эфир бороновой кислоты 281 (выход 15%).
Пример 188
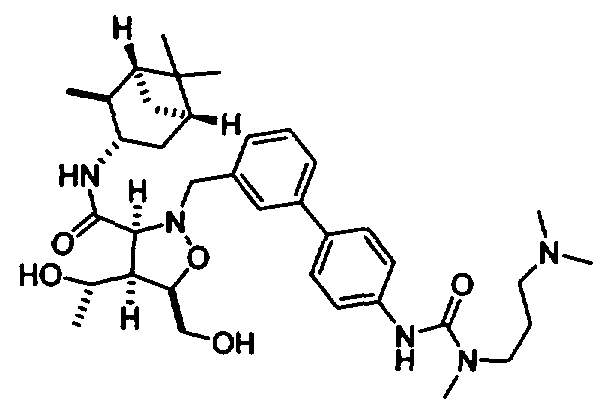
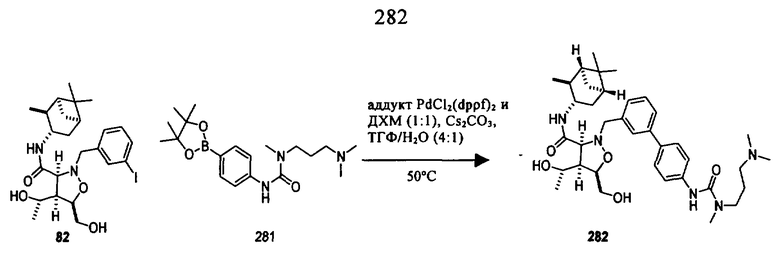
Соединение 82 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на эфир бороновой кислоты 281 (выход 70%).
МС (ESI(+)) (m/e): 650,4 (М+Н)+.
Пример 189
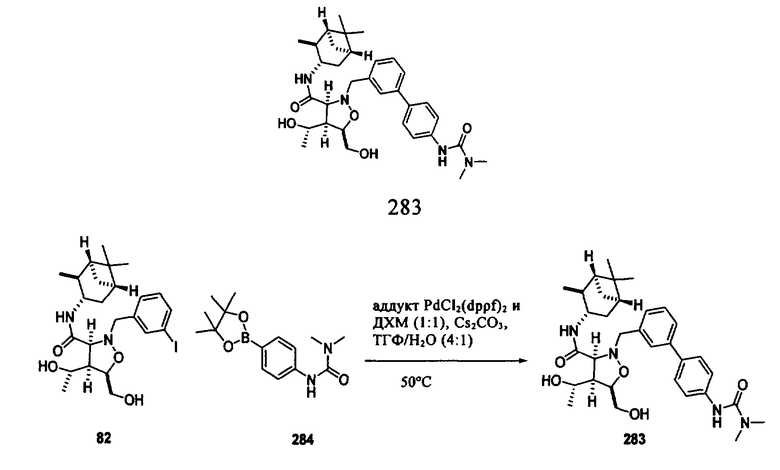
Соединение 82 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на эфир бороновой кислоты 284 (выход 47%).
МС (ESI(+)) (m/e): 579,3 (М+Н)+.
Пример 190
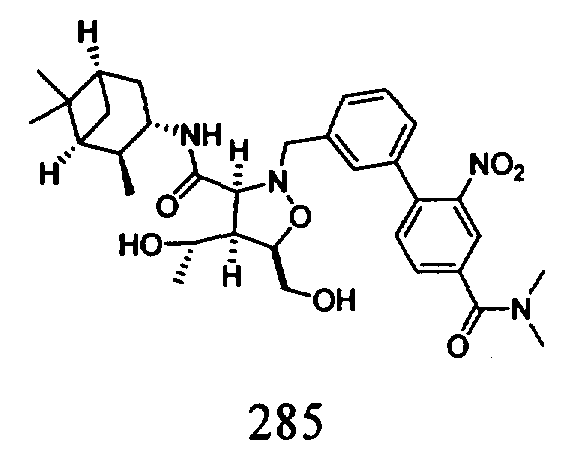
Стадия А
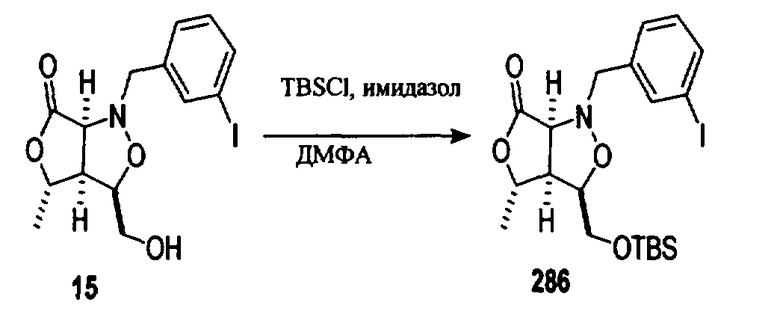
Соединение 15 (1 ммоль) добавляли в безводный ДМФА (1 мл), затем добавляли TBSCl (1,6 ммоля) и имидазол (2,0 ммоля). Полученный раствор перемешивали в атмосфере аргона в течение 8 ч. Затем добавляли воду (5 мл), водный слой отделяли и экстрагировали EtOAc (3×10 мл). Органические экстракты объединяли, сушили над MgSO4, фильтровали и концентрировали. Неочищенный материал очищали хроматографией на силикагеле, при этом получали соединение 286 (98%).
Стадия В
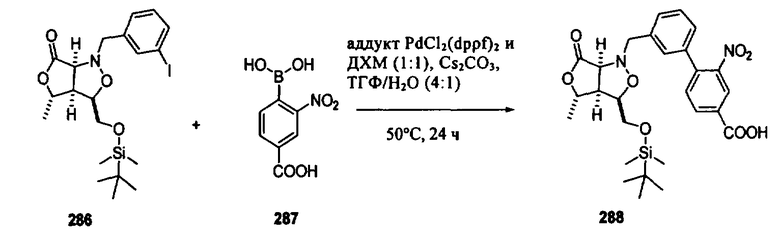
Соединение 288 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на бороновую кислоту 287 (выход 56%).
Стадия С

В раствор соединения 288 (0,33 г, 0,60 ммоля) и HATU (0,68 г, 1,8 ммоля) в ДМФА (2 мл) при КТ добавляли диметиламин в ТГФ (2 М, 1 мл). Полученный раствор перемешивали в течение 14 ч, распределяли между водой и EtOAc (1:1, 200 мл), органический слой отделяли, промывали Н2О (2×50 мл), водные слои объединяли и повторно экстрагировали EtOAc (2×50 мл). Органические экстракты объединяли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: ДХМ, затем ДХМ/EtOAc, 2:1), при этом получали требуемый продукт (элюент: ДХМ/EtOAc, 4:1, Rf 0,4, 67 мг, 20%) в виде полутвердого вещества коричневого цвета.
Стадия D
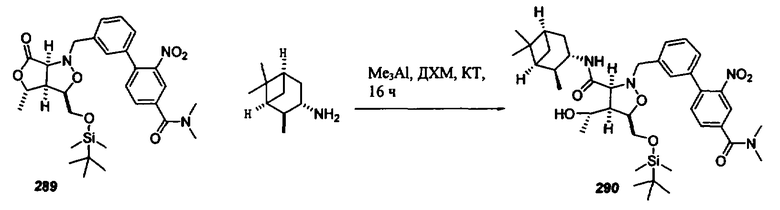
Амин (30 мг, 0,2 ммоля) растворяли в ДХМ (0,3 мл), затем при КТ добавляли раствор триметилалюминия в гексане (2,0 М, 0,1 мл), полученный раствор перемешивали при КТ в течение 10 мин, добавляли раствор лактона 289 (56 мг, 0,1 ммоля) в ДХМ (0,2 мл). Реакционную смесь инкубировали при КТ в течение 16 ч, разбавляли насыщенным водным раствором тартрата калий-натрия (15 мл, сегнетова соль), который добавляли одной порцией. Полученную смесь интенсивно перемешивали при КТ в течение 2 ч, при этом получали прозрачную двухфазную смесь. Водный слой отделяли и экстрагировали ДХМ (2×15 мл), органические фазы объединяли, промывали водой (15 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали на силикагеле SGC (элюент: гексан/EtOAc, 1:1), при этом получали требуемый продукт (40 мг, 56%) в виде масла бледно-желтого цвета.
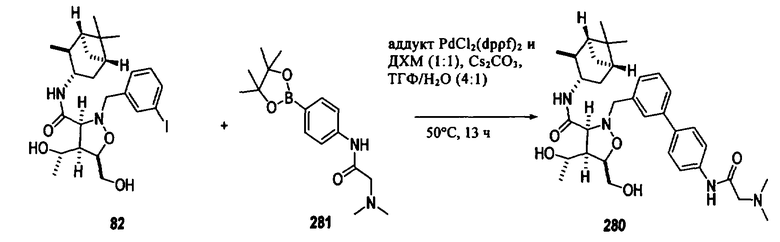
Соединение 290 получали по методике, описанной в примере 183. но при замене бороновой кислоты 271 на эфир бороновой кислоты 281 (выход 15%).
Пример 191
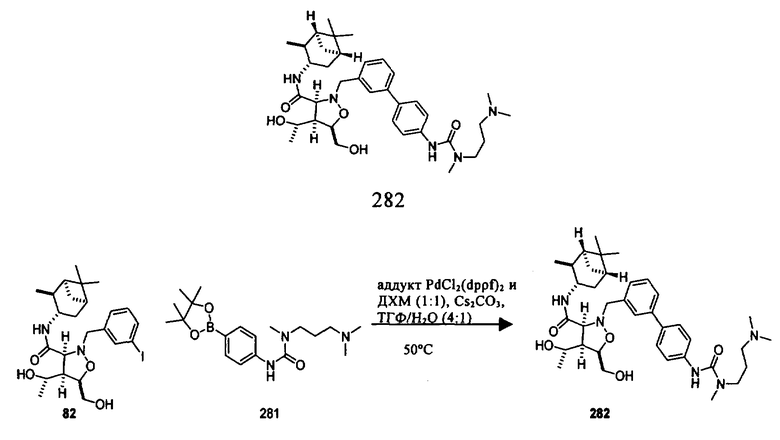
Соединение 82 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на эфир бороновой кислоты 281 (выход 70%).
МС (ESI(+)) (m/e): 650,4 (М+Н)+.
Пример 192
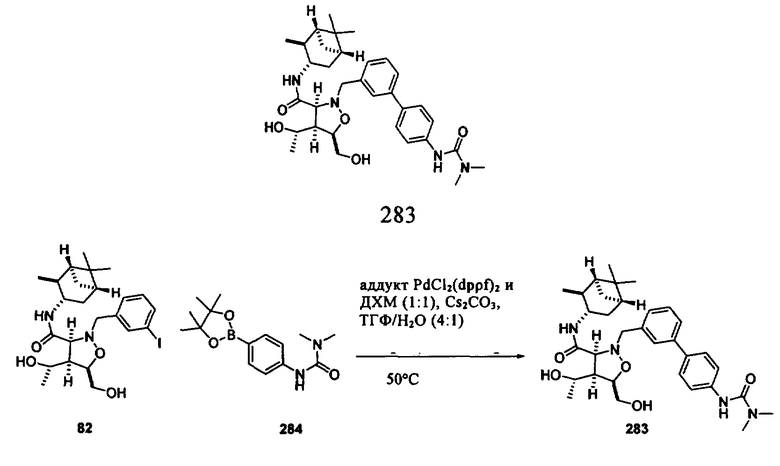
Соединение 82 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на эфир бороновой кислоты 284 (выход 47%).
МС (ESI(+)) (m/e): 579,3 (М+Н)+.
Пример 193
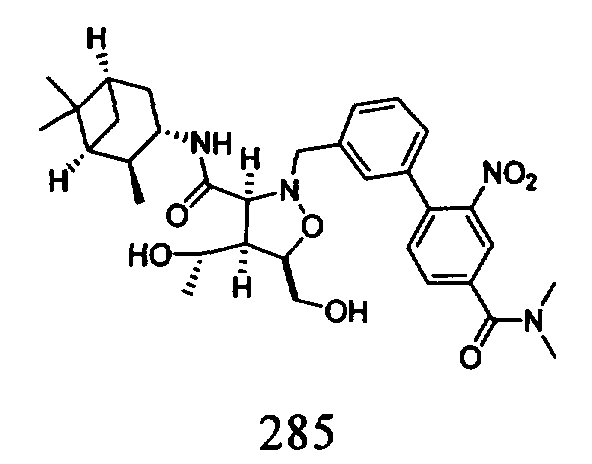
Стадия А
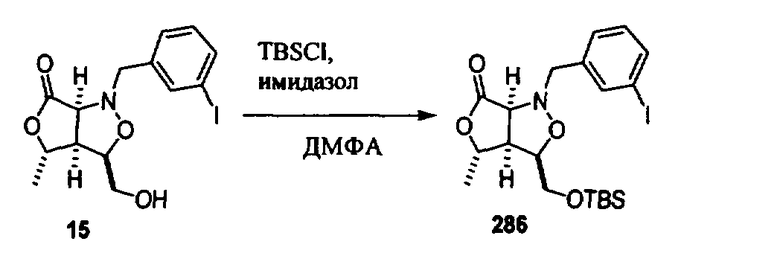
Спирт 15 (0,4 г, 1 ммоль) добавляли в ДМФА (1 мл), затем добавляли TBSCl (0,24 г, 1,6 ммоля) и имидазол (0,13 г, 2,0 ммоля). Полученный раствор перемешивали в атмосфере аргона в течение 8 ч. Затем добавляли воду (5 мл), водный слой отделяли и экстрагировали EtOAc (3×10 мл). Органические экстракты объединяли, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный материал очищали хроматографией на силикагеле (элюент: Et2O/CH2Cl2, 1:29), при этом получали соединение 286 (4,9 г, 98%).
Стадия В
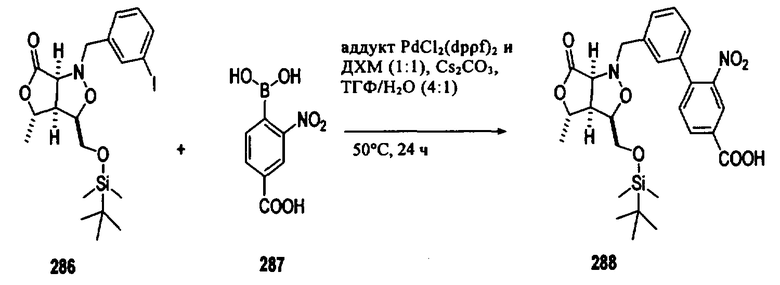
Соединение 288 получали по методике, описанной в примере 183, но при замене бороновой кислоты 271 на бороновую кислоту 287 (выход 56%).
Стадия С

В раствор соединения 288 (0,33 г, 0,6 ммоля) и HATU (0,68 г, 1,8 ммоля) в ДМФА (2 мл) добавляли диметиламин в ТГФ (1 мл, 2 М раствор, 3,1 ммоля). Полученный раствор перемешивали при КТ в течение 14 ч, разбавляли смесью вода/EtOAc (1:1, 200 мл), водный слой экстрагировали EtOAc (2×50 мл). Органические экстракты объединяли, промывали водой (2×50 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: CH2Cl2/EtOAc, 2:1), при этом получали соединение 289 (67 мг, 20%) в виде полутвердого вещества коричневого цвета.
Стадия D
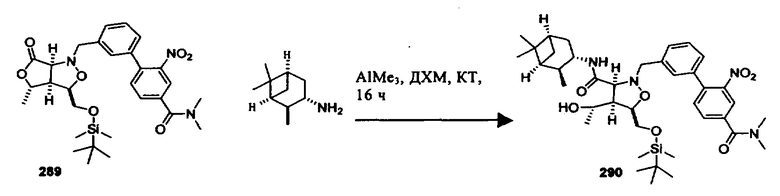
В раствор (+)-изопинокамфеиламина (30 мг, 0,2 ммоля) в CH2Cl2 (0,3 мл) добавляли AlMe3 (0,1 мл, 2 М раствор в гексане, 0,2 ммоля). После перемешивания в течение 10 мин добавляли раствор лактона 289 (56 мг, 0,1 ммоля) в CH2Cl2 (0,2 мл), и смесь перемешивали в течение 16 ч. Реакцию останавливали добавлением насыщенного водного раствора сегнетовой соли (15 мл) и интенсивно перемешивали при КТ в течение 2 ч, при этом получали прозрачную двухфазную смесь. Водный слой отделяли и экстрагировали CH2Cl2 (2×15 мл), органические экстракты объединяли, промывали водой (15 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: гексан/EtOAc, 1:1), при этом получали соединение 290 (40 мг, 56%) в виде масла бледно-желтого цвета.
Стадия Е
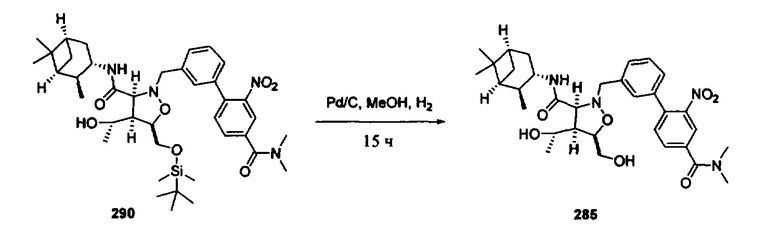
В раствор соединения 290 (15 мг, 0,02 ммоля) в МеОН (0,4 мл) в атмосфере аргона одной порцией добавляли Pd/C (1,5 мг). Полученную смесь инкубировали в атмосфере водорода (1 атм) при КТ в течение 15 ч. Твердое вещество отделяли фильтрованием, фильтрат концентрировали в вакууме. Неочищенный продукт очищали хроматографией на силикагеле (элюент: EtOAc), при этом получали соединение 285 (8 мг, 63%) в виде масла бледно-желтого цвета.
МС (ESI(+)) (m/e): 609,3 (М+Н)+.
Пример 194
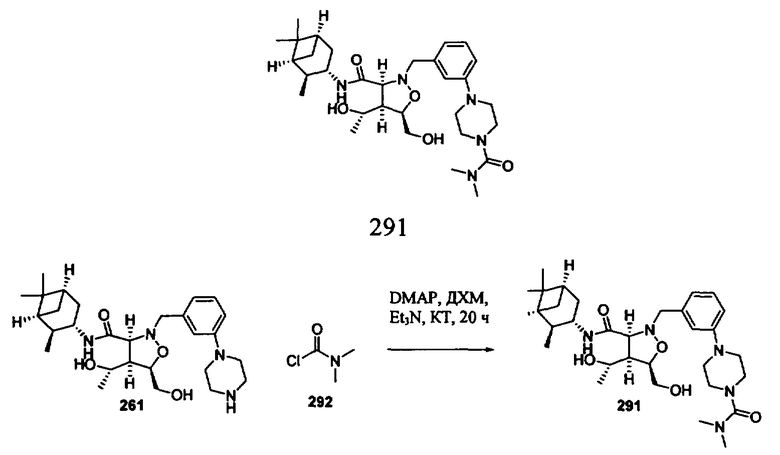
Соединение 261 (3 мг, 0,006 ммоля), ацилхлорид 292 (1,2 мкл, 0,012 ммоля), DMAP (один кристалл) и Et3N (0,012 ммоля) растворяли в CH2Cl2 (0,2 мл) при КТ. Полученный раствор перемешивали в течение 20 ч и концентрировали в вакууме. Полученный остаток очищали хроматографией на силикагеле (элюент: CH2Cl2/MeOH, 30:1), при этом получали соединение 291 (2 мг, 58%) в виде твердого вещества светло-желтого цвета.
МС (ESI(+)) (m/e): 572,3 (М+Н)+.
Пример 195
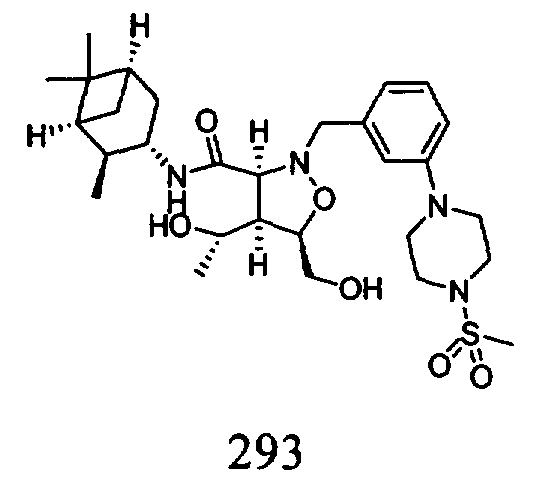
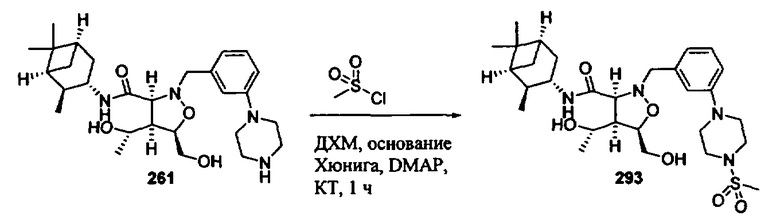
В раствор амина 261 (3 мг, 0,006 ммоля) в CH2Cl2 (0,2 мл) добавляли метансульфонилхлорид (2,6 мг, 0,012 ммоля), DMAP (один кристалл) и диизопропилэтиламин (1,5 мг, 2 мкл, 0,012 ммоля). Раствор перемешивали в течение 20 ч и концентрировали в вакууме. Полученный остаток очищали хроматографией, при этом получали соединение 293 (1,2 мг, 37%).
МС (ESI(+)) (m/e): 579,2 (M+H)+.
Пример 196
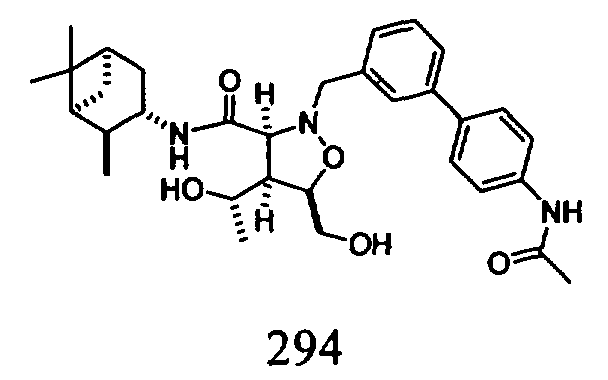
Стадия А
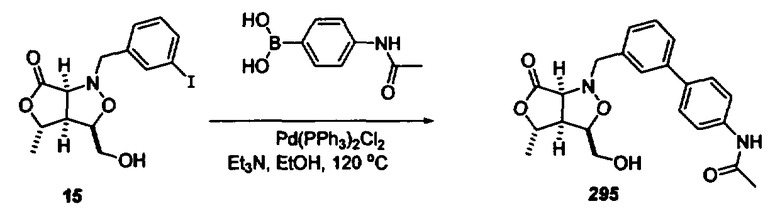
В смесь арилиодида 15 (100 мг, 0,26 ммоля), бороновой кислоты (46 мг, 0,26 ммоля), Et3N (77 мг, 0,11 мл, 0,77 ммоля), Pd(PPh3)2Cl2 (4 мг, 2 мол.%) добавляли EtOH (1,5 мл). Полученную смесь нагревали при 80°С в течение 10 мин, при этом получали практически гомогенную смесь, которую затем нагревали при 120°С в течение 30 мин в микроволновом реакторе. Реакционную смесь концентрировали в вакууме, а остаток очищали хроматографией на силикагеле (элюент: градиент CH2Cl2/MeOH, от 60:1 до 10:1), при этом получали соединение 295 (86 мг, 84%) в виде масла бледно-желтого цвета.
Стадия В

В раствор (+)-изопинокамфеиламина (3 мг, 0,02 ммоля) в CH2Cl2 (0,4 мл) добавляли АlMe3 (42 мкл, 2 М раствор в гексане, 0,042 ммоля). После перемешивания в течение 10 мин добавляли раствор лактона 295 (8 мг, 0,02 ммоля) в CH2Cl2 (0,4 мл), и реакционную смесь перемешивали в течение 1 ч. Раствор разбавляли CH2Cl2, реакцию останавливали добавлением насыщенного водного раствора сегнетовой соли (25 мл). Раствор интенсивно перемешивали в течение 2 ч, при этом получали прозрачную двухфазную смесь. Слои разделяли, органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (элюент: градиент CH2Cl2 в МеОН, от 40:1 до 15:1), при этом получали соединение 294 (3 мг, 27%) в виде полутвердого вещества.
МС (ESI(+)) (m/e): 550,4 (M+H)+.
Пример 197
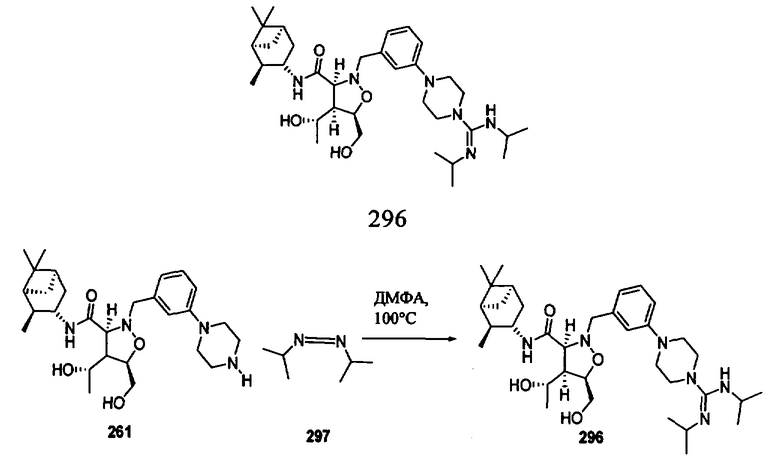
В раствор амина 261 (7 мг, 0,01 ммоля) в ДМФА (0,2 мл) добавляли диизопропилкарбодиимид 297 (2 мг, 0,02 ммоля). Реакционную смесь нагревали при 110°С в течение 12 ч. Раствор охлаждали до комнатной температуры, разбавляли водой (0,5 мл), водную фазу отделяли и экстрагировали EtOAc (0,5 мл). Органические экстракты объединяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали масло, которое очищали методом обращенно-фазовой хроматографии (колонка С18, элюент: 20% ацетонитрил в воде, содержащий 0,1% бикарбонат аммония), при этом получали соединение 296 (1,5 мг, 24%).
МС (ESI(+)) (m/e): 627,4 (M+H)+.
Пример 198
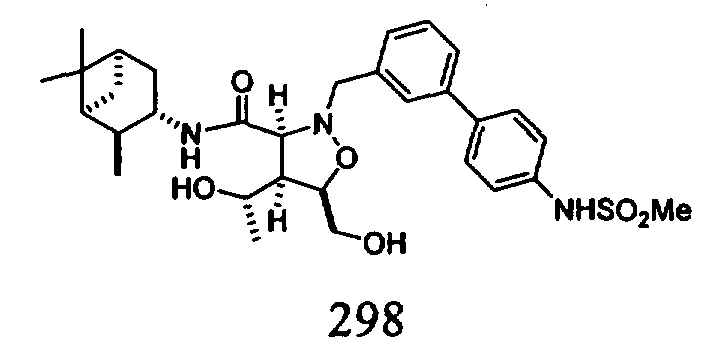
Соединение 298 получали по методике, описанной в примере 183, но при замене 4-ацетамидофенилбороновой кислоты на 4-(метилсульфонамидо)фенилбороновую кислоту (выход 78%).
МС (ESI(+)) (m/e): 586,3 (М+Н)+.
Пример 199
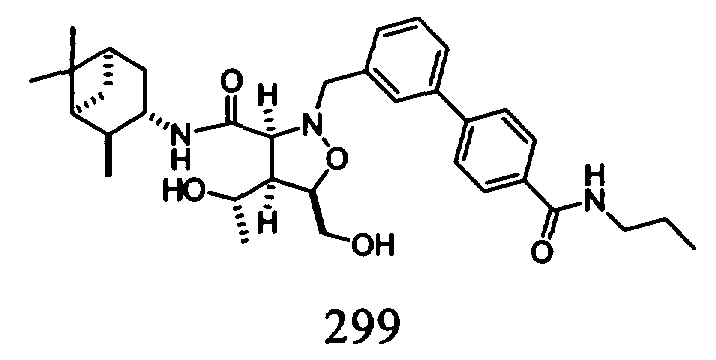
Соединение 299 получали по методике, описанной в примере 148, но при замене соединения 189 на пропиламин (выход 50%). МС (ESI(+)) (m/e): 578,4 (М+Н)+.
Пример 200
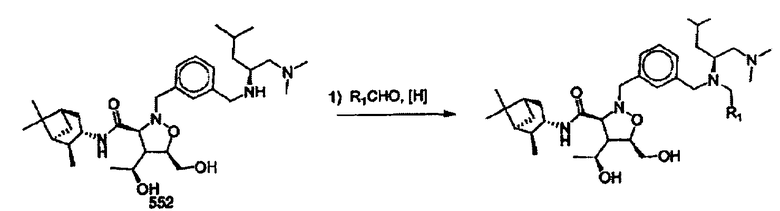
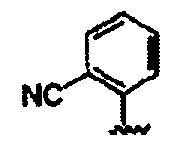
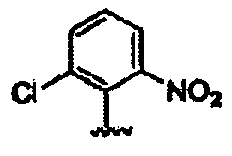
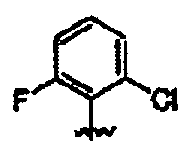
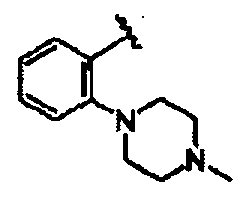
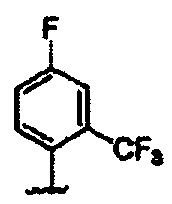
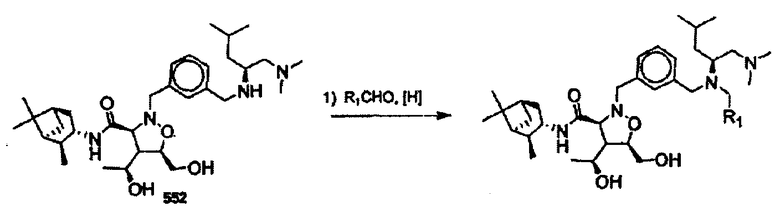
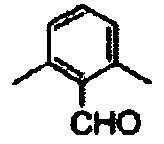
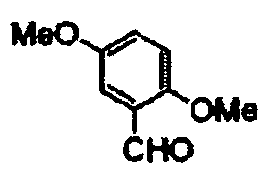
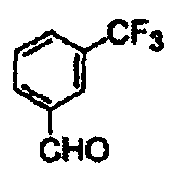
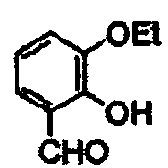
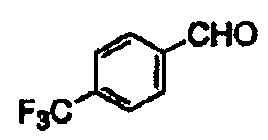
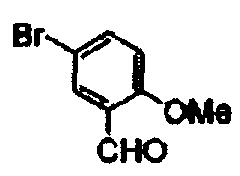
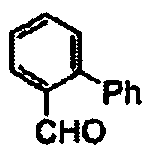
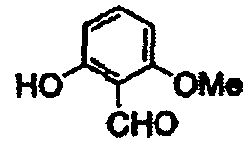
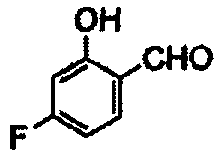
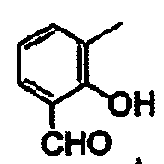
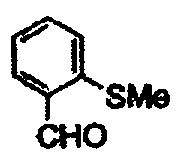
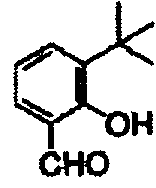
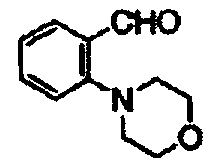
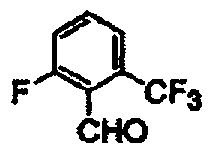
Соединения 300-339 синтезировали аналогично тому, как описано в примере 149. Для соединений 300-339 данные МС представлены в следующей таблице.
Пример 201
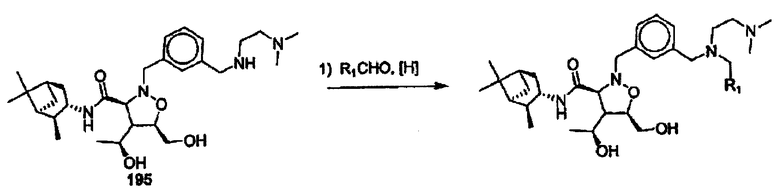
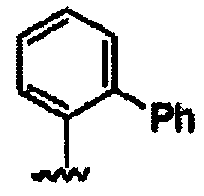
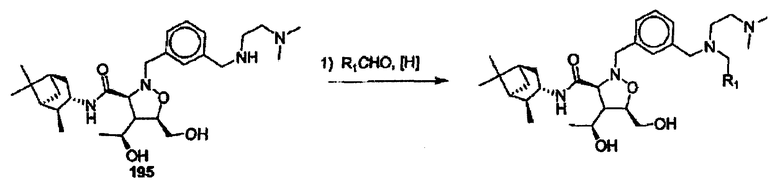
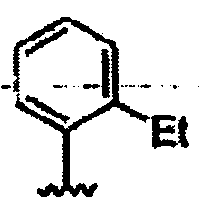
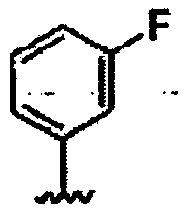
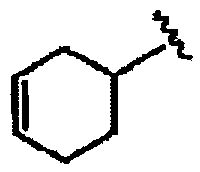
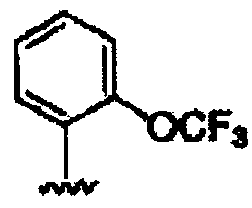
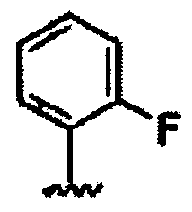
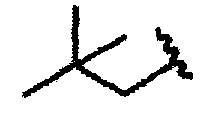
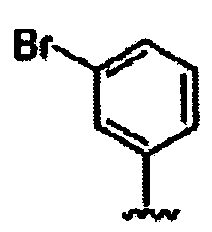

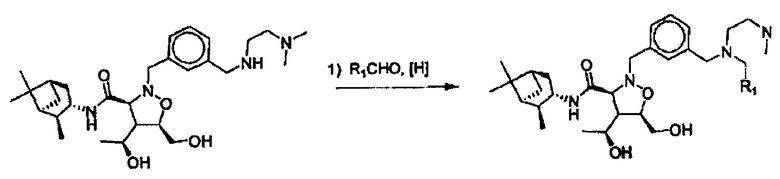
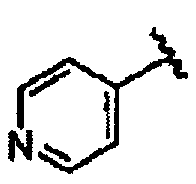
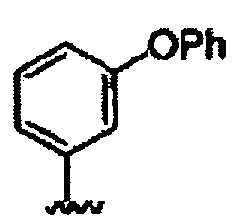
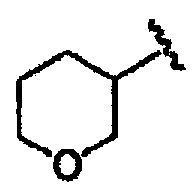
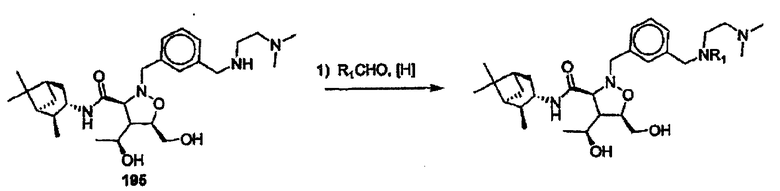
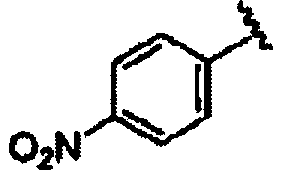
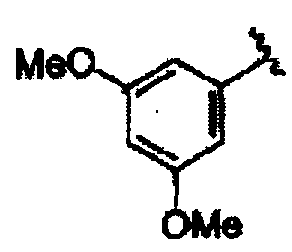
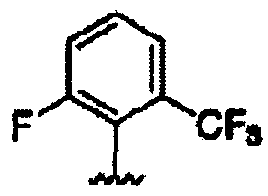
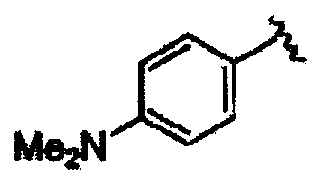
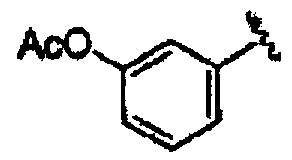
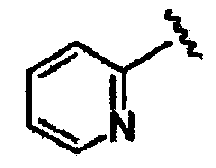
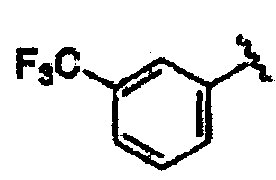
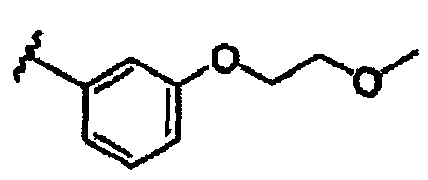
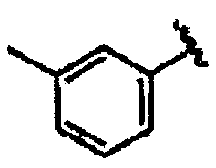
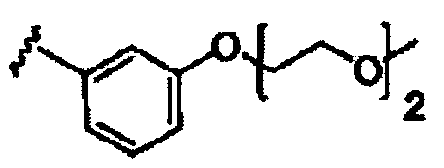
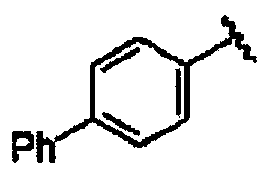


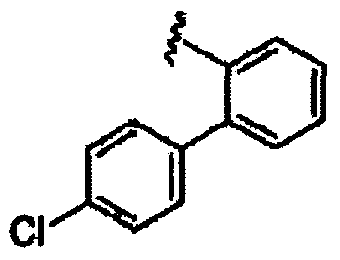
Соединения 340-413 синтезировали аналогично тому, как описано в примере 149. Для соединений 340-413 данные МС представлены в следующей таблице.
Пример 202
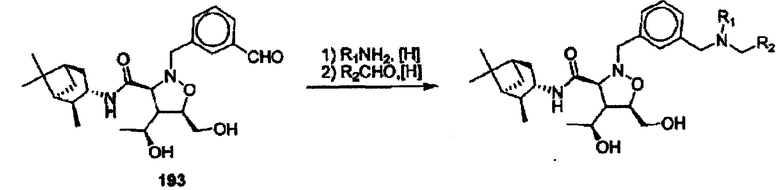
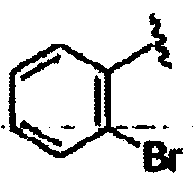
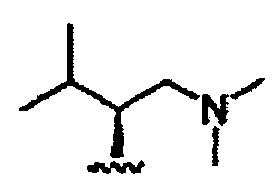
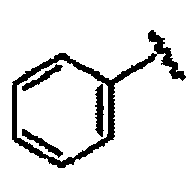
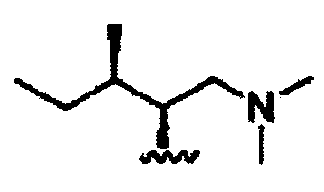
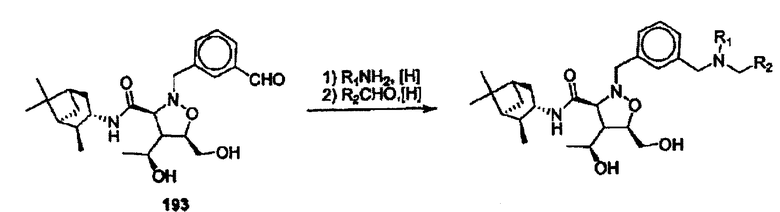
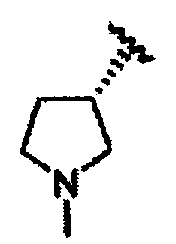
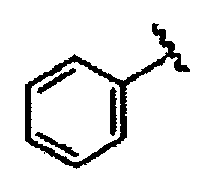
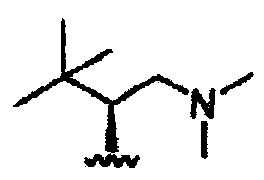
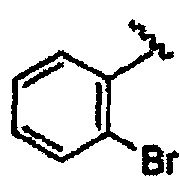
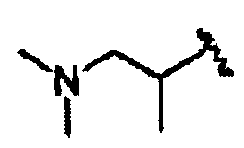
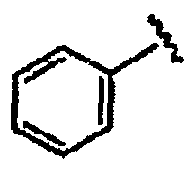
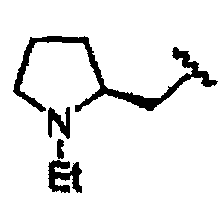
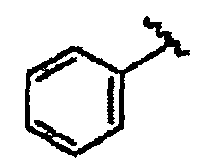
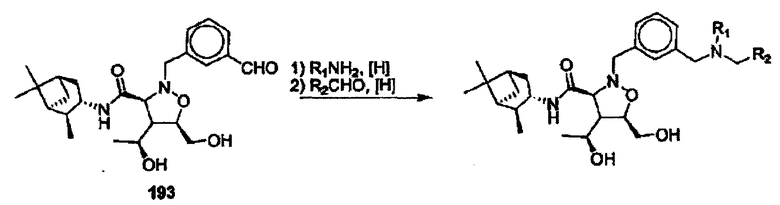
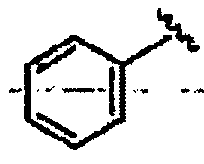
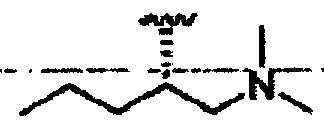
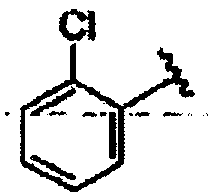
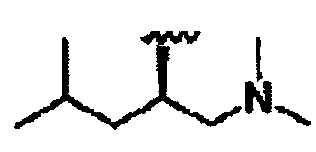
Соединения 414-440 синтезировали аналогично тому, как описано в примере 149. Для соединений 414-440 данные МС представлены в следующей таблице.
Пример 203
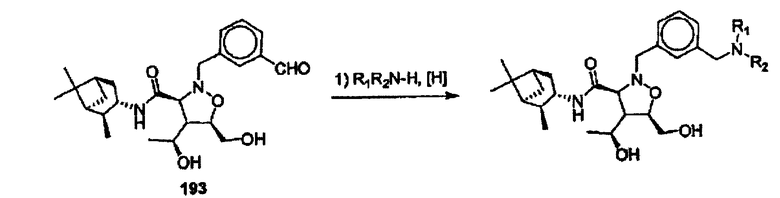
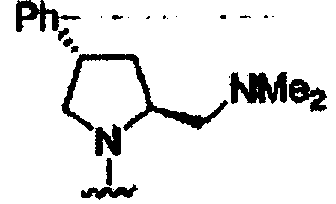
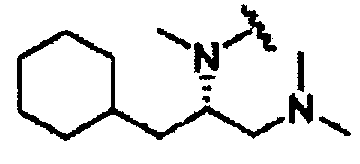
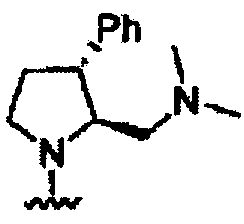
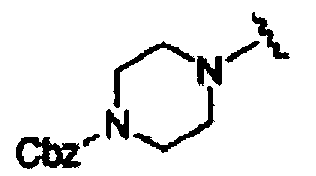
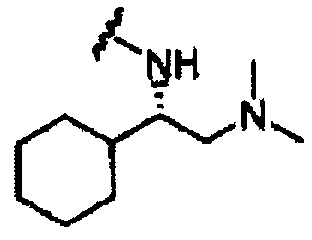
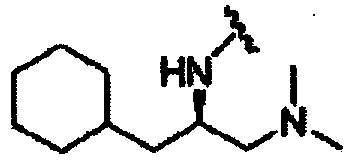
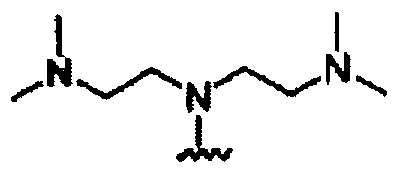
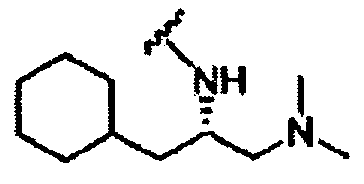
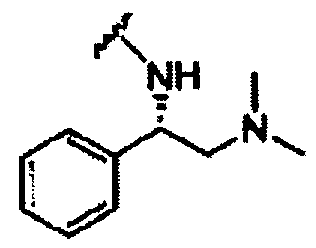
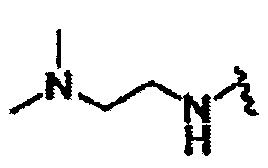
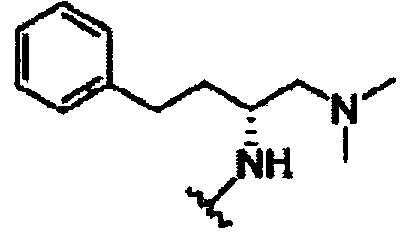
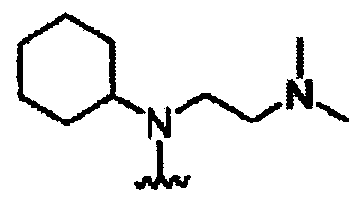
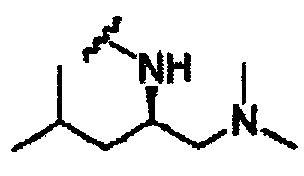
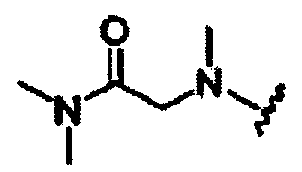
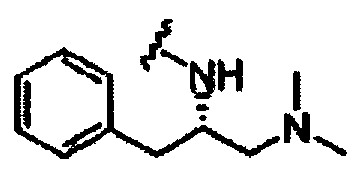
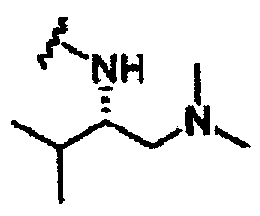
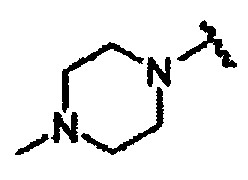
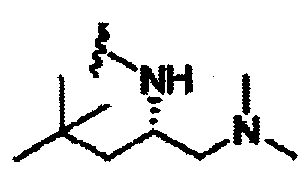
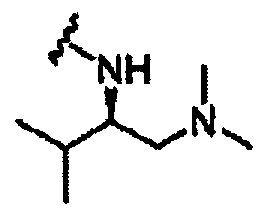
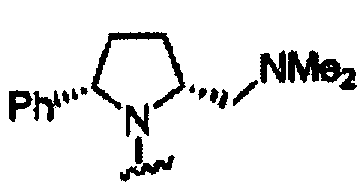
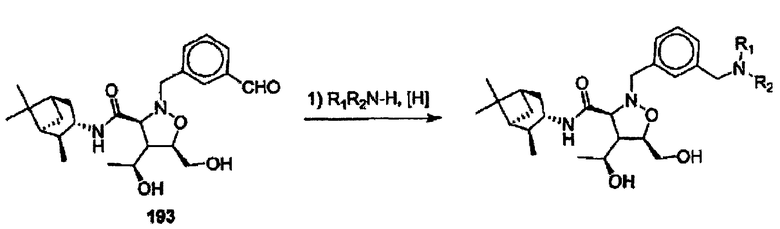
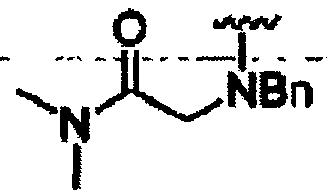
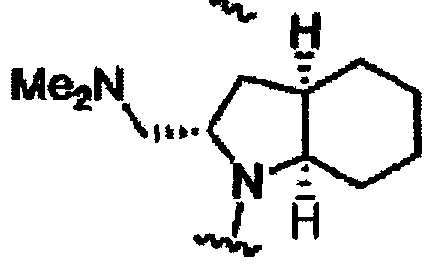
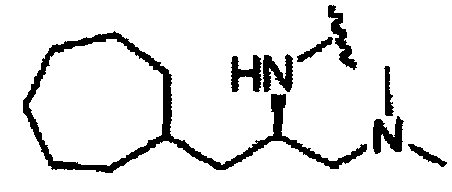
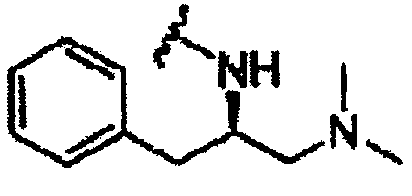
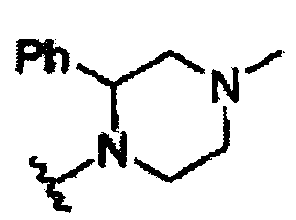
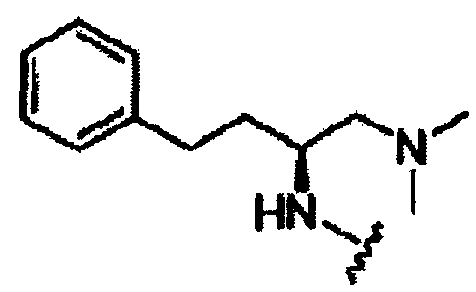
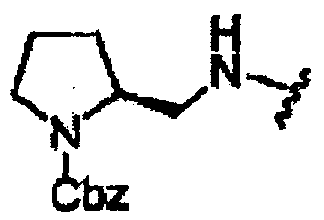
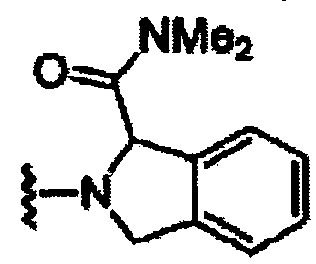
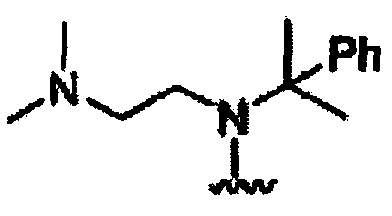
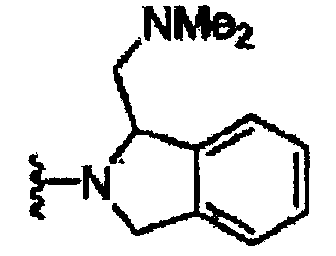
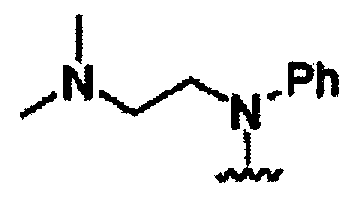
Соединения 441-476 синтезировали аналогично тому, как описано в примере 149. Для соединений 441-476 данные МС представлены в следующей таблице.
Пример 204
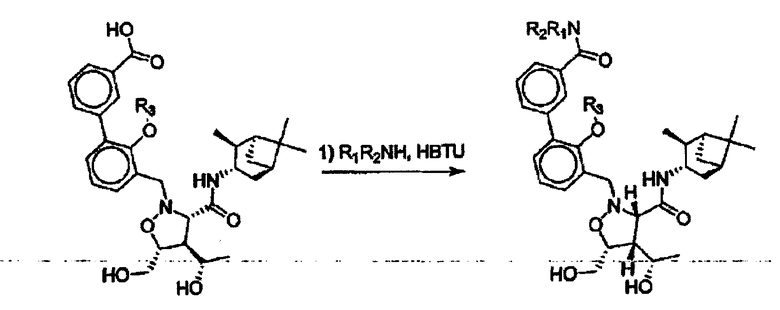
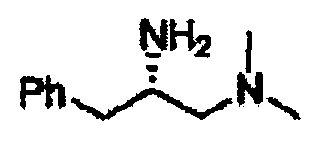
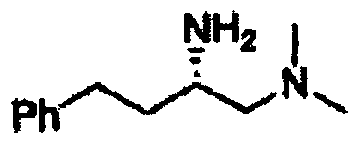
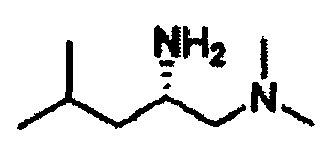
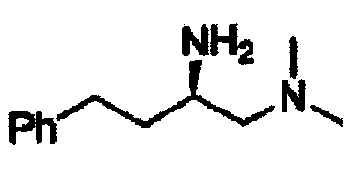
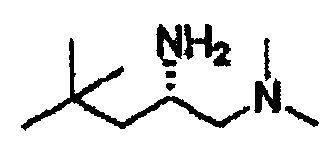
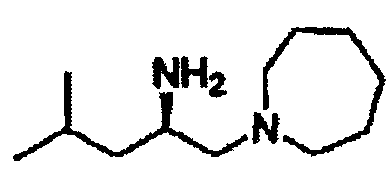
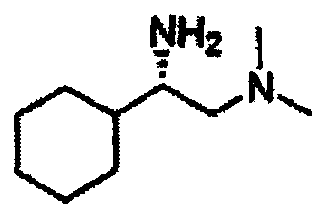
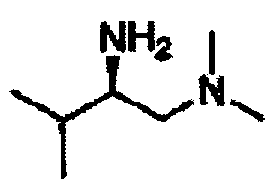
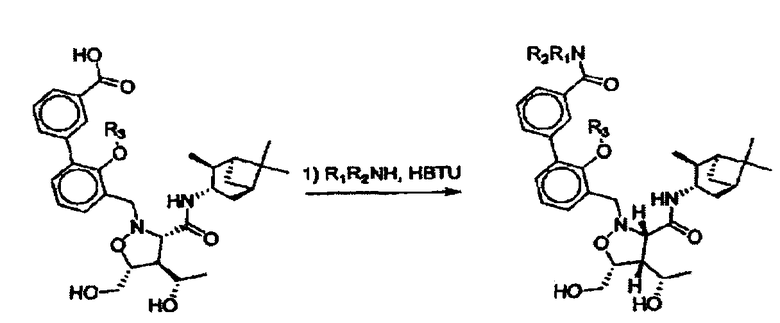
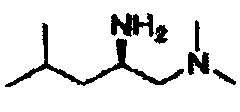
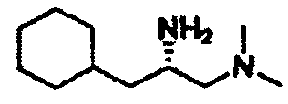
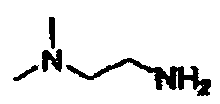
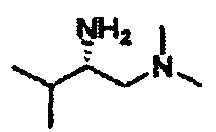


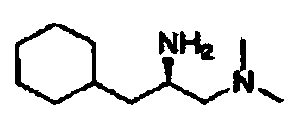
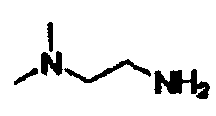
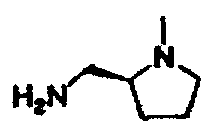
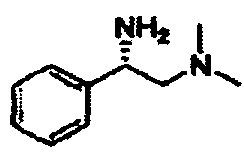

Соединения 477-497 синтезировали аналогично тому, как описано в примере 149. Для соединений 477-497 данные МС представлены в следующей таблице.
Пример 205
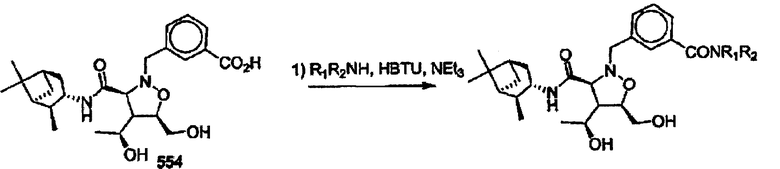
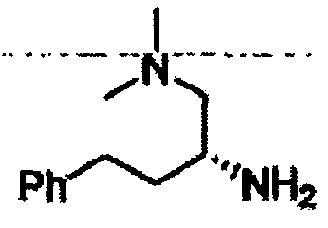
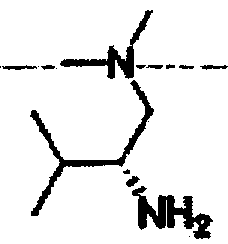
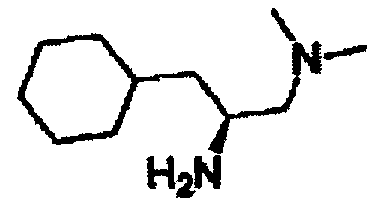
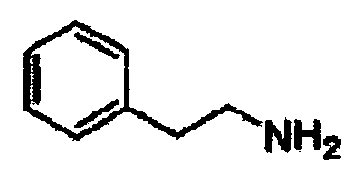
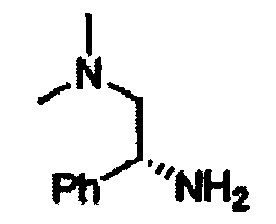
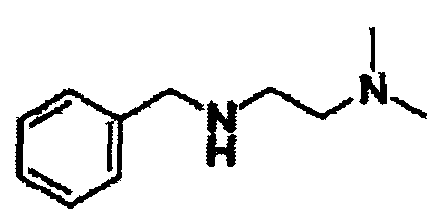
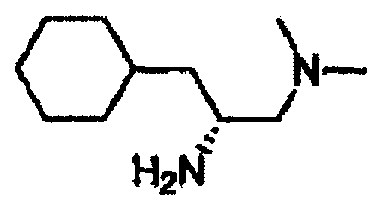
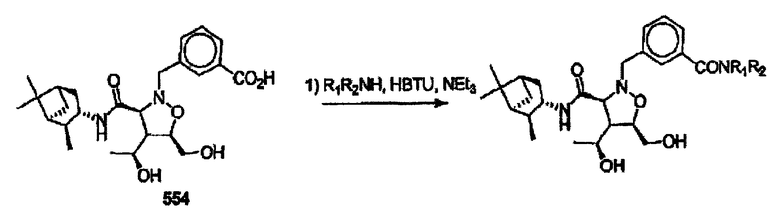
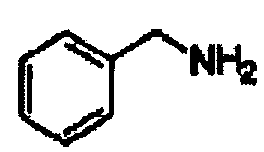
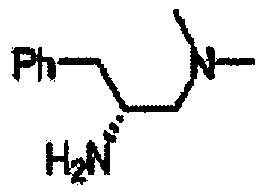
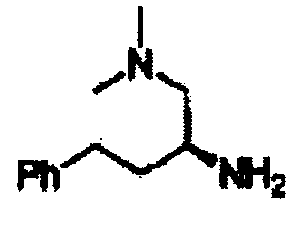
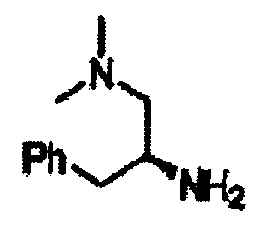
Соединение 554 синтезировали по следующей методике.
Раствор альдегида 554 (60 мг, 1 экв.) в ТГФ (1,5 мл) помещали во флакон, снабженный перемешивающим стержнем, и добавляли изопрен (170 мкл, 15 экв.), 2,7 М фосфатный буферный раствор (225 мкл, 4,5 экв.) и раствор NaClO2 (490 мкл, 3,6 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем добавляли воду (30 мл) и полученный раствор подкисляли 6 М HCl до рН 1. Раствор экстрагировали CH2Cl2 (3×15 мл). Органический слой отделяли и сушили над Na2SO4, концентрировали, при этом получали пену белого цвета (74 мг), которую использовали на следующей стадии без дальнейшей очистки.
Соединения 498-511 синтезировали аналогично тому, как описано в примере 170. Данные МС для соединений 498-511 представлены в следующей таблице.
Пример 206
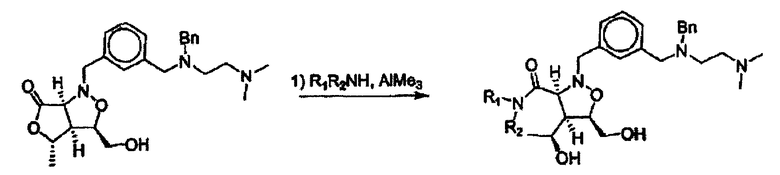
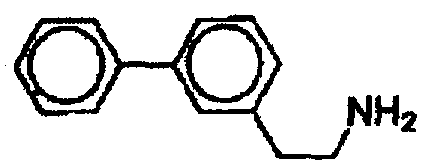
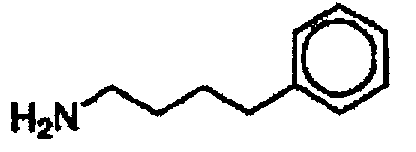
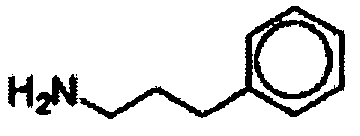

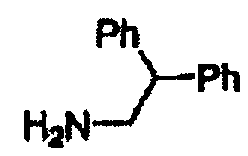
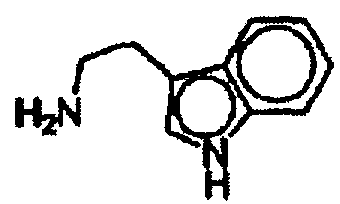
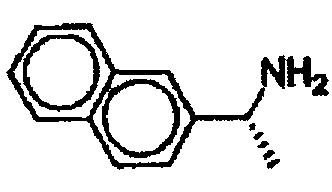
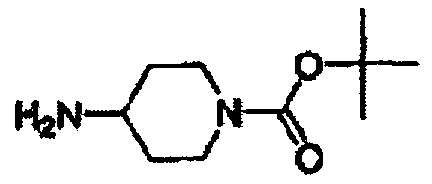
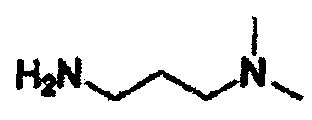
Соединения 512-528 синтезировали аналогично тому, как описано в примере 149 и в примере 177, стадия F. Данные МС для соединений 512-528 представлены в следующей таблице.
Пример 207
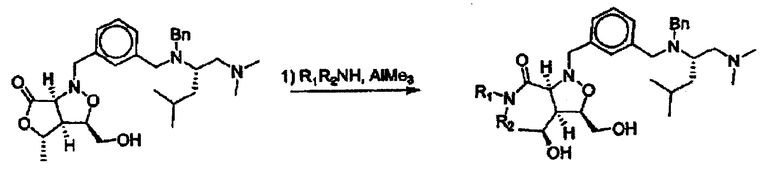
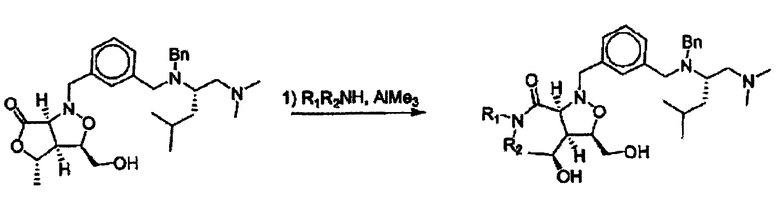
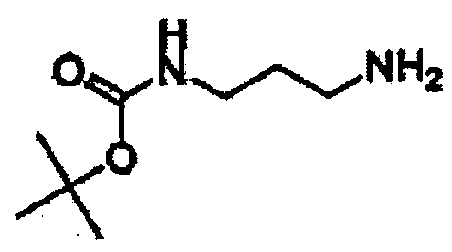
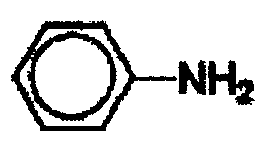

Соединения 529-544 синтезировали аналогично тому, как описано в примере 149 и в примере 177, стадия F. Данные МС для соединений 529-544 представлены в следующей таблице.
Пример 208
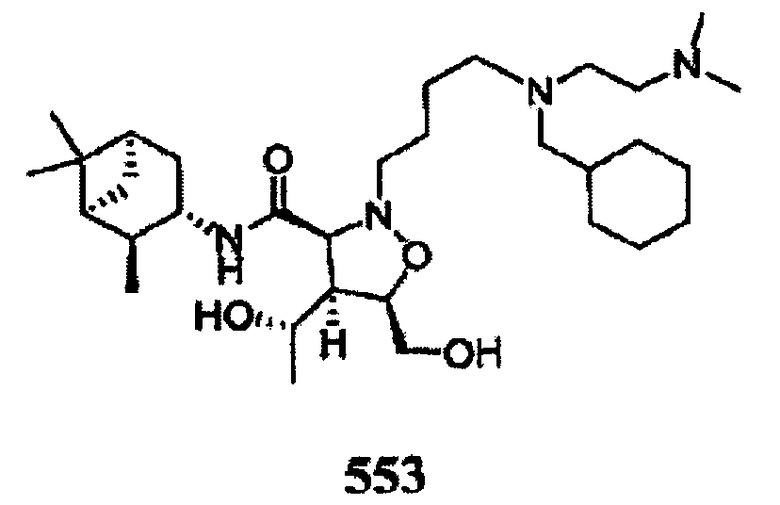
Стадия А

В раствор 4-гептаналя (3,2 г) в МеОН/ТГФ (3:1, 16 мл) добавляли одной порцией водный раствор гидрохлорида гидроксиламина (2,6 г, 4 мл). Величину рН доводили 6 н. KOH до 9 и смесь перемешивали при комнатной температуре в течение 2 ч, при этом данные ТСХ свидетельствовали о полном потреблении альдегида. Затем добавляли NaBH3CN (3,6 г, 2 экв.) и рН раствора доводили до 2-3, используя раствор HCl в МеОН (20 об.%) и раствор перемешивали в течение ночи. Величину рН раствора доводили до 11, используя 2 н. KOH. Слои разделяли и водный слой экстрагировали CH2Cl2 (3×50 мл), объединенные органические экстракты сушили, фильтровали и концентрировали в вакууме, при этом получали гидроксиламин в виде жидкости белого цвета (3 г).
Стадия В
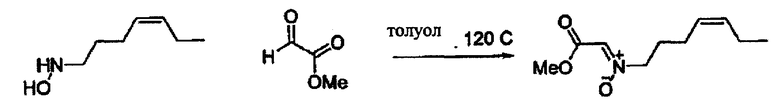
Гидроксиламин (0,9 г) и метиловый эфир глиоксиловой кислоты (0,6 г) растворяли в толуоле (10 мл). Смесь нагревали при 120°С в течение 3 ч. Смесь концентрировали до 6 мл и использовали на следующей стадии.
Стадия С
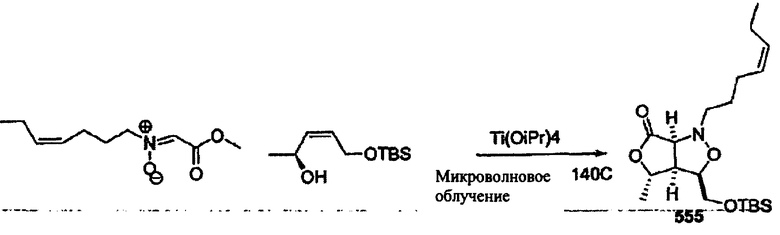
Эфир нитроновой кислоты (0,4 г, 2 ммоля), TBS аллиловый спирт (0,5 г, 2 ммоля) и изопропоксид титана (0,9 г, 3 ммоля) помещали во флакон для микроволнового реактора. Смесь нагревали при 140°С в течение 15 мин при перемешивании. Смесь охлаждали и разбавляли этилацетатом. Смесь промывали водой, солевым раствором, сушили и концентрировали. После экспресс-хроматографии на силикагеле (элюент: гексан, 10% EtOAc в гексане) получали 300 мг частично очищенного продукта.
Стадия D
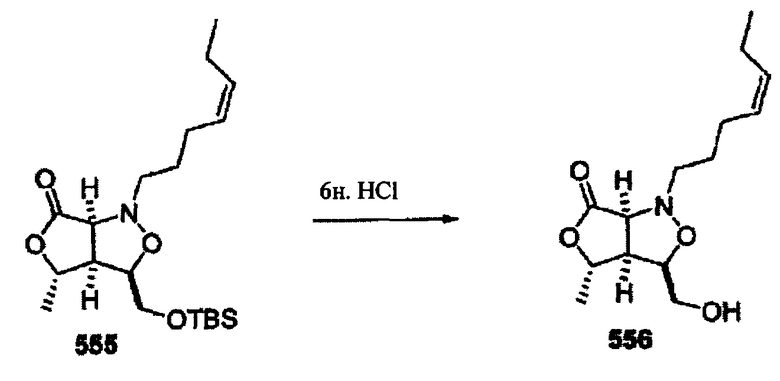
Изоксазолидин растворяли в ТГФ (14 мл), добавляли 6 н. HCl (1 мл) и смесь перемешивали при комнатной температуре в течение 1,5 ч. Смесь разбавляли EtOAc (3×50 мл) и слои разделяли. Водный слой экстрагировали EtOAc и объединенные экстракты промывали солевым раствором, сушили, фильтровали и концентрировали, при этом получали 0,2 г требуемого продукта.
Стадия Е
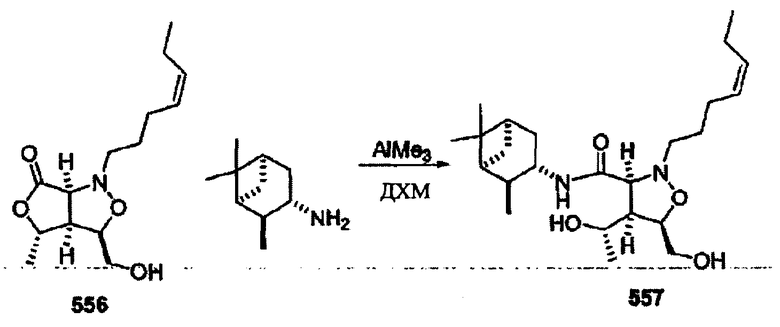
Изопинокамфеиламин (0,6 г, 4 ммоля) растворяли в безводном CH2Cl2 (3 мл) и в раствор по каплям добавляли Al(Ме)3 (2 М, 4 ммоля). Раствор перемешивали при комнатной температуре в течение 20 мин. Исходный лактон (0,7 г, 3 ммоля) растворяли в безводном CH2Cl2 (3 мл), полученный раствор медленно добавляли в раствор амина и Al(Ме)3. Реакционную смесь перемешивали при комнатной температуре в течение 12 ч и разбавляли CH2Cl2 (60 мл), добавляли насыщенный раствор сегнетовой соли (5 мл) и два слоя перемешивали при комнатной температуре в течение 2 ч. Органический слой отделяли от водного слоя и промывали водой и солевым раствором, сушили (Na2SO4), фильтровали и концентрировали, при этом получали неочищенный продукт (400 мг). Неочищенный продукт очищали экспресс-хроматографией (силикагель, 50% EtOAc в гексане) и получали требуемый продукт (260 мг).
Стадия F
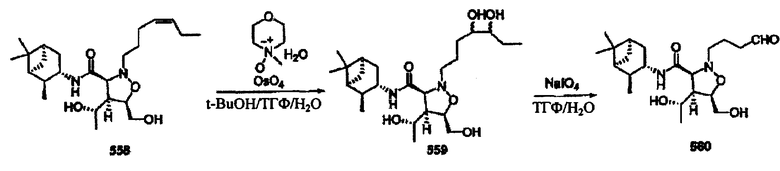
В раствор соединения 558 (0,25 г, 0,59 ммоля) в 2-метил-2-пропаноле (6 мл), ТГФ (3 мл) и Н2О (1 мл) добавляли NMO (0,1 г, 1,5 экв.), затем по каплям добавляли OsO4 (0,37 мл, 2,5% раствор в 2-метил-2-пропаноле). Через 1,5 ч реакционную смесь концентрировали до 3 мл, разбавляли CH2Cl2, солевым раствором и 10% раствором Na2S2O3, экстрагировали CH2Cl2 (2х), сушили над MgSO4, фильтровали и концентрировали, получали пену грязно-белого цвета. Полученное вещество использовали на следующей стадии без дополнительной очистки.
Пену грязно-белого цвета растворяли в ТГФ/Н2О (6 мл ТГФ, 0,6 мл Н2О, 10:1), добавляли одной порцией периодат натрия (0,19 г). Полученную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь разбавляли CH2Cl2, солевом раствором и 10% раствором Na2S2O3, экстрагировали CH2Cl2 (2х), сушили над MgSO4, фильтровали и концентрировали, получали пену грязно-белого цвета. Неочищенный продукт очищали на колонке (силикагель SG60: 20 г, элюент: 80% EtOAc/гексан), при этом получали 0,12 г твердого вещества белого цвета.
Стадия G
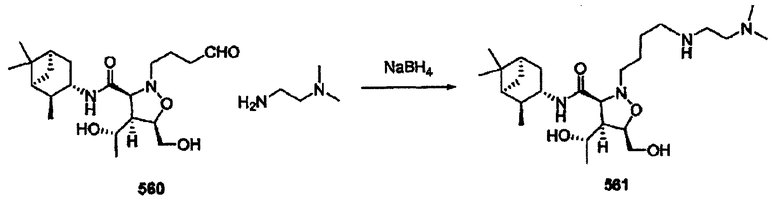
В раствор соединения 560 (0,1 г, 0,3 ммоля) в МеОН (2 мл) добавляли N1,N1-диметилэтан-1,2-диамин (0,030 г, 0,36 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 3,5 ч. Одной порцией добавляли NaBH4 (0,01 г, 0,3 ммоля) и реакционную смесь перемешивали в течение еще 2 ч. Реакцию останавливали добавлением уксусной кислоты (10 мкл) и реакционную смесь концентрировали до 0,5 мл. Смесь разбавляли CH2Cl2, добавляли концентрированный раствор NaHCO3 и смесь экстрагировали CH2Cl2 (2x), сушили над MgSO4, фильтровали и концентрировали, при этом получали требуемый продукт (100 мг).
Стадия Н
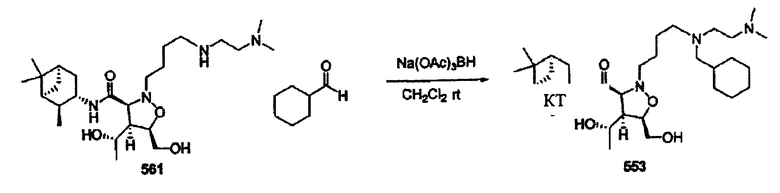
Соединение 561 (15 г) растворяли в безводном ДХМ (1 мл) и добавляли циклогексанкарбальдегид (7 мг). Раствор перемешивали в течение 15 мин, затем добавляли триацетоксиборгидрид натрия (17 мг). Через 1 ч в реакционную смесь добавляли метанол (0,5 мл) и раствор очищали ЖХВР (элюент градиент: бикарбонат аммония-ацетонитрил). Фракции объединяли, замораживали и лиофилизировали, при этом получали 7 мг требуемого продукта. МС (ESI(+)): m/e 565,75 (М+Н)+.
Пример 209
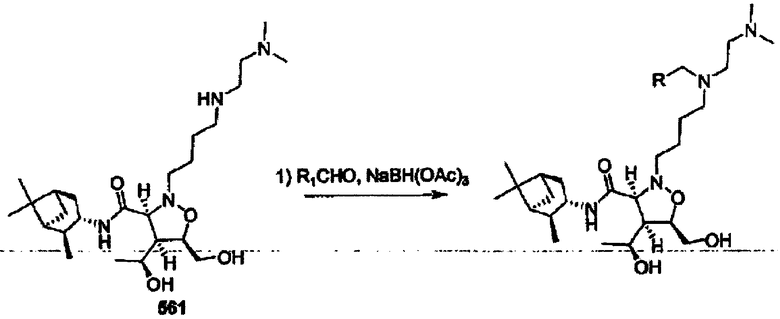
Соединения 545-551 синтезировали аналогично тому, как описано в примере 207. Данные МС для соединений 545-551 представлены в следующей таблице.
Пример 210
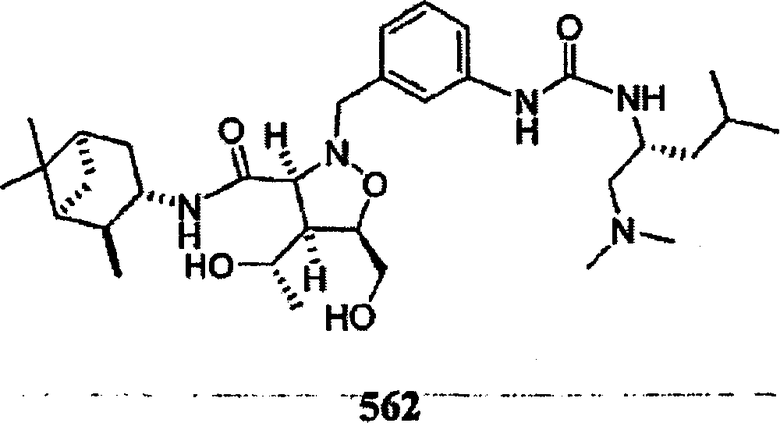
Стадия А
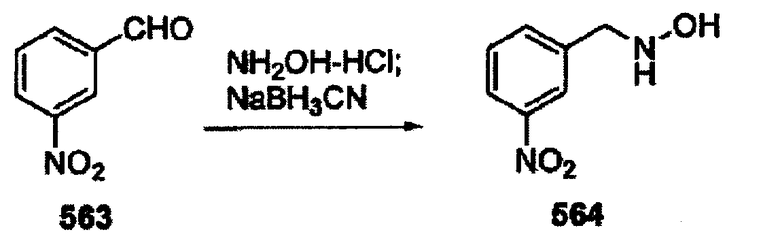
В раствор 3-нитробензилальдегида (20 г) в МеОН/ТГФ (3:1, 400 мл) одной порцией добавляли водный раствор гидроксиламина (13 г в 60 мл воды). Величину рН доводили до 11 добавлением 6 н. KOH и перемешивали при комнатной температуре в течение 2 ч, затем добавляли NaBH3CN (17 г, 2 экв.) и раствор подкисляли до рН 2-3 добавлением раствора HCl в МеОН (20 об.%). В ходе реакции величину рН реакционной смеси поддерживали при 3 добавлением небольших количеств раствора HCl в метаноле. Смесь перемешивали в течение 12 ч, при этом рН 3 поддерживали добавлением раствора HCl в метаноле. Через 8 ч раствор подщелачивали водным раствором 2 н. KOH до рН 11 и экстрагировали CH2Cl2 (3 раза), сушили над MgSO4, концентрировали в вакууме, при этом получали соединение 564 (20 г, 91%).
Стадия В
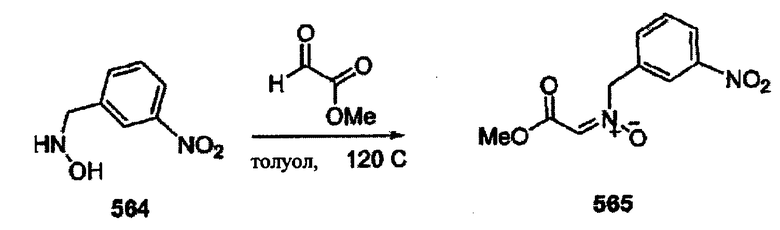
Соединение 564 (7 г) и эфир глиоксиловой кислоты (4 г) растворяли в 100 мл толуола. Смесь нагревали при 120°С при перемешивании. Через 3 ч растворитель упаривали до 15 мл. Полученный раствор использовали на следующей стадии без дополнительной очистки.
Стадия С
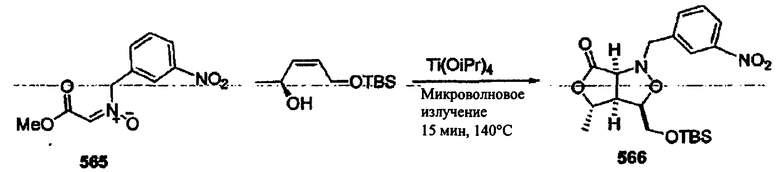
Раствор соединения 565 (5 мл, полученного на стадии А) добавляли в пробирку для микроволнового реактора, добавляли аллиловый спирт (1 г) и Ti(OiPr)4 (2 г). Смесь нагревали при 140°С в течение 15 мин в микроволновом реакторе. Смесь охлаждали до комнатной температуры. Неочищенную смесь разбавляли этилацетатом и промывали водой, солевым раствором, сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией на колонке (силикагель элюент: 0% EtOAc в гексане), при этом получали 0,8 г соединения 566.
Стадия D
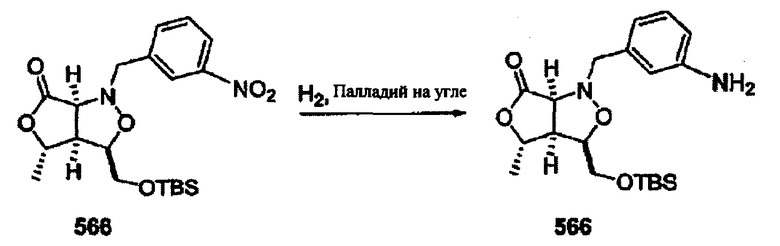
Соединение 566 (1 г) растворяли в МеОН (10 мл). Реакционную смесь продували N2, добавляли палладий на угле (10%, 0,7 г) и к реакционной колбе присоединяли баллон с Н2. Реакционную смесь перемешивали при комнатной температуре в течение 45 мин, затем смесь фильтровали через тонкий слой целита.
Слой целита промывали двумя порциями EtOAc. Органические растворы объединяли и концентрировали, при этом получали 0,9 г неочищенного продукта. После очистки экспресс-хроматографией (силикагель, элюент: гексан, 30% EtOAc в гексане) получали 0,8 г соединения 566.
Стадия Е
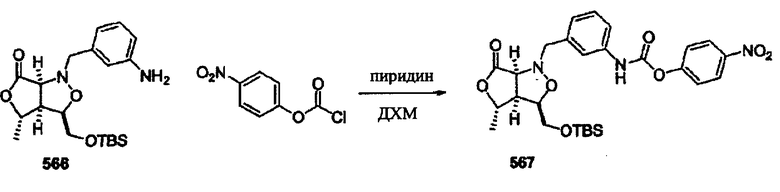
Пара-нитрофенилхлорформит (300 мг) растворяли в CH2Cl2 (2 мл), в реакционную смесь добавляли пиридин (0,1 мл), полученную суспензию белого цвета охлаждали до 0°С и одной порцией добавляли раствор соединения 566 (450 мг) в CH2Cl2 (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Смесь разбавляли CH2Cl2, промывали водой, солевым раствором и сушили над MgSO4 и концентрировали, при этом получали неочищенное соединение 567 (0,65 г), которое использовали без дополнительной очистки на стадии Е.
Стадия Е
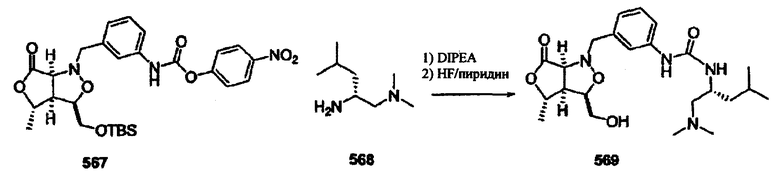
Соединение 567 (20 мг) растворяли в CH2Cl2 (1 мл). В реакционную смесь добавляли соединение 568 (10 мг) и DIPEA (10 мкл). Раствор перешивали при комнатной температуре в течение 12 ч, затем раствор концентрировали и снова растворяли в 1 мл сухого ТГФ, добавляли HF/пиридин (150 мкл) и смесь перемешивали при комнатной температуре в течение 1,5 ч. Реакцию останавливали добавлением TMSOMe (1,5 мл) и смесь концентрировали. Неочищенное соединение очищали методом ЖХВР и получали соединение 569 (15 мг).
Стадия F
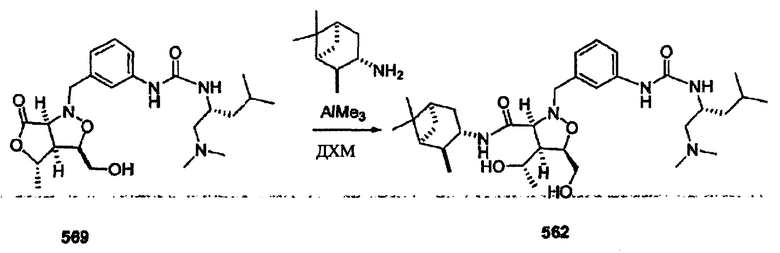
Изопинокамфеиламин (5 мг) растворяли в безводном CH2Cl2 (1 мл). В реакционную смесь по каплям добавляли AlMe3 (2 М, 20 мкл). Смесь перемешивали при комнатной температуре в течение 20 мин. Раствор соединения 569 (7 мг в безводном CH2Cl2 (1 мл)) медленно добавляли в реакционную смесь, которую перемешивали при комнатной температуре в течение 12 ч, затем разбавляли CH2Cl2 (25 мл), добавляли насыщенный водный раствор сегнетовой соли (5 мл) и полученную смесь перемешивали при комнатной температуре в течение 2 ч. Органический слой отделяли от водного слоя и промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали, при этом получали неочищенный продукт (10 мг), который очищали ЖХВР, и получали соединение 562 (3 мг). МС (ESI(+)): m/e 602,45 (М+Н)+.
Пример 211
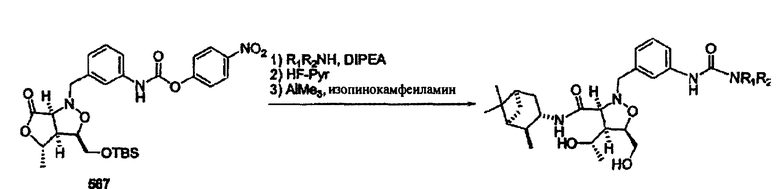
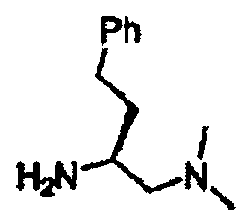
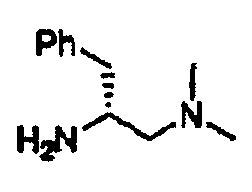
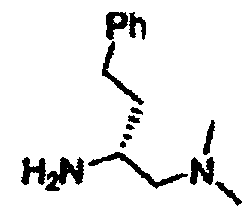
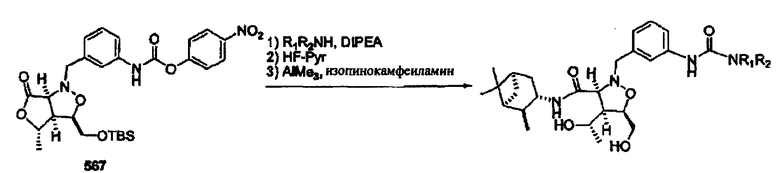
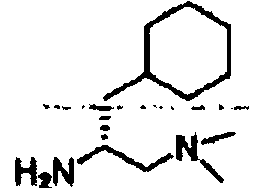
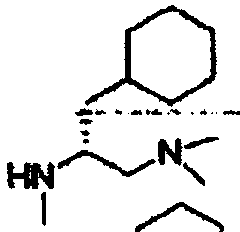
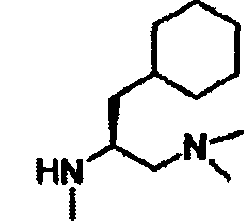
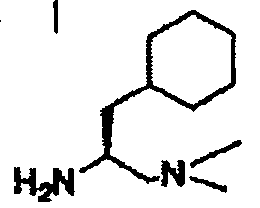
Соединения 570-577 синтезировали аналогично тому, как описано в примере 208. Данные МС для соединений 570-577 представлены в следующей таблице.
Пример 212
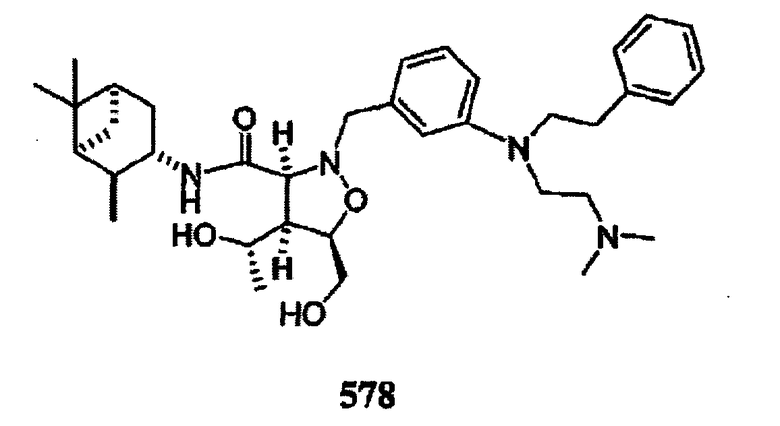
Стадия А
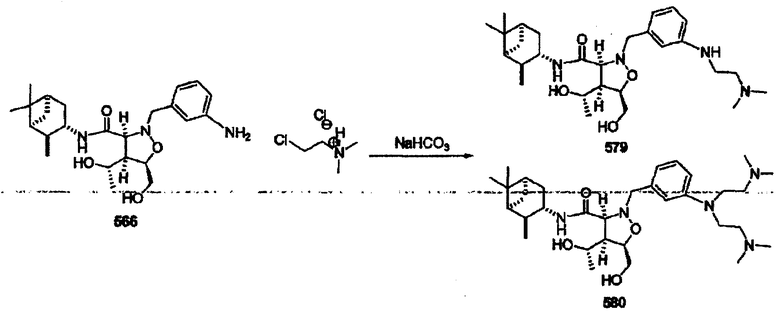
Бикарбонат натрия (200 мг), 2-хлор-N,N-диметилэтанамин (200 мг) добавляли в раствор соединения 566 (200 мг) в EtOH (1 мл) в атмосфере N2. Реакционную смесь перемешивали при 60°С в течение 14 ч и разбавляли в EtOAc (50 мл), промывали солевым раствором, сушили над Na2SO4 и концентрировали, при этом получали 300 мг неочищенного продукта, который очищали методом ЖХВР, и получали соединение 568 (20 мг) и соединение 567 (90 мг).
Стадия В
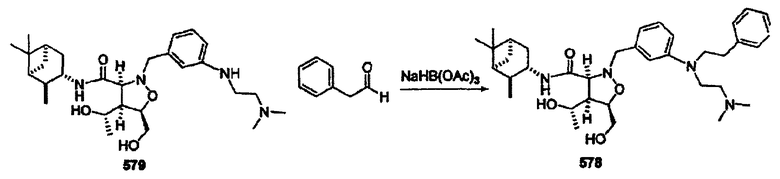
Соединение 579 (15 мг), фенилацетальдегид (11 мг) и NaHB(OAc)3 (20 мг) растворяли в безводном ТГФ в атмосфере N2. Смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь очищали методом ЖХВР и получали соединение 578 (4 мг).
МС (ESI(+)): 607,74 m/e (М+Н)+.
Пример 213
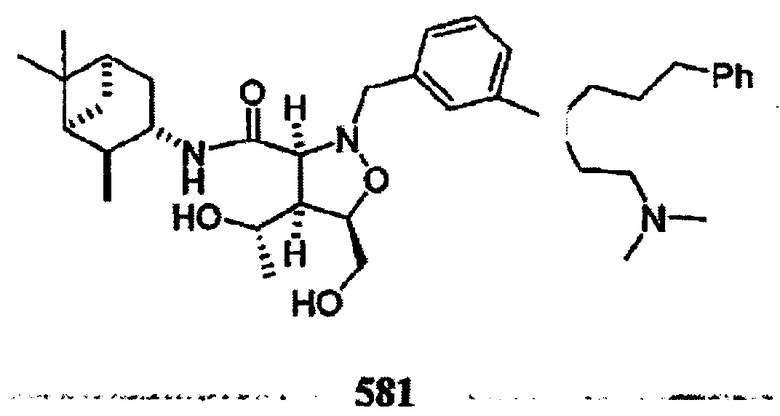
Соединение 581 синтезировали аналогично тому, как описано в примере 210, но при замене фенилацетальдегида на 3-фенилпропаналь.
МС (ESI(+)): m/e 621,44 (М+Н)+.
Пример 214
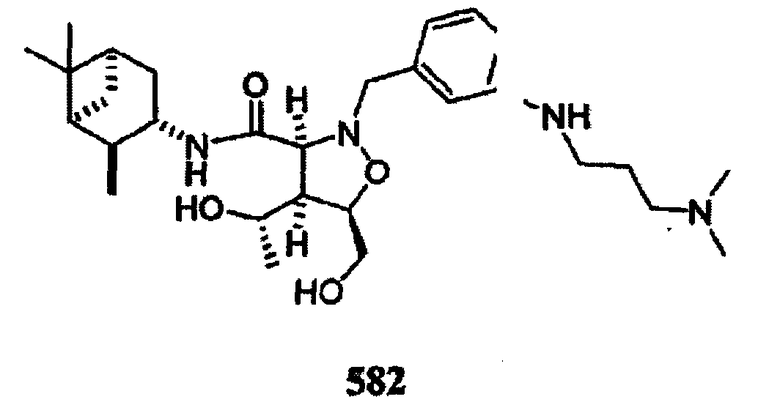
Соединение 582 синтезировали аналогично тому, как описано в примере 210, но при замене 2-хлор-N,N-диметилэтанамина на 3-хлор-N,N-диметилпропанамин.
МС (ESI(+)): m/e 517,3 (М+Н)+.
Пример 215
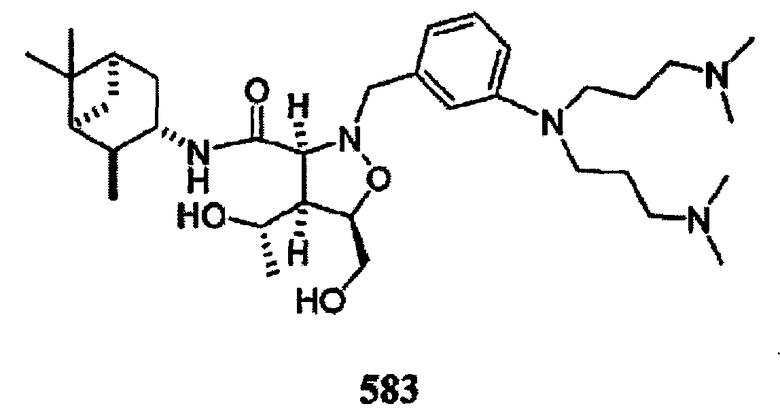
Соединение 583 синтезировали аналогично тому, как описано в примере 210, но при замене 2-хлор-N,N-диметилэтанамина на 3-хлор-N,N-диметилпропанамин.
МС (ESI(+)): m/e 602,43 (М+Н)+.
Пример 216
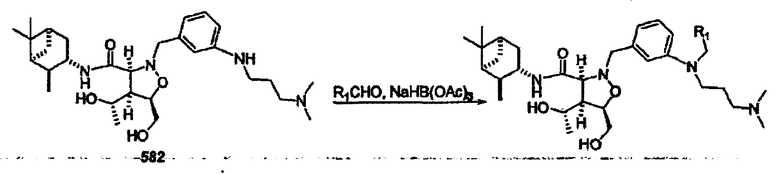

Пример 217
Результаты анализа связывания соединений по настоящему изобретению с Bcl-2 и Bcl-XL представлены ниже. Знак «****» означает, что Ki<0,8 мкМ, «***» означает, что Ki составляет от 0,8 до 6 мкМ, «**» означает, что Ki составляет от 6 до 50 мкМ, «*» означает, что Ki>50 мкМ. Знак «†» означает, что Ki>100 мкМ и «††» означает, что Ki>200 мкМ. Знак «н/о» означает, что величина не определена.
Пример 218
Данные анализа на цитотоксичность с использованием клеточной линии фолликулярной лимфомы RL человека
1) Готовили серийные разведения соединений в ДМСО в интервале концентрации от 9 мМ до 4 мкМ в 96-луночных планшетах.
2) В планшетах для анализа готовили разведения образцов (310-кратные в 150 мкл среды, содержащей 30000 клеток/лунку), при этом конечная концентрация ДМСО составляла 0,3%, концентрация каждого соединения в интервале от 29 мкМ до 0,013 мкМ.
3) Планшеты инкубировали при 37°С в атмосфере 5% СО2 в течение 72 ч.
4) Через 72 ч в каждую лунку добавляли аламаровый синий и планшет считывали по инструкции фирмы-производителя.
Результаты анализа на цитотоксичность с использованием клеток RL приведены ниже для различных соединений по настоящему изобретению. Символ "***" означает, что величина IC50<2 мкМ, символ "**" означает, что величина IC50 равна 2-5 мкМ, а символ "*" означает, что IC50>5. Термин «н/о» означает, что цитотоксичность не определена.
Пример 219
Анализ гибели клеток с использованием клеточной линии Panc1 рака поджелудочной железы человека
1) Готовили серийные разведения соединений в ДМСО от исходной концентрации от 9 мМ до конечной концентрации 0,004 мМ в 96-луночных планшетах.
2) В планшетах для анализа готовили разведения образцов (310-кратные в 150 мкл среды, содержащей 10000 клеток/лунку), в присутствии или отсутствии 3 мкМ камптотецина, при этом получали конечную концентрацию ДМСО 0,3% и концентрацию соединения в интервале от 29 мкМ до 0,013 мкМ для каждого соединения.
3) Планшеты инкубировали при 37°С в атмосфере 5% СО2 в течение 72 ч.
4) Через 72 ч в каждую лунку добавляли иодид пропидиния и краситель Хохста по инструкции фирмы-производителя.
5) Планшеты считывали в флуоресцентном микроскопе при длинах волн λвозб.560/λисп.650 (иодид пропидия) и λвозб.360/λисп.465 (краситель Хохста).
6) Для определения гибели клеток в % использовали программное обеспечение MetaMorph при делении числа прокрашенных иодидом пропидия клеток (мертвые клетки) на число прокрашенных красителем Хохста клеток (все клетки) и умножении на 100.
Результаты анализа с использованием клеток Panc1 in vitro для различных соединений по настоящему изобретению представлены ниже. Символ "****" означает, что величина IC50<1,3 мкМ, символ "***" означает, что величина IC50 равна 1,3-1,8 мкМ, символ "**" означает, что IC50 равна 1,8-2,5 мкМ, а символ "*"означает, что IC50>2,5. Термин «н/о» означает, что данная величина не определена.
Пример 217
Анализ in vivo
Действие соединения 221 (пример 166) оценивали с использованием линии клеток фолликулярной лимфомы человека RL и ксенотранстплантанта у самок мышей SCID/NOD. Самке мыши SCID/NOD подкожно имплантировали клетки RL (2×106 клеток в матричном геле 1:1). После развития опухоли до среднего размера 150 мм3 животных рандомизировали по экспериментальным группам (8 особей в группе), т.е. группа для введения только носителя и группа для введения исследуемого соединения в соответствии с приведенной ниже схемой. Исследуемое соединение или носитель вводили внутривенным способом (в/в) через хвостовую вену в объеме 0,1 мл в течение приблизительно 10 с. Животных забивали через 38 сут и сравнивали размеры опухолей.
Мыши: самки мышей SCID/NOD, n=8/группа
Группы с различными дозами:
1. Соединение 221 50 мг/кг в/в (3 сут подряд ПВС)
2. Соединение 221 50 мг/кг в/в (через сутки ПСПт)
3. Соединение 221 50 мг/кг внутрибрюшинно (в/б) (1 раз в сутки в течение 7 сут/1 недели ПВСЧПтСбВ)
4. Носитель в/в (3 сут подряд ПВС)
Носитель: 10% циклодекстрин в 0,1 М Na2HPO4 рН 6,0
Схема лечения:
П - понедельник
В - вторник
С - среда
Ч - четверг
Пт - пятница
Сб - суббота
В - Воскресенье
Введение лекарственного средства: Соединение 221 (рН 6,8) вводили через хвостовую вену внутривенно или внутрибрюшинно в количестве 5 мл/кг или 125 мкл на 25 г массы мыши в течение приблизительно 20 с.
Обследование животных: Мышам имплантировали подкожным способом клетки линии фолликуллярной лимфомы человека RL (2×10 клеток 1:1 в матричном геле) со стороны спины в правую заднюю ногу с использованием иглы №25. После развития опухоли до среднего размера приблизительно 150 мм3 животных рандомизировали по экспериментальным группам. Животных распределяли по 4 группам (n=8 мышей/группа). Состояние животных регистрировали через 30 мин после введения дозы, а затем ежедневно оценивали развитие клинических симптомов. Измеряли размеры опухолей, определяли массу тела и полученные данные регистрировали 2 раза в неделю. Размеры опухолей измеряли 2 раза в неделю с помощью цифрового штангенциркуля, а объем опухоли рассчитывали по формуле (ширина2×длина)/2. Состояние мышей регистрировали до тех пор, пока объем опухоли в контрольной группе (носитель) не достигал величины, указанной в IACUC.
Продолжительность испытаний: Животным вводили дозы в течение 4 недель или до тех пор, пока размер опухоли в контрольной группе (носитель) не достигал 3500 мм3. Затем 50% выживших животных забивали, опухоли удаляли, быстро замораживали и хранили при -80°С. Остальные 50% мышей использовали для оценки дальнейшего роста опухолей и продолжительности противоопухолевого действия.
Результаты испытаний:
Через 4 ч после введения дозы:
Животных забивали по 4 особи из каждой группы
Кровь отбирали и опухоли замораживали
Группы: носитель, (ПСПт) в/в, 1 раз в сут в/б
Через 24 ч после введения дозы:
Животных забивали по 4 мыши из каждой группы
Кровь отбирали и опухоли замораживали
Группы: носитель, (ПСПт) в/в, 1 раз в сут в/б
Результаты анализа биологической активности соединений по настоящему изобретению показаны на чертеже.
Включение в качестве ссылки
Все патенты США и опубликованные заявки на выдачу патента США, цитированные в настоящем описании, всключены в качестве ссылок.
Эквиваленты
Специалистам в данной области техники представляется очевидным, что можно модифицировать множество эквивалентов специфических вариантов осуществления настоящего изобретения, описанных в данном контексте. Такие эквиваленты включены в объем пунктов формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И СПОСОБЫ ИНГИБИРОВАНИЯ ВЗАИМОДЕЙСТВИЯ БЕЛКОВ BCL С КОМПОНЕНТАМИ СВЯЗЫВАНИЯ | 2006 |
|
RU2424230C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| ИНГИБИТОРЫ IAP | 2006 |
|
RU2451025C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ИНГИБИРОВАНИЯ ВЗАИМОДЕЙСТВИЯ БЕЛКОВ Bcl С ПАРТНЕРАМИ СВЯЗЫВАНИЯ | 2007 |
|
RU2449996C2 |
| ТРИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ ТРОМБИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2329264C9 |
| АНАЛОГИ ЦИКЛОПАМИНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2403256C2 |
| ИНГИБИТОРЫ IAP | 2008 |
|
RU2491276C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2015 |
|
RU2733405C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ И ОНКОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2009 |
|
RU2545023C9 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
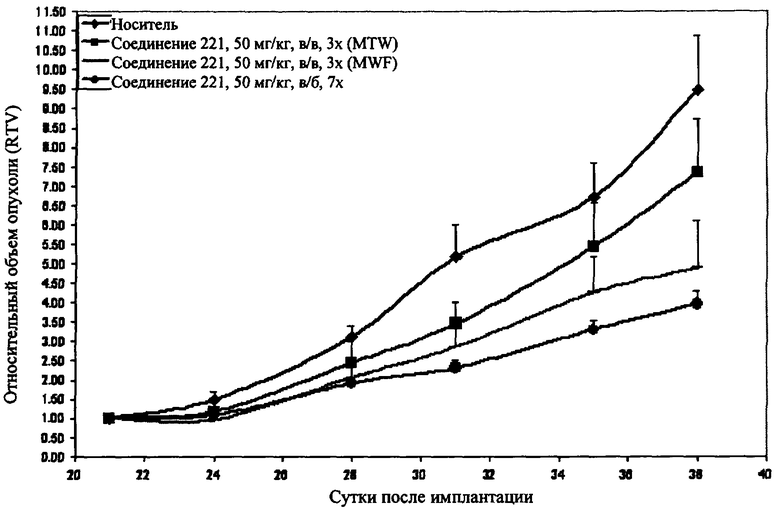
Настоящее изобретение относится к соединениям формулы 1, к соединениям формулы 5 и к их фармацевтически приемлемым солям. В формулах

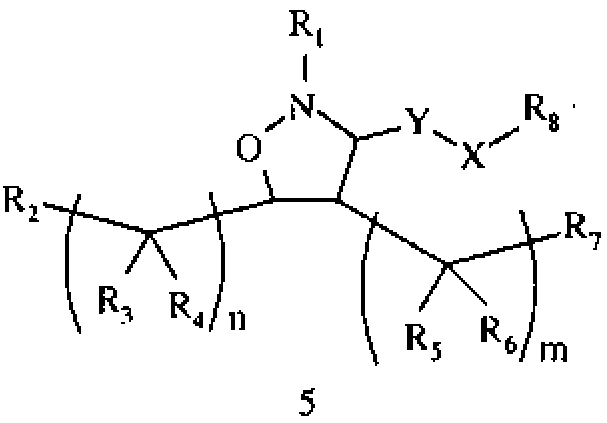
Y означает -С(О)-, X означает -N(R11)-, R1 означает остаток формулы 1а или 1b - для формулы 1 или остаток формул 5а или 5b - для формулы 5
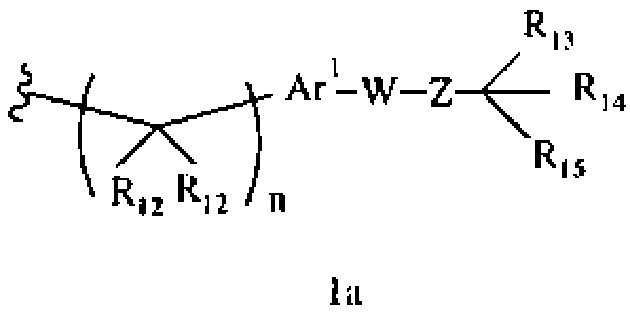
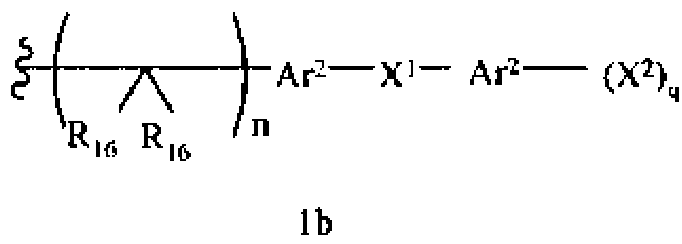
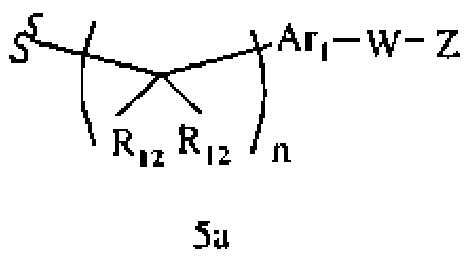
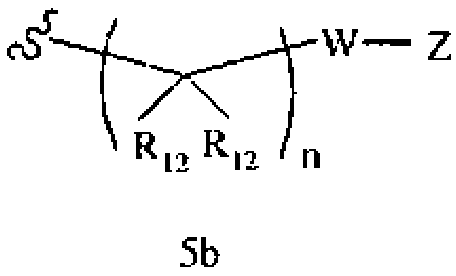
R2 и R7 независимо означают Н, гидроксил или (С1-С6)алкил; R3 и R6 каждый независимо означает Н, гидроксил или (С1-С6)алкил; R4 и R5 каждый независимо означает Н или (C1-С6)алкил; значения остальных радикалов указаны в формуле изобретения. Также изобретение относится к индивидуальным соединениям, представленным в формуле изобретения, к фармацевтической композиции, обладающей свойствами ингибировать апоптозную активность Bcl связанного белка, включающей терапевтически эффективное количество соединения изобретения, к способу лечения нарушения, опосредованного bcl, включающего введение терапевтически эффективного количества соединения изобретения, и к способу лечения нарушения, опосредованного bcl, включающего совместное введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества камптотецина и терапевтически эффективного количества соединения изобретения. 8 н. и 76 з.п. ф-лы, 15 табл., 1 ил.
1. Соединение формулы 1
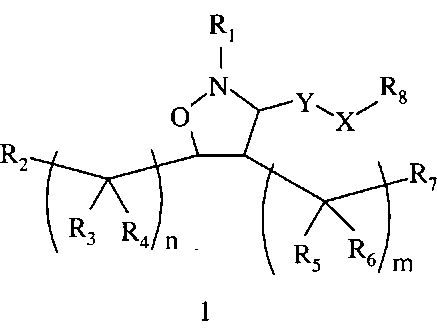
или его фармацевтически приемлемые соли, где
Y означает -С(О)-,
X означает -N(R11)-,
X′ каждый независимо означает О или N(R10) или S,
m равно 0, 1, 2, 3, 4, 5 или 6,
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6,
R1 означает остаток формулы 1а или 1b
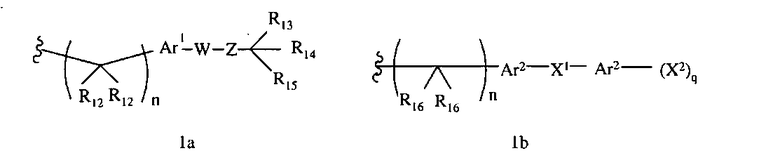
где R12 каждый независимо означает Н;
Ar1 означает моноциклический или бициклический (С6-С14)арил;
W означает двухвалентный (С2-С7)алкенил или (С2-С7)алкинил;
Z означает химическую связь, -(C(R12)2)n- или -X′(C(R12)2)n-;
R13 и R14 независимо означают Н, (С1-С6)алкил, (С5-С7)арил, (С3-С10)циклоалкил, (С5-С7)арил(С1-С6)алкил, (С5-С7)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, (С5-С7)гетероарил(С1-С6)алкил, где в гетероариле один, два или три кольцевых атома независимо представляют собой N, О или S, или -А1-А2-А3; или R13 и R14 вместе образуют (С5-С17)моноциклическое или полициклическое кольцо; или R13 и R14 вместе с R15 образуют (С5-С7)арильное кольцо или (С5-С7)гетероарильное кольцо, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
R15 означает галоген, гидроксил, (С5-С7)арил, -N(R10)2, (С1-С6)ациламино, -N(R10)SO2R19, -N(R10)C(X′)N(R19)2, -N(R10)(C(R9)2)n-A1-A2-A3 или -(С(R9)2)n-галоген; или R15 вместе с R13 и R14 образуют (С5-С7)арильное кольцо или (С5-С7)гетероарильное кольцо, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
R16 каждый независимо означает Н или (С1-С6)алкил;
Ar2 каждый независимо означает моноциклический или бициклический (С6-С14)арил; или моноциклический или бициклический (С5-С14)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
X1 каждый независимо означает химическую связь, О, (С2-С7)алкенильный бирадикал или (С2-С7)алкинильный бирадикал;
X2 каждый независимо означает Н, галоген, гидроксил, амино, (С1-С6)ациламино, нитро, карбоксамид, карбоксил, -CO2R10, -COR10, -N(R10)SO2R10 или -N(R10)C(X′)N(R10)2; и
q в каждом случае независимо равно 1, 2, 3, 4 или 5;
R2 и R7 независимо означают Н, гидроксил или (С1-С6)алкил;
R3 и R6 каждый независимо означает Н, гидроксил или (С1-С6)алкил;
R4 и R5 каждый независимо означает Н или (С1-С6)алкил;
R8 означает разветвленный или неразветвленный (С1-С10)алкил или (С2-С10)алкенил, (С3-С10)циклоалкил, (С7-С10)бициклоалкил замещенный (С1-С6)алкилом, (С3-С10)гетероциклоалкил замещенный (С5-С7)арил(С1-С6)алкилом, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, или остаток формулы 1с
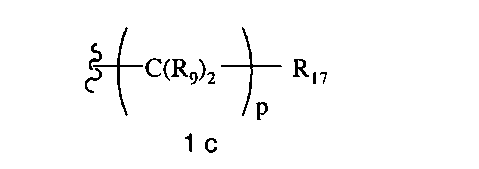
где р равно 0, 1, 2, 3, 4, 5 или 6; и
R17 означает (С5-С7)арил, (С5-С7)арил(С1-С6)алкил, (С3-С10)циклоалкил, (С3-С10)циклоалкенил, (С9-С10)бициклогетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, -N(R18)2 или -CO2R10;
где R18 каждый независимо означает (С1-С6)алкил, (С5-С7)арил, (С5-С7)арил(С1-С6)алкил или -CR9=CR9(C(R9)2)nCR9=C(R9)2;
R9 каждый независимо означает Н или (С1-С6)алкил,
R10 каждый независимо означает Н, (С1-С6)алкил, (С5-С7)арил, (С5-С7)арил(С1-С6)алкил или (С9-С10)бициклогетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
R11 каждый независимо означает Н или (С1-С6)алкил;
R19 каждый независимо означает Н, (С5-С7)арил, (С5-С7)арил(С1-С6)алкил, (С5-С7)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, (С7-С10)бициклогетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, или -А1-А2-А3;
А1 и А3 каждый независимо означает (С1-С6)алкил, (С3-С10)циклоалкил, (С5-С7)арил, (С5-С7)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, или (С7-С10)бициклогетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
А2 каждый независимо означает О или химическую связь; и
стереохимическая конфигурация в любом стереоцентре соединения формулы 1 означает R-, S-конфигурацию или смесь этих конфигураций.
2. Соединение по п.1, в котором Y означает -С(О)-, X означает -N(R11)-, R2 и R7 означают гидроксил, R6 означает метил, этил или пропил, a R3, R4 и R5 означают Н.
3. Соединение по п.1, в котором соединение имеет формулу 1d
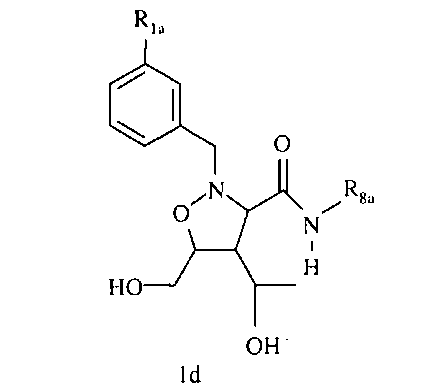
где R1a соответствует формулам 1е и 1f:
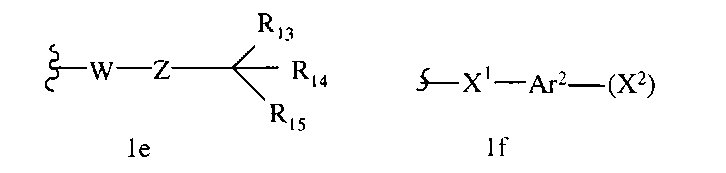
где W означает (С2-С7)алкенильную или (С2-С7)алкинильную цепь;
Z означает химическую связь, -(C(R12)2)n- или -O(C(R12)2)n-;
R13 и R14 независимо означают Н, (С1-С6)алкил, (С5-С7)арил, (С3-С10)циклоалкил, (С5-С7)арил(С1-С6)алкил, (С5-С7)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, (С5-С7)гетероарил(С1-С6)алкил, где в гетероариле один, два или три кольцевых атома независимо представляют собой N, О или S, или -А1-А2-А3; или R13 и R14 вместе образуют (С5-С17)моноциклическое или полициклическое кольцо; или R13 и R14 вместе с R15 образуют (С5-С7)арильное кольцо или (С5-С7)гетероарильное кольцо, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
R15 означает галоген, гидроксил, (С5-С7)арил, -N(R10)2, (С1-С6)ациламино, -N(R10)SO2R19, -N(R10)C(O)N(R19)2, -N(R10)(C(R9)2)n-A1-A2-A3 или -(С(R9)2)n-галоген; или R15 вместе с R13 и R14 образуют (С5-С7)арильное кольцо или (С5-С7)гетероарильное кольцо, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
Ar2 каждый независимо означает моноциклический или бициклический (С6-С14)арил; или моноциклический или бициклический (С5-С14)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
X1 каждый независимо означает химическую связь, (С2-С7)алкенильный бирадикал или (С2-С7)алкинильный бирадикал;
X2 каждый независимо означает Н, галоген, гидроксил, амино, нитро, карбоксамид, карбоксил, -COR10, -N(R10)SO2R10 или -N(R10)C(O)N(R10)2; и
q равно 1, 2, 3, 4 или 5;
R8a означает разветвленный или неразветвленный (С1-С10)алкил или (С2-С10)алкенил, (С3-С10)циклоалкил, (С7-С10)бициклоалкил замещенный (С1-С6)алкилом, (С3-С10)гетероциклоалкил, замещенный (С5-С7)арил(С1-С6)алкилом, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, или остаток формулы 1g
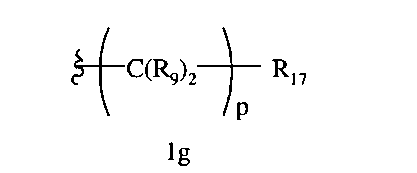
где р равно 0, 1, 2, 3, 4, 5 или 6; и
R17 означает (С5-С7)арил, (С5-С7)арил(С1-С6)алкил, (С3-С10)циклоалкил, (С3-С10)циклоалкенил, (С9-С10)бициклогетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, -N(R18)2 или -CO2R10;
где R18 каждый независимо означает (С1-С6)алкил, (С5-С7)арил, (С5-С7)арил(С1-С6)алкил или -CR9=CR9(C(R9)2)nCR9=C(R9)2.
4. Соединение по п.3, в котором
R13 и R14 независимо означают Н, (С1-С6)алкил, (С5-С7)арил или -А1-А2-А3; или R13 и R14 вместе образуют (С5-С17)моноциклическое или полициклическое кольцо; или R13 и R14 вместе с R15 образуют (С5-С7)гетероарильное кольцо, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
R15 означает галоген, гидроксил, (С5-С7)арил, -N(R10)2; (С1-С6)ациламино, (С5-С7)арил(С1-С6)алкил, -N(R10)SO2R19 или -N(R10)C(O)N(R19)2.
5. Соединение по п.3, в котором
R8a означает (С7-С10)бициклоалкил замещенный (С1-С6)алкилом, (С3-С10)гетероциклоалкил, замещенный (С5-С7)арил(С1-С6)алкилом, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, или остаток формулы 1g
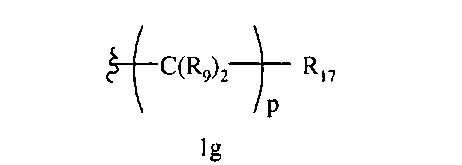
где р равно 0, 1, 2, 3, 4, 5 или 6; и
R17 означает (С5-С7)арил, (С5-С7)арил(С1-С6)алкил, (С3-С10)циклоалкил, (С3-С10)циклоалкенил, (С9-С10)бициклогетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, или -N(R18)2;
где R18 каждый независимо означает (С1-С6)алкил, (С5-С7)арил или (С5-С7)арил(С1-С6)алкил.
6. Соединение по п.3, в котором где W означает (С2-С7)алкинильную цепь, a Z означает химическую связь.
7. Соединение по п.3, в котором R13 и R14 означают Н, a R15 означает (С1-С6)ациламино.
8. Соединение по п.3, в котором R13 и R14 вместе образуют циклогексил, a R15 означает аминогруппу.
9. Соединение по п.3, в котором R8a означает (С7-С10)бициклоалкил замещенный (C1-С6)алкилом.
10. Соединение по п.3, в котором R8a означает остаток формулы Ig, a R17 означает N(CH3)Ph.
11. Соединение по п.3, в котором R1a означает остатки формул

или  .
.
12. Соединение по п.3, в котором R8a означает остатки формул
 или
или 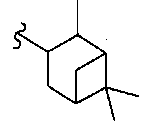 .
.
13. Соединение по п.3, в котором
R8a означает 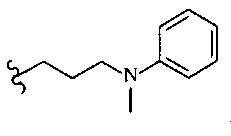 , а R1a означает:
, а R1a означает: 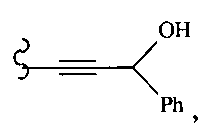
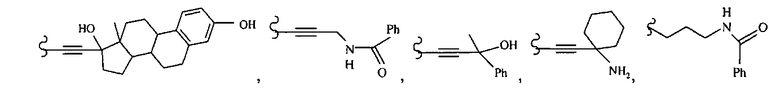 или
или 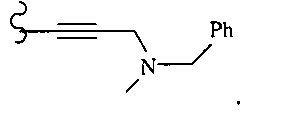
14. Соединение по п.3, в котором R1a означает 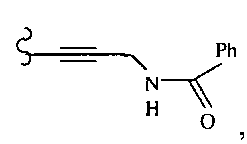 а R8a означает:
а R8a означает: 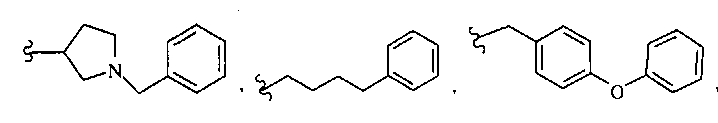 или
или 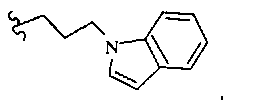
15. Соединение по п.3, в котором R8a означает 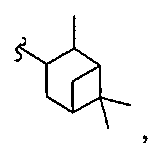 а R1a означает:
а R1a означает: 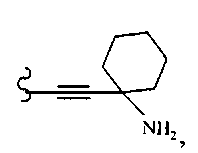
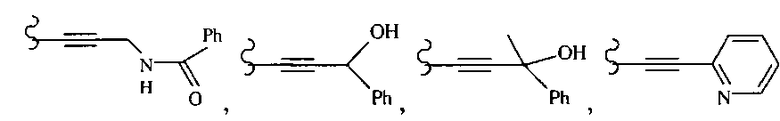 или
или 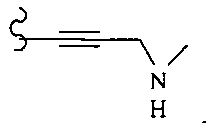
16. Соединение по п.3, в котором R1a означает 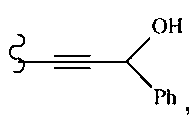 , а R8a означает:
, а R8a означает: 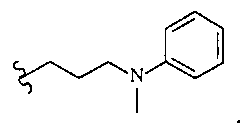
17. Соединение по п.3, в котором R1a означает 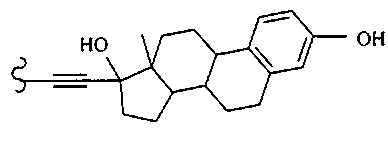 , а R8a означает
, а R8a означает 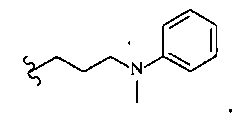
18. Соединение по п.3, в котором R1a означает 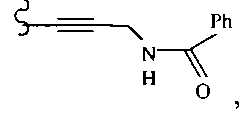 а R8a означает
а R8a означает 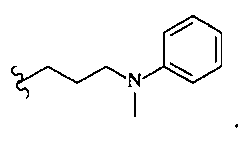
19. Соединение по п.3, где R1a означает 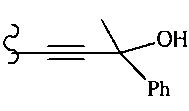 , R8a означает
, R8a означает 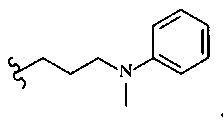 .
.
20. Соединение по п.3, в котором R1a означает 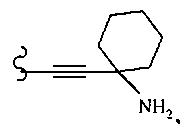 , а R8a означает
, а R8a означает 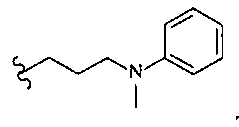
21. Соединение по п.3, в котором R1a означает 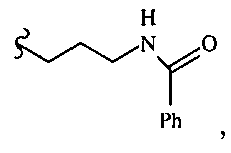 a R8a означает
a R8a означает 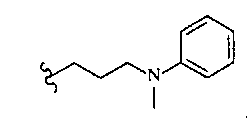 .
.
22. Соединение по п.3, в котором R1a означает 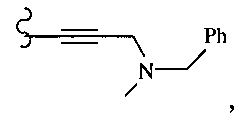 a R8a означает .
a R8a означает .
23. Соединение по п.3, в котором R1a означает 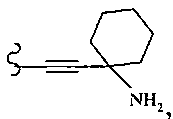 a R8a означает
a R8a означает 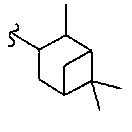 .
.
24. Соединение по п.3, в котором R1a означает 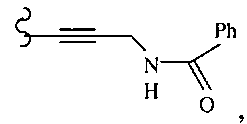 a R8a означает .
a R8a означает .
25. Соединение по п.3, в котором R1a означает 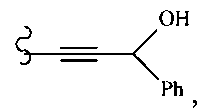 a R8a означает .
a R8a означает .
26. Соединение по п.3, в котором R1a означает 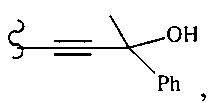 a R8a означает
a R8a означает 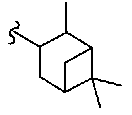 .
.
27. Соединение по п.3, в котором R1a означает 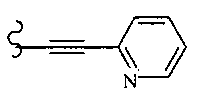 , а R8a означает
, а R8a означает 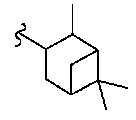 .
.
28. Соединение по п.3, в котором R1a означает 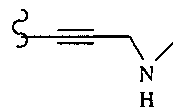 , a R8a означает
, a R8a означает 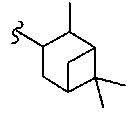 .
.
29. Соединение по п.3, в котором R1a означает 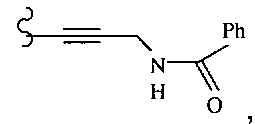 a R8a означает
a R8a означает 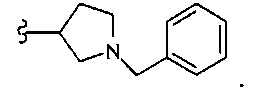
30. Соединение по п.3, в котором R1a означает 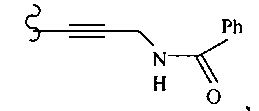 a R8a означает
a R8a означает 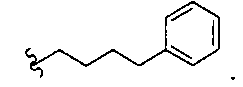
31. Соединение по п.3, в котором R1a означает 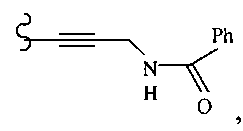 a R8a означает
a R8a означает  .
.
32. Соединение по п.3, в котором R1a означает  a R8a означает
a R8a означает  .
.
33. Соединение, выбранное из группы включающей:
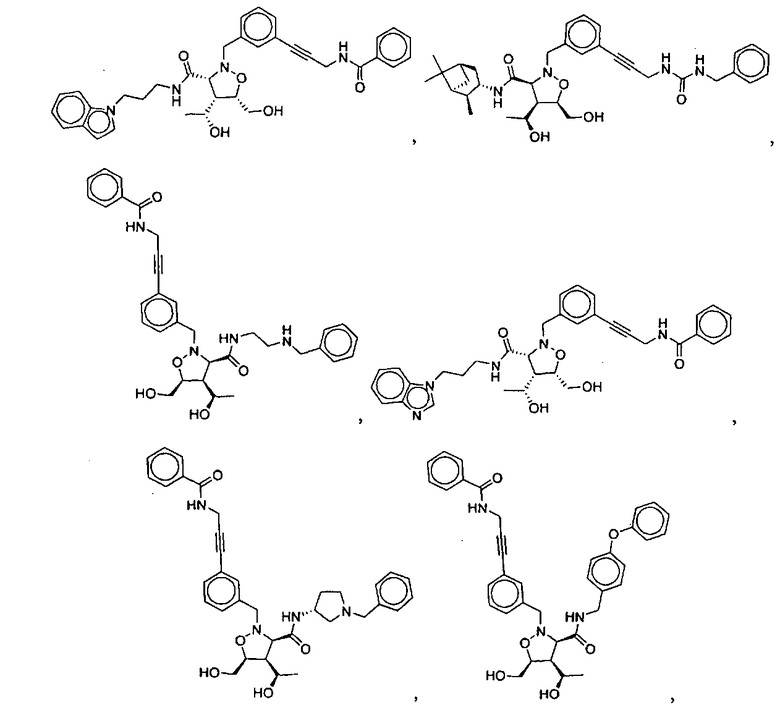
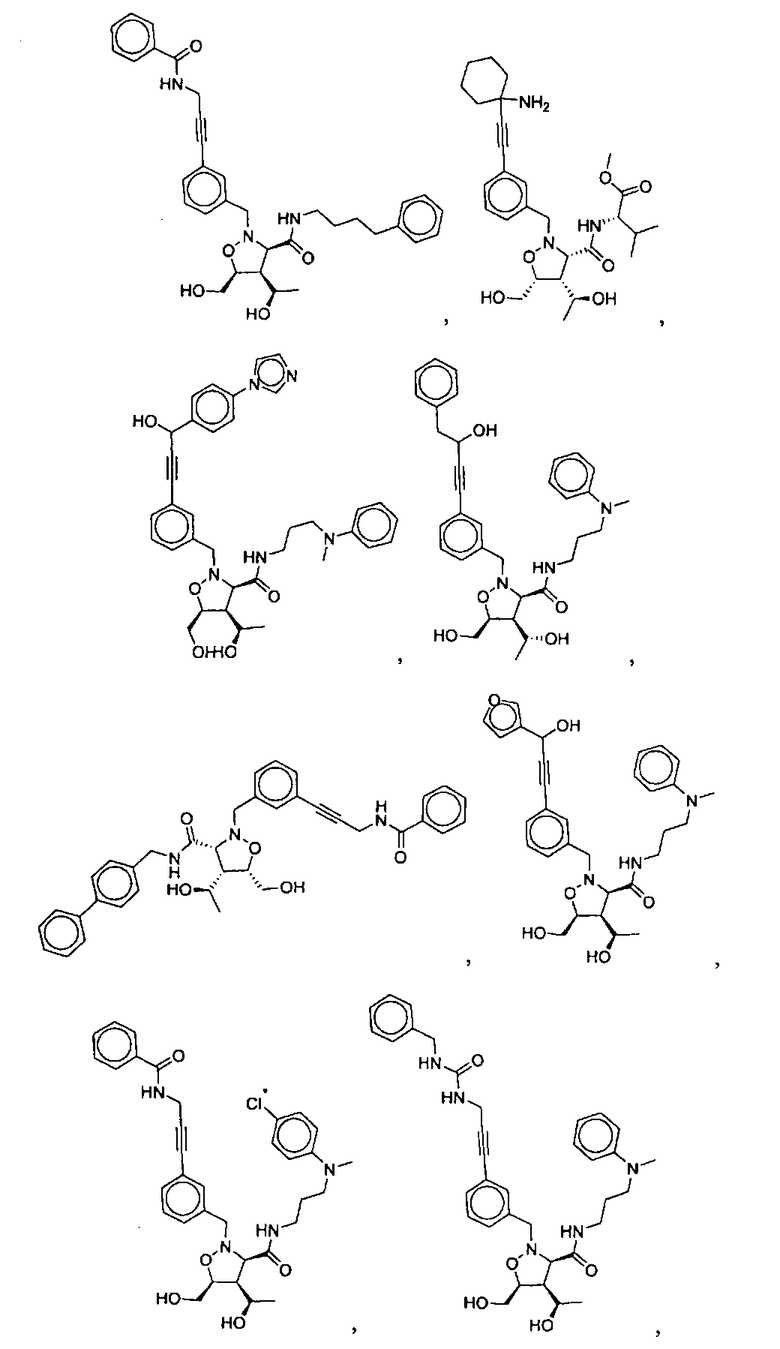
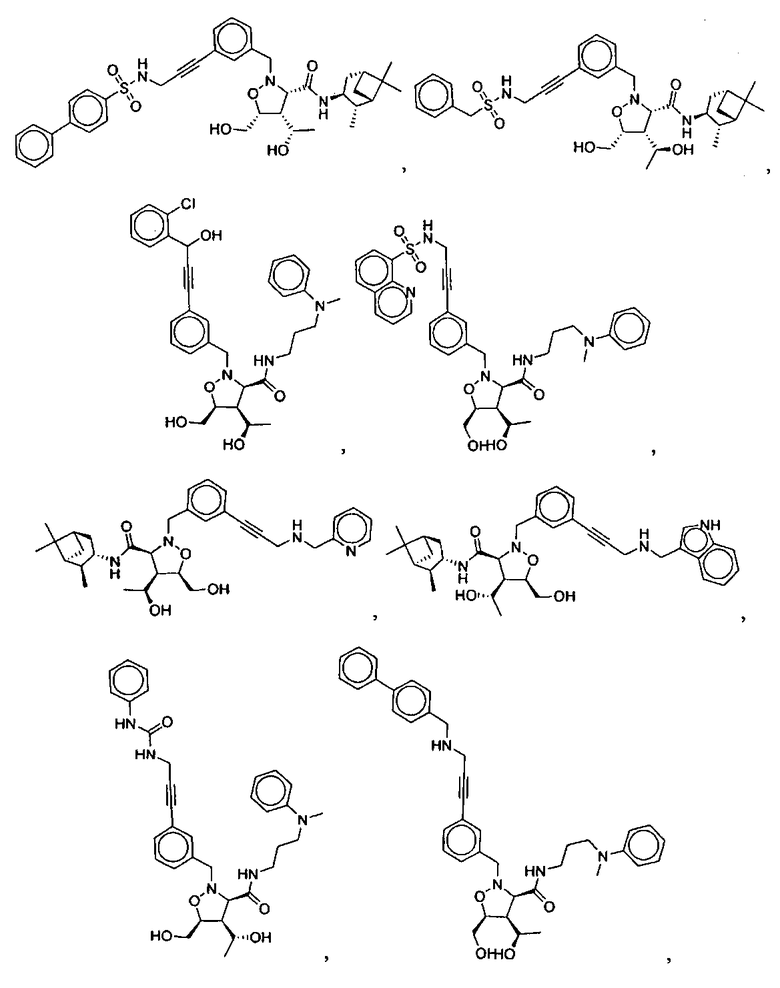
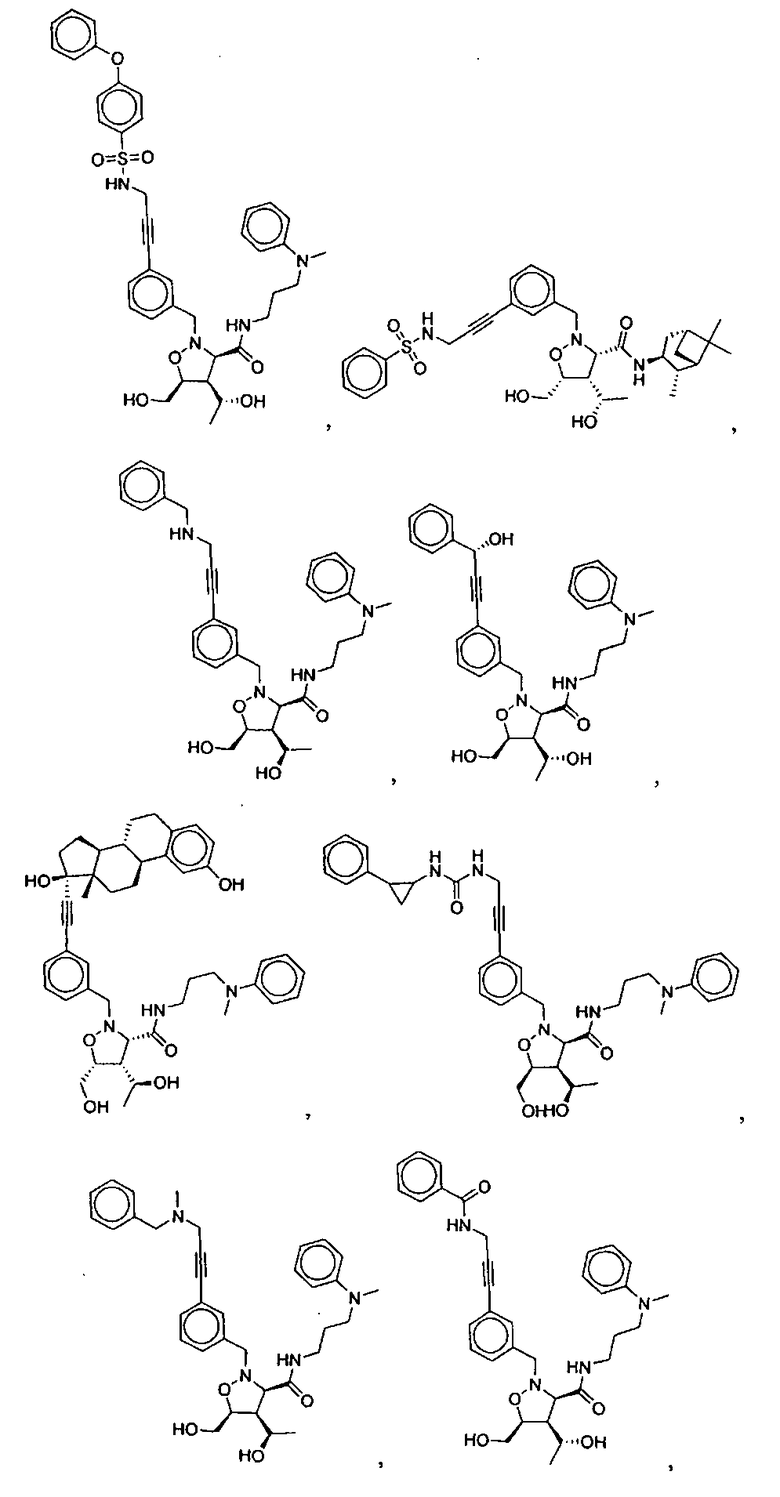
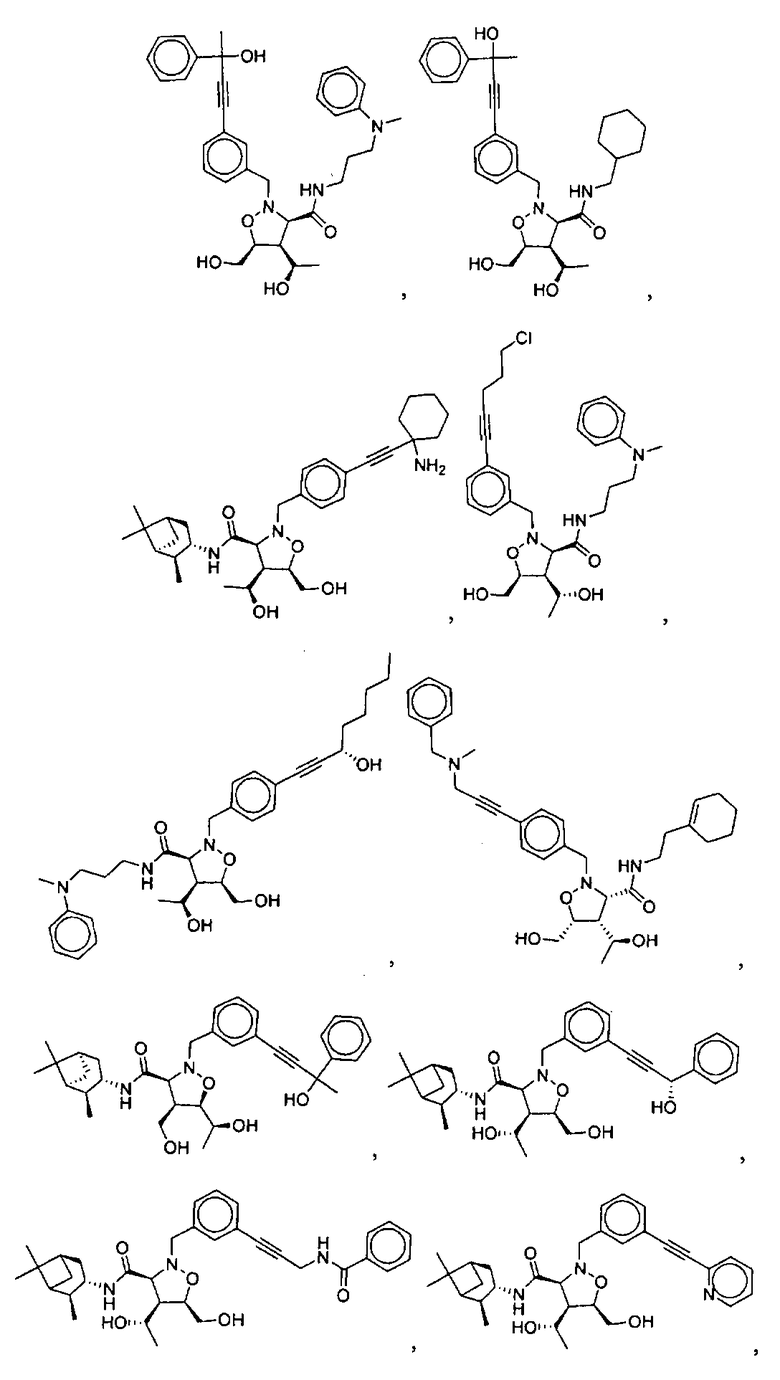
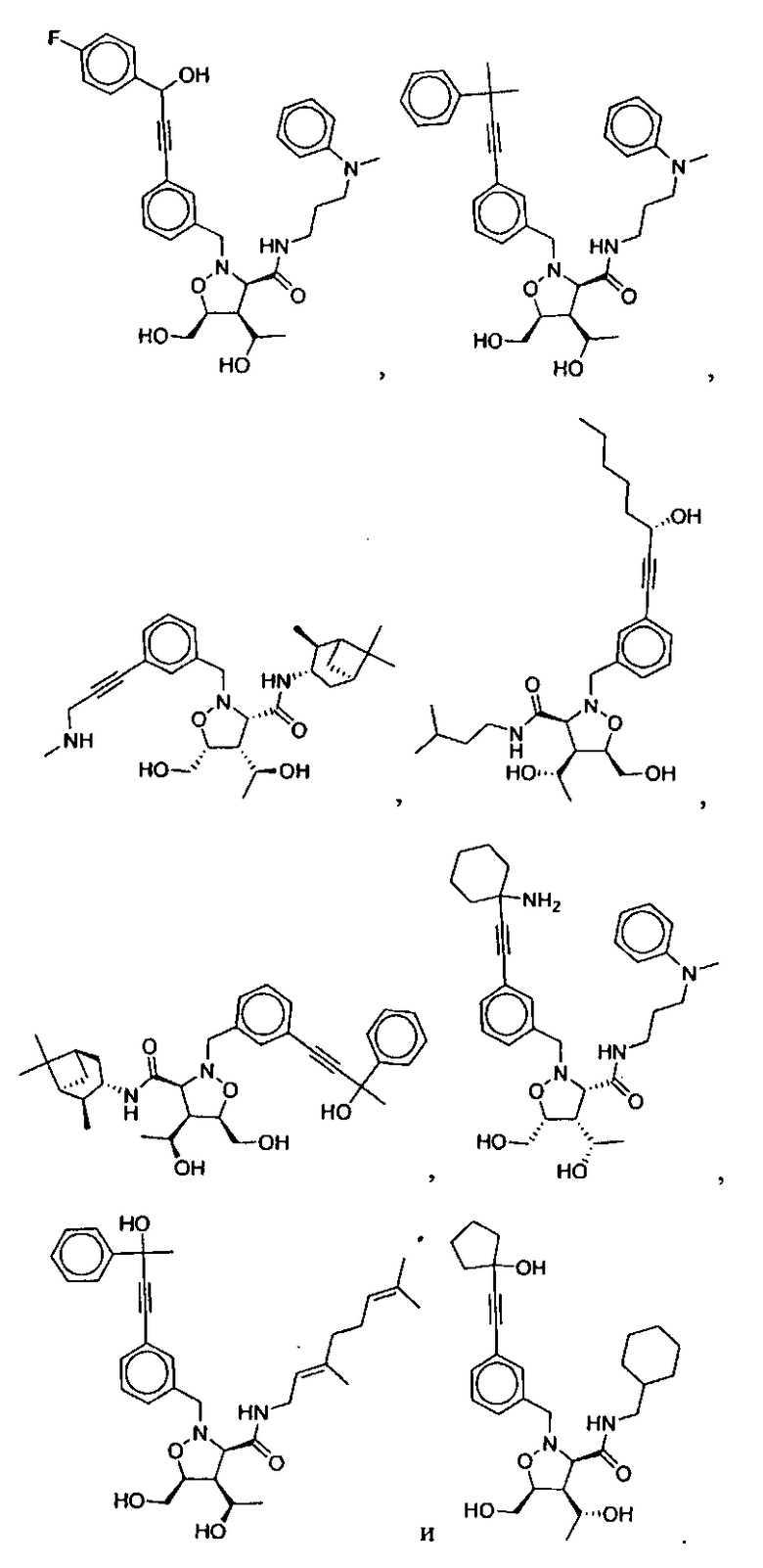
34. Соединение по п.1, где
Z означает химическую связь, -(C(R12)2)n- или -O(C(R12)2)n-;
R15 означает галоген, гидроксил, (С5-С7)арил, -N(R10)2, (С1-С6)ациламино, -N(R10)SO2R19, -N(R10)C(O)N(R19)2, -N(R10)(C(R9)2)n-A1-A2-A3 или -(C(R9)2)n-галоген; или R15 вместе с R13 и R14 образуют (С5-С7)гетероарильное кольцо, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
X1 означает химическую связь;
X2 каждый независимо означает Н, галоген, гидроксил или амино;
q равно 1 или 2; и
R3 и R6 каждый независимо означает Н или (С1-С6)алкил.
35. Соединение по п.34, в котором R2 и R7 означают гидроксил.
36. Соединение по п.34, в котором R2 и R7 означают гидроксил, a R4, R5 и R6 означают Н.
37. Соединение по п.34, в котором R2 и R7 означают гидроксил, R4, R5 и R6 означают Н, а m и n равны 1.
38. Соединение по п.34, в котором R2 и R7 означают гидроксил, R4, R5 и R6 означают Н, m и n равны 1, a R3 означает метил.
39. Соединение формулы 5
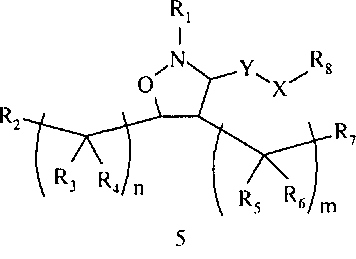
или его фармацевтически приемлемые соли,
где Y означает -С(О)-;
X означает -N(R11)-;
m равно 0, 1, 2, 3, 4, 5 или 6;
n каждое независимо равно 0, 1, 2, 3, 4, 5 или 6;
R1 означает остаток формул 5а или 5b
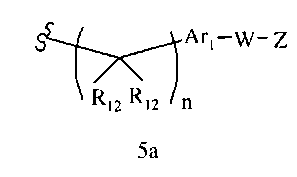 или
или 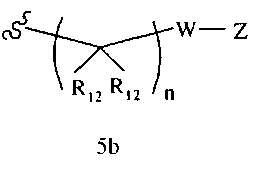
где R12 каждый независимо означает Н;
Ar1 означает моноциклический или бициклический (С6-С14)арил; или
Ar1 означает остаток формулы 5с
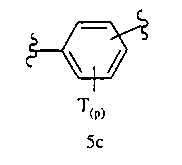
где Т каждый независимо означает Н, разветвленный или неразветвленный (С1-С6)алкил, (С3-С10)циклоалкил, (С3-С10)циклоалкен, (С7-С14)бициклоалкил, (С7-С14)бициклоалкен, (C1-С6)алкокси, (С3-С10)циклоалкил(С1-С6)алкокси, (С5-С7)арил, (С5-С7)арил(С1-С6)алкил, (С5-C7)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, (С3-С10)гетероциклил замещенный (С5-С7)арилом, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, (С3-С10)гетероциклоалкил, в котором один, два или три кольцевых атома независимо представляют собой N, О или S, бис-(С5-С7)арил, простой эфир бис-(С5-С7)арила, простой эфир (С5-С7)арил(С1-С6)алкила замешенный (С1-С6)алкокси; и
p равно 0, 1, 2, 3 или 4;
W означает химическую связь или двухвалентный (С1-С6)алкил, (С5-С7)арил или (С3-С10)гетероциклил, в котором один, два или три кольцевых атома независимо представляют собой N, O или S;
Z означает химическую связь, H, -S(O)2R, -N(R)2, -C(O)N(R)2, -C(S)N(R)2, -NRC(O)R, -NRC(O)(C(R9)2)nN(R)2; или моно- или бициклический (С6-С14)арил или моно- или бициклический (С5-С14)гетероарил, в котором один, два или три кольцевых атома независимо представляют собой N, O или S;
где R каждый независимо означает H, разветвленный или неразветвленный (С1-С6)алкил, (С1-С6)алкокси, (С5-С7)арил(С1-С6)алкил или -(C(R9)2)nT;
R2 и R7 каждый независимо означает Н, гидроксил или (С1-С6)алкил;
R3 и R6 каждый независимо означает Н, гидроксил или (С1-С6)алкил;
R4 и R5 каждый независимо означает Н или (С1-С6)алкил; и
R8 означает разветвленный или неразветвленный амино(С1-С10)алкил, (С7-С10)бициклоалкил замещенный (С1-С6)алкилом, или (С3-С10)гетероциклоалкил замещенный (С5-С7)арил(С1-С6)алкилом, в котором один, два или три кольцевых атома независимо представляют собой N, О или S;
R9 и R11 каждый независимо означает Н или (С1-С6)алкил;
при условии, что Ar1, W и Z могут быть дополнительно замещены одной или более группами, выбранными из галогена, (С5-С7)арила, (С1-С6)алкиламино, (С1-С6)алкокси, -NO2, гидроксила, -NR2 или -CN; и где Ar1, W и Z могут быть связаны друг с другом в орто-, мета- или пара-положении; и
стереохимическая конфигурация в любом стереоцентре соединения формулы 5 означает R-, S-конфигурацию или смесь этих конфигураций.
40. Соединение по п.39, в котором R2 означает OH.
41. Соединение по п.39, в котором R6 означает метил или этил, а R7 означает гидроксил.
42. Соединение по п.39, в котором Y означает -C(O)-, X означает -N(R11)-, а R8 означает (С7-С10)бициклоалкил замещенный (С1-С6)алкилом.
43. Соединение по п.39, в котором R1 означает остаток формулы 5а.
44. Соединение по п.39, в котором R1 означает остаток формулы 5а, a R12 означает Н или метил.
45. Соединение по п.39, в котором R1 означает остаток формулы 5а, а Ar1 означает бензольное кольцо.
46. Соединение по п.39, в котором R1 означает остаток формулы 5а, a W означает химическую связь, -СН2- или бензольное кольцо.
47. Соединение по п.39, в котором R1 означает остаток формулы 5b, a R12 означает Н или метил.
48. Соединение по п.39, в котором R1 означает остаток формулы 5b, где n равно 4.
49. Соединение по п.39, в котором R1 означает остаток формулы 5b, a Z означает N(R)2.
50. Соединение по п.39, где соединение имеет формулу 5d или 5е
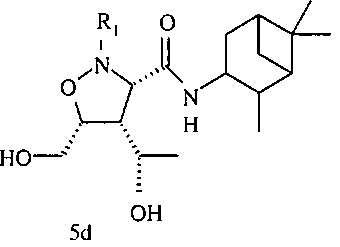 или
или 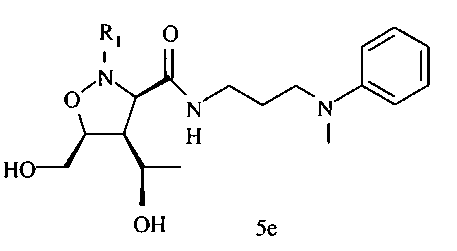 ,
,
где R1 имеет формулу 5f
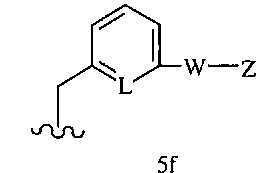
где L означает CR.
51. Соединение по п.39, в котором L означает СН, W означает бензольное кольцо, a Z означает -C(O)N(R)2.
52. Соединение по п.39, в котором L означает CR, R означает (C1-C6)алкокси, W означает бензольное кольцо, a Z означает -C(O)N(R)2.
53. Соединение по п.39, в котором L означает СОМе, W означает бензольное кольцо, a Z означает -C(O)N(R)2.
54. Соединение по п.39, в котором L означает COEt, W означает бензольное кольцо, a Z означает -C(O)N(R)2.
55. Соединение по п.39, в котором L означает СОСН2(циклопропил), W означает бензольное кольцо, a Z означает -C(O)N(R)2.
56. Соединение по п.39, в котором L означает СН, W означает бензольное кольцо, a Z означает Н.
57. Соединение по п.39, в котором L означает СН, W означает -СН2-, a Z означает -N(R)2.
58. Соединение по п.39, в котором L означает СОМе, W означает -СН2-, a Z означает -N(R)2.
59. Соединение по п.39, в котором L означает СН, W означает пиперазин, a Z означает -C(S)N(R)2.
60. Соединение по п.39, в котором L означает СН, W означает пиперазин, a Z означает -C(O)N(R)2.
61. Соединение по п.39, в котором L означает СН, W означает химическую связь, a Z означает N(R)2.
62. Соединение по п.39, в котором L означает СН, W означает химическую связь, a Z означает -C(O)N(R)2.
63. Соединение по п.39, в котором L означает СН, W означает химическую связь, a Z означает -NRC(O)(C(R9)2)nN(R)2.
64. Соединение формулы
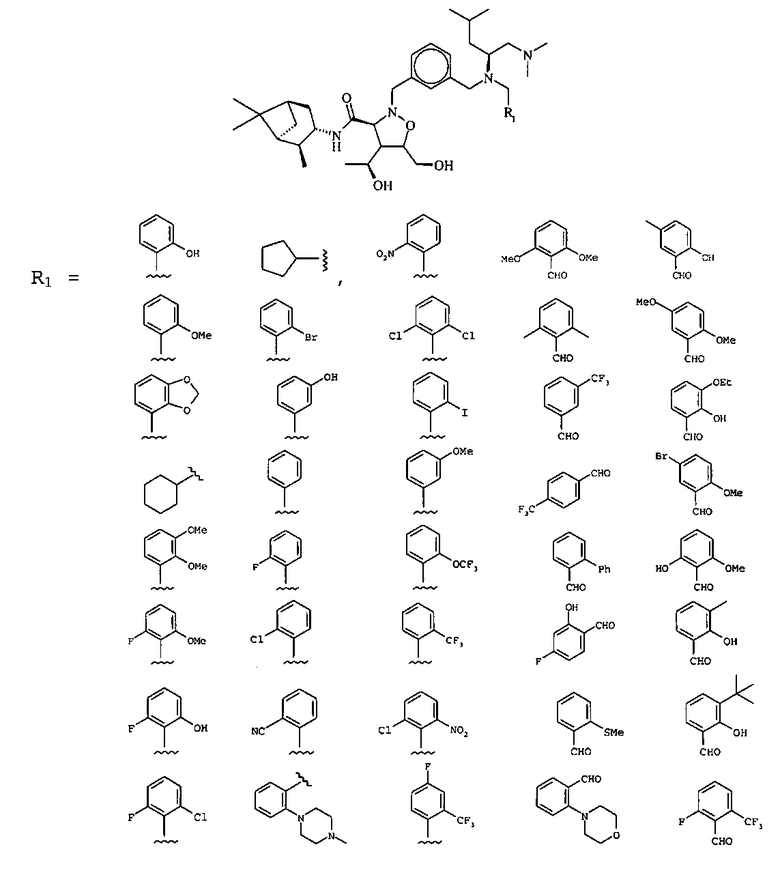
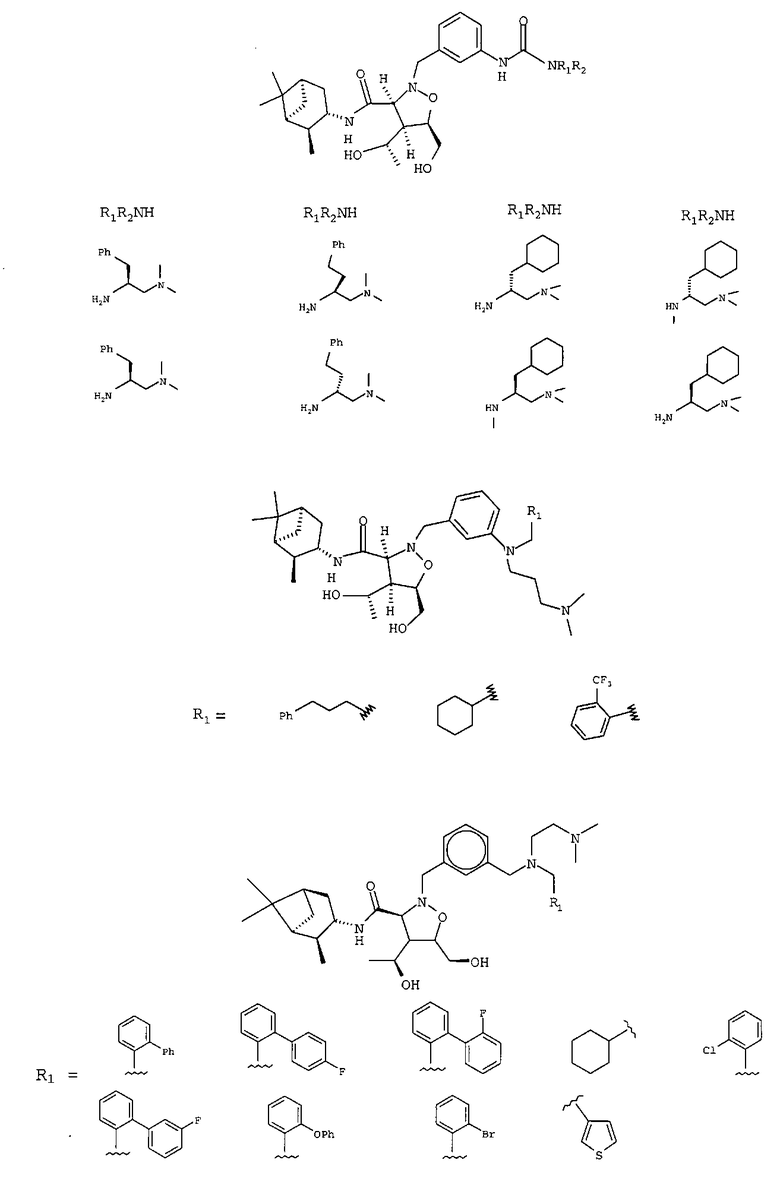
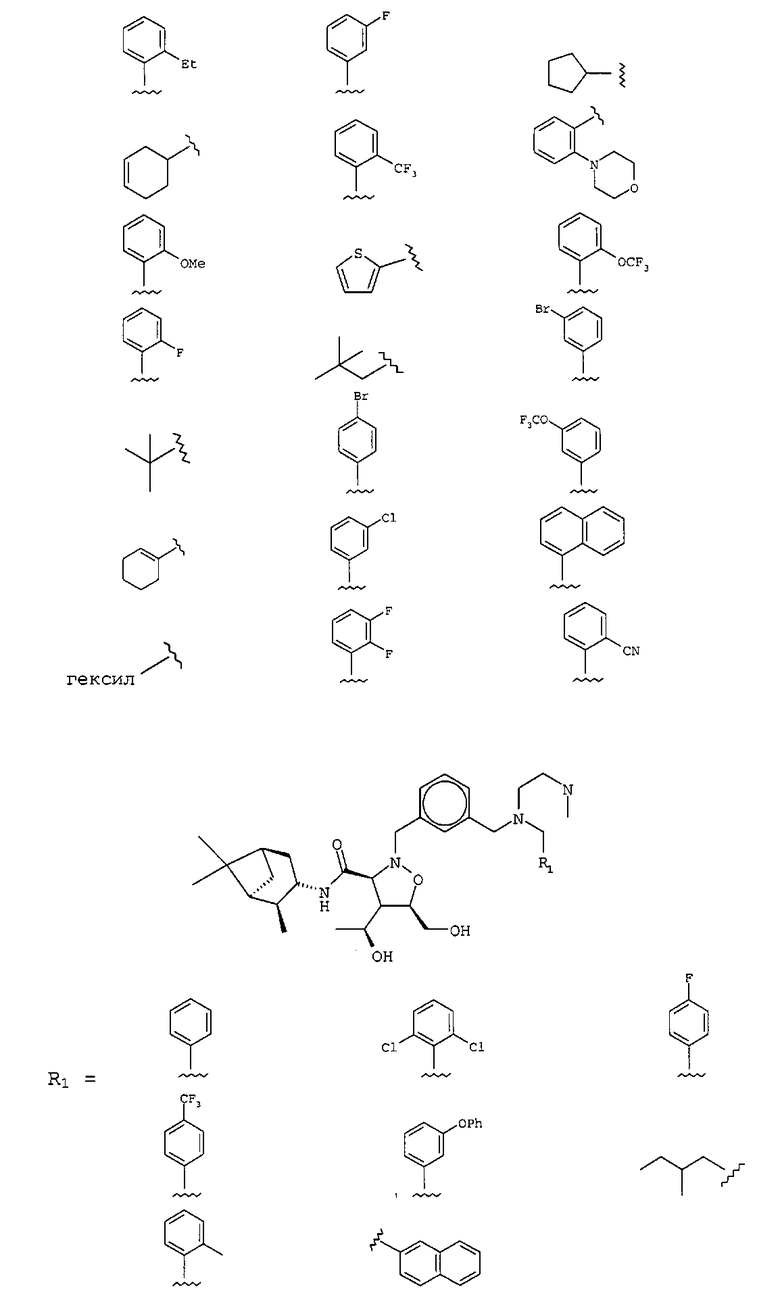
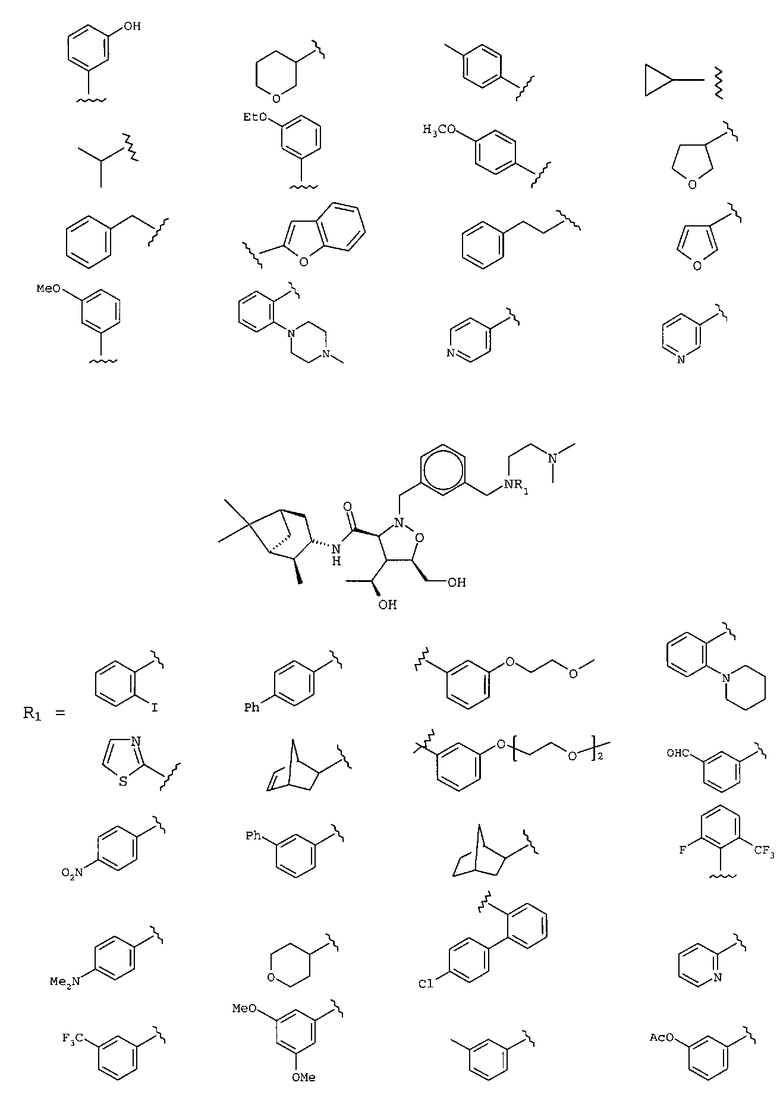
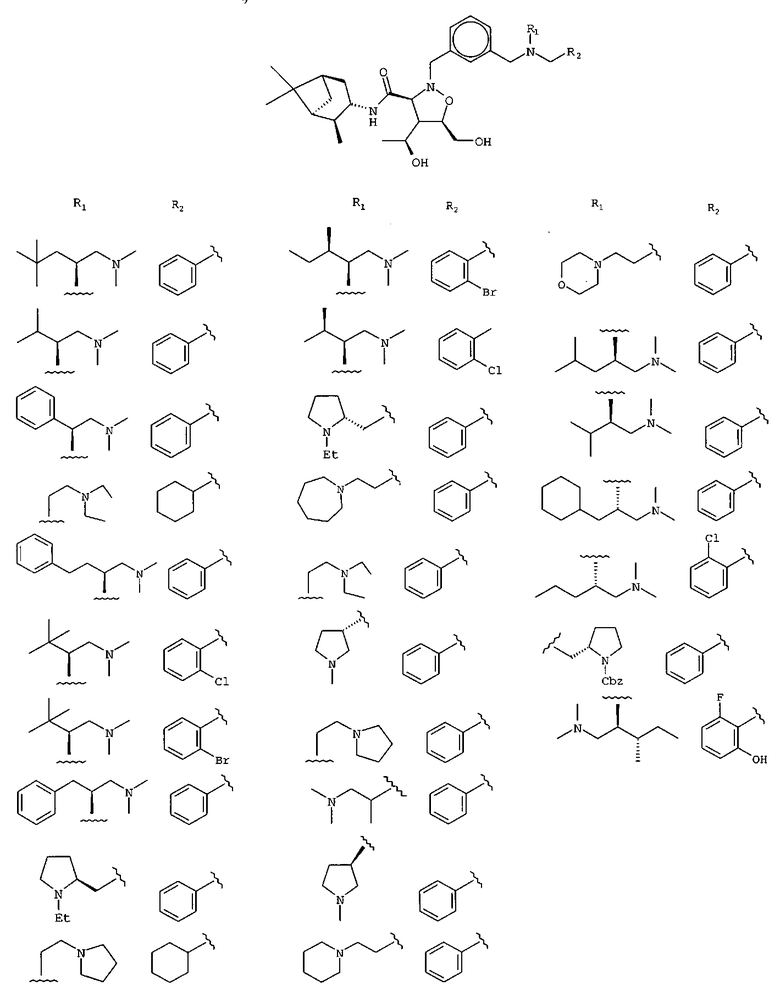
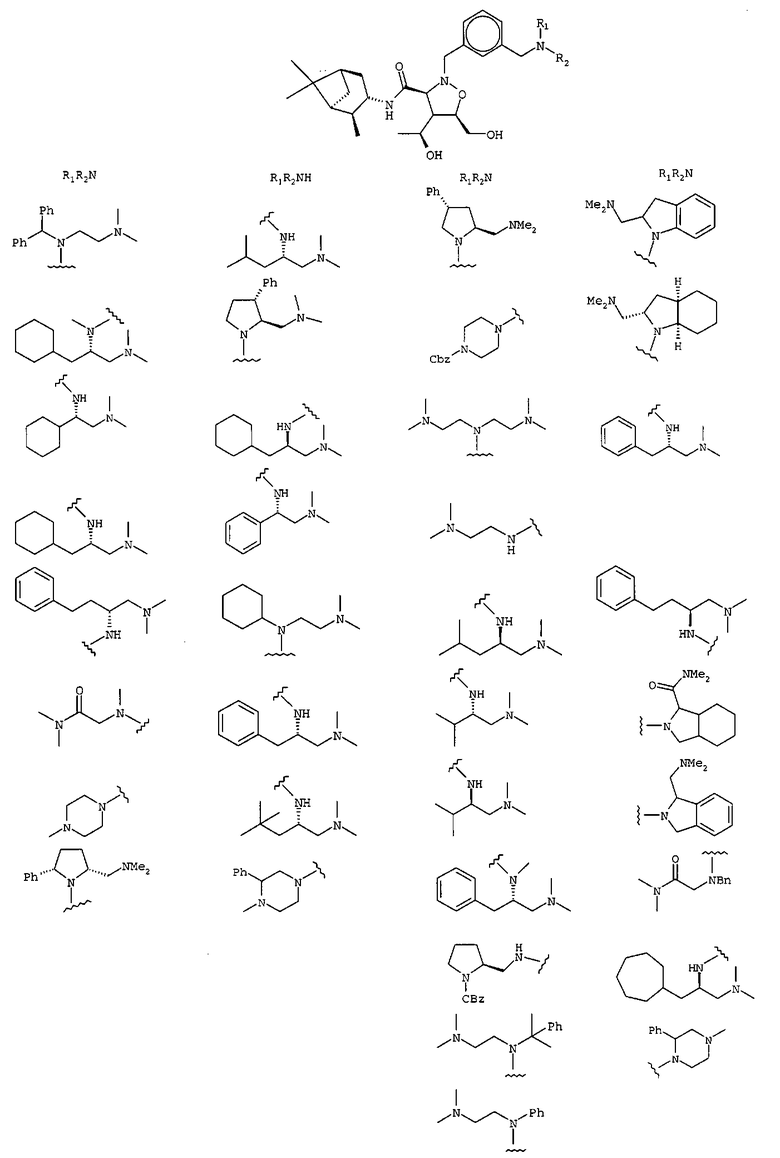
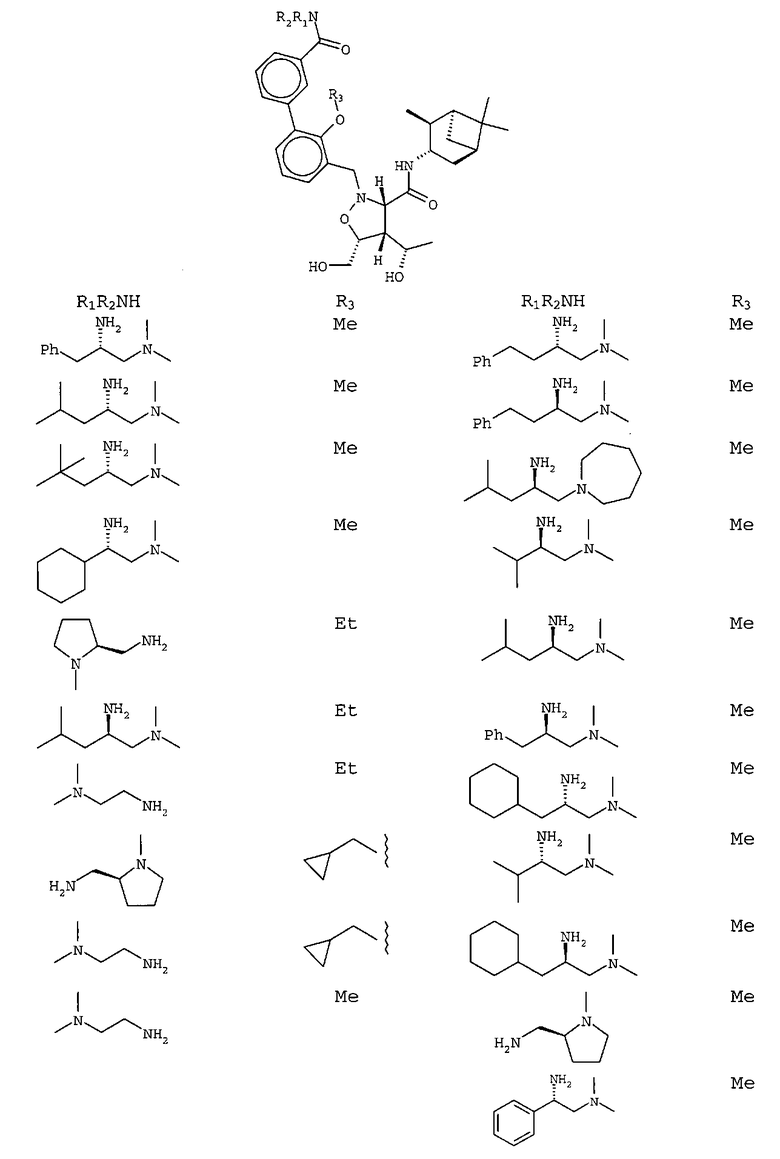
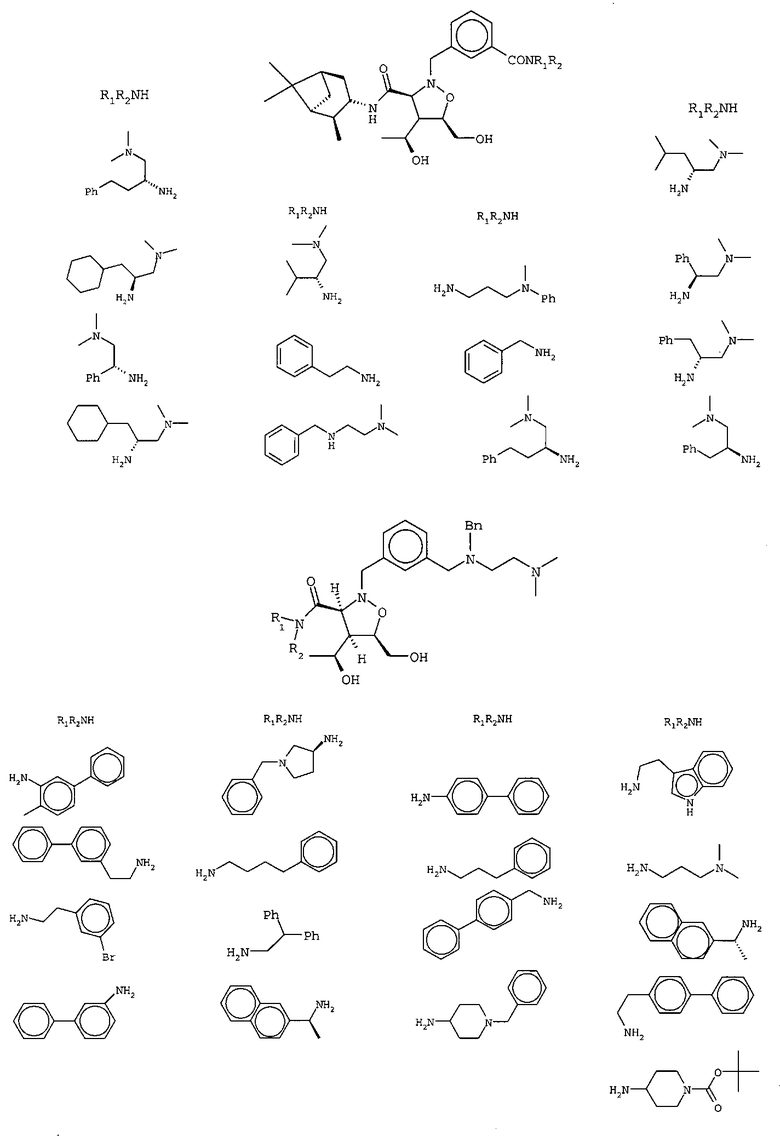
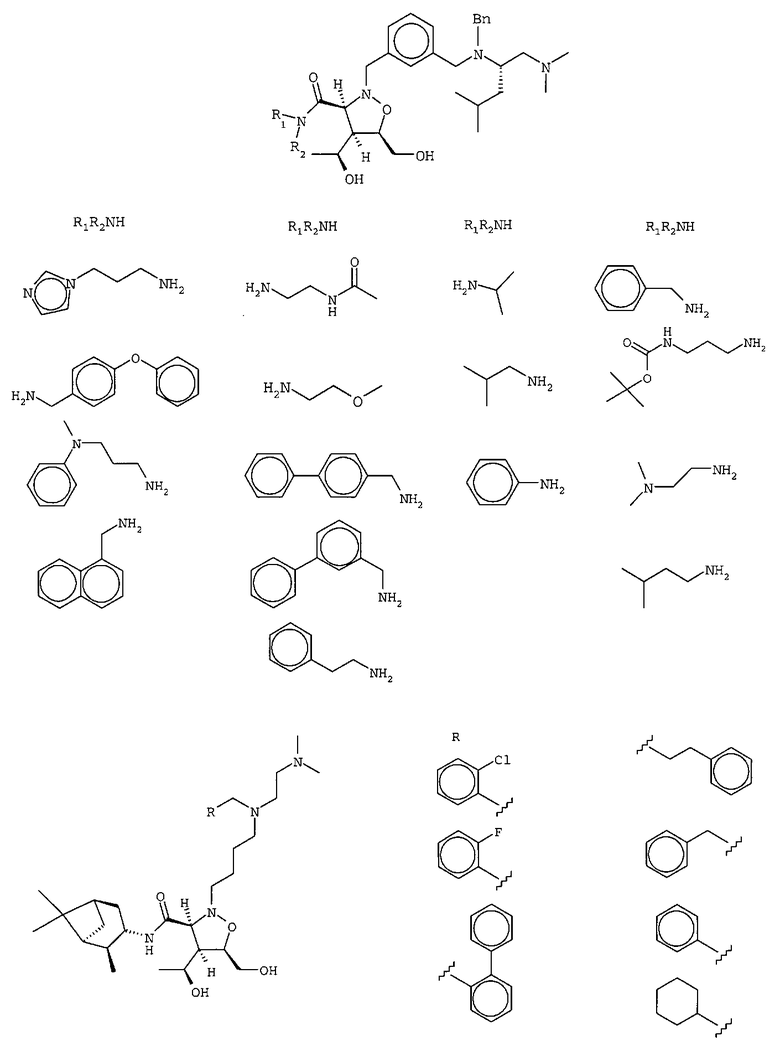
65. Соединение, которое выбирают из следующих соединений:
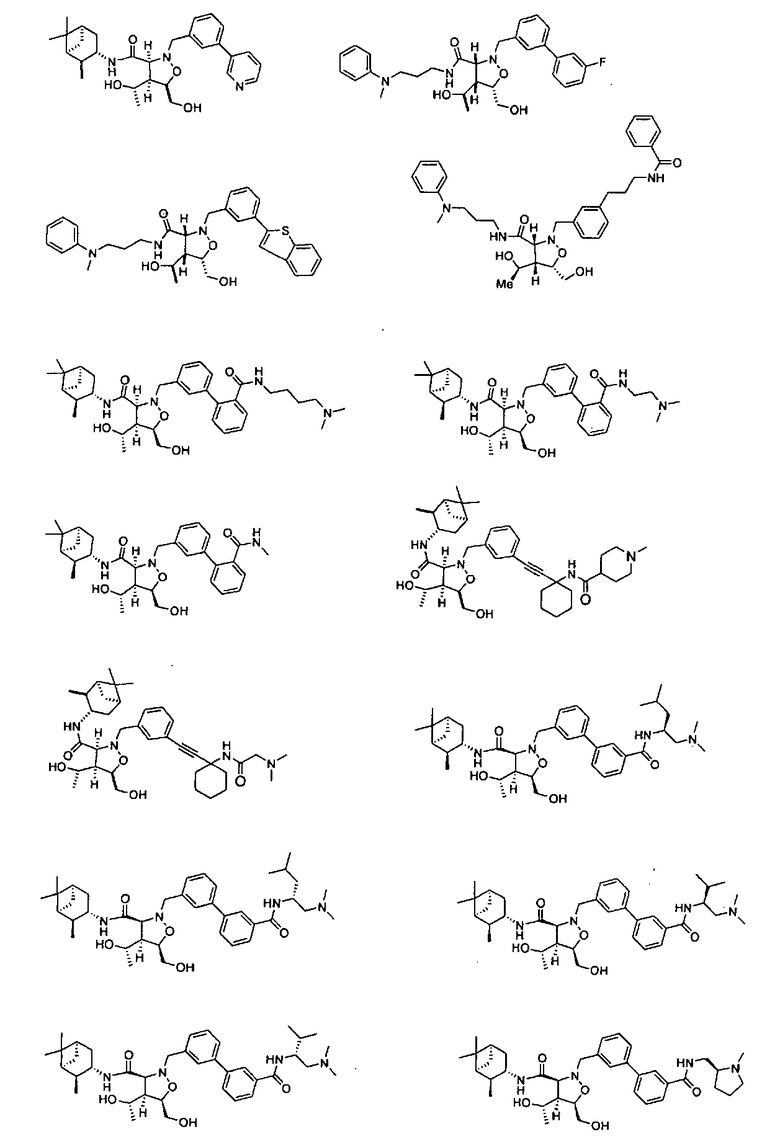
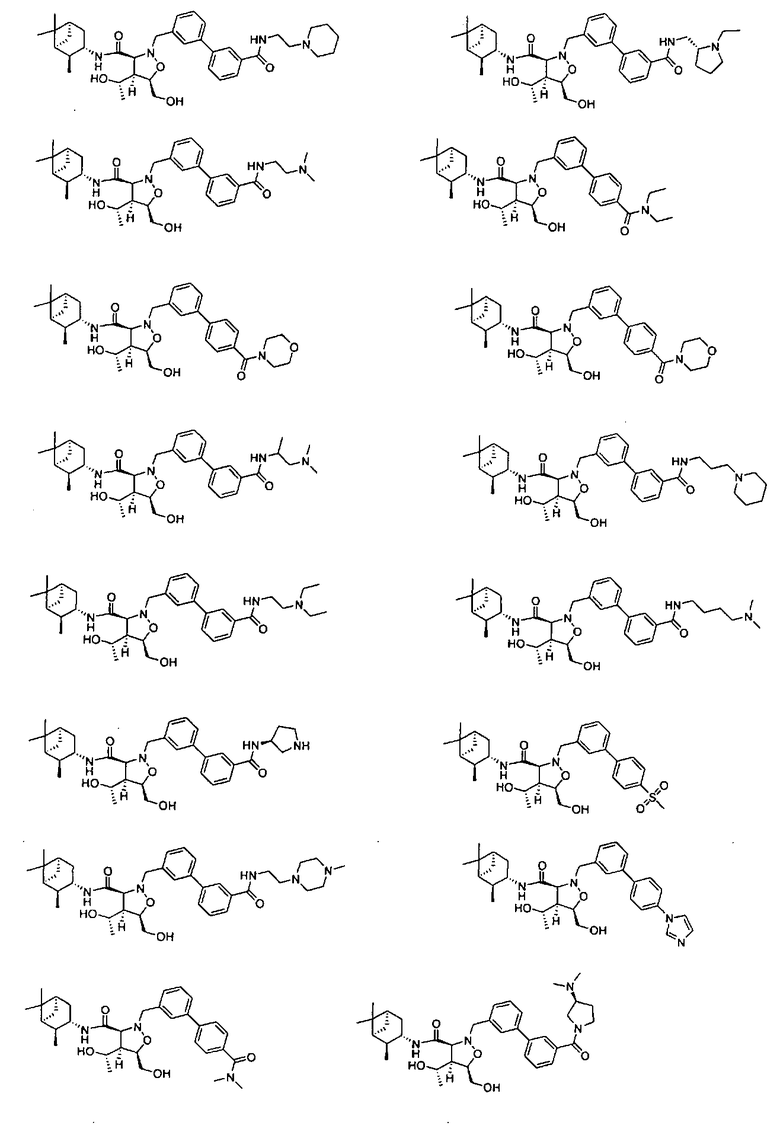
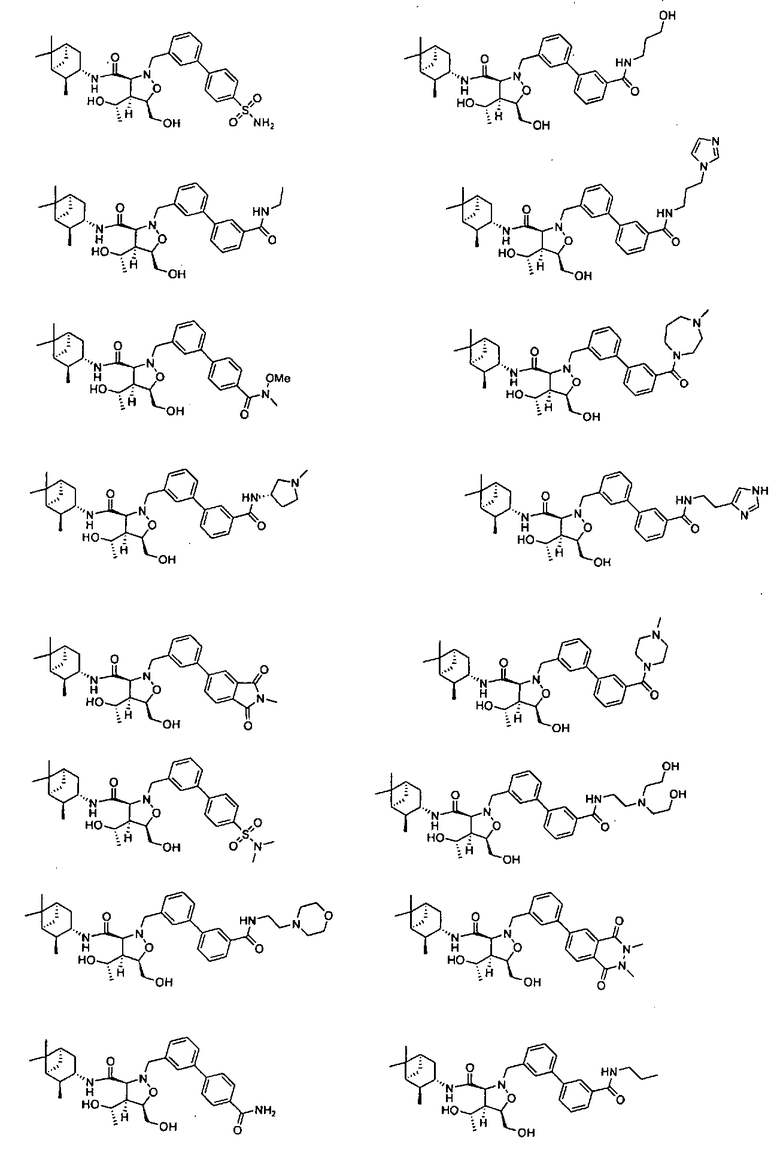
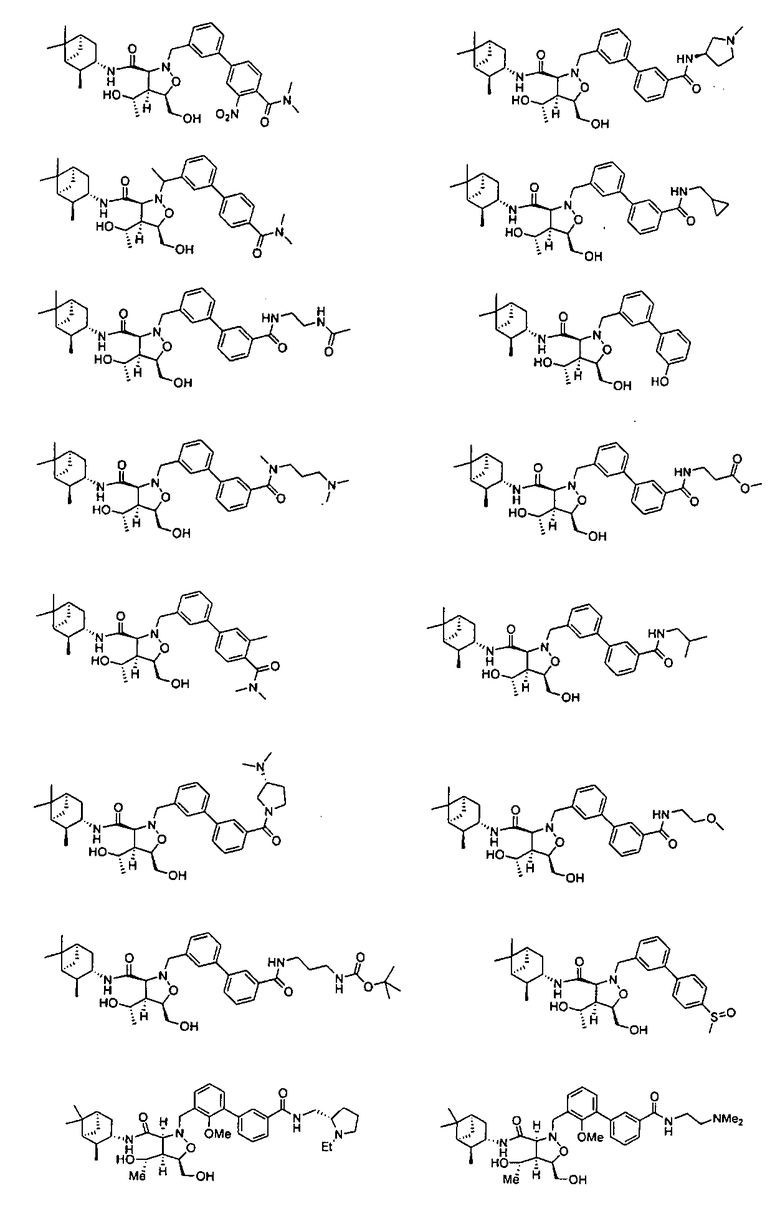
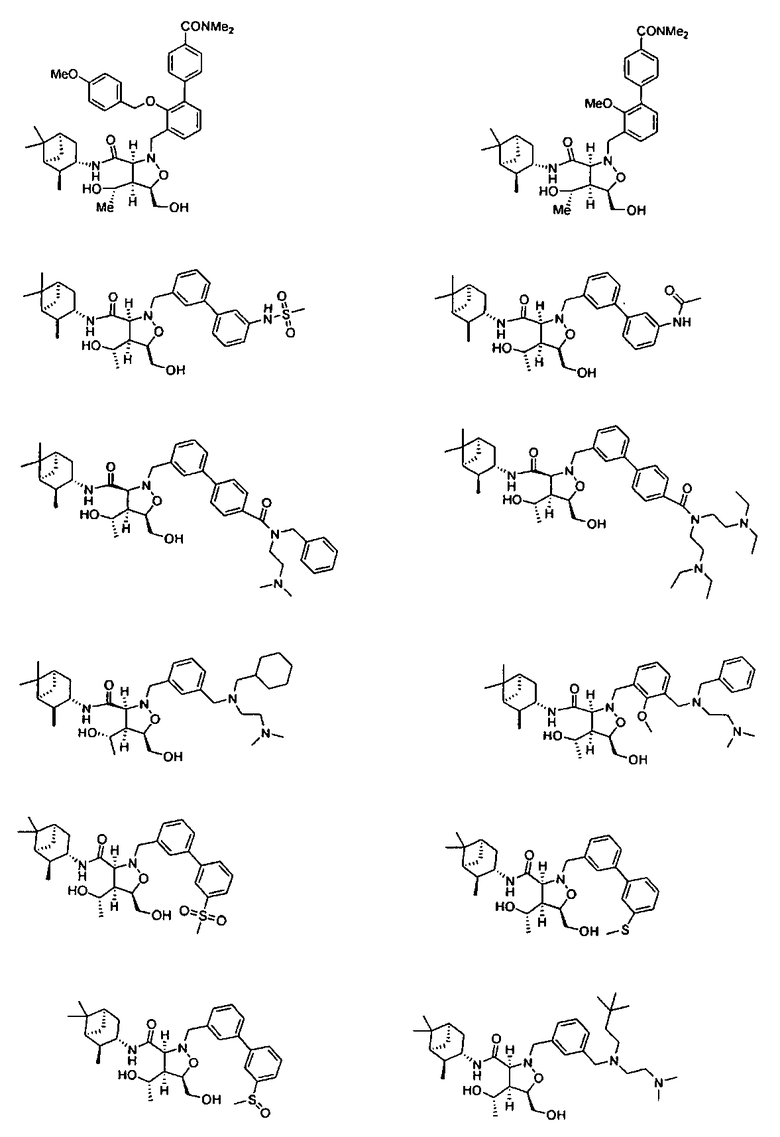
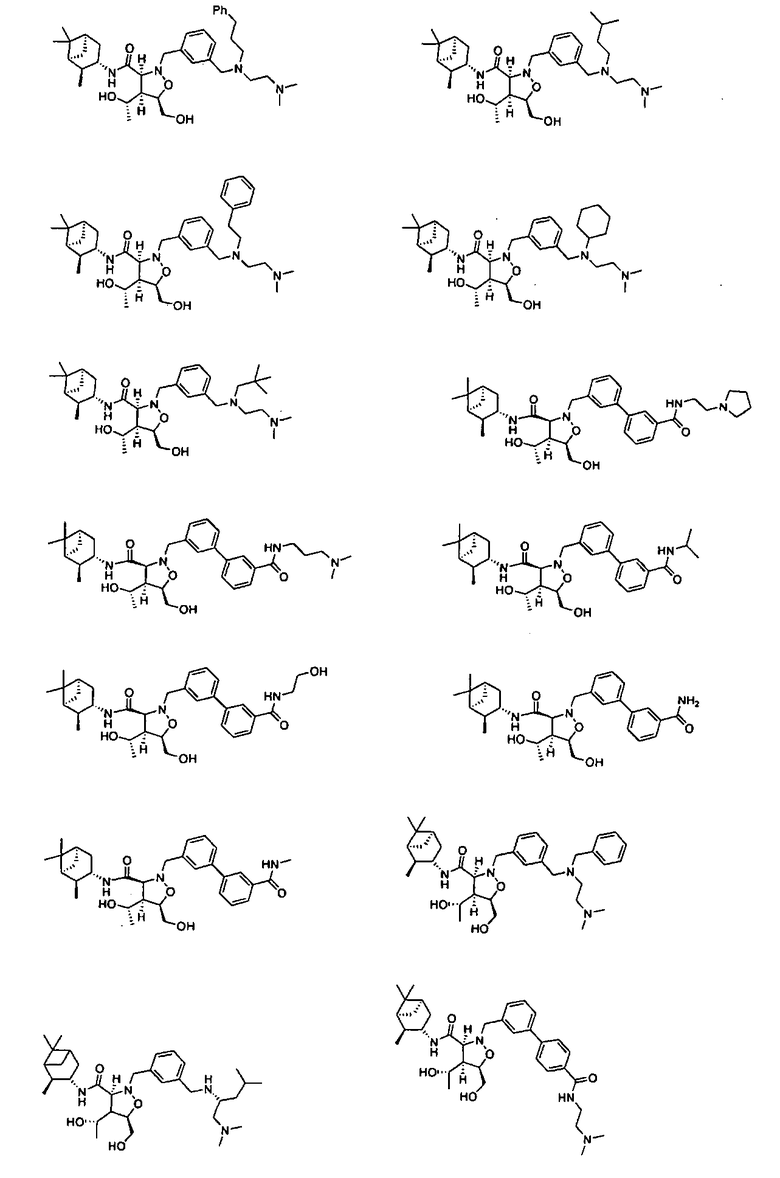
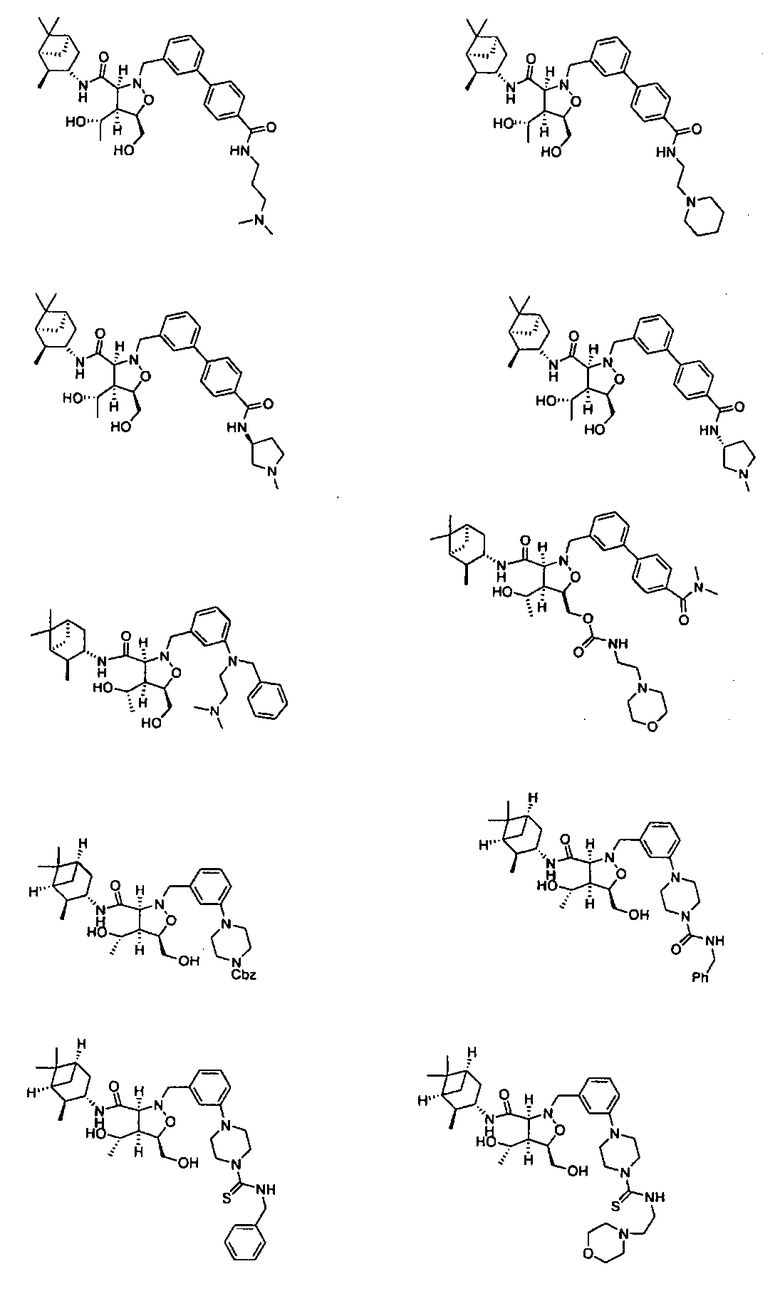
66. Фармацевтическая композиция, обладающая свойствами ингибировать апоптозную активность Bcl связанного белка, включающая терапевтически эффективное количество соединения по любому из пп.1-65 и по меньшей мере один фармацевтически приемлемый эксиципиент.
67. Способ лечения нарушения, опосредованного bcl, включающий введение терапевтически эффективного количества соединения по любому из пп.1-65.
68. Способ по п.67, в котором нарушением, опосредованным bcl, является рак или неопластическое заболевание.
69. Способ по п.68, в котором при раке наблюдается сверхэкспрессия белка Bcl.
70. Способ по п.68, в котором прогрессирование рака зависит от белка Bcl.
71. Способ по п.69, в котором упомянутым белком Bcl является Bcl-2.
72. Способ по п.69, в котором упомянутым белком Bcl является Bcl-xL.
73. Способ лечения нарушения, опосредованного bcl, включающий совместное введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества химиотерапевтического агента и терапевтически эффективного количества соединения по любому из пп.1-65, где указанным химиотерапевтическим агентом является камптотецин.
74. Способ по п.73, в котором нарушением, опосредованным bcl, является рак или неопластическое нарушение.
75. Способ по п.74, в котором при раке наблюдается сверхэкспрессия белка Bcl.
76. Способ по п.74, в котором прогрессирование рака зависит от белка Bcl.
77. Способ по п.75, в котором упомянутым белком Bcl является Bcl-2.
78. Способ по п.75, в котором упомянутым белком Bcl является Bcl-xL.
79. Способ по п.67 или 73, в котором упомянутое соединение или соединения вводят парентеральным способом.
80. Способ по п.67 или 73, в котором упомянутое соединение или соединения вводят внутримышечным, внутривенным, подкожным, пероральным, местным или интраназальным способом.
81. Способ по п.67 или 73, в котором упомянутое соединение или соединения вводят системно.
82. Способ по п.67 или 73, в котором упомянутым пациентом является млекопитающее.
83. Способ по п.67 или 73, в котором упомянутым пациентом является примат.
84. Способ по п.67 или 73, в котором упомянутым пациентом является человек.
| ПНЕВМАТИЧЕСКИЙ НАСОС ДЛЯ ПОДЪЕМА ЖИДКОСТЕЙ ИЗ ГЛУБОКИХ КОЛОДЦЕВ | 1924 |
|
SU924A1 |
| RU 94022252 А1, 27.06.1996 | |||
| US 6747050 В1, 08.06.2004. | |||

