Изобретение относится к индазол-карбоксамидным соединениям, которые могут быть использованы в качестве агонистов 5-НТ4-рецептора. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, к способам применения таких соединений для лечения или предупреждения патологических состояний, опосредуемых активностью 5-НТ4-рецептора, и к способам и промежуточным соединениям, используемым для получения указанных соединений.
Серотонин (5-гидрокситриптамин, 5-HT) представляет собой нейромедиатор, который широко распространен во всех тканях организма, а также в центральной нервной системе и в периферических системах. Было идентифицировано, по меньшей мере, семь подтипов рецепторов серотонина, и взаимодействие серотонина с указанными различными рецепторами связано с физиологическими функциями широкого ряда. Поэтому, разработка терапевтических средств, нацеленных на 5-НТ-рецепторы специфических подтипов, представляет особый интерес.
В частности, в последнее время центром повышенного внимания научных исследований является характеризация 5-НТ4-рецепторов и поиск фармацевтических средств, взаимодействующих с этими рецепторами (см., например, обзор Langlois и Fischmeister, J. Med. Chem. 2003, 46, 319-344). Агонисты 5-HT4 - рецептора могут быть использованы для лечения расстройств, связанных с пониженной перистальтикой желудочно-кишечного тракта. Такими расстройствами являются синдром раздраженной толстой кишки (СРТК), хронический запор, функциональная диспепсия, замедленное опорожнение желудка, рефлюкс-эзофагит (GERD), парез желудка, послеоперационный илеит, псевдообструкция тонкого кишечника и индуцированное лекарственными средствами замедленное прохождение содержимого через желудок. Кроме того, было высказано предположение, что некоторые соединения-агонисты 5-HT4-рецептора могут быть использованы для лечения расстройств центральной нервной системы, включая нарушение познавательной способности, поведенческие расстройства, расстройства настроения и нарушения регуляции функций вегетативной нервной системы.
Несмотря на широкое применение фармацевтических средств, модулирующих активность 5-HT4-рецептора, в настоящее время клиническое применение находят лишь несколько соединений-агонистов 5-НТ4-рецептора.
Поэтому необходимость в разработке новых агонистов 5-НТ4-рецептора, которые позволяли бы достигать нужных эффектов при минимальных побочных эффектах, остается актуальной. Предпочтительные средства могут, помимо других свойств, обладать повышенной селективностью и эффективностью, улучшенными фармакокинетическими свойствами и/или увеличенной продолжительностью действия.
Описание сущности изобретения
Настоящее изобретение относится к новым соединениям, обладающим агонистической активностью по отношению к 5-НТ4-рецептору. Было обнаружено, что соединения согласно изобретению, помимо других свойств, обладают эффективностью и селективностью агонистов 5-НТ4-рецептора. Кроме того, было обнаружено, что соединения согласно изобретению обладают ценными фармакокинетическими свойствами, которые, как предполагается, могут обеспечивать хорошую биологическую доступность при пероральном введении.
В соответствии с этим, настоящее изобретение относится к соединению формулы (I):

где:
R1 представляет собой водород, галоген, гидрокси, C1-4алкил или C1-4алкокси;
R2 представляет собой C3-4алкил или C3-6циклоалкил;
R3 представляет собой гидрокси, C1-3алкокси, гидрокси-замещенный C1-4алкил или -OC(O)NRaRb;
R4 представляет собой водород или C1-4алкил;
W выбран из:
(a) Y, где Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10, -N(R8a)C(O)OR12, -N(R8a)C(O)NR13R14 и -N(R8a)S(O)2NR13R14; и
(b) группы формулы (b):

где:
X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -S(R11)O2, -N(R8)C(O)OR12, -N(R8)C(O)NR13R14, -N(R8)S(O)2NR13R14, -C(O)NR13R14, -OC(O)NR13R14, -C(O)OR12, -OR15, -NR8R16, циано, -SR15, CF3, пиридинила, пирролила, пиримидинила, тиоморфолинила, тиазолидинила, 1,1-диоксоизотиазолидинила, имидазолила, индолила, тетрагидрофуранила, пирролидинила и пиперидинила, где пирролидинил необязательно замещен оксо, а пиперидинил необязательно замещен 1-3 атомами галогена;
R5 представляет собой водород или C1-4алкил, где C1-4алкил необязательно замещен гидрокси, C1-3алкокси или циано;
R6 и R7, в каждом случае, независимо выбраны из водорода, гидрокси, галогена, циано и C1-4алкила, где C1-4алкил необязательно замещен 1-2 заместителями, выбранными из гидрокси, C1-3алкокси, галогена и циано;
R8 и R8a представляют собой водород или C1-4алкил;
или R5 и R8, R5 и R6 или R6 и R8, взятые вместе, образуют C2-5алкиленил, где C2-5алкиленил необязательно замещен гидрокси, галогеном, гидрокси-замещенным C1-3алкилом или C1-3алкокси;
или R3 и R5 или R3 и R8a, взятые вместе, образуют -OCH2CH2-;
или R5 и R6, взятые вместе, образуют -(CH2)q-Q-(CH2)q, где Q представляет собой кислород или серу, а q независимо равно 0, 1 или 2;
или R7 и X, взятые вместе, образуют -NHC(O)NHC(O)- или -C(O)NHC(O)NH-;
R9 выбран из водорода, фуранила, тетрагидрофуранила, пиридинила или C1-4алкила, где C1-4алкил необязательно замещен гидрокси или 1-3 атомами галогена;
R10 выбран из водорода, C1-4алкила, пиридинила и имидазолила, где C1-4алкил необязательно замещен -S(O)2Rc, C3-6циклоалкилом или 1-3 атомами галогена, а имидазолил необязательно замещен C1-3алкилом;
или R8 и R10, взятые вместе, образуют С3алкиленил;
R11 представляет собой -NRaRb или C1-4алкил, где C1-4алкил необязательно замещен 1-3 атомами галогена;
или R5 и R11 или R6 и R11, взятые вместе, образуют C2-5алкиленил;
R12 представляет собой C1-4алкил;
R13 и R14 независимо представляют собой водород или C1-4алкил;
R15 представляет собой водород или C1-4алкил, где C1-4алкил необязательно замещен гидрокси;
или, если X представляет собой -SR15, то R5 и R15, взятые вместе, образуют C1-4алкиленил;
R16 представляет собой -(CH2)r-R17, где r равно 0, 1, 2 или 3; и R17 выбран из водорода, гидрокси, C1-3алкила, C1-3алкокси, -C(O)NRaRb, -C(O)-морфолинила, пиридинила, пирролила, пиримидинила, морфолинила и тетрагидрофуранила, где C1-3алкокси необязательно замещен гидрокси; при условии, что если r равно 0, то R17 выбран из водорода, C1-3алкила, пиридинила и пиримидинила; и если r равно 1, то R17 представляет собой водород или R17 образует углерод-углеродную связь с -(CH2)r-углеродным атомом;
R18 представляет собой -C(O)O-C1-3алкил, -S(O)2-C1-3алкил или -C(O)-C1-3алкил;
Ra, Rb и Rc независимо представляют собой водород или C1-3алкил;
a равно 0 или 1; и
n равно целому числу 1, 2, 3, 4 или 5; при условии, что если n равно 1, то X представляет собой -SR15 или X образует углерод-углеродную связь с атомом углерода, несущим заместители R6 и R7;
или к его фармацевтически приемлемой соли, сольвату или стереоизомеру.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение согласно изобретению и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способу лечения заболевания или состояния, ассоциированного с активностью 5-HT4-рецептора, например, лечения расстройства, связанного с пониженной перистальтикой желудочно-кишечного тракта, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения согласно изобретению.
Кроме того, настоящее изобретение относится к способу лечения заболевания или состояния, ассоциированного с активностью 5-HT4-рецептора у млекопитающего, где указанный способ включает введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции согласно изобретению и фармацевтически приемлемого носителя.
Соединения согласно изобретению могут быть также использованы в качестве экспериментальных средств, т.е., для исследования биологических систем или образцов, или для исследования активности других химических соединений. В соответствии с этим, в другом своем аспекте, настоящее изобретение относится к способу применения соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или стереоизомера в качестве экспериментального средства для исследования биологической системы или образца, или для поиска новых агонистов 5-HT4-рецептора, где указанный способ включает контактирование биологической системы или образца с соединением согласно изобретению и определение влияния указанного соединения на указанную биологическую систему или образец.
В своих отдельных и конкретных аспектах, настоящее изобретение также относится к способам синтеза соединений и к описанным здесь промежуточным соединениям, которые могут быть использованы для получения соединений согласно изобретению.
Настоящее изобретение также относится к описанному здесь соединению согласно изобретению, применяемому в терапии лекарственными средствами, а также к применению соединения согласно изобретению в целях получения композиции или лекарственного средства для лечения заболевания или состояния, ассоциированного с активностью 5-HT4-рецептора, например, расстройства, связанного с пониженной перистальтикой желудочно-кишечного тракта у млекопитающего.
Подробное описание изобретения
Настоящее изобретение относится к новым индазол-карбоксамидным соединениям-агонистам 5-HT4-рецептора формулы (I) или к их фармацевтически приемлемым солям, сольватам или стереоизомерам. Нижеследующие репрезентативные и предпочтительные значения для радикалов, заместителей и интервалов значений приводятся лишь в целях иллюстрации, но при этом, могут быть использованы и другие определенные значения или другие значения в определенных интервалах, указанных для радикалов и заместителей.
В конкретном аспекте изобретения, R1 представляет собой водород, галоген или C1-4алкил.
В других конкретных аспектах изобретения, R1 представляет собой водород, фтор, хлор, бром или метил; R1 представляет собой водород или галоген; или R1 представляет собой водород.
В конкретном аспекте изобретения, R2 представляет собой C3-4-алкил. Репрезентативными группами R2 являются н-пропил, изопропил, н-бутил, втор-бутил и трет-бутил.
В другом конкретном аспекте изобретения, R2 представляет собой изопропил или C4-5циклоалкил.
В другом конкретном аспекте изобретения, R2 представляет собой изопропил.
В конкретном аспекте изобретения, R3 представляет собой гидрокси, C1-3алкокси или -OC(O)NRaRb.
В другом конкретном аспекте изобретения, R3 представляет собой гидрокси, метокси, гидроксиметил, -OC(O)NHCH3 или -OC(O)N(CH3)2.
В конкретных аспектах изобретения, R4 представляет собой водород или метил; или R4 представляет собой водород.
В одном из аспектов изобретения, R1 представляет собой водород или галоген, R2 представляет собой изопропил или C4-5циклоалкил, а R4 представляет собой водород.
В конкретных аспектах изобретения, R3 представляет собой гидрокси, C1-3алкокси, C1-3алкил, замещенный в концевом положении гидрокси, или -OC(O)NRaRb; R3 представляет собой гидрокси, C1-3алкокси или -OC(O)NRaRb; или R3 представляет собой гидрокси.
В другом конкретном аспекте изобретения, R3 представляет собой гидрокси, метокси, гидроксиметил, -OC(O)NHCH3 или -OC(O)N(CH3)2, или R3 и R5 или R3 и R8a, взятые вместе, образуют -OCH2CH2-.
В конкретном аспекте изобретения, R5 представляет собой водород, C1-3алкил или C1-3алкил, замещенный в концевом положении гидрокси, C1-3алкокси или циано. Репрезентативными группами R5 являются, но не ограничиваются ими, гидрокси, метил, этил, 2-гидроксиэтил, 2-метоксиэтил, цианометил и 2-цианоэтил.
В других конкретных аспектах изобретения, R5 представляет собой водород, C1-3алкил или C1-3алкил, замещенный в концевом положении гидрокси; R5 представляет собой водород или C1-3алкил; или R5 представляет собой водород или метил.
В другом конкретном аспекте изобретения, R5 представляет собой водород или C1-3алкил, или R5 и R8 образуют C2-3алкиленил.
В конкретном аспекте изобретения, R3 и R5, взятые вместе, образуют -OCH2CH2-.
В конкретном аспекте изобретения, n равно 1, 2 или 3, и R5 и R6, взятые вместе, образуют -CH2CH2O- или -CH2CH2OCH2-.
Альтернативно, в другом конкретном аспекте изобретения, n равно 2, а R5 и R6, взятые вместе, образуют C2-3алкиленил.
В конкретном аспекте изобретения, R6 и R7 независимо представляют собой водород, гидрокси, галоген, C1-3алкил или гидрокси-замещенный C1-3алкил. Репрезентативными группами R6 и R7 являются, но не ограничиваются ими, водород, гидрокси, фтор, хлор, гидроксиэтил и гидроксиметил.
В других конкретных аспектах изобретения, R6 и R7 в каждом случае независимо представляют водород, гидрокси, галоген или циано; или каждый из R6 и R7 представляет собой водород.
В конкретных аспектах изобретения, R8 представляет собой водород или C1-3алкил или R8 представляет собой водород или метил.
В другом конкретном аспекте изобретения, R8 представляет собой водород или C1-3алкил, либо R5 и R8, взятые вместе, образуют C2-3алкиленил.
Альтернативно, в другом конкретном аспекте изобретения, R5 и R8, взятые вместе, образуют C2-3алкиленил.
В конкретном аспекте изобретения, n равно 2, а R5 и R8, взятые вместе, образуют С2алкиленил.
В конкретном аспекте изобретения, n равно 2, а R5 и R8, взятые вместе, образуют С3алкиленил.
Альтернативно, в другом конкретном аспекте изобретения, R6 и R8, взятые вместе, образуют C2-3алкиленил.
В конкретных аспектах изобретения, R8a представляет собой водород или C1-3алкил, или R3 и R8a, взятые вместе, образуют -OCH2CH2-; R8a представляет собой водород или C1-3алкил; или R8a представляет собой водород или метил.
В конкретном аспекте изобретения, R9 представляет собой водород, тетрагидрофуранил, пиридинил или C1-3алкил, где C1-3алкил необязательно замещен гидрокси. Репрезентативными группами R9 являются, но не ограничиваются ими, водород, тетрагидрофуран-2-ил, метил, этил, пропил, изопропил и 1-гидроксиэтил.
В других конкретных аспектах изобретения, R9 представляет собой водород, тетрагидрофуранил, пиридинил или C1-3алкил, такой как метил; или R9 представляет собой водород или метил.
В конкретном аспекте изобретения, R10 представляет собой водород или C1-3алкил, где C1-3алкил необязательно замещен -SO2Rc, C3-6циклоалкилом или 1-3 атомами галогена, где Rc представляет собой C1-3алкил. Репрезентативными группами R10 являются, но не ограничиваются ими, метил, этил, пропил, изопропил и метансульфонилметил.
В другом конкретном аспекте изобретения, R10 представляет собой C1-3алкил, где C1-3алкил необязательно замещен -S(O)2C1-3алкилом или 1-3 атомами галогена.
В других конкретных аспектах изобретения, R10 представляет собой C1-3алкил; R10 представляет собой метил или этил; или R10 представляет собой метил.
В конкретном аспекте изобретения, R11 представляет собой -NRaRb или C1-3алкил, где C1-3алкил необязательно замещен 1-3 атомами галогена.
В других конкретных аспектах изобретения, R11 представляет собой -NH2, -NH(CH3), -N(CH3)2, метил, этил или -CF3; или R11 представляет собой метил.
В другом конкретном аспекте изобретения, n равно 2, а R5 и R11, взятые вместе, образуют С2алкиленил.
В еще одном конкретном аспекте изобретения, n равно 2 или 3, а R6 и R11, взятые вместе, образуют С2алкиленил.
В конкретных аспектах изобретения, R12 представляет собой C1-3алкил; или R12 представляет собой метил или этил.
В конкретных аспектах изобретения, R13 и R14 независимо представляют собой водород или C1-3алкил; или R13 и R14 независимо представляют собой водород, метил или этил.
В конкретном аспекте изобретения, R15 представляет собой водород, C1-3алкил или C1-3алкил, замещенный в концевом положении гидрокси. Репрезентативными группами R15 являются, но не ограничиваются ими, водород, метил, этил и 2-гидроксиэтил, а именно, водород, метил и этил.
В других конкретных аспектах изобретения, R15 представляет собой водород или C1-3алкил; либо R15 представляет собой водород, метил или этил; либо R15 представляет собой водород или метил.
В конкретном аспекте изобретения, R16 представляет собой -(CH2)r-R17, где r равно 0, 1 или 2. В другом конкретном аспекте изобретения, R16 представляет собой -(CH2)r-R17, где r равно 1 или 2. Репрезентативными группами R16 являются, но не ограничиваются ими, -CH2-C(O)NRaRb, -CH2-C(O)-морфолинил, -CH2-пиридинил, -CH2-пиримидинил и -CH2-тетрагидрофуранил.
В конкретном аспекте изобретения, R17 выбран из гидрокси, C1-3алкокси, -C(O)NRaRb, -C(O)-морфолинила, пиридинила, пиримидинила, морфолинила и тетрагидрофуранила.
В конкретных аспектах изобретения, R18 представляет собой -C(O)OCH3, -S(O)2CH3 или -C(O)CH3; либо R18 представляет собой -C(O)OCH3.
В конкретном аспекте изобретения, Ra, Rb и Rc независимо представляют собой водород, метил или этил; или Ra, Rb и Rc независимо представляют собой водород или метил.
В конкретном аспекте изобретения, a равно 0. В другом конкретном аспекте изобретения, a равно 1.
В конкретном аспекте изобретения, n равно целому числу 1, 2, 3 или 4, включая 1, 2 или 3, а именно, 2 или 3. В другом конкретном аспекте изобретения, n равно 2.
В конкретном аспекте изобретения, W выбран из:
(a) Y, где Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10 и -N(R8a)C(O)NR13R14; и
(b) группы формулы (b), где X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -N(R8)C(O)OR12, -N(R8)C(O)NR13R14, -N(R8)S(O)2NR13R14, -C(O)NR13R14, -OR15 и циано.
В еще одном конкретном аспекте изобретения, W представляет собой Y.
В конкретных аспектах изобретения, W выбран из Y, где Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10, -N(R8a)C(O)OR12 и -N(R8a)C(O)NR13R14; либо Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10 и -N(R8a)C(O)NR13R14. В других конкретных аспектах изобретения, W выбран из Y, где Y представляет собой -N(R8a)S(O)2R10; Y представляет собой -N(R8a)C(O)R9; либо Y представляет собой -N(R8a)S(O)2R10.
В другом аспекте изобретения, W выбран из Y, где Y имеет значение, определенное выше; R8a представляет собой водород или метил; R9 представляет собой водород, тетрагидрофуранил, пиридинил или метил; R10 и R12 представляют собой метил или этил; и R13 и R14 независимо представляют собой водород или метил.
Альтернативно, W представляет собой группу формулы (b). В конкретном аспекте изобретения, W представляет собой группу формулы (b), где (i) X представляет собой циано; или (ii) a равно 0, n равно 2, R6 и R7 представляют собой водород, R5 и R8, взятые вместе, образуют С2алкиленил и X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10 и -N(R8)C(O)NR13R14.
В другом конкретном аспекте изобретения, W представляет собой группу формулы (b), где a равно 0, n равно 2, R6 и R7 представляют собой водород, и R5 и R8, взятые вместе, образуют С2алкиленил. В этом аспекте изобретения, репрезентативными значениями X являются, но не ограничиваются ими, -N(R8)C(O)R9, -N(R8)S(O)2R10 и -N(R8)C(O)NR13R14.
В еще одном аспекте изобретения, W представляет собой группу формулы (b); a равно 0 или 1; n равно 1, 2 или 3; R5 представляет собой водород или метил; либо R5 и R8, взятые вместе, образуют C2-5алкилен; или R3 и R5, взятые вместе, образуют -OCH2CH2-; каждый из R6 и R7 представляет собой водород; либо R5 и R6, взятые вместе, образуют C2-5алкилен; а X имеет значения, определенные выше.
В еще одном конкретном аспекте изобретения, W выбран из -NHC(O)H, -N(CH3)C(O)H, -NHC(O)CH3, -N(CH3)C(O)CH3, -N(CH3)S(O)2CH3, -N(CH3)C(O)NHCH3, -N(CH3)CH2CH2CN, 1-метансульфонилпиперазин-4-ила, 1-диметиламинокарбонил-пиперазин-4-ила, 1-(тетрагидрофуран-2-ил)карбонилпиперазин-4-ила, 3-(метоксикарбонил-амино)пирролидин-1-ила и 2-(метоксиметилен)пирролидин-1-ила.
В конкретном аспекте изобретения, W представляет собой группу формулы (b), где X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -S(R11)O2, -N(R8)C(O)OR12, -N(R8)C(O)NR13R14, -N(R8)S(O)2NR13R14, -C(O)NR13R14, -OC(O)NR13R14, -C(O)OR12, -OR15, -NR8R16, циано, -SR15, -CF3, пиридинила, пирролила, 1,1-диоксоизотиазолидинила, имидазолила и пирролидинила, где указанный пирролидинил необязательно замещен оксо.
В другом конкретном аспекте изобретения, W представляет собой группу формулы (b), где X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -S(R11)O2, -N(R8)C(O)OR12, -N(R8)C(O)NR13R14, -N(R8)SO2NR13R14, -C(O)NR13R14, -OC(O)NR13R14, -C(O)OR12, -OR15 и циано.
В других конкретных аспектах изобретения, W представляет собой группу формулы (b), где X выбран из -N(R8)C(O)R9; -N(R8)S(O)2R10, -S(R11)O2, -N(R8)C(O)NR13R14, -C(O)NR13R14, -OC(O)NR13R14, -OR15 и циано; либо X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -N(R8)C(O)NR13R14 и циано.
В других конкретных аспектах изобретения, W представляет собой группу формулы (b), где X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10 и -N(R8)C(O)NR13R14; либо X представляет собой -N(R8)S(O)2R10.
В одном из аспектов изобретения, R1, R2, R3, R4 и a определены выше;
W выбран из Y и группы формулы (b), и
R5 представляет собой водород, C1-3алкил или C1-3алкил, замещенный в концевом положении гидрокси;
R6 и R7, в каждом случае, независимо представляют собой водород, гидрокси, галоген или циано;
R8 и R8a представляют собой водород или C1-3алкил;
или R3 и R5 или R3 и R8a , взятые вместе, образуют -OCH2CH2-;
или R5 и R6, взятые вместе, образуют a C2-5алкилен;
или R5 и R8, взятые вместе, образуют a C2-5алкилен;
R9 представляет собой водород, тетрагидрофуранил, пиридинил или C1-3алкил;
R10 представляет собой C1-3алкил, где C1-3алкил необязательно замещен -S(O)2C1-3алкилом или 1-3 атомами галогена;
R11 представляет собой -NRaRb или C1-4алкил, где C1-4алкил необязательно замещен 1-3 атомами галогена;
либо R5 и R11 или R6 и R11, взятые вместе, образуют C2-5алкиленил;
R12 представляет собой C1-3алкил;
R13, R14 и R15 независимо представляют собой водород
или C1-3алкил;
R16 представляет собой -CH2-C(O)NRaRb, -CH2-C(O)-морфолинил, -CH2-пиридинил, -CH2-пиримидинил или -CH2-тетрагидрофуран;
R18 представляет собой -C(O)OCH3, -S(O)2CH3 или -C(O)CH3; и
n равно целому числу 1, 2 или 3.
В другом своем аспекте, настоящее изобретение относится к соединению формулы (I), которое представляет собой соединение формулы (I-a):
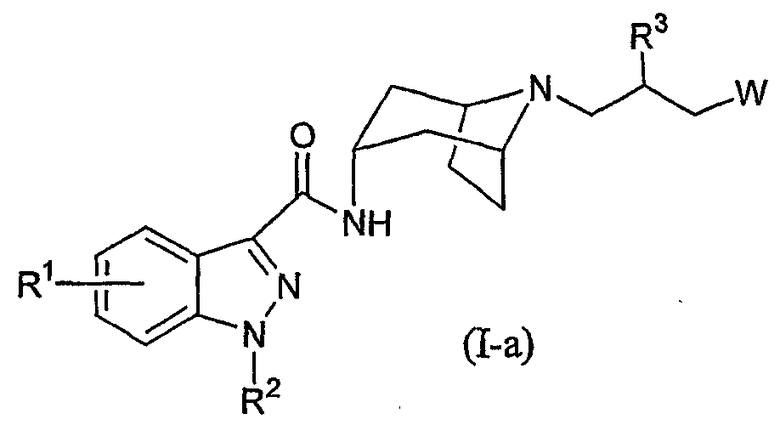
где:
R1 представляет собой водород, галоген или C1-4алкил;
R2 представляет собой изопропил или C4-5циклоалкил;
R3 представляет собой гидрокси, C1-3алкокси или OC(O)NRaRb;
W выбран из
(a) Y, где Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10, -N(R8a)C(O)OR12, -N(R8a)C(O)NR13R14 и -N(R8a)S(O)2NR13R14; и
(b) группу формулы (b):

где
X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -S(R11)O2, -N(R8)C(O)OR12, -N(R8)C(O)NR13R14, -N(R8)S(O)2NR13R14, -C(O)NR13R14, -OC(O)NR13R14, -C(O)OR12, -OR15, -NR8R16, циано, -SR15, CF3, пиридинила, пирролила, 1,1-диоксо-изотиазолидинила, имидазолила и пирролидинила, где указанный пирролидинил необязательно замещен оксо;
R5 представляет собой водород, C1-3алкил или C1-3алкил, замещенный в концевом положении гидрокси;
R6 и R7, в каждом случае, независимо представляют собой водород, гидрокси, галоген или циано;
R8 и R8a представляют собой водород или C1-3алкил;
либо R5 и R8 или R5 и R6, взятые вместе, образуют C2-5алкилен;
либо R3 и R5 или R3 и R8a, взятые вместе, образуют -OCH2CH2-;
R9 представляет собой водород, тетрагидрофуранил, пиридинил или метил;
R10 представляет собой C1-3алкил, где C1-3алкил необязательно замещен -S(O)2C1-3алкилом или 1-3 атомами галогена;
R11 представляет собой -NRaRb или C1-3алкил, где C1-3алкил необязательно замещен 1-3 атомами галогена;
либо R5 и R11 или R6 и R11, взятые вместе, образуют C2-5алкиленил;
R12 представляет собой C1-3алкил;
R13, R14 и R15 независимо представляют собой водород или C1-3алкил;
R16 представляет собой -CH2-C(O)NRaRb, -CH2-C(O)-морфолинил, -CH2-пиридинил, -CH2-пиримидинил или -CH2-тетрагидрофуранил;
R18 представляет собой -C(O)OCH3, -S(O)2CH3 или -C(O)CH3;
Ra и Rb независимо представляют собой водород или C1-3алкил;
a равно 0 или 1; и
n равно целому числу 1, 2 или 3; при условии, что, если n равно 1, то X представляет собой -SR15, либо X образует углерод-углеродную связь с атомом углерода, несущим заместители R6 и R7;
или к его фармацевтически приемлемой соли, сольвату или стереоизомеру.
Настоящее изобретение также относится к соединению формулы (I-b):

где R1, R2, R3, R4, n и X имеют любые значения, определенные выше.
Настоящее изобретение также относится к соединению формулы (I-c):

где W' выбран из -C(O)R9, -S(O)2R10, -C(O)OR12, -C(O)NR13R14, -S(O)2NR13R14 и -(CR6R7)n-X; и R1, R2, R6, R7, R9, R10, R12, R13 и R14 имеют любые значения, определенные выше.
Настоящее изобретение также относится к соединению формулы (I-d):

где n' равно целому числу 0, 1 или 2, и R1, R2, R3, R4 и X имеют любые значения, определенные выше.
В еще одном своем аспекте, настоящее изобретение относится к соединениям, перечисленным в нижеприведенных таблицах I-X.
Используемая здесь общепринятая химическая номенклатура проиллюстрирована на соединении примера 1:

которое имеет название: {(1S,3R,5R)-8-[2-гидрокси-3-((S)-2-метоксиметилпирролидин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты, данное в соответствии с программным обеспечением AutoNom, поставляемым MDL Information Systems, GmbH (Frankfurt, Germany). Обозначение (1S,3R,5R) указывает на относительную ориентацию связей, ассоциированных с бициклической системой, которые показаны сплошными и пунктирными клинообразными линиями. Альтернативно, указанное соединение имеет название N-[(3-эндо)-8-(3-((S)-2-метоксиметилпирролидин-1-ил)-2-гидроксипропил)-8-азабицикло[3.2.1]окт-3-ил]-1-(1-метилэтил)-1H-индазол-3-карбоксамид.
В этой связи могут быть упомянуты нижеследующие конкретные соединения:
{(1S,3R,5R)-8-[2-гидрокси-3-((S)-2-метоксиметилпирролидин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
((1S,3R,5R)-8-{3-[(2-циано-этил)метиламино]-2-гидроксипропил}-8-азабицикло[3.2.1]окт-3-ил)амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
((1S,3R,5R)-8-{2-гидрокси-3-[4-(тетрагидрофуран-2-карбонил)пиперазин-1-ил]пропил}-8-азабицикло[3.2.1]окт-3-ил)амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
[(1S,3R,5R)-8-(4-карбамоилметилморфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(4-диметилкарбамоилпиперазин-1-ил)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(4-метансульфонилпиперазин-1-ил)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
метиловый эфир [1-(2-гидрокси-3-{(1S,3R,5R)-3-[(1-изопропил-1H-индазол-3-карбонил)амино]-8-азабицикло[3.2.1]окт-8-ил}пропил)пирролидин-3-ил]карбаминовой кислоты;
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(ацетил-метиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(формил-метиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(1,3-диметилуреидо)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
((1S,3R,5R)-8-{2-гидрокси-3-[(пиридин-4-карбонил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)амид 1-изопропил-1H-индазол-3-карбоновой кислоты;
[(1S,3R,5R)-8-(3-формиламино-2-гидроксипропил)-8-азабицикло[3.2.1]окт-3-ил]-амид 1-изопропил-1H-индазол-3-карбоновой кислоты и
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты.
Как проиллюстрировано выше, соединения согласно изобретению могут содержать один или несколько хиральных центров. В соответствии с этим, настоящее изобретение включает рацемические смеси, чистые стереоизомеры и обогащенные стереоизомерами смеси таких изомеров, если это не оговорено особо. Если указан конкретный стереоизомер, то для специалиста в данной области очевидно, что в композициях согласно изобретению могут присутствовать небольшие количества других стереоизомеров, если это не оговорено особо, при условии, что присутствие таких других изомеров, в целом, не оказывает негативного влияния на эффективность данной композиции.
Определения
При описании соединений, композиций и способов согласно изобретению используются термины, определение которых приводится ниже, если это не оговорено особо.
Термин “алкил” означает одновалентную насыщенную углеводородную группу, которая может быть прямой группой или разветвленной группой, или может представлять собой их комбинацию. Если это не оговорено особо, то такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Репрезентативными алкильными группами являются, например, метил, этил, н-пропил (n-Pr), изопропил (i-Pr), н-бутил (n-Bu), втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и т.п.
Термин "алкиленил" или "алкилен" означает двухвалентную насыщенную углеводородную группу, которая может быть прямой группой или разветвленной группой, или может представлять собой их комбинацию. Репрезентативными "алкиленильными" группами являются, например, метиленил, этиленил, н-пропиленил, изопропиленил, н-бутиленил, втор-бутиленил, трет-бутиленил и т.п.
Термин "алкокси" означает одновалентную группу -O-алкил, где алкил определен выше. Репрезентативными алкоксигруппами являются, например, метокси, этокси, пропокси, бутокси и т.п.
Термин “соединение" означает соединение, которое было получено путем синтеза или любым другим методом, например, в результате метаболизма.
Термин “циклоалкил” означает одновалентную насыщенную карбоциклическую группу, которая может быть моноциклической или полициклической. Если это не оговорено особо, то такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода. Репрезентативными циклоалкильными группами являются, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и т.п.
Термин “галоген” означает фтор, хлор, бром или йод.
Термин “терапевтически активное количество” означает количество, достаточное для эффективного лечения при его введении пациенту, нуждающемуся в таком лечении.
Используемый здесь термин “лечение” означает лечение заболевания, расстройства или патологического состояния у пациента, такого как млекопитающее (а в частности, человек), где такое лечение предусматривает:
(a) предупреждение развития заболевания, расстройства или патологического состояния, то есть, профилактическое лечение пациента;
(b) снижение тяжести заболевания, расстройства или патологического состояния, то есть, устранение указанного заболевания, расстройства или патологического состояния у пациента или стимуляцию наступления их ремиссии;
(c) подавление симптомов заболевания, расстройства или патологического состояния, то есть, замедление или прекращение развития указанного заболевания, расстройства или патологического состояния у пациента; или
(d) ослабление симптомов указанного заболевания, расстройства или патологического состояния у пациента.
Термин “фармацевтически приемлемая соль” означает соль, полученную из кислоты или основания, которые являются пригодными для введения пациенту, такому как млекопитающее. Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических кислот и из фармацевтически приемлемых оснований. Обычно, фармацевтически приемлемые соли соединений согласно изобретению получают из кислот.
Солями, полученными из фармацевтически приемлемых кислот, являются, но не ограничиваются ими, соли уксусной, бензолсульфоновой, бензойной, камфорсульфоновой, лимонной, этансульфоновой, фумаровой, глюконовой, глутаминовой, бромистоводородной, хлористоводородной, молочной, малеиновой, яблочной, миндальной, метансульфоновой, слизевой, азотной, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой, ксинафойной (1-гидрокси-2-нафтойной) и нафталин-1,5-дисульфоновой кислот и т.п.
Термин “сольват” означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, то есть, соединения согласно изобретению или его фармацевтически приемлемой соли, и одной или несколькими молекулами растворителя. Такими сольватами обычно являются кристаллические твердые вещества, имеющие, в основном, фиксированное молярное отношение растворенного вещества и растворителя. Репрезентативными растворителями являются, например, вода, метанол, этанол, изопропанол, уксусная кислота и т.п. Если растворителем является вода, то образованным сольватом является гидрат.
Следует отметить, что термин “или его фармацевтически приемлемая соль, или сольват, или стереоизомер” включает все модификации солей, сольватов и стереоизомеров, таких как сольват фармацевтически приемлемой соли стереоизомера соединения формулы (I).
Термин “аминозащитная группа” означает защитную группу, подходящую для предотвращения нежелательных реакций у атома азота аминогруппы. Репрезентативными аминозащитными группами являются, но не ограничиваются ими, формил; ацильные группы, например, алканоильные группы, такие как ацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr) и 1,1-ди-(4'-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBDMS) и т.п.
Общие процедуры синтеза
Соединения согласно изобретению могут быть получены из легко доступных исходных соединений в соответствии с нижеследующими общими методами и процедурами. Хотя конкретный аспект согласно изобретению представлен в нижеследующих схемах, однако, для специалистов в данной области очевидно, что все соединения согласно изобретению могут быть получены описанными здесь методами или другими методами с использованием других реагентов и исходных веществ, известных специалистам. Также очевидно, что в тех случаях, где описаны типичные или предпочтительные условия реакций (то есть, температуры реакций, время проведения реакций, молярные отношения реагентов, растворители, давление и т.п.), могут быть также применены и другие условия реакций, если это не оговорено особо. Оптимальные условия реакций могут варьироваться в зависимости от конкретно используемых реагентов или растворителей, и такие условия могут быть определены специалистом путем проведения обычных процедур оптимизации.
Кроме того, как очевидно для специалистов в данной области, может оказаться необходимым введение подходящих защитных групп для предупреждения некоторых функциональных групп от нежелательных реакций. Выбор подходящей защитной группы для конкретной функциональной группы, а также выбор подходящих условий реакций защиты и снятия защиты может быть легко осуществлен самим специалистом. Так, например, различные защитные группы и их введение и удаление описаны в руководстве T. W. Greene и G. M. Wuts, Protecting Groups в Organic Синтез, Third Edition, Wiley, New York, 1999, и в цитируемых там работах.
Заместители и значения символов, указанные в нижеследующих схемах, являются такими, как они были определены выше, если это не оговорено особо.
В одном из методов синтеза, соединения формулы (I), где R3 определен как гидрокси или гидрокси-замещенный C1-4алкил, получают как показано на схеме A:
Схема A

посредством реакции взаимодействия промежуточного соединения 1 с промежуточным соединением 2 и промежуточным соединением 3, где L представляет собой уходящую группу, такую как хлор, бром, йод, метансульфонилокси, п-толуолсульфонилокси или трифторметансульфонилокси, с получением соединения формулы (I).
Реакцию обычно проводят посредством взаимодействия промежуточного соединения 1 примерно с 1-3 эквивалентами каждого из промежуточных соединений 2 и 3 в инертном растворителе, таким как метанол или этанол, в присутствии избытка основания, например, приблизительно 3-6 эквивалентов основания, такого как N,N-диизопропилэтиламин. Такую реакцию обычно проводят при температуре примерно 50-80°С в течение приблизительно 12-24 часов, или до тех пор, пока реакция не будет, по существу, завершена. При этом, но необязательно, могут быть добавлены порциями равные молярные эквиваленты промежуточных соединений 2 и 3.
Продукт формулы (I) выделяют и очищают в соответствии со стандартными процедурами. Так, например, полученный продукт может быть концентрирован досуха при пониженном давлении, растворен в водном растворе слабой кислоты и очищен с помощью ВЭЖХ.
Следует отметить, что в реакции, описанной на схеме A, и в других описанных ниже реакциях, проводимых с использованием промежуточного соединения 1, промежуточное соединение 1 может быть получено в форме свободного основания или соли, с соответствующей, если это необходимо, корректировкой условий реакции, известной специалистам.
На схеме A, реакцию взаимодействия промежуточного соединения 1 с промежуточными соединениями 2 и 3 проводят в одну стадию. Альтернативно, такая реакция может быть проведена в несколько стадий. В условиях реакции, аналогичных условиям, описанным выше, промежуточные соединения 1 и 3 могут быть сначала подвергнуты реакции сочетания с получением промежуточного соединения 5:
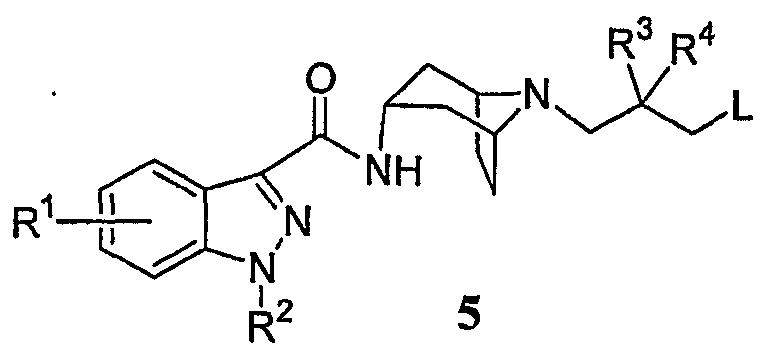
которое затем подвергают реакции взаимодействия с промежуточным соединением 2, в результате чего получают соединение формулы (I). Альтернативно, промежуточное соединение 2 может быть сначала подвергнуто реакции сочетания с промежуточным соединением 3 с получением промежуточного соединения 10:

которое затем подвергают реакции взаимодействия с индазол-карбоксамид-тропановым промежуточным соединением 1, в результате чего получают соединение формулы (I).
Соединения формулы (I) могут быть также получены путем N-алкилирования соединения формулы (I), в котором R2 определен как водород и которое может быть получено, как показано на схеме A. Реакцию N-алкилирования обычно проводят путем взаимодействия соединения формулы (I), в котором R2 представляет собой водород, приблизительно с 1-4 эквивалентами соединения формулы L'-R2, где L' представляет собой уходящую группу, такую как йод или бром. Такую реакцию обычно проводят в полярном апротонном растворителе, таком как диметилформамид, в присутствии приблизительно 2-4 эквивалентов сильного основания, такого как трет-бутоксид калия или гидрид натрия. Указанную реакцию обычно проводят при температуре примерно 60-100°C в течение примерно 6-24 часов, или до тех пор, пока реакция не будет, по существу, завершена.
В еще одном альтернативном варианте, соединения формулы (I), в которых R1 не является водородом, получают стандартными методами, такими как ароматическое галогенирование из соединений формулы (I), в которых R1 представляет собой водород.
В другом методе синтеза, соединения формулы (I), в которых R3 представляет собой гидрокси, C1-3алкокси или -OC(O)NRaRb, а атом углерода, несущий заместитель R3, не является хиральным, могут быть получены путем взаимодействия азетидинового промежуточного соединения 11, как показано на схеме B:
Схема B

где L' представляет собой противоион, такой как галогенид, например, Cl-, Br- или трифторацетат, с промежуточным соединением 2, H-W, с получением соединения формулы (I).
Такую реакцию обычно проводят путем взаимодействия промежуточного соединения 11 приблизительно с 1-4 эквивалентами промежуточного соединения 2 в инертном растворителе, таком как этанол, метанол или диметилформамид, в присутствии избытка основания, например, приблизительно 2-4 эквивалентов основания, такого как N,N-диизопропилэтиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или триэтиламин. Указанную реакцию обычно проводят при температуре примерно 50-80°С в течение приблизительно 1-16 часов, или до тех пор, пока реакция не будет, по существу, завершена. Полученный продукт выделяют и очищают стандартными методами.
В еще одном методе синтеза, соединения формулы (I), где R3 представляет собой гидрокси, могут быть получены как показано на схеме С, где звездочка означает хиральный центр, посредством взаимодействия промежуточного соединения 1 с промежуточным соединением 12, с получением соединения формулы (I).
Схема C

Если используется оптически чистый энантиомер промежуточного соединения формулы 12, то посредством проведения реакции, показанной на схеме С, могут быть получены оптически чистые энантиомеры соединений формулы (I), имеющие хиральный центр у атома углерода, несущего заместитель R4. Обычно, в реакции, показанной на схеме С, промежуточное соединение 1 подвергают реакции взаимодействия приблизительно с 1-1,2 эквивалентами эпоксида 12 в инертном разбавителе, таком как этанол или толуол. Такую реакцию обычно проводят при температуре примерно 50-100°С в течение приблизительно 12-24 часов, или до тех пор, пока реакция не будет, по существу, завершена. Полученный продукт выделяют и очищают стандартными методами.
Промежуточные соединения, используемые в вышеописанных схемах A, B и C, получают из легко доступных исходных веществ. Так, например, если R3 представляет собой гидрокси, то азетидиновое промежуточное соединение формулы 13 может быть получено в соответствии с процедурой, проиллюстрированной на схеме D.
Схема D
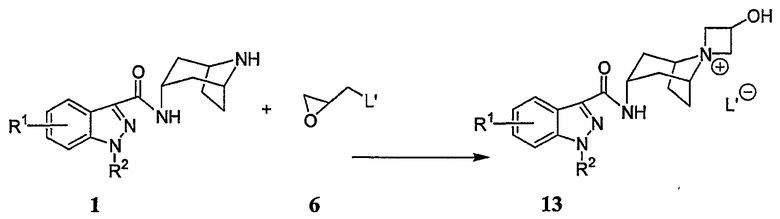
Промежуточное соединение 1 подвергают реакции взаимодействия с промежуточным соединением 6, т.е., с оксирановым соединением, в котором L' представляет собой галогеновую уходящую группу, такую как бром, хлор или йод (например, L' представляет собой бром, а оксирановым соединением является 2-бромметилоксиран, часто называемый эпибромгидрином), с получением промежуточного соединения 13, то есть, соли азетидина. Такую реакцию обычно проводят посредством взаимодействия промежуточного соединения 1 приблизительно с 2-4 эквивалентами оксиранового соединения в полярном разбавителе, таком как этанол. Указанную реакцию обычно проводят при температуре окружающей среды в течение приблизительно 24-48 часов, или до тех пор, пока реакция не будет, по существу, завершена.
Для получения азетидинового промежуточного соединения 11, в котором R3 представляет собой C1-3алкокси, вышеописанное промежуточное соединение 13 подвергают реакции взаимодействия с С1-3алкилгалогенидом в количестве, составляющем в пределах от приблизительно менее чем одного эквивалента до приблизительно одного эквивалента, в инертном разбавителе, в присутствии приблизительно 1-3 эквивалентов сильного основания, такого как трет-бутоксид калия или гидрид натрия. Такую реакцию обычно проводят при температуре окружающей среды в течение периода времени, составляющего примерно от 15 минут до одного часа, или до тех пор, пока реакция не будет, по существу, завершена. Подходящими инертными разбавителями являются тетрагидрофуран, толуол, диметилформамид и т.п.
Промежуточное соединение 11, в котором R3 представляет собой группу карбаминовой кислоты формулы -OC(O)NRaRb, может быть получено из промежуточного соединения формулы 13, в котором R3 представляет собой гидрокси. Так, например, для получения соединения формулы 11, в котором R3 представляет собой -OC(O)N(H)CH3 или -OC(O)N(CH3)2, промежуточное соединение 13 подвергают взаимодействию приблизительно с 1-3 эквивалентами метилизоцианата или диметилкарбамилхлорида, соответственно, в инертном разбавителе, в присутствии приблизительно 1-3 эквивалентов основания, такого как N,N-диизопропилэтиламин, и каталитического количества сильного основания, такого как трет-бутоксид калия или гидрид натрия. Такую реакцию обычно проводят при температуре окружающей среды в течение приблизительно 4-24 часов, или до тех пор, пока реакция не будет, по существу, завершена.
Альтернативно, соединения формулы (I), в которых R3 представляет собой группу карбаминовой кислоты формулы -OC(O)NRaRb, могут быть получены из соединения формулы (I), в котором R3 представляет собой гидрокси. Так, например, для получения соединения формулы (I), в котором R3 представляет собой -OC(O)N(H)CH3 или -OC(O)N(CH3)2, соединение формулы (I), в котором R3 представляет собой гидрокси, подвергают взаимодействию приблизительно с 1-3 эквивалентами метилизоцианата или диметилкарбамилхлорида, соответственно, в условиях, аналогичных описанным выше условиям получения соединения формулы (I), в котором R3 представляет собой -OC(O)NRaRb.
Способ получения промежуточных соединений формулы 1 проиллюстрирован на схеме E.
Схема E

Промежуточное соединение 1 может быть получено посредством взаимодействия промежуточного соединения 14 с промежуточным соединением 15, где P1 представляет собой аминозащитную группу.
Такую реакцию обычно проводят сначала путем превращения соединения 14 в хлорангидрид посредством взаимодействия промежуточного соединения 14, по меньшей мере, с одним эквивалентом, а предпочтительно, примерно с 1-2 эквивалентами активирующего агента, такого как тионилхлорид или оксалилхлорид, в ароматическом разбавителе, таком как толуол, бензол, ксилол или т.п. Указанную реакцию обычно проводят при температуре примерно 80-120°С в течение периода времени, составляющего приблизительно от 15 минут до 4 часов, или до тех пор, пока реакция не будет, по существу, завершена.
К двухфазной смеси примерно 1 эквивалента аминотропана 15 обычно добавляют раствор хлорангидрида, в результате чего получают защищенное промежуточное соединение, которое затем экстрагируют в соответствии со стандартными процедурами. Двухфазную смесь соединения 15 обычно получают путем растворения соединения 15 в ароматическом разбавителе, таком как толуол, бензол, ксилол или т.п., и добавления водного раствора, содержащего избыток основания, такого как гидроксид натрия или гидроксид калия, а предпочтительно, примерно 2-5 эквивалентов основания. Такую реакцию обычно проводят при температуре примерно 80-120°С в течение периода времени, составляющего примерно от 15 минут до 4 часов, или до тех пор, пока реакция не будет, по существу, завершена.
Альтернативно, реакция сочетания амида промежуточного соединения 15 с карбоновой кислотой 14 может быть проведена в присутствии агента сочетания, такого как 1,3-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC) или гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония (PyBop), необязательно, в комбинации с 1-гидрокси-7-азабензотриазолом (HOAt). В еще одном альтернативном способе, реакция сочетания амида промежуточного соединения 15 с карбоновой кислотой 14 может быть проведена путем превращения карбоновой кислоты 14 в активированный сложный эфир.
Защитную группу P1 удаляют стандартными методами с получением промежуточного соединения 1. Так, например, если защитной группой является Boc, то такое удаление обычно проводят путем обработки кислотой, такой как трифторуксусная кислота, с получением соли кислоты промежуточного соединения. Соль кислоты промежуточного соединения 1 может быть превращена в свободное основание путем стандартной обработки основанием. В качестве другого примера, защитную группу Cbz обычно удаляют путем гидрогенолиза на подходящем металлическом катализаторе, таком как палладий на угле.
Защищенный аминотропан 15, используемый в реакциях, описанных в настоящей заявке, получают из легко доступных исходных веществ. Так, например, если защитной группой P1 является Boc, то защищенный аминотропан 16 может быть получен в соответствии с процедурой, проиллюстрированной на схеме F.
Схема F
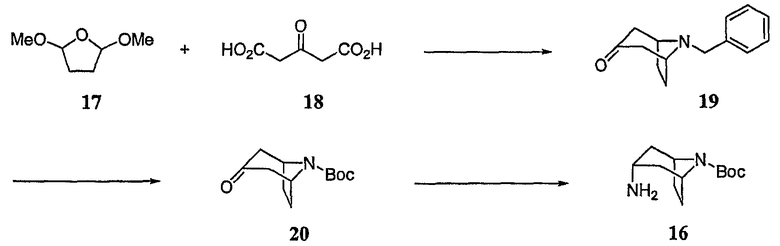
Так, например, защищенное аминотропановое промежуточное соединение 16 может быть получено посредством взаимодействия 2,5-диметокситетрагидрофурана 17 с 1,3-ацетондикарбоновой кислотой 18, с образованием 8-бензил-8-азабицикло[3.2.1]октан-3-она 19, обычно называемого N-бензилтропаноном. Затем N-бензилтропанон подвергают реакции взаимодействия с небольшим избытком ди-трет-бутилдикарбоната в присутствии катализатора на основе переходного металла, в результате чего получают Вос-защищенное промежуточное соединение 20, которое затем восстанавливают с получением защищенного аминотропанового промежуточного соединения 16.
Сначала 2,5-диметокситетрагидрофуран 17 подвергают взаимодействию примерно с 1-2 эквивалентами, а предпочтительно, примерно с 1,5 эквивалента бензиламина, и небольшого избытка, например, примерно 1,1 эквивалента 1,3-ацетондикарбоновой кислоты 18 в водном растворе кислоты в присутствии забуферивающего агента, такого как бифосфат натрия. Полученную реакционную смесь нагревают до температуры примерно 60-100°C для гарантии декарбоксилирования всех карбоксилированных промежуточных соединений в данном продукте, а именно, в 8-бензил-8-азабицикло[3.2.1]октан-3-оне 19, обычно называемом N-бензилтропаноном.
Промежуточное соединение 19 обычно подвергают реакции взаимодействия с небольшим избытком ди-трет-бутилдикарбоната, например, приблизительно с 1,1 эквивалента, в атмосфере водорода в присутствии катализатора на основе переходного металла, с получением Вос-защищенного промежуточного соединения 20, а именно, трет-бутилового эфира 3-оксо-8-азабицикло[3.2.1]октан-8-карбоновой кислоты. Такую реакцию обычно проводят при температуре окружающей среды в течение примерно 12-72 часов.
И, наконец, промежуточное соединение 20 подвергают взаимодействию с большим избытком, например, более чем 25 эквивалентов формиата аммония, в инертном разбавителе, таком как метанол, в присутствии катализатора на основе переходного металла, с получением промежуточного соединения 16 в эндо-конфигурации. Продукт 16 может быть очищен стандартными методами, такими как экстракция щелочью.
1H-индазолкарбоновая кислота, промежуточное соединение 14, может быть легко получена методами, аналогичными методам, описанным в публикации Harada et al. Chem. and Pharm Bull. 1995, 43, 1912-30, и в нижеследующих примерах.
Оксирановое промежуточное соединение 12, используемое в схеме С, может быть получено, как показано на схеме G:
Схема G
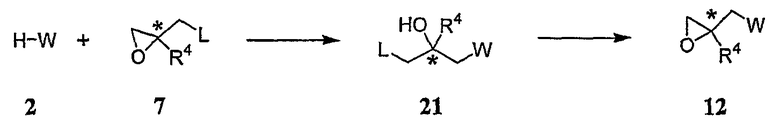
где промежуточное соединение 2 подвергают реакции взаимодействия с промежуточным соединением 7, замещенным оксираном, где L представляет собой галогеновую уходящую группу, с получением промежуточного соединения 21, которое затем снова подвергают реакции циклизации, в результате чего получают промежуточное соединение 12. Такую реакцию обычно проводят посредством взаимодействия аминового промежуточного соединения 2 приблизительно с 1-2 эквивалентами промежуточного соединения 7 в полярном разбавителе, таком как этанол. Такую реакцию обычно проводят при температуре окружающей среды в течение приблизительно 12-24 часов или до тех пор, пока реакция не будет, по существу, завершена. Промежуточное соединение 21 с прямой цепью обычно выделяют в виде твердого вещества стандартными методами. Твердое вещество 21 обычно растворяют в инертном разбавителе, таком как тетрагидрофуран, в присутствии молярного избытка основания, например, гидроксида натрия, с получением циклического промежуточного соединения 12.
В еще одном альтернативном методе синтеза, соединения формулы (I) могут быть получены путем проведения реакции сочетания замещенной 1H-индазолкарбоновой кислоты 14 с промежуточным соединением формулы 22, как показано на схеме H.
Схема H
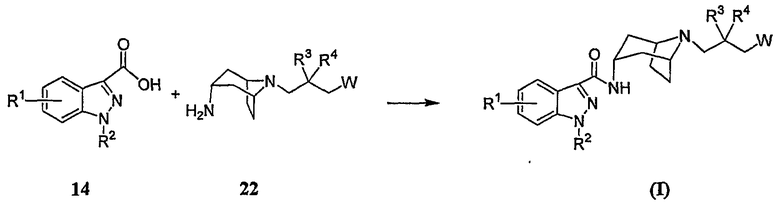
Реакцию взаимодействия карбоновой кислоты 14 с промежуточным соединением 22 обычно проводят в условиях реакции сочетания с амидом, как показано выше на схеме E.
Промежуточные соединения формулы 22 могут быть получены путем проведения реакции снятия защиты с промежуточного соединения формулы 23:

где P2 представляет собой аминозащитную группу.
Промежуточные соединения формулы 23 могут быть получены из легко доступных исходных веществ в соответствии с процедурами реакций, аналогичных реакциям, описанным выше, и/или путем проведения альтернативных реакций, хорошо известных специалистам. Так, например, промежуточное соединение 23 может быть получено с использованием промежуточного соединения 24:

которое может быть получено посредством защиты атома азота аминогруппы аминоазабициклооктана 15 с использованием аминозащитной группы P2 с последующим удалением P1 у атома азота азабициклооктановой группы. Защитные группы P1 и P2 выбирают так, чтобы они могли быть удалены в различных условиях. Так, например, если в качестве P1 была выбрана группа Boc, то в качестве P2 может быть использована группа Cbz. В результате замещения промежуточного соединения 1 защищенным аминотропаном 24 в реакциях, описанных на схемах A и C, получают промежуточные соединения формулы 23.
В еще одном альтернативном методе синтеза, соединение формулы (I), в котором W представляет собой группу формулы (b) и a равно 1, может быть получено, как описано ниже на схеме J:
Схема J

где промежуточное соединение 25 подвергают реакции взаимодействия с промежуточным соединением 26, где L представляет собой уходящую группу, такую как галоген, например, хлор, или этокси, либо L-R18 представляет собой карбоновую кислоту, то есть, L представляет собой гидроксигруппу, в результате чего получают соединение формулы (I).
Репрезентативными реагентами промежуточного соединения 26 являются метансульфонилхлорид и ацетилхлорид, и т.п. Оптимальные условия реакций, описанных на схеме J, могут варьироваться в зависимости от химических свойств указанных реагентов, как хорошо известно специалистам.
Так, например, если L представляет собой галогеновую уходящую группу, такую как хлор, то реакцию обычно проводят посредством взаимодействия промежуточного соединения 25 примерно с 1-4 эквивалентами промежуточного соединения 26 в инертном разбавителе, таком как дихлорметан, в присутствии избытка основания, например, приблизительно 3-6 эквивалентов основания, такого как N,N-диизопропилэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). Подходящими инертными разбавителями также являются N,N-диметилформамид (ДМФ), трихлорметан, 1,1,2,2-тетрахлорэтан, тетрагидрофуран и т.п. Эту реакцию обычно проводят при температуре примерно от -100°С до 30°С в течение периода времени, составляющего примерно от четверти часа до 2 часов, или до тех пор, пока данная реакция не будет, по существу, завершена.
Если промежуточным соединением 26 является карбоновая кислота, то реакцию, представленную на схеме J как реакцию сочетания с амидом, обычно проводят посредством взаимодействия промежуточного соединения 25 примерно с 1-4 эквивалентами карбоновой кислоты в инертном разбавителе, например, N,N-диметилформамиде, в присутствии агента сочетания, такого как гексафторфосфат бензотриазол-1-илокситрипирролидино-фосфония (PyBOP). Эту реакцию обычно проводят при комнатной температуре в течение периода времени, составляющего примерно от четверти часа до двух часов, или до тех пор, пока данная реакция не будет, по существу, завершена. Подходящими альтернативными агентами сочетания являются 1,3-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC) и PyBOP, объединенный с 1-гидрокси-7-азабензотриазолом (HOAt).
Альтернативно, реакция сочетания амида промежуточного соединения 25 с карбоновой кислотой может быть осуществлена путем превращения карбоновой кислоты в активированный сложный эфир, такой как сложный эфир N-гидроксисукцинимида (NHS) или сложный п-нитрофениловый эфир, или кислотное производное имидазола, которое затем подвергают реакции взаимодействия с промежуточным соединением 25 с получением соединения формулы (I).
Если промежуточное соединение 26 представляет собой жидкость, например, этилформиат, то эта реакция может быть осуществлена путем растворения промежуточного соединения 25 в большом избытке промежуточного соединения 26 и нагревания до температуры примерно от 50 до 100°С в течение периода времени примерно от 12 до 24 часов с получением соединения формулы (I). Этот продукт, то есть, соединение формулы (I), может быть затем выделен и очищен стандартными методами.
Промежуточное соединение 25 может быть получено из легко доступных исходных веществ в соответствии с описанными здесь схемами A, B, C и H или посредством альтернативных реакций, хорошо известных специалистам.
В другом альтернативном синтезе, соединение формулы (I), где, в случае, если a равно 0, R5 представляет собой водород или необязательно замещенный C1-4алкил, или R3 и R5, взятые вместе, образуют -OCH2CH2-, может быть получен, как показано на схеме K:
Схема K

в котором промежуточное соединение 27 (где R19 представляет собой R5, R8a или R18) подвергают реакции взаимодействия с промежуточным соединением 28 (где L представляет собой уходящую группу, такую как галоген, и W” представляет собой остаток, который, вместе с концевым атомом азота промежуточного соединения 27 образует W), в результате чего получают соединение формулы (I). Если R19 представляет собой R8a, то W” представляет собой -C(O)R9, -S(O)2R10, -C(O)OR12, -C(O)NR13R14 или -S(O)2NR13R14. Если R19 представляет собой R18, то W” представляет собой -(CH2)2-N(R5)(CR6R7)n-X. Если a равно 0, то R19 представляет собой R5, W” представляет собой -(CR6R7)n-X, а R5 представляет собой водород или C1-4алкил, где C1-4алкил необязательно замещен гидрокси, C1-3алкокси или циано, либо R3 и R5, взятые вместе, образуют -OCH2CH2-.
Эту реакцию обычно проводят посредством взаимодействия промежуточного соединения 27 приблизительно с 1-3 эквивалентами промежуточного соединения 28 в инертном разбавителе, таком как дихлорметан или т.п., в присутствии основания, такого как N,N-диизопропилэтиламин или т.п. Такую реакцию обычно проводят при температуре примерно от 0 до 100°С в течение приблизительно 6-24 часов, или до тех пор, пока реакция не будет, по существу, завершена. Полученный продукт выделяют и очищают стандартными методами с получением соединения формулы (I).
Промежуточные соединения формулы 27 могут быть получены посредством реакции взаимодействия азетидинового промежуточного соединения 11 с амином 30:
H2N-R19
30
где R19 представляет собой R5, R8a или R18, с получением промежуточного соединения 27. Так, например, промежуточным соединением 30 может быть метиламин или т.п.
Альтернативно, промежуточные соединения формулы 27, в которой R3 представляет собой -OH, могут быть получены посредством реакции взаимодействия промежуточного соединения 1 с оксиранилметиловым соединением, имеющим защищенный атом азота, а затем посредством реакции снятия защиты. Одним из подходящих реагентов является 2-оксиранилметил-изоиндол-1,3-дион, обычно называемый эпоксипропилфталимидом, который подвергают реакции взаимодействия с промежуточным соединением 1, в результате чего получают промежуточное соединение, в котором фталимидил-замещенная 2-гидроксипропильная группа:

связана с атомом азота азабицклооктанового кольца 1. Затем фталимидильную группу удаляют путем кипячения соединения с обратным холодильником в гидразине, в результате чего получают промежуточное соединение 27, в котором R3 представляет собой -OH, и R4 и R19 представляют собой водород.
Помимо синтеза, описанного на схеме К, соединение формулы (I), в котором R3 и R5, либо R3 и R8a, взятые вместе, образуют -OCH2CH2-, и W выбран из Y или группы формулы (b), где a равно 0, может быть получено, как показано на схеме L:
Схема L

где промежуточное соединение 31 подвергают реакции взаимодействия с промежуточным соединением 32 (где L представляет собой уходящую группу, такую как галоген, а W' выбран из -C(O)R9, -S(O)2R10, -C(O)OR12, -C(O)NR13R14, -S(O)2NR13R14 и -(CR6R7)n-X, так, что W' представляет собой остаток, который вместе с атомом азота морфолинового кольца промежуточного соединения 31 образует W, как было определено выше), в результате чего получают соединение формулы (I).
Такую реакцию обычно проводят посредством взаимодействия промежуточного соединения 31 приблизительно с 1-3 эквивалентами промежуточного соединения 32 в инертном разбавителе, таком как метанол или этанол, в присутствии основания, такого как N,N-диизопропилэтиламин. Указанную реакцию обычно проводят при температуре примерно 60-95°С в течение приблизительно 6-24 часов, или до тех пор, пока реакция не будет, по существу, завершена. Продукт формулы (I) выделяют и очищают стандартными методами. Так, например, полученный продукт может быть концентрирован досуха при пониженном давлении, растворен в водном растворе слабой кислоты и очищен с помощью ВЭЖХ.
Способ получения промежуточных соединений формулы 31 представлен на схеме M:
Схема M

где промежуточное соединение 1 подвергают реакции взаимодействия с промежуточным соединением 34, в котором P3 представляет собой аминозащитную группу, такую как BOC, в результате чего получают промежуточное соединение 35, которое затем подвергают реакции снятия защиты с получением промежуточного соединения 31. Такую реакцию обычно проводят посредством взаимодействия промежуточного соединения 1 приблизительно с 1-3 эквивалентами промежуточного соединения 34 в инертном разбавителе, таком как ацетонитрил, в присутствии карбоната калия, при температуре примерно от 60 до 90°С в течение приблизительно 12-24 часов, или до тех пор, пока реакция не будет, по существу, завершена. После выделения промежуточного соединения 35, группу P3 удаляют и получают промежуточное соединение 31.
Первичные или вторичные амины промежуточных соединений 2, 30, 32 и H-W являются коммерчески доступными, либо они могут быть легко синтезированы из хорошо известных исходных веществ в соответствии со стандартными протоколами, описанными в литературе или в справочной литературе, такой как публикация J. March, Advanced Organic Chemistry, Fourth Edition, Wiley, New York, 1992, и проиллюстрированными в настоящей заявке.
Более подробное описание конкретных условий реакций и других процедур получения репрезентативных соединений согласно изобретению или их промежуточных соединений приводится ниже в разделе “Примеры”.
В соответствии с этим, в одном из своих аспектов, настоящее изобретение относится к способу получения соединения формулы (I), где R1, R2, R3, R4 и W имеют значения, определенные в формуле (I), или его соли или стереоизомера, где указанный способ включает:
(a) реакцию взаимодействия соединения формулы (II):

с соединением формулы (III):
 или
или
(b) реакцию взаимодействия соединения формулы (IV):

с соединением формулы (V):

с получением соединения формулы (I) или его соли или стереоизомера.
Настоящее изобретение также относится к способу получения соединения формулы (I), где R3 представляет собой гидрокси, а R1, R2, R4 и W являются такими, как они были определены в формуле (I), или его соли или стереоизомера, где указанный способ включает:
стадию (a) или стадию (b), как определено выше, или
(c) реакцию взаимодействия соединения формулы (VI):

или его соли, с соединением формулы (III):
и с соединением формулы (VII):

где L представляет собой уходящую группу; или
(d) реакцию взаимодействия соединения формулы (VI) с соединением формулы (VIII):

с получением соединения формулы (I) или его соли или стереоизомера.
Настоящее изобретение также относится к способу получения соединения формулы (I), где R1, R2, R3, R4, R5, R6, R7, R8a, R9, R10, R12, R13, R14, R18, a, n, W и X являются такими, как они были определены в формуле (I), при условии, что если a равно 0, то R5 представляет собой водород или C1-4алкил, где C1-4алкил необязательно замещен гидрокси, C1-3алкокси или циано, либо, R3 и R5, взятые вместе, образуют -OCH2CH2-; или его соли или стереоизомер, где указанный способ включает:
реакцию взаимодействия соединения формулы (IX):

где R19 представляет собой R5, R8a или R18;
с соединением формулы (X):
L-W' (X)
где L представляет собой уходящую группу; и
(a) если R19 представляет собой R8a, то W” выбран из -C(O)R9, -S(O)2R10, -C(O)OR12, -C(O)NR13R14 и -S(O)2NR13R14;
(b) если R19 представляет собой R18, то W” представляет собой -(CH2)2-N(R5)(CR6R7)n-X; и
(c) если a равно 0, то R19 представляет собой R5, W” представляет собой -(CR6R7)n-X, и R5 представляет собой водород или C1-4алкил, где C1-4алкил необязательно замещен гидрокси, C1-3алкокси или циано, либо, R3 и R5, взятые вместе, образуют -OCH2CH2-;
с получением соединения формулы (I).
Помимо вышеописанного способа, настоящее изобретение также относится к способу получения соединения формулы (I), где R3 и R5 или R3 и R8a, взятые вместе, образуют -OCH2CH2-; W выбран из Y и группы формулы (b), где a равно 0; и R1, R2, R4, R6, R7, R9, R10, R12, R13, R14, n и X являются такими, как они были определены в формуле (I); или его соли или стереоизомера, где указанный способ включает:
реакцию взаимодействия соединения формулы (XI):
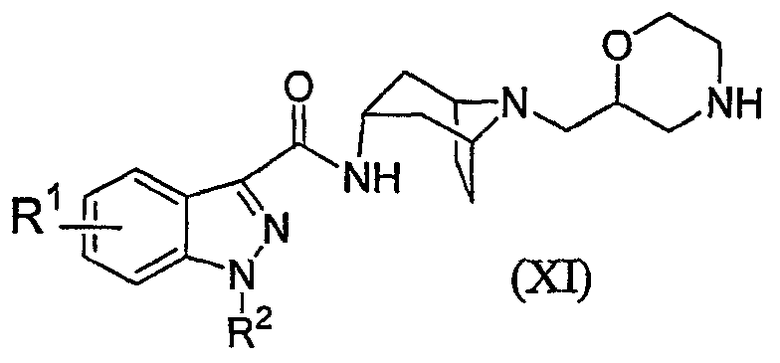
с соединением формулы (XII):
L-W' (XII)
где L представляет собой уходящую группу, а W' выбран из -C(O)R9, -S(O)2R10, -C(O)OR12, -C(O)NR13R14, -S(O)2NR13R14 и -(CR6R7)n-X;
с получением соединения формулы (I).
Настоящее изобретение также относится к способу получения соединения формулы (I), где W представляет собой группу формулы (b) и a равно 1; и R1, R2, R4, R5, R6, R7, R18, n и X являются такими, как они были определены в формуле (I); или его соли или стереоизомера, где указанный способ включает:
реакцию взаимодействия соединения формулы (XIII):
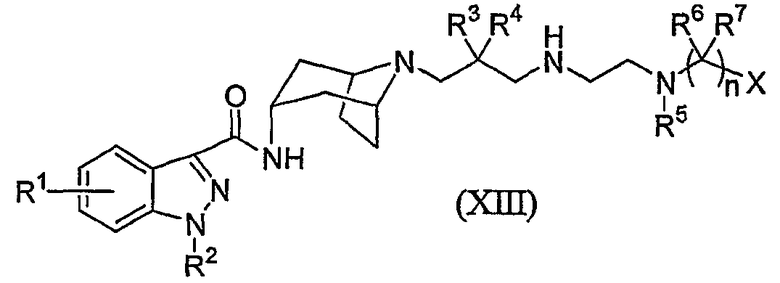
с соединением формулы (XIV):
L-R18 (XIV)
где L представляет собой уходящую группу;
с получением соединения формулы (I).
В других своих вариантах, настоящее изобретение относится к другим описанным здесь способам и к продуктам, полученным любыми из описанных здесь способов.
Фармацевтические композиции
Индазол-карбоксамидные соединения согласно изобретению обычно вводят пациенту в виде фармацевтической композиции. Такие фармацевтические композиции могут быть введены пациенту любым приемлемым способом, включая, но не ограничиваясь ими, пероральное введение, ректальное введение, вагинальное введение, назальное введение, введение путем ингаляции, местное введение (включая трансдермальное введение) и парентеральное введение.
В соответствии с этим, в одном из своих аспектов, относящихся к композициям, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель или наполнитель и терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. Такие фармацевтические композиции, если это необходимо, но необязательно, могут содержать другие терапевтические и/или лекарственные средства.
Фармацевтические композиции согласно изобретению обычно содержат терапевтически эффективное количество соединения согласно изобретению или его фармацевтически приемлемой соли. Обычно, такие фармацевтические композиции содержат примерно 0,1-95 мас.% активного вещества, предпочтительно, примерно 5-70 мас.% активного вещества, а более предпочтительно, примерно 10-60 мас.% указанного активного вещества.
В фармацевтических композициях согласно изобретению может быть использован любой подходящий носитель или наполнитель. Выбор конкретного носителя или наполнителя, либо комбинаций носителей или наполнителей, зависит от способа введения, применяемого для лечения конкретного пациента или типа патологического процесса или патологического состояния. В этой связи следует отметить, что подходящая фармацевтическая композиция для конкретного способа введения может быть получена специалистом-фармацевтом. Кроме того, ингредиенты для таких композиций являются коммерчески доступными и поставляются, например, фирмой Sigma, Р.О. Вох 14508, St. Louis, MO 63178. Так, например, в качестве дополнительной информации, можно отметить, что стандартные методы получения фармацевтических композиций описаны в руководстве Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceuticals Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Репрезентативными примерами материалов, которые могут служить в качестве фармацевтически приемлемых носителей, являются, но не ограничиваются ими, (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза, такая как микрокристаллическая целлюлоза и ее производные, такие как натрий-содержащая карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразная трагакантовая камедь; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, масло из семян хлопчатника, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) забуференные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатно-буферные растворы и (21) другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции согласно изобретению обычно получают путем интенсивного и тщательного перемешивания или смешивания соединения согласно изобретению с фармацевтически приемлемым носителем и одним или несколькими вспомогательными ингредиентами. Если необходимо или желательно, полученная равномерно размешанная смесь может быть затем сформована в таблетки, капсулы, драже и т.п. в соответствии со стандартными процедурами с использованием соответствующего оборудования.
Фармацевтические композиции согласно изобретению, предпочтительно, изготавливают в виде унифицированной лекарственной формы. Термин “унифицированная лекарственная форма” означает физически дискретную форму, подходящую для введения соответствующей дозы пациенту, где каждая такая форма содержит предварительно определенное количество активного ингредиента, которое, взятое отдельно или в комбинации с одной или несколькими дополнительными лекарственными формами, является достаточным для продуцирования нужного терапевтического эффекта. Так, например, такими унифицированными лекарственными формами могут быть капсулы, таблетки, драже и т.п.
В предпочтительном аспекте изобретения, фармацевтические композиции согласно изобретению являются подходящими для перорального введения. Фармацевтические композиции, подходящие для перорального введения, могут быть получены в форме капсул, таблеток, пилюль, пастилок, саше, драже, порошков, гранул; или в форме раствора или суспензии в водной или в безводной жидкости; или в форме эмульсии типа “масло в воде” или “вода в масле”; либо в форме эликсира или сиропа или т.п.; при этом, каждая такая форма содержит предварительно определенное количество соединения согласно изобретению в качестве активного ингредиента.
Когда для перорального введения используется твердая лекарственная форма (то есть, капсулы, таблетки, драже и т.п.), фармацевтические композиции согласно изобретению обычно содержат соединение согласно изобретению в качестве активного ингредиента и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия или дикальцийфосфат. Необязательно или альтернативно, такие твердые лекарственные формы могут также содержать: (1) наполнители или разбавители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремневая кислота; (2) связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; (3) увлажнители, такие как глицерин; (4) дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и/или карбонат натрия; (5) вещества, замедляющие растворение, такие как парафин; (6) ускорители абсорбции, такие как четвертичные аммониевые соединения; (7) смачивающие агенты, такие как цетиловый спирт и/или моностеарат глицерина; (8) абсорбенты, такие как каолин и/или бентонитовая глина; (9) лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; (10) красящие агенты и (11) забуферивающие агенты.
В фармацевтических композициях согласно изобретению могут также присутствовать вещества, модулирующие высвобождение, смачивающие вещества, вещества для нанесения покрытий, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты. Примерами фармацевтически приемлемых антиоксидантов являются: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и т.п.; (2) растворимые в масле антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) вещества, образующие хелатные комплексы с металлами, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбит, винная кислота, фосфорная кислота и т.п. Веществами для нанесения покрытий на таблетки, капсулы, драже и т.п., являются вещества, используемые для нанесения энтеросолюбильных покрытий, такие как ацетат-фталат целлюлозы (САР), поливинилацетат-фталат (PVAP), гидроксипропилфталат метилцеллюлозы, сополимеры метакриловой кислоты - эфира метакриловой кислоты, ацетат-тримеллитат целлюлозы (САТ), карбоксиметилэтилцеллюлоза (СМЕС), ацетат-сукцинат гидроксипропилметилцеллюлозы (HPMCAS) и т.п.
Если необходимо, то фармацевтические композиции согласно изобретению могут быть также приготовлены в виде композиций с замедленным или регулируемым высвобождением активного ингредиента, с использованием, например, гидроксипропилметилцеллюлозы в различных соотношениях; или других полимерных матриц, липосом и/или микросфер.
Кроме того, фармацевтические композиции согласно изобретению могут, но необязательно, содержать вещества, вызывающие помутнение, и могут быть приготовлены так, чтобы активный ингредиент высвобождался только, или предпочтительно, в некоторых отделах желудочно-кишечного тракта, необязательно, в замедленном режиме. Примерами заливочных композиций, которые могут быть использованы, являются полимерные вещества и воски. Активный ингредиент может быть также приготовлен в микроинкапсулированной форме, если это необходимо, вместе с одним или несколькими вышеописанными наполнителями.
Подходящими жидкими лекарственными формами для перорального введения могут служить, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Такие жидкие лекарственные формы обычно содержат активный ингредиент и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (такие как, например, масло из семян хлопчатника, арахисовое масло, кукурузное масло, масло из проросших семян, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, и их смеси. Суспензии, помимо активного ингредиента, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакантовая камедь, и их смеси.
Альтернативно, фармацевтические композиции согласно изобретению могут быть приготовлены в виде препаратов для введения путем ингаляции. Подходящие фармацевтические композиции для введения путем ингаляции обычно приготавливают в форме аэрозоля или порошка. Такие композиции обычно вводят с помощью хорошо известных устройств, используемых для их доставки, таких как ингалятор с дозирующим клапаном, инсуффлятор, распылитель или аналогичное устройство для доставки.
Если данная композиция предназначена для введения путем ингаляции с использованием контейнера под давлением, то такие фармацевтические композиции согласно изобретению обычно содержат активный ингредиент и подходящий пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, двуокись углерода или другой подходящий газ.
Кроме того, фармацевтическая композиция может быть приготовлена в форме капсулы или картрижда (изготовленного, например, из желатина), содержащего соединение согласно изобретению и порошок, подходящий для использования в инсуффляторе. Подходящими порошковыми основами являются, например, лактоза или крахмал.
Соединения согласно изобретению могут быть также введены чрескожно с использованием известных систем и наполнителей, подходящих для чрескожной доставки. Так, например, соединение согласно изобретению может быть смешано с усилителями проницаемости, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-оны и т.п., и введено в пластырь или аналогичную систему для доставки. Если необходимо, то в таких композициях для чрескожного введения могут быть использованы дополнительные наполнители, включая гелеобразующие агенты, эмульгаторы и буферы.
Ниже, в качестве иллюстрации, приводятся репрезентативные фармацевтические композиции согласно изобретению:
Композиция примера А
Твердые желатиновые капсулы для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем загружают в твердую желатиновую капсулу (260 мг композиции на капсулу).
Композиция примера В
Твердые желатиновые капсулы для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем пропускают через сито № 45, США, загружают в твердую желатиновую капсулу (200 мг композиции на капсулу).
Композиция примера С
Капсулы для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем загружают в твердую желатиновую капсулу (310 мг композиции на капсулу).
Композиция примера D
Таблетки для перорального введения получают следующим образом:
Репрезентативная процедура: Активный ингредиент, крахмал и целлюлозу пропускают через сито № 45, США, тщательно смешивают. Раствор поливинилпирролидона смешивают с полученным порошком, после чего эту смесь пропускают через сито № 45, США. Полученные таким образом гранулы сушат при 50-60°С и пропускают через сито № 18, США. Затем к этим гранулам добавляют натрий-содержащий карбоксиметилированный крахмал, стеарат магния и тальк (предварительно пропущенный через сито № 45, США). После перемешивания, смесь спрессовывают на таблетировочной машине и получают таблетку массой в 100 мг.
Композиция примера Е
Таблетки для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем спрессовывают в таблетки (440 мг композиции на таблетку).
Композиция примера F
Таблетки с одной насечкой, предназначенные для перорального введения, получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем спрессовывают в таблетки с одной насечкой (215 мг композиции на таблетку).
Композиция примера G
Суспензию для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты смешивают до образования суспензии, содержащей 10 мг активного ингредиента на 10 мл суспензии.
Композиция примера Н
Сухой порошок для введения путем ингаляции получают следующим образом:
Репрезентативная процедура: Активный ингредиент измельчают в порошок, а затем смешивают с лактозой. Затем эту размешиваемую смесь загружают в желатиновый картридж для ингаляции. Содержимое этого картриджа вводят с помощью инсуффлятора.
Композиция примера I
Сухой порошок, предназначенный для введения путем ингаляции с помощью ингалятора с дозирующим клапаном, получают следующим образом:
Репрезентативная процедура: Суспензию, содержащую 5 мас.% соединения согласно изобретению и 0,1 мас.% лецитина, получают путем диспергирования 10 г активного соединения в виде тонкодисперсного порошка с частицами, имеющими средний размер менее 10 мкм в растворе, образованном из 0,2 г лецитина, растворенного в 200 мл деминерализованной воды. Затем суспензию сушат распылением, и полученный материал измельчают до получения частиц, имеющих средний диаметр менее чем 1,5 мкм. Эти частицы загружают в картриджи с 1,1,1,2-тетрафторэтаном под давлением.
Композиция примера J
Препарат для инъекций получают следующим образом:
Репрезентативная процедура: Вышеуказанные ингредиенты смешивают, и рН доводят до 4±0,5 с использованием 0,5 н. HCl или 0,5 н. NaOH.
Композиция примера К
Капсулы для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем загружают в желатиновую капсулу (размер #1, белая, непрозрачная) (264 мг композиции на капсулу).
Композиция примера L
Капсулы для перорального введения получают следующим образом:
Репрезентативная процедура: Эти ингредиенты тщательно смешивают, а затем загружают в желатиновую капсулу (размер #1, белая, непрозрачная) (148 мг композиции на капсулу).
Следует отметить, что в обсуждаемых выше фармацевтических композициях могут быть использованы соединения согласно изобретению в любой форме (то есть, в форме свободного основания, фармацевтической соли или сольвата), подходящей для конкретного способа введения.
Применение
Имидазол-карбоксамидные соединения согласно изобретению представляют собой агонисты 5-НТ4-рецептора, поэтому, предполагается, что они могут быть использованы для лечения патологических состояний, опосредуемых 5-НТ4-рецепторами или ассоциированных с активностью 5-НТ4-рецептора, то есть, патологических состояний, симптомы которых могут быть ослаблены путем лечения агонистом 5-НТ4-рецептора. Такими патологическими состояниями являются, но не ограничиваются ими, синдром раздраженной кишки (СРК), хронический запор, функциональная диспепсия, замедленное опорожнение желудка, рефлюкс-эзофагит (GERD), парез желудка, диабетическая и идиопатическая гастропатия, послеоперационный илеит, псевдообструкция тонкого кишечника и индуцированное лекарственными средствами замедленное прохождение содержимого через желудок. Кроме того, было высказано предположение, что некоторые соединения-агонисты 5-HT4-рецептора могут быть использованы для лечения расстройств центральной нервной системы, включая нарушение познавательной способности, поведенческие расстройства, расстройства настроения и нарушения регуляции функций вегетативной нервной системы.
В частности, предполагается, что соединения согласно изобретению способствуют улучшению перистальтики желудочно-кишечного тракта (ЖКТ), а поэтому, предполагается, что они могут быть использованы для лечения расстройств желудочно-кишечного тракта, вызываемых пониженной перистальтикой у млекопитающих, включая человека. Примерами таких нарушений перистальтики желудочно-кишечного тракта могут служить, в качестве иллюстрации, синдром раздраженной толстой кишки, хронический запор, функциональная диспепсия, диабетический парез желудка и идиопатический парез желудка.
Соединения согласно изобретению, используемые для лечения расстройств, ассоциированных с пониженной перистальтикой желудочно-кишечного тракта, или других состояний, опосредуемых 5-НТ4-рецепторами, в основном, вводят перорально в виде одной суточной дозы в день или в виде дробных доз несколько раз в день, хотя могут быть использованы и другие схемы введения. Обычно, количество активного вещества, вводимое в качестве одной дозы, или общее суточное количество вводимого активного вещества, в каждом конкретном случае, определяется лечащим врачом в зависимости от состояния, подвергаемого лечению, выбранного способа введения, конкретно вводимого соединения и его относительной активности, а также от возраста и массы пациента, его индивидуальной восприимчивости к лечению, тяжести симптомов заболевания и т.п.
Подходящие дозы активного вещества, предназначенные для лечения расстройств, ассоциированных с пониженной перистальтикой желудочно-кишечного тракта или других расстройств, опосредуемых 5-НТ4-рецепторами, могут составлять примерно от 0,0007 до 20 мг/кг/день, а предпочтительно, от 0,0007 до 1 мг/кг/день. Для человека со средней массой 70 кг, такая доза активного вещества должна составлять примерно от 0,05 до 70 мг в день.
В одном из аспектов изобретения, соединения согласно изобретению используются для лечения хронического запора. Соединения согласно изобретению, используемые для лечения хронического запора, в основном, вводят перорально в виде одной суточной дозы в день или в виде дробных доз несколько раз в день. Предпочтительная доза для лечения хронического запора будет составлять примерно от 0,05 до 70 мг в день.
В другом аспекте изобретения, соединения согласно изобретению используются для лечения синдрома раздраженной кишки. Если соединения согласно изобретению применяются для лечения синдрома раздраженной кишки, то эти соединения, в основном, вводят перорально в виде одной суточной дозы в день или в виде дробных доз несколько раз в день. Предпочтительная доза для лечения хронического запора будет составлять примерно от 0,05 до 70 мг в день.
В другом аспекте изобретения, соединения согласно изобретению используются для лечения диабетического пареза желудка. Если соединения согласно изобретению применяются для лечения диабетического пареза желудка, то эти соединения, в основном, вводят перорально в виде одной суточной дозы в день или в виде дробных доз несколько раз в день. Предпочтительная доза для лечения хронического запора будет составлять примерно от 0,05 до 70 мг в день.
В другом аспекте изобретения, соединения согласно изобретению используются для лечения идиопатического пареза желудка. Если соединения согласно изобретению применяются для лечения идиопатического пареза желудка, то эти соединения, в основном, вводят перорально в виде одной суточной дозы в день или в виде дробных доз несколько раз в день. Предпочтительная доза для лечения хронического запора будет составлять примерно от 0,05 до 70 мг в день.
В другом аспекте изобретения, соединения согласно изобретению используются для лечения функциональной диспепсии. Если соединения согласно изобретению применяются для лечения функциональной диспепсии, то эти соединения, в основном, вводят перорально в виде одной суточной дозы в день или в виде дробных доз несколько раз в день. Предпочтительная доза для лечения хронического запора будет составлять примерно от 0,05 до 70 мг в день.
Настоящее изобретение также относится к способу лечения млекопитающего, страдающего заболеванием или состоянием, ассоциированным c активностью 5-НТ4-рецептора, где указанный способ включает введение указанному млекопитающему терапевтически эффективного количества соединения согласно изобретению или фармацевтической композиции, содержащей соединение согласно изобретению.
Поскольку соединениями согласно изобретению являются агонисты 5-НТ4-рецептора, то такие соединения также могут быть использованы в качестве экспериментальных средств для исследования или изучения биологических систем или образцов, содержащих 5-НТ4-рецепторы, или для поиска новых агонистов 5-НТ4-рецептора. Кроме того, поскольку соединения согласно изобретению обладают селективностью связывания с 5-НТ4-рецепторами, но не с 5-НТ-рецепторами других подтипов, а в частности, с 5-НТ3-рецепторами, то такие соединения могут быть, в частности, использованы для исследования эффектов селективного агонистического действия 5-НТ4-рецепторов в биологической системе или в образце. В таких исследованиях, которые могут быть проведены либо in vitro, либо in vivo, могут быть использованы любые подходящие биологические системы или образцы, содержащие 5-НТ4-рецепторы. Репрезентативными биологическими системами или образцами, подходящими для таких исследований, являются, но не ограничиваются ими, клетки, клеточные экстракты, плазматические мембраны, образцы тканей, млекопитающие (такие как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и т.п.
В этом аспекте изобретения, биологическую систему или образец, содержащие 5-НТ4-рецептор, подвергают контакту с соединением согласно изобретению в количестве, достаточном для стимуляции 5-НТ4-рецептора. Затем действие, способствующее активации 5-НТ4-рецептора, определяют с использованием стандартных процедур и оборудования, таких как анализы на связывание с радиоактивным лигандом и функциональные анализы. Такими функциональными анализами являются анализы на опосредуемые лигандом изменения уровней внутриклеточного циклического аденозинмонофосфата (сАМР); анализы на опосредуемые лигандом изменения активности фермента аденилил-циклазы (которая синтезирует сАМР); анализы на опосредуемые лигандом изменения уровня включения аналогов гуанозинтрифосфата (GTP), такого как [35S]GTPγS (гуанозин-5'-О-(γ-тио)трифосфат) или GTP-Eu, в выделенные мембраны посредством катализируемой рецептором замены аналогов GTP аналогами GDP; анализы на опосредуемые лигандом изменения уровней свободных внутриклеточных ионов кальция (измеренных, например, на визуализирующем флуоресцентном планшет-ридере или FLIPR®, Molecular Devices, Inc.) и измерение уровня активации протеинкиназы, активированной митогеном (МАРК). Соединение согласно изобретению может стимулировать или повышать активацию 5-НТ4-рецепторов в любом из вышеописанных функциональных анализов или анализов аналогичного типа. 5-НТ4-рецептор-стимулирующее количество соединения согласно изобретению составляет, в основном, примерно от 1 до 500 наномоль.
Кроме того, соединения согласно изобретению могут быть использованы в качестве экспериментальных средств для поиска новых агонистов 5-НТ4-рецептора. В этом аспекте, уровни связывания с 5-НТ4-рецептором или функциональные свойства тест-соединения или группы тест-соединений сравнивают с уровнями связывания с 5-НТ4-рецептором или с функциональными свойствами соединения согласно изобретению в целях идентификации тест-соединений, обладающих наилучшей активностью связывания или функциональной активностью, если она присутствует. Этот аспект настоящего изобретения включает, в качестве отдельных аспектов, получение данных для сравнения (с использованием соответствующих анализов) и анализ этих экспериментальных данных для идентификации представляющих интерес тестируемых соединений.
В анализе на связывание с радиоактивным лигандом было обнаружено, что соединения согласно изобретению, помимо других свойств, обладают сильной агонистической активностью по отношению к 5-НТ4-рецептору, а также обладают селективностью к рецептору подтипа 5-НТ4, но не подтипа 5-НТ3. Кроме того, в исследованиях на крысиной модели, предпочтительные соединения согласно изобретению обнаруживали нужные фармакокинетические свойства.
Эти свойства, а также эффективность соединений согласно изобретению могут быть продемонстрированы с применением различных in vitro и in vivo анализов, хорошо известных специалистам. Репрезентативные анализы более подробно описаны в нижеследующих примерах.
Примеры
Нижеследующие примеры синтеза и биологические примеры приводятся лишь в целях иллюстрации настоящего изобретения и не должны рассматриваться как ограничения его объема. В нижеследующих примерах используются сокращения, которые имеют указанные ниже значения, если это не оговорено особо. Сокращения, которые не определены ниже, имеют свои обычные общепринятые значения.
Boc=трет-бутоксикарбонил
(Boc)2O=ди-трет-бутилдикарбонат
DCM (ДХМ)=дихлорметан
DMF (ДМФ)=N,N-диметилформамид
DMSO (ДМСО)=диметилсульфоксид
EtOAc=этилацетат
mCPBA=м-хлорпербензойная кислота
MeCN=ацетонитрил
MTBE=бутил-трет-метиловый эфир
PyBop=гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония
Rf=время удерживания
RT (КТ)=комнатная температура
TFA=трифторуксусная кислота
THF (ТГФ)=тетрагидрофуран
Реагенты (включая вторичные амины) и растворители были закуплены у коммерческих фирм-поставщиков (Aldrich, Fluka, Sigma, etc.) и были использованы без дополнительной очистки. Реакции проводили в атмосфере азота, если это не оговорено особо. Мониторинг реакционных смесей проводили с помощью тонкослойной хроматографии (ТСХ), аналитической высокоэффективной жидкостной хроматографии (аналит. ВЭЖХ) и масс-спектрометрии, подробное описание которых приводится ниже и отдельно в конкретных примерах реакций. Реакционные смеси обрабатывали, как указано в описании каждой конкретной реакции. В основном, реакционные смеси очищают путем экстракции и другими методами очистки, такими как зависимая от температуры и растворителя кристаллизация, и осаждение. Кроме того, реакционные смеси подвергали рутинной очистке с помощью препаративной ВЭЖХ. Общий протокол проведения препаративной ВЭЖХ описан ниже. Характеризацию продуктов реакции проводили общепринятым методом с помощью масс- и 1Н-ЯМР-спектрометрии. Для измерения ЯМР, образцы растворяют в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6), а 1Н-ЯМР-спектры регистрировали с помощью оборудования Varian Gemini 2000 (300 МГц) в стандартных условиях наблюдений. Масс-спектрометрическую идентификацию соединений проводили методом ионизации электрораспылением (ESMS) с помощью оборудования Perkin Elmer (PE SCIEX API 150EX).
Общий протокол аналитической ВЭЖХ
Неочищенные соединения растворяют в 50% MeCN/H2O (c 0,1% TFA) при концентрации 0,5-1,0 мг/мл и анализировали в следующих условиях:
Общий протокол очистки с помощью препаративной ВЭЖХ
Неочищенные соединения растворяют в 50% уксусной кислоте в воде при концентрации 50-100 мг/мл, фильтровали и фракционировали в соответствии с нижеследующей процедурой:
Получение вторичных аминов
Тиоморфолин-1,1-диоксид получают из тиоморфолина посредством реакции защиты вторичного амина N-Boc-тиоморфолином ((Boc)2O, MeOH), окисления до сульфона (mCPBA, CH2Cl2, 0°C) и удаления N-Boc-группы с получением свободного амина (CF3CO2H, CH2Cl2). (m/z): [М+H]+ для C4H9NO2S: вычислено 136,04; найдено, 135,9.
N-сульфонильные производные пиперазина получают из N-Boc-пиперазина посредством реакции взаимодействия с соответствующим сульфонилхлоридом (iPr2NEt, CH2Cl2, 0°C) и удаления N-Boc-группы (CF3CO2H, CH2Cl2). 1-метансульфонил-пиперазин: 1H-ЯМР (CDCl3; нейтральный): δ (м.д.) 3,1 (т, 4H), 2,9 (т, 4H), 2,7 (с, 3H). 1-(метилсульфонил)метансульфонил-пиперазин: 1Н-ЯМР (CD3OD): δ (м.д.) 2,90 (с, 3H), 3,02 (м, 4H), 3,38 (м, 4H), 4,61 (с, 2H).
Рацемические формы или индивидуальные хиральные изомеры 3-ацетиламинопирролидина получают путем обработки N1-Boc-3-аминопирролидина (рацемата, 3R, или 3S) ацетилхлоридом (iPr2NEt, CH2Cl2, 0°C) и удаления N-Boc-группы (CF3CO2H, CH2Cl2). 3-(ацетамидо)пирролидин: 1H-ЯМР (ДМСО-d6; TFA-соль): δ (м.д.) 4,2 (квинт., 1H), 3,3-3,1 (м, 3H), 2,9 (м, 1H), 2,0 (м, 1H), 1,8 (шир. с, 4H).
3-((R)-2-гидроксипропионамидо)пирролидин получают после амидирования N1-Boc-3-аминопирролидина (L-молочной кислоты, PyBOP, ДМФ, к.т.) и удаления N-Boc-группы (CF3CO2H, CH2Cl2). (m/z): [М+H]+ для C7H14N2O2: вычислено, 159,11; найдено, 159,0. 1H-ЯМР (CD3OD; TFA-соль): δ (м.д.) 4,4 (квинт., 1H), 4,1 (кв., 1H), 3,5-3,4 (м, 2H), 3,3-3,2 (м, 2H), 2,3 (м, 1H), 2,0 (м, 1H), 1,3 (д, 3H).
N3-алкансульфонильные производные (3R)-аминопирролидина получают путем обработки N1-Boc-(3R)-аминопирролидина пропионилсульфонилхлоридом или циклогексилметилсульфонилхлоридом (i-Pr2NEt, CH2Cl2, 0°C) и удаления N-Boc-группы (CF3CO2H, CH2Cl2).
3-(N-ацетил-N-метиламидо)пиперидин получают из N3-Cbz защищенного трет-бутилового эфира 3-амино-пиперидин-1-карбоновой кислоты (De Costa, B., et al. J. Med. Chem. 1992, 35, 4334-43) после проведения четырех стадий синтеза: (i) MeI, n-BuLi, ТГФ, от -78°С до комнатной температуры; (ii) H2 (1 атм), 10% Pd/C, EtOH; (iii) AcCl, i-Pr2NEt, CH2Cl2; (iv) CF3CO2H, CH2Cl2, m/z: [М+H]+ для C8H16N2O: вычислено 157,13; найдено, 157,2. 1H-ЯМР (CD3OD; TFA-соль): δ (м.д.) 4,6 (м, 1H), 3,3 (м, 1H), 3,2 (м, 1H), 3,0 (м, 1H), 2,9 (с, 3H), 2,8 (м, 1H), 2,0 (с, 3H), 1,9-1,7 (м, 4H).
3-(N-ацетиламидо)пиперидин получают из трет-бутилового эфира 3-амино-пиперидин-1-карбоновой кислоты после N-ацетилирования и удаления N-Boc-группы: (i) AcCl, i-Pr2NEt, CH2Cl2; (ii) CF3CO2H, CH2Cl2. 1H-ЯМР (CD3OD; TFA-соль): δ (м.д.) 3,9 (м, 1H), 3,3 (дд, 1H), 3,2 (м, 1H), 2,9 (дт, 1H), 2,75 (дт, 1H), 2,0-1,9 (м, 2H), 1,9 (с, 3H), 1,8-1,4 (м, 2H).
N3-алкансульфонильные производные 3-аминопиперидина синтезировали посредством реакции хиральных или рацемических форм трет-бутилового эфира 3-амино-пиперидин-1-карбоновой кислоты с соответствующим алкансульфонилхлоридом (i-Pr2NEt, CH2Cl2) и удаления N-Boc-группы (CF3CO2H, CH2Cl2). (3S)-3-(этансульфониламидо)пиперидин: 1H-ЯМР (CD3OD): δ (м.д.) 1,29 (т, 3H, J1=7,4 Гц), 1,50-1,80 (м, 2H), 1,90-2,10 (м, 2H), 2,89 (м, 2H), 3,05 (кв., 2H, J1=7,4 Гц), 3,27 (м, 2H), 3,40 (дд(шир.), 1H), 3,52 (м, 1H). 3S-метилсульфонилметансульфониламидо-пиперидин: 1H-ЯМР (CD3OD): δ (м.д.) 2,13-2,30 (м, 2H), 2,40-2,57 (м, 2H), 2,98 (м, 2H), 3,15 (с, 3H), 3,21 (м, 2H), 3,30 (шир. д, 1H), 3,74 (м, 1H).
3-(метиламино)-1-ацетилпирролидин получают из 3-(метиламино)-1-бензилпирролидина (TCI America) после проведения четырех стадий: (i) (Boc)2O, MeOH, при комнатной температуре; (ii) H2 (1 атм), 10% Pd/C, EtOH; (iii) AcCl, i-Pr2NEt, CH2Cl2; iv) CF3CO2H, CH2Cl2. (m/z): [М+H]+ для C7H14N2O: Вычислено, 143,12; найдено, 143,0,
3-(метиламино)-1-(метансульфонил)пирролидин получают из 3-(метиламино)-1-бензилпирролидин после проведения четырех стадий: (i) (Boc)2O, MeOH, при комнатной температуре; (ii) H2 (1 атм), 10% Pd/C, EtOH; (iii) CH3S(O)2Cl, i-Pr2NEt, CH2Cl2; (iv) CF3CO2H, CH2Cl2. (m/z): [М+H]+ для C6H14N2O2S: вычислено, 179,08; найдено, 179,2. 3R-метиламино-1-(метансульфонил)пирролидин получают аналогичным образом из (3R)-(метиламино)-1-бензилпирролидина.
Производные тетрагидро-3-тиофенамин-1,1-диоксида получают в соответствии с протоколом, описанным Loev, B. J. Org. Chem. 1961, 26, 4394-9, посредством реакции взаимодействия 3-сульфолена с требуемым первичным амином в метаноле (кат. KOH, к.т.). N-метил-3-тетрагидротиофен-амин-1,1-диоксид (TFA-соль): 1H-ЯМР (ДМСО-d6): δ (м.д.) 9,4 (шир. с, 2H), 4,0-3,8 (квинт., 1H), 3,6-3,5 (дд, 1H), 3,4-3,3 (м, 1H), 3,2-3,1 (м, 2H), 2,5 (с, 3H), 2,4 (м, 1H), 2,1 (м, 1H). N-2-(1-гидрокси)этил-3-тетрагидротиофенамин-1,1-диоксид: (m/z): [М+H]+ для C6H13NO3S: вычислено, 180,07; найдено, 180,2.
N-метил-тетрагидро-2H-тиопиран-4-амин-1,1-диоксид получают из тетрагидро-4H-тиопиран-4-она: (i) MeNH2, NaBH4; (ii) (Boc)2O, MeOH; (iii) mCPBA, CH2Cl2, 0°C; (iv) CF3CO2H, CH2Cl2. (m/z): [М+H]+ для C6H13NO2S: вычислено 164,07; найдено, 164,9. 1H-ЯМР (CD3OD; TFA-соль): δ (м.д.) 3,4-3,1 (м, 5H), 2,7 (с, 3H), 2,4 (шир. д, 2H), 2,1 (шир. м, 2H).
1-ацетил-3-(метиламино)пиперидин получают из N3-Cbz защищенного 3-метиламино-пиперидина: (i) AcCl, i-Pr2NEt, CH2Cl2; (ii) H2 (1 атм), 10% Pd/C, EtOH. 1H-ЯМР (CD3OD): δ (м.д.) 4,0 (м, 1H), 3,6 (м, 1H), 3,4-3,2 (м, 2H), 3,0 (м, 1H), 2,6 (с, 3H), 2,1 (с, 3H), 1,8-1,6 (м, 4H).
1-(метансульфонил)-3-(метиламино)пиперидин получают из N3-Cbz-защищенного 3-метиламино-пиперидина: (i) CH3S(O)2Cl, I-Pr2NEt, CH2Cl2; (ii) H2 (1 атм), 10% Pd/C, EtOH. (m/z): [М+H]+ для C7H16N2O2S: вычислено, 193,10; найдено, 193,0. 1H-ЯМР (ДМСО-d6; TFA-соль): δ (м.д.) 3,4 (дд, 1H), 3,2 (м, 2H), 3,10 (с, 3H), 3,0-2,9 (м, 2H), 2,8 (с, 3H), 1,85-1,75 (м, 2H), 1,6-1,4 (м, 2H).
Диметиламид пролина и иминодиацетонитрил закупали у фирм Bachem и Aldrich, соответственно.
N-производные пиперазина, такие как (метоксикарбонил)пиперазин, 1-(диметиламинокарбонил)пиперазин и 1-(диметиламиносульфонил)пиперазин получают посредством реакции взаимодействия пиперазина с метилхлорформиатом, диметиламинохлорформиатом или диметиламиносульфамоилхлоридом, соответственно.
1-метиламино-2-метилсульфонилэтан получают посредством реакции взаимодействия метиламина с метилвинилсульфоном в метаноле. N-[2-(2-метоксиэтиламино)этил]-N-метил-метансульфонамид синтезировали из частично N-Boc-защищенного этандиамина после проведения четырех стадий реакций в нижеследующей последовательности: (i) метилсульфонилхлорид, триэтиламин; (ii) MeI, Cs2CO3; (iii) NaH, 1-бром-2-метоксиэтан; (iv) CF3CO2H.
Изонипекотамид (пиперидин-4-карбоксамид) и амид пролина закупали у Aldrich. 2-гидроксиметилморфолин поставлялся фирмой Tyger Scientific Product.
Метил-4-пиперидинилкарбамат получают посредством реакции взаимодействия N1-Boc-защищенного 4-аминопиперидина с метилхлорформиатом, с последующим удалением N-Boc-группы.
4-пиперидинол-диметилкарбамат и N-диметил-N'-(3-пиперидинил)мочевину получают посредством реакции взаимодействия диметилкарбамоилхлорида с N-Boc-защищенным 4-пиперидинолом или N1-Boc-3-аминопиперидином, соответственно.
3-(метиламино)-1-(диметиламиносульфонил)пирролидин получают посредством реакции взаимодействия 3-(N-метил-N-Boc-амино)пирролидина с диметилсульфамоилхлоридом.
2-(3-пирролидинил)изотиазолидин-1,1-диоксид синтезировали путем обработки N1-Boc-защищенного 3-аминопирролидина 3-хлорпропилсульфонилхлоридом в присутствии триэтиламина, с последующей обработкой TFA для удаления Boc-группы.
Пример 1: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-((S)-2-метоксиметилпирролидин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение 8-бензил-8-азабицикло[3.2.1]октан-3-она
Концентрированную хлористоводородную кислоту (30 мл) добавляют, перемешивая, к гетерогенному раствору 2,5-диметокси- тетрагидрофурана (82,2 г, 0,622 моль) в воде (170 мл). В отдельной колбе, охлажденной до 0°C (на ледяной бане), концентрированную хлористоводородную кислоту (92 мл) медленно добавляют к раствору бензиламина (100 г, 0,933 моль) в воде (350 мл). 2,5-диметокситетрагидрофурановый раствор перемешивают примерно 20 минут, разбавляют водой (250 мл), а затем добавляют раствор бензиламина, после чего добавляют раствор 1,3-ацетондикарбоновой кислоты (100 г, 0,684 моль) в воде (400 мл) с последующим добавлением бифосфата натрия (44 г, 0,31 моль) в воде (200 мл). pH доводят до значения от 1 до ~4,5 с использованием 40% NaOH. Полученный мутный бледно-желтый раствор перемешивают в течение ночи. Затем раствор подкисляют до pH 3-7,5 с использованием 50% хлористоводородной кислоты, нагревают до 85°C и перемешивают в течение 2 часов. Раствор охлаждают до комнатной температуры, подщелачивают до pH 12 с использованием 40% NaOH, и экстрагируют ДХМ (3×500 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении с получением указанного в заголовке промежуточного соединения в виде вязкого коричневого масла с количественным выходом (чистота ~95%, на что указывает аналитическая ВЭЖХ) 1H-ЯМР (CDCl3) δ (м.д.) 7,5-7,2 (м, 5H, C6H5), 3,7 (с, 2H, CH2Ph), 3,45 (шир. с, 2H, CH-NBn), 2,7-2,6 (дд, 2H, CH2CO), 2,2-2,1 (дд, 2H, CH2CO), 2,1-2,0 (м, 2H, CH2CH2), 1,6 (м, 2H, CH2CH2). (m/z): [М+H]+ для C14H17NO: вычислено, 216,14; найдено, 216,0.
b. Получение трет-бутилового эфира 3-оксо-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
К раствору 8-бензил-8-азабицикло[3.2.1]октан-3-она (75 г, 0,348 моль) в EtOAc (300 мл) добавляют раствор ди-трет-бутил-дикарбоната (83,6 г, 0,383 моль, 1,1 экв.) в EtOAc (300 мл). Полученный раствор и промывку (100 мл EtOAc) добавляют, в потоке азота, в 1-литровый гидрогенизатор Парра, содержащий 23 г гидроксида палладия (20 мас.% Pd, из расчета сухого вещества, на угле, ~50% влажность; например, катализатор Pearlman). Реакционный сосуд дегазируют (поочередно пять раз в вакууме и пять раз в атмосфере N2) и подают газообразный H2 под давлением 60 фунт/кв.дюйм. Реакционный раствор перемешивают в течение двух дней и снова подают H2, необходимый для поддержания давления H2 при 60 фунт/кв.дюйм до тех пор, пока реакция не будет завершена, на что указывает тонкослойная хроматография на двуокиси кремния. Затем раствор фильтруют через слой целита® и концентрируют при пониженном давлении с получением указанного в заголовке промежуточного соединения с количественным выходом в виде вязкого желто-оранжевого масла. Это вещество используют в следующей стадии без дополнительной обработки. 1H-ЯМР (CDCl3) δ (м.д.) 4,5 (шир., 2H, CH-NBoc), 2,7 (шир., 2H, CH2CO), 2,4-2,3 (дд, 2H, CH2CH2), 2,1 (шир. м, 2H, CH2CO), 1,7-1,6 (дд, 2H, CH2CH2), 1,5 (с, 9H, (CH3)3COCON)).
c. Получение трет-бутилового эфира (1S,3R,5R)-3-амино-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
К раствору продукта, полученного в предыдущей стадии (75,4 г, 0,335 моль) в метаноле (1 л), добавляют формиат аммония (422,5 г, 6,7 моль), воду (115 мл) и 65 г палладия на активированном угле (10% из расчета сухого вещества ~50% влажность; типа Degussa E101NE/W) в потоке N2, перемешивают с помощью механической мешалки. Через каждые 24 и 48 часов добавляют дополнительные порции формиата аммония (132 г, 2,1 моль). После прекращения реакции, на что указывает аналитическая ВЭЖХ, добавляют целит® (>500 г), после чего полученную густую суспензию фильтруют и собранное твердое вещество промывают метанолом (~500 мл). Фильтраты объединяют и концентрируют при пониженном давлении до тех пор, пока не будет удален весь метанол. Затем полученный мутный двухфазный раствор разбавляют 1М фосфорной кислотой до конечного объема ~1,5-2,0 л при рН 2 и промывают дихлорметаном (3×700 мл). Водный слой подщелачивают до pH 12 с использованием 40% вод. NaOH и экстрагируют дихлорметаном (3×700 мл). Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют на роторном испарителе, а затем в условиях высокого вакуума, в результате чего получают 52 г (70%) указанного в заголовке промежуточного соединения, обычно называемого N-Boc-эндо-3-аминотропаном, в виде твердого вещества от белого до светло-желтого цвета. Отношение изомеров эндо:экзо-амина указанного продукта составляет >99, на что указывает 1H-ЯМР-анализ (чистота >96%, в соответствии с аналитической ВЭЖХ). 1H-ЯМР (CDCl3) δ (м.д.) 4,2-4,0 (шир. д, 2H, CHNBoc), 3,25 (т, 1H, CHNH2), 2,1-2,05 (м, 4H), 1,9 (м, 2H), 1,4 (с, 9H, (CH3)3OCON), 1,2-1,1 (шир., 2H). (m/z): [М+H]+ для C12H22N2O2: вычислено, 227,18; найдено, 227,2. Аналитическая ВЭЖХ (изократический метод; 2:98 (A:B) - 90:10 (A:B) в течение 5 мин): время удерживания=3,68 мин.
d. Получение 1-изопропил-1H-индазол-3-карбоновой кислоты
В соответствии с процедурой, описанной Harada et al., Chemical & Pharmaceutical Bulletin 1995, 43, 1912-30, сначала, индазол-1H-3-карбоновую кислоту превращают в метиловый эфир (с чистотой >95%) путем кипячения с обратным холодильником кислоты в метаноле, содержащем несколько капель концентрированной серной кислоты. (m/z): [М+H]+ 177,0; 1H-ЯМР-спектроскопия (CD3OD; δ (м.д.) 8,0 (1H, д), 7,5 (1H, д), 7,4 (1H, т), 7,2 (1H, т), 3,9 (3H, с)). Этот сложный эфир обрабатывают изопропилйодидом и трет-бутоксидом калия в кипящем ТГФ, и получают метиловый эфир 1-изопропил-1H-индазол-3-карбоновой кислоты. ТСХ (Rf=0,45 в смеси гексан/EtOAc, 3/1). 1H-ЯМР (CD3OD): δ (м.д.) 8,1-8,0 (1H, д), 7,6 (1H, д), 7,4 (1H, т), 7,2 (1H, т), 5,0 (1H, квинт.), 3,9 (с, 3H), 1,5 (6H, д). N1-изопропилметиловый эфир гидролизируют в 1M NaOH/ТГФ при комнатной температуре с получением указанного в заголовке промежуточного соединения. (m/z): [М+Na]+ 226,6. 1H-ЯМР (CD3OD): δ (м.д.) 8,1-8,0 (1H, д), 7,6 (1H, д), 7,4 (1H, т), 7,2 (1H, т), 5,0 (1H, квинт.), 1,5 (6H, д).
e. Получение трет-бутилового эфира (1S,3R,5R)-3-[1-изопропил-1H-индазол-3-карбонил)амино]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
Суспензию 1-изопропил-1H-индазол-3-карбоновой кислоты (56,35 г; 0,276 моль) в толуоле (500 мл) перемешивают и нагревают в течение 5 минут, а затем добавляют тионилхлорид (30,2 мл; 0,414 моль). После нагревания при 100°С в течение 15 минут, смесь превращается в гомогенный раствор, который перемешивают при той же температуре в течение еще 90 минут. В отдельной реакционной колбе, трет-бутиловый эфир (1S,3R,5R)-3-амино-8-азабицикло[3.2.1]октан-8-карбоновой кислоты, полученный на стадии c (62,43 г; 0,276 моль), растворяют в 250 мл толуола, а затем добавляют гидроксид натрия (66,3 г), растворенный в 250 мл воды. Эту двухфазную смесь охлаждают на ледяной бане. Раствор хлорангидрида индазоловой кислоты, полученный как описано выше, охлаждают до комнатной температуры и в течение 15 минут добавляют к двухфазному раствору, который затем интенсивно перемешивают на ледяной бане. После перемешивания в течение 1,5 часа, реакционную смесь переносят в делительную воронку. Сначала, водный слой отделяют от толуолового слоя (сохраняют) и экстрагируют EtOAc (2×500 мл). Толуоловый слой концентрируют при пониженном давлении, и полученный остаток растворяют в органическом экстракте (1 л; EtOAc). Раствор промывают 1М H3PO4 (400 мл) и нас. NaHCO3 (400 мл), а затем насыщенным раствором соли (400 мл). После сушки над MgSO4, органический раствор упаривают досуха при пониженном давлении, в результате чего получают 119,2 г указанного в заголовке промежуточного соединения. 1H-ЯМР (ДМСО-d6): δ (м.д.) 1,41 (с, 9H), 1,51 (д, 6H), 1,82 (м, 2H), 1,97 (шир. с, 4H), 2,09 (м, 2H), 4,10 (м, 3H), 5,10 (септ., 1H), 7,23 (т, 1H), 7,42 (т, 1H), 7,79 (д, 1H), 7,82 (д, 1H), 8,18 (д, 1H). (m/z): [М+H]+ для C23H32N4O3, вычислено 413,26; найдено, 413,1. Время удерживания (анал. ВЭЖХ: 2-95% MeCN/H2O в течение 6 мин)=4,85 мин.
f. Получение {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
Трет-бутиловый эфир (1S,3R,5R)-3-[1-изопропил-1H-индазол-3-карбонил)амино]-8-азабицикло-[3.2.1]октан-8-карбоновой кислоты, продукт стадии (e), солюбилизируют в дихлорметане (200 мл), охлаждают на ледяной бане, а затем смешивают с 200 мл трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Затем эту смесь по каплям добавляют к этиловому эфиру (2 л), перемешиваемому в колбе, в результате чего получают 102,7 г указанного в заголовке промежуточного соединения в виде соли моно(трифторуксусной кислоты) (выход 87% для двух стадий). 1H-ЯМР (ДМСО-d6): δ (м.д.) 1,54 (д, 6H), 2,05 (м, 2H), 2,24 (м, 6H), 4,03 (с, 2H), 4,12 (кв., 1H), 5,09 (септ., 1H), 7,28 (т, 1H), 7,45 (т, 1H), 7,81 (д, 1H), 8,00 (д, 1H), 8,11 (д, 1H), 8,54 (шир. д, 2H). (m/z): [М+H]+ для C18H24N4O: вычислено, 313,20; найдено, 313,1. Время удерживания (анал. ВЭЖХ: 2-95% MeCN/H2O в течение 6 минут)=2,65 минут.
g. Синтез {(1S,3R,5R)-8-[2-гидрокси-3-((S)-2-метоксиметил-пирролидин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
(S)-2-Метоксиметилпирролидин (0,15 ммоль) добавляют к перемешиваемому раствору монотрифторацетатной соли {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты (64 мг, 0,15 ммоль) и диизопропилэтиламина (0,078 мл, 0,45 ммоль) в метаноле (1,5 мл). Затем добавляют 1,3-дибромпропанол (0,015 мл, 0,15 ммоль) и реакционную смесь нагревают при 65°C в течение 16 ч. Реакционную смесь концентрируют в вакууме, разбавляют смесью уксусная кислота/вода, 1:1 (1 мл) и очищают препаративной ВЭЖХ, в результате чего получают бистрифторацетатную соль указанного в заголовке соединения. (m/z): [М+H]+ для C27H41N5O3, вычислено, 484,33; найдено 484,3. Время удерживания (анал. ВЭЖХ)=2,04 мин.
Примеры 2-4
Соединения примеров 2-4 получают способами, аналогичными способам, описанным в примере 1, за исключением того, что в стадии (g) вместо (S)-2-метоксиметилпирролидина используют соответствующий реагент.
Пример 2:
Синтез ((1S,3R,5R)-8-{2-гидрокси-3-[4-(тетрагидрофуран-2-карбонил)пиперазин-1-ил]пропил}-8-аза-бицикло[3.2.1]окт-3-ил)амида 1-изопропил-1H-индазол-3-карбоновой кислоты; (m/z): [М+H]+ для C30H44N6O4: вычислено 553,35; найдено 553,7. Время удерживания (анал. ВЭЖХ)=2,02 мин.
Пример 3:
Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло-[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты; (m/z): [М+H]+ для C26H40N6O4S: вычислено, 533,29; найдено 533,3. Время удерживания (анал. ВЭЖХ)=2,03 мин.
Пример 4:
Синтез ((1S,3R,5R)-8-{3-[(2-циано-этил)метиламино]-2-гидроксипропил}-8-азабицикло[3.2.1]окт-3-ил)амид 1-изопропил-1H-индазол-3-карбоновой кислоты; (m/z): [М+H]+ для C25H36N6O2: вычислено 453,30; найдено 453,2. Время удерживания (анал. ВЭЖХ)=2,00 мин.
Пример 4: Альтернативный синтез ((1S,3R,5R)-8-{3-[(2-циано-этил)метиламино]-2-гидроксипропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение 3-гидрокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана
Эпибромгидрин (0,06 мл, 0,70 ммоль) добавляют к перемешиваемому раствору трифторацетатной соли {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты, полученной как описано в стадии (f) примера 1 (200 мг, 0,47 ммоль), и DIPEA (0,082 мл, 0,47 ммоль), в этаноле (4 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 12 ч, а затем снова добавляют эпибромгидрин (2×0,06 мл, 0,70 ммоль). Еще через 72 ч, реакционную смесь концентрируют в вакууме и получают неочищенное масло, которое используют без дополнительной очистки.
Альтернативно, 3-гидрокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октан получают следующим образом. Эпибромгидрин (1,65 мл, 19,23 ммоль) добавляют к перемешиваемому раствору трифторацетатной соли {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты, полученной как описано в стадии (f) примера 1 (3,00 г, 9,62 ммоль), в этаноле (50 мл), при комнатной температуре. Реакционную смесь перемешивают в течение 96 ч, а затем снова добавляют эпибромгидрин (0,165 мл, 1,92 ммоль). Еще через 4 ч реакционную смесь концентрируют в вакууме с получением неочищенного масла (4,29 г), которое использовали без дополнительной очистки. (m/z): [М+H]+ для C21H29N4O2: вычислено 369,23; найдено 368,9. [М]+1Н-ЯМР (ДМСО): 1,47 (д, 6H), 2,19-2,40 (м, 8H), 3,85-4,05 (м, 3H), 4,21-4,43 (м, 3H), 4,57-4,73 (м, 2H), 5,10 (септ, 1H), 6,22 (д, 1H), 7,31 (м, 1H), 7,43 (м, 1H), 7,82 (д, 1H), 8,03 (м, 1H), 8,16 (д, 1H).
b. Синтез ((1S,3R,5R)-8-{3-[(2-циано-этил)метиламино]-2-гидроксипропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1H-индазол-3-карбоновой кислоты
3-метиламинопропаннитрил (0,066 мл, 0,71 ммоль) добавляют к 3-гидрокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}-спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октану, продукт предыдущей стадии (0,47 ммоль), и диизопропилэтиламину (0,245 мл, 1,41 ммоль) в метаноле (4 мл). Реакционную смесь встряхивают в нагревательном блоке при 65°C в течение 16 ч. Затем добавляют 3-метиламинопропаннитрил (2×0,066 мл, 0,71 ммоль) и через 20 часов реакционную смесь охлаждают, концентрируют в вакууме, разбавляют смесью уксусная кислота/вода, 1:1 (3 мл) и очищают препаративной ВЭЖХ, в результате чего получают бис-трифторацетатную соль указанного в заголовке соединения. (m/z): [М+H]+ для C25H36N6O2: вычислено 453,30; найдено 453,2. Время удерживания (анал. ВЭЖХ)=2,00 мин.
Пример 5: Синтез {(1S,3R,5R)-8-[3-(4-диметилкарбамоилпиперазин-1-ил)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
Пиперазин-4-диметилмочевину (23 мг, 0,15 ммоль) растворяют в растворе 3-гидрокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана, полученного как описано в примере 4, стадии (a) (42 мг, 0,10 ммоль), и диизопропилэтиламина (0,017 мл, 0,15 ммоль) в этаноле (2 мл). Реакционную смесь встряхивают в нагревательном блоке при 80°C в течение 16 ч. Реакционную смесь концентрируют в вакууме, разбавляют 50% уксусной кислотой в воде (1,5 мл) и очищают препаративной ВЭЖХ (в градиенте 5-75%) с получением бис-трифторацетатной соли указанного в заголовке соединения (56 мг) в виде белого твердого вещества. (m/z): [М+H]+ для C28H43N7O3: вычислено 526,35; найдено 526,4. Время удерживания (анал. ВЭЖХ)=1,92 мин.
Пример 6: Синтез метилового эфира [1-(2-гидрокси-3-{(1S,3R,5R)-3-[(1-изопропил-1H-индазол-3-карбонил)амино]-8-азабицикло[3.2.1]окт-8-ил}пропил)пирролидин-3-ил]карбаминовой кислоты
Пирролидин-3-NH-метилкарбамат (моно-TFA-соль; 39 мг, 0,15 ммоль) растворяют в растворе 3-гидрокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана, полученного как описано в примере 4, стадии (a) (32 мг, 0,075 ммоль), и диизопропилэтиламина (0,065 мл, 0,37 ммоль) в ДМФ (2 мл). Реакционную смесь встряхивают в нагревательном блоке при 80°C в течение 16 ч. Реакционную смесь концентрируют в вакууме, разбавляют 50% уксусной кислотой в воде (1,5 мл) и очищают препаративной ВЭЖХ (в градиенте 5-75%) с получением бис-трифторацетатной соли указанного в заголовке соединения (35 мг) в виде белого твердого вещества. (m/z): [М+H]+ для C27H40N6O4: вычислено 513,32; найдено 513,4. Время удерживания (анал. ВЭЖХ)=1,98 мин.
Пример 7: Синтез {(1S,3R,5R)-8-[3-(4-метансульфонилпиперазин-1-ил)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение 3-метокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана
Эпибромгидрин (2,81 мл, 32,86 ммоль) добавляют к перемешиваемому раствору TFA-соли {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты, полученной как описано в примере 1, стадии (f) (7,00 г, 16,43 ммоль), и диизопропилэтиламина (2,86 мл, 16,43 ммоль) при комнатной температуре. Реакционную смесь перемешивают в течение 72 ч и концентрируют в вакууме с получением неочищенного масла (11,7 г, содержание предполагаемого продукта составляло 63 мас.%), содержащего в качестве примесей соли диизопропилэтиламина. Образец неочищенного масла (1,5 г) суспендируют в дихлорметане (45 мл) и охлаждают до 0°С. Затем добавляют трет-бутоксид калия (472 мг, 4,21 ммоль), после чего добавляют метилйодид (0,145 мл, 2,32 ммоль) и реакционную смесь оставляют для нагревания до комнатной температуры. Через 30 минут, реакционную смесь охлаждают до 0°С и добавляют дополнительное количество трет-бутоксида калия (2×236 мг, 2,10 ммоль) и метилиодида (2×0,145 мл, 2,32 ммоль). Через 1 час, аналитическая ВЭЖХ указывала приблизительно на 90% превращение исходного вещества в основной продукт. Реакцию гасят добавлением воды (20 мл) и смесь разбавляют дихлорметаном (50 мл). Слои разделяют, и водный слой снова экстрагируют дихлорметаном (50 мл) и хлороформом (25 мл). Органические слои объединяют, сушат (MgSO4), фильтруют и концентрируют в вакууме с получением неочищенного продукта, который используют без дополнительной очистки (1 г, чистота 85% по ВЭЖХ).
b. Синтез {(1S,3R,5R)-8-[3-(4-метансульфонилпиперазин-1-ил)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
N-метилсульфонилпиперазин (70 мг, 0,252 ммоль) добавляют к раствору неочищенного продукта, полученного в предыдущей стадии (64 мг), и диизопропилэтиламина (0,131 мл, 0,755 ммоль) в этаноле (1,5 мл). Реакционную смесь встряхивают в нагревательном блоке при 80°С в течение 16 ч. Реакционную смесь концентрируют в вакууме, разбавляют смесью уксусная кислота/вода, 1:1 (1 мл), и очищают препаративной ВЭЖХ с получением бис-трифторацетатной соли указанного в заголовке соединения. (m/z): [М+H]+ для C27H42N6O4S: вычислено 547,31; найдено 547,2.
Пример 8: Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение 1-хлор-(R)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропана
(R)-эпихлоргидрин (3,10 мл, 39,5 ммоль) добавляют к перемешиваемому раствору TFA-соли пиперазинсульфонамида (10,0 г, 35,9 ммоль) и диизопропилэтиламина (6,26 мл, 35,9 ммоль) в этаноле (150 мл) при комнатной температуре. Реакционную смесь перемешивают течение 18 ч, добавляют дополнительное количество (R)-эпихлоргидрина (0,28 мл, 3,6 ммоль) и перемешивают еще 3 часа. Реакционную смесь концентрируют в вакууме, и белое твердое вещество суспендируют в этаноле (150 мл) и перемешивают в течение 2 дней. Твердое вещество собирают фильтрацией и промывают холодным этанолом, в результате чего получают указанное в заголовке соединение (5,69 г) в виде белого твердого вещества, которое используют без дополнительной очистки. (m/z): [М+H]+ для C8H17ClN2O3S: вычислено 257,07; найдено 257,2. 1H-ЯМР (ДМСО-d6): δ (м.д.) 2,37 (дд, 1H), 2,45 (дд, 1H), 2,50-2,58 (м, 4H), 2,86 (с, 3H), 3,09 (м, 4H), 3,55 (дд, 1H), 3,65 (дд, 1H), 3,84 (м, 1H), 5,09 (д, 1H).
b. Получение 1-[(2R)-оксиранилметил]-4-метансульфонилпиперазина
Гидроксид натрия (1,07 г, 26,7 ммоль) добавляют к интенсивно перемешиваемому раствору 1-хлор-(R)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропана, продукта предыдущей стадии (5,69 г, 22,2 ммоль), в ТГФ/воде (150 мл/35 мл). Реакционную смесь перемешивают в течение 35 мин, концентрируют в вакууме до объема приблизительно 50 мл, разбавляют хлороформом (200 мл) и промывают 1M NaOH (2×70 мл) и насыщенным раствором соли (70 мл). Органический слой сушат (MgSO4), фильтруют и концентрируют в вакууме с получением белого кристаллического твердого вещества, которое затем перекристаллизовывают из горячего EtOAc/гексана, 1:1 (800 мл), в результате чего получают указанное в заголовке промежуточное соединение (2,62 г) в виде белого кристаллического твердого вещества. (Фильтрат может быть дополнительно перекристаллизован с получением дополнительного количества продукта). ESMS (C8H16N2O3S): вычислено 221,10; найдено 221,3 (m/z): [М+H]+; 1H-ЯМР (ДМСО-d6): δ (м.д.) 2,22 (дд, 1H), 2,45-2,60 (м, 5H), 2,69-2,75 (м, 2H), 2,87 (с, 3H), 3,02 (м, 1H), 3,11 (м, 4H).
c. Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
Раствор 1-[(2R)-оксиранилметил]-4-метансульфонилпиперазина, продукта предыдущей стадии (585 мг, 2,66 ммоль) в толуоле (12 мл) добавляют к {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амиду 1-изопропил-1H-индазол-3-карбоновой кислоты (830 мг, 2,66 ммоль), и смесь перемешивают при 98°C в течение 16 ч. Реакционную смесь охлаждают, концентрируют в вакууме, разбавляют 50% уксусной кислотой в воде, содержащей 10% TFA (12 мл), и очищают препаративной ВЭЖХ, в результате чего получают бис-трифторацетатную соль указанного в заголовке соединения (525 мг) в виде белого твердого вещества. (m/z): [М+H]+ для C26H40N6O4S: вычислено 533,29; найдено 533,5. Время удерживания (анал. ВЭЖХ: 2-40% MeCN/H2O в течение 6 мин)=4,03 мин; 1H-ЯМР (CD3OD): δ (м.д.) 1,55 (д, 6H), 2,41 (м, 6H), 2,55-2,63 (м, 2H), 2,92 (с, 3H), 3,10-3,40 (м, 6H), 3,82 (шир. с, 4H), 4,16-4,24 (м, 3H), 4,66 (м, 1H), 4,99 (септ, 1H), 7,22 (т, 1H), 7,39 (т, 1H), 7,62 (д, 1H), 8,12 (д, 1H).
Пример 9: Синтез {(1S,3R,5R)-8-[(S)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]-окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение 1-хлор-(S)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропана
(S)-Эпихлоргидрин (48,0 мл, 0,612 моль) добавляют к перемешиваемому раствору пиперазинсульфонамида (87,3 г, 0,532 моль) в этаноле (1,33 л) при комнатной температуре. Реакционную смесь перемешивают в течение 18 ч и образовавшийся белый твердый осадок собирают фильтрацией и промывают этанолом, в результате чего получают указанное в заголовке промежуточное соединение (107,76 г) в виде белого твердого вещества, которое используют без дополнительной очистки. (m/z): [М+H]+ для C8H17ClN2O3S: вычислено 257,07; найдено 257,2. 1H-ЯМР (ДМСО-d6): δ (м.д.) 2,37 (дд, 1H), 2,45 (дд, 1H), 2,50-2,58 (м, 4H), 2,86 (с, 3H), 3,09 (м, 4H), 3,55 (дд, 1H), 3,65 (дд, 1H), 3,84 (м, 1H), 5,09 (д, 1H).
b. Получение 1-[(2S)-оксиранилметил]-4-метансульфонилпиперазина
Гидроксид натрия (22,15 г, 0,534 моль) добавляют к интенсивно перемешиваемому раствору продукта предыдущей стадии (118,13 г, 0,461 моль) в ТГФ/воде (1200 мл/300 мл) при 0°С. Реакционную смесь перемешивают в течение 90 минут и слои разделяют. Органический слой концентрируют в вакууме и разбавляют дихлорметаном (1500 мл), а затем промывают смесью предварительно отделенного водного слоя и 1М NaOH (500 мл). Затем органический слой дополнительно промывают 1M NaOH (500 мл) и насыщенным раствором соли (500 мл), после чего сушат (MgSO4), фильтруют и концентрируют в вакууме с получением белого кристаллического твердого вещества, которое перекристаллизовывают из горячего EtOAc/гексана, 1:1 (800 мл), в результате чего получают указанное в заголовке соединение (43,33 г) в виде белого кристаллического твердого вещества. (Фильтрат может быть дополнительно перекристаллизован с получением дополнительного количества продукта). (m/z): [М+H]+ для C8H16N2O3S: вычислено 221,10; найдено 221,3. 1H-ЯМР (ДМСО-d6): δ (м.д.) 2,22 (дд, 1H), 2,45-2,60 (м, 5H), 2,69-2,75 (м, 2H), 2,87 (с, 3H), 3,02 (м, 1H), 3,11 (м, 4H).
c. Синтез {(1S,3R,5R)-8-[(S)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]-окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
Раствор 1-[(2S)-оксиранилметил]-4-метансульфонилпиперазина, продукта предыдущей стадии (0,44 г, 9,10 ммоль) в толуоле (30 мл) добавляют к {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амиду 1-изопропил-1H-индазол-3-карбоновой кислоты (2,83 г, 9,10 ммоль) и смесь перемешивают при 98°C в течение 16 ч. Реакционную смесь охлаждают, концентрируют в вакууме, разбавляют 50% уксусной кислотой в воде, содержащей 10% TFA (20 мл), и очищают препаративной ВЭЖХ, в результате чего получают бис-трифторацетатную соль указанного в заголовке соединения (1,83 г) в виде белого твердого вещества. (m/z): [М+H]+ для C26H40N6O4S: вычислено 533,29; найдено 533,5. Время удерживания (анал. ВЭЖХ: 2-40% MeCN/H2O в течение 6 минут)=4,03 мин; 1H-ЯМР (CD3OD): δ (м.д.) 1,55 (д, 6H), 2,41 (м, 6H), 2,55-2,63 (м, 2H), 2,92 (с, 3H), 3,10-3,40 (м, 6H), 3,82 (шир. с, 4H), 4,16-4,24 (м, 3H), 4,66 (м, 1H), 4,99 (септ, 1H), 7,22 (т, 1H), 7,39 (т, 1H), 7,62 (д, 1H), 8,12 (д, 1H).
Пример 10: Синтез [(1S,3R,5R)-8-(4-карбамоилметилморфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение 2-хлорметилморфолина
2-хлорметилморфолин синтезируют в соответствии с процедурами, описанными в литературе (Araki, K. et al., J. Med. Chem. 1993, 36, 1356-63; Nishimura, Y. et al., Chem. & Pharm. Bull. 1990, 38, 2190-6), и его вторичный амин защищают N-Boc-группой путем обработки (Boc)2O (растворенным в метаноле, при комнатной температуре).
b. Получение [(1S,3R,5R)-8-(4-(трет-бутоксикарбонил)морфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К раствору монотрифторацетатной соли {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты, полученной как описано в примере 1, стадии (f) (2 г; 4,7 ммоль), и растворенной в 50 мл ацетонитрила, добавляют карбонат калия (1,3 г, 9,4 ммоль) и 2-хлорметилморфолин, продукт предыдущей стадии (1,2 г, 5,1 ммоль). Реакционную смесь перемешивают при 85°С в течение 24 ч и охлаждают до комнатной температуры, а затем реакцию гасят водой (100 мл). Смесь разбавляют дихлорметаном (300 мл) и переносят в делительную воронку. После встряхивания, органический слой собирают и промывают насыщенным раствором соли (100 мл). Этот слой сушат над MgSO4 и упаривают в вакууме с получением указанного в заголовке промежуточного соединения в виде бледно-желтого масла, которое используют без дополнительной очистки. (m/z): [М+H]+ для C28H41N5O4: вычислено 512,33; найдено 512,4.
c. Получение [(1S,3R,5R)-8-(морфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1H-индазол-3-карбоновой кислоты
[(1S,3R,5R)-8-(4-(трет-бутокси-карбонил)морфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амид 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт предыдущей стадии, растворяют в дихлорметане (20 мл), а затем добавляют трифторуксусную кислоту. После перемешивания в течение 30 минут при комнатной температуре, смесь упаривают в вакууме с получением бледно-желтого масла. Это масло растворяют в 50% водном ацетонитриле (5% TFA) и фракционируют с помощью препаративной ВЭЖХ, в результате чего получают трифторацетатную соль указанного в заголовке промежуточного соединения. (m/z): [М+H]+ для C23H33N5O2: вычислено 412,27; найдено 412,6. Время удерживания (анал. ВЭЖХ: 10-40% MeCN/H2O за 6 мин)=3,9 мин.
d. Синтез [(1S,3R,5R)-8-(4-карбамоилметилморфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1H-индазол-3-карбоновой кислоты
[(1S,3R,5R)-8-(морфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амид 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт предыдущей стадии (0,1 г, 0,19 ммоль), растворяют в этаноле (10 мл), содержащем диизопропилэтиламин (0,066 мл, 0,38 ммоль), а затем добавляют бромацетамид (26 мг, 0,21 ммоль). Смесь нагревают при 90°С в течение 12 ч и концентрируют в вакууме с получением светло-желтого масла. Полученное масло фракционируют и получают бис-трифторацетатную соль указанного в заголовке соединения. (m/z): [М+H]+ для C25H36N6O3: вычислено 469,29; найдено 469,2. Время удерживания (анал. ВЭЖХ)=2,28 мин.
Пример 11: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение {(1S,3R,5R)-8-[2-гидрокси-3-метиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К раствору этанола (5 мл), содержащего 3-гидрокси-3'-{[1-изопропил-1H-индазол-3-ил)карбонил]амино}спиро-азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октан, полученного как описано в примере 4, в стадии (a) (0,365 г, 0,8 ммоль), добавляют метиламин в ТГФ (41%, 0,27 мл). Смесь перемешивают при 90°С в течение трех дней и концентрируют в вакууме с получением указанного в заголовке промежуточного соединения. Полученный продукт используют в следующей стадии без дополнительной очистки.
b. Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К холодному раствору дихлорметана (5 мл), содержащего {(1S,3R,5R)-8-[2-гидрокси-3-метиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт предыдущей стадии (0,4 г, 1 ммоль), добавляют 1,8-диазабицикло[5,4,0]ундец-7-ен (0,18 мл, 1,2 ммоль), а затем метансульфонилхлорид (0,082 мл, 1,05 ммоль). Смесь перемешивают на ледяной бане в течение 1 часа, а затем реакцию гасят водой, и смесь концентрируют в вакууме. Остаток растворяют в 50% водной уксусной кислоте и очищают препаративной ВЭЖХ с получением монотрифторацетатной соли указанного в заголовке соединения. (m/z): [М+H]+ для C23H35N5O4S: вычислено 478,25; найдено 478,2. Время удерживания (анал. ВЭЖХ)=3,0 мин.
Пример 12: Синтез {(1S,3R,5R)-8-[3-(ацетил-метиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К холодному раствору ДМФ (5 мл), содержащему {(1S,3R,5R)-8-[2-гидрокси-3-метиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт примера 11, стадии (a) (0,4 г, 1 ммоль), добавляют диизопропилэтиламин (0,21 мл, 1,2 ммоль), а затем ацетилхлорид (0,075 мл, 1,05 ммоль). Смесь перемешивают на ледяной бане в течение 1 ч, а затем реакцию гасят водой и смесь концентрируют в вакууме. Остаток растворяют в 50% водной уксусной кислоте и очищают препаративной ВЭЖХ, в результате чего получают монотрифторацетатную соль указанного в заголовке соединения. (m/z): [М+H]+ для C24H35N5O3: вычислено 442,28; найдено 442,2. Время удерживания (анал. ВЭЖХ)=2,9 мин.
Пример 13: Синтез {(1S,3R,5R)-8-[3-(формил-метиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
Раствор чистого этилформиата (5 мл), содержащего {(1S,3R,5R)-8-[2-гидрокси-3-метиламино)пропил]-8-азабицикло-[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт примера 11, стадии (a) (0,4 г, 1 ммоль), перемешивают при 80°C в течение 2 ч. Смесь концентрируют в вакууме, растворяют в 50% водной уксусной кислоте и очищают препаративной ВЭЖХ с получением монотрифторацетатной соли указанного в заголовке соединения. (m/z): [М+H]+ для C23H33N5O3: вычислено 428,27; найдено 428,2. Время удерживания (анал. ВЭЖХ)=1,4 мин.
Пример 14: Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение (S)-1-(бензил-метил-амино)-3-хлорпропан-2-ола
N-Бензилметиламин (13,95 мл, 108,1 ммоль) и (S)-2-хлорметилоксиран, обычно называемый (S)-эпихлоргидрином, (8,48 мл, 108,1 ммоль) растворяют в гексане (40 мл) и перемешивают в течение 16 ч. Затем раствор подвергают флеш-хроматографии (SiO2, элюируя смесью 10% метанола/90% дихлорметана). Фракции, содержащие продукт, концентрируют с получением указанного в заголовке промежуточного соединения в виде масла (19,7 г). 1H-ЯМР (ДМСО-d6, 299,96 МГц): δ (м.д.) 2,01 (с, 3H), 2,2-2,4 (м, 2H), 3,21-3,5 (м, 3H), 3,53-3,6 (м, 1H), 3,65-3,75 (м, 1H), 4,95 (д, 1H), 7,0-7,25 (м, 5H). (m/z): [М+H]+ для C11H16ClNO: вычислено 214,10; найдено 214,1.
b. Получение трет-бутилового эфира ((S)-3-хлор-2-гидроксипропил)метилкарбаминовой кислоты
(S)-1-(бензил-метил-амино)-3-хлорпропан-2-ол, продукт предыдущей стадии (8,4 г, 39,3 ммоль), растворяют в этилацетате (75 мл), а затем добавляют ди-трет-бутилдикарбонат (9,3 г, 43,23 ммоль) и гидроксид палладия (2,5 г). Смесь встряхивают в течение 12 ч в атмосфере водорода (60 атм). Затем смесь фильтруют через слой целита® и концентрируют досуха при пониженном давлении. Полученное масло фильтруют через двуокись кремния, элюируют гексаном, затем дихлорметаном, а после этого диэтиловым эфиром. Эфирный слой концентрируют с получением указанного в заголовке промежуточного соединения (7,1 г) в виде масла. 1H-ЯМР (ДМСО-d6, 299,96 МГц): δ (м.д.) 1,35-1,46 (с, 9H), 2,81-2,85 (с, 3H), 2,95-3,1 (м, 1H), 3,3-3,6 (м, 3H), 3,67-3,85 (м, 1H), 5,25-5,4 (м, 1H). (m/z): [М+H-Boc]+ для C9H18ClNO3: вычислено 123,10; найдено 123,1,
c. Получение {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(N-трет-бутоксикарбонил-N-метиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К раствору монотрифторацетатной соли {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты (1,3 г, 3,0 ммоль), растворенной в 12 мл метанола, добавляют диизопропилэтиламин (1,57 мл, 9,0 ммоль) и трет-бутиловый эфир ((S)-3-хлор-2-гидроксипропил)метилкарбаминовой кислоты, продукт предыдущей стадии (1 г, 4,49 ммоль). Смесь перемешивают при 90°С в течение 32 ч и упаривают в вакууме, в результате чего получают бледно-желтое масло. Это масло очищают флеш-хроматографией на колонке с двуокисью кремния, элюируя 10% метанолом в дихлорметане, в результате чего получают указанное в заголовке соединение (1,2 г).
d. Получение {(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(N-трет-бутоксикарбонил-N-метиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт предыдущей стадии, растворяют в 10 мл дихлорметана, а затем добавляют трифторуксусную кислоту (10 мл). Смесь перемешивают в течение 10 минут и концентрируют в вакууме. Концентрат промывают эфиром, сушат в вакууме и получают бис-трифторацетатную соль указанного в заголовке промежуточного соединения, которое используют в следующей стадии без дополнительной обработки.
e. Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К холодному раствору дихлорметана (4 мл), содержащего продукт предыдущей стадии (0,54 г, 0,86 ммоль), добавляют 1,8-диазабицикло[5,4,0]ундец-7-ен (0,448 мл, 3 ммоль), а затем метансульфонилхлорид (0,07 мл, 0,9 ммоль). Смесь перемешивают на ледяной бане в течение 1 часа, после чего реакцию гасят водой и концентрируют в вакууме. Остаток растворяют в 50% водной уксусной кислоте и очищают препаративной ВЭЖХ, в результате чего получают монотрифторацетатную соль указанного в заголовке соединения. (m/z): [М+H]+ для C23H35N5O4S: вычислено 478,25; найдено 478,2. Время удерживания (анал. ВЭЖХ)=3,0 мин.
Пример 15: Синтез ((1S,3R,5R)-8-{2-гидрокси-3-[(пиридин-4-карбонил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1H-индазол-3-карбоновой кислоты
a. Получение {(1S,3R,5R)-8-[3-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-2-гидрокси-пропил]-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты
Этаноловый раствор (100 мл) {(1S,3R,5R)-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты (3,15 г, 10 ммоль) перемешивают при 22°C в 250 мл колбе. Затем добавляют 2-оксиранилметил-изоиндол-1,3-дион, обычно называемый эпоксипропилфталимидом (3,07 г, 15 ммоль), и раствор нагревают при 80°C в течение 48 ч. Реакционную смесь охлаждают до комнатной температуры и добавляют этилацетат (200 мл). После завершения промывок насыщенным NaHCO3 и насыщенным раствором NaCl, органические слои объединяют и сушат над MgSO4. После фильтрации, летучие вещества удаляют в вакууме с получением указанного в заголовке промежуточного соединения (5,5 г) в виде желтого твердого вещества. (m/z): [М+H]+ для C29H39N5O4: вычислено 516,26; найдено 516,5. Время удерживания (анал. ВЭЖХ: 10-70% MeCN/H2O за 6 минут)=3,18 мин.
b. Получение [(1S,3R,5R)-8-(2-гидрокси-3-аминопропил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1Н-индазол-3-карбоновой кислоты
Этаноловый раствор (80 мл) {(1S,3R,5R)-8-[3-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-2-гидрокси-пропил]-8-аза-бицикло[3.2.1]окт-3-ил}амида 1-изопропил-1H-индазол-3-карбоновой кислоты, продукт предыдущей стадии, (3,0 г, 5,8 ммоль) перемешивают при 22°С в 250 мл колбе. Затем добавляют чистый раствор гидразина (3,07 г, 15 ммоль) и смесь нагревают при 80°С в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры и фильтруют для удаления нерастворившегося вещества. После выпаривания фильтрата в вакууме получают указанное в заголовке промежуточное соединение (2,5 г) в виде желтого твердого вещества, (m/z): [М+Н]+ для C29H39N5O4: вычислено 386,26; найдено 386,2. Время удерживания (анал. ВЭЖХ: 10-70% MeCN/H2O за 6 минут)=1,85 мин.
с. Синтез ((1S,3R,5R)-8-{2-гидрокси-3-[(пиридин-4-карбонил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К размешиваемому дихлорметановому раствору (5 мл) [(1S,3R,5R)-8-(2-гидрокси-3-аминопропил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1Н-индазол-3-карбоновой кислоты, продукта предыдущей стадии, (42 мг, 0,10 ммоль), охлажденному при 0°С, добавляют хлорангидрид пиридин-4-карбоновой кислоты (21 мг, 0,12 ммоль) в виде твердого вещества и диизопропилэтиламин (0,021 мл, 0,12 ммоль) путем микропипетирования. Реакционную смесь перемешивают при 0°С в течение 5 минут, а затем нагревают до комнатной температуры и перемешивают в течение 16 часов. Смесь концентрируют в вакууме, разбавляют 50% водной уксусной кислотой (1,5 мл) и очищают препаративной ВЭЖХ (элюируя градиентом 5-75%), в результате чего получают монотрифторацетатную соль указанного в заголовке соединения (22 мг) в виде белого твердого вещества. (m/z): [М+H]+ для C27H34N6O3: вычислено 491,28; найдено 491,1. Время удерживания (анал. ВЭЖХ)=1,91 мин.
Пример 16: Синтез [(1S,3R,5R)-8-(3-формиламино-2-гидроксипропил)-8-азабицикло[3.2.1]окт-3-ил]-амида 1-изопропил-1H-индазол-3-карбоновой кислоты
К ДМФ-раствору (5 мл) ((1S,3R,5R)-8-{2-гидрокси-3-[(пиридин-4-карбонил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1H-индазол-3-карбоновой кислоты, продукта примера 15, (42 мг, 0,10 ммоль) добавляют этилформиат (0,01 мл, 1,2 ммоль) путем микропипетирования. Реакционную смесь герметично закрывают и встряхивают при 80°С в течение 16 часов. Затем смесь концентрируют в вакууме и разбавляют 50% водной уксусной кислотой (1,5 мл) и очищают препаративной ВЭЖХ (градиентом 5-75%), в результате чего получают монотрифторацетатную соль указанного в заголовке соединения (35 мг) в виде белого твердого вещества. (m/z): [М+H]+ для C22H31N5O3: вычислено 414,25; найдено 414,1. Время удерживания (анал. ВЭЖХ)=2,04 мин.
Пример 17: Соединения согласно изобретению
В соответствии с процедурами, описанными в примерах 1-16, и их вариантами получают соединения, представленные в таблицах I-X, которые были охарактеризованы с помощью масс-спектрометрии. В таблицах, содержащих соединения, полученные в виде чистых стереомеров, хиральность атома углерода, отмеченного звездочкой, указана в столбце, озаглавленном звездочкой (*). В соединениях таблиц I-X, индазол-карбоксамидная группа присутствует в эндо-конфигурации по отношению к азабицикло-октановой группе.
Таблица I

[M+H]
[M+H]




Таблица II

Таблица III

Таблица IV

Таблица V




Таблица VII
Таблица VIII

Таблица IX
Таблица Х












Пример 18: Анализ на связывание радиоактивного лиганда с человеческими 5-HT4(c)-рецепторами
a. Мембранный препарат 5-HT4(c)
Клетки НЕК 293 (клетки почек человеческого эмбриона), стабильно трансфецированные кДНК человеческого 5-HT4(c)-рецептора (Bmax=~6,0 пмоль/мг белка, как было определено в анализе на связывание с радиоактивным мембранным лигандом [3H]-GR113808), культивируют в колбах T-225 в модифицированной по способу Дульбекко среде Игла (DMEM), которая содержит 4500 мг/л D-глюкозы и гидрохлорида пиридоксина (GIBCO-Invitrogen Corp., Carlsbad CA: Cat #11965), и в которую добавлены 10% фетальная бычья сыворотка (FBS) (GIBCO-Invitrogen Corp.: Cat #10437), 2 мМ L-глутамин и смесь пенициллина (100 единиц)-стрептомицина (100 мкг)/мл (GIBCO-Invitrogen Corp.: Cat #15140) в 5% CO2, в инкубаторе с повышенной влажностью, при 37°С. Клетки культивируют под непрерывным давлением отбора путем добавления в среду 800 мкг/мл генетицина (GIBCO-Invitrogen Corp.: Cat #10131).
Клетки культивируют приблизительно до 60-80%-ной конфлюентности (<35 пассажей субкультуры). За 20-22 часов до сбора, клетки два раза промывают и подпитывают бессывороточной DMEM. Все стадии получения мембранного препарата проводят на льду. Затем клеточный монослой встряхивают путем легкого механического перемешивания и гомогенизации 25-миллилитровой пипеткой. Клетки собирают путем центрифугирования при 1000 об/мин (5 мин).
Для получения мембранного препарата, клеточные осадки ресуспендируют в охлажденной льдом 50 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоте (HEPES), pH 7,4 (буфер для мембранного препарата) (40 мл/общий выход клеток от 30-40 колб T225) и гомогенизируют с использованием политронного дезинтегратора (c установленными параметрами 19,2×10 с) на льду. Полученные гомогенаты центрифугируют при 1200×g в течение 5 мин при 4°C. Осадок отбрасывают, и супернатант центрифугируют при 40000×g (20 мин). Осадок один раз промывают путем ресуспендирования в буфере для мембранных препаратов и центрифугировали при 40000×g (20 мин). Конечный осадок ресуспендируют в 50 мМ HEPES, pH 7,4 (аналитический буфер) (1 эквивалент, колба T225/1 мл). Концентрацию белка в мембранной суспензии определяют методом Брэдфорда (Bradford, 1976). Мембраны хранят в замороженном виде в аликвотах при -80°C.
b. Анализы на связывание с радиоактивным лигандом
Анализы на связывание с радиоактивным лигандом осуществляют в 1,1 - миллилитровых глубоких 96-луночных полипропиленовых аналитических планшетах (Axygen) в общем аналитическом объеме 400 мл, содержащем 2 мкг мембранного белка в 50 мм буфере HEPES pH 7,4, включающем 0,025% альбумина бычьей сыворотки (BSA). Исследования на связывание с насыщением, проводимые в целях определения величин Kd для радиоактивных лигандов, осуществляют с использованием [3H]-GR113808 (Amersham Inc., Bucks, UK: Cat #TRK944; удельная активность ~82 Ки/ммоль) при 8-12 различных концентрациях в пределах от 0,001 нМ до 5,0 нМ. Анализы на вытеснение метки, проводимые для определения величин pKi для соединений, осуществляют с использованием [3H]-GR113808 в концентрации 0,15 нМ и одиннадцати различных концентраций соединений в пределах от 10 пМ-100 мМ.
Тестируемые соединения получают в виде 10 мм маточных растворов в ДМСО и разводят до 400 мМ в 50 мм HEPES pH 7,4 при 25°С, содержащем 0,1% BSA, и в том же самом буфере делают серийные разведения (1:5). Неспецифическое связывание определяют в присутствии 1 мМ немеченного GR113808. Аналитические смеси инкубируют в течение 60 минут при комнатной температуре, а затем реакции связывания прекращают путем быстрой фильтрации на 96-луночных фильтровальных планшетах GF/B из стекловолокна (Packard BioScience Co., Meriden, CT), предварительно смоченных в 0,3% полиэтиленимине. Эти фильтровальные планшеты три раза промывают фильтрующим буфером (охлажденным льдом 50 мМ HEPES, pH7,4) для удаления несвязанной радиоактивности. Планшеты сушат, а затем в каждую лунку добавляют 35 мкл сцинтилляционной жидкости Microscint-20 (Packard BioScience Co., Meriden, CT), и планшеты считывают на жидкостном сцинтилляционном счетчике Packard Topcount (Packard BioScience Co., Meriden, CT).
Данные по связыванию анализируют с помощью нелинейного регрессионного анализа с помощью программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и с использованием 3-параметрической модели конкуренции за моносайтовое связывание. BOTTOM (минимальное значение на кривой) представляет собой фиксированную величину неспецифического связывания, определенную в присутствии 1 мкМ GR113808. С помощью Prism были вычислены величины Ki для тестируемых соединений, исходя из величин наилучшего соответствия IC50, а величину Kd для радиоактивного лиганда вычисляют по уравнению Ченга-Прусова (Cheng и Prusoff, Biochemical Pharmacology, 1973, 22, 3099-108): Ki=IC50/(1+[L]/Kd), где [L]=концентрация [3H]-GR113808. Результаты выражают в виде отрицательных десятичных логарифмов величин Ki, pKi.
Тестируемые соединения, имеющие более высокое значение pKi в этом анализе, имеют более высокую аффинность связывания с 5-НТ4-рецептором. Соединения согласно изобретению, которые были протестированы в этом анализе, имеют величину pKi в пределах примерно от 6,9 до 9,5, а обычно, в пределах примерно от 7,0 до 8,6.
Пример 19: Анализ на связывание радиоактивного лиганда с человеческими 5-HT3A-рецепторами: определение селективности по отношению к рецептору данного подтипа
a. Мембранный препарат 5-HT3A
Клетки НЕК 293 (клетки почек человеческого эмбриона), стабильно трансфецированные кДНК человеческого 5-HT3A-рецептора, получают от Dr. Michael Bruess (Боннский Университет, Германия) (Bmax=~9,0 пмоль/мг белка, как было определено в анализе на связывание с радиоактивным мембранным лигандом [3H]-GR65630). Клетки культивируют в колбах T-225 или в клеточных фабриках в 50% модифицированной по способу Дульбекко среде Игла (DMEM), (GIBCO-Invitrogen Corp., Carlsbad CA: Cat #11965), и в 50% среде Хэмса F12 (GIBCO-Invitrogen Corp.: Cat #11765), в которую добавляют 10% термоинактивированную фетальную бычью сыворотку (FBS) (Hyclone, Logan, UT: Cat #SH3007003) и смесь (50 единиц) пенициллина - (50 мкг) стрептомицина/мл (GIBCO-Invitrogen Corp.: Cat #15140) в 5% CO2, в инкубаторе с повышенной влажностью, при 37°С.
Клетки культивируют приблизительно до 70-80%-ной конфлюентности (<35 пассажей субкультуры). Все стадии получения мембранного препарата проводят на льду. Для сбора клеток, среду отсасывают, и клетки промывают забуференным фосфатом физиологическим раствором Дульбекко, не содержащим Ca2+, Mg2+ (dPBS). Клеточный монослой отделяют путем легкого механического перемешивания. Клетки собирают путем центрифугирования при 1000 об/мин (5 мин). Последующие стадии получения мембранного препарата проводят в соответствии с протоколом, описанным выше для мембран, экспрессирующих 5-HT4(c)-рецепторы.
b. Анализы на связывание с радиоактивным лигандом
Анализы на связывание с радиоактивным лигандом осуществляют в 96-луночных полипропиленовых аналитических планшетах в общем аналитическом объеме 200 мл, содержащем 1,5-2 мкг мембранного белка в 50 мм буфере HEPES pH 7,4, включающим аналитический буфер с 0,025% BSA. Исследования на связывание с насыщением, проводимые в целях определения величин Kd для радиоактивных лигандов, осуществляют с использованием [3H]-GR65630 (PerkinElmer Life Sciences Inc., Boston, MA: Cat #NET1011, удельная активность ~85 Ки/ммоль) при двенадцати различных концентрациях в пределах от 0,005 нМ до 20 нМ. Анализы на вытеснение метки, проводимые в целях определения величин pKi для соединений, осуществляют с использованием [3H]-GR65630 при концентрации 0,50 нМ и при одиннадцати различных концентрациях соединений от 10 пМ до 100 мкМ. Соединения получают в виде 10 мМ маточных растворов в ДМСО (см. раздел 3.1) и разводят до 400 мкМ в 50 мм HEPES pH 7,4 при 25°С, содержащим 0,1% BSA, и в том же самом буфере делают серийные разведения (1:5). Неспецифическое связывание определяют в присутствии 10 мкМ немеченного MDL72222. Аналитические смеси инкубируют в течение 60 минут при комнатной температуре, а затем реакции связывания прекращают путем быстрой фильтрации на 96-луночных фильтровальных планшетах GF/B из стекловолокна (Packard BioScience Co., Meriden, CT), предварительно смоченных в 0,3% полиэтиленимине. Эти фильтровальные планшеты три раза промывают фильтрующим буфером (охлажденным льдом 50 мМ HEPES, pH7,4) для удаления несвязанной радиоактивности. Планшеты сушат; в каждую лунку добавляют 35 мкл сцинтилляционной жидкости Microscint-20 (Packard BioScience Co., Meriden, CT), и планшеты считывают на жидкостном сцинтилляционном счетчике Packard Topcount (Packard BioScience Co., Meriden, CT).
Данные по связыванию анализируют с помощью нелинейного регрессионного анализа, описанного выше для определения величин Ki. BOTTOM (минимальное значение на кривой) представляет собой фиксированную величину неспецифического связывания, определенную в присутствии 10 мкМ MDL72222. Количество [L] в уравнении Ченга-Пруссова определяют как концентрацию [3H]-GR65630.
Селективность к рецептору подтипа 5-HT4 по сравнению с селективностью к рецептору подтипа 5-HT3 вычисляют как отношение Ki(5-HT3A)/Ki(5-HT4(c)). Соединения согласно изобретению, которые были протестированы в этом анализе, обычно, имеют отношение селективностей к рецепторам указанных подтипов Ki(5-HT3A)/Ki(5-HT4(c)) в пределах примерно от 10 до 40000, а обычно примерно от 100 до 4000.
Пример 20: Анализ на аккумуляцию cAMP в интактных клетках, проводимый с использованием планшетов Flashplate с клетками HEK-293, экспрессирующими человеческие 5-HT4(c)-рецепторы
В этом анализе, функциональную эффективность тестируемых соединений определяют путем измерения количества циклического AMP, продуцируемого клетками HEK-293, экспрессирующими рецепторы 5-HT4, при их контактировании с различными концентрациями тестируемого соединения.
a. Клеточная культура
Были получены клетки НЕК 293 (клетки почек человеческого эмбриона), стабильно трансфецированные клонированной кДНК человеческого 5-HT4(c)-рецептора и экспрессирующие рецептор, при двух различных плотностях: (1) при плотности примерно 0,5-0,6 пмоль/мг белка, как было определено в анализе на связывание с радиоактивным мембранным лигандом [3H]-GR113808), и (2) при плотности примерно 6,0 пмоль/мг белка. Эти клетки культивируют в колбах T-225 в модифицированной по способу Дульбекко среде Игла (DMEM), которая содержит 4500 мг/л D-глюкозы (GIBCO-Invitrogen Corp., Cat #11965) и в которую добавляют 10% фетальную бычью сыворотку (FBS) (GIBCO-Invitrogen Corp.: Cat #10437) и смесь пенициллина (100 единиц) - стрептомицина (100 мкг)/мл (GIBCO-Invitrogen Corp.: Cat #15140) в 5% CO2, в инкубаторе с повышенной влажностью, при 37°С. Клетки культивируют под непрерывным давлением отбора путем добавления в среду 800 мкг/мл генетицина (GIBCO-Invitrogen Corp.: Cat #10131).
b. Клеточный препарат
Клетки культивируют приблизительно до 60-80%-ной конфлюентности. За 20-22 часа до анализа, клетки два раза промывают и подпитывают бессывороточной DMEM, содержащей 4500 мг/л D-глюкозы (GIBCO-Invitrogen Corp., Cat #11965). Для сбора клеток, среду отсасывают, и в каждую колбу Т-225 добавляют 10 мл версена (GIBCO-Invitrogen Corp.: Cat #15040). Клетки инкубируют в течение 5 минут при комнатной температуре, а затем их вынимают из колбы путем механического перешивания. Клеточную суспензию переносят в центрифужную пробирку, содержащую равный объем предварительно нагретого (37°С) dPBS, и центрифугируют в течение 5 минут при 1000 об/мин. Затем супернатант отбрасывают, и осадок снова суспендируют в предварительно нагретом (37°С) буфере для стимуляции (10 мл эквивалента на 2-3 колбы T-225). Это время было зарегистрировано как время 0. Клетки подсчитывают на счетчике Coulter (число, превышающее 8 мкм, давало 1-2×107 клеток/колбу). Клетки ресуспендируют при концентрации 5×105 клеток/мл в предварительно нагретом (37°С) буфере для стимуляции (как рекомендовано в инструкции к набору Flashplate) и предварительно инкубируют при 37°C в течение 10 минут.
Анализы на уровни cAMP осуществляют в формате радиоиммуноанализа с помощью аналитической системы активации аденилилциклазы на планшетах Flashplate с использованием 125I-cAMP (SMP004B, PerkinElmer Life Sciences Inc., Boston, MA), в соответствии с инструкциями производителя.
Клетки культивируют и приготавливают как описано выше. Конечные концентрации клеток в этом анализе составляют 25×103 клеток/лунку, а конечный аналитический объем составляет 100 мкл. Тестируемые соединения получают в виде 10 мМ маточных растворов в ДМСО и разводят до 400 мМ в 50 мм HEPES pH 7,4 при 25°С, содержащем 0,1% BSA, и в том же самом буфере делают серийные разведения (1:5). Анализы на аккумуляцию циклического AMP проводят с использованием 11 различных концентраций соединения в пределах от 10 пМ до 100 мкМ (конечные аналитические концентрации). Для каждого планшета строят кривую зависимости “концентрация 5-HT - ответ” (10 пМ-100 мкМ). Клетки инкубируют со встряхиванием при 37°С в течение 15 минут, и реакцию завершают путем добавления в каждую лунку 100 мкл охлажденного льдом буфера для детекции (как указано в инструкции к набору Flashplate). Затем, эти планшеты герметично закрывают и инкубируют при 4°C в течение ночи. Связанную радиоактивность количественно оценивают путем сцинтилляционной проксимальной спектроскопии с использованием счетчика Topcount (Packard BioScience Co., Meriden, CT).
Количество cAMP, продуцируемого на один миллилитр реакционной смеси, экстраполируют исходя из стандартной кривой для cAMP в соответствии с рекомендациями производителя, представленными в руководстве для пользователя. Полученные данные анализируют с помощью нелинейного регрессионного анализа и программного обеспечения GraphPad Prism с использованием 3-параметрической сигмоидальной модели доза-ответ (тангенс угла наклона не превышает 1). Данные, относящиеся к эффективности соединений, выражают как величины pEC50 в виде отрицательных десятичных логарифмов величин EC50, где EC50 представляет собой эффективную концентрацию, дающую 50% максимального ответа.
Тестируемые соединения, имеющие более высокое значение pKi в этом анализе, имеют более высокую аффинность связывания с 5-НТ4-рецептором. Соединения согласно изобретению, которые были протестированы в этом анализе, например, в данной клеточной линии (1), имеющей плотность примерно от 6,5 до 9,0, давали величину pEC50 в пределах примерно от 6,9 до 9,5, а обычно, в пределах примерно от 7,5 до 8,5.
Пример 21: In vitro модель пероральной биологической доступности: анализ на проницаемость Caco-2
Анализ на проницаемость клеток Caco-2 осуществляют для моделирования способности тестируемых соединений проходить через тонкий кишечник и поступать в кровоток после перорального введения. Для этого определяют скорость, при которой тестируемые соединения, присутствующие в растворе, проникают в клеточный монослой, созданный для имитации плотного контакта клеточных монослоев тонкого кишечника человека.
Клетки Caco-2 (клетки аденокарциномы толстой кишки человека) получали из ATCC (Американской коллекции типовых культур; Rockville, MD). Для исследования такой проницаемости, клетки высевали при плотности 63000 клеток/см2 на предварительно смоченные поликарбонатные фильтры Transwell (Costar; Cambridge, MA). Клеточный монослой образовывался на 21-й день после начала культивирования. После культивирования клеток в планшете Transwell, мембранный фильтр, содержащий клеточный монослой, отделяют от планшета Transwell и вводят в диффузионную камеру (Costar; Cambridge, MA). Эту диффузионную камеру помещают в нагревательный блок, снабженный внешней системой с циркулирующей водой, регулируемой термостатом при 37°С для температурного контроля. В каждый отсек диффузионной камеры с помощью воздушного распределительного трубопровода доставляется 95% O2/5% CO2, в результате чего, в клеточном монослое создается ламинарный поток, который эффективно способствует уменьшению неперемешивающегося пограничного слоя.
Исследования на проницаемость проводят с использованием тестируемого соединения в концентрации 100 мкМ и 14C-маннита для мониторинга целостности монослоя. Все эксперименты проводят при 37°C в течение 60 минут. Образцы берут через 0, 30 и 60 минут из выходного и приемного отверстий камеры. Образцы анализируют на концентрации тестируемого соединения и маннита с помощью ВЭЖХ или жидкостного сцинтилляционного счетчика. Затем вычисляют коэффициент проницаемости (Kp) в см/сек.
В этом анализе, величина Kp, превышающая примерно 10×10-6 см/сек, является показателем хорошей биологической доступности. Соединения согласно изобретению, которые были протестированы в данном анализе, обычно имели величины Kp, составляющие примерно от 5×10-6 см/сек до 50×10-6 см/сек, а, в основном, примерно от 15×10-6 см/сек до 40×10-6 см/сек.
Пример 22: Фармакокинетичские исследования на крысах
Препараты, полученные в виде водных растворов тестируемых соединений, приготавливают в 0,1% молочной кислоте при pH примерно 5-6. Самцам крыс Sprague-Dawley (штамм CD, Charles River Laboratories, Wilmington, MA) вводят тестируемые соединения внутривенно (i.v.) в дозе 2,5 мг/кг или перорально через зонд (р.о.) в дозе 5 мг/кг. Объем дозы для внутривенного введения составляет 1 мл/кг, а для перорального введения - 2 мл/кг. Пробы крови для серийного разведения берут у животных перед введением дозы и через 2 (только при i.v.), 5, 15 и 30 минут, и через 1, 2, 4, 8, и 24 часов после введения дозы. Концентрации тестируемых соединений в плазме крови определяют с помощью жидкостной хроматографии-масс-спектрометрии (ЖХ-МС/МС) (MDS SCIEX, API 4000, Applied Biosystems, Foster City, CA) с нижним пределом количественной оценки 1 нг/мл.
Стандартные фармакокинетические параметры оценивают с помощью не-системного анализа (Model 201 для IV и Model 200 для PO) с использованием WinNonlin (Version 4,0,1, Pharsight, Mountain View, CA). Максимум на кривой зависимости концентраций тестируемых соединений в плазме крови от времени обозначают Cmax. Площадь под кривой зависимости концентраций от времени введения доз до введения последней измеряемой концентрации (AUC(0-t)), включительно, вычисляют по линейной формуле трапеций. Пероральная биодоступность (F(%)), то есть, нормализованное по дозе отношение AUC(0-t), для введения п.о., к AUC(0-т) для введения i.v., вычисляют по формуле:
F(%)=AUCPO/AUCIV×дозаIV/дозаPO×100%
При этом предполагается, что при пероральном введении, тестируемые соединения, имеющие более высокие значения параметров Cmax, AUC(0-t) и F(%) в данном анализе, обладают лучшей биологической доступностью. Соединения согласно изобретению, которые были протестированы в данном анализе, обычно имели значения Cmax в пределах примерно от 0,05 до 0,45 мкг/мл, а в основном, примерно от 0,10 до 0,35 мкг/мл; а величины AUC(0-t) составляли в пределах примерно от 0,05 до 0,9 мкг·ч/мл, а обычно, примерно от 0,25 до 0,8 мкг·ч/мл. Так, например, соединение примера 2 имело значение Cmax=0,055 мкг/мл, величину AUC(0-t)=0,427 мкг·ч/мл и биологическую доступность при пероральном введении F(%) в крысиной модели, составляющую примерно 30%.
Хотя настоящее изобретение было описано со ссылками на конкретные варианты его осуществления, однако, для специалиста в данной области очевидно, что в него могут быть внесены различные изменения и могут быть использованы различные эквиваленты, не выходящие за рамки существа и объема изобретения. Кроме того, для адаптации конкретной ситуации, материала, композиции, способа, стадии или стадий данного способа к целям, существу и объему настоящего изобретения, в него могут быть внесено множество модификаций. Все указанные модификации не должны выходить за рамки объема прилагаемой формулы изобретения. Кроме того, все публикации, патенты и патентные документы, цитируемые выше, во всей своей полноте вводятся в настоящее описание посредством ссылки так, как если бы они были отдельно введены посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| СОЕДИНЕНИЯ-АГОНИСТЫ РЕЦЕПТОРА 5-НТ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ ПОЗНАВАТЕЛЬНОЙ СПОСОБНОСТИ | 2010 |
|
RU2569056C2 |
| СОЕДИНЕНИЯ 8-АЗАБИЦИКЛО[3.2.1]ОКТАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ МЮ-ОПИОИДОВ | 2007 |
|
RU2423362C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛООКТАНА, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, СОЕДИНЕНИЯ | 2000 |
|
RU2262505C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
Изобретение относится к новым индазол-карбоксамидным соединениям формулы (I-а), в которой радикалы и группы имеют определения, приведенные в п.1 формулы изобретения. Указанные соединения являются агонистами 5-НТ4-рецептора. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, к способам применения таких соединений для лечения заболеваний, опосредуемых активностью 5-НТ4-рецептора, и к способам получения указанных соединений. 8 н. и 8 з.п. ф-лы, 10 табл.

1. Соединение по п.1, которое представляет собой соединение формулы (I-а)
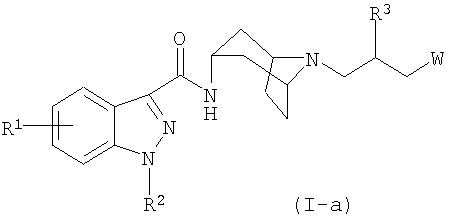
где R1 представляет собой водород;
R2 представляет собой изопропил;
R3 представляет собой гидрокси, С1-3алкокси или OC(O)NRaRb;
W выбран из
(a) Y, где Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10, -N(R8a)C(O)OR12,
-N(R8a)C(O)NR13R14 и -N(R8a)S(O)2NR13R14; и
(b) группу формулы (b)

где X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10, -S(R11)O2, -N(R8)C(O)OR12,
-N(R8)C(O)NR13R14, -N(R8)S(O)2NR13R14, -C(O)NR13R14, -OC(O)NR13R14, -C(O)OR12,
-OR15, -NR8R16, циано, -SR15, CF3, пиридинила, пирролила, 1,1-диоксо-изотиазолидинила, и пирролидинила, где указанный пирролидинил необязательно замещен оксо;
R5 представляет собой водород, С1-3алкил или С1-3алкил, замещенный в концевом положении гидрокси;
R6 и R7 в каждом случае независимо представляют собой водород, гидрокси, галоген или циано;
R8 и R8a представляют собой водород или С1-3алкил;
либо R5 и R8 или R5 и R6, взятые вместе, образуют С2-5алкилен;
либо R3 и R5 или R3 и R8a, взятые вместе, образуют -ОСН2СН2-;
R9 представляет собой водород, тетрагидрофуранил, пиридинил или С1-3алкил;
R10 представляет собой С1-3алкил, где С1-3алкил необязательно замещен
-S(O)2С1-3алкилом или 1-3 атомами галогена;
R11 представляет собой водород, -NRaRb или С1-3алкил, где С1-3алкил необязательно замещен 1-3 атомами галогена;
либо R5 и R11 или R6 и R11, взятые вместе, образуют С2-5алкилен;
R12 представляет собой С1-3алкил;
R13, R14 и R15 независимо представляют собой водород или С1-3алкил;
R16 представляет собой -CH2-C(O)NRaRb, -СН2-С(O)-морфолинил, -СН2-пиридинил,
-СН2-пиримидинил или -СН2-тетрагидрофуранил;
R18 представляет собой -С(O)ОСН3 или -С(O)СН3;
Ra и Rb независимо представляют собой водород или С1-3алкил;
а равно 0 или 1 и
n равно 1, 2 или 3; при условии, что если n равно 1, то X представляет собой -SR15,
или его фармацевтически приемлемая соль или стереоизомер.
2. Соединение по п.1, где W выбран из:
(a) Y, где Y выбран из -N(R8a)C(O)R9, -N(R8a)S(O)2R10 и -N(R8a)C(O)NR13R14; и
(b) группы формулы (b), где X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10,
-N(R8)C(O)OR12, -N(R8)C(O)NR13R14, -N(R8)S(O)2NR13R14, -C(O)NR13R14, -OR15 и циано.
3. Соединение по п.1 или 2, где W выбран из Y, где R8a представляет собой водород или метил; R9 представляет собой водород, тетрагидрофуранил, пиридинил или метил; R10 и R12 представляют собой метил или этил и R13 и R14 независимо представляют собой водород или метил.
4. Соединение по п.1 или 2, где W представляет собой группу формулы (b), где
(i) X представляет собой циано или
(ii) а равно 0, n равно 2, R6 и R7 представляют собой водород, R5 и R8, взятые вместе, образуют С2алкиленил и X выбран из -N(R8)C(O)R9, -N(R8)S(O)2R10 и
-N(R8)C(O)NR13R14.
5. Соединение по п.1 или 2, где W выбран из -NHC(O)H, N(CH3)C(O)H, -NHC(O)CH3, -N(CH3)C(O)CH3, -N(CH3)S(O)2CH3, N(CH3)C(O)NHCH3, -N(CH3)CH2CH2CN, 1-метансульфонилпиперазин-4-ила, 1-диметиламинокарбонилпиперазин-4-ила, 1-(тетрагидрофуран-2-ил)карбонилпиперазин-4-ила, 3-(метоксикарбониламино)пирролидин-1-ила и 2-(метоксиметилен)пирролидин-1-ила.
6. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из:
{(1S,3R,5R)-8-[2-гидрокси-3-((S)-2-метоксиметилпирролидин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
((1S,3R,5R)-8-{3-[(2-циано-этил)метиламино]-2-гидроксипропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
((1S,3R,5R)-8-{2-гидрокси-3-[4-(тетрагидрофуран-2-карбонил)пиперазин-1-ил]пропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
[(1S,3R,5R)-8-(4-карбамоилметилморфолин-2-илметил)-8-азабицикло[3.2.1]окт-3-ил]амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{{1S,3R,5R)-8-[3-(4-диметилкарбамоилпиперазин-1-ил)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(4-метансульфонилпиперазин-1-ил)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
метилового эфира[1-(2-гидрокси-3-{(1S,3R,5R)-3-[(1-изопропил-1Н-индазол-3-карбонил)амино]-8-азабицикло[3.2.1]окт-8-ил}пропил)пирролидин-3-ил]карбаминовой кислоты;
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(4-метансульфонилпиперазин-1-ил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)-пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(ацетил-метиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(формил-метиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(1,3-диметилуреидо)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
((1S,3R,5R)-8-{2-гидрокси-3-[(пиридин-4-карбонил)амино]-пропил}-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-1Н-индазол-3-карбоновой кислоты;
[(1S,3R,5R)-8-(3-формиламино-2-гидроксипропил)-8-азабицикло[3.2.1]окт-3-ил]-амида 1-изопропил-1Н-индазол-3-карбоновой кислоты; и
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-1Н-индазол-3-карбоновой кислоты; и
их фармацевтически приемлемых солей и стереоизомеров.
7. Фармацевтическая композиция, обладающая агонистической активностью по отношению к 5-НТ4-рецептору, содержащая терапевтически эффективное количество соединения по любому из пп.1-6 и фармацевтически приемлемый носитель.
8. Соединение по любому из пп.1-6, которое может быть использовано в терапии для лечения патологического состояния у млекопитающего, ассоциированного с активностью 5-НТ4-рецептора.
9. Применение соединения по любому из пп.1-6 для приготовления лекарственного средства для лечения патологического состояния у млекопитающего, ассоциированного с активностью 5-НТ4-рецептора.
10. Применение по п.9, где указанным заболеванием или состоянием является расстройство, ассоциированное с пониженной перистальтикой желудочно-кишечного тракта.
11. Способ лечения млекопитающего, страдающего патологическим состоянием, ассоциированным с активностью 5-НТ4-рецептора, где указанный способ включает введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по любому из пп.1-6.
12. Способ по п.11, где указанное патологическое состояние выбрано из группы, состоящей из синдрома раздраженной толстой кишки, хронического запора, функциональной диспепсии, замедленного опорожнения желудка, рефлюкс-эзофагита, пареза желудка, послеоперационного илеита, псевдообструкции тонкого кишечника и индуцированного лекарственными средствами замедленного прохождения содержимого через желудок.
13. Способ лечения расстройства, ассоциированного с пониженной перистальтикой желудочно-кишечного тракта у млекопитающего, где указанный способ включает введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по любому из пп.1-6.
14. Способ получения соединения формулы (I-а), где R1, R2, R3 и W имеют значения, определенные в п.1, или его соли, или стереоизомера, где указанный способ включает: взаимодействие соединения формулы (II)
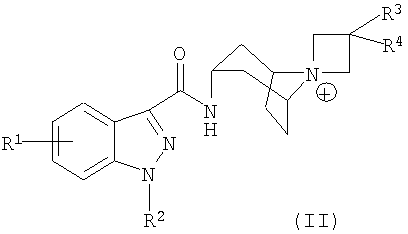
где R4 представляет собой водород,
с соединением формулы (III)

с получением соединения формулы (I-а), или его соли, или стереоизомера.
15. Способ получения соединения формулы (I-а), где R3 представляет собой гидрокси, a R1, R2 и W определены в п.1, или его соли, или стереоизомера, где указанный способ включает:
взаимодействие соединения формулы (VI)
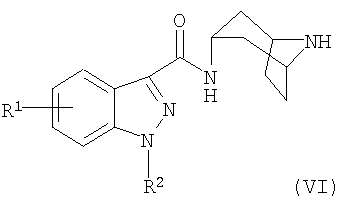
с соединением формулы (VIII)

где R4 представляет собой водород, с получением соединения формулы (I-а), или его соли, или стереоизомера.
16. Способ исследования биологической системы или образца, содержащего 5-НТ4-рецептор, где указанный способ включает:
(a) контактирование указанной биологической системы или указанного образца с соединением по любому из пп.1-6 и
(b) определение влияния указанного соединения на указанную биологическую систему.
| Способ центрирования немагнитной электропроводной полосы | 1980 |
|
SU908459A1 |
| ЕР 0230718 А1, 05.08.1978 | |||
| ПРОИЗВОДНЫЕ 5-АРИЛ-3-(8-АЗАБИЦИКЛО[3.2.1]ОКТ-3-ИЛ)-1,3,4-ОКСАДИАЗОЛ-2(3H)-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2186066C2 |

