Область техники, к которой относится изобретение
Изобретение относится к соединениям 8-азабицикло[3.2.1]октана, которые могут быть использованы в качестве антагонистов рецепторов мю-опиоидов. Настоящее изобретение относится также к фармацевтическим композициям, содержащим такие соединения, и способам применения таких соединений для лечения или уменьшения интенсивности заболеваний, опосредованных активностью рецепторов мю-опиоидов, к промежуточным соединениям, пригодным для получения таких соединений, и способам получения таких промежуточных соединений.
Уровень техники
В настоящее время общепризнанным фактом является то, что эндогенные опиоиды оказывают комплексное воздействие на физиологию желудочно-кишечного тракта. Рецепторы опиоидов экспрессированы во всем организме, как в центральной нервной системе, так и в периферических областях, включая желудочно-кишечный (GI) тракт.
Соединения, действующие в качестве агонистов рецепторов опиоидов, типичным примером которых является морфин, составляют основу аналгезирующей терапии при лечении боли от умеренной до сильной. К сожалению, применение опиоидных анальгетиков часто связано с вредным воздействием на желудочно-кишечный тракт, известным как вызванная опиоидами дисфункция кишечника (OBD). OBD сопровождается такими симптомами, как запор, плохое опорожнение желудка, боли и дискомфорт в брюшной полости, вздутие живота, тошнота и гастроэзофагеальный рефлюкс. Как центральные, так и периферические рецепторы опиоидов, по-видимому, замедляют желудочно-кишечный транзит после введения опиоидов. Однако имеющиеся данные позволяют предположить, что вредное воздействие на функционирование желудочно-кишечного тракта оказывают главным образом периферические рецепторы опиоидов, экспрессированные в желудочно-кишечном тракте.
Так как побочные эффекты опиоидов обусловлены главным образом периферическими рецепторами, в то время как аналгезия является центральной по своей природе, то избирательный антагонист, воздействующий на периферические рецепторы, потенциально может блокировать нежелательные побочные эффекты в желудочно-кишечном тракте, не влияя при этом на благоприятные центральные эффекты аналгезии или не вызывая абстиненцию центральной нервной системы.
Считается, что из трех основных подтипов рецепторов опиоидов, именуемых мю, дельта и каппа, наиболее широко применяемые в клинической практике опиоидные аналгетики активируют рецепторы мю-опиоидов, вызывая аналгезию и изменяя перистальтику желудочно-кишечного тракта. Поэтому можно предположить, что периферически воздействующие избирательные антагонисты мю-опиоидов должны быть пригодны для лечения вызванной опиоидами дисфункции кишечника. Предпочтительные средства демонстрируют значительное связывание с рецепторами мю-опиоидов in vitro и являются активными in vivo в животных моделях желудочно-кишечного тракта.
Послеоперационная непроходимость кишечника (POI) является нарушением, обусловленным пониженной перистальтикой желудочно-кишечного тракта после операции на брюшной полости или другой операции. Симптомы POI подобны вышеуказанным симптомам OBD. Кроме того, поскольку оперируемые субъекты часто получают опиоидые аналгетики во время и после операции, то продолжительность POI может быть увеличена вследствие пониженной перистальтики желудочно-кишечного тракта, вызванной применениеим опиоидов. Поэтому можно также предположить, что антагонисты мю-опиоидов, пригодные для лечения OBD, могут оказывать благоприятное воздействие при лечении POI.
Сущность изобретения
Настоящее изобретение относится к новым соединениям, обладающим активностью антагонистов рецепторов мю-опиоидов.
Таким образом, настоящее изобретение относится к соединению формулы (I):

где R1 выбирают из -ORa, -C(O)NRaRb, -NHS(O)2Rc, -NRaRb, -C(O)ORa и -СН2ОН;
А означает С1-4алкиленил;
R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним -ORа, одним или двумя атомами галогена, одним или двумя С1-3алкилами, замещенными двумя или тремя атомами галогена, или одним, двумя, тремя или четырьмя С1-3алкилами;
G означает С1-4алкиленил;
R3 выбирают из водорода, -C(O)R4, -C(O)NHR5, -S(O)2Rс и -S(O)2NRаRb;
R4 означает С3-6циклоалкил или С1-6алкил, при этом
С3-6циклоалкил необязательно замещен одним -ORа, и
С1-6алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORа, -C(O)ORа, -S(O)2R6, -C(O)NRаRb, -NRаRb, -NHC(O)NRaRb, -CN,
С3-6циклоалкила и фенила; или одним -D-(CH2)j-R7,
где D означает  или
или  ,
,
j равно 1, 2 или 3, n равно 1 или 2 и р равно 1 или 2;
R6 означает С1-3алкил, необязательно замещенный R7;
R7 означает -С(O)ORа, -C(O)NRаRb, -NRаRb или -NHC(O)NRаRb;
R5 означает С1-6алкил, бензо[1.3]диоксол или -(СН2)q-фенил;
где фенил необязательно замещен одним или двумя заместителями, выбираемыми из галогена, -ORа, С1-3алкила и С1-3алкокси, где С1-3алкил и С1-3алкокси необязательно замещены 2 или 3 атомами галогена и q равно 0, 1 или 2;
Rа и Rb независимо означают водород или С1-4алкил; и
Rс означает С1-3алкил;
при условии, что, когда R2 означает фенил, замещенный в положении 4, R3 не является -С(O)R4, где R4 означает С1-4алкил, замещенный -С(O)ORa;
к его фармацевтически приемлемой соли или сольвату.
Настоящее изобретение относится также к фармацевтической композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемый носитель.
Настоящее изобретение относится также к способу лечения заболевания или болезненного состояния, обусловленного активностью рецептора мю-опиоида, например, нарушения, связанного с пониженной перистальтикой желудочно-кишечного тракта, такого как вызванная опиоидами дисфункция кишечника и послеоперационная непроходимость кишечника, который включает введение млекопитающему терапевтически эффективного количества соединения или фармацевтической композиции по настоящему изобретению.
Соединения по настоящему изобретению могут быть также использованы в качестве научных инструментов, то есть для исследования биологических систем или образцов либо для изучения активности других химических соединений. Таким образом, настоящее изобретение относится к способу применения соединения формулы (I), его фармацевтически приемлемой соли или сольвата в качестве научного инструмента для исследования биологической системы или образца либо для поиска новых соединений, обладающих активностью в отношении рецепторов мю-опиоидов, который включает осуществление контактирования биологической системы или образца с соединением по настоящему изобретению и определение воздействия, оказываемого данным соединением на указанную биологическую систему или образец.
Отдельными объектами настоящего изобретения являются также промежуточные соединения, пригодные для получения соединений по настоящему изобретению, и способы синтеза промежуточных соединений, представленных в настоящем описании изобретения.
Настоящее изобретение относится также к соединению по настоящему изобретению, предназначенному для применения в лекарственной терапии, а также к применению соединения по настоящему изобретению для приготовления препарата или лекарственного средства для лечения у млекопитающего заболевания или болезненного состояния, обусловленного активностью рецепторов мю-опиоидов, например, нарушения, связанного с пониженной перистальтикой желудочно-кишечного тракта.
Краткое описание чертежей
На фиг.1 показана порошковая рентгенограмма кристаллического гликолята 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидрокси-пропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по настоящему изобретению.
На фиг.2 показана порошковая рентгенограмма кристаллического оксалата 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидрокси-пропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по настоящему изобретению.
На фиг.3 показана порошковая рентгенограмма кристаллического фосфата 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)-(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида по настоящему изобретению.
Подробное описание изобретения
Настоящее изобретение относится к антагонистам рецепторов мю-опиоидов, которые представляют собой соединение 8-азабицикло[3.2.1]октана формулы (I), его фармацевтически приемлемые соли или сольваты. Приведенные ниже заместители и значения являются типичными примерами разных вариантов осуществления настоящего изобретения. Указанные типичные значения далее определяют такие варианты осуществления изобретения, не исключая при этом других значений и не ограничивая объем изобретения.
В одном конкретном варианте осуществления изобретения R1 выбирают из -ORа, -C(O)NRаRb, -NHS(O)2Rс, -NRаRb, -C(O)ORа и -СН2ОН.
В других конкретных вариантах осуществления изобретения R1 выбирают из -ORа, -C(O)NRаRb и -NHS(O)2Rс, либо R1 означает -ORа или -С(О)NRаRb, либо R1 означает -ОН или -С(О)NRаRb.
В другом конкретном варианте осуществления изобретения R1 означает -ОН или -С(О)NH2.
В другом конкретном варианте осуществления изобретения R1 означает -С(О)NH2.
В одном конкретном варианте осуществления изобретения А означает С1-4алкиленил.
В других конкретных вариантах осуществления изобретения А означает -(СН2)2-, -СН(СН3)- или -СН2-; либо А означает -(СН2)2- или -СН2; либо А означает -СН2-.
В одном конкретном варианте осуществления изобретения G означает С1-4алкиленил.
В других конкретных вариантах осуществления изобретения G означает -(СН2)3-, -(СН2)2- или -СН2-; либо G означает -(СН2)2- или -СН2-; либо G означает -СН2-.
В одном конкретном варианте осуществления изобретения R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним -ORа, одним или двумя атомами галогена, одним или двумя С1-3алкилами, замещенными двумя или тремя атомами галогена; либо одним, двумя, тремя или четырьмя С1-3алкилами.
В другом конкретном варианте осуществления изобретения R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним -ORа, одним или двумя атомами галогена, одним или двумя С1-3алкилами, необязательно замещенными двумя или тремя атомами галогена.
В другом конкретном варианте осуществления изобретения R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним или двумя атомами галогена, одним или двумя С1-3алкилами, необязательно замещенными 2 или 3 атомами галогена. Типичные группы R2 в данном варианте осуществления изобретения включают, не ограничиваясь ими, циклопентил, циклогексил, циклогептил, адамантил, фенил и нафтил, причем циклогексил, фенил и нафтил необязательно замещены одним или двумя атомами галогена или С1-3алкилом, замещенным двумя или тремя атомами галогена.
В другом конкретном варианте осуществления изобретения R2 означает циклобутил, циклопентил, циклогексил, адамантил или фенил, причем циклогексил и фенил необязательно замещены одним или двумя атомами галогена или С1-3алкилом, замещенным двумя или тремя атомами галогена.
В другом конкретном варианте осуществления изобретения R2 означает циклогексил или фенил, которые необязательно замещены одним или двумя атомами галогена или С1-3алкилом, замещенным двумя или тремя атомами галогена; либо R2 означает циклогексил или фенил, которые необязательно замещены одним или двумя атомами галогена.
В другом конкретном варианте осуществления изобретения R2 означает циклогексил, необязательно замещенный одним или двумя атомами галогена.
В другом конкретном варианте осуществления изобретения R2 означает циклогексил.
В другом конкретном варианте осуществления изобретения R2 означает фенил, необязательно замещенный одним или двумя атомами галогена.
В другом конкретном варианте осуществления изобретения R2 означает фенил.
В одном конкретном варианте осуществления изобретения R3 выбирают из водорода, -C(O)R4, -C(O)NHR5, -S(O)2Rс или -S(O)2NRаRb.
В другом конкретном варианте осуществления изобретения R3 выбирают из водорода, -C(O)R4, -S(O)2Rс и -S(O)2NRаRb.
В одном конкретном варианте осуществления изобретения R3 выбирают из водорода, -С(O)R4 и -C(O)NHR5.
В других конкретных вариантах осуществления изобретения R3 означает -C(O)R4 или -C(O)NHR5, либо R3 означает -С(O)R4.
В другом конкретном варианте осуществления изобретения R3 означает -C(O)R4, где R4 означает С3-6циклоалкил или С1-6алкил, причем С1-6алкил необязательно замещен одним или двумя -ORа или одним заместителем, выбираемым из -C(O)ORа, -S(O)2R6, -C(O)NRаRb, -NRаRb и С3-6циклоалкила, и R6 означает С1-3алкил, необязательно замещенный R7, где R7 означает -С(O)ORа.
В другом конкретном варианте осуществления изобретения R3 означает -С(O)R4, где R4 означает С3-6циклоалкил или С1-6алкил, причем С3-6циклоалкил необязательно замещен одним -ORа, и С1-6алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORа, -C(O)ORа, -S(O)2R6, -C(O)NRаRb, -NRаRb, -CN,
C3-6циклоалкила и фенила, где R6 означает С1-3алкил, необязательно замещенный R7, где R7 означает -С(O)ORа.
В другом конкретном варианте осуществления изобретения R3 означает -C(O)R4, где R4 означает С5-6циклоалкил, необязательно замещенный одним -ОН.
В других конкретных вариантах осуществления изобретения R3 означает -С(O)R4, где R4 означает С1-4алкил, необязательно замещенный одним или двумя заместителями, выбираемыми из -ORа, -S(O)2R6, -NRаRb, -CN, С3-6циклоалкила и фенила, где R6 означает С1-3алкил; либо R4 означает С1-4алкил, необязательно замещенный одним или двумя заместителями, выбираемыми из -ОН, -ОСН3, -S(O)2CH3, -NH2, -NHCH3, -NH(CH3)2 и фенила. Типичные значения R4 в данном варианте осуществления изобретения включают, не ограничиваясь ими, -СН2ОН, -СН(ОН)СН2ОН, -СН2SO2CH3, -CH2SO2CH2C(O)OH, -CH2CN, -CH2OCH3, -C(CH3)2OH, -CH(CH3)OH, -CH(OH)CH(CH3)OH, -CH(OH)CH3, -(CH2)N(CH3)2 и СН(NHCH3)CH2OH.
В другом конкретном варианте осуществления изобретения R3 означает -С(O)R4, где R4 выбирают из -СН2ОН, -СН(ОН)СН2ОН, -СН(ОН)СН3 и -СН2SO2СН3.
В другом конкретном варианте осуществления изобретения R3 означает -С(О)NHR5.
В другом конкретном варианте осуществления изобретения R3 означает -С(O)NHR5, где R5 означает С1-6алкил, бензо[1.3]диоксол или -(СН2)q-фенил, где q равно 0 или 1 и фенил необязательно замещен одним или двумя заместителями, выбираемыми из хлора, фтора, -ОН и -OCF2.
В других конкретных вариантах осуществления изобретения R3 означает -С(О)NHR5, где R5 означает С1-6алкил или бензо[1.3]диоксол; либо R5 означает -СН(СН3)2 или бензо[1.3]диоксол; либо R5 означает -СН(СН3)2.
Настоящее изобретение далее относится к соединению формулы (I), в котором:
R1 означает -ORа или -С(O)NRаRb;
А означает -(СН2)2- или -СН2-;
G означает -(СН2)2- или -СН2-;
R2 выбирают из циклобутила, циклопентила, циклогексила, адамантила и фенила, причем циклогексил и фенил необязательно замещены 1 или 2 атомами галогена или С1-3алкилом, замещенным 2 или 3 атомами галогена;
R3 выбирают из -C(O)R4, -S(O)2Rс, -S(O)2NRаRb и -C(O)NHR5;
R4 означает С3-6циклоалкил или С1-6алкил, причем С3-6циклоалкил необязательно замещен одним -ORа и С1-6алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORа, -C(O)ORа, -S(O)2R6, -C(O)NRаRb, -NRаRb, -CN,
C3-6циклоалкила и фенила, где R6 означает С1-3алкил, необязательно замещенный R7, где R7 означает -С(O)ORа;
R5 означает С1-4алкил, бензо[1.3]диоксол или -(СН2)q-фенил, где q равно 0 или 1 и фенил необязательно замещен одним или двумя заместителями, выбираемыми из хлора, фтора, -ОН и -ОCF2;
Rа и Rb независимо означают водород или С1-3алкил; и
Rс означает С1-3алкил;
при условии, что, когда R2 означает фенил, замещенный в положении 4, R3 не является -С(O)R4, где R4 означает С1-4алкил, замещенный -С(О)ОН;
к его фармацевтически приемлемой соли или сольвату.
Другой вариант осуществления настоящего изобретения относится к соединению формулы (I), в котором:
R1 означает -ОН или -С(О)NH2;
А означает -(СН2)2- или -СН2-;
G означает -(СН2)2- или -СН2-;
R2 означает циклогексил или фенил, причем циклогексил необязательно замещен 1 или 2 атомами галогена;
R3 означает -С(O)R4 или -C(O)NHR5;
R4 выбирают из -СН2ОН, -СН(ОН)СН2ОН, -СН(ОН)СН3 и -СН2SO2CH3;
R5 означает -СН(СН3)2 или бензо[1.3]диоксол;
к его фармацевтически приемлемой соли или сольвату.
Настоящее изобретение далее относится к соединению формулы (I'):
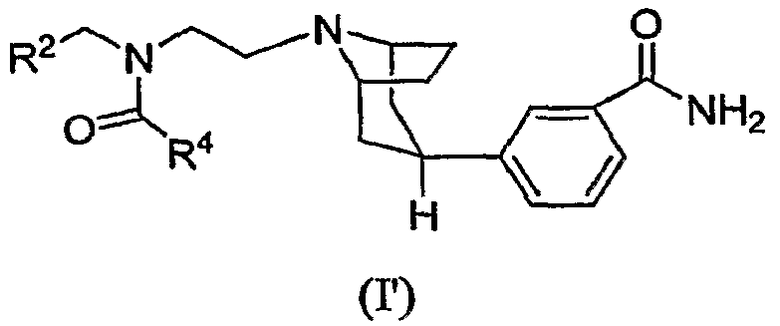
где
R2 означает циклогексил или фенил, которые необязательно замещены одним или двумя атомами галогена; и
R4 означает С3-6циклоалкил или С1-4алкил, при этом
С3-6циклоалкил необязательно замещен одним -ORа и
С1-4алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORа, -S(O)2R6, -NRаRb, -CN и С3-6циклоалкила;
Rа и Rb независимо означают водород или С1-3алкил; и
R6 означает С1-3алкил;
или к его фармацевтически приемлемой соли или сольвату.
Данный вариант осуществления настоящего изобретения относится к соединению формулы (I'), в котором R4 означает С1-4алкил, необязательно замещенный одним или двумя заместителями, выбираемыми из -ОН, -ОСН3, -S(О)2CH3, -NH2, -NHCH3 и -NH(CH3)2.
Данный вариант осуществления настоящего изобретения относится также к соединению формулы (I'), в котором R2 означает циклогексил или 4,4-дифторциклогексил и R4 означает С1-4алкил, замещенный одним или двумя -ОН.
Настоящее изобретение далее относится к соединениям по примерам 1-204, приведенным в настоящем описании изобретения.
Стандартное обозначение химической номенклатуры, использованное в настоящем описании изобретения, проиллюстрировано для соединения по примеру 1:

которое является N-бензил-2-гидрокси-N-{2-[3-эндо(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамидом. Альтернативно при использовании стандартных обозначений IUPAC, применяемых в программном обеспечении AutoNom (MDL Information Systems, GmbH, Frankfurt, Germany), данное соединение обозначено как N-бензил-2-гидрокси-N-{2-[(1R,3R,5S)-3-(3-гидроксифенил)-8-азабицикло[3.2.1]-окт-8-ил]этил}ацетамид. Таким образом названия, использованные в настоящем описании изобретения, соответствуют номенклатуре IUРAC с эндо-ориентацией замещенной фенильной группы относительно четко выраженной 8-азабицикло[3.2.1]октановой группы. Все соединения по настоящему изобретению имеют эндо-ориентацию. В значении, использованном для удобства в настоящем описании изобретения, термин “8-азабициклооктан” означает 8-азабицикло[3.2.1]октан.
Помимо эндо-стереохимии относительно бициклогруппы, соединения по настоящему изобретению могут содержать хиральный центр в заместителях R4, R5 или А. Поэтому за исключением особо оговоренных случаев в объем настоящего изобретения входят рацемические смеси, чистые стереоизомеры и смеси таких изомеров с высоким содержанием стереоизомеров. При указании стереохимии соединения, включая ориентацию относительно 8-азабициклооктановой группы и хиральность в любых заместителях R4, R5 или А, специалистам в данной области должно быть понятно, что за исключением особо оговоренных случаев в композициях по настоящему изобретению могут присутствовать незначительные количества других стереоизомеров при условии, что наличие таких других изомеров не препятствует любому применению указанной композиции в целом.
Определения терминов
При описании соединений, композиций и способов по настоящему изобретению приведенные ниже термины имеют следующие значения за исключением особо оговоренных случаев.
Термин “алкил” означает одновалентную насыщенную углеводородную группу с линейной, разветвленной или комбинированной цепью. За исключением особо оговоренных случаев такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Типичные алкильные группы включают в виде примера метил, этил, н-пропил (н-Pr), изопропил (изо-Pr), н-бутил (н-Bu), втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и тому подобные.
Термин “алкиленил” означает двухвалентную насыщенную углеводородную группу с линейной, разветвленной или комбинированной цепью. За исключением особо оговоренных случаев такие алкиленильные группы обычно содержат от 1 до 10 атомов углерода. Типичные алкиленильные группы включают в виде примера метилен, этилен, н-пропилен, н-бутилен, пропан-1,2-диил(1-метилэтилен), 2-метилпропан-1,2-диил(1,1-диметилэтилен) и тому подобные.
Термин “алкокси” означает одновалентную -О-алкильную группу, в которой алкил имеет указанные выше значения. Типичные алкоксильные группы включают в виде примера метокси, этокси, пропокси, бутокси и тому подобные.
Термин “циклоалкил” означает одновалентную насыщенную карбоциклическую группу, которая может быть моноциклической или полициклической. За исключением особо оговоренных случаев такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода. Типичные циклоалкильные группы включают в виде примера циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, адамантил и тому подобные.
Термин “арил” означает одновалентную ароматическую углеводородную группу с одним кольцом (то есть фенил) или конденсированными кольцами (то есть нафталин). За исключением особо оговоренных случаев такие арильные группы обычно содержат от 6 до 10 атомов углерода в кольце. Типичные арильные группы включают в виде примера фенил, нафталин-1-ил, нафталин-2-ил и тому подобные.
Термин “галоген” означает фтор, хлор, бром или иод.
Термин “соединение” означает соединение, полученное путем синтеза или любым другим способом, таким как метаболизм.
Термин “терапевтически эффективное количество” означает количество, достаточное для оказания лечебного действия при введении субъекту, нуждающемуся в лечении.
Термин “лечение” в используемом здесь значении означает лечение заболевания, нарушения или болезненного состояния у субъекта, такого как млекопитающее (в частности, человек), которое включает:
(а) предотвращение возникновения заболевания, нарушения или болезненного состояния, то есть профилактическое лечение субъекта;
(b) уменьшение интенсивности заболевания, нарушения или болезненного состояния, то есть устранение или регресс заболевания, нарушения или болезненного состояния у субъекта, которое включает противодействие воздействию других лекарственных средств;
(с) подавление заболевания, нарушения или болезненного состояния, то есть замедление или прекращение развития заболевания, нарушения или болезненного состояния у субъекта; или
(d) облегчение симптомов заболевания, нарушения или болезненного состояния у субъекта.
Термин “фармацевтически приемлемая соль” означает соль, полученную из кислоты или основания, которая пригодна для введения субъекту, такому как млекопитающее. Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических кислот и из фармацевтически приемлемых оснований. Фармацевтически приемлемые соли соединений по настоящему изобретению обычно получают из кислот.
Соли, получаемые из фармацевтически приемлемых кислот, включают, не ограничиваясь ими, уксусную, адипиновую, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, гликолевую, бромистоводородную, хлористоводородную, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, щавелевую, пантотеновую, фосфорную, янтарную, серную, винную, паратолуолсульфоновую, ксинафоевую (1-гидрокси-2-найтойную кислоту), нафталин-1,5-дисульфоновую кислоту и тому подобные.
Термин “сольват” означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, то есть соединения по настоящему изобретению или его фармацевтически приемлемой соли, и одной или несколькими молекулами растворителя. Такие сольваты обычно являются кристаллическими твердыми веществами, имеющими по существу фиксированное молярное отношение растворенного вещества и растворителя. Типичные растворители включают в виде примера воду, метанол, этанол, изопропанол, уксусную кислоту и тому подобные. Когда растворителем является вода, образованный сольват представляет собой гидрат.
Очевидно, что термин “его фармацевтически приемлемая соль или сольват” означает все соли и сольваты, такие как сольват фармацевтически приемлемой соли соединения формулы (I).
Термин “аминозащитная группа” означает защитную группу, пригодную для предотвращения нежелательных реакций в положении азота аминогруппы. Типичные аминозащитные группы включают, не ограничиваясь ими, формил; ацильные группы, например, алканоильные группы, такие как ацетил и трифторацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr) и 1,1-ди(4'-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBDMS); и тому подобные.
Общие способы синтеза
Соединения по настоящему изобретению могут быть получены из легко доступных исходных веществ при помощи нижеследующих общих способов и методов. Несмотря на то что на приведенных ниже схемах проиллюстрирован конкретный вариант осуществления настоящего изобретения, специалистам в данной области должно быть понятно, что все соединения по настоящему изобретению могут быть получены способами, рассмотренными в настоящем описании изобретения, или другими способами с использованием реагентов и исходных веществ, известных специалистам в данной области. Кроме того, очевидно, что помимо указанных типичных или предпочтительных условий реакций (то есть температуры реакций, время, молярные отношения реагентов, растворители, давления и т.д.) могут быть также использованы другие условия реакций за исключением особо оговоренных случаев. Оптимальные условия реакций могут изменяться в зависимости от используемых конкретных реагентов или растворителей, но такие условия могут быть определены специалистом в данной области в соответствии со стандартными методами оптимизации.
Кроме того, специалистам в данной области должно быть понятно, что для защиты определенных функциональных групп от нежелательных реакций могут быть необходимы стандартные защитные группы. В данной области хорошо известны методы выбора приемлемой защитной группы для определенной функциональной группы, а также приемлемых условий для введения и удаления защитной группы. Например, многочисленные защитные группы, способы их введения и удаления описаны в публикации T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и в ссылках, приведенных в указанной публикации.
Один способ синтеза соединений по настоящему изобретению показан на схеме А. (Заместители и переменные элементы, указанные на нижеследующих схемах, имеют приведенные выше определения, за исключением особо оговоренных случаев).
Схема А
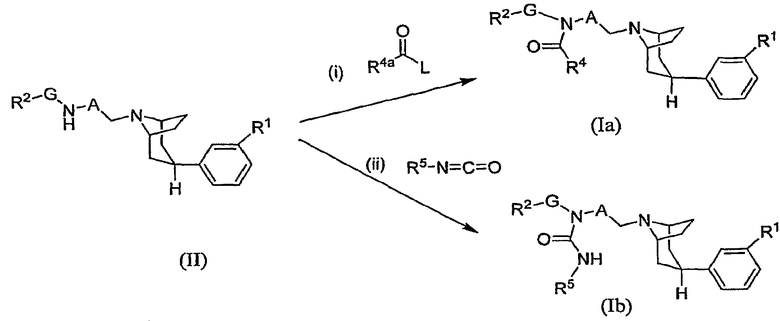
На схеме А R4а означает R4 или защищенную форму R4 и L означает удаляемую группу, такую как хлор или бром, либо R4аC(O)-L означает карбоновую кислоту или карбоксилат. Например, при получении соединения, в котором R4а означает -СН2ОН, приемлемым реагентом является ацетоксиацетилхлорид, в котором R4а означает -СН2ОС(О)СН3 и L означает хлор. Когда R4а представляет собой защищенную форму R4, реакция (i) включает также стадию удаления защитной группы, которая не показана. При получении соединения, в котором R1 означает аминогруппу, для R1 в промежуточном соединении (II) предпочтительно используют защищенную аминогруппу и последовательность реакций включает конечную стадию удаления защитной группы.
Оптимальные условия реакции (i) на схеме А при получении соединений формулы (Ia) могут изменяться в зависимости от химических свойств реагента R4C(O)-L, как хорошо известно специалистам в данной области. Например, когда L означает галоген в качестве удаляемой группы, такой как хлор, реакцию (i) обычно выполняют, осуществляя контактирование промежуточного соединения (II) примерно с 1-2 эквивалентами соединения формулы R4аC(O)-L в инертном разбавителе, таком как дихлорметан, в присутствии избытка основания, например, около 3-6 эквивалентов основания, такого как N,N-диизопропилэтиламин или триэтиламин. Приемлемые инертные разбавители включают также 1,1,2,2-тетрахлорэтан, тетрагидрофуран, диметилацетамид и тому подобные. Указанную реакцию обычно выполняют при температуре в интервале от около -50°С до около 30°С в течение периода времени от около четверти часа до около 16 часов или до полного осуществления реакции.
Когда реагент R4аC(O)-L представляет собой карбоновую кислоту или карбоксилат, реакцию (i) обычно выполняют, осуществляя контактирование промежуточного соединения (II) примерно с 1-5 эквивалентами кислоты R4аC(O)OH или карбоксилата, например, R4аC(O)OLi, в инертном разбавителе и в присутствии избытка основания, описанных выше, и в присутствии примерно 1-6 эквивалентов активирующего агента, такого как N,N-карбонилдиимидазол (CDI), гексафторфосфат N,N,N',N'-тетраметил-О-(7-азабензотриазол-1-ил)урония (HATU) или 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC). Указанную реакцию обычно выполняют при температуре в интервале от около 25°С до около 100°С в течение периода времени от около 2 часов до около 16 часов или до полного осуществления реакции.
Как описано в приведенных ниже примерах, определенные соединения формулы (Ia) могут быть получены в результате осуществления реакции сочетания промежуточного соединения формулы (II) с альтернативными реагентами, такими как циклические ангидриды или диоксоланкарбоновая кислота.
Получение соединений мочевины формулы (Ib) показано в реакции (ii) на схеме А. Указанную реакцию обычно выполняют, осуществляя контактирование промежуточного соединения (II) примерно с 1-2 эквивалентами соединения изоцианата R5-N=C=O в присутствии от около 3 до около 6 эквивалентов основания, такого как N,N-диизопропилэтиламин. Указанную реакцию обычно выполняют при комнатной температуре в течение периода времени от около 1 часа до около 16 часов или до полного осуществления реакции.
Общий способ получения промежуточного соединения формулы (II) показан на схеме В1, где Р1 означает аминозащитную группу.
Схема В1
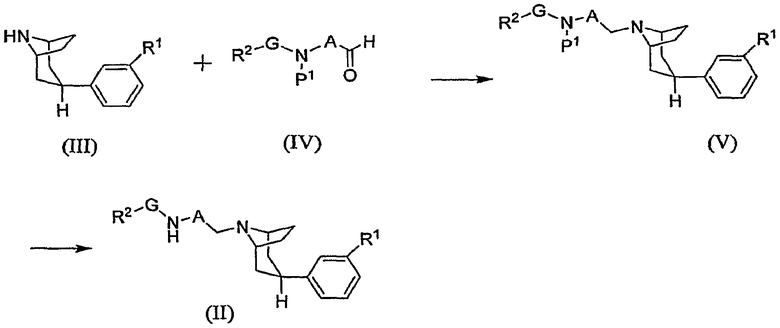
Промежуточное соединение формулы (III), определяемое в настоящем описании изобретения как “фенилтропан”, подвергают восстановительному N-алкилированию, осуществляя взаимодействие с альдегидом формулы (IV), и получают защищенное промежуточное соединение (V), из которого удаляют защитную группу стандартными методами, получая при этом промежуточное соединение (II).
Начальную реакцию обычно выполняют, осуществляя контактирование промежуточного соединения (III) примерно с 1-2 эквивалентами альдегида формулы (IV) в приемлемом разбавителе, таком как инертный разбавитель, в присутствии от около 0,9 до около 2 эквивалентов восстановителя. Указанную реакцию обычно выполняют при температуре в интервале от около 0°С до комнатной температуры в течение периода времени от около получаса до около 3 часов или до полного осуществления реакции. Приемлемые инертные разбавители включают дихлорметан и вышеуказанные разбавители. Кроме того, в качестве разбавителя может быть использован метанол или этанол. Типичные восстановители включают триацетоксиборгидрид натрия, боргидрид натрия и цианоборгидрид натрия. Соединение (V) выделяют стандартными методами. Защитную группу из соединения (V) удаляют стандартными методами. Например, когда защитной группой Р1 является Вос, соединение (V) обычно обрабатывают кислотой, такой как трифторуксусная кислота, получая при этом промежуточное соединение (II). При выполнении реакции, показанной на схеме В1, промежуточное соединение (III) может быть получено в виде свободного основания или соли. В последнем случае при выполнении данной реакции может быть необязательно использован примерно 1 эквивалент основания.
Другой общий способ получения промежуточного соединения формулы (II) показан на схеме В2.
Схема В2
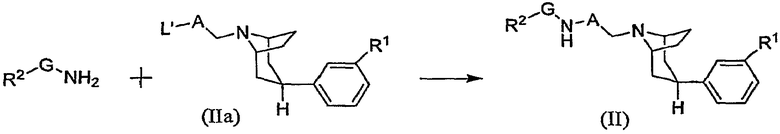
где L' означает сульфонатную удаляемую группу, такую как мезилат или тозилат. Указанную реакцию обычно выполняют, осуществляя контактирование промежуточного соединения (IIa) примерно с 1-2 эквивалентами R2-G-NH2 амина в инертном растворителе, таком как диметилформамид или спирт, в присутствии примерно 1-2 эквивалентов основания, такого как N,N-диизопропилэтиламин или тому подобного. Указанную реакцию обычно выполняют при температуре в интервале от около 25°С до около 80°С в течение периода времени от около получаса до около 2 часов или до полного осуществления реакции.
Промежуточные соединения формулы (IIa) могут быть получены стандартными методами. Например, промежуточное соединение формулы (IIa), в котором L' означает мезилат, может быть получено в соответствии с описанием, приведенным в примере 130. Галогензамещенный спирт формулы НО-А-СН2-Х, где Х означает галоген, подвергают взаимодействию с фенилтропаном (III), получая при этом промежуточное соединение в виде спирта, в котором НО-А-СН2- связан с атомом азота тропана и которое далее подвергают взаимодействию с метансульфонилхлоридом, получая при этом промежуточное соединение (IIa).
Третий способ получения промежуточного соединения (II) показан на схеме В3.
Схема В3

где Gа имеет такие значения, при которых Gа-CH2 является переменным элементом G, то есть Gа означает С1-3алкиленил или Gа означает ковалентную связь. Промежуточное соединение (IIb) подвергают восстановительному N-алкилированию, осуществляя взаимодействие с альдегидом R2-Gа-C(O)H, и получают промежуточное соединение (II). Указанную реакцию обычно выполняют в условиях, описанных выше для реакции N-алкилирования соединения (III) на схеме В1. Промежуточное соединение (IIb) может быть получено в результате восстановительного N-алкилирования фенилтропана (III) защищенным аминоальдегидом формулы N(HP1)-A-C(O)H с последующим удалением защитной группы.
В другом альтернативном способе получения промежуточного соединения (II) карбоновую кислоту подвергают реакции сочетания с фенилтропаном (III) в присутствии амида, являющегося связующим веществом, с образованием промежуточного соединения в виде амида, который затем восстанавливают, получая при этом промежуточное соединение (II) в соответствии с описанием, приведенным, например, в нижеследующем препаративном примере 22.
В еще одном альтернативном способе промежуточное соединение формулы (II), в котором переменный элемент А является метиленом, получают способом, показанным на схеме С.
Схема С

Как показано на схеме С, промежуточное соединение (III) подвергают восстановительному N-алкилированию, осуществляя взаимодействие с диметоксиацетальдегидом, и получают промежуточное соединение (VI) в виде ацеталя в вышеописанных условиях N-алкилирования. Затем промежуточное соединение (VI), являющееся ацеталем, гидролизуют в водном растворе сильной кислоты, таком как 3 н. или 6 н. раствор HCl, получая при этом промежуточное соединение (VII), являющееся альдегидом, в виде хлористоводородной соли. Указанную реакцию обычно выполняют при температуре в интервале от около 20°С до около 40°С в течение периода времени от около 3 до около 72 часов или до полного осуществления реакции.
И наконец, в результате восстановительного аминирования промежуточного соединения (VII) амином формулы R2-G-NH2 получают промежуточное соединение формулы (II'). Альдегид (VII) в инертном разбавителе обычно подвергают взаимодействию примерно с 1-2 эквивалентами амина в присутствии примерно 1-2 эквивалентов восстановителя и примерно одного эквивалента основания. Указанную реакцию обычно выполняют при комнатной температуре в течение периода времени от около 15 минут до 2 часов или до полного осуществления реакции.
Промежуточные соединения (III) и (IV) могут быть получены из легко доступных исходных веществ. Например, один способ получения фенилтропана (III'), в котором R1 означает гидрокси, показан на схеме D.
Схема D

где Bn означает бензильную аминозащитную группу. Защищенный тропанон (VIII) можно получить, осуществляя взаимодействие 2,5-диметокситетрагидрофурана с бензиламином и 1,3-ацетондикарбоновой кислотой в кислом водном растворе в присутствии буферного агента в соответствии с описанием, приведенным в заявке на патент США 2005/0228014. (См. также патент США № 5753673).
Тропанон (VIII) сначала добавляют к раствору примерно 1-2 эквивалентов реагента Гриньяра, являющегося бромидом 3-метоксифенилмагния, в инертном разбавителе. Указанную реакцию обычно выполняют при температуре от около 0°С до около 10°С в течение периода времени от около 1 часа до около 3 часов или до полного осуществления реакции. Повторное метилирование рагента Гриньяра до его использования для замены магния цезием в результате осуществления взаимодействия с эквивалентным количеством хлорида цезия обеспечивает хороший выход промежуточного соединения (IX). Гидроксильный заместитель удаляют из промежуточного соединения (IX) путем обработки 6 н. водным раствором HCl, получая при этом хлористоводородную соль промежуточного соединения (Х). Указанную реакцию обычно выполняют при температуре в интервале от около 50°С до около 100°С в течение периода времени от около 1 часа до около 3 часов или до полного осуществления реакции.
Гидрирование промежуточного соединения (Х) насыщает двойную связь алкеновой группы и удаляет бензильную защитную группу с образованием промежуточного соединения (XI). Указанную реакцию обычно выполняют, помещая соль HCl соединения (Х), растворенную в этаноле, в атмосферу водорода в присутствии катализатора на основе переходного металла. И наконец, метильную группу удаляют из промежуточного соединения (XI), осуществляя контактирование охлажденного раствора промежуточного соединения (XI) в инертном разбавителе примерно с 1-2 эквивалентами трибромида бора, бромоводорода или трихлорида бора. Указанную реакцию обычно выполняют при температуре в интервале от около -80°С до около 0°С в течение периода времени от около 12 часов до около 36 часов или до полного осуществления реакции. Альтернативно промежуточное соединение (XI) может быть выделено в виде хлористоводородной соли, которую обрабатывают примерно 1-2 эквивалентами водного раствора бромистоводородной кислоты с образованием промежуточного соединения (III'), являющегося фенилтропаном.
Промежуточное соединение (III') может быть выделено стандартными методами в виде свободного основания или бромистоводородной соли. В результате кристаллизации бромистоводородной соли образуется промежуточное соединение (III') с высокой стереоспецифичностью в эндо-конфигурации (соотношение эндо-изомеров и экзо-изомеров превышает 99,1:0,8).
Как указано в приведенных ниже примерах, для получения промежуточного соединения (III') могут быть альтернативно использованы определенные варианты вышеуказанного способа. Например, другие реагенты могут быть использованы для удаления гидроксильной группы из соединения (IX) при получении промежуточного соединения (Х), которое может быть выделено в виде свободного основания вместо соли. В другом альтернативном способе обработка промежуточного соединения (IX) трибромидом бора или HBr позволяет удалить метильную группу, так как данная группа элиминирует гидроксильный заместитель.
Один способ получения промежуточного соединения (III”), в котором переменный элемент R1 означает -С(О)NH2 и в качестве исходного вещества использовано соединение (III'), показан на схеме Е.
Схема Е
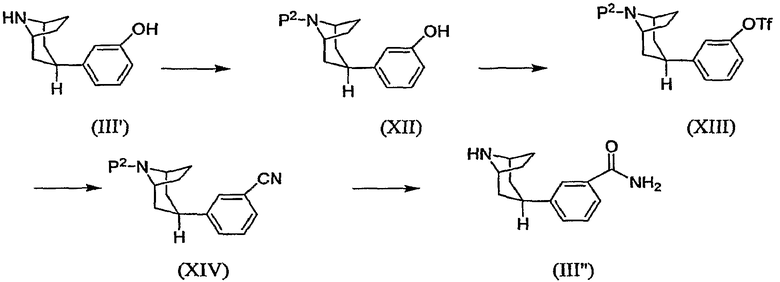
где -OTf означает трифторметансульфонат (обычно трифлат) и Р2 означает аминозащитную группу.
Например, при использовании Вос в качестве защитной группы, фенилтропан (III') обычно сначала подвергают взаимодействию примерно с 1 эквивалентом ди-трет-бутилдикарбоната (обычно Вос2О), получая при этом Вос-защищенное промежуточное соединение (XII). Реагенты обычно охлаждают примерно до 0°С и затем оставляют нагреваться до комнатной температуры в течение периода времени от около 12 часов до около 24 часов. При использовании трифторацетила в качестве защитной группы, соединение (III') обычно подвергают взаимодействию примерно с 2 эквивалентами трифторацетилангидрида с образованием защищенного промежуточного соединения (XII). Затем промежуточное соединение (XII) в инертном разбавителе подвергают взаимодействию с небольшим избытком, например, примерно 1,1 эквивалента трифторметансульфонилхлорида в присутстувии примерно 1-2 эквивалентов основания с образованием промежуточного соединения (XIII), которое может быть выделено стандартными методами. Осуществление взаимодействия соединения (XIII) с цианидом цинка в присутствии катализатора на основе переходного металла позволяет получить промежуточное соединение (XIV). Указанную реакцию обычно выполняют при температуре в интервале от около 60°С до 120°С в инертной атмосфере в течение периода времени от около 2 часов до около 12 часов или до полного осуществления реакции.
И наконец, промежуточное соединение (XIV), являющееся нитрилом, гидролизуют и удаляют защитную группу, получая при этом промежуточное соединение (III”) в виде карбоксамида. При выполнении данной реакции, когда Р2 означает Вос, промежуточное соединение (XIV) в кислотном растворителе, таком как трифторуксусная кислота, подвергают взаимодействию примерно с 4-6 эквивалентами концентрированной серной кислоты. Указанную реакцию обычно выполняют при температуре в интервале от около 50°С до около 80°С в течение периода времени от около 8 часов до около 24 часов или до полного осуществления реакции. Продукт обычно выделяют в виде свободного основания. Альтернативно трансформацию соединения (XIV) в соединение (III”) выполняют в два этапа, при выполнении которых нитрильный заместитель промежуточного соединения (XIV) сначала гидролизуют с образованием карбоксамида в результате осуществления взаимодействия с карбонатом калия и пероксидом водорода, после чего защитную группу Вос удаляют, производя обработку кислотой, например, трифторуксусной кислотой.
При использовании трифторацетильной защитной группы промежуточное соединение, являющееся нитрилом, сначала гидролизуют с образованием карбоксамида в концентрированной серной кислоте в соответствии с приведенным выше описанием. Реакцию гидролиза гасят, добавляя основание, что позволяет также удалить защитную группу. Продукт обычно выделяют в виде хлористоводородной соли. В приведенных ниже примерах описана альтернативная последовательность реакций с использованием защищенного промежуточного соединения, являющегося цианофенилпропаном.
Промежуточное соединение формулы (III), в котором R1 означает -NHS(O)2R6, может быть получено из промежуточного соединения (XIII), показанного на схеме Е. Как указано, например, в приведенном ниже препаративном примере 23, промежуточное соединение формулы (III), в котором R1 означает -NHS(O)2CH3, может быть получено в результате осуществления взаимодействия промежуточного соединения (XIII) с бензофенонимином в присутствии палладиевого катализатора с образованием промежуточного соединения, являющегося защищенным 8-азабициклооктаном, замещенным 3-аминофенилом, которое в свою очередь подвергают взаимодействию с метансульфонилхлоридом для получения защищенного промежуточного соединения, в котором R1 означает -NHS(O)2CH3. Защитную группу затем удаляют стандартными методами, получая при этом промежуточное соединение формулы (III).
Замещенное трифлатом промежуточное соединение (XIII) также может быть использовано в качестве исходного вещества для получения других промежуточных соединений, используемых при получении соединений по настоящему изобретению. Промежуточное соединение формулы (III), в котором R1 означает сложный эфир, -С(О)ОRа, где Rа означает С1-3алкил, может быть получено карбонилированием соединения (XIII) с использованием палладиевого катализатора в присутствии спиртового растворителя RаOH с последующим удалением защитной группы. Промежуточное соединение (III), в котором R1 означает кислоту, -С(О)ОН, может быть получено путем гидролиза защищенной формы промежуточного соединения (III), в котором R1 означает сложный эфир, в присутствии неорганического основания с последующим удалением защитной группы. Восстановление защищенного кислотного промежуточного соединения при помощи восстановителя, такого как боргидрид натрия, после удаления защитной группы позволяет получить промежуточное соединение формулы (III), в котором R1 означает -СН2ОН.
Промежуточные соединения формулы (IV), представленные на схеме В1, могут быть получены из спирта формулы (XV) способом, показанным на схеме F.
Схема F
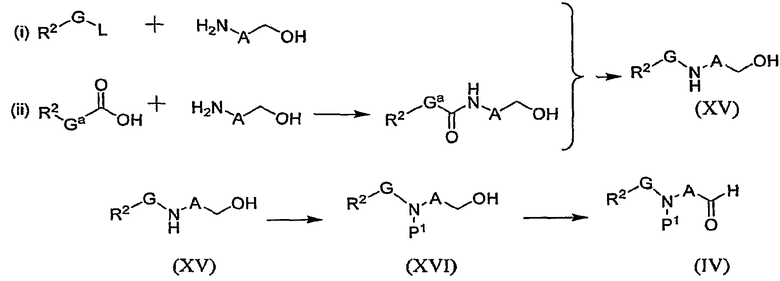
где Gа имеет такое значение, при котором Gа-CH2 означает G и L означает удаляемую группу. Спирт (XV) может быть получен в результате выполнения реакции (i), при осуществлении которой спирт формулы Н2N-A-CH2-OH подвергают взаимодействию с замещенным алкилгалогенидом формулы R2-G-L в условиях, аналогичных описанным для реакции, показанной на схеме В2. Спирт (XV) может быть также получен в результате выполнения реакции (ii) в типичных условиях сочетания с кислотой, описанных выше, с образованием промежуточного соединения амида, которое восстанавливают, например, бораном, с образованием промежуточного соединения спирта формулы (XV). Затем, добавляя аминозащитную группу стандартными методами, получают промежуточное соединение (XVI), которое окисляют с образованием промежуточного соединения формулы (IV). Промежуточное соединение (IV) можно получить и хранить в виде аддукта бисульфита, из которого альдегид высвобождают перед использованием.
В альтернативном способе получения соединений по настоящему изобретению формулы (Ia) промежуточное соединение (III), являющееся фенилтропаном, подвергают взаимодействию с промежуточным соединением формулы (XVII)
Схема G

в условиях, аналогичных описанным для начальной реакции, показанной на схеме В1. Когда R4а является защищенной формой R4, конечную стадию удаления защитной группы выполняют с образованием соединения (Ia). Промежуточное соединение (XVII) может быть получено в результате осуществления взаимодействия спирта (XV) с реагентом R4аC(O)-L для добавления -С(О)R4а к атому азота соединения (XV) с последующим окислением полученного спирта в альдегид (XVII). Соединения мочевины по настоящему изобретению формулы (Ib) могут быть получены способами, аналогичными показанным на схеме G.
Более подробно конкретные условия выполнения реакций и другие способы получения типичных соединений по настоящему изобретению или промежуточных соединений описаны в приведенных ниже примерах.
Таким образом, настоящее изобретение относится к способу получения соединения формулы (I), его соли или защищенного производного, который включает (а) осуществление взаимодействия соединения формулы (II) с (i) соединением формулы R4аC(O)-L или (ii) соединением формулы R5-N=C=O; или (b) осуществление взаимодействия соединения формулы (III) с соединением формулы (XVII); и необязательно удаление защитной группы или групп из R4а, в резульате чего образуется соединение формулы (I), его соль или защищенное производное.
Отдельные варианты осуществления настоящего изобретения далее относятся к соединению формулы (II) и соединению формулы (III), в которых переменные элементы R1, R2, G и А имеют любые значения, указанные в вышеописанных вариантах осуществления настоящего изобретения. В частности, настоящее изобретение относится к соединению формулы (II), в котором R1 означает -С(О)NH2, R2 означает циклогексил или фенил, причем циклогексил и фенил могут быть необязательно замещены одним или двумя атомами галогена, G означает -СН2- и А означает -СН2-. Кроме того, еще один конкретный вариант осуществления настоящего изобретения относится к соединению формулы (III), в котором R1 означает -ORа или -C(O)NRаRb либо R1 означает -ОН или -С(О)NH2.
Фармацевтические композиции
Соединения 8-азабициклооктана по настоящему изобретению обычно вводят субъекту в виде фармацевтической композиции или препарата. Такие фармацевтические композиции можно вводить субъекту любыми приемлемыми способами введения, которые включают, не ограничиваясь ими, пероральный, ректальный, вагинальный, назальный, ингаляционный, местный (в том числе чрескожный) и парентеральные способы введения.
Таким образом, один из вариантов осуществлеия настоящего изобретения относится к фармацевтическим композициям, содержащим фармацевтически приемлемый носитель или наполнитель и терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. Такие фармацевтические композиции при желании могут необязательно содержать другие лекарственные и/или фармацевтические агенты. При рассмотрении композиций термин “соединение по настоящему изобретению” может также определяться в настоящем описании изобретения как “активный агент”. В используемом здесь значении термин “соединение по настоящему изобретению” означает соединения формулы (I), а также варианты, определяемые формулой (I'). Термин “соединение по настоящему изобретению” означает также фармацевтически приемлемые соли и сольваты данного соединения за исключением особо оговоренных случаев.
Фармацевтические композиции по настоящему изобретению обычно содержат терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли. Такие фармацевтические композиции обычно содержат от около 0,1 до около 95 мас.% активного агента; предпочтительно от около 5 до около 70 мас.% и более предпочтительно от около 10 до около 60 мас.% активного агента.
В фармацевтических композициях по настоящему изобретению могут быть использованы любые стандартные носители или наполнители. Выбор конкретного носители или наполнителя либо комбинаций носителей или наполнителей зависит от способа введения, применяемого для лечения конкретного субъекта, типа болезненного состояния или тяжести заболевания. В данной связи получение приемлемой фармацевтической композиции для конкретного способа введения находится в компетенции специалистов в области фармакологии. Кроме того, носители или наполители, используемые в фармацевтических композициях по настоящему изобретению, являются коммерчески доступными. В качестве дальнейшей иллюстрации можно отметить, что стандартные методы приготовления лекарственных средств описаны в публикациях Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); and H. C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типичные примеры веществ, которые могут быть использованы в качестве фармацевтически приемлемых носителей, включают, не ограничиваясь ими, следующие вещества: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу, такую как микрокристаллическая целлюлоза и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический физиологический раствор; раствор Рингера; этиловый спирт; растворы фосфатного буфера и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции обычно получают путем тщательного и однородного смешивания или перемешивания активного агента с фармацевтически приемлемым носителем и одним или несколькими необязательными ингредиентами. Полученную однородно смешанную смесь затем используют для изготовления таблеток, капсул, пилюль и тому подобных при помощи стандартных методов и оборудования.
Фармацевтические композиции по настоящему изобретению предпочтительно помещают в упаковку, содержащую лекарственное средство в дозах на один прием. Термин “стандартная лекарственная форма” означает физически раздельную форму с дозой на один прием, вводимую субъекту, то есть каждая форма содержит заранее определенное количество активного агента, обеспечивающее достижение желаемого терапевтического эффекта при отдельном использовании или в комбинации с одной или несколькими дополнительными лекарственными формами. Например, такие стандартные лекарственные формы могут представлять собой капсулы, таблетки, пилюли и тому подобные или упаковки, содержащие лекарственное средство в дозах на один прием для парентерального введения.
В одном варианте осуществления изобретения фармацевтические композиции по настоящему изобретению предназначены для перорального введения. Фармацевтические композиции, пригодные для перорального введения, могут иметь форму капсул, таблеток, пилюль, лепешек, крахмальных облаток, драже, порошков, гранул; растворов или суспензий в водной или неводной жидкости; жидкой эмульсии масла в воде или воды в масле; эликсира или сиропа и тому подобные, причем указанные композиции содержат заранее определенное количество соединения по настоящему изобретению в качестве активного ингредиента.
Фармацевтические композиции по настоящему изобретению, предназначенные для перорального введения в виде твердой лекарственной формы (то есть капсул, таблеток, пилюль и тому подобных), обычно содержат активный агент и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия и дикальцийфосфат. Необязательно или альтернативно такие твердые лекарственные формы могут также содержать: наполнители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; связывающие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; увлажнители, такие как глицерин; дезинтеграторы, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из кассавы, альгиновая кислота, определенные силикаты и/или карбонат натрия; замедлители растворения, такие как парафин; ускорители абсорбции, такие как соединения четвертичного аммония; смачивающие вещества, такие как цетиловый спирт и/или моностеарат глицерина; абсорбенты, такие как каолин и/или бентонит; смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красители и буферные агенты.
В фармацевтических композициях по настоящему изобретению могут также присутствовать релизинг-агенты, смачивающие вещества, агенты, образующие покрытие, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты. Примеры фармацевтически приемлемых антиоксидантов включают: водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и тому подобные; антиоксиданты, растворимые в масле, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и тому подобные; и хелатообразователи металлов, такие как лимонная кислота, этилендиаминотетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и тому подобные. Агенты, образующие покрытие для таблеток, капсул, пилюль и тому подобных, включают агенты, используемые для образования энтеросолюбильных покрытий, такие как фталат ацетата целлюлозы, фталат поливинилацетата, фталат гидроксипропилметилцеллюлозы, сополимеры метакриловой кислоты и сложного эфира метакриловой кислоты, тримеллитат ацетата целлюлозы, карбоксиметилэтилцеллюлоза, сукцинат ацетата гидроксипропилметилцеллюлозы и тому подобные.
Фармацевтические композиции по настоящему изобретению могут также характеризоваться замедленным или регулируемым высвобождением активного агента при использовании в качестве примера гидроксипропилметилцеллюлозы в разных пропорциях, или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции по настоящему изобретению могут необязательно содержать агенты, делающие вещество непрозрачным, и могут обеспечивать высвобождение активного ингредиента только или предпочтительно в определенном отделе желудочно-кишечного тракта, необязательно с замедленным действием. Примеры заливающих композиций включают полимерные вещества и воски. Активный агент может также быть в микроинкапсулированной форме при необходимости с одним или несколькими вышеуказанными наполнителями.
Жидкие лекарственные формы, приемлемые для перорального введения, включают в качестве примера фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно содержат активный агент и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое, арахисовое, кукурузное, проростковое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли, сложные эфиры жирных кислот сорбита и их смеси. Суспензии, помимо активного ингредиента, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар, трагант и их смеси.
Соединения по настоящему изобретению можно также вводить парентерально (например, в виде внутривенных, подкожных, внутримышечных или внутрибрюшинных инъекций). Активный агент, предназначенный для парентерального введения, обычно смешивают с приемлемым наполнителем для парентерального введения, включающим в качестве примера стерильные водные растворы, физиологический раствор, низкомолекулярные спирты, такие как пропиленгликоль, полиэтиленгликоль, растительные масла, желатин, сложные эфиры жирных кислот, такие как этилолеат, и тому подобные. Препараты для парентерального введения могут также содержать один или несколько антиоксидантов, солюбилизаторов, стабилизиторов, консервантов, смачивающих веществ, эмульгаторов, буферных агентов или диспергаторов. Указанные препараты могут быть стерилизованы при помощи стерильной инъекционной среды, стерилизующего агента, фильтрации, облучения или нагревания.
Альтернативно фармацевтические композиции по настоящему изобретению могут быть получены в форме, предназначенной для ингаляции. Фармацевтические композиции, пригодные для введения ингаляцией, обычно имеют форму аэрозоля или порошка. Такие композиции обычно вводят при помощи хорошо известных доставляющих устройств, таких как дозирующий ингалятор, ингалятор сухого порошка, распылитель или подобное доставляющее устройство.
При введении путем ингаляции с использованием находящегося под давлением баллона фармакологические композиции по настоящему изобретению обычно содержат активный ингредиент и приемлемый пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой приемлемый газ. Кроме того, фармацевтическая композиция может быть в форме капсулы или картриджа (изготовленного, например, из желатина), содержащего соединение по настоящему изобретению и порошок, приемлемый для использования в ингаляторе порошка. Приемлемые порошкообразные основы включают в качестве примера лактозу или крахмал.
Соединения по настоящему изобретению можно также вводить чрескожно с использованием известных систем для чрескожной доставки и наполнителей. Например, активный агент может быть смешан с усилителями проникновения, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-оны и тому подобные, и введен в пластырь или подобную систему доставки. При желании такие композиции для чрескожного введения могут содержать дополнительные наполнители, включающие гелеобразующие агенты, эмульгаторы и буферы.
Соединения по настоящему изобретению при желании можно вводить в комбинации с одним или несколькими другими лекарственными средствами. В одном варианте осуществления изобретения соединение по настоящему изобретению физически смешивают с другим лекарственным агентом с образованием композиции, содержащей оба агента, или каждый агент находится в отдельных композициях, которые вводят субъекту одновременно или последовательно.
Например, соединение формулы I можно объединить со вторым лекарственным агентом при помощи стандартных методов и оборудования, получив при этом композицию, содержащую соединение формулы I и второй лекарственный агент. Кроме того, лекарственные агенты могут быть объединены с фармацевтически приемлемым носителем с образованием фармацевтической композиции, содержащей соединение формулы I, второй лекарственный агент и фармацевтически приемлемый носитель. В данном варианте осуществления изобретения компоненты композиции обычно смешивают или перемешивают с образованием физической смеси. Физическую смесь затем вводят в терапевтически эффективном количестве любым из описанных способов. Альтернативно лекарственные агенты могут находиться в раздельных формах до введения субъекту. В данном варианте осуществления изобретения указанные агенты физически не смешивают друг с другом до введения, а вводят одновременно или в разное время в виде отдельных композиций. Такие композиции могут быть упакованы отдельно или вместе в виде набора. Два лекарственных агента в наборе можно вводить одинаковыми или разными способами введения.
В качестве второго лекарственного агента может быть использован любой лекарственный агент, совместимый с соединениями по настоящему изобретению. В частности, в комбинации с соединениями по настоящему изобретению могут быть использованы прокинетические агенты, действующие в соответствии с механизмами, отличными от антагонизма в отношении рецепторов мю-опиоидов. Например, в качестве второго лекарственного агента могут быть использованы агонисты рецепторов 5-НТ4, такие как тегасерод, рензаприд, мозаприд, прукалоприд, {(1S,3R,5R)-8-[2-(4-ацетилпиперазин-1-ил)этил]-8-азабицикло[3.2.1]-окт-3-ил}амид 1-изопропил-1Н-индазол-3-карбоновой кислоты, {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты или метиловый эфир 4-(4-{[(2-изопропил-1Н-бензоимидазол-4-карбонил)амино]метил}пиперидин-1-илметил)пиперидин-1-карбоновой кислоты.
Дополнительные пригодные прокинетические агенты включают, не ограничиваясь ими, агонисты рецепторов 5-НТ3 (например, пумосетраг), антагонисты рецепторов 5-НТ1А (например, AGI001), альфа-2-дельта-лиганды (например, PD-217014), средства, открывающие хлоридные каналы (например, лубипростон), антагонисты допамина (например, итоприд, метаклопрамид, домперидон), агонисты GABA-B (например, баклофен, AGI 006), агонисты каппа-опиоидов (например, азимадолин), антагонисты М1 и М2 мускарина (например, акотиамид), агонисты мотилина (например, митемцинал), активаторы гуанилатциклазы (например, MD-1100) и агонисты грелина (например, Tzp 101, RC 1139).
Кроме того, соединения по настоящему изобретению могут быть объединены с опиоидными лекарственными средствами. Такие опиоидные средства включают, не ограничиваясь ими, морфин, петидин, кодеин, дигидрокодеин, оксиконтин, оксикодон, гидрокодон, суфентанил, фентанил, ремифентанил, бупренорфин, метадон и героин.
В данной области известны многочисленные дополнительные примеры таких лекарственных средств, и в комбинации с соединениями по настоящему изобретению могут быть использованы любые такие известные лекарственные средства. Вторичные средства, в случае их использования, присутствуют в терапевтически эффективном количестве, то есть в любом количестве, обеспечивающем терапевтически благоприятное действие при совместном введении с соединением по настоящему изобретению. Приемлемые дозы других лекарственных средств, вводимых в комбинации с соединением по настоящему изобретению, обычно находятся в пределах от около 0,05 мкг/сутки до около 100 мг/сутки.
Таким образом, фармацевтические композиции по настоящему изобретению необязательно содержат второе лекарственное средство, описанное выше.
Приведенные ниже примеры иллюстрируют типичные фармацевтические композиции по настоящему изобретению.
Пример приготовления лекарственного средства А. Твердые желатиновые капсулы для перорального введения
Соединение по настоящему изобретению (50 г), высушенную распылением лактозу (200 г) и стеарат магния (10 г) тщательно перемешивают. Полученную композицию вводят в твердую желатиновую капсулу (260 мг композиции в одной капсуле).
Пример приготовления лекарственного средства В. Твердые желатиновые капсулы для перорального введения
Соединение по настоящему изобретению (20 мг), крахмал (89 мг), микрокристаллическую целлюлозу (89 мг) и стеарат магния (2 мг) тщательно перемешивают и пропускают через сито с размером ячеек США № 45. Полученную композицию вводят в твердую желатиновую капсулу (200 мг композиции в одной капсуле).
Пример приготовления лекарственного средства С. Желатиновые капсулы для перорального введения
Соединение по настоящему изобретению (10 мг), моноолеат полиоксиэтиленсорбита (50 мг) и порошкообразный крахмал (250 мг) тщательно перемешивают и вводят в желатиновую капсулу (310 мг композиции в одной капсуле).
Пример приготовления лекарственного средства D. Таблетки для перорального введения
Соединение по настоящему изобретению (5 мг), крахмал (50 мг) и микрокристаллическую целлюлозу (35 мг) пропускают через сито с размером ячеек США № 45 и тщательно смешивают. Раствор поливинилпирролидона (10 мас.% в воде, 4 мг) смешивают с полученным порошком и полученную смесь пропускают через сито с размером ячеек США № 14. Полученные таким образом гранулы сушат при 50-60°С и пропускают через сито с размером ячеек США № 18. К гранулам добавляют карбоксиметиловый крахмал натрия (4,5 мг), стеарат магния (0,5 мг) и тальк (1 мг), которые предварительно пропускают через сито с размером ячеек США № 60. Все компоненты смешивают и смесь прессуют в таблетирующей машине, получая таблетку массой 100 мг.
Пример приготовления лекарственного средства Е. Таблетки для перорального введения
Соединение по настоящему изобретению (25 мг), микрокристаллическую целлюлозу (400 мг), выпаренный диоксид кремния (10 мг) и стеариновую кислоту (5 мг) тщательно перемешивают и прессуют с образованием таблеток (440 мг композиции в одной таблетке).
Пример приготовления лекарственного средства F. Таблетки с одной риской для перорального введения
Соединение по настоящему изобретению (15 мг), кукурузный крахмал (50 мг), натрий-кроскармеллозу (25 мг), лактозу (120 мг) и стеарат магния (5 мг) тщательно перемешивают и прессуют с образованием таблетки с одной риской (215 мг композиции в одной таблетке).
Пример приготовления лекарственного средства G. Суспензия для перорального введения
Нижеследующие ингредиенты тщательно смешивают с образованием суспензии для перорального введения, содержащей 100 мг активного ингредиента в 10 мл суспензии.
Пример приготовления лекарственного средства Н. Композиция в виде сухого порошка
Микроизмельченное соединение по настоящему изобретению (1 мг) смешивают с лактозой (25 мг) и вводят в желатиновый картридж для ингаляции. Содержимое картриджа вводят при помощи ингалятора порошка.
Пример приготовления лекарственного средства J. Инъецируемый препарат
Соединение по настоящему изобретению (0,1 г) смешивают с 0,1 М раствором буфера на основе цитрата натрия (15 мл). Показатель рН полученного раствора доводят до рН 6, добавляя 1 н. водный раствор хлористоводородной кислоты или 1 н. водный раствор гидроксида натрия. Затем добавляют стерильный нормальный физиологический раствор в цитратном буфере, получая общий объем, равный 20 мл.
Очевидно, что в вышеуказанных фармацевтических композициях может быть использована любая форма соединений по настоящему изобретению (то есть свободное основание, фармацевтическая соль или сольват), пригодная для конкретного способа введения.
Полезность
Соединения 8-азабициклооктана по настоящему изобретению являются антагонистами рецепторов мю-опиоидов, поэтому предполагается, что указанные соединения должны быть пригодны для лечения болезненных состояний, опосредованных рецепторами мю-опиоидов или ассоциированных с активностью рецепторов мю-опиоидов, то есть болезненных состояний, которые могут быть облегчены в результате лечения антагонистом рецепторов мю-опиоидов. В частности, предполагается, что соединения по настоящему изобретению пригодны для лечения вредных эффектов, связанных с использованием опиоидных аналгетиков, то есть таких симптомов, как запор, плохое опорожнение желудка, боли в брюшной полости, вздутие живота, тошнота и гастроэзофагеальный рефлюкс, которые в совокупности определяются как вызванная опиоидами дисфункция кишечника. Антагонисты рецепторов мю-опиоидов по настоящему изобретению могут быть также пригодны для лечения послеоперационной непроходимости кишечника (POI), нарушения, обусловленного пониженной перистальтикой желудочно-кишечного тракта после операции на брюшной полости или другой операции. Кроме того, считается, что антагонисты рецепторов мю-опиоидов могут быть использованы для устранения вызванной опиоидами тошноты и рвоты. Антагонисты рецепторов мю-опиоидов, характеризующиеся некоторым проникновением в центральную нервную систему, могут быть пригодны для лечения наркотической и алкогольной зависимости или пристрастия к азартным играм либо для профилактики, лечения и/или уменьшения ожирения.
Так как соединения по настоящему изобретению повышают перистальтику желудочно-кишечного (GI) тракта в животных моделях, то указанные соединения могут быть использованы для лечения у млекопитающих, включая человека, нарушений желудочно-кишечного тракта, вызванных пониженной перистальтикой. Такие нарушения перистальтики желудочно-кишечного тракта включают в качестве примера хронический запор, вызванный запором синдром раздраженной толстой кишки (С-IBS), диабетический и идиопатический гастропарез и функциональную диспепсию.
Поэтому один вариант осуществления настоящего изобретения относится к способу повышения перистальтики желудочно-кишечного тракта у млекопитающего, который включает введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по настоящему изобретению.
При использовании для лечения нарушений, связанных с пониженной перистальтикой желудочно-кишечного тракта, или других состояний, опосредованных рецепторами мю-опиоидов, соединения по настоящему изобретению обычно вводят перорально в виде однократной суточной дозы или нескольких доз в сутки, хотя могут быть использованы другие формы введения. Например, при использовании для лечения послеоперационной непроходимости кишечника соединения по настоящему изобретению могут быть введены парентерально. Количество активного агента, вводимого в одной дозе, или общее количество, вводимое в сутки, обычно определяет лечащий врач с учетом всех значимых факторов, включающих состояние, подлежащее лечению, выбранный способ введения, вводимое соединение и его относительную активность, возраст, массу тела и реакцию конкретного субъекта, тяжесть симптомов у субъекта и тому подобные.
Приемлемые дозы для лечения нарушений, связанных с пониженной перистальтикой желудочно-кишечного тракта, или других нарушений, опосредованных рецепторами мю-опиоидов, находятся в диапазоне от около 0,0007 до около 20 мг/кг/сутки активного агента, в том числе от около 0,0007 до около 1,4 мг/кг/сутки. Для человека со средней массой тела 70 кг доза должна составлять от около 0,05 до около 100 мг/сутки активного агента.
В одном варианте осуществления изобретения соединения по настоящему изобретению используют для лечения вызванной опиоидами дисфункции кишечника. При использовании для лечения вызванной опиоидами дисфункции кишечника соединения по настоящему изобретению обычно вводят перорально в виде однократной суточной дозы или нескольких доз в сутки. Доза для лечения вызванной опиоидами дисфункции кишечника предпочтительно составляет от около 0,05 до около 100 мг/сутки.
В другом варианте осуществления изобретения соединения по настоящему изобретению используют для лечения послеоперационной непроходимости кишечника. При использовании для лечения послеоперационной непроходимости кишечника соединения по настоящему изобретению обычно вводят перорально или внутривенно в виде однократной суточной дозы или нескольких доз в сутки. Доза для лечения послеоперационной непроходимости кишечника предпочтительно составляет от около 0,05 до около 100 мг/сутки.
Настоящее изобретение относится также к способу лечения млекопитающего, страдающего заболеванием или нарушением, ассоциированным с активностью рецепторов мю-опиоидов, который включает введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению или фармацевтической композиции, содержащей соединение по настоящему изобретению.
Как было указано выше, соединения по настоящему изобретению являются антагонистами рецепторов мю-опиоидов. Поэтому настоящее изобретение далее относится к способу антагонистического воздействия на рецептор мю-опиоида у млекопитающего, который включает введение указанному млекопитающему соединения по настоящему изобретению.
Антагонисты рецепторов мю-опиоидов по настоящему изобретению необязательно вводят в комбинации с другим лекарственным средством или средствами, в частности, в комбинации с прокинетическими агентами, действие которых не связано с механизмами воздействия на мю-опиоиды. Таким образом, другой вариант осуществления изобретения относится к композициям по настоящему изобретению, содержащим терапевтически эффективное количество другого прокинетического агента, и способам их применения.
Кроме того, соединения по настоящему изобретению могут быть также использованы в качестве научных инструментов для исследования или изучения биологических систем или образцов, имеющих рецепторы мю-опиоидов, или для поиска новых соединений, обладающих активностью в отношении рецепторов мю-опиоидов. В таких исследованиях, выполняемых in vitro или in vivo, может быть использована любая приемлемая биологическая система или образец, имеющие рецепторы мю-опиоидов. Типичные биологические системы или образцы, пригодные для таких исследований, включают, не ограничиваясь ими, клетки, клеточные экстракты, плазматические мембраны, образцы ткани, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и тому подобные. Эффекты контактирования биологической системы или образца, содержащих рецепторы мю-опиоидов, с соединением по настоящему изобретению определяют при помощи стандартных методов и оборудования, таких как анализ связывания радиоактивных лигандов и функциональный анализ, рассмотренные в настоящем описании изобретения, или другие функциональные анализы, известные в данной области. Такие функциональные анализы включают, не ограничиваясь ими, опосредуемые лигандами изменения внутриклеточного циклического аденозинмонофосфата (сАМР), опосредуемые лигандами изменения активности фермента аденилилциклазы, опосредуемые лигандами изменения включения аналогов гуанозинтрифосфата (GTP), таких как [35S]GTPγS (гуанозин-5'-O-(γ-тио)трифосфат) или GTP-Eu, в выделенные мембраны в результате катализируемой рецепторами замены аналогов GDP аналогами GTP и опосредуемые лигандами изменения свободных внутриклеточных ионов кальция. Приемлемая концентрация соединения по настоящему изобретению для таких исследований обычно составляет от около 1 наномоля до около 500 наномолей.
При использовании соединений по настоящему изобретению в качестве научных инструментов для поиска новых соединений, обладающих активностью в отношении рецепторов мю-опиоидов, данные связывания или функциональные данные для испытуемого соединения или группы соединений сравнивают с данными связывания с рецепторами мю-опиоидов или функциональными данными для соединения по настоящему изобретению для выявления испытуемых соединений, обладающих более высокой активностью связывания или функциональной активностью, если таковая имеется. Данный объект изобретения включает в качестве отдельных вариантов осуществления изобретения получение сравнительных данных (при помощи соответствующих анализов) и анализ экспериментальных данных для выявления представляющих интерес испытуемых соединений.
Установлено, что наряду с другими свойствами соединения по настоящему изобретению характеризуются сильным связыванием с рецепторами мю-опиоидов и незначительным агонизмом или отсутствием агонизма в отношении указанных рецепторов при выполнении функциональных анализов рецепторов мю-опиоидов. Поэтому соединения по настоящему изобретению являются сильными антагонистами рецепторов мю-опиоидов. Кроме того, соединения по настоящему изобретению демонстрировали в животных моделях главным образом периферическую активность по сравнению с активностью в центральной нервной системе. Поэтому можно предположить, что указанные соединения должны восстанавливать вызванное опиоидами снижение перистальтики желудочно-кишечного тракта, не влияя при этом на благоприятные центральные эффекты аналгезии. Указанные свойства, а также полезность соединений по настоящему изобретению можно продемонстрировать при выполнении разных анализов in vitro и in vivo, хорошо известных специалистам в данной области. Репрезентативные анализы подробно описаны в приведенных ниже примерах.
Примеры
Нижеследующие примеры синтеза и биологические примеры приведены для иллюстрации настоящего изобретения и не ограничивают объем изобретения. Аббревиатуры, использованные в приведенных ниже примерах, имеют следующие значения за исключением особо оговоренных случаев. Не указанные ниже аббревиатуры имеют общепринятые значения.
Реагенты (включая вторичные амины) и растворители были приобретены у коммерческих поставщиков (Aldrich, Fluka, Sigma и т.д.) и использованы без дальнейшей очистки. Реакции выполняли в атмосфере азота за исключением особо оговоренных случаев. Изменение реакционных смесей контролировали при помощи тонкослойной хроматографии (ТСХ), аналитической высокоэффективной жидкостной хроматографии (анал. ВЭЖХ) и масс-спектрометрии, которые подробно описаны ниже в соответствующих примерах реакций. Реакционные смеси обрабатывали в соответствии с описанием каждой реакции; как правило, реакционные смеси очищали экстракцией и другими методами очистки, такими как кристаллизация в зависимости от температуры и растворителя и осаждение. Кроме того, реакционные смеси очищали препаративной ВЭЖХ, общий метод выполнения которой описан ниже. Продукты реакции исследовали при помощи масс-спектрометрии и 1Н-ЯМР-спектроскопии. Для измерения ЯМР образцы растворяли в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6); спектры 1Н-ЯМР были получены в приборе Varian Gemini 2000 (300 МГц) в стандартных условиях наблюдения. Масс-спектрометрию соединений выполняли методом электрораспылительной ионизации (ESMS) в приборе модели АРI 150 EX Applied Biosystems (Foster City, CA) или в приборе модели 1100 LC/MSD Agilent (Palo Alto, CA).
Препаративный пример 1. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
а. Получение 8-бензил-8-азабицикло[3.2.1]октан-3-она
Смесь 2,5-диметокситетрагидрофурана (20,5 г, 155,1 ммоль), воды (42,5 мл) и концентрированной HCl (30 мл) перемешивали при комнатной температуре в течение примерно 30 минут. К перемешиваемой смеси последовательно добавляли воду (62,5 мл), предварительно смешанный раствор бензиламина (17,9 мл, 162,9 ммоль), воды (87,5 мл) и концентрированной HCl (23 мл), предварительно смешанный раствор 1,3-ацетондикарбоновой кислоты (25 г) и воды (100 мл) и раствор Na2HPO4 (16,5 г) в воде (50 мл). Полученную смесь доводили до рН 4-5, добавляя 40% водный раствор NaOH, и перемешивали в течение ночи при комнатной температуре. Смесь подкисляли до рН 3 концентрированной HCl и нагревали до 85°С в течение 3 часов. Затем смесь охлаждали до комнатной температуры и подщелачивали 20% водным раствором NaOH, насыщали твердым хлоридом натрия и экстрагировали дихлорметаном. Органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали с образованием темного масла, которое очищали флэш-хроматографией на колонке. Продукт элюировали смесью 20% этилацетата/гексанов. Требуемые фракции объединяли и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла (16,17 г).
b. Получение 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]-октан-3-ола
Раствор 8-бензил-8-азабицикло[3.2.1]октан-3-она (7,28 г, 33,8 ммоль) в сухом ТГФ (113 мл) охлаждали до -78°С в атмосфере азота. К холодному раствору через капельную воронку добавляли 1,0 М раствор бромида 3-метоксифенилмагния в ТГФ (44 мл). Полученную смесь нагревали до комнатной температуры и перемешивали в течение примерно 20 минут. Реакционную смесь охлаждали до 0°С и добавляли дополнительное количество бромида 3-метоксифенилмагния (30 мл, 30,0 ммоль) в ТГФ. После добавления реакционную смесь снова нагревали до комнатной температуры и перемешивали в течение 30 минут. Реакционную смесь гасили насыщенным раствором хлорида аммония и продукт экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на колонке и элюировали смесью 5% (200 мл), 10% (200 мл), 15% (200 мл), 20% (200 мл), 30% (200 мл) и 100% этилацетата/гексанов. Требуемые фракции объединяли и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде светло-желтого масла (4,0 г). Выделяли исходное вещество (3,87 г). (m/z): [M+H]+, вычислено для С21Н25NO2 324,20; найдено 324,5.
с. Получение 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)фенола
К раствору 8-бензил-3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ола (4,65 г, 14,4 ммоль) в дихлорметане (70 мл) при 0°С добавляли 1,0 М раствор трибромида бора в дихлорметане (28 мл). Полученную смесь перемешивали при 0°С в течение одного часа, оставляли нагреваться до комнатной температуры и перемешивали при указанной температуре в течение ночи. Реакционную смесь концентрировали и трижды упаривали с метанолом. Образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (20 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (4,6 г). (m/z): [M+H]+, вычислено для С20Н21NO 292,17; найдено 292,3.
d. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
К раствору соли ТФУ 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)фенола (2,0 г) в этаноле (200 мл) при комнатной температуре добавляли гидроксид палладия на угле (50 мас.% воды, 20 мас./мас.% на угле, 800 мг). Полученную суспензию дегазировали и обрабатывали в течение ночи в атмосфере водорода. Реакционную смесь фильтровали через целит и промывали этанолом. Фильтрат концентрировали, получая при этом соль ТФУ указанного в заголовке соединения (1,2 г). (m/z): [M+H]+, вычислено для С13Н17NO 204,14; найдено 204,3.
Препаративный пример 2. Синтез 3-эндо-[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]фенола
а. Получение 3-эндо-[8-(2,2-диметоксиэтил)-8-азабицикло[3.2.1]-окт-3-ил]фенола
К перемешиваемой суспензии 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола (2,0 г, 6,3 ммоль) в СН2Cl2 (25 мл) при комнатной температуре последовательно добавляли N,N-диизопропилэтиламин (814 мг, 6,3 ммоль), 2,2-диметоксиацетальдегид (1,31 г, 12,6 ммоль) и триацетоксиборгидрид натрия (1,73 г, 8,19 ммоль). Полученную смесь обрабатывали ультразвуком для ускорения растворения и перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь разбавляли дихлорметаном. Органический слой промывали насыщенным раствором бикарбоната натрия и затем насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом желтоватое масло, которое использовали на следующей стадии без очистки. (m/z): [M+H]+, вычислено для С17Н25NO3 292,19; найдено 292,3.
b. Получение [3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]ацетальдегида
Маслянистый продукт, полученный на предыдущей стадии, обрабатывали 6 н. водным раствором HCl (30 мл) при комнатной температуре в течение двух дней. Растворители удаляли в вакууме. Остаток растворяли в воде и сушили вымораживанием, получая при этом указанное в заголовке промежуточное соединение в виде его хлористоводородной соли (1,3 г). (m/z): [M+H]+, вычислено для С15Н19NO2 246,15; найдено 246,1.
с. Синтез 3-эндо-[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]фенола
К перемешиваемому раствору хлористоводородной соли [3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]ацетальдегида (384 мг, 1,36 ммоль) в дихлорметане (4,5 мл) при комнатной температуре добавляли триацетоксиборгидрид натрия (374 мг, 1,76 ммоль), затем N,N-диизопропилэтиламин (176 мг, 1,36 ммоль) и бензиламин (175 мг, 1,63 ммоль). Раствор перемешивали при комнатной температуре в течение 30 минут и реакционную смесь гасили насыщенным раствором бикарбоната натрия. Водный слой экстрагировали дихлорметаном. Образовавшийся органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали с образованием маслянистого остатка. Остаток повторно растворяли в 50% растворе уксусной кислоты в воде, фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли бистрифторуксусной кислоты (191 мг). (m/z): [M+H]+, вычислено для С22Н28N2О 337,23; найдено 337,3.
Препаративный пример 3
В соответствии со способом по препаративному примеру 2, стадия с, при замене бензиламина соответствующим амином были получены соли бистрифторуксусной кислоты следующих соединений:
3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]-окт-3-ил}фенол; (m/z): [M+H]+, вычислено для С22Н34N2О 343,28; найдено 343,5.
3-эндо-{8-[2-(3-фторбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенол; (m/z): [M+H]+, вычислено для С22Н27FN2О 355,22; найдено 355,5.
3-эндо-{8-[2-(2,6-дифторбензиламино)этил]-8-азабицикло[3.2.1]-окт-3-ил}фенол; (m/z): [M+H]+, вычислено для С22Н26F2N2O 373,21; найдено 373,3.
3-эндо-{8-[2-(4-фторбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенол; (m/z): [M+H]+, вычислено для С22Н27FN2O 355,22; найдено 355,3.
3-эндо-{8-[2-(4-хлорбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенол; (m/z): [M+H]+, вычислено для С22Н27ClN2O 371,19; найдено 371,4.
Препаративный пример 4. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
а. Получение 8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-илового эфира трифторметансульфоновой кислоты
К раствору 8-бензил-8-азабицикло[3.2.1]октан-3-она (12,8 г, 59,5 ммоль) в безводном тетрагидрофуране (200 мл) при -78°С по каплям добавляли 1,0 М раствор гексаметилдисилазана натрия (77 мл, 77,4 ммоль) в ТГФ. Полученную смесь перемешивали при -78°С в течение тридцати минут и добавляли раствор (100 мл) N-фенилтрифторметансульфонимида (PhNHTf2) (25 г, 69,9 ммоль) в ТГФ. Примерно через сорок минут результаты тонкослойной хроматографии показали, что реакция не закончена. Добавляли дополнительное количество PhNНTf2 (2,0 г) в ТГФ. Через 30 минут реакционную смесь гасили насыщенным раствором хлорида аммония. Слои разделяли и органический слой дважды промывали насыщенным раствором хлорида аммония и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток далее очищали флэш-хроматографией и элюировали смесью от 0% (500 мл) до 5% (500 мл), до 10% (500 мл), до 15% (500 мл), до 20% (100 мл) этилацетата/гексанов. Требуемые фракции объединяли и концентрировали, получая при этом желтоватое масло (25,6 г, загрязненное N-фенилтрифторметан-сульфонамидом (PhNHTf). (m/z): [M+H]+, вычислено для С15Н16F3NO3S 348,09; найдено 348,0.
b. Получение 8-бензил-3-(3-бензилоксифенил)-8-азабицикло[3.2.1]-окт-2-ена
К раствору продукта, полученного на предыдущей стадии, (24,6 г, загрязненного PhNHTf) в ТГФ (120 мл) и DMA (120 мл) при комнатной температуре добавляли 3-бензилоксифенилбороновую кислоту (16,46 г), карбонат калия (19,9 г) и тетракис(трифенилфосфин)палладий(0) (5,0 г). Полученную смесь дегазировали, продували азотом и перемешивали в атмосфере азота в течение ночи. Реакционную смесь фильтровали через подушку целита и фильтрат концентрировали с образованием темного густого масла, которое очищали флэш-хроматографией (и элюировали 40% этилацетата в гексанах), получая при этом указанное в заголовке промежуточное соединение (6,9 г). (m/z): [M+H]+, вычислено для С27Н27NO 382,22; найдено 382,5.
с. Получение 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
К гидроксиду палладия (3,5 г, 20 мас./мас.% на угле) добавляли продукт, полученный на предыдущей стадии, (6,9 г) в этаноле (50 мл). Суспензию интенсивно перемешивали в атмосфере водорода в течение 12 часов. Реакционную смесь фильтровали через подушку целита и фильтрат концентрировали, получая при этом указанное в заголовке промежуточное соединение (5,6 г). (m/z): [M+H]+, вычислено для С13Н17NO 204,14; найдено 204,3.
d. Получение трет-бутилового эфира 3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
Продукт, полученный на предыдущей стадии, (5,6 г) растворяли в тетрагидрофуране (75 мл) и добавляли N,N-диизопропилэтиламин (3,6 мл). К перемешиваемому раствору по каплям добавляли ди-трет-бутилдикарбонат (5,4 г), растворенный в ТГФ (20 мл), и реакционную смесь перемешивали в течение 1 часа. Реакционную смесь гасили метанолом, концентрировали в вакууме, разбавляли дихлорметаном (100 мл), промывали 1,0 н. раствором HCl (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Органический слой обрабатывали безводным сульфатом натрия и растворитель сушили в вакууме. Неочищенный продукт очищали флэш-хроматографией (и элюировали 20% этилацетата в гексанах) с образованием указанного в заголовке неочищенного промежуточного соединения (3,4 г). Образовавшееся твердое вещество растворяли в этилацетате (10 мл), нагревали до 50°С и добавляли гептан (50 мл). Раствор оставляли охлаждаться до комнатной температуры в течение 2 часов. Образовавшиеся кристаллы фильтровали, получая при этом указанное в заголовке промежуточное соединение (2,2 г).
е. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
Трет-бутиловый эфир 3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (2,5 г) обрабатывали дихлорметаном (10 мл) и ТФУ (10 мл) при комнатной температуре в течение 10 минут. Реакционную смесь концентрировали, четыре раза упаривали с этилацетатом с образованием белого твердого вещества и сушили в вакууме, получая при этом указанное в заголовке соединение в виде его соли ТФУ (3,5 г). (m/z): [M+H]+, вычислено для С13Н17NO 204,14; найдено 204,3.
Препаративный пример 5. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 8-бензил-3-экзо-(3-бромфенил)-8-азабицикло[3.2.1]-октан-3-ола
К раствору 1,3-дибромбензола (7,4 г, 31,3 ммоль) в безводном ТГФ (80 мл) при -78°С в атмосфере азота по каплям добавляли раствор 1,6 М раствора н-бутиллития в гексанах (20 мл, 31,4 ммоль). Полученную смесь перемешивали при -78°С в течение 30 минут и по каплям добавляли раствор 8-бензил-8-азабицикло[3.2.1]октан-3-она (4,5 г, 20,9 ммоль) в безводном ТГФ (20 мл). Реакционную смесь оставляли медленно нагреваться до -40°С в течение одного часа и затем до комнатной температуры в течение 30 минут. Реакционную смесь гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали, концентрировали и очищали флэш-хроматографией. Продукт элюировали смесью от 20% (300 мл) до 30% (600 мл) этилацетата/гексанов. Требуемые фракции объединяли и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла (6,1 г). (m/z): [M+H]+, вычислено для С20Н22BrNO 372,10; найдено 372,3, 374,2 (изотоп).
b. Получение 3-экзо-(8-бензил-3-гидрокси-8-азабицикло[3.2.1]окт-3-ил)бензонитрила
К раствору продукта, полученного на предыдущей стадии (6,1 г, 16,4 ммоль), в безводном ДМФА (82 мл) добавляли цианид цинка (2,89 г, 24,6 ммоль). Суспензию дегазировали, продували азотом и добавляли тетракис(трифенилфосфин)палладий(0) (2,84 г, 2,5 ммоль). Полученную реакционную смесь нагревали до 85°С в атмосфере азота в течение ночи. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и промывали этилацетатом. Органический слой трижды промывали водой. Водный слой подвергали обратной экстракции этилацетатом. Органические слои объединяли, концентрировали примерно до 25 мл и четыре раза экстрагировали 1 н. водным раствором HCl. Объединенные органические слои дважды подвергали обратной экстракции диэтиловым эфиром и подщелачивали до рН 10, добавляя NaOH (гранула). Основный раствор трижды экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла (4,0 г). (m/z): [M+H]+, вычислено для С21Н22N2O 319,18; найдено 319,3.
с. Получение 3-экзо-(8-бензил-3-гидрокси-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору 3-экзо-(8-бензил-3-гидрокси-8-азабицикло[3.2.1]окт-3-ил)бензонитрила (3,74 г, 11,76 ммоль) в ДМСО (80 мл) при комнатной температуре добавляли карбонат калия (243 мг) и затем по каплям добавляли 30% водный раствор пероксида водорода (6 мл). Ход реакции контролировали при помощи масс-спектрометрии. Примерно через 2,5 часа реакция закончилась. Реакционную смесь гасили водой (140 мл). Водный слой трижды экстрагировали этилацетатом. Органические слои объединяли и промывали полунасыщенным раствором соли (5×40 мл) до полного отсутствия пероксида по результатам теста с помощью полоски иодированного крахмала. Органический слой промывали насыщенным рствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соеднение в виде белого твердого вещества (3,15 г). (m/z): [M+H]+, вычислено для С21Н24N2O2 337,19; найдено 337,3.
d. Получение 3-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида
Продукт, полученный на предыдущей стадии (3,15 г), обрабатывали ТФУ (30 мл) при 75°С в течение четырех часов. Реакционную смесь концентрировали, остаток растворяли в 50% растворе уксусной кислоты в воде (15 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке промежуточного соединения в виде белого твердого вещества. (m/z): [M+H]+, вычислено для С21Н22N2O 319,18; найдено 319,3.
е. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
Соль ТФУ 3-эндо-(8-бензил-8-азабицикло[3.2.1]окт-2-ен-3-ил)бензамида (3,0 г) растворяли в этаноле (50 мл) при комнатной температуре и обрабатывали гидроксидом палладия на угле (50 мас.% воды, 20% Pd на сухой основе, 300 мг). Полученную суспензию дегазировали, трижды продували азотом и выдерживали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали через целит и промывали этанолом. Фильтрат концентрировали с образованием светло-желтого масла, которое превращалось в пену при сушке в вакууме, и получали указанное в заголовке соединение в виде его соли ТФУ (2,2 г). (m/z): [M+H]+, вычислено для С14Н18N2O 231,15; найдено 231,3.
Препаративный пример 6. Синтез 3-эндо-[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
а. Получение 3-эндо-[8-(2,2-диметоксиэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
К раствору соли ТФУ 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида, полученного способом по препаративному примеру 5 (1,03 г, 2,98 ммоль), в дихлорметане (15,0 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (385 мг, 2,98 ммоль), 2,2-диметоксиацетальдегид (621 мг, 5,96 ммоль) и триацетоксиборгидрид натрия (821 мг, 3,87 ммоль). Полученную смесь обрабатывали ультразвуком для ускорения растворения. Примерно через 30 минут реакционную смесь концентрировали. Образовавшийся остаток дважды упаривали с метанолом, затем растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке промежуточное соединение в виде его соли ТФУ (577 мг). (m/z): [M+H]+, вычислено для С18Н26N2O3 319,20; найдено 319,3.
b. Получение 3-эндо-[8-(2-оксоэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
Продукт, полученный на предыдущей стадии (577 мг), обрабатывали 6 н. раствором HCl (20 мл) при комнатной температуре в течение ночи. Реакционную смесь концентрировали, остаток разбавляли водой и сушили вымораживанием, получая при этом указанное в заголовке промежуточное соединение в виде его хлористоводородной соли (554 мг). (m/z): [M+H]+, вычислено для С16Н20N2O2 273,16; найдено 273,1.
с. Синтез 3-эндо-[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
К суспензии хлористоводородной соли 3-эндо-[8-(2-оксоэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида (220 мг, 0,71 ммоль) в смеси дихлорметана (3 ил) и ДМФА (1 мл) при комнатной температуре добавляли триацетоксиборгидрид натрия (196 мг, 0,92 ммоль), бензиламин (91 мг, 0,85 ммоль) и N,N-диизопропилэтиламин (92 мг, 0,71 ммоль). Через тридцать минут результаты масс-спектрометрии (электронораспылительной) показали, что реакция закончилась. Реакционную смесь разбавляли дихлорметаном. Органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, фильтровали и концентрировали. Остаток повторно растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли бистрифторуксусной кислоты (27 мг). (m/z): [M+H]+, вычислено для С23Н29N3O 364,24; найдено 364,3.
Препаративный пример 7. Синтез 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
К раствору хлористоводородной соли 3-эндо-[8-(2-оксоэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида (554 мг, 1,80 ммоль) в дихлорметане (9,0 мл) при 0°С добавляли триацетоксиборгидрид натрия (496 мг, 2,34 ммоль), циклогексилметиламин (244 мг, 2,16 ммоль) и N,N-диизопропилэтиламин (233 мг, 1,80 ммоль). Охлаждающую баню со смесью воды со льдом после добавления удаляли, реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при указанной температуре в течение одного часа. Затем реакционную смесь концентрировали. Остаток повторно растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли бистрифторуксусной кислоты (400 мг). (m/z): [M+H]+, вычислено для С23Н35N3O 370,29; найдено 370,5.
Препаративный пример 8. Синтез трет-бутилового эфира циклогексилметил-(2-оксоэтил)карбаминовой кислоты
а. Получение 2-(циклогексилметиламино)этанола
Смесь циклогексилметилбромида (23,2 г, 131 ммоль) и этаноламина (47,9 г, 786 ммоль) в EtOH (131 мл) нагревали при 75°С в течение 2 часов. Результаты анализа ЯМР аликвоты реакционной смеси показали, что реакция закончилась. Реакционную смесь концентрировали для удаления этанола и образовавшийся остаток разбавляли DCM. Органический слой последовательно промывали водой (3×100 мл) и насыщенным раствором соли (100 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде светло-желтого масла (10,57 г). (m/z): [M+H]+, вычислено для С9Н19NO 158,16; найдено 158,2. 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 3,59 (т, J=5,4 Гц, 2H), 2,73 (т, J=5,4 Гц, 2H), 2,42 (д, J=6,6 Гц, 2H), 1,6-1,77 (м, 6H), 1,36-1,6 (м, 1H), 1,10-1,28 (м, 2H), 0,82-0,95 (м, 2H).
b. Получение трет-бутилового эфира циклогексилметил(2-гидроксиэтил)карбаминовой кислоты
К раствору продукта, полученного на предыдущей стадии (10,57 г, 67,3 ммоль), в DCM (200 мл) при 0°С по каплям добавляли раствор ди-трет-бутилдикарбоната (13,2 г, 60,57 ммоль) в DCM (100 мл). Полученную смесь оставляли медленного нагреваться до комнатной температуры в течение ночи. Смесь последовательно промывали 1 н. водным раствором HCl (3×100 мл), насыщенным раствором бикарбоната натрия (100 мл) и насыщенным раствором соли (100 мл). Реакционную смесь сушили над сульфатом натрия, органический слой фильтровали и концентрировали, получая при этом указанное в заголовке соединение в виде светло-желтого масла (16,5 г). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 3,71-3,73 (м, 2H), 3,37 (ушир.с, 2H), 3,03-3,05 (м, 2H), 1,61-1,72 (м, 6H), 1,3-1,5 (м, 1H), 1,48 (с, 9H), 1,14-1,21(м, 2H), 0,86-0,91 (м, 2H).
с. Синтез трет-бутилового эфира циклогексилметил(2-оксоэтил)карбаминовой кислоты
К продукту, полученному на предыдущей стадии (16,5 г, 64,2 ммоль), в DCM (256 мл) при 0°С последовательно добавляли диметилсульфоксид (7,52 г, 96,3 ммоль), N,N-диизопропилэтиламин (20,74 г, 160,5 ммоль) и комплекс пиридина с триоксидом серы (25,5 г, 160,5 ммоль). Через тридцать минут результаты анализа ЯМР аликвоты реакционной смеси показали, что реакция закончилась. Затем смесь последовательно промывали 1 н. водным раствором HCl (3×100 мл), насыщенным раствором бикарбоната натрия и насыщенным раствором соли, фильтровали через подушку силикагеля и элюировали DCM. Реакционную смесь концентрировали, получая при этом указанное в заголовке соединение в виде светло-желтого масла (10,46 г). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 9,55 (с, 1H), 3,88 (с, 1H), 3,79 (с, 1H), 3,07-3,15 (м, 2H), 1,56-1,72 (м, 6H), 1,3-1,5 (м, 1H), 1,4 (с, 9H), 1,1-1,25 (м, 2H), 0,87-0,98 (м, 2H).
Препаративный пример 9. Синтез 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
а. Получение трет-бутилового эфира {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметилкарбаминовой кислоты
К перемешиваемому раствору 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида (1,16 г, 5,0 ммоль), полученного способом по препаративному примеру 13, в DCM (20 мл) при 0°С добавляли раствор трет-бутилового эфира циклогексилметил(2-оксоэтил)карбаминовой кислоты (1,53 г, 6,0 ммоль) в DCM (5 мл) и триацетоксиборгидрид натрия (1,27 г, 6,0 ммоль). После добавления полученную смесь нагревали до комнатной температуры и перемешивали при указанной температуре в течение 30 минут до окончания реакции по результатам масс-спектрометрии. Смесь разбавляли DCM, дважды промывали насыщенным раствором бикарбоната натрия и затем насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом желтоватое масло, которое использовали на следующей стадии без дальнейшей очистки. (m/z): [M+H]+, вычислено для С28Н43N3O3 470,34; найдено 470,6.
b. Синтез 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
Маслянистый остаток, полученный на предыдущей стадии, растворяли в DCM (12 мл) и обрабатывали ТФУ (12 мл) при комнатной температуре в течение примерно 40 минут. Окончание реакции определяли по результатам масс-спектрометрии. Затем смесь концентрировали и трижды упаривали с этилацетатом, разбавляли DCM и подщелачивали до рН 8,0 насыщенным раствором бикарбоната натрия. Слои разделяли и водный cлой еще раз экстрагировали DCM. Объединенный органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали с образованием коричневатого масла. Продукт сушили в вакууме, получая при этом светло-коричневую пену (1,34 г). (m/z): [M+H]+, вычислено для С23Н35N3O 370,29; найдено 370,4. 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 7,89 (с, 1H), 7,70-7,72 (м, 1H), 7,54-7,56 (м, 1H), 7,39-7,44 (м, 1H), 3,43 (ушир.с, 2H), 3,16-3,21 (м, 1H), 3,06-3,10 (м, 2H), 2,91 (д, J=7,2 Гц, 2H), 2,65-2,69 (м, 2H), 2,05-2,51 (м, 2H), 2,01-2,05 (м, 2H), 1,79-1,91 (м, 8H), 1,60-1,63 (м, 2H), 1,27-1,42 (м, 3H), 1,09-1,17 (м, 2H).
Препаративный пример 10. Синтез трет-бутилового эфира (2-оксоэтил)-(4-трифторметилбензил)карбаминовой кислоты
а. Получение 2-(4-трифторметилбензиламино)этанола
В соответствии со способом по препаративному примеру 8, стадия а, 4-трифторметилбензилбромид (664 мг, 2,78 ммоль) нагревали с этаноламином (1,02 г, 16,7 ммоль) в этаноле (3 мл) при 75°С в течение ночи. Продукт выделяли, получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла (585 мг). (m/z): [M+H]+, вычислено для С10Н12F3NO 220,10; найдено 220,3. 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 7,59 (д, J=7,8 Гц, 2H), 7,45 (д, J=7,8 Гц, 2H), 3,88 (с, 2H), 3,66-3,70 (м, 2H), 2,80-2,83 (м, 2H).
b. Получение трет-бутилового эфира (2-гидроксиэтил)(4-трифторметилбензил)карбаминовой кислоты
В соответствии со способом по препаративному примеру 8, стадия b, продукт, полученный на предыдущей стадии (585 мг, 2,65 ммоль), обрабатывали ди-трет-бутилдикарбонатом (525 мг, 2,41 ммоль), получая при этом указанное в заголовке промежуточное соединение в виде светло-желтого масла (796 мг). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 7,57 (д, J=7,8 Гц, 2H), 7,32 (д, J=7,8 Гц, 2H), 4,52 (ушир.с, 2H), 3,71 (ушир.с, 2H), 3,40 (ушир.с, 2H), 1,44 (ушир.с, 9H).
с. Синтез трет-бутилового эфира (2-оксоэтил)(4-трифторметилбензил)карбаминовой кислоты
В соответствии со способом по препаративному примеру 8, стадия с, продукт, полученный на предыдущей стадии (796 мг, 2,49 ммоль), окисляли комплексом пиридина с триоксидом серы (990 мг, 6,22 ммоль), получая при этом указанное в заголовке соединение в виде светло-желтого масла (538 мг).
Препаративный пример 11. Синтез 3-эндо-{8-[2-(4-трифторметилбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола
а. Получение трет-бутилового эфира {2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}(4-трифторметилбензил)карбаминовой кислоты
В соответствии со способом по препаративному примеру 9, стадия а, трет-бутиловый эфир (2-оксоэтил)(4-трифторметилбензил)карбаминовой кислоты (253 мг, 1,84 ммоль) подвергали взаимодействию с солью ТФУ 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола, полученного способом по препаративному примеру 4 (126 мг, 0,92 ммоль), получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла, которое использовали на следующей стадии без очистки.
b. Синтез 3-эндо-{8-[2-(4-трифторметилбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола
В соответствии со способом по препаративному примеру 9, стадия b, продукт, полученный на предыдущей стадии, обрабатывали ТФУ (1,5 мл) и дихлорметаном (1,5 мл) и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли бистрифторуксусной кислоты (142 мг). (m/z): [M+H]+, вычислено для С23Н27F3N2O 405,22; найдено 405,2. 1H ЯМР (CD3OH, 300 МГц) δ (м.д.): 7,75 (д, J=7,8 Гц, 2H), 7,68 (д, J=7,8 Гц, 2H), 7,11-7,16 (м, 1H), 6,91-6,94 (м, 1H), 6,87 (с, 1H), 6,62-6,65 (м, 1H), 4,34 (с, 2H), 4,04 (ушир.с, 2H), 3,52 (т, J=6,3 Гц, 2H), 3,31-3,39 (м, 2H), 3,13-3,20 (м, 1H, 2,52-2,59 (м, 4H), 2,02-2,06 (м, 2H), 1,87-1,90 (м, 2H).
Препаративный пример 12. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 8-бензил-3-(3-метоксифенил)-8-азабицикло[3.2.1]окт-2-ена
В 500 мл колбу, оснащенную магнитной мешалкой, вводили 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ол (21,44 г, 66,2 ммоль) и ангидрид уксусной кислоты (150 мл). Затем добавляли трифлат иттербия (20,51 г, 33,1 моль) в виде твердого вещества и реакционную смесь отверждали. Добавляли дополнительное количество ангидрида уксусной кислоты (100 мл) для суспендирования твердого вещества. Реакционную смесь нагревали до 60°С в течение 4 часов. Реакционную смесь прекращали перемешивать, разбавляли этилацетатом и осторожно гасили 1 н. раствором NaOH. Органический слой промывали насыщенным раствором соли и сушили над сульфатом магния. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде липкого желтого масла (9,9 г, выход 48%). (m/z): [M+H]+, вычислено для С21Н23NO 306,19; найдено 306,3.
b. Получение 3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октана
В маленькую колбу Парра, содержащую продукт, полученный на предыдущей стадии (9,9 г, 32,5 ммоль), добавляли этанол (70 мл). Смесь перемешивали при комнатной температуре до полного растворения реагента. К раствору порциями осторожно добавляли гидроксид палладия (4,45 г, ~50 мас.%) в виде твердого вещества. Реакционный сосуд продували сухим азотом и помещали в атмосферу водорода (3,85 атм. (55 фунтов/кв.дюйм)) на ночь. После окончания реакции по результатам ВЭЖХ реакционную смесь продували азотом и фильтровали через целит. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде желтого масла (6,9 г, выход 98%). (m/z): [M+H]+, вычислено для С14Н19NO 218,16; найдено 218,3.
с. Получение 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
В 1 л круглодонную колбу, оснащенную магнитной мешалкой, вводили продукт, полученный на предыдущей стадии (6,9 г, 31,79 ммоль), и дихлорметан (200 мл). Реакционную смесь охлаждали на бане со смесью сухого льда с ацетоном до -78°С в течение 15 минут. К охлажденному реакционному раствору быстро добавляли трибромид бора в виде 1 М раствора в дихлорметане (64 мл, 63,59 ммоль). Реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение 20 часов. Реакционную смесь гасили, осторожно добавляя метанол. Мешалку удаляли и растворитель удаляли в вакууме с образованием хрустящего коричневого твердого вещества. Твердое вещество растворяли в метаноле. Растворитель удаляли в вакууме с образованием хрустящего коричневого твердого вещества. Твердое вещество снова растворяли в метаноле. Растворитель удаляли в вакууме с образованием хрустящего коричневого твердого вещества, которое сушили в вакууме. Высушенное твердое вещество растворяли в дихлорметане и раствор промывали 1 н. раствором NaOH и насыщенным раствором хлорида натрия. Органический слой отделяли и сушили над безводным сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде желтого масла. (m/z): [M+H]+, вычислено для С13Н17NO 204,14; найдено 204,3.
d. Получение трет-бутилового эфира 3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В 500 мл реакционную колбу, содержащую 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенол (2,5 г, 12,3 ммоль), добавляли дихлорметан (100 мл) в атмосфере сухого азота и тетрагидрофуран (70 мл). К суспензии одной порцией добавляли N,N-диизопропилэтиламин (3 мл) и ди-трет-бутилдикарбонат (3 мл, 12,3 ммоль) в виде расплавленной жидкости. Реакционную смесь продолжали перемешивать при комнатной температуре в течение 16 часов. После окончания реакции по результатам ВЭЖХ реакционную смесь переносили к колбу большего размера и удаляли большую часть растворителя. Образовавшийся остаток растворяли в этилацетате и органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и растворитель удаляли в вакууме с образованием указанного в заголовке неочищенного промежуточного соединения. Неочищенное вещество хроматографировали на силикагеле, используя смесь 20-25% этилацетата/гексанов в качестве подвижной фазы. Фракции объединяли и растворитель удаляли в вакууме, получая при этом 2,4 г очищенного продукта. Очищенное вещество растворяли в дихлорметане (~10 мл) и добавляли гексаны (150 мл). Дихлорметан удаляли в роторном испарителе. Раствор переносили в колбу Эрленмейера и добавляли несколько затравочных кристаллов, полученных тем же способом в предыдущем препаративном примере. Раствор оставляли кристаллизоваться в течение ночи. Кристаллы выделяли фильтрованием и промывали гексанами. Продукт сушили в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде белых игл (1,01 г, выход 27%). В маточной жидкости начинали расти кристаллы, которые собирали, промывали гексанами и сушили в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде белых игл (850 мг, выход 23%). (m/z): [M+H]+, вычислено для С18Н25NO3 304,19; найдено 304,3, 248,3 (исходное соединение - трет-бутил).
е. Получение трет-бутилового эфира 3-эндо-(3-трифторметансульфонил-оксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В 50 мл реакционную колбу, оснащенную магнитной мешалкой и продуваемую сухим азотом, добавляли трет-бутиловый эфир 3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (587 мг, 1,94 ммоль) и диметилформамид (10 мл). Реакционную смесь перемешивали до образования раствора и одной порцией в виде твердых веществ вместе добавляли карбонат калия (0,40 г, 2,90 ммоль) и N-фенилтрифторметансульфонимид (1,03 г, 2,90 ммоль). Реакционную смесь нагревали до 50°С в течение ночи. Реакционную смесь разбавляли смесью этилацетата:гексанов в отношении 1:1 и водой и органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде бесцветного масла (856 мг, выход >100%). (m/z): [M+H]+, вычислено для С19Н24F3NO5S 436,14; найдено 436,2, 380,2 (исходное вещедство - трет-бутил).
f. Получение трет-бутилового эфира 3-эндо-(3-цианофенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В 1 л круглодонную колбу, оснащенную магнитной мешалкой и продуваемую сухим азотом, вводили трет-бутиловый эфир 3-эндо-(3-трифторметансульфонилоксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (38,4 г, 88,3 ммоль) и диметилформамид (320 мл). Раствор перемешивали в течение 5 минут для растворения всего исходного вещества и дегазировали в вакууме. Затем снова подавали сухой азот. К дегазированному раствору одной порцией в виде твердых веществ вместе добавляли цианид цинка (15,5 г, 132 ммоль) и тетракис(трифенилфосфин)палладий(0) (5,1 г, 4,41 ммоль). Реакционную смесь снова дегазировали в вакууме для удаления случайного кислорода и подавали сухой азот. Реакционную смесь нагревали до 80°С в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли изопропилацетатом (500 мл). Образовавшийся мутный раствор фильтровали через целит (10 г). Полученный органический раствор переносили в делительную воронку и промывали насыщенным водным раствором бикарбоната натрия (400 мл) и насыщенным водным раствором хлорида натрия (400 мл). Органический слой отделяли и сушили над безводным сульфатом натрия (30 г). Осушитель удаляли фильтрованием и растворитель удаляли в вакууме, получая при этом указанное в заголовке неочищенное промежуточное соединение в виде воскообразных коричневых кристаллов (29,9 г, выход >100%). (m/z): [M+H]+, вычислено для С19Н24N2O2 313,19; найдено 313,3, 257,3 (исходное соединение - трет-бутил).
g. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
Трет-бутиловый эфир 3-эндо-(3-цианофенил)-8-азабицикло[3.2.1]-октан-8-карбоновой кислоты (4,8 г, 15,36 ммоль) вводили в 500 мл круглодонную колбу и разбавляли ДМСО (105 мл). Добавляли карбонат калия (3,18 г, 23,04 ммоль) в виде твердого вещества и затем за противовзрывным экраном осторожно добавляли 30% раствор пероксида водорода в воде (8 мл). Реакционную смесь перемешивали на открытом воздухе в течение ночи при комнатной температуре. После окончания реакции по результатам ВЭЖХ добавляли воду (160 мл) и реакционную смесь экстрагировали в этилацетат (3×150 мл). Объединенные органические слои промывали сульфитом натрия (пероксид во всех водных слоях гасили сульфитом натрия) и насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом защищенное промежуточное соединение, являющееся трет-бутиловым эфиром 3-(3-карбамоилфенил)-8-азабицикло[3.2.1]-октан-8-карбоновой кислоты, в виде бесцветного масла (4,8 г, выход 95%). (m/z): [M+H]+, вычислено для С19Н26N2O3 331,20; найдено 331,4, 275,0 (исходное вещество - трет-бутил).
Защищенное промежуточное соединение обрабатывают дихлорметаном и трифторуксусной кислотой способом по препаративному примеру 4, стадия е, получая при этом соль ТФУ указанного в заголовке соединения.
Препаративный пример 13. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ола
В 3 л трехгорлую колбу, оснащенную верхней мешалкой и продуваемую сухим азотом, вводили порошкообразный хлорид церия (88,2 г, 0,35 моль). Твердое вещество разжижали безводным тетрагидрофураном (500 мл) и охлаждали до 0°С. К суспензии по каплям добавляли 1 М раствор бромида 3-метоксифенилмагния в ТГФ (360 мл, 0,36 моль), поддерживая температуру ниже 10°С. Полученный раствор перемешивали при 0°С в течение 1,5 часа. Затем по каплям добавляли раствор 8-бензил-8-азабицикло[3.2.1]октан-3-она (54,5 г, 0,25 моль) в тетрагидрофуране (50 мл), поддерживая внутреннюю температуру ниже 5°С. Полученный раствор перемешивали при 0°С в течение 2 часов. Реакционную смесь гасили 10% водным раствором уксусной кислоты (400 мл) и перемешивали в течение 30 минут при комнатной температуре. Затем добавляли насыщенный раствор хлорида натрия (400 мл) и полученную суспензию перемешивали при комнатной температуре в течение 20 часов до полной кристаллизации продукта в виде соли уксусной кислоты. Кристаллы фильтровали и промывали холодной водой (200 мл) и изопропилацетатом (200 мл) и сушили в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде белого кристаллического порошка (91,1 г, выход 93%). (m/z): [M+H]+, вычислено для С21Н25NO2 324,20; найдено 324,5.
b. Получение 8-бензил-3-(3-метоксифенил)-8-азабицикло[3.2.1]окт-2-ена
В 1 л колбу, оснащенную магнитной мешалкой, вводили 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ол в виде соли уксусной кислоты (80,4 г, 0,209 моль) и 6 М водный раствор хлористоводородной кислоты (300 мл). Реакционную смесь нагревали до 70°С в течение 2 часов. Реакционную смесь прекращали перемешивать и разбавляли дихлорметаном (200 мл). Смесь переносили в делительную воронку, слои смешивали и оставляли для осаждения. Органический слой удаляли и сохраняли. Водный слой экстрагировали дихлорметаном (2×200 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (400 мл) и сушили над безводным сульфатом натрия (30 г). Растворитель удаляли в вакууме, получая при этом хлористоводородную соль указанного в заголовке промежуточного соединения в виде липкого желтого масла (65,4 г, выход 91%). (m/z): [M+H]+, вычислено для С21Н23NO 306,19; найдено 306,3.
с. Получение 3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октана
В 1 л круглодонную колбу, содержащую продукт, полученный на предыдущей стадии (65,4 г, 0,191 моль), добавляли этанол (300 мл). Смесь перемешивали при комнатной температуре до полного растворения промежуточного соединения. К раствору осторожно порциями добавляли гидроксид палладия (6,7 г, ~10 мас.%) в виде твердого вещества. Реакционный сосуд продували сухим азотом и при помощи баллона с иглой осторожно вводили водород. Водород барботировали через раствор в течение 10 минут, после чего раствор перемешивали в течение ночи в атмосфере водорода. После окончания реакции по результатам ВЭЖХ водород удаляли из реакционной смеси и сосуд продували сухим азотом в течение 10 минут. Реакционную смесь фильтровали через целит (5 г) и слой целита промывали этанолом (100 мл). Объединенный раствор в этаноле упаривали в вакууме и образовавшийся остаток растворяли в дихлорметане (400 мл). Органический слой промывали 3 н. раствором гидроксида натрия (300 мл). Слои разделяли и водный слой экстрагировали дихлорметаном (2×200 мл). Объединенные органические слои промывали водным раствором хлорида натрия (300 мл) и сушили над карбонатом калия (30 г). Осушитель удаляли фильтрованием и растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде желтого масла (27,6 г, выход 66%). (m/z): [M+H]+, вычислено для С14Н19NO 218,16; найдено 218,3.
d. Получение 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
В 1 л круглодонную колбу, оснащенную магнитной мешалкой и капельной воронкой, вводили продукт, полученный на предыдущей стадии, (27,6 г, 0,127 моль) и дихлорметан (300 мл). Реакционную смесь охлаждали на бане со смесью сухого льда с ацетоном до -78°С. К охлажденной реакционной смеси добавляли трибромид бора (1 М раствор в дихлорметане, 152 мл, 0,152 моль). Реакционную смесь оставляли медленно нагреваться до комнатной температуре в течение 20 часов. Реакционную смесь помещали на баню со льдом и гасили, осторожно добавляя метанол (100 мл). Растворитель удаляли в вакууме с образованием хрустящего бежевого твердого вещества. Твердое вещество повторно растворяли в метаноле (100 мл). Растворитель удаляли в вакууме с образованием хрустящего бежевого твердого вещества. Твердое вещество снова повторно растворяли в метаноле (100 мл). Растворитель удаляли в вакууме с образованием хрустящего бежевого твердого вещества, которое затем сушили в вакууме в течение 2 часов. Высушенное твердое вещество суспендировали в этаноле (110 мл) и раствор нагревали на масляной бане до 80°С. К горячему раствору добавляли достаточное количество метанола для растворения всего твердого вещества (72 мл). Раствор медленно охлаждали до комнатной температуры с целью образования белых кристаллов бромистоводородной соли указанного в заголовке промежуточного соединения. Затем раствор охлаждали до -20°С в морозильной камере в течение одного часа. Кристаллизованный раствор нагревали до комнатной температуры и кристаллы собирали фильтрованием. Белые кристаллы промывали холодным этанолом (35 мл) и сушили в вакууме с образованием бромистоводородной соли указанного в заголовке промежуточного соединения в виде белого порошка (19,5 г, выход 54%). Маточный раствор упаривали с образованием хрустящего бежевого твердого вещества. Указанное твердое вещество повторно растворяли в этаноле (30 мл) и нагревали до 80°С. Образовался прозрачный коричневый раствор. Раствор охлаждали до комнатной температуры и затем до -20°С в течение одного часа. Кристаллы собирали фильтрованием, промывали холодным этанолом (10 мл) и сушили в вакууме, получая при этом второй сбор кристаллов (5,5 г, выход 15%). (m/z): [M+H]+, вычислено для С13Н17NO 204,14; найдено 204,4.
е. Получение трет-бутилового эфира 3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В 500 мл реакционную колбу, содержащую бромистоводородную соль 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола (24,8 г, 0,087 моль), добавляли дихлорметан (200 мл) в атмосфере сухого азота. Суспензию охлаждали до 0°С. К суспензии одной порцией в виде твердого вещества добавляли N,N-диизопропилэтиламин (22,75 мл, 0,13 моль) и ди-трет-бутилдикарбонат (19,03 г, 0,087 моль). Реакционную смесь оставляли нагреваться до комнатной температуры в течение 16 часов. После окончания реакции по результатам ВЭЖХ реакционную смесь (теперь прозрачный светло-коричневый раствор) переносили в делительную воронку и разбавляли изопропилацетатом (200 мл). Органическую смесь промывали насыщенным водным раствором бикарбоната натрия (300 мл). Органический слой удаляли и водный слой экстрагировали изопропилацетатом (200 мл). Объединенные органические слои промывали водным раствором хлорида натрия (300 мл), слои разделяли и органический слой сушили над безводным сульфатом натрия (20 г). Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде белого твердого вещества (27,1 г, выход >100%). (m/z): [M+H]+, вычислено для С18Н25NO3 304,19; найдено 304,3, 248,3 (исходное соединение - трет-бутил).
f. Получение трет-бутилового эфира 3-эндо-(3-трифторметансульфонилоксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В 500 мл реакционную колбу, оснащенную магнитной мешалкой и продуваемую сухим азотом, вводили продукт, полученный на предыдущей стадии (27,1 г, 0,089 моль), и дихлорметан (250 мл). Раствор охлаждали до 0°С на бане со льдом. К холодному раствору по каплям добавляли триэтиламин (12,4 мл, 0,097 моль) и трифторметансульфонилхлорид (9,43 мл, 0,097 моль), поддерживая внутреннюю температуру ниже 10°С. К реакционной смеси одной порцией добавляли твердый 4-N,N-диметиламинопиридин (0,544 г, 4,46 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. Конечный раствор переносили в делительную воронку. Органический слой промывали насыщенным водным раствором бикарбоната натрия (200 мл) и насыщенным водным раствором хлорида натрия (200 мл). Органический слой отделяли и сушили над безводным сульфатом натрия (20 г). Осушитель удаляли фильтрованием и растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде прозрачного масла (38,4 г, выход 98%). (m/z): [M+H]+, вычислено для С19Н24F3NO5S 436,14; найдено 436,2, 380,3 (исходное соединение - трет-бутил).
g. Получение трет-бутилового эфира 3-эндо-(3-цианофенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В 1 л круглодонную колбу, оснащенную магнитной мешалкой и продуваемую сухим азотом, вводили продукт, полученный на предыдущей стадии (38,4 г, 88,3 ммоль), и диметилформамид (320 мл). Раствор перемешивали в течение 5 минут для растворения всего исходного вещества и дегазировали в вакууме. Затем снова подавали сухой азот. К дегазированному раствору одной порцией в виде твердых веществ вместе добавляли цианид цинка (15,5 г, 132 ммоль) и тетракис(трифенилфосфин)палладий(0) (5,1 г, 4,41 ммоль) в виде твердых веществ. Реакционную смесь снова дегазировали в вакууме и подавали сухой азот. Реакционную смесь нагревали до 80°С в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли изопропилацетатом (500 мл). Образовавшийся мутный раствор фильтровали через целит (10 г). Полученный органический раствор промывали насыщенным водным раствором бикарбоната натрия (400 мл) и насыщенным водным раствором хлорида натрия (400 мл). Органический слой отделяли и сушили над безводным сульфатом натрия (30 г). Осушитель удаляли фильтрованием и растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение в виде воскообразных коричневых кристаллов (29,9 г, выход >100%). (m/z): [M+H]+, вычислено для С19Н24N2O2 313,19; найдено 313,3, 257,3 (исходное соединение - трет-бутил).
h. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
В 15 мл круглодонную колбу, оснащенную магнитной мешалкой и обратным холодильником, вводили трет-бутиловый эфир 3-эндо-(3-цианофенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (500 мг, 1,60 ммоль) в виде твердого вещества и трифторуксусную кислоту (4 мл). К полученному раствору добавляли концентрированную серную кислоту (440 мкл, 5,0 экв.). Реакционную смесь нагревали до 65°С в течение 10 часов. Реакционную смесь выливали в насыщенный водный раствор хлорида натрия (70 мл) и переносили в делительную воронку. Водный слой промывали изопропилацетатом (50 мл) для удаления остаточного оксида трифенилфосфина, полученного на предыдущей стадии. К водному слою добавляли 3 н. водный раствор гидроксида натрия (15 мл), чтобы довести рН до 14. Водный слой экстрагировали тетрагидрофураном (2×50 мл). Объединенные органические слои сушили над безводным сульфатом натрия (3 г). Осушитель удаляли фильтрованием и растворитель удаляли в вакууме, получая при этом указанное в заголовке соединение в виде хрустящей, частично кристаллической пены (300 мг, выход 79%). (m/z): [M+H]+, вычислено для С14Н18N2O 231,15; найдено 231,2.
Препаративный пример 14. Синтез метоксикарбонилметансульфонил-уксусной кислоты
а. Получение трет-бутилового эфира метоксикарбонилметилсульфонил-уксусной кислоты
К раствору метилового эфира меркаптоуксусной кислоты (1,0 г, 9,42 ммоль) в диметилформамиде (10 мл) при комнатной температуре добавляли карбонат калия (1,69 г) и трет-бутиловый эфир бромуксусной кислоты (1,84 г, 9,42 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение ночи и разбавляли гексанами. Органический слой трижды промывали водой и один раз насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом бесцветное масло, которое использовали на следующей стадии без очистки. 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 3,72 (с, 3H), 3,37 (с, 2H), 3,27 (с, 2H), 1,45 (с, 9H).
b. Получение трет-бутилового эфира метоксикарбонилметансульфонил уксусной кислоты
К раствору продукта, полученного на предыдущей стадии (830 мг, 3,75 ммоль), в дихлорметане (25 мл) при 0°С добавляли 3-хлорпероксибензойную кислоту (1,94 г, 11,25 ммоль). После добавления полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 2,5 часа. Реакционную смесь гасили насыщенным раствором сульфита натрия (30 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Слои разделяли и органический слой последовательно промывали 1 н. водным раствором NaOH, насыщенным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде бесцветного масла (683 мг), которое использовали на следующей стадии без очистки. 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 4,32 (с, 2H), 4,21 (с, 2H), 3,81 (с, 3H), 1,48 (с, 9H).
с. Синтез метоксикарбонилметансульфонилуксусной кислоты
Продукт, полученный на предыдущей стадии (683 мг), обрабатывали трифторуксусной кислотой (10 мл) при комнатной температуре в течение двух часов. Смесь концентрировали, повторно растворяли в этилацетате и органический слой промывали водой и насыщенным раствором соли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке соединение в виде бесцветного масла, которое превращалось в воск после сушки в вакууме (345 мг). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 4,39 (с, 2H), 4,33 (с, 2H), 3,83 (с, 3H).
Препаративный пример 15. Синтез (S)-2,2-диметил[1,3]диоксолан-4-карбоновой кислоты
К раствору метилового эфира α,β-изопропилиден-1-глицериновой кислоты (2,2 г, 13,7 ммоль) в МеОН (20 мл) при комнатной температуре добавляли моногидрат гидроксида лития (1,15 г, 27,4 ммоль) в воде (5,0 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали, остаток подкисляли 10% водным раствором HCl (20 мл) и трижды экстрагировали дихлорметаном. Объединенный органический слой дважды промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке соединение в виде бесцветного масла (819 мг). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 4,60 (дд, J=4,5, 7,2 Гц, 1H), 4,28 (дд, J=7,2, 8,7 Гц, 1H), 4,18 (дд, J=4,5, 8,7 Гц, 1H), 1,51 (с, 3H), 1,40 (с, 3H).
Препаративный пример 16. Синтез [бензил(2-оксоэтил)карбамоил]метилового эфира уксусной кислоты
а. Синтез [бензил(2-гидроксиэтил)карбамоил]метилового эфира уксусной кислоты
Бензилэтаноламин (1,78 г, 11,8 ммоль) вводили в 25 мл круглодонную колбу и разбавляли дихлорметаном. При помощи шприца быстро добавляли N,N-диизопропилэтиламин (2,66 мл, 15,3 ммоль) и реакционную смесь охлаждали до 0°С. Смесь перемешивали при 0°С в течение 10 минут и при помощи шприца по каплям добавляли ацетоксиацетилхлорид (1,26 мл, 11,8 ммоль). Реакционную смесь перемешивали в течение ночи и оставляли нагреваться до комнатной температуры. Реакционную смесь разбавляли этилацетатом и промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Органический слой отделяли и сушили над безводным сульфатом магния. Осушитель удаляли фильтрованием и растворитель удаляли в вакууме с образованием указанного в заголовке неочищенного промежуточного соединения в виде желтого масла. Неочищенное вещество хроматографировали на силикагеле, используя этилацетат в качестве подвижной фазы. Фракции объединяли и растворитель удаляли в вакууме, получая при этом указанное в заголовке чистое промежуточное соединение в виде прозрачного масла (1,75 г, выход 59%). (m/z): [M+H]+, вычислено для С13Н17NO4 252,13; найдено 252,3.
b. Синтез [бензил(2-оксоэтил)карбамоил]метилового эфира уксусной кислоты
[Бензил(2-гидроксиэтил)карбамоил]метиловый эфир уксусной кислоты (1,28 г, 5,10 ммоль) вводили в 200 мл круглодонную колбу и продували азотом. Добавляли дихлорметан (50 мл) и реакционную смесь охлаждали до -15°С в течение 10 минут. Затем при -15°С последовательно добавляли диметилсульфоксид (3,61 мл, 51,0 ммоль), N,N-диизопропилэтиламин (4,43 мл, 25,5 ммоль) и комплекс пиридина с триоксидом серы (4,06 г, 25,5 ммоль). Реакционную смесь оставляли медленно нагреваться до комнатной температуры и перемешивали в течение ночи. После окончания реакции по результатам тонкослойной хроматографии реакционную смесь разбавляли этилацетатом. Органический раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Органический слой отделяли и сушили над безводным сульфатом магния. Осушитель удаляли фильтрованием и растворитель удаляли в вакууме с образованием указанного в заголовке неочищенного соединения в виде желтого масла. Неочищенное вещество хроматографировали на силикагеле с использованием смеси этилацетата:дихлорметана в отношении 1:1 в качестве подвижной фазы. Фракции объединяли и растворитель удаляли в вакууме, получая при этом указанное в заголовке соединение в виде бесцветного масла (0,72 г, выход 57%). (m/z): [M+H]+, вычислено для С13Н15NO4 250,11; найдено 250,0.
Препаративный пример 17. Синтез 3-эндо-[8-(2-фенетиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
а. Получение 2-фенетиламиноэтанола
Смесь 2-бромэтилбензола (2,0 г, 10,8 ммоль) и этаноламина (3,96 г, 64,8 ммоль) в этаноле (11 мл) нагревали при 75°С в течение 16,5 часа, когда по результатам ЖХ/МС реакция закончилась. Реакционную смесь концентрировали для удаления этанола и образовавшийся остаток разбавляли DCM (100 мл). Органический слой отделяли водой (100 мл) и водный слой экстрагировали DCM (50 мл). Объединенные органические слои промывали водой (2×5 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке соединение в виде светло-желтого масла (1,5 г). (m/z): [M+H]+, вычислено для С10Н15NO 166,13; найдено 166,2. 1H ЯМР (d 6-DMSO, 300 МГц) δ (м.д.): 7,13-7,29 (м, 5H), 4,4 (ушир., 1H), 3,42 (т, J=5,7 Гц, 2H), 2,61-2,76 (м, 4H), 2,55-2,59 (т, J=5,7 Гц, 2H), 1,55 (ушир., 1H).
b. Получение трет-бутилового эфира (2-гидроксиэтил)фенетил-карбаминовой кислоты
В соответствии со способом по препаративному примеру 8, стадия b, продукт, полученный на предыдущей стадии (1,5 г, 9,09 ммоль), подвергали взаимодействию с ди-трет-бутилдикарбонатом (1,78 г, 8,2 ммоль) в DCM (14 мл), получая при этом указанное в заголовке промежуточное соединение (2,26 г) в виде светло-желтого масла.
с. Получение трет-бутилового эфира (2-оксоэтил)фенетилкарбаминовой кислоты
В соответствии со способом по препаративному примеру 8, стадия с, продукт, полученный на предыдущей стадии (2,26 г, 8,5 ммоль), превращали в указанное в заголовке промежуточное соединение, которое было получено в виде желтоватого масла (1,27 г). 1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 9,37 (с, 1H), 7,21-7,28 (м, 2H), 7,17-7,20 (м, 3H), 3,93 (с, 2H), 3,41 (т, 2H), 2,74 (т, 2H), 1,30 (с, 9H).
d. Получение трет-бутилового эфира {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}фенетилкарбаминовой кислоты
В соответствии со способом по препаративному примеру 9, стадия а, 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамид (60 мг, 0,28 ммоль), полученный способом по препаративному примеру 13, подвергали взаимодействию с трет-бутиловым эфиром (2-оксоэтил)-фенетилкарбаминовой кислоты (87 мг, 0,34 ммоль), получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла. (m/z): [M+H]+, вычислено для С29Н39N3O3 478,30; найдено 478,4.
е. Получение 3-эндо-[8-(2-фенетиламиноэтил)-8-азабицикло[3.2.1]-окт-3-ил]бензамида
В соответствии со способом по препаративному примеру 9, стадия b, продукт, полученный на предыдущей стадии, обрабатывали ТФУ, получая при этом указанное в заголовке соединение в виде темного масла. (m/z): [M+H]+, вычислено для С24Н31N3O 378,25; найдено 378,2.
Препаративный пример 18. Синтез 3-эндо-[8-(2-(3-фенилпропиламино)этил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
В соответствии со способом по препаративному примеру 17 при замене 2-бромэтилбензола 1-бром-3-фенилпропаном были получены следующие промежуточные соединения:
а. 2-(3-фенилпропиламино)этанол
1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 7,14-7,28 (м, 5H), 4,4 (ушир., 1H), 3,42 (т, J=5,7 Гц, 2H), 2,46-2,58 (м, 6H), 1,61-1,71 (p, 2H), 1,65 (ушир., 1H).
b. трет-бутиловый эфир (2-гидроксиэтил)(3-фенилпропил)-карбаминовой кислоты
с. трет-бутиловый эфир (2-оксоэтил)(3-фенилпропил)карбаминовой кислоты
1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 9,44 (с, 1H), 7,24-7,39 (м, 2H), 7,15-7,19 (м, 3H), 3,97 (с, 2H), 3,24 (т, 2H), 2,49 (т, 2H), 1,69-1,74 (м, 2H), 1,34 (с, 9H).
d. трет-бутиловый эфир {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}(3-фенилпропил)карбаминовой кислоты (m/z): [M+H]+, вычислено для С30Н41N3O3 492,31; найдено 492,4.
е. 3-эндо-{8-[2-(3-фенилпропиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (m/z): [M+H]+, вычислено для С25Н33N3O 392,26; найдено 392,4.
Препаративный пример 19. Синтез 3-эндо-{8-(2-(2-циклогексилэтиламино)этил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
В соответствии со способом по препаративному примеру 17 при замене 2-бромэтилбензола 1-бром-2-циклогексилэтаном были получены следующие промежуточные соединения:
а. 2-(2-циклогексилэтиламино)этанол
1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 4,4 (ушир., 1H), 3,38-3,42 (т, J=5,7 Гц, 2H), 2,46-2,54 (м, 4H), 1,58-1,65 (м, 5H), 1,06-1,29 (м, 6H), 0,82-0,89 (м, 2H).
b. трет-бутиловый эфир (2-циклогексилэтил)(2-гидроксиэтил)-карбаминовой кислоты
с. трет-бутиловый эфир (2-циклогексилэтил)(2-оксоэтил)-карбаминовой кислоты
1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 9,43 (с, 1H), 3,93 (с, 2H), 3,20 (т, 2H), 1,64-1,68 (м, 4H), 1,38 (с, 9H), 1,30-1,37 (м, 4H), 1,14-1,27 (м, 3H), 0,83-0,87 (м, 2H).
d. трет-бутиловый эфир {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}(2-циклогексилэтил)карбаминовой кислоты (m/z): [M+H]+, вычислено для С29Н45N3O3 484,35; найдено 484,4.
е. 3-эндо-{8-[2-(2-циклогексилэтиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (m/z): [M+H]+, вычислено для С24Н37N3O 384,29; найдено 384,4.
Препаративный пример 20. Синтез 3-эндо-{8-(2-(3-циклогексилпропиламино)этил)-8-азабицикло[3.2.1]окт-3-ил}бензамида
а. Получение 3-циклогексилпропиональдегида
3-Циклогексил-1-пропанол (3,96 г, 27,8 ммоль) растворяли в DCM (90 мл) при 0°С и последовательно обрабатывали диметилсульфоксидом (3,25 г, 41,7 ммоль), N,N-диизопропилэтиламином (8,98 г, 69,6 ммоль) и комплексом пиридина с триоксидом серы (11 г, 69,6 ммоль). Через один час реакционную смесь разбавляли DCM (100 мл) и промывали 1 н. водным раствором HCl (3×50 мл), насыщенным раствором бикарбоната натрия (3×50 мл) и насыщенным раствором соли. Органический слой сушили над MgSO4, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде светло-желтого масла (3,8 г).
1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 9,72 (с, 1H), 3,65-3,72 (м, 2H), 2,46 (т, 2H), 1,65-1,73 (м, 3H), 1,51-1,56 (м, 2H), 1,14-1,48 (м, 4H), 0,85-0,96 (м, 2H).
b. Получение 2-(3-циклогексилпропиламино)этанола
К раствору этаноламина (0,44 г, 7,1 ммоль) в DCM (15 мл) при 0°С добавляли раствор 3-циклогексилпропиональдегида (1,0 г, 7,1 ммоль) в DCM (10 мл) и триацетоксиборгидрид натрия (1,67 г, 7,86 ммоль). Полученную смесь нагревали до комнатной температуры. Через 2,5 часа требуемый продукт был обнаружен при выполнении масс-спектрометрического анализа. Реакционную смесь перемешивали в течение ночи, разбавляли DCM (50 мл), промывали насыщенным раствором бикарбоната натрия (2×50 мл) и насыщенным раствором соли (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом прозрачное масло (1,0 г), которое использовали на следующей стадии без дальнейшей очистки. (m/z): [M+H]+, вычислено для С11Н23NO 186,20; найдено 186,0.
В соответствии со способом по препаративному примеру 17, стадии b-е, при замене 2-фенетиламиноэтанола по препаративному примеру 17, стадия b, 2-(3-циклогексилпропиламино)этанолом были получены следующие промежуточные соединения:
с. трет-бутиловый эфир (3-циклогексилпропил)(2-гидроксиэтил)карбаминовой кислоты
d. трет-бутиловый эфир (3-циклогексилпропил)(2-оксоэтил)-карбаминовой кислоты
1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 9,43 (с, 1H), 3,94 (с, 2H), 3,14 (т, 2H), 1,621 (м, 4H), 1,41-1,46 (м, 5H), 1,38 (с, 9H), 1,34-1,36 (м, 4H), 0,86-0,88 (м, 2H).
е. трет-бутиловый эфир {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}(3-циклогексилпропил)карбаминовой кислоты (m/z): [M+H]+, вычислено для С30Н47N3O3 498,36; найдено 498,6.
f. 3-эндо-{8-[2-(3-циклогексилпропиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (m/z): [M+H]+, вычислено для С25Н39N3O 398,31; найдено 398,4.
Препаративный пример 21. Синтез 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение трет-бутилового эфира (4,4-дифторциклогексилметил)карбаминовой кислоты
К раствору трет-бутилового эфира (4-оксоциклогексилметил)-карбаминовой кислоты (2,0 г, 8,81 ммоль) в дихлорметане (50 мл) при 0°С в атмосфере азота по каплям добавляли трифторид бис(2-метоксиэтил)аминосеры (Deoxo-Fluor®) (3,90 г, 17,62 ммоль). После добавления реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при указанной температуре в течение ночи. Затем реакционную смесь медленно гасили насыщенным раствором бикарбоната натрия. Добавляли дополнительное количество дихлорметана (300 мл) и полученную смесь фильтровали через подушку целита. Слои фильтрата разделяли и органический слой трижды промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли. Остаток сушили над сульфатом натрия, фильтровали и концентрировали с образованием коричневатого масла, которое очищали флэш-хроматографией. Соединение элюировали от 25% (400 мл) до 30% (200 мл) и 40% (200 мл) смесью этилацетата/гексанов. Требуемые фракции объединяли и концентрировали, получая при этом желтоватое масло, которое отверждалось после сушки в вакууме (694 мг).
b. Получение (4,4-дифторциклогексил)метиламина
Продукт, полученный на предыдущей стадии (694 мг), обрабатывали смесью дихлорметана и трифторуксусной кислоты в отношении 1:1 (6 мл) при комнатной температуре в течение тридцати минут. Реакционную смесь концентрировали и трижды упаривали вместе с этилацетатом. Образовавшийся остаток сушили в вакууме, получая при этом соль трифторуксусной кислоты указанного в заголовке промежуточного соединения в виде коричневатого масла.
с. Синтез 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору 3-эндо-[8-(2-оксоэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида (185 мг, 0,6 ммоль) в дихлорметане (3 мл) при комнатной температуре добавляли триацетоксиборгидрид натрия (165 мг, 1,8 ммоль) и соль ТФУ (4,4-дифторциклогексил)метиламина (315 мг, 1,2 ммоль) в дихлорметане (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение примерно полутора часов и концентрировали. Образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (73 мг). (m/z): [M+H]+, вычислено для С23Н33F2N3O 406,27; найдено 406,2.
Препаративный пример 22. Синтез 3-эндо-[8-(2-бензиламинопропил)-8-азабицикло[3.2.1]окт-3-ил]фенола
а. Получение N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]-окт-8-ил]-1-метил-2-оксоэтил}бензамида
2-Бензоиламинопропионовую кислоту (319 мг, 1,65 ммоль) и гексафторфосфат бензотриазол-1-илокси)трис(диметиламино)фосфония (ВОР) (731 мг, 1,65 ммоль) добавляли к перемешиваемому раствору соли ТФУ 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола (полученного способом по препаративному примеру 4) (524 мг, 1,65 ммоль) и N,N-диизопропилэтиламина (0,86 мл, 4,96 ммоль) в ТГФ (14 мл) при комнатной температуре в атмосфере азота. Через 90 минут реакционную смесь гасили, добавляя воду (1 мл), разбавляли этилацетатом (60 мл) и промывали 1 М раствором HCl (20 мл), насыщенным водным раствором бикарбоната натрия (20 мл), насыщенным раствором соли (20 мл), водой (20 мл) и насыщенным раствором соли (20 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (EtOAc:гексаны, от 7:3 до 4:1), получая при этом указанное в заголовке промежуточное соединение (597 мг) в виде белого твердого вещества.
b. Синтез 3-эндо-[8-(2-бензиламинопропил)-8-азабицикло[3.2.1]окт-3-ил]фенола
Комплекс диметилсульфида борана (10-10,2 М раствор, 2,16 мл, 21,6 ммоль) по каплям добавляли к перемешиваемому раствору продукта, полученного на предыдущей стадии (544 мг, 1,44 ммоль), в ТГФ (15 мл) при -20°С в атмосфере азота. После добавления реакционную смесь нагревали до температуры кипения с обратным холодильником. Через 3 часа реакционную смесь охлаждали до -20°С, осторожно добавляли метанол (30 мл) и перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме, разбавляли 4 М раствором HCl в диоксане (10 мл) и перемешивали в течение 2 часов. Реакционную смесь снова концентрировали в вакууме, разбавляли метанолом и добавляли гидроксид калия (10 экв.). Через 2 часа реакционную смесь концентрировали в вакууме, разбавляли водой (10 мл) и экстрагировали смесью дихлорметана:ТГФ, 3:1 (2×20 мл). Органические слои объединяли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая при этом указанное в заголовке соединение (509 мг) в виде белого твердого вещества, которое использовали без дальнейшей очистки. (m/z): [M+H]+, вычислено для С23Н30N2O 351,25; найдено 351,5.
Препаративный пример 23. Синтез N-[3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенил]метансульфонамида
а. Получение трет-бутилового эфира 3-эндо-(3-аминофенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
К раствору трет-бутилового эфира 3-эндо-(3-трифторметан-сульфонилоксифенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (400 мг, 0,92 ммоль), полученного способом по препаративному примеру 13, в тетрагидрофуране (9,0 мл) добавляли бензофенонимин (216,7 мг, 1,2 ммоль), трет-бутоксид калия (154,8 мг, 1,38 ммоль) и рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP) (51,5 мг, 0,08 ммоль). Полученную смесь дегазировали, продували азотом и добавляли ацетат палладия(II) (19,3 мг, 0,08 ммоль). Затем смесь нагревали до 78°С в течение двух часов. Реакционную смесь охлаждали до комнатной температуры, обрабатывали 2 н. раствором HCl (5,0 мл) в течение трех часов и подщелачивали до рН 8 5% водным раствором гидроксида натрия. Водный слой экстрагировали этилацетатом. Полученный органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали, концентрировали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке промежуточного соединения. (m/z): [M+H]+, вычислено для С18Н26N2O2 303,41; найдено 303,2.
b. Получение трет-бутилового эфира 3-эндо-(3-метансульфониламинофенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
К перемешиваемой смеси соли ТФУ трет-бутилового эфира 3-эндо-(3-аминофенил)-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (114 мг, 0,37 ммоль), N,N-диизопропилэтиламина (146 мг, 1,13 ммоль) и 4-диметиламинопиридина (DMAP) (9 мг, 0,075 ммоль) в DCM (2,0 мл) при 0°С добавляли раствор метансульфонилхлорида (45 мг, 0,39 ммоль) в DCM (0,2 мл). Через тридцать минут результаты аналитической ВЭЖХ показали, что реакция закончилась. Добавляли дополнительное количество метансульфонилхлорида (17 мг, 0,15 ммоль), смесь перемешивали при 0°С в течение еще тридцати минут и затем гасили насыщенным раствором бикарбоната натрия. Водный слой экстрагировали DCM. Органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде желтоватого масла (140 мг).
с. Синтез N-[3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенил]метансульфонамида
Маслянистый продукт, полученный на предыдущей стадии, обрабатывали DCM (2 мл) и ТФУ (20 мл) при комнатной температуре в течение тридцати минут, после чего результаты аналитической ВЭЖХ показали, что реакция закончилась. Реакционную смесь концентрировали и образовавшийся остаток трижды упаривали с этилацетатом и сушили в вакууме, получая при этом соль ТФУ указанного в заголовке промежуточного соединения в виде желтоватого масла, которое использовали без дальнейшей очистки.
Препаративный пример 24. Синтез аддукта бисульфита бензилового эфира N-циклогексилметил(2-оксоэтил)карбаминовой кислоты
а. Получение N-циклогексилметил(2,2-диэтоксиэтил)амина
К смеси 2,2-диэтоксиэтиламина (209 мл, 1,43 моль) и MeTHF (1050 л) добавляли циклогексанкарбальдегид (107 мл, 0,89 моль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре и охлаждали до 0°С. В течение 40 минут добавляли триацетоксиборгидрид натрия (378 г, 1,79 моль), реакционную смесь перемешивали в течение 2 часов и охлаждали до 0°С. Добавляли 1 М раствор NaOH (1 л). Органический слой промывали насыщенным раствором соли в воде (1:1, 2×1 л) и объем уменьшали до ~20%. Добавляли МеTHF (1 л) и объем уменьшали до ~20%. Раствор указанного в заголовке неочищенного промежуточного соединения использовали на следующей стадии без очистки.
b. Получение бензилового эфира N-циклогексилметил(2,2-диэтоксиэтил)карбаминовой кислоты
К продукту, полученному на предыдущей стадии (~213 г, ~0,9 моль), добавляли MeTHF (2 л) и DIPEA (233 мл, 1,34 моль). Реакционную смесь охлаждали до 0°С и по каплям добавляли бензилхлорформиат (140 мл, 0,98 моль). Реакционную смесь перемешивали в течение 30 минут при 0°С, в течение 2 часов при температуре от 0°С до комнатной температуры и затем в течение 1 часа при комнатной температуре. Добавляли воду (1,6 л) и реакционную смесь перемешивали в течение 10 минут. Фазы разделяли и органический слой промывали бикарбонатом натрия (1,6 л) и водой (1,6 л). Слои разделяли и органический слой уменьшали примерно до 20%. Добавляли MeTHF (1 л) и объем уменьшали до ~20%. Раствор указанного в заголовке неочищенного промежуточного соединения использовали на следующей стадии без очистки.
с. Синтез аддукта бисульфита бензилового эфира N-циклогексилметил(2-оксоэтил)карбаминовой кислоты
К продукту, полученному на предыдущей стадии (~302 г, ~0,62 моль), и ацетонитрилу (2 л) добавляли 1 М раствор HCl (2 л) и реакционную смесь перемешивали при 30°С в течение 7 часов. Добавляли этилацетат (2 л) и реакционную смесь перемешивали в течение 10 минут. Фазы разделяли, органический слой промывали 1 М раствором HCl (1,5 л), фазы снова разделяли и органический слой промывали 0,5 М раствором HCl (1 л). Добавляли бисульфит натрия (71,4 г, 0,69 моль), реакционную смесь перемешивали в течение ночи и затем фильтровали. Реактор и фильтровальную лепешку промывали этилацетатом (1 л). Полученный раствор сушили на воздухе в течение 2 часов и в вакууме в течение ночи, получая при этом указанное в заголовке соединение в виде белого твердого вещества (199 г, чистота >99% по результатам ВЭЖХ). Фильтрат подвергали аналогичной обработки, в результате чего была получена вторая порция указанного в заголовке соединения (30 г).
Препаративный пример 25. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 8-бензил-3-(3-метоксифенил)-8-азабицикло[3.2.1]окт-2-ена
В 3 л колбу вводили гидрохлорид 8-бензил-3-экзо-(3-метоксифенил)-8-азабицикло[3.2.1]октан-3-ола (383,9 г, 1,06 моль), 6 М раствор HCl (800 мл) и MeTHF (200 мл). Полученную суспензию нагревали в атмосфере азота при 70°С в течение 2,5 часа. Реакционную смесь переносили в 12 л реактор и охлаждали до 10°С. Реакционную колбу промывали МеTHF (1 л), который добавляли в 12 л реактор. Добавляли NaOH (50 мас.% в воде, 200 мл) и порциями добавляли дополнительное количество NaOH (50 мас.%, 150 мл) до достижения рН ~13. Фазы разделяли, водный слой экстрагировали МеTHF (1 л) и объединенные слои МeTHF промывали насыщенным раствором соли (1 л). Растворитель удаляли в роторном испарителе при 30-40°С, получая при этом указанное в заголовке промежуточное соединение (360 г) в виде густого масла. Добавляли EtOH (1,5 л), объем уменьшали до ~500 мл и затем доводили до 1,8 л.
b. Получение 3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октана
К 8-бензил-3-(3-метоксифенил)-8-азабицикло[3.2.1]окт-2-ену (в 95% EtOH, 400 мл, 0,20 моль), полученному на предыдущей стадии, добавляли 6 М раствор HCl (45 мл) и MeTHF (50 мл). Реакционную смесь продували азотом, нагревали до 40°С и добавляли палладий на угле (10 мас.%, 8 г). В реакторе повышали давление, подавая водород (3×1,4 атм (20 фунтов/кв.дюйм)), и гидрировали под давлением 1,4 атм (20 фунтов/кв.дюйм) при 40°С в течение 18 часов. Реакционную смесь фильтровали через целит, концентрировали, промывали MeTHF (2×100 мл), фильтровали через стеклянный фильтр грубой очистки, промывали MeTHF (10 мл) и сушили на фильтре, получая при этом хлористоводородную соль указанного в заголовке промежуточного соединения в виде белого твердого вещества (31 г, один изомер (экзо-изомер не обнаружен ВЭЖХ)). Из маточного раствора было дополнительно выделено 5,2 г продукта.
с. Получение 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола
В 500 мл колбу вводили гидрохлорид 3-эндо-(3-метоксифенил)-8-азабицикло[3.2.1]октана (115 г, 0,45 моль) и бромистоводородную кислоту (48 мас.% в воде, 100 мл, 0,88 моль). Смесь нагревали до 120°С и при перемешивании поддерживали указанную температуру в течение 24 часов. Добавляли дополнительное количество раствора бромистоводородной кислоты (25 мл), реакционную смесь нагревали при перемешивали в течение 6 часов и затем охлаждали до 70°С. Добавляли ацетонитрил (200 мл), образовавшуюся суспензию охлаждали до 10°С и фильтровали, фильтровальную лепешку промывали ацетонитрилом (50 мл), получая при этом бромистоводородную соль указанного в заголовке промежуточного соединения (99 г, чистота >99%) в виде белого гранулированного твердого вещества.
d. Получение 2,2,2-трифтор-1-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этанона
К раствору гидробромида 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола (54,4 г, 0,19 моль), толуола (210 мл) и триэтиламина (40 мл, 0,29 моль) добавляли ангидрид трифторуксусной кислоты (54 мл, 0,38 моль) в течение 20 минут. Реакционную смесь перемешивали при 40°С в течение 2 часов. Добавляли этилацетат (370 мл) и насыщенный раствор соли в воде (1:1, 265 мл). Реакционную смесь перемешивали в течение 15 минут и фазы разделяли. К органической фазе добавляли насыщенный раствор бикарбоната натрия (300 мл) и смесь интенсивно перемешивали в течение ночи. Фазы разделяли и органический слой промывали насыщенным раствором соли в воде (1:1, 265 мл), сушили над сульфатом натрия и большую часть растворителя удаляли в роторном испарителе. Добавляли толуол (100 мл) и растворитель удаляли в роторном испарителе, получая при этом указанное в заголовке неочищенное промежуточное соединение.
е. Получение 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]фенилового эфира трифторметансульфоновой кислоты
В 500 мл колбу вводили раствор этилацетата (220 мл) промежуточного соединения, полученного на предыдущей стадии (32,8 г, 0,11 моль), и триэтиламин (23 мл, 0,17 моль). Раствор охлаждали до 5°С и по каплям добавляли трифторметансульфонилхлорид (14 мл, 0,13 моль). Смесь оставляли нагреваться до 25°С и перемешивали при указанной температуре в течение 1 часа. Добавляли насыщенный раствор бикарбоната натрия (200 мл), слои разделяли, к органическому слою добавляли насыщенный раствор соли (150 мл), слои снова разделяли и растворитель удаляли из органического слоя, получая при этом указанное в заголовке неочищенное промежуточное соединение.
f. Получение 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]бензонитрила
В 100 мл колбу вводили 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]окт-3-ил]фениловый эфир трифторметансульфоновой кислоты (25,3 г, 58,7 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,81 г, 0,9 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (1,01 г, 1,8 ммоль) и цианид цинка (4,2 г, 35,8 ммоль). Колбу три раза продували азотом в течение 5 минут и затем помещали в вакуум на 5 минут. В колбу добавляли ДМФА (150 мл) и дистиллированную воду (2,5 мл). Раствор продували азотом при перемешивании в течение 10 минут, нагревали до 120°С и перемешивали при 120°С в атмосфере азота в течение 4 часов. После окончания реакции добавляли 20 г продукта из предыдущей порции, полученной аналогичным способом, и перемешивали в течение 20 минут.
Большую часть растворителя удаляли перегонкой и раствор охлаждали до 22°С. К раствору добавляли этилацетат (445 мл) и полученный раствор фильтровали через целит. Добавляли бикарбонат натрия (450 мл) и раствор перемешивали в течение 15 минут. Слои разделяли, органический слой промывали разбавленным насыщенным раствором соли (2×95 мл) и фильтровали через сульфат натрия. Объем уменьшали примерно до 50 мл, удаляя этилацетат. Добавляли изопропиловый спирт (150 мл) и раствор перемешивали при 22°С в течение 1 часа. Твердые вещества отделяли фильтрованием и промывали изопропиловым спиртом (2×25 мл), получая при этом указанное в заголовке промежуточное соединение (33,5 г, чистота 100% по результатам ВЭЖХ) в виде не совсем белого/светло-коричневого твердого вещества. Вторую порцию продукта (6,3 г, чистота >98% по результатам ВЭЖХ) выделяли из фильтрата.
g. Синтез 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида
Раствор 3-эндо-[8-(2,2,2-трифторацетил)-8-азабицикло[3.2.1]-окт-3-ил]бензонитрила (10 г, 32 ммоль) в серной кислоте (96%, 12 мл) нагревали до 50°С при перемешивании и выдерживали при указанной температуре, перемешивая в течение 2 часов. Реакционную смесь охлаждали до 22°С и медленно вводили в 500 мл колбу, содержащую 5 н раствор NaOH (90 мл) и метанол (100 мл), охлажденный до 10°С. Осадок соли фильтровали и фильтрат перемешивали при 22°С в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли MeTHF (150 мл) и реакционную смесь перемешивали при 22°С в течение 5 минут. Слои разделяли и к водному слою добавляли MeTHF (100 мл). Слои разделяли и к объединенным органическим слоям добавляли насыщенный раствор соли (150 мл). Слои разделяли, органический слой сушили над карбонатом калия и фильтровали, растворитель удаляли. К остатку при перемешивании добавляли смесь EtOH (25 мл) и концентрированной HCl (2,6 мл), затем добавляли МТВЕ (25 мл) и раствор перемешивали при 22°С. Осажденные твердые вещества фильтровали и подвергали воздушной сушке, получая при этом хлористоводородную соль указанного в заголовке соединения (8 г, чистота 97% по результатам ВЭЖХ) в виде белого твердого вещества.
Пример 1. Синтез N-бензил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
3-эндо-[8-(2-Бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]фенол, полученный способами по препаративным примерам 1 и 2 (246 мг, 0,73 ммоль), растворяли в дихлорметане (3,6 мл) и N,N-диизопропилэтиламине (123 мг, 0,95 ммоль) при комнатной температуре. Полученную смесь охлаждали до 0°С. Добавляли ацетоксиацетилхлорид (119 мг, 0,87 ммоль) и смесь перемешивали при 0°С в течение примерно 30 минут. Реакционную смесь гасили насыщенным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали с образованием желтоватого масла. Маслянистую смесь растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соли ТФУ моноацилированного продукта (62 мг) и бисацилированного побочного продукта (160,9 мг). Соль ТФУ бисацилированного побочного продукта (160,9 мг) растворяли в этаноле (2 мл) при комнатной температуре и обрабатывали 1 н. водным раствором гидроксида натрия (1,0 мл) в течение примерно 30 минут. Реакционную смесь концентрировали, остаток растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения в виде белого твердого вещества (46,5 мг). (m/z): [M+H]+, вычислено для С24Н30N2O3 395,24; найдено 395,3.
Пример 2. Синтез N-бензил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
К раствору соли ТФУ 3-эндо-[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]фенола, полученного способами по препаративным примерам 1 и 2 (25 мг, 0,044 ммоль), в дихлорметане (0,2 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (23 мг, 0,177 ммоль) и затем ацетилхлорид (0,044 ммоль). Примерно через 10 минут реакционную смесь концентрировали. Остаток повторно растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали, очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (10 мг). (m/z): [M+H]+, вычислено для С24Н30N2O2 379,23; найдено 379,2.
Примеры 3-5
Соли ТФУ соединений по примерам 3-5 были получены способами, аналогичными способу по примеру 2, за исключением замены ацетилхлорида 0,044 ммоль соответствующего ацилхлорида.
Пример 3 бензил-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]-окт-8-ил]этил}амид циклопентанкарбоновой кислоты (18,3 мг) (m/z): [M+H]+, вычислено для С28Н36N2O2 433,29; найдено 433,2.
Пример 4 N-бензил-2-этил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}бутиламид (15,1 мг) (m/z): [M+H]+, вычислено для С28Н38N2O2 435,30; найдено 435,2.
Пример 5 метиловый эфир N-бензил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовой кислоты (19,6 мг) (m/z): [M+H]+, вычислено для С27Н34N2O2 451,26; найдено 451,2.
Пример 6. Синтез N-бензил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовой кислоты
К раствору соли ТФУ 3-эндо-[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]фенола, полученного способами по препаративным примерам 1 и 2 (110 мг, 0,195 ммоль), в дихлорметане (1,0 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (101 мг, 0,78 ммоль) и 3-карбометоксипропионилхлорид (44 мг, 0,29 ммоль). Примерно через тридцать минут реакционную смесь концентрировали. Остаток повторно растворяли в этаноле (2 мл), обрабатывали моногидратом гидроксида лития (33 мг, 0,78 ммоль) в воде (1 мл) в течение примерно тридцати минут и затем концентрировали. Остаток повторно растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (46,5 мг). (m/z): [M+H]+, вычислено для С26Н32N2O2 437,25; найдено 437,12.
Пример 7. Синтез N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
К раствору соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способами по препаративным примерам 1 и 3 (25 мг, 0,044 ммоль), в дихлорметане (0,2 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (34 мг, 0,26 ммоль) и ацетилхлорид (0,044 ммоль). Реакционную смесь концентрировали в роторном испарителе. Остаток растворяли в этаноле (0,2 мл) и гидролизовали 1 н. водным раствором NaOH (0,1 мл) при комнатной температуре в течение 30 минут. Растворители удаляли в роторном испарителе, образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (12 мг). (m/z): [M+H]+, вычислено для С24Н36N2O2 385,29; найдено 385,2.
Примеры 8-14
Соли ТФУ соединений по примерам 3-5 были получены способами, аналогичными способу по примеру 7, за исключением замены ацетилхлорида 0,044 ммоль соответствующего ацилхлорида.
Пример 8 N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}пропионамид (7,1 мг) (m/z): [M+H]+, вычислено для С25Н38N2O2 399,30; найдено 399,2.
Пример 9 циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}амид циклопентанкарбоновой кислоты (16,4 мг) (m/z): [M+H]+, вычислено для С28Н42N2O2 439,34; найдено 439,2.
Пример 10 циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}амид циклогексанкарбоновой кислоты (14 мг) (m/z): [M+H]+, вычислено для С29Н44N2O2 453,35; найдено 453,4.
Пример 11 N-циклогексилметил-3-циклопентил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-пропионамид (11,4 мг) (m/z): [M+H]+, вычислено для С30Н46N2O2 467,37; найдено 467,4.
Пример 12 N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-3-метилбутирамид (14,1 мг) (m/z): [M+H]+, вычислено для С27Н42N2O2 427,33; найдено 427,4.
Пример 13 N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамид (10,8 мг) (m/z): [M+H]+, вычислено для С24Н36N2O3 401,28; найдено 401,2.
Пример 14 N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-2-фенилацетамид (16,8 мг) (m/z): [M+H]+, вычислено для С30Н40N2O2 461,32; найдено 461,2.
Пример 15. Синтез {1-[(циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбамоил)метил]циклогексил}уксусной кислоты
К раствору соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-илфенола, полученного способом по препаративным примерам 1 и 3 (24 мг, 0,042 ммоль), в дихлорметане (0,4 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (22 мг, 0,17 ммоль) и ангидрид 1,1-циклогександиуксусной кислоты (0,042 ммоль). Через 20 минут реакция заканчивалась, и реакционную смесь концентрировали. Остаток растворяли в метаноле и гидролизовали 1 н. водным раствором NaOH (0,1 мл) при комнатной температуре в течение 30 минут. Растворители удаляли и образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения. (m/z): [M+H]+, вычислено для С32Н48N2O4 525,37; найдено 525,2.
Примеры 16-17
Соли ТФУ соединений по примерам 16 и 17 были получены способами, аналогичными способу по примеру 15, за исключением замены ангидрида 1,1-циклогександиуксусной кислоты 0,042 ммоль указанного ангидрида.
Пример 16 {1-[(циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбамоил)метил]-циклопентил}уксусная кислота; реагент: ангидрид 3,3-тетраметиленглутаровой кислоты; (m/z): [M+H]+, вычислено для С31Н46N2O4 511,36; найдено 511,2.
Пример 17 2-(циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбамоил)циклогексанкарбоновая кислота; реагент: ангидрид 1,2-циклогександикарбоновой кислоты; (m/z): [M+H]+, вычислено для С30Н44N2O4 497,34; найдено 497,2.
Пример 18А. N-Циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовая кислота
К раствору 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 и превращенного в свободное основание (83 мг, 0,24 ммоль), в дихлорметане (2,5 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (124 мг, 0,96 ммоль) и затем 3-карбометоксипропионилхлорид (72 мг, 0,48 ммоль). Полученную смесь перемешивали при комнатной температуре в течение примерно 10 минут и концентрировали, растворяли в этаноле (2 мл) и гидролизовали моногидратом гидроксида лития (61 мг) в воде (2 мл) в течение примерно 30 минут. Реакционную смесь концентрировали, растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения. (m/z): [M+H]+, вычислено для С26Н38N2O4 443,29; найдено 443,2.
Пример 18В. N-Циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовая кислота
К раствору соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративному примеру 4 и аналогично способу по препаративному примеру 11 (70 мг, 0,12 ммоль), в дихлорметане (0,4 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (62 мг, 0,48 ммоль) и янтарный ангидрид (0,16 ммоль). Полученную смесь перемешивали при комнатной температуре в течение примерно 10 минут и затем концентрировали. Остаток повторно растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой. Остаток сушили вымораживанием, твердое вещество растворяли в смеси МеОН (1,0 мл) и воды (2,0 мл) и обрабатывали моногидратом гидроксида лития (30 мг) при комнатной температуре в течение тридцати минут. Продукт концентрировали, растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (28,9 мг). (m/z): [M+H]+, вычислено для С26Н38N2O4 443,29; найдено 443,5. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 7,10-7,155 (м, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,85 (с, 1H), 6,63 (д, J=8,1 Гц, 1H), 4,01 (ушир.с, 2H), 3,71-3,79 (м, 2H), 3,26-3,28 (м, темный 2H), 3,10-3,12 (м, 3H), 2,67 (с, 4H), 2,48-2,52 (м, 4H), 2,04-2,08 (м, 2H), 1,706-1,875 (м, 8H), 1,238-1,30 (м, 3H), 0,98-1,03 (м, 2H).
Пример 19. Синтез 4-(циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбамоил)-3,3-диметилмасляной кислоты
К раствору соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 (25 мг, 0,044 ммоль), в дихлорметане (0,4 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (23 мг, 0,18 ммоль) и ангидрид 3,3-диметилглутаровой кислоты (9 мг, 0,07 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали в роторном испарителе, растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (13,3 мг). (m/z): [M+H]+, вычислено для С29Н44N2O4 485,34; найдено 485,4.
Пример 20. Синтез [(циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбамоил)метансульфонил]уксусной кислоты
К раствору метоксикарбонилметансульфонилуксусной кислоты (10 мг, 0,525 ммоль) в диметилацетамиде (1 мл) при комнатной температуре добавляли 1,1'-карбонилдиимидазол (114 мг, 0,7 ммоль). Через два часа к перемешиваемой смеси добавляли раствор соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 (100 мг, 0,175 ммоль), и N,N-диизопропилэтиламина (91 мг, 0,7 ммоль) в диметилацетамиде (1 мл). Реакционную смесь нагревали до 65°С в течение 3 часов и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этанолом (2,0 мл) и обрабатывали раствором моногидрата гидроксида лития (150 мг) в воде (1,5 мл) в течение примерно 30 минут. Растворители удаляли и остаток растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения в виде белого твердого вещества (28,3 мг). (m/z): [M+H]+, вычислено для С26Н38N2O6S 507,26; найдено 507,2. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 7,1-7,17 (м, 1H), 6,87-6,94 (м, 2H), 6,63-6,72 (м, 1H), 4,63 (с, 2H), 4,44 (с, 2H), 4,07 (ушир.с, 2H), 3,80 (т, J=5,7 Гц, 2H), 3,34 (д, J=7,2 Гц, темный 2H), 3,13-3,2 (м, 3H), 2,51-2,53 (м, 4H), 1,69-1,88 (м, 8H), 1,19-1,33 (м, 3H), 0,92-1,06 (м, 2H).
Пример 21. Синтез N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}малонамовой кислоты
К раствору моно-трет-бутилового эфира малоновой кислоты (84 мг, 0,53 ммоль) в диметилацетамиде (1,0 мл) при комнатной температуре добавляли 1,1'-карбонилдиимидазол (114 мг, 0,7 ммоль). Через два часа к перемешиваемой смеси добавляли раствор соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 (100 мг, 0,175 ммоль), и N,N-диизопропилэтиламина (91 мг, 0,7 ммоль) в диметилацетамиде (1 мл). Полученную реакционную смесь нагревали до 65°С в течение 3 часов и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и последовательно промывали водой и насыщенным раствором соли. Реакционную смесь сушили над сульфатом натрия, фильтровали, концентрировали с образованием желтоватого масла и сушили в вакууме в течение 30 минут. Остаток обрабатывали трифторуксусной кислотой (5 мл) при комнатной температуре в течение 10 минут. Реакционную смесь разбавляли водой (5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения в виде белого твердого вещества (51,5 мг). (m/z): [M+H]+, вычислено для С25Н36N2O4 429,28; найдено 429,2. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 7,11-7,17 (м, 1H), 6,87-6,94 (м, 2H), 6,63-6,66 (м, 1H), 4,08 (ушир.с, 2H), 3,77-3,81 (м, 2H), 3,58 (деформированный с, 1H), 3,24 (д, J=6,9 Гц, темный 2H), 3,14-3,18 (м, 3H), 2,51-2,54 (м, 4H), 2,05-2,09 (м, 2H), 1,69-1,89 (м, 8H), 1,24-1,31 (м, 3H), 0,98-1,03 (м, 2H).
Пример 22. Синтез 3-втор-бутил-1-циклогексилметил-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевины
К раствору соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 (30 мг, 0,053 ммоль), в диметилформамиде (0,2 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (27 мг, 0,21 ммоль) и втор-бутилизоцианат (0,079 ммоль). Полученную смесь встряхивали при комнатной температуре в течение ночи, концентрировали, растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (17,8 мг). (m/z): [M+H]+, вычислено для С27Н43N3O2 442,35; найдено 442,4.
Примеры 23-28
Соли ТФУ соединений по примерам 23-28 были получены способами, аналогичными способу по примеру 22, за исключением замены втор-бутилизоцианата 0,079 ммоль указанного изоцианата.
Пример 23 3-бензил-1-циклогексилметил-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевина (13,2 мг); реагент: бензилизоцианат; (m/z): [M+H]+, вычислено для С30Н41N3O2 476,33; найдено 476,2.
Пример 24 3-бензо[1,3]диоксол-5-ил-1-циклогексилметил-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевина (12,0 мг); реагент: 3,4-метилендиоксифенилизоцианат; (m/z): [M+H]+, вычислено для С30Н39N3O4 506,30; найдено 506,2.
Пример 25 1-циклогексилметил-3-(3-фторфенил)-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевина (14,3 мг); реагент: 3-фторфенилизоцианат; (m/z): [M+H]+, вычислено для С29Н38FN3O2 480,30; найдено 480,2.
Пример 26 1-циклогексилметил-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-3-пентилмочевина (14,6 мг); реагент: пентилизоцианата; (m/z): [M+H]+, вычислено для С28Н45N3O2 456,36; найдено 456,4.
Пример 27 1-циклогексилметил-3-(4-фторбензил)-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевина (13,6 мг); реагент: 4-фторбензилизоцианат; (m/z): [M+H]+, вычислено для С30Н40FN3O2 494,34; найдено 494,2.
Пример 28 1-циклогексилметил-3-(4-дифторметоксифенил)-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевина (21,8 мг); реагент: 4-дифторметоксифенилизоцианат; (m/z): [M+H]+, вычислено для С30Н39F2N3O3 528,31; найдено 528,2.
Пример 29А. Синтез метилового эфира N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовой кислоты
К раствору 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 (114 мг, 0,20 ммоль), в дихлорметане (1 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (103 мг, 0,870 ммоль) и раствор 3-карбометоксипропионилхлорида (0,20 ммоль) в дихлорметане (0,3 мл). Полученную смесь перемешивали при комнатной температуре в течение примерно 10 минут, концентрировали, повторно растворяли в 50% растворе уксусной кислоты в воде (5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения в виде белой соли (46,4 мг). (m/z): [M+H]+, вычислено для С27Н40N2O4 457,31; найдено 457,3.
Пример 29В. Синтез метилового эфира N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовой кислоты
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративному примеру 4 и аналогично способу по препаративному примеру 11 (88 мг, 0,15 ммоль), в дихлорметане (0,4 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (80 мг, 0,62 ммоль) и раствор (0,15 мл) 3-карбометоксипропионилхлорида (0,18 ммоль) в DCM. Полученную смесь перемешивали при комнатной температуре в течение примерно 10 минут. Результаты масс-спектрометрии (электронораспылительной) показали наличие исходного вещества. Добавляли дополнительное количество 3-карбометоксипропионилхлорида (0,05 ммоль). Когда результаты аналитической ВЭЖХ показали, что реакция закончена, реакционную смесь концентрировали, растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения в виде белого твердого вещества (44,7 мг). (m/z): [M+H]+, вычислено для С27Н40N2O4 457,31; найдено 457,5. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 7,10-7,15 (м, 1H), 6,90 (д, J=7,8 Гц, 1H), 6,85 (с, 1H), 6,63 (д, J=7,8 Гц, 1H), 4,02 (ушир.с, 2H), 3,70-3,71 (м, 2H), 3,63 (с, 3H), 3,26-3,28 (м, темный 2H), 3,08-3,11 (м, 3H), 2,67-2,71 (м, 4H), 2,47-2,49 (м, 4H), 2,04-2,1 (м, 2H), 1,708-1,878 (м, 8H), 1,21-1,38 (м, 3H), 0,95-1,03 (м, 2H).
Пример 30. Синтез N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-2-метансульфонилацетамида
К раствору метансульфонилуксусной кислоты (0,10 ммоль) в диметилформамиде (0,2 мл) при комнатной температуре добавляли 1,1'-карбонилдиимидазол (21 мг, 0,13 ммоль). Реакционную смесь встряхивали в течение примерно одного часа и добавляли смесь соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола (полученного способом по препаративным примерам 1 и 3) (30 мг, 0,053 ммоль) и N,N-диизопропилэтиламина (0,10 ммоль) в диметилформамиде (0,3 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, концентрировали, разбавляли 50% раствором уксусной кислоты в воде (8 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (7,5 мг). (m/z): [M+H]+, вычислено для С25Н38N2O4S 463,27; найдено 463,5.
Пример 31. Синтез N-циклогексилметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамида
К раствору N-циклогексилметил-N-{2-[3-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовой кислоты, продукта по примеру 18 (95 мг, 0,17 ммоль), в диметилацетамиде (0,2 мл) при комнатной температуре добавляли 1,1'-карбонилдиимидазол (165 мг, 1,02 ммоль). Через один час добавляли ацетат аммония (79 мг, 1,02 ммоль) и N,N-диизопропилэтиламин (132 мг, 1,02 ммоль). Полученную смесь встряхивали при комнатной температуре в течение ночи, растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (5,6 мг). (m/z): [M+H]+, вычислено для С26Н39N3O3 442,31; найдено 442,5.
Пример 32. Синтез 1-циклогексилметил-3-(3,4-диметоксифенил)-1-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}мочевины
К раствору соли ТФУ 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способом по препаративным примерам 1 и 3 (30 мг, 0,05 ммоль), в диметилформамиде (0,2 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (27 мг, 0,21 ммоль) и 3,4-диметоксифенилизоцианат (14 мг, 0,078 ммоль). Полученную смесь оставляли выстаиваться при комнатной температуре в течение ночи, концентрировали в роторном испарителе, повторно растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (13,5 мг). (m/z): [M+H]+, вычислено для С31Н43N3O4 522,34; найдено 522,2.
Примеры 33-38
Соли ТФУ соединений по примерам 33-38 были получены общим способом по примеру 7 за исключением замены промежуточного соединения циклогексиламина соответствующим замещенным бензиламином, полученным способом по препаративному примеру 3, с использованием промежуточного соединения 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола, полученного способом по препаративному примеру 1, и замены ацетилхлорида соответствующим ацилхлоридом.
Пример 33 N-(3-фторбензил)-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамид (9,6 мг); (m/z): [M+H]+, вычислено для С24Н29FN2O3 413,23; найдено 413,2.
Пример 34 N-(3-фторбензил)-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовая кислота (12,0 мг); (m/z): [M+H]+, вычислено для С26Н31FN2O4 455,24; найдено 455,2.
Пример 35 N-(3-фторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-3-метилбутирамид (9,9 мг); (m/z): [M+H]+, вычислено для С27Н35FN2O2 439,28; найдено 439,2.
Пример 36 N-(2,6-дифторбензил)-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамид (9,0 мг); (m/z): [M+H]+, вычислено для С24Н28F2N2O3 431,22; найдено 431,2.
Пример 37 N-(2,6-дифторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-3-метилбутирамид (13,1 мг); (m/z): [M+H]+, вычислено для С27Н34F2N2O2 457,27; найдено 457,2.
Пример 38 N-(2,6-дифторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамид (11,7 мг); (m/z): [M+H]+, вычислено для С24Н28F2N2O2 415,22; найдено 415,2.
Примеры 39-42
Соли ТФУ соединений по примерам 39-42 были получены общим способом по примеру 21 за исключением замены промежуточного соединения циклогексиламина соответствующим замещенным бензиламином, полученным способом по препаративному примеру 3, с использованием промежуточного соединения 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенола, полученного способом по препаративному примеру 1, или замены моно-трет-бутилового эфира малоновой кислоты метансульфонилуксусной кислотой в примерах 40 и 42.
Пример 39 N-(3-фторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}малонамовая кислота (11,4 мг); (m/z): [M+H]+, вычислено для С25Н29FN2O4 441,22; найдено 441,2.
Пример 40 N-(3-фторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-2-метансульфонилацетамид (25,0 мг); (m/z): [M+H]+, вычислено для С25Н31FN2O4S 475,21; найдено 475,2.
Пример 41 N-(2,6-дифторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}малонамовая кислота (11,1 мг); (m/z): [M+H]+, вычислено для С25Н28F2N2O4 459,21; найдено 459,2.
Пример 42 N-(2,6-дифторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-2-метансульфонилацетамид (11,1 мг); (m/z): [M+H]+, вычислено для С25Н30F2N2O4S 493,20; найдено 493,2.
Пример 43. Синтез N-(4-фторбензил)-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[2-(4-фторбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола, полученного способами по препаративным примерам 4 и 3 (30 мг, 0,05 ммоль), в дихлорметане (0,2 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (26 мг, 0,2 ммоль) и затем ацетоксиацетилхлорид (0,075 ммоль). После окончания реакции, определяемого по результатам масс-спектрометрического анализа, смесь концентрировали в роторном испарителе, остаток растворяли в EtOH (0,2 мл) и гидролизовали моногидратом гидроксида лития (17 мг) в воде (0,2 мл) при комнатной температуре в течение 30 минут. Растворители удаляли в роторном испарителе, образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (16,6 мг). (m/z): [M+H]+, вычислено для С24Н29FN2O3 413,23; найдено 413,2.
Пример 44. Синтез N-(4-хлорбензил)-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
Соль ТФУ указанного в заголовке соединения (19,1 мг) была получена способом по примеру 43 с использованием соответствующего замещенного бензиламина по препаративному примеру 3. (m/z): [M+H]+, вычислено для С24Н29ClN2O3 429,20; найдено 429,2.
Пример 45. Синтез 2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-N-(4-трифторметилбензил)ацетамида
Соль ТФУ указанного в заголовке соединения (19,5 мг) была получена способом по примеру 43 с использованием замещенного бензиламина, полученного способом по препаративному примеру 11. (m/z): [M+H]+, вычислено для С25Н29F3N2O3 463,22; найдено 463,2.
Пример 46А. Синтез 3-эндо-(8-{2-[бензил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору соли бистрифторуксусной кислоты 3-эндо[8-(2-бензиламиноэтил)-8-азабицикло[3.2.1]окт-3-ил]бензамида, полученного способом по препаративным примерам 5 и 6 (111 мг, 0,188 ммоль), в дихлорметане (0,94 мл) при -20°С добавляли N,N-диизопропилэтиламин (97 мг, 0,75 ммоль) и ацетоксиацетилхлорид (27 мг, 0,29 ммоль) в дихлорметане (0,5 мл). Реакционную смесь перемешивали при температуре от -20°С до -10°С в течение примерно 30 минут, гасили насыщенным раствором бикарбоната натрия и экстрагировали DCM. Органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали с образованием желтоватого масла, которое растворяли в EtOH (1,0 мл) и обрабатывали моногидратом гидроксида лития (24 мг, 0,56 ммоль) в воде (0,5 мл) при комнатной температуре в течение 30 минут. Растворители удаляли в роторном испарителе и образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (58,7 мг). (m/z): [M+H]+, вычислено для С25Н31N3O3 422,25; найдено 422,3.
Пример 46В. Синтез 3-эндо-(8-{2-[бензил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида, полученного способом по препаративному примеру 13 (102 мг, 0,44 ммоль), в дихлорметане (2 мл) при комнатной температуре добавляли раствор [бензил(2-оксоэтил)карбамоил]метилового эфира уксусной кислоты (164 мг, 0,66 ммоль) в дихлорметане (2 мл) и затем триацетоксиборгидрид натрия (131 мг). Реакционную смесь перемешивали при комнатной температуре в течение примерно тридцати минут и по результатам масс-спектрометрического анализа определяли, что реакция закончена. Реакционную смесь концентрировали, повторно растворяли в EtOH (6 мл) и обрабатывали моногидридом гидроксида лития (111 мг) в воде (4 мл) в течение примерно тридцати минут. Затем смесь концентрировали, повторно растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (115,7 мг). (m/z): [M+H]+, вычислено для С25Н31N3O3 422,25; найдено 422,25. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.) 8,0 (с, 1H), 7,68-7,75 (м, 2H), 7,26-7,47 (м, 6H), 4,57 (с, 2H), 4,37 (с, 2H), 4,08 (ушир.с, 2H), 3,72 (т, J=5,7 Гц, 2H), 3,05 (т, J=5,4 Гц, 2H), 2,59-2,62 (м, 4H), 1,99-2,03 (м, 2H), 1,74-1,81 (м, 2H).
Пример 47. Синтез N-{2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-N-циклогексилметилсукцинамовой кислоты
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 5 и 7 (114 мг, 0,19 ммоль), в дихлорметане (0,95 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (98 мг, 0,76 ммоль). Затем полученную смесь охлаждали до -30°С и добавляли раствор 3-карбометоксипропионилхлорида (30 мг, 0,20 ммоль) в DCM (0,5 мл). После окончания реакции смесь концентрировали. Остаток повторно растворяли в EtOH (2 мл) и обрабатывали моногидратом гидроксида лития (32 мг) в воде (1 мл) в течение примерно 30 минут. Смесь концентрировали, растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (63,2 мг). (m/z): [M+H]+, вычислено для С27Н39N3O4 470,30; найдено 470,5.
Пример 48. Синтез 3-эндо-(8-{2-[циклогексилметил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 13 и 9 (734 мг, 1,23 ммоль), в дихлорметане (5 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (635 мг, 4,9 ммоль). Полученную смесь охлаждали до -20°С и добавляли раствор ацетоксиацетилхлорида (184 мг, 1,35 ммоль) в DCM (2 мл). Через пять минут результаты масс-спектрометрического анализа показали, что реакция закончена. Реакционную смесь концентрировали, растворяли в EtOH (15 мл) и обрабатывали моногидратом гидроксида лития (309 мг) в воде (5 мл) в течение примерно 30 минут. Затем реакционную смесь концентрировали, растворяли в 50% растворе уксусной кислоты в воде (15 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (585,0 мг). (m/z): [M+H]+, вычислено для С25Н37N3O3 428,29; найдено 428,2. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 8,0 (с, 1H), 7,68-7,74 (м, 2H), 7,42-7,47 (т, J=8,1 Гц, 1H), 4,27 (с, 2H), 4,09 (ушир.с, 2H), 3,72 (т, J=5,7 Гц, 2H), 3,2-3,35 (м, темный 1H), 3,09-3,14 (м, 4H), 2,59-2,62 (м, 4H), 2,07-2,12 (м, 2H), 1,62-1,83 (м, 8H), 1,15-1,35 (м, 3H), 0,87-1,16 (м, 2H).
Пример 49. Синтез 3-эндо-{8-[2-(циклогексилметилфенилацетиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 5 и 7 (112 мг, 0,19 ммоль), в дихлорметане (1 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (97 мг, 0,75 ммоль). Полученную смесь охлаждали до -40°С и добавляли раствор фенилацетилхлорида (32 мг, 0,21 ммоль) в DCM (0,1 мл). Полученную смесь перемешивали при температуре от -40°С до -20°С в течение примерно 30 минут. Результаты масс-спектрометрического анализа показали, что реакция закончена. Реакционную смесь концентрировали, остаток повторно растворяли в 50% растворе уксусной кислоты в воде (10 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (27,5 мг). (m/z): [M+H]+, вычислено для С31Н41N3O2 488,33; найдено 488,8.
Пример 50. Синтез метилового эфира N-{2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-N-циклогексилметилсукцинамовой кислоты
К раствору соли бистрифторуксусной кислоты 3-эндо{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 5 и 7 (97 мг, 0,16 ммоль), в дихлорметане (0,8 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (83 мг, 0,64 ммоль). Полученную смесь охлаждали до -20°С, добавляли раствор 3-карбометоксипропионилхлорида (29 мг, 0,19 ммоль) в DCM (0,1 мл) и затем еще одну порцию 3-карбометоксипропионилхлорида (0,20 ммоль) в DCM (0,3 мл). Через 30 минут результаты анализа ЖХ-МС аликвоты реакционной смеси показали наличие требуемой молекулярной массы. Реакционную смесь концентрировали, повторно растворяли в 50% растворе уксусной кислоты в воде (8 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения в виде белого твердого вещества (46,4 мг). (m/z): [M+H]+, вычислено для С28Н41N3O4 484,32; найдено 484,5.
Пример 51. Синтез 3-эндо-{8-[2-(3-втор-бутил-1-циклогексилметилуреидо)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
В соответствии со способом по примеру 32 соль бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 5 и 9 (30 мг, 0,05 ммоль), подвергали взаимодействию с втор-бутилизоцианатом (0,075 ммоль). Реакционную смесь очищали, получая при этом соль ТФУ указанного в заголовке соединения (24,2 мг). (m/z): [M+H]+, вычислено для С28Н44N4O2 469,35; найдено 469,4.
Пример 52. Синтез 3-эндо-{8-[2-(1-циклогексилметил-3-пентилуреидо)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 5 и 9 (30 мг, 0,05 ммоль), в ДМФА (0,4 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (27 мг, 0,20 ммоль) и пентилизоцианат (0,075 ммоль). Полученную смесь встряхивали при комнатной температуре в течение ночи, концентрировали, растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (23,3 мг). (m/z): [M+H]+, вычислено для С29Н46N4O2 483,37; найдено 483,4.
Примеры 53-55
Соли ТФУ соединений по примерам 53-55 были получены общим способом по примеру 52 при замене пентилизоцианата соответствующим изоцианатом.
Пример 53 3-эндо-(8-{2-[1-циклогексилметил-3-(4-фторбензил)-уреидо]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (30,3 мг); (m/z): [M+H]+, вычислено для С31Н41FN4O2 521,33; найдено 521,2.
Пример 54 3-эндо-{8-[2-(3-бензо[1,3]диоксол-5-ил-1-циклогексилметилуреидо)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (24,5 мг); (m/z): [M+H]+, вычислено для С31Н40N4O4 533,31; найдено 533,2. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.) 7,97 (с, 1H), 7,71 (д, J=7,8 Гц, 1H), 7,66 (д, J=8,1 Гц, 1H), 7,42 (дд, J=7,8 Гц, 1H), 3,90-3,99 (м, 3H), 3,60(т, J=4,8 Гц, 2H), 3,25-3,29(м, 1H, перекрывание растворителем), 3,02-3,09 (м, 4H), 2,55-2,58 (м, 4H), 2,03-2,07 (м, 2H), 1,63-1,78 (м, 8H), 1,07-1,25 (м, 9H), 0,92-1,01 (м, 2H).
Пример 55 3-эндо-{8-[2-(1-циклогексилметил-3-изопропилуреидо)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (24,5 мг); (m/z): [M+H]+, вычислено для С27Н42N4O2 455,33; найдено 455,4. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.) 7,97(с, 1H), 7,71(д, J=7,8 Гц, 1H), 7,66(д, J=8,1 Гц, 1H), 7,42(дд, J=7,8 Гц, 1H), 3,90-3,99 (м, 3H), 3,60 (т, J=4,8 Гц, 2H), 3,25-3,29 (м, 1H, перекрывание растворителем), 3,02-3,09 (м, 4H), 2,55-2,58 (м, 4H), 2,03-2,07 (м, 2H), 1,63-1,78 (м, 8H), 1,07-1,25 (м, 9H), 0,92-1,01 (м, 2H).
Пример 56. Синтез 3-эндо-(8-{2-[циклогексилметил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору метансульфонилуксусной кислоты (90 мг, 0,65 ммоль) в ДМФА (0,2 мл) добавляли 1,1'-карбонилдиимидазол (105 мг, 0,65 ммоль). Через один час добавляли 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид, полученный способом по препаративным примерам 12 и 9 (60 мг, 0,16 ммоль), и затем N,N-диизопропилэтиламин (84 мг, 0,65 ммоль). Полученную смесь нагревали до 60°С в течение двух часов и охлаждали до комнатной температуры в течение 60 часов. Затем реакционную смесь концентрировали, растворяли в 50% растворе уксусной кислоты в воде (3 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (35,1 мг). (m/z): [M+H]+, вычислено для С26Н39N3O4S 490,27; найдено 490,2. 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 7,98 (с, 1H), 7,66-7,74 (м, 2H), 7,41-7,46 (м, 1H), 4,77 (с, 2H), 4,38 (ушир.с, 2H), 3,78-3,82 (м, 2H), 3,04-3,33 (м, темный 5H), 3,21 (с, 3H), 2,57-2,61 (м, 4H), 2,07-2,10 (м, 2H), 1,66-1,79 (м, 8H), 1,22-1,29 (м, 3H), 0,94-1,01 (м, 2H).
Пример 57. Синтез 3-эндо-(8-{2-[(2-аминоацетил)циклогексилметиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору н-трет-бутоксикарбонилглицина (34 мг, 0,20 ммоль) в ДМФА (0,2 мл) при комнатной температуре добавляли 1,1'-карбонилдиимидазол (32 мг, 0,2 ммоль). Реакционную смесь встряхивали при комнатной температуре в течение 2 часов и добавляли соль бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 12 и 9 (30 мг, 0,05 ммоль), и N,N-диизопропилэтиламин (26 мг, 0,2 ммоль). Полученную реакционную смесь встряхивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали, остаток обрабатывали 50% раствором ТФУ в DCM (1 мл). Смесь концентрировали и остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (15,9 мг). (m/z): [M+H]+, вычислено для С25Н38N4O2 427,31; найдено 427,2.
Пример 58А. Синтез 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору (S)-2,2-диметил[1,3]диоксолан-4-карбоновой кислоты (98 мг, 0,67 ммоль) в ДМФА (0,2 мл) при комнатной температуре добавляли 1,1'-карбонилдиимидазол (109 мг, 0,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа и добавляли соль бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 12 и 9 (100 мг, 0,167 ммоль), и N,N-диизопропилэтиламин (87 мг, 0,67 ммоль). Реакционную смесь нагревали при 60°С в течение двух часов и затем при комнатной температуре в течение 72 часов. Смесь разбавляли водой и экстрагировали этилацетатом. Органический слой последовательно промывали водой (2×2 мл), 1 н, раствором NaOH (2 мл), насыщенным раствором соли (2 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Образовавшийся остаток растворяли в уксусной кислоте (1,5 мл) и воде (0,5 мл) и нагревали до 65°С в течение ночи. Смесь концентрировали в роторном испарителе, повторно растворяли в 50% растворе уксусной кислоты в воде (3,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (15,5 мг). (m/z): [M+H]+, вычислено для С26Н39N3O2 458,30; найдено 458,2. 1H ЯМР (CD3OD, 300 MГц) δ (м.д.): 7,98 (с, 1H), 7,67-7,73 (м, 2H), 7,40-7,45 (м, 1H), 4,57 (т, J=5,4 Гц, 1H), 3,94-4,12 (м, 3H), 3,69-3,72 (м, 2H), 3,50-3,58 (м, 1H), 3,35-3,43 (м, 1H), 3,20-3,27 (темный 2H, частичное перекрывание растворителем), 3,12-3,15 (м, 2H), 2,52 (ушир.с, 4H), 1,98-2,02 (м, 2H), 1,61-1,70 (м, 8H), 1,09-1,22 (м, 3H), 0,88-0,95 (м, 2H).
Соль ТФУ указанного в заголовке соединения (4,45 г, 7,78 ммоль), полученного вышеописанным способом с использованием в качестве реагентов 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида, полученного способом по препаративным примерам 13 и 9, и (4S)-2,2-диметил-1,3-диоксолан-4-карбоксилата лития, растворяли в метаноле (<10мл) и разбавляли DCM (400 мл). Органический раствор промывали 1 М раствором NaOH (500 мл). Основный водный слой экстрагировали DCM (2×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (500 мл). Органический слой сушили над карбонатом калия. Раствор фильтровали и растворитель удаляли в вакууме, получая при этом указанное в заголовке соединение (3,09 г, выход 87%) в виде стекловидного твердого вещества. (m/z): [M+H]+, вычислено для С26Н39N3O2 458,30; найдено 458,5. 1H ЯМР (300 МГц, d 6-ДМСО): 7,81-7,83 (с, 1H), 7,61-7,65 (ушир. с, 1H), 7,55-7,60 (д, 1H), 7,30-7,35 (д, 1H), 7,18-7,22 (м, 2H), 4,80-4082 (д, 0,8H), 4,62-4,65 (д, 0,64H), 4,50-4,60 (м, 1,1H), 4,22-4,38 (м, 0,83H), 4,10-4,20 (м, 0,65H), 3,00-3,50 (м, 8H), 2,70-2,99 (м, 2H), 2,00-2,30 (м, 4H), 1,60-1,80 (м, 2H), 1,43-1,60 (м, 4H), 1,22-1,40 (м, 3H), 0,93-1,19 (м, 3H), 0,82-0,94 (м, 2H).
Пример 58В. Синтез кристаллического сульфата 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение бензилового эфира N-циклогексилметил(2-оксоэтил)карбаминовой кислоты
В 100 мл колбу вводили аддукт бисульфита бензилового эфира N-циклогексилметил(2-оксоэтил)карбаминовой кислоты (3,94 г, 1 ммоль) и МеTHF (35 мл) и воду (25 мл). Полученную суспензию перемешивали при комнатной температуре в течение 5 минут и добавляли 1 М раствор NaOH (8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 45 минут. Слои разделяли и объем органического слоя уменьшали до ~8 мл, получая при этом указанное в заголовке неочищенное промежуточное соединение.
b. Получение бензилового эфира {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметилкарбаминовой кислоты
К продукту, полученному на предыдущей стадии, добавляли ДМФА (15 мл) и гидрохлорид 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида (2,67 г, 1 ммоль), полученного способом по препаративному примеру 25, и ДМФА (10 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, охлаждали до 10°С и добавляли триацетоксиборгидрид натрия (4,25 г, 2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 90 минут и охлаждали до 10°С. Добавляли изопропилацетат (100 мл) и 1 М раствор NaOH (50 мл). Смесь перемешивали в течение 15 минут и фазы разделяли. Органический слой промывали насыщенным раствором соли в воде (1:1, 2×50 мл) и объем органического слоя уменьшали до ~10 мл, получая при этом указанное в заголовке неочищенное промежуточное соединение.
с. Получение 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
К продукту, полученному на предыдущей стадии, добавляли EtOH (30 мл) и концентрированную HCl (1,5 мл). Раствор продували азотом, добавляли 10% палладий на угле (470 мг), смесь продували азотом в течение 5 минут и гидрировали под давлением 2,1 атм (30 фунтов/кв.дюйм) в течение ночи. Реакционную смесь продували азотом в течение 2 минут, раствор фильтровали через целит и растворитель удаляли до ~10 мл. Добавляли изопропилацетат (40 мл) и 1 М раствор NaOH (20 мл). Слои разделяли и органический слой промывали насыщенным раствором соли (20 мл), фазы разделяли и органический растворитель удаляли до 5-10 мл. Добавляли изопропилацетат (20 мл), объем уменьшали до ~8 мл и добавляли изопропилацетат (20 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Продукт выделяли фильтрованием, реакционную колбу и фильтровальную лепешку промывали изопропилацетатом (10 мл), получая при этом указанное в заголовке промежуточное соединение (2,4 г, чистота 98%) в виде не совсем белого твердого вещества.
d. Получение сульфата 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (в форме гидрата)
В 500 мл колбу вводили 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (31 г, 83,9 ммоль) и ДМФА (150 мл). Смесь перемешивали в течение 10 минут, добавляли гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония (56,8 г, 109 ммоль) и (4S)-2,2-диметил-1,3-диоксолан-4-карбоксилат лития (15,6 г, 92,3 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли этилацетат (600 мл) и 0,5 М раствор NaOH (300 мл) и фазы разделяли. Органический слой содержал неочищенный {2-[3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметиламид (S)-2,2-диметил[1,3]диоксолан-4-карбоновой кислоты (~84 ммоль), который не был выделен.
Органический слой промывали насыщенным раствором соли в воде (1:1, 2×300 мл) и фазы разделяли. К органическому слою добавляли 2 М раствор Н2SO4 (42 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли ацетонитрил (300 мл) и образовавшуюся суспензию перемешивали в течение 2-6 часов. Продукт выделяли фильтрованием, фильтровальную лепешку промывали ацетонитрилом (200 мл), сушили на воздухе в течение 2 часов и затем в вакууме при комнатной температуре в течение 20 часов, получая при этом указанное в заголовке соединение (40 г, чистота 97% по результатам ВЭЖХ) в виде белого порошка.
е. Синтез кристаллического сульфата 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
В 100 мл колбу вводили сульфат 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида в виде гидрата (2 г) и МеОН (40 мл). Полученную суспензию нагревали до 65°С в атмосфере азота в течение 20 минут до полного растворения исходного вещества. Раствор охлаждали до комнатной температуры при перемешивании. Примерно 20 мл растворителя удаляли при немного пониженном давлении и полученную суспензию перемешивали при комнатной температуре в течение ночи. Продукт выделяли фильтрованием, колбу и фильтровальную лепешку промывали ацетонитрилом (2×5 мл). Фильтровальную лепешку сушили на воздухе в течение 2 часов и затем в вакууме при комнатной температуре в течение ночи, получая при этом указанное в заголовке соединение (1,71 г, чистота >99% по результатам ВЭЖХ, выход ~85%) в виде белого порошка.
Образец, полученный вышеуказанным способом, исследовали при помощи 1Н ЯМР-спектроскопии. 1H ЯМР (400 МГц, ДМСО d 6): δ (м.д.) 9,08 и 8,94 (две серии ушир.с, 1H), 7,99-8,04 (м, 2H), 7,74-7,76 (м, 1H), 7,68-7,70 (м, 1H), 7,41-7,45 (м, 2H), 4,81, 5,00 и 5,30 (три серии ушир.с, 2H), 4,34 (деформированный м, 1H), 4,00 и 4,05 (деформированный м, 2H), 3,01-3,25 и 3,47-3,55 и 3,75-3,82 (три серии м, 10H), 2,50-2,55 (м, 2H), 1,99 (деформированный м, 2H), 1,56-1,70 (м, 8H), 1,15-1,19 (м, 3H), 0,89-0,99 (м, 2H).
Пример 58С. Синтез кристаллического гликолята 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
3-эндо-(8-{2-[Циклогексилметил-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (35 мг) растворяли в водном растворе ацетона (2% воды, 98% ацетона, 0,46 мл). К полученному раствору добавляли 0,78 М раствор гликолевой кислоты в ацетонитриле (0,10 мл). Быстро образовывался осадок, который в течение 2 часов превращался в двупреломляющее вещество. Маточный раствор сливали и оставшееся твердое вещество сушили с образованием указанного в заголовке соединения. Порошковая рентгенограмма (XRPD) кристаллического вещества показана на фигуре 1. Дифракционные пики были обнаружены при значениях 2θ 8,00±0,2, 12,50±0,2, 16,19±0,2, 16,91±0,2, 18,41±0,2, 20,69±0,2, 22,04±0,2, 23,03±0,2, 25,44±0,2, 25,85±0,2 и 28,76±0,2.
Все данные XRPD, представленные в настоящем описании изобретения, были получены в дифрактометре Ригаку с использованием излучения Сu Kα (30,0 кВ, 15,0 мА), действующем в режиме непрерывного сканирования 3° в минуту с величиной шага 0,03°.
Пример 58D. Синтез кристаллического оксалата 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
3-эндо-(8-{2-[Циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (25,4 мг) растворяли в ацетоне (0,34 мл). К указанному раствору добавляли 0,4 М раствор щавелевой кислоты в ацетонитриле (0,14 мл) и воду (0,24 мл). Полученную дисперсию обрабатывали ультразвуком в течение 30 секунд и добавляли воду (0,045 мл) и DCM (0,015 мл). Через 4 дня указанное в заголовке соединение выделяли путем фильтрации под вакуумом в виде кристаллического твердого вещества (19,8 мг). XRPD кристаллического вещества показана на фигуре 2. Дифракционные пики были обнаружены при значениях 2θ 5,84±0,2, 13,80±0,2, 17,03±0,2, 23,00±0,2 и 28,85±0,2.
Пример 59. Синтез 3-эндо-(8-{2-[(2-гидроксиацетил)фенетиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Продукт по препаративному примеру 17 (~0,26 ммоль) растворяли в DCM (0,5 мл) и охлаждали до 0°С. Реакционную смесь обрабатывали N,N-диизопропилэтиламином (100 мг, 0,78 ммоль) и ацетоксиацетилхлоридом (39 мг, 0,29 ммоль). Реакционную смесь разбавляли дихлорметаном и промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное масло растворяли в этаноле (1 мл) и обрабатывали моногидратом гидроксида лития (66 мг, 1,2 ммоль) в воде (0,5 мл). Через один час реакционную смесь концентрировали и остаток растворяли в 50% растворе уксусной кислоты в воде (1,2 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (38,7 мг). (m/z): [M+H]+, вычислено для С26Н33N3O3 436,25; найдено 436,4.
Примеры 60-62
Соли ТФУ соединений по примерам 60-62 были получены способом по примеру 59 при замене продукта по препаративному примеру 17 продуктами по препаративным примерам 18, 19 и 20.
Пример 60 3-эндо-(8-{2-[(2-гидроксиацетил)(3-фенилпропил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (66,7 мг); (m/z): [M+H]+, вычислено для С27Н35N3O3 450,27; найдено 450,4.
Пример 61 3-эндо-(8-{2-[(2-циклогексилэтил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (68,6 мг); (m/z): [M+H]+, вычислено для С26Н39N3O3 442,30; найдено 442,6.
Пример 62 3-эндо-(8-{2-[3-циклогексилпропил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (37,1 мг); (m/z): [M+H]+, вычислено для С27Н41N3O3 456,31; найдено 456,4.
Пример 63А. Синтез 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
В соответствии со способом по примеру 48 продукт по препаративному примеру 21, 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (72,5 мг, 1 экв.) обрабатывали ацетоксиацетилхлоридом (1,3 экв.), гидролизовали и очищали ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (14,4 мг). (m/z): [M+H]+, вычислено для С25Н35F2N3O3 464,27; найдено 464,2.
Пример 63В. Синтез 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида, полученного способом по препаративному примеру 13 (2,15 г, 9,34 ммоль), в DCM (45,0 мл) при 0°С добавляли уксусную кислоту (0,56 г, 9,34 ммоль), раствор [(4,4-дифторциклогексилметил)(2-оксоэтил)карбамоил]метилового эфира уксусной кислоты (2,59 г, 8,9 ммоль) в DCM (10,0 мл) и триацетоксиборгидрид натрия (2,26 г, 10,7 ммоль). Полученную смесь перемешивали при 0°С в течение 30 минут и разбавляли DCM (40,0 мл). Органический слой последовательно промывали насыщенным раствором бикарбоната натрия (20,0 мл) и насыщенным раствором соли (20,0 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом [{2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}(4,4-дифторциклогексилметил)карбамоил]метиловый эфир уксусной кислоты в виде светло-желтой пены.
Продукт, полученный на предыдущей стадии, растворяли в метаноле (20,0 мл) при комнатной температуре и обрабатывали моногидратом гидроксида лития (0,56 г, 13,4 ммоль) в воде (5,0 мл) в течение 30 минут. Реакционную смесь концентрировали. Остаток растворяли в 25% растворе уксусной кислоты в воде (48,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой. Требуемые фракции объединяли и сушили вымораживанием, получая при этом указанное в заголовке соединение в виде соли ТФУ (2,1 г). 1H ЯМР (CD3OD, 300 МГц) δ (м.д.) 8,07 (с, 1H), 7,75-7,81 (м, 2H), 7,48 (дд, J=7,8 Гц, 1H), 4,35 (с, 2H), 4,17 (ушир.с, 2H), 3,81 (т, J=6,0 Гц, 2H), 3,35-3,38 (темный, 1H, перекрывание растворителем), 3,25 (д, J=6,9 Гц, 2H), 3,20 (т, J=5,4 Гц, 2H), 2,66-2,72 (м, 4H), 2,10-2,19 (м, 4H), 1,77-1,90 (м, 7H), 1,33-1,41 (м, 2H).
Пример 63С. Синтез кристаллического фосфата 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
3-эндо-(8-{2-[(4,4-Дифторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (20 мг), находящийся в 4 мл стеклянном флаконе, растворяли в метаноле (0,172 мл) при комнатной температуре. К полученному раствору добавляли 1,0 М раствор фосфорной кислоты в метаноле (0,043 мл) и ацетон (0,228 мл). Смесь осторожно перемешивали в течение 16 часов при комнатной температуре. Указанное в заголовке соединение выделяли фильтрацией под вакуумом в виде кристаллического порошка (13,4 мг). XRPD кристаллического вещества показана на фигуре 3. Дифракционные пики были обнаружены при значениях 2θ 5,51±0,20, 7,27±0,20, 17,30±0,20, 18,05±0,20, 19,94±0,20, 20,39±0,20, 21,89±0,20, 24,62±0,20, 26,66±0,20, 27,38±0,20, 28,52±0,20, 29,21±0,20 и 32,87±0,20.
Примеры 64-74
Раствор продукта по препаративному примеру 22, 3-эндо-[8-(2-бензиламинопропил)-8-азабицикло[3.2.1]окт-3-ил]фенола (509 мг, 1,45 ммоль) и N,N-диизопропилэтиламина (0,76 мл) в дихлорметане (12 мл) распределяли в виде 12 одинаковых порций во флаконы, содержащие соответствующий хлорангидрид кислоты (0,16 ммоль). Флаконы встряхивали при комнатной температуре в течение 45 минут и реакционные смеси концентрировали в вакууме. Каждый остаток растворяли в этаноле (1 мл), добавляли раствор гидроксида лития (6 экв.) в воде (0,2 мл) и флаконы встряхивали при 40°С в течение 30 минут. Содержимое флаконов концентрировали в вакууме, разбавляли смесью уксусной кислоты:воды в отношении 1:1 (1 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соли ТФУ соединений по примерам 64-74.
Пример 64 (R)-бензил-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}амид циклопропанкарбоновой кислоты (7,9 мг); (m/z): [M+H]+, вычислено для С27Н34N2O2 419,26; найдено 419,2.
Пример 65 N-бензил-3-циклопентил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}пропионамид (1,9 мг); (m/z): [M+H]+, вычислено для С31Н42N2O2 475,32; найдено 475,2.
Пример 66 N-бензил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}-2-фенилацетамид (3,9 мг); (m/z): [M+H]+, вычислено для С31Н36N2O2 469,28; найдено 469,2.
Пример 67 N-бензил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}-3-метилбутирамид (6,5 мг); (m/z): [M+H]+, вычислено для С28Н38N2O2 435,29; найдено 435,2.
Пример 68 N-бензил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}-3-метилбутирамид (3,5 мг); (m/z): [M+H]+, вычислено для С25Н32N2O3 409,24; найдено 409,2.
Пример 69 N-бензил-2-циклопентил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}ацетамид (2,6 мг); (m/z): [M+H]+, вычислено для С30Н40N2O2 461,31; найдено 461,2.
Пример 70 (R)-бензил-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}амид циклогексанкарбоновой кислоты (3,8 мг); (m/z): [M+H]+, вычислено для С30Н40N2O2 461,31; найдено 461,2.
Пример 71 N-бензил-2-этил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}бутирамид (3,8 мг); (m/z): [M+H]+, вычислено для С29Н40N2O2 449,31; найдено 449,2.
Пример 72 N-бензил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}сукцинамовая кислота (5,7 мг); (m/z): [M+H]+, вычислено для С27Н34N2O4 451,25; найдено 451,2.
Пример 73 (R)-бензил-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}амид циклопентанкарбоновой кислоты (5,2 мг); (m/z): [M+H]+, вычислено для С29Н38N2O2 447,29; найдено 447,2.
Пример 74 N-бензил-N-{(R)-2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]-1-метилэтил}ацетамид (4,6 мг); (m/z): [M+H]+, вычислено для С25Н32N2O2 393,25; найдено 393,2.
Пример 75. Синтез N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-метансульфониламинофенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
а. Получение трет-бутилового эфира циклогексилметил{2-[3-эндо-(3-метансульфониламинофенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбаминовой кислоты
К раствору соли ТФУ N-[3-эндо-(8-азабицикло[3.2.1]окт-3-ил)фенил]метансульфонамида, продукта по препаративному примеру 23 (140 мг, 0,35 ммоль), в DCM (2 мл) при комнатной температуре добавляли раствор трет-бутилового эфира циклогексилметил(2-оксоэтил)карбаминовой кислоты (116 мг, 0,455 ммоль) и триацетоксиборгидрид натрия (96 мг, 0,455 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи и разбавляли DCM. Органический слой промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде маслянистого остатка, который использовали на следующей стадии без очистки. (m/z): [M+H]+, вычислено для С28Н45N3O4S 520,31; найдено 520,4.
b. Получение N-(3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенил)метансульфонамида
Маслянистый продукт, полученный на предыдущей стадии, обрабатывали DCM (1,5 мл) и ТФУ (1,5 мл) при комнатной температуре в течение тридцати минут. Затем реакционную смесь концентрировали, повторно растворяли в смеси уксусной кислоты и воды в отношении 1:1 (6 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке промежуточное соединение в виде его соли бистрифторуксусной кислоты (38,9 мг). (m/z): [M+H]+, вычислено для С23Н37N3O2S 420,26; найдено 420,4.
с. Синтез N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-метансульфониламинофенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида
К раствору продукта, полученного на предыдущей стадии (39 мг, 0,06 ммоль), в DCM (0,2 мл) при комнатной температуре добавляли N,N-диизопропилэтиламин (31 мг, 0,24 ммоль) и ацетоксиацетилхлорид (12 мг, 0,09 ммоль). Через пять минут реакционную смесь концентрировали, повторно растворяли в этаноле (0,2 мл) и обрабатывали моногидратом гидроксида лития (15 мг, 0,36 ммоль) в воде (0,2 мл) при комнатной температуре в течение тридцати минут. Затем реакционную смесь повторно концентрировали и образовавшийся остаток растворяли в смеси уксусной кислоты и воды в отношении 1:1 (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли ТФУ (16,9 мг). (m/z): [M+H]+, вычислено для С25Н39N3O4S 478,27; найдено 478,2.
Примеры 76-204
В приведенных ниже примерах промежуточное соединение 8-азабициклооктанфенола или 8-азабициклооктанбензамида было получено способом по препаративному примеру 13 при наличии следующих исключений: препаративный пример 1: пример 137; препаративный пример 12, стадии а-с: примеры 106-108 и 112; препаративный пример 12: примеры 91, 101, 109-111 и 131.
Пример 76. Синтез N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-2-(S)-фенилацетамида
К раствору 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}фенола (30 мг, 0,087 ммоль) в ДМФА (0,4 мл) добавляли HATU (39,6 мг, 0,10 ммоль) и (S)-гидроксифенилуксусную кислоту (15,2 мг, 0,1 ммоль). Реакционную смесь концентрировали в роторном испарителе и остаток растворяли в 50% растворе уксусной кислоты в воде (1,2 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом указанный в заголовке продукт в виде соли ТФУ (8,6 мг). (m/z): [M+H]+, вычислено для С30Н40N2O3 477,30; найдено 477,4.
Примеры 77-84
Соли ТФУ соединений по примерам 77-84 были получены способами, аналогичными способу по примеру 76, за исключением замены (S)-гидроксифенилуксусной кислоты соответствующей карбоновой кислотой.
Пример 77 (S)-N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-3-фенилпропионамид (15,9 мг); (m/z): [M+H]+, вычислено для С31Н42N2O3 491,68; найдено 491,4.
Пример 78 (R)-N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-4-фенилбутирамид (17,8 мг); (m/z): [M+H]+, вычислено для С32Н44N2O3 505,34; найдено 505,4.
Пример 79 циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}амид 1-гидроксициклопропанкарбоновой кислоты (5,7 мг); (m/z): [M+H]+, вычислено для С26Н38N2O3 427,29; найдено 427,4.
Пример 80 циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}амид (S)-2-гидрокси-4-метилпентановой кислоты (12,7 мг); (m/z): [M+H]+, вычислено для С28Н44N2O3 457,34; найдено 457,4.
Пример 81 (S)-N-циклогексилметил-2-диметиламино-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-3-фенилпропионамид (9,4 мг); (m/z): [M+H]+, вычислено для С33Н47N3O2 518,37; найдено 518,6.
Пример 82 циклогексилметил{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}амид 2-гидроксигексановой кислоты (10,6 мг); (m/z): [M+H]+, вычислено для С28Н44N2O3 457,34; найдено 457,5.
Пример 83 (R)-2-циклогексил-N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамид (9,8 мг); (m/z): [M+H]+, вычислено для С30Н46N2O3 483,35; найдено 483,2.
Пример 84 (S)-2-циклогексил-N-циклогексилметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамид (14,5 мг); (m/z): [M+H]+, вычислено для С30Н46N2O3 483,35; найдено 483,4.
Примеры 85-89
Соли ТФУ соединений по примерам 85-89 были получены способами, аналогичными способу по примеру 76 при замене азабициклооктанфенола 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамидом и использовании соответствующей карбоновой кислоты.
Пример 85 3-эндо-(8-{2-[циклогексилметил((S)-2-гидрокси-3-фенилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (15,2 мг); (m/z): [M+H]+, вычислено для С32Н43N3O3 518,33; найдено 518,4.
Пример 86 3-эндо-(8-{2-[циклогексилметил((S)-2-гидрокси-4-метилпентаноил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (19 мг); (m/z): [M+H]+, вычислено для С29Н45N3O3 484,35; найдено 484,4.
Пример 87 3-эндо-(8-{2-[циклогексилметил(1-гидроксициклопропанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (15,5 мг); (m/z): [M+H]+, вычислено для С27Н39N3O3 454,30; найдено 454,4.
Пример 88 3-эндо-(8-{2-[циклогексилметил((S)-2-диметиламино-3-фенилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (17,4 мг); (m/z): [M+H]+, вычислено для С34Н48N4O2 545,38; найдено 545,4.
Пример 89 3-эндо-(8-{2-[циклогексилметил(3-гидрокси-2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид (5,3 мг); (m/z): [M+H]+, вычислено для С28Н43N3O3 470,33; найдено 470,4.
Пример 90. Синтез 3-эндо-(8-{2-[циклогексилметил(4-диметиламинобутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
В флакон, содержащий хлористоводородную соль 4-диметиламиномасляной кислоты (19,7 мг, 0,15 ммоль), добавляли ДМФА (0,3 мл) и HATU (57,0 мг, 0,15 ммоль). Реакционную смесь перемешивали в течение 1 часа и обрабатывали DIPEA (25,8 мг, 0,2 ммоль) и солью бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (30,0 мг, 0,05 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 часов и нагревали при 65°С в течение ночи. Реакционную смесь концентрировали, образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл) и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде соли бистрифторуксусной кислоты (4,9 мг). (m/z): [M+H]+, вычислено для С29Н46N4O2 483,36; найдено 483,4.
Примеры 91-92
Соединения по примерам 91-92 были получены способами, аналогичными способу по примеру 90, за исключением замены 4-диметиламиномасляной кислоты соответствующей карбоновой кислотой.
Пример 91 соль ТФУ 3-эндо-(8-{2-[циклогексилметил(1-гидрокси-циклопропанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (18,8 мг); (m/z): [M+H]+, вычислено для С27Н39N3O3 454,30; найдено 454,2.
Пример 92 соль бистрифторуксусной кислоты 3-эндо-(8-{2-[циклогексилметил((S)-3-гидрокси-2-метиламинопропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (24,0 мг); (m/z): [M+H]+, вычислено для С27Н42N4O3 471,33; найдено 471,4.
Пример 93. Синтез 3-эндо-(8-{2-[циклогексилметил(3-гидрокси-2-гидроксиметил-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 5-метил-2-фенил-1,3-диоксинан-5-карбоновой кислоты
В круглодонную колбу последовательно вводили 3-гидрокси-2-гидроксиметил-2-метилпропионовую кислоту (10,0 г, 74,5 ммоль), ацетон (75,0 мл), диметилацеталь бензальдегида (17,02 г, 111,0 ммоль) и моногидрат паратолуолсульфоновой кислоты (0,71 г, 3,7 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 часов и фильтровали. Фильтровальную лепешку промывали холодным ацетоном и сушили в вакууме, получая при этом указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 7,46-7,48 (м, 2H), 7,34-7,36 (м, 3H), 5,49 (с, 1H), 4,65 (д, J=10,8 Гц, 2H), 3,70 (д, J=11,4 Гц, 2H), 1,11 (с, 3H).
b. Синтез 3-эндо-(8-{2-[циклогексилметил(3-гидрокси-2-гидроксиметил-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (55,7 мг, 0,25 ммоль), в ДМФА (0,5 мл) при комнатной температуре добавляли HATU (95,0 мг, 0,25 ммоль). Реакционную смесь перемешивали в течение 2 часов и обрабатывали солью бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (75,0 мг, 0,13 ммоль) и DIPEA (64,9 мг, 0,50 ммоль). Реакционную смесь нагревали при 40°С в течение ночи. Смесь концентрировали, остаток обрабатывали смесью уксусной кислоты (2,1 мл) и воды (0,7 мл) при 70°С в течение 2 часов и повторно концентрировали. Образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл) и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (24,6 мг). (m/z): [M+H]+, вычислено для С28Н43N3O4 486,33; найдено 486,4.
Пример 94. Синтез 3-эндо-(8-{2-[циклогексилметил((S)-4-диметиламино-2-гидроксибутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение (S)-4-трет-бутоксикарбониламино-2-гидроксибутирата лития
Раствор метилового эфира (S)-4-трет-бутоксикарбониламино-2-гидроксимасляной кислоты (1,52 г, 6,52 ммоль) в метаноле (20,0 мл) обрабатывали моногидратом гидроксида лития (273,8 мг, 6,52 ммоль) и водой (2,0 мл) в течение 30 минут, концентрировали и сушили в вакууме, получая при этом белое твердое вещество (1,26 г).
b. Получение 3-эндо-(8-{2-[((S)-4-амино-2-гидроксибутирил)-циклогексилметиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Продукт, полученный на предыдущей стадии (150 мг, 0,67 ммоль), растворяли в ДМФА (1,5 мл) при комнатной температуре. К полученному раствору последовательно добавляли соль бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (200,0 мг, 0,335 ммоль), HATU (253,3 мг, 0,67 ммоль) и DIPEA (173,2 мг, 1,34 ммоль). Реакционную смесь перемешивали в течение 2 часов, разбавляли EtOAc (100,0 мл), последовательно промывали полунасыщенным раствором бикарбоната натрия (20,0 мл), насыщенным раствором бикарбоната натрия (15,0 мл) и насыщенным раствором соли (15,0 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом промежуточное соединение трет-бутилового эфира [((S)-3-{2-[3-(3-эндо-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметилкарбамоил)-3-гидроксипропил]карбаминовой кислоты в виде желтоватого масла. (m/z): [M+H]+, вычислено для С32Н50N4O5 571,4; найдено 571,6.
Промежуточное соединение затем обрабатывали DCM (1,5 мл) и ТФУ (2,5 мл) при комнатной температуре в течение тридцати минут. Реакционную смесь концентрировали, остаток растворяли в 25% растворе уксусной кислоты в воде (8,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (194,4 мг). (m/z): [M+H]+, вычислено для С27Н42N4O3 471,3; найдено 471,6.
с. Синтез 3-эндо-(8-{2-[((S)-4-диметиламино-2-гидроксибутирил)циклогексилметиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (65,3 мг, 0,09 ммоль), в метаноле (0,3 мл) добавляли 37% водный раствор формальдегида (0,02 мл, 0,27 ммоль) и цианоборгидрид натрия (14,0 мг, 0,27 ммоль). Полученную смесь перемешивали при комнатной температуре в течение двадцати минут и концентрировали. Остаток повторно растворяли в 25% растворе уксусной кислоты в воде (6,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (33,0 мг). 1H ЯМР (CD3OD, 400M Гц) δ (м.д.): 8,06 (с, 1H), 7,74-7,81 (м, 2H), 7,49-7,51 (м, 1H), 4,66-4,69 (м, 1H), 4,14-4,18 (м, 2H), 3,84-3,89 (м, 1H), 3,73-3,78 (м, 1H), 3,37-3,44 (темный, 3H, частичное перекрывание растворителем), 3,28-3,30 (темный 2H, частичное перекрывание растворителем), 3,20-3,22 (м, 2H), 2,96 (с, 6H), 2,61-2,70 (м, 4H), 2,09-2,18 (м, 4H), 1,74-1,88 (м, 8H), 1,25-1,36 (м, 3H), 1,04-1,12 (м, 2H); (m/z): [M+H]+, вычислено для С29Н46N4O3 499,36; найдено 499,6.
Пример 95. Синтез 3-эндо-(8-{2-[((S)-4-трет-бутиламино-2-гидроксибутирил)циклогексилметиламино]этил}-8-азабицикло[3.2.1]окт-3-илбензамида
К раствору соли бистрифторуксусной кислоты 3-эндо-(8-{2-[((S)-4-амино-2-гидроксибутирил)циклогексилметиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (29,0 мг, 0,04 ммоль) в ДМФА (0,5 мл) добавляли DIPEA (20,7 мг, 0,16 ммоль) и трет-бутилиодид (14,7 мг, 0,08 ммоль). Полученную смесь нагревали при 75°С в течение 2 часов. Реакционную смесь концентрировали, остаток растворяли в 25% растворе уксусной кислоты в воде и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (4,5 мг). (m/z): [M+H]+, вычислено для С31Н50N4O3 526,38; найдено 526,6.
Пример 96. Синтез 3-эндо-(8-{2-[циклогексилметил((S)-4-диэтиламино-2-гидроксибутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Соль бистрифторуксусной кислоты указанного в заголовке соединения была получена способом, аналогичным способу по примеру 95, при замене трет-бутилиодида этилиодидом (3,0 экв.). (m/z): [M+H]+, вычислено для С31Н50N4O3 527,39; найдено 527,2.
Пример 97. Синтез 3-эндо-(8-{2-[циклогексилметил(3-диметиламино-2-гидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 9Н-флуорен-9-илметилового эфира [2-({2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметилкарбамоил)-2-гидроксиэтил]карбаминовой кислоты
Указанное в заголовке соединение было получено в виде желтоватого масла способом, аналогичным способу по примеру 94(b), при замене (S)-4-трет-бутоксикарбониламино-2-гидроксибутирата лития 3-(9Н-флуорен-9-илметоксикарбониламино)-2-гидроксипропионовой кислотой (163,7 мг, 0,5 ммоль, 2,0 экв.). (m/z): [M+H]+, вычислено для С41Н50N4O5 679,4; найдено 679,6.
b. Получение 3-эндо-(8-{2-[(3-амино-2-гидроксипропионил)циклогексилметиламино]этил}-8-азабицикло]3.2.1]окт-3-ил)бензамида
Продукт, полученный на предыдущей стадии, обрабатывали ДМФА (2,0 мл) и пиперидином (0,4 мл) при комнатной температуре в течение 5 минут и концентрировали. Остаток очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (93,7 мг). (m/z): [M+H]+, вычислено для С26Н40N4O3 457,3; найдено 457,4.
с. Синтез 3-эндо-(8-{2-[циклогексилметил(3-диметиламино-2-гидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
В соответствии со способом по примеру 94с была получена соль бистрифторуксусной кислоты указанного в заголовке соединения. (m/z): [M+H]+, вычислено для С28Н44N4O3 485,34; найдено 485,4.
Пример 98. Синтез 3-эндо-(8-{2-[циклогексилметил(4-гидроксибутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору γ-бутиролактона (170,0 мг, 1,98 ммоль) в метаноле (0,5 мл) добавляли воду (0,2 мл) и моногидрат гидроксида лития (83 мг, 2,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и остаток сушили в вакууме, получая при этом промежуточное соединение 4-гидроксибутирата лития в виде белого твердого вещества. Указанное промежуточное соединение (18,3 мг, 0,17 ммоль) добавляли к смеси соли бистрифторуксусной кислоты 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (50 мг, 0,083 ммоль) и DIPEA (58,3 мкл, 0,33 ммоль) в ДМФА (0,4 мл). Затем добавляли HATU (63,1 мг, 0,17 ммоль) и полученную реакционную вмесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали, остаток повторно растворяли в 50% растворе уксусной кислоты в воде (6 мл), фильтровали и очищали препаративной ВЭЖХ. Требуемые фракции объединяли и сушили вымораживанием, получая при этом белое твердое вещество. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,05 (с, 1H), 7,79 (д, J=8,0 Гц, 1H), 7,74 (д, J=7,6 Гц, 1H), 7,51 (дд, J=7,6, 8,0 Гц, 1H), 4,13 (ушир.с, 2H), 3,78 (т, J=5,6 Гц, 2H), 3,69 (т, J=6,0 Гц, 2H), 3,31-3,35 (темный 3H, перекрывание растворителем), 3,17 (т, J=5,6 Гц, 2H), 2,43-2,53 (м, 6H), 2,01-2,05 (м, 2H), 1,61-1,84 (м, 10H), 1,12-1,26 (м, 3H), 0,89-0,98 (м, 2H); (m/z): [M+H]+, вычислено для С27Н41N3O3 456,31; найдено 456,4.
Примеры 99-100
Соединения по примерам 99-100 были получены способами, аналогичными способу по примеру 98, при замене γ-бутиролактона соответсвующим лактоном.
Пример 99 соль ТФУ 3-эндо-(8-2-[циклогексилметил((S)-2,4-дигидроксибутирил)амино]этил-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O4 472,31; найдено 472,4.
Пример 100 соль ТФУ 3-эндо-(8-2-[циклогексилметил((S)-3,4-дигидроксибутириламино]этил-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O4 472,31; найдено 472,4.
Пример 101. Синтез 3-эндо-(8-{2-[циклогексилметил(2-диметиламиноацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 3-эндо-{(8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
В соответствии со способом по препаративным примерам 9 и 12 3-эндо(8-азабицикло[3.2.1]окт-3-ил)бензамид (1,18 г) подвергали взаимодействию с трет-бутиловым эфиром циклогексилметил(2-оксоэтил)карбаминовой кислоты (1,57 г) с образованием трет-бутилового эфира {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}циклогексилметилкарбаминовой кислоты, который затем обрабатывали DCM и ТФУ. Полученный неочищенный продукт растворяли в 50% растворе уксусной кислоты в воде (15,0 мл) и очищали препаративной ВЭЖХ с обращенной фазой. Требуемые фракции объединяли и сушили вымораживанием, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (1,65 г). 1H ЯМР (CD3OD, 300 МГц) δ (м.д.): 8,03 (с, 1H), 7,71-7,77 (м, 2H), 7,44-7,50 (м, 1H), 4,11 (ушир.с, 2H), 3,52 (т, J=6,0 Гц, 2H), 3,33-3,40 (темный, 3H, перекрывание растворителем), 2,96 (д, J=6,6 Гц, 2H), 2,67-2,67 (м, 4H), 2,05-2,12 (м, 2H), 1,70-1,84 (м, 8H), 1,20-1,39 (м, 3H), 1,03-1,10 (м, 2H); (m/z): [M+H]+, вычислено для С23Н35N3O 370,28; найдено 370,2.
b. Синтез 3-эндо-(8-{2-[циклогексилметил(2-диметиламиноацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (33,0 мг, 0,055 ммоль), в DCM (0,3 мл) при комнатной температуре добавляли DIPEA (28,4 мг, 0,22 ммоль) и хлористоводородную соль диметиламиноацетилхлорида (12,6 мг, 0,08 ммоль). Реакционную смесь перемешивали в течение 10 минут и концентрировали. Образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (16,1 мг). (m/z): [M+H]+, вычислено для С27Н42N4O2 455,33; найдено 455,2.
Примеры 102-103
Соединения по примерам 102-103 были получены способами, аналогичными способу по примеру 101, при замене диметиламино-ацетилхлорида соответствующим хлоридом.
Пример 102 соль ТФУ 3-эндо-(8-(2-((циклогексилметил)(N,N-диметилсульфамоил)амино)этил)-8-азабицикло[3.2.1]окт-3-ил)бензамида (21,2 мг); (m/z): [M+H]+, вычислено для С25Н40N4O3S 477,28; найдено 477,2.
Пример 103 соль ТФУ 3-эндо-(8-{2-[циклогексилметил(2-метоксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н39N3O3 442,30; найдено 442,4.
Пример 104. Синтез 3-эндо-(8-{2-[((S)-2,3-дигидроксипропионил)-(3-гидроксиадамантан-1-илметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 3-аминометиладамантан-1-ола
К интенсивно перемешиваемой смеси концентрированной серной кислоты (22,7 мл) и 65% раствору азотной кислоты (2,3 мл) при 0°С по каплям добавляли С-адамантан-1-илметиламин (2,0 г, 12,12 ммоль). Реакционную смесь перемешивали в течение 2 часов при 0°С, нагревали до комнатной температуры и перемешивали при указанной температуре в течение 24 часов, охлаждали до 0°С и медленно гасили льдом (10,8 г). Смесь оставляли нагреваться до комнатной температуры по мере таяния льда в течение ночи, снова охлаждали до 0°С и обрабатывали небольшими порциями гидроксида натрия (50 г). Образовавшуюся коричневатую пасту фильтровали и фильтровальную лепешку промывали DCM (200 мл). Слои разделяли, органический слой промывали насыщенным раствором соли (2×20 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом белое твердое вещество (1,09 г). (m/z): [M+H]+, вычислено для С11Н19N 182,2; найдено 182,2.
b. Получение 3-эндо-(8-{2-[(3-гидроксиадамантан-1-илметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору хлористоводородной соли 3-эндо-[8-(2-оксоэтил)-8-азабицикло[3.2.1]окт-3-ил)бензамида (108,6 мг, 0,60 ммоль) в DCM (5,0 мл) добавляли триацетоксиборгидрид натрия (137,8 мг, 0,65 ммоль) и продукт, полученный на предыдущей стадии (190,0 мг, 0,50 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток растворяли в 25% растворе уксусной кислоты в воде (6,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль бистрифторуксусной кислоты указанного в заголовке соединения (91,0 мг). (m/z): [M+H]+, вычислено для С27Н39N3O2 438,3; найдено 438,4.
с. Синтез 3-эндо-(8-{2-[((S)-2,3-дигидроксипропионил)(3-гидрокси-адамантан-1-илметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К суспензии (4S)-2,2-диметил-1,3-диоксолан-4-карбоксилата лития (28,0 мг, 0.17 ммоль) в ДМФА (0,5 мл) добавляли HATU (62,5 мг, 0,164 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком для ускорения растворения. Через 1 час добавляли DIPEA (87,9 мг, 0,68 ммоль) и продукт, полученный на предыдущей стадии (60,0 мг, 0,085 ммоль). Полученную смесь перемешивали в течение 2 часов и затем концентрировали. Остаток обрабатывали смесью уксусной кислоты (2,1 мл) и воды (0,7 мл) при 70°С в течение 1 часа. Реакционную смесь концентрировали, маслянистый остаток растворяли в 50% растворе уксусной кислоты в воде (3,0 мл) и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (6,2 мг). (m/z): [M+H]+, вычислено для С30Н43N3O5 526,32; найдено 526,4.
Пример 105. Синтез соли 3-эндо-(8-{2-[(2-гидроксиацетил)(3-гидроксиадамантан-1-илметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору соли бистрифторуксусной кислоты 3-эндо-{2-[(3-гидроксиадамантан-1-илметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида (132,2 мг, 0,20 ммоль) в DCM (1,0 мл) добавляли DIPEA (0,14 мл, 0,79 ммоль) и хлорид ацетоксиацеталя (54,6 мг, 0,40 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 5 минут и концентрировали. Образовавшийся остаток повторно растворяли в метаноле (2,0 мл) и обрабатывали моногидратом гидроксида лития (50,0 мг, 1,2 ммоль) в течение 30 минут и повторно концентрировали. Остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом соль ТФУ указанного в заголовке соединения (23,4 мг). 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,16 (с, 1H), 7,88 (д, J=7,6 Гц, 1H), 7,85 (д, J=8,0 Гц, 1H), 7,60 (дд, J=7,6, 8,0 Гц, 1H), 4,46 и 4,44 (две серии с, всего 2H), 4,25 & 4,20 (две серии ушир.с, всего 2H), 3,92 (т, J=5,6 Гц, 2H), 3,44-3,46 (м, 1H), 3,31 (т, J=5,6 Гц, 2H), 3,25 (с, 2H), 2,75-2,82 (м, 4H), 2,36 (ушир.с, 2H), 2,24-2,30 (м, 2H), 1,92-1,98 (м, 2H), 1,62-1,85 (м, 12H). (m/z): [M+H]+, вычислено для С29Н41N3O4 496,31; найдено 496,4.
Примеры 106-112
Соединения по примерам 106-112 были получены способами, аналогичными способу по примерам 104b и 105, с использованием соответствующего оксоэтил-8-азабициклооктана и соответствующего хлорида.
Пример 106 соль ТФУ N-адамантан-1-илметил-2-гидрокси-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}ацетамида; (m/z): [M+H]+, вычислено для С28Н40N2O3 453,31; найдено 453,2.
Пример 107 соль ТФУ N-адамантан-1-илметил-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-ацетамида; (m/z): [M+H]+, вычислено для С28Н40N2O2 437,31; найдено 437,2.
Пример 108 соль ТФУ N-адамантан-1-илметил-N-{2-[3-эндо-3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-сукцинамовой кислоты; (m/z): [M+H]+, вычислено для С30Н42N2O4 495,31; найдено 495,2.
Пример 109 соль ТФУ 3-эндо-(8-{2-[адамантан-1-илметил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С29Н41N3O3 480,31; найдено 480,2.
Пример 110 соль ТФУ N-адамантан-1-илметил-N-{2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-сукцинамовой кислоты; (m/z): [M+H]+, вычислено для С31Н43N3O4 522,33; найдено 522,2.
Пример 111 соль ТФУ 3-эндо-{8-[2-(ацетиладамантан-1-илметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида; (m/z): [M+H]+, вычислено для С29Н41N3O2 464,32; найдено 464,2.
Пример 112 соль ТФУ N-(2,6-дифторбензил)-N-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}-сукцинамовой кислоты; (m/z): [M+H]+, вычислено для С26Н30F2N2O4 473,22; найдено 473,2.
Пример 113. Синтез 3-эндо-(8-{2-[(2,6-дифторбензил)(2-гидрокси-ацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида N-(2,6-дифторбензил)-N-{2-[3-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}сукцинамовой кислоты
а. Получение 2-(2,6-дифторбензиламино)этанола
Смесь 2,6-дифторбензилбромида (3,7 г, 17,8 ммоль) и этаноламина (6,46 мл, 107 ммоль) в этаноле (18 мл) нагревали при 75°С в течение 16 часов. Реакционную смесь концентрировали и образовавшийся остаток разбавляли дихлорметаном (50 мл). Органический слой распределяли водой (75 мл) и водный слой экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали, получая при этом указанное в заголовке соединение в виде желтого твердого вещества (3,25 г). (m/z): [M+H]+, вычислено для С9Н11F2NO 188,08; найдено 188,1. 1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 7,37-7,34 (м, 1H), 7,10-7,05 (м, 2H), 4,47 (т, J=5,4 Гц, 1H), 3,75 (с, 2H), 3,42 (кв, J=5,5 Гц, 2H), 2,25 (т, J=5,7 Гц, 2H), 1,82 (ушир. с, 1H).
b. Получение трет-бутилового эфира (2,6-дифторбензил)(2-гидроксиэтил)карбаминовой кислоты
К раствору продукта, полученного на предыдущей стадии (3,25 г, 17,4 ммоль), в дихлорметане (20 мл) при 0°С в течение 5 минут шприцем по каплям добавляли раствор ди-трет-бутилдикарбоната (3,40 г, 15,6 ммоль). Полученную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи в атмосфере азота. Неочищенную реакционную смесь разбавляли DCM (50 мл) и последовательно промывали 1 н. водным раствором HCl (2×50 мл), насыщенным раствором NaHCO3 (3×50 мл) и насыщенным раствором соли (50 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали, получая при этом указанное в заголовке соединение (4,46 г). (m/z): [M+H]+, вычислено для С14Н19F2NO3 288,13; найдено 288,2.
с. Получение трет-бутилового эфира (2,6-дифторбензил)(2-оксоэтил)карбаминовой кислоты
К раствору продукта, полученного на предыдущей стадии (4,46 г, 15,5 ммоль), в DCM (50 мл) при 0°С последовательно добавляли диметилсульфоксид (1,79 г, 23 ммоль), DIPEA (5,01 г, 38,9 ммоль) и комплекс пиридина с триоксидом серы (6,20 г, 38,9 ммоль). Через 30 минут реакционную смесь последовательно промывали 1 н водным раствором HCl (3×100 мл), насыщенным раствором NaHCO3 (100 мл) и насыщенным раствором соли (100 мл), фильтровали и элюировали DCM. Реакционную смесь концентрировали, получая при этом указанное в заголовке соединение в виде желтого масла (2,31 г). 1H ЯМР (d 6-ДМСО, 300 МГц) δ (м.д.): 9,42 (с, 1H), 7,40 (м, 1H), 7,09 (м, 2H), 4,49 (с, 2H), 4,00 (д, J=24,6, 2H), 1,31 (с, 9H).
d. Получение трет-бутилового эфира {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-3-ил]этил}(2,6-дифторбензил)карбаминовой кислоты
К раствору 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида (120 мг, 0,56 ммоль) в DCM (2 мл) при 0°С добавляли раствор продукта, полученного на предыдущей стадии (193 мг, 0,68 ммоль), в DCM (1 мл) и триацетоксиборгидрид натрия (144 мг, 0,68 ммоль). Полученную смесь нагревали до комнатной температуры и оставляли для взаимодействия в течение 1 часа. Реакционную смесь разбавляли DCM, промывали насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом желтоватое масло, которое использовали на следующей стадии без дальнейшей очистки. (m/z): [M+H]+, вычислено для С28Н35F2N3O3 500,26; найдено 500,1.
е. Получение 3-эндо-{8-[2-(2,6-дифторбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
Маслянистый остаток, полученный на предыдущей стадии, растворяли в DCM (2 мл) и обрабатывали ТФУ (2 мл) при комнатной температуре в течение 2 часов. Смесь концентрировали и трижды упаривали с этилацетатом, разбавляли DCM и подщелачивали до рН 8,0 насыщенным раствором бикарбоната натрия. Слои разделяли и водный слой экстрагировали DCM. Объединенный органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом темное масло (200 мг). (m/z): [M+H]+, вычислено для С23Н27F2N3O 400,21; найдено 400,4.
f. Синтез 3-эндо-(8-{2-[(2,6-дифторбензил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору соли монотрифторуксусной кислоты 3-эндо-{8-[2-(2,6-дифторбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (132 мг, 0,26 ммоль) в DCM (1 мл) при 0°С добавляли DIPEA (132 мг, 1,0 ммоль). Затем реакционную смесь обрабатывали ацетоксиацетилхлоридом (46 мг, 0,34 ммоль) в течение 30 минут. Реакционную смесь концентрировали, неочищенное масло растворяли в этаноле (0,5 мл) и обрабатывали моногидратом гидроксида лития (66 мг, 1,65 ммоль) в воде (0,2 мл). Растворитель концентрировали, остаток растворяли в 50% растворе уксусной кислоты в воде (5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (30,8 мг). (m/z): [M+H]+, вычислено для С25Н29F2N3O3 458,22; найдено 458,2. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,025 (с, 1H), 7,75 (д, J=7,6 Гц, 1H), 7,72 (д, J=8,4 Гц, 1H), 7,49-7,45 (м, 2H), 7,103 (т, J=8,8 Гц, 2H), 4,67 (с, 2H), 4,52 (с, 2H), 4,10 (ушир. с, 2H), 3,71 (т, J=6,0 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,08 (т, J=5,6, 2H), 2,63-2,61 (м, 4H), 2,06-2,03 (м, 2H), 1,83-1,79 (м, 2H).
Примеры 114-118
Соединения по примерам 114-118 были получены способами, аналогичными способу по примеру 113(f), с использованием соответствующего хлорида.
Пример 114 соль ТФУ 3-эндо-(8-(2-((2,6-дифторбензил)(N,N-диметилсульфамоил)амино)этил)-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н32F2N4O3S 507,22; найдено 507,4.
Пример 115 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)(2-гидрокси-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н33F2N3O3 486,25; найдено 486,4.
Пример 116 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)(2-метоксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н31F2N3O3 472,23; найдено 472,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,023 (с, 1H), 7,75 (д, J=8,0 Гц, 1H), 7,72 (д, J=8,8 Гц, 1H) 7,49-7,45 (м, 2H), 7,103 (т, J=8,4 Гц, 2H), 4,72 (с, 2H), 4,45 (с, 2H), 4,08 (ушир. с, 2H), 3,71 (т, J=6,0 Гц, 2H), 3,48 (с, 3H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,05 (т, J=5,6, 2H), 2,62-2,59 (м, 4H), 2,05-2,02 (м, 2H), 1,81-1,79 (м, 2H).
Пример 117 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)(2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С28Н35F2N3O2 484,27; найдено 484,4.
Пример 118 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)метансульфониламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н29F2N3O2S 478,19; найдено 478,2. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,00 (с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,70 (д, J=8,4 Гц, 1H) 7,49-7,45 (м, 2H), 7,10 (т, J=8,4 Гц, 2H), 4,67 (с, 2H), 4,10 (ушир. с, 2H), 3,65 (т, J=6,0 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,10 (т, J=5,6 Гц, 2H), 2,99 (с, 3H), 2,62-2,59 (м, 4H), 2,04-2,02 (м, 2H), 1,83-1,81 (м, 2H).
Пример 119. Синтез 3-эндо-(8-{2-[(2,6-дифторбензил)(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору метансульфонилуксусной кислоты (80,8 мг, 0,58 ммоль) в ДМФА (1,0 мл) добавляли HATU (220 мг, 0,58 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и обрабатывали солью монотрифторуксусной кислоты 3-эндо-{8-[2-(2,6-дифторбензиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (150 мг, 0,29 ммоль) и DIPEA (74,8 мг, 0,58 ммоль) при 45°С в течение 16 часов. Растворитель концентрировали и остаток растворяли в 50% растворе уксусной кислоты в воде (5 мл), фильтровали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (52,4 мг). (m/z): [M+H]+, вычислено для С26Н31F2N3O4S 520,20; найдено 520,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,01 (с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,70 (д, J=8,0 Гц, 1H) 7,51-7,45 (м, 2H), 7,11 (т, J=8,4 Гц, 2H), 4,89 (с, 2H), 4,68 (с, 2H), 4,10 (ушир. с, 2H), 3,78 (т, J=6,0 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем, 3,26 (с, 3H), 3,15 (т, J=5,6 Гц, 2H), 2,63-2,60 (м, 4H), 2,09-2,06 (м, 2H), 1,83-1,80 (м, 2H).
Примеры 120-129
Соединения по примерам 120-129 были получены способами, аналогичными способу по примеру 119, с использованием соответствующей карбоновой кислоты или карбоксилата.
Пример 120 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)((S)-2-гидрокси-3-фенилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С32Н35F2N3O3 548,26; найдено 548,4.
Пример 121 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)((S)-2-гидрокси-4-метилпентаноил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С29Н37F2N3O3 514,28; найдено 514,4.
Пример 122 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н31F2N3O4 488,23; найдено 488,4.
Пример 123 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н31F2N3O4 488,23; найдено 488,2.
Пример 124 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)((S)-2-гидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н31F2N3O3 472,23; найдено 472,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,02 (с, 1H), 7,77-7,70 (м, 2H), 7,50-7,45 (м, 2H), 7,10 (т, J=8,0 Гц, 2H), 4,91 (с, 2H), 4,08-4,00 (м, 2H), 3,87-3,83 (м, 1H), 3,61-3,60 (м, 1H), 3,01 (т, J=6,0 Гц, 2H), 2,63-2,61 (м, 4H), 2,04-1,99 (м, 2H), 1,81-1,79 (м, 2H), 1,40 (д, J=6,4 Гц, 3H).
Пример 125 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)(3-гидрокси-2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С28Н35F2N3O3 500,26; найдено 500,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,01 (с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,70 (д, J=7,6 Гц, 1H), 7,49 (м, 2H), 7,09 (т, J=8,4 Гц, 2H), 5,02 (с, 2H), 4,04 (ушир. с, 2H), 3,78 (с, 2H), 3,66 (т, J=6,4 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 2,98 (т, J=6,0 Гц, 2H), 2,66-2,54 (м, 4H), 2,02-1,99 (м, 2H), 1,80-1,76 (м, 2H), 1,41 (с, 6H). Реагент 3-гидрокси-2,2-диметилпропионовый карбоксилат лития, полученный путем обработки метилового эфира 3-гидрокси-2,2-диметилпропионовой кислоты (5,0 г, 37,7 ммоль) в метаноле (45 мл) моногидратом гидроксида лития (1,6 г, 37,8 ммоль).
Пример 126 соль ТФУ 3-эндо-(8-{2-[(2-цианоацетил)(2,6-дифторбензил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н28F2N4O2 467,22; найдено 467,2. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,02 (с, 1H), 7,76 (д, J=8,0 Гц, 1H), 7,72 (д, J=7,6 Гц, 1H), 7,50-7,48 (м, 2H), 7,12 (т, J=8,4 Гц, 2H), 4,72 (с, 2H), 4,11 (ушир. с, 2H), 3,70 (т, J=6,4 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,09 (т, J=6 Гц, 2H), 2,65-2,59 (м, 4H), 2,06-2,05 (м, 2H), 1,82-1,79 (м, 2H).
Пример 127 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)(1-гидроксициклопропанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н31F2N3O3 484,23; найдено 484,4.
Пример 128 соль ТФУ 3-эндо-(8-{2-[(2-трет-бутоксиацетил)(2,6-дифторбензил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С29Н37F2N3O3 514,28; найдено 514,4.
Пример 129 соль ТФУ 3-эндо-(8-{2-[(2,6-дифторбензил)(транс-4-гидроксициклогексанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С30Н37F2N3O3 526,28; найдено 526,4.
Пример 130. Синтез 3-эндо-(8-{3-[циклогексилметил(2-гидроксиацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение 3-эндо-[8-(гидроксипропил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
Смесь 3-(8-азабицикло[3.2.1]окт-3-ил)бензамида (326,7 мг, 1,42 ммоль) и 3-бром-1-пропанола (217,1 мг, 1,56 ммоль) в EtOН (1,0 мл) нагревали при 70°С в течение одного часа. Реакционную смесь концентрировали, остаток трижды упаривали с DCM и сушили в вакууме, получая при этом указанное в заголовке соединение в виде светло-желтой пены. (m/z): [M+H]+, вычислено для С17Н24N2O2 289,2; найдено 289,0.
b. Получение 3-эндо-[3-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]пропилового эфира метансульфоновой кислоты
Продукт, полученный на предыдущей стадии, растворяли в DCM (7,0 мл). К полученному раствору добавляли DIPEA (367,1 мг, 2,84 ммоль), метансульфонилхлорид (275,2 мг, 2,41 ммоль) и DMAP (24,2 мг, 0,20 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов и выдерживали при 4°С в течение ночи. Реакционную смесь концентрировали, маслянистый остаток сушили в вакууме, получая при этом масло оранжевого цвета, которое использовали на следующей стадии без очистки.
с. Получение 3-эндо-{8-[2-(циклогексилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
Половину неочищенного продукта, полученного на предыдущей стадии, растворяли в ДМФА (0,8 мл) и обрабатывали DIPEA (183,2 мг, 1,42 ммоль) и циклогексилметиламином (200,6 мг, 1,77 ммоль) при 75°С в течение 1 часа. Реакционную смесь концентрировали, остаток растворяли в 50% растворе уксусной кислоты в воде (8,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли бистрифторуксусной кислоты (103,9 мг). (m/z): [M+H]+, вычислено для С24Н37N3O 384,3; найдено 384,4.
d. Синтез 3-эндо-(8-{3-[циклогексилметил(2-гидроксиацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (100,0 мг, 0,164 ммоль), в DCM (0,6 мл) добавляли DIPEA (84,4 мг, 0,65 ммоль) и ацетоксиацетилхлорид (24,5 мг, 0,18 ммоль). Через пять минут реакционную смесь концентрировали. Остаток повторно растворяли в метаноле (0,6 мл) и обрабатывали моногидратом гидроксида лития (41,32 мг, 0,984 ммоль) в воде (0,6 мл) в течение тридцати минут. Реакционную смесь концентрировали, образовавшийся остаток растворяли в смеси 50% раствора уксусной кислоты в воде (6,0 мл) и воды (2,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли ТФУ (22,1 мг). (m/z): [M+H]+, вычислено для С26Н39N3O3 442,30; найдено 442,6. 1H ЯМР (CD3OD, 400M Гц) δ (м.д.): 8,08 (с, 1H), 7,76-7,80 (м, 2H), 7,48-7,55 (м, 6H), 4,28 (с, 2H), 4,08 (ушир.с, 2H), 3,36-3,38 (темный 1H, частичное перекрывание растворителем), 3,20 (т, J=7,2 Гц, 2H), 3,12 (т, J=8,4 Гц, 2H), 2,62-2,78 (м, 4H), 2,21-2,29 (м, 2H), 2,09-2,12 (м, 2H), 1,78-1,84 (м, 2H).
Пример 131. Синтез 3-эндо-{8-[3-(ацетилциклогексилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}бензамида
К раствору соли бистрифторуксусной кислоты 3-эндо-{8-[3-(циклогексилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (30,0 мг, 0,05 ммоль) в DCM (0,2 мл) добавляли DIPEA (25,8 мг, 0,20 ммоль) и ангидрид уксусной кислоты (7,65 мг, 0,075 ммоль). Через тридцать минут реакционную смесь концентрировали. Остаток растворяли в этаноле (0,4 мл) и обрабатывали моногидратом гидроксида лития (12,6 мг, 0,30 ммоль) в воде (0,4 мл) в течение тридцати минут. Реакционную смесь концентрировали, образовавшийся остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли ТФУ (26,7 мг). (m/z): [M+H]+, вычислено для С26Н39N3O2 426,30; найдено 426,2.
Пример 132. Синтез 3-эндо-(8-{3-[циклогексилметил(2-метансульфонилацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору метансульфонилуксусной кислоты (20,7 мг, 0,15 ммоль) в ДМФА (0,2 мл) добавляли N,N-карбонилдиимидазол (24,3 мг, 0,15 ммоль) при комнатной температуре. Через тридцать минут к перемешиваемой смеси добавляли DIPEA (25,8 мг, 0,20 ммоль) и соль бистрифторуксусной кислоты 3-эндо-{8-[3-(циклогексилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}бензамида (30,0 мг, 0,05 ммоль). Полученную реакционную смесь нагревали при 65°С в течение 3 часов, концентрировали, растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли ТФУ (16,0 мг). (m/z): [M+H]+, вычислено для С27Н41N3O4S 504,28; найдено 504,2.
Пример 133. Синтез 3-эндо-(8-{3-[бензил(2-гидроксиацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
В соответствии со способом по примеру 130 соль бистрифторуксусной кислоты 3-эндо-[8-(3-бензиламинопропил)-8-азабицикло[3.2.1]окт-3-ил]бензамида (100 мг, 0,165 ммоль) подвергали взаимодействию с ацетоксиацетилхлоридом (24,5 мг, 0,18 ммоль), получая при этом соль ТФУ указанного в заголовке соединения (22,6 мг). 1H ЯМР (CD3OD, 400M Гц) δ (м.д.): 8,08 (с, 1H), 7,77-7,81 (м, 2H), 7,51 (т, J=7,6, 8,0 Гц, 1H), 7,31-7,46 (м, 5H). (m/z): [M+H]+, вычислено для С26Н33N3O3 436,25; найдено 436,4.
Пример 134. Синтез 3-эндо-(8-{3-[бензил-((S)-2-гидроксипропионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение трет-бутилового эфира бензил(3-гидроксипропил)карбаминовой кислоты
К смеси хлористоводородной соли 3-бензиламино-1-пропанола (2,0 г, 9,9 ммоль) в DCM (49,0 мл) при 0°С добавляли DIPEA (1,72 мл, 9,9 ммоль) и раствор ди-трет-бутилдикарбоната (1,94 г, 8,91 ммоль) в DCM (20,0 мл). Реакционную смесь перемешивали в течение 3 часов при 0°С, последовательно промывали 1 н. раствором HCl (2×10,0 мл), насыщенным раствором бикарбоната натрия (10,0 мл) и насыщенным раствором соли (10,0 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая при этом желтоватое масло (2,22 г). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 7,20-7,39 (м, 5H), 4,40 (с, 2H), 3,55 (ушир.с, 2H), 3,38 (ушир.с, 2H), 1,61 (ушир.с, 2H), 1,44 (с, 9H).
b. Получение трет-бутилового эфира бензил(3-оксопропил)карбаминовой кислоты
К раствору продукта, полученного на предыдущей стадии (2,22 г, 7,38 ммоль), в DCM (38,0 мл) при 0°С добавляли DIPEA (2,38 г, 18,45 ммоль), ДМСО (0,86 г, 11,07 ммоль) и комплекс пиридина с триоксидом серы (2,93 г, 18,45 ммоль). Реакционную смесь перемешивали в течение 30 минут при 0°С, разбавляли DCM (20,0 мл) и последовательно промывали 1 н. раствором HCl (10,0 мл), насыщенным раствором бикарбоната натрия (10,0 мл) и насыщенным раствором соли (10,0 мл), сушили над сульфатом натрия, фильтровали и концентрировали с образованием желтоватого масла, которое очищали хроматографией с нормальной фазой, получая при этом указанное в заголовке соединение в виде бесцветного масла (1,54 г). 1H ЯМР (CDCl3, 300M Гц) δ (м.д.): 9,73 (с, 1H), 7,24-7,36 (м, 5H), 4,45 (с, 2H), 3,51 (ушир.с, 2H), 2,65 (ушир.с, 2H), 1,46 (с, 9H).
с. Получение 3-эндо-[8-(3-бензиламинопропил)-8-азабицикло[3.2.1]окт-3-ил]бензамида
К раствору продукта, полученного на предыдущей стадии (972,6 мг, 3,25 ммоль), в DCM (16,0 мл) при комнатной температуре добавляли 3-(8-азабицикло[3.2.1]окт-3-ил)бензамид (747,5 мг, 3,25 ммоль) и триацетоксиборгидрид натрия (826,6 мг, 3,9 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и разбавляли DCM (40,0 мл). Органический слой промывали насыщенным раствором бикарбоната натрия (20,0 мл) и насыщенным раствором соли (20,0 мл), сушили над сульфатом натрия, фильтровали и концентрировали с образованием желтоватого масла. Маслянистый остаток обрабатывали смесью ТФУ (5,0 мл) и DCM (5,0 мл) при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали, образовавшийся остаток растворяли в смеси воды (4,0 мл) и ацетонитрила (3,0 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли бистрифторуксусной кислоты (1,22 г). 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,06 (с, 1H), 7,74-7,78 (м, 2H), 7,46-7,53 (м, 6H), 4,27 (с, 2H), 4,08 (ушир. с, 2H), 3,35-3,38 (темный 1H, частичное перекрывание растворителем), 3,20 (т, J=8,0 Гц, 2H), 3,12 (т, J=8,4 Гц, 2H), 2,62-2,78 (м, 4H), 2,21-2,29 (м, 2H), 2,09-2,12 (м, 2H), 1,79-1,84 (м, 2H). (m/z): [M+H]+, вычислено для С24Н31N3O 378,25; найдено 378,4.
d. Синтез 3-эндо-(8-{3-[бензил((S)-2-гидроксипропионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (30,0 мг, 0,06 ммоль), в DCM (0,5 мл) добавляли DIPEA (32,4 мг, 0,24 ммоль) и ацетоксиацетилхлорид (9,8 мг, 0,072 ммоль) при комнатной температуре. Реакция сразу же заканчивалась. Реакционную смесь концентрировали, образовавшийся остаток повторно растворяли в метаноле (0,5 мл) и обрабатывали моногидратом гидроксида лития (15,1 мг, 0,36 ммоль) в течение ночи. Реакционную смесь концентрировали и остаток растворяли в 50% растворе уксусной кислоты в воде (1,5 мл), фильтровали и очищали препаративной ВЭЖХ с обращенной фазой, получая при этом указанное в заголовке соединение в виде его соли ТФУ (23,4 мг). (m/z): [M+H]+, вычислено для С27Н35N3O3 450,27; найдено 450,4.
Примеры 135-199
Приведенные ниже дополнительные соединения были получены способами, описанными в вышеуказанных примерах, в результате осуществления взаимодействия соответствующего промежуточного соединения формулы (II) с приемлемым хлорангидридом кислоты, карбоновой кислотой или карбоксилатом, как показано на схеме А(i), с последующим выполнением в конкретных случаях стадии N-алкилирования. Промежуточные соединения формулы (II) были получены в результате осуществления взаимодействия альдегида формулы (IV) с азабициклооктилбензамидом способом по примеру 134(с), как показано на схеме В. Альдегиды формулы (IV) были получены общим способом, показанным на схеме F, как описано, например, в примере 134(в) и (b).
Пример 135 соль ТФУ 3-эндо-(8-{2-[((S)-2,3-дигидроксипропионил)фенетиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O4 466,27; найдено 466,4.
Пример 136 соль ТФУ 3-эндо-(8-{2-[(2-метансульфонилацетил)-фенетиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O4S 498,23; найдено 498,4.
Пример 137 соль ТФУ (бензил-{2-[3-эндо-(3-гидроксифенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбамоил)метилового эфира уксусной кислоты; (m/z): [M+H]+, вычислено для С26Н32N2O4 437,24; найдено 437,4.
Пример 138 соль ТФУ 3-эндо-(8-{2-[бензил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,05 (с, 1H), 7,79 (д, J=7,6 Гц, 1H), 7,74 (д, J=8,0 Гц, 1H), 7,37-7,52 (м, 6H), 4,90-4,95 (м, 1H), 4,77-4,83 (м, 1H), 4,72 (т, J=5,2 Гц, 1H), 4,10 (ушир.с, 2H), 3,85-3,91 (м, 3H), 3,63-3,69 (м, 1H), 3,31-3,34 (темный 1H, частичное перекрывание растворителем), 3,09-3,12 (м, 2H), 2,64 (ушир.с, 4H), 2,04-2,07 (м, 2H), 1,80-1,83 (м, 2H); (m/z): [M+H]+, вычислено для С26Н33N3O4 452,25; найдено 452,2.
Пример 139 соль ТФУ 3-эндо-(8-{3-[бензил((S)-2,3-дигидроксипрпионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O4 466,26; найдено 466,4.
Пример 140 соль ТФУ 3-эндо-(8-{3-[бензил((R)-2,3-дигидроксипропионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O4 466,26; найдено 466,4.
Пример 141 соль ТФУ 3-эндо-(8-{3-[бензил(2,3-дигидроксибутирил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С28Н37N3O4 480,28; найдено 480,4.
Пример 142 соль ТФУ 3-эндо-(8-{3-[бензил(2-метансульфонилацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,04 (с, 1H), 7,72-7,77 (м, 2H), 7,48 (т, J=7,6 Гц, 1H), 7,31-7,37 (м, 5H), 4,79 (с, 2H), 4,49 (с, 2H), 3,99 и 3,92 (две серии ушир.с, 2H), 3,61 (т, J=6,4 Гц, 2H), 3,34-3,38 (м, 1H), 3,22 (с, 3H), 2,98 (т, J=8,0 Гц, 2H), 2,53-2,72 (м, 4H), 1,96-2,11 (м, 4H), 1,74-1,80 (м, 2H). (m/z): [M+H]+, вычислено для С27Н35N3O4S 498,23; найдено 498,2.
Пример 143 соль ТФУ 3-эндо-(8-{2-[бензил(2-гидрокси-2-метилпропионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С28Н37N3O3 464,28; найдено 464,4.
Пример 144 соль ТФУ 3-эндо-(8-{3-[бензил(2,2-диметилпропионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С29Н39N3O2 462,30; найдено 462,4.
Пример 145 соль ТФУ 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)-(3-гидрокси-2-гидроксиметил-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С28Н41F2N3O4 522,31; найдено 522,2.
Пример 146 соль ТФУ 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н37F2N3O4 494,28; найдено 494,4.
Пример 147 соль ТФУ 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)-((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н37F2N3O4 494,28; найдено 494,4.
Пример 148 соль ТФУ 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)-(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,06 (с, 1H), 7,79 (д, J=7,6 Гц, 1H), 7,75 (д, J=8,8 Гц, 1H), 7,51 (дд, J=7,6, 8,0 Гц, 1H), 4,51 (с, 2H), 4,18 (деформированный м, 2H), 3,89 (т, J=6,0 Гц, 2H), 3,48 (д, J=7,2 Гц, 2H), 3,28 (с, 3H), 3,24 (т, J=6,0 Гц, 2H), 2,65-2,68 (м, 4H), 2,10-2,19 (м, 4H), 1,75-1,90 (м, 7H), 1,36-1,45 (м, 2H); (m/z): [M+H]+, вычислено для С26Н37F2N3O4S 526,25; найдено 526,4.
Пример 149 соль бистрифторуксусной кислоты 3-эндо-(8-{2-[(4,4-дифторциклогексилметил)((S)-4-диметиламино-2-гидроксибутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С29Н44F2N4O3 535,34; найдено 535,4.
Пример 150 соль ТФУ 3-эндо-(8-2-[(4,4-дифторциклогексилметил)-((S)-2-гидроксипропионил)амино]этил-8-азабицикло[3.2.1]окт-3-ил)бензамида; 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,05 (с, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,74 (д, J=7,6 Гц, 1H), 7,49 (дд, J=7,6 Гц, 1H), 4,64 (кв., J=6,0 Гц, 1H), 4,10-4,14 (м, 2H), 3,72-3,86 (м, 1H), 3,32-3,46 (темный м, 3H, частичное перекрывание растворителем), 3,18 (т, J=5,6 Гц, 2H), 2,64-2,67 (м, 4H), 2,11-2,16 (м, 4H), 1,78-1,88 (м, 7H), 1,37-1,43 (м, 5H); (m/z): [M+H]+, вычислено для С26Н37F2N3O3 478,28; найдено 478,4.
Пример 151 соль ТФУ 3-эндо-(8-{2-[(4-фторциклогексилметил)((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н38FN3O4 476,28; найдено 476,4.
Пример 152 соль ТФУ 3-эндо-(8-{2-[(4-фторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н36FN3O3 446,27; найдено 446,4.
Пример 153 соль ТФУ 3-эндо-(8-{2-[циклопентилметил(3-гидрокси-2-гидроксиметил-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O4 472,31; найдено 472,4.
Пример 154 соль ТФУ 3-эндо-(8-{2-[циклопентилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н37N3O4 444,28; найдено 444,4.
Пример 155 соль ТФУ 3-эндо-(8-{2-[циклопентилметил((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н37N3O4 444,28; найдено 444,4.
Пример 156 соль ТФУ 3-эндо-(8-{2-[циклопентилметил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н37N3O4S 476,25; найдено 476,2.
Пример 157 3-эндо-(8-{2-[циклопентилметил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н35N3O3 414,27; найдено 414,4.
Пример 158 соль ТФУ 3-эндо-(8-{2-[циклобутилметил(3-гидрокси-2-гидроксиметил-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н39N3O4 458,29; найдено 458,4.
Пример 159 соль ТФУ 3-эндо-(8-{2-[циклобутилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н35N3O4 430,26; найдено 430,4.
Пример 160 соль ТФУ 3-эндо-(8-{2-[циклобутилметил((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н35N3O4 430,26; найдено 430,4.
Пример 161 соль ТФУ 3-эндо-(8-{2-[циклобутилметил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н35N3O4S 462,23; найдено 462,2.
Пример 162 соль ТФУ 3-эндо-(8-{2-[циклобутилметил(2-метансульфонил-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н39N3O4S 490,27; найдено 490,2.
Пример 163 соль ТФУ 3-эндо-(8-{2-[циклобутилметил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С23Н33N3O3 400,25; найдено 400,4.
Пример 164 соль ТФУ 3-эндо-(8-{2-[циклобутилметил((S)-2-гидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н35N3O3 414,27; найдено 414,4.
Пример 165 соль ТФУ 3-эндо-(8-{2-[бензил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н33N3O4S 484,22; найдено 484,2. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,01 (с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,70 (д, J=8,0 Гц, 1H), 7,49-7,35 (м, 6H), 4,80 (с, 2H), 4,57 (с, 2H) 4,07 (ушир. с, 2H), 3,80 (т, J=6,0 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,25 (с, 3H), 3,08 (т, J=6 Гц, 2H), 2,61-2,60 (м, 4H), 2,03-2,00 (м, 2H), 1,82-1,79 (м, 2H).
Пример 166 соль ТФУ 3-эндо-(8-{2-[бензил(2-гидрокси-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O3 450,27; найдено 450,2.
Пример 167 соль ТФУ 3-эндо-(8-{2-[бензил((S)-2-гидрокси-1-оксопропил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н33N3O3 436,25; найдено 436,5. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,02 (с, 1H), 7,77-7,70 (м, 2H), 7,50-7,32 (м, 6H), 4,83-4,71 (м, 2H), 4,07-4,01 (м, 2H), 3,81 (м, 1H), 3,64 (м, 1H), 3,03 (т, J=6,0 Гц, 2H), 2,63-2,60 (м, 4H), 2,03-1,94 (м, 2H), 1,80-1,77 (м, 2H), 1,40 (д, J=6,4 Гц, 3H).
Пример 168 соль ТФУ 3-эндо-(8-{2-[бензил(2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил}бензамида; (m/z): [M+H]+, вычислено для С28Н37N3O2 448,29; найдено 448.
Пример 169 соль ТФУ 3-эндо-{8-[2-(бензилметансульфониламино)этил]-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С24Н31N3O3S 442,21; найдено 442,2.
Пример 170 соль ТФУ 3-эндо-(8-{2-[бензил(2-метоксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н33N3O3 436,25; найдено 436,5.
Пример 171 соль ТФУ 3-эндо-(8-{2-[бензил(3-гидрокси-2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С28Н37N3O3 464,28; найдено 464,2.
Пример 172 соль ТФУ 3-эндо-(8-{2-[бензил(1-гидроксициклопропанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н33N3O3 448,25; найдено 448,3.
Пример 173 соль ТФУ 3-эндо-(8-{2-[бензил(2-цианоацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н30N4O2 431,24; найдено 431,5.
Пример 174 соль ТФУ 3-эндо-(8-{2-[бензил((R)-3-гидрокси-2-(S)-гидроксибутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O4 466,26; найдено 466,4.
Пример 175 соль ТФУ 3-эндо-(8-{2-[бензил((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н33N3O4 452,25; найдено 452,2.
Пример 176 соль ТФУ 3-эндо-(8-{2-[циклопентилметил(3-гидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н37N3O3 428,28; найдено 428,4.
Пример 177 соль ТФУ 3-эндо-(8-{2-[циклопентилметил(3-гидрокси-2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O3 456,31; найдено 456,5.
Пример 178 соль ТФУ 3-эндо-(8-{2-[циклопентилметил(транс-4-гидроксициклогексанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С29Н43N3O3 482,33; найдено 482,5.
Пример 179 соль ТФУ 3-эндо-(8-{2-[циклопентилметил(2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O 440,32; найдено 440,4.
Пример 180 соль ТФУ 3-эндо-{8-[2-(циклопентилметилметансульфониламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамида; (m/z): [M+H]+, вычислено для С23Н35N3O3S 434,24; найдено 434,2.
Пример 181 соль ТФУ 3-эндо-(8-{2-[(2-гидроксиацетил)(4-трифторметилбензил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н30F3N3O3 490,22; найдено 490,4.
Пример 182 соль ТФУ 3-эндо-(8-(2-((4-трифторметилбензил)(N,N-диметилсульфамоил)аминоэтил)-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н33F3N4O3S 539,22; найдено 539,5.
Пример 183 соль ТФУ 3-эндо-(8-{2-[(2-метансульфонилацетил)(4-трифторметилбензил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н32F3N3O4S 552,21; найдено 552,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,02 (с, 1H), 7,77-7,70 (м, 4H), 7,56-7,54 (м, 3H)) 4,94 (с, 2H), 4,56 (с, 2H), 4,12 (ушир. с, 2H), 3,80 (т, J=5,6 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,26 (с, 3H), 3,17 (т, J=5,6 Гц, 2H), 2,63-2,61 (м, 4H), 2,10-2,06 (м, 2H), 1,84-1,81 (м, 2H).
Пример 184 соль ТФУ 3-эндо-(8-{2-[(4-фторбензил)(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н32FN3O4S 502,21; найдено 502,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,01 (с, 1H), 7,77-7,68 (м, 2H), 7,46 (т, J= 8,0 Гц, 1H), 7,40-7,36 (м, 2H), 7,15 (т, J=8,8 Гц, 2H), 4,79 (с, 2H) 4,10 (ушир. с, 2H), 3,78 (т, J=6,0 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,25 (с, 3H), 3,11 (т, J=5,6 Гц, 2H), 2,61-2,60 (м, 4H), 2,06-2,03 (м, 2H), 1,80-1,78 (м, 2H).
Пример 185 соль ТФУ 3-эндо-(8-{2-[(4-фторбензил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н30FN3O3 440,23; найдено 440,4.
Пример 186 соль ТФУ 3-эндо-(8-(2-((4-фторметилбензил)(N,N-диметилсульфамоил)амино)этил)-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н33FN4O3S 489,23; найдено 489,5.
Пример 187 соль ТФУ 3-эндо-(8-{2-[[2-(3-фторфенил)этил](2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н32FN3O3 454,24; найдено 454,2.
Пример 188 соль ТФУ 3-эндо-(8-{2-[[2-(4-фторфенил)этил](2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н32FN3O3 454,24; найдено 454,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,03 (с, 1H), 7,77-7,71 (м, 2H), 7,47 (т, J= 8,0 Гц, 1H), 7,30-7,26 (м, 2H), 7,08-7,04 (м, 2H), 4,13 (ушир. с, 2H) 4,08 (с, 2H), 3,78 (т, J=5,2 Гц, 2H), 3,52 (т, J=7,2 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,17 (т, J=5,6 Гц, 2H), 2,91 (т, J=7,6 Гц, 2H), 2,65-2,61 (м, 4H), 2,14-2,11 (м, 2H), 1,85-1,82 (м, 2H).
Пример 189 соль ТФУ 3-эндо-(8-{2-[[2-(4-фторфенил)этил](2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н34F2N3O4S 516,23; найдено 516,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,01 (с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,70 (д, J=8,0 Гц, 1H) 7,49-7,45 (м, 1H), 7,32 (м, 2H), 7,08 (м, 2H), 4,21 (с, 2H), 4,11 (ушир. с, 2H), 3,79-3,73 (м, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,19 (с, 3H), 3,16 (т, J=5,6 Гц, 2H), 2,97 (т, J=7,2 Гц, 2H), 2,63-2,60 (м, 4H), 2,12-2,09 (м, 2H), 1,83-1,80 (м, 2H).
Пример 190 соль ТФУ 3-эндо-[8-(2-{(2-гидроксиацетил)[2-[(4-гидроксифенил)этил]амино}этил)-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н33N3O4 452,25; найдено 452,2.
Пример 191 соль ТФУ 3-эндо-(8-{2-[[2-(4-гидроксифенил)этил](2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н35N3O5S 514,23; найдено 514,3.
Пример 192 соль ТФУ 3-эндо-(8-{2-[(3-фторбензил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С25Н30FN3O3 440,23; найдено 440,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,03 (с, 1H), 7,77-7,71 (м, 2H), 7,49-7,43 (м, 2H), 7,13-7,06 (м, 3H), 4,62 (с, 2H), 4,37 (с, 2H) 4,12 (ушир. с, 2H), 3,75 (т, J=5,6 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,12 (т, J=5,6 Гц, 2H), 2,64-2,62 (м, 4H), 2,09-2,06 (м, 2H), 1,84-1,81 (м, 2H).
Пример 193 соль ТФУ 3-эндо-(8-{2-[(3-фторбензил)(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н32FN3O4S 502,21; найдено 502,4. 1H ЯМР (CD3OD, 400 МГц) δ (м.д.): 8,02 (с, 1H), 7,77-7,70 (м, 2H), 7,49-7,45 (м, 2H), 7,19-7,11 (м, 3H), 4,8-4,9 (темный, 2H, перекрывание растворителем), 4,56 (с, 2H) 4,10 (ушир. с, 2H), 3,80 (т, J=5,6 Гц, 2H), 3,4-3,3 (темный, 1H, перекрывание растворителем), 3,25 (с, 3H), 3,13 (т, J=6,0 Гц, 2H), 2,63-2,60 (м, 4H), 2,08-2,05 (м, 2H), 1,83-1,80 (м, 2H).
Пример 194 соль ТФУ 3-эндо-(8-{2-[(2,2-дифтор-2-фенилэтил)((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н33F2N3O4 502,24; найдено 502,4.
Пример 195 соль ТФУ 3-эндо-(8-{2-[(2-метансульфонилацетил)(4-метилциклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O4S 504,28; найдено 504,4.
Пример 196 соль ТФУ 3-эндо-(8-{2-[((S)-2,3-дигидроксипропионил)-(4-метилциклогексилметил)амино]этил}-8-азабицикло[3.2.1]-окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н41N3O4 472,31; найдено 472,4.
Пример 197 соль ТФУ 3-эндо-(8-{2-[(2-гидроксиацетил)(4-трифторметилциклогексилметил)амино]этил}-8-азабицикло[3.2.1]-окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С26Н36F3N3O3 496,27; найдено 496,4.
Пример 198 соль ТФУ 3-эндо-(8-{2-[(2-метансульфонилацетил)(4-трифторметилциклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н38F3N3O4S 558,25; найдено 558,4.
Пример 199 соль ТФУ 3-эндо-8-{2-[((S)-2,3-дигидроксипропионил)-(4-трифторметилциклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида; (m/z): [M+H]+, вычислено для С27Н38F3N3O4 526,28; найдено 526,4.
Пример 200. Синтез 3-эндо-(8-{2-[циклогексилметил((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение (R)-2,2-диметил-1,3-диоксолан-4-карбоксилата лития
К раствору метилового эфира (R)-2,2-диметил-1,3-диоксолан-4-карбоновой кислоты (5,0 г, 31,25 ммоль) в МеОН (32,0 мл) при комнатной температуре добавляли раствор моногидрата гидроксида лития (1,31 г, 31,25 ммоль) в воде (10,0 мл). Полученную смесь перемешивали в течение тридцати минут и затем концентрировали. Образовавшееся белое твердое вещество сушили в вакууме, получая при этом указанное в заголовке соединение (4,59 г).
b. Получение {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил)этил}циклогексилметиламида (R)-2,2-диметил{1,3]диоксолан-4-карбоновой кислоты
3-эндо-{8-[2-(Циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензамид (600 мг, 1,6 ммоль) растворяли в ДМФА (5 мл) и добавляли (R)-2,2-диметил[1,3]диоксолан-4-карбоксилат лития (270 мг, 1,78 ммоль) в виде твердого вещества. Раствор перемешивали при комнатной температуре до растворения твердого вещества и добавляли HATU (687 мг, 1,78 ммоль) в виде твердого вещества. Ярко-желтый раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом (100 мл). Органический раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Органический раствор сушили над безводным сульфатом натрия и растворитель удаляли в вакууме, получая при этом указанное в заголовке соединение (781 мг) в виде неочищенного желтого масла.
с. Синтез 3-эндо-(8-{2-[циклогексилметил((R)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Неочищенный продукт, полученный на предыдущей стадии, растворяли в ацетонитриле (10 мл) и добавляли 1 н. раствор HCl (водный) (10 мл). Желтый раствор перемешивали при комнатной температуре в течение 2 часов, затем реакционную смесь концентрировали в вакууме. Неочищенное вещество растворяли в ацетонитриле/воде/ТФУ и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения в виде белого порошка (386 мг, чистота 99,4%). (m/z): [M+H]+, вычислено для С26Н39N3O4 458,30; найдено 458,4.
Пример 201. Синтез 3-эндо-(8-{2-[(2-гидроксиацетил)(2,2,3,3-тетраметилциклопропилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение (2,2,3,3-тетраметилциклопропил)метанола
К перемешиваемому раствору 2,2,3,3-тетраметилциклопентанкарбоновой кислоты (500 мг, 3,5 ммоль) в ТГФ (25 мл) добавляли 2,0 М раствор комплекса диметилсульфида борана в ТГФ (1,8 мл, 3,5 ммоль) при 0°С. Реакционную смесь нагревали и перемешивали при 50°С в течение 3 часов. Раствор охлаждали до комнатной температуры и осторожно добавляли метанол (10 мл). Реакционную смесь концентрировали и фильтровали. Фильтрат концентрировали, получая при этом указанное в заголовке промежуточное соединение в виде масла (250 мг, 56%). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 4,16 (т, J=4,8 Гц, 1H), 3,39 (дд, J=4,5, 7,5 Гц, 2H), 1,03 (с, 6H), 0,93 (с, 6H), 0,38 (т, J=7,5 Гц, 1H).
b. Получение 2,2,3,3-тетраметилциклопропанкарбальдегида
К перемешиваемому раствору продукта, полученного на предыдущей стадии, и DIPEA (680 мкл, 4,0 ммоль) в DCM (5 мл) добавляли раствор комплекса пиридина с триоксидом серы (620 мг, 3,9 ммоль) в ДМСО (5 мл) при -20°С. Через 2 часа реакционную смесь нагревали до комнатной температуры, разбавляли DCM (20 мл) и промывали 1,0 н. раствором HCl (25 мл) и водой (25 мл). Органическую часть отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество очищали хроматографией на силикагеле, получая при этом указанное в заголовке промежуточное соединение (45 мг, 18%).
с. Получение бензилового эфира (2-гидроксиэтил)карбаминовой кислоты
К перемешиваемому раствору этаноламина (4,0 г, 66 ммоль) в DCM (4 мл) добавляли бензилхлорформиат (4,6 мл, 33 ммоль) при 0°С. Через 1 час реакционную смесь нагревали до комнатной температуры и промывали 1,0 н. раствором HCl (40 мл) и водой (40 мл). Органическую часть отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. К раствору неочищенного продукта в этилацетате (30 мл) добавляли гексаны (30 мл). Образовавшиеся кристаллы фильтровали и сушили, получая при этом указанное в заголовке промежуточное соединение в виде белого твердого вещества (4,5 г, 70%). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 7,4-7,29 (м, 5H), 5,00 (с, 2H), 4,64 (т, J=5,5 Гц, 1H), 3,38 (кв., J=6,0 Гц, 2H), 3,07 (т, J=6,0 Гц, 2H).
d. Получение бензилового эфира (2-оксоэтил)карбаминовой кислоты
К перемешиваемому раствору бензилового эфира (2-гидроксиэтил)карбаминовой кислоты (1,0 г, 5,1 ммоль) и DIPEA (1,78 мл, 10,2 ммоль) в DCM (15 мл) добавляли раствор комплекса пиридина с триоксидом серы (1,63 г, 10,2 ммоль) в ДМСО (15 мл) при -20°С. Через 1 час реакционную смесь нагревали до комнатной температуры, разбавляли дихлорметаном (50 мл) и промывали 1,0 н. раствором HCl (50 мл) и насыщенным раствором соли. Органическую часть отделяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали хроматографией на силикагеле, получая при этом указанное в заголовке промежуточное соединение (810 мг, 82%). 1H ЯМР (CDCl3, 300 МГц) δ (м.д.): 9,5 (с, 1H) 7,4-7,2 (м, 5H), 5,1 (с, 2H), 3,9 (д, J=5,8 Гц, 2H), 2,9-3,3 (ушир., 1H).
е. Получение бензилового эфира {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбаминовой кислоты
Суспензию 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамида и бензилового эфира (2-оксоэтил)карбаминовой кислоты (0,99 г, 6,2 ммоль) в DCM (20 мл) обрабатывали ультразвуком в течение 5 минут. К перемешиваемой суспензии добавляли триацетоксиборгидрид натрия (1,3 г, 6,1 ммоль). Реакционную смесь перемешивали в течение 30 минут и концентрировали. Неочищенную реакционную смесь разбавляли этилацетатом (50 мл) и промывали 1,0 н. раствором NaOH (50 мл) и водой (50 мл). Органическую часть отделяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали хроматографией на силикагеле, получая при этом указанное в заголовке промежуточное соединение (1,4 г, 57%). (m/z): [M+H]+, вычислено для С24Н29N3O3 408,22; найдено 408,5.
f. Получение 3-эндо-[8-(2-аминоэтил)-8-азабицикло[3.2.1]окт-3-ил)бензамида
Раствор продукта, полученного на предыдущей стадии (1,4 г, 3,4 ммоль), в метаноле (20 мл) добавляли к гидроксиду палладия на угле (50 мас.% воды, 20% Pd на сухой основе, 140 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Раствор фильтровали через целит и концентрировали, получая при этом масло (1,0 г), которое использовали на следующей стадии без очистки. (m/z): [M+H]+, вычислено для С16Н23N3O 274,19; найдено 274,5.
g. Получение 3-эндо-(8-{2-[(2,2,3,3-тетраметилциклопропилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (100 мг, 0,37 ммоль), и продукта, полученного на стадии b (45 мг, 0,37 ммоль), в дихлорметане (2 мл) и метаноле (0,2 мл) добавляли триацетоксиборгидрид натрия (93 мг, 0,44 ммоль). Реакционную смесь перемешивали в течение 1 часа и концентрировали. Неочищенную реакционную смесь разбавляли этилацетатом (20 мл) и промывали 1,0 н. раствором NaOH (20 мл). Органическую часть отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт использовали на следующей стадии без очистки. (m/z): [M+H]+, вычислено для С24Н37N3O 384,30; найдено 384,3.
h. Синтез 3-эндо-(8-{2-[(2-гидроксиацетил)(2,2,3,3-тетраметилциклопропилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
К раствору продукта, полученного на предыдущей стадии (0,366 ммоль), в DCM (2 мл) добавляли ацетоксиацетилхлорид (50 мкл, 0,44 ммоль). Через 1 час реакционную смесь концентрировали и образовавшееся неочищенное масло перемешивали в метаноле (2,0 мл) и 6,0 н. растворе NaOH (35 мкл) в течение ночи. Реакционную смесь концентрировали и очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (30,9 мг). (m/z): [M+H]+, вычислено для С26Н39N3O3 442,30; найдено 442,4.
Примеры 202-203
Промежуточные соединения по примерам 202 и 203 были получены следующим образом.
К раствору этилового эфира 4-гидроксициклогексанкарбоновой кислоты в виде смеси цис- и тринс-изомеров (8,7 г, 50,51 ммоль) в ТГФ (300 мл) при 0°С добавляли имидазол (4,8 г, 70,72 ммоль), DMAP (20 мол.%, 1,23 г, 10,10 ммоль) и трет-бутилдиметилхлорсилан (9,1 г, 60,61 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение ночи, разбавляли этилацетатом и насыщенным водным раствором хлорида аммония. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли, неочищенное масло фильтровали через силикагель и растворитель удаляли, получая при этом этиловый эфир 4-(трет-бутилдиметилсиланилокси)циклогексанкарбоновой кислоты (11,1 г, 77%) в виде смеси диастереомеров.
К раствору продукта, полученного на предыдущей стадии (11,1 г, 38,7 ммоль), в МТВЕ (150 мл) и метаноле (2,35 мл, 58,11 ммоль) добавляли боргидрид лития (1,27 г, 58,11 ммоль). Реакционную смесь нагревали до 50°С в течение 2 часов, охлаждали до комнатной температуры и гасили метанолом. Раствор разбавляли этилацетатом, промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом магния. Растворитель удаляли в вакууме с образованием неочищенного [4-(трет-бутилдиметилсиланилокси)циклогексил]метанола в виде смеси диастереомеров. Неочищенное вещество очищали хроматографией на силикагеле (10-40% этилацетата/гексаны), разделяя цис- и транс-диастереомеры. Верхнее пятно (цис-изомер) 1H ЯМР (300 МГц, CD3OD): 3,91-3,95 (м, 1H), 3,31-3,32 (д, 2H), 1,61-1,63 (м, 2H), 1,36-1,46 (м, 7H), 0,85-0,86 (с, 9H), 0,00 (с, 6H). Нижнее пятно (транс-изомер) 1H ЯМР (300 МГц, CD3OD): 3,49-3,53 (м, 1H), 3,27-3,28 (д, 2H), 1,81-1,84 (м, 2H), 1,71-1,74 (м, 2H), 1,28-1,36 (м, 1H), 1,18-1,26 (м, 2H), 0,93-0,97 (м, 2H), 0,82-0,84 (с, 9H), 0,00 (с, 6H).
Пример 202. Синтез 3-эндо-(8-{2-[(транс-4-гидроксициклогексилметил)-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
а. Получение транс-4-(трет-бутилдиметилсиланилокси)циклогексилметилового эфира толуол-4-сульфоновой кислоты
К раствору транс-[4-(трет-бутилдиметилсиланилокси)циклогексил]метанола (555 мг, 2,24 ммоль) в DCM (20 мл) при 0°С добавляли DIPEA (0,59 мл, 3,37 ммоль), DMAP (20 мол.%, 54 мг) и паратолуолсульфонилхлорид (570 мг, 2,68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и добавляли DABCO (250 мг). Реакционную смесь перемешивали в течение ночи. Раствор разбавляли DCM, промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение. 1H ЯМР (300 МГц, CD3OD): 7,75-7,77 (д, 2H), 7,40-7,42 (д, 2H), 3,80-3,81 (д, 1H), 3,45-3,51 (м, 1H), 2,40 (с, 3H) 1,78-1,82 (м, 2H), 1,62-1,71 (м, 2H), 1,48-1,58 (м, 1H), 1,30-1,33 (м, 1H), 1,15-1,21 (м, 2H), 0,85-1,00 (м, 2H), 0,85-0,90 (с, 9H), 0,00 (с, 6H).
b. Получение 2-{(транс-4-(трет-бутилдиметилсиланилокси)циклогексилметил]амино}этанола
К раствору транс-4-(трет-бутилдиметилсиланилокси)циклогексил метилового эфира толуол-4-сульфоновой кислоты (600 мг, 1,50 ммоль) в ацетонитриле (15 мл) добавляли этаноламин (2,0 мл, 25 экв.). Раствор нагревали до 50°С в течение ночи. Реакционную смесь разбавляли DCM (200 мл), промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над карбонатом калия. Растворитель удаляли, получая при этом указанное в заголовке неочищенное промежуточное соединение (0,44 г) в виде желтого масла. (m/z): [M+H]+, вычислено для С15Н33NSiO2 288,3; найдено 288,2.
с. Получение трет-бутилового эфира [транс-4-(трет-бутилдиметилсиланилокси)циклогексилметил](2-гидроксиэтил)карбаминовой кислоты
К раствору продукта, полученного на предыдущей стадии (0,44 г, 1,50 ммоль), в DCM (30 мл) добавляли ди-трет-бутилдикарбонат (324 мг, 1,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, разбавляли DCM (100 мл), промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над карбонатом калия. Растворитель удаляли, получая при этом указанное в заголовке промежуточное соединение (0,496 г) в виде желтого масла.
d. Получение трет-бутилового эфира [транс-4-(трет-бутилдиметилсиланилокси)циклогексилметил](2-оксоэтил)карбаминовой кислоты
К раствору продукта, полученного на прудыдущей стадии (496 мг, 1,27 ммоль), в DCM (20 мл) при -15°С добавляли ДМСО (12,7 ммоль, 0,905 мл) и DIPEA (1,103 мл, 6,35 ммоль). Реакционную смесь перемешивали в течение 10 минут и добавляли комплекс пиридина с триоксидом серы (1,01 г, 6,35 ммоль). Реакционную смесь перемешивали в течение 1 часа, нагревали до комнатной температуры, разбавляли DCM (50 мл), промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение (480 мг) в виде желтого масла.
е. Получение трет-бутилового эфира [транс-4-(трет-бутилдиметилсиланилокси)циклогексилметил]{2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}карбаминовой кислоты
К раствору продукта, полученного на предыдущей стадии (480 мг, 1,24 ммоль), в DCM (20 мл) добавляли 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензамид (343 мг, 1,48 ммоль). Реакционную смесь перемешивали в течение 30 минут и добавляли триацетоксиборгидрид натрия (525 мг, 2,48 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, разбавляли DCM (100 мл), промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке неочищенное промежуточное соединение (700 мг, 94%) в виде хрустящего желтого твердого вещества. (m/z): [M+H]+, вычислено для С34Н58N3SiO4 600,4; найдено 600,6.
f. Получение 3-эндо-(8-{2-[(транс-4-гидроксициклогексилметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Продукт, полученный на предыдущей стадии (700 мг, 1,16 ммоль), растворяли в DCM (15 мл) и добавляли трифторуксусную кислоту (7 мл). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, разбавляли DCM (100 мл) и 1 н. раствором NaOH (100 мл). Водный слой экстрагировали дихлорметаном (2×25 мл) и объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке неочищенное промежуточное соединение (425 мг, выход 94%). (m/z): [M+H]+, вычислено для С23Н36N3O2 386,3; найдено 386,5.
g. Получение {2-[3-эндо-(3-карбамоилфенил)-8-азабицикло[3.2.1]окт-8-ил]этил}(транс-4-гидроксициклогексилметил)амида (S)-2,2-диметил[1,3]диоксолан-4-карбоновой кислоты
К раствору продукта, полученного на предыдущей стадии (425 мг, 1,10 ммоль), в ДМФА (15 мл) добавляли (S)-2,2-диметил[1,3]диоксолан-4-карбоксилат лития (0,19 г, 1,21 ммоль) и HATU (0,46 г, 1,21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, разбавляли DCM (100 мл), промывали водой, смесью воды : насыщенного водного раствора бикарбоната натрия в отношении 1:1 и насыщенным раствором соли и сушили над карбонатом калия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке неочищенное промежуточное соединение в виде желтого масла. (m/z): [M+H]+, вычислено для С29Н43N3O5 514,3; найдено 514,5.
h. Синтез 3-эндо-(8-{2-[(транс-4-гидроксициклогексилметил)((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Продукт, полученный на предыдущей стадии, растворяли в ацетонитриле (15 мл) и 1 н. растворе HCl (15 мл) и раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли DCM (200 мл) и 1 н. раствором NaOH (150 мл). Водный слой экстрагировали дихлорметаном (2×50 мл), объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме и неочищенное вещество очищали препаративной ВЭЖХ, получая при этом соль ТФУ указанного в заголовке соединения (45 мг) в виде белого порошка. (m/z): [M+H]+, вычислено для С26Н39N2O5 474,29; найдено 474,4. 1H ЯМР (300 МГц, CD3OD): 8,00-8,02 (с, 1H), 7,65-7,80 (м, 2H), 7,40-7,45 (т, 1H), 4,59-4,62 (м, 1H), 3,95-4,20 (м, 3H) 3,65-3,79 (м, 2H), 3,40-3,60 (м, 2H), 3,10-3,20 (м, 2H), 2,05-2,19 (м, 2H), 1,95-2,00 (м, 2H), 1,40-1,85 (м, 4H), 1,20-1,35 (м, 2H), 1,01-1,20 (м, 2H).
Пример 203. Синтез 3-эндо-(8-{2-[(цис-4-гидроксициклогексилметил)-((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида
Указанное в заголовке соединение было получено способом по примеру 202 с использованием на стадии “а” соответствующего цис-изомера, цис-[4-(трет-бутилдиметилсиланилокси)циклогексил]метанола. (m/z): [M+H]+, вычислено для С26Н39N2O5 474,29; найдено 474,4. 1H ЯМР (300 МГц, CD3OD): 8,00-8,02 (с, 1H), 7,65-7,80 (м, 2H), 7,40-7,50 (т, 1H), 4,60-4,62 (м, 1H), 3,95-4,20 (м, 3H) 3,65-3,79 (м, 2H), 3,40-3,60 (м, 2H), 3,10-3,20 (м, 2H), 2,05-2,19 (м, 2H), 1,40-1,85 (м, 6H), 1,20-1,30 (м, 4H).
Пример 204. Синтез 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензойной кислоты
а. Получение метилового эфира 3-эндо-(8-азабицикло[3.2.1]окт-3-ил)бензойной кислоты
3-эндо-(8-Азабицикло[3.2.1]окт-3-ил)бензамид (2,5 г, 9,36 ммоль) вводили в 200 мл колбу и продували азотом. Добавляли метанол (100 мл) и раствор хлористоводородной кислоты в диоксане (7 мл 4,0 н. раствора, 28 ммоль). Раствор нагревали до температуры кипения с обратным холодильником и перемешивали в течение ночи. Метанол удаляли выпариванием и реакционную смесь разбавляли DCM (200 мл) и 1 н. раствором NaOH (150 мл). Водный слой экстрагировали DCM (2Ч50 мл), объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли и неочищенный остаток очищали препаративной ВЭЖХ. Чистые фракции объединяли, получая при этом указанное в заголовке промежуточное соединение (1,66 г) в виде белого порошка. (m/z): [M+H]+, вычислено для С15Н20NO2 246; найдено 246,3.
b. Получение метилового эфира 3-эндо-{8-[2-(бензилоксикарбонилциклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензойной кислоты
К раствору продукта, полученного на предыдущей стадии (1,66 г, 4,61 ммоль), в DCM (20 мл) и метаноле (20 мл) добавляли бензиловый эфир циклогексилметил(2-оксоэтил)карбаминовой кислоты (1,27 г, 4,61 ммоль). Реакционную смесь перемешивали в течение 20 минут и добавляли триацетоксиборгидрид натрия (1,95 г, 9,22 ммоль). Реакционную смесь перемешивали в течение 2 часов, разбавляли DCM (100 мл), промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение (2,65 г, выход >100%) в виде хрустящего желтого твердого вещества, которое использовали на следующей стадии без очистки.
с. Получение метилового эфира 3-эндо-{8-[2-(циклогексилметиламино)этил]-8-азабицикло[3.2.1]окт-3-ил}бензойной кислоты
К раствору продукта, полученного на предыдущей стадии (2,65 г), в этаноле (20 мл) и 1 н. водном растворе HCl (10 мл) добавляли палладий на угле (10 мас.%, 270 мг). Реакционную смесь продували газообразным водородом и перемешивали в атмосфере водорода в течение ночи. Катализатор удаляли фильтрованием и реакционную смесь разбавляли DCM (200 мл) и 1 н. раствором NaOH (150 мл). Водный слой экстрагировали DCM (2×50 мл), объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли, получая при этом указанное в заголовке промежуточное соединение (2,0 г) в виде воскообразного желтого масла. (m/z): [M+H]+, вычислено для С24Н36N2O2 385,3; найдено 385,5.
d. Получение метилового эфира 3-эндо-(8-{2-[циклогексилметил((S)-2,2-диметил[1,3]диоксолан-4-карбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензойной кислоты
К раствору продукта, полученного на предыдущей стадии (2,0 г, 5,2 ммоль), в ДМФА (35 мл) добавляли (S)-2,2-диметил[1,3]диоксолан-4-карбоксилат лития (0,72 г, 5,7 ммоль) и HATU (2,18 г, 5,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, разбавляли DCM (100 мл), промывали водой, смесью воды:насыщенного водного раствора бикарбоната натрия в отношении 1:1 и насыщенным раствором соли и сушили над карбонатом калия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке неочищенное промежуточное соединение в виде коричневого масла, которое использовали на следующей стадии без очистки.
е. Получение метилового эфира 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензойной кислоты
Продукт, полученный на предыдущей стадии, растворяли в ацетонитриле (15 мл) и 1 н. растворе HCl (15 мл) и раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли DCM (200 мл) и 1 н. раствором NaOH (150 мл). Водный слой экстрагировали DCM (2×50 мл), объединенные органические слои промывали насыщенным раствором соли и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая при этом указанное в заголовке промежуточное соединение (2,2 г) в виде желтого масла. (m/z): [M+H]+, вычислено для С27Н40N2O5 473,3; найдено 473,3.
f. Синтез 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензойной кислоты
К раствору продукта, полученного на предыдущей стадии (2,2 г, 5,2 ммоль), в ТГФ (5 мл) добавляли раствор гидроксида лития (1,31 г, 31,2 ммоль) в воде (5 мл). Раствор интенсивно перемешивали при комнатной температуре. После окончания гидролиза сложного эфира ТГФ удаляли в вакууме и остаток очищали препаративной ВЭЖХ. Чистые фракции объединяли, получая при этом указанное в заголовке соединение (0,32 г) в виде белого порошка. (m/z): [M+H]+, вычислено для С26Н38N2O5 459,28; найдено 459,5.
Анализ 1. Анализ связывания радиоактивного лиганда с рецепторами мю-опиоидов человека, дельта-опиоидов человека и каппа-опиоидов морской свинки
а. Приготовление мембран
Клетки СНО-К1 (яичника китайского хомячка), устойчиво трансфецированные кДНК рецепторов мю-опиоидов человека или каппа-опиоидов морской свинки, выращивали во влажной камере с 5% СО2 при 37°С в среде, состоящей из среды F12 Хэма, содержащей 10% FBS, 100 единиц/мл пенициллина - 100 мкг/мл стрептомицина и 800 мкг/мл генетицина. Уровни экспрессии рецепторов (Вmax соответственно равно ~2,0 и ~0,414 пмоль/мг белка) определяли, используя [3H]-дипренорфин (удельная активность ~50-55 Ci/ммоль), при выполнении анализа связывания радиоактивного лиганда с мембраной.
Клетки выращивали до 80-95% слияния (<25 пассажей). Для пассирования линии клеток монослой клеток инкубировали в течение 5 минут при комнатной температуре и собирали при механическом взбалтывании в 10 мл физиологического раствора с фосфатным буфером (PBS), содержащего 5 мМ EDTA. Клетки ресуспендировали, переносили в 40 мл свежей питательной среды, центрифугировали в течение 5 минут со скоростью 1000 оборотов/мин и ресуспендировали в свежей питательной среде при соответствующем отношении расщепления.
Для приготовления мембран клетки собирали при осторожном механическом взбалтывании с 5 мМ EDTA в PBS и центрифугировали (2500 g в течение 5 минут). Осадки ресуспендировали в аналитическом буфере (50 мМ 4-(2-гидроксиэтил)пиперазин-1-этансульфоновой кислоты, N-(2-гидроксиэтил)пиперазин-N'-(2-этансульфоновой кислоты) (HEPES)), рН 7,4, и гомогенизировали в дезинтеграторе Polytron на льду. Полученные гомогенаты центрифугировали (1200 g в течение 5 минут), осадки удаляли и супернатант центрифугировали (40000 g в течение 20 минут). Осадки промывали один раз, ресуспендируя в аналитическом буфере, и подвергали дополнительному центрифугированию (40000 g в течение 20 минут). Конечные осадки ресуспендировали в аналитическом буфере (колба Т-225 для 1 эквивалента/1 мл аналитического буфера). Концентрацию белка определяли, используя набор для анализа белка Bio-Rad Bradford, и мембраны хранили до использования в замороженных аликвотах при -80°С.
Мембраны рецепторов дельта-опиоидов человека (hDOP) были приобретены в компании Perkin Elmer. Значения Кd и Вmax для указанных мембран, определенные с помощью анализов насыщения при выполнении анализов связывания радиоактивного лиганда [3H]-натриндола, были равны соответственно 0,14 нМ (рКd = 9,85) и 2,2 пмоль/мг белка. Концентрацию белка определяли при помощи набора для анализа белка Bio-Rad Bradford. Мембраны хранили до использования в замороженных аликвотах при -80°С.
b. Анализы связывания радиоактивного лиганда
Анализы связывания ридоактивного лиганда выполняли на 96-луночном полипропиленовом аналитическом планшете с 1,1 мл лунками Axygen при общем анализируемом объеме 200 мкл, содержащем соответствующее количество мембранного белка (соответственно ~3, ~2 и ~20 мкг для мю-, дельта- и каппа-опиоидов) в аналитическом буфере, включающем 0,025% бычьего сывороточного альбумина (BSA). Анализы связывания в насыщенном состоянии для определения значений Кd радиоактивного лиганда выполняли, используя [3H]-дипренорфин в 8-12 разных концентрациях от 0,001 нМ до 5 нМ. Анализы замещения для определения значений рКi соединений выполняли с использованием [3H]-дипренорфина в количестве 0,5, 1,2 и 0,7 нМ соответственно для мю-, дельта- и каппа-опиоидов и одиннадцати концентраций соединений от 10 пМ до 100 мкМ.
Данные связывания анализировали при помощи нелинейного регрессионного анализа с использованием пакета программ GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и трехмерной модели для локальной конкуренции. Минимальное значение кривой было задано в соответствии со значением неспецифического связывания, определенным в присутствии 10 мкМ налоксона. Значения Кi для испытуемых соединений вычисляли в программе Prism на основании значений IC50 с наилучшим приближением, и значение Kd радиоактивного лиганда определяли из уравнения Ченга-Прусоффа (Кi=IC50/(1+([L]/Kd)), где [L] означает концентрацию [3H]-дипренорфина. Результаты выражены в виде отрицательного десятичного логарифма значений Кi, рКi.
Испытуемые соединения, характеризующиеся более высоким значением pKi при выполнении указанных анализов, обладают более высоким сродством связывания с рецепторами мю-, дельта- или каппа-опиоидов. С помощью вышеуказанных анализов были испытаны соединения по примерам 1-204. За исключением соединения по примеру 204, которое характеризовалось связыванием с рецепторами мю-опиоидов на микромолярном уровне, все соединения имели значение рКi от около 8,0 до до 10,5 для рецепторов мю-опиоидов человека. Например, соединения по примерам 1, 46В, 58, 59 и 136 имели значения рКi, равные соответственно 10,1, 10,0, 9,9, 9,2 и 9,8. Соединения по примерам 1-203 также имели значения рКi от около 7,0 до около 10,5 для рецепторов дельта-опиоидов человека и каппа-опиоидов морской свинки.
Анализ 2. Опосредованная агонистом активация рецепторов мю-опиоидов в мембранах, полученных из клеток СНО-К1, экспрессирующих рецепторы мю-опиоидов человека
При выполнении данного анализа определяли значения эффективности и собственной активности испытуемых соединений, измеряя количество связанного GTP-Eu после активации рецепторов в мембранах, приготовленных из клеток СНО-К1, экспрессирующих рецепторы мю-опиоидов человека.
а. Приготовление мембран, экспрессирующих рецепторы мю-опиоидов
Мембраны, экспрессирующие рецепторы мю-опиоидов человека (hMOP), были получены вышеописанным способом или приобретены в компании Perkin Elmer. Значения pKd и Вmax для приобретенных мембран, определенные при помощи анализов насыщения в процессе выполнения анализов связывания радиоактивного лиганда [3H]-дипренорфина, были равны соответственно 10,06 и 2,4 пмоль/мг белка. Концентрацию белка определяли при помощи набора для анализа белка Bio-Rad Bradford. Мембраны хранили до использования в замороженных аликвотах при -80°С. Лиофилизованные GTP-Eu и GDP разводили соответственно до 10 мкМ и 2 мМ в дважды дистиллированной Н2О, смешивали, оставляли выстаиваться при комнатной температуре в течение 30 минут и переносили в отдельные аликвоты образцов для хранения при -20°С.
b. Анализ замены нуклеотидов GTP-Eu мю-опиоидов человека
Анализы замены нуклеотидов GTP-Eu выполняли при помощи набора для связывания GTP DELPHIA (Perkin/Elmer) на 96-луночных фильтровальных планшетах Acro Well в соответствии с инструкциями изготовителя. Аликвоты, содержащие мембраны, полученные вышеописанным способом, до начала анализа разводили до концентрации 200 мкг/мл в аналитическом буфере (50 мМ HEPES, рН 7,4 при 25°С) и гомогенизировали в течение 10 секунд в гомогенизаторе Polytron. Испытуемые соединения, полученные в виде 10 мМ исходных растворов в ДМСО, разводили до 400 мкМ в аналитическом буфере, содержащем 0,1% BSA, и затем последовательно разводили (1:5) для получения десяти концентраций соединения от 40 пМ до 80 мкМ, GDP и GTP-Eu разводили в аналитическом буфере соответственно до 4 мкМ и 40 нМ. Данный анализ выполняли в общем объеме 100 мкл, содержащем 5 мкг мембранного белка, при этом испытуемые соединения имели значения от 10 пМ до 20 мкМ, 1 мкМ GDP и 10 нМ GTP-Eu разводили в 10 мМ MgCl2, 50 мМ NaCl и 0,0125% BSA (конечные анализируемые концентрации). Для каждого планшета строили кривую зависимости от концентрации DAMGO (Tyr-D-Ala-Gly-(metil)Phe-Gly-ol) (в пределах от 12,8 пМ до 1 мкМ).
Аналитические планшеты готовили непосредственно перед выполнением анализа, добавляя 25 мкл аналитического буфера, 25 мкл испытуемого соединения и 25 мкл GDP и GTP-Eu. Анализ инициировали, добавляя 25 мкл мембранного белка, который инкубировали в течение 30 минут. Аналитические планшеты фильтровали при помощи фильтрационного устройства Waters, присоединенного к вакуумной установке, создающей давление до 250-300 мм Hg (10-12 дюймов Hg), и промывали промывным раствором GTP (2×300 мл) при комнатной температуре. Основания планшетов промокали для удаления избытка жидкости. Затем планшеты сразу же считывали для определения количества связанного GTP-Eu путем измерения флуоресценции с разрешением по времени (TRF) на носителе спектрофотометра для прочтения планшетов Packard, при этом количество ДМСО не превышало 1% от конечной анализируемой концентрации.
Количество связанного GTP-Eu пропорционально степени активации рецепторов мю-опиоидов испытуемым соединением. Собственную активность (IA), выраженную в процентах, определяли в виде отношения количества связанного GTP-Eu, необходимого для активации испытуемым соединением, к количеству, необходимому для активации DAMGO, который считается полным агонистом (IA = 100). Соединения по примерам 1-204 характеризовались собственной активностью в данном анализе менее примерно 40, обычно менее примерно 25. Например, соединения по примерам 1, 46В, 58, 59 и 136 имели значения IA, равные соответственно 6, -3, -3, -2 и 14. Таким образом, установлено, что соединения по настоящему изобретению являются антагонистами рецепторов мю-опиоидов человека.
Анализ 3. Мышиная модель эффективности in vivo
При выполнении указанных анализов определяли эффективность испытуемых соединений в модели желудочно-кишечного транзита, которая позволяет оценить периферическую активность, а также в модели исследования аналгезии у грызунов с использованием горячей пластинки, которая позволяет оценить активность центральной нервной системы. Результаты, полученные при помощи двух указанных моделей, позволяют вычислить относительную периферическую избирательность испытуемых соединений. Указанные исследования были утверждены Комитетом по охране и использованию животных в лечебных учреждениях в компании Theravance, Inc. и соответствовали руководству по охране и использованию лабораторных животных, опубликованному Национальной академией наук (© 1996).
а. Анализ кишечного транзита у мышей
Испытуемые соединения исследовали при выполнении анализа желудочно-кишечного транзита у мышей для определения их способности восстанавливать замедленный желудочно-кишечный транзит, вызванный морфином. Мышей не кормили в течение 24 часов до введения испытуемых соединений или носителя внутривенным, подкожным, внутримышечным или пероральным способами введения в дозах от 0,001 до около 10 миллиграммов/килограмм (мг/кг). Вслед за введением испытуемого соединения подкожно вводили морфин в дозе 3 мг/кг или носитель. Через пять минут после введения морфина или носителя с помощью желудочного зонда вводили непитательную, неабсорбируемую угольную муку и животным предоставляли свободный доступ к воде на протяжении шестидесяти минут эксперимента. Затем животных умерщвляли путем асфиксии, вызванной диоксидом углерода, производили торакотомию и осторожно иссекали желудочно-кишечный тракт от желудка до слепой кишки. Желудок лигировали у нижней части сфинктера пищевода и сфинктера привратника для предотвращения дополнительного опорожнения в тонкую кишку во время выполнения измерений. Кишечный транзит определяли как расстояние, пройденное передним фронтом муки, относительно общей длины кишечника (у илеоцекального соединения).
b. Исследование мышей методом горячей пластинки
Активность соединений исследовали в мышиной модели методом горячей пластинки (модель № 7406 Letica Scientific Instruments; Panlab, S.L., Barcelona, Spain) для определения их способности реверсировать действие морфина, опосредуемое центральной нервной системой. Соединения исследовали в отношении их способности устранять аналгезию, вызванную морфином, по результатам сокращения латентного периода облизывания лап по сравнению с контрольными животными, которым вводили морфин. Испытуемые соединения вводили внутривенно, подкожно, внутримышечно или перорально в дозах от 0,1 до 30 мг/кг с последующим подкожным введением морфина в дозе 10 мг/кг или носителя. Животных возвращали в их клетки, где они проводили остальные тридцать минут времени эксперимента. Затем животных помещали в аппарат с горячей пластинкой (53°С), и сотрудник, не осведомленный о том, к которой экспериментальной группе относится данное животное, регистрировал время облизывания лапы мышью. У животных, которые не облизывали лапу ранее предельного времени, равного 35 секундам, автоматически регистрировали латентный период, равный 35 секундам.
с. Анализ данных и результаты исследования
Данные анализировали с помощью пакета программ Prism GraphPad (GraphPad Software, Inc., San Diego, CA). Кривые процентного значения реверсии строили методом нелинейного регрессионного анализа с использованием модели сигмоидальной реакции на дозу (переменный наклон) и вычисляли значения ID50 с наилучшим приближением. Минимальные и максимальные значения кривой были заданы в соответствии с контрольными значениями морфина (0% реверсия) и контрольными значениями носителя (100% реверсия). Результаты выражены в виде ID50, дозы, необходимой для 50% реверсии действия морфина, в миллиграммах на килограмм. В указанной модели были исследованы отобранные соединения по настоящему изобретению. Соединения по настоящему изобретению, вводимые подкожно или перорально, характеризовались значениями ID50 в модели кишечного транзита от около 0,1 до около 3 мг/кг.
Периферическую избирательность вычисляли для каждого соединения на основании значения ID50, полученного методом горячей пластинки (измерение центрального действия), которое делили на значение ID50 кишечного транзита (измерение периферического действия). Соединения по настоящему изобретению, которые были исследованы при выполнении указанных анализов, характеризовались периферической избирательностью в пределах от около 2-кратного до 500-кратного значения. В частности, соединения по примерам 46В, 48В, 58, 59 и 63 характеризовались периферической избирательностью, равной соответственно 15, 30, 300, 43 и 22 при подкожном введении и соответственно 19, 42, 35, 6 и 22 при пероральном введении.
Несмотря на то что настоящее изобретение было описано со ссылкой на конкретные варианты его осуществления, специалистам в данной области должно быть понятно, что в изобретение могут быть внесены разные изменения и использованы эквиваленты, не выходящие за пределы духа и объема изобретения. Кроме того, могут быть произведены многие модификации для приведения конкретной ситуации, вещества, композиции, способа, одной или нескольких стадий способа в соответствие с целями, духом и объемом настоящего изобретения. Все такие модификации входят в объем прилагаемой формулы изобретения. Все публикации, патенты и патентные документы, приведенные в настоящем описании изобретения, полностью включены в качестве ссылки, как если бы каждый из указанных документов был отдельно включен в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ 8-АЗАБИЦИКЛО[3.2.1]ОКТАНА | 2008 |
|
RU2458061C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА АНТАГОНИСТОВ мю-РЕЦЕПТОРА ОПИОИДОВ | 2008 |
|
RU2469035C2 |
| ИНДАЗОЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2404179C2 |
| АЗАБИЦИКЛИЧЕСКИЕ АЛКАНОВЫЕ ПРОИЗВОДНЫЕ, ЗАМЕЩЕННЫЕ КОНДЕНСИРОВАННЫМ БИЦИКЛОГЕТЕРОЦИКЛОМ | 2007 |
|
RU2437884C2 |
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2016 |
|
RU2720678C2 |
| СРЕДСТВА, ИНДУЦИРУЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА, ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2568611C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
| ПРОИЗВОДНОЕ АМИНОБЕНЗОЙНОЙ КИСЛОТЫ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1993 |
|
RU2067979C1 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛООКТАНА, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, СОЕДИНЕНИЯ | 2000 |
|
RU2262505C2 |
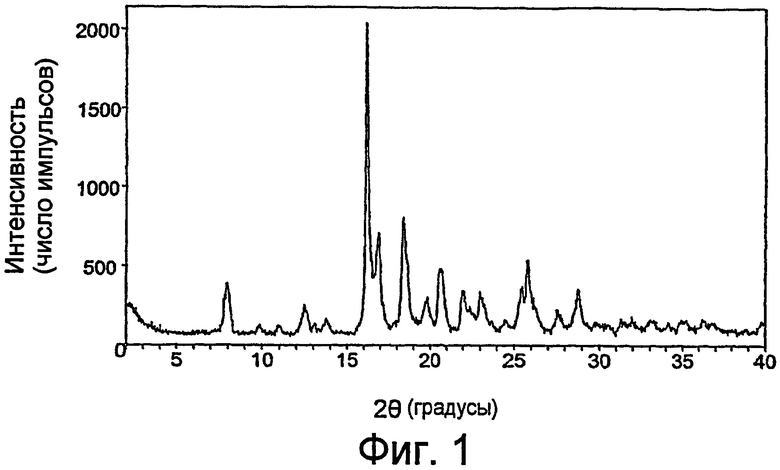

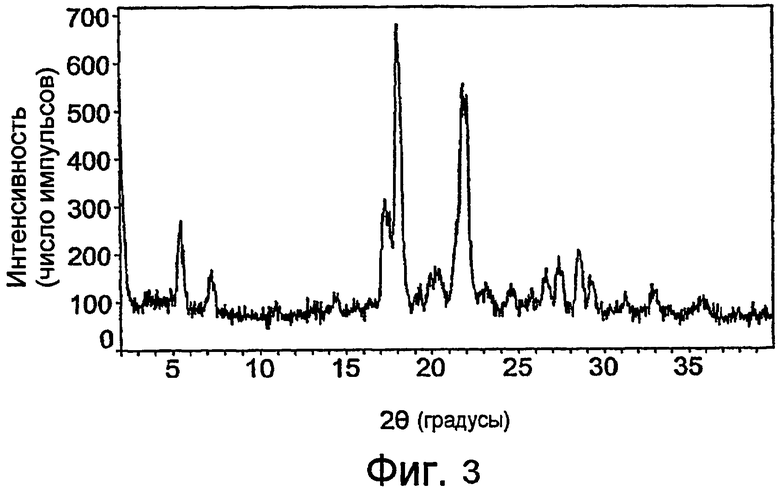
Изобретение относится к новым соединениям формулы (I):

где R1 выбирают из -ORa, -C(O)NRaRb, -NHS(O)2Rc, -C(O)ORa; А означает С1-4алкиленил; R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним -ORa, одним или двумя атомами галогена, одним или двумя C1-3алкилами, замещенными двумя или тремя атомами галогена; или одним, двумя, тремя или четырьмя C1-3алкилами; G означает С1-4алкиленил; R3 выбирают из водорода, -C(O)R4, -C(O)NHR5, -S(O)2Rc и -S(O)2NRaRb; R4 означает С3-6циклоалкил или C1-6алкил, при этом С3-6циклоалкил необязательно замещен одним -ORa, и C1-6алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORa, -C(O)ORa, -S(O)2R6, -C(O)NRaRb, -NRaRb, -CN, С3-6циклоалкила и фенила; или одним -D-(CH2)j-R7, где D означает  , j равно 1, n равно 1 или 2; R6 означает C1-3алкил, необязательно замещенный R7; R7 означает - C(O)ORa; R5 означает C1-6алкил, бензо[1.3]диоксол или -(СН2)q-фенил; где фенил необязательно замещен одним или двумя заместителями, выбираемыми из галогена, -ORa, C1-3алкила и C1-3алкокси, где C1-3алкил и C1-3алкокси необязательно замещены 2 или 3 атомами галогена, и q равно 0, 1; Ra и Rb независимо означают водород или С1-4алкил; и Rc означает С1-3алкил; при условии, что, когда R2 означает фенил, замещенный в положении 4, R3 не является -C(O)R4, где R4 означает С1-4алкил, замещенный -С(O)ОRa; к их фармацевтически приемлемым солям. Изобретение также относится к фармацевтической композиции, к способу получения соединения формулы (I), к соединениям формулы (II), (III), к применению соединений по любому из п.п.1-14, к способу исследования биологической системы или образца, содержащего рецептор мю-опиоида, а также к способу лечения млекопитающего, страдающего заболеванием, обусловленным активностью рецептора мю-опиоида. Технический результат - получение новых биологически активных соединений, обладающих антагонистической активностью в отношении рецепторов мю-опиоидов. 8 н. и 18 з.п. ф-лы, 3 ил.
, j равно 1, n равно 1 или 2; R6 означает C1-3алкил, необязательно замещенный R7; R7 означает - C(O)ORa; R5 означает C1-6алкил, бензо[1.3]диоксол или -(СН2)q-фенил; где фенил необязательно замещен одним или двумя заместителями, выбираемыми из галогена, -ORa, C1-3алкила и C1-3алкокси, где C1-3алкил и C1-3алкокси необязательно замещены 2 или 3 атомами галогена, и q равно 0, 1; Ra и Rb независимо означают водород или С1-4алкил; и Rc означает С1-3алкил; при условии, что, когда R2 означает фенил, замещенный в положении 4, R3 не является -C(O)R4, где R4 означает С1-4алкил, замещенный -С(O)ОRa; к их фармацевтически приемлемым солям. Изобретение также относится к фармацевтической композиции, к способу получения соединения формулы (I), к соединениям формулы (II), (III), к применению соединений по любому из п.п.1-14, к способу исследования биологической системы или образца, содержащего рецептор мю-опиоида, а также к способу лечения млекопитающего, страдающего заболеванием, обусловленным активностью рецептора мю-опиоида. Технический результат - получение новых биологически активных соединений, обладающих антагонистической активностью в отношении рецепторов мю-опиоидов. 8 н. и 18 з.п. ф-лы, 3 ил.
1. Соединение формулы (I)
где R1 выбирают из -ORa, -C(O)NRaRb, -NHS(O)2Rc, -C(O)ORa;
А означает С1-4алкиленил;
R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним -ORa, одним или двумя атомами галогена, одним или двумя C1-3алкилами, замещенными двумя или тремя атомами галогена; или одним, двумя, тремя или четырьмя С1-3алкилами;
G означает С1-4алкиленил;
R3 выбирают из водорода, -C(O)R4, -C(O)NHR5, -S(O)2Rc и -S(O)2NRaRb;
R4 означает С3-6циклоалкил или С1-6алкил, при этом
С3-6диклоалкил необязательно замещен одним -ORa, и
C1-6алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORa, -C(O)ORa, -S(O)2R6, -C(O)NRaRb, -NRaRb, -CN, С3-6циклоалкила и фенила; или одним -D-(CH2)j-R7,
где D означает , j равно 1, n равно 1 или 2;
R6 означает С1-3алкил, необязательно замещенный R7;
R7 означает -C(O)ORa;
R5 означает C1-6алкил, бензо[1.3]диоксол или -(СН2)q-фенил;
где фенил необязательно замещен одним или двумя заместителями, выбираемыми из галогена, -ORa, C1-3алкила и С1-3алкокси, где C1-3алкил и С1-3алкокси необязательно замещены 2 или 3 атомами галогена, и q равно 0, 1;
Ra и Rb независимо означают водород или С1-4алкил; и
Rc означает C1-3алкил;
при условии, что, когда R2 означает фенил, замещенный в положении 4, R3 не является -C(O)R4, где R4 означает С1-4алкил, замещенный -C(O)ORa;
его фармацевтически приемлемая соль.
2. Соединение по п.1,
в котором R2 означает С3-12циклоалкил или С6-10арил, которые необязательно замещены одним -ORa, одним или двумя атомами галогена или одним или двумя C1-3алкилами, необязательно замещенными 2 или 3 атомами галогена;
R3 выбирают из водорода, -C(O)R4, -C(O)NHR5;
R4 означает С3-6циклоалкил или С1-6алкил,
где C1-6алкил необязательно замещен одним или двумя -ORa либо одним заместителем, выбираемым из -C(O)ORa, -S(O)2R6, -С(O)NRaRb, -NRaRb, С3-6циклоалкила, -D-(CH2)j-R7 и фенила; и
Ra и Rb независимо означают водород или С1-3алкил.
3. Соединение по п.1, в котором R1 означает -ORa или -C(O)NRaRb.
4. Соединение по п.1, в котором R2 выбирают из циклобутила, циклопентила, циклогексила, адамантила и фенила, при этом циклогексил и фенил необязательно замещены одним или двумя атомами галогена или С1-3алкилом, замещенным двумя или тремя атомами галогена.
5. Соединение по п.1, в котором А означает -(СН2)2- или -СН2- и G означает
-(СН2)2- или -СН2-.
6. Соединение по п.1,
в котором R3 выбирают из -C(O)R4, -S(O)2Rc и -S(O)2NRaRb;
R4 означает С3-6циклоалкил или С1-6алкил, при этом
С3-6циклоалкил необязательно замещен одним -ORa, и
C1-6алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORa,- C(O)ORa, -S(O)2R6, -C(O)NRaRb, -NRaRb, -CN, С3-6циклоалкила и фенила, где R6 означает C1-3алкил, необязательно замещенный R7, где R7 означает -C(O)ORa.
7. Соединение по п.1,
в котором R3 означает -C(O)NHR5 и
R5 означает С1-4алкил, бензо[1.3]диоксол или -(CH2)q-фeнил, где q равно 0 или 1 и фенил необязательно замещен одним или двумя заместителями, выбираемыми из хлора, фтора, -ОН и -OCF2.
8. Соединение по п.1, которое является соединением формулы (I'):
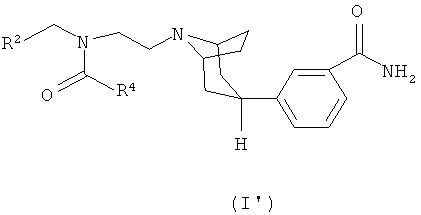
где R2 означает циклогексил или фенил, которые необязательно замещены одним или двумя атомами галогена; и
R4 означает С3-6циклоалкил или С1-4алкил, при этом
С3-6циклоалкил необязательно замещен одним -ORa и
С1-4алкил необязательно замещен одним или двумя заместителями, выбираемыми из -ORa, -S(O)2R6, -NRaRb, -CN и С3-6циклоалкила, где Ra и Rb независимо означают водород или C1-3алкил и R6 означает C1-3алкил;
его фармацевтически приемлемой солью.
9. Соединение по п.8, в котором R4 означает С1-4алкил, необязательно замещенный одним или двумя заместителями, выбираемыми из -ОН, -ОСН3, -S(O)2СН3, -NH2, -NHCH3 и -NН(СН3)2.
10. Соединение по п.8, в котором R2 означает циклогексил или 4,4-дифторциклогексил и R4 означает С1-2алкил, замещенный одним или двумя -ОН.
11. Соединение по п.1, которое выбирают из группы, включающей:
3-эндо-(8-{2-[циклогексилметил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[бензил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2-гидроксиацетил)фенетиламино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(4,4-дифторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1] окт-3-ил)бензамид;
3-эндо-{8-[2-(3-бензо[1,3]диоксол-5-ил-1-циклогексилметилуреидо)этил]-8-азабицикло [3.2.1] окт-3-ил)бензамид;
3-эндо-{8-[2-(1-циклогексилметил-3-изопропилуреидо)этил]-8-азабицикло [3.2.1] окт-3-ил}бензамид;
3-эндо-(8-{3-[циклогексилметил(2-гидроксиацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил(4-диметиламинобутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[бензил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2,1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2,6-дифторбензил)(2-гидроксиацетил)амино]этил}-8-азабицикло [3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2-метансульфонилацетил)(4-трифторметилбензил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2,6-дифторбензил)(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{3-[бензил(2-гидроксиацетил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(4,4-дифторциклогексилметил)(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[[2-(4-фторфенил)этил](2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(3-фторбензил)(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[[2-(4-фторфенил)этил](2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(4-фторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2-гидроксиацетил)(5-гидрокситрицикло[3.3.1.1(3,7)]декан-2-илметил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил((S)-3-гидрокси-2-метиламино-пропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2,6-дифторбензил)(2-гидрокси-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2,6-дифторбензил)(2-метоксиацетил)амино]этил}-8-азабицикло [3.2.1] окт-3-ил)бензамид;
3-эндо-(8-{2-[(2,6-дифторбензил)((S)-2-гидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(2-цианоацетил)(2,6-дифторбензил)амино]этил}-8-азабицикло [3.2.1] окт-3-ил)бензамид;
3-эндо-(8-{2-[(2,6-дифторбензил)(транс-4-гидроксициклогексанкарбонил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклопентилметил(2-метансульфонилацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклопентилметил(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2,1]окт-3-ил)бензамид;
3-эндо-(8-{2-[бензил((R)-3-гидрокси-2-(S)-гидроксибутирил)-амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[бензил(2,2-диметилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[бензил(2-гидрокси-2-метилпропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[бензил((S)-2-гидрокси-1-оксопропил)амино]этил}-8-азабицикло [3.2.1] окт-3-ил)бензамид;
3-эндо-(8-{3-[бензил((S)-2-гидроксипропионил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил((S)-4-диметиламино-2-гидрокси-бутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил(3-диметиламино-2-гидрокси-пропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-2-[(4,4-дифторциклогексилметил)((8)-2-гидрокси-пропионил)амино]этил-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-2-[циклогексилметил((S)-2,4-дигидроксибутирил)амино]этил-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[циклогексилметил(4-гидроксибутирил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
его фармацевтически приемлемую соль.
12. Соединение по п.11, которое выбирают из группы, включающей:
3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-{2-[(4,4-дифторциклогексилметил)(2-гидроксиацетил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамид;
3-эндо-(8-2-[(4,4-дифторциклогексилметил)((S)-2-гидрокси-пропионил)амино]этил-8-азабицикло [3.2.1]окт-3-ил)бензамид;
его фармацевтически приемлемую соль.
13. Соединение по п.12, которое является гликолятом 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида.
14. Соединение по п.12, которое является оксалатом 3-эндо-(8-{2-[циклогексилметил((S)-2,3-дигидроксипропионил)амино]этил}-8-азабицикло[3.2.1]окт-3-ил)бензамида.
15. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецепторов мю-опиоидов, содержащая соединение по любому из пп.1-14 и фармацевтически приемлемый носитель.
16. Способ получения соединения формулы (I)
где R1, R2, R3, А и G имеют значения, указанные в п.1, его фармацевтически приемлемой соли или защищенного производного, включающий:
(а) взаимодействие соединения формулы (II)

с соединением формулы R4aC(O)-L или соединением формулы R5-N=C=O, где R4a означает R4 или защищенную форму R4a и L означает удаляемую группу или R4aC(O)-L
означает карбоновую кислоту или карбоксилат; или (b) взаимодействие соединения формулы (III)

с соединением формулы (XVII)
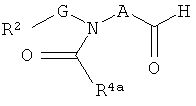 ; и
; и
(с) необязательное удаление защитной группы или групп из R4a с образованием соединения формулы (I), его фармацевтически приемлемой соли или защищенного производного.
17. Соединение формулы (II)
где R1, R2, А и G имеют значения, указанные в п.1;
или его соль.
18. Соединение по п.17,
в котором R1 означает -C(O)NH2;
R2 означает циклогексил или фенил, которые необязательно замещены одним или двумя атомами галогена;
G означает -СН2-; и
А означает -СН2-.
19. Соединение формулы (III)
где R1 означает -ORa или -C(O)NRaRb.
20. Соединение по п.19, в котором R1 означает -ОН или -C(O)NH2.
21. Соединение по любому из пп.1-14, обладающее антагонистической активностью в отношении рецепторов мю-опиоидов.
22. Применение соединения по любому из пп.1-14 для приготовления лекарственного средства для лечения заболевания или болезненного состояния у млекопитающего, ассоциированного с активностью рецептора мю-опиоида.
23. Применение по п.22, в котором заболевание или болезненное состояние представляет собой вызванную опиоидами дисфункцию кишечника или послеоперационную непроходимость кишечника.
24. Способ исследования биологической системы или образца, содержащего рецептор мю-опиоида, который включает:
(a) осуществление контактирования биологической системы или образца с соединением по любому из пп.1-14; и
(b) определение эффекта, оказываемого данным соединением на биологическую систему или образец.
25. Способ лечения млекопитающего, страдающего заболеванием, обусловленным активностью рецептора мю-опиоида, который включает введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по любому из пп.1-14.
26. Способ по п.25, в котором заболевание выбирают из вызванной опиоидами дисфункции кишечника и послеоперационной непроходимости кишечника.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ПРОИЗВОДНЫЕ 8-АЗАБИЦИКЛО[3.2.1] ОКТАН-3-МЕТАНАМИНА В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРОВ ДОФАМИНА D И D И РЕЦЕПТОРОВ СЕРОТОНИНА 5HT И 5HT И СОДЕРЖАЩЕЕ ИХ ЛЕКАРСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2203281C2 |

