Область техники, к которой относится изобретение
Изобретение относится к хинолинонкарбоксамидным соединениям, которые применимы в качестве агонистов 5-HT4 рецепторов. Изобретение также относится к фармацевтическим композициям, включающим такие соединения, способам применения таких соединений для лечения и профилактики патологических состояний, связанных с активностью 5-HT4 рецепторов, а также процессам и промежуточным продуктам, применимым для получения таких соединений.
Уровень техники
Серотонин (5-гидрокситриптамин, 5-HT) является нейромедиатором, который широко распространен в организме человека, как в центральной нервной системе, так и в периферических тканях. Идентифицировано, по крайней мере, семь подтипов серотониновых рецепторов и взаимодействие серотонина с этими разными рецепторами связано с широким разнообразием физиологических функций. Поэтому имеется существенная заинтересованность в разработке терапевтических агентов, мишенями которых являются специфические подтипы 5-HT рецепторов.
В частности, характеристика 5-HT4 рецепторов и идентификация фармацевтических агентов, которые с ними взаимодействуют, находятся в фокусе внимания в последнее время (см., например, обзор Langlois и Fischmeister, J. Med. Chem. 2003, 46, 319-344). Агонисты рецепторов 5-HT4 применимы для лечения расстройств сниженной моторной активности желудочно-кишечного тракта. Такие расстройства включают синдром раздраженной кишки (IBS), хронический запор, функциональную диспепсию, замедленное опорожнение желудка, гастроэзофагеальную рефлюксную болезнь (ГЭРБ), парез желудка, послеоперационную кишечную непроходимость, кишечную псевдообструкцию, лекарственное замедление транзита по ЖКТ. Кроме того, предполагается, что некоторые соединения - агонисты рецепторов 5-HT4 могут быть использованы в лечении расстройств центральной нервной системы, включая когнитивные расстройства, расстройства поведения, расстройства настроения и расстройства контроля вегетативных функций.
Несмотря на широкое применение фармацевтических агентов, влияющих на активность рецепторов 5-HT4, только некоторые агонисты рецепторов 5-HT4 в настоящее время применяются в клинической практике. Один из препаратов, цисаприд, который широко применялся для лечения расстройств моторики желудочно-кишечного тракта, был отозван с рынка, как сообщалось, из-за побочного действия на сердце. Последняя фаза клинических исследований другого средства, прукалоприда, приостановлена.
Таким образом, существует потребность в новых агонистах 5-HT4 рецепторов, которые оказывают желаемый эффект при минимальном побочном действии. Предпочтительные средства могут обладать помимо других свойств улучшенной избирательностью, сильнодействием, фармакокинетическими свойствами и/или длительностью действия.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к новым соединениям, которые обладают активностью агонистов 5-HT4 рецепторов. Среди других свойств соединения по изобретению, как было найдено, являются сильнодействующими и избирательными агонистами 5-HT4 рецепторов. Кроме того, соединения по изобретению, как было найдено, демонстрируют благоприятные фармакокинетические свойства, что прогнозирует хорошую биодоступность при оральном введении.
Таким образом, изобретение относится к соединению формулы (I):

где:
R1 представляет собой водород, галоген, гидрокси, C1-4алкил или C1-4алкокси;
R2 представляет собой C3-4алкил, или C3-6циклоалкил;
R3 представляет собой водород или C1-3алкил;
R4 представляет собой -S(O)2R6 или -C(O)R7;
R5 представляет собой водород, C1-3алкил, C2-3алкил, замещенный с помощью -OH или C1-3алкокси, или -CH2-пиридил;
R6 представляет собой C1-3алкил;
или, R5 и R6, взятые вместе, образуют C3-4алкиленил; и
R7 представляет собой водород, C1-3алкил или пиридил;
или к его фармацевтически приемлемой соли или сольвату или стереоизомеру.
Изобретение также относится к фармацевтической композиции, содержащей соединение по изобретению и фармацевтически приемлемый носитель.
Изобретение также относится к способу лечения заболевания или патологического состояния, связанного с действием 5-HT4 рецепторов, например расстройства сниженной моторной активности желудочно-кишечного тракта, к способу, включающему введение млекопитающему терапевтически эффективного количества соединения по изобретению.
Кроме того, изобретение относится к способу лечения заболевания или патологического состояния, связанного с активностью 5-HT4 рецепторов у млекопитающего, к способу, включающему введение млекопитающему терапевтически эффективного количества фармацевтической композиции по изобретению.
Соединения по изобретению могут также быть использованы в качестве инструментов для исследования, то есть для изучения биологических систем или объектов, или для изучения активности других химических соединений. Таким образом, в других аспектах указанного способа, изобретение обеспечивает способ применения соединения формулы (I), или его фармацевтически приемлемой соли или сольвата или его стереоизомера, как инструмента для исследования биологической системы или образца или для раскрытия новых агонистов 5-HT4 рецепторов, способ, включающий контактирование биологической системы или образца с соединением по изобретению и определением воздействия, вызванного соединением, на биологическую систему или объект.
В отдельных и определенных аспектах, изобретение также раскрывает методики синтеза и промежуточные соединения, описанные в настоящей заявке, которые являются пригодными для получения соединений по изобретению.
Изобретение также относится к соединениям по изобретению, как описано в настоящей заявке, для применения в медицинской терапии, так же как для применения соединения по изобретению для производства состава или лекарственного препарата для лечения заболевания или состояния, связанного с действием 5-HT4 рецепторов, например расстройства сниженной моторики желудочно-кишечного тракта у млекопитающего.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение обеспечивает новые хинолинонкарбоксамидные агонисты 5-HT4 рецепторов формулы (I), или их фармацевтически приемлемые соли или их сольваты или их стереоизомеры. Следующие заместители и обозначения предназначены для раскрытия типичных примеров различных аспектов по настоящему изобретению. Эти типичные значения предназначены для дальнейшего определения таких аспектов и не предназначены для исключения других значений или ограничений области изобретения.
В конкретном аспекте изобретения, R1 представляет собой водород, галоген, С1-4алкил, или C1-4алкокси.
В других конкретных аспектах, R1 представляет собой водород, галоген, или C1-4алкил; или R1 представляет собой водород или галоген; или R1 представляет собой фтор; или R1 представляет собой бром.
В еще другом конкретном аспекте, R1 представляет собой водород.
В конкретном аспекте, R2 представляет собой C3-4алкил или C3-6циклоалкил.
В другом конкретном аспекте, R2 представляет собой C3-4алкил. Типичные R2 группы включают н-пропил, изoпропил, н-бутил, втор.-бутил и трет-бутил.
В другом конкретном аспекте, R2 представляет собой изoпропил.
В еще других конкретных аспектах, R2 представляет собой C3-4алкил или C4-5циклоалкил; или R2 представляет собой изoпропил или C4-5циклоалкил.
В конкретном аспекте, R3 представляет собой водород или C1-3алкил.
В других конкретных аспектах, R3 представляет собой водород, или R3 представляет собой метил.
В конкретном аспекте, R4 представляет собой -S(O)2R6, где R6 представляет собой C1-3алкил.
В другом конкретном аспекте, R4 представляет собой -S(O)2CH3.
В конкретном аспекте, R4 представляет собой -C(O)R7, где R7 представляет собой водород, C1-3алкил или пиридил.
В других конкретных аспектах, R4 представляет собой -C(O)R7, где R7 представляет собой водород или C1-3алкил;
или R4 представляет собой -C(O)R7, где R7 представляет собой водород или метил; или R4 представляет собой -C(O)R7, где R7 представляет собой водород; или R4 представляет собой -C(O)R7, где R7 представляет собой метил.
В еще другом конкретном аспекте, R4 представляет собой -C(O)R7, где R7 представляет собой 3-пиридил или 4-пиридил.
В конкретном аспекте, R5 представляет собой водород; C1-3алкил; C2-3алкил, замещенный с помощью -OH или C1-3алкокси; или -CH2-пиридил.
В других конкретных аспектах, R5 представляет собой водород, C1-3алкил или -CH2-пиридил; или R5 представляет собой водород или C1-3алкил.
В еще других конкретных аспектах, R5 представляет собой -CH2-3-пиридил; или R5 представляет собой водород или метил; или R5 представляет собой водород; или R5 представляет собой метил.
В еще других конкретных аспектах, R5 и R6, взятые вместе, образуют -(CH2)3- или -(CH2)4; или R5 и R6, взятые вместе, образуют -(CH2)3-.
В одном аспекте, изобретение обеспечивает соединение формулы (I), где R3 представляет собой водород.
В другом аспекте, изобретение обеспечивает соединение формулы (I), где R4 представляет собой -S(O)2R6.
В другом аспекте, изобретение обеспечивает соединение формулы (I), где R4 представляет собой -C(O)R7.
Изобретение, кроме того, обеспечивает соединение формулы (I), где R1 представляет собой водород или галоген; R2 представляет собой изoпропил или C4-5циклоалкил; и R3, R4, R5, R6 и R7 определены как в формуле (I).
В еще другом аспекте, изобретение обеспечивает соединение формулы (I), где:
R1 представляет собой водород;
R2 представляет собой C3-4алкил или C4-5циклоалкил;
R3 представляет собой водород;
R4 представляет собой -S(O)2R6 или -C(O)R7;
R5 представляет собой водород или C1-3алкил;
R6 представляет собой C1-3алкил; и
R7 представляет собой водород или C1-3алкил.
В еще другом аспекте, изобретение обеспечивает группу соединений формулы (II):

где R1 представляет собой водород, R2 представляет собой изoпропил, и R3, R4, R5 и R6, или R3, R4, R5 и R7 принимают значения, показанные в Таблице I и II соответственно.
Химические названия обозначений, используемых в настоящем описании, проиллюстрированы для соединения по Примеру 1:

которое обозначает {(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты, что предложено программой AutoNom, выпущенной MDL Information Systems, GmbH (Frankfurt, Germany). Определение (1S,3R,5R) описывает относительную ориентацию связей, связанных с бициклической кольцевой системой, которую определяют как сплошные и пунктирные клинья. Соединение альтернативно определяют как N-[(3-эндо)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил]-1-(1-метилэтил)-2-оксо-1,2-дигидро-3-хинолинкарбоксамид. Во всех соединениях по изобретению, обозначенных выше, хинолинонкарбоксамид находится в эндо положении по отношению к азабициклооктановой группе.
Можно перечислить следующие конкретные соединения
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты; и
{(1S,3R,5R)-8-[(R)-3-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты.
Как видно на примере типичных соединений, приведенных выше, соединения по изобретению могут содержать хиральный центр, в особенности, при атоме углерода в формулах (I) или (II), несущем заместитель -OR3. Таким образом, изобретение включает рацемические смеси, чистые стереоизомеры, и смеси, обогащенные стереоизомерами, если иное не обозначено. Когда указано на конкретный стереоизомер, среднему специалисту понятно, что в композициях по изобретению могут присутствовать незначительные количества других стереоизомеров, если иное не обозначено, при условии, что любое применение композиции в целом, не ограничивается из-за присутствия таких других изомеров.
Определения
Когда описывают соединения, композиции и способы по изобретению, нижеследующие термины имеют следующие значения, если иное не обозначено.
Термин "алкил" использован для обозначения одновалентной насыщенной углеводородной группы, которая может быть линейной или разветвленной или их комбинации. Если иное не определено, такая алкильная группа, как правило, содержит от 1 до 10 атомов углерода. Представители алкильных групп включают, в качестве примера, метил, этил, н-пропил (n-Pr), изoпропил (i-Pr), н-бутил (n-Bu), втор.-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и им подобные.
Термин "алкиленил" использован для обозначения двухвалентной насыщенной углеводородной группы, которая может быть линейной или разветвленной или их комбинациями. Если иное не определено, такая алкиленильная группа, как правило, содержит от 1 до 10 атомов углерода. Представители алкиленильных групп включают, в качестве примера, метилен, этилен, н-пропилен, н-бутилен, пропан-1,2-диил(1-метилэтилен), 2-метилпропан-1,2-диил (1,1-диметилэтилен) и им подобные.
Термин "алкокси" использован для обозначения одновалентной группы -O-алкил, где алкил определен как указано выше. Представители алкоксигрупп включают, в качестве примера, метокси, этокси, пропокси, бутокси, и им подобные.
Термин "циклоалкил" использован для обозначения одновалентной насыщенной карбоциклической группы, которая может быть моноциклической или полициклической. Если иное не определено, такая циклоалкильная группа, как правило, содержит от 3 дo 10 атомов углерода. Представители циклоалкильных групп включают, в качестве примера, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, и им подобные.
Термин "галоген" использован для обозначения фтора, хлора, брома или иода.
Термин "соединение" использован для обозначения соединения, которое получено синтетически или получено любым другим путем, таким как метаболизм.
Термин "терапевтически эффективное количество" использован для обозначения количества, достаточного для эффективного лечения, которое вводят пациенту, нуждающемуся в лечении.
Термин "лечение" использован в настоящем описании для обозначения лечения заболевания, нарушения или патологического состояния пациента, такого как млекопитающее (особенно человека), который включает:
(а) профилактику заболевания, нарушения или возникновения патологического состояния, то есть профилактическое лечение пациента;
(b) уменьшение интенсивности симптомов заболевания, нарушения или патологического состояния, то есть ликвидация или вызов регрессии заболевания, нарушения или патологического состояния у пациента;
(c) подавление заболевания, нарушения или патологического состояния, то есть замедление или остановку развития заболевания, нарушения или патологического состояния у пациента; или
(d) облегчение симптомов заболевания, нарушения или патологического состояния у пациента.
Термин "фармацевтически приемлемая соль" использован для обозначения соли, полученной из кислоты или основания, которая приемлема для введения пациенту, такому как млекопитающее. Такие соли могут быть получены из фармацевтически приемлемых минеральных или органических кислот и из фармацевтически приемлемых оснований. Как правило, фармацевтически приемлемые соли соединений по настоящему изобретению получают из кислот.
Соли, полученные из фармацевтически приемлемых кислот, включают, но без ограничения, уксусную, адипиновую, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, молочную, малеиновую, янтарную, миндальную, метансульфоновую, слизевую, азотную, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую, ксинафоновую (1-гидрокси-2-нафтойная кислота), нафталин-1,5-дисульфоновую кислоту и им подобные.
Термин "сольват" использован для обозначения комплекса или агрегата, образованного с помощью одной или большего количества молекул в растворе, то есть соединения по изобретению или его фармацевтически приемлемой соли, и одной или большего количества молекул растворителя. Такие сольваты являются, как правило, кристаллическими твердыми веществами, имеющими по существу постоянное мольное соотношение растворенного вещества и растворителя. Представители растворителей включают в качестве примера, воду, метанол, этанол, изопропанол, уксусную кислоту, и им подобные. Когда растворителем является вода, образующийся сольват представляет собой гидрат.
Будет понятно, что термин "или фармацевтически приемлемая соль или сольват его стереоизомера", как предполагается, включает все комбинации солей, сольватов и стереоизомеров, такие как сольват фармацевтически приемлемой соли стереоизомера соединения формулы (I).
Термин "аминозащитная группа" использован для обозначения защитной группы, подходящей для предупреждения нежелательных реакций по азоту аминогруппы. Представители аминозащитных групп включают, но без ограничения, формил; ацильные группы, например алканоильные группы, такие как ацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr), и 1,1-ди-(4'-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBDMS); и им подобные.
Общие способы получения
Соединения по изобретению могут быть получены из легко доступных исходных веществ, используя следующие общие способы и методики. Несмотря на то, что конкретный аспект настоящего изобретения представлен на схемах, указанных ниже, средним специалистом будет оценено, что все аспекты настоящего изобретения могут быть осуществлены, используя способы, описанные в настоящем изобретении, или используя другие способы, реагенты и исходные вещества, известные среднему специалисту. Также должно быть учтено, там, где проведены обычные или предпочтительные условия для осуществления процесса (то есть температура реакционной смеси, время, мольное соотношение реагентов, растворители, давление, и так далее), могут также быть использованы другие условия, если иное не указано. Оптимальные реакционные условия могут изменяться при использовании конкретных реагентов или растворителя, но такие условия могут быть определены средним специалистом в данной области с помощью рутинной оптимизации способа.
Кроме того, как очевидно среднему специалисту, обычные защитные группы могут быть необходимы для защиты некоторых функциональных групп от протекания нежелательных реакций. Выбор подходящей защитной группы для конкретной функциональной группы, так же как подходящих условий для защиты и снятия защиты, хорошо известны среднему специалисту. Например, многочисленные защитные группы, и их введение и удаление описаны у T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и приведены как ссылки в настоящем описании.
В одном из способов синтеза соединение формулы (I) получают как представлено на Схеме A. (Заместители и переменные, представленные в последующих схемах, имеют определения, указанные выше, если иное не обозначено).
Схема A
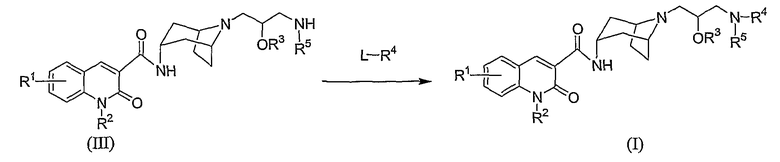
На Схеме A, L представляет собой уходящую группу, такую как хлор, бром, иод, или этокси, или реагент L-R4 представляет собой карбоновую кислоту HO-C(O)R7, то есть L формально представляет собой гидроксильную группу.
Оптимальные условия реакции для реакционной Схемы A могут изменяться в зависимости от химических свойств реагента L-R4, как хорошо известно среднему специалисту.
Например, когда L представляет собой галогеновую уходящую группу, такую как хлор, реакцию, как правило, проводят путем взаимодействия промежуточного соединения формулы (III), со взятым в количестве от около 1 и до около 4 эквивалентов соединения формулы L-R4 в инертном растворителе, таком как дихлорметан, в присутствии избытка основания, например, взятого в количестве от около 3 и до около 6 эквивалентов основания, такого как N,N-диизoпропилэтиламин или 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU). Подходящие инертные растворители также включают N,N-диметилформамид, трихлорметан, 1,1,2,2-тетрахлорэтан, тетрагидрофуран, и им подобные. Реакцию, как правило, проводят при температуре в интервале от около -100°C дo около 30°С в течение времени от около четверти часа дo около 2 часов, или пока реакция в основном не завершится. Примеры реагентов формулы L-R4, в которой L представляет собой хлор, включают метансульфонилхлорид и ацетилхлорид.
Когда реагент формулы L-R4 представляет собой карбоновую кислоту, Схема A представляет собой реакцию амидного связывания, которую, как правило, проводят путем взаимодействия промежуточного соединения формулы (III) со взятым в количестве от около 1 и до около 4 эквивалентов соединением карбоновой кислоты L-R4 в инертном растворителе, например, N,N-диметилформамиде, в присутствии агента связывания, такого как гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония (PyBop). Реакцию, как правило, проводят при комнатной температуре, в течение времени от около четверти часа дo около 2 часов, или пока реакция в основном не завершится. Подходящие альтернативные агенты связывания включают 1,3-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC), и PyBop, комбинированный с 1-гидрокси-7-азабензотриазолом (НОАt).
Амидное связывание промежуточного соединения формулы (III) с карбоновой кислотой L-R4 альтернативно может быть выполнено путем превращения соединения формулы L-R4 в активированный эфир, такой как N-гидроксисукцинимидный эфир (NHS) или п-нитрофенильный эфир, или имидазольную кислоту, которые затем взаимодействуют с промежуточным соединением формулы (III).
Альтернативно, когда реагент L-R4 представляет собой жидкость, например этилформиат, реакция может быть выполнена путем растворения соединения формулы (III) в значительном избытке реагента формулы L-R4, и нагревания дo температуры в интервале от между около 50°C и до около 100°С в течение времени от около от 12 дo около 24 часов.
Продукт формулы (I) выделяют и очищают с помощью обычных способов. Например, продукт может быть сконцентрирован досуха при пониженном давлении, перенесен в слабый водный раствор кислоты и очищен с помощью ВЭЖХ.
Альтернативно, соединение формулы (I) может быть получено путем N-алкилирования соединения формулы (I), в котором R2 представляет собой водород, который может быть получен в соответствии со Схемой A. Реакцию N-алкилирования, как правило, проводят путем взаимодействия соединения формулы (I), в котором R2 представляет собой водород, со взятым в количестве от около 1 и до около 4 эквивалентов соединением формулы L'-R2, в котором L' представляет собой уходящую группу, такую как иод или бром. Указанную реакцию, как правило, проводят в полярном апротонном растворителе, таком как диметилформамид в присутствии взятого от около 2 и до около 4 эквивалентов сильного основания, такого как трет-бутоксид калия. Как правило, реакцию выполняют в интервале температур от около 60°C и примерно до 100°С в течение времени от около 6 и около 24 часов, или пока реакция в основном не завершится.
В еще другом альтернативном способе, соединение формулы (I), в котором R1 является иным, чем водород, получают с помощью обычных методик из соединений формулы (I), в которых R1 представляет собой водород.
Промежуточные соединения формулы (III) получают из готовых доступных исходных продуктов. Например, когда углерод, несущий заместитель -OR3, не является хиральным, промежуточное соединение формулы (III) получают с помощью методики, представленной на Схеме B.
Схема B
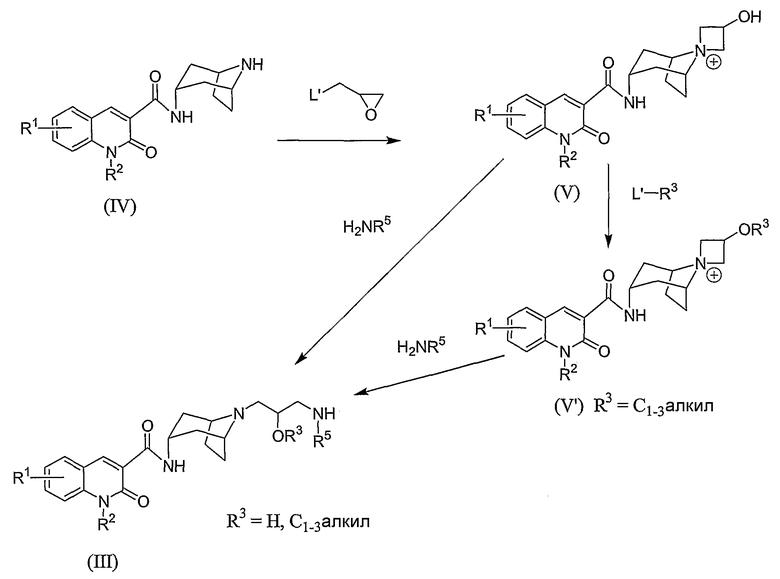
где L1 независимо представляет собой галогеновую уходящую группу, такую как бром, хлор или иод. Отрицательно заряженный противоион также присутствует, связанный с положительно заряженным промежуточным соединением формулы (V) или (V').
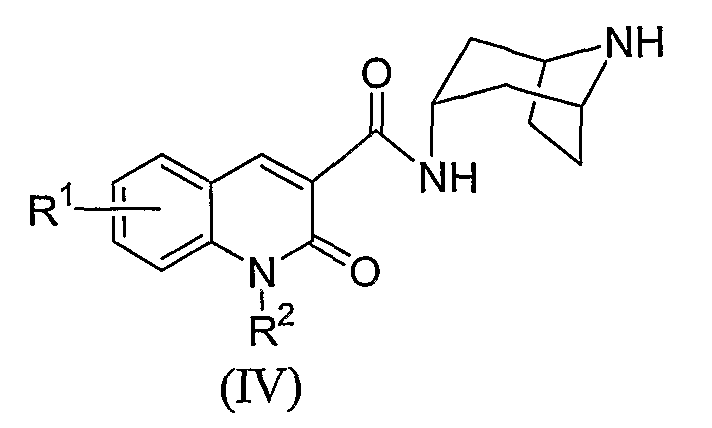
Вначале, промежуточное соединение формулы (IV) взаимодействует с оксирановым соединением, например, 2-бромметилоксираном (как правило, эпибромгидрином), чтобы образовать азетидиновую соль формулы (V). Эту реакцию, как правило, проводят путем взаимодействия соединения формулы (IV), со взятым от около 2 и до около 4 эквивалентов 2-бромметилоксирана в полярном растворителе, таком как этанол. Реакцию, как правило, проводят при комнатной температуре в течение времени от около 24 и до около 48 часов, или пока реакция в основном не завершится.
Должно быть понятно, что в соответствии с методикой на Схеме B и в соответствии с другими методиками, описанными ниже, в случае необходимости, применение промежуточного соединения формулы (IV), как известно среднему специалисту, может быть обеспечено в форме свободного основания или в виде соли с соответствующей корректировкой условий реакции, если это необходимо.
Промежуточное соединение формулы (V'), в котором R3 представляет собой C1-3алкил, может быть получено путем контактирования промежуточного соединения формулы (V) со взятым не менее, чем от одного до примерно одного эквивалента соединения формулы L'-R3, где R3 представляет собой C1-3алкил в инертном растворителе в присутствии от около 1 и до около 3 эквивалентов сильного основания, такого как трет-бутоксид калия или гидрид натрия. Реакцию, как правило, проводят при комнатной температуре в течение времени от около четверти часа до часа, или пока реакция в основном не завершится. Подходящие инертные растворители включают дихлорметан, трихлорметан, 1,1,2,2-тетрахлорэтан, и им подобные.
Кроме того, азетидиновое промежуточное формулы (V) или (V') взаимодействует с амином формулы H2NR5, чтобы обеспечить промежуточное соединение формулы (III). Как правило, промежуточный азетидин растворяют в инертном растворителе, таком как этанол, и взаимодействие осуществляют со взятым от около 1 и до около 8 эквивалентов амина H2NR5. Например, когда амин H2NR5 представляет собой летучий реагент, такой как метиламин, предпочтительно, используют амин в количестве, взятый от около 5 и до около 7 эквивалентов. Реакцию, как правило, проводят при температуре от примерно 50°C и до примерно 100°С в течение времени от около 12 и около 24 часов, или пока реакция в основном не завершится.
Промежуточное соединение формулы (III), в котором R5 представляет собой водород, может быть получено из азетидинового промежуточного формулы (V) или (V'), используя формиат аммония вместо аммиака, то есть вместо реагента формулы H2NR5, обозначенного на Схеме B. Альтернативно, для получения промежуточного соединения формулы (III), где R5 представляет собой водород, азетидиновое кольцо формулы (V) или (V') может быть раскрыто реакцией с азидом, таким как азид натрия, с последующим осуществлением реакции восстановления, чтобы обеспечить промежуточное соединение формулы (III), или кольцо может быть раскрыто с помощью реакции с гидроксидом аммония.
Как описано подробно в Примере 4a, когда R3 и R5 представляют собой водород и углерод, несущий заместитель -OR3 не является хиральным, промежуточное соединение формулы (III) может быть получено путем взаимодействия промежуточного соединения формулы (IV) с оксиранилметильным соединением, имеющим защищенный атом азота, с последующим снятием защиты. Одним из пригодных реагентов является 2-оксиранилметилизоиндол-1,3-дион, как правило, эпоксипропилфталимид, который взаимодействует с промежуточным соединением формулы (IV), чтобы образовать промежуточное соединение, в котором фталимидилзамещенная 2-гидроксипропильная группа:
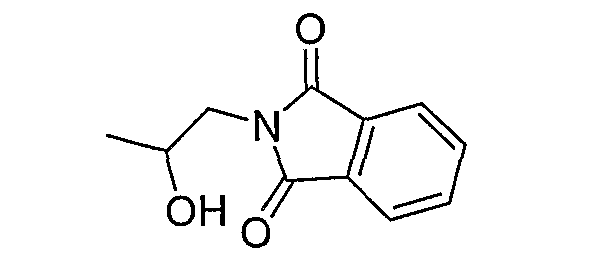
связана с атомом азота aзабициклооктанового кольца формулы (IV). Фталимидильную группу затем удаляют при нагревании до кипения с обратным холодильником в гидразине с образованием промежуточного соединения формулы (III), в котором R3 и R5 представляют собой водород.
Промежуточное соединение формулы (III), в котором R3 и R5 представляют собой водород, может также быть получено с помощью реакции азетидина формулы (V) с анионом фталимида и последующей обработкой гидразином.
В альтернативном способе синтеза, промежуточное соединение формулы (III), в котором R3 представляет собой водород, может быть получено с помощью реакции промежуточного соединения формулы (IV) с защищенным промежуточным соединением формулы (VI):

с последующей стадией снятия защиты. В формуле (VI), P1 представляет собой аминозащищенную группу, L' представляет собой галогеновую уходящую группу, и звездочка обозначает хиральный центр. Способ с использованием промежуточного соединения формулы (VI) является применимым для получения различных форм промежуточного соединения формулы (III), в котором стереохимия на центре, отмеченном с помощью звездочки, конкретно является (R) или (S), так же как для получения нехиральных форм промежуточного соединения формулы (III).
Как правило, промежуточное соединение формулы (IV) взаимодействуют со взятым от около 1 и до около 2 эквивалентов промежуточным соединением формулы (VI) в полярном растворителе, таком как метанол, в присутствии большего, чем один эквивалент количества основания, такого как N,N-диизoпропилэтиламин. Реакцию, как правило, проводят при температуре от примерно 60°C и до примерно 100°С в течение времени от около 12 и до около 24 часов, или пока реакция в основном не завершится. Защитную группу P1 удаляют с помощью обычной методики, чтобы обеспечить промежуточное соединение формулы (III). Пригодной защитной группой P1 является Boc, которую, как правило, удаляют обработкой кислотой, такой как трифторуксусная кислота.
В еще другом альтернативном процессе получения промежуточного соединения формулы (III), промежуточное соединение формулы (VI) может вначале быть превращено в цикличную форму формулы (VII):
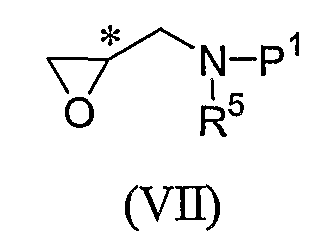
перед выполнением реакции с промежуточным соединением формулы (IV), чтобы обеспечить промежуточное соединение формулы (III). Промежуточное соединение формулы (VII), как правило, получают путем растворения промежуточного соединения формулы (VI) в инертном растворителе, например, тетрагидрофуране, в присутствии основания, например гидроксида натрия. Реакцию соединения формулы (VII) с соединением формулы (IV), чтобы обеспечить промежуточное соединение формулы (III), как правило, выполняют путем взаимодействия промежуточного соединения формулы (IV) со взятым от около 1 и до около 4 эквивалентов промежуточным соединением формулы (VII) в полярном растворителе, таком как метанол. Реакцию, как правило, проводят при температуре от примерно 60°C дo примерно 100°С в течение времени от около часа и до около 4, часов, или пока реакция в основном не завершится. Защитную группу P1 удаляют с помощью обычной методики, чтобы обеспечить промежуточное соединение формулы (III).
Защищенное промежуточное соединение формулы (VI) может быть получено из оксирана, как представлено на Схеме С для конкретного примера образования Boc-защищенного хирального промежуточного соединения формулы (VI'), используя хиральный оксиран. Реакция является одинаково пригодной для получения нехиральных соединений формулы (VI).
Схема C

Как показано на Схеме C, бензиламин 2 контактирует с по крайней мере одним эквивалентом хирального оксирана формулы 1 в неполярном растворителе, таком как гексан или толуол, чтобы образовать 2-гидроксипропиламин формулы 3. Реакцию, как правило, проводят при комнатной температуре в течение времени от около 12 и до около 24 часов, или пока реакция в основном не завершится. Промежуточное соединение формулы 3, как правило, взаимодействует с небольшым избытком ди-трет-бутилдикарбоната (как правило, (Boc)2O, например, около 1,1 эквивалента, в атмосфере водорода в присутствии катализатора из переходного металла, чтобы обеспечить Boc-защищенное промежуточное соединение формулы (VI'). Реакцию, как правило, проводят при комнатной температуре в течение времени от около 8 дo около 24 часов.
Способ получения промежуточного соединения формулы (IV) показан на Схеме D.
Схема D

Защищенный аминоазабициклооктан, или как правило, аминотропан формулы 5, вначале взаимодействует с замещенной хинолинонкарбоновой кислотой формулы (VIII). Как правило, эту реакцию проводят вначале с помощью превращения соединения формулы (VIII) в хлорангидрид кислоты, путем взаимодействия соединения формулы (VIII) с, по крайней мере, одним эквивалентом, предпочтительно взятым от около 1 и до около 2 эквивалентов активного агента, такого как тионилхлорид или оксалилхлорид в ароматическом растворителе, таком как толуол, бензол, ксилол, или им подобные. Реакцию, как правило, проводят в интервале температур от примерно 80°C дo примерно 120°С в течение времени от около 15 минут дo около 4 часов, или пока реакция в основном не завершится.
Раствор хлорангидрида, как правило, добавляют в двухфазную смесь из примерно 1 эквивалента аминотропана формулы 5, чтобы образовать защищенное промежуточное соединение, которое экстрагируют с помощью обычной методики. Двухфазную смесь соединения формулы 5, как правило, получают путем растворения соединения формулы 5 в ароматическом растворителе, таком как указанные выше, и добавлением водного раствора, содержащего избыток основания, такого как гидроксид натрия или гидроксид калия, предпочтительно от около 2 и дo 5 эквивалентов основания.
Альтернативно, амидное связывание промежуточного соединения формулы 5 с карбоновой кислотой формулы (VIII) может быть выполнено в присутствии агента связывания, такого как 1,3-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC) или гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония (PyBop), необязательно в сочетании с 1-гидрокси-7-азабензотриазолом (НОАt), как описано выше для амидного связывания промежуточного соединения формулы (III) с карбоновой кислотой. В еще другой альтернативной методике, амидное связывание промежуточного соединения формулы 5 с карбоновой кислотой формулы (VIII) может быть осуществлено с помощью превращения соединения формулы (VIII) в активированный эфир, такой как описанный выше.
Защитную группу P1 удаляют с помощью обычной методики, чтобы обеспечить промежуточное соединение формулы (IV). Например, когда защитная группа представляет собой Boc, как правило, удаление осуществляют обработкой кислотой, такой как трифторуксусная кислота, обеспечивая получение кислой соли промежуточного соединения. Кислая соль промежуточного соединения формулы (IV) может быть превращена в свободное основание, при необходимости, путем обычной обработки основанием. Защитную группу Cbz, для другого примера, легко удаляют с помощью гидрогенолиза над подходящим металлическим катализатором, таким как палладий на угле.
Защищенный аминотропан формулы 5, применяемый в реакциях, описанных в настоящей заявке, получают из легко доступных исходных веществ. Например, когда защитная группа P1 представляет собой Boc, защищенный аминотропан формулы 5' получают с помощью методики, представленной на Схеме E.
Схема E
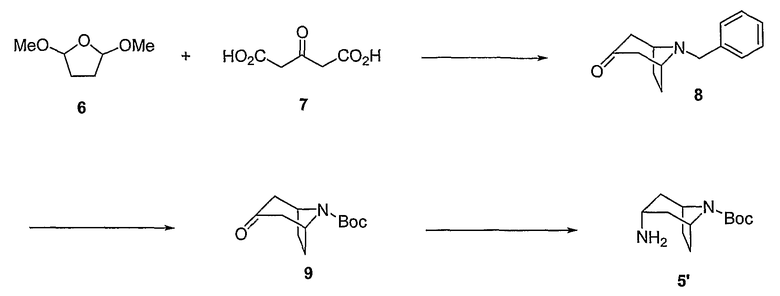
Как описано подробно в Примере 1a, представленном ниже, для получения защищенного промежуточного соединения формулы 5', вначале 2,5-диметокситетрагидрофуран формулы 6 взаимодействуют со взятым примерно 1 и 2 эквивалентами, предпочтительно примерно 1,5 эквивалентами бензиламина и небольшим избытком, например, около 1,1 эквивалентом, 1,3-ацетондикарбоновой кислоты формулы 7 в кислом водном растворе в присутствии буферного агента, такого как гидрофосфат натрия. Реакционную смесь нагревают до температуры от приблизительно 60 и до приблизительно 100°C, чтобы обеспечить декарбоксилирование любых карбоксилированных промежуточных соединений в продукт, 8-бензил-8-азабицикло[3.2.1]октан-3-он формулы 8, как правило, N-бензилтропанон.
Промежуточное соединение формулы 8, как правило, взаимодействует с небольшим избытком ди-трет-бутилдикарбоната (как правило, (Boc)2O), например, взятым в количестве, около 1,1 эквивалентов, в атмосфере водорода в присутствии катализатора переходного металла, чтобы обеспечить Boc-защищенное промежуточное соединение формулы 9, трет-бутиловый эфир 3-оксо-8-азабицикло[3.2.1]октан-8-карбоновой кислоты. Реакцию, как правило, проводят при комнатной температуре в течение времени от около 12 дo около 72 часов. В конечном счете, промежуточное соединение формулы 9 взаимодействует со значительным избытком, например, по крайней мере около 25 эквивалентами формиата аммония в инертном растворителе, таком как метанол, в присутствии катализатора переходного металла, чтобы обеспечить продукт соединения формулы 5' в эндо конфигурации с высокой стереоспецифичностью, например, при соотношении эндо к экзo >99:1. Реакцию, как правило, проводят при комнатной температуре в течение времени от около 12 дo около 72 часов, или пока реакция в основном не завершится. Целесообразно добавлять порциями формиат аммония. Например, промежуточное соединение формулы 9 взаимодействует с первоначальной порцией формиата аммония, взятом в количестве от около 15 дo около 25 эквивалентов. После интервала времени от около 12 дo около 36 часов добавляют дополнительную порцию формиата аммония в количестве от около 5 дo около 10 эквивалентов. Последующее добавление может быть повторено после завершения аналогичного интервала времени. Продукт соединения формулы 5' может быть очищен с помощью обычной методики, такой как щелочная экстракция.
При альтернативном способе синтеза соединение формулы (I) получают путем связывания замещенной хинолинонкарбоновой кислоты формулы (VIII) с промежуточным соединением формулы (IX) как представлено на Схеме F.
Схема F

Реакцию по Схеме F, как правило, проводят в условиях амидного связывания, описанного выше для реакции карбоновой кислоты формулы (VIII) с промежуточным соединением формулы 5.
Промежуточное соединение формулы (IX) может быть получено путем снятия защиты с промежуточного соединения формулы (Х):

где P2 представляет собой аминозащитную группу.
Промежуточные соединения формулы (X) могут быть получены из легко доступных исходных продуктов, используя методики, аналогичные алкилированию и другим реакциям, описанных выше, и/или используя альтернативные реакции, хорошо известные среднему специалисту. Например, промежуточное соединение формулы (X) может быть получено, используя промежуточное соединение формулы 10

которое может быть образовано путем защиты азота аминогруппы аминоазобициклооктана формулы 5 с помощью аминозащитной группы P2 и последующего удаления P1 с азота азабициклооктановой группы. Защитные группы P1 и P2 выбраны такими, что их удаляют в соответствии с указанными ниже различными условиями. Например, когда P1 выбран как Boc, то затем Cbz может быть использован как P2. Замещение защищенного аминотропана формулы 10 для промежуточного соединения формулы (III) в реакциях, описанных выше для получения промежуточного соединения формулы (III), обеспечивает промежуточные соединения формулы (X).
В еще другом способе синтеза соединения формулы (I), в которых R3 представляет собой водород, представленные ниже в виде формулы (I'), могут быть получены как представлено на Схеме G.
Схема G

Промежуточное соединение формулы (XI) может содержать хиральный центр, как показано детально для защищенного оксиранового промежуточного соединения формулы (VII).
Как правило, промежуточное соединение формулы (IV) взаимодействуют в количестве, взятом около 1 и около 2 эквивалентов оксиранового промежуточного формулы (XI) в полярном растворителе, таком как этанол, чтобы образовать продукт соединения формулы (I'). Промежуточное соединение формулы (IV) может быть использовано в виде соли, и в этом случае небольшой мольный избыток щелочного основания содержится в реакционной смеси перед добавлением оксирана. Реакцию, как правило, проводят при температуре от приблизительно 60°C дo приблизительно 100°С в течение времени от около часа и около 3 часов, или пока реакция в основном не завершится. Продукт может быть выделен путем кристаллизации из инертного разбавителя в виде свободного основания или в виде соли кислоты.
Промежуточные соединения формулы (XI) могут быть получены с помощью реакции промежуточного оксирана формулы 1, представленного на Схеме C, с вторичным амином формулы HNR4R5. Как правило, водный раствор амина формулы HNR4R5, содержащий около 1 эквивалента основания, такого как гидроксид натрия, гидроксид лития, гидроксид цезия или гидроксид калия, взаимодействует со взятом в количестве от около 1,5 и до около 2,5 эквивалентами промежуточного оксирана формулы 1. Реакцию, как правило, проводят при температуре от примерно 0°C и до примерно 10°С в течение времени от около 12 и до около 30 часов, или пока реакция в основном не завершится.
Хинолинонкарбоновую кислоту формулы (VIII) легко получают с помощью методики, аналогично той, которая представлена в уровне техники у Suzuki et al., Heterocycles, 2000, 53, 2471-2485 и описана в примерах, представленных ниже.
Реагенты L'-R2, L'-R3, L-R4, H2NR5 и HNR4R5 являются коммерчески доступными или их легко получают с помощью обычной методики из общих исходных продуктов.
Кроме того, детали, касающиеся конкретных условий реакции и других методик получения типичных соединений по изобретению или их промежуточных соединений, описаны в примерах, представленных ниже.
Таким образом, в аспекте способа изобретение обеспечивает методику получения соединения формулы (I), или его соли или стереоизомера, включающую:
(а) взаимодействие соединения формулы (III):

с соединением формулы L-R4, где L представляет собой уходящую группу, или L-R4 представляет собой HO-C(O)R7; или
(b) взаимодействие соединения формулы (VIII):

с соединением формулы (IX):

чтобы обеспечить соединение формулы (I), или его соль или стереоизомер.
Изобретение, кроме того, обеспечивает соединение формулы (III), или его соль или стереоизомер или его защищенное производное, где R1, R2, R3 и R5 определены как в формуле (I).
В дополнительном аспекте способа, изобретение обеспечивает методику получения соединения формулы (I'), где R1, R2, R4 и R5 определены как в формуле (I), или его соли или стереоизомера, включающей взаимодействие соединения формулы (IV):
или его соли с соединением формулы (XI):

чтобы обеспечить соединение формулы (I') или его соль или стереоизомер.
Фармацевтические композиции
Хинолинонкарбоксамидные соединения настоящего изобретения обычно вводят пациенту в форме фармацевтической композиции. Такие фармацевтические композиции могут быть введены пациенту любым приемлемым путем введения, включая, помимо прочего, пероральный, ректальный, вагинальный, интраназальный, ингаляционный, местный (включая трансдермальный) и парентеральные пути введения.
Соответственно, в одном из аспектов, относящегося к композиции, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель или наполнитель и терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемую соль. При необходимости такие фармацевтические композиции могут содержать другие терапевтические и/или вспомогательные агенты.
Фармацевтические композиции изобретения обычно содержат терапевтически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемую соль. Обычно такие фармацевтические композиции содержат приблизительно от 0,1 до 95% активного агента; предпочтительно, от 5 до примерно 70%; более предпочтительно примерно от 10 до 60% по массе активного агента.
Любой традиционный носитель или наполнитель может быть использован в создании фармацевтических композиций настоящего изобретения. Выбор конкретного носителя или наполнителя, или комбинации носителей и наполнителей, будет зависеть от пути введения, применяемого у конкретного пациента, или вида патологического состояния или болезни. В этом отношении, приготовление подходящей фармацевтической композиции для конкретного способа введения находится в компетенции специалиста-фармацевта. Кроме того, ингредиенты таких композиций могут быть приобретены, например, в компании Sigma (P.O. Box 14508, St. Louis, MO 63178). Дальнейшая иллюстрация традиционной методики приготовления препаратов, описана у Remington: Tlie Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типичные примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей включают помимо прочего следующие: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу, такую как микрокристаллическая целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) измельченный трагакант; (5) солод; (6) желатин; (7) тальк; (8) наполнители, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннитол и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновая кислота; (16) апирогенная вода; (17) изотонический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатно-буферные растворы и (21) другие нетоксичные сочетающиеся вещества, применяемые в фармацевтических композициях.
Фармацевтические композиции данного изобретения обычно готовят посредством тщательного смешивания или добавления вещества настоящего изобретения с фармацевтически приемлемым носителем и одним или более необязательных ингредиентов. Если необходимо или требуется, полученной однородно перемешанной смеси придают форму или наполняют ею таблетки, капсулы, пилюли и подобные лекарственные формы, используя обычные методики и оборудование.
Фармацевтические композиции данного изобретения предпочтительно выпускают в виде единичной дозированной формы. Термин "единичная дозированная форма" относится к физически дискретной единице, подходящей для дозирования пациенту, т.е. каждая единица, содержащая заданное количество действующего агента, рассчитана, как обеспечивающая желаемый терапевтический эффект, как в случае монотерапии, так и в комбинации с одним или более дополнительными единицами. Например, такие единичные дозированные формы могут представлять собой капсулы, таблетки, пилюли и т.п.
В предпочтительном воплощении фармацевтические композиции данного изобретения подходят для перорального введения. Пригодные для перорального введения фармацевтические композиции могут быть представлены капсулами, таблетками, пилюлями, ромбовидными пластинками, крахмальными капсулами, драже, порошками, гранулами; или быть в виде раствора или суспензии в водной или безводной жидкости; или в виде жидкой эмульсии по типу "масло-в-воде" или "вода-в-масле"; или в виде эликсира или сиропа; и т.п.; каждая форма содержит заданное количество соединения настоящего изобретения в качестве активного ингредиента.
Для введения внутрь в виде твердой лекарственной формы (т.е. капсул, таблеток, пилюль и т.п) фармацевтические композиции данного изобретения в типичном случае содержат соединение настоящего изобретения в качестве активного ингредиента и один или более фармацевтически приемлемых носителей, таких как цитрат натрия или дикальций фосфат. Необязательно или альтернативно такие твердые лекарственные формы могут также содержать: (1) наполнители или разбавители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннитол и/или кремниевая кислота; (2) связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или гуммиарабик; (3) смачивающие агенты, такие как глицерин; (4) разрыхлители, такие как агар-агар, карбонат кальция, картофельный и маниоковый крахмал, альгиновая кислота, некоторые силикаты и/или карбонат натрия; (5) замедляющие растворение агенты, такие как парафин; (6) ускорители всасывания, такие как четвертичные соединения аммония; (7) увлажняющие агенты, такие как цетиловый спирт и/или моностеарат глицерина; (8) абсорбенты, такие как каолин и/или бентонитовая глина; (9) смызывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смесь; (10) красящие агенты; и (11) буферные агенты.
Высвобождающие агенты, увлажняющие агенты, покровные средства, подсластители, ароматизаторы и отдушки, консервирующие агенты и антиоксиданты могут также присутствовать в фармацевтических композициях данного изобретения. Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия и сульфит натрия и т.п.; (2) жирорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и т.п; и (3) металл-хелатирующие агенты, такие как лимонная кислота, этилендиаминотетрауксусная кислота (ЭДТА), сорбит, винная кислота, фосфорная кислота и т.п. Покровные вещества для таблеток, капсул, пилюль и т.п. включают вещества для кишечнорастворимых оболочек, такие как ацетатфталат целлюлозы (CAP), поливинилацетатфталат (PVAP), фталат гидроксипропилметилцеллюлозы, сополимер метакриловой кислоты и сложного эфира метакриловой кислоты; тримеллитат ацетатцеллюлозы (CAT), карбоксиметилэтилцеллюлоза (CMEC), гидроксипропилметилцеллюлозы ацетат сукцинат (HPMCAS) и т.п.
При необходимости фармацевтические композиции настоящего изобретения могут также быть изготовлены для обеспечения медленного или контролируемого высвобождения активного ингредиента с использованием, в качестве примера, гидроксипропилметилцеллюлозы в различных пропорциях; или другой полимерной матрицы, липосом и/или микросфер.
Кроме того, фармацевтические композиции настоящего изобретения могут по необходимости содержать рентгеноконтрастные средства и могут быть приготовлены таким образом, что они будут высвобождать активный ингредиент исключительно или главным образом в конкретном отделе желудочно-кишечного тракта, по необходимости, замедленным образом. Примеры встраиваемых композиций, которые могут быть использованы, включают полимеры и воски. Активный ингредиент может быть в форме микрокапсул, если это необходимо, с одним или более из вышеописанных наполнителей.
Подходящие жидкие лекарственные формы для приема внутрь включают для примера фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Такие жидкие лекарственные формы обычно включают активный ингредиент и инертный разбавитель, такой как, например, вода или другие растворители, повышающие растворимость агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, угольноэтиловый эфир, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, а также их смеси. Суспензии наряду с активным ингредиентом могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и сложные эфиры сорбитана, микрокристаллическая целлюлоза, алюминия метагидроксид, бентонит, агар-агар и трагакант, а также их смеси.
Альтернативно, фармацевтические композиции данного изобретения выпускаются для ингаляционного пути введения. Подходящие фармацевтические композиции для ингаляционного введения в типичном случае будут в виде аэрозоля или порошка. Такие композиции обычно вводятся с помощью хорошо известных устройств доставки, таких как дозированный ингалятор, порошковый ингалятор, небулайзер или сходные с ними устройства. В случае, когда препарат вводится ингаляционно под давлением, фармацевтические композиции изобретения обычно содержат активный ингредиент и подходящий пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, углекислый газ или другой подходящий газ.
Кроме того, фармацевтическая композиция может быть в форме капсулы или картриджа (сделанного, например, из желатина), содержащего соединение изобретения и порошок, подходящий для применения в порошковом ингаляторе. Подходящая порошковая основа включает, к примеру, лактозу или крахмал.
Соединения по изобретению могут также вводиться чрескожно с помощью известных систем трансдермального введения и наполнителей. Например, к соединению изобретения могут быть добавлены средства, улучшающие проницаемость, такие как пропиленгликоль, полиэтиленгликоля монолаурат, азациклоалкан-2-оны и им подобные, и включены в состав пластыря или подобной системы доставки. Дополнительные наполнители, включающие загустители, эмульгаторы и буферы, могут применяться в таких композициях для чрескожного введения при необходимости.
Нижеследующие препараты иллюстрируют типичные фармацевтические композиции настоящего изобретения:
Пример препарата А
Твердые желатиновые капсулы для приема внутрь готовят следующим образом:
Типичная методика: ингредиенты тщательно смешивают и затем помещают в твердые желатиновые капсулы (260 мг композиции на капсулу).
Пример препарата B
Твердые желатиновые капсулы для приема внутрь готовят следующим образом:
Типичная методика: Ингредиенты тщательно смешивают и пропускают через сито № 45 U.S., помещают в твердые желатиновые капсулы (по 200 мг композиции на капсулу).
Пример препарата C:
Капсулы для приема внутрь готовят следующим образом:
Типичная методика: Ингредиенты тщательно смешивают и помещают в желатиновые капсулы (по 310 мг композиции на капсулу).
Пример препарата D
Таблетки для перорального введения были приготовлены следующим образом:
Типичная методика: активный ингредиент, крахмал и целлюлозу пропускают через сито № 45 U.S. и тщательно перемешивают. Раствор поливинилпирролидона смешивают с полученным порошком, эту смесь затем пропускают через сито № 14 U.S. Таким образом полученные гранулы высушивают при температуре 50-60°С, затем их пропускают через сито №18 U.S. Затем кабоксиметилкрахмал натрия, стеарат магния и тальк (предварительно просеянные через сито U.S. №60) добавляют к гранулам. После перемешивания смесь прессуют на таблетировочной машине для получения таблетки весом 100 мг.
Пример препарата E
Таблетки для приема внутрь изготавливают следующим образом:
Типичная методика: Ингредиенты тщательно перемешивают и затем прессуют для получения таблеток (440 мг композиции на таблетку).
Пример препарата F
Таблетки с насечкой для приема внутрь изготавливают следующим образом:
Типичная методика: ингредиенты тщательно смешивают, затем прессуют до получения таблеток с насечкой (215 мг композиции на таблетку).
Пример препарата G
Суспензию для приема внутрь получают следующим образом:
Типичная методика: ингредиенты тщательно смешивают до получения суспензии, содержащей 10 мг активного ингредиента на 10 мл суспензии.
Пример препарата H
Порошок для ингаляционного введения получают следующим образом:
Типичная методика: активный ингредиент микронизируют, затем смешивают с лактозой. Эту полученную смесь затем помещают в желатиновый картридж для ингаляций. Содержимое картриджа вводят с помощью порошкового ингалятора.
Пример препарата I
Порошок для введения посредством дозированного ингалятора получают следующим образом:
Типичная методика: Суспензию, содержащую 5 мас.% соединения по изобретению и 0,1 мас.% лецитина, готовят путем диспергирования 10 г активного ингредиента в виде микронизированных частиц со средним диаметром менее 10 мкм в растворе, полученным из 0,2 г лецитина, растворенного в 200 мл деминерализованной воды. Суспензию высушивают распылением и полученное вещество микронизируют до частиц со средним диаметром менее 1,5 мкм. Эти частицы помещают в картриджи, где под давлением находится 1,1,1,2-тетрафторэтан.
Пример препарата J
Препарат для внутривенного введения получают следующим образом:
Типичная методика: вышеперечисленные ингредиенты смешивают, pH доводят до уровня 4±0,5 с помощью 0,5 н. HCl или 0,5 н. NaOH.
Пример препарата K
Капсулы для приема внутрь получают следующим образом:
Типичная методика: ингредиенты тщательно смешивают и затем помещают в желатиновые капсулы (размер #1, белые, непрозрачные) (264 мг композиции на капсулу).
Пример препарата L
Капсулы для приема внутрь получают следующим образом:
Типичная методика: ингредиенты тщательно смешивают и затем помещают в желатиновые капсулы (размер #1, белые, непрозрачные) (148 мг композиции на капсулу).
Необходимо понимать, что любая форма соединений изобретения (т.е. свободное основание, фармацевтическая соль или сольват), которая подходит для конкретного способа введения, может быть использована в описанных выше фармацевтических композициях.
Применение
Хинолинонкарбоксамидные соединения данного изобретения являются агонистами 5-HT4 рецепторов и, следовательно, ожидается их пригодность для лечения патологических состояний, опосредованных 5-HT4 рецепторами или связанных с активностью 5-HT4 рецепторов, т.е. патологических состояний, которые разрешаются при лечении агонистами 5-HT4 рецепторов. Такие патологические состояния включают помимо прочих синдром раздраженного кишечника (СРК), хронический запор, функциональную диспепсию, замедленное опорожнение желудка, гастроэзофагеальную рефлюксную болезнь (ГЭРБ), парез желудка, послеоперационную кишечную непроходимость, кишечную псевдообструкцию и лекарственное нарушение транзита по ЖКТ. Кроме того, предполагается, что некоторые соединения агонисты рецепторов 5-HT4 могут быть использованы в лечении расстройств центральной нервной системы, включая когнитивные расстройства, расстройства поведения, расстройства настроения и расстройства контроля вегетативных функций.
В частности, соединения данного изобретения усиливают моторику желудочно-кишечного тракта (ЖКТ), и, поэтому, ожидается, что они будут полезны в лечении расстройств ЖКТ, вызванных сниженной моторикой у млекопитающих, включая человека. Такие расстройства моторики ЖКТ включают, например, хронический запор, синдром раздраженного кишечника с преобладанием запоров, диабетический и идиопатический парез желудка и функциональную диспепсию.
Таким образом, с одной стороны изобретение описывает способ усиления моторики желудочно-кишечного тракта у млекопитающих, способ, включающий введение млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение изобретения.
При использовании для лечения расстройств со сниженной моторикой ЖКТ или других состояний, опосредованных 5-HT4 рецепторами, соединения по изобретению обычно назначают внутрь один или несколько раз в день, хотя могут применяться и другие формы введения. Количество активного соединения, вводимого в каждый прием, или общее количество за день в типичных случаях определяется лечащим врачом, исходя из частных обстоятельств, включающих характер заболевания, выбранный путь введения, конкретное назначенное соединение и его относительную активность, возраст, вес и индивидуальную реакцию пациента, тяжесть симптомов заболевания и т.п.
Подходящие дозы для лечения расстройств с ослабленной моторикой ЖКТ или других расстройств, связанных с 5-HT4 рецепторами, находятся в пределах от около 0,0007 до около 20 мг/кг/день активного соединения, включая диапазон от 0,0007 до 1 мг/кг/день. Для обычного человека 70 кг доза активного соединения составляет от около 0,05 до около 70 мг/день.
В одном аспекте изобретения соединения по настоящему изобретению используют для лечения хронического запора. В случае лечения хронического запора соединения по изобретению обычно вводятся внутрь один или несколько раз в день. Предпочтительно, доза препарата для лечения хронического запора находится в пределах от около 0,05 до около 70 мг в день.
В другом аспекте изобретения, соединения по настоящему изобретению используют для лечения синдрома раздраженного кишечника. Для лечения синдрома раздраженного кишечника с преобладанием запоров соединения по изобретению обычно вводятся внутрь один или несколько раз в день. Предпочтительно, доза препарата для лечения синдрома раздраженного кишечника с преобладанием запоров находится в пределах от около 0,05 до около 70 мг в день.
В еще одном аспекте изобретения, соединения по настоящему изобретению используют для лечения диабетического пареза желудка. В случае лечения диабетического пареза желудка соединения по изобретению обычно вводятся внутрь один или несколько раз в день. Предпочтительно, доза препарата для лечения диабетического пареза желудка находится в пределах от около 0,05 до около 70 мг в день.
В еще другом аспекте изобретения, соединения по настоящему изобретению используют для лечения функциональной диспепсии. В случае лечения функциональной диспепсии соединения по изобретению обычно вводятся внутрь один или несколько раз в день. Предпочтительно, доза препарата для лечения функциональной диспепсии находится в пределах от около 0,05 до около 70 мг в день.
Изобретение также обеспечивает способ лечения млекопитающих, страдающих от заболевания или состояния, связанного с активностью 5-HT4 рецепторов, способ включает введение млекопитающему терапевтически эффективного количества соединения данного изобретения или фармацевтической композиции, содержащей соединение данного изобретения.
Как сказано выше, соединения данного изобретения являются агонистами 5-HT4 рецепторов. Изобретение также описывает способ снижения активности 5-HT4 рецепторов в организме млекопитающего, способ, включающий введение соединения по изобретению млекопитающему. Кроме того, соединения данного изобретения также применимы в качестве инструментов исследования для выявления или изучения биологических систем или объектов, имеющих 5-HT4 рецепторы или для открытия новых агонистов 5-HT4 рецепторов. Более того, поскольку соединения настоящего изобретения проявляют избирательность связывания с 5-HT4 рецепторами при сравнении со связыванием 5-HT рецепторов других подтипов, в частности, 5-HT3 рецепторов, такие соединения особенно подходят для изучения эффектов избирательного воздействия на 5-HT4 рецепторы в биологических системах и объектах. Любые подходящие биологические система или объект, имеющие 5-HT4 рецепторы, могут быть использованы в таких исследованиях, которые можно проводить как in vitro, так и in vivo. Типичные биологические системы и объекты, подходящие для таких исследований, включают помимо прочего клетки, экстракты клеток, плазматические мембраны, образцы тканей, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.п) и подобные им.
В этом аспекте, биологическая система или объект, содержащий 5-HT4 рецептор вступает во взаимодействие с количеством соединения по изобретению, подавляющим 5-HT4 рецепторы. Эффекты воздействия на 5-HT4 рецепторы затем определяют с помощью обычных методик и оборудования, таких как исследование радиолигандного связывания и функциональные исследования. Такие функциональные исследования включают лиганд-опосредованные изменения во внутриклеточном циклическом аденозинмонофосфате (цАМФ), лиганд-опосредованные изменения активности фермента аденилилциклазы (который синтезирует цАМФ), лиганд-опосредованные изменения включения аналогов гуанозинтрифосфата (ГТФ), таких как [35S]GTPγS (гуанозин 5'-О-(γ-тио)трифосфат) или ГТФ-Eu, в изолированные мембраны посредством катализированного рецептором перехода аналогов ГТФ в аналоги ГДФ, лиганд-опосредованные изменения в свободных внутриклеточных ионах кальция (измеренных, например, с помощью устройства для считывания флуоресцентных сигналов с планшетов - FLIPR® от Molecular Devices, Inc.) и измерение активации киназы митогенактивируемого белка (MAPK). Соединение по изобретению может подавлять или увеличивать активацию 5-HT4 рецепторов в любых из перечисленных функциональных исследованиях или сходных исследованиях. Количество соединения по изобретению, подавляющего 5-HT4 рецепторы, обычно находится в пределах от около 1 нМ до примерно 500 нМ.
Кроме того, соединения настоящего изобретения могут быть использованы в качестве инструментов исследования для открытия новых агонистов 5-HT4 рецепторов. В этом воплощении данные связывания 5-HT4 рецепторов и функциональные данные для тестируемого соединения или группы тестируемых соединений сравнивают с данными связывания 5-HT4 рецепторов и функциональными данными для соединения настоящего изобретения для выявления тестируемых соединений, которые обладают большей функциональной активностью и большим связыванием, если таковые есть. Эта сторона изобретения включает, как отдельное воплощение, и получение сравнительных данных (с помощью соответствующих исследований), и анализ данных исследования для выявления представляющих интерес тестируемых соединений.
Среди прочих свойств соединения по изобретению, как было установлено, являются мощными агонистами 5-HT4 рецепторов и проявляют значительную избирательность в отношении подтипа 5-HT4 рецепторов по сравнению с подтипом 5-HT3 в исследовании с радиолигандным связыванием. Кроме того, соединения настоящего изобретения демонстрируют хорошие фармакокинетические свойства на крысиной модели. Таким образом ожидается, что соединения данного изобретения имеют высокую биодоступность при пероральном введении. Кроме того, эти соединения не демонстрируют неприемлемый уровень ингибирования потока ионов калия в in vitro модели с фикскацией потенциала с использованием изолированных цельных клеток, экспрессирующих hERG кардиальные калиевые каналы. Исследование с фиксацией потенциала является приемлемым доклиническим способом оценивания потенциала фармацевтических средств изменять паттерн реполяризации сердца, особенно вызывать так называемое удлинение QT, которое связано с сердечными аритмиями (Cavero et al., Opinion on Pharmacotherapy, 2000,1, 947-73, Fermini et al., Nature Reviews Drug Discovery, 2003, 2, 439-447). Соответственно, фармацевтические композиции, содержащие соединения настоящего изобретения, как ожидается, имеют приемлемый набор характеристик для сердца.
Эти свойства, как и применимость соединений изобретения, могут быть продемонстрированы с использованием различных in vitro и in vivo исследований, хорошо известных специалисту данной области. Показательные исследования детально описаны в нижеследующих примерах.
ПРИМЕРЫ
Последующие синтетические и биологические примеры предложены для иллюстрации изобретения, и не должны никаким образом ограничить область изобретения. В представленных ниже примерах последующие сокращения имеют следующие значения, если иное не обозначено. Сокращения, не определенные ниже, имеют, как правило, общепринятые значения.
Boc = трет-бутоксикарбонил
(Boc)2O = ди-трет-бутилдикарбонат
DCM = дихлорметан
ДМФА = N,N-диметилформамид
DMSO = диметилсульфоксид
EtOAc = этилацетат
mCPBA = м-хлорбензойная кислота
MeCN = ацетонитрил
MTBE = трет-бутилметиловый эфир
PyBop = гексафторфосфат бензотриазол-1-илокситрипирролидинфосфония
Rf = фактор задержки
RT = комнатная температура
TFA = трифторуксусная кислота
ТГФ = тетрагидрофуран
Реагенты (в том числе вторичные амины) и растворители приобретают из коммерческих источников (Aldrich, Fluka, Sigma и так далее), и используют без дальнейшей очистки. Реакции проводят в атмосфере азота, если иное не отмечено. Ход реакции контролируют с помощью тонкослойной хроматографии (TLC), аналитической высокоэффективной жидкостной хроматографии (аналитическая ВЭЖХ), и масс-спектрометрии, которые в деталях раскрыты ниже и отдельно в конкретных примерах синтеза. Реакционные смеси обрабатывают, как описано конкретно в каждом синтезе; как правило, их очищают путем экстрагирования и другими способами очистки, такими как зависимая от температуры и растворителя кристаллизация и осаждение. Кроме того, реакционные смеси рутинно очищают с помощью препаративной ВЭЖХ: общее правило описано ниже. Характеристику реакционных продуктов рутинно осуществляют с помощью масс- и 1H-ЯМР спектрометрии. Для ЯМР измерения, образцы растворяют в дейтерированном растворителе (CD3OD, CDCl3, или DMSO-d 6), и 1H-ЯМР спектр получают с помощью Varian Gemini 2000 инструмента (300 МГц) в обычных условиях наблюдения. Масс-спектрометрическую идентификацию соединений выполняют с помощью электроспрейного ионизационного способа (ESMS) на приборе Applied Biosystems (Foster City, CА) модели API 150 EX или Agilent (Palo Alto, CА) модели 1100 LC/MSD. Содержание воды определяют с помощью методики титрирования по Карлу Фишеру, используя кулонометр Brinkmann (Westbury, NY) Metrohm Karl Fischer Model 813.
Пример 1: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
а. Получение 8-бензил-8-азабицикло[3.2.1]октан-3-она
Концентрированную соляную кислоту (30 мл) добавляют в гетерогенный раствор 2,5-диметокситетрагидрофурана (82,2 г, 0,622 моль) в воде (170 мл) при перемешивании. В отдельную колбу, охлажденную до температуры 0°C (баня со льдом), к раствору бензиламина (100 г, 0,933 моль) в воде (350 мл) медленно добавляют концентрированную соляную кислоту (92 мл). Раствор 2,5-диметокситетрагидрофурана перемешивают в течение приблизительно 20 минут, затем разбавляют водой (250 мл), затем добавляют раствор бензиламина, а затем добавляют раствор 1,3-ацетондикарбоновой кислоты (100 г, 0,684 моль) в воде (400 мл) и добавляют гидрофосфат натрия (44 г, 0,31 моль) в воде (200 мл). Значение рН доводят до значения от 1 до ~4,5, используя 40% раствор NaOH. Полученный мутный и бледно-желтого цвета раствор перемешивают в течение ночи. Раствор затем подкисляют до значения рН, от 3 до 7,5, используя 50% раствор соляной кислоты, нагревают до температуры 85°C и перемешивают в течение 2 часов. Раствор охлаждают до комнатной температуры, подщелачивают до значения рН, равного 12, используя 40% раствор NaOH, и экстрагируют DCM (3×500 мл). Объединенные органические слои промывают рассолом, сушат (MgSО4), фильтруют и концентрируют при пониженном давлении, чтобы получить неочищенное указанное промежуточное соединение в виде густого масла коричневого цвета (52 г).
К раствору неочищенного промежуточного соединения в метаноле (1000 мл) добавляют ди-трет-бутилдикарбонат (74,6 г, 0,342 моль) при температуре 0°C. Раствору дают возможность нагреться дo комнатной температуры, а затем его перемешивают в течение ночи. Метанол удаляют при пониженном давлении и полученное масло растворяют в дихлорметане (1000 мл). Промежуточное соединение экстрагируют в 1 M H3PO4 (1000 мл) и промывают дихлорметаном (3×250 мл). Водный слой подщелачивают до значения рН, равного 12, используя водный раствор NaOH, и экстрагируют дихлорметаном (3×500 мл). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют при пониженном давлении, чтобы получить указанное промежуточное соединение в виде густого масла светло-коричневого цвета.
1H-ЯМР (CDCl3) δ (ч.н.м.) 7,5-7,2 (м, 5H, C6H5), 3,7 (с, 2H, CH2Ph), 3,45 (шир.с, 2H, CH-NBn), 2,7-2,6 (дд, 2H, CH2CO), 2,2-2,1 (дд, 2H, CH2CO), 2,1-2,0 (м, 2H, CH2CH2), 1,6 (м, 2H, CH2CH2).
(m/z): [M+H]+ Вычислено для: C14H17NO 216,14; Найдено: 216,0.
b. Получение трет-бутилового эфира 3-оксо-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
К раствору 8-бензил-8-азабицикло[3.2.1]октан-3-она (75 г, 0,348 моль) в EtOAc (300 мл) добавляют раствор ди-трет-бутилдикарбоната (83,6 г, 0,383 моль, 1,1 экв.) в EtOAc (300 мл). Полученный раствор и смывку (100 мл EtOAc) добавляют в сосуд Парра для гидрогенизации, объемом 1 л, содержащий 23 г гидроксида палладия (20 мас.% Pd, сухую массу на угле, ~50% влажности; например, катализатор Перльмана) в токе азота. Реакционный сосуд дегазируют (альтернативно вакуум и N2 пять раз) и создают давление 60 пси с помощью газа H2. Реакционный раствор перемешивают в течение двух дней и вновь продувают H2, при необходимости, чтобы поддержать давление H2 на уровне 60 пси, пока реакция не закончится, ход которой контролируют с помощью тонкослойной хроматографии на силикагеле. После чего полученный раствор черного цвета фильтруют через таблетку из Целита® и концентрируют при пониженном давлении, получая на выходе указанное промежуточное соединение с количественным выходом в виде густого, от желтого дo оранжевого цвета масла. Его используют на следующей стадии без дополнительной обработки.
1H-ЯМР (CDCl3) δ (ч.н.м.) 4,5 (шир., 2H, CH-NBoc), 2,7 (шир., 2H, CH2CO), 2,4-2,3 (дд, 2H, CH2CH2), 2,1 (шир. м, 2H, CH2CO), 1,7-1,6 (дд, 2H, CH2CH2), 1,5 (с, 9H, (CH3)3COCON)).
c. Получение трет-бутилового эфира (1S,3R,5R)-3-амино-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
К раствору продукта, полученного на предыдущей стадии (75,4 г, 0,335 моль) в метаноле (1 л), добавляют формиат аммония (422,5 г, 6,7 моль), воду (115 мл) и 65 г палладия на активированном угле (10% сухой массы, ~50% влажность; Компания Degussa тип E101NE/W) в токе N2 во время перемешивания с помощью механической мешалки. Через 24 и 48 часов, дополнительно каждый раз добавляют порции формиата аммония (132 г, 2,1 моль). После прекращения реакции, ход которой контролируют с помощью аналитической ВЭЖХ добавляют Celite® (>500 г), и полученную густую суспензию фильтруют, а затем собранное твердое вещество промывают метанолом (~500 мл). Фильтраты объединяют и концентрируют при пониженном давлении до полного удаления метанола. После чего, полученный мутный двухфазный раствор разбавляют 1М фосфорной кислотой дo конечного объема от ~1,5 дo 2,0 л при значении рН, равного 2, и промывают дихлорметаном (3×700 мл). Водный слой подщелачивают до значения рН, равного 12, используя 40% водный раствор NaOH, и экстрагируют дихлорметаном (3×700 мл). Объединенные органические слои сушат над MgSО4, фильтруют, и концентрируют на роторном испарителе после полного вакуума, остается 52 г (70%) указанного промежуточного соединения, как правило, N-Boc-эндо-3-аминотропана, в виде твердого вещества от белого дo бледно-желтого цвета. Соотношение изомеров эндо к экзо амина продукта составляет >99:1, исходя из 1H-ЯМР анализа (>96% чистота, определяемая с помощью аналитической ВЭЖХ).
1H-ЯМР (CDCl3) δ (ч.н.м.) 4,2-4,0 (шир. д, 2H, CHNBoc), 3,25 (т, 1H, CHNH2), 2,1-2,05 (м, 4H), 1,9 (м, 2H), 1,4 (с, 9H, (CH3)3OCON), 1,2-1,1 (шир., 2H). (m/z): [M+H]+ Вычислено: C12H22N2O2) 227,18; Найдено: 227,2. Аналитическая ВЭЖХ (изократический способ; от 2:98 (A:B) дo 90:10 (A:B) в течение 5 минут): время удерживания = 3,68 минут.
d. Получение 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
Вначале, ацетон (228,2 мл, 3,11 моль) добавляют к перемешиваемой суспензии 2-аминофенилметанола (255,2 г, 2,07 моль) и уксусной кислоты (3,56 мл, 62 ммоль) в воде (2 л) при комнатной температуре. Через 4 часа суспензию охлаждают до температуры 0°C и перемешивают в течение еще 2,5 часов, а затем фильтруют. Твердое вещество собирают и промывают водой и влажное твердое вещество охлаждают и сушат путем лиофилизации, получая на выходе 2,2,-диметил-1,4-дигидро-2Н-бензо[1,3]оксазин (332,2 г, 98%) в виде грязно-белого цвета.
1H-ЯМР (CDCl3; 300 МГц): 1,48 (с, 6H, C(CH 3)2), 4,00 (шир. с, 1H, NH), 4,86 (с, 2H, СН 2), 6,66 (д, 1H, ArH), 6,81 (т, 1H, ArH), 6,96 (д, 1H, ArH), 7,10 (т, 1H, ArH).
Раствор 2,2,-диметил-1,4-дигидро-2Н-бензо[1,3]оксазина (125 г, 0,77 моль) в ТГФ (1 л) фильтруют через сцинтилляционную воронку, а затем в перемешиваемый раствор добавляют по каплям через делительную воронку в течение 2,5 часов 1,0 M LiAlН4 в ТГФ (800 мл) при температуре 0°C. Реакцию гасят путем медленного порционного добавления Na2SО4·10H2O (110 г), в течение 1,5 часов, при температуре 0°C. Реакционную смесь перемешивают в течение ночи, фильтруют, и твердое вещество солей промывают тщательно с помощью ТГФ. Фильтрат концентрируют при пониженном давлении, получая на выходе 2-изoпропиламинофенилметанол (120 г, 95%) в виде масла желтого цвета.
1H-ЯМР (CDCl3; 300 МГц): 1,24 (д, 6H, CH(CH 3)2), 3,15 (шир. с, 1H, OH), 3,61 (сeпт, 1H, CH(CH 3)2), 4,57 (с, 2H, CH 2), 6,59 (т, 1H, ArH), 6,65 (д, 1H, ArH), 6,99 (д, 1H, ArH), 7,15 (т, 1H, ArH).
Диоксид магния (85% 182,6 г, 1,79 моль) добавляют к перемешиваемому раствору 2-изoпропиламинофенилметанола (118 г, 0,71 моль) в толуоле (800 мл), и реакционную смесь нагревают до температуры 117°С в течение 4 часов. Реакционной смеси дают возможность остыть до комнатной температуры в течение ночи, а затем фильтруют через слой целита, который элюируют толуолом. Фильтрат концентрируют при пониженном давлении, получая на выходе 2-изoпропиламинобензальдегид (105 г, 90%) в виде масла оранжевого цвета.
1H-ЯМР (CDCl3; 300 МГц): 1,28 (д, 6H, CH(CH 3)2), 3,76 (септ, 1H, CH(CH3)2), 6,65 (т, 1H, ArH), 6,69 (д, 1H, ArH), 7,37 (д, 1H, ArH), 7,44 (т, 1H, ArH), 9,79 (с, 1Н, СНО).
2,2-Диметил-[1,3]диоксан-4,6-дион, как правило, кислоту Мельдрума, (166,9 г, 1,16 моль) добавляют к перемешиваемому раствору 2-изoпропиламинобензальдегида (105 г, 0,64 моль), уксусной кислоте (73,6 мл, 1,29 моль) и этилендиамину (43,0 мл, 0,64 моль) в метаноле (1 л) при температуре 0°C. Реакционную смесь перемешивают в течение часа при температуре 0°C, а затем при комнатной температуре в течение ночи. Полученную суспензию фильтруют, и твердое вещество промывают метанолом и собирают, получая на выходе указанное промежуточное соединение 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновую кислоту (146 г, 98%) в виде грязно-белого цвета.
1H-ЯМР (CDCl3; 300 МГц): 1,72 (д, 6H, CH(CH 3)2), 5,50 (шир. с, 1H, CH(CH3)2), 7,44 (т, 1H, ArH), 7,75-7,77 (м, 2H, ArH), 7,82 (д, 1H, ArH), 8,89 (с, 1H, CH).
e. Получение трет-бутилового эфира (1S,3R,5R)-3-[1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
Тионилхлорид (36,6 мл, 0,52 моль) добавляют к перемешиваемой суспензии 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (80 г, 0,35 моль) в толуоле (600 мл) при температуре 85°C, а затем реакционную смесь нагревают до температуры 95°С в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры, а затем добавляют в течение более 25 минут к энергично перемешиваемому двухфазному раствору трет-бутилового эфира (1S,3R,5R)-3-амино-8-азабицикло[3,2,1]октан-8-карбоновой кислоты (78,2 г, 0,35 моль) и гидроксида натрия (69,2 г, 1,73 моль) в смеси толуол/вода (1:1) (1 л) при температуре °C. Через час слоям дают возможность разделиться, и органическую фазу концентрируют при пониженном давлении. Водную фазу промывают EtOAc (1 л) и затем (500 мл), а затем используют объединенные органические экстракты, чтобы растворить концентрированный органический остаток. Этот раствор промывают 1М H3PO4 (500 мл), насыщенным водным раствором NaHCO3 (500 мл) и рассолом (500 мл), сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, получая на выходе указанное промежуточное соединение (127,9 г, приблизительно 84%) в виде твердого вещества желтого цвета.
1H-ЯМР (CDCl3): 1,47 (с, 9H), 1,67 (д, 6H), 1,78-1,84 (м, 2H), 2,04-2,18 (м, 6H), 4,20-4,39 (м, 3H), 5,65 (шир. с, 1H), 7,26 (дд, 1H), 7,63 (м, 2H), 7,75 (дд, 1H), 8,83 (с, 1H), 10,63 (д, 1H).
f. Получение {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
TFA (300 мл) добавляют к перемешиваемому раствору продукта, полученного на предыдущей стадии (127,9 г) в CH2Cl2 (600 мл), при температуре 0°C. Реакционную смесь нагревают дo комнатной температуры и перемешивают в течение часа, а затем концентрируют при пониженном давлении. Затем маслянистый коричневого цвета осадок выливают в энергично перемешиваемый раствор эфира (3 л), при этом немедленно образуется твердый осадок. Суспензию перемешивают в течение ночи, а затем твердое вещество собирают фильтрованием и промывают эфиром, получая на выходе указанное промежуточное соединение как его соль с трифторуксусной кислотой (131,7 г, 86% в течение двух стадий) в виде твердого вещества светло-желтого цвета.
1H-ЯМР (CDCl3): 1,68 (д, 6H), 2,10 (д, 2H), 2,33-2,39 (м, 4H), 2,44-2,61 (м, 2H), 4,08 (шир. с, 2H), 4,41 (м, 1H), 5,57 (шир. с, 1H), 7,31 (м, 1H), 7,66 (м, 2H), 7,77 (д, 1H), 8,83 (с, 1H), 9,38 (шир. д, 2H), 10,78 (д, 1H).
g. Получение 3-гидрокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана (Промежуточное соединение (V) с R 1 = H, R 2 = изопропил)
2-Бромметилоксиран (10,72 мл, 129,5 ммоль) добавляют к перемешиваемому раствору соли трифторуксусной кислоты {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (14,65 г, 43,2 ммоль) в этаноле (150 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 36 часов, в течение которых образуется твердый осадок. Твердое вещество собирают фильтрованием и промывают этанолом (70 мл), получая на выходе указанное промежуточное соединение в виде бромидной соли (8,4 г). (m/z): [M]+ Вычислено: C23H30N3O3 396,23; Найдено: 396,5. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,13 минут.
h. Получение {(1S,3R,5R)-8-[2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
3-Гидрокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октанбромид (678 мг, 1,4 ммоль) растворяют в этаноле (10 мл), а затем добавляют метиламин (41% раствор в воде) (510 мкл, 8,0 ммоль). Смесь нагревают при температуре 80°С в течение 16 часов, а затем концентрируют при пониженном давлении, что дает указанное промежуточное соединение в виде неочищенного масла, которое используют непосредственно на следующей стадии.
i. Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
Продукт, полученный на предыдущей стадии, растворяют в дихлорметане (10 мл), а затем добавляют 1,8-диазабицикло[5.4.0]ундек-7-ен (763 мкл, 5,1 ммоль), и смесь перемешивают в атмосфере азота, после чего охлаждают до температуры 0°C. Добавляют метансульфонилхлорид (132 мкл, 1,7 ммоль) и смесь вновь перемешивают при температуре 0°С в течение 30 минут. Реакцию гасят путем добавления воды, и концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (10 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, получая на выходе указанное соединение в виде соли трифторуксусной кислоты (340 мг). (m/z): [M+H]+ Вычислено: C25H36N4O5S, 505,25; Найдено: 505,4. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,17 минуты.
Пример 2: Синтез {(1S,3R,5R)-8-[2-метокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
а. Получение 3-метокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана (Промежуточное соединение (V') с R 1 = H, R 2 = изопропил, R 3 = метил)
трет-Бутоксид калия (1,63 г, 14,5 ммоль) добавляют к перемешиваемой суспензии 3-гидрокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октанбромида (3,45 г, 7,25 в моль) в дихлорметане (100 мл) при комнатной температуре. Через 2 минуты в реакционную смесь добавляют метилиодид (0,477 мл, 7,61 ммоль). Через 30 минут добавляют воду (2 мл), чтобы погасить реакцию, и реакционную смесь концентрируют при пониженном давлении. Осадок растворяют в небольшом объеме смеси уксусная кислота/вода (1:1) и очищают с помощью препаративной ВЭЖХ, получая на выходе указанное промежуточное соединение в виде соли с трифторуксусной кислотой (2,1 г). (m/z): [M]+ Вычислено: C24H32N3O3, 410,24; Найдено: 410,5. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,36 минуты.
b. Получение {(1S,3R,5R)-8-[2-метокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
Соль трифторуксусной кислоты 3-метокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октана (410 мг, 0,84 ммоль) растворяют в этаноле (10 мл), а затем добавляют метиламин (41% раствор в воде, 320 мкл, 5 ммоль). Смесь нагревают при температуре 80°С в течение 16 часов, а затем концентрируют при пониженном давлении, что дает продукт в виде неочищенного масла, который используют непосредственно на последующей стадии.
c. Синтез {(1S,3R,5R)-8-[2-метокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
Продукт, полученный на предыдущей стадии (53,6 мг, 0,12 ммоль), растворяют в дихлорметане (1,0 мл), а затем добавляют 1,8-диазабицикло[5.4.0]ундек-7-ен (89,7 мкл, 0,6 ммоль), и смесь перемешивают в атмосфере азота, а затем охлаждают до температуры 0°C. Добавляют метансульфонилхлорид (18,6 мкл, 0,24 ммоль), и смесь перемешивают при температуре 0°С в течение 30 минут. Смесь гасят путем добавления воды, и концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (45,8 мг). (m/z): [M+H]+ Вычислено: C26H38N4O5S, 519,27; Найдено: 519,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,72 минуты.
Пример 3: Синтез {(1S,3R,5R)-8-[3-(метансульфонилпиридин-3-илметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
а. Получение {(1S,3R,5R)-8-{2-метокси-3-[(пиридин-3-илметил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
3-Метокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октан (410 мг, 0,84 ммоль) растворяют в этаноле (10 мл), а затем добавляют 3-аминометилпиридин (153 мкл, 1,5 ммоль). Смесь нагревают при температуре 60°С в течение 16 часов, а затем концентрируют при пониженном давлении, что дает указанное промежуточное соединение в виде неочищенного масла, которое используют непосредственно на следующей стадии.
b. Синтез {(1S,3R,5R)-8-[3-(метансульфонилпиридин-3-илметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
Продукт, полученный на предыдущей стадии (102,6 мг, 0,2 ммоль), растворяют в дихлорметане (1,0 мл), а затем добавляют 1,8-диазабицикло[5.4.0]ундек-7-ен (119,6 мкл, 0,8 ммоль), и смесь перемешивают в атмосфере азота и охлаждают до температуры 0°C. Добавляют метансульфонилхлорид (30,1 мкл, 0,4 ммоль), после чего смесь вновь перемешивают при температуре 0°С в течение 30 минут. Реакцию гасят путем добавления воды, и смесь концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (63,5 мг). (m/z): [M+H]+ Вычислено: C31H41N5O5S, 596,29; Найдено: 596,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,35 минуты.
Пример 4: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
а. Получение {(1S,3R,5R)-8-[3-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-Азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (3,39 г, 10 ммоль) растворяют в этаноле (40 мл), и затем добавляют 2-оксиранилметилизоиндол-1,3-дион, как правило, эпоксипропилфталимид, (3,05 г, 15 ммоль). Смесь нагревают при температуре 80°С в течение 36 часов, охлаждают, а затем концентрируют при пониженном давлении. Полученное масло подвергают флеш-хроматографии (SiO2, элюируя 9:1 раствором дихлорметан/метанол), что дает указанное промежуточное соединение (4,95 г) в виде твердого вещества белого цвета, которое используют непосредственно на следующей стадии.
b. Получение {(1S,3R,5R)-8-[2-гидрокси-3-аминопропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
Продукт, полученный на предыдущей стадии (4,95 г, 9,13 ммоль), растворяют в этаноле (40 мл), а затем добавляют гидразин (860 мкл, 27,4 ммоль). Смесь нагревают при температуре кипения с обратным холодильником в течение 16 часов, а затем охлаждают до комнатной температуры. Смесь фильтруют, и фильтрат концентрируют, что дает указанное промежуточное соединение в виде неочищенного масла, которое используют непосредственно на следующей стадии без дальнейшей очистки.
c. Синтез {(1S,3R,5R)-8-[2-гидрокси-3-метансульфониламино-пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
Продукт, полученный на предыдущей стадии (82 мг, 0,2 ммоль), растворяют в дихлорметане (1,0 мл), затем добавляют 1,8-диазабицикло[5.4.0]ундек-7-ен (60 мкл, 0,4 ммоль), смесь перемешивают в атмосфере азота и охлаждают до температуры -78°C. Добавляют метансульфонилхлорид (15,5 мкл, 0,2 ммоль), смесь перемешивают и дают ей возможность нагреться до комнатной температуры в течение 30 минут. Реакцию гасят путем добавления воды, и полученную смесь концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (70,7 мг). (m/z): [M+H]+ Вычислено: C24H34N4O5S, 491,23; Найдено: 491,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,43 минуты.
Пример 5: Синтез {(1S,3R,5R)-8-[3-(метансульфонилпиридин-3-илметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
а. Получение {(1S,3R,5R)-8-{2-гидрокси-3-[(пиридин-3-илметил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
3-Гидрокси-3'-{[1-изoпропил-2-оксо-1,2-дигидрохинолин-3-ил)карбонил]амино}спиро[азетидин-1,8'-(1S,3R,5R)-8-азабицикло[3.2.1]октанбромид (505 мг, 1,27 ммоль) растворяют в этаноле (10 мл), а затем добавляют 3-(аминометил)-пиридин (193 мкл, 1,9 ммоль). Смесь нагревают при температуре 80°С в течение 16 часов, а затем концентрируют при пониженном давлении, что дает указанное промежуточное соединение в виде неочищенного масла, которое используют непосредственно в следующей стадии.
b. Синтез {(1S,3R,5R)-8-[3-(метансульфонилпиридин-3-илметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
Продукт, полученный на предыдущей стадии (42,5 мг, 0,08 ммоль) растворяют в дихлорметане (1,0 мл), добавляют 1,8-диазабицикло[5.4.0]ундек-7-ен (74,8 мкл, 0,5 ммоль), и смесь перемешивают в атмосфере азота и затем охлаждают до температуры 0°C. Добавляют метансульфонилхлорид (6,1 мкл, 0,08 ммоль), и смесь вновь перемешивают при температуре 0°С в течение 30 минут. Реакцию гасят путем добавления воды, и концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (22,8 мг). (m/z): [M+H]+ Вычислено: C30H39N5O5S, 582,28; Найдено: 582,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,78 минуты.
Пример 6: Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
а. Получение (S)-(бензилметиламино)-3-хлорпропан-2-ола
N-Бензилметиламин (13,95 мл, 108,1 ммоль) и (S)-2-хлорметилоксиран, как правило, (S)-эпихлоргидрин (8,48 мл, 108,1 ммоль) растворяют в гексане (40 мл), и перемешивают в течение 16 часов. Раствор затем подвергают флеш-хроматографии (SiO2, элюируя смесью 10% метанол/90% дихлорметан). Фракции, содержащие продукт, концентрируют, что дает указанное промежуточное соединение в виде масла (19,7 г).
1H-ЯМР (DMSO d6, 299,96 МГц): δ (ч.н.м.) 2,01 (с, 3H), 2,2-2,4 (м, 2H), 3,21-3,5 (м, 3H), 3,53-3,6 (м, 1H), 3,65-3,75 (м, 1H), 4,95 (д, 1H), 7,0-7,25 (м, 5H). (m/z): [M+H]+ Вычислено: C11H16ClNO, 214,10; Найдено: 214,1.
b. Получение трет-бутилового эфира ((S)-3-хлор-2-гидроксипропил)метилкарбаминовой кислоты
(S)-1-(бензилметиламино)-3-хлорпропан-2-ол (8,4 г, 39,3 ммоль) растворяют в этилацетате (75 мл), после чего добавляют ди-трет-бутилдикарбонат (9,3 г, 43,23 ммоль) и гидроксид палладия (2,5 г). Смесь встряхивают в течение 12 часов в атмосфере водорода (60 атм.). Смесь фильтруют через подушку из Целита®, и концентрируют досуха при пониженном давлении. Полученное масло фильтруют через силикагель, элюируя гексаном, затем дихлорметаном, затем диэтиловым эфиром. Слой эфира концентрируют, что дает указанное промежуточное соединение в виде масла (7,1 г).
1H-ЯМР (DMSO d6, 299,96 МГц): δ (ч.н.м.) 1,35-1,46 (с, 9H), 2,81-2,85 (с, 3H), 2,95-3,1 (м, 1H), 3,3-3,6 (м, 3H), 3,67-3,85 (м, 1H), 5,25-5,4 (м, 1H). (m/z): [M+H-Boc]+ Вычислено: C9H18ClNO3, 123,10; Найдено: 123,1.
c. Получение трет-бутилового эфира ((R)-2-гидрокси-3-{3-[(1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-(1S,3R,5R)-8-азабицикло[3.2.1]окт-8-ил}пропил)метилкарбаминовой кислоты
трет-Бутиловый эфир ((S)-3-хлор-2-гидроксипропил)метилкарбаминовой кислоты (335 мг, 1,5 ммоль) и {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (339 мг, 1,0 ммоль), растворяют в метаноле (5 мл), а затем добавляют N,N-диизoпропилэтиламин (523 мкл, 3,0 ммоль). Смесь нагревают при температуре 80°С в течение 16 часов, а затем концентрируют при пониженном давлении. Полученное масло подвергают флеш-хроматографии (SiO2, элюируя смесью 10% метанол/90% дихлорметан). Фракции, содержащие продукт концентрируют, что дает указанное промежуточное соединение в виде твердого вещества белого цвета (0,5 г). (m/z): [M+H]+ Вычислено: C29H42N4O, 527,33; Найдено: 527,6. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,75 минуты.
d. Получение {(1S,3R,5R)-8-[(S)-2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
трет-Бутиловый эфир ((R)-2-гидрокси-3-{3-[(1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-(1S,3R,5R)-8-азабицикло[3.2.1]окт-8-ил}пропил)метилкарбаминовой кислоты (575 мг, 1,09 ммоль) растворяют в дихлорметане (5 мл), а затем медленно добавляют трифторуксуснкю кислоту (5 мл). Смесь перемешивают в течение 30 минут, а затем концентрируют при пониженном давлении. Полученное масло растирают в порошок с диэтиловым эфиром, а затем фильтруют. Осадок сушат в вакууме, что дает указанное промежуточное соединение в виде соли трифторуксусной кислоты (0,68 г). (m/z): [M+H] Вычислено: C24H34N4O3, 427,27; Найдено: 427,2. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 3,40 минуты.
e. Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (1,03 г, 1,57 ммоль) растворяют в дихлорметане (6,0 мл), добавляют 1,8-диазабицикло[5.4.0]ундек-7-ен (747 мкл, 5,0 ммоль), смесь перемешивают в атмосфере азота и охлаждают до температуры 0°C. Добавляют метансульфонилхлорид (124,4 мкл, 1,6 ммоль), и смесь вновь перемешивают при температуре 0°С в течение 30 минут. Реакцию гасят путем добавления воды, и смесь концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (0,38 г).
(m/z): [MH+H]+ Вычислено: C25H36N4O5S, 505,25; Найдено: 505,4. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,17 минуты. Свободное основание: 1H-ЯМР (DMSO d6, 299,96 МГц): δ (ч.н.м.) 1,40-1,68 (д, 6H), 1,81-2,02 (шир. с, 4H), 2,02-2,18 (шир. м, 2H), 2,22-2,36 (д, 2H), 2,78-2,90 (2 с, 6H), 2,91-3,04 (м, 1H), 3,10-3,30 (м, 4H), 3,61-3,78 (шир. с, 1H), 4,02-4,17 (м, 1H), 4,71-4,79 (шир. с, 1H), 5,2-5,8 (шир. с, 1H), 7,3-7,4 (т, 1H), 7,67-7,78 (т, 1H), 7,82-7,94 (д, 1H), 7,98-8,02 (д, 1H), 8,80 (с, 1H), 10,37-10,40 (д, NH).
Пример 7: Синтез {(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
В соответствии с методикой, аналогичной методике получения соединения по Примеру 6, с заменой (R)-2-хлорметилоксирана на (S)-2-хлорметилоксиран на стадии а, получают следующее промежуточные соединения и указанное соединение.
(R)-1-(бензилметиламино)-3-хлорпропан-2-ол: 1H-ЯМР (DMSO d6, 299,96 МГц): δ (ч.н.м.) 2,01 (с, 3H), 2,2-2,4 (м, 2H), 3,21-3,5 (м, 3H), 3,53-3,6 (м, 1H), 3,65-3,75 (м, 1H), 4,95 (д, 1H), 7,0-7,25 (м, 5H). (m/z): [M+H]+ Вычислено: C11H16ClNO, 214,10; Найдено: 214,1.
трет-Бутиловый эфир ((R)-3-хлор-2-гидроксипропил)метилкарбаминовой кислоты: 1H-ЯМР (DMSO d6, 299,96 МГц): δ (ч.н.м.) 1,35-1,46 (с, 9H), 2,81-2,85 (с, 3H), 2,95-3,1 (м, 1H), 3,3-3,6 (м, 3H), 3,67-3,85 (м, 1H), 5,25-5,4 (м, 1H). (m/z): [M+H-Boc]+ Вычислено: C9H18ClNO3, 123,10; Найдено: 123,1.
трет-Бутиловый эфир ((S)-2-гидрокси-3-{3-[(1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-(1S,3R,5R)-8-азабицикло[3.2.1]окт-8-ил}пропил)метилкарбаминовой кислоты: (m/z): [M+H]+ Вычислено: C29H42N4O5 527,33; Найдено: 527,6. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,75 минуты.
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты: (m/z): [M+H]+ Вычислено: C24H34N4O3, 427,27; Найдено: 427,2. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 3,40 минуты.
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты: (m/z): [M+H]+ Вычислено: C25H36N4O5S, 505,25; Найдено: 505,4. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,17 минуты.
Пример 8: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-[метил-(пиридин-4-карбонил)амино]пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (71 мг, 0,17 ммоль) растворяют в ДМФА (0,5 мл), а затем добавляют N,N-диизoпропилэтиламин (88,9 мкл, 0,51 ммоль). Затем добавляют раствор изоникотиновой кислоты (41,8 мг, 0,34 ммоль) и PyBOP (177 мг, 0,34 ммоль) в ДМФА. Полученную смесь встряхивают при комнатной температуре в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (30,8 мг). (m/z): [M+H]+ Вычислено: C30H37N5O4, 532,29; Найдено: 532,7. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,11 минуты.
Пример 9: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-[(пиридин-4-карбонил)амино]пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-гидрокси-3-аминопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (82,4 мг, 0,2 ммоль) растворяют в ДМФА (0,5 мл), а затем добавляют N,N-диизoпропилэтиламин (69,7 мкл, 0,4 ммоль). Затем добавляют раствор изоникотиновой кислоты (49,2 мг, 0,4 ммоль), и добавляют PyBOP (208,2 мг, 0,4 ммоль) в ДМФА. Полученную смесь встряхивают при комнатной температуре в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (82,1 мг). (m/z): [M+H]+ Вычислено: C29H35N5O4, 518,28; Найдено: 518,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,20 минуты.
Пример 10: Синтез {(1S,3R,5R)-8-[3-(ацетилметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-метокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (53,6 мг, 0,12 ммоль) растворяют в ДМФА (1,0 мл), а затем добавляют N,N-диизoпропилэтиламин (104 мкл, 0,6 ммоль). Смесь перемешивают в атмосфере азота и охлаждают до температуры 0°C. Добавляют ацетилхлорид (17,1 мкл, 0,24 ммоль), и смесь перемешивают при температуре 0°С в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (40,4 мг). (m/z): [M+H]+ Вычислено: C27H38N4O4, 483,30; Найдено: 483,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,54 минуты.
Пример 11. Синтез {(1S,3R,5R)-8-[2-метокси-3-[метил-(пиридин-4-карбонил)амино]пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-метокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (53,6 мг, 0,12 ммоль) растворяют в ДМФА (1,0 мл), а затем добавляют N,N-диизoпропилэтиламин (104,5 мкл, 0,6 ммоль). Смесь перемешивают в атмосфере азота и охлаждают до температуры 0°C. Добавляют изоникотинилхлорид (42,7 мг, 0,24 ммоль), и смесь перемешивают при температуре 0°С в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (54,4 мг). (m/z): [M+H]+ Вычислено: C31H39N5O4, 546,31; Найдено: 546,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,29 минуты.
Пример 12. Синтез {(1S,3R,5R)-8-[3-(ацетилпиридин-3-илметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
(8-{2-Метокси-3-[(пиридин-3-илметил)амино]пропил}-8-азабицикло[3.2.1]окт-3-ил)амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (102,6 мг, 0,2 ммоль) растворяют в ДМФА (1,0 мл), а затем добавляют N,N-диизoпропилэтиламин (139,4 мкл, 0,8 ммоль). Смесь перемешивают в атмосфере азота и охлаждают до температуры 0°C. Добавляют ацетилхлорид (28,5 мкл, 0,4 ммоль), и смесь перемешивают при температуре 0°С в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (33,2 мг). (m/z): [M+H]+ Вычислено: C32H41N5O4, 560,33; Найдено: 560,4. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,27 минуты.
Пример 13. Синтез {(1S,3R,5R)-8-[3-ацетиламино-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-гидрокси-3-аминопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (82,4 мг, 0,2 ммоль) растворяют в ДМФА (0,5 мл), а затем добавляют N,N-диизoпропилэтиламин (69,7 мкл, 0,4 ммоль). Затем добавляют раствор уксусной кислоты (22,7 мкл, 0,4 ммоль) и PyBOP (208,2 мг, 0,4 ммоль) в ДМФА. Полученную смесь встряхивают при комнатной температуре в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное соединение в виде соли трифторуксусной кислоты (79,2 мг). (m/z): [M+H]+ Вычислено: C25H34N4O4, 455,27; Найдено: 455,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,33 минуты.
Пример 14. Синтез {(1S,3R,5R)-8-[3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (43 мг, 0,1 ммоль) растворяют в ДМФА (1,0 мл), а затем добавляют N,N-диизoпропилэтиламин (87,1 мкл, 0,5 ммоль). Смесь перемешивают в атмосфере азота и охлаждают до температуры 0°C. Добавляют ацетилхлорид (17,8 мкл, 0,25 ммоль), и смесь перемешивают при температуре 0°С в течение 30 минут. Смесь концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное промежуточное соединение в виде соли трифторуксусной кислоты (17,1 мг). (m/z): [M+H]+ Вычислено: C26H36N4O4, 469,28; Найдено: 469,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,49 минуты.
Пример 15. Синтез {(1S,3R,5R)-8-[3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (43 мг, 0,1 ммоль) растворяют в этилформиате (1,0 мл). Смесь нагревают при температуре 65°С в течение 16 часов, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное промежуточное соединение в виде соли трифторуксусной кислоты (22,7 мг). (m/z): [M+H]+ Вычислено: C25H34N4O4, 455,27; Найдено: 455,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,37 минуты.
Пример 16. Синтез {(1S,3R,5R)-8-[3-формиламино-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[2-гидрокси-3-аминопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (41,2 мг, 0,1 ммоль) растворяют в этилформиате (1,0 мл). Смесь нагревают при температуре 65°С в течение 16 часов, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное промежуточное соединение в виде соли трифторуксусной кислоты (37,5 мг). (m/z): [M+H]+ Вычислено: C24H32N4O4, 441,25; Найдено: 441,2. Время удерживания (аналитическая ВЭЖХ: 5-40% MeCN/H2O в течение 4 минут) = 2,89 минуты.
Пример 17. Синтез {(1S,3R,5R)-8-[(R)-3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (200 мг, 0,3 ммоль) растворяют в ДМФА (1,0 мл), а затем добавляют N,N-диизoпропилэтиламин (160,3 мкл, 0,92 ммоль). Затем добавляют раствор уксусной кислоты (17,3 мкл, 0,3 ммоль) и PyBOP (159 мг, 0,3 ммоль) в ДМФА. Полученную смесь встряхивают при комнатной температуре в течение 30 минут, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное промежуточное соединение в виде соли трифторуксусной кислоты (130 мг). (m/z): [M+H]+ Вычислено: C26H36N4O4, 469,28; Найдено: 469,5. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 3,94 минуты.
Пример 18. Синтез {(1S,3R,5R)-8-[(R)-3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-метиламинопропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (235 мг, 0,36 ммоль) растворяют в этилформиате (5,0 мл). Смесь нагревают при температуре 65°С в течение 16 часов, а затем концентрируют досуха при пониженном давлении. Продукт переносят в смесь уксусной кислоты/воды (1:1) (1,5 мл) и очищают с помощью ВЭЖХ. Очищенные фракции лиофилизуют, что дает указанное промежуточное соединение в виде соли трифторуксусной кислоты (97,5 мг). (m/z): [M+H]+ Вычислено: C25H34N4O4, 455,27; Найдено: 455,2. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 3,87 минуты.
Пример 19: Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 5-бром-1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
К раствору {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (500 мг, 0,99 ммоль) растворяют в смеси ацетонитрила (5 мл) и уксусной кислоты (10 мл) добавляют бром (0,30 мл, 5,9 ммоль). Смесь перемешивают при комнатной температуре в течение 12 часов, и концентрируют при пониженном давлении, получая на выходе масляный осадок бледно-желтого цвета. Осадок растворяют в 20% ацетонитриле в воде (0,5% TFА) (5 мл), и очищают с помощью ВЭЖХ. Указанное соединение получают в виде основного продукта и выделяют в виде соли трифторуксусной кислоты (200 мг).
1H-ЯМР (CD3OD): δ (ч.н.м.) 8,64 (с, 1H), 7,97 (с, 1H), 7,72-7,70 (м, 2H), 4,18 (шир. м, 2H), 4,0 (шир. с, 1H), 3,2-3,0 (м), 2,88 (с, 3H), 2,79 (с, 3H), 2,5-2,2 (м, 2H), 1,56 (д, 6H). (m/z): [M+H]+ Вычислено: C25H35BrN4O5S, 583,16; Найдено: 583,4. Время удерживания (аналитическая ВЭЖХ: 10-50% MeCN/H2O в течение 6 минут) = 3,90 минуты.
Пример 20: Альтернативный синтез трет-бутилового эфира ((R)-2-гидрокси-3-{3-[(1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-(1S,3R,5R)-8-азабицикло[3.2.1]окт-8-ил}пропил)метилкарбаминовой кислоты
а. Получение трет-бутилового эфира метил-(S)-1-оксиранилметилкарбаминовой кислоты
трет-Бутиловый эфир ((S)-3-хлор-2-гидроксипропил)-метилкарбаминовой кислоты (2,23 г, 10 ммоль) растворяют в ТГФ (30 мл), и затем добавляют водный раствор гидроксида натрия (0,48 г в 10 мл вода). Смесь перемешивают в течение 2 часов. Затем смесь концентрируют при пониженном давлении, чтобы удалить большую часть ТГФ, и остатки водного раствора экстрагируют в этилацетате, а затем промывают водой. Продукт сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, что дает указанное промежуточное соединение в виде масла (1,6 г). (m/z): [M+Nа]+ Вычислено: C9H17NO3, 210,10; Найдено: 210,1.
1H-ЯМР (DMSO d6, 299,96 МГц): δ (ч.н.м.) 1,32-1,42 (с, 9H), 2,69-2,73 (м, 1H), 2,75-2,85 (с, 3H), 2,95-3,05 (шир. с, 1H), 3,10-3,15 (дм, 1H), 3,16-3,21 (д, 1H), 3,37-3,51 (м, 2H).
b. Синтез трет-бутилового эфира ((R)-2-гидрокси-3-{3-[(1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-(1S,3R,5R)-8-азабицикло[3.2.1]окт-8-ил}пропил)метилкарбаминовой кислоты
трет-Бутиловый эфир метил-(S)-1-оксиранилметилкарбаминовой кислоты (9,53 г, 51,1 ммоль) и {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрoхинолин-3-карбоновой кислоты (8,7 г, 25,6 ммоль) растворяют в метаноле (100 мл). Смесь нагревают при температуре 80°С в течение 2 часов, а затем концентрируют при пониженном давлении. Полученное масло переносят в этилацетат и промывают насыщенным водным раствором бикарбоната натрия, затем насыщенным водным раствором хлорида натрия. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученное масло подвергают флеш-хроматографии (SiO2, элюируя смесью 10% метанол/90% дихлорметан). Фракции, содержащие продукт, концентрируют, что дает указанное промежуточное соединение в виде твердого вещества белого цвета (13,5 г). (m/z): [M+H]+ Вычислено: C29H42N4O, 527,33; Найдено: 527,6. Время удерживания (аналитическая ВЭЖХ: 2-50% MeCN/H2O в течение 5 минут) = 4,75 минуты.
Пример 21: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилэтиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
В соответствии с методикой, аналогичной методике получения соединения по Примеру 1, заменяя метиламин на этиламин в стадии h, получают указанное соединение (m/z): [M+H]+ Вычислено: C26H38N4O5S, 519,28; Найдено: 519,2. Время удерживания (аналитическая ВЭЖХ: 10-70% MeCN/H2O в течение 6 минут) = 2,91 минуты.
Пример 22: Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфониламино)-пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
а1. Получение (S)-2-оксиранилметилизоиндол-1,3-диона
В холодный раствор (S)-1-оксиранилметанола (5 г, 67,5 ммоль) и фталимида (9,9 г, 67,3 ммоль) в тетрагидрофуране (200 мл), охлажденный на бане со льдом, добавляют трифенилфосфин (17,9 г, 68,2 ммоль) и диэтилазодикарбоксилат (12,3 г, 70,6 ммоль). Смесь перемешивают при температуре 0°С в течение 2 часов и при комнатной температуре в течение 48 часов. Смесь концентрируют в вакууме, и маслянистый осадок очищают с помощью флеш-хроматографии на колонке с силикагелем, получая на выходе желаемый продукт (10,1 г) в виде твердого вещества бледно-желтого цвета: Rf=0,51 в смеси 1:1 EtOAc/гексан.
1H-ЯМР (CDCl3, 300 МГц): δ (ч.н.м.) 7,76-7,64 (м, 2H), 7,63-7,60 (м, 2H), 3,9-3,8 (дд, 1H), 3,70-3,65 (дд, 1H), 3,15 (м, 1H), 2,70-2,67 (дд, 1H), 2,58-2,55 (дд, 1H).
b1. Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфониламино)пропил]-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
В соответствии с методикой, аналогичной методике получения соединения по Примеру 4, заменяя рацемический 2-оксиранилметилизоиндол-1,3-дион на хиральное промежуточное соединение, полученное на предыдущей стадии, получают трифторацетатную соль названного соединения (m/z): [M+H]+ Вычислено: C24H34N4O5S, 491,24; Найдено: 491,4. Время удерживания (аналитическая ВЭЖХ: 10-50% MeCN/H2O в течение 6 минут) = 3,69 минуты.
1H-ЯМР (CD3OD, 300 МГц): δ (ч.н.м.) 8,67 (с, 1H), 7,74-7,73 (м, 3H), 7,7-7,6 (дт, 1H), 7,3-7,2 (т, 1H), 4,2 (шир. с, 2H), 4,0 (шир. м, 2H), 3,2-2,9 (м, 4H), 2,8 (с, 3H), 2,6-2,3 (шир. м, 6H), 2,2-2,1 (шир. м, 2H), 1,57-1,55 (д, 6H).
Указанное соединение также получают с помощью следующей методики.
a2. Получение N-((S)-оксиранилметил)этансульфонамида
В холодный раствор метансульфонамида (10 г, 0,105 моль) в воде (100 мл) на бане со льдом добавляют гидроксид натрия в виде гранул (8,4 г, 0,21 моль) и затем (S)-2-хлорметилоксиран (12,4 г, 0,158 моль). Смесь перемешивают при той же самой температуре в течение 2 часов, и при комнатной температуре в течение 12 часов, а затем добавляют концентрированную соляную кислоту (18 мл). Продукт выделяют путем экстрагирования водного слоя дихлорметаном (2×300 мл). Органический слой сушат над MgSO4, а затем выпаривают досуха, получая на выходе бесцветную жидкость (2,5 г), которую используют непосредственно на следующей стадии.
b2. Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
К раствору {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (TFА соль; 4 г, 8,8 ммоль) в метаноле (150 мл) добавляют N,N-диизoпропилэтиламин (1,7 мл, 9,5 ммоль) и продукт, полученный на предыдущей стадии (2,5 г, 18,2 ммоль). Смесь перемешивают при температуре 8°С в течение 2 дней. После концентрации в вакууме остаток очищают с помощью препаративной ВЭЖХ, получая на выходе трифторацетатную соль указанного соединения (1,3 г). (m/z): [M+H]+ Вычислено: C24H34N4O5S, 491,24; Найдено: 491,4. Время удерживания (аналитическая ВЭЖХ: 10-50% MeCN/H2O в течение 6 минут) = 3,69 минуты.
Пример 23: Синтез {(1S,3R,5R)-8-[2-гидрокси-3-(1,1-диоксо-2-изотиазолидинил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
а. Получение {(1S,3R,5R)-8-[2-гидрокси-3-(3-хлорпропансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
В холодный раствор TFА соли {(1S,3R,5R)-8-[2-гидрокси-3-аминопропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (0,125 г, 0,195 ммоль) в дихлорметане (2 мл) на бане со льдом добавляют N,N-диизoпропилэтиламин (0,119 мл, 0,683 ммоль) и 3-хлорпропансульфонилхлорид (0,025 мл, 0,205 ммоль). После перемешивания при температуре 0°C в течение 2 часов смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют дихлорметаном (50 мл) и промывают рассолом, а затем насыщенным раствором NaHCO3. После высушивания над MgSO4, органический раствор выпаривают досуха, получая на выходе маслянистый осадок. Сырой продукт используют непосредственно на следующей стадии.
b. {(1S,3R,5R)-8-[2-гидрокси-3-(1,1-диоксо-2-изотиазолидинил)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты
К раствору {(1S,3R,5R)-8-[2-гидрокси-3-(3-хлорпропансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты (100 мг, 0,18 ммоль) в безводном ДМФА (3 мл) добавляют карбонат калия (75 мг, 0,56 ммоль). Реакционную смесь встряхивают при температуре 85°C в течение 12 часов, и концентрируют в вакууме. Осадок растворяют в дихлорметане (50 мл), и промывают насыщенным раствором NaHCO3. После высушивания над MgSO4, фильтрат выпаривают досуха и осадок очищают с помощью препаративной ВЭЖХ, получая на выходе указанное соединение, (m/z): [M+H]+ Вычислено: C26H36N4O5S, 517,26; Найдено: 517,3.
Пример 24: Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
а. Получение трет-бутилового эфира (1S,3R,5R)-3-[1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбонил)амино]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты
В колбе, объемом 3 л, 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновую кислоту (112,4 г, 0,486 моль, 1,1 экв.) суспендируют в толуоле (1 л). Смесь нагревают до температуры 85°C и добавляют по каплям тионилхлорид (86,74 г, 0,729 моль) в течение 70 минут. Смесь нагревают при температуре 95°С в течение 1,5 часов при перемешивании, а затем ей дают возможность остыть до комнатной температуры.
В отдельной колбе, объемом 12 л, трет-бутиловый эфир (1S,3R,5R)-3-амино-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (100,0 г, 0,442 моль, 1 экв.) суспендируют в толуол (1 л) и добавляют 3 M NaOH (4 экв.). Смесь перемешивают при комнатной температуре в течение 10 минут, а затем охлаждают до температуры около 5°C. Медленно добавляют раствор хлорангидрида при перемешивании в течение 40 минут, сохраняя внутреннюю температуру смеси ниже 10°C. Смесь перемешивают при температуре 3-5°С в течение 30 минут, и слоям дают возможность разделиться в течение ночи. Слой толуола (~2,5 л) собирают, концентрируют до около половины объема (~1,2 л) упаривания на роторном испарителе, и используют непосредственно на следующей стадии.
b. Получение {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
К раствору толуола, полученного на предыдущей стадии (~1,2 л), добавляют при перемешивании трифторуксусную кислоту (200 мл) в течение 20 минут при температуре 20°C. Смесь перемешивают при температуре 20°С в течение 2 часов. Затем добавляют воду (1,55 л), и смесь перемешивают в течение 30 минут при температуре 20°C. Через 30 минут смесь разделяют на три слоя. Нижний слой (~350 мл), вязкое масло коричневого цвета, содержит неочищенное промежуточное соединение.
В колбу, объемом 12 л, с загруженным MTBE (2,8 л), добавляют при перемешивании неочищенное масло коричневого цвета в течение более часа при температуре 1-2°C. Суспензию перемешивают при той же самой температуре в течение часа, а затем фильтруют. Фильтрат промывают MTBE (2×300 мл) и сушат в вакууме при комнатной температуре в течение 4 дней, чтобы обеспечить трифторацетатную соль названного промежуточного соединения (163,3 г) в виде порошка бледно-желтого цвета.
c. Получение N-метил-N-[(S)-2-оксиран-2-илметил]метансульфонамида
Колбу, объемом 12 л, загружают водой (1 л), затем добавляют NaOH (50% в воде, 146,81 г, 1,835 моль). Химический стакан, содержащий NaOH, промывают водой (2×500 мл), и в колбу добавляют смывки. Смесь перемешивают при комнатной температуре в течение 10 минут и охлаждают до температуры ~8°C. Добавляют (N-метил)метансульфонамид (200,2 г, 1,835 моль) в воде (500 мл) в течение более 5 минут. Смесь перемешивают в течение часа при температуре ~4°C и затем добавляют (S)-2-хлорметилоксиран (339,6 г, 3,67 моль). Смесь перемешивают в течение 20 часов при температуре 3-4°C. Добавляют дихлорметан (2 л), и смесь перемешивают в течение 30 минут при температуре 5-10°C. Двум слоям дают возможность разделиться в течение 10 минут, и затем их собирают. Органический слой (~2,5 л) помещают обратно в колбу, объемом 12 л, и промывают 1 M H3PO4 (800 мл) и рассолом (800 мл). Дихлорметан удаляют путем упаривания на роторном испарителе. К сырому продукту добавляют толуол (400 мл) и затем его удаляют путем упаривания на роторном испарителе. После трех дополнительных циклов толуольного процесса получают указанное промежуточное соединение (228,2 г), которое используют без дальнейшей очистки на следующей стадии.
d. Синтез {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
В колбе, объемом 3 л {(1S,3R,5R)-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты, трифторацетат (105,0 г, 0,232 моль) суспендируют в абсолютном этаноле (400 мл). К этой суспензии добавляют NaOH (50% в воде, 0,243 моль, 1,05 экв.), растворенный в абсолютном этаноле (100 мл), при комнатной температуре. Химический стакан, содержащий NaOH, промывают этанолом (2×50 мл) и добавляют промывки в реакционную смесь. Через 30 минут перемешивания добавляют раствор N-метил-N-[(S)-2-оксиран-2-илметил]метансульфонамида (62,0 г, 1,5 экв.) в абсолютном этаноле (100 мл). Смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов, охлаждают до комнатной температуры и добавляют затравку в виде кристаллов названного соединения. Примерно через 5 минут перемешивания образуется твердое вещество белого цвета. Смесь охлаждают до температуры 3-5°C и перемешивают в течение 2 часов. Твердое вещество белого цвета фильтруют, и влажный отжатый фильтрованный осадок промывают холодным абсолютным этанолом (3×50 мл). Твердое вещество сушат в вакууме при температуре 30°С в течение 60 часов, чтобы обеспечить указанное соединение (93,8 г, содержание воды, определяемое с помощью методики по Карлу Фишеру, составляет 2,03%).
1Н-ЯМР (CDCl3) δ (ч.м.п.) 10,52 (д, 1H), 8,83 (c, 1H), 7,75 (д, 2H), 7,64-7,60 (м, 2H), 7,28-7,26 (м, 1H), 4,33-4,26 (м, 2H), 3,78-3,75 (м, 1H), 3,27-3,20 (м, 4H), 3,01 (с, 3H), 2,88 (с, 3H), 2,58-2,53 (м, 1H), 2,30-1,81 (м, 11H), 1,68 (д, 6H).
Кристаллы затравки получают из предыдущего примера получения указанного соединения по указанному примеру в меньшем масштабе, в котором кристаллизация происходит самопроизвольно.
Пример 25: Синтез гидрохлорида {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
В колбе, объемом 1 л {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (34,7 г, 0,069 моль) суспендируют в абсолютном этаноле (210 мл). Затем добавляют при перемешивании концентрированную HCl (1,1 экв.) при комнатной температуре. Смесь перемешивают при температуре кипения с обратным холодильником в течение 30 минут, а затем охлаждают до комнатной температуры и перемешивают в течение 2 часов. Твердое вещество фильтруют и влажный отжатый фильтрованный осадок промывают холодным абсолютным этанолом (3×50 мл). Твердое вещество сушат в вакууме при температуре 30°С в течение 48 часов, чтобы обеспечить указанное соединение (34,5 г, 93,7% выходом, содержание воды, определяемое с помощью методики по Карлу Фишеру, составляет 0,13%).
Пример 26: Синтез цитрата {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (0,1 г, 0,2 ммоль) суспендируют в этаноле (1 мл). К этой суспензии добавляют 1М раствор лимонной кислоты в этаноле (0,072 мл, 0,072 ммоль, 0,33 экв.). Смесь быстро обрабатывают ультразвуком, пока не наступит прозрачность, сосуд со смесью закрывают крышкой, а затем смеси дают возможность отстояться в течение ночи. Затем крышку удаляют, и смеси дают возможность выпариваться в условиях окружающей среды до появления твердых веществ. Сосуд со смесью затем повторно закрывают крышкой и дают возможность отстояться в течение 72 часов. Полученное твердое вещество фильтруют и промывают холодным этанолом, что дает указанное соединение в виде твердого вещества (74,3 мг).
Пример 27: Синтез кислых солей {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
В соответствии с методикой, аналогичной методике получения соединения по Примеру 26, кислые соли {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты, представленные ниже в Таблице III, получают в виде твердого вещества, используя указанные эквиваленты кислоты.
Соли кислоты
Пример 28: Синтез метансульфонатной соли {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
К раствору {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты (0,1 г, 0,2 ммоль) в 50% смеси ацетонитрила/воды (1 мл) добавляют 1М раствор метансульфоновой кислоты в этаноле (0,2 мл, 0,2 ммоль, 1 экв.). Смесь затем замораживают и лиофилизуют досуха в течение ночи. Полученное твердое вещество растворяют в изопропаноле (1 мл) с помощью легкого нагревания и затем ей дают возможность остыть. Затем полученное твердое вещество собирают фильтрованием и промывают холодным изопропанолом, что дает указанное соединение в виде твердого вещества (90 мг).
Пример 29: Синтез кислых солей {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты
В соответствии с методикой, аналогичной методике получения соединения по Примеру 28, кислые соли {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изoпропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты, представленные ниже в Таблице IV, получают в виде твердого вещества, используя указанные эквиваленты кислоты.
Соли кислоты
Анализ 1: Реакция радиолигандного связывания c 5-HT 4(C) рецепторами человека
a. Получение мембран 5-HT 4(с)
HEK-293 (человеческая эмбриональная почка) клетки, стабильно трансфицированные человеческим 5-HT4(C) рецептором cDNA (Bmax = ~6,0 пмoл/мг белка, как определено с использованим [3Н]-GR113808 реакции мембранного радиолигандного связывания) был культивирован в T-225 колбах в питательной среде Игла, модифицированной по Дульбекко (DMEM), содержащей 4500 мг/л D-глюкозы и гидрохлорида пиридоксина (GIBCO-Invitrogen Corp., Carlsbad CA: Сat#11965), c добавленной 10%-ной эмбриональной бычьей сывороткой (FBS) (GIBCO-Invitrogen Corp.: Сat#10437), 2 мМ L-глутамина и (100 единиц) пенициллина-(100 мкг) стрептомицина/мл (GIBCO - Invitrogen Corp.: Сat#15140) в 5%-ном CO2 в увлажненном инкубаторе при температуре 37°C. Клетки были культивированы под непрерывно выбираемым давлением с добавлением 800 мкг/мл генетицина (GIBCO-Invitrogen Corp.: Cat#10131) к среде.
Клетки были культивированы до смыкания монослоя примерно на 60-80% (<35 посевов культур). За 20-22 часов до сбора клетки были промыты дважды и питались бессывороточным DMEM. Все стадии получения мембраны были осуществлены на льду. Монослой клеток был поднят с помощью осторожного механического перемешивания и растирания пипеткой на 25 мл. Клетки были собраны центрифугированием при 1000 об/мин (5 минут).
Для получения мембраны дебрис ресуспендировали в ледяной 50 ммоль 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоте (НEPES), pH 7,4 (мембранный буфер) (40 мл/общий выход клеток на 30-40 T225 колб) и гомогенизировали с использованием гомогенизатора типа "политрон" на льду. Полученные гомогенаты центрифугировали в 1200 г в течение 5 минут при температуре 4°C. Дебрис был отделен, и супернатант центрифугировали в 40000 г (20 минут). Дебрис был промыт один раз ресуспендированием буфером получения мембраны и центрифугированием в 40000 г (20 минут). Полученный дебрис был ресуспендирован в 50 мМ HEPES, pH 7,4 (реакционный буфер) (1 эквивалент T225 колбы/1 мл). Концентрация белка мембранной суспензии была определена методом Брэдфорда (Bradford, 1976). Мембраны хранились замороженными в аликвотах при -80°C.
b. Реакции радиолигандного связывания
Реакции радиолигандного связывания были осуществлены в 1,1 мл глубоких лунках полипропиленового аналитического планшета (Axygen) во всем реакционном объеме 400 мкл, cодержащем 2 мкг белка мембраны в 50 мМ HEPES pH 7,4, содержащем 0,025% бычьего сывороточного альбумина (BSA). Исследование связывания методом насыщения для определения Kd значения радиолиганда было проведено с использованием [3H]-GR113808 (Amersham Inc., Bucks, UK: Cat #TRK944; удельная активность ~82 Ки/ммоль) при 8-12 различных концентрациях в пределах от 0,001 нМ - 5,0 нМ. Анализы методом вытеснения для определения значений pKi соединений проводились с [3H]-GR113808 при 0,15 нМ и одиннадцати различных концентрациях соединения в пределах от 10 пM - 100 мкM.
Испытуемые соединения были получены как 10 мМ маточные растворы в DMSO и разбавлены до 400 мкM в 50 мМ HEPES pH 7,4 при температуре 25°C, содержащем 0,1% BSA, и затем последовательные разведения (1:5) были сделаны в том же самом буфере. Неспецифическое связывание было определено в присутствии 1 мкM немеченного GR113808. Реакции были инкубированы в течение 60 минут при комнатной температуре, и затем реакции связывания были завершены быстрой фильтрацией через 96-луночные GF/B стекловолоконные фильтровальные планшеты (Packard BioScience Co., Meriden, СТ), предварительно замоченные в 0,3%-ном полиэтиленимине. Фильтровальные планшеты были промыты три раза фильтрационным буфером (охлажденный льдом 50 мM HEPES, pH 7,4), чтобы удалить свободную радиоактивность. Планшеты были высушены, 35 мкл сцинтилляторной жидкости Microscint-20 (Packard BioScience Co., Meriden, СТ) были добавлены к каждой лунке, и планшеты были посчитаны в сцинтилляционном счетчике Topcount фирмы Packard (Packard BioScience Co., Meriden, СТ).
Данные связывания были проанализированы методом нелинейной регрессии с помощью программного обеспечения GraphPad Prism Software package (GraphPad Software, Inc., San Diego, CA) c использованием модели с 3 параметрами для конкуренции на одном сайте. Нижний уровень (минимум кривой) был установлен для значения неспецифического связывания, как определено при наличии 1 мкм GR113808. Значения Кi для испытуемых соединений были рассчитаны с помощью программы Prism, методом наибольшего подобия IC50 значения, и значение Kd радиолиганда, рассчитано, используя уравнение Гена-Прусова (Cheng and Prusoff, Biochemical Pharmacology, 1973, 22, 3099-108): Ki = IC50/(1+[L]/Kd), где [L] = концентрация [3H]-GR113808. Результаты выражены в виде десятичного логарифма значения Ki, pKi.
Испытуемые соединения, имеющие более высокое значение pKi в этом анализе, имеют более высокое связывание, сродство к 5-HT4 рецепторам. Соединения изобретения, которые были протестированы в этом анализе, имели значения pKi в пределах от приблизительно 6,3 до приблизительно 9,0, обычно в пределах от приблизительно 6,5 до приблизительно 8,5.
Анализ 2: Реакция радиолигандного связывания на рецепторах человека 5-HT 3A : Определение селективности подтипа рецептора
a. Получение мембран 5-HT 3А
HEK-293 (человеческая эмбриональная почка) клетки, стабильно трансфицированные человеческим 5-HT3A рецептором c DNA, были получены от Dr. Michael Bruess (University of Bonn, GDR). (Bmax = ~9,0 пмоль/мг белка, как определено с использованием [Н]-GR65630 реакции лигандного связывания). Клетки были культивированы в T-225 колбах или устройствах для культивирования клеток в 50%-ной питательной среде Игла, модифицироанной по Дульбекко (DMEM) (GIBCO-Invitrogen Corp., Сarlsbad, CA: Сat #11965) и 50%-ной Ham's F12 (GIBCO-Invitrogen Corp.: Cat#11765), дополненной 10%-ной термоинактивированной эмбриональной бычьей сывороткой (FBS) (Hyclone, Logan, UT: Cat #SH30070.03) и (50 единиц) пенициллина-(50 мкг) стрептомицина/мл (GIBCO-Invitrogen Corp.: Сat#15140) в 5%-ном CO2 в увлажненном инкубаторе при температуре 37°C.
Клетки были культивированы до смыкания монослоя примерно на 70-80% (<35 посевов культур). Все стадии получения мембран выполнялись на льду. Для сбора клеток среда была аспирирована, и клетки были промыты фосфатным солевым буфером Дульбекко (dPBS), не содержащим Ca2+, Mg2+. Клеточный монослой был поднят осторожным механическим перемешиванием. Клетки были собраны центрифугированием при 1000 об/мин (5 минут). Последующие стадии получения мембраны следовали за методикой, описанной выше для мембран, экспрессирующих 5-HT4(C) рецепторы.
b. Реакция радиолигандного связывания
Реакция радиолигандного связывания была осуществлена в 96-луночных полипропиленовых планшетах для анализа во всем объеме 200 мкл, содержащем 1,5-2 мкг белков мембраны в 50 мМ HEPES pH 7,4, содержащем 0,025% BSA реакционного буфера. Исследование связывания методом насыщения для определения значения Кd радиолиганда осуществлялось с использованием [3H]-GR65630 (PerkinElmer Life Sciences Incю, Boston, MA: Cat #NET1011, специфическая активность 85 Ки/ммоль) при двенадцати различных концентрациях в пределах от 0,005 нМ до 20 нМ. Анализы методом вытеснения для определения значения pKi соединений были осуществлены с [3Н]-GR65630 при 0,50 нМ и одиннадцати различных концентрациях соединения в пределах от 10 пМ до 100 мкм. Соединения были получены как 10 мМ маточные растворы в диметилсульфоксиде (см. раздел 3.1), разбавлены до 400 мкM в 50 мM HEPES pH 7,4 при температуре 25°C, содержащем 0,1% BSA, и затем последовательные (1:5) разбавления были сделаны в том же самом буфере. Неспецифическое связывание было определено в присутствии 10 мкM немеченного MDL72222. Реакции были инкубированы в течение 60 минут при комнатной температуре, затем реакции связывания были завершены быстрой фильтрацией через 96-луночные GF/B стекловолоконные фильтровальные планшеты (Packard BioScience Co., Meriden, СТ), предварительно замоченные в 0,3%-ном полиэтиленимине. Фильтровальные пластины были промыты три раза фильтровальным буфером (охлажденный льдом 50 мM HEPES, pH 7,4), для удаления свободной радиоактивности. Планшеты были высушены, 35 мкл сцинтилляторной жидкости Microscint-20 (Packard BioScience Co., Meriden, СТ) было добавлено в каждую лунку, и планшеты были посчитаны в сцинтилляционном счетчике типа Topcount фирмы Packard (Packard BioScience Co., Meriden, СТ).
Данные связывания были проанализированы, используя нелинейную регрессию, описанную выше, для определения значений Ki. Нижний уровень (минимум кривой) был установлен для значения неспецифического связывания, как определено в присутствии 10 мкM MDL72222. Количество [L] в уравнении Гена-Прусова было определено как концентрация [3H]-GR65630. Селективность для 5-HT4 подтипа рецептора в сравнении с 5-HT3 подтипом рецептора была рассчитана как отношение Ki(5-HT3A)/Ki(5-HT4(c)). Соединения изобретения, которые были исследованы в этом анализе, имели селективность 5-HT4/5-HT3 подтипа рецептора в пределах от приблизительно 50 до приблизительно 8000, обычно в пределах от приблизительно 100 до приблизительно 4000.
Анализ 3: Анализ накопления клеточного цАМФ Flashplate с HEK-293 клетками, экспрессирующими человеческие 5-HT 4(C) pецепторы
В этом анализе функциональная возможность тестируемого соединения была определена с помощью измерения количества циклической АТФ, полученной, когда HEK-293 клетки, экспрессирующие 5-HT4 рецепторы, контактировали с тестируемым соединением в различных концентрациях.
a. Клеточная культура
HEK-293 (человеческая эмбриональная почка) клетки, стабильно трансфицированные с клонированным человеческим 5-HT4(C) рецептором cDNA были получены экспрессией рецептора в двух различных удельных весах: (1) при плотности приблизительно 0,5-0,6 пмоль/мг белка, как определено с использованием [3H]-GR113808 при реакции радиолигандного связывания, и (2) при плотности приблизительно 6,0 пмоль/мг белка. Клетки были выращены в T-225 колбах в питательной среде Игла, модифицированной по Дульбекко (DMEM), содержащей D-глюкозы на 4,500 мг/л (GIBCO-Invitrogen Corp.: Сat#11965) c добавленной 10%-ной эмбриональной бычьей сывороткой (FBS) (GIBCO-Invitrogen Corp.: Сat#10437) и (100 единиц) пенициллина-(100 мкг) стрептомицина/мл (GIBCO-Invitrogen Corp.: Сat#15140) в 5% CO2, в увлажненном инкубаторе при температуре 37°C. Клетки были культивированы при непрерывном давлении отбора добавлением генетицина (800 мкг/мл: GIBCO-Invitrogen Corp.: Сat#10131) к питательной среде.
b. Получение клеток
Клетки были культивированы до смыкания монослоя примерно на 60-80%. За двадцать - двадцать два часа до анализа клетки были промыты дважды и питались с бессывороточным DMEM, содержащим D-глюкозу в количестве 4,500 мг/л (GIBCO-Invitrogen Corp.: Cat#11965). Для сбора клеток среда была аспирирована и 10 мл Versene (GIBCO-Invitrogen Corp.: Cat#15040) было добавлено к каждой колбе T-225. Клетки были культивированы в течение 5 минут при комнатной температуре и затем удалены из колбы при механическом перемешивании. Клеточная суспензия была перемещена в центрифужную пробирку, содержащую равный объем предварительно нагретого (температура 37°C) dPBS, и центрифугировалась в течение 5 минут при 1000 об/мин. Супернатант был удален, и дебрис был ресуспендирован в предварительно нагретом (температура 37°C) стимулирующем буфере (10 мл эквивалент в 2-3 T-225 колбах). Это время было записано и отмечено как нулевое время. Клетки считали с помощью счетчика Coulter (отсчет около 8 мкм, выход был 1-2×107 клеток/колба). Клетки были ресуспендированы при концентрации 5×105 клеток/мл в предварительно нагретом (температура 37°C) стимуляционном буфере (как предусмотрено во флашплайт (flashplate) методе) и предварительно инкубированы при температуре 37°C в течение 10 мин.
ЦAMФ анализ был осуществлен методом радиоиммуноанализа, используя систему анализа Активации Аденилциклазы с 125I-цАМФ (SMP004B, PerkinElmer Life Sciences Inc., Boston, MA), согласно инструкциям производителя.
Клетки были культивированы и получены, как описано выше. Результирующие концентрации клеток в анализе были 25×103 клетки/лунка и результирующий объем при анализе был 100 мкл. Исследуемые соединения были получены как основные растворы 10 мМ в DMSO, разбавлены до 400 мкм в 50 мМ HEPES pH 7,4 при 25°C, содержащем 0,1% BSA, и затем были сделаны последовательные разбавления (1:5) в том же самом буфере. Анализ накопления циклической АМФ выполнялся при 11-ти различных концентрациях соединения в пределах от 10 пМ до 100 мкм (результитующие концентрации анализа). 5-HT концентрационная кривая (от 10 пМ до 100 мкм) была построена для каждой планшеты. Клетки были культивированы с колебанием, при температуре 37°C в течение 15 минут, и реакция завершена добавлением 100 мкл ледяного буфера обнаружения (как предусмотрено в наборе флэш-планшет) в каждую лунку. Планшеты были герметизированы и выдержаны в термостате при температуре 4°C в течение ночи. Удержанная радиоактивность была определена количественно спектроскопией сцинтилляции, используя устройство Topcount (Packard BioScience Co., Meriden, СТ).
Количество цАМФ, произведенного в мл реакционного объема, экстраполировалось от цАМФ- калибровочной кривой, согласно инструкциям, обеспеченным в пользовательском руководстве изготовителя. Данные были проанализированы анализом нелинейной регрессии с помощью программы GraphPad Prism Software package, используя сигмоидальную модель зависимости от дозы с 3 параметрами (тангенс угла, стремящийся к единице). О данных потенции сообщают как значение pEC50, десятичный логарифм значения EC50, где EC50 - эффективная концентрация для 50%-ной максимальной ответной реакции.
Тестируемые соединения, показывающие более высокое значение pEC50 в этом анализе, имеют более высокую мощность для подавления активности в отношении 5-HT4 рецептора. Соединения изобретения, которые были исследованы в этом анализе, например, на линии клеток (1), имеющих плотность приблизительно 0,5-0,6 пмоль/мг белка, имели значение pEC50 в пределах от приблизительно 7,0 до приблизительно 9,0, обычно в пределах от приблизительно 7,5 до приблизительно 8,5.
Анализ 4: In vitro Способ фиксации напряжения ингибирования калиевого канала во всех клетках, экспрессирующих hERG кардиальный калиевый канал
CHO-K1 клетки, стабильно трансфектированные hERG cDNA, были получены от Гейла Робертсона из Университета Висконсина. Клетки криогенно хранились до необходимости в них. Клетки были выращены и посеяны в питательной среде Игла, модифицированной по Дульбекко/F12, с добавлением 10%-ной эмбриональной бычьей сыворотки и 200 мкг/мл генетицина. Клетки были посеяны на поли-D-лизине (100 мкг/мл), покрытом покровным стеклом, на 35 мм2 планшета (содержащего 2 мл питательной среды) при плотности, позволяющей изолировать клетки, которые будут отобраны для исследования по методу фиксации напряжения. Планшеты поддерживали в увлажненном 5%-ном CO2 при температуре 37°C.
Внеклеточный раствор получали по крайней мере каждые 7 дней и сохраняли при температуре 4°C, когда не использовали. Внеклеточный раствор содержит (мМ): NaCl (137), KCl (4), CaCl2 (1,8), MgCl2 (1), Глюкоза (10), 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновую кислоту (HEPES) (10), pH 7,4 с NaOH. Внеклеточный раствор в отсутствии или при наличии тестируемого соединения содержался в резервуарах, из которых перетекал в прибор регистрации приблизительно в количестве 0,5 мл в минуту. Внутриклеточный раствор был получен и сохранен в аликвотах при температуре -20°C до использования. Внутриклеточный раствор содержит (мМ): KCl (130), MgCl2 (1), соль этиленгликоль-бис(бета-аминоэтиловый эфир)-N,N,N',N'-тетрауксусной кислоты (EGTA) (5), MgATP (5), 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты (HEPES) (10), pH 7,2 с KOH. Все эксперименты были осуществлены при комнатной температуре (20-22°C).
Покровные стекла, на которых клетки были посеяны, были помещены в прибор регистрации и непрерывно перфузированы. Герметизирующий слой был сформирован между клеткой и проводящим электродом. Как только соединение было достигнуто, в способе фиксации напряжения начинали вести запись при исходном потенциале -80 мВ. После достижения устойчивой клеточной проводимости клетки контактировали с тестируемым соединением. Стандартный протокол напряжения был: перепад от исходного потенциала от -80 мВ до +20 мВ за 4,8 сек, реполяризация до -50 мВ за 5 сек, и затем возврат к исходному потенциалу (-80 мВ). Этот протокол напряжения запускался один раз в 15 сек (0,067 Гц). Максимум амплитуды тока в течение фазы реполяризации был определен с использованием программного обеспечения pClamp. Тестируемые соединения при концентрации 3 мкм были перфузированы через клетки в течение 5 минут с последующей 5-ти минутной стадией перфузии в отсутствии соединения. Наконец, контрольный образец (цисасприд, 20 нМ) был добавлен к перфузату для тестирования функции клетки. Перепад напряжения от-80 мВ до +20 мВ активизирует канал hERG, суммирующийся в выходящем токе. Перепад до -50 мВ результируется в выходящем токе, как канал возвращают с инактивации и дезактивируют.
Максимум амплитуды тока в течение фазы реполяризации был определен с использованием программного обеспечения pClamp. Контрольные и испытательные данные были направлены в прибор Origin® (OriginLab Corp., Нортхемптон MA), где индивидуальные амплитуды тока были приведены к начальной амплитуде тока в отсутствие соединения. Текущее значение тока и стандартные отклонения для каждого условия были рассчитаны, и подготовлены диаграммы с указанием времени эксперимента.
Сравнения были сделаны между определяемым K текущего ингибирования после пятиминутного воздействия на испытуемый образец или контрольный (обычно 0,3%-ный DMSO). Статистические сравнения между экспериментальными группами были осуществлены с использованием независимого t-теста с двумя совокупностями (Microcal Происхождение v. 6.0). Различия считали значительными при p<0,05.
Чем меньше в процентах ингибирование калиевого канала в этом анализе, тем меньше потенциал для тестируемых соединений для замены паттерна кардиальной реполяризации, при использовании их в качестве терапевтических средств. Соединения изобретения, которые были тестированы в этом анализе при концентрации 3 мкM, показали ингибирование калиевого канала менее, чем приблизительно 20%, обычно, менее чем приблизительно 15%.
Анализ 5: In vitro Модель биодоступности при пероральном введении: Caco-2 анализ проницаемости
Caco-2 анализ проницаемости осуществляется на модели способности испытуемых соединений для прохождения через кишечный тракт и проникновения в кровоток после перорального приема. Соотношение, при котором тестируемые соединения в растворе проникают в монослой клеток, имитирующий запирающую зону монослоя клеток кишечника человека, было определено.
Caco-2 (толстая кишка, аденокарцинома; человек) клетки были получены от ATCC (American Type Culture Collection; Rockville, MD). Для изучения проницаемости клетки были посеяны с плотностью 63000 клеток/см2 на предварительно увлажненных поликарбонатных фильтрах (Costar; Кембридж, MA). Клеточный монослой формировался после 21 дня при посеве. При следующем клеточном посеве в планшете мембрансодержащий клеточный монослой был отделен от планшеты и помещен в диффузионную камеру (Costar; Кембридж, MA). Диффузионная камера была вставлена в нагревательный блок, который был оборудован внешней циркуляцией, термостатически регулируемой 37°C-ной водой для контроля температуры. Воздушный трубопровод снабжал 95% О2/5% СО2 к каждой половине диффузионной камеры и создавал структуру ламинарного потока через клеточный монослой, который был эффективен при диспергировании невозмутимого пограничного слоя.
Изучение проникновения осуществляли при концентрациях испытуемых соединений 100 мкM и с 14C-маннитолом, чтобы контролировать целостность монослоя. Все эксперименты проводились при температуре 37°C в течение 60 мин. Пробы были взяты при 0, 30 и 60 минут и от донора, и от сторон приемника камеры. Образцы были проанализированы с помощью ВЭЖХ или сцинтилляторного счетчика для тестируемого соединения и концентраций маннитола. Коэффициент проникания (Kp) в см/сек был рассчитан.
В этом анализе значение Kp больше, чем приблизительно 10×10-6 см/сек считают показателем хорошей биодоступности. Соединения изобретения, которые были проверены в этом анализе, показали значения Кp приблизительно между 10×10-6 см/cек и приблизительно 50×10-6 см/сек, обычно между приблизительно 20×10-6 см/сек и приблизительно 40×10-6 см/сек.
Анализ 6: Фармакокинетическое исследование на крысах
Водные композиции растворов испытуемых соединений были получены в 0,1%-ной молочной кислоте при pH между приблизительно 5 и приблизительно 6. Самцам крыс линии Sprague-Dawley (CD strain, Charles River Laboratories, Wilmington, MA) дозировали тестируемые соединения через внутривенное введение (IV) в дозе 2,5 мг/кг или пероральным питанием (PO) в дозе 5 мг/кг. Объем дозирования был 1 мл/кг для IV и 2 мл/кг для PO введений. Последовательные пробы крови были собраны у животных перед введением, и через промежутки времени 2 (только IV), 5, 15 и 30 минут, и 1, 2, 4, 8 и 24 часа после введения. Концентрации тестируемых соединений в плазме крови были определены анализом жидкостной хроматографии - масс-спектрометрии (LC-MS/MS) (MDS SCffiX, API 4000, Applied Biosystems, Foster City, CA) с нижним пределом количественной оценки 1 нг/мл.
Стандартные фармакокинетические параметры были оценены c помощью неблочного анализа (Модель 201 для FV и Модели 200 для РО) с использованием программы WinNonlin (Версия 4.0.1, Pharsight, Mountain View, CA). Максимум концентрации тестируемого соединения в плазме крови на кривой зависимости от времени обозначен Cmax. Область под кривой зависимости концентрации от времени дозирования до последней измеряемой концентрации (AUC(0-t)) была рассчитана по правилу трапеций. Биодоступность при пероральном введении (F(%)), то есть отношение AUC(0-t) для PO введения к AUC(0-t) для FV введения, была рассчитана как:
F(%) = AUCPO/AUCIV × DoseIV/DosePO × 100%
Тестируемые соединения, которые показывают большие значения параметров Cmax, AUC(0-t), и F(%) в этом анализе, будут иметь большую биодоступность при пероральном применении. Соединения изобретения, которые были проверены в этом анализе, имели значения Сmax обычно в пределах от приблизительно 0,1 до приблизительно 0,25 мкг/мл, и AUC(0-t) значения обычно в пределах от приблизительно 0,4 до приблизительно 0,9 мкг·ч/мл. Например, соединение Примера 1 имело значение Сmax 0,17 (мкг/мл, при значении AUC(0-t) 0,66 мкг·ч/мл и биодоступности при пероральном введении (F(%)) в модели на крысе приблизительно 35%.
В то время, как настоящее изобретение было описано со ссылкой на определенные его воплощения, квалифицированному в данной области специалисту должно быть понятно, что очевидны различные модификации настоящего изобретения с эквивалентной заменой, не выходя за рамки объема и области настоящего изобретения. Кроме того, возможно множество модификаций, чтобы частные случаи, вещество, состав вещества, способ, стадии или стадии способа соответствовали цели и объему настоящего изобретения. Предполагается, что все такие модификации находятся в области изобретения, охарактеризованной пунктами формулы изобретения, приведенными далее. Кроме того, все публикации, патенты, и патентные документы, процитированные выше, включены здесь в качестве ссылки в полном объеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ-АГОНИСТЫ РЕЦЕПТОРА 5-НТ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ ПОЗНАВАТЕЛЬНОЙ СПОСОБНОСТИ | 2010 |
|
RU2569056C2 |
| ИНДАЗОЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2404179C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| КОНДЕНСИРОВАННОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ АМИНОПИРРОЛИДИНА | 2007 |
|
RU2443698C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
Изобретение относится к новым соединениям формулы (I):

где:
R1 представляет собой водород, галоген; R2 представляет собой С3-4алкил, или С3-6циклоалкил; R3 представляет собой водород или C1-3алкил; R4 представляет собой -S(O)2R6 или -C(O)R7; R5 представляет собой водород, С1-3алкил, С2-3алкил, замещенный -ОН или C1-3алкокси, или -СН2-пиридил; R6 представляет собой C1-3алкил; или, R5 и R6, взятые вместе, образуют С3-4алкиленил; и R7 представляет собой водород, C1-3алкил или пиридил; или к их фармацевтически приемлемым солям или сольватам или стереоизомерам. Изобретение также относится к фармацевтической композиции, к применению соединений, к способу лечения млекопитающего, к способу лечения расстройства со сниженной моторикой желудочно-кишечного тракта у млекопитающего, к способу получения соединений по любому из пп.1-12, к способу получения соединений формулы (I'), а также к соединениям формулы (III). Технический результат - получение новых биологически активных соединений, обладающих активностью в качестве 5-НТ4 рецепторов. 8 н. и 16 з.п. ф-лы, 4 табл.
1. Соединение формулы (I):

где R1 представляет собой водород, галоген;
R2 представляет собой С3-4алкил или С3-6циклоалкил;
R3 представляет собой водород или С1-3алкил;
R4 представляет собой -S(O)2R6 или -C(O)R7;
R5 представляет собой водород, С1-3алкил, С2-3алкил, замещенный -ОН или C1-3алкокси, или -СН2-пиридил;
R6 представляет собой C1-3алкил;
или R5 и R6, взятые вместе, образуют С3-4алкиленил; и
R7 представляет собой водород, С1-3алкил или пиридил;
или его фармацевтически приемлемая соль, или сольват, или стереоизомер.
2. Соединение по п.1, где
R5 представляет собой водород, С1-3алкил, С2-3алкил, замещенный -ОН или
C1-3алкокси, или -СН2-пиридил; и
R6 представляет собой C1-3алкил.
3. Соединение по п.2, где R2 представляет собой С3-4алкил.
4. Соединение по п.2, где R4 представляет собой -S(O)2R6.
5. Соединение по п.5, где R4 представляет собой -S(O)2СН3.
6. Соединение по п.2, где R4 представляет собой -C(O)R7.
7. Соединение по п.6, где R7 представляет собой водород или метил.
8. Соединение по п.2, где R5 представляет собой водород или метил.
9. Соединение по п.2, где
R1 представляет собой водород;
R2 представляет собой С3-4алкил или С4-5циклоалкил;
R3 представляет собой водород;
R4 представляет собой -S(O)2R6 или -C(O)R7;
R5 представляет собой водород или C1-3алкил;
R6 представляет собой C1-3алкил; и
R7 представляет собой водород или С1-3алкил.
10. Соединение по п.1, где соединение выбрано из:
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-метокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(метансульфонил-пиридин-3-илметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(метансульфонил-пиридин-3-илметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-[метил-(пиридин-4-карбонил)амино]пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-[(пиридин-4-карбонил)амино]пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(ацетилметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-метокси-3-[метил-(пиридин-4-карбонил)амино]пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(ацетилпиридин-3-илметиламино)-2-метоксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-ацетиламино-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-ацетилметиламино-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-формиламино-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 5-бром-1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилэтиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты и {(1S,3R,5R)-8-[2-гидрокси-3-(1,1-диоксо-2-изотиазолидинил)пропил]-8-азабицикло[3.2.1] окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты;
и из его фармацевтически приемлемых солей и сольватов и стереоизомеров.
11. Соединение по п.2, где соединение выбрано из:
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло [3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил)амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло [3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-3-(ацетилметиламино)-2-гидроксипропил]-8-азабицикло [3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-3-(формилметиламино)-2-гидроксипропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолинон-3-карбоновой кислоты; и
и его фармацевтически приемлемых солей и сольватов и стереоизомеров.
12. Соединение по п.11, где соединение выбрано из:
{(1S,3R,5R)-8-[2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(S)-2-гидрокси-3-(метансульфонилметиламино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты;
{(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфониламино)пропил]-8-азабицикло[3.2.1]оксо-3-ил}амида 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты; и
и его фармацевтически приемлемых солей и сольватов и стереоизомеров.
13. Фармацевтическая композиция, которая обладает активностью в качестве 5-НТ4 рецепторов, содержащая терапевтически эффективное количество соединения по любому одному из пп.1-12 и фармацевтически приемлемый носитель.
14. Соединение по любому одному из пп.1-12 для применения в терапии, обладающее активностью в качестве 5-НТ4 рецепторов.
15. Применение соединения по любому одному из пп.1-12 для производства лекарственного средства, обладающего активностью в качестве 5-НТ4 рецепторов.
16. Применение по п.15, где лекарственное средство используют для лечения патологического состояния у млекопитающего, связанного с активностью 5-НТ4 рецепторов.
17. Применение по п.16, где заболеванием или состоянием является расстройство сниженной моторики желудочно-кишечного тракта.
18. Способ лечения млекопитающего, имеющего патологическое состояние, связанное с активностью 5-НТ4 рецепторов, который включает введение млекопитающему, терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по любому одному из пп.1-12.
19. Способ по п.18, отличающийся тем, что патологическое состояние выбрано из группы, включающей синдром раздраженной кишки, хронический запор, функциональную диспепсию, замедленное опорожнение желудка, гастроэзофагеальную рефлюксную болезнь, парез желудка, послеоперационную кишечную непроходимость, кишечную псевдообструкцию, лекарственное замедление транзита.
20. Способ лечения расстройства со сниженной моторикой желудочно-кишечного тракта у млекопитающего, который включает введение млекопитающему терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по любому одному из пп.1-12.
21. Способ по п.20, отличающийся тем, что патологическое состояние, при котором снижается моторика, выбрано из группы, включающей хронический запор, синдром раздраженного кишечника с преобладанием запоров, диабетический и идиопатический парез желудка и функциональную диспепсию.
22. Способ получения соединения по любому одному из пп.1-12, включающий:
взаимодействие соединения формулы (III):

с соединением формулы L-R4, где L представляет собой уходящую группу, или L-R4 представляет собой HO-C(O)R7, где R1, R2, R4, R5 определены в любом из пп.1-9,
с получением соединения формулы (I) или его фармацевтически приемлемой соли или сольвата или стереоизомера.
23. Способ получения соединения формулы (I'):

где R1, R2, R4 и R5 определены в п.1; или его фармацевтически приемлемой соли, или сольвата, или стереоизомера, включающий взаимодействие соединения формулы (IV):

или его соли с соединением формулы (XI):

с получением соединения формулы (I') или его фармацевтически приемлемой соли или сольвата или стереоизомера.
24. Соединение формулы (III):
где R1 представляет собой водород, галоген;
R2 представляет собой С1-4алкил или С3-6циклоалкил;
R3 представляет собой водород или C1-3алкил; и
R5 представляет собой водород, С1-3алкил, С2-3алкил, замещенный -ОН или C1-3алкокси, или -СН2-пиридил; или
его соль, или стереоизомер, или защищенное производное.
| CHEMICAL AND PHARMACEUTICAL BULLETIN, vol.49, №1, 2001, p.29-39 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| JOURNAL OF MEDICINAL CHEMISTRY, vol.46, №3, 2003, p.319-344 | |||
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ АДДИТИВНЫЕ СОЛИ КИСЛОТЫ, ИЛИ N-ОКСИД ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ, ИЛИ ЕГО АДДИТИВНАЯ СОЛЬ КИСЛОТЫ | 1992 |
|
RU2047614C1 |

