Всюду по тексту настоящего описания сделаны ссылки на различные публикации, патентные заявки и опубликованные патенты. Описание таких публикаций, таким образом, включено полностью в виде ссылки в настоящее описание для более полного описания состояния области техники, к которой принадлежит настоящее изобретение.
Область изобретения
Настоящее изобретение относится к композициям энантиомера R(+)-N-пропаргил-1-аминоиндана (называемого далее в настоящем описании как R(+) PAI или разагилин), который является селективным необратимым ингибитором В-формы фермента моноаминоксидазы, используемым, например, для лечения болезни Паркинсона. Фермент моноаминоксидазу называют в настоящем описании МАО, а его В-форму - МАО-В.
Предшествующий уровень техники изобретения
В патенте Соединенных Штатов номер 5532415 описан R(+)-N-пропаргил-1-аминоиндан, его получение и его различные фармацевтически приемлемые соли. В патенте Соединенных Штатов номер 6126968 описаны фармацевтические композиции, включающие R(+) PAI. Было показано, что R(+) PAI и его соли являются селективными ингибиторами МАО-В, применимыми в лечении болезни Паркинсона и различных других состояний.
Хотя ранее и не указывалось в качестве проблемы R(+) PAI, лекарственное вещество может состоять из частиц, которые препятствуют, например, из-за размера и формы однородному распределению лекарственного вещества в смеси для получения таблеток. Если такое лекарственное вещество, без дополнительной обработки, используют в производстве таблеток, полученные таким образом таблетки не будут иметь однородного содержимого и обладать приемлемым содержанием лекарственного средства (например, патент США № 5622720). Было показано, что плохая однородность содержимого вызывает заметное снижение биодоступности. Также плохая однородность содержимого может вызывать токсичность, если количество лекарственного вещества слишком высоко.
Из-за повышенного внимания к биодоступности и безопасности нормативные органы, такие как Фармакопея Соединенных Штатов (USP), внедрили многостадийное исследование однородности содержимого, которое включает: 1) оценку десяти таблеток для подтверждения, что относительное стандартное отклонение (RSD) содержания активного компонента составляет менее чем или равно 6,0%, и отсутствуют значения вне 85-115%, и 2) оценку еще двадцати таблеток для подтверждения, что RSD для всех тридцати таблеток менее или равно 7,8%, не более чем одно значение находится вне 85-115%, и отсутствуют значения вне диапазона 75-125% указанного содержимого.
Сущность изобретения
В соответствии с изобретением было неожиданно обнаружено, что определенные распределения частиц по размерам обладают благоприятным действием на однородность состава твердой фармацевтической композиции R(+) PAI. Перемалывание, а также другие способы, могут быть использованы для изменения распределения частиц по размерам (далее ″PSD″) R(+) PAI с целью обеспечения большей однородности содержимого лекарственного продукта.
Задачей настоящего изобретения является обеспечение однородности содержимого лекарственных продуктов, включающих R(+) PAI, включая измельчение частиц R(+) для уменьшения размера частиц.
Объект изобретения относится к смеси частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана, где более чем 90% общего количества объема частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 250 микрон.
Объект изобретения также относится к способу получения композиции, который включает уменьшение размера частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана до размера частиц менее чем 250 микрон.
Краткое описание чертежей
На фиг.1 изображены частицы мезилата R(+) PAI перед измельчением. Микрофотографию получали в виде суспензии в парафиновом масле и принимали с 80-кратным увеличением.
На фиг.2 изображены частицы мезилата R(+) PAI после измельчения. Микрофотографию получали в виде суспензии в парафиновом масле и принимали с 80-кратным увеличением.
Подробное описание изобретения
Объект изобретения относится к смеси частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана, где более чем 90% общего количества объема частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 250 микрон.
В дополнительном варианте осуществления изобретения более чем 90% общего объемного количества частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 240 микрон.
В дополнительном варианте осуществления изобретения более чем 90% общего объемного количества частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 230 микрон.
В дополнительном варианте осуществления изобретения более чем 90% общего объемного количества частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 220 микрон.
В дополнительном варианте осуществления изобретения более чем 90% общего объемного количества частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 210 микрон.
В дополнительном варианте осуществления изобретения более чем 90% общего объемного количества частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер менее чем 200 микрон.
В дополнительном варианте осуществления изобретения более чем 90% общего объемного количества частиц соли R(+)-N-пропаргил-1-аминоиндана имеют размер более чем 6 микрон.
В дополнительном варианте осуществления изобретения фармацевтически приемлемой солью является тартратная, эзилатная, мезилатная или сульфатная соль.
В дополнительном варианте осуществления изобретения фармацевтически приемлемой солью является мезилатная соль.
Объект изобретения также относится к твердой композиции, включающей некоторое количество частиц соли R(+)-N-пропаргил-1-аминоиндана и носитель.
В следующем варианте осуществления изобретения твердая композиция включает терапевтически эффективное количество смеси частиц и фармацевтически приемлемый носитель.
В следующем варианте осуществления изобретения смесь частиц и носителя составляет гранулят.
В следующем варианте осуществления изобретения композиция находится в твердой лекарственной форме.
В следующем варианте осуществления изобретения композиция находится в пероральной лекарственной форме.
В следующем варианте осуществления изобретения пероральной лекарственной формой является таблетка.
В следующем варианте осуществления изобретения относительное стандартное отклонение (RSD) содержания соли R(+)-N-пропаргил-1-аминоиндана между твердыми лекарственными формами составляет менее чем 4%.
В следующем варианте осуществления изобретения относительное стандартное отклонение (RSD) содержания соли R(+)-N-пропаргил-1-аминоиндана между твердыми лекарственными формами составляет менее чем 3%.
В следующем варианте осуществления изобретения относительное стандартное отклонение (RSD) содержания соли R(+)-N-пропаргил-1-аминоиндана между твердыми лекарственными формами составляет менее чем 2%.
В следующем варианте осуществления изобретения однородность содержимого составляет от 95% до 105%.
Объект изобретения также относится к способу лечения пациента, пораженного болезнью Паркинсона, включающему введение пациенту любой из вышеуказанных композиций.
Объект изобретения также относится к способу получения композиции, который включает уменьшение размеров частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана до размера частиц менее чем 250 микрон.
В следующем варианте осуществления изобретения размер частиц составляет менее чем 200 микрон.
В следующем варианте осуществления изобретения стадия уменьшения включает измельчение частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана.
В следующем варианте осуществления изобретения способ дополнительно включает смешивание частиц R(+)-N-пропаргил-1-аминоиндана с носителем для получения гранулята.
В следующем варианте осуществления изобретения однородность смеси гранулята составляет от 90% до 110% и относительное стандартное отклонение (RSD) смеси составляет менее чем 2%.
В следующем варианте осуществления изобретения однородность смеси гранулята составляет от 95% до 105% и относительное стандартное отклонение (RSD) смеси составляет менее чем 2%.
В следующем варианте осуществления изобретения способ дополнительно включает прессование гранулята, содержащего частицы фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана, имеющего размер частиц менее чем 250 микрон в твердую лекарственную форму.
В следующем варианте осуществления изобретения твердой лекарственной формой является таблетка.
В следующем варианте осуществления изобретения способ получения твердой композиции включает:
а) измельчение партии частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана;
b) смешивание продукта стадии а) с носителем для получения гранулята;
с) определение однородности смеси гранулята, и
d) преобразование гранулята в композицию из гранулята, только если однородность смеси гранулята удовлетворяет заранее определенным критериям так, чтобы получить композицию.
В следующем варианте осуществления изобретения заранее определенными критериями являются однородность смеси между 90% и 110% и относительное стандартное отклонение менее чем 2%.
В следующем варианте осуществления изобретения заранее определенными критериями являются однородность смеси между 95% и 105% и относительное стандартное отклонение менее чем 2%.
В следующем варианте осуществления изобретения стадия образования с) включает получение твердой лекарственной формы.
В следующем варианте осуществления изобретения стадия образования с) включает получение пероральной лекарственной формы.
В следующем варианте осуществления пероральной лекарственной формой является таблетка.
В следующем варианте осуществления изобретения способ дополнительно включает определение однородности содержимого твердой лекарственной формы.
В следующем варианте осуществления изобретения способ дополнительно включает стадию классификации твердой лекарственной формы как приемлемой композиции, только если однородность содержимого удовлетворяет заранее определенным критериям.
В следующем варианте осуществления изобретения заранее определенным критерием является однородность содержимого между 95% и 105%.
В следующем варианте осуществления изобретения заранее определенным критерием является относительное стандартное отклонение однородности содержимого менее чем 4%.
В следующем варианте осуществления изобретения заранее определенным критерием является относительное стандартное отклонение однородности содержимого менее чем 3%.
В следующем варианте осуществления изобретения заранее определенным критерием является относительное стандартное отклонение однородности содержимого менее чем 2%.
В следующем варианте осуществления изобретения способ, следующий за стадией с), включает стадию уменьшения размера частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана любой партии, которая не соответствует заранее определенным критериям.
В следующем варианте осуществления изобретения стадия b) включает уменьшение размеров частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана.
В следующем варианте осуществления изобретения стадия уменьшения включает измельчение частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана.
Объект изобретения также относится к твердой фармацевтической композиции, включающей терапевтически эффективное количество фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана, полученной любым из вышеуказанных способов.
Объект изобретения также относится к способу лечения пациента, пораженного болезнью Паркинсона, включающему введение пациенту фармацевтической композиции по изобретению в количестве, эффективном для лечения болезни Паркинсона у пациента.
Фармацевтические композиции, составляющие объект изобретения, могут быть использованы отдельно для лечения болезни Паркинсона или альтернативно они могут быть использованы в качестве дополнения к обычной терапии L-ДОПА.
Способы лечения болезни Паркинсона, в которых комбинируют применение фармацевтических композиций по изобретению с другими лекарственными средствами, такими как агонисты допамина, бромокриптин, перголид, лизурид, а также ингибиторы катехоламиноксидазы метилтрансферазы, также находятся в рамках предмета изобретения.
Такие композиции могут включать соединение R(+) PAI или его фармацевтически приемлемые аддитивные соли кислот вместе с фармацевтически приемлемыми носителями и/или эксципиентами. В практике настоящего изобретения фармацевтически приемлемые соли включают, но не ограничиваются ими, мезилат, малеат, фумарат, тартрат, гидробромид, эзилат, п-толуолсульфонат, бензоат, ацетат, фосфат и сульфат. Частицы солей R(+) PAI называют в настоящем описании как «частицы R(+) PAI» или активный ингредиент, или лекарственное вещество.
Композиции могут быть получены как лекарственные препараты для введения перорально, парентерально, ректально или чрескожно. Подходящие формы для перорального введения включают таблетки, прессованные или покрытые оболочкой пилюли, драже, пакеты-саше, твердые или мягкие желатиновые капсулы, сублингвальные таблетки, сиропы и суспензии; для парентерального введения изобретение относится к ампулам или флаконам, которые содержат водный или безводный раствор или эмульсию; для ректального введения используют суппозитории с гидрофильными или гидрофобными носителями; и для тонического нанесения в виде мазей и чрескожной доставки используют подходящие системы доставки, как известно в области техники.
Частные примеры фармацевтически приемлемых носителей и эксципиентов, которые могут быть использованы для рецептирования пероральных лекарственных форм по настоящему изобретению, описаны, например, в патенте США № 6126968, на имя Peskin et al., выданном 3 октября 2000. Методики и композиции для получения лекарственных форм, применимых в настоящем изобретении, описаны в следующих ссылках: 7 Modern Pharmaceutics, Chapters 9 and 10 (Banker & Rhodes, Editors, 1979); Pharmaceutical Dosage Forms: Tablets (Lieberman et al., 1981); Ansel, Introduction to Pharmaceutical Dosage Forms 2nd Edition (1976); Remington's Pharmaceutical Sciences, 17th. Ed. (Mack Publishing Company, Easton, Pa., 1985); Advances in Pharmaceutical Sciences (David Ganderton, Trevor Jones, Eds., 1992); Advances in Pharmaceutical Sciences Vol. 7. (David Ganderton, Trevor Jones, James McGinity, Eds., 1995); Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms (Drugs and the Pharmaceutical Sciences, Series 36 (James McGinity, Ed., 1989); Pharmaceutical Particulate Carriers: Therapeutic Applications: Drugs and the Pharmaceutical Sciences, Vol. 61 (Alain Rolland, Ed., 1993); Drug Delivery to the Gastrointestinal Tract (Ellis Horwood Books in the Biological Sciences. Series in Pharmaceutical Technology; J.G. Hardy, S.S. Davis, Clive G. Wilson, Eds.); Modem Pharmaceutical Drugs and the Pharmaceutical Sciences, Vol. 40 (Gilbert S. Banker, Christopher T. Rhodes, Eds.).
Таблетки могут содержать подходящие связующие лубриканты, дезинтеграторы, красящие вещества, ароматизирующие вещества, вещества, индуцирующие текучесть, и плавящие вещества. Например, для перорального введения в стандартной лекарственной форме таблетки или капсулы активный лекарственный компонент может быть объединен с пероральным, нетоксичным, фармацевтически приемлемым, инертным носителем, таким как лактоза, желатин, агар, крахмал, сахароза, глюкоза, метилцеллюлоза, фосфат дикальция, сульфат кальция, манит, сорбит, микрокристаллическая целлюлоза и подобные. Подходящие связующие включают крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, кукурузный крахмал, природные или синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия, повидон, карбоксиметилцеллюлоза, полиэтиленгликоль, воска и подобные. Лубриканты, используемые в таких лекарственных формах, включают олеат натрия, стеарат натрия, бензоат натрия, ацетат натрия, хлорид натрия, стеариновую кислоту, стеарилфумарат натрия, тальк и подобные. Дезинтеграторы включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь, кроскармеллозу натрия, гликолат крахмала натрия и подобные.
R(+) PAI предназначен для составления в виде пероральной твердой лекарственной формы, особенно в форме таблетки. Составление таблеток наиболее часто включает прессование отмеренного объема лекарственного продукта, в форме гранулированного порошка, двумя штампами в полости матрицы. Таблетка принимает размер и форму штампов и полости матрицы (Rudnic et al., Chpt. 45, Remington's Pharmaceutical Sciences, 20th Edition, Lippincott Williams & Wilkins, Baltimore, Md. (2000)).
Главным вопросом, который возникал в процессе составления R(+) PAI, была трудность сохранения однородности содержимого лекарственного продукта. Стандартное количество R(+) PAI относительно невелико по отношению к общей массе таблетки; типичная композиция, например, включает 1 мг R(+) PAI в таблетке с общей массой более 200 мг. Как таковое, небольшое колебание в количестве R(+) PAI из-за проблем текучести или разделения может приводить к большому процентному отклонению от 1 мг.
Другой причиной такой проблемы является крупная и неправильная форма частиц R(+) PAI, что возникает в результате кристаллизации соли. Хотя лекарственное вещество обычно измеряется по массе, объем также играет важную роль в процессе получения таблеток. Следовательно, крупные и неправильной формы частицы могут легко уменьшить однородность содержимого. Следовательно, размер частиц уменьшают и делают более однородным с целью обеспечить однородность содержимого.
Уменьшение размера частиц достигают посредством измельчения или механического процесса уменьшения размера частиц, который включает любое одно или более из нарезания, выкрашивания, дробления, измельчения, перемалывания, микронизации и растирания. Обычно наиболее общепринятой причиной для уменьшения размера частиц посредством измельчения является увеличение растворимости. Скорость растворения мелких частиц обычно больше, чем крупных частиц, из-за того, что большая площадь поверхности лекарственного вещества находится в контакте с жидкой средой. Этот эффект подчеркивается большей скоростью растворения, наблюдаемой после микронизации плохо растворимых лекарственных средств (Abdou et al., Chpt. 35, Remington's, infra). Следовательно, при рецептировании лекарственного средства с низкой скоростью растворения желательно уменьшить размер частиц с целью увеличения растворимости и таким образом облегчить быструю абсорбцию в желудочно-кишечном тракте или ротовой полости.
Мезилат R(+) PAI, однако, является легко растворимым в воде (приблизительно 617 мг/мл при 25°С). Такое быстрое поведение при растворении минимизирует проблемы, связанные с абсорбцией в желудочно-кишечном тракте и биодоступностью. Следовательно, увеличение растворимости не является определяющим фактором для измельчения R(+) PAI.
В таких случаях, когда лекарственные вещества уже обладают высокой растворимостью, уменьшение размера частиц может быть нежелательным и даже вредным. Увеличение площади поверхности путем измельчения может увеличить скорость разложения активного вещества. Как обсуждается в патенте США № 6126968, R(+) PAI подвержен разложению. Лекарственные вещества также могут подвергаться нежелательной полиморфной трансформации во время измельчения. В результате измельчение может влиять на эффективность и безопасность лекарственного вещества.
Недостатки измельчения иллюстрируются лекарственным средством Нитрофурантоин, антибактериальным лекарственным средством, используемым в лечении неосложненных инфекций нижних мочевых путей. Его скорость абсорбции зависит от размера кристаллов. Макрокристаллическая форма Нитрофурантоина имеет меньшую скорость растворения и абсорбции, давая более низкие концентрации в сыворотке и более длительное достижение пиковой концентрации в моче, чем микрокристаллическая форма (Мартиндал). Однако 18-летнее исследование показало, что микрокристаллическая форма дает отрицательные побочные эффекты («нежелательные явления»), такие как тошнота и желудочно-кишечные расстройства. Такие отрицательные побочные эффекты не проявляются у пациентов, которые принимают макрокристаллическую лекарственную форму (Brumfitt, W. And J.M.T. Hamilton-Miller, J. Antimicrobial Chemotherapy 42: 363-371 (1998)).
Несмотря на тот факт, что измельчение необязательно для увеличения растворимости легко растворимого R(+) PAI, неожиданно было обнаружено, что уменьшение размера частиц, посредством измельчения, улучшает однородность содержимого таблетки.
Способ измельчения определяют на основании характеристик вещества, таких как исходный размер частиц и желаемый размер частиц, а также температура плавления, ломкость, жесткость и содержание влаги (O'Conner et al., Chpt. 37, Remington's, infra). Перемалывание было определено как подходящее для изменения PSD R(+) PAI с целью обеспечения однородности содержимого лекарственного продукта.
Множество аналитических средств доступно для определения PSD. На ранней стадии разработки анализ PSD проводили посредством микроскопического и гранулометрического анализа. Лазерная дифракция была выбрана как окончательный аналитический метод для измерения PSD. Так как крупные частицы были наибольшей проблемой, важными характеристиками PSD были d (0,9), который представляет собой размер, в микронах, ниже которого обнаруживаются 90% частиц по объему, и d(0,1), который является размером, в микронах, ниже которого обнаруживаются 10% частиц по объему.
«Однородность смеси», как используется в настоящем описании, относится к однородности гранулята, включающего частицы R(+) PAI до рецептирования таблеток, и может представлять собой или один образец, или среднее более чем одного образца.
«Однородность содержимого», как используется в настоящем описании, относится к однородности содержимого R(+) PAI в лекарственных формах, например таблетках, после рецептирования.
«Частица», как используется в настоящем описании, относится к агрегированной физической единице соединения R(+) PAI, т.е. куску или грануле R(+) PAI. Например, фиг. 1 и 2 обеспечивают фотографии различных частиц R(+) PAI.
«Относительное стандартное отклонение» или «RSD», как используется в настоящем описании, относится к оценке насколько точно каждое измерение однородности смеси или однородности содержимого, т.е. насколько каждая отдельная единица отклоняется от группы.
Детали экспериментов
Пример 1
Образцы мезилата R(+) PAI перед измельчением содержали крупные, неправильной пластинчатой формы частицы различных размеров (см. фиг.1). PDS четырех различных партий измеряли с использованием Лазерной Дифракции Malvern с использованием модели Mastersizer S. Лазерная дифракция опирается на факт, что угол дифракции света является обратно пропорциональным размеру частиц. Свойства частиц измеряли и интерпретировали как меру сферы (сфера является единственной формой, которая может быть описана одним уникальным числом). Кроме того, лазерная дифракция рассчитывает распределение размеров частиц на основании околообъемных терминов, таким образом, выделяя количество частиц из определения размера частиц. Модель Mastersizer S измеряет частицы с использованием единственной методики и единственного диапазона установок.
Значения в таблице представляют собой среднее двух измерений. Каждое измерение получали через 3 минуты рециркуляции с контролем скорости в положении 14 часов. Средой для разведения был 1% раствор соли диоктилсульфосукцината натрия в н-гексане. Концентрация лекарственного вещества, используемая в измерениях составила от 0,05% до 0,35% по объему. Результаты были подтверждены микроскопическими наблюдениями. Для световой микроскопии эмульсию или суспензию, разведенную или неразведенную, помещали на стекло или рифленую ячейку. Окуляр микроскопа снабжали микрометром, посредством которого может быть оценен размер частиц. Результаты эксперимента показаны в таблице 1.
D (0,1) представляет собой размер частиц, в микронах, ниже которого обнаруживается 10% объемного распределения популяции, и d(0,9) представляет собой размер частиц, в микронах, ниже которого обнаруживается 90% объемного распределения популяции.
Результаты
Из примера 1 очевидно, что перемалывание лекарственного вещества изменяет PSD и, следовательно, получают более мелкие частицы.
Пример 2
Получали композиции в соответствии со следующим способом, с использованием нескольких партий R(+) PAI с регулируемым PSD, определяемым способом, используемым в примере 1:
Все эксципиенты, за исключением лубрикантов, смешивали с мезилатом R(+) PAI и во время перемешивания добавляли воду. Когда гранулят становился однородным, его сушили в сушилке с псевдоожиженным слоем. Высушенный гранулят затем перемалывали в осцилляционном грануляторе. Затем получали смазанную смесь с использованием барабанного смесителя. Затем смесь прессовали в таблетки весом 210,0 г.
Определяли среднюю однородность смеси и однородность содержимого. Среднюю однородность смеси каждой партии гранулята определяли взятием 10 образцов, которые представляли собой верхний, средний и нижний слои каждой партии окончательной смеси (перед таблетированием), проводя ВЭЖХ анализ для оценки количества активного ингредиента в образцах, и сравнением количества активного ингредиента в каждом образце с меченным количеством активного ингредиента. Затем определяли стандартное отклонение и относительное стандартное отклонение в соответствии со следующей формулой:
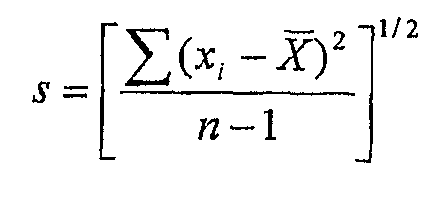
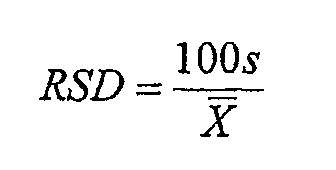
В вышеуказанных формулах s представляет собой стандартное отклонение; RSD представляет собой относительное стандартное отклонение; x1, x2, x3 … xn являются отдельными количествами тестируемых образцов, выраженными в процентах меченого количества лекарственного вещества в каждом образце; X (полоска) представляет собой среднее значение, полученное из тестируемого образца, выраженное в процентах от меченного количества лекарственного вещества в каждом образце; и n является количеством тестируемых единиц.
Однородность содержимого таблеток определяли с использованием 10 случайных таблеток, проведением ВЭЖХ анализа для оценки количества активного ингредиента в каждой таблетке, и сравнения количества активного ингредиента в каждой таблетке с меченым количеством активного ингредиента. Стандартное отклонение и относительное стандартное отклонение определяли как описано выше.
d(0,1) и d(0,9) определяли как в примере 1. Результаты показаны в таблице 2.
Результаты
Однородность содержимого тестируемых партий варьировала от 98,6% до 100,6%. RSD (относительное стандартное отклонение, выраженное как процент от среднего) было ниже, чем 2,0% для всех партий таблеток, показывая, что однородность таблеток была высокой, несмотря на небольшое количество активного ингредиента в каждой таблетке. Следовательно, такие результаты пройдут критерии приемлемости, указанные первой стадией теста однородности содержимого USP.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОСТАВЫ РАЗАГИЛИНА С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2734632C2 |
| СОСТАВЫ РАЗАГИЛИНА С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2607595C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ АТОРВАСТАТИНА | 2004 |
|
RU2325903C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ АТОРВАСТАТИН, ИЗГОТОВЛЕННЫЙ БЕЗ ГРАНУЛИРОВАНИЯ | 2004 |
|
RU2314804C2 |
| ТАБЛЕТКИ ТАМЗУЛОСИНА | 2002 |
|
RU2311903C2 |
| ТАБЛЕТИРУЕМЫЙ ПРЕПАРАТ (4'-ТРИФТОРМЕТИЛФЕНИЛ)АМИДА (Z)-2-ЦИАНО-3-ГИДРОКСИ-БУТ-2-ЕНОЕВОЙ КИСЛОТЫ С УЛУЧШЕННОЙ УСТОЙЧИВОСТЬЮ | 2010 |
|
RU2681079C2 |
| ОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ТОФАЦИТИНИБА С НЕПРЕРЫВНЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2674345C2 |
| ОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ТОФАЦИТИНИБА С НЕПРЕРЫВНЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2790166C2 |
| СТАБИЛЬНЫЕ КОМПОЗИЦИИ АТОРВАСТАТИНА, ПОЛУЧАЕМЫЕ ВЛАЖНЫМ ГРАНУЛИРОВАНИЕМ | 2004 |
|
RU2332211C2 |
| СТАБИЛЬНЫЕ СОСТАВЫ ШИПУЧЕГО БИСФОСФОНАТА СО СВОЙСТВАМИ БЫСТРОЙ СОЛЮБИЛИЗАЦИИ | 2011 |
|
RU2608724C2 |

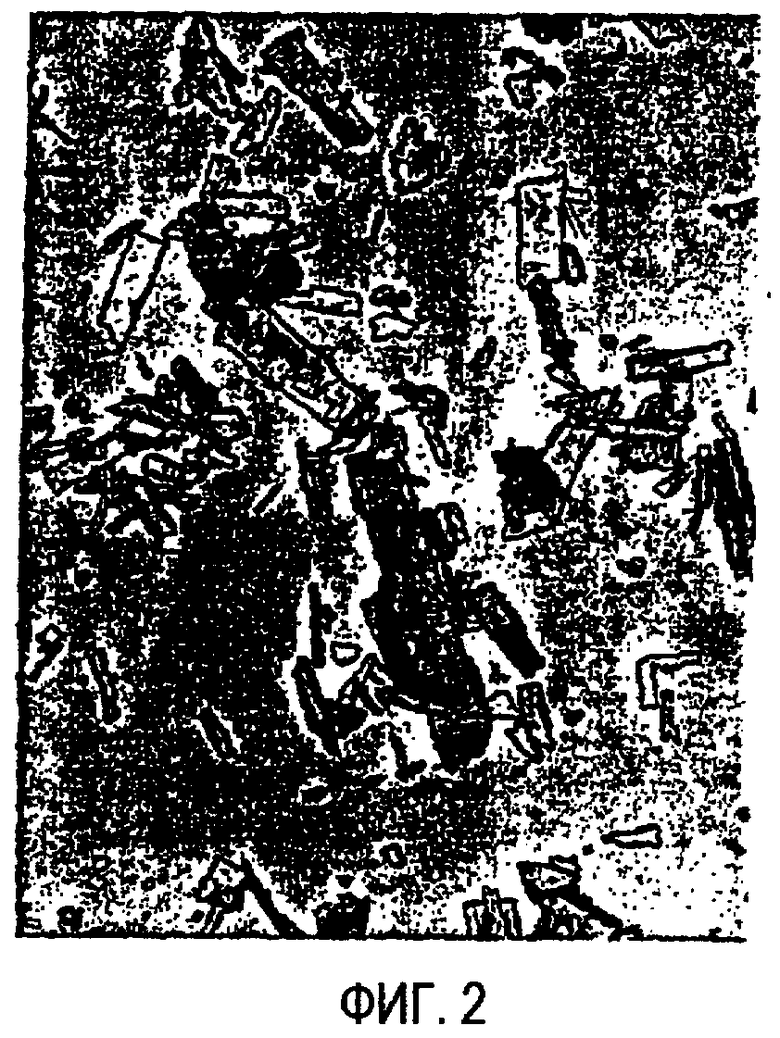
Изобретение относится к медицине и касается фармацевтических препаратов солей R(+)-N-пропаргил-1-аминоиндана. Изобретение обеспечивает улучшенную однородность содержимого лекарственного продукта. 5 н. и 27 з.п. ф-лы, 2 ил., 2 табл.
1. Смесь частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана, где более чем 90% общего количества по объему частиц соли R(+)-N-пропаргил-1-аминоиндана имеет размер менее 250 мкм.
2. Смесь по п.1, где более чем 90% общего количества по объему частиц соли R(+)-N-пропаргил-1-аминоиндана имеет размер менее 220 мкм.
3. Смесь по п.2, где более чем 90% общего количества по объему частиц соли R(+)-N-пропаргил-1-аминоиндана имеет размер менее 200 мкм.
4. Смесь по любому из пп.1-3, где по меньшей мере 90% общего количества по объему частиц соли R(+)-N-пропаргил-1-аминоиндана имеет размер более 6 мкм.
5. Смесь по п.1, где фармацевтически приемлемой солью является соль тартрат, эзилат, мезилат или сульфат.
6. Смесь по п.5, где фармацевтически приемлемой солью является мезилатная соль.
7. Твердая композиция, включающая терапевтически эффективное количество смеси частиц по любому из пп.1-6 и носитель.
8. Твердая композиция по п.7, где носитель представляет собой фармацевтически приемлемый носитель.
9. Твердая композиция по любому из пп.7 или 8, где смесь частиц и носитель составляют гранулят.
10. Композиция по п.9 в твердой лекарственной форме.
11. Композиция по п.10 в пероральной лекарственной форме.
12. Композиция по п.11, где пероральной лекарственной формой является таблетка.
13. Композиция по любому из пп.10-12, где относительное стандартное отклонение (RSD) содержания соли R(+)-N-пропаргил-1-аминоиндана между твердыми лекарственными формами составляет менее 4%.
14. Композиция по п.13, где относительное стандартное отклонение (RSD) содержимого соли R(+)-N-пропаргил-1-аминоиндана составляет менее 3%.
15. Композиция по п.14, где относительное стандартное отклонение (RSD) содержимого соли R(+)-N-пропаргил-1-аминоиндана составляет менее 2%.
16. Композиция по любому из пп.10-12 и 14 и 15, где однородность содержимого составляет от 95 до 105%.
17. Применение композиции по любому из пп.7-16 в получении лекарственного средства для лечения пациента, пораженного болезнью Паркинсона.
18. Способ получения композиции по п.7, который включает уменьшение размера частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана до размера частиц менее 250 мкм,
где стадия уменьшения включает измельчение частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана,
способ дополнительно включает смешивание частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана с носителем для получения гранулята,
способ дополнительно включает следующие стадии:
i) определение однородности смеси гранулята и
ii) образование композиции из гранулята, только если однородность смеси гранулята удовлетворяет заранее определенным критериям, так чтобы получить композицию.
19. Способ по п.18, где размер частиц составляет менее 200 мкм.
20. Способ по п.18 или 19, где однородность смеси гранулята составляет от 90 до 110% и относительное стандартное отклонение (RSD) однородности смеси составляет менее 2%.
21. Способ по п.20, где однородность смеси гранулята составляет от 95 до 105% и относительное стандартное отклонение (RSD) однородности смеси составляет менее 2%.
22. Способ по п.18, где стадия образования ii) включает получение твердой лекарственной формы.
23. Способ по п.22, где стадия образования ii) включает получение пероральной лекарственной формы.
24. Способ по п.23, где пероральной лекарственной формой является таблетка.
25. Способ по любому из пп.22-24, дополнительно включающий определение однородности содержимого твердой лекарственной формы.
26. Способ по п.25, дополнительно включающий стадию количественного определения твердой лекарственной формы в качестве приемлемой композиции, только если однородность содержимого удовлетворяет заранее определенным критериям.
27. Способ по п.26, где заранее определенным критерием является однородность содержимого от 95 до 105%.
28. Способ по п.26, где заранее определенным критерием является относительное стандартное отклонение однородности содержимого менее 4%.
29. Способ по любому из пп.18, 21, 23, 24 и 26-28, дополнительно после стадии i) включающий стадию уменьшения размера частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана любой партии, которая не соответствует заранее определенным критериям.
30. Способ по любому из пп.18, 21, 23, 24 и 26-28, дополнительно до 1 стадии i) включающий уменьшение размера частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана.
31. Способ по п.30, где стадия уменьшения включает измельчение частиц фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана.
32. Твердая фармацевтическая композиция, включающая терапевтически эффективное количество фармацевтически приемлемой соли R(+)-N-пропаргил-1-аминоиндана, полученная способом по любому из пп.18-31.
| US 6126968 A, 03.10.2000 | |||
| US 5622720 A, 22.04.1997 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |

