ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к фармацевтическим композициям, приготовленным в форме для пролонгированного высвобождения активных соединений, подходящих для лечения нейродегенеративных заболеваний, в частности, болезни Паркинсона, и повреждений нервной системы.
УРОВЕНЬ ТЕХНИКИ
Было показано, что некоторые производные пропаргил амина селективно ингибируют активность моноаминоксидаз (МАО)-Б и/или МАО-А, которые инактивируют моноаминергические нейромедиаторы, такие как допамин, и, таким образом, подходят для лечения нейродегенеративных заболеваний, таких как болезнь Паркинсона (БП) и болезнь Альцгеймера (БА), которые характеризуются низкими уровнями допамина. Также было показано, что данные соединения защищают от нейродегенерации путем предотвращения апоптоза.
Первым соединением, которое, как было обнаружено, селективно ингибирует МАО-Б, был R-(-)-N-метил-N-(проп-2-инил)-2-аминофенилпропан, также известный как L-(-)-депренил, R-(-)-депренил или селегилин (selegiline). Помимо БП, селегилин, как было показано, может применяться также при лечении, других заболеваний и патологических состояний, которые включают синдром отмены лекарственного средства (WO 92/21333, включая синдром отмены психостимуляторов, опиатов, наркотических средств и барбитуратов); депрессию (US 4861800); БА; дегенерацию желтого пятна (US 5242950); возрастные дегенерации, включая дегенерацию функции почек и дегенерацию когнитивной функции, на основании данных о способности к пространственному обучению (US 5151449); болезнь Кушинга гипофизарного происхождения у людей и животных (US 5192808); дисфункцию иммунной системы как у людей (US 5387615), так и у животных (US 5276057); возрастную потерю массы тела у млекопитающих (US 5225446); шизофрению (US 5151419); и различные неопластические состояния, включая раковые опухоли, такие как рак молочной железы и гипофиза. В WO 92/17169 описано применение селегилина для лечения нейромышечных и нейродегенеративных заболеваний и для лечения повреждений ЦНС вследствие гипоксии, гипогликемии, ишемического инсульта или травмы. Кроме того, было широко изучено биохимическое действие селегилина в отношении нервных клеток (см., например, Tatton, 1993; и Tatton and Greenwood, 1991). В US 6562365 описано применение десметилселегилина для лечения чувствительных к селегилину заболеваний и патологических состояний.
Разагилин (rasagiline), R(+)-N-пропаргил-1-аминоиндан, высокоактивный селективный необратимый ингибитор МАО-Б, был разрешен для лечения БП в Европе, Израиле и в США под торговым названием АЗИЛЕКТ® (AZILECT®) или АГИЛЕКТ® (AGILECT®) (Teva Pharmaceutical Industries Ltd., Петах-Тиква, Израиль). При исследованиях на культурах клеток и in vivo было показано, что разагилин обладает нейропротекторной активностью и антиапоптотическим действием в отношении различных поражений (Youdim and Weinstock, 2002а). Механизм, лежащий в основе нейрозащиты разагилином, был изучен на допаминергических клетках SH-SY5Y и PC 12 в культуре в отношении апоптоза, индуцированного N-метил(R)салсолинолом, донором пероксинитрита N-морфолино-сиднонимином (SIN-1), 6-гидроксидопамином, и удалением сыворотки и фактора роста нервов (Youdim et al, 2001b; Akao et al., 1999, 2002; Marayama et al., 2001a, 2001b, 2002).
Разагилин и его фармацевтически приемлемые соли были впервые описаны в патентах США №5387612, 5453446, 5457133, 5576353, 5668181, 5786390, 5891923 и 6630514 в качестве подходящих для лечения БП, нарушений памяти, деменции альцгеймеровского типа, депрессии и синдрома гиперактивности. Производные 4-фтор-, 5-фтор- и 6-фтор-N-пропаргил-1-аминоиндана были описаны в US 5486541 для тех же целей. В патентах США №5519061, 5532415, 5599991, 5744500, 6277886, 6316504, 5576353, 5668181, 5786390, 5891923 и 6630514 описан разагилин и его фармацевтически приемлемые соли в качестве подходящих для лечения при дополнительных терапевтических показаниях, в частности, для лечения аффективного расстройства, неврологической гипоксии или аноксии, нейродегенеративных заболеваний, нейротоксического поражения, инсульта, ишемии головного мозга, повреждения при травме головы, повреждения при травме позвоночника, шизофрении, синдрома дефицита внимания, рассеянного склероза и синдромов отмены.
В US 6251938 описаны соединения N-пропаргилфенилэтиламина, а в патентах США №6303650, 6462222 и 6538025 описаны соединения N-пропаргил-1-аминоиндана и N-пропаргил-1-аминотетралина в качестве подходящих для лечения депрессии, синдрома дефицита внимания, синдрома дефицита внимания и гиперактивности, синдрома Туретта, БА и другой деменции, такой как сенильная деменция, деменция паркинсоновского типа, сосудистая деменция и деменция с тельцами Леви.
В предыдущей работе было сделано предположение, что разагилин и родственные производные пропаргиламина подавляют каскад апоптотической гибели, начинающийся в митохондриях, за счет предотвращения предапоптотического снижения мембранного потенциала митохондрий (ΔΨm) вследствие изменения проницаемости и активации каспазы 3, ядерную транслокацию глицеральдегид-3-фосфатдегидрогеназы и апоптотические процессы нуклеосомной фрагментации ДНК (Youdim and Weinstock, 2002b). При контролируемой монотерапии и в качестве вспомогательного средства к L-допе (L-dopa) разагилин продемонстрировал активность в отношении болезни Паркинсона.
Были синтезированы два аналога разагилина, содержащие карбаматную группу, в попытке комбинации ингибирующих свойств в отношении МАО и нейропротекторных свойств разагилина с ингибирующей холинэстеразу (ChE) активностью ривастигмина (rivastigmine), лекарственного средства с доказанной эффективностью у пациентов с БА. Данные аналоги представляют собой (N-пропаргил-(3R)-аминоиндан-5-ил)-этилметилкарбамат (TV3326), обладающий ингибирующей активностью в отношении как ChE, так и МАО-А и Б, и его S-изомер, TV3279, являющийся ингибитором ChE, но не МАО (Weinstock, 1999; Grossberg and Desai, 2001). Подобно разагилину, TV3326 и TV3279 обладают нейропротекторными свойствами в отношении различных поражений, которые не зависят от ингибирующей активности в отношении ChE и МАО, а могут возникать вследствие некоторой фармакологической активности, свойственной пропаргиламинному фрагменту (Youdim and Weinstock, 2002а). Кроме того, данные соединения стимулируют высвобождение нейротрофического/нейропротекторного неамилоидогенного растворимого белка-предшественника амилоида (sAPPP) путем активации протеинкиназы С и путей митоген-активируемой протеинкиназы (Yogev-Falach, 2002). Таким образом, данные лекарственные средства могут влиять на образование потенциально амилоидогенных производных и могут иметь клиническое значение для лечения БА.
В US 5169868, US 5840979 и US 6251950 описаны алифатические пропаргиламины в качестве селективных ингибиторов МАО-Б, нейропротекторных агентов и агентов «спасения» клеток. Было показано, что основное соединение, (R)-N-(2-гептил)метилпропаргиламин, является мощным ингибитором МАО-Б и антиапоптотическим агентом (Durden et al., 2000).
Много лет тому назад сообщалось, что пропаргиламин является суицидным ингибитором медьсодержащей аминоксидазы бычьей плазмы (BPAO), хотя его активность была невысокой. В US 6395780 пропаргиламин описан в качестве слабого ингибитора системы расщепления глицина.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению было обнаружено, что введение разагилина с обеспечением его замедленного высвобождения, вследствие чего воздействие лекарственного средства заметно продлевается по сравнению с воздействием после разового введения, может иметь решающее значение для достижения оптимального нейропротекторного действия при различных поражениях ЦНС. В частности, в то время как разовое введение возрастающих доз разагилина (0,1, 0,12 или 0,15 мг/кг) в модели болезни Паркинсона (БП) у мышей, индуцированной N-метил-4-фенил-1,2,3,6-тетрагидропиридином (МРТР), оказывало практически сходное действие на уровни допамина у мышей, приводя к повышению содержания допамина до примерно 60% по сравнению с мышами, не получавшими лекарственное средство, введение таких же трех доз лекарственного средства с замедленным высвобождением в течение 24 часов приводило к значительному ответу на дозу, при котором уровни допамина составляли 57%, 74% и 88%, соответственно, по сравнению с мышами, не получавшими лекарственное средство, что указывает на выраженный благоприятный эффект введения с замедленным высвобождением по сравнению с немедленным высвобождением в отношении уровней допамина в головном мозге мышей, подвергнутых воздействию МРТР. Интересным является тот факт, что схожие результаты получали после введения метаболита разагилина, 1-аминоиндана, с замедленным высвобождением, что приводило к значительному восстановлению уровней допамина по сравнению с мышами, которым вводили такую же дозу лекарственного средства один раз в сутки в течение такого же периода времени.
Также, как было обнаружено в модели индуцированной 6-гидроксидопамином (6-OHDA) БП у крыс, заметно улучшенный эффект при чистом вращении, индуцированном амфетамином, наблюдали у крыс, получавших лечение разагилином, вводимым способом с замедленным высвобождением, по сравнению с крысами, получавшими лечение тем же лекарственным средством путем ежедневных инъекций.
Таким образом, в соответствии с одним из аспектов настоящего изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, приготовленная в форме для пролонгированного высвобождения указанного активного агента. В предпочтительном варианте реализации активный агент, содержащийся в указанной фармацевтической композиции, представляет собой R(+)-N-пропаргил-1-аминоиндан (разагилин) или его фармацевтически приемлемую соль.
В соответствии с другим аспектом настоящего изобретения предложена пеллета с пролонгированным высвобождением, содержащая:
(i) инертное ядро пеллеты;
(ii) слой лекарственного средства, покрывающий указанное ядро пеллеты, при этом указанный слой лекарственного средства содержит активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, возможно подходящим образом смешанные со связующим веществом и/или пленкообразующим полимером, и возможно дополнительно смешанные с веществом, способствующим скольжению;
(iii) возможно, изолирующий/защитный подслой покрытия, покрывающий указанный слой лекарственного средства; и
(iv) слой покрытия с замедленным высвобождением, покрывающий указанный подслой покрытия (при его наличии) или указанный слой лекарственного средства.
В соответствии с еще одним аспектом настоящего изобретения предложена фармацевтическая композиция для перорального введения, содержащая пеллеты с пролонгированным высвобождением, как определено выше.
Различные фармацевтические композиции согласно настоящему изобретению подходят для лечения нейродегенеративных заболеваний, предпочтительно болезни Паркинсона, и повреждений нервной системы.
Таким образом, в соответствии с другим аспектом настоящее изобретение относится к способу лечения нейродегенеративного заболевания или повреждения нервной системы у нуждающегося в этом индивидуума, включающему введение указанному индивидууму фармацевтической композиции, определенной выше.
В соответствии с еще одним аспектом настоящее изобретение относится к способу получения состава в форме для пролонгированного высвобождения, содержащего активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, включающему этапы:
(i) растворения указанного активного агента, возможно подходящим образом смешанного со связующим веществом и/или веществом, способствующим скольжению, в подходящей системе растворителей с получением однородной суспензии;
(ii) нанесения слоя указанной суспензии, полученной на этапе (i), на инертные пеллеты, такие как инертные гранулы нонпарель (nonpareil seeds);
(iii) возможно, нанесения на пеллетысодержащие активный агент, полученные на этапе (ii), изолирующего/защитного подслоя покрытия;
(iv) нанесения на пеллеты, полученные на этапе (ii) или (iii), слоя покрытия с пролонгированным высвобождением, который обеспечивает пролонгированное высвобождение указанного активного агента, с получением тем самым указанного состава с пролонгированным высвобождением; и
(v) возможно, смешивания покрытых пеллет, полученных на этапе (iv), с подходящим наполнителем.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фиг. 1 показано влияние комбинаций разагилин-прамипексол (pramipexole) (обозначенных Комб. 1, 2 и 3), в которых доза прамипексола является постоянной (0,5 мг/кг), а доза разагилина варьируется (0,1, 0,12 или 0,15 мг/кг, соответственно) на уровни допамина (DA) в головном мозге. В частности, как показано, введение МРТР без лекарственной терапии (физиологический раствор интраперитонеально и с замедленным высвобождением) вызывало снижение уровней допамина более чем на 80% по сравнению с уровнями у мышей, ранее не подвергнутых экспериментам (мыши, ранее не подвергнутые экспериментам, интраперитонеальное и введение способом с замедленным высвобождением). Лечение (интраперитонеальное введение) комбинациями разагилин-прамипексол вызывало восстановление уровней допамина до примерно 60% у мышей, ранее не подверженных экспериментам, - эффект, практически одинаковый для всех трех комбинаций; однако те же три комбинации при их введении способом с замедленным высвобождением (ЗВ) с использованием насоса Alzet, приводило к значительному дозозависимому повышению уровней допамина до 57%, 74% и 88% в соответствии с повышенными дозами разагилина.
На Фиг. 2 показано влияние метаболита разагилина, аминоиндана, при введении ЗВ на уровни допамина (DA) в головном мозге. В частности, воздействие МРТР вызывало снижение уровней допамина более чем на 90% по сравнению с уровнями у мышей, ранее не подвергнутых экспериментам. Лечение аминоинданом, введенным способом с медленным высвобождением (ЗВ), вызывало значительное восстановление уровней допамина по сравнению с уровнями у мышей, получавших лечение разбавителем или аминоинданом, вводимым путем ежедневных интраперитонеальных инъекций (IP).
На Фиг. 3 показано чистое вращение, индуцированное амфетамином, которое представляет собой вращение по часовой стрелке за вычетом вращения против часовой стрелки (CW-CCW), измеренное у крыс, получавших лечение разагилином, как описано в Примере 3. Значительно улучшенный эффект в чистом вращении показан у крыс, получавших лечение разагилином с пролонгированным высвобождением (ЗВ) с использованием насоса Alzet, по сравнению с крысами, получавшими лечение разагилином с быстрым высвобождением (БВ) путем ежедневных интраперитонеальных инъекций.
На Фиг. 4 приведены данные растворения in vitro для пеллет мезилата разагилина (1,0 мг) со слоем покрытия с пролонгированным высвобождением (ПВ) из Примеров 4-6 (15% ПВ, 22% ПВ и 28% ПВ, соответственно) в буферном растворе кишечной жидкости (IFS).
На Фиг. 5 приведены данные растворения in vitro для пеллет мезилата разагилина (1,0 мг) со слоем покрытия с пролонгированным высвобождением (ПВ), содержащие подслой покрытия, из Примеров 7-8 (15% ПВ и 16% ПВ, соответственно) в буфере IFS.
На Фиг. 6 приведены данные растворения in vitro для пеллет мезилата разагилина (1,0 мг) со слоем покрытия с пролонгированным высвобождением (ПВ), содержащие подслой покрытия, из Примера 7 (15% ПВ) в (i) буфере IFS (рН 6,8), имитирующем условия в кишечнике; (ii) буферном растворе желудочного сока (GFS) (рН 1,2), имитирующем условия в пустом желудке; (iii) буфере GFS в течение 2 часов, а затем буфере IFS в течение еще 20 часов; (iv) ацетатном буфере (рН 4,5), имитирующем условия в полном желудке; и (v) дистиллированной воде (DI).
На Фиг. 7 приведены данные исследования стабильности in vitro в буфере IFS для пеллет мезилата разагилина (1,0 мг) со слоем покрытия с пролонгированным высвобождением (ПВ), содержащих подслой покрытия, из Примера 7 (15% ПВ) в нулевой момент времени (сразу после получения), через 1 месяц при 40°C и 75% влажности (1М Acc.) и через 2 и 3 месяца при 40°C и 75% влажности (2М Acc. и 3М Асе, соответственно).
На Фиг. 8 приведены данные растворения in vitro для пеллет мезилата разагилина со слоем покрытия с пролонгированным высвобождением (ПВ), содержащих подслой покрытия, из Примера 7 (27% ПВ) в буфере IFS.
На Фиг. 9 показан график зависимости концентрации разагилина в плазме крови (нг/мл) от времени, вводимого способом внутривенного болюсного введения, дуоденального болюсного введения или болюсного введения в толстую кишку.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Основной причиной ингибирования моноаминоксидазы Б (МАО-Б) при болезни Паркинсона является повышение активности стриарного допамина, приводящее к симптоматическим полезным эффектам в отношении двигательной функции. Поскольку МАО-Б ответственна, среди прочего, за гидролиз допамина, ингибирование МАО-Б вызывает увеличение уровня допамина. Согласно описанному механизму действия, активность разагилина отделена от его фармакокинетики вследствие того, что ингибирование МАО-Б разагилином является необратимым и, таким образом, эффект, обусловленный указанным ингибированием, сохраняется до образования новой МАО-Б, т.е. в течение примерно 2-3 недель. Следовательно, можно предположить, что не будет никакого полезного эффекта от введения разагилина способом с замедленным высвобождением. Тем не менее, недавно полученные данные показывают, что разагилин может индуцировать нейропротективное действие по альтернативному механизму - через ингибирование апоптоза или других путей. Также известно, что разагилин подвергается значительному метаболизму, и его основной метаболит, 1-аминоиндан, обладает нейропротекторной активностью, не связанной с ингибированием МАО-Б (Bar-Am et al., 2007; Weinreb et al., 2010).
Разагилин, селегилин и другие структурно родственные производные пропаргиламина увеличивают выживаемость нейронов независимо от ингибирования МАО-Б, отчасти за счет уменьшения апоптоза (Tatton et al., 2002). Данный эффект наиболее вероятно модулируется за счет изменения уровней или субклеточной локализации белков, влияющих на проницаемость мембраны митохондрий, удаляющих окислительные радикалы или участвующих в конкретных сигнальных путях апоптоза. Было подтверждено, что как разагилин, так и селегилин, а также другие производные пропаргил амина защищают нейроны от клеточной гибели, индуцируемой различными поражениями в моделях нейродегенеративных нарушений в клетках и у животных, таких как болезнь Паркинсона и болезнь Альцгеймера. Цепь пропаргиламина придает дозозависимые антиоксидантные и антиапоптотические эффекты, которые ассоциировали с нейропротекторным действием во многих экспериментальных моделях. Согласно недавним публикациям, нейропротекторное действие разагилина может быть связано с действием, обусловленным комбинацией разагилина и его метаболита 1-аминоиндана (Tazik et al., 2009; Bar-Am, 2010).
В соответствии с одним из аспектов настоящего изобретения предложена фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, приготовленная в форме для пролонгированного высвобождения указанного активного агента.
Идея, лежащая в основе настоящего изобретения, основана на результатах, приведенных в разделе Примеры далее. В Примере 1 показано, что в то время как однократное введение возрастающих доз разагилина (0,1, 0,12 или 0,15 мг/кг) в модели БП, индуцированной МРТР, у мышей оказывало практически схожее действие на уровни допамина, приводя к повышению содержания допамина до примерно 60% по сравнению с мышами, не получавшими лекарственное средство (подвергнутые воздействию МРТР), введение таких же трех доз разагилина способом с замедленным высвобождением в течение 24 часов приводило к значительному эффекту дозы, при котором уровни допамина составляли 57%, 74% и 88%, соответственно, по сравнению с мышами, не получавшими лекарственное средство, что указывает на высоко благоприятный эффект введения способом с замедленным высвобождением по сравнению со способом с быстрым высвобождением в отношении уровней допамина в головном мозге мышей, подверпгутых воздействию МРТР. В Примере 2 описано исследование с использованием такой же модели БП у мышей, в котором мышей лечили метаболитом разагилина - 1-аминоинданом, и показано, что лечение 1-аминоинданом, вводимым способом с замедленным высвобождением, вызывает значительное восстановление уровней допамина по сравнению с уровнями у мышей, получавшими лечение разбавителем (физиологическим раствором) или тем же лекарственным средством, вводимым путем ежедневных инъекций. Эти выводы также подтверждены исследованием, описанным в Примере 3, показывающим, что в модели БП, индуцированной 6-OHDA, у крыс наблюдают значительно улучшенный эффект при чистом вращении (CW-CCW), индуцированном амфетамином, у крыс, получавших лечение разагилином, вводимым способом с замедленным высвобождением, по сравнению с крысами, получавшими лечение тем же лекарственным средством путем ежедневных инъекций.
Как в действительности показано в настоящем описании в первый раз, когда разагилин вводят способом с пролонгированным высвобождением, воздействие указанного лекарственного средства или его активного метаболита, 1-аминоиндана, значительно продлевается, обеспечивая, таким образом, гораздо более эффективное нейропротективное действие, которое может заметно улучшать состояние пациента. В соответствии с данной идей, как разагилин, так и селегилин, являющиеся ингибиторами МАО-Б, показанными для лечения болезни Паркинсона, а также другие производные пропаргиламина, можно считать «пролекарствами», непрерывно высвобождающими активный агент, или «доставляющими разбавителями» пропаргиламина/аминоиндана. Данные пролекарства или доставляющие разбавители независимо от их ингибирующей активности в отношении МАО, защищают нервные клетки на разных стадиях процесса апоптоза за счет постоянного замедленного воздействия активного агента, определенного выше, т.е. активного агента, содержащего пропаргиламинный фрагмент, аминоиндановый фрагмент, или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемой соли.
Согласно настоящему изобретению можно использовать любую фармацевтически приемлемую соль активного агента. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, мезилатную соль, эзилатную соль, тозилатную соль, сульфатную соль, сульфонатную соль, фосфатную соль, карбоксилатную соль, малеатную соль, фумаратную соль, тартратную соль, бензоатную соль, ацетатную соль, гидрохлоридную соль и гидробромидную соль.
В некоторых вариантах реализации активный агент, содержащийся в фармацевтической композиции согласно настоящему изобретению, представляет собой N-пропаргил-1-аминоиндан, его энантиомер, его метаболит, его аналог или фармацевтически приемлемую соль любого из вышеуказанных соединений.
В одном из конкретных вариантов реализации активный агент представляет собой N-пропаргил-1-аминоиндан в рацемической форме, как описано, например, в US 6630514, или его фармацевтически приемлемую соль.
В других конкретных вариантах реализации активный агент представляет собой R(+)-N-пропаргил-1-аминоиндан (разагилин), его S-энантиомер S-(-)-N-пропаргил-1-аминоиндан или его фармацевтически приемлемую соль. В более конкретных вариантах реализации активный агент представляет собой мезилатную соль, эзилатную соль, тозилатную соль, сульфатную соль, сульфонатную соль, фосфатную соль, карбоксилатную соль, малеатную соль, фумаратную соль, тартратную соль, бензоатную соль, ацетатную соль, гидрохлоридную соль или гидробромидную соль либо разагилина, либо S-(-)-N-пропаргил-1-аминоиндана. В предпочтительных вариантах реализации активный агент представляет собой мезилат разагилина, описанный, например, в US 5532415; эзилат разагилина или сульфат разагилина, описанные, например, в US 5599991; или гидрохлорид разагилина, описанный, например, в US 6630514, и, более предпочтительно, мезилат разагилина.
В другом конкретном варианте реализации активный агент представляет собой метаболит разагилина - 1-аминоиндан, или его фармацевтически приемлемую соль.
В еще других конкретных вариантах реализации активный агент представляет собой аналог N-пропаргил-1-аминоиндана, его энантиомера или его фармацевтически приемлемую соль. Примеры указанных аналогов включают соединения, описанные в US 5486541, такие как, без ограничения ими, 4-фтор-N-пропаргил-1-аминоиндан, 5-фтор-N-пропаргил-1-аминоиндан и 6-фтор-N-пропаргил-1-аминоиндан; соединения, описанные в US 6251938, такие как, без ограничения ими, 3-(N-метил-N-пропилкарбамилокси)-α-метил-N’-пропаргилфенэтиламин, 3-(N,N-диметилкарбамилокси)-α-метил-N’-метил-N’-пропаргилфенэтиламин; 3-(N-метил-N-гексилкарбамилокси)α-метил-N’-метил-N’-пропаргилфенэтиламин; 3-(N-метил-N-циклогексилкарбамилокси)-α-метил-N’-метил-N’-пропаргилфенэтиламин и 3-(N-метил-N-гексилкарбамилокси-α-метил-N’-метил-N’-пропаргилфенэтиламин; соединения, описанные в US 6303650, такие как, без ограничения ими, 6-(N-метил-N-этилкарбамилокси)-N’-пропаргил-1-аминоиндан; 6-(N,N-диметилкарбамилокси)-N’-метил-N’-пропаргил-1-аминоиндан; 6-(N-метил-N-этилкарбамилокси-N’-пропаргил-1-аминотетралин; 6-(N,N-диметилтиокарбамилокси)-1-аминоиндан; 6-(N-пропилкарбамилокси)-N’-пропаргил-1-аминоиндан; 5-хлор-6-(N-метил-N-пропилкарбамилокси)-N’-пропаргил-1-аминоиндан и 6-(N-метил)-N-пропилкарбамилокси)-N’-пропаргил-1-аминоиндан; и соединения, описанные в US 6462222, такие как, без ограничения ими, 6-(N-метил-N-этилкарбамилокси)-N’-метил-N’-пропаргил-1-аминоиндан.
В некоторых других вариантах реализации активный агент, содержащийся в фармацевтической композиции согласно настоящему изобретению, представляет собой пропаргиламин, алифатический пропаргиламин или его фармацевтически приемлемую соль.
В одном из конкретных вариантов реализации активный агент представляет собой пропаргиламин или его фармацевтически приемлемую соль.
В других конкретных вариантах реализации активный агент представляет собой алифатический пропаргиламин, описанный в US 5169868, US 5840979 или US 6251950, такой как, не ограничиваясь ими, N-(1-гептил)пропаргиламин; N-(1-октил)пропаргиламин; N-(1-нонил)пропаргиламин; N-(1-децил)пропаргиламин; N-(1-ундецил)пропаргиламин; N-(1-додецил)пропаргиламин; N-(2-бутил)пропаргиламин; N-(2-пентил)пропаргиламин; N-(2-гексил)пропаргиламин; N-(2-гептил)пропаргиламин; N-(2-октил)пропаргиламин; N-(2-нонил)пропаргиламин; N-(2-децил)пропаргиламин; N-(2-ундецил)пропаргиламин; N-(2-додецил)пропаргиламин; N-(1-бутил)-N-метилпропаргиламин; N-(2-бутил)-N-метилпропаргиламин; N-(2-пентил)-N-метилпропаргиламин; (1-пентил)-N-метилпропаргиламин; N-(2-гексил)-N-метилпропаргиламин; (2-гептил)-N-метилпропаргиламин; N-(2-децил)-N-метилпропаргиламин; (2-додецил)-N-метилпропаргиламин; энантиомер указанных соединений; или фармацевтически приемлемую соль указанных соединений.
В некоторых других вариантах реализации активный агент, содержащийся в фармацевтической композиции согласно настоящему изобретению, представляет собой селегилин, десметилселегилин, паргилин или хлоргилин.
В другом варианте реализации активный агент, содержащийся в фармацевтической композиции согласно настоящему изобретению, представляет собой (N-метил-N-пропаргил)-10-аминометилдибензо[b,f]оксепин, также известный как CGP 3466 и описанный в публикации Zimmermann et al. (1999).
Полное содержание всех патентов США и других публикаций, указанных выше, включено в настоящее описание посредством ссылки так, как если бы было полностью приведено в настоящем документе.
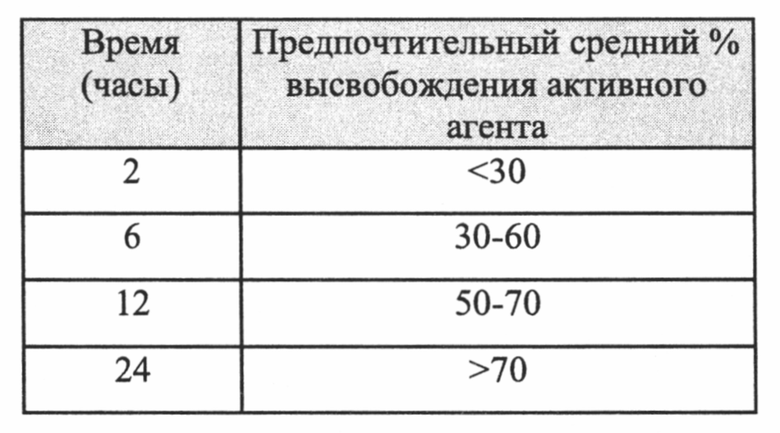
Термины «пролонгированное высвобождение» (ПВ), «контролируемое высвобождение» или «замедленное высвобождение» (ЗВ), являющиеся взаимозаменяемыми в настоящем описании, относятся к такому режиму высвобождения активного агента из его состава, при котором он всасывается в организме в течение определенного периода времени. Состав активного агента с пролонгированным высвобождением может быть получен, например, путем заключения указанного активного агента в каркас вещества, которое организм медленно растворяет, так, чтобы активный ингредиент медленно и постоянно «просачивался» из слоя покрытия, или за счет набухания активного агента с образованием геля с почти непроницаемой поверхностью, при этом лекарственное средство медленно выходит из полупроницаемого слоя.
Основные принципы получения продукта с контролируемым высвобождением заключаются в высвобождении вещества в определенном месте (прицельное воздействие); с постоянной скоростью; и в пределах необходимого терапевтического окна. Механизмы, основанные на принципе Системы контроля растворителями (Solvent Controlled System), такой как системы набухания и осмоса, поддерживающие постоянную концентрацию активного вещества в крови в течение длительных периодов времени, позволяют достигать более эффективных уровней лекарственного средства с меньшими побочными эффектами. Иными словами, терапевтическое окно представляет собой дозу лекарственного средства между количеством, оказывающим эффект (эффективная доза), и количеством, оказывающим больше побочных эффектов, чем желаемых эффектов. Поэтому, характеристики растворения каждого лекарственного средства должны быть соответствовать индивидуальной биодоступности, месту воздействия и всасываемости каждого соединения.
Фармацевтическая композиция согласно настоящему изобретению должна обеспечивать контролируемое высвобождение лекарственного средства, т.е. активного агента. В некоторых вариантах реализации лекарственное средство высвобождается из фармацевтической композиции способом с контролируемым высвобождением согласно кривой высвобождения нулевого, первого, второго или любого другого порядка (N порядка). Контролируемое высвобождение лекарственного средства предпочтительно должно быть медленными, и в некоторых вариантах реализации фармацевтическую композицию готовят в форме для непрерывного замедленного высвобождения лекарственного средства, пульсирующего высвобождения лекарственного средства, многоэтапного высвобождения лекарственного средства или их комбинацию.
Фармацевтические композиции согласно настоящему изобретению могут быть получены традиционными способами, например, описанными в Remington: The Science and Practice of Pharmacy, 19th Ed., 1995, могут находиться в любой традиционной форме и могут быть представлены в различных дозировках.
Композиции могут быть приготовлены в форме для любого подходящего способа введения, например, внутривенного, внутриартериального, внутримышечного, подкожного или интраперитонеального введения, но, предпочтительно, их готовят в форме для перорального введения.
Доза будет зависеть от состояния пациента и будет определяться по усмотрению лечащего врача. В конкретных вариантах реализации доза составляет 0,1-2,0, предпочтительно, 0,2-1,5, более предпочтительно, 0,5-1,0 мг в сутки для взрослого с массой тела 60 кг. Композиции согласно настоящему изобретению можно вводить, например, непрерывно, ежедневно, два раза в сутки, три раза в сутки или четыре раза в сутки, в течение периодов различной длительности, например, недель, месяцев, лет или десятилетий.
Фармацевтическая композиция согласно настоящему изобретению может, например, находиться в форме стерильной водной или масляной суспензии для инъекций, которая может быть приготовлена в соответствии с известными в данной области способами с применением подходящих диспергирующих, смачивающих или суспендирующих агентов. Стерильный лекарственный препарат для инъекций также может представлять собой стерильный раствор или суспензию для инъекций в приемлемом для парентерального введения нетоксичном разбавителе или растворителе. Приемлемые для использования разбавители и растворители включают, но не ограничиваются ими, воду, раствор Рингера и изотонический раствор хлорида натрия.
Фармацевтические композиции согласно настоящему изобретению, приготовленные для перорального введения, могут быть в форме таблеток, пастилок, леденцов, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов или эликсиров. Фармацевтические композиции, предназначенные для перорального применения, могут быть приготовлены в соответствии с любым способом, известным в данной области техники для приготовления фармацевтических композиций, и могут дополнительно содержать один или более агентов, выбранных из подсластителей, ароматизаторов, красителей и консервантов, с целью получения фармацевтически привлекательных и приятных на вкус препаратов. Таблетки содержат активный агент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, подходящими для изготовления таблеток. Данные наполнители могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие агенты и вещества, улучшающие распадаемость таблеток, такие как кукурузный крахмал или альгиновая кислота; связующие вещества; и смазывающие вещества. На таблетки предпочтительно наносят покрытие с использованием известных способов для задерживания распада и всасывания в желудочно-кишечном тракте и, тем самым, обеспечения пролонгированного высвобождения лекарственного средства в течение более длительного периода времени. Например, может быть использовано вещество с замедленным распадом, такое как глицерилмоностеарат или глицерилдистеарат. На них также может быть нанесено покрытие с использование способов, описанных в патентах США US 4256108, US 4166452 и US 4265874, с получением осмотических терапевтических таблеток для контроля высвобождения. Фармацевтическая композиция согласно настоящему изобретению также может быть в форме двухслойных таблеток, в которых два или более различных слоев гранулята спрессованы вместе и отдельными слоями лежат один поверх другого, при этом каждый отдельный слой получен для обеспечения разных режимов высвобождения лекарственного средства. Фармацевтическая композиция для перорального применения согласно настоящему изобретению также может быть в форме эмульсии «масло-в-воде».
Фармацевтические композиции согласно настоящему изобретению также могут быть изготовлены в виде матрицы с контролируемым высвобождением, например, в виде матричных таблеток с контролируемым высвобождением, в которых высвобождение растворимого активного агента контролируется за счет наличия активной диффузии через гель, образованный после набухания гидрофильного полимера, приведенного в контакт с растворяющей жидкостью (in vitro) или желудочно-кишечной жидкостью (in vivo). Было описано множество полимеров, способных образовывать такой гель, например, производные целлюлозы, в частности, простые эфиры целлюлозы, такие как гидроксипропилцеллюлоза, гидроксиметилцеллюлоза, метилцеллюлоза или гидроксипропилметилцеллюлоза, и среди разных коммерческих доступных таких эфиров есть соединения с довольно высокой вязкостью. В других вариантах реализации указанные композиции содержат активный агент, находящийся в микроинкапсулированной лекарственной форме с контролируемым высвобождением, представляющей собой мелкие вкрапления активного агента, покрытые оболочкой или мембраной с образованием частиц размером от нескольких микрометров до нескольких миллиметров.
Другая предполагаемая лекарственная форма представляет собой депо-системы на основе биоразлагаемых полимеров, в которых по мере распада полимера медленно высвобождается активный агент. Наиболее распространенной группой биоразлагаемых полимеров являются гидролитически лабильные полиэфиры, полученные из молочной кислоты, гликолевой кислоты или их комбинаций.
Фармацевтическая композиция согласно настоящему изобретению может содержать один или более фармацевтически приемлемых наполнителей. Например, таблетка может содержать по меньшей мере один наполнитель, например, лактозу, этилцеллюлозу, микрокристаллическую целлюлозу, силицированную микрокристаллическую целлюлозу; по меньшей мере один разрыхлитель, например, поперечно-сшитый поливинилпирролидон; по меньшей мере одно связующее вещество, например, поливинилпиридон, гидроксипропилметилцеллюлозу; по меньшей мере одно поверхностно-активное вещество, например, лаурилсульфат натрия; по меньшей мере одно вещество, способствующее скольжению, например, коллоидный диоксид кремния; и по меньшей мере одно смазывающее вещество, например, стеарат магния.
В Примерах 4-6 далее описано получение трех типов пеллет мезилата разагилина со слоем покрытия с пролонгированным высвобождением (ПВ), содержащих слой лекарственного средства, покрывающий инертные пеллеты, и слой покрытия с пролонгированным высвобождением, т.е. функциональный слой, покрывающий указанный слой лекарственного средства (слой ПВ - 15%, 22% и 28%). Для получения слоя лекарственного средства повидон USP (PVP К29/32) растворяли в смеси дистиллированной воды и этанола; лекарственное средство растворяли в полученном растворе; затем диспергировали сверхтонкий тальк и добавляли его к указанному раствору с образованием однородной суспензии, которую наносили на сахарные сферические частицы размером 600-710 мкм (диаметр). Для получения покрывающих пленок ПВ различной толщины готовили один раствор, из которого во время процесса нанесения покрытия в различные моменты времени отбирали пробы, соответствующие различным количествам распыляемого раствора, в результате нанесения которых формируются слои различной толщины. Указанный раствор состоял из Ethocel 45 сПз (этилцеллюлоза; полимер, контролирующий высвобождение), растворенной в смеси ацетона и этанола; и полиэтиленгликоля (ПЭГ) 4000, растворенного в дистиллированной воде, которые затем смешивали с образованием гомогенного раствора. Функциональный раствор наносили на содержащие лекарственное средство пеллеты, описанные выше, с образованием пленок ПВ различной толщины. Характеристики растворения пеллет с разными покрытиями ПВ оценивали в Аппарате 1 (корзина) согласно Фармакопее США (USP) при скорости вращения шпинделя 100 об/мин и температуре 37°C с использованием раствора кишечной жидкости (IFS, рН 6,8), имитирующего условия в кишечнике, и, как показано, скорость высвобождения зависела от толщины пленки, и при большей толщине функционального слоя высвобождение происходило более медленно.
В Примерах 7-8 описано получение двух типов пеллет мезилата разагилина со слоем покрытия ПВ, содержащих слой лекарственного средства, покрывающий инертные пеллеты, подслой покрытия, покрывающий указанный слой лекарственного средства, и функциональный слой, покрывающий указанный подслой покрытия (слой ПВ - 15% и 18%). Для получения слоя лекарственного средства повидон (PVP К25) растворяли в смеси дистиллированной воды и этанола; лекарственное средство растворяли в полученном растворе; затем диспергировали сверхтонкий тальк и добавляли к полученному раствору с образованием однородной суспензии, которую затем наносили на сахарные сферические частицы размером 600-710 мкм. Раствор для подслоя покрытия готовили растворением PVP К25 в смеси дистиллированной воды и этанола, а затем наносили на загруженные лекарственным средством пеллеты. Для получения покрывающих пленок ПВ различной толщины готовили один раствор, из которого во время процесса нанесения покрытия в различные моменты времени отбирали пробы, соответствующие различным количествам распыляемого раствора, в результате нанесения которых формируются слои различной толщины. Указанный раствор состоял из Ethocel 45 сПз, растворенной в смеси ацетона и этанола; и ПЭГ 3000, растворенного в дистиллированной воде, которые затем смешивали с образованием гомогенного раствора. Сверхтонкий тальк диспергировали в дистиллированной воде и добавляли к раствору с образованием однородной суспензии, которую затем наносили на пеллеты с подслоем покрытия с образованием пленок ПВ различной толщины. Затем полученные пеллеты смешивали в сухом виде с Аэросилом 200 (Aerosil 200). Характеристики растворения полученных пеллет с двумя разными покрытиями ПВ оценивали в Аппарате 1 USP при 100 об/мин и температуре 37°C с использованием (i) IFS (рН 6,8), имитирующего условия в кишечнике; (ii) раствора желудочного сока (GFS, рН 1,2), имитирующего условия в пустом желудке, в течение 2 часов, а затем IFS в течение еще 20 часов; и (iii) ацетатного буфера (рН 4,5), имитирующего условия в полном желудке, и, как показано, скорость высвобождения оставалась постоянной (в пределах приемлемого диапазона ±10% для тестирования растворимости) в диапазоне рН 1,2-6,8 за счет присутствия в слое ПВ рН-зависимых полимеров, и оставалась стабильной в течение 3 месяцев несмотря на то, что пеллеты выдерживали в условиях ускоренного старения для оценки стабильности. Скорость высвобождения активного агента из пеллет с покрытием 15% ПВ была выше, чем из пеллет с покрытием 18% ПВ вследствие разной толщины функционального слоя.
В Примере 9 описано получение третьего типа пеллет мезилата разагилина со слоем покрытия ПВ, содержащих слой лекарственного средства, покрывающий инертные пеллеты, подслой покрытия и внешний функциональный слой с более высоким процентным содержанием (27%) слоя покрытия ПВ, которые смешивали в сухом виде с диоксидом кремния вместо Аэросила 200, используемого в Примерах 7-8. Характеристики растворения таких пеллет определяли в Аппарате 1, USP, при 100 об/мин и температуре 37°C с использованием IFS (рН 6,8), и, как показано, высвобождение в данном случае происходило медленнее, чем в случае пеллет из Примеров 7-8, вследствие разной толщины функционального слоя.
Дополнительные пеллеты мезилата разагилина ПВ с подслоем покрытия или без него, имеющие другие профили высвобождения лекарственного средства, описаны в Примере 10.
При создании продукта 24-часового пролонгированного высвобождения для перорального введения необходимо, чтобы лекарственное средство абсорбировалось в течение всего периода высвобождения, т.е. во всех отделах желудочно-кишечного тракта, включая двенадцатиперстную кишку и толстую кишку. В Примере 11 описано фармакокинетическое исследование, в котором однократную болюсную дозу разагилина вводили в виде водного раствора в толстую кишку, двенадцатиперстную кишку или яремную вену крыс, образцы крови забирали у животных за 5 минут до введения дозы и через 5, 15, 30, 50, 90, 150 и 200 минут после введения дозы, и измеряли содержание как разагилина, так и его метаболита в плазме крови. Как видно, исходный 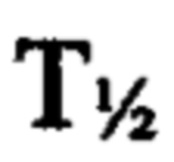 для групп введения в толстую и двенадцатиперстную кишку был больше по сравнению с для внутривенного введения. Кроме того, рассчитанные значения площади под кривой (ППК) для внутривенной дозы и дозы для двенадцатиперстной кишки оказались близкими, что позволяет предположить полную абсорбцию при пероральном введении, тогда как значение ППК после введения в толстую кишку составляло приблизительно 28% от значения ППК для внутривенной дозы, что подтверждает возможность абсорбции в толстой кишке.
для групп введения в толстую и двенадцатиперстную кишку был больше по сравнению с для внутривенного введения. Кроме того, рассчитанные значения площади под кривой (ППК) для внутривенной дозы и дозы для двенадцатиперстной кишки оказались близкими, что позволяет предположить полную абсорбцию при пероральном введении, тогда как значение ППК после введения в толстую кишку составляло приблизительно 28% от значения ППК для внутривенной дозы, что подтверждает возможность абсорбции в толстой кишке.
С учетом характеристик растворения, приведенных для различных типов пеллет мезилата разагилина с покрытием ПВ, описанных выше, и исследования, описанного выше, демонстрирующего абсорбцию разагилина в различных отделах желудочно-кишечного тракта, в некоторых вариантах реализации фармацевтическую композицию согласно настоящему изобретению изготавливают в форме для перорального введения. В конкретных вариантах реализации фармацевтическая композиция может быть твердой в форме частиц, зерен, шариков или пеллет, которые смешивают и заключают в капсулы или саше или прессуют в таблетки любым традиционным способом, известным в данной области техники, как показано для некоторых пеллет мезилата разагилина ПВ, описанных в Примере 10. Например, предложена таблетка, в которой активный агент присутствует по меньшей мере в двух отдельных слоях, т.е. двухслойная или многослойная таблетка, при этом указанные слои возможно разделены промежуточным неактивным слоем, например, слоем, содержащим один или более разрыхлителей. Фармацевтическая композиция также может представлять собой полужидкую или жидкую систему.
В некоторых вариантах реализации фармацевтическая композиция согласно настоящему изобретению, изготовленная в форме для перорального введения, находится в форме монолитной матрицы, т.е. структуры, включающей пространственно стабильный матричный материал, имеющий дискретный размер и форму; таблетки, такой как двухслойная или многослойная таблетка, матричная таблетка, распадающаяся таблетка, растворяющаяся таблетка или жевательная таблетка; или капсулы или саше, например, заполненного гранулами, зернами, шариками или пеллетами. В некоторых других вариантах реализации фармацевтическая композиция согласно настоящему изобретению, изготовленная в форме для перорального введения, находится в форме депо-системы на основе биоразлагаемых полимеров, в которой по мере распада полимера медленно высвобождается активный агент. Наиболее распространенной группой биоразлагаемых полимеров являются гидролитически лабильные полиэфиры, полученные из молочной кислоты, гликолевой кислоты или их комбинаций. Примеры биоразлагаемых полимеров, полученных из данных конкретных мономеров, включают, но не ограничиваются ими, поли(D,L-лактид) (PLA), полигликолид (полигликолевую кислоту; PGA) и сополимер поли(D,L-лактид-ко-гликолид) (PLGA).
В некоторых конкретных вариантах реализации согласно настоящему изобретению предложена фармацевтическая композиция, определенная выше, т.е. фармацевтическая композиция, содержащая активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, при этом указанная композиция имеет следующие характеристики растворения в Аппарате 1 (корзина), USP, при 50-150, предпочтительно 100 об/мин при значении рН до 7,4, предпочтительно 1,2-6,8, при 37°C:

Предпочтительно фармацевтические композиции представляют собой композиции, имеющие следующие характеристики растворения в Аппарате 1 (корзина), USP, при 50-150, предпочтительно 100 об/мин при значении рН до 7,4, предпочтительно 1,2-6,8, при 37°C:
В более конкретных вариантах реализации активный агент, содержащийся в данной композиции, представляет собой N-пропаргил-1-аминоиндан; его энантиомер, т.е. разагилин или S-(-)-N-пропаргил-1-аминоиндан; его метаболит, в частности, 1-аминоиндан; его аналог или фармацевтически приемлемую соль любого из вышеуказанных соединений. В более конкретных вариантах реализации активный агент, содержащийся в данной композиции, представляет собой разагилин или его фармацевтически приемлемую соль.
В некоторых вариантах реализации фармацевтические композиции согласно настоящему изобретению при их введении обеспечивают более низкую Cmax и меньший индекс колебаний, и приводят к меньшим нежелательным побочным эффектам по сравнению с лекарственной формой с быстрым высвобождением. В настоящем описании термин «Cmax» относится к максимальной концентрации терапевтического лекарственного средства в плазме крови; и термин «индекс колебаний» в настоящем описании относится к изменениям концентрации терапевтического лекарственного средства в сыворотке в зависимости от времени после введения лекарственного средства. Нежелательные побочные эффекты от приема разагилина включают, но не ограничиваются ими, тяжелые аллергические реакции (сыпь; крапивницу; зуд; затруднение дыхания; сдавленность в груди; отек рта, лица, губ или языка); стул черного цвета или кровянистый стул; кровь в моче; затуманенное зрение; изменения половой функции или желания; боль в груди; спутанность сознания; депрессию; расширенные зрачки; частое или нерегулярное сердцебиение; лихорадку; галлюцинации; неспособность сидеть спокойно; онемение или покалывание рук или ног; одностороннюю слабость; припадки; чувствительность к свету; сильную головную боль; изменения кожи; боль или ригидность затылочных мышц; тремор; затруднение мышления или ходьбы; необъяснимую тошноту или рвоту; несвойственную потливость; проблемы со зрением или речью; диарею; головокружение; сонливость; сухость во рту; гриппоподобные симптомы; головную боль; боль в суставах; дурноту; бессонницу; расстройство желудка; и заложенность носа.
В соответствии с другим аспектом настоящего изобретения предложена пеллета с пролонгированным высвобождением, содержащая:
(i) инертное ядро пеллеты;
(ii) слой лекарственного средства, покрывающий указанное ядро пеллеты, при этом указанный слой лекарственного средства содержит активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, возможно подходящим образом смешанные со связующим веществом и/или пленкообразующим полимером и возможно смешанные с веществом, способствующим скольжению;
(iii) возможно, изолирующий/защитный подслой покрытия, покрывающий указанный слой лекарственного средства; и
(iv) слой покрытия с замедленным высвобождением, покрывающий указанный подслой покрытия (при его наличии) или указанный слой лекарственного средства.
Пеллета ПВ согласно настоящему изобретению может возможно содержать изолирующий/защитный подслой покрытия, покрывающий указанный слой лекарственного средства. Роль данного подслоя покрытия заключается в изолировании слоя активного вещества от внешнего покрытия ПВ и защите от возможных взаимодействий с активным агентом, которые могут влиять на его стабильность и приводить к образованию продуктов распада активной фармацевтической субстанции (АФС). В некоторых вариантах реализации подслой покрытия содержит пленкообразующий полимер и, возможно, вещество, способствующее скольжению.
Пеллета ПВ согласно настоящему изобретению содержит внешний слой покрытия ПВ, также называемый в настоящем описании «функциональным слоем», покрывающий подслой покрытия (при его наличии) либо слой лекарственного средства.
В некоторых вариантах реализации слой покрытия ПВ содержит по меньшей мере один рН-независимый полимер, т.е. набухающий в воде/водонерастворимый/гидрофобный полимер и возможно порообразующий агент, при этом пеллета с пролонгированным высвобождением характеризуется рН-независимым высвобождением in vitro. В других вариантах реализации функциональный слой содержит рН-независимый полимер, гидрофильный полимер-модулятор высвобождения, выступающий в качестве порообразующего агента, и возможно гидрофобный или гидрофильный пластификатор и/или вещество, способствующее скольжению. В некоторых других вариантах реализации слой покрытия ПВ содержит смесь рН-зависимого полимера для энтеросолюбильного покрытия и рН-независимого полимера, при этом при значениях рН не более 7,4 пеллета с пролонгированным высвобождением имеет характеристику высвобождения in vitro, порядок которого близок к нулевому порядку.
Связующие вещества для применения в фармацевтике представляют собой гидрофильные, вещества, такие как сахара и полимеры природного и синтетического происхождения, используемые в изготовлении твердых лекарственных форм вследствие их адгезионных и когезионных свойств. Роль связующих веществ заключается в способствовании увеличения размера частиц за счет придания порошкам когезионной способности, таким образом, обеспечивая гранулы и таблетки необходимой прочностью сцепления. Хотя связующие вещества улучшают внешний вид, твердость и прочность на истирание данных препаратов, они не влияют на скорость распада или растворения активных веществ. Связующие вещества природного происхождения, которые широко использовали раньше, включают гуммиарабик, желатин, крахмал и гидролизованный крахмал. Эти вещества заменили связующие вещества синтетического происхождения, наиболее важными из которых являются повидон и различные производные целлюлозы. Примеры связующих веществ, которые могут быть смешаны с активным агентом в слое лекарственного средства, покрывающем пеллету ПВ согласно настоящему изобретению, включают, но не ограничиваются ими, поливинилпирролидон (PVP), гидроксипропилметилцеллюлозу (ГПМЦ), гидроксипропилцеллюлозу (ГПЦ), микрокристаллическую целлюлозу и их комбинации. Связующее вещество может присутствовать в количестве от примерно 0,5% до примерно 20%, предпочтительно от примерно 0,5% до примерно 10% от массы всей пеллеты.
В настоящем описании термин «пленкообразующий полимер» относится к полимерам, способным затвердевать с образованием адгезивных пленок. Кроме того, физическим свойством данных полимеров, необходимым для нанесения в качестве покрытия, является способность образовывать пленки или некоторая адгезионная способность в отношении материала, на которое наносят покрытие. Примеры пленкообразующих полимеров включают, но не ограничиваются ими, PVP, ГПМЦ, ГПЦ, микрокристаллическую целлюлозу и их комбинации. Пленкообразующий полимер, содержащийся в слое лекарственного средства, может присутствовать в количестве до 90% от массы всего слоя лекарственного средства, предпочтительно от примерно 0,5% до примерно 20% от массы всей пеллеты. Количество пленкообразующего полимера в подслое покрытия может составлять до 100% от массы всего подслоя покрытия, предпочтительно от примерно 0,5% до примерно 10% от массы всей пеллеты.
Вещества, способствующие скольжению, как правило, добавляют в фармацевтические композиции для повышения текучести гранулятов и порошков за счет уменьшения трения и поверхностного заряда. Кроме того, их используют в качестве веществ, препятствующих слипанию, во время процесса нанесения покрытия. Определенные вещества, способствующие скольжению, такие как тальк и глицерилмоностеарат, обычно используют в составах для нанесения покрытий в качестве веществ, препятствующих слипанию, уменьшающих склонность к слипанию частиц при более низких температурах продукта. Другие вещества, способствующие скольжению, такие как коллоидный диоксид кремния, обеспечивают желаемые характеристики текучести, которые используют для улучшения сыпучести сухих порошков в ряде таких процессов, как таблетирование и капсулирование, из-за малого размера их частиц и большой удельной поверхности. Не ограничивающие примеры веществ, способствующих скольжению, включают тальк, в частности, сверхтонкий тальк, коллоидный диоксид кремния, глицерилмоностеарат и их комбинации.
Вещество, способствующее скольжению, когда оно содержится в слое лекарственного средства, может присутствовать в количестве до 30% от массы всего слоя лекарственного средства, предпочтительно от примерно 0,5% до примерно 5% от массы всей пеллеты. Количество вещества, способствующего скольжению при содержании его в подслое покрытия может составлять до примерно 10% от массы всего подслоя покрытия, предпочтительно от примерно 0,5% до примерно 5% от массы всей пеллеты.
Примеры рН-независимых полимеров, которые могут содержаться в пеллете ПВ согласно настоящему изобретению, включают, но не ограничиваются ими, этилцеллюлозу, Surelease®, сополимеры сложных эфиров акриловой и метакриловой кислот, такие как Эудрагит® RL (Eudragit® RL) (поли(этилакрилат, метилметакрилат, триметиламмониоэтилметакрилата хлорид), 1:2:0,2), Эудрагит® RS (поли(этилакрилат, метилметакрилат, триметиламмониоэтилметакрилата хлорид), 1:2:0,1), Эудрагит® NE (поли(этилакрилат, метилметакрилат), 2:1) и их комбинации. рН-независимый полимер может присутствовать в количестве от примерно 10% до примерно 50%, предпочтительно от примерно 10% до примерно 30% от массы всей пеллеты.
Примеры рН-зависимых полимеров для энтеросолюбильного покрытия, которые могут содержаться в пеллете ПВ согласно настоящему изобретению, включают, но не ограничиваются ими, Эудрагит® S (поли(метакриловая кислота, метилметакрилат), 1:2), Эудрагит® L 55 (поли(метакриловая кислота, этилакрилат), 1:1), Колликоат® (Kollicoat®) (поли(метакриловая кислота, этилакрилат), 1:1), фталат гадроксипропилметилцеллюлозы (ФГПМЦ), альгинаты, карбоксиметилцеллюлозу и их комбинации. рН-зависимый полимер для энтеросолюбильного покрытия может присутствовать в количестве от примерно 10% до примерно 50%, предпочтительно от примерно 10% до примерно 30% от массы всей пеллеты.
В настоящем описании термин «порообразующий агент» относится к веществу, которое растворяется в среде организма, в результате чего в матрице образуются открытые поры, увеличивающие скорость диффузии активного агента через слой покрытия. Размер образуемых пор в некоторой степени можно регулировать с помощью размера используемого зернистого твердого материала. Для однородности размера пор зернистый материал может быть просеян через ряд сит с последовательным уменьшением размера ячеек с получением желаемого диапазона размеров частиц. Порообразующий агент, который может содержаться в пеллетах ПВ согласно настоящему изобретению, представляет собой неорганическое либо органическое вещество, включая, например, поливинилпирролидон (PVP), полиэтиленгликоль (ПЭГ), ГПМЦ, ГПЦ, метилцеллюлозу, 1,2-пропиленгликоль, лактозу, сахарозу, тальк, в частности, сверхтонкий тальк, и их комбинации. Порообразующий агент может присутствовать в количестве от примерно 0,1% до примерно 20%, предпочтительно от примерно 0,1% до примерно 10% от массы всей пеллеты.
В настоящем описании термин «гидрофильный полимер-модулятор высвобождения» относится к водорастворимому полимеру, контролирующему высвобождение активного агента. Тем не менее, в некоторых вариантах реализации гидрофильный полимер-модулятор высвобождения, содержащийся в слое покрытия ПВ пеллеты ПВ согласно настоящему изобретению, на самом деле выступает в качестве порообразующего агента. Примеры гидрофильных полимеров-модуляторов высвобождения включают, но не ограничиваются ими, PVP, ПЭГ, ГПМЦ, ГПЦ и их комбинации. Гидрофильный полимер-модулятор высвобождения может присутствовать в количестве от примерно 0,1% до примерно 20%, предпочтительно от примерно 0,1% до примерно 10% от массы всей пеллеты.
В настоящем описании термин «пластификатор» включает любое соединение или комбинацию соединений, способных пластифицировать или смягчать полимер, используемый в пеллете ПВ согласно настоящему изобретению. В ходе изготовления слоя покрытия ГШ пластификатор может понижать температуру плавления или температуру стеклования (температуру размягчения) используемого полимера или комбинации полимеров; может увеличивать среднюю молекулярную массу указанного полимера или комбинации полимеров, и дополнительно может уменьшать вязкость указанного полимера или комбинации полимеров для удобства обработки раствора для нанесения покрытия. Не ограничивающие примеры пластификаторов включают дибутилсебацинат; дибутилфталат; эфиры лимонной кислоты, такие как триэтилцитрат и триацетин; пропиленгликоль; поли(алкиленоксиды) небольшой молекулярной массы, такие как ПЭГ, поли(пропиленгликоли) и поли(этилен/пропиленгликоли); и их комбинации. Пластификаторы могут присутствовать в количестве от примерно 0,1% до примерно 20%, предпочтительно от примерно 0,1% до примерно 10% от массы всей пеллеты.
Слой покрытия лекарственного средства пеллеты ПВ согласно настоящему изобретению может содержать любой активный агент, как определено выше, т.е. любой активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль. В конкретных вариантах реализации указанный активный агент выбран из N-пропаргил-1-аминоиндана, его энантиомера, его метаболита, его аналога или его фармацевтически приемлемой соли. В более конкретных вариантах реализации активный агент представляет собой разагилин или его фармацевтически приемлемую соль.
Пеллета ПВ согласно настоящему изобретению может содержать дополнительные неактивные ингредиенты, такие как агент, регулирующий осмотическое давление/тоничность. Указанные агенты обычно используют для контролируемого по времени распада, когда необходима пульсирующая доставка лекарственного средства. Примеры подходящих наполнителей, регулирующих осмотическое давление/тоничность, которые могут быть использованы при получении пеллеты ПВ, включают, но не ограничиваются ими, хлорид натрия и маннит. Агент, регулирующий осмотическое давление/тоничность, содержащийся в пеллете ПВ, может присутствовать в количестве до 20%, предпочтительно от примерно 0,5% до примерно 10% от массы всей пеллеты.
В конкретном варианте реализации, приведенном в настоящем описании в качестве примера, пеллеты ПВ, приведенные в качестве примера в настоящем описании, содержат инертное ядро пеллеты; слой лекарственного средства, содержащий активный агент, смешанный с PVP в качестве пленкообразующего полимера/связующего вещества и со сверхтонким тальком в качестве вещества, способствующего скольжению; и слой покрытия ПВ, содержащий этилцеллюлозу в качестве рН-независимого полимера и ПЭГ в качестве порообразующего агента; при этом количество указанного пленкообразующего полимера/связующего вещества составляет до 90% от массы всего слоя лекарственного средства или от примерно 0,5% до примерно 20% от массы всей пеллеты; количество указанного вещества, способствующего скольжению, составляет до 30% от массы всего слоя лекарственного средства или от примерно 0,1% до примерно 10% от массы всей пеллеты; количество указанного рН-независимого полимера составляет от примерно 50% до примерно 90% от массы всего слоя покрытия ПВ или от примерно 10% до примерно 30% от массы всей пеллеты; и количество указанного порообразующего агента составляет от примерно 1% до примерно 20% от массы всего слоя покрытия ПВ или от примерно 0,1% до примерно 10% от массы всей пеллеты.
В других конкретных вариантах реализации, приведенных в качестве примера в настоящем описании, пеллета ПВ согласно настоящему изобретению содержит инертное ядро пеллеты; слой лекарственного средства, содержащий указанный активный агент, смешанный с PVP в качестве пленкообразующего полимера/связующего вещества и со сверхтонким тальком в качестве вещества, способствующего скольжению; изолирующий/защитный подслой покрытия, содержащий PVP в качестве пленкообразующего полимера; и слой покрытия ПВ, содержащий этилцеллюлозу в качестве рН-независимого полимера, ПЭГ в качестве порообразующего агента и сверхтонкий тальк в качестве вещества, способствующего скольжению; при этом количество указанного пленкообразующего полимера/связующего вещества в указанном слое лекарственного средства составляет до 90% от массы всего слоя лекарственного средства или от примерно 0,5% до примерно 20% от массы всей пеллеты; количество указанного вещества, способствующего скольжению, в указанном слое лекарственного средства составляет до 30% от массы всего слоя лекарственного средства или от примерно 0,1% до примерно 10% от массы всей пеллеты; количество указанного пленкообразующего полимера в указанном подслое покрытия составляет до 100% от массы всего подслоя покрытия или от примерно 0,5% до примерно 20% от массы всей пеллеты; количество указанного рН-независимого полимера составляет от примерно 50% до примерно 90% от массы всего слоя покрытия ПВ или от примерно 10% до примерно 30% от массы всей пеллеты; количество указанного порообразующего агента составляет от примерно 1% до примерно 20% от массы всего слоя покрытия ПВ или от примерно 0,1% до примерно 10% от массы всей пеллеты; и количество указанного вещества, способствующего скольжению, в указанном слое покрытия ПВ составляет от примерно 0,1% до примерно 20% от массы всего слоя покрытия ПВ или от примерно 0,1% до примерно 10% от массы всей пеллеты.
В соответствии с другим аспектом настоящего изобретения предложена фармацевтическая композиция для перорального введения, содержащая пеллеты ПВ, как определено выше. В некоторых вариантах реализации пеллеты ПВ, содержащиеся в данной композиции, смешивают с одним или несколькими подходящими наполнителями и заключают в капсулы либо прессуют в таблетки. Приготовление таких капсул или таблеток можно осуществлять с использованием любой подходящей технологии, известной в данной области техники.
Примеры подходящих наполнителей, которые могут быть использованы при приготовлении фармацевтической композиции для перорального введения, включают, но не ограничиваются ими, диоксиды кремния, а также другие вещества, способствующие скольжению, известные в данной области техники, как определено выше.
Наполнители таблеток заполняют объем таблетки или капсулы, что делает удобным процесс их получения и является удобным для использования потребителем. За счет увеличения суммарного объема наполнители обеспечивают конечному продукту объем, удобный для использования пациентом. Хороший наполнитель должен быть инертным, совместимым с другими компонентами лекарственной формы, негигроскопичным, относительно дешевым, способным к уплотнению и предпочтительно безвкусным или приятным на вкус. Растительная целлюлоза (чистый растительный наполнитель) представляет собой популярный наполнитель в таблетках или твердых желатиновых капсулах. Двухосновный фосфат кальция представляет собой другой широко используемый наполнитель для таблеток. В мягких желатиновых капсулах можно использовать ряд растительных жиров и масел. Наполнители для таблеток включают, например, лактозу, маннит/Parteck®, сорбит, крахмал и их комбинации.
Разрыхлитель расширяется и растворяется при контакте с влагой, вызывая распад таблетки в пищеварительном тракте с высвобождением активных ингредиентов для их абсорбции. Типы разрыхлителей включают вещества, облегчающие поглощение воды, и активаторов разрушения таблетки. Они обеспечивают быстрый распад таблетки при контакте ее с водой на более мелкие фрагменты, облегчая ее растворение. Не ограничивающие примеры разрыхлителей включают поперечноно-сшитый поливинилпирролидон (кросповидон), карбоксиметилцеллюлозу (КМЦ) натрия/кальция, кроскармеллозу натрия, гидроксипропилцеллюлозу с низкой степенью замещения, бикарбонат натрия, крахмал, натрия крахмала гликолят и их комбинации.
Смазывающие вещества добавляют в небольших количествах в лекарственные формы таблеток и капсул для улучшения некоторых технологических характеристик. В частности, данные агенты предотвращают слипание и прилипание ингредиентов к пуансонам для изготовления таблеток или машине для заполнения капсул. Смазывающие вещества также обеспечивают возможность формирования и выталкивания таблеток с малым трением между твердым веществом и стенкой матрицы. Примеры смазывающих веществ включают, но не ограничиваются ими, глицерин бегенат, стеариновую кислоту, тальк, стеарат цинка, стеарат кальция и их комбинации.
Как показано в разделе Примеры далее, в частности, в отношении разагилина и его метаболита 1-аминоиндана, фармацевтические композиции согласно настоящему изобретению подходят для лечения болезни Паркинсона и, кроме того, любого другого нейродегенеративного заболевания или состояния, а также повреждений нервной системы, для лечения которых активный агент, содержащийся в данной композиции, был описан как подходящий. Такие нейродегенеративные заболевания или состояния включают, не ограничиваясь ими, болезнь Альцгеймера; синдром отмены лекарственного средства, включая синдром отмены психостимуляторов, опиатов, наркотических средств и барбитуратов; депрессию; возрастные дегенерации, включая дегенерацию функции почек и дегенерацию когнитивной функции, о которой свидетельствует способность к пространственному обучению; болезнь Кушинга гипофизарного происхождения у людей и животных; дисфункцию иммунной системы как у людей, так и у животных; возрастную потерю массы тела у млекопитающих; шизофрению; различные неопластические состояния, включая раковые опухоли, такие как рак молочной железы и гипофиза; нейромышечное и нейродегенеративное заболевание; деменцию, такую как сенильная деменция, например, деменция паркинсоновского и альцгеймеровского типа, сосудистая деменция и деменция с тельцами Леви; синдром гиперактивности; аффективное расстройство; синдром дефицита внимания; синдром гиперактивности; рассеянный склероз; и синдром Туретта. Конкретные повреждения нервной системы, которые можно лечить фармацевтической композицией согласно настоящему изобретению, включают, не ограничиваясь ими, повреждение ЦНС вследствие повреждений при травме головы, гипоксию, аноксию, гипогликемию, нейротоксическое поражение, ишемический инсульт и травму, а также другие поражения нервов, при которых протекает апоптотический процесс.
Таким образом, в соответствии с другим аспектом настоящее изобретение относится к способу лечения нейродегенеративного заболевания или повреждения нервной системы у нуждающегося в этом индивидуума, включающему введение указанному индивидууму фармацевтической композиции, определенной выше.
В некоторых вариантах реализации фармацевтическую композицию, применяемую в способе согласно настоящему изобретению, изготавливают в форме для перорального введения. В конкретных вариантах реализации способ согласно настоящему изобретению включает введение фармацевтической композиции для перорального применения, изготовленной в форме для пролонгированного высвобождения активного агента, как определено выше, в частности, в которой активный агент выбран из N-пропаргил-1-аминоиндана, его энантиомера, его метаболита, его аналога или его фармацевтически приемлемой соли, предпочтительно, в которой активный агент представляет собой разагилин или его фармацевтически приемлемую соль.
Способ согласно настоящему изобретению можно применять для лечения любого нейродегенеративного заболевания или повреждения нервной системы, как определено выше. В конкретных вариантах реализации указанное нейродегенеративное заболевание представляет собой болезнь Паркинсона или болезнь Альцгеймера, и указанное повреждение нервной системы представляет собой острое повреждение головного мозга, такое как инсульт или травматическое повреждение головного мозга.
В соответствии с другим аспектом настоящее изобретение относится к способу получения состава в форме для пролонгированного высвобождения, содержащего активный агент, определенный выше, т.е. активный агент, содержащий пропаргиламинный фрагмент, аминоиндановый фрагмент или пропаргиламинный и аминоиндановый фрагменты, или его фармацевтически приемлемую соль, при этом указанный способ включает этапы:
(i) растворения указанного активного агента, возможно подходящим образом смешанного со связующим веществом и/или веществом, способствующим скольжению, в подходящей системе растворителей с получением однородной суспензии;
(ii) нанесения слоя указанной суспензии, полученной на этапе (i), на инертные пеллеты, такие как инертные гранулы нонпарель (nonpareil seeds);
(iii) возможно, нанесения на пеллеты, содержащие активный агент, полученные на этапе (ii), изолирующего/защитного подслоя покрытия;
(iv) нанесения на пеллеты, полученные на этапе (ii) или (iii), слоя покрытия с пролонгированным высвобождением, т.е. полимерного слоя, обеспечивающего пролонгированное высвобождение указанного активного агента, с получением тем самым указанного состава с пролонгированным высвобождением; и
(v) возможно, смешивания покрытых пеллет, полученных на этапе (iv), с подходящим наполнителем.
Способ, описанный в настоящем тексте, получения состава с пролонгированным высвобождением, содержащего активный агент, определенный выше, может быть осуществлен с использованием любой подходящей технологии, известной в данной области техники, например, подробно описанной в разделе Примеры далее. В некоторых вариантах реализации один или несколько: этапов (ii) и (iv) этого способа, а также этапов (iii) и (v) при условии их проведения, осуществляют с использованием сушилки с псевдоожиженным слоем.
В некоторых вариантах реализации состав с пролонгированным высвобождением, полученный в соответствии с данным способом, затем заключают в капсулы или прессуют в таблетки.
Далее настоящее изобретение будет проиллюстрировано следующими не ограничивающими Примерами.
ПРИМЕРЫ
Экспериментальная часть
Модели болезни Паркинсона
Экспериментальные модели болезни Паркинсона (БП) нужны для понимания возможных патологических механизмов указанного заболевания, а также необходимы для разработки и тестирования новых терапевтических стратегий, будь то фармакологические или иные стратегии.
Модель МРТР у мыши
Значительный объем биохимических данных секционных патологоанатомических исследований головного мозга человека и животных указывает на постоянный процесс окислительного стресса в черной субстанции, который может инициировать допаминергическую нейродегенерацию. Хотя и не известно, является ли окислительный стресс первичным или вторичным явлением, окислительный стресс, индуцированный нейротоксином МРТР (N-метил-4-фенил-1,2,3,6-тетрагадропиридин), использовали в животных моделях для исследования процесса нейродегенерации с целью разработки антиоксидантных нейропротекторных лекарственных средств.
МРТР превращается в головном мозге в положительно заряженную молекулу МРР+ (1-метил-4-фенилпиридиний) под действием фермента моноаминоксидазы МАО-Б, вызывая паркинсонизм у приматов вследствие уничтожения определенных допамин-продуцирующих нейронов в черной субстанции. МРР+ вмешивается в процесс окислительного фосфорилирования в митохондриях, что вызывает истощение АТФ и гибель клеток. Он также ингибирует синтез катехоламинов, снижает уровни допамина и сердечного норэпинефрина и инактивирует тирозингидроксилазу.
Модель 6-OHDA у крысы
При моделировании БП развитие заболевания в основном происходило с введением нейротоксина катехоламинов, 6-гидроксидопамина (6-OHDA). Данная молекула переносится в клеточные тела и волокна как допаминергических, так и норадренергических нейронов, и вызывает дегенерацию нервных окончаний, а также может влиять на клеточные тела, в частности, при введении в участки клеточного тела. Нейротоксичность 6-OHDA связана с его мощным ингибирующим действием по отношению к дыхательным ферментам митохондрий (комплексы цепи I и IV). Вследствие недостаточного метаболизма из-за блокады данных ферментов, нейроны больше не могут осуществлять нормальные физиологические функции и, следовательно, гибнут. Поскольку при БП именно допаминергический нигростриарный путь подвергается дегенерации, были разработаны животные модели, в которых поражения 6-OHDA допаминергической системы осуществляли путем односторонней инъекции указанного токсина напрямую в основную эфферентную проекцию нигростриарного волокна.
Подготовка образцов для ВЭЖХ анализа допамина и его метаболитов
Образцы ткани стриатума гомогенизировали на льду в 500 мкл буфера для гомогенизации (0,1 М хлорная кислота, 0,02% ЭДТА и 1% ЕТОН) с использованием набора для гомогенизации OMNI Tip от OMNI International (промежуточная скорость, 3×10 секунд с 5-секундными интервалами). Гомогенаты обрабатывали ультразвуком в течение 5 минут, а затем центрифугировали при 15000 об/мин при 4°C в течение 15 мин. Надосадочные жидкости переносили в чистые пробирки, и определяли содержание допамина методом ВЭЖХ.
Пример 1. Исследование комбинаций разагилин-прамипексол in vivo в модели БП, индуцированной МРТР, у мышей
Указанное исследование включало 10 групп по примерно 7-9 мышей каждая, которые получали лечение в соответствии с Таблицей 1. В частности, мышам интраперитонеально (ГР) вводили МРТР для индуцирования модели болезни Паркинсона (БП), и лечили комбинациями лекарственных средств, содержащими постоянную дозу прамипексола, неэрголинового агониста допамина, показанного для лечения БП на ранней стадии, и различные дозы разагилина. МРТР вводили инъекционно каждый день в течение первых 5 дней (дни 0-4) и комбинации лекарственных средств вводили в дни 0-11 либо интраперитонеально, либо с использованием насоса ALZET (микроосмотический насос ALZET, модель 1002 со скоростью 0,25 мкл/ч, DURECT Corporation, Cupertino, США) для имитации замедленного высвобождения. Группам 5-7 вводили комбинацию лекарственных средств за 30 минут до введения МРТР каждый день в течение первых пяти дней (дни 0-4), а в последующие дни (5-11) лекарственные средства вводили приблизительно в одно и то же время каждый день введения доз. Насос ALZET имплантировали интраперитонеально за 15-17 часов перед первым введением МРТР (группы 8-10) и общее количество лекарственных средств, вводимых посредством указанного насоса в течение периода введения доз, было равным количеству, вводимому в группах интраперитонеальных инъекций. Контроли представляли собой не получавших лекарственное средство мышей, которым инъекционно вводили физиологический раствор, и подвергнутых воздействию МРТР мышей, которым инъекционно вводили физиологический раствор.
Массу тела измеряли до введения доз и ежедневно во время введения доз, и рассчитывали индивидуальные изменения массы тела. Клинические признаки фиксировали два раза в неделю на протяжении всего исследования. В 12 день, день окончания исследования, всех животных подвергали эвтаназии посредством асфиксии СО2. Головной мозг быстро извлекали, помещали на охлажденную пластину и рассекали на части. Извлекали левый и правый стриатум, определяли их массу, мгновенно замораживали в жидком азоте и хранили при -70°C до дальнейшей обработки. Образцы тканей стриатума подготавливали для ВЭЖХ, как описано в Экспериментальной части.
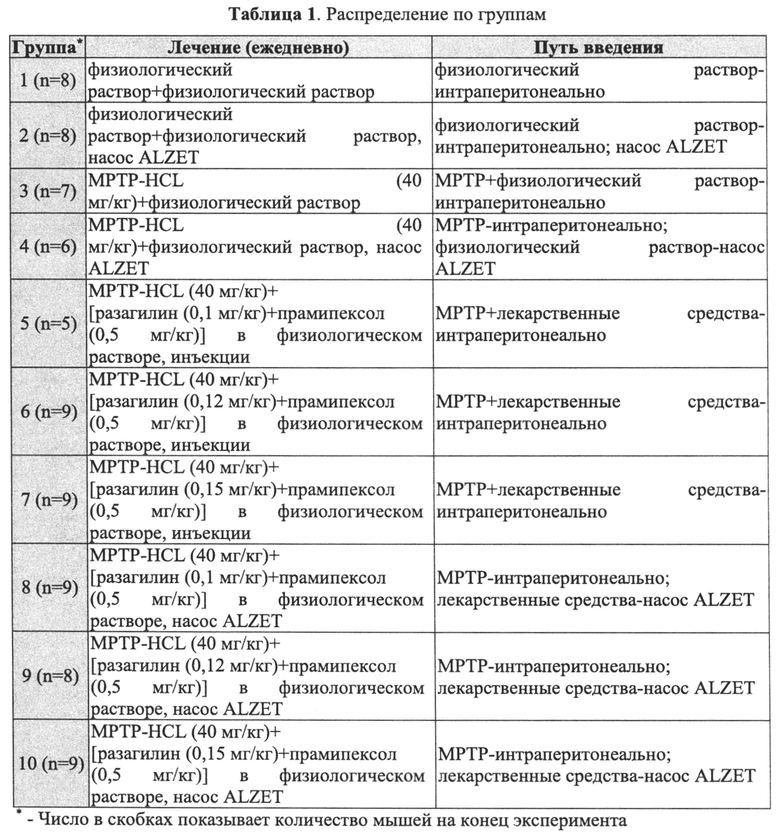
Как показано на Фиг. 1, при интраперитонеальном введении трех комбинаций разагилин + прамипексол их влияние на уровни допамина у мышей было практически одинаковым, они вызывали повышение содержания допамина до примерно 60% по сравнению с мышами, не получавшими лекарственное средство. Однако при введении указанных трех комбинаций способом с замедленным высвобождением (ЗВ) с использованием насоса ALZET в том же количестве в течение 24 часов наблюдали значительный эффект дозы, при котором уровни допамина повышались в соответствии с увеличением доз разагилина. Поскольку количество прамипексола было одинаковым во всех комбинациях, наблюдаемый эффект, должно быть, обусловлен возрастающими дозами разагилина, что указывает на крайне полезный эффект способа введения с замедленным высвобождением по сравнению со способом с быстрым высвобождением в отношении уровней допамина в головном мозге мышей, которым вводили МРТР.
Пример 2. Исследование метаболита разагилина, аминоиндана, in vivo в модели БП, индуцированной МРТР, у мышей
Самцов мышей С57В1/6 массой 20+/-1 г использовали во всех экспериментах (по 10 мышей в группе). МРТР вводили путем интраперитонеальной инъекции в дозе 40 мг/кг в сутки в течение 5 дней. Контроли представляли собой не получавших лекарственное средство мышей, которым инъекционно вводили физиологический раствор, и подвергнутых воздействию МРТР мышей, которым инъекционно вводили физиологический раствор. Аминоиндан вводили в течение 12 дней либо путем ежедневной интраперитонеальной инъекции, либо способом с замедленным высвобождением с использованием насоса ALZET, имплантированного интраперитонеально. Эффект лечения оценивали путем измерения содержания допамина и его метаболитов (дигидроксифенилуксусной кислоты и гомованилиновой кислоты) в левом и правом стриатуме, извлеченном у мышей в конце эксперимента. Образцы тканей стриатума подготавливали для анализа методом ВЭЖХ, как описано в Экспериментальной части.
Как показано на Фиг. 2, воздействие МРТР вызывало более чем 90% истощение уровней допамина по сравнению с уровнями контрольных мышей, ранее не подверженных экспериментам. Лечение метаболитом разагилина, аминоинданом, вводимым способом с замедленным высвобождением (ЗВ) с использованием насоса ALZET, вызывало значительное восстановление уровней допамина по сравнению с уровнями у мышей, получавших лечение разбавителем (физиологическим раствором) или тем же лекарственным средством, вводимым путем ежедневных интраперитонеальных инъекций
Пример 3. Исследование разагилина in vivo в модели БП, индуцированной 6-OHDA, у крыс
Одностороннее поражение медиального пучка переднего мозга (MFB) 6-OHDA вызывает одностороннее разрушение допаминергических нейронов нигростриарного пути, приводя к асимметрии моторного поведения у крыс. Когда пораженным крысам вводят лекарственные средства, действующие на систему допамина, они демонстрируют активное вращательное поведение. В частности, введение DA-высвобождающего агента, D-амфетамина, создает дисбаланс допамина в направлении непораженной нигростриарной проекции и, таким образом, приводит к вращениям по часовой стрелке. Эффект лечения заключается в том, что большее количество DA становится доступным и ожидается большее число вращений по часовой стрелке (CW). Индуцированные лекарственным средством вращения измеряют с использованием автоматизированного измерителя вращения (rotameter), состоящего из чаши для вращения и шарнира, прикрепленного к туловищу крысы.
В дополнение к результатам, представленным в Примерах 1-2, демонстрирующим явное преимущество способа лечения разагилином или его метаболитом, аминоинданом, с замедленным высвобождением (ЗВ) в отношении биохимического конечного содержания допамина в модели БП, индуцированной МРТР, у мышей, в данном исследовании терапевтический эффект введения разагилина способом ЗВ в отношении конечного поведения тестировали с использованием модели БП, индуцированной 6-OHDA, у крыс.
У взрослых самцов крыс линии Sprague-Dawley массой 250-300 г медиальный пучок переднего мозга (MFB) поражали 6-OHDA. Крыс анестезировали кетамином-ксилазином (85:15) 0,1 мл/100 г и закрепляли в стереотаксическом аппарате. 6-OHDA вводили путем инъекций в односторонний MFB в соответствии со следующими стереотаксическими координатами: АР-2,8 мм, ML-2 мм относительно брегмы и DV-9 относительно твердой мозговой оболочки. Скорость введения составляла 1 мкл/мин с использованием инъекционного насоса и микрошприца Hamilton. После инъекции микрошприц оставляли на 5 мин в месте инъекции, и отверстие закрывали костным воском.
Такую же дозу разагилина вводили либо путем ежедневных интраперитонеальных инъекций в течение 28 дней (начиная за 7 дней до введения 6-OHDA до 21 дня после), либо вводили с постоянной скоростью в течение 24 часов каждый день, всего 28 дней, с использованием насоса ALZET, трансплантированного за 7 дней до введения 6-OHDA, и продолжали в течение еще 21 дня. В обоих случаях за лечением следовало вымывание лекарственного средства в течение 10 дней перед умерщвлением крыс. В конце исследования, т.е. на 32 день после введения 6-OHDA, оценивали моторную асимметрию с использованием Rota Meter с помощью амфетамина в качестве индуктора. Амфетамин представляет собой допамин-высвобождающий агент и, следовательно, зависит от количества допаминергических (DAegic) нейронов, оставшихся жизнеспособными и функционирующими после введения 6-OHDA. Амфетамин вводили интраперитонеально путем инъекций в однократной дозе, составляющей 1,5 мг/кг, с последующей фиксацией вращения в течение 60 минут на Rota Meter.
Как показано на Фиг. 3, значительно улучшенный эффект в чистом вращении, т.е. вращении по часовой стрелке за вычетом вращения против часовой стрелки (CW-CCW), наблюдали у крыс, получавших лечение разагилином с использованием насоса ALZET, демонстрирующего замедленное высвобождение, по сравнению с крысами, получавшими лечение разагилином путем ежедневных интраперитонеальных инъекций, демонстрирующих быстрое высвобождение.
Примеры 4-6. Пеллеты разагилина с покрытием с пролонгированным высвобождением без подслоя покрытия
Получали пеллеты мезилата разагилина с пролонгированным высвобождением (ПВ) без подслоя покрытия, имеющие состав, представленный в Таблице 2. В частности, для получения слоя лекарственного средства повидон USP (PVP К29/32) растворяли в смеси дистиллированной воды и 96% этанола, а затем в полученном растворе мезилат разагилина растворяли. Диспергировали сверхтонкий тальк и добавляли к полученному раствору с образованием однородной суспензии, которую затем наносили на сахарные сферические гранулы размером 600-710 мкм с использованием установки для нанесения покрытия с псевдоожиженным слоем. Суспензию для нанесения функционального покрытия готовили путем растворения Ethocel 45 сПз (этилцеллюлоза; полимер, контролирующий высвобождение) в смеси ацетона и 96% этанола, а затем полиэтиленгликоль (ПЭГ) 4000 растворяли в дистиллированной воде и добавляли к полученному раствору. Полученную суспензию наносили на содержащие лекарственное средство пеллеты с использованием установки для нанесения покрытия с псевдоожиженным слоем.
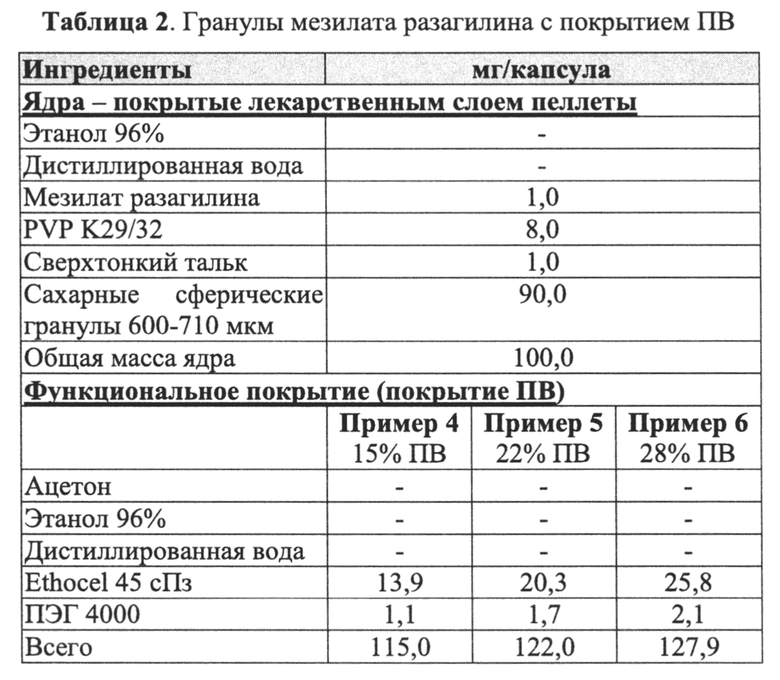
Характеристики растворения пеллет с разными покрытиями ПВ оценивали в следующих условиях: Аппарат 1, USP (Фармакопея США) использовали для перемешивания среды для растворения (900 мл раствора кишечной жидкости, EFS, рН 6,8) со скоростью вращения шпинделя 100 об/мин и при температуре 37°C. Характеристики растворения представлены в Таблице 3 и на Фиг. 4.
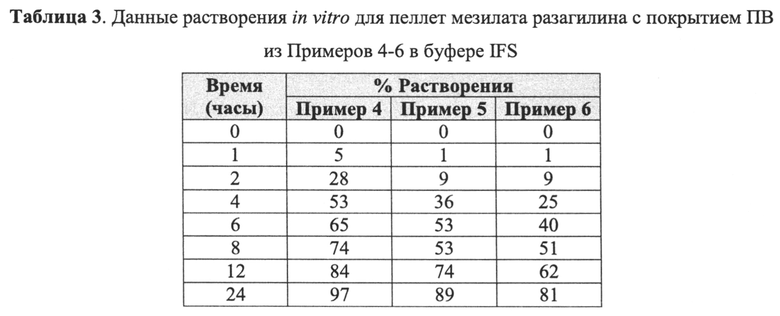
Примеры 7-8. Пеллеты разагилина с покрытием с пролонгированным высвобождением с подслоем покрытия
Получали пеллеты мезилата разагилина ПВ с подслоем покрытия, имеющие состав, представленный в Таблице 4. В частности, для получения слоя лекарственного средства повидон (PVP К25) растворяли в смеси дистиллированной воды и 96% этанола, а затем в полученном растворе растворяли мезилат разагилина. Диспергировали сверхтонкий тальк и добавляли его к полученному раствору с образованием однородной суспензии, которую затем наносили на сахарные сферические гранулы размером 600-710 мкм с использованием установки для нанесения покрытия с псевдоожиженным слоем. Раствор для нанесения подслоя покрытия готовили путем растворения PVP К25 в смеси дистиллированной воды и 96% этанола, а затем полученный раствор наносили на содержащие лекарственное средство пеллеты с использованием установки для нанесения покрытия с псевдоожиженным слоем. Суспензию для нанесения функционального покрытия готовили путем растворения Ethocel 45 сПз в смеси ацетона и 96% этанола, а затем ПЭГ 3000 растворяли в дистиллированной воде и добавляли к полученному раствору. Диспергировали сверхтонкий тальк и добавляли к полученному раствору с образованием однородной суспензии, которую затем наносили на пеллеты с подслоем покрытия с помощью установки для нанесения покрытия с псевдоожиженным слоем. Сухую смесь пеллет разагилина ПВ и Аэросила 200 готовили с использованием смесителя Tumbler Bin Blender.
Характеристики растворения пеллет с разными покрытиями ПВ оценивали в условиях, описанных в Примерах 4-6, и полученные результаты представлены в Таблице 5 и на Фиг. 5. Данные растворения in vitro для пеллет мезилата разагилина с покрытием ПВ из Примера 7 (15% ПВ) в (i) буфере IFS (рН 6,8), имитирующем условия в кишечнике; (ii) буфере GFS (раствор желудочного сока) (рН 1,2), имитирующем условия в пустом желудке; (iii) буфере GFS в течение 2 часов, а затем в буфере IFS в течение еще 20 часов; и (iv) ацетатном буфере (рН 4,5), имитирующем условия в полном желудке, представлены на Фиг. 6. Данные изучения стабильности in vitro в буфере IFS для тех же пеллет с покрытием ПВ в нулевой момент времени (сразу после получения), через 1 месяц в условиях ускоренного старения (40°C, влажность 75%) и через 2 и 3 месяца в тех же условиях ускоренного старения представлены на Фиг. 7.


Пример 9. Капсулы разагилина с пролонгированным высвобождением с подслоем покрытия
Пеллеты мезилата разагилина ПВ с подслоем покрытия, имеющие состав, представленный в Таблице 6, получали, как описано в Примерах 7-8; за исключением того, что для приготовления сухой смеси использовали коллоидный диоксид кремния вместо Аэросила 200. Характеристики растворения данных полученных пеллет с покрытием ПВ оценивали в условиях, приведенных в Примерах 4-8, и полученные данные представлены в Таблице 7 и на Фиг. 8.
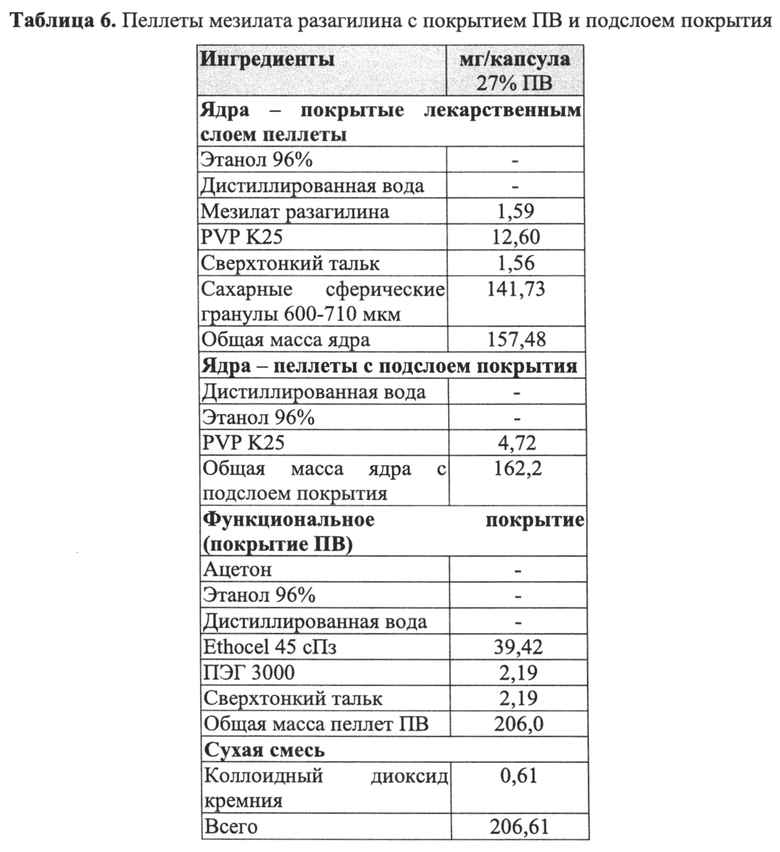

Пример 10. Пеллеты разагилина с покрытием с пролонгированным высвобождением с/без подслоем(я) покрытия
Дополнительные пеллеты мезилата разагилина ПВ с подслоем покрытия или без него, имеющие состав, представленный в Таблицах 8-12, могут быть получены в соответствии с процедурой, описанной в Примерах 4-9. Сноски, указанные в каждой из Таблиц, включенных в данный Пример, представлены под Таблицей 16.
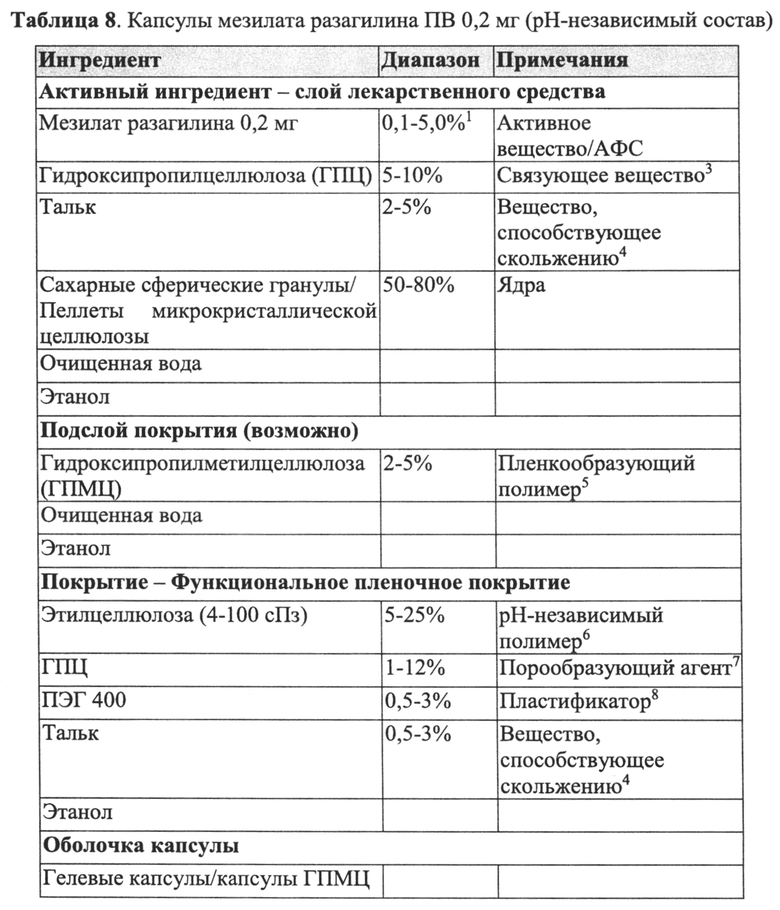
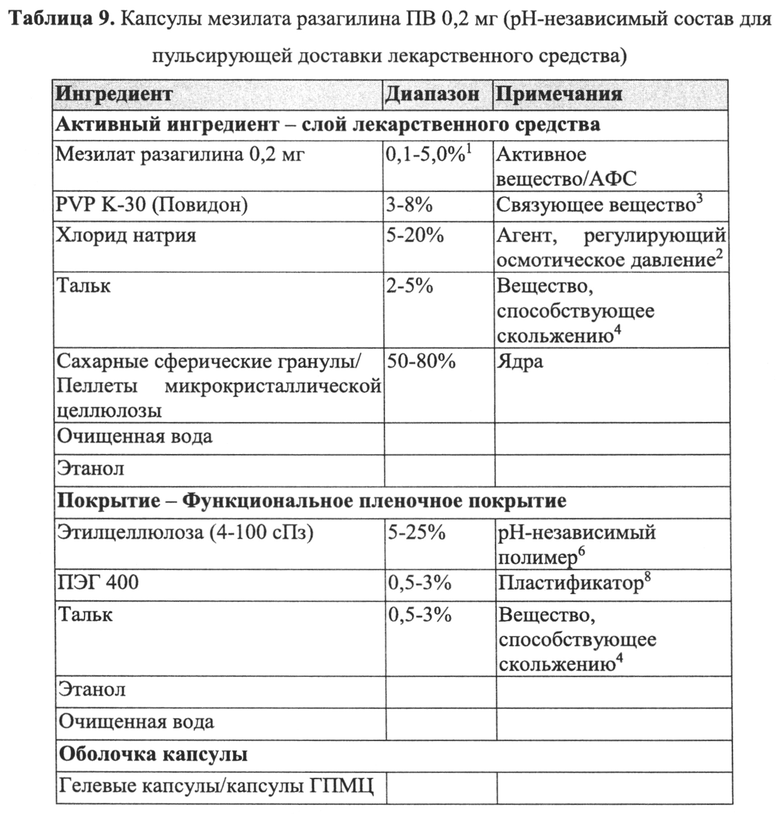
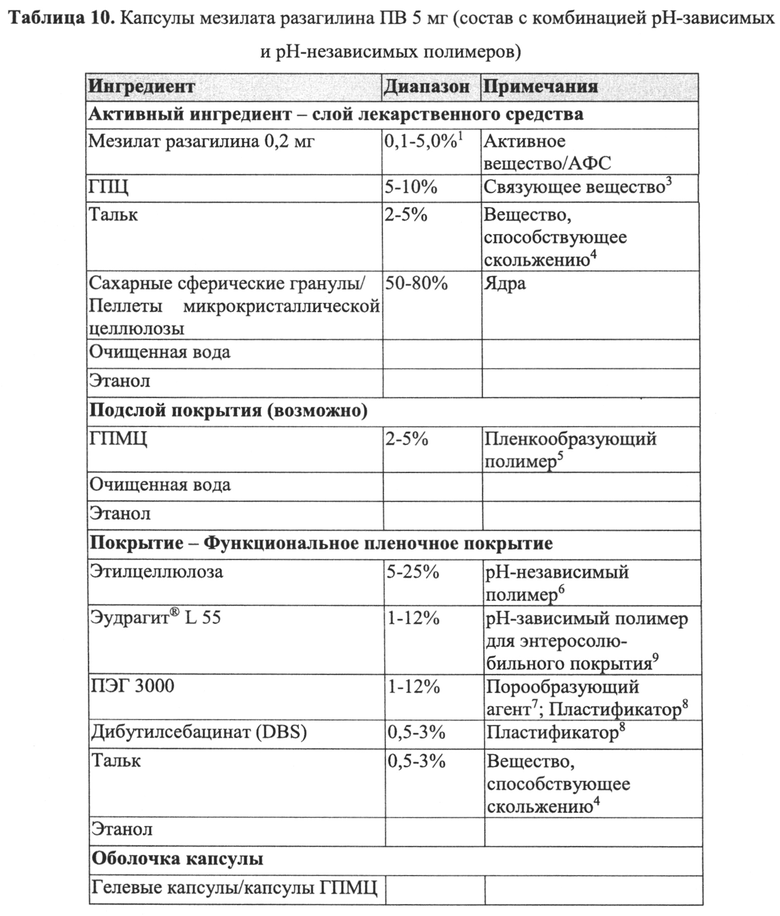
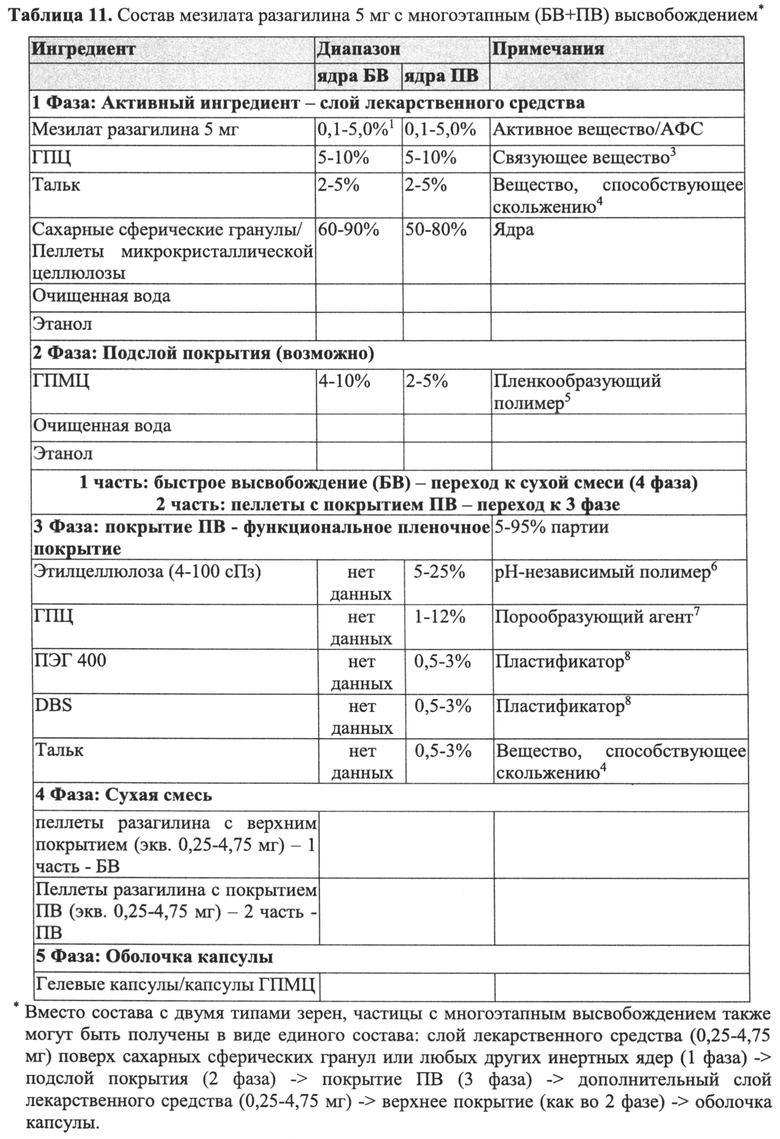
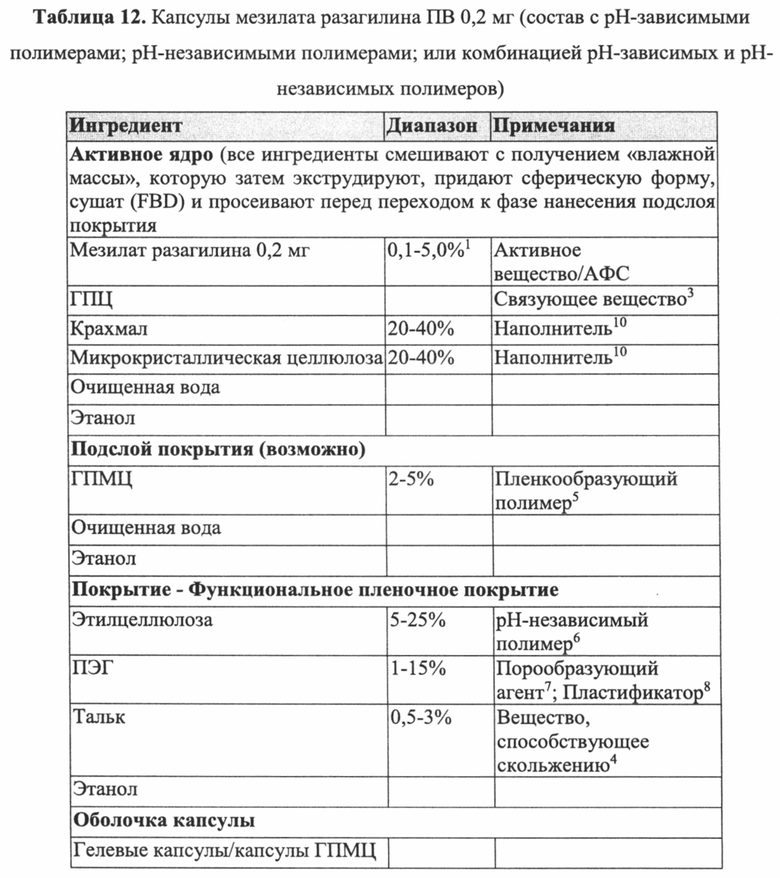
Составы мезилата разагилина, описанные в Таблицах 2, 4 и 6, а также составы, описанные в Таблицах 8-12 выше, можно прессовать в таблетки для изготовления также таблеток разагилина с покрытием ПВ. Для этой цели пеллеты разагилина с покрытием ПВ в сухом виде смешивают с дополнительными наполнителями с получением гомогенной смеси, которую затем прессуют в таблетки, на которые наносят верхний/косметический/нефункциональный слой покрытия (см., например, Таблицу 13).
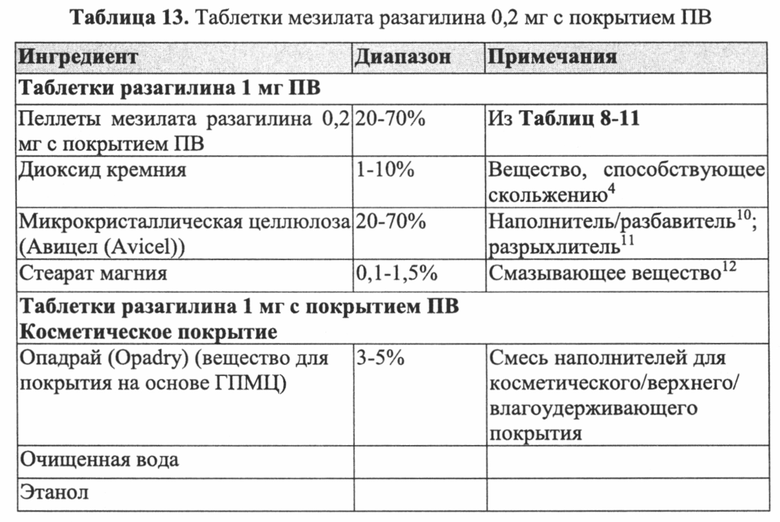
В Таблице 14 представлен состав таблеток разагилина 0,2 мг с покрытием ПВ, получаемый из влажного гранулята, который затем сушат, измельчают, смешивают в сухом виде, таблетируют и, наконец, на который наносят покрытие ПВ.
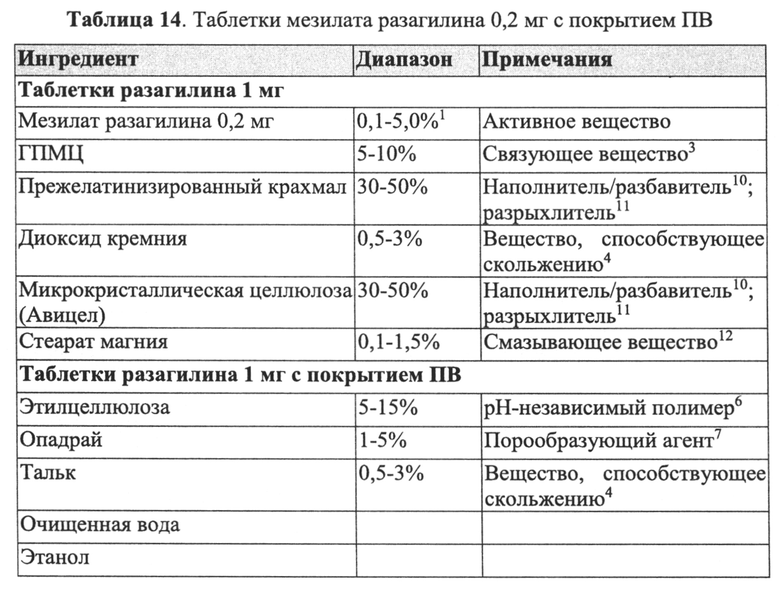
В Таблице 15 представлен состав таблеток разагилина 0,2 мг с покрытием ПВ, получаемый из влажного гранулята, содержащего полимеры, контролирующие высвобождение, который затем сушат, измельчают, смешивают в сухом виде, таблетируют и, наконец, на который наносят верхнее покрытие.
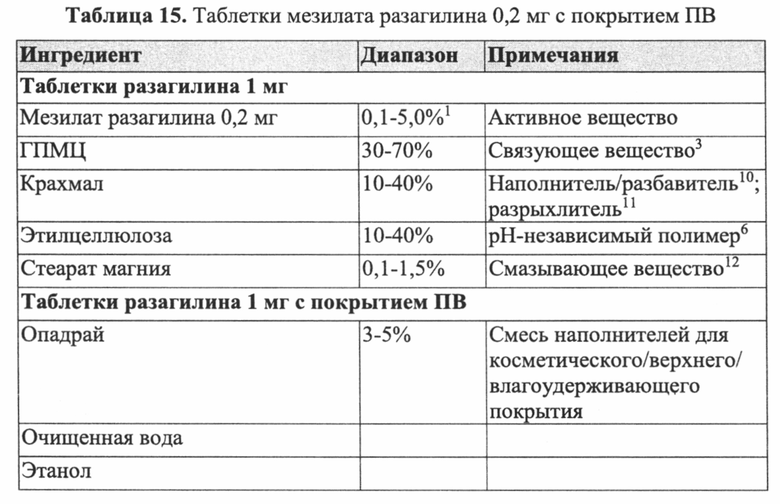
В Таблице 16 представлен состав таблеток разагилина 5 мг с покрытием ПВ, получаемый из влажного гранулята, который затем сушат, измельчают, смешивают в сухом виде, таблетируют с образованием двух слоев и, наконец, на который наносят верхнее покрытие.
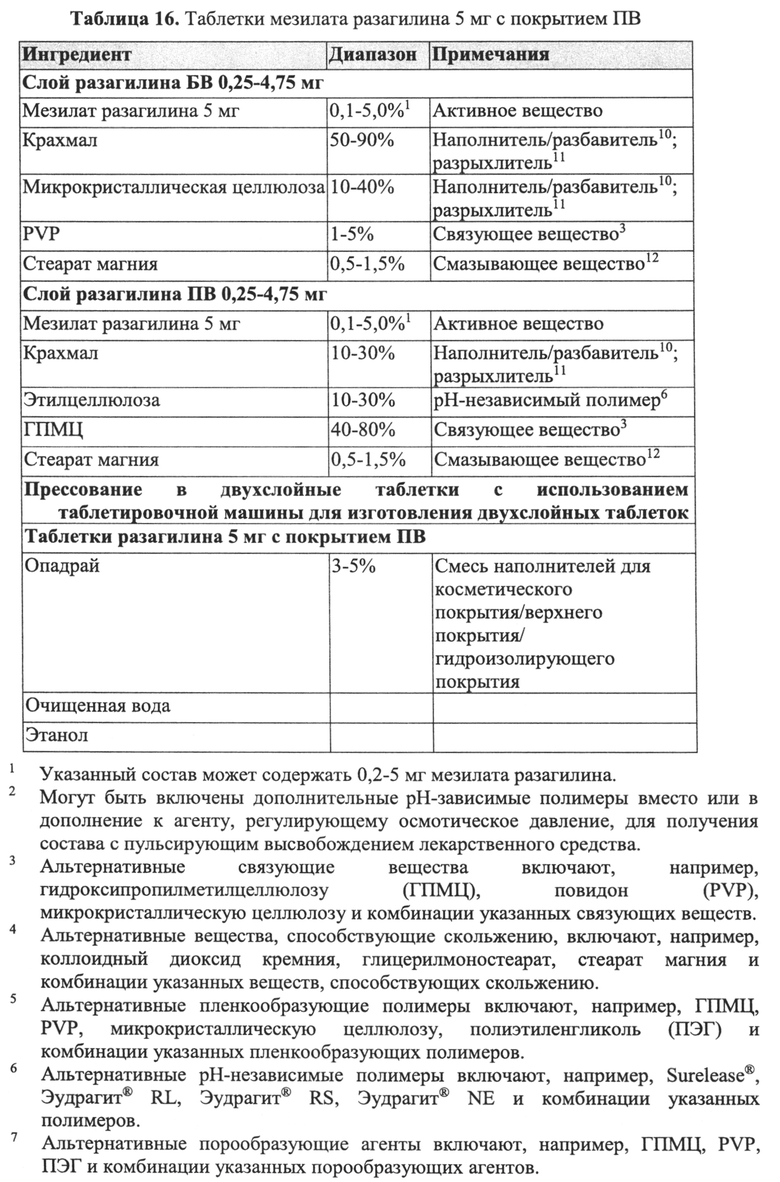

Пример 11. Абсорбция разагилина в различных отделах желудочно-кишечного тракта
Лекарственные средства по-разному абсорбируются из различных отделов желудочно-кишечного тракта. Для создания продукта для перорального введения 24-часового замедленного высвобождения необходимо, чтобы лекарственное средство абсорбировалось на протяжении всего времени, т.е. из всех отделов желудочно-кишечного тракта. Известно, что большинство лекарственных средств хорошо абсорбируются из двенадцатиперстной кишки; однако многие лекарственные средства не эффективно абсорбируются из толстой кишки. Поскольку лекарственное средство находится в течение значительного количества времени в толстой кишке, прежде чем оно будет выведено из организма, важно оценить его абсорбцию из толстой кишки для того, чтобы эффективно изобразить кривую высвобождения.
В данном исследовании разагилин (1,5 мг/кг) вводили в виде водного раствора 0,5 мг/мл посредством полиэтиленового катетера, имплантированного за один день до начала фармакокинетического эксперимента свободно перемещающимся самцам крыс линии Вистар. Указанные катетеры помещали в толстую кишку, либо двенадцатиперстную кишку, либо яремную вену для болюсного введения в толстую кишку, болюсного введения в двенадцатиперстную кишку или внутривенного болюсного введения, соответственно. Осуществляли введение однократной болюсной дозы в каждый участок. Кроме того, второй постоянный катетер помещали в правую вену каждого животного для систематического забора крови. Образцы крови (0,5 мл) забирали за 5 минут до введения дозы и через 5, 15, 30, 50, 90, 150 и 200 минут после введения дозы. Для предотвращения обезвоживания крысам вводили равные объемы физиологического раствора после каждого забора образца крови. Плазму отделяли путем центрифугирования с последующим аналитическим определением количества разагилина и его основного метаболита, 1-аминоиндана, с использованием тройной квадрупольной системы ЖХ-МС-МС. Фармакокинетический анализ без учета компартментов проводили с использованием программного обеспечения Excel. Площадь под кривой (ППК) рассчитывали на основе анализа без учета компартментов конечной измеряемой выборки с использованием линейно-логарифмического метода трапеций. Биодоступность разагилина при пероральном введении (F) рассчитывали в виде процентного соотношения: ППК(двенадцатиперстная кишка)/ППК(внутривенное) или ППК(толстая кишка)/ППК(внутривенное).
В Таблице 17 и на Фиг. 9 показаны различия максимальной (или пиковой) концентрации в плазме (Cmax) и ППК между группами введении в двенадцатиперстную и толстую кишку (данные представлены в виде среднего значения ±SE, n=4-5). В частности, исходный 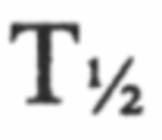 был больше для групп введения в толстую и двенадцатиперстную кишку по сравнению с после внутривенного введения. Рассчитанные значения ППК для внутривенной дозы и дозы для двенадцатиперстной кишки оказались схожими, что позволяет предположить полную абсорбцию при пероральном введении. ППК после введения в толстую кишку составляла приблизительно 28% от ППК внутривенной дозы, что подтверждает возможность абсорбции в толстой кишке. В соответствии с данными результатами, создание системы доставки разагилина с контролируемым высвобождением осуществимо и имеет практическое значение.
был больше для групп введения в толстую и двенадцатиперстную кишку по сравнению с после внутривенного введения. Рассчитанные значения ППК для внутривенной дозы и дозы для двенадцатиперстной кишки оказались схожими, что позволяет предположить полную абсорбцию при пероральном введении. ППК после введения в толстую кишку составляла приблизительно 28% от ППК внутривенной дозы, что подтверждает возможность абсорбции в толстой кишке. В соответствии с данными результатами, создание системы доставки разагилина с контролируемым высвобождением осуществимо и имеет практическое значение.
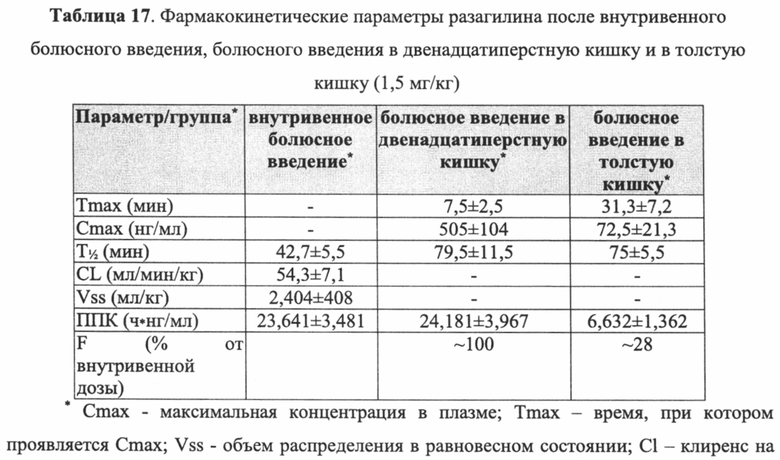
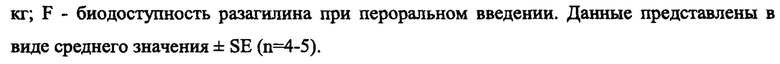
ССЫЛКИ
Akao Y., Nakagawa Y., Maruyama W., Takahashi Т., Naoi M., Apoptosis induced by an endogenous neurotoxin, N-methyl(R)salsolinol, is mediated by activation of caspase-3, Neurosci. Lett, 1999, 267, 153-156
Akao Y., Maruyama W., Shimizu S., Yi H., Nakagawa Y., Shamoto-Nagai M., Youdim M.B.H., Tsujimoto Y., Naoi M., Mitochondrial permeability transition mediates apoptosis induced by N-methyl(R)salsolinol, an endogenous neurotoxin, and is inhibited by Bcl-2 and Rasagiline, N-Propargyl-1(R)-arrunoindan, J. Neurochem., 2002a, 82, 913-923
Akao Y., Maruyama W., Yi FL, Shamoto-Nagai M., Youdim M.B.H., Naoi M., An anti-Parkinson’s disease drug, N-propargyl-1(R)-aminoindan (rasagiline), enhances expression of anti-apoptotic Bcl-2 in human dopaminergic SH-SY5Y cells, Neurosci. Lett., 2002b, 326, 105-108
Bar-Am O., Amit Т., Youdim M.B., Aminoindan and hydroxyaminoindan, metabolites of rasagiline and ladostigil, respectively, exert neuroprotective properties in vitro, J. Neurochem., 2007, 103(2), 500-508
Bar-Am O., Weinreb O., Amit Т., Youdim M.B., The neuroprotective mechanism of 1-(R)-ammoindan, the major metabolite of the antiparkinsonian drug rasagiline, J. Neurochem., 2010, 112, 1131-1137
Durden D.A., Dyck L.E., Davis B.A., Liu Y.D., Boulton A.A., Metabolism and pharmacokinetics, in the rat, of (R)-N-(2-heptyl)methyl-propargylamine (R-2HMP), a new potent monoamine oxidase inhibitor and antiapoptotic agent, Drug Metab Dispos., 2000, 28, 147-154
Grossberg G., Desai A., Review of rivastigmine and its clinical applications in Alzheimer’s disease and related disorders, Expert Opin. Pharmacother., 2000, 2, 653-666
Maruyama W., Boulton A.A., Davis B.A., Dostert P., Naoi M., Enantio-specific induction of apoptosis by an endogenous neurotoxin, N-methyl(R)salsolinol, in dopaminergic SH-SY5Y cells: suppression of apoptosis by N-(2-hep1yl)-N-memylpropargylamine, J. Neural Transm., 2001a, 108, 11-24
Maruyama W., Akao Y., Youdim M.B.H., Boulton A.A., Davis B.A., Naoi M., Transfection-enforced Bcl-2 overexpression and an anti-Parkinson drug, rasagiline, prevent nuclear accumulation of glyceraldehyde-3 phosphate dehydrogenase induced by an endogenous dopaminergic neurotoxin, N-methyl(R)salsolinol, J. Neurochem., 2001b, 78, 727-735
Maruyama W., Takahashi Т., Youdim, M.B.H., Naoi M., The anti-Parkinson drag, rasagiline, prevents apoptotic DNA damage induced by peroxynitrite in human dopaminergic neuroblastoma SH-SY5Y cells, J. Neural Transm., 2002, 109, 467-481
Tazik S., Johnson S., Lu D., Johnson C., Youdim M.B., Stockmeier C.A., Ou X.M., Comparative neuroprotective effects of rasagiline and aminoindan with selegiline on dexamethasone-induced brain cell apoptosis, Neurotoxicity Research, 2009, 15, 284-290
Tatton W.G., Chalmers-Redman R.M., Ju W.J., Mammen M., Carlile G.W., Pong A.W., Tatton N.A., Propargylamines induce antiapoptotic new protein synthesis in serum- and nerve growth factor (NGF)-withdrawn, NGF-differentiated PC-12 cells, J Pharmacol Exp Пег., 2002, 301, 753-764
Tatton W.G., Greenwood C.E., Rescue of dying neurons: a new action for deprenyl in MPTP parkinsonism, J Neurosci Res., 1991, 30, 666-672
Tatton W.G., Selegiline can mediate neuronal rescue rather than neuronal protection, Movement Disorders 8 (Supp.1), 1993, S20-S30
Weinreb O., Amit Т., Bar-Am O., Yousim M.B., Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity, Prog Neurobiol., 2010, 92(3), 330-344
Weinstock M., Selectivity of cholinesterase inhibition: Clinical implications for the treatment of Alzheimer’s disease, CNS Drugs, 1999, 12, 307-323
Yogev-Falach M., Amit Т., Bar-Am O., Sagi Y., Weinstock M., Youdim M.B.H., The involvement of mitogen-activated protein (MAP) kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline, FASEB J., 2002, 16, 1674-1676
Youdim M.B.H., Weinstock M., ovel neuroprotective anti-Alzheimer drugs with antidepressant activity derived from the anti-Parkinson drug, rasagiline, Mechanisms of Ageing & Developments, 2002a, 123, 1081-1086
Youdim M.B.H., Gross A., Finberg J.P.M., Rasagiline [N-Propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B, Br. J. Pharmacol., 2001a, 132, 500-506
Youdim M.B.H., Wadia A., Tatton N.A., Weinstock M., The anti-Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo, Ann N Y Acad Sci, 2001b, 939, 450-458
Zimmermann K, Waldmeier P.C., Tatton W.G., Dibenzoxepines as treatments for neurodegenerative diseases, Pure Appl Chem, 1999, 71, 2039-2046
| название | год | авторы | номер документа |
|---|---|---|---|
| СОСТАВЫ РАЗАГИЛИНА С ПРОЛОНГИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2607595C2 |
| ТЕРАПИЯ БОЛЕЗНИ ПАРКИНСОНА С ПРИМЕНЕНИЕМ КОМБИНАЦИИ С ФИКСИРОВАННЫМИ ДОЗАМИ | 2013 |
|
RU2642962C9 |
| ТЕРАПИЯ БОЛЕЗНИ ПАРКИНСОНА С ПРИМЕНЕНИЕМ КОМБИНАЦИИ С ФИКСИРОВАННЫМИ ДОЗАМИ | 2013 |
|
RU2771522C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2009 |
|
RU2540470C9 |
| КОМПОЗИЦИИ РАЗАГИЛИНА С УЛУЧШЕННОЙ ОДНОРОДНОСТЬЮ СОДЕРЖИМОГО | 2006 |
|
RU2404746C2 |
| ПРИМЕНЕНИЕ РАЗАГИЛИНА ДЛЯ ЛЕЧЕНИЯ ОБОНЯТЕЛЬНОЙ ДИСФУНКЦИИ | 2011 |
|
RU2587330C2 |
| СОСТАВЫ L-ОРНИТИН ФЕНИЛАЦЕТАТА | 2016 |
|
RU2755904C2 |
| СПОСОБ ЛЕЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 1999 |
|
RU2207856C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2004 |
|
RU2342929C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ИНГИБИТОРА ГИСТОНДИАЦЕТИЛАЗЫ В КОМБИНАЦИИ С БЕНДАМУТИНОМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609833C2 |
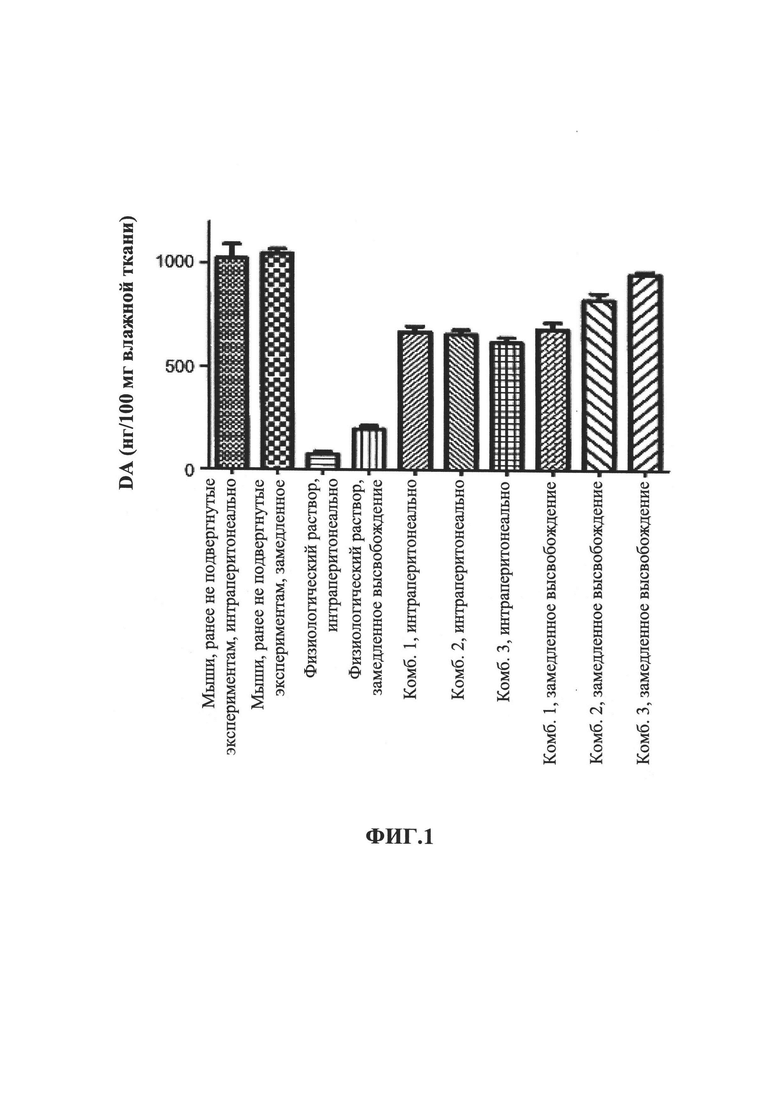
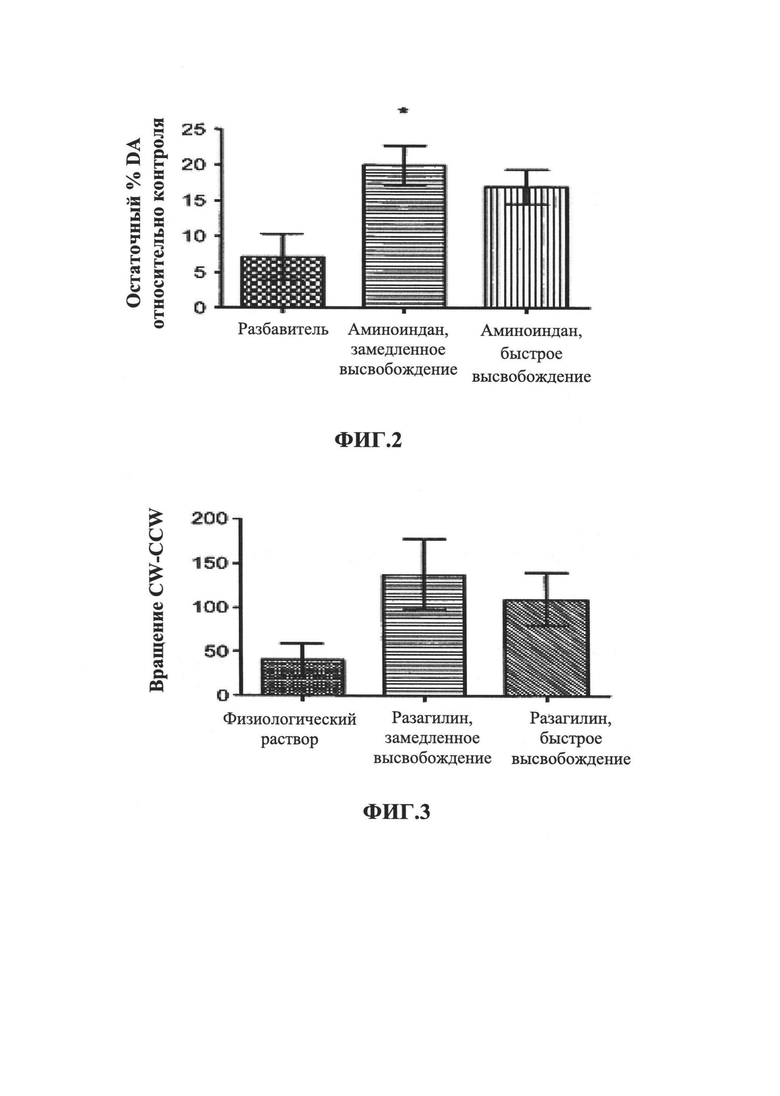
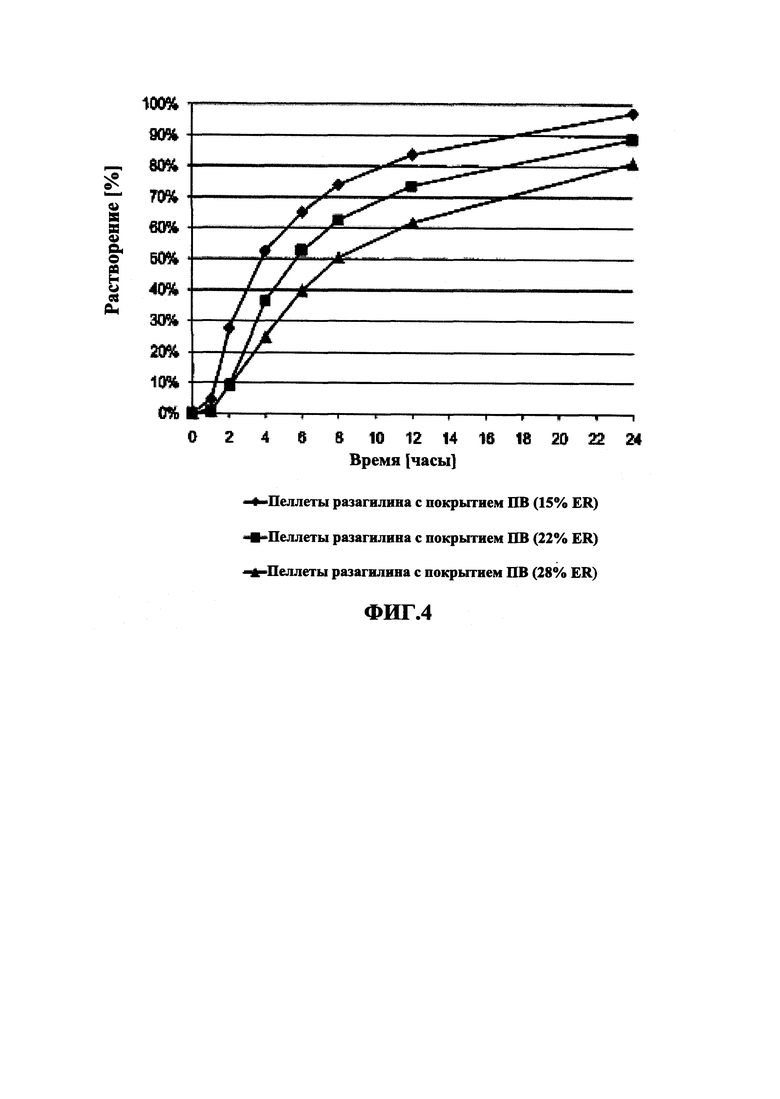
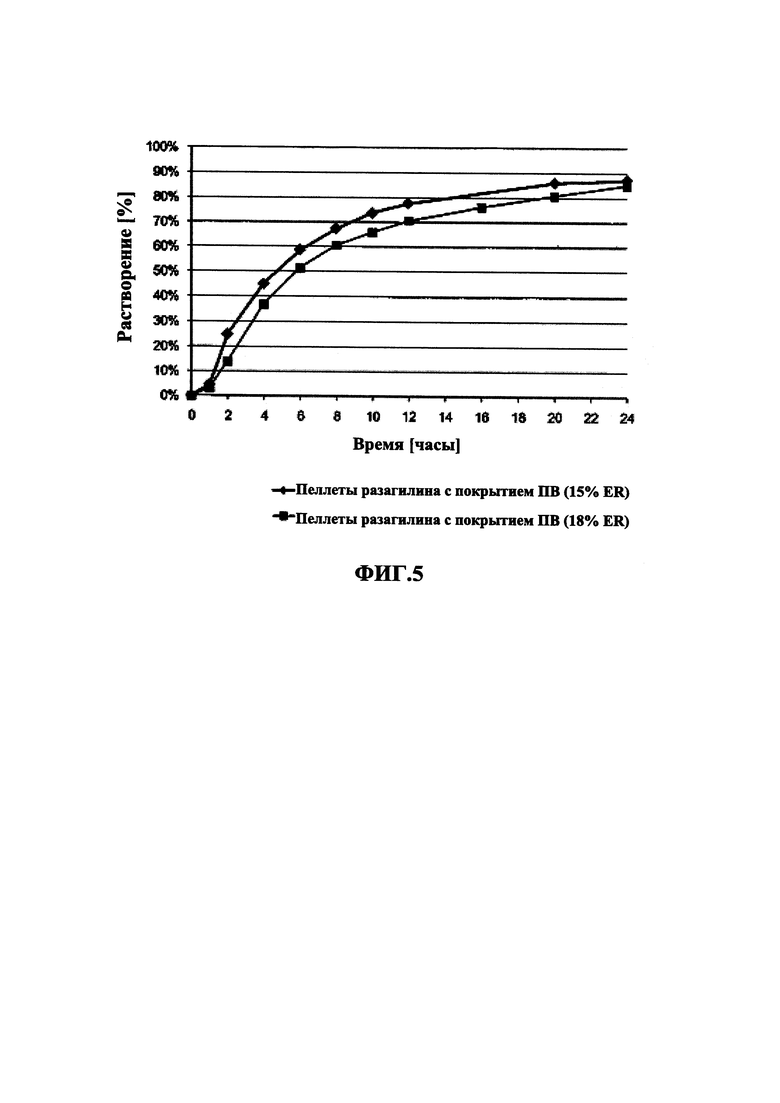
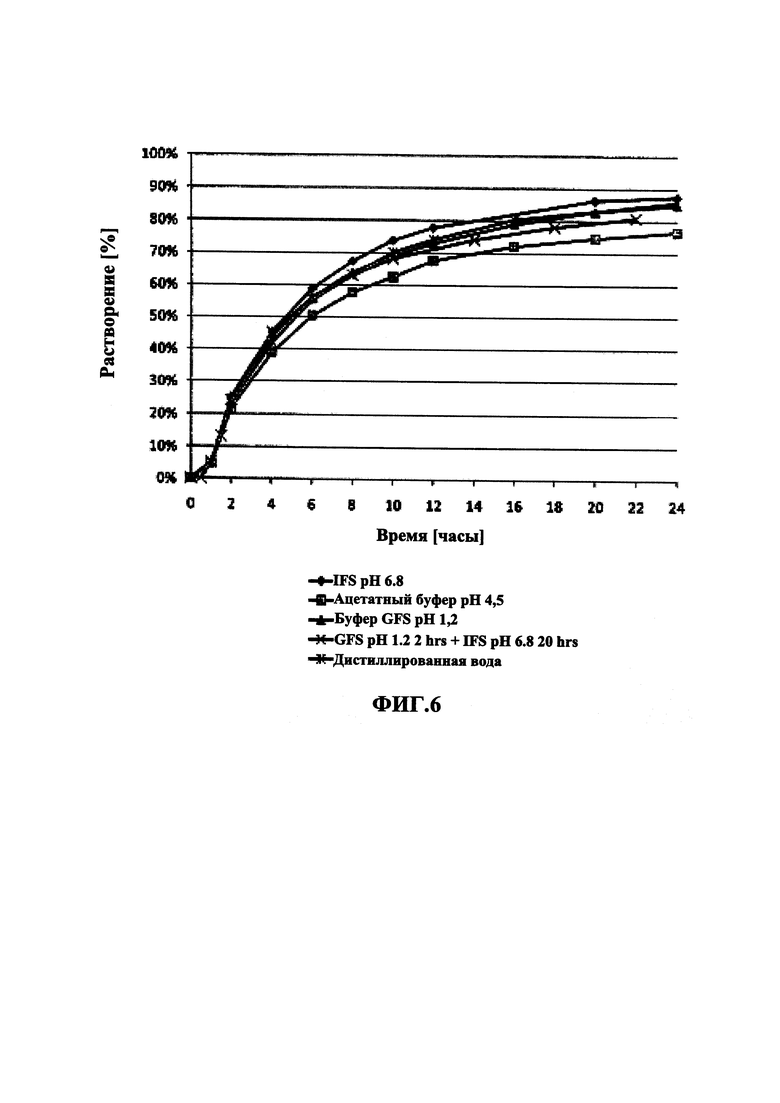
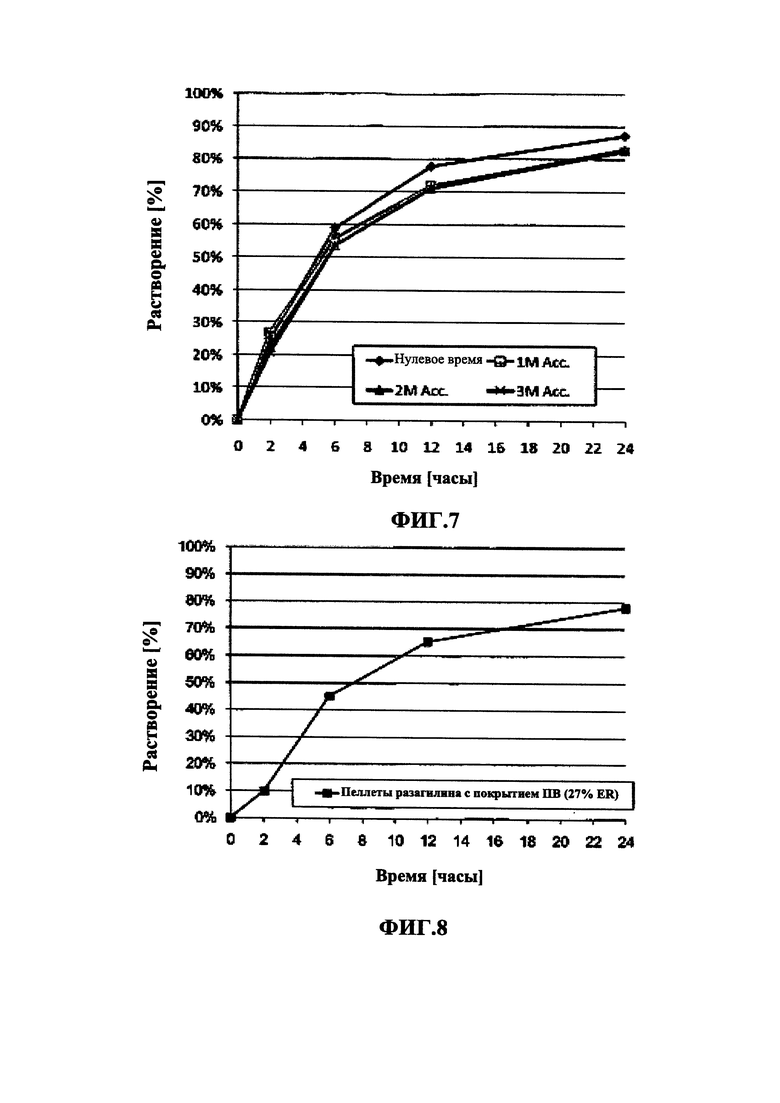
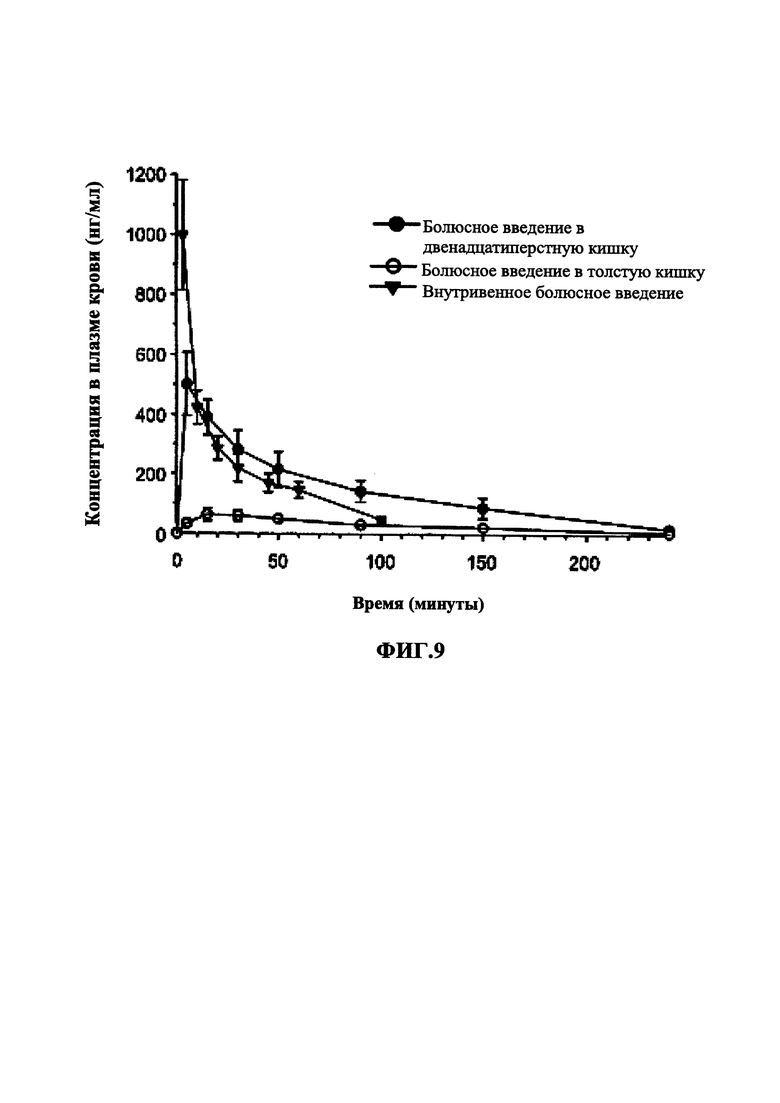
Настоящее изобретение относится к применению пероральной фармацевтической композиции с пролонгированным высвобождением, содержащей R-(+)-N-пропаргил-1-аминоиндан (разагилин) или его фармацевтически приемлемую соль в качестве активного агента, для лечения болезни Паркинсона, при этом доза активного агента составляет от 0,2 до 2,0 мг в сутки, и указанная композиция имеет характеристики растворения в Аппарате 1 (корзина) согласно Фармакопее США (USP) при 50-150 об/мин при значении pH вплоть до 7,4, при 37 °С, при которых 30-70% указанного активного агента высвобождается в течение первых шести часов. Также настоящее изобретение относится к способу лечения болезни Паркинсона у нуждающегося в этом индивидуума. Техническим результатом настоящего изобретения является получение фармацевтической композиции, приготовленной в форме для пролонгированного высвобождения активного соединения, подходящего для лечения нейродегенеративных заболеваний, в частности болезни Паркинсона, а также повреждений нервной системы. 2 н. и 10 з.п. ф-лы, 9 ил., 17 табл., 11 пр.
1. Применение пероральной фармацевтической композиции с пролонгированным высвобождением, содержащей R-(+)-N-пропаргил-1-аминоиндан (разагилин) или его фармацевтически приемлемую соль в качестве активного агента, для лечения болезни Паркинсона, при этом доза активного агента составляет от 0,2 до 2,0 мг в сутки, и указанная композиция имеет характеристики растворения в Аппарате 1 (корзина) согласно Фармакопее США (USP) при 50-150 об/мин при значении pH вплоть до 7,4, при 37 °С, при которых 30-70% указанного активного агента высвобождается в течение первых шести часов.
2. Применение по п. 1, отличающееся тем, что доза активного агента составляет от 0,2 до 1,5 мг в сутки.
3. Применение по п. 2, отличающееся тем, что доза активного агента составляет от 0,2 до 1,0 мг в сутки.
4. Применение по п. 3, отличающееся тем, что доза активного агента составляет от 0,5 до 1,0 мг в сутки.
5. Применение по п. 1, отличающееся тем, что указанная фармацевтически приемлемая соль выбрана из мезилатной соли, эзилатной соли, тозилатной соли, сульфатной соли, сульфонатной соли, фосфатной соли, карбоксилатной соли, малеатной соли, фумаратной соли, тартратной соли, бензоатной соли, ацетатной соли, гидрохлоридной соли или гидробромидной соли R(+)-N-пропаргил-1-аминоиндана, предпочтительно мезилатной соли R(+)-N-пропаргил-1-аминоиндана.
6. Применение по любому из пп. 1-5, отличающееся тем, что указанная фармацевтическая композиция имеет следующие характеристики растворения в Аппарате 1 (корзина) согласно Фармакопее США (USP) при 50-150 об/мин при значении pH вплоть до 7,4, при 37 °С:
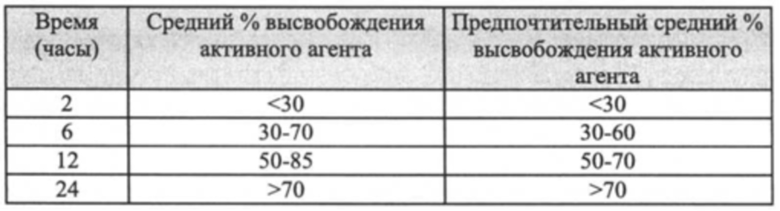
7. Способ лечения болезни Паркинсона у нуждающегося в этом индивидуума, включающий введение указанному индивидууму пероральной фармацевтической композиции, содержащей активный агент, выбранный из R-(+)-N-пропаргил-1-аминоиндана (разагилина) или его фармацевтически приемлемой соли, причем указанная композиция приготовлена в форме для пролонгированного высвобождения и имеет характеристики растворения в Аппарате 1 (корзина) согласно Фармакопее США (USP) при 50-150 об/мин при значении pH вплоть до 7,4, при 37 °С, при которых 30-70% указанного активного агента высвобождается в течение первых шести часов, и при этом доза активного агента составляет от 0,2 до 2,0 мг в сутки.
8. Способ по п. 7, отличающийся тем, что доза активного агента составляет от 0,2 до 1,5 мг в сутки.
9. Способ по п. 8, отличающийся тем, что доза активного агента составляет от 0,2 до 1,0 мг в сутки.
10. Способ по п. 9, отличающийся тем, что доза активного агента составляет от 0,5 до 1,0 мг в сутки.
11. Способ по п. 7, отличающийся тем, что указанная фармацевтически приемлемая соль выбрана из мезилатной соли, эзилатной соли, тозилатной соли, сульфатной соли, сульфонатной соли, фосфатной соли, карбоксилатной соли, малеатной соли, фумаратной соли, тартратной соли, бензоатной соли, ацетатной соли, гидрохлоридной соли или гидробромидной соли R(+)-N-пропаргил-1-аминоиндана, предпочтительно мезилатной соли R(+)-N-пропаргил-1-аминоиндана.
12. Способ по любому из пп. 7-11, отличающийся тем, что указанная фармацевтическая композиция имеет следующие характеристики растворения в Аппарате 1 (корзина) согласно Фармакопее США (USP) при 50-150 об/мин при значении pH до 7,4, при 37 °С:
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ТРАНСДЕРМАЛЬНЫЙ ПЛАСТЫРЬ, СОДЕРЖАЩИЙ РАЗАГИЛИН, ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ НЕРВНОЙ СИСТЕМЫ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2007 |
|
RU2379029C1 |
| ХАРЕНКО А.В | |||
| и др | |||
| Основные факторы, контролирующие кинетику высвобождения теофиллина из полимерных композиций на основе интерполимерных комплексов | |||
| Проблемы качества лекарственных средств | |||
| НПО "Биотехнология" М., 1991, с | |||

