ОТРАСЛЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к оральным композициям 3-((3R, 4R)-4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)-амино]-пиперидин-1-ил)-3-оксопропионитрила (далее по тексту тофацитиниб) с замедленным высвобождением, который является ингибитором протеинкиназ, таких как фермент Янус киназа (JAK), и таким образом применяется в терапии в качестве иммуносупрессивных агентов при следующих заболеваниях: трансплантация органов, ксенотрансплантация, волчанка, рассеянный склероз, ревматоидный артрит, псориаз, псориатический артрит, диабет типа I и диабетические осложнения, рак, астма, атопический дерматит, аутоиммунные расстройства щитовидной железы, язвенный колит, анкилозирующий спондилоартрит, ювенильный идиопатический артрит, болезнь Крона, болезнь Альцгеймера, лейкемия, и других показаниях, где желательно подавление иммунного ответа. Изобретение относится к композициям с замедленным высвобождением, содержащим тофацитиниб или его фармацевтически приемлемые соли. Композиции, описанные в данном документе, имеют желаемые фармакокинетические характеристики. Примеры включают соотношения AUC, Cmax, дозо-корригированная AUC, дозо-корригированный Cmax, и соотношение AUC и Cmax после еды/натощак.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Тофацитиниб, 3-((3R,4R)-4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)-амино]-пиперидин-1-ил)-3-оксопропионитрил, имеет химическую формулу C16H20N6O и следующую структурную формулу:
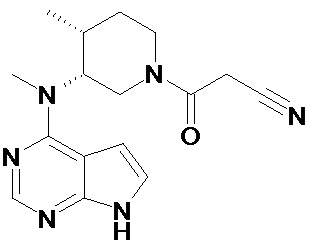
Термин “тофацитиниб” следует понимать, пока не указано обратное, как включающий любую фармацевтически приемлемую форму и соль соединения. Тофацитиниб может существовать в кристаллической или аморфной формах. Тофацитиниб, соли тофацитиниба, способы синтеза тофацитиниба, определенные полиморфы тофацитиниба, и применения тофацитиниба описаны в WO01/42246, WO02/096909 и WO03/048162.
Тофацитиниб широко известен в качестве ингибитора протеинкиназ, таких как фермент Янус киназа (JAK), и таким образом, является пригодным в качестве иммуносупрессивных агентов при следующих состояниях: трансплантация органов, ксенотрансплантация, волчанка, рассеянный склероз, ревматоидный артрит, псориаз, псориатический артрит, диабет типа I и диабетические осложнения, рак, астма, атопический дерматит, аутоиммунные расстройства щитовидной железы, язвенный колит, болезнь Крона, болезнь Альцгеймера, лейкемия и другие показания, где желательно подавление иммунитета.
Тофацитиниб разрабатывали в виде таблетированной формы с мгновенным высвобождением с дозами в диапазоне от 5 мг дo 10 мг вводимыми дважды в день. Тофацитиниб, в виде цитратной соли тофацитиниба, одобрен в США под торговой маркой XELJANZ™. Фармацевтические дозированные формы тофацитиниба известны и описаны в WO01/42246, WO02/096909 и WO03/048162. Кроме того, WO2012/100949 описывает композицию тофацитиниба с модифицированным высвобождением. Хотя WO2012/100949 упоминает, что тофацитиниб может быть формулирован в композицию с модифицированным высвобождением, желаемые фармакокинетические характеристики описаны не были.
Хотя коммерческая таблетированная дозированная форма с мгновенным высвобождением обеспечивает эффективные уровни тофацитиниба у пациентов (продиктованные средней концентрацией тофацитиниба в плазме крови, Cave, на протяжении 24 часов), есть возможность уменьшить количество доз до одного раза в день благодаря дозированной форме тофацитиниба с замедленным высвобождением, при этом поддерживая тот же терапевтический эффект, таким образом, улучшая комфорт и потенциально улучшая податливость лечению.
Дозированные формы с замедленным высвобождением обычно разработаны для обеспечения наиболее длительного периода высвобождения, для минимизации: 1) флуктуаций концентрации в плазме крови на протяжении интервала дозирования (то есть, соотношение максимальной концентрации в плазме крови, Cmax,ss, к минимальной концентрации в плазме крови, Cmin,ss, на протяжении интервала дозирования), и 2) количества лекарственного средства, необходимого для достижения желаемого терапевтического эффекта, с целью улучшения профиля безопасности и толерантности. Например, WO2012/100949 описывает композицию тофацитиниба с модифицированным высвобождением с таким преимуществом, что тофацитиниб постепенно высвобождается на протяжении сравнительно длительного периода с постоянной концентрацией, что приводит к незначительным флуктуациям в крови пациента.
Однако неожиданно было обнаружено, что биодоступность тофацитиниба уменьшается с увеличением периода высвобождения, таким образом, требуя увеличения количества требуемого тофацитиниба в дозированной форме с замедленным высвобождением для обеспечения эффективного уровня в крови пациента.
Кроме того, ФК профиль таблеток с мгновенным высвобождением принимаемых дважды в день, содержит периоды на протяжении 24 часов ниже IC50 для гетеродимера JAK1/3 (“отдых от лекарства”), вследствие комбинации всех адсорбированных лекарств и соотношения максимальной концентрации в плазме крови, Cmax,ss, к минимальной концентрации в плазме крови, Cmin,ss, на протяжении интервалов дозирования. Тофацитиниб является селективным ингибитором семейства Янус киназ (JAK), которые обладают высоким уровнем селективности, по сравнению с другими киназами в человеческом геноме. В исследованиях киназ, тофацитиниб ингибирует JAK1, JAK2, JAK3, и в меньшей мере тирозинкиназу (TyK2). В наборах клеток, где JAK киназы сигнализируют по парам, тофацитиниб преимущественно ингибирует цитокины, которые сигнализируют через JAK3 и/или JAK1, включая интерлейкин (IL)-2, -4, -6, -7, -9, -15, -21, и интерфероны типа I и II. Эти цитокины являются про-воспалительными и неотъемлемыми для функционирования лимфоцитов. Ингибирование их сигнализирования может привести к модулированию многочисленных аспектов иммунного ответа. Ингибирование сигнализирования через JAK3 и/или JAK1 может нарушить иммунную систему организма.
Неожиданно обнаружили, что период отдыха от лекарства тофацитиниба относительно IC50 для сигнализирования JAK1/3 на протяжении 24 часов увеличивается с увеличением продолжительности высвобождения из дозированной формы с замедленным высвобождением. По сути дозированные формы с замедленным высвобождением, как описано в уровне техники, содержащие тофацитиниб, не обеспечивают период отдыха от лекарства сопоставимый с ФК профилем таблеток с мгновенным высвобождением, принимаемых дважды в день, вследствие пониженной концентрации в плазме крови тофацитиниба, обеспеченной дозированными формами с замедленным высвобождением, как описано в уровне техники. Соответственно, неожиданно обнаружили, что для обеспечения оптимального ФК профиля (то есть, оптимальной экспозиции и оптимального соотношения Cmax,ss/Cmin,ss, при этом избегая повышенного уровня максимальной концентрации в плазме крови) для приема тофацитиниба один раз в день, предпочтительными являются дозированные формы с более коротким периодом замедленного высвобождения. Неожиданно так же обнаружили, что для минимизации общей дозы тофацитиниба, введенной пациентам, и в то же время для обеспечения эффективного уровня в крови пациентов, предпочтительными являются дозированные формы с более коротким периодом замедленного высвобождения.
Краткое изложение сущности изобретения
Данное изобретение относится к оральным композициям тофацитиниба с замедленным высвобождением для лечения противовоспалительных и аутоиммунных заболеваний, и особенно ревматоидного артрита (RA). Замедленное высвобождение тофацитиниба можно осуществлять известными в отрасли способами, включая, но не ограничиваясь применением осмотических дозированных форм, дозированных форм с матрицей, дозированных форм с многочисленными частицами, дозированных форм герметичных в желудке, и пульсирующих дозированных форм.
Данное изобретение относится к фармацевтической дозированной форме с приемом один раз в день, которая содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где указанная дозированная форма является дозированной формой с замедленным высвобождением, и которая при введении субъекту имеет среднюю площадь под кривой концентрации в плазме против время после введения, от приблизительно 27 нг-час/мл на мг введенного тофацитиниба до приблизительно 42 нг-час/мл на мг введенного тофацитиниба, и среднее соотношение Cmax - Cmin в плазме от приблизительно 10 до приблизительно 100, желательно среднее соотношение Cmax - Cmin в плазме от приблизительно 20 до приблизительно 40, и более желательно от приблизительно 20 до приблизительно 30. Фармацевтическая дозированная форма может содержать от приблизительно 10 мг до приблизительно 12 мг тофацитиниба, желательно 11 мг тофацитиниба. В другом варианте осуществления, фармацевтическая дозированная форма может содержать от приблизительно 20 до приблизительно 24 мг тофацитиниба, желательно 22 мг тофацитиниба. Фармацевтическая дозированная форма изобретения также обеспечивает у субъекта единое пролонгированное время с концентрацией выше приблизительно 17 нг/мл в диапазоне от приблизительно 6 до приблизительно 15 часов, и единое пролонгированное время с концентрацией ниже приблизительно 17 нг/мл в диапазоне от приблизительно 9 до приблизительно 18 часов в интервале дозирования в 24 часа. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией выше приблизительно 17 нг/мл в диапазоне от приблизительно 6 до приблизительно 9 часов. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией ниже приблизительно 17 нг/мл в диапазоне от приблизительно 15 до приблизительно 18 часов. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией выше приблизительно 17 нг/мл в диапазоне от приблизительно 11 до приблизительно 15 часов. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией ниже приблизительно 17 нг/мл в диапазоне от приблизительно 9 до приблизительно 13 часов. В другом варианте осуществления, фармацевтическая дозированная форма данного изобретения обеспечивает пациенту значение максимальной концентрации в плазме (Cmax) от приблизительно 3 нг/мл на мг до приблизительно 6 нг/мл на мг введенного тофацитиниба.
Данное изобретение так же обеспечивает фармацевтическую дозированную форму для введения один раз в день, которая содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где указанная дозированная форма является дозированной формой с замедленным высвобождением, и которая при введении субъекту имеет среднюю площадь под кривой концентрации в плазме против время после введения от приблизительно 17 нг-час/мл на мг введенного тофацитиниба до приблизительно 42 нг-час/мл на мг введенного тофацитиниба, и среднее соотношение Cmax - Cmin в плазме от приблизительно 10 до приблизительно 100, желательно среднее соотношение Cmax - Cmin в плазме от приблизительно 20 дo 40, и более желательно приблизительно 20 - 30. Фармацевтическая дозированная форма может содержать от приблизительно 10 мг до приблизительно 12 мг тофацитиниба, желательно 11 мг тофацитиниба. В другом варианте осуществления, фармацевтическая дозированная форма может содержать от приблизительно 20 до приблизительно 24 мг тофацитиниба, желательно 22 мг тофацитиниба. Фармацевтическая дозированная форма изобретения так же обеспечивает единое пролонгированное время с концентрацией выше приблизительно 17 нг/мл в диапазоне от приблизительно 6 до приблизительно 15 часов, и единое пролонгированное время с концентрацией ниже приблизительно 17 нг/мл в диапазоне от приблизительно 9 до приблизительно 18 часов на протяжении интервала дозирования в 24 часа. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией выше приблизительно 17 нг/мл в диапазоне от приблизительно 6 до приблизительно 9 часов. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией ниже приблизительно 17 нг/мл в диапазоне от приблизительно 15 до приблизительно 18 часов. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией выше приблизительно 17 нг/мл в диапазоне от приблизительно 11 до приблизительно 15 часов. В другом варианте осуществления изобретения, пациент имеет единое пролонгированное время с концентрацией ниже приблизительно 17 нг/мл в диапазоне от приблизительно 9 до приблизительно 13 часов. В другом варианте осуществления, фармацевтическая дозированная форма данного изобретения может обеспечивать пациенту значение максимальной концентрации в плазме (Cmax) от приблизительно 3 нг/мл на мг до приблизительно 6 нг/мл на мг введенного тофацитиниба.
Данное изобретение так же обеспечивает фармацевтическую дозированную форму для введения один раз в день, которая содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где указанная дозированная форма является дозированной формой с замедленным высвобождением, и при введении субъекту орально имеет значение постоянной минимальной концентрации (Cmin) в плазме менее чем приблизительно 0,3 нг/мл на мг введенного тофацитиниба.
В другом варианте осуществления, данное изобретение обеспечивает фармацевтическую дозированную форму для введения один раз в день, которая содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где указанная дозированная форма является дозированной формой с замедленным высвобождением, и при введении субъекту орально имеет среднее соотношение после еды/натощак площади под кривой концентрации в плазме против времени на уровне от приблизительно 0,7 до приблизительно 1,4, и среднее соотношение после еды/натощак максимальной плазменной концентрации (Cmax) на уровне от приблизительно 0,7 до приблизительно 1,4, желательно от приблизительно 0,8 до приблизительно 1,25.
В другом варианте осуществления, данное изобретение обеспечивает фармацевтическую дозированную форму, которая содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где указанная дозированная форма является дозированной формой с замедленным высвобождением, и при добавлении в исследуемую среду, содержащую 900 мл 0,05M pH 6,8 буфера фосфата калия при 37°C в стандартной USP мешалке с вращающимися лопастями, со скоростью вращения лопастей 50 об/мин, растворяется не более чем 30% лекарственного средства в час, и не менее чем 35% и не более чем 75% лекарственного средства за 2,5 часа, и не менее чем 75% тофацитиниба за 5 часов; желательно не более чем 25% лекарственного средства в час, и не менее чем 40% и не более чем 70% лекарственного средства за 2,5 часа.
В другом варианте осуществления, данное изобретение обеспечивает фармацевтическую дозированную форму, которая содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где дозированная форма является дозированной формой с замедленным высвобождением и при введении субъекту орально обеспечивает AUC в диапазоне от 80% дo 125% от AUC количества тофацитиниба введенного в композиции с мгновенным высвобождением дважды в день, и где дозированная форма с замедленным высвобождением обеспечивает среднее соотношение Cmax - Cmin в плазме от приблизительно 10 до приблизительно 100, желательно AUC находится в диапазоне от 90% дo 110% и среднее соотношение Cmax - Cmin в плазме составляет от приблизительно 20 до приблизительно 40 и более желательно от приблизительно 20 до приблизительно 30.
В другом варианте осуществления, фармацевтическая дозированная форма данного изобретения так же может обеспечивать среднее Cmax в плазме в диапазоне от 70% дo 125% от среднего значения Cmax в плазме тофацитиниба, введенного в композиции в мгновенным высвобождением дважды в день в стационарном состоянии, при оральном введении пациенту. В другом варианте осуществления, фармацевтическая дозированная форма данного изобретения обеспечивает отдых от лекарства в диапазоне от 80% дo 110% отдыха от лекарства тофацитиниба, введенного в композиции в мгновенным высвобождением дважды в день за период в 24 часа, при оральном введении пациенту. Фармацевтическая дозированная форма данного изобретения может содержать от приблизительно 10 мг до приблизительно 12 мг тофацитиниба, а эквивалентное количество тофацитиниба, введенное в композиции в мгновенным высвобождением дважды в день составляет 5 мг, желательно фармацевтическая дозированная форма содержит 11 мг тофацитиниба. Фармацевтическая дозированная форма данного изобретения может содержать от приблизительно 20 мг до приблизительно 24 мг тофацитиниба, а эквивалентное количество тофацитиниба, введенное в композиции в мгновенным высвобождением дважды в день составляет 10 мг, желательно фармацевтическая дозированная форма может содержать 22 мг тофацитиниба. В другом варианте осуществления, фармацевтическая дозированная форма данного изобретения обеспечивает отдых от лекарства от приблизительно 15 до приблизительно 18 часов на протяжении 24-го периода. В другом варианте осуществления, фармацевтическая дозированная форма данного изобретения обеспечивает отдых от лекарства от приблизительно 9 до приблизительно 13 часов на протяжении 24-го периода.
Данное изобретение так же обеспечивает фармацевтические композиции для получения указанных композиций с замедленным высвобождением. В одном варианте осуществления, фармацевтическая дозированная форма с замедленным высвобождением данного изобретения содержит ядро, которое содержит тофацитиниб или его фармацевтически приемлемую соль, и полупроницаемое мембранное покрытие, где указанное покрытие, по сути, состоит из нерастворимого в воде полимера. Дозированная форма с замедленным высвобождением данного изобретения может доставлять тофацитиниб в основном благодаря осмотическому давлению. В другом варианте осуществления данного изобретения, дозированная форма с замедленным высвобождением данного изобретения может содержать систему доставки, выбранную из группы, состоящей из следующих: система экструдируемого ядра, система разбухающего ядра, или ассиметричная мембранная технология.
В другом варианте осуществления, нерастворимый в воде полимер содержит производную целлюлозы, желательно ацетат целлюлозы. В другом варианте осуществления данного изобретения, покрытие дополнительно содержит растворимый в воде полимер, имеющий среднюю молекулярную массу 2000-100,000 дальтон. В другом варианте осуществления данного изобретения, растворимый в воде полимер выбирают из группы, состоящей из следующих: растворимые в воде производные целлюлозы, акация, декстрин, гуаровая смола, мальтодекстрин, альгинат натрия, крахмал, полиакрилаты и поливиниловые спирты. В другом варианте осуществления данного изобретения, растворимые в воде производные целлюлозы включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу или гидроксиэтилцеллюлозу.
В другом варианте осуществления данного изобретения, ядро содержит сахар, желательно, сорбитол.
В другом варианте осуществления, фармацевтическая дозированная форма с замедленным высвобождением данного изобретения, содержит тофацитиниб или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, где указанный тофацитиниб погружен в матрицу, высвобождающую тофацитиниб путем диффузии. В одном варианте осуществления, часть внешней поверхности матрицы покрыта непроницаемым покрытием, а оставшаяся часть внешней поверхности остается без покрытия.
В другом варианте осуществления данного изобретения, дозированная форма имеет форму таблетки, а поверхность без покрытия представляет собой отверстие через непроницаемое покрытие.
В другом варианте осуществления данного изобретения, дозированная форма имеет форму таблетки, и поверхность без покрытия представляет собой проход, проникающий сквозь всю таблетку.
В другом варианте осуществления данного изобретения, дозированная форма имеет форму таблетки, а поверхность без покрытия представляет собой один или больше прорезов через указанное непроницаемое покрытие, или удаленных от туда одну или больше полос.
В другом варианте осуществления данного изобретения, матрица дозированной формы остается по сути интактной на протяжении высвобождения тофацитиниба.
В другом варианте осуществления данного изобретения, фармацевтически приемлемый носитель, содержащий материал матрицы, выбирают из группы, состоящей из следующих: воски, спирта с длинной цепью, сложные эфиры жирных кислот, гликолизированные сложные эфиры жирных кислот, фосфоглицериды, эфиры полиоксиэтилен алкила, карбоновые кислоты с длинной цепью, сахарные спирты, и их смеси.
В другом варианте осуществления данного изобретения, внешняя поверхность указанной матрицы покрыта кишечным покрытием. Матрица может быть сформирована в виде расплавленного-замороженного ядра.
В другом варианте осуществления данного изобретения, матрица дозированной формы содержит гидроксипропилметилцеллюлозу.
В другом варианте осуществления данного изобретения, тофацитиниб погружен в матрицу, высвобождающую тофацитиниб путем эрозии.
В другом варианте осуществления данного изобретения, матрица дозированной формы содержит гидроксипропилметилцеллюлозу.
В другом варианте осуществления данного изобретения, матрица дозированной формы содержит поли(этиленоксид).
В другом варианте осуществления данного изобретения, матрица дозированной формы содержит полиакриловую кислоту.
В другом варианте осуществления данного изобретения, резервуар тофацитиниба заключен в мембрану, ограничивающую скорость высвобождения тофацитиниба путем диффузии.
В другом варианте осуществления, фармацевтическая дозированная форма с замедленным высвобождением данного изобретения обеспечивает дозированную форму в виде таблетки, покрытой мембраной.
В другом варианте осуществления, фармацевтическая дозированная форма с замедленным высвобождением данного изобретения обеспечивает дозированную форму в виде многочисленных частиц, содержащая частицы, независимо покрытые мембраной, ограничивающей скорость высвобождения тофацитиниба путем диффузии.
Данное изобретение также относится к способу лечения иммунологических расстройств у субъекта, включающему введение субъекту, нуждающемуся в этом, фармацевтической дозированной формы данного изобретения с замедленным высвобождением в количестве, эффективном для лечения таких расстройств. Иммунологическое расстройство выбирают из группы, состоящей из следующих: трансплантация органов, ксенотрансплантация, волчанка, рассеянный склероз, ревматоидный артрит, псориаз, диабет типа I и диабетические осложнения, рак, астма, атопический дерматит, аутоиммунные расстройства щитовидной железы, язвенный колит, анкилозирующий спондилоартрит, ювенильный идиопатический артрит болезнь Крона, псориатический артрит, болезнь Альцгеймера, и лейкемия, желательно, иммунологическое расстройство выбирают из группы, состоящей из следующих: трансплантация органов, ревматоидный артрит, псориаз, псориатический артрит, язвенный колит, анкилозирующий спондилоартрит, ювенильный идиопатический артрит и болезнь Крона. В другом варианте осуществления данного изобретения, способ дополнительно включает один или больше дополнительных агентов, модулирующих иммунную систему млекопитающих, или противовоспалительных агентов. Дополнительный агент может быть выбран из группы, состоящей из следующих: небиологические DMARD, метотрексат, глюкокортикоид, агонист рецептора глюкокортикоида, лефлуномид, нестероидные противовоспалительные лекарственные средства, 6-мекраптопурин, азатиоприн, сульфазалазин, и 5-аминосалицилаты, желательно, дополнительный агент выбирают из группы, состоящей из небиологических DMARD и агониста рецептора глюкокортикоида, более желательно, дополнительный агент означает метотрексат.
Данное изобретение также относится к способу лечения атеросклероза у субъекта, включающему введение субъекту, нуждающемуся в этом, фармацевтической формы данного изобретения с замедленным высвобождением, в количестве, эффективном для лечения атеросклероза. В другом варианте осуществления данного изобретения, способ также включает введение ингибитора HMG-CoA редуктазы, желательно, ингибитором HMG-CoA редуктазы является аторвастатин или его фармацевтически приемлемая соль.
Термин “тофацитиниб” следует понимать, пока не указано обратное, как включающий любую фармацевтически приемлемую форму и соли соединения. Тофацитиниб может находиться в кристаллической или аморфной формах. Данное изобретение относится к дозированной форме тофацитиниба с замедленным высвобождением для обеспечение введения один раз в день и определенных ФК характеристик с целью: 1) минимизировать количество тофацитиниба в дозированной форме с замедленным высвобождением, необходимого для получения эффективного уровня в крови пациента, 2) оптимизации величины связывания тофацитиниба с JAK 1/3 гетеродимерами (определено с помощью IC50, что происходит у людей с концентрацией лекарства в плазме крови на уровне приблизительно 17 нг/мл или 56 нM, как описано в Meyer DM, Jesson MI, Xiong L, et al. Anti-inflammatory activity and neutrophil reduction mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis J. of Inflammation 2010;7:41, включенный в данный документ в качестве ссылки), которая регулирует иммунный ответ, для обеспечения желаемого уровня эффективности (на основании средней Cave) на протяжении интервала дозирования в 24 часа. Дозированная форма с замедленным высвобождением данного изобретения обеспечивает вышеуказанные желаемые ФК характеристики, и особенно свойства для дозированной формы с приемом один раз в день. Желательно, дозированная форма изобретения с замедленным высвобождением значительно не изменяет ФК профиль тофацитиниба при введении после еды (то есть не имеет пищевого эффекта), так как это минимизирует отклонение от оптимального охвата JAK 1/3 гетеродимеров.
Под термином “замедленное высвобождение” понимают, что тофацитиниб высвобождается из оральной дозированной формы с меньшей скоростью, чем мгновенное высвобождение. Оральная дозированная форма включает таблетки, капсулы, множественные частицы или гранулы. “Замедленное высвобождение” включает оральную композицию, состоящую из одного или комбинации следующих:
а) только компонент с контролируемым высвобождением;
b) компонент с контролируемым или отсроченным высвобождением;
c) компонент с отсроченным и мгновенным высвобождением.
Под термином "фармацевтически приемлемая форма" понимают любую фармацевтически приемлемую форму, включая, сольваты, гидраты, изоморфы, полиморфы, co-кристаллы, псевдоморфы, нейтральные формы, формы кислотно-аддитивных солей, и пролекарства. Фармацевтически приемлемые кислотно-аддитивные соли тофацитиниба получают обычным путем обработкой раствора или суспензии свободного основания одним или двумя химическими эквивалентами фармацевтически приемлемой кислоты. Для выделения солей использовали стандартные техники концентрирования и перекристаллизации. Примерами приемлемых кислот являются уксусная, молочная, янтарная, малеиновая, виннокаменная, лимонная, глюконовая, аскорбиновая, мезиловая, тозиловая, бензойная, коричная, фумаровая, серная, фосфорная, соляная, бромистоводородная, йодистоводородная, сульфаминовая, сульфоновая, такая как метансульфоновая, бензолсульфоновая и подобные кислоты. Некоторые предпочтительные формы тофацитиниба включают свободное основание и цитрат тофацитиниба.
Термины “субъект”, “пациент” и “индивидуум” используются взаимозаменяемо для обозначения позвоночных, желательно, млекопитающих, более желательно человека.
“Твердая оральная дозированная форма” данного изобретения означает фармацевтически приемлемую твердую оральную дозированную форму, что означает, что дозированная форма является безопасной для введения людям, и все наполнители в дозированной форме являются фармацевтически приемлемыми, другими словами, безопасными для приема людьми.
Термин “натощак”, как использовано в данном документе, означает следующее: состояние дозирования, которое наступает после ночи без приема пищи (где принимается 0 калорий) на протяжении как минимум 10 часов. Пациентам дают дозированную форму с 240 мл воды. Есть не разрешают на протяжении, как минимум, 4 часов после приема дозы. В случае необходимости можно пить воду, кроме периода в 1 час до и после приема лекарства.
Термин “после еды” как использовано в данном документе, означает следующее: состояние дозирования, которое наступает после ночи без приема пищи (где принимается 0 калорий) на протяжении как минимум 10 часов, после чего пациенту назначают высококалорийную пищу за 30 мин. до приема лекарственного средства. Пациент должен съесть эту еду за 30 мин или меньше; однако, лекарство должно быть введено через 30 мин. после начала приема пищи. Лекарство принимают с 240 мл воды. Есть не разрешают на протяжении, как минимум, 4 часов после приема дозы. В случае необходимости можно пить воду, кроме периода в 1 час до и после приема лекарства. Еда с высоким содержанием жира (приблизительно 50% всех калорий пищи происходят от жира) и высококалорийная еда (приблизительно 800-1000 калорий) должна использоваться в качестве тест еды, в условиях приема лекарства после еды. В этой тест пище приблизительно 150, 250 и 500-600 калорий происходят от протеина, углевода и жира, соответственно. Примером тест пищи являются два яйца, поджаренных на масле, два ломтика бекона, два ломтика хлеба с маслом, 4 унции картофельных оладий и 8 унций цельного молока.
Расчет средней площади под кривой концентрации в сыворотке против времени (AUC) является известной процедурой в фармацевтике, и описан, например, в Welling, "Pharmacokinetics Processes and Mathematics," ACS Monograph 185 (1986). AUC как использовано в данном документе, включает площадь под кривой концентрация-время, начиная с нулевого времени и экстраполируя на бесконечность при введении разовой дозы, или площадь под кривой концентрация-время начиная с нулевого времени и до времени окончания интервала дозирования при введении устойчивой многократной дозы. Кроме того, расчеты для Cmax, Cmin,ss, Tmax, и выделение периода полураспада (t ), так же известны специалистам в отрасли, и описаны, например, в Shargel, Wu-Pong, and Yu, Applied Biopharmaceutics and Pharmacokinetics (2005). Для определения среднего соотношения после еды/натощак, сначала рассчитывали индивидуальное соотношение средней площади под кривой концентрация-время для тофацитиниба (то есть AUC0-inf) в состоянии после еды, к средней площади под кривой концентрация-время для тофацитиниба (например, AUC0-inf) в состоянии натощак, а потом соответствующие индивидуальные соотношения усредняли. Таким образом, определяют среднее для соотношений каждого индивидуума.
), так же известны специалистам в отрасли, и описаны, например, в Shargel, Wu-Pong, and Yu, Applied Biopharmaceutics and Pharmacokinetics (2005). Для определения среднего соотношения после еды/натощак, сначала рассчитывали индивидуальное соотношение средней площади под кривой концентрация-время для тофацитиниба (то есть AUC0-inf) в состоянии после еды, к средней площади под кривой концентрация-время для тофацитиниба (например, AUC0-inf) в состоянии натощак, а потом соответствующие индивидуальные соотношения усредняли. Таким образом, определяют среднее для соотношений каждого индивидуума.
“Тест на растворимость 1” относится к следующему тестированию дозированных форм тофацитиниба. Тест на растворимость проводили в стандартной USP мешалке с вращающимися лопастями, как описано в Фармакопее США (USP), Тест на растворимость, раздел 711, аппарат 2. Лопасти вращались со скоростью 50 об/мин, и дозированную форму добавляли в 900 мл 0,05M pH 6,8 буфера фосфата калия при 37°C. Через соответствующее время после начала теста (например, введения дозированной формы в аппарат), отфильтрованные аликвоты (обычно 1,5 мл) из тест-среды анализировали на тофацитиниб с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Результаты растворимости представлены как процент от общей дозы тестированного тофацитиниба, растворенный за время.
детальное описание изобретения
Данное изобретение относится к оральным композициям тофацитиниба с замедленным высвобождением для лечения противовоспалительных и аутоиммунных заболеваний, и особенно, ревматоидного артрита (RA). Замедленное высвобождение тофацитиниба может осуществляться известными в отрасли способами, включая, но не ограничиваясь применением осмотических дозированных форм, дозированных форм с матрицей, дозированных форм с многочисленными частицами, дозированных форм герметичных в желудке, и пульсирующих дозированных форм.
Матричные системы с замедленным высвобождением (Таблетки)
В одном варианте осуществления, тофацитиниб включен в таблетку с эродируемой или неэродируемой полимерной матрицей. Под эродируемой матрицей понимают эродируемую в воде или разбухающую в воде, или растворимую в воде матрицу, в смысле эродируемую, разбухающую или растворимую в чистой воде, или нуждающуюся в присутствии кислоты или основания для ионизации полимерной матрицы, в количестве достаточном для вызывания эрозии или растворения. При контакте с водной средой, эродируемая полимерная матрица впитывает воду и образует разбухший в воде гель или "матрицу", которая захватывает тофацитиниб. Разбухшая в воде гелевая матрица постепенно эродирует, разбухает, дезинтегрирует, диспергирует или растворяется в использованной среде, таким образом, контролируя высвобождение тофацитиниба в использованную среду. Примеры таких дозированных форм широко известны в отрасли. Смотреть, например, Remington: The Science and Practice of Pharmacy, 20th Edition, 2000.
Ключевым ингредиентом разбухающей в воде матрицы является разбухающий в воде, эродируемый или растворимый полимер, который можно в целом описать как осмополимер, гидрогель или разбухающий в воде полимер. Такие полимеры могут быть линейными, разветвленными или поперечно сшитыми. Они могут быть гомополимерами или coполимерами. Примеры полимеров включают присутствующие в природе полисахариды, такие как хитин, хитозан, декстран и пуллулан; агаровая смола, гуммиарабик, камедь карайи, камедь плодов рожкового дерева, трагакантовая камедь, каррагенаны, камедь гхатти, гуаровая смола, ксантановая смола и склероглюкан; крахмалы, такие как декстрин и мальтодекстрин; гидрофильные коллоиды, такие как пектин; альгинаты, такие как альгинат аммония, альгинат натрия, калия или кальция, альгинат пропиленгликоля; желатин; коллаген; и целлюлозы. Под “целлюлозами” понимают полимер целлюлозы, модифицированный взаимодействием как минимум части гидроксильных групп на повторяющихся единицах сахарида с соединением для образования эстер-связанного или этер-связанного заместителя. Например, целлюлозная этилцеллюлоза имеет этер-связанный этиловый заместитель, присоединенный к повторяющейся единице сахарида, в то время как целлюлозный целлюлозы ацетат имеет эстер-связанный ацетатный заместитель.
Целлюлозы для эродируемой матрицы включают растворимые в воде и эродируемые в воде целлюлозы, такие как этилцеллюлоза (ЭЦ), метилэтилцеллюлоза (MЭЦ), карбоксиметилцеллюлоза (КMЦ), карбоксиметилэтилцеллюлоза (КМЭЦ), гидроксиэтилцеллюлоза (ГЭЦ), гидроксипропилцеллюлоза (ГПЦ), целлюлозы ацетат фталат (ЦАФ), ацетат тримеллитат целлюлозы (АЦT), гидроксипропилметилцеллюлоза (ГПМЦ), гидроксипропилметилцеллюлозы фталат (ГПМЦФ), гидроксипропилметил целлюлозы ацетат сукцинат (ГПМЦAС), гидроксипропилметил целлюлозы ацетат тримеллитат (ГПМЦAT) и этилгидроксиэтилцеллюлоза (ЭГЭЦ).
Особенно предпочтительный класс таких целлюлоз включает ГПМЦ с разными степенями низкой вязкости (ММ менее чем или равно 50,000 дальтон) и высокой вязкости (ММ более чем 50,000 дальтон). Коммерчески доступные полимеры ГПМЦ с низкой вязкостью включают Dow METHOCEL™ серии E3, E5, E15LV, E50LV и K100LV, в то время как ГПМЦ с высокой вязкостью включают E4MCR, E10MCR, K4M, K15M и K100M; особенно предпочтительными в этой группе являются METHOCEL™ серии K. Другие коммерчески доступные виды ГПМЦ включают Shin Etsu METOLOSE™ серии 90SH. В одном варианте осуществления, ГПМЦ имеет низкую вязкость, что означает, что вязкость 2% (м./о.) раствора ГПМЦ в воде равняется менее чем приблизительно 120 сП. Предпочтительной ГПМЦ является ГПМЦ с вязкостью 2% (м./о.) раствора ГПМЦ в воде в диапазоне от 80 дo 120 сП (например, METHOCEL™ K100LV).
Другие материалы, приемлемые в качестве эродируемого материала матрицы включают, но не ограничиваются следующими: пуллулан, поливинилпирролидон, поливиниловый спирт, поливинилацетат, сложные эфиры жирных кислот глицерина, полиакриламид, полиакриловую кислоту, coполимеры этакриловой кислоты или метакриловой кислоты (EUDRAGIT®, Rohm America, Inc., Piscataway, New Jersey) и производные другие акриловых кислот, такие как гомополимеры и coполимеры бутилметакрилата, метилметакрилата, этилметакрилата, этилакрилата, (2-диметиламиноэтил)метакрилата и хлорида (триметиламиноэтил) метакрилата.
Эродируемый материал матричного полимера также может содержать добавки и наполнители, известные в фармацевтике, включая осмополимеры, осмагены, агенты, улучшающие или замедляющие растворимость и наполнители, стимулирующие стабильность или обрабатываемость дозированной формы.
В неэродируемой матричной системе, тофацитиниб распределен в инертной матрице. Лекарство высвобождается путем диффузии через инертную матрицу. Примеры материалов, приемлемых для инертной матрицы, включают нерастворимый пластик, например, coполимеры этилена и винилацетата, coполимеры метилакрилата-метилметакрилата, поливинилхлорид и полиэтилен; гидрофильные полимеры, такие как этилцеллюлоза, целлюлозы ацетат и поперечно сшитый поливинилпирролидон (так же известный как кросповидон); и жирные соединения, такие как карнаубский воск, микрокристаллический воск и триглицериды. Такие дозированные формы описаны в: The Science and Practice of Pharmacy, 20th edition (2000).
Матричные системы с замедленным высвобождением (Многочисленные частички)
В другом варианте осуществления, матрица из многочисленных частиц, содержит множество частиц, содержащих тофацитиниб, где каждая частица, содержащая смесь тофацитиниба с одним или более наполнителями, отобрана для формирования матрицы, способной ограничивать скорость растворения тофацитиниба в водной среде. Материалы матрицы, приемлемые для этого варианта осуществления обычно являются нерастворимыми в воде, такие как воски, целлюлоза, или другие нерастворимые в воде полимеры. В случае необходимости, материалы матрицы необязательно могут быть формулированы с растворимыми в воде материалами, которые могут быть использованы в качестве связующих или агентов модифицирующих проницаемость. Материалы матрицы, приемлемые для изготовления этих дозированных форм включают микрокристаллическую целлюлозу, такую как Avicel (торговая марка FMC Corp., Philadelphia, Pa.), включая классы микрокристаллической целлюлозы, куда добавляют связывающие вещества, такие как гидроксипропилметилцеллюлоза, воски, такие как парафин, модифицированные растительные масла, карнаубский воск, гидрогенизированное касторовое масло, пчелиный воск, и т.д., а так же искусственные полимеры, такие как поли(винилхлорид), поли(винилацетат), coполимеры винилацетата и этилена, полистирол, и т.д. Растворимые в воде связующие или агенты, модифицирующие проницаемость, которые необязательно включают в матрицу, включают растворимые в воде полимеры, такие как гидроксипропилцеллюлоза (ГПЦ), гидроксипропилметилцеллюлоза (ГПМЦ), метилцеллюлоза, поли(N-винил-2-пирролидинон) (ПВП), поли(этиленоксид) (ПЭO), поли(виниловый спирт) (ПВС), ксантановая смола, каррагенин, и другие такие природные и синтетические материалы. Кроме того, материалы, функционирующие как агенты, модифицирующие высвобождение, включают растворимые в воде материалы, такие как сахара или соли. Желаемыми растворимыми в воде материалами являются лактоза, сахароза, глюкоза, и маннит, а так же ГПЦ, ГПМЦ и ПВП.
Способ получения матрицы из множественных частиц включает процесс экструзии/ окатывания. Для этого процесса, тофацитиниб во влажном состоянии соединяют со связующим, экструдируют через перфорированную пластину или матрицу, и помещают на вращающийся диск. Экструдат идеально распадается на частицы, которые закругляют в сферы, сфероиды или закругленные бруски на вращающейся пластине. Другой процесс и композиция для этого способа включает использование воды для смачивания смеси, содержащей приблизительно 20-75% микрокристаллической целлюлозы, смешанной с, соответственно, приблизительно 80-25% тофацитиниба.
Способ получения матрицы из множественных частиц включает получение восковых гранул. В этом процессе, желательное количество тофацитиниба перемешивают с жидким воском с образованием гомогенной смеси, охлаждают и затем пропускают через сито с образованием гранул. Желательными матричными материалами являются восковые вещества. Некоторые восковые вещества включают гидрогенизированное касторовое масло и карнаубский воск и стеариловый спирт.
Другой способ получения матрицы из множественных частиц включает использование органического растворителя для улучшения перемешивания тофацитиниба с материалом матрицы. Эта техника может использоваться, когда желательно использовать материал матрицы с неподходяще высокой температурой плавления, который в случае использования материала в расплавленном состоянии, вызовет разложение лекарственного средства или материала матрицы, или приведет к неприемлемой вязкости расплава, таким образом, предотвращая перемешивание тофацитиниба с материалом матрицы. Тофацитиниб и материал матрицы могут быть объединены с незначительным количеством растворителя с образованием пасты, и затем пропущены через сито с образованием гранул, из которых затем удаляют растворитель. Альтернативно, тофацитиниб и материал матрицы могут быть объединены с достаточным количеством растворителя для завершения растворения материала матрицы, и полученный раствор (который может содержать твердые частицы лекарства) подвергали распылительной сушке с образованием дозированной формы из частиц. Эта техника является предпочтительной, когда материалом матрицы является высокомолекулярный синтетический полимер, такой как эфир целлюлозы или сложный эфир целлюлозы. Растворители, которые обычно используют для этого процесса, включают ацетон, этанол, изопропанол, этилацетат, и смеси двух или более.
В одном варианте осуществления, множественные частицы матрицы образованы распылением расплава и затвердеванием. Расплавленное-затвердевшее ядро содержит материал матрицы. Материал матрицы имеет две функции. Первая, материал матрицы позволяет образовываться относительно гладким круглым ядрам, восприимчивым к нанесению покрытия. Вторая, материал матрицы связывает необязательные наполнители и/или лекарственные вещества, которые могут быть включены в ядро. Материал матрицы имеет следующие физические свойства: достаточно низкую вязкость в расплавленном состоянии для образования множественных частиц, как описано ниже; и быстрого застывания в твердые вещества при охлаждении ниже их точки плавления. Для множественных частиц, включающих лекарство в ядро, матрица желательно имеет температуру плавления ниже, чем температура плавления или температура разложения лекарственного средства, и по сути не растворяет лекарственное вещество.
Расплавленное-затвердевшее ядро состоит, по сути, из непрерывной фазы материала матрицы и необязательно других наполнителей, с необязательными инкапсулированными в него частицами лекарственного вещества и необязательными частицами разбухающего агента. Вследствие этого должно присутствовать достаточное количество материала матрицы для образования гладких ядер, достаточно больших для нанесения покрытия. В случае ядер, содержащих твердые частицы, такие как лекарственное вещество или разбухающий агент, ядро должно содержать достаточное количество материала матрицы для инкапсулирования лекарственного вещества и разбухающего агента для образования относительно гладких и сферических ядер, на которые легче наносить покрытие с помощью стандартного распыления, чем на ядра с нестандартной формой. Материал матрицы может присутствовать в ядре в количестве от как минимум приблизительно 30 мас.%, как минимум приблизительно 50 мас.%, как минимум приблизительно 70 мас.%, как минимум приблизительно 80 мас.%, как минимум приблизительно 90 мас.%, и до 100 мас.% от массы ядра без покрытия.
Для формирования маленьких гладких круглых ядер, материал матрицы должен быть способен к расплавлению и затем распылению. Материал матрицы или смесь материалов является твердой при 25°C. Однако материал матрицы плавится или способен к расплавлению с добавлением необязательного вспомогательного вещества, при температуре ниже чем 200°С, для пригодности использования в процессе плавления-отвердения, описанном ниже. Желательно, материал матрицы имеет температура плавления 50°C-150°C. Хотя термин "плавление" обычно относится к переходу кристаллического материала из кристаллического в жидкое состояние, что происходит при достижении температуры его плавления, и термин "расплавленный" обычно относится к такому кристаллическому материалу в жидком состоянии, как использовано в данном документе, эти термины использованы в более широком смысле. В случае "плавления", термин используется для нагревания любого материала или смеси материалов до достижения ими жидкого состояния, в том смысле что он может быть накачан или распылен подобно кристаллическому материалу в жидком состоянии. Подобно "расплавленный" относится к любому материалу или смеси материалов, находящихся в жидком состоянии.
Материал матрицы выбирают из группы, состоящей из следующих: воски, спирта с длинной цепью (Ci2 или большей), сложные эфиры жирных кислот, гликолизированные сложные эфиры жирных кислот, фосфоглицериды, эфиры полиоксиэтилен алкила, карбоновые кислоты с длинной цепью (Ci2 или большей), сахарные спирты, и их смеси. Примеры материала матрицы включают высокоочищенные формы восков, такие как карнаубский воск, белый и желтый пчелиный воск, церезиновый воск, микрокристаллический воск, и парафиновый воск; спирты с длинной цепью, такие как стеариловый спирт, цетиловый спирт и полиэтиленгликоль; сложные эфиры жирных кислот (также известные как жиры или глицериды), такие как изoпропилпальмитат, изoпропилмиристат, глицерил моноолеат, глицерил моностеарат, глицерил пальмитостеарат, смеси моно-, ди- и триалкилглицеридов, включая смеси глицерил моно-моно-, ди- и трибегената, глицерил тристеарата, глицерил трипальмитата, и гидрогенезированные растительные масла, включая гидрогенезированное хлопковое масло; гликолизированные сложные эфиры жирных кислот, такие как полиэтиленгликоль стеарат и полиэтиленгликоль дистеарат; эфиры полиоксиэтиленалкила; полиэтоксилированные производные касторового масла; карбоновые кислоты с длинной цепью, такие как стеариновая кислота; и сахарные спирты, такие как маннит и эритритол. Материал матрицы может содержать смеси материалов, такие как смеси любого из перечисленных.
Ядро также может содержать различные наполнители, присутствующие в ядре в количестве от 0 дo 40 мас.%, на основе массы ядра без покрытия. Одним из желательных наполнителей является усилитель растворения, используемый для увеличения скорости захвата воды ядром и как следствие расширение разбухающего агента. Усилитель растворения имеет другой материал, чем материал матрицы. Усилитель растворения может находиться в отдельной или одной фазе с материалом матрицы. Желательно, как минимум часть усилителя растворения разделена фазой с материалом матрицы. Во время попадания воды в ядро, усилитель растворения растворяется, оставляя каналы, позволяющие воде более быстро попадать в ядро. В целом, усилители растворения являются амфифильными соединениями, и как правило являются более гидрофильными, чем материалы матрицы. Примеры усилителей растворения включают: поверхностно-активные вещества, такие как полоксамеры, соли докузата, производные полиоксиэтилен касторового масла, полисорбаты, лаурилсульфат натрия, и моноэстеры сорбитана; сахара, такие как глюкоза, ксилитол, сорбитол и малтитол; соли, такие как хлорид натрия, хлорид калия, хлорид лития, хлорид кальция, хлорид магния, сульфат натрия, сульфат калия, карбонат натрия, сульфат магния и фосфат калия; и аминокислоты, такие как аланин и глицин; и их смеси. Одним усилителем растворения поверхностно-активного типа является полоксамбетар (коммерчески доступный как LUTROL или PLURONIC производства BASF Corp.).
Ядро также может содержать другие необязательные наполнители, такие как агенты, ингибирующие или задерживающие высвобождение лекарственного вещества из множественных частиц. Такие агенты, ингибирующие растворение обычно являются гидрофобными и включают диалкилфталаты, такие как дибутилфталат, и углеводородные воски, такие как микрокристаллический воск и парафиновый воск. Другой приемлемый класс наполнителей содержит материалы, которые можно использовать для корректировки вязкости расплавленного материала для формирования ядер. Такие наполнители для корректировки вязкости обычно составляют от 0 дo 25 мас.% ядра. Вязкость расплавленного материала является ключевой переменной при получении ядер с примерно одинаковым размером частиц. Например, при применении распылителя с вращающим диском, желательно чтобы вязкость расплавленной смеси была как минимум приблизительно 1 сП и менее чем приблизительно 10,000 cП, желательно как минимум 50 сП и менее чем приблизительно 1000 cП. Если расплавленная смесь имеет вязкость, не попадающую в эти диапазоны, можно добавить агент, регулирующий вязкость, для получения расплавленной смеси в указанном диапазоне вязкости. Примеры наполнителей понижающих вязкость включают стеариловый спирт, цетиловый спирт, низкомолекулярный полиэтиленгликоль (то есть менее чем приблизительно 1000 дальтон), изoпропиловый спирт и вода. Примеры наполнителей повышающих вязкость включают микрокристаллический воск, парафиновый воск, искусственный воск, высокомолекулярные полиэтиленгликоли (то есть более чем приблизительно 5000 дальтон), этилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, метилцеллюлозу, диоксид кремния, микрокристаллическую целлюлозу, силикат магния, сахара и соли.
Для тех вариантов осуществления, где лекарственное вещество содержится в ядре, можно добавлять другие наполнители для корректировки характеристик высвобождения лекарственного средства из ядер. Например, основа или кислота могут быть включены в композицию с целью модифицирования скорости, при которой лекарственное вещество высвобождается в водную среду. Примеры кислот или основ, которые могут быть включены в композицию, включают следующие: лимонная кислота, адипиновая кислота, яблочная кислота, фумаровая кислота, янтарная кислота, виннокаменная кислота, ди- и трехосновный фосфат натрия, ди- и трехосновный фосфат кальция, моно-, ди-, и триэтанолaмин, бикарбонат натрия и дигидроцитрат натрия. Такие наполнители могут составлять от 0 до 25 мас.% ядра, на основе общей массы ядра.
Другие наполнители добавляют для улучшения обработки, такие как наполнители, уменьшающие статический заряд на ядрах, или уменьшающие температуру плавления материала матрицы. Примеры таких антистатических агентов включают тальк и диоксид кремния. Ароматизаторы, красители и другие наполнители так же добавляют в обычных количествах для обычных целей. Такие наполнители могут составлять от 0 дo 25 мас.% ядра, на основе общей массы ядра.
Множественные частицы получают с помощью процесса плавления-отвердения, включающего стадии: (a) образование расплавленной смеси, содержащей лекарственное вещество, глицерид (или другие воски), и любой агент, модифицирующий высвобождение; (b) доставка расплавленной смеси стадии (a) в распылитель с образованием капель расплавленной смеси; и (c) отвердевание капель со стадии (b) с образованием множественных частиц.
Условия обработки выбирают для поддержания кристалличности лекарственного средства. Температуру расплавленной смеси удерживают ниже температуры плавления лекарственного средства. Желательно, как минимум 70 мас.% лекарственного средства остается кристаллическим в расплавленной смеси, более желательно, как минимум 80 мас.% и наиболее желательно как минимум 90 мас.%.
Термин "расплавленная смесь" как использовано в данном документе, относится к смеси лекарственного вещества, глицерида (или другие воски), и других агентов, модифицирующих высвобождение, которая нуждается в значительном нагревании для перехода в по сути жидкое состояние, так чтобы можно было образовывать капли или распылять. Распыление расплавленной смеси можно осуществлять, используя любой из нижеописанных способов распыления. Обычно, смесь расплавляют в том смысле, что она будет течь при подвергании одному или более усилиям, таким как давление, усилие сдвига, и центробежная сила, как, например, происходит в распылителе с центрифугой или вращающимся диском. Таким образом, смесь лекарственное вещество/глицерид/агент, модифицирующий высвобождение можно считать "расплавленной" когда любая часть смеси лекарственного средства/глицерид/агент, модифицирующий высвобождение становится достаточно жидкой, что смесь в целом может быть распылена. Обычно, смесь является достаточно жидкой для распыления, когда вязкость расплавленной смеси составляет менее чем приблизительно 20,000 cП. Часто, смесь расплавляется, когда смесь нагревают выше температуры плавления смеси глицерид/агент, модифицирующий высвобождение, в случаях, где смесь глицерид/агент, модифицирующий высвобождение является достаточно кристаллической, чтобы иметь относительно высокую температуру плавления; или, когда смесь глицерид/ агент, модифицирующий высвобождение является аморфной, выше температуры размягчения смеси глицерид/агент, модифицирующий высвобождение. Расплавленная смесь, таким образом, часто является суспензией твердых частиц в жидкой матрице. В одном предпочтительном варианте осуществления, расплавленная смесь содержит смесь частиц по сути кристаллического лекарственного вещества, суспендированных в по сути жидкой смеси глицерид/агент, модифицирующий высвобождение. В таких случаях, часть лекарственного средства может быть растворена в смеси глицерид/агент, модифицирующий высвобождение, а часть смеси глицерид/ агент, модифицирующий высвобождение может оставаться твердой.
Фактически любой процесс может быть использован для образования расплавленной смеси. Один из способов включает нагревание смеси глицерид/агент, модифицирующий высвобождение в резервуаре до перехода в жидкое состояние, и затем добавление лекарственного вещества в расплавленную смесь глицерид/агент, модифицирующий высвобождение. Обычно смесь глицерид/ агент, модифицирующий высвобождение нагревают до температуры приблизительно 10°C, или выше температуры перехода в жидкое состояние. Когда один или больше компонентов смеси глицерид/агент, модифицирующий высвобождение является кристаллическим, температура составляет на приблизительно 10°C или выше температуры плавления наиболее низкой температуры плавления материала смеси. Процесс осуществляют так, чтобы как минимум часть смеси оставалась жидкой до распыления. Как только смесь глицерид/агент, модифицирующий высвобождение становится жидкой, лекарственное вещество может быть добавлено к жидкому носителю или "расплаву." Хотя термин "расплав" обычно конкретно относится к переходу кристаллического материала из кристаллического в жидкое состояние, что происходит при температуре плавления, и термин "расплавленный" обычно относится к такому кристаллическому материалу в его жидком состоянии, как использовано в данном документе, эти термины используют более широко, относя случай "плавления" к нагреванию любого материла или смеси материалов до достижения жидкого состояния, то есть возможности быть накаченным или распыленным подобно кристаллическому материалу в жидком состоянии. Подобным образом, "расплавленный" относится к любого материалу или смеси материалов, находящихся в таком жидком состоянии. Альтернативно, лекарственное вещество, глицерид (или другой воск) и агент, модифицирующий высвобождение, могут быть добавлены в резервуар, и смесь нагревают до достижения смеси жидкого состояния.
Как только смесь глицерид/агент, модифицирующий высвобождение стала жидкой и добавили лекарственное вещество, расплавленная смесь смешивается для обеспечения равномерного распределения лекарственного вещества в ней. Перемешивание обычно осуществляется, используя механические средства, такие как накладные миксеры, магнитные миксеры и магнитные мешалки, орбитальные миксеры и гомогенизаторы. Необязательно, содержимое резервуара может быть выкачено из него и пропущено через поточный статический миксер или экструдер, и затем возвращено обратно в резервуар. Усилие сдвига, использованное для перемешивания расплавленной смеси, должно быть достаточно высоким для обеспечения равномерного распределения лекарственного средства в расплавленном носителе. Усилие сдвига поддерживают на достаточно низком уровне, чтобы не изменить форму лекарственного средства, то есть так, чтобы вызвать увеличение количества аморфного лекарственного вещества, или изменений в кристаллической форме лекарственного средства. Также является предпочтительным, чтобы усилие сдвига не было настолько высоким, чтобы уменьшить размер частиц кристаллов лекарственного средства. Расплавленная смесь может быть перемешана за время от нескольких минут до нескольких часов, время перемешивания зависит от вязкости смеси и растворимости лекарственного вещества и любого необязательного наполнителя в носителе.
Альтернативным способом получения расплавленной смеси является использование двух резервуаров, плавление глицерида (или других восков) или агента, модифицирующего высвобождение, в одном резервуаре, а другого компонента во втором резервуаре. Лекарственное вещество добавляют в один из этих резервуаров и перемешивают, как описано выше. Два расплава затем пропускают через поточный статический миксер или экструдер с получением одной расплавленной смеси, которую направляют на распыление, как описано ниже.
Другим способом получения расплавленной смеси является использование системы резервуаров с непрерывным перемешиванием. В этой системе, лекарственное вещество, глицерид (или других восков), и агент, модифицирующий высвобождение, непрерывно добавляются в нагретый резервуар, оснащенный средствами для непрерывного перемешивания, в то время как расплавленную смесь непрерывно удаляют из резервуара. Содержимое резервуара нагревают так, что температура содержимого выше на приблизительно 10°C или больше температуры плавления носителя. Лекарственное вещество, глицерид (или другие воска) и агент, модифицирующий высвобождение добавляют в таких пропорциях, что расплавленная смесь, которую удаляют из резервуара, имеет желаемый состав. Лекарственное вещество обычно добавляют в твердой форме и его могут предварительно нагреть перед добавлением в резервуар. Глицерид (или другие воска) и агент, модифицирующий высвобождение, так же могут быть предварительно нагреты или даже предварительно расплавлены перед добавлением в систему резервуаров с непрерывным перемешиванием.
Другим способом получения расплавленной смеси является использование экструдера. Под "экструдером" понимают устройство или набор устройств, образующее расплавленный экструдат путем нагревания и/или усилия сдвига, и/или образующее однородно перемешанный экструдат из твердой и/или жидкой (например, расплавленной) смеси. Такие устройства включают, но не ограничиваются следующими: одношнековые экструдеры; двухшнековые экструдеры, включая экструдеры с одинаковым направлением вращения, с противоположным направлением вращения, с перекрестным вращением, и не перекрестным вращением; многошнековые экструдеры; поршневые экструдеры, состоящие из нагретого цилиндра и поршня для экструзии расплавленной смеси; экструдеры с насосом шестереночного типа, состоящие из нагретого насоса шестереночного типа, обычно с противоположным направлением вращения, который одновременно нагревает и накачивает расплавленную смесь; и конвейерные экструдеры. Конвейерные экструдеры содержат конвейер для транспортировки твердой и/или порошкообразной смеси, такой как винтовой или пневматический конвейер, и насос.
Как минимум часть конвейера нагревают до достаточно высокой температуры, чтобы получить расплавленную смесь. Расплавленная смесь может необязательно быть направлена в аккумулирующий резервуар, перед направлением в насос, который направляет расплавленную смесь в распылитель. Необязательно, поточный миксер может быть использован перед или после насоса для обеспечения гомогенности расплавленной смеси. В каждом из этих экструдеров расплавленную смесь перемешивают с образованием однородно перемешанного экструдата. Такое перемешивание может быть осуществлено различными механическими и технологическими средствами, включая элементы перемешивания, элементы растирания и перемешивание усилием сдвига в противотоке. Таким образом, в таких устройствах композиция подается в экструдер, производящий расплавленную смесь, которую затем направляют в распылитель.
В одном варианте осуществления, композиция подается в экструдер в виде твердого порошка. Порошкообразную смесь получают, используя известные в отрасли способы получения порошкообразных смесей с высокой однородностью. Обычно предпочтительно, чтобы размеры частиц лекарственного средства, глицерида (или других восков) и агент, модифицирующего высвобождение, были одинаковыми для получения однородной смеси. Однако это не является существенным для благополучного осуществления данного изобретения.
Приведены следующие примеры способов получения по сути однородной смеси. Сначала, глицерид (или другие воска) и агент, модифицирующий высвобождение, перемалывают так, чтобы размер частиц был примерно одинаковым с размером частиц лекарственного средства; далее лекарственное вещество, глицерид (или другие воска), и агент, модифицирующий высвобождение, перемешивают в V-блендере на протяжении 20 мин; полученную смесь затем протирают для удаления больших частиц; полученную смесь в конце перемешивают на протяжении дополнительных 4 мин. в некоторых случаях, тяжело перемолоть глицерид (или другие воска) и агент, модифицирующий высвобождение, до желательного размера частиц, в виду того, что многие их этих материалов являются воскообразными, а тепло, образованное на протяжении перемалывания, может склеить перемалывающее оборудование. В таких случаях, маленькие частицы глицерида (или других восков), и агента, модифицирующего высвобождение, могут образовываться, используя процесс плавление- или распыление-отвердение, как описано ниже. Полученные отвердевшие частицы глицерида (или других восков), и агента, модифицирующего высвобождение, могут быть затем перемешаны с лекарственным веществом для получения смеси для экструдера.
Другой способ получения смеси для экструдера включает плавление глицерида (или других восков) и агента, модифицирующий высвобождение, в резервуаре, перемешивание с лекарственным веществом, как описано выше для системы резервуаров, и затем охлаждение расплавленной смеси с получением отвердевшей смеси лекарственного вещества и носителя. Эта отвердевшая смесь затем может быть перемолота до однородного размера частиц и направлена в экструдер.
Система экструдера с двумя подачами так же может быть использована для получения расплавленной смеси. В этой системе, лекарственное вещество, глицерид (или другие воска) и агент, модифицирующий высвобождение, все в порошкообразном виде, подаются в экструдер через один или разные каналы подачи смеси. В этом случае, отпадает потребность в перемешивании компонентов.
Альтернативно, глицерид (или другие воска) и агент, модифицирующий высвобождение, в порошкообразном виде, подаются в экструдер в одной точке, позволяя экструдеру плавить глицерид (или другие воска) и агент, модифицирующий высвобождение. Лекарственное вещество добавляют потом в расплавленный глицерид (или другие воска) и агент, модифицирующий высвобождение, через второй канал подачи по ходу длины экструдера, таким образом, сводя к минимуму время контакта лекарственного средства с расплавленным глицеридом (или другими восками) и агентом, модифицирующим высвобождение. Чем ближе второй канал подачи к выходу из экструдера, тем меньше времени проводит лекарственное вещество в экструдере. Экструдеры с множественными подачами могут быть использованы, когда необязательные наполнители включены в многочисленные частицы.
В другом способе, композиция находится скорее в виде твердых частиц большого размера или твердой массы, чем в виде порошка, при подаче в экструдер. Например, отвержденная смесь может быть получена, как описано выше, и затем расплавлена для попадания в цилиндр поршневого экструдера и использована напрямую без помола.
В другом способе, глицерид (или другие воска) и агент, модифицирующий высвобождение, могут быть сначала расплавлены в, например, резервуаре, и поданы в экструдер в расплавленном виде. Лекарственное вещество, обычно в порошкообразном виде, затем может быть введено в экструдер через тот же или другой канал доставки, который использовали для доставки глицерида (или других восков) и агента, модифицирующего высвобождение, в экструдер. Эта система имеет преимущество разделения стадии плавления для глицерида (или других восков) и агента, модифицирующего высвобождение, от стадии перемешивания, сводя к минимуму время контакта лекарственного средства с расплавленным глицеридом (или другими восками) и агентом, модифицирующим высвобождение.
В каждом из вышеприведенных способов, экструдер должен быть разработан так, чтобы он производил расплавленную смесь с кристаллами лекарственного вещества, однородно распределенными в смеси глицерид/агент, модифицирующий высвобождение. Обычно температура экструдата должна быть на приблизительно 10°C или больше выше температуры, при которой лекарственное вещество и смесь носителя становятся жидкими. Различные зоны в экструдере должны быть нагреты до подходящей температуры, чтобы получить желаемую температуру экструдата, а также желательную степень перемешивания или деформации, используя известные в отрасли средства. Как было описано выше для механического перемешивания, должно быть применено минимальное усилие сдвига для получения однородно расплавленной смеси, так чтобы кристаллическая форма лекарственного средства осталась неизменной, а растворение или образование аморфного лекарственного вещества сводилось к минимуму.
Смесь желательно расплавляют перед отвердением в течении как минимум 5 сек, более желательно как минимум 10 сек, и наиболее желательно как минимум 15 сек, для обеспечения адекватной гомогенности плавления лекарственного средства/глицерида/агента, модифицирующего высвобождение. Также желательно чтобы расплавленная смесь оставалась расплавленной в течение не более 20 мин для ограничения воздействия лекарственного средства на расплавленную смесь. Как описано выше, в зависимости от активности выбранной смеси глицерид/агент, модифицирующий высвобождение, может быть желательно еще уменьшить время, при котором смесь находится в расплавленном состоянии до менее 20 мин, для ограничения разложения лекарственного вещества до приемлемого уровня. В таких случаях, такие смеси поддерживают в расплавленном состоянии менее 15 мин, и в некоторых случаях, даже менее 10 мин. Когда экструдер используют для получения расплавленной смеси, вышеуказанное время означает среднее время от введения материала в экструдер до затвердения расплавленной смеси. Такое среднее время можно определить с помощью известных процедур. В одном способе, небольшое количество красителя или другого подобного соединения добавляли в смесь, при работе экструдера в номинальных условиях. Отвердевшие множественные частицы затем собирали со временем и анализировали на краситель, откуда получали среднее время.
Как только образовалась расплавленная смесь, ее доставляют в распылитель, который разбивает расплавленную смесь на мелкие капли. Фактически любой способ можно использовать для доставки расплавленной смеси в распылитель, включая применение насосов и пневматических устройств различного типа (например, баллоны под давлением, помповые контейнеры). Когда экструдер используют для формирования расплавленной смеси, экструдер сам по себе может быть использован для доставки расплавленной смеси в распылитель. Обычно расплавленную смесь поддерживают при повышенной температуре во время доставки смеси в распылитель для предотвращения отвердения смеси и для поддержания текучести расплавленной смеси.
Обычно распыление производят по одному из нескольких путей, включая (1) "давление" или пульверизаторы с одной жидкостью; (2) пульверизаторы с двумя жидкостями; (3) центробежные распылители или распылители с вращающим диском, (4) ультразвуковые пульверизаторы; и (5) механические вибрирующие пульверизаторы. Детальное описание процессов распыления можно найти в Lefebvre, Atomization and Sprays (1989) or in Perry's Chemical Engineers' Handbook (7th Ed., 1997). Желательно, используют центробежные распылители или распылители с вращающим диском, такие как роторный распылитель FX1 100-мм производства Niro A/S (Себорг, Дания).
После распыления расплавленной смеси, капли застывают, обычно при контакте с газом или жидкостью при температуре ниже температуры отвердения капель. Обычно желательно, чтобы капли застывали менее чем через приблизительно 60 сек, желательно менее чем через приблизительно 10 сек, более желательно менее чем через приблизительно 1 сек. Часто, застывание при температуре окружающей среды приводит к достаточно быстрому отвердению капель. Однако стадия застывание часто происходит в закрытом пространстве для облегчения сбора множественных частиц. В таких случаях, температура застывания среды (газа или жидкости) будет увеличиваться со временем попадания капель в замкнутое пространство, потенциально влияя на образование множественных частиц или химическую стабильность лекарственного средства. Таким образом, охлаждающий газ или жидкость часто циркулируют в замкнутом пространстве для поддержания постоянной температуры застывания. Если желательно минимизировать время подвергания лекарственного вещества высоким температурам, например, чтобы предотвратить разложение, охлаждающий газ или жидкость могут быть охлаждены до температуры ниже температуры окружающей среды для стимулирования быстрого застывания, таким образом, сводя к минимуму образование разложенных веществ.
После образования множественных частиц, желательно подвергнуть множественные частицы обработке для улучшения кристалличности лекарственного вещества и/или стабильности множественных частиц.
Множественные частицы также могут быть смешаны или купажированы с одним или больше фармацевтически приемлемым материалом с образованием приемлемой дозированной формы. Приемлемые дозированные формы включают таблетки, капсулы, саше, оральные порошки для разбавления, и т.д.
После образования множественных частиц распылительной кристаллизацией, множественные частицы могут необязательно быть покрыты дополнительным внешним покрытием. Внешнее покрытие может быть представлено любым стандартным покрытием, таким как защитное пленочное покрытие, покрытие, обеспечивающее отсроченное или замедленное высвобождение лекарственного средства, или маскирующее вкус.
В одном варианте осуществления, покрытием является кишечное покрытие, обеспечивающее отсроченное высвобождение лекарственного средства. Под "кишечным покрытием" понимают покрытие резистентное к кислотам, которое остается интактным и не растворяется при pH менее чем приблизительно 4. Кишечное покрытие окружает многочисленные частицы, так что твердый аморфный дисперсионный слой не растворяется или не эродирует в желудке. Кишечное покрытие может включать кишечные полимеры. Кишечные полимеры включают поликислоты с pKa приблизительно 3-5. Примеры кишечных полимеров включают: производные целлюлозы, такие как ацетат фталат целлюлозы, ацетат тримеллитат целлюлозы, гидроксипропил метилацетат сукцинат целлюлозы, ацетат сукцинат целлюлозы, карбоксиметилэтилцеллюлоза, метилцеллюлозы фталат и этилгидрокси целлюлозы фталат; полимеры винила, такие как поливинилацетат фталат, coполимер винилацетат-малеиновый ангидрид; полиакрилаты; и полиметакрилаты, такие как coполимер метилакрилат-метакриловая кислота, coполимер метакрилат-метакриловая кислота-октилакрилат; и coполимер стирол-малеиновый моноэфир. Указанные соединения могут быть использованы как по отдельности, так и в комбинации, или вместе с другими полимерами, не описанными выше.
Один класс материалов кишечного покрытия представлен фармацевтически приемлемыми coполимерами метакриловой кислоты, являющиеся coполимерами, анионного характера, на основе метакриловой кислоты и метилметакрилата. Некоторые из этих полимеров известны и продаются как кишечные полимеры, например, с растворимостью в водной среде при pH 5,5 и выше, такие как коммерчески доступные кишечные полимеры EUDRAGIT, например, Eudragit L 30, полимер, синтезированный из диметиламиноэтилметакрилата, и Eudragit S и Eudragit FS.
Внешние покрытия могут включать стандартные пластификаторы, включая дибутилфталат; дибутил себацинат; диэтил фталат; диметил фталат; триэтил цитрат; бензилбензоат; бутиловый и гликолевые сложные эфиры жирных кислот; минеральное масло; олеиновая кислота; стеариновая кислота; цетиловый спирт; стеариловый спирт; касторовое масло; кукурузное масло; кокосовое масло; и масло камфоры; и другие наполнители, такие как антиадгезивы, глиданты, и т.д. Для пластификаторов, триэтил цитрат, кокосовое масло и дибутил себацинат являются особенно предпочтительными.
Внешние покрытия могут быть сформированы, используя способы на основе растворителя и горячее-плавление. В способах нанесения покрытия на основе растворителя, покрытие получают сначала образованием раствора или суспензии, содержащей растворитель, материал покрытия и необязательные добавки покрытия. Материалы покрытия могут полностью растворяться в покрывающем растворителе, или только диспергировать в растворителе как эмульсия или суспензия или их комбинация. Латексные дисперсии являются примером эмульсии или суспензии, приемлемой для способов нанесения покрытия на основе растворителя. В одном аспекте, растворитель является жидким при комнатной температуре.
Нанесение покрытия можно осуществить с помощью традиционных технологий, таких как дражировочные котлы, роторные грануляторы и устройства для нанесения покрытия в кипящем слое, такие как верхние брызгала, тангенциальные брызгала или нижние брызгала (Wurster покрытие). Способ верхнего брызгала также используют для нанесения покрытий. В этом способе, покрывающий раствор распыляют вниз на флюидизированные ядра. Растворитель испаряется из покрытых ядер, и покрытые ядра повторно флюидизируют в аппарате. Нанесение продолжают до достижения покрытия нужной толщины. Композиции и способы получения множественных частиц этого варианта осуществления детально описаны в патентных заявках США, US 2005-0181062, US 2005-0181062, US 2008-0199527, US 2005-0186285A1, включенных в данный документ с помощью ссылок.
Множественные частицы изобретения обычно имеют средний диаметр от приблизительно 40 до приблизительно 3,000 микрон, с желательным диапазоном 50-1,000 микрон, и наиболее желательно от приблизительно 100 дo 300 микрон. Хотя множественные частицы могут иметь любую форму и текстуру, желательно чтобы они были сферическими, с гладкой поверхностью. Эти физические характеристики множественных частиц улучшают их свойства текучести, позволяют им быть однородно покрытыми (в случае необходимости). Как указанно в данном документе, термин "приблизительно" означает ±10% от значения.
Множественные частицы данного изобретения являются особенно приемлемыми для контролируемого высвобождения или отсроченного высвобождения, или любой комбинации этих профилей высвобождения при введении в среду использования. Как указанно в данном документе, "среда использования" может быть in vivo средой желудочно-кишечного тракта (ЖК), или теста на растворимость in vitro, описанного в данном документе. Информация о скоростях высвобождения in vivo можно получить из ФК профиля, используя стандартную деконволюцию или обработку данных Вагнера-Нельсона, широко известную специалистам в отрасли.
После формирования матрицы тофацитиниба из множественных частиц с помощью вышеуказанных способов, их смешивают со сжимаемыми наполнителями, такими как лактоза, микрокристаллическая целлюлоза, дифосфат кальция, и т.д., и смесь прессуют с образованием таблетки или капсулы. Также используют дезинтегранты, такие как натрия крахмал гликолят или поперечно сшитый поли(винилпирролидон). Таблетки или капсулы, полученные этим способом, дезинтегрируют при попадании в водную среду (такую как ЖК тракт), таким образом, воздействуя на матрицу многочисленных частиц, высвобождающую тофацитиниб.
Другие стандартные наполнители композиций применяют при контролируемом высвобождении изобретения, включая известные в отрасли наполнители, например, как описано в Remington: The Science and Practice of Pharmacy, 20th edition (2000). Обычно наполнители, такие как поверхностно-активные вещества, модификаторы pH, наполнители, материалы матрицы, комплексообразователи, солюбилизаторы, пигменты, лубриканты, глиданты, ароматизаторы и т.д. могут быть использованы для обычных нужд и в типичных количествах, при этом не оказывая негативного влияния на свойства композиций.
Примеры материалов матрицы, наполнителей или разбавителей включают следующие: лактоза, маннит, ксилитол, декстроза, сахароза, сорбитол, прессованный сахар, микрокристаллическая целлюлоза, порошкообразная целлюлоза, крахмал, прежелатинизированный крахмал, декстраты, декстран, декстрин, декстроза, мальтодекстрин, карбонат кальция, двухосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, карбонат магния, полоксамеры, полиэтиленоксид, гидроксипропилметилцеллюлоза и их смеси.
Осмотические системы с замедленным высвобождением
В другом варианте осуществления, тофацитиниб включен в устройства осмотической доставки или "осмотические насосы", как они известны в отрасли. Осмотические насосы содержат ядро, которое содержит осмотически эффективную композицию, окруженную полупроницаемой мембраной. Термин "полупроницаемый" в этом контексте означает, что вода может легко диффундировать через мембрану, но растворенные в воде вещества обычно не могут легко диффундировать через мембрану, по сравнению со скоростью диффузии воды через мембрану. В употреблении, при помещении в водную окружающую среду, устройство впитывает воду благодаря осмотической активности ядра композиции. Благодаря полупроницаемой природе окружающей мембраны, содержимое устройства (включая тофацитиниб и любые наполнители) не может пройти через непористые участки мембраны, а направляется благодаря осмотическому давлению покинуть устройство через отверстия или каналы, предварительно сконструированные в дозированной форме, или, альтернативно, образованные in situ в ЖК тракте в результате разрыва преднамеренно включенных слабых мест в покрытии под влиянием осмотического давления. Осмотически эффективная композиция включает растворимые в воде виды, образующие коллоидное осмотическое давление, и разбухающий в воде полимеры. Примеры таких дозированные форм хорошо известны в отрасли. Смотреть, например, Remington: The Science and Practice of Pharmacy, 21st Edition, 2006 Chapter 47; page 950-1, включенный с помощью ссылки.
В одном варианте осуществления данного изобретения, тофацитиниб включен в двухслойное устройство осмотической доставки, так, что композиция, содержащая тофацитиниб, должна содержать захватывающий агент в форме разбухающего в воде полимера, и второй отталкивающий слой или разбухающий в воде слой, содержащий разбухающие в воде полимеры и/или осмотически активные агенты, но не содержащий никакого активного агента. Двухслойная таблетка или капсула окружена полупроницаемой мембраной, содержащей одно или больше отверстий, сделанных в дозированной форме посредством такой техники как лазерное сверление. Такие разбухающие в воде полимеры часто называют в фармацевтике "осмополимерами" или "гидрогелями". Захватывающий агент суспендирует или захватывает лекарственное вещество для облегчения доставки лекарственного средства через каналы доставки. Не привязываясь к определенной теории, считается, что во время всасывания воды в дозированную форму, захватывающий агент имеет достаточную вязкость, чтобы суспедировать или захватить лекарственное вещество, при этом оставаясь достаточно жидким для прохождения через канал доставки вместе с лекарственным веществом. Количество захватывающего агента в композиции, содержащей тофацитиниб, находится в диапазоне от приблизительно 20 мас.% до приблизительно 95 мас.%. Захватывающий агент может быть представлен одним материалом или смесью материалов. Не-поперечно сшитый полиэтиленоксид (ПЭO) может быть использован в качестве захватывающего агента. Другие приемлемые захватывающие агенты включают гидроксипропилцеллюлозу (ГПЦ), гидроксипропилметилцеллюлозу (ГПМЦ), метилцеллюлозу (MC), гидроксиэтилцеллюлозу (ГЭЦ) и поливинилпирролидон (ПВП), а так же смеси этих полимеров с ПЭO.
Выбор молекулярной массы для ПЭO зависит от части от того или ПЭO составляет основную массу части, не содержащей тофацитиниб, в композиции, содержащей тофацитиниб, или того включено ли значительное количество других низкомолекулярных водорастворимых наполнителей; то есть, выбор молекулярной массы ПЭO зависит от фракции композиции, содержащей тофацитиниб, состоящей из ПЭO. Композиция, содержащая тофацитиниб, не должна быстро становиться жидкой, дозированная форма может разбухнуть или разрушить покрытие, окружающее ядро, потенциально вызывая повреждение дозированной формы. Когда наполнители композиции, содержащей тофацитиниб, в основном представлены ПЭO (например, ПЭO составляет приблизительно 60 мас.% или больше не-тофацитинибных компонентов композиции, содержащая тофацитиниб), обычно желательно чтобы ПЭO имел среднюю молекулярную массу от приблизительно 100,000 до 300,000 дальтон (как указанно в данном документе, ссылка на молекулярные массы полимеров должна пониматься как средняя молекулярная масса).
Альтернативно, другой вариант осуществления данного изобретения использует высокомолекулярный ПЭO от приблизительно 500,000 до 800,000 дальтон в нижней фракции не-тофацитинибных наполнителей, часть ПЭO замещена пластификатором. Обычно, когда ПЭO составляет приблизительно 60 мас.% или больше не-тофацитинибных компонентов композиции, содержащей тофацитиниб, ПЭO с молекулярной массой 500,000 дальтон или больше делает композицию, содержащую тофацитиниб, слишком вязкой, и может привести к разрушению покрытия или как минимум задержке высвобождения тофацитиниба. Однако выяснили, что такой высокомолекулярный ПЭO является предпочтительным, когда не-тофацитинибные компоненты композиции, содержащей тофацитиниб, включают менее чем приблизительно 60 мас.% ПЭO, а также включают пластификатор. Используя высокомолекулярный ПЭO, количество пластификатора, присутствующего в композиции, содержащей тофацитиниб, находится в диапазоне от приблизительно 5 до приблизительно 50 мас.%, желательно 10-30 мас.% композиции, содержащей тофацитиниб. Желательными пластификаторами являются низкомолекулярные, растворимые в воде растворенные вещества, такие как не-восстанавливаемые сахара и органические кислоты с растворимостью в воде 30 мг/мл или больше. Приемлемые сахара включают ксилитол, маннит, сорбитол и малтитол. Соли, приемлемые в качестве пластификатора включают хлорид натрия, лактат натрия и ацетат натрия. Органические кислоты, приемлемые в качестве пластификатора включают следующие: адипиновая кислота, лимонная кислота, яблочная кислота, фумаровая кислота, янтарная кислота и виннокаменная кислота.
Присутствие пластификатора, наряду с относительно низким уровнем высокомолекулярного ПЭO (например, от приблизительно 500,000 до приблизительно 800,000 дальтон) позволяет композиции, содержащей тофацитиниб, быстро достигать низкой вязкости при поглощении воды. Кроме того, выяснили, что такой вариант осуществления способен доставить относительно высокое количество тофацитиниба.
Композиция, содержащая тофацитиниб, также может содержать другие разбухающие в воде полимеры. Например, композиция, содержащая тофацитиниб, может содержать относительно небольшое количество разбухающих в воде полимеров, которые значительно увеличиваются в объеме в присутствии воды. Такие разбухающие в воде полимеры включают натрия крахмал гликолят, продающийся под торговой маркой EXPLOTAB, и кросскармелоза натрия, продающаяся под торговой маркой AC-DI-SOL. Такие полимеры могут присутствовать в количестве от 0 мас.% дo 10 мас.% композиции, содержащей тофацитиниб.
Композиция, содержащая тофацитиниб, может необязательно включать осмотически эффективные растворенные вещества, которые называются "осмогены" или "осмагенты." Количество осмагента в композиции, содержащей тофацитиниб, находится в диапазоне от приблизительно 0 мас.% до приблизительно 50 мас.%, желательно 10 мас.% - 30 мас.% композиции, содержащей тофацитиниб. Типичные классы приемлемых осмагентов включают растворимые в воде соли, сахара, органические кислоты и другие низкомолекулярные органические соединения, способные поглощать воду, таким образом устанавливая градиент осмотического давления поперек барьера окружающего покрытия. Типичные приемлемые соли включают сульфат магния, хлорид магния, хлорид кальция, хлорид натрия, хлорид лития, сульфат калия, карбонат натрия, сульфит натрия, сульфат лития, хлорид калия и сульфат натрия. Условно, хлоридные соли, такие как хлорид натрия используют в качестве осмагентов.
Композиция, содержащая тофацитиниб, также может включать агенты, улучшающие растворимость, или солюбилизаторы, которые стимулируют растворение лекарственного средства в воде, в количестве от приблизительно 0 до приблизительно 30 мас.% композиции, содержащей тофацитиниб. Солюбилизаторы, используемые с тофацитинибом, включают органические кислоты и соли органических кислот, частичные глицериды, например, частично эстерифицированные производные глицерина, включая глицериды, моноглицериды, диглицериды, производные глицеридов, сложные эфиры полиэтиленгликоля, сложные эфиры полипропиленгликоля, сложные эфиры многоатомного спирта, эфиры полиоксиэтилена, сложные эфиры сорбитана, сложные эфиры полиоксиэтилен и сорбитана, и карбонатные соли.
Предпочтительным классом солюбилизаторов являются органические кислоты. В виду того, что тофацитиниб является основанием, которое солюбилизируется протонированием, и что его растворимость в водной окружающей среде с pH 5 или выше является пониженной, считается, что добавление органической кислоты в композиции, содержащую тофацитиниб, поможет солюбилизации и как следствие абсорбции тофацитиниба. Даже незначительное снижение pH водного раствора при высокой pH приведет к резкому увеличению растворимости тофацитиниба. Органические кислоты также могут стимулировать стабильность на протяжении хранения до введения в среду применения, благодаря их склонности поддерживать протонированное состояние тофацитиниба.
При выборе соответствующей органической кислоты для применения в качестве солюбилизатора с тофацитинибом в осмотической дозированной форме, принимают во внимание многочисленные факторы. Кислота должна неблагоприятно не взаимодействовать с тофацитинибом, должна иметь приемлемую растворимость в воде, и должна обеспечивать хорошие производственные характеристики.
Соответственно, выяснили, что предпочтительный подкласс органических кислот, отвечающий таким критериям, включает лимонную, янтарную, фумаровую, адипиновую, яблочную и виннокаменную кислоты. Лимонная, яблочная и виннокаменная кислота имеют преимущество в высокой растворимости в воде и высоком осмотическом давлении. Янтарная и фумаровая кислота дают как умеренную растворимость, так и умеренное осмотическое давление.
Разбухающая в воде композиция также может необязательно содержать краситель. Целью красителя является обеспечение идентификации стороны поверхности таблетки, содержащей лекарственного средство, с целью обеспечения канала доставки, например, с помощью лазерного сверления через покрытие. Приемлемые красители включают, но не ограничиваются следующими: Красный No. 40, FD C голубой 2 и FD C желтый 6.
Слой, содержащий тофацитиниб и/или разбухающий в воде слой и/или мембрана, контролирующая функциональную скорость, могут необязательно содержать антиоксидант, включая, но не ограничиваясь следующими: BHT, BHA, метабисульфит натрия, пропил галат, глицерин, витамин E, лимонная кислота или аскорбил пальмитат. Антиоксидант может присутствовать в количестве в диапазоне от 0 до 10 мас.% композиции, содержащей тофацитиниб, слоя и/или слоя, разбухающего в воде и/или мембраны, контролирующей функциональную скорость. Дополнительные примеры антиоксидантов приведены в C.-M. Andersson, A. Hallberg, and T. Hoegberg. Advances in the development of pharmaceutical antioxidantes. Advances in Drug Research. 28:65-180, 1996.
Разбухающая в воде композиция также может содержать другие обычные фармацевтически приемлемые наполнители, такие как связующее, включая ГПЦ, ГПМЦ, ГЭЦ, MC и ПВП, таблетирующая добавка, такая как микрокристаллическая целлюлоза, и лубрикант, такой как стеарат магния.
Разбухающую в воде композицию получают перемешиванием разбухающего в воде полимера и других наполнителей с образованием однородной смеси. Для получения однородной смеси, желательно чтобы в сухом или влажном грануляте или смеси сухих ингредиентов был одинаковый размер частиц, используя способы известные в отрасли.
Таблетирование
Ядро получают сначала помещением смеси композиции, содержащей тофацитиниб, в таблеточный пресс, а затем выравниванием смеси аккуратным прессованием. Композицию, разбухающую в воде, затем помещали поверх композиции, содержащей тофацитиниб, и прессовали для завершения формирования ядра. Альтернативно, сначала в таблеточный пресс можно было поместить композицию, разбухающую в воде, а затем композицию, содержащую тофацитиниб.
Соответствующее количество композиции, содержащей тофацитиниб, и разбухающей в воде композиции выбирают так, чтобы обеспечить удовлитворительное высвобождение тофацитиниба. Когда желательно обеспечить значительную дозу тофацитиниба в дозированной форме относительно небольшого размера, желательно максимально увеличить количество композиции, содержащей тофацитиниб, и минимизировать количество разбухающей в воде композиции, при этой сохраняя хорошее высвобождение. В дозированных формах данного изобретения, когда разбухающий в воде полимер в композиции, разбухающей в воде, представлен только ПЭO, композиция, содержащая тофацитиниб, может составлять от приблизительно 50 до приблизительно 85 мас.% ядра, и желательно от приблизительно 60 до приблизительно 70 мас.%. Эти величины соответствуют массовому соотношению композиции, содержащей тофацитиниб, и разбухающей в воде композиции от 1 до приблизительно 5,7. Когда весь или часть разбухающего в воде полимера в разбухающей в воде композиции содержит натрия крахмал гликолят или кросскармелозу натрия, композиция, содержащая тофацитиниб, может составлять от 50 дo 90 мас.% ядра, и желательно от приблизительно 75 до приблизительно 85 мас.%. Эти величины соответствуют массовому соотношению композиции, содержащей тофацитиниб, и разбухающей в воде композиции от 1 дo 9. Абсолютная величина диаметра и высоты таблетки данного изобретения может варьировать в широком диапазоне.
Покрытие
После формирования ядра, наносят полупроницаемое покрытие. Покрытие должно иметь высокую проницаемость для воды и значительную прочность, и тоже время его должно быть легко получать и наносить. Высокая проницаемость для воды требуется для проникновения воды в ядро в достаточном объеме. Значительная прочность требуется для уверенности в том, что покрытие не распадется при разбухании ядра во время поглощения воды, что приведет к неконтролируемой доставке содержимого ядра. В конце концов, покрытие должно иметь высокую воспроизводимость и выход.
Существенно чтобы покрытие имело, как минимум, один канал доставки в сообщении с внутренней и внешней поверхностью покрытия для доставки композиции, содержащей тофацитиниб. Более того, покрытие не должно растворяться или эродировать на протяжении высвобождения композиции, содержащей тофацитиниб, что обычно означает, что покрытие должно быть нерастворимым в воде, так что тофацитиниб по сути полностью доставляется через каналы доставки, в отличии от доставки путем проникновения через покрытие.
Покрытия с такими характеристиками можно получить, используя гидрофильные полимеры, такие как пластифицированные и непластифицированные сложные эфиры, простые эфиры и сложные эфиры - простые эфиры целлюлозы. Особенно приемлемые полимеры включают целлюлозы ацетат (АЦ), целлюлозы ацетат бутират (АЦБ), и этилцеллюлозу (ЭЦ). Один набор полимеров включает ацетаты целлюлозы, имеющие содержание ацетила 25-42%. Один типичный полимер представлен АЦ, имеющим содержание ацетила 39,8%, а именно, АЦ 398-10 (Eastman Fine Chemicals, Kingsport, Tenn.). АЦ 398-10 имеет среднюю молекулярную массу приблизительно 40,000 дальтон. Другой типичный АЦ с содержанием ацетила 39,8% представлен высокомолекулярным АЦ со средней молекулярной массой более чем приблизительно 45,000, а именно, АЦ 398-30 (Eastman Fine Chemical) со средней молекулярной массой 50,000 дальтон.
Нанесение покрытия производят обычным образом, сначала образованием покрывающего раствора, а затем его нанесением путем погружения, нанесением в псевдоожиженном слое, или смазыванием форм. Для осуществления указанного, покрывающий раствор формируют с содержанием полимера и растворителя. Типичные растворители приемлемые для целлюлозных полимеров включают ацетон, метил ацетат, этилацетат, изoпропил ацетат, н-бутил ацетат, метил изoбутил кетон, метил пропил кетон, моноэтиловый эфир этиленгликоля, этиленгликоля моноэтилацетат, метилeн дихлорид, этилен дихлорид, пропилeн дихлорид, нитроэтан, нитропропан, тетрахлорэтан, 1,4-диоксан, тетрагидрофуран, диглим, и их смеси. Покрывающий раствор обычно содержит 2-15 мас.% полимера.
Покрывающий раствор также может содержать порообразователи или нерастворители в любом количестве, до тех пор пока полимер остается растворимым в условиях, использованных для формирования покрытия, и до тех пор пока покрытие остается проницаемым для воды и имеет достаточную прочность. Порообразователи и их применение в получении покрытий описано в патентах США № 5,698,220 и 5,612,059, описание которых включено в данный документ с помощью ссылки. Термин "порообразователь," как использовано в данном документе, относится к материалу добавляемому к покрывающему раствору, который является нелетучим, по сравнению с растворителем, так что он остается частью покрытия после нанесения покрытия, но он значительно разбухает в воде или растворяется в воде, так что в водной среде он образует заполненный водой или набухший водой канал или "пору" для прохождения воды, таким образом улучшая проницаемость покрытия для воды. Приемлемые порообразователи включают, но не ограничиваются гидроксипропилцеллюлозой (ГПЦ), полиэтиленгликолем ("ПЭГ"), ПВП и ПЭO. Для получения комбинации высокой проницаемости для воды и высокой прочности при использовании ПЭГ или ГПЦ в качестве порообразователя, массовое соотношение АЦ:ПЭГ или АЦ:ГПЦ должно находиться в диапазоне от приблизительно 6:4 до приблизительно 9:1.
Добавление нерастворителя, такого как вода, к покрывающему раствору приводит к исключительному результату. Под "нерастворителем" понимают любой материал, добавляемый к покрывающему раствору, который существенно растворяется в покрывающем растворе и понижает растворимость полимерного покрытия или полимеров в растворителе. В целом, функцией нерастворителя является придание пористости полученному материалу. Как описано ниже, пористые покрытия имею более высокую проницаемость для воды, чем эквивалентная масса покрытия той же композиции, но без пор, и эта пористость указывает на уменьшение плотности покрытия (масса/объем). Несмотря на то что исследователи не хотят привязываться к какому-то конкретному механизму образования пор, обычно считают, что добавление нерастворителя придает пористость покрытию на протяжении выпаривания растворителя, вызывая разделения жидкости - жидкой фазы покрывающего раствора перед отвердением. Пригодность и количество определенного материала кандидата для использования в качестве нерастворителя может быть определена постепенным добавлением кандидата нерастворителя к покрывающему раствору до появления мутности. Если этого не случается при добавлении количества, составляющего до приблизительно 50 мас.% покрывающего раствора, считается, что данный кандидат не пригоден в качестве нерастворителя. При наблюдении помутнения, называется "температура помутнения", приемлемым уровнем нерастворителя для максимальной пористости является количество ниже температуры помутнения. Для растворов ацетона, содержащих 7 мас.% АЦ и 3 мас.% ПЭГ, температура помутнения составляет приблизительно 23 мас.% воды. Если желательна низкая пористость, количество нерастворителя можно уменьшить до желаемого уровня.
Приемлемые нерастворители являются любыми материалами с приемлемой растворимостью в растворителе, и эта растворимость меньше растворимости полимерного покрытия в растворителе. Предпочтительный нерастворитель зависит от растворителя и покрывающего полимера. В случае использования летучего полярного покрывающего растворителя, такого как ацетон, приемлемые нерастворители включают воду, глицерин, спирты, такие как метанол или этанол.
Используя АЦ 398-10, массовые соотношения покрывающего раствора АЦ:ПЭГ3350:вода составляют 2,4:1,6:5, 2,8:1,2:5, 3,2:0,8:5 и 3,6:0,4:5, с остатком раствора, содержащим растворитель, такой как ацетон. Таким образом, например, в растворе с массовым соотношением АЦ:ПЭГ3350:вода равным 2,8:1,2:5, АЦ составляет 2,8 мас.% раствора, ПЭГ 3350 составляет 1,2 мас.% раствора, вода составляет 5 мас.% раствора, и ацетон составляет оставшиеся 91 мас.%. Также, покрывающий раствор с массовым соотношением АЦ:ГПЦ:вода равным 1,2:0,8:9,8, 2,4:1,6:19,6, 1,6:0,4:4,9 и 3,2:0,8:9,8, с остатком раствора, содержащим растворитель, такой как ацетон. Таким образом, например, в растворе с массовым соотношением АЦ:ГПЦ:вода равным 1,2:0,8:10, АЦ составляет 1,2 мас.% раствора, ГПЦ составляет 0,8 мас.% раствора, вода составляет 10 мас.% раствора, и ацетон составляет оставшиеся 88 мас.%. Также, покрывающий раствор с массовым соотношением АЦ:ГПЦ:метанол равным 1,8:1,2:19,6, 2,4:1,6:19,6, 1,6:0,4:4,9 и 3,2:0,8:9,8, с остатком раствора, содержащим растворитель, такой как ацетон. Таким образом, например, в растворе с массовым соотношением АЦ:ГПЦ:метанол на уровне 1,8:1,2:19,6, АЦ составляет 1,8 мас.% раствора, ГПЦ составляет 1,2 мас.% раствора, метанол составляет 19,6 мас.% раствора, и ацетон составляет оставшиеся 77,4 мас.%.
При включении антиоксидантов в покрывающий раствор, может появиться необходимость в третьем растворителе для обеспечения хорошего диспергирования антиоксиданта в покрытии. Например, композиция АЦ:ПЭГ:вода в соотношении 2,4:1,6:5 содержащая 0,05 мас.% раствора антиоксиданта требует 5 мас.% метанола и 86% ацетона.
Покрытия, образованные из таких покрывающих растворов обычно пористые. Под "пористыми" понимают, что покрытие в сухом состоянии имеет плотность, меньшую плотности такого же материала в непористой форме. Под "непористой формой" понимают материал покрытия, образованный используя покрывающий раствор, не содержащий нерастворитель, или содержащий минимальное количество нерастворителя, необходимое для производства гомогенного покрывающего раствора. Плотность сухого состояния покрытия может быть рассчитана путем деления массы покрытия (определенной из прибавки таблетки в массе перед и после нанесения покрытия) на объем покрытия (рассчитанный путем умножения толщины покрытия, определенной оптической или сканирующей электронной микроскопией, на площадь поверхности таблетки). Пористость покрытия является одним из факторов, приводящих к комбинации высокой проницаемости для воды и высокой прочности покрытия.
Масса покрытия вокруг ядра зависит от композиции и пористости покрытия, но обычно находится в диапазоне от 3 до 30 мас.% от массы непокрытого ядра. Масса покрытия как минимум приблизительно 8 мас.%, обычно является предпочтительной для достаточной прочности, хотя более низкая масса покрытия может быть использована для получения высокого уровня поглощения воды и, как следствие, высокую скорость высвобождения тофацитиниба из дозированной формы.
Хотя пористые покрытия на основе АЦ, ПЭГ или ГПЦ и воды, описанные выше, показывают прекрасные результаты, другие фармацевтически приемлемые материалы могут быть использованы в покрытии, при условии обеспечения требуемой комбинации высокой проницаемости для воды, высокой прочности и легкости производства и нанесения. Также, такие покрытия могут быть плотными, пористыми или "асимметричными," имеющими один или больше плотных слоев, и один или больше пористых слоев, такими как описано в патентах США № 5,612,059 и 5,698,220, включенных в данный документ с помощью ссылок.
Покрытие должно содержать как минимум один канал доставки в сообщении с внутренней и внешней поверхностью покрытия для высвобождения композиции, содержащей лекарственное средство, из дозированной формы. Размер входного отверстия может разниться от приблизительно размера частиц лекарственного средства, таким образом, может быть таким маленьким как 1-100 микрон в диаметре и называться порой, и до приблизительно 5000 микрон в диаметре. Форма канала может быть по сути круговой, иметь форму щели или иметь другую приемлемую форму для легкости производства и обработки. Каналы могут быть образованы после нанесения покрытия механической или термической обработкой или лучом света (например, лазером), пучком частиц или другим высокоэнергетическим источником, или могут быть образованы in situ разрушением небольшой части покрытия. Такой разрыв можно контролировать намеренным введением относительно небольшой части в покрытие. Каналы доставки также могут быть образованы in situ путем эрозии пробки из растворимого в воде материала или путем разрушения части растворителя покрытия над впадиной в ядре. Каналы доставки могут быть образованы путем покрытия ядра таким образом, что один или больше небольших участком остаются без покрытия. Кроме того, канал доставки может быть представлен большим количеством отверстий или пор, образованных на протяжении нанесения покрытия, как в случае покрытий из асимметричных мембран, детально описанных в данном документе, и покрытий, описанных в патентах США № 5,612,059 и 5,698,220, включенных в данный документ с помощью ссылок. Когда каналы доставки представлены порами, возможно присутствие многочисленных пор размером от 1 микрона дo более чем 100 микрон. На протяжении использования, одна или больше таких пор могут увеличиться под влиянием гидростатического давления, вырабатываемого на протяжении использования. Как минимум один канал доставки должен быть сформирован на стороне покрытия, смежным с композицией, содержащей тофацитиниб, так что композиция, содержащая тофацитиниб, будет экструдирована из канала доставки вследствие разбухания разбухающей в воде композиции. Выяснили, что в процессе образования каналов доставки также могут образовываться отверстия или поры в покрытии, смежном с разбухающей в воде композицией.
Покрытие может необязательно содержать канал с сообщением с разбухающей в воде композицией. Такой канал доставки обычно не изменяет характеристики дозированной формы тофацитиниба, и может обеспечить производственные преимущества. Считается, что разбухающие в воде композиции, такие как композиции, содержащие ПЭO с молекулярной массой 3,000,000-8,000,000 дальтон, являются слишком вязкими для значительного выхода из канала. В дозированных формах, где каналы доставки высверлены механически или лазером, таблетка должна быть ориентирована так, чтобы как минимум один канал доставки был сформирован в покрытии, смежном с композицией, содержащей тофацитиниб. Краситель в разбухающей в воде композиции используют для ориентации ядра дозированной формы на протяжении стадии сверления при производстве. Путем обеспечения канала доставки на обоих сторонах дозированной формы, потребность в ориентировки дозированной формы отпадает, и краситель может быть удален из разбухающей в воде композиции.
В другом варианте осуществления, тофацитиниб включен в вариации осмотических устройств доставки, описанных выше, ассиметричная мембранная технология (AMT). Эти устройства описаны в Herbig, et al., J. Controlled Release, 35, 1995, 127-136, и патентах США № 5,612,059 т 5,698,220, как покрытия в осмотических системах доставки лекарственного вещества. Эти AMT системы обеспечивают главное преимущество осмотических устройств с контролируемым высвобождением (надежную доставку лекарственного вещества независимо от расположения в ЖК тракте), а также не требуют стадии сверления отверстия в покрытии, что требуют многие другие осмотические системы. При формировании таких пористых покрытий, нерастворимый в воде полимер объединяют с растворимым в воде порообразующим материалом. Смесь наносят на ядро осмотической таблетки из комбинации воды и растворителя. После высыхания покрытия, инверсия фазы осуществляется посредством образования пористой асимметричной мембраны. Применение AMT систем контролируемого высвобождения лекарственного вещества со сходными физико-химическими характеристиками описано в публикации патентной заявки США № US2007/0248671 и включенной в данный документ с помощью ссылки.
Хотя было описано много материалов для применения в качестве порообразователей для производства асимметричных мембран, все вышеописанные материалы обеспечивают химическую или физическую стабильность системы. А именно, многие из материалов уровня техники являются жидкостями, которые потенциально могут мигрировать на поверхность покрытия на протяжении хранения. Среди твердых материалов описаны как полимерные, так и неорганические материалы. Неорганические материалы могут быть трудными в употреблении по нескольким причинам. А именно, они часто имеют тенденцию к кристаллизации и/или адсорбируют влагу на протяжении хранения. Конкретные полимерные материалы включают производные поливинилпирролидона (ПВП) и полиэтиленгликоля (ПЭГ). Оба этих материала имеют сильную тенденцию к формированию пероксидов и/или формальдегида на протяжении хранения (смотреть, например, Waterman, et al., "Impurities in Drug Products" in Handbook of Isolation and Characterization of Impurities in Pharmaceuticals, S. Ajira and K. M. Alsante, Eds. 2003, pp. 75-85). Многие лекарственные вещества взаимодействуют с такими продуктами разложения полимера, вследствие их внутренней реактивности, а также их тенденции к миграции на протяжении хранения. Однако этот интервал композиций достаточно узок. Патент США 4,519,801 описывает широкий перечень растворимых в воде полимерных компонентов, приемлемых для покрытий в осмотических системах, но не раскрывает, как правильно подобрать растворимые в воде компоненты для систем AMT. Таким образом, остается потребность в новых порообразующих материалах для AMT систем, где порообразующие материалы не вырабатывают реактивные побочные продукты, не кристаллизуются или мигрируют из покрытия на протяжении хранения.
Один аспект данного изобретения относится к дозированной форме, которая содержит (a) ядро, которое содержит как минимум один фармацевтически активный ингредиент и (b) как минимум одно покрытие в виде ассиметричной мембраны, где указанное покрытие включает:
a. один или больше по сути нерастворимых в воде полимера, и
b. один или больше твердых, растворимых в воде полимерных материалов, не содержащих перекись водорода или формальдегид в количестве более чем приблизительно 0,01% м:м после хранения при 40°C/75% ОВ на протяжении 12 недель.
Один аспект данного изобретения также относится к дозированной форме, где дозированная форма доставляет лекарственное вещество в основном благодаря осмотическому давлению. В конкретных вариантах осуществления, данное изобретение относится к дозированной форме, где фармацевтически активный ингредиент представлен тофацитинибом или его фармацевтически приемлемой солью. Нерастворимый в воде полимер, как использовано в данном изобретении, желательно содержит производную целлюлозы, более желательно, ацетат целлюлозы. Твердый, растворимый в воде полимерный материал, как использовано в данном изобретении, содержит полимер, имеет среднемассовую молекулярную массу от 2000 до 50,000 дальтон. В желательных вариантах осуществления, твердый растворимый в воде полимерный материал выбирают из группы, состоящей из следующих: растворимые в воде производные целлюлозы, акация, декстрин, гуаровая смола, мальтодекстрин, альгинат натрия, крахмал, полиакрилаты, поливиниловые спирты и зеин. В конкретных вариантах осуществления, растворимые в воде производные целлюлозы включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и гидроксиэтилцеллюлозу. В определенных вариантах осуществления, твердый растворимый в воде полимерный материал имеет вязкость 5% м:м водного раствора, ниже чем 400 мПа сек. В определенных вариантах осуществления, твердый растворимый в воде полимерный материал имеет вязкость 5% м:м водного раствора, ниже чем 300 мПа сек. В других вариантах осуществления, твердый растворимый в воде полимерный материал имеет температуру размягчения более чем 55°C.
Дозированная форма данного изобретения может иметь форму таблетки или многочисленных частиц. В определенных вариантах осуществления, ядро данного изобретения содержит сахар. Более желательно, сахар означает сорбитол. В определенных вариантах осуществления, нерастворимый в воде полимер означает ацетат целлюлозы, и указанный твердый растворимый в воде полимерный материал означает гидроксипропилцеллюлозу. В определенных желательных вариантах осуществления, дозированная форма изобретения содержит тофацитиниб или его фармацевтически приемлемую соль, в виде фармацевтически активного ингредиента, в то время как нерастворимый в воде полимер представлен ацетатом целлюлозы, а твердый растворимый в воде полимерный материал представлен гидроксипропилцеллюлозой.
Способ данного изобретения охватывает способ, где покрытие наносят из смеси ацетона и воды, используя дражирование. Способ данного изобретения также охватывает способ, где асимметричная мембрана содержит целлюлозы ацетат и гидроксипропилцеллюлозу, которую наносят из смеси ацетона и воды в соотношении приблизительно 9:1 - 6:4, м:м, и более желательно приблизительно 7:3 - 6:4, м:м, используя дражировочный котел. А именно, способ данного изобретения охватывает способ, где ядро содержит тофацитиниб или его фармацевтически приемлемую соль.
В получении покрытия из асимметричной мембраны данного изобретения, нерастворимый в воде компонент асимметричной мембраны предпочтительно получают из производных целлюлозы. А именно, эти производные включают сложные и простые эфиры целлюлозы, конкретно, моно-, ди- и триацил сложные эфиры, где ацильная группа состоит из 2-4 атомов углерода, и простые эфиры низших алкилов целлюлозы, где алкил содержит 1-4 атомов углерода. Сложные эфиры целлюлозы также могут быть смешаны со сложными эфирами, такими как целлюлозы ацетат бутират, или смесь сложных эфиров целлюлозы. Такие же комбинации подходят и для простых эфиров целлюлозы, и включают смеси сложных эфиров целлюлозы и простых эфиров целлюлозы. Другие производные целлюлозы, которые могут быть использованы для получения асимметричных мембран данного изобретения включают нитрат целлюлозы, ацетальдегид диметилцеллюлозы, целлюлозы ацетат этилкарбамат, ацетат фталат целлюлозы, целлюлозы ацетат метилкарбамат, ацетат сукцинат целлюлозы, целлюлозы ацетат диметаминоацетат, целлюлозы ацетат этилкарбонат, целлюлозы ацетат диметаминоацетат, целлюлозы ацетат этилкарбонат, целлюлозы ацетат хлорацетат, целлюлозы ацетат этилоксалат, целлюлозы ацетат метилсульфонат, целлюлозы ацетат бутилсульфонат, целлюлозы ацетат п-толуолсульфонат, цианоацетаты целлюлозы, ацетат тримеллитат целлюлозы, метакрилаты целлюлозы и гидроксипропилметилацетат сукцинат целлюлозы. Особенно желаемый нерастворимый в воде компонент означает ацетат целлюлозы. Особенно желаемые ацетаты целлюлозы включают соединения с содержанием ацетила приблизительно 40%, а гидроксила приблизительно 3,5%. Другие материалы также могут быть использованы для получения ассиметричных мембран, обеспечивая такие материалы, которые, по сути, нерастворимы в воде, образуют пленку и безопасны для применения в фармацевтике.
При получении асимметричных мембран данного изобретения, растворимый в воде полимерный компонент данного изобретения содержит твердые полимерные материалы не образующие перекись водорода или формальдегид на протяжении хранения в течении 12 недель при 40°C./75% относительной влажности, в количестве более чем приблизительно 0,01% м./м. (100 частей на миллион, млн.ч.). Касательно растворимости в воде, твердый полимерный растворимый в воде материал предпочтительно имеет растворимость в воде более чем 0,5 мг/мл; более желательно, более чем 2 мг/мл; и еще более желательно, более чем 5 мг/мл.
Твердый полимерный растворимый в воде материал имеет температуру плавления или размягчения выше комнатной температуры. Предпочтительно, твердый материал имеет температуру плавления или размягчения выше 30°C; более предпочтительно, выше 40°C; и наиболее предпочтительно, выше 50°C. Температуру плавления или размягчения можно определить визуально, используя устройство для определения температуры плавления, или альтернативно, может быть измерена, используя дифференциальную сканирующую калориметрию (ДСК), известную в отрасли. Полимер может быть гомополимером или coполимером. Такие полимеры могут быть природными полимерами, или производными природных продуктов, или полностью синтетическими. Молекулярная масса таких материалов предпочтительно достаточно высока для предотвращения миграции и помощи в образовании пленки, и при этом достаточно мала для обеспечения нанесения покрытия (как описано ниже). Предпочтительный диапазон молекулярной массы данного изобретения составляет от 2000 до 50,000 дальтон (средняя масса). Предпочтительные полимеры, приемлемые как растворимые в воде компоненты ассиметричной мембраны данного изобретения, включают замещенные, растворимые в воде производные целлюлозы, акацию, декстрин, гуаровую смолу, мальтодекстрин, альгинат натрия, крахмал, полиакрилаты, поливиниловые спирты и зеин. Особенно предпочтительные растворимые в воде полимеры включают гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу и поливиниловый спирт.
Тяжело получить асимметричные мембраны, если вязкость покрывающего раствора слишком высокая, поэтому одним из подходов решения данной проблемы является использование более разведенных растворов полимера. В результате поведения фаз покрывающего раствора, который содержит как растворимые в воде, так и растворимые в органике компоненты, существует минимальная граница концентрации растворимых в воде полимеров для обеспечения коммерциализированного процесса. По этой причине, желательно чтобы растворимые в воде полимеры не имели слишком большую вязкость. Вязкость определяют при температуре 25°C, используя вискозиметр Brookfield LVF (производства Brookfield Engineering Corp., Middleboro, Mass.) с комбинациями оси и скорости, в зависимости от уровня вязкости 5% (м:м) водных растворов. Предпочтительные растворимые в воде полимеры имеют вязкость 5% (м:м) растворов ниже чем 400 мПа сек; более желательно, менее 300 мПа сек.
Используя вышеуказанные критерии, особенно предпочтительные растворимые в воде полимеры включают гидроксипропилцеллюлозу и гидроксиэтилцеллюлозу с вязкостью для 5% (м:м) ниже чем 300 мПа сек, коммерчески доступные примеры таких полимеров включают Klucel EF.TM. и Natrasol LR.TM., оба производства Aqualon Division of Hercules Corp., Хоупвелл, Вирджиния.
Стабильность растворимых в воде твердых полимерных материалов к образованию перекиси водорода можно измерить хранением полимера в печи с температурой и относительной влажностью (ОВ) 40°C и 75% ОВ, соответственно. Полимер должен храниться в печи в "открытых" условиях. Полимер должен храниться как минимум 12 недель. Уровень перекиси водорода можно определить, как описано в G. M. Eisenberg, "Colorimetric determination of hydrogen peroxide" in Ind. Eng. Chem. (Anal. Ed.), 1943, 15, 327-328. В таких условиях хранения, приемлемые полимерные материалы данного изобретение имеют уровень перекиси водорода ниже 100 частей на миллион (млн.ч.); более желательно, ниже 50 млн.ч.; и наиболее желательно, ниже 10 млн.ч.
Аналогично, стабильность растворимых в воде полимеров к образованию формальдегида можно измерить хранением полимера в печи с температурой 40°C и 75% ОВ. Полимер должен храниться в герметичном контейнере для предупреждения потери летучего формальдегида. Полимер должен храниться как минимум 12 недель. Уровень формальдегида можно определить, как описано в M. Ashraf-Khorassani, et al., "Purification of pharmaceutical excipients with supercritical fluid extraction" in Pharm. Dev. Tech. 2005, 10, 1-10. В таких условиях хранения, приемлемые растворимые в воде полимерные материалы данного изобретение имеют уровень формальдегида ниже 100 млн.ч., более желательно, ниже 50 млн.ч., и наиболее желательно, ниже 10 млн.ч.
Специалисту в отрасли понятно, что композиция ассиметричной мембраны может содержать небольшое количество других материалов без значительного изменения ее функции или изменения природы данного изобретения. Такие добавки включают глиданты (например, тальк и оксид кремния) и пластификаторы (например, триэтилцитрат и триацетин), которые обычно добавляют в случае необходимости в количестве менее чем приблизительно 5 % (м:м) от покрытия.
Специалисту в отрасли понятно, что активные фармацевтические ингредиенты могут находиться в форме фармацевтически приемлемых солей. Ядра данного изобретения также могут содержать солюбилизаторы. Такие добавки включают pH-буферизирующие добавки для поддержания ядра на уровне pH, при котором активный фармацевтический ингредиент имеет достаточно высокую растворимость для выкачивания из дозированной формы в раствор. Активный фармацевтический ингредиент может находиться в ядре в количестве от приблизительно 0,1% (м:м) до приблизительно 75% (м:м).
Ядро может содержать осмотические агенты, которые помогают обеспечить движущую силу для доставки лекарственного вещества. Такие осмотические агенты включают растворимые в воде сахара и соли. Особенно предпочтительный осмотический агент включает маннит или хлорид натрия.
Ядро системы AMT может содержать другие добавки для обеспечения таких преимуществ как стабильность, обрабатываемость и производительность системы. Стабилизирующие наполнители включают pH-модифицирующие ингредиенты, антиоксиданты, хелатирующие агенты, и другие подобные добавки, известные в отрасли. Наполнители, улучшающие обрабатываемость включают агенты, помогающие текучести, прессованию или экструзии. Текучесть можно улучшить такими агентами как тальк, стеараты и оксид кремния. Текучесть также можно улучшить грануляцией лекарственного средства и наполнителей, как известно в отрасли. Такая грануляция часто выигрывает от добавления связующих, таких как гидроксипропилцеллюлоза, крахмал и поливинилпироллидон (повидон). Прессование можно улучшить добавлением разбавителей к композиции. Примеры разбавителей включают лактозу, маннит, микрокристаллическую целлюлозу и т.д., как известно в отрасли. Для ядер, полученных экструзией, важны характеристики плавления наполнителей. Обычно, желательно чтобы такие наполнители имели температуру плавления ниже приблизительно 100°C. Примеры соответствующих наполнителей для процесса плавления включают эстерифицированные глицерины и стеариловый спирт. Для прессованных дозированных форм, обрабатываемость улучшают добавлением лубрикантов. Особенно предпочтительным лубрикантом является стеарат магния.
Ядра можно получить, используя стандартные способы таблетирования, как известно в отрасли. Такие способы включают наполнение пресс формы порошком, затем прессование, используя подходящие пуансоны. Ядра также можно получить экструзией. Экструзия хорошо подходит для получения маленьких ядер (множественных частиц). Предпочтительной экструзией является плавление-распыление-отвердение как описано в WO2005/053653A1, включенной с помощью ссылки. Ядра также можно получить нанесением слоев лекарственного вещества на ядра. Такие ядра предпочтительно сделаны из сахара или микрокристаллической целлюлозы. Лекарство может быть нанесено на ядра распылением, предпочтительно в кипящем слое, как известно в отрасли.
В практике данного изобретения, ядра покрыты асимметричной мембраной с помощью любой техники, которая может обеспечить асимметричная мембрана как покрытие поверх всего ядра. Предпочтительными способами нанесения покрытия являются дражирование и нанесения покрытия в кипящем слое. В обоих способах, нерастворимый в воде полимер и растворимый в воде полимер, а также любые другие добавки сначала растворяют или диспергируют в подходящем растворителе или комбинации растворителей. Для получения приемлемой пористой мембраны, покрывающий растворитель должен быть оптимизирован для продуктивности. Обычно, растворители выбирают таким образом, чтоболее летучий растворитель является предпочтительным для нерастворимого в воде полимерного компонента. В результате в процессе нанесения покрытия, нерастворимый в воде полимерный компонент осаждается из раствора. Предпочтительные растворители и соотношение растворителей определяют путем исследования многокомпонентной растворимости системы. Предпочтительной смесью растворителей является ацетон и вода, в соотношении от приблизительно 9:1 до 6:4, м:м.
В предпочтительном варианте осуществления данного изобретения, тофацитиниб включен в монолитное устройство осмотической доставки, известное как система экструдируемого ядра, так что композиция, содержащая тофацитиниб, должна содержать увеличивающие вязкость полимеры и осмотически активные агенты, и может необязательно включать агенты, улучшающие растворимость и/или антиоксиданты. Монолитная таблетка или капсула окружены полупроницаемой мембраной, содержащей одно или больше отверстий, сделанных в дозированной форме посредством таких технологий, как лазерное сверление. Увеличивающие вязкость полимеры подвешивают или охватывают лекарственное вещество для облегчения доставки лекарственного средства через каналы доставки. Не привязываясь к определенной теории, считается, что при впитывании воды внутрь дозированной формы, увеличивающий вязкость полимер имеет достаточную вязкость для подвешивания или охватывания лекарственного вещества, при этом оставаясь достаточно жидким для прохождения увеличивающего вязкость полимера через каналы доставки вместе с лекарственным веществом. Количество увеличивающего вязкость полимера, содержащегося в композиции, содержащей тофацитиниб, находится в диапазоне от приблизительно 2 мас.% до приблизительно 20 мас.%, желательно от приблизительно 3 до приблизительно 15%, и более желательно от приблизительно 4 мас.% до приблизительно 10 мас.%. Увеличивающий вязкость полимер может состоять из одного материала или смеси материалов. Не-поперечно сшитый полиэтиленоксид (ПЭO) и гидроксиэтилцеллюлоза (ГЭЦ) могут быть использованы в качестве увеличивающего вязкость полимера. ГЭЦ является предпочтительным в качестве увеличивающего вязкость полимер. Молекулярная масса ГЭЦ может составлять от приблизительно 300,000 до приблизительно 2,000,000, более желательно от приблизительно 700,000 до приблизительно 1,500,000.
Композиция, содержащая тофацитиниб, также включает осмотически эффективные растворенные вещества, которые называют "осмогены" или "осмагенты." Количество осмагента в композиции, содержащей тофацитиниб, находится в диапазоне от приблизительно 15 мас.% до приблизительно 95 мас.%, желательно от приблизительно 40 мас.% до приблизительно 90 мас.%, более желательно от приблизительно 60% до приблизительно 85%, и наиболее желательно от приблизительно 70% до приблизительно 85%, композиции, содержащей тофацитиниб. Типичные классы приемлемых осмагентов включают растворимые в воде соли, сахара, органические кислоты, и другие низкомолекулярные органические соединения, способные поглощать воду, образуя градиент осмотического давления поперек барьера окружающего покрытия. Типичные соли включают сульфат магния, хлорид магния, хлорид кальция, хлорид натрия, хлорид лития, сульфат калия, карбонат натрия, сульфит натрия, сульфат лития, хлорид калия и сульфат натрия. Предпочтительные соли включают как хлорид натрия, так и хлорид калия. Предпочтительные органические кислоты включают следующие: аскорбиновая кислота, 2-бензолкарбоновая кислота, бензойная кислота, фумаровая кислота, лимонная кислота, малеиновая кислота, сербациновая кислота, сорбиновая кислота, эдипиновая кислота, эдитовая кислота, глутаминовая кислота, толуолсульфоновая кислота, и виннокаменная кислота. Предпочтительные сахара включают маннит, сахарозу, сорбитол, ксилитол, лактозу, декстрозу и трегалозу. Более предпочтительным сахаром является сорбитол. Осмагенты могут быть использованы по одному или в комбинации с двумя или больше осмагентами.
Композиция, содержащая тофацитиниб, также может включать агенты, улучшающие растворимость, или солюбилизаторы, стимулирующие растворимость лекарственного средства в воде, присутствующие в диапазоне от приблизительно 0 до приблизительно 30 мас.% композиции, содержащей тофацитиниб. Солюбилизаторы, приемелемые с тофацитинибом, включают органические кислоты и соли органических кислот, частичные глицериды, например, частично эстерифицированные производные глицерина, включая глицериды, моноглицериды, диглицериды, производные глицеридов, сложные эфиры полиэтиленгликоля, сложные эфиры полипропиленгликоля, сложные эфиры многоатомного спирта, эфиры полиоксиэтилена, сложные эфиры сорбитана, полиоксиэтилен сложные эфиры сорбитана и карбонатные соли.
Предпочтительным классом солюбилизаторов являются органические кислоты. В виду того, что тофацитиниб является основанием, которое солюбилизируют протонированием, и виду того, что его растворимость в водной окружающей среде с pH=5 или выше понижена, считается, что добавление органической кислоты к композиции, содержащей тофацитиниб, помогает солюбилизации и улучшает абсорбцию тофацитиниба. Даже небольшое снижение pH водного раствора при высоких pH приводит к существенному росту растворимости тофацитиниба. Органические кислоты также могут стимулировать стабильность на протяжении хранения перед введением в среду использования, благодаря их способности поддерживать тофацитиниб в протонированном состоянии.
Существует много факторов, которые необходимо учитывать при выборе соответствующей органической кислоты для применения в качестве солюбилизатора с тофацитинибом в осмотической дозированной форме. Кислота не должна неблагоприятно взаимодействовать с тофацитинибом, должна быть соответствующе растворима в воде, и должна обеспечивать хорошие производственные характеристики.
Соответственно, выяснили, что предпочтительный подкласс органических кислот, соответствующий таким критериям, включает лимонную, янтарную, фумаровую, адипиновую, яблочную и виннокаменную кислоты. Лимонная, яблочная и виннокаменная кислота имеют преимущество в высокой растворимости в воде и высоком осмотическом давлении. Янтарная и фумаровая кислота дают комбинацию умеренной растворимости и умеренного осмотического давления.
Слой и/или мембрана, контролирующая функциональную скорость, композиции, содержащей тофацитиниб, может необязательно содержать антиоксидант, такой как, но не ограничиваясь следующими: BHT, BHA, метабисульфит натрия, пропил галат, глицерин, витамин E, лимонная кислота или аскорбил пальмитат. Антиоксидант может присутствовать в диапазоне от 0 дo 10 мас.% слоя и/или разбухающего в воде слоя и/или мембрана, контролирующая функциональную скорость, композиции, содержащей тофацитиниб. Дополнительные примеры антиоксидантов смотреть в C.-M. Andersson, A. Hallberg, and T. Hoegberg. Advances in the development of pharmaceutical antioxidantes. Advances in Drug Research. 28:65-180, 1996.
Композицию, содержащую тофацитиниб, получают смешиванием увеличивающего вязкость полимера и других наполнителей с образованием однородной смеси. Для получения однородной смеси, желательно подвергнуть компоненты влажной или сухой грануляции, или сухому перемешиванию, используя известные в отрасли способы.
Таблетирование
Ядро получают, сначала помещением смеси композиции, содержащей тофацитиниб, в таблеточный пресс и прессованием для формирования ядра. Форма таблетки может быть любой, известной в отрасли формой. Предпочтительные формы таблетки включают SRC (стандартная круглая вогнутая), овальная, модифицированная овальная, капсула, таблетка в виде капсулы и миндальная. Более предпочтительные формы таблетки включают овальную, модифицированную овальную, таблетку в виде капсулы и капсулу.
Нанесение покрытия
После образования ядра наносят полупроницаемое покрытие. Покрытие должно иметь высокую проницаемость для воды и высокую прочность, и в тоже время его должно быть легко изготовлять и наносить. Высокая проницаемость для воды требуется для попадания воды в ядро в достаточном количестве. Высокая прочность требуется для того чтобы удостовериться, что покрытие не лопнет при разбухании ядра во время впитывания воды, что приведет к неконтролируемой доставки содержимого ядра. Наконец, покрытие должно иметь высокую воспроизводимость и выход.
Существенно чтобы покрытие имело как минимум один канал доставки в сообщении с внешней и внутренней поверхностью покрытия для доставки композиции, содержащей тофацитиниб. Более того, покрытие не должно растворяться и эродировать на протяжении высвобождения композиции, содержащей тофацитиниб, обычно имеется в идее, что оно должно быть водо-нерастворимым, так что тофацитиниб по сути полностью доставляется через каналы доставки, в отличии от доставки посредством проникновения через покрытие.
Покрытия с такими характеристиками получают, используя гидрофильные полимеры, такие как пластифицированные и непластифицированные сложные эфиры целлюлозы, простые эфиры и сложные эфиры-простые эфиры. Особенно приемлемые полимеры включают целлюлозы ацетат (АЦ), целлюлозы ацетат бутират (АЦБ), и этилцеллюлозу (ЭЦ). Один набор полимеров включает ацетаты целлюлозы с содержанием ацетила 25-42%. Один типичный полимер представлен АЦ с содержанием ацетила 39,8%, а именно, АЦ 398-10 (Eastman Fine Chemicals, Kingsport, Tenn.). АЦ 398-10 имеет среднюю молекулярную массу приблизительно 40,000 дальтон. Другой типичный АЦ с содержанием ацетила 39,8% является высокомолекулярным АЦ со средней молекулярной массой более чем приблизительно 45,000, а именно, АЦ 398-30 (Eastman Fine Chemical), который имеет среднюю молекулярную массу 50,000 дальтон.
Нанесение покрытия осуществляют обычным образом, сначала образованием покрывающего раствора, а затем нанесением покрытия путем погружения, нанесения покрытия в кипящем слое, или дражированием. Для осуществления указанного, покрывающий раствор одержит полимер и растворитель. Типичные растворители, приемлемые для целлюлозных полимеров включают ацетон, метил ацетат, этилацетат, изoпропил ацетат, н-бутил ацетат, метил изoбутил кетон, метил пропил кетон, моноэтиловый эфир этиленгликоля, этиленгликоля моноэтилацетат, метилeн дихлорид, этилен дихлорид, пропилeн дихлорид, нитроэтан, нитропропан, тетрахлорэтан, 1,4-диоксан, тетрагидрофуран, диглим, и их смеси. Покрывающий раствор обычно содержит 2-15 мас.% полимера.
Покрывающий раствор также может содержать порообразователи или нерастворители в любом количестве, пока полимер остается растворимым в условиях, использованных для формирования покрытия, и пока покрытие остается проницаемым для воды и имеет достаточную прочность. Порообразователи и их применение в получении покрытий описано в патентах США № 5,698,220 и 5,612,059, включенных в данный документ с помощью ссылок. Термин "порообразователь", как использовано в данном документе, относится к материалу, добавляемому к покрывающему раствору, который не летуч или слабо летуч, по сравнению с растворителем, так что он остается как часть покрытия после нанесения покрытия, но преимущественно разбухает или растворяется в воде, так что в водной среде использования обеспечивает образование наполненного водой или разбухшего в воде канала или "поры" для прохождения воды, таким образом, улучшая проницаемость покрытия для воды. Приемлемые порообразователи включают, но не ограничиваются гидроксипропилцеллюлозой (ГПЦ), полиэтиленгликолем ("ПЭГ"), ПВП и ПЭO. Для получения комбинации высокой проницаемости для воды и высокой прочности при использовании ПЭГ или ГПЦ в качестве порообразователя, массовое соотношение АЦ:ПЭГ или АЦ:ГПЦ должно находиться в диапазоне от приблизительно 6:4 до приблизительно 9:1. АЦ:ГПЦ является предпочтительной покрывающей композицией. Предпочтительное соотношение АЦ:ГПЦ находится в диапазоне от 6:4 дo 7:3. Предпочтительное массовое соотношение АЦ:ПЭГ находится в диапазоне от 6:4 дo 7:3.
Добавление нерастворителя, такого как вода, к покрывающему раствору приводит к исключительному результату. Под "нерастворителем" понимают любой материал, добавляемый к покрывающему раствору, который существенно растворяется в покрывающем растворе и понижает растворимость полимерного покрытия или полимеров в растворителе. В целом, функцией нерастворителя является придание пористости полученному материалу. Как описано ниже, пористые покрытия имею более высокую проницаемость для воды, чем эквивалентная масса покрытия той же композиции, но без пор, и эта пористость указывает на уменьшение плотности покрытия (масса/объем). Не смотря на то, что исследователь не хотят привязываться к какому-то конкретному механизму образования пор, обычно считают, что добавление нерастворителя придает пористости покрытию на протяжении выпаривания растворителя, вызывая разделения жидкости - жидкой фазы покрывающего раствора перед отвердением. Пригодность и количество определенного материала кандидата для использования в качестве нерастворителя может быть определена постепенным добавлением кандидата нерастворителя к покрывающему раствору до появления мутности. Если этого не случается при добавлении количества, составляющего до приблизительно 50 мас.% покрывающего раствора, считается, что данный кандидат не пригоден в качестве нерастворителя. При наблюдении помутнения, называется "температура помутнения", приемлемым уровнем нерастворителя для максимальной пористости является количество ниже температуры помутнения. Для растворов ацетона, содержащих 7 мас.% АЦ и 3 мас.% ПЭГ, температура помутнения составляет приблизительно 23 мас.% воды. Если желательна низкая пористость, количество нерастворителя можно уменьшить до желаемого уровня.
Приемлемые нерастворители являются любыми материалами с приемлемой растворимостью в растворителе, и растворимостью ниже, чем растворимость полимерного покрытия в растворителе. Предпочтительный нерастворитель зависит от растворителя и покрывающего полимера. В случае использования летучего полярного покрывающего растворителя, такого как ацетон, приемлемые нерастворители включают воду, глицерин, спирты, такие как метанол или этанол.
Используя АЦ 398-10, массовые соотношения покрывающего раствора АЦ:ПЭГ3350:вода составляют 2,4:1,6:5, 2,8:1,2:5, 3,2:0,8:5 и 3,6:0,4:5, с остатком раствора, содержащим растворитель, такой как ацетон. Таким образом, например, в растворе с массовым соотношением АЦ:ПЭГ3350:вода равным 2,8:1,2:5, АЦ составляет 2,8 мас.% раствора, ПЭГ 3350 составляет 1,2 мас.% раствора, вода составляет 5 мас.% раствора, и ацетон составляет оставшиеся 91 мас.%. Также, покрывающий раствор с массовым соотношением АЦ:ГПЦ:вода равным 1,2:0,8:9,8, 2,4:1,6:19,6, 1,6:0,4:4,9 и 3,2:0,8:9,8, с остатком раствора, содержащим растворитель, такой как ацетон. Таким образом, например, в растворе с массовым соотношением АЦ:ГПЦ:вода равным 1,2:0,8:10, АЦ составляет 1,2 мас.% раствора, ГПЦ составляет 0,8 мас.% раствора, вода составляет 10 мас.% раствора, и ацетон составляет оставшиеся 88 мас.%. Также покрывающий раствор с массовым соотношением АЦ:ГПЦ:метанол равным 1,8:1,2:19,6, 2,4:1,6:19,6, 1,6:0,4:4,9 и 3,2:0,8:9,8, с остатком раствора, содержащим растворитель, такой как ацетон. Таким образом, например, в растворе с массовым соотношением АЦ:ГПЦ:метанол на уровне 1,8:1,2:19,6, АЦ составляет 1,8 мас.% раствора, ГПЦ составляет 1,2 мас.% раствора, метанол составляет 19,6 мас.% раствора, и ацетон составляет оставшиеся 77,4 мас.%.
При включении антиоксидантов в покрывающий раствор, может появиться необходимость в третьем растворителе для обеспечения хорошего диспергирования антиоксиданта в покрытии. Например, композиция АЦ:ПЭГ:вода в соотношении 2,4:1,6:5 содержащая 0,05 мас.% раствора требует 5 мас.% метанола и 86% ацетона.
Покрытия, образованные из таких покрывающих растворов обычно пористые. Под "пористыми" понимают, что покрытие в сухом состоянии имеет плотность, меньшую плотности такого же материала в непористой форме. Под "непористой формой" понимают материал покрытия, образованный используя покрывающий раствор, не содержащий нерастворитель, или содержащий минимальное количество нерастворителя, необходимое для производства гомогенного покрывающего раствора. Плотность сухого состояния покрытия может быть рассчитана путем деления массы покрытия (определенной из прибавки таблетки в массе перед и после нанесения покрытия) на объем покрытия (рассчитанный путем умножения толщины покрытия, определенной оптической или сканирующей электронной микроскопией, на площадь поверхности таблетки). Пористость покрытия является одним из факторов, приводящих к комбинации высокой проницаемости для воды и высокой прочности покрытия.
Масса покрытия вокруг ядра зависит от композиции и пористости покрытия, но обычно находится в диапазоне от 3 до 30 мас.% от массы непокрытого ядра. Масса покрытия как минимум приблизительно 8 мас.%, обычно является предпочтительной для достаточной прочности, хотя более низкая масса покрытия может быть использована для получения высокого уровня поглощения воды и, как следствие, высокую скорость высвобождения тофацитиниба из дозированной формы. Для дозированных форм, содержащих тофацитиниб, прирост массы покрытия в 5-10% является предпочтительным для обеспечения желаемого высвобождения.
Хотя пористые покрытия на основе АЦ, ПЭГ или ГПЦ и воды, описанные выше, показывают прекрасные результаты, другие фармацевтически приемлемые материалы могут быть использованы в покрытии, при условии обеспечения требуемой комбинации высокой проницаемости для воды, высокой прочности и легкости производства и нанесения. Также, такие покрытия могут быть плотными, пористыми или "асимметричными", имеющими один или больше плотных слоев, и один или больше пористых слоев, такими как описано в патентах США № 5,612,059 и 5,698,220, включенных в данный документ с помощью ссылок.
Покрытие должно содержать как минимум один канал доставки в сообщении с внутренней и внешней поверхностью покрытия для высвобождения композиции, содержащей лекарственное средство, из дозированной формы. Размер входного отверстия может разниться от приблизительно размера частиц лекарственного средства, таким образом, может быть таким маленьким как 1-100 микрон в диаметре и называться порой, и до приблизительно 5000 микрон в диаметре. Форма канала может быть по сути круговой, иметь форму щели или иметь другую приемлемую форму для легкости производства и обработки. Каналы могут быть образованы после нанесения покрытия механической или термической обработкой или лучом света (например, лазером), пучком частиц или другим высокоэнергетическим источником, или могут быть образованы in situ разрушением небольшой части покрытия. Такой разрыв можно контролировать намеренным введением относительно небольшой части в покрытие. Каналы доставки также могут быть образованы in situ путем эрозии пробки из растворимого в воде материала или путем разрушения части растворителя покрытия над впадиной в ядре. Каналы доставки могут быть образованы путем покрытия ядра таким образом, что один или больше небольших участком остаются без покрытия. Кроме того, канал доставки может быть представлен большим количеством отверстий или пор, образованных на протяжении нанесения покрытия, как в случае покрытий из асимметричных мембран, детально описанных в данном документе, и покрытий, описанных в патентах США № 5,612,059 и 5,698,220, включенных в данный документ с помощью ссылок. Когда каналы доставки представлены порами, возможно присутствие многочисленных пор размером от 1 микрона дo более чем 100 микрон. На протяжении использования, одна или больше таких пор могут увеличиться под влиянием гидростатического давления, вырабатываемого на протяжении использования. Расположение каналов доставки может быть где угодно на поверхности таблетки. Предпочтительное расположение каналов доставки включает лицевую поверхность таблетки и таблеточную ленту. Более предпочтительное расположение включает приблизительно центр таблеточной ленты для круглых таблеток SRC-формы, и приблизительно центр таблеточной ленты вдоль главной оси и/или приблизительно центр таблеточной ленты вдоль малой оси таблеточной ленты для капсулы, таблетки в виде капсулы, овальной или модифицированной овальной таблетки. Наиболее предпочтительное расположение каналов доставки включает приблизительный центр таблеточной ленты вдоль главной оси таблеточной ленты для капсулы, таблетки в виде капсулы, овальной или модифицированной овальной таблетки.
Система резервуаров с замедленным высвобождением
Другой класс дозированных форм тофацитиниба с замедленным высвобождением данного изобретения включает мембрана-модерируемые или резервуарные системы. В этом классе, резервуар тофацитиниба окружен мембраной, ограничивающей скорость высвобождения. Тофацитиниб перемещается по мембране с помощью хорошо известного механизма переноса массы, включая, но не ограничиваясь растворением в мембране, после чего диффузией через мембрану или диффузией через наполненные жидкостью поры внутри мембраны. Эти дозированные формы с индивидуальной системой резервуаров могут быть большими, как в случае таблетки, содержащей один большой резервуар, или многочисленные частицы, как в случае капсулы, содержащей множество частиц, каждая из которых покрыта мембраной. Покрытие может быть непористым, но проницаемым для тофацитиниба (например, тофацитиниб может диффундировать напрямую через мембрану), или пористым. В других вариантах осуществления изобретения, определенный механизм транспорта не является критичным.
Покрытия с замедленным высвобождением, известные в отрасли, могут использоваться для получения мембраны, особенно полимерные покрытия, такие как сложные или простые эфиры целлюлозы, акриловый полимер, или смесь полимеров. Предпочтительные материалы включают этилцеллюлозу, целлюлозы ацетат и целлюлозы ацетат бутират. Полимер может быть нанесен в виде раствора в органическом растворителе, или в виде водной дисперсии или латекса. Процедура нанесения покрытия может быть проведена на стандартном оборудовании, таком как аппарат для нанесения покрытий в кипящем слое, установка Вюрстера или ротационная установка для нанесения покрытий.
В случае необходимости, проницаемость покрытия может быть скорректирована смешиванием двух или больше материалов. Приемлемый способ подгонки пористости покрытия включает добавление заранее установленного количества хорошо измельченного растворимого в воде материала, такого как сахара или соли или растворимые в воде полимеры, к раствору или дисперсии (например, водному латексу) полимера, образующего мембрану. Когда дозированная форма вводиться в водную среду ЖК тракта, эти растворимые в воде добавки в мембране вымываются из мембраны, оставляя поры, что способствует высвобождению лекарственного средства. Мембранное покрытие также может быть модифицировано добавлением известных в отрасли пластификаторов.
Приемлемый вариант способа нанесения мембранного покрытия включает растворение покрывающего полимера в смеси растворителей, выбранных таким образом, что при высыхании покрытия, в нанесенном покрывающем растворе происходит обращение фазы, что приводит к образованию мембраны с пористой структурой. Многочисленные примеры системы нанесения покрытия такого типа приведены в Европейском патенте 0 357 369 B1, опубликованном 07.03.1990, включенном с помощью ссылки.
Морфология мембраны не имеет критической важности, до тех пор пока сохраняются характеристики проницаемости, приведенные выше. Мембрана может быть аморфной или кристаллической. Она может иметь любую категорию морфологии, полученную любым способом, и может быть, например, межлицевой-полимеризованной мембраной (которая содержит тонкую ограничивающую оболочку на пористой подложке), пористой гидрофильной мембраной, пористой гидрофобной мембраной, мембраной из гидрогеля, ионной мембраной, и может состоять из других таких материалов, характеризующих контролированной проницаемостью для тофацитиниа.
Приемлемой резервуарной системой является капсула, имеющая оболочку, содержащую материал ограничивающей скорость мембраны, включая любые материалы мембран, описанные ранее, и заполненная композицией тофацитиниба. Особенным преимуществом такой конфигурации является то, что капсула может быть получена независимо от композиции лекарственного средства, таким образом, условия производства, которые могли негативно сказаться на лекарственном веществе, могут быть использованы для получения капсулы. Один вариант осуществления представлен капсулой, имеющей оболочку, сделанную из пористого или проницаемого полимера, полученного термической обработкой. Другой вариант осуществления представлен оболочкой капсулы в виде асимметричной мембраны; например, мембрана, имеющая тонкий поверхностный слой на одной поверхности, и большая часть ее толщины состоит из проницаемого пористого материала. Способ получения капсулы с асимметричной мембраной включает инверсию фаз с заменой растворителя, где раствор полимера, нанесенный на форму в виде капсулы, делится на фазы путем замены растворителя смешивающимся нерастворителем. Примеры асимметричных мембран, приемлемых для данного изобретения, описаны в вышеуказанном Европейском патенте 0 357 369 B1.
Другой вариант осуществления класса резервуарной системы включает многочисленные частицы, где каждая частица покрыта полимером, разработанным для обеспечения замедленного высвобождения тофацитиниба. Многочисленные частицы содержат тофацитиниб и один или больше наполнителей, что необходимо для изготовления и технических характеристик. Размер индивидуальных частиц, как было указано ранее, обычно составляет приблизительно 50 микрон - 3 мм, хотя небольшие отклонения от указанных размеров также подходят. В целом, частицы содержат тофацитиниб и один или больше связующих. Обычно желательно производить дозированные формы маленького размера, чтобы их было легко глотать, предпочтительными являются частицы, содержащие большую фракцию тофацитиниба, по сравнению с наполнителем. Связующие, приемлемые для изготовления таких частиц включают микрокристаллическую целлюлозу (например, Avicel.RTM., FMC Corp.), гидроксипропилцеллюлозу (ГПЦ), гидроксипропилметилцеллюлозу (ГПМЦ), и похожие материалы или их комбинации. В целом, связующие, приемлемые для грануляции и таблетирования, такие как крахмал, прежелатинизированный крахмал и поли(N-винил-2-пирролидинон) (ПВП) также можно использовать для формирования множественных частиц.
Резервуарная система множественных частиц тофацитиниба может быть получена, используя известные в отрасли технологии, включая, но не ограничиваясь технологиями экструзии и окатывания, влажной грануляции, грануляции в кипящем слое, и роторной грануляции. Кроме того, частицы также могут быть получены созданием композиции тофацитиниба (лекарственное вещество + наполнители) поверх ядра (например, зерно нонпарель) путем нанесения слоя лекарственного вещества, например, нанесением порошка или нанесением композиции тофацитиниба распылением раствора или дисперсии тофацитиниба в подходящем растворе связующего на ядра в кипящем слое, с помощью установки Вюрстера или роторного устройства. Примером приемлемой композиции и способа является распыление дисперсии композиции тофацитиниб/гидроксипропилцеллюлоза в воде. Преимущественно, тофацитиниб может быть загружен в водную композицию сверх предела его растворимости в воде.
Способом производства ядер многочисленных частиц этого варианта осуществления является экструзия/окатывание, как описывалось ранее для множественных частиц матрицы. Другой процесс и композиция для этого способа включает использование воды для влажной смеси из приблизительно 5-75% микрокристаллической целлюлозы и соответственно приблизительно 95-25% тофацитиниба. В другом варианте осуществления, способ включает применение воды для влажной смеси из приблизительно 5-30% микрокристаллической целлюлозы и соответственно приблизительно 5-70% тофацитиниба.
Покрытия с замедленным высвобождением известные в отрасли, особенно полимерные покрытия, могут применяться для производства мембраны, как описывалось ранее для резервуарных систем. Приемлемые и предпочтительные полимерные покрытия, оборудование и способы нанесения покрытий также включают ранее описанные.
Скорость высвобождения тофацитиниба из покрытых множественных частиц также может контролироваться такими факторами, как композиция и содержание связующего в ядре, содержащем лекарственное средство, толщина и проницаемость покрытия, и соотношение площади поверхности к объему множественных частиц. Специалисту в отрасли понятно, что увеличение толщины покрытия уменьшит скорость высвобождения, в то время как увеличение проницаемости покрытия или соотношения площади поверхности к объему множественных частиц увеличит скорость высвобождения. В случае необходимости, проницаемость покрытия может корректироваться смешиванием двух или более материалов. Приемлемые серии покрытий включают смеси нерастворимых в воде и растворимых в воде полимеров, например, этилцеллюлоза и гидроксипропилметилцеллюлоза, соответственно. Приемлемая модификация покрытия включает добавление хорошо измельченного растворимого в воде материала, такого как сахара или соли. При помещении в водную среду, эти растворимые в воде добавки вымываются из мембраны, оставляя поры, которые улучшают доставку лекарственного средства. Мембранное покрытие также может быть модифицировано добавлением пластификаторов, известных специалистам в отрасли. Другим приемлемым вариантом мембраны является смесь растворителей, выбранная таким образом, что при высыхании покрытия, в нанесенном покрывающем растворе происходит обращение фазы, что приводит к образованию мембраны с пористой структурой.
Другой вариант осуществления представлен многочисленными частицами, содержащими приблизительно 5-50 % тофацитиниба, где индивидуальны частицы покрыты водной дисперсией этилцеллюлозы, которая высыхает с образованием непрерывной пленки.
Другой вариант осуществления представлен ситуацией, когда частицы тофацитиниба имеют размер менее чем приблизительно 400 микрон и покрыты фазоинверсной мембраной из этилцеллюлозы или ацетата целлюлозы.
Другой вариант осуществления представлен ситуацией, когда частицы тофацитиниба имеют размер менее чем приблизительно 400 микрон и покрыты водной дисперсией этилцеллюлозы, которая высыхает с образованием непрерывной пленки.
Другой вариант осуществления представлен ситуацией, когда частицы тофацитиниба имеют размер менее чем приблизительно 300 микрон и покрыты водной дисперсией этилцеллюлозы, которая высыхает с образованием непрерывной пленки.
Компоненты с отсроченным и контролированным высвобождением
Другой класс дозированных форм включает формы, предоставляют отсрочку перед началом контролированного высвобождения тофацитиниба. Один вариант осуществления представлен таблеткой, содержащей ядро, которое содержит тофацитиниб, покрытое первым покрытием из полимерного материала, пригодного для контролированного высвобождения тофацитиниба, и вторым покрытием, пригодным для отсроченного высвобождения лекарственного вещества, при глотании дозированной формы. Первое покрытие наносят поверх и вокруг таблетки. Второе покрытие поверх и вокруг первого покрытия.
Таблетка может быть получена известными в отрасли способами, и содержит терапевтически приемлемое количество тофацитиниба плюс наполнители, необходимые для формирования таблетки с помощью выбранной техники.
Первое покрытие может быть покрытием с контролированным высвобождением известное в отрасли, особенно полимерным покрытием, для получения мембраны, как обсуждалось ранее для резервуарных систем. Приемлемые полимерные материалы покрытия, оборудование и способы нанесения покрытия также включают вышеуказанные.
Материалы, приемлемые для получения второго покрытия на таблетке включают полимеры известные в отрасли как кишечные покрытия с отсроченным высвобождением лекарственных веществ. Они представлены в основном материалами чувствительными к pH, такими как ацетат фталат целлюлозы, ацетат тримеллитат целлюлозы, гидроксипропилметилцеллюлозы фталат, поли(винилацетат фталат), и акриловые coполимеры, такие как Eudragit L-100 (RohmPharma), Eudragit L 30 D-55, Eudragit S 100, Eudragit FS 30D, и подобные материалы, более детально описаны ниже в отделе "отсроченное высвобождение". Толщину и тип покрытия с отсроченным высвобождением корректируют для получения желаемой отсрочки. В целом, более тонкие покрытия более резистентны к эрозии и, как следствие, обеспечивают более длительную отсрочку, как и покрытия, растворяющиеся при pH выше 7. Предпочтительные покрытия обычно имеют толщину от приблизительно 10 микрон до приблизительно 3 мм, и более желательно 10 мкм - 500 мкм.
При проглатывании, дважды покрытая таблетка проходит через желудок, где второе покрытие предотвращает высвобождение тофацитиниба в кислотных условиях. При прохождении таблетки из желудка в тонкий кишечник, где pH выше, второе покрытие эродирует или растворяется согласно физико-химическим свойствам выбранного материала. При эрозии или растворении второго покрытия, первое покрытие предотвращает мгновенное или быстрое высвобождение тофацитиниба и модулирует высвобождение таким образом, чтобы избежать высоких концентраций, таким образом, сводя к минимум побочные эффекты.
Другой вариант осуществления включает многочисленные частицы, где каждая частица покрыта двойным покрытием, как описано выше для таблетки, сначала полимером, разработанным для контролируемого высвобождения тофацитиниба, а затем покрыта полимером с отсроченным высвобождением в среде ЖК тракта при проглатывании дозированной формы. Частицы содержат тофацитиниб и могут содержать один или более наполнителей, необходимых для изготовления и рабочих характеристик. Многочисленные частицы, содержащие значительную фракцию тофацитиниба, по сравнению со связующим, являются предпочтительными. Многочисленные частицы могут иметь композицию и быть полученными как описано ранее для множественных частиц, используемых для получения резервуарных систем (включая экструзию и окатывание, влажную грануляцию, грануляцию в кипящем слое, и роторную грануляцию, получение зерен, и т.д.).
Покрытия с контролируемым высвобождением известны в отрасли, особенно полимерные покрытия для образования мембран, как обсуждалось ранее для резервуарных систем. Приемлемые полимерные материалы покрытия, оборудование и способы нанесения покрытия также включают вышеуказанные.
Скорость высвобождения тофацитиниба из множественных частиц с контролируемым высвобождением (например, множественные частицы перед нанесением покрытия с отсроченным высвобождением) и способы модифицирования покрытия контролируются факторами, ранее описанными для резервуарных систем с множественными частицами тофацитиниба.
Вторая мембрана или покрытие для множественных частиц с двойным покрытием представлена покрытием с отсроченным высвобождением, которое наносят поверх первого покрытия с контролируемым высвобождением, как было описано для таблетки, и может быть образована из тех же материалов. Следует отметить, что применение так называемых "кишечных" материалов для данного изобретения существенно отличается от их применения для получения обычных кишечных дозированных форм. Для стандартных кишечных форм, целью является отсрочить высвобождение лекарственного средства пока дозированная форма не пройдет желудок, а потом для доставки дозы вскоре после выхода из желудка. Высвобождение тофацитиниба непосредственно и полностью в двенадцатиперстной кишке нежелательно, однако, вследствие локального метаболизма, который пытаются свести к минимуму или избежать в данном изобретении. Поэтому, если обычные кишечные полимеры используются в данном изобретении, необходимо наносить их значительно более толстым слоем, чем обычно, для обеспечения отсрочки высвобождения лекарственного вещества до попадания дозированной формы в кишечник. Однако желательно обеспечить контролируемую доставку тофацитиниба после растворения или эродирования покрытия с отсроченным высвобождением, таким образом, преимущество данного изобретения реализуется подходящей комбинацией отсроченного высвобождения с контролируемым высвобождением, и часть с отсроченным высвобождением сама по себе может необязательно отвечать кишечным критерия Фармакопеи США. Толщина покрытия с отсроченным высвобождением устанавливается для обеспечения необходимой отсрочки. В целом, более толстые покрытия являются более устойчивыми к эрозии и, как следствие, обеспечивают более длительную отсрочку.
Также необходимо отметить, что описанные выше осмотические системы с замедленным высвобождением, также могут относиться к категории отсроченного высвобождения скорее, чем контролируемого высвобождения. Типичные осмотические системы с замедленным высвобождением обеспечивают начальную отсрочку в 0,5-6 часов перед высвобождением лекарственного вещества в контролируемой манере. В такой манере, типичные осмотические монолитные или двухслойные системы с замедленным высвобождением воплощают определение отсрочки с последующим контролируемым высвобождением.
Разрывающиеся осмотические гранулы и ядра (пульсирующая доставка)
В другом варианте воплощения ("устройство для разрывающегося осмотического ядра"), тофацитиниб включен в осмотическое разрывающееся устройство, содержащее ядро таблетки или ядро гранулы, содержащее тофацитиниб и, необязательно, один или больше осмагентов. Устройства такого типа были описаны в Baker, патент США № 3,952,741, включенный в данный документ в качестве ссылки. Примеры осмагентов включают сахара, такие как глюкоза, сахароза, маннит, лактоза, и т.д.; и соли, такие как хлорид натрия, хлорид калия, карбонат натрия, и т.д.; растворимые в воде кислоты, такие как виннокаменная кислота, фумаровая кислота, и т.д.. Ядро таблетки или ядро гранулы, содержащее тофацитиниб, покрыто полимером, образующим полупроницаемую мембрану, то есть мембрану проницаемую для воды, но по сути непроницаемую для тофацитиниба. Примеры полимеров, обеспечивающих полупроницаемой мембраной включают ацетат целлюлозы, целлюлозы ацетат бутират и этилцеллюлозу, желательно ацетат целлюлозы. Полупроницаемая мембрана может альтернативно состоять из одного или больше восков, таких как животный воск и воск насекомых, такой как пчелиный воск, и растительных восков, таких как карнаубский воск и гидрогенезированные растительные масла. Расплавленная смесь полиэтиленгликоля, например, полиэтиленгликоль-6000, и гидрогенезированного масла, например, гидрогенезированное касторовое масло, могут быть использованы в качестве покрытий, как описано для ионизированной таблетки Yoshino (Capsugel Symposia Series; Current Status on Targeted Drug Delivery to the Gastrointestinal Tract; 1993; pp.185-190). Некоторые предпочтительные полупроницаемые мембраны включают сложные и простые эфиры целлюлозы, производные полиакриловой кислоты, такие как полиакрилаты и сложные эфиры полиакрилатов, и поливиниловые спирты и полиалкены, такие как coполимер этилена и винилового спирта. Другие полупроницаемые мембраны включают целлюлозы ацетат и целлюлозы ацетат бутират.
Когда таблетка или гранула "разрывающегося осмотического ядра" с покрытием этого изобретения помещена в водную окружающую среду, вода проходит сквозь полупроницаемую мембрану внутрь ядра, растворяя часть тофацитиниба и осмагент, генерируя коллоидное осмотическое давление, что приводит к разрыву полупроницаемой мембраны и высвобождению тофацитиниба в водную среду. Выбором размера и формы ядра гранулы или таблетки, представителя и количества осмагента, и толщины полупроницаемой мембраны, может быть выбрано время отсрочки между помещением дозированной формы в водную среду и высвобождением тофацитиниба. Специалисту в отрасли понятно, что увеличение соотношения площади поверхности к объему дозированной формы, и увеличение осмотической активности осмагента уменьшает время отсрочки, в то время как увеличение толщины покрытия будет увеличивать время отсрочки. Осмотически-разрывающиеся устройства данного изобретения практически не высвобождают тофацитиниб из дозированной формы до тех пор, пока дозированная форма не выйдет из желудка и попадет в тонкий кишечник за приблизительно 15 мин. или больше. Некоторые осмотически-разрывающиеся устройства практически не высвобождают тофацитиниб из дозированной формы до тех пор, пока дозированная форма не выйдет из желудка и попадет в тонкий кишечник за приблизительно 30 или больше. Другие осмотически-разрывающиеся устройства практически не высвобождают тофацитиниб из дозированной формы до тех пор, пока дозированная форма не выйдет из желудка и попадет в тонкий кишечник за приблизительно 90 или больше. Другие осмотически-разрывающиеся устройства практически не высвобождают тофацитиниб из дозированной формы до тех пор, пока дозированная форма не выйдет из желудка и попадет в тонкий кишечник желательно за приблизительно 3 часа или больше, таким образом, обеспечивая минимальное высвобождение тофацитиниба в двенадцатиперстной кишке и верхнем отделе тонкого кишечника. Осмотическая таблетка или гранула с разрывающимся ядром имеет ядро, содержащее от приблизительно 10-95% тофацитиниба, приблизительно 0-60% осмагента, как описано выше, и приблизительно 5-20% других фармацевтических добавок, таких как связующие и лубриканты. Полупроницаемая мембрана на таблетке, например ацетат целлюлозы, составляет от приблизительно 2% до приблизительно 30%, желательно от приблизительно 3% до приблизительно 10% массы ядра таблетки. Полупроницаемая мембрана на грануле, например, ацетат целлюлозы, составляет от приблизительно 2% до приблизительно 80% массы ядра гранулы. В другом варианте осуществления, полупроницаемое покрытие на грануле составляет 3-30% массы ядра гранулы.
Осмотическое устройство с разрывающимся ядром не обладает механизмом для "обнаружения" того или вышло устройство из желудка и вошло в двенадцатиперстную кишку. Таким образом, устройства такого типа высвобождают тофацитиниб в заранее определенное время после попадания в водную среду, например, после проглатывания. Натощак неперевариваемые неразлагающиеся твердые вещества, такие как "осмотическое устройство с разрывающимся ядром" изобретения, выходят из желудка на протяжении фазы III желудочного мигрирующего миоэлектрического комплекса (IMMC), что происходит приблизительно каждые 2 часа у человека. В зависимости от стадии IMMC во время дозировки натощак, осмотическое устройство с разрывающимся ядром может выйти из желудка практически сразу же после дозировки, или через 2 после дозировки. В состоянии после еды, неперевариваемые неразлагающиеся твердые вещества, диаметром <11 мм, медленно выходят из желудка в содержимым пищи (Khosla and Davis, Int. J. Pharmaceut. 62 (1990) R9-R11). Если неперевариваемые неразлагающиеся твердые вещества имеют диаметр более чем приблизительно 11 мм, например, размер обычной таблетки, они задержатся в желудке на протяжении переваривания пищи, и попадут в двенадцатиперстную кишку на протяжении фазы III комплекса IMMC, после полного переваривания пищи и ее выхода из желудка. Высвобождение тофацитиниба может быть отсрочено до приблизительно 15 мин или больше. Высвобождение тофацитиниба может быть отсрочено до 30 мин или больше. Высвобождение тофацитиниба может быть отсрочено до приблизительно 90 мин или больше. Высвобождение тофацитиниба может быть отсрочено до приблизительно 3 часов или больше после выхода дозированной формы из желудка. Осмотическое устройство с разрывающимся ядром начинает высвобождать тофацитиниб через приблизительно 2,5 часа после попадания в водную среду, например, после проглатывания, для большей надежности, что устройство высвободит тофацитиниб дистально от двенадцатиперстной кишки, при приеме натощак. Другое "осмотическое устройство с разрывающимся ядром" начинает высвобождать тофацитиниб через приблизительно 4 часа после попадания в водную среду. Эти 4 часа отсрочки позволяют принимать дозу в сытом состоянии, и дают приблизительно 3,5 часа отсрочки в полном желудке, после чего приблизительно 30 мин. отсрочки после того как дозированная форма выйдет из желудка. Таким образом, высвобождение тофацитиниба в наиболее чувствительном участке ЖК тракта, в двенадцатиперстной кишке, сводится к минимуму.
В другом варианте воплощения, "разрывающееся разбухающее ядро с покрытием", таблетку или гранулу, содержащую тофацитиниб, получают таким образом, что она также содержит 25-70% разбухающего материала, такого как разбухающий коллоид (например, желатин), как описано в Milosovich, патент США 3,247,066, включенный с помощью ссылки. Разбухающие материалы ядра представлены гидрогелями, например, гидрофильные полимеры, поглощающие воду и разбухающие, такие как полиэтиленоксиды, производные полиакриловой кислоты, такие как полиметилметакрилат, полиакриламиды, поливиниловый спирт, поли-N-винил-2-пирролидон, карбоксиметилцеллюлоза, крахмалы, и т.д. Разбухающие гидрогели для данного варианта воплощения включают полиэтиленоксиды, карбоксиметилцеллюлозу и кросскармелозу натрия. Ядро таблетки или гранулы, содержащее коллоид/гидрогель и тофацитиниб, покрыто, как минимум частично, полупроницаемой мембраной. Примеры полимеров, обеспечивающих полупроницаемую мембрану, включают целлюлозы ацетат и целлюлозы ацетат бутират, и этилцеллюлозу. Полупроницаемая мембрана может альтернативно состоять из одного или больше восков, таких как животный воск и воск насекомых, такой как пчелиный воск, и растительных восков, таких как карнаубский воск и гидрогенезированные растительные масла. Расплавленная смесь полиэтиленгликоля, например, полиэтиленгликоль-6000, и гидрогенизированного масла, например, гидрогенизированное касторовое масло, могут быть использованы в качестве покрытия, как описано для ионизированной таблетки Yoshino (Capsugel Symposia Series; Current Status on Targeted Drug Delivery to the Gastrointestinal Tract; 1993; pp.185-190). Некоторые полупроницаемые мембраны включают сложные и простые эфиры целлюлозы, производные полиакриловой кислоты, такие как полиакрилаты и сложные эфиры полиакрилатов, и поливиниловые спирты и полиалкены, такие как coполимер этилена и винилового спирта, целлюлозы ацетат и целлюлозы ацетат бутират.
Когда покрытая таблетка или гранула, содержащая разрывающееся разбухающее ядро с покрытием, помещена в водную окружающую среду, вода проходит сквозь полупроницаемую мембрану внутрь ядра, ядро разбухает, что приводит к разрыву полупроницаемой мембраны и высвобождению тофацитиниба в водную среду. Выбором размера и формы ядра гранулы или таблетки, представителя и количества разбухающего агента, и толщины полупроницаемой мембраны, может быть выбрано время отсрочки между помещением дозированной формы в водную среду и высвобождением тофацитиниба. Предпочтительные устройства с разрывающимся разбухающим ядром с покрытием этого изобретения включают устройства, по сути, не высвобождающие тофацитиниба из дозированной формы, до тех пор пока дозированная форма не выйдет из желудка и попадет в тонкий кишечник через приблизительно 15 мин. или больше, желательно приблизительно 30 мин или больше, таким образом сводя к минимум высвобождение тофацитиниба в двенадцатиперстной кишке.
Таблетка или гранула, содержащая разрывающееся разбухающее ядро с покрытием, содержит от приблизительно 10-70% тофацитиниба; приблизительно 15-60% разбухающего материала, например, гидрогеля; приблизительно 0-15% необязательного осмагента; и приблизительно 5-20% других фармацевтических добавок, таких как связующие и лубриканты. Полупроницаемая мембрана на таблетке, желательно ацетат целлюлозы, составляет от приблизительно 2% до приблизительно 30%, желательно 3-10% массы ядра таблетки. Полупроницаемая мембрана на грануле, желательно ацетат целлюлозы, составляет от приблизительно 2% до приблизительно 80%, желательно 3-30% массы ядра гранулы.
Устройство с разрывающимся разбухающим ядром с покрытием не обладает механизмом для "обнаружения" того или вышло устройство из желудка и вошло в двенадцатиперстную кишку. Таким образом, устройства такого типа высвобождают тофацитиниб в заранее определенное время после попадания в водную среду, например, после проглатывания, как ранее описывалось для осмотического устройства с разрывающимся ядром, и те же соображения и преимущества относятся к устройствам с разрывающимся разбухающим ядром с покрытием. Устройства с разрывающимся разбухающим ядром с покрытием могут быть объединены с устройствами с мгновенным высвобождением для получения дозированной формы, высвобождающей лекарственное вещество как мгновенно после введения, так и через один или больше заранее установленных промежутков времени после дозирования.
В другом варианте воплощения, "pH-запускаемое осмотическое разрывающееся устройство", тофацитиниб включен в устройство, описанное в патенте США 5,358,502, опубликованном 25.10.1994, включенный с помощью ссылки. Устройство содержит тофацитиниб и необязательно один или больше осмагентов, окруженные как минимум частично полупроницаемой мембраной. Полупроницаемая мембрана является проницаемой для воды и по сути непроницаемой для тофацитиниба и осмагента. Приемлемые осмагенты являются такими же, как описаны выше для осмотического устройства с разрывающимся ядром. Приемлемые материалы полупроницаемой мембраны являются такими же, как описаны выше для осмотического устройства с разрывающимся ядром. Средства pH-запускания присоединены к полупроницаемой мембране. Средства pH-запускания активируются уровнем pH выше 5,0, и запускают неожиданную доставку тофацитиниба. В этом варианте воплощения, средства pH-запускания включают мембрану или полимерное покрытие, окружающее полупроницаемое покрытие. pH-запускаемое покрытие содержит полимер, по сути непроницаемый и нерастворимый в среде с pH желудка, но который становится проницаемым и растворимым при pH двенадцатиперстной кишки, приблизительно pH 6,0.
Примеры pH-чувствительных полимеров включают полиакриламиды, производные фталата, такие как кислые фталаты углеводородов, амилозы ацетат фталат, ацетат фталат целлюлозы, другие фталаты сложных эфиров целлюлозы, фталаты простых эфиров целлюлозы, гидроксипропилцеллюлозы фталат, гидроксипропилэтилцеллюлозы фталат, гидроксипропилметилцеллюлозы фталат, метилцеллюлозы фталат, поливинилацетат фталат, поливинилацетат гидрофталат, натрия ацетат фталат целлюлозы, крахмал кислоты фталат, coполимер стирол-малеиновой кислоты и дибутилфталата, coполимер стирол-малеиновой кислоты поливинилацетат фталата, coполимеры стирола и малеиновой кислоты, производные полиакриловой кислоты, такие как coполимеры акриловой кислоты и акрилового сложного эфира, полиметакриловая кислота и ее сложные эфиры, coполимеры полиакриловой и метакриловой кислоты, шеллак, и coполимеры винилацетата и кротоновой кислоты.
Предпочтительные pH-чувствительные полимеры включают шеллак; производные фталата, особенно ацетат фталат целлюлозы, поливинилацетат фталат, и гидроксипропилметилцеллюлозы фталат; производные полиакриловой кислоты, особенно полиметилметакрилат смешанный с акриловой кислотой, и coполимеры акрилового сложного эфира; и coполимеры винилацетата и кротоновой кислоты. Как описано выше ацетат фталат целлюлозы доступен в виде латекса под торговой маркой Aquateric.RTM. (торговая марка FMC Corp., Филадельфия), и акриловые coполимеры доступны под торговой маркой Eudragit-R.RTM и Eudragit-L.RTM. Для соответствующего нанесения в этом варианте воплощения, эти полимеры должны быть пластифицированы, используя пластификаторы, описанные выше. pH-запускаемое покрытие также может содержать смесь полимеров, например целлюлозы ацетат и ацетат фталат целлюлозы. Другая приемлемая смесь содержит Eudragit-L.RTM и Eudragit-S.RTM; их соотношение и толщина покрытия определяют чувствительность "запуска", например, pH при котором внешнее pH-запускаемое покрытие ослабляется и растворяется.
pH-регулируемое осмотическое разрывающееся устройство обычно работает следующим образом. После орального проглатывания, pH-регулируемое покрытие, окружающее полупроницаемое покрытие, которое в свою очередь окружает ядро таблетки или гранулы, содержащее тофацитиниб, остается нерастворенным и интактным в желудке. В желудке, вода может начать проникать, сквозь pH-регулируемое покрытие и полупроницаемое покрытие, таким образом, начиная гидратацию ядра, содержащего тофацитиниб и необязательный осмагент. После того как устройство вышло из желудка и попало в тонкий кишечник, pH-регулируемое покрытие быстро дезинтегрирует и растворяется, и вода проходит сквозь полупроницаемое покрытие, растворяя тофацитиниб и необязательный осмагент внутри ядра. После того как коллоидное осмотическое давление поперек полупроницаемого покрытия превысит определенную пороговую величину, полупроницаемое покрытие разрушается, и устройство разрывается, высвобождая тофацитиниб. Предпочтительно, чтобы этот разрыв и высвобождение тофацитиниба происходило через приблизительно 15 мин. или больше, желательно 30 мин. или больше, после выхода pH-регулируемого осмотического разрывающегося устройства из желудка и попадания в двенадцатиперстную кишку, таким образом, сводя к минимуму влияние тофацитиниба на чувствительную двенадцатиперстную кишку.
Для pH-регулируемое осмотическое разрывающееся устройство, период задержки или период отсрочки контролируется выбором и количеством осмагента в ядре, выбором полупроницаемого покрытия, и толщиной полупроницаемого покрытия. Специалист в отрасли понимает, что, например, более толстое полупроницаемое покрытие обеспечит более длительную отсрочку после выхода устройства из желудка. Предпочтительным pH-регулируемым осмотически разрывающимся устройством является ядро таблетки или гранулы, содержащее тофацитиниб с необязательным осмагентом, покрытое 3-20 мас.% мембраной ацетата целлюлозы, покрытое 3-20 мас.%, мембраной, содержащей приблизительно ацетат целлюлозы/ацетат фталат целлюлозы в соотношении 1:1. Другое предпочтительное pH-регулируемое осмотическое разрывающееся устройство представлено ядром таблетки или гранулы, содержащей тофацитиниб с необязательным осмагентом, покрытым 3-20 мас.% мембраной ацетата целлюлозы, покрытым 3-20 мас.% мембраной, содержащая от приблизительно 9:1 до приблизительно 1:1 Eudragit-L.RTM./Eudragit-S.RTM.
Преимущественно, благодаря тому, что pH-регулируемое осмотическое разрывающееся устройство имеет механизм, определяющий выход устройства из желудка, индивидуальная вариабельность в опорожнении желудка не имеет значения.
В другом варианте воплощения, "pH-регулируемое разрывающееся разбухающее ядро с покрытием", ядро таблетки или гранулы, содержащей тофацитиниб, и разбухающий материал покрыты полупроницаемым покрытием, которое само покрыто pH-чувствительным покрытием. Композиция ядра, включая выбор разбухающего материала, является таким, как описано выше для разрывающегося разбухающего ядро с покрытием. Выбор полупроницаемого материала покрытия и pH-чувствительного материала покрытия является таким как описано выше для "pH-регулируемого осмотического ядра". Это устройство детально описано в патентной заявке США 08/023,227, поданной 25.02.1993, включенной с помощью ссылки.
pH-регулируемое разрывающееся разбухающее ядро обычно работает следующим образом. После орального проглатывания, pH-регулируемое покрытие, окружающее полупроницаемое покрытие, которое в свою очередь окружает ядро таблетки или гранулы, содержащее тофацитиниб, остается нерастворенным и интактным в желудке. В желудке, вода может начать проникать, сквозь pH-регулируемое покрытие и полупроницаемое покрытие, таким образом, начиная гидратацию ядра, содержащего тофацитиниб и разбухающий в воде материал, желательно гидрогель. Когда pH-регулируемое разрывающееся разбухающее ядро вышло из желудка и попало в тонкий кишечник, pH-регулируемое покрытие быстро дезинтегрирует и растворяется, и вода проходит сквозь полупроницаемое покрытие, растворяя тофацитиниб и разбухающий в воде материал внутри ядра. После того как разбухающее давление поперек полупроницаемого покрытия превысит определенную пороговую величину, полупроницаемое покрытие разрушается, и устройство разрывается, высвобождая тофацитиниб. Предпочтительно, чтобы этот разрыв и высвобождение тофацитиниба происходило через приблизительно 15 мин. или больше, желательно 30 мин. или больше, после выхода pH-регулируемого осмотического разрывающегося устройства из желудка и попадания в двенадцатиперстную кишку, таким образом сводя к минимуму влияние тофацитиниба на чувствительную двенадцатиперстную кишку.
Для "pH-регулируемого разрывающегося разбухающего ядра ", период задержки или период отсрочки контролируется выбором и количеством разбухающего материала в ядре, выбором полупроницаемого покрытия, и толщиной полупроницаемого покрытия. Специалист в отрасли понимает, что, например, более толстое полупроницаемое покрытие обеспечит более длительную отсрочку после выхода устройства из желудка. pH-регулируемое разрывающееся разбухающее ядро содержит ядро таблетки или гранулы, содержащее тофацитиниб с синтетическим гидрогелем, желательно карбоксиметилцеллюлозой, покрытое 3-20 мас.% мембраны из ацетата целлюлозы, покрыто 3-20 мас.% мембраны, состоящей из ацетат целлюлозы/ацетат фталат целлюлозы в соотношении 1:1. Другое pH-регулируемое разрывающееся разбухающее ядро содержит ядро таблетки или гранулы, содержащее тофацитиниб с синтетическим гидрогелем, желательно карбоксиметилцеллюлозой, покрытое 3-20 мас.% мембраны из ацетата целлюлозы, покрытое 3-20 мас.% мембраны, состоящей из от приблизительно 9:1 до приблизительно 1:1 Eudragit-L.RTM./Eudragit-S.RTM.
Преимущественно, благодаря тому, что pH-регулируемое разрывающееся разбухающее ядро имеет механизм, определяющий выход устройства из желудка, индивидуальная вариабельность в опорожнении желудка не имеет значения. pH-регулируемые разрывающиеся разбухающие ядра могут быть объединены с устройствами мгновенного высвобождения с образованием дозированной формы, высвобождающей лекарственное вещество как мгновенно после введения, так и в одном или больше предварительно обозначенных местах в ЖК тракте после приема дозы.
Текущий обзор такой технологии разрыва представлен в журнале Journal of Controlled Release; 134 (2009) 74-80, который включен с помощью ссылки.
Варианты осуществления изобретения с отсроченным высвобождением представлены твердыми дозированными формами для орального введения, содержащими тофацитиниб и фармацевтически приемлемый носитель, высвобождающими не более чем 10% включенного тофацитиниб в желудок млекопитающих, и высвобождающие не более чем еще 10% на протяжении первых 15 минут после попадания в двенадцатиперстную кишку млекопитающих. Тайминг высвобождения тофацитиниба в желудке или двенадцатиперстной кишке можно протестировать, используя разные подходы, включая, но не ограничиваясь следующими: рентгеновское определение, магнитно-резонансная визуализация, гамма сцинтиграфия, или прямой отбор образцов содержимого желудка или двенадцатиперстной кишки путем интубации. Эти тесты, хотя возможны, но весьма проблематичны для проведения на людях. Более подходящим тестом для отсроченного высвобождения данного изобретения является двухстадийный тест на растворение in vitro.
Изобретение проиллюстрировано следующими неограничивающими примерами.
Примеры
Пример 1. Осмотическая таблетка с экструдируемым ядром
22 мг ядро таблетки
Половину размера партии сорбитола, 2663,01 г (также смотреть Таблицу 1 ниже), добавляли в бункер объемом 28 л. Размер партии Коповидона, 420,00 г, затем добавляли в бункер объемом 28 л. Размер партии Тофацитиниба, 623,98 г, затем добавляли в бункер объемом 28 л. Размер партии Гидроксицеллюлозы, 560,00 г, затем добавляли в бункер объемом 28 л. Оставшуюся половину размера партии сорбитола, 2663,01 г добавляли в бункер объемом 28 л. Все компоненты перемешивали в бункере на протяжении 15 мин. при 12 +/- 1 об./мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 950 об/мин. Смесь собирали во второй бункер объемом 28 л. Содержимое бункера перемешивали на протяжении 10 мин. при 15 +/- 1 об/мин.
Стеарат магния, 70 г, пропускали через сито с размером меш 850-микрон и добавляли в бункер, и содержимое перемешивали на протяжении 5,5 мин при 12 +/- 1 об/мин. Финальную смесь переносили в приемную воронку ротационного таблеточного пресса Fette. Таблетки прессовали, используя 0,2620” × 0,5240” модифицированные овальные инструменты, до средней целевой массы 400 мг +/- 5%, средней целевой толщины 5,35 мм +/- 0,05 мм, и целевой прочности 13 кПа. Таблетки пропускали через пылеуловитель и металлодетектор.
11 мг ядро таблетки.
Половину размера партии сорбитола, 2819,01 г (также смотреть Таблицу 2 ниже), добавляли в бункер объемом 28 л. Размер партии Коповидона, 420,00 г, затем добавляли в бункер объемом 28 л. Размер партии Тофацитиниба, 311,99 г, затем добавляли в бункер объемом 28 л. Размер партии Гидроксицеллюлозы, 560,00 г, затем добавляли в бункер объемом 28 л. Оставшуюся половину размера партии сорбитола, 2819,0 г добавляли в бункер объемом 28 л. Все компоненты перемешивали в бункере на протяжении 15 мин при 12 +/- 1 об/мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 950 об/мин. Смесь собирали во второй бункер объемом 28 л. Содержимое бункера перемешивали на протяжении 10 мин при 15 +/- 1 об/мин.
Стеарат магния, 70 г, пропускали через сито с размером меш 850-микрон и добавляли в бункер, и содержимое перемешивали на протяжении 5,25 мин при 12 +/- 1 о/мин. Финальную смесь переносили в приемную воронку ротационного таблеточного пресса Fette. Таблетки прессовали, используя 0,2620” × 0,5240” модифицированные овальные инструменты, до средней целевой массой 400 мг +/- 5%, средней целевой толщиной 5,35 мм +/- 0,05 мм, и целевой прочности 15 кПа. Таблетки пропускали через пылеуловитель и металлодетектор.
Нанесение покрытия и сверление таблетки
Раствор для покрытия (4,049 кг) получали согласно следующим стадиям: сначала, все 396,0 г воды (также смотреть Таблицу 3 ниже) и 1464,0 г ацетона добавляли в сосуд объемом 5 л и перемешивали на протяжении 5 мин. Гидроксипропилцеллюлозу (32,4 г) добавляли к смеси и перемешивали на протяжении 5 мин. Ацетат целлюлозы (48,6 г) добавляли к смеси и перемешивали на протяжении 5 мин. Оставшиеся 2108 г ацетона добавляли к смеси и перемешивали на протяжении 3 часов. Эта процедура дала 2% твердый раствор (м./м.).
Овальные таблетки (900 г) массой по 400 мг покрывали в глазировочном аппарате Vector LDCS-5 с 1,5 л частично перфорированным поддоном, работающим при 20 об/мин и потоком воздуха 30 куб.ф./мин с температурой выпуска 40°C. 2% (м./м.). Твердый раствор наносили до достижения влажной массы уровня 6,2%. После этого таблетки извлекали из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Единственное отверстие (1000 микрон) просверливали в конце полосы овальной таблетки. Отверстие может быть просверлено механическим путем или лазером. Покрытие 6% обеспечило следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 4):
% растворенного лекарства
% растворенного лекарства
Пример 2. Осмотические таблетки с экструдируемым ядром с покрывающим раствором Ацетон:Метанол (200 мг)
11 мг ядро таблетки
Половину размера партии сорбитола, 38,014 кг (также смотреть Таблицу 5 ниже), добавляли в бункер объемом 300 л. Размер партии Коповидона, 6,00 кг, затем добавляли в бункер объемом 300 л. Размер партии Тофацитиниба, 8,914 кг, затем добавляли в бункер объемом 300 л. Размер партии Гидроксицеллюлозы, 8,00 кг, затем добавляли в бункер объемом 300 л. Оставшуюся половину размера партии сорбитола, 38,014 г добавляли в бункер объемом 300 л. Все материалы добавляли с помощью вакуумной системы переноса и пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 1400 об./мин. Все компоненты перемешивали в бункере на протяжении 20 мин. при 12 +/- 1 об./мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 1400 об./мин. Смесь собирали во втором бункере объемом 300 л. Содержимое бункера перемешивали на протяжении 20 мин. при 12 +/- 1 об./мин.
Стеарат магния, 1,00 кг, пропускали через сито с размером меш 850-микрон и добавляли в бункер, и содержимое перемешивали на протяжении 5 мин. при 12 +/- 1 об./мин. Таблетки прессовали, используя 0,2080” x 0,4160” модифицированные овальные инструменты на роторном таблеточном прессе Manesty Mark IV, до средней целевой массы 200 мг +/- 5%, средней целевой толщины 4,17 мм +/- 0,05 мм, и целевой прочности 10 кПа. Таблетки пропускали через пылеуловитель и металлодетектор
100 кг
22 мг ядро таблетки
Половину размера партии сорбитола, 33,086 кг (также смотреть Таблицу 6 ниже), добавляли в бункер объемом 300 л. Размер партии коллоидного диоксида кремния, 1,00 кг, затем добавляли в бункер объемом 300 л. Размер партии Коповидона, 6,00 кг, затем добавляли в бункер объемом 300 л. Размер партии Тофацитиниба, 8,914 кг, затем добавляли в бункер объемом 300 л. Размер партии Гидроксицеллюлозы, 8,00 кг, затем добавляли в бункер объемом 300 л. Оставшуюся половину размера партии сорбитола, 33,086 г добавляли в бункер объемом 300 л. Все материалы добавляли с помощью вакуумной системы переноса и пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 1400 об./мин. Все компоненты смешивали в бункере на протяжении 20 мин. при 12 +/- 1 об./мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем работающим со скоростью приблизительно 1400 об./мин. Смесь собирали во втором бункере объемом 300 л. Содержимое бункера перемешивали на протяжении 20 мин. при 12 +/- 1 об./мин.
Стеарат магния, 1,00 кг, пропускали через сито с размером меш 850-микрон и добавляли в бункер, и содержимое перемешивали на протяжении 5 мин. при 12 +/- 1 об./мин. Таблетки прессовали, используя 0,2080” x 0,4160” модифицированные овальные инструменты на роторном таблеточном прессе Manesty Mark IV, до средней целевой массы 200 мг +/- 5%, средней целевой толщины 4,17 мм +/- 0,05 мм, и целевой прочности 11 кПа. Таблетки пропускали через пылеуловитель и металлодетектор.
Покрывающий раствор (750 кг) получали согласно следующим стадиям (смотреть также Таблицу 7): сначала, все 147,0 кг метанола и 580,5 г ацетона добавляли в колбу объемом 250 галлон. Ацетат целлюлозы (13,5 кг) добавляли к смеси. Гидроксипропилцеллюлозу (9,0 кг) добавляли к смеси. Содержимое контейнера перемешивали на протяжении часа. Эта процедура дала 3% (м./м.) твердый раствор.
200 мг
Овальные таблетки (250 кг) массой по 200 мг покрывали в глазировочном аппарате Vector HC-130, работающем при 8 об./мин. и потоком воздуха 1000 куб.ф./мин. с температурой выпуска 28°C. Твердый раствор 3% (м./м.) наносили до достижения увеличения влажной массы уровня 6,8%. Затем таблетки извлекали из дражировочного котла и высушивали при 45°C на протяжении 24 часов.
Единственное отверстие (600 микрон) просверливали в конце полосы овальной таблетки. Отверстие может быть просверлено механическим путем или лазером. Покрытие 6,6% обеспечило следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об./мин. на основании теста на растворимость 1 (Таблица 8):
% растворенного лекарства
% растворенного лекарства
Пример 3. Осмотические таблетки с экструдируемым ядром и покрывающей мембраной из Ацетата целлюлозы и Полиэтиленгликоля (200 мг)
Таблетки-ядра 11 мг и 22 мг с замедленным высвобождением тофацитиниба получали, как описано в Примере 2.
Покрывающий раствор 1200 г получали согласно следующим стадиям (смотреть также Таблицу 9): сначала, 60 г воды и 19,2 г полиэтиленгликоля добавляли в сосуд объемом 5 л и перемешивали до достижения прозрачности раствора. Метанол (60 г) и 0,504 г БГА добавляли к раствору и перемешивали до достижения прозрачности. Ацетон (1031,496 г) и 28,8 г ацетата целлюлозы добавляли к смеси. Содержимое контейнера перемешивали на протяжении 3 часов. Эта процедура дала твердый раствор 4% (м./м.).
Овальные таблетки (240 г) массой по 200 мг покрывали в глазировочном аппарате Vector LDCS-5, работающем при 30 об/мин и потоком воздуха 40 куб.ф./мин с температурой выпуска 28°C. Твердый раствор 3% (м./м.) наносили до достижения увеличения влажной массы уровня 9,2%. Затем таблетки извлекали из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Единственное отверстие (600 микрон) просверливали в конце полосы овальной таблетки. Отверстие может быть просверлено механическим путем или лазером. Покрытие 9% обеспечивает следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 10):
% растворенного лекарства
% растворенного лекарства
Пример 4. Таблетка с гидрофильной матрицей и контролируемым высвобождением
Металлические поверхности бункера объемом 10л предварительно покрывали путем добавления размера партии (также смотреть Таблицу 11 ниже) 1484,85 г лактозы Fast Flo 316 и перемешиванием на протяжении 2 мин при 12 +/- 1 об/мин. Размер партии Тофацитиниба, 171,15 г, добавляли в бункер объемом 10 л и загружали на моногидрат лактозы. Контейнер с Тофацитинибом промывали некоторым количеством моногидрата лактозы из бункера объемом 10 л. Размер партии Гипромелозы, 720 г, добавляли в бункер объемом 10 л. Все компоненты перемешивали в бункере на протяжении 10 мин при 12 +/- 1 об/мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 1400 об/мин. Смесь собирали во втором бункере объемом 10 л. Содержимое бункера перемешивали на протяжении 10 мин при 12 +/- 1 об/мин.
Внутрикристаллитный стеарат магния, 6 г, добавляли в бункер и перемешивали на протяжении 3 мин при 12 +/- 1 об/мин. Смазанную смесь пропускали через роликовый пресс Gerteis, оснащенный роликами с насеченной головкой, съемными бортами обода и линейной вибромельницей, содержащей портативный ротор и пемзовальную пластину толщиной 1 мм. Требуемую слоистую твердую фракцию 0,7 (0,67-0,73) и гранулы собирали в первоначальный бункер объемом 10 л.
Внутрикристаллитный стеарат магния, 18 г, добавляли в бункер и содержимое перемешивали на протяжении 3 мин при 12 +/- 1 об/мин. Финальную смесь пропускали через роторный таблеточный пресс Kilian T-100. Таблетки прессовали, используя инструменты 13/32” SRC, до средней целевой массы 500 мг +/- 5% и целевой прочности 15 кПа. Таблетки пропускали через пылеуловитель и металлодетектор.
Композиция таблетки Тофацитиниба (22 мг) с гидрофильной матрицей;
масса всех таблеток 500 мг
Fast Flo 316
Прессованные таблетки обеспечивают следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 12).
Пример 5. Двухслойная осмотическая таблетка (20 мг)
Размеры партий Тофацитиниба и Полиэтиленоксида N80 (смотреть также Таблицу 13) пропускали через сито 30 меш и добавляли в склянку из темного стекла объемом 500 см3. Смесь перемешивали на протяжении 10 мин с помощью блендера Turbula. Стеарат магния (0,2 г) пропускали через сито 30 меш и добавляли в колбу с активной смесью и перемешивали на протяжении 3 мин.
Размеры партий полиэтиленоксида (класс коагулянтов), голубого лакообразующего красителя и хлорида натрия пропускали через сито 20 или 30 меш, и добавляли в этом порядке в колбу объемом 500 см3. Смесь перемешивали на протяжении 10 мин. с помощью блендера Turbula. Стеарат магния (0,5 г) пропускали через сито 30 меш и добавляли в колбу с разбухающим слоем и перемешивали на протяжении 3 мин.
Таблетки прессовали, используя 9 мм стандартные круглые выпуклые инструменты, до средней целевой массы 400 мг +/- 5%, средней целевой толщины 7 мм +/- 0,05 мм, и целевой прочности 15 кПа.
(мг)/единицу:
(мг)/единицу:
№ 2d
Покрывающий раствор получали согласно следующим стадиям (смотреть также Таблицу 14 ниже): сначала все 194,6 г воды и 800 г ацетона добавляли в резервуар объемом 5 л и перемешивали на протяжении 5 мин. Гидроксипропилцеллюлозу (24 г) добавляли к смеси и перемешивали на протяжении 5 мин. Ацетат целлюлозы (36 г) добавляли к смеси и перемешивали на протяжении 5 мин. Оставшиеся 946,3 г ацетона добавляли к смеси и перемешивали на протяжении 3 часов. Эта процедура дала 3% твердый раствор (м./м.).
Таблетки SRC (250 г) по 400 мг покрывали с помощью системы покрытия Vector LDCS-5 с полуперфорированным поддоном объемом 0,5 л, работающим со скоростью 30 об/мин и потоком воздуха 30 куб.ф./мин с температурой выпуска 40°C. Твердый раствор 3% (м./м.) наносили до достижения увеличения влажной массы до 6,2%. После этого таблетки извлекали из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Единственное отверстие (1000 микрон) просверливали в конце полосы овальной таблетки. Отверстие может быть просверлено механическим путем или лазером. Нужное покрытие 6% обеспечило следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 15):
(% растворенного лекарства)
Пример 6. Двухслойная осмотическая таблетка (11 мг)
Размер партии Полиэтиленоксида N80 (активный слой) (смотреть также Таблицу 16) пропускали через сито 30 меш. Большие частицы, оставшиеся на сите, отбрасывали. Полиэтиленоксид добавляли в склянку из темного стекла объемом 500 см3 и вручную перемешивали для покрытия внутренней части склянки. Добавляли размер партии Тофацитиниба и перемешивали на протяжении 10 мин. с помощью блендера Turbula. Стеарат магния (1,0 г) добавляли в склянку с активной смесью и перемешивали на протяжении 3 мин.
Размер партии Полиэтиленоксида класса коагулянт (разбухающий слой) и хлорида натрия пропускали через сито 30 меш. Полиэтиленоксид, размер партии микрокристаллической целлюлозы, размер партии голубого лакообразующего красителя и порошка хлорида натрия добавляли в этом порядке в колбу объемом 950 см3. Смесь перемешивали на протяжении 10 мин. с помощью блендера Turbula. Стеарат магния (1,0 г) добавляли в колбу с разбухающим слоем и перемешивали на протяжении 3 мин.
Двухслойные таблетки прессовали, используя стандартные круглые выпуклые инструменты 9/32 дюйма, до средней целевой массы 180,0 мг +/- 5% и средней целевой толщины 5,0 мм +/- 0,1 мм.
(мг)/ед.:
(мг)/ед.:
№ 2d
Покрывающий раствор получали согласно следующим стадиям (смотреть также Таблицу 17): сначала, 180,0 г воды добавляли к 48,6 г ПЭГ 3350 в колбе для смешивания объемом 4 л и перемешивали или взбалтывали вручную до полного растворения ПЭГ. Затем 131,4 г ацетата целлюлозы добавляли в колбу для смешивания объемом 4 л, содержащую раствор ПЭГ-вода. АЦ разбрасывали в виде суспензии или влажного осадка. Используя колбу для смешивания объемом 4 л, оснащенную роторным импеллером, 2,640,0 г ацетона добавляли к смеси ПЭГ-вода-АЦ. Содержимое колбы для смешивания взбалтывали до растворения всех твердых веществ.
Двухслойные таблетки ядра (250 г) по 180 мг покрывали с помощью системы покрытия Vector LDCS-5 с полностью перфорированным дражировочным котлом объемом 0,5 л, работающим со скоростью 30 об/мин и потоком воздуха 35 куб.ф./мин с температурой выпуска 32°C. Твердый раствор 6% (м./м.) наносили до достижения прироста влажной массы в процессе нанесения покрытия в размере 25,2 мг на таблетку. После этого таблетки извлекали из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Единственный впускной канал диаметром 1,0 мм образовывали через покрывающую мембрану по центру внешней стороны слоя лекарственного средства двухслойной таблетки. Впускной канал может быть образован механически или лазером. Нужное покрытие 23,4 мг или 13% массы целевого двухслойного ядра обеспечило контролируемое высвобождение, показывающее доставку 80% лекарственного средства за 3,5 час в середе с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1. Дополнительные данные по растворимости приведены в Таблице 18.
Пример 7. Двухслойная осмотическая таблетка с антиоксидантами (11 мг)
Композицию Примера 7 получали следующим образом (смотреть также Таблицу 19): Полиэтиленоксид N80 пропускали через сито 30 меш. Большие частицы полиэтиленоксида N80, оставшиеся на сите удаляли. Отдельно, первичный размер частиц метабисульфита натрия и бутилированного гидроксианизола уменьшали, используя ступку и пестик. Четверть размера партии полиэтиленоксида объединяли с размерами партий метабисульфита натрия и бутилированного гидроксианизола, и добавляли в склянку с затемненным стеклом объемом 950 см3 и перемешивали на протяжении 5 мин. в блендере Turbula. Оставшийся полиэтиленоксид N80 и размер партии цитрата тофацитиниба добавляли в склянку с затемненным стеклом объемом 950 см3 и перемешивали в блендере Turbula на протяжении 10 мин. Смесь пропускали через мини мельницу Co-mil, используя сито с размером меш 0,8 мм для улучшения смешивания и распределения компонентов. Смесь затем перемешивали на протяжении дополнительных 10 мин в блендере Turbula. Размер партии стеарата магния затем добавляли к предыдущей смеси в склянку с затемненным стеклом объемом 950 см3 и перемешивали на протяжении 3 мин в блендере Turbula.
Полиэтиленоксид класса коагулянт и хлорид натрия пропускали через сито 30 меш, и большие частицы, оставшиеся на сите, отбрасывали. Отдельно, первичный размер частиц метабисульфита натрия и бутилированного гидроксианизола уменьшали, используя ступку и пестик. Половину размера партии полиэтиленоксида объединяли с размерами партий метабисульфита натрия и бутилированного гидроксианизола, и добавляли в склянку с затемненным стеклом объемом 950 см3 и перемешивали на протяжении 5 мин в блендере Turbula. Оставшееся количество полиэтиленоксида класса коагулянт, микрокристаллическую целлюлозу, голубой лакообразующий краситель и порошок хлорида натрия добавляли в этом порядке в склянку с затемненным стеклом объемом 950 см3 и перемешивали на протяжении 10 мин. с помощью блендера Turbula. Смесь пропускали через мини мельницу Co-mil, используя сито с размером меш 0,8 мм для улучшения смешивания и распределения компонентов. Стеарат магния (1,0 г) добавляли в склянку и перемешивали на протяжении 3 мин.
Двухслойные таблетки прессовали, используя стандартные круглые выпуклые инструменты 9/32 дюйма, до средней целевой массы 180,0 мг +/- 5% и средней целевой толщины 5,0 мм +/- 0,1 мм.
№ 2
Покрывающий раствор получали согласно следующим стадиям (смотреть также Таблицу 20): сначала, 150,0 г воды добавляли к 40,5 г ПЭГ 3350 в колбе для смешивания объемом 4 л и перемешивали или взбалтывали вручную до полного растворения ПЭГ. Далее целлюлозы ацетат (109,5 г) добавляли в колбу для смешивания объемом 4 л, содержащую раствор ПЭГ-вода. АЦ разбрасывали в виде суспензии или влажного осадка. Используя колбу для смешивания объемом 4 л, оснащенную роторным импеллером, 2198,1 г ацетона добавляли к смеси ПЭГ-вода-АЦ. Содержимое колбы для смешивания взбалтывали до растворения всех твердых веществ.
Двухслойные таблетки-ядра тофацитиниба (250 г) покрывали в Vector LDCS-5 с полностью перфорированным дражировочным котлом объемом 0,5 л, работающим со скоростью 30 об/мин и потоком воздуха 35 куб.ф./мин с температурой выпуска 32°C. Твердый раствор 6% (м./м.) наносили до увеличения массы покрытия до 25,2 мг на таблетку. После этого таблетки извлекали из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Единственное входное отверстие диаметром 1,0 мм формировали через покрывающую мембрану, по центру внешнего слоя лекарственного средства двухслойной таблетки. Входное отверстие формировали механически или лазером. Нужное покрытие (23,7 мг или 13% массы двухслойного ядра) обеспечило контролируемое высвобождение лекарственного средства, которое отвечало 80% лекарственного средства доставленного за 2,8 часов в среде с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1. Дополнительные даны по растворимости приведены в Таблице 21.
Пример 8. Двухслойная осмотическая таблетка 22 мг
Композицию Примера 8 получали следующим образом (смотреть также Таблицу 22): Размер партии Полиэтиленоксида N80 пропускали через сито 30 меш. Большие частицы, оставшиеся на сите, отбрасывали. Полиэтиленоксид N80 добавляли в склянку из темного стекла объемом 500 см3 и перемешивали вручную для покрытия внутренней поверхности склянки. Размер партии Тофацитиниба добавляли и перемешивали на протяжении 10 мин. с помощью блендера Turbula. Стеарат магния (1,0 г) добавляли в склянку с активной смесью и перемешивали на протяжении 3 мин.
Размер партии Полиэтиленоксида класса коагулянт и хлорид натрия пропускали через сито 30 меш. Полиэтиленоксид, размер партии микрокристаллической целлюлозы, размер партии голубого лакообразующего красителя и порошок хлорида натрия добавляли в этом порядке в колбу объемом 950 см3. Смесь перемешивали на протяжении 10 мин с помощью блендера Turbula. Стеарат магния (1,0 г) добавляли в колбу разбухающего слоя и перемешивали на протяжении 3 мин.
Таблетки прессовали, используя стандартные круглые выпуклые инструменты 5/16 дюймов, до средней целевой массы 250,0 мг +/- 5% и средней целевой толщины 5,6 мм +/- 0,1 мм.
(мг)/ед.:
(мг)/ед.:
Покрывающий раствор получали согласно следующим стадиям (смотреть также Таблицу 23): сначала, 180,0 г воды добавляли к 48,6 г ПЭГ 3350 в колбе для смешивания объемом 4 л, и перемешивали или взбалтывали вручную до полного растворения ПЭГ. Далее, 131,4 г ацетата целлюлозы добавляли в колбу для смешивания объемом 4 л, содержащую раствор ПЭГ-вода. АЦ диспергировали в виде суспензии или влажного осадка. Используя колбу для смешивания объемом 4 л, оснащенную роторным импеллером, 2,640,0 г ацетона добавляли к смеси ПЭГ-вода-АЦ. Содержимое колбы для смешивания взбалтывали до растворения всех твердых веществ.
Двухслойные таблетки-ядра 250 г покрывали с помощью системы покрытия Vector LDCS-5 с полностью перфорированным дражировочным котлом (0,5 л) или барабаном, работающим со скоростью 30 об/мин и потоком воздуха 35 куб.ф./мин с температурой выпуска 32°C. Твердый раствор 6% (м./м.) наносили до достижения массы покрытия 30,0 мг на таблетку. После этого таблетки извлекали из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Единственное входное отверстие диаметром 1,0 мм формировали через покрывающую мембрану по центру внешнего слоя лекарственного средства двухслойной таблетки. Входное отверстие формировали механически или лазером. Нужное покрытие (27,5 мг или 11% массы двухслойного ядра) обеспечивало контролируемое высвобождение лекарственного средства, которое представляло доставку 80% лекарственного средства за 3,7 часов в среде с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1. Дополнительные данные по растворимости приведены в Таблице 24.
Пример 9. Композиция AMT 20 мг
Композицию Примера 9 готовили следующим образом (смотреть также Таблицу 25). Пропускали размеры партий Тофацитиниба, Маннитола, Микрокристаллической целлюлозы и Двухосновного фосфата кальция через сито 30 меш и добавляли в склянку из темного стекла объемом 500 см3. Перемешивали смесь на протяжении 10 мин. с помощью блендера Turbula. Пропускали 0,3 г стеарата магния через сито 30 меш и добавляли в склянку с активной смесью и перемешивали на протяжении 3 мин.
Прессовали смесь в прессовки, содержащие твердую фракцию ~0,70. Дробили прессовки с образованием гранулята. Пропускали 0,2 г стеарата магния через сито 30 меш, добавляли в склянку с активным гранулятом и перемешивали на протяжении 3 мин.
Прессовали таблетки, используя стандартные круглые выпуклые инструменты 9 мм, до средней целевой массы 400 мг +/- 5%, средней целевой толщины 7 мм +/- 0,05 мм, и целевой прочности 15 кПа.
(мг)/единицу:
Готовили покрывающий раствор согласно следующим стадиям (смотреть также Таблицу 26): сначала, добавляли 115 г воды и 150 г ацетона в колбу объемом 2 л и перемешивали на протяжении 5 мин. Добавляли 12 г гидроксипропилцеллюлозы к смеси и перемешивали на протяжении 5 мин. Добавляли 28 г ацетата целлюлозы к смеси и перемешивали на протяжении 5 мин. Добавляли оставшиеся 195 г ацетата целлюлозы к смеси и перемешивали на протяжении 3 часов. Эта процедура дала твердый раствор 8% (м./м.).
Покрывали 250 г таблеток SRC массой по 400 мг с помощью системы покрытия Vector LDCS-5 с полуперфорированным поддоном объемом 0,5 л, работающим со скоростью 30 об/мин и потоком воздуха 30 куб.ф./мин. с температурой выпуска 40°C. Распыляли твердый раствор 8% (м./м.) до увеличения влажной массы 7,5%. Удаляли таблетки из дражировочного котла и высушивали при 40°C на протяжении 16 часов.
Пример 10. Двухслойная осмотическая капсула 20 мг
Предварительная смесь
98,94 г полиэтиленоксида (Polyox WSR N80 LEO) и 1,06 г стеарата магния пропускали через сито 30-меш и добавляли в склянку из темного стекла объемом 250 мл. Смесь перемешивали, используя миксер Turbula (модель T2F), с рабочими параметрами 49 циклов/мин, на протяжении 2 мин.
Активный слой - масса 600 мг
283,71 мг предварительной смеси добавляли в мензурку 1 и взбалтывали вручную для предварительного покрытия внутренней части стеклянной колбы. Цитрат тофацитиниба (32,57 мг) пропускали через сито 20 или 30 меш и добавляли в мензурку 1. Дополнительные 283,71 мг предварительной смеси затем добавляли в мензурку 1. Содержимое мензурки перемешивали, используя миксер Turbula (модель T2F), с рабочими параметрами 49 циклов/мин на протяжении 5 мин. Смесь затем переносили в гидравлический таблеточный пресс Natoli с отдельной станцией и прессовали до нужной толщины 15,6 мм, используя 5,500” B-тип 0,3051” верхний пуансон с модифицированной головкой и 4.755” B-тип 0,3051” нижний пуансон с плоской поверхностью и конусным краем.
Разбухающий слой - масса 300 мг
Разбухающий слой для композиции Примера 10 получали следующим образом (смотреть также Таблицу 27): размеры партий полиэтиленоксида класса коагулянт, голубого лакообразующего красителя, хлорида натрия и микрокристаллической целлюлозы пропускали через сито 20 или 30 меш, добавляли в указанном порядке в бункерный смеситель объемом 10 л. Содержимое блендера перемешивали на протяжении 10 мин при 12 об/мин. Смесь затем пропускали через просеиватель Comil 197S с круглым импеллером и 0,055” круглым ситом, со скоростью 1000 об/мин. Размер партии стеарата магния добавляли в середину протертой смеси в бункерный смеситель. Содержимое блендера перемешивали на протяжении 5 мин при 12 об/мин. Смесь затем переносили в роторный таблеточный пресс Kilian T-100 и прессовали до заданной массы в 300 мг и заданной толщины в 6,65 мм, используя 5,500” B-тип 0,3051” верхний пуансон с модифицированной головкой и 4.755” B-тип 0,3051” нижний пуансон с плоской поверхностью и конусным краем.
(мг)/единицу:
Оболочка капсулы
Раствор для предварительного покрытия (2,5 кг) получали объединением 25 г полисорбата 80 с 2475 г ацетона и перемешиванием на протяжении 10 мин или до растворения с получением 1% (м./м.) раствора.
Функциональный покрывающий раствор (15 кг) получали согласно следующим стадиям (смотреть также Таблицу 28): сначала, все 375 г воды и 120 г ПЭГ 3350 добавляли в приемлемую колбу и перемешивали. Ацетон (14,325 г) добавляли к смеси и перемешивали. Ацетат целлюлозы (180 г) добавляли к смеси и перемешивали до получения однородного раствора. Эта процедура дала твердый раствор 2% (м./м.).
1 кг HDPE капсульных пресс-форм (колпачков или тел) покрывали с помощью системы покрытия Vector LDCS-5 с полуперфорированным поддоном объемом 1,5 л, с рабочими параметрами 18 об/мин и потоком воздуха 40 куб.ф./мин. и температурой выпуска 40°C. После быстрого покрытия пресс-форм 1% м./м. раствором для предварительного покрытия, 2% (м./м.) функциональный твердый покрывающий раствор распыляли со скоростью 20 г/мин с давлением распыления 10 psi и расстоянием от распылителя до объекта 3 дюйма до увеличения влажной массы на 12,5%. Затем капсульные пресс-формы удаляли из дражировочного котла и высушивали при 40°C на протяжении 24 часов. Оболочки капсул убирали из пресс-форм и зачищали.
Единственное отверстие (2000 микрон) просверливали в конце тела капсул. Отверстие может быть просверлено механическим путем или лазером.
Комплект
Активный слой вставляли в половину оболочки капсулы через предварительно просверленное отверстие. Разбухающий слой вставляли в туже половину оболочки капсулы, сначала плоской стороной, располагая вплотную к активному слою. Эти компоненты вставляли в другую половину оболочки капсулы для закрытия капсулы. Полученные и объединенные таким образом, эти компоненты обеспечивают следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 29).
80% тофацитиниба растворили за приблизительно 6 часов в тесте на растворимость 1.
Пример 11. Двухслойная Осмотическая капсула 20 мг
Предварительная смесь
Полиэтиленоксид (Polyox WSR N80 LEO) (98,94 г) и 1,06 г стеарата магния пропускали через сито 30-меш и добавляли в склянку с темным стеклом объемом 250 мл. Смесь перемешивали, используя миксер Turbula (модель T2F), работающий со скоростью 49 циклов/мин., на протяжении 2 мин.
Активный слой - масса 600 мг
Предварительную смесь (283,71 мг) добавляли в мензурку 1 и взбалтывали вручную для предварительного покрытия внутренней поверхности мензурки. Цитрат тофацитиниба (32,57 мг) пропускали через сито 20 или 30 меш и добавляли в мензурку 1. Предварительную смесь (283,71 мг) затем добавляли в мензурку 1. Содержимое мензурки затем перемешивали, используя миксер Turbula (модель T2F), работающий со скоростью 49 циклов/мин., на протяжении 5 мин. Смесь затем переносили в гидравлический таблеточный пресс Natoli с отдельной станцией и прессовали до требуемой толщины 15,6 мм, используя 5,500” B-тип 0,3051” верхний пуансон с модифицированной головкой и 4.755” B-тип 0,3051” нижний пуансон с плоской поверхностью и конусным краем.
Разбухающий слой
Разбухающий слой для композиции Примера 11 получали следующим образом (смотреть также Таблицу 30). Размеры партий полиэтиленоксида класса коагулянт, голубого лакообразующего красителя, хлорида натрия и микрокристаллической целлюлозы пропускали через сито размером 20 или 30 меш, добавляли в указанном порядке в бункерный смеситель объемом 10 л. Содержимое блендера перемешивали на протяжении 10 мин со скоростью 12 об/мин. Смесь затем пропускали через просеиватель Comil 197S с круглым импеллером и 0,055” круглым ситом со скоростью 1000 об/мин. Размер партии стеарата магния добавляли в середину просеянной смеси в бункерный смеситель. Содержимое блендера перемешивали на протяжении 5 мин со скоростью 12 об/мин. Смесь затем переносили в роторный таблеточный пресс Kilian T-100 и прессовали до заданной массы таблетки 300 мг и толщины 6,65 мм, используя 5,500” B-тип 0,3051” верхний пуансон с модифицированной головкой и 4.755” B-тип 0,3051” нижний пуансон с плоской поверхностью и конусным краем.
(мг)/ед.:
Оболочка капсулы
Раствор для предварительного покрытия (2,5 кг) получали объединением 25 г полисорбата 80 с 2475 г ацетона и перемешиванием на протяжении 10 мин или до растворения с получением 1% (м./м.) раствора.
Функциональный покрывающий раствор (15 кг) получали согласно следующим стадиям (смотреть также Таблицу 31): сначала, 375 г воды и 61,5 г ПЭГ 3350 добавляли в приемлемую колбу и перемешивали. Ацетон (14,325 г) добавляли к смеси и перемешивали. Ацетат целлюлозы (225 г) добавляли к смеси и перемешивали. TЭЦ 13,5 г добавляли к смеси и перемешивали до получения однородного раствора. Эта процедура дала 2% твердый раствор (м./м.).
1 кг HDPE капсульных пресс-форм (крышечек или тел) покрывали с помощью системы покрытия Vector LDCS-5 с полуперфорированным поддоном объемом 1,5 л, с рабочими параметрами 18 об/мин и потоком воздуха 40 куб.ф./мин с температурой выпуска 40°C. После быстрого покрытия пресс-форм 1% м./м. раствором для предварительного покрытия, 2% (м./м.) функциональный твердый покрывающий раствор распыляли со скоростью 20 г/мин. с давлением распыления 10 psi и расстоянием от распылителя до объекта 3 дюйма до увеличения влажной массы на 12,5%. Затем пресс-формы капсулы удаляли из дражировочный котла и высушивали при 40°C на протяжении 24 часов. Оболочки капсул убирали из пресс-форм и зачищали.
Единственное отверстие (2000 микрон) просверливали в конце тела капсул. Отверстие может быть просверлено механическим путем или лазером.
Комплект
Активный слой вставляли в половину оболочки капсулы через предварительно просверленное отверстие. Разбухающий слой вставляли в туже половину оболочки капсулы, сначала плоской стороной, располагая плотную к активному слою. Эти компоненты вставляли в другую половину оболочки капсулы для закрытия капсулы. Полученные и объединенные таким образом, эти компоненты обеспечивают следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 32).
80% тофацитиниба растворялось за приблизительно 14 часов в способе растворения 1.
Пример 12. Таблетка с гидрофильной матрицей и контролируемым высвобождением
Металлические поверхности бункера объемом 10л предварительно покрывали добавлением размера партии (также смотреть Таблицу 33 ниже), 963г лактозы Fast Flo 316 и перемешивали на протяжении 2 мин при 12 +/- 1 об/мин. Размер партии Тофацитиниба (164г) добавляли в бункер объемом 10 л и добавляли к моногидрату лактозы. Контейнер с Тофацитинибом промывали некоторым количеством моногидрата лактозы из бункера объемом 10 л. Размер партии Гипромелозы, 1150 г, добавляли в бункер объемом 10 л. Все компоненты перемешивали в бункере на протяжении 10 мин. при 12 +/- 1 об/мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающим со скоростью приблизительно 1400 об/мин. Смесь собирали во втором бункере объемом 10 л. Содержимое бункера перемешивали на протяжении 10 мин при 12 +/- 1 об/мин.
Внутрикристаллитный стеарат магния, 5,75 г, добавляли в бункер и перемешивали на протяжении 3 мин. при 12 +/- 1 об/мин. Смазанную смесь пропускали через роликовый пресс Gerteis, оснащенный роликами с насеченной головкой, съемными бортами обода, и линейной вибромельницей, включающей портативный ротор и 1 мм пемзовальную пластину. Требуемую ленту твердой фракции 0,7 (0,67-0,73) и гранулы собирали в первоначальный бункер объемом 10 л.
Внутрикристаллитный стеарат магния, 17,25 г, добавляли в бункер и содержимое перемешивали на протяжении 3 мин при 12 +/- 1 об/мин. Финальную смесь пропускали через роторный таблеточный пресс Kilian T-100. Таблетки прессовали, используя инструменты 13/32” SRC, до средней целевой массы 500 мг +/- 5% и целевой прочности 15 кПа. Таблетки пропускали через пылеуловитель и металлодетектор.
Композиция таблетки Тофацитиниба с гидрофильной матрицей (22 мг); общая масса таблеток 500 мг
Fast Flo 316
Прессованные таблетки обеспечивают следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 об/мин на основании теста на растворимость 1 (Таблица 34).
Пример 13. Таблетка с мгновенным высвобождением (10 мг)
Композицию таблетки Примера 13 получали согласно следующему способу. Компоненты 1-4 объединяли и обрабатывали, используя процедуру перемешивание-помол-перемешивание. Компонент 5 затем добавляли в смесь и объединяли, используя процедуру смешивания. Такую смазанную смесь затем поддавали сухому гранулированию. Компонент 6 затем добавляли к сухому грануляту и объединяли, используя процедуру смешивания. Смазанный гранулят прессовали в таблетки массой 400 мг, используя роторный таблеточный пресс. Таблетки затем покрывали, используя устройство для нанесения пленочного покрытия, распыляющее раствор, содержащий Компоненты 7 и 8 до нанесения на таблетки покрытия массой 12 мг.
Пример 14. Таблетка с мгновенным высвобождением (5 мг)
Таблетку получали согласно следующему способу. Компоненты 1-4 объединяли и обрабатывали, используя процедуру перемешивание-помол-перемешивание. Компонент 5 затем добавляли в смесь и объединяли, используя процедуру смешивания. Такую смазанную смесь затем поддавали сухому гранулированию. Компонент 6 затем добавляли к сухому грануляту и объединяли, используя процедуру смешивания. Смазанный гранулят прессовали в таблетки массой 500 мг, используя роторный таблеточный пресс. Таблетки затем покрывали, используя устройство для нанесения пленочного покрытия, распыляющее раствор, содержащий Компоненты 7 и 8 до нанесения на таблетки покрытия массой 20 мг.
Пример 15. Исследование A
Определяли относительную биодоступность одной дозы 2-х разных оральных композиций тофацитиниба (20 мг) с замедленным высвобождением по отношению к одной дозе таблеток тофацитиниба (10 мг) с мгновенным высвобождением (МВ), и определяли следующие предельные показатели для тофацитиниба: Cmax, Tmax, AUCinf, AUClast. Дополнительные предельные показатели определяли для относительной биодоступности (%RBA) тофацитиниба каждой композиции с замедленным высвобождением, по отношению к композиции с мгновенным высвобождением (МВ).
Исследование было рандомизированным, открытым, с разовой дозой, 3-мя периодами, 3-мя обработками, с 6-ю пересекающимися последовательностями на 12-ти здоровых пациентах мужского пола (смотреть Таблицу 37). Пациенты получали две разные композиции тофацитиниба с замедленным высвобождением, и таблетку с мгновенным высвобождением с периодом вымывания 3 дня между дозами. Композиции с замедленным высвобождением давали в виде разовой дозы 20 мг, а композицию с мгновенным высвобождением давали в виде двух таблеток с разовой дозой 5 мг.
A: Таблетка с мгновенным высвобождением, 10 мг;
B: Пример 10 двухслойная осмотическая капсула, 20 мг;
C: Пример 11 двухслойная осмотическая капсула, 20 мг.
Пациентов держали натощак на протяжении ночи, как минимум 8 часов перед введением исследуемого лекарства. Утром первого дня в каждом периоде, все пациенты получали одну оральную дозу исследуемого лекарства с 240 мл воды. Пациентам давали стандартизированный ланч через 4 часа после введения дозы.
Введенные дозированные формы:
Таблетка Тофацитиниба (10 мг) с мгновенным высвобождением (ссылка): получена в Примере 13.
Двухслойная осмотическая капсула Тофацитиниба (20 мг): получена в Примере 10.
Двухслойная осмотическая капсула Тофацитиниба (20 мг): получена в Примере 11.
На протяжении всех периодов исследования, образцы крови для обеспечения плазмой для фармакокинетического анализа собирали периодично. ФК образцы анализировали, используя стандартные аналитические способы. Нормализованные по дозе природные логарифмически преобразованные AUCinf, AUClast и Cmax анализировали для тофацитиниба, используя модель со смешанным эффектом: с последовательностью, периодом и обработкой в качестве фиксированных эффектов, и субъектом в последовательности в качестве случайного эффекта. Оценку выравненных стандартных отклонений (Сравнительный Тест) и соответствующие 90% доверительные интервалы получали благодаря этой модели. Выравненные стандартные отклонения и 90% доверительные интервалы для разниц возводили в степень для оценки соотношения выравненных среднегеометрических значений (Тест/Сравнение) и 90% доверительные интервалы для коэффициентов. Контрольная композиция таблетки с мгновенным высвобождением была Сравнительным лечением, а композиции с замедленным высвобождением были Тест лечением.
Относительную биодоступность тофацитиниба оценивали как соотношение дозо-нормализованых выравненных среднегеометрических значений Теста и Сравнительного теста для AUCinf.
ФК параметры AUCinf, AUClast, Cmax, Tmax, и t1/2 суммировали описательно путем обработки и анализа (где применимо). Для AUCinf и Cmax, индивидуальные параметры субъекта были нанесены на график путем обработки каждого анализируемого вещества по отдельности (где применимо). Концентрации включали в перечень и суммировали описательно указанием времени взятия ФК образцов, обработки и анализируемого вещества (где применимо). Индивидуальный, средний и медианные профили данных концентрация-время были нанесены на график путем обработки и анализа (где применимо). Для обзора статистики, для средних и медианных графиков по времени отбора проб использовали номинальное время отбора ФК образцов, для графиков времени отдельных пациентов использовали фактическое время отбора ФК образцов.
Прогнозируемые статические значения получали, используя метод суперпозиции, используя программное обеспечение WinNonLin (Pharsight Corp). Суперпозицию использовали на каждом индивидуальном фармакокинетическом профиле для получения статического фармакокинетического профиля каждого пациента. Обозначения и способ определения ФК параметров приведены в Таблице 38. Результаты исследования показаны на Таблице 39.
концентрация в плазме
концентрация в плазме, поделенная на введенную дозу
(время ниже
17 нг/мл)
10 мг
(Сравнительный)
(Тест)
(Тест)
Вышеприведенные значения указаны как среднегеометрические значения (% коэффициента вариации (CV)) для всех, кроме: средний (диапазон) для Tmax; среднеарифметическое (%CV) для t½. Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
МВ дважды в день
(Сравнительный)
20 мг, QD
(Тест)
25 мг, QD*
(Тест)
20 мг, QD
(Тест)
33 мг, QD*
(Тест)
(30%)
(24%)
(24%)
(30%)
(30%)
(39%)
(37%)
(37%)
(76%)
(76%)
(39%)
(37%)
(37%)
(76%)
(76%)
(128-215%)
(160-268%)
(123-581%)
(203-958%)
(2 x 6,3 ч)
Вышеприведенные значения указаны как среднегеометрические значения (%CV). Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
* - необходимая корректировка дозы для достижения 100% RBA с теми длительностями модифицированного высвобождения.
Результаты этого исследования показывают, что дозированные формы с замедленным высвобождением, которым необходимо 6 часов или больше для высвобождения и растворения 80% тофацитиниба, не отвечают желаемым ФК характеристикам для дозированных форм тофацитиниба с замедленным высвобождением. А именно, дозированная форма с замедленным высвобождением, которой необходимо 6 часов или больше для высвобождения и растворения 80% тофацитиниба, и которая содержит необходимое количество тофацитиниба для обеспечения значения AUC эквивалентного дозированной форме с мгновенным высвобождением, обеспечивает большее время с концентрацией 17 нг/мл (JAK 1/3 рецептор значения IC50), чем время с концентрацией 17 нг/мл дозированной формы с мгновенным высвобождением. Также дозированная форма с замедленным высвобождением, которой необходимо 14 часов для высвобождения и растворения 80% тофацитиниба, имеет более высокое дозо-нормализованное значение Cmin,ss, более низкую дозо-нормализованную AUC, и низкую относительную биодоступность, по сравнению с дозированной формой с мгновенным высвобождением, что требует увеличения концентрации тофацитиниба для получения AUC эквивалентной AUC дозированной формы с мгновенным высвобождением. Эти результаты поддерживают требование для дозированной формы тофацитиниба с замедленным высвобождением, чтобы высвобождение и растворение 80% тофацитиниба длилось менее 6 часов.
Пример 16. Исследование B
Определяли относительную биодоступность 3-х разных оральных композиций тофацитиниба (22 мг) с замедленным высвобождением, по отношению к одной дозе таблеток тофацитиниба (10 мг) с мгновенным высвобождением (МВ), и определяли следующие предельные показатели для тофацитиниба: Cmax, Tmax, AUCinf, AUClast. Дополнительные предельные показатели определяли для относительной биодоступности (%RBA) тофацитиниба каждой композиции с замедленным высвобождением, по отношению к композиции с мгновенным высвобождением (МВ).
Исследование было рандомизированным, открытым, с разовой дозой, 4-мя периодами, 6-ю обработками, с 6-ю частично пересекающимися исследованиями на 30-ти здоровых пациентах мужского пола (смотреть Таблицу 40). В первом периоде, пациенты получали одну из двух разных композиций тофацитиниба с замедленным высвобождением в состоянии после еды. Во втором и третьем периодах, пациенты получали две из трех разных композиций тофацитиниба с замедленным высвобождением. В четвертом периоде, пациенты получали таблетку с мгновенным высвобождением. Период вымывания составлял 3 дня между дозами. Три композиции с замедленным высвобождением давали в виде разовой дозы 22 мг, а композицию с мгновенным высвобождением давали в виде двух таблеток с разовой дозой 5 мг.
A: 4-часа, Таблетка с экструдируемым ядром, 22 мг, после еды;
B: Пример 4, Таблетка с матрицей, 22 мг, после еды;
C: 4-часа, Таблетка с экструдируемым ядром, 22 мг, натощак;
D: Пример 4 Таблетка с матрицей, 22 мг, натощак;
E: Пример 12 Таблетка с матрицей, 22 мг, натощак;
F: Таблетка с мгновенным высвобождением, 2 × 5 мг, натощак.
В 1-м периоде, после ночного голодания на протяжении как минимум 8 часов, пациентам давали стандартный завтрак с высоким содержанием жира FDA за 30 мин перед введением исследуемого лекарства. Завтрак употребляли в течении 30 мин или меньше. Пациенты получали Лечение A или Лечение B через 30 мин (+/-5 мин) после начала завтрака. Нельзя было употреблять пищу в течение как минимум 4 часов после приема дозы. Нельзя было пить воду за час до и в течение часа после приема дозы. Прием дозы в периоды 1, 2 и 3 производили с минимальным временем вымывания в 72 часа. Следующий период исследования (периоды 2, 3 и 4) начинался сразу же после завершения 72-часового перерыва и отбора ФК образцов на 4-й день предыдущего периода (периоды 1, 2 и 3 соответственно). В периодах 2, 3 и 4, исследуемое лекарство вводили после ночного голодания на протяжении как минимум 8 часов. Есть разрешали только через 4 часа после приема дозы. Нельзя было пить воду за час до и в течение часа после приема исследуемого лекарства.
Введенные дозированные формы:
Таблетка Тофацитиниба с мгновенным высвобождением, 5 мг, (сравнительный): получена в Примере 14 выше.
Таблетка Тофацитиниба с экструдируемым ядром, 22 мг: получена в Примере 1 выше.
Таблетки Тофацитиниба с матрицей, 22 мг: получены в Примере 4 и 12 выше.
На протяжении всех периодов исследования, периодично собирали образцы крови для обеспечения плазмой для ФК анализов. Результаты исследования приведены в Таблице 41. ФК образцы анализировали, используя стандартные аналитические методы.
Нормализованные по дозе природные логарифмически преобразованные AUCinf, AUClast и Cmax анализировали для тофацитиниба, используя модель со смешанным эффектом: с последовательностью, периодом и обработкой в качестве фиксированных эффектов, и субъектом в последовательности в качестве случайного эффекта. Оценку выравненных стандартных отклонений (Сравнительный Тест) и соответствующие 90% доверительные интервалы получали благодаря этой модели. Выравненные стандартные отклонения и 90% доверительные интервалы для разниц возводили в степень для оценки соотношения выравненных среднегеометрических значений (Тест/Сравнение) и 90% доверительные интервалы для коэффициентов. Контрольная композиция таблетки с мгновенным высвобождением была Сравнительным лечением, а композиции с замедленным высвобождением были Тест лечением.
Относительную биодоступность тофацитиниба оценивали как соотношение дозо-нормализованых выравненных среднегеометрических значений Теста и Сравнительного теста для AUCinf.
ФК параметры AUCinf, AUClast, Cmax, Tmax, и t1/2 суммировали описательно путем обработки и анализа (где применимо). Для AUCinf и Cmax, индивидуальные параметры субъекта были нанесены на график путем обработки каждого анализируемого вещества по отдельности (где применимо). Концентрации включали в перечень и суммировали описательно указанием времени взятия ФК образцов, обработки и анализируемого вещества (где применимо). Данные среднего и медиана профилей отдельного пациента концентрация-время были нанесены на график обработки и анализа (где применимо). Для обзора статистики, для средних и медианных графиков по времени отбора проб использовали номинальное время отбора ФК образцов, для графиков времени отдельных пациентов использовали фактическое время отбора ФК образцов.
Прогнозируемые статические значения получали, используя метод суперпозиции, используя программное обеспечение WinNonLin (Pharsight Corp). Суперпозицию использовали на каждом индивидуальном фармакокинетическом профиле для получения статического фармакокинетического профиля каждого пациента.
(Тест)
(Тест)
(Пример 12)
(Тест) (ч)
(ч)
(82%-104%)
(75%-94%)
(50%-62%)
(87%-96%)
(92%-101%)
(85%-94%)
Вышеприведенные значения указаны как среднегеометрические значения (% коэффициента вариации (CV)) для всех, кроме: средний (диапазон) для Tmax; среднеарифметическое (%CV) для t. Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
(Сравнит.)
(Тест)
(Сравнит.)
(Тест) (ч)
Вышеприведенные значения указаны как среднегеометрические значения (% коэффициента вариации (CV)) для всех, кроме: средний (диапазон) для Tmax; среднеарифметическое (%CV) для t. Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
МВ дважды в день
ECS QD
(Пример 4)
(Пример 12)
(Сравнит.)
(Тест)
(Тест)
(Тест)
(нг/мл)
(30%)
(30%)
(32%)
(27%)
(нг/мл)
(66%)
(92%)
(89%)
(60%)
(нг/мл/мг)
(66%)
(92%)
(89%)
(60%)
соотношение (%)
(59-92%)
(51-81%)
(134-190%)
(2 x 6,7 ч)
Вышеприведенные значения указаны как среднегеометрические значения (%CV). Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
Дозированные формы с замедленным высвобождением, содержащие 22 мг тофацитиниба, которые высвобождают и растворяют 80% тофацитиниба за 4-5 часов, обеспечивают дозо-пропорциональные ФК характеристики и отвечают желаемым ФК требованиям при приеме натощак. Дозированные формы с замедленным высвобождением, содержащие 22 мг тофацитиниба, которые высвобождают и растворяют 80% тофацитиниба с помощью осмотического давления за 4 час, обеспечивают аналогичные ФК характеристики при приеме как натощак, так и после еды. Дозированные формы с замедленным высвобождением, содержащие 22 мг тофацитиниба, которые высвобождают и растворяют 80% тофацитиниба путем диффузии и эрозии через матрицу за 5 часов, не обеспечивают аналогичные Cmax характеристики при приеме как натощак, так и после еды.
Пример 17. Исследование C
Определяли относительную биодоступность одной дозы оральной композиции тофацитиниба (11 мг) с замедленным высвобождением, по отношению к одной дозе таблеток тофацитиниба (22 мг) с замедленным высвобождением, и определяли следующие предельные показатели для тофацитиниба: Cmax, Tmax, AUCinf, AUClast. Дополнительный предельный показатель определяли для относительной биодоступности (%RBA) тофацитиниба в композиции с замедленным высвобождением (11 мг), по отношению к композиции с замедленным высвобождением (22 мг).
Исследование было рандомизированным, открытым, с разовой дозой, 2-мя периодами, 2-мя обработками, с 2-мя пересекающимися последовательностями на 20-ти здоровых пациентах мужского пола (смотреть Таблицу 42). Пациенты получали две разные композиций тофацитиниба с замедленным высвобождением с периодом вымывания 3 дня между дозами. Композиции с замедленным высвобождением давали в виде разовой дозы 11 или 22 мг.
A: Таблетка с экструдируемым ядром, 11 мг, получена в Примере 1 выше.
B: Таблетка с экструдируемым ядром, 22 м, получена в Примере 1 выше.
Пациентов держали натощак ночью в течении как минимум 8 часов перед приемом исследуемого лекарства. Утром 1-го дня каждого периода, все пациенты получали единоразовую оральную дозу исследуемого лекарства с 240 мл воды. Пациентам давали стандартный ланч через 4 часа после приема дозы.
Введенные дозированные формы:
Дозированные формы с замедленным высвобождением Тофацитиниба, 22 мг: получены в Примере 1 выше.
Дозированные формы с замедленным высвобождением Тофацитиниба, 11 мг: получены в Примере 1 выше.
На протяжении всех периодов исследования, периодично собирали образцы крови для обеспечения плазмой для ФК анализов. Результаты исследования приведены в Таблице 43. ФК образцы анализировали, используя стандартные аналитические методы.
Нормализованные по дозе природные логарифмически преобразованные AUCinf, AUClast и Cmax анализировали для тофацитиниба, используя модель со смешанным эффектом: с последовательностью, периодом и обработкой в качестве фиксированных эффектов, и субъектом в последовательности в качестве случайного эффекта. Оценку выравненных стандартных отклонений (Сравнительный Тест) и соответствующие 90% доверительные интервалы получали благодаря этой модели. Выравненные стандартные отклонения и 90% доверительные интервалы для разниц возводили в степень для оценки соотношения выравненных среднегеометрических значений (Тест/Сравнение) и 90% доверительные интервалы для коэффициентов. Композиция таблетки с мгновенным высвобождением, 22 мг, была Сравнительным лечением, а композиция с замедленным высвобождением, 11 мг, была Тест лечением.
Относительную биодоступность тофацитиниба оценивали как соотношение дозо-нормализованых выравненных среднегеометрических значений Теста и Сравнительного теста для AUCinf.
ФК параметры AUCinf, AUClast, Cmax, Tmax, и t суммировали описательно путем обработки и анализа (где применимо). Для AUCinf и Cmax, индивидуальные параметры субъекта были нанесены на график путем обработки каждого анализируемого вещества по отдельности (где применимо). Концентрации включали в перечень и суммировали описательно указанием времени взятия ФК образцов, обработки и анализируемого вещества (где применимо). Данные среднего и медиана профилей отдельного пациента концентрация-время были нанесены на график обработки и анализа (где применимо). Для обзора статистики, для средних и медианных графиков по времени отбора проб использовали номинальное время отбора ФК образцов, для графиков времени отдельных пациентов использовали фактическое время отбора ФК образцов.
суммировали описательно путем обработки и анализа (где применимо). Для AUCinf и Cmax, индивидуальные параметры субъекта были нанесены на график путем обработки каждого анализируемого вещества по отдельности (где применимо). Концентрации включали в перечень и суммировали описательно указанием времени взятия ФК образцов, обработки и анализируемого вещества (где применимо). Данные среднего и медиана профилей отдельного пациента концентрация-время были нанесены на график обработки и анализа (где применимо). Для обзора статистики, для средних и медианных графиков по времени отбора проб использовали номинальное время отбора ФК образцов, для графиков времени отдельных пациентов использовали фактическое время отбора ФК образцов.
Прогнозируемые статические значения получали, используя метод суперпозиции, используя программное обеспечение WinNonLin (Pharsight Corp). Суперпозицию использовали на каждом индивидуальном фармакокинетическом профиле для получения статического фармакокинетического профиля каждого пациента.
ФК параметры
(Тест)
(Сравнит.) (ч)
Вышеприведенные значения указаны как среднегеометрические значения (%CV) для всех, кроме: средний (диапазон) для Tmax; среднеарифметическое (%CV) для t. Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
(Тест)
(Сравнит.)
Вышеприведенные значения указаны как среднегеометрические значения (%CV). Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
Дозированные формы с замедленным высвобождением, содержащие 11 мг и 22 мг тофацитиниба, которые высвобождают и растворяют тофацитиниб согласно формуле (на основе теста на растворимость 1) обеспечивают дозо-пропорциональные ФК характеристики и отвечают желаемым ФК требованиям.
Пример 18 Исследование D
Определяли относительную биодоступность таблеток тофацитиниба с замедленным высвобождением, 11 мг, по отношению к разовой дозе двух таблеток тофацитиниба с мгновенным высвобождением, 5 мг (МВ), и определяли следующие предельные показатели для тофацитиниба: Cmax, Tmax, AUCinf, AUClast. Дополнительный предельный показатель определяли для относительной биодоступности (%RBA) тофацитиниба для каждой композиции с замедленным высвобождением по отношению к композиции МВ.
Исследование было рандомизированным, открытым, с разовой дозой, 2-мя периодами, 2-мя обработками, с 2-мя пересекающимися последовательностями на 26-ти здоровых пациентах мужского пола (смотреть Таблицу 44). Пациенты получали композиции цитрата тофацитиниба (11 мг) с замедленным высвобождением или две композиции цитрата тофацитиниба (5 мг) с мгновенным высвобождением, с периодом вымывания 3 дня между дозами.
A: Таблетка с экструдируемым ядром, 11 мг, получена следующим образом:
300 кг
Половину размера партии сорбитола, 114,172 кг, добавляли в бункер объемом 800 л. Размер партии Коповидона, 18,000 кг, затем добавляли в бункер объемом 800 л. Размер партии Тофацитиниба, 26,656 кг, затем добавляли в бункер объемом 800 л. Размер партии Гидроксицеллюлозы, 24,000 кг, затем добавляли в бункер объемом 800 л. Оставшуюся половину размера партии сорбитола, 114,172 г добавляли бункер объемом 800 л. Все материалы добавляли с помощью вакуумной системы переноса и пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающую со скоростью приблизительно 1400 об/мин. Все компоненты перемешивали в бункере на протяжении 20 мин при 12 +/- 1 об/мин.
Смесь пропускали через ротационную мельницу Comil, оснащенную фильтром 0,032” и импеллером с круглым краем, работающую со скоростью приблизительно 1400 об/мин. Смесь собирали во втором бункере объемом 800 л. Содержимое бункера перемешивали на протяжении 20 мин при 12 +/- 1 об/мин.
Стеарат магния, 3,000 кг, пропускали через сито с размером меш 850-микрон и добавляли в бункер и содержимое перемешивали на протяжении 5 мин при 12 +/- 1 об/мин. Таблетки прессовали, используя 0,2080” × 0,4160” модифицированные овальные инструменты на роторном таблеточном прессе Manesty Mark IV, до средней целевой массы 200 мг +/- 5%, средней целевой толщины 4,17 мм +/- 0,05 мм, и целевой прочности 11 кПа. Таблетки пропускали через пылеуловитель и металлодетектор.
200 мг
Покрывающий раствор 750 кг получали согласно следующим стадиям. Сначала, все 147,0 кг метанола и 580,5 г ацетона добавляли в резервуар объемом 250 галлон. Ацетат целлюлозы (13,5 кг) добавляли к смеси. Гидроксипропилцеллюлозу (9,0 кг) добавляли к смеси. Содержимое контейнера перемешивали на протяжении часа. Эта процедура дала 3% твердый раствор (м./м.).
Овальные таблетки (250 кг) по 200 мг покрывали в устройстве для нанесения покрытия Vector HC-130, со скоростью 8 об/мин и потоком воздуха 1500 куб.ф./мин. с температурой выпуска 28°C. 3% твердый раствор (м./м.) наносили до достижения увеличения влажной массы уровня 7%. Затем таблетки извлекали из дражировочного котла и высушивали при 45°C на протяжении 24 часов.
Единственное отверстие (600 микрон) просверливали в конце полосы овальной таблетки. Отверстие может быть просверлено механическим путем или лазером. Покрытие в 7% обеспечило следующее высвобождение в среду с pH 6,8, при скорости вращения лопастей 50 о./мин на основании теста на растворимость 1 (Таблица 47):
% растворенного лекарства
B: Тофацитиниб 2 × 5 мг
Таблетку с мгновенным высвобождением (сравнит.) готовили следующим образом:
Таблетку получали согласно следующему способу. Компоненты 1-4 объединяли и обрабатывали, используя процедуру перемешивание-дробление-перемешивание. Компонент 5 затем добавляли в смесь и объединяли, используя процедуру смешивания. Такую смазанную смесь затем поддавали сухому гранулированию. Компонент 6 затем добавляли к сухому грануляту и объединяли, используя процедуру смешивания. Смазанный гранулят прессовали в таблетки массой 200 мг, используя роторный таблеточный пресс. Таблетки затем покрывали, используя устройство для нанесения пленочного покрытия, распыляющее раствор, содержащий Компоненты 7 и 8 до нанесения 6 мг покрытия на таблетки.
Пациентов держали натощак ночью на протяжении как минимум 8 часов перед приемом исследуемого лекарства. На утро 1-го дня каждого периода, все пациенты получали единоразовую дозу исследуемого лекарства с 240 мл воды. Пациентам давали стандартизированный ланч через 4 часа после приема дозы.
Введенные дозированные формы:
Таблетка с мгновенным высвобождением Тофацитиниба 5 мг (сравнит.): получена, как описано выше.
Дозированные формы Тофацитиниба (11 мг) с замедленным высвобождением: получены, как описано выше.
На протяжении всех периодов исследования, периодично собирали образцы крови для обеспечения плазмой для ФК анализов. Результаты исследования приведены в Таблице 49. ФК образцы анализировали, используя стандартные аналитические методы. Нормализованные по дозе природные логарифмически преобразованные AUCinf, AUClast и Cmax анализировали для тофацитиниба, используя модель со смешанным эффектом: с последовательностью, периодом и обработкой в качестве фиксированных эффектов, и субъектом в последовательности в качестве случайного эффекта. Оценку выравненных стандартных отклонений (Сравнительный Тест) и соответствующие 90% доверительные интервалы получали благодаря этой модели. Выравненные стандартные отклонения и 90% доверительные интервалы для разниц возводили в степень для оценки соотношения выравненных среднегеометрических значений (Тест/Сравнение) и 90% доверительные интервалы для коэффициентов. Композиция с мгновенным высвобождением 2 × 5 мг была сравнительным лечением, а композиция с замедленным высвобождением 11 мг была Тест лечением.
Относительную биодоступность тофацитиниба оценивали как соотношение дозо-нормализованых выравненных среднегеометрических значений Теста и Сравнительного теста для AUCinf.
ФК параметры AUCinf, AUClast, Cmax, Tmax, и t1/2 суммировали описательно путем обработки и анализа (где применимо). Для AUCinf и Cmax, индивидуальные параметры субъекта были нанесены на график путем обработки каждого анализируемого вещества по отдельности (где применимо). Концентрации включали в перечень и суммировали описательно указанием времени взятия ФК образцов, обработки и анализируемого вещества (где применимо). Данные среднего и медиана профилей отдельного пациента концентрация-время были нанесены на график обработки и анализа (где применимо). Для обзора статистики, для средних и медианных графиков по времени отбора проб использовали номинальное время отбора ФК образцов, для графиков времени отдельных пациентов использовали фактическое время отбора ФК образцов.
Прогнозируемые статические значения получали, используя метод суперпозиции, используя программное обеспечение WinNonLin (Pharsight Corp). Суперпозицию использовали на каждом индивидуальном фармакокинетическом профиле для получения статического фармакокинетического профиля каждого пациента.
ФК параметры
(Тест)
(сравнит.) (ч)
(100%-107%)
Вышеприведенные значения указаны как среднегеометрические значения (%CV) для всех, кроме: средний (диапазон) для Tmax; среднеарифметическое (%CV) для t. Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
(Тест)
(сравнит.)
Вышеприведенные значения указаны как среднегеометрические значения (%CV). Вышеприведенные соотношения представлены как соотношения среднегеометрических значений (90% доверительные интервалы).
Дозированные формы с замедленным высвобождением, содержащие 11 мг тофацитиниба, которые высвобождают и растворяют 80% тофацитиниба за 4-5 часов, обеспечивают ФК параметры аналогичные параметрам дозированной формы с мгновенным высвобождением, содержащей 10 мг тофацитиниба, и соответствуют желаемым ФК требованиям при приеме натощак.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ТОФАЦИТИНИБА С НЕПРЕРЫВНЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2674345C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ, ВКЛЮЧАЮЩАЯ ФЕБУКСОСТАТ | 2011 |
|
RU2602188C2 |
| СПОСОБЫ И ЛЕКАРСТВЕННЫЕ ФОРМЫ ДЛЯ КОНТРОЛИРУЕМОЙ ДОСТАВКИ ПАЛИПЕРИДОНА | 2003 |
|
RU2321391C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ ЗИПРАЗИДОНА | 2004 |
|
RU2351316C2 |
| НОВАЯ КОМПОЗИЦИЯ ЗАМЕДЛЕННОГО ВЫСВОБОЖДЕНИЯ ТОФАЦИТИНИБА, ЕГО ПРОИЗВОДНЫХ И СОЛЕЙ | 2021 |
|
RU2839277C1 |
| СПОСОБЫ ЛЕЧЕНИЯ НАРУШЕНИЙ ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА НЕЗАВИСИМО ОТ ПОТРЕБЛЕНИЯ ПИЩИ | 2014 |
|
RU2563993C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ МЕТФОРМИНА С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2006 |
|
RU2433821C2 |
| ФОРМА ПРОЛОНГИРОВАННОГО ВЫСВОБОЖДЕНИЯ ВЕНЛАФАКСИНА ГИДРОХЛОРИДА | 2004 |
|
RU2340331C2 |
| КОМПОЗИЦИИ ГИДРОКОДОНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2001 |
|
RU2253452C2 |
| ТАКРОЛИМУС ДЛЯ УЛУЧШЕННОГО ЛЕЧЕНИЯ ПАЦИЕНТОВ С ТРАНСПЛАНТАТАМИ | 2009 |
|
RU2574006C2 |
Данное изобретение относится к оральным композициям с непрерывным высвобождением тофацитиниба и его фармацевтически приемлемых солей. Композиции, описанные в данном документе, имеют желательные фармакокинетические характеристики. 3 н. и 22 з.п. ф-лы, 49 табл., 18 пр.
1. Одноразовая фармацевтическая дозированная форма, которая содержит ядро, содержащее 22 мг тофацитиниба или эквивалентное количество тофацитиниба в виде его фармацевтически приемлемой соли и осмоген,
и полупроницаемое мембранное покрытие, окружающее ядро, где указанное покрытие содержит не растворимый в воде полимер,
где указанная дозированная форма является дозированной формой с замедленным высвобождением и при добавлении в исследуемую среду, содержащую 900 мл 0.05 М буфера фосфата калия с рН 6,8, при 37°С в стандартном согласно фармакопее США аппарате с вращающимися лопастями, где лопасти вращаются со скоростью 50 об/мин, растворяется не более чем 30% тофацитиниба или его фармацевтически приемлемой соли за 1 час, и не менее чем 35%, и не более чем 75% тофацитиниба или его фармацевтически приемлемой соли за 2,5 часа, и не менее чем 75% тофацитиниба или его фармацевтически приемлемой соли за 5 часов, и где указанная дозированная форма доставляет тофацитиниб или его фармацевтически приемлемую соль субъекту за счет осмотического давления, и где не растворимый в воде полимер представляет собой производное целлюлозы, которое поддерживает высвобождение тофацитиниба или его фармацевтически приемлемой соли.
2. Одноразовая фармацевтическая дозированная форма, которая содержит ядро, содержащее 22 мг тофацитиниба или эквивалентное количество тофацитиниба в виде его фармацевтически приемлемой соли и осмоген,
и полупроницаемое мембранное покрытие, окружающее ядро, где указанное покрытие содержит не растворимый в воде полимер,
где дозированная форма является дозированной формой с замедленным высвобождением, и при введении субъекту орально обеспечивает AUC в диапазоне от 80 до 125% от AUC 10 мг тофацитиниба или эквивалентного количества тофацитиниба в форме его фармацевтически приемлемой соли, вводимого в виде композиции с мгновенным высвобождением дважды в день (BID), и обеспечивает среднегеометрическое соотношение Cmax к Cmin в плазме от 10 до 100, и где дозированная форма доставляет тофацитиниб или его фармацевтически приемлемую соль субъекту за счет осмотического давления, и где не растворимый в воде полимер представляет собой производное целлюлозы, которое поддерживает высвобождение тофацитиниба или его фармацевтически приемлемой соли.
3. Фармацевтическая дозированная форма по п. 2, где диапазон AUC составляет 90-110% и среднегеометрическая концентрация от Cmax до Cmin в плазме составляет от 20 до 40.
4. Фармацевтическая дозированная форма по п. 3, где среднегеометрическая концентрация от Cmax до Cmin в плазме составляет от 20 до 30.
5. Фармацевтическая дозированная форма по п. 2, где при введении дозированной формы субъекту орально обеспечивается среднее значение Cmax в плазме в диапазоне от 70 до 125% от среднего значения Cmax в плазме тофацитиниба, введенного в композиции с мгновенным высвобождением дважды в день в стационарном состоянии.
6. Фармацевтическая дозированная форма по п. 2, где при введении дозированной формы субъекту орально обеспечивается отдых от лекарственного средства в диапазоне от 80 до 110% от отдыха от лекарственного средства тофацитиниба, введенного в композиции с мгновенным высвобождением дважды в день на протяжении 24 часового периода.
7. Фармацевтическая дозированная форма по п. 2, где отдых от лекарственного средства составляет от 15 до 18 часов на протяжении 24-часового периода.
8. Одноразовая фармацевтическая дозированная форма, которая содержит ядро, содержащее 22 мг тофацитиниба или эквивалентное количество тофацитиниба в виде его фармацевтически приемлемой соли и осмоген,
и полупроницаемое мембранное покрытие, окружающее ядро, где указанное покрытие содержит не растворимый в воде полимер,
где указанная дозированная форма является дозированной формой с замедленным высвобождением, и которая при введении субъекту имеет среднюю площадь под кривой концентрации в плазме по отношению ко времени после введения от 17 нг-час/мл на мг тофацитиниба до 42 нг-час/мл на мг тофацитиниба, и среднегеометрическое соотношение Cmax к Cmin в плазме составляет от 10 до 100, и при этом дозированная форма высвобождает тофацитиниб, или его фармацевтически приемлемую соль, у субъекта за счет осмотического давления, и при этом не растворимый в воде полимер представляет собой производное целлюлозы, которое поддерживает высвобождение тофацитиниба, или его фармацевтически приемлемой соли.
9. Фармацевтическая дозированная форма по п. 8, где среднегеометрическое соотношение Cmax к Cmin в плазме составляет от 20 до 40.
10. Фармацевтическая дозированная форма по п. 9, где среднегеометрическое соотношение Cmax к Cmin в плазме составляет от 20 до 30.
11. Фармацевтическая дозированная форма по п. 8, где субъект имеет единое пролонгированное время при дозе выше 17 нг/мл от 6 до 15 часов, и единое пролонгированное время при дозе ниже 17 нг/мл от 9 до 18 часов в течение 24 часового интервала дозирования.
12. Фармацевтическая дозированная форма по п. 11, где субъект имеет единое пролонгированное время при дозе выше 17 нг/мл от 6 до 9 часов.
13. Фармацевтическая дозированная форма по п. 11, где субъект имеет единое пролонгированное время при дозе ниже 17 нг/мл от 15 до 18 часов.
14. Фармацевтическая дозированная форма по п. 11, где субъект имеет единое пролонгированное время при дозе выше 17 нг/мл от 11 до 15 часов.
15. Фармацевтическая дозированная форма по п. 11, где субъект имеет единое пролонгированное время при дозе ниже 17 нг/мл от 9 до 13 часов.
16. Фармацевтическая дозированная форма по п. 8, где субъект имеет среднюю максимальную концентрацию в плазме (Cmax) от 3 до 6 нг/мл на мг дозированного тофацитиниба.
17. Фармацевтическая дозированная форма по п. 8, где указанная дозированная форма доставляет тофацитиниб или его фармацевтически приемлемую соль с помощью системы, выбранной из группы, состоящей из системы экструдируемого ядра, системы разбухающего ядра, и асимметричной мембранной технологии.
18. Фармацевтическая дозированная форма по п. 8, где указанное производное целлюлозы представляет собой ацетат целлюлозы.
19. Фармацевтическая дозированная форма по п. 8, где указанное покрытие дополнительно содержит растворимый в воде полимер, который имеет среднюю молекулярную массу от 2000 до 100000 дальтон.
20. Фармацевтическая дозированная форма по п. 19, где указанный растворимый в воде полимер выбирают из группы, состоящей из: растворимых в воде производных целлюлозы, гуммиарабика, декстрина, гуаровой смолы, мальтодекстрина, альгината натрия, крахмала, полиакрилатов и поливиниловых спиртов.
21. Фармацевтическая дозированная форма по п. 20, где указанные растворимые в воде производные целлюлозы представляют собой гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу или гидроксиэтилцеллюлозу.
22. Фармацевтическая дозированная форма по п. 8, где осмоген содержит сахар.
23. Фармацевтическая дозированная форма по п. 22, где сахаром является сорбит.
24. Фармацевтическая дозированная форма по п. 8, где субъект имеет среднюю минимальную концентрацию в состоянии равновесия (Cmin) в плазме менее чем 0,3 нг/мл на мг дозированного тофацитиниба.
25. Фармацевтическая дозированная форма для приема один раз в день по п. 8, где при введении субъекту орально дозированная форма имеет среднее соотношение площади под кривой концентрации в плазме после еды/натощак против времени от 0,7 до 1,4 и среднее соотношение максимальной концентрации (Cmax) в плазме после еды/натощак от 0,7 до 1,4.
| WO 2012100949 A1, 2012.08.02 | |||
| US 20070248671 A1, 25.10.2007 | |||
| Tan, H | |||
| et al., Dose Response and Pharmacokinetics of Tofacitinib (CP-690,550), an Oral Janus Kinase Inhibitor, in the Treatment of Chronic Plaque Psoriasis | |||
| CPT: Pharmacometrics & Systems Pharmacology, 2013, 2 (5), e44, найдено онлайн, найдено в Интернете: |

