УРОВЕНЬ ТЕХНИКИ
Изобретение относится к обладающим покрытием абразивным частицам и материалам и к способу их изготовления.
Абразивные зернистые материалы, такие как частицы алмаза и кубического нитрида бора, широко применяются для распиливания, сверления, шлифования, полирования и других абразивных операций и операций резания. В таких случаях применения зернистый материал обычно окружен матрицей, состоящей из металлов, таких как Fe, Co, Ni, Cu и их сплавы (связывание металлом). Альтернативно можно использовать матрицы из смолы (связывание смолой) или стекла (связывание стеклом), и выбор матрицы зависит от конкретной области применения абразива.
Применение абразивного зернистого материала для изготовления абразивных инструментов связано с затруднениями. Связанные стеклом шлифовальные круги и инструменты, содержащие сверхтвердые абразивные частицы, такие как алмаз и кубический нитрид бора, широко применяются в обычных операциях шлифования. Обычно абразивные частицы удерживаются в пористой стеклянной матрице. Инструменты изготавливают путем смешивания или объединения сверхтвердых абразивных частиц со стеклообразной фриттой и/или стеклообразующими исходными материалами, уплотнения или придания необходимой формы шлифовальному кругу или компонентам указанного круга с последующей термической обработкой при температуре, достаточной для спекания стекла в необходимой степени, так чтобы образовалась измельчающаяся пористая матрица для сверхтвердых абразивных частиц.
Имеются различные затруднения, которые ограничивают изготовление и применение таких шлифовальных кругов и изделий.
Во-первых, в случае, когда необходимые сверхтвердые абразивные частицы представляют собой алмаз, использующиеся или идеально необходимые температуры, длительность термической обработки и среда в печи являются такими, при которых может происходить значительное разрушение частиц алмаза вследствие окисления. Хорошо известно, что реакции окисления алмаза могут в заметной степени начаться на воздухе при температуре, равной лишь 550°С, и могут стать очень быстрыми при температуре, превышающей 800°С. Это ограничивает технологии изготовления в том отношении, что используются приводящие к затруднениям и иногда дорогостоящие газовые среды. Кроме того, реакции окисления алмаза зависят от площади поверхности и становятся чрезвычайно быстрыми, когда размер абразивных частиц алмаза становится небольшим. Это ограничивает обычное применение алмаза в связанных стеклом системах крупными зернами, такими как обладающими диаметром примерно от 100 до 150 мкм, хотя для некоторых случаев применения могут потребоваться мелкие частицы алмаза размером от 1 до 10 мкм.
В стеклянные спрессованные элементы часто необходимо включать органические соединения и агенты, чтобы путем пиролиза и термического разложения таких органических соединений можно было регулируемым образом придать пористость. Хотя можно использовать инертные газовые среды, такой пиролиз органических компонентов приводит к обладающим высокой окислительной способностью продуктам, которые могут окислить и разрушить абразивные частицы алмаза.
Во-вторых, когда необходимым сверхтвердым абразивом являются частицы кубического нитрида бора, некоторые стеклообразующие компоненты или соединения могут нежелательным образом взаимодействовать с кубическим нитридом бора с выделением большого количества газа и вспениванием, что может разрушить или повредить шлифовальный круг или изделие. Примерами таких стеклообразующих компонентов являются оксиды щелочных металлов, такие как оксид лития (Li2O), оксид натрия (Na2O) и оксид калия (K2O). Эти компоненты могут являться флюсами, необходимыми для спекания и образования стекла. Известно, что оксид лития легко реагирует с кубическим нитридом бора при повышенных температурах с выделением азота (N2). Такое выделение газа и вызванное этим вспенивание может разрушить заготовку связанного стеклом шлифовального круга или изделие. Поэтому выбор связующих стекол ограничивается такими, которые не содержат значительных количеств соединений, которые могут энергично взаимодействовать с кубическим нитридом бора.
Это затруднение усиливается, когда частицы кубического нитрида бора становятся мельче, поскольку значительно увеличивается площадь поверхности и образующейся реакционноспособной поверхности, и поэтому наблюдается тенденция не использовать кубический нитрид бора, обладающий широким распределением частиц по размерам.
В-третьих, когда механические смеси комбинаций сверхтвердых частиц и стеклянной фритты и/или исходных материалов для изготовления стекла обрабатывают при условиях спекания и образования стекла, связывание и закрепление абразивных частиц в стеклообразной матрице может быть затруднительным вследствие недостаточного смачивания и контактирования абразивных частиц и стекла.
В-четвертых, во время изготовления связанных стеклом инструментов часто необходимы низкие скорости охлаждения, чтобы свести к минимуму вызванные растрескиванием повреждения, которые могут происходить вследствие несогласованного термического расширения абразивных зерен и пористой связывающей стеклянной матрицы.
В предшествующем уровне техники рассматривали такие затруднения. В ЕР 0400322 (так же опубликованном, как US 4951427) заявлены абразивные частицы, включая алмаз и cBN, содержащие огнеупорный оксид металла, в основном покрывающий поверхность указанных частиц. Заявлено, что покрытия из оксидов металлов способны в основном устранить воздействие связывающей стеклянной матрицы на частицы cBN в шлифовальных кругах во время их изготовления. Предпочтительными тугоплавкими оксидами металлов являются оксиды титана, циркония, алюминия и кремния. Наиболее предпочтительным является диоксид титана.
Рассматриваемый способ изготовления шлифовальных кругов включает нанесение на частицы покрытия из металла в элементной форме с последующим превращением указанного покрытия в оксиды посредством термической обработки, предпочтительно - во время обжига в окислительной атмосфере. Хотя в одном примере описан альтернативный способ для TiO2, включающий образование взвеси с металлоорганическим соединением, а именно с тетраизопропилтитанатом, с последующим разложением указанного металлоорганического соединения путем нагревания, приведенный пример является неосуществимым, подробно не описан и не предоставляет средства для нанесения на отдельные мелкие частицы покрытия из выбранных фаз диоксида титана.
Кроме того, эти методики неприемлемы, когда становятся меньше размеры частиц необходимых исходных компонентов, в особенности в случае микрометровых и субмикрометровых измельченных материалов и в еще большей степени - в случае нанометровых измельченных материалов, что обусловлено значительной трудностью нанесения равномерного покрытия на каждую очень мелкую частицу и склонностью к образованию агломератов мелких частиц. Таким образом, применение таких методик налагает ограничения на нанесение покрытий на мелкие частицы измельченных абразивных материалов.
В US 4011064 показано, что шероховатые зернистые прилипающие покрытия можно нанести на абразивные частицы cBN путем размола частиц вместе с соединениями металлов на шаровой мельнице, проводимого таким образом, чтобы соединение металла могло размазаться по поверхностям частиц. Затем соединение металла можно разложить путем нагревания при температуре примерно от 800 до 1400°С в инертной или восстановительной атмосфере с превращением соединения металла в металл. Типичным рассмотренным соединением металла является сульфид вольфрама, WS2, который приводит к гранулированному покрытию из металлического вольфрама на частицах cBN размером от 125 до 149 мкм.
Предполагается, что эту методику весьма затруднительно использовать для более мелких частиц, таких как размером, равным 10 мкм или менее, и что она совершенно неприменима для частиц субмикрометрового и нанометрового размера вследствие того, что сам размазывающийся материал должен быть в достаточной степени измельчен в частицы, намного меньшие, чем частицы, на которые наносится покрытие. Кроме того, соединения металлов, применимые в этой методике, ограничиваются такими, которые обладают механическими характеристиками, обеспечивающими размазывание.
Большая часть исследований предшествующего уровня техники, относящихся к включению абразивных частиц в связанные инструменты и круги, посвящена нанесению на абразивные частицы покрытий из металлов, керамики и комбинации таких материалов. В этих исследованиях предшествующего уровня техники для образования таких покрытий используются различные методики химического осаждения из паровой фазы или физического осаждения из паровой фазы. Кроме того, предполагается, что такие методики в ограниченной степени и с трудом можно применять для мелких абразивных частиц, в особенности микрометрового, субмикрометрового или нанометрового размера. Предполагается, что методики предшествующего уровня техники в целом обладают тем недостатком, что для всех и каждой частиц затруднительно создать одинаковые условия проведения реакций и нанесения покрытия, что неизбежно приведет к неодинаковым покрытиям на разных частицах.
Сохраняется необходимость в эффективных способах нанесения на абразивные частицы покрытий из материалов, которые защищают абразив от химического взаимодействия со многими необходимыми связующими материалами шлифовальных кругов и инструментов, связывающих стеклом или металлом, и др. В частности, необходимы способы, которые позволяют использовать мелкозернистые абразивы микрометрового, субмикрометрового и даже нанометрового размера.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящего изобретения способ нанесения покрытия на сверхтвердые абразивные частицы включает стадии использования множества сверхтвердых абразивных частиц, обладающих витреофильными поверхностями, нанесения на сверхтвердые абразивные частицы покрытия из оксидного материала-предшественника и термической обработки обладающих покрытием сверхтвердых абразивных частиц для высушивания и очистки покрытий.
Затем подвергнутые термической обработке обладающие покрытием сверхтвердые абразивные частицы дополнительно обрабатывают для превращения материала-предшественника в оксид, нитрид, карбид, оксинитрид, оксикарбид или карбонитрид оксидного материала-предшественника, или в элементную форму оксидного материала-предшественника, или в их комбинации.
Оксидный материал-предшественник предпочтительно представляет собой аморфный или нанокристаллический оксид, гидроксид или оксогидроксид.
Сверхтвердые абразивные частицы предпочтительно выбраны из группы, включающей алмаз, кубический нитрид бора, карбид кремния, нитрид кремния, карбид бора, субоксид бора (В6О) и т.п.
Предпочтительно, если сверхтвердые абразивные частицы представляют собой алмаз или кубический нитрид бора или комбинацию этих материалов и в этом случае частицы должны быть подвергнуты поверхностной обработке, чтобы сделать их поверхности витреофильными. Это образует другой объект настоящего изобретения, согласно которому находящиеся на поверхности химические частицы выбираются и генерируются путем соответствующей обработки для того, чтобы образовавшиеся таким образом на поверхности химические частицы могли быть совместимыми с последующими мокрыми химическими реакциями и средствами нанесения покрытия на сверхтвердые частицы и участвовали в них. Поверхностные химические частицы такого рода можно описать как витреофильные или склонные взаимодействовать со стеклом, в том отношении, что они могут образовывать связи с оксидными компонентами, типичными для стекла и стеклоподобных аморфных материалов. В этом случае материалы покрытия, вероятно, химически свяжутся с поверхностью сверхтвердых частиц.
Подвергнутые превращению материалы-предшественники матрицы обычно выбраны из числа обладающих зернами микрометрового, субмикрометрового или нанометрового размера оксидов, нитридов, карбидов, оксинитридов, оксикарбидов, карбонитридов или элементных форм материалов-предшественников, или их комбинаций. Они обычно включают оксиды, нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды алюминия, титана, кремния, ванадия, циркония, ниобия, гафния, тантала, хрома, молибдена и вольфрама и любые подходящие комбинации этих материалов. Предпочтительно, если эти оксидные материалы-предшественники являются аморфными или обладают зернами нанометрового размера.
Некоторые оксидные материалы-предшественники с помощью соответствующей обработки можно восстановить в элементные формы. Примерами этого класса материалов-предшественников являются оксиды молибдена и вольфрама.
Оксидные материалы-предшественники предпочтительно наносят на сверхтвердые абразивные частицы с помощью так называемой золь-гелевой методики. Сверхтвердые частицы суспендируют в жидких средах, в которые введены подходящие химические реагенты, предпочтительно - один или большее количество алкоксидов, так чтобы могли образоваться коллоидные частицы, которые связываются с поверхностями и включаются в покрытия, находящиеся на указанных частицах. Образованные таким образом покрытия преимущественно представляют собой микропористые оксиды, гидроксиды или оксогидроксиды указанных выше металлов или металлоидов.
Для удаления летучих веществ и нежелательных химических веществ, присоединенных к большим участкам поверхности микропористых аморфных покрытий, таких как гидроксилсодержащие частицы, в особенности -ОН, предпочтительно проводить нагревание на воздухе, в вакууме или инертном газе с регулированием температуры.
Для кристаллизации покрытий с образованием мелкозернистых или нанометровых оксидных керамик можно использовать дополнительную термическую обработку или прокаливание.
Поскольку некоторые оксидные керамики в некоторых температурных диапазонах подвергаются фазовым превращениям, выбор конкретных кристаллических фаз путем использования соответствующих температуры и длительности является другим объектом настоящего изобретения.
Реакции с регулированием температуры в реакционноспособных газах также можно использовать для превращения аморфных оксидов или кристаллических оксидных керамик в кристаллические неоксидные керамики. В частности, по реакции покрытий с аммиаком образуются нитриды. Карбиды можно получить по реакции покрытий со смесями углеродсодержащих газов с водородом, например со смесями метана или этана с водородом. Если некоторые оксидные покрытия восстанавливаются водородом, то их можно превратить в обладающие зернами микрометрового и нанометрового размера элементы или металлы.
Отличительной особенностью настоящего изобретения является то, что вследствие аморфного или микрокристаллического характера оксидных предшественников покрытий температуры, необходимые для их превращения в соответствующие керамики или металлы по реакции с газами, намного ниже температур, необходимых для обычных оксидных керамик, получаемых с помощью обычного прокаливания и плавления.
Способ, предлагаемый в настоящем изобретении, также предоставляет возможность изготовления множества обладающих покрытием сверхтвердых абразивных материалов, предпочтительно - обладающих микрометровым или меньшим диаметром, более предпочтительно - обладающих субмикрометровым и нанометровым размером. Однако также можно изготовить специфические материалы из обладающего покрытием алмаза и кубического нитрида бора диаметром от нескольких десятков микрометров до нескольких сотен микрометров, которые включают керамические покрытия, обладающие специфическими фазами, структурой и размером зерен, и, в частности, обладающие зернами нанометрового размера керамики. Примеры таких оксидных керамик включают диоксид циркония, ZrO2 в метастабильной тетрагональной фазе, структуры диоксида циркония, стабилизированные путем изменения состава, такие как содержащие от 3 до 8% оксида иттрия, и моноклинной фазы диоксида циркония, и диоксида титана, TiO2, преимущественно в фазе анатаза или рутила. Также являются новыми многие из неоксидных керамических покрытий, получаемых способом, предлагаемым в настоящем изобретении, включая нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды переходных металлов, таких как ванадий, ниобий, тантал, гафний, молибден и вольфрам. Кроме того, некоторые оксидные материалы покрытий не кристаллизуются в широких диапазонах температур и поэтому по механизмам спекания стекол могут образовывать плотные стекла. Сверхтвердые абразивы, полностью покрытые плотными стеклами, предпочтительно на основе диоксида кремния, SiO2, толщиной от нанометров до нескольких микрометров, являются новыми и их можно получить способами, предлагаемыми в настоящем изобретении.
Обладающие покрытием сверхтвердые абразивные частицы, которые являются очень мелкими, обладают микрометровым, субмикрометровым и нанометровым размером, обладающие покрытием из оксидных керамик, неоксидных керамик, таких как нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды, и из металлов, таких как молибден и вольфрам, являются уникальными только вследствие небольших размеров сверхтвердых частиц. Кроме того, дополнительная уникальность обусловлена специфическими структурами и тем, что материалы покрытий обладают зернами нанометрового размера.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Настоящее изобретение ниже только в качестве примера будет более подробно описано со ссылкой на прилагаемые чертежи, на которых представлено следующее:
На фиг.1 приведена блок-схема стадий способа, предлагаемого в настоящем изобретении.
На фиг.2 приведена рентгенограмма частиц cBN, обладающих покрытием из диоксида титана, промежуточного материала в предпочтительном варианте осуществления способа, предлагаемого в настоящем изобретении.
На фиг.3 приведена рентгенограмма частиц cBN, обладающих покрытием из нитрида титана, полученного термической обработкой частиц cBN, обладающих покрытием из диоксида титана, охарактеризованных на фиг.2.
На фиг.4 приведена рентгенограмма частиц cBN, обладающих покрытием из диоксида титана, полученного в соответствии с другим предпочтительным вариантом осуществления способа, предлагаемого в настоящем изобретении, после термической обработки при 475°С (А) и после термической обработки при 800°С (В).
На фиг.5 приведена рентгенограмма частиц алмаза, обладающих покрытием из нитрида титана, полученных в соответствии с другим предпочтительным вариантом осуществления способа, предлагаемого в настоящем изобретении.
На фиг.6 приведена рентгенограмма частиц cBN, обладающих покрытием из диоксида циркония, полученного в соответствии с еще одним предпочтительным вариантом осуществления способа, предлагаемого в настоящем изобретении, после сушки (А), после термической обработки при 475°С (В) и после термической обработки при 800°С (С).
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Настоящее изобретение относится к обладающим покрытием абразивным частицам и материалам, предназначенным для применения для распиливания, сверления, шлифования, полирования и других абразивных операций и операций резания.
Настоящее изобретение позволяет преодолеть множество из затруднений, имеющихся в предшествующем уровне техники, относящихся к возможности эффективного нанесения покрытий на более мелкие абразивные частицы, предпочтительно - на частицы размером несколько микрометров и менее, и еще более предпочтительно - на абразивы субмикрометрового и даже нанометрового размера. В частности, обнаружено, нанесение на частицы покрытия по золь-гелевой методике в суспензии может быть все более эффективным и полезным по мере того, как частицы становятся более мелкими и приобретают все более и более значительную площадь поверхности, и при этом нанесению покрытий может способствовать химическая обработка поверхности. Кроме того, среда из химикатов вокруг всех частиц в динамически перемешиваемой суспензии может быть практически одинаковой, что приводит к предположению о том, что покрытие на всех частицах будет в основном идентичным. Таким образом можно свести к минимуму различия материалов покрытий на всех частицах.
Сверхтвердые абразивные частицы предпочтительно выбраны из группы, включающей алмаз, кубический нитрид бора, карбид кремния, нитрид кремния, карбид бора, субоксид бора (B6O) и т.п.
Предпочтительными сверхтвердыми абразивными частицами являются алмаз и кубический нитрид бора (cBN), обладающие размерами от нанометрового (нм) до миллиметрового (мм).
Керамические покрытия могут быть пористыми или не содержать пор.
Керамические материалы покрытий включают аморфные и кристаллические фазы оксидных керамик. Они включают оксиды титана, кремния, циркония, алюминия, ванадия, ниобия, гафния, тантала, хрома, молибдена и вольфрама и т.п. и любые подходящие комбинации этих материалов. Предпочтительными оксидами являются оксиды титана, циркония, кремния и алюминия.
Неоксидные керамики включают нитриды металлов, карбиды металлов, карбонитриды металлов. Предпочтительными нитридами являются нитриды титана, ванадия, ниобия, тантала, молибдена и вольфрама.
Диапазон толщин керамических покрытий находится в интервале от нанометрового (нм) до микрометрового (мкм).
В настоящем изобретении субмикрометровые частицы или зерна определяются как обладающие наибольшим диаметром, равным от 1 мкм (1000 нм) до 0,1 мкм (100 нм), и нанометровые частицы или зерна определяются как обладающие наибольшим диаметром, равным менее 0,1 мкм (100 нм).
Способ, предлагаемый в настоящем изобретении, обычно включает три технологические стадии, а именно: 1) использование сверхтвердых абразивных частиц, обладающих витреофильными поверхностями, или, если это является целесообразным, химическую обработку поверхностей сверхтвердых абразивных частиц для придания им витреофильности; 2) использование методик коллоидных суспензионных реакций для нанесения на сверхтвердые частицы покрытия из оксидного материала-предшественника; 3) термическую обработку обладающих нанесенным таким образом покрытием сверхтвердых частиц в газовых средах для высушивания и очистки покрытий с последующим превращением в выбранные оксиды (включая стекла), нитриды, карбиды, оксинитриды, оксикарбиды, карбонитриды и металлы выбранной фазы и состава.
На первой стадии на поверхности сверхтвердого измельченного материала проводят химические реакции для придания частицам витреофильной природы. Витреофильная, склонная взаимодействовать со стеклом, определяется, как обладающая такой природой, что легко может образовывать химические связи с оксидными материалами. Виды обработки, которые могут привести к образованию на поверхности химических соединений, необходимых для проявления витреофильности сверхтвердых частиц, включают, но не ограничиваются только ими, кипячение в кислотах-окислителях, таких как концентрированная азотная кислота, если это является подходящим, или обработка сильными окислительными реагентами, такими как растворы пероксида водорода, или нагревание на воздухе или в кислороде. Образованные таким образом поверхности обеспечивают образование и рост покрытий на основе оксидов или гидроксидов на измельченном материале и хорошую адгезию с образованными таким образом предшественниками покрытий на основе оксидов.
На второй стадии используется коллоидное суспензионное нанесение на сверхтвердые абразивные частицы покрытия из аморфных и/или обладающих зернами нанометрового размера гидратированных оксидных материалов-предшественников. Обнаружено, что модификация некоторых коллоидных методик позволяет аккуратно наносить покрытия на микрометровые, субмикрометровые и даже нанометровые частицы сверхтвердых материалов. Имеются две общие коллоидные методики, с помощью которых можно получить подходящие покрытия, в одной из которых используют водные растворы неорганических солей, а в другой используют металлоорганические соединения. Для этого предпочтительным подходом является указанная золь-гелевая методика, более предпочтительными - золь-гелевые методики с использованием гидролиза и поликонденсации алкоксидов или алкоголятов. Предшественники покрытий, сформированные по этой методике, являются микропористыми, аморфными или обладающими зернами нанометрового размера гидратированными оксидами с большой площадью поверхности. Золь-гелевые методики, в частности, являются весьма универсальными и пригодными для регулирования гетерогенного зародышеобразования и роста чрезвычайно правильных покрытий из гидратированных оксидных материалов-предшественников на поверхностях витреофильных суспендированных частиц, размер которых может составлять лишь 10 нм или даже менее.
Предпочтительной золь-гелевой методикой является медленное прибавление спиртового раствора алкоксида металла или комбинации алкоксидов металлов к суспензии частиц сверхтвердого материала в аликвоте раствора воды низкой концентрации в том же спирте. Алкоксиды металлов гидролизуются водой с образованием мономеров гидроксидов металлов, которые, в свою очередь, вступают в реакцию поликонденсации, которая постепенно приводит к образованию гидратированных микропористых оксидов, которые в настоящем изобретении называют оксидными материалами-предшественниками или покрытиями. Путем соответствующего выбора типа спирта, который обычно содержит такие же алкильные группы, как и алкоксид(ы), концентрации суспендированных сверхтвердых частиц, концентрации раствора алкоксида в спирте, соотношения алкоксид/вода, температуры и наличия или отсутствия других реагентов, таких как кислоты или основания, можно регулировать образование покрытия из оксидного предшественника на суспендированных сверхтвердых частицах. Для нанесения на суспендированный сверхтвердый измельченный материал необходимого покрытия в случае каждого типа использованного алкоксида необходимы специальные условия.
Важной особенностью этого подхода является то, что побочными продуктами реакций гидролиза алкоксидов и поликонденсации являются вода, спирты и гидроксидные соединения, находящиеся на части свободных поверхностей покрытия. Все эти побочные продукты легко удаляются путем сушки и термической обработки при низкой температуре. Кроме того, сами алкоксиды легко доступны в виде продуктов высокой чистоты. Таким образом, золь-гелевая методика приводит к очень чистым незагрязненным оксидам.
Еще одной очень важной особенностью способа, предлагаемого в настоящем изобретении, является то, что путем одновременного использования более одного типа алкоксида разных металлов можно получить большое количество смешанных оксидных материалов-предшественников. При этом подходе полученный таким образом оксидный материал-предшественник будет представлять собой смешанный оксид, в котором различные металлы распределены в молекулярном масштабе. Альтернативно известно, что можно получить алкоксидные комплексы, содержащие более одного металла. Эти алкоксидные комплексы можно использовать в способе, предлагаемом в настоящем изобретении. Следовательно, оксиды, нитриды и карбиды, полученные при полном применении способа, предлагаемого в настоящем изобретении, могут включать смешанные и легированные фазы. Кроме того, известно, что можно получить смешанные структуры алкоксидов металлов. Использование таких смешанных алкоксидов металлов также приводит к смешанным предшественникам оксидов металлов и затем к смешанным фазам покрытий.
Применение смесей алкоксидов или смешанных алкоксидов также позволяет легировать матричные материалы-предшественники и последующие материалы агентами, модифицирующими спекание и структуру, такими как оксид иттрия, оксид магния и т.п. Альтернативно такие модифицирующие структуру агенты можно ввести с помощь растворимых солей или во время проведения реакций с алкоксидами, или после их завершения. Примеры таких солей предпочтительно включают ацетаты и нитраты иттрия и магния. При получении материалов покрытий способом, предлагаемым в настоящем изобретении, можно использовать большое количество информации, имеющейся в области керамики, керметов и металлургии.
После извлечения из суспензии и промывки обладающие покрытием частицы медленно сушат, например, путем нагревания в вакууме при температуре ниже 100°С. Микропористые, аморфные покрытия можно дополнительно очистить путем нагревания в диапазоне температур от 300 до 400°С, обычно на воздухе или в инертной атмосфере для удаления остаточного спирта и воды из микропор и в особенности для преимущественного удаления содержащих гидроксильные группы (-ОН) поверхностных частиц, которые обычно находятся на больших участках поверхностей пор. При необходимости формирования относительно толстых покрытий иногда обнаруживается, что вследствие капиллярных сил, проявляющихся при испарении спирта и воды из пор, при сушке происходит усадка и растрескивание. Этот эффект можно свести к минимуму путем медленной сушки и путем использования так называемых регулирующих сушку добавочных химикатов, РСДХ.
Эти химикаты делают поры в оксидном предшественнике покрытия более широкими и однородными и тем самым уменьшают капиллярные силы, что приводит к меньшей склонности к растрескиванию. Примерами таких РСДХ являются щавелевая кислота и диметилформамид, ДМФ. Последний из них является предпочтительным.
Микропористые, аморфные или обладающие зернами нанометрового размера структуры материалов-предшественников покрытий делают их идеальными для реакционной термической обработки с программированием температуры в газообразных реагентах или средах с получением необходимых мелкозернистых и обладающих зернами нанометрового размера керамических фаз или стеклообразных фаз в качестве материала покрытия. В действительности, если оксидное покрытие способно восстанавливаться водородом, то можно получить металлические покрытия.
На третьей стадии реакционную термическую обработку с программированием температуры предшественников обладающих покрытием сверхтвердых частиц в выбранной газовой среде используют для частичного уплотнения покрытия и для его превращения в выбранный мелкозернистый или обладающий зернами нанометрового размера керамический материал. Термическую обработку на воздухе, в кислороде или в инертном газе можно использовать для прокаливания, уплотнения покрытия и кристаллизации покрытия в виде требующейся оксидной фазы. Выбор скорости нагрева, максимальной температуры и длительности нагревания при максимальной температуре зависит от структуры, фазы и типа требующегося оксида.
Если покрытие необходимо превратить в нитрид, то высушенный или прокаленный на воздухе обладающий покрытием материал можно нагреть в сухом аммиаке при температурах, обычно достигающих 1100°С, хотя в некоторых случаях может потребоваться использование температур, достигающих примерно 1400°С включительно. Обнаружено, что эта реакционная термическая обработка с программированием температуры приводит к постепенному восстановлению материала покрытия и может превратить оксидные основные покрытия в стехиометрические и нестехиометрические нитриды и оксинитриды. И в этом случае выбор скорости нагрева, скоростей потоков газов, максимальной температуры и длительности нагревания при максимальной температуре зависит от структуры, фазы и типа требующегося нитрида. Также обнаружено, что путем соответствующего выбора условий можно получить оксинитридные фазы.
Если покрытие необходимо превратить в карбид, то высушенный или прокаленный на воздухе обладающий покрытием материал можно нагреть в смеси углеродсодержащих газов, таких как метан или этан, с водородом при температурах, обычно ниже 1200°С, хотя в некоторых случаях может потребоваться использование температур, достигающих примерно 1500°С включительно. И в этом случае выбор скорости нагрева, скоростей потоков газов, максимальной температуры и длительности нагревания при максимальной температуре зависит от структуры, фазы и типа требующегося карбида. Также обнаружено, что путем соответствующего выбора условий можно получить оксикарбидные фазы. Альтернативно, обнаружено, что нитридные покрытия, полученные так, как описано выше, можно превратить в карбиды путем соответствующей термической обработки в смесях метана или этана с водородом. Путем соответствующего выбора условий можно получить карбонитридные фазы.
Некоторые оксидные покрытия можно легко восстановить в соответствующий элементарный металл путем восстановления в чистом водороде. Примерами таких покрытий являются оксиды вольфрама и молибдена, WO3 и МоО3, которые можно легко восстановить в металлы при низких температурах, обычно в диапазоне от 500 до 700°С.
Основной особенностью стадии реакции с программированием температуры способа, предлагаемого в настоящем изобретении, является то, что обнаружено, что размеры всех зерен полученных оксидных, нитридных, карбидных покрытий на сверхтвердых частицах часто являются нанометровыми. Кроме того, другой важной особенностью этой термической обработки является то, что температуры и времена, необходимые для превращения, являются низкими и непродолжительными соответственно по сравнению с температурами и временами, необходимыми для аналогичных превращений обычных оксидных материалов, проводимых по методикам плавления или сплавления. В некоторых случаях в способе, предлагаемом в настоящем изобретении, температуры образования нитридов ниже температур образования нитридов обычных оксидных материалов на величину, достигающую 400°С. Кроме того, обладающие покрытием сверхтвердые частицы можно отделить в неагломерированном виде.
Указанные выше стадии способа будут подробнее описаны ниже со ссылкой на фиг.1.
1. Обработка поверхности сверхтвердых частиц для придания им витреофильности.
В случае обладающего зернами микрометрового, субмикрометрового или нанометрового размера алмаза с помощью таких методик, как нагревание в концентрированных окисляющих кислотах, таких как смеси азотной и/или серной кислоты, можно сделать так, чтобы концевые поверхностные функциональные группы в основном представляли собой группы С-ОН, С-О-С, С=O и O=С-O-. Альтернативно, газовая термическая обработка в смеси 20% водород/аргон при 900°С для образования на поверхности концевых Н с последующей обработкой в смеси 20% кислород/аргон при 480°С приводит к тому, что на поверхности преобладают кислородсодержащие частицы. Также можно использовать другие методики образования кислородсодержащих функциональных групп, присоединенных к поверхности алмаза. Окисление поверхности алмаза делает ее витреофильной, т.е. способной к образованию химических связей с оксидами, включая, в частности, гидратированные оксидные структуры.
Предполагается, что в случае частиц cBN термическая обработка на воздухе при температуре выше 600°С приведет к увеличению концентрации борокислородных и азоткислородных частиц на поверхности, и это можно обнаружить с помощью инфракрасной Фурье-спектроскопии отражения. Такая поверхность обладает витреофильностью при последующем коллоидном нанесении покрытия на оксиды, полученные по золь-гелевой методике. Многие другие хорошо известные сверхтвердые материалы, такие как карбид кремния и нитрид кремния и т.п., содержат на своей поверхности окисленные химические группы, что обычно делает их витреофильными и пригодными для использования в способе, предлагаемом в настоящем изобретении.
2. Коллоидное нанесение покрытия на абразивные частицы.
В части 2(а) блок-схемы используются обычные золь-гелевые методики получения предшественников гидратированных оксидных материалов для необходимых матричных материалов. Один пример такого подхода включает гидролиз растворов сульфата алюминия при повышенных температурах, таких как равная 100°С, в присутствии органических соединений, таких как мочевина, для нанесения покрытия на частицы в суспензии. Таким образом можно получить покрытия из водного оксида алюминия.
Однако предпочтительным более общим подходом является применение реакций гидролиза и поликонденсации алкоксидов металлов в спиртовых растворах. Алкоксиды или алкоголяты металлов обладают общей формулой вида Mn+[OR]n, где М обозначает металл валентности n, О обозначает кислород и R обозначает алкильную группу. Металл связан с алкильными группами через атомы кислорода. Большинство алкоксидов металлов растворимы в спиртах и могут легко гидролизоваться водой в спиртовом растворе с образованием гидроксидов:
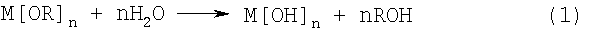
Затем можно провести реакции поликонденсации, такие как представленные приведенным ниже уравнением (2), и образовать связи М-О-М.
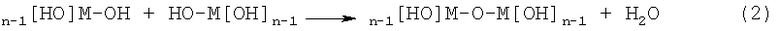
Последовательное проведение этих реакций приводит к трехмерной сетке -М-O-М-O-М-. Образовавшийся таким образом оксидный материал-предшественник обычно является аморфным или обладающим зернами нанометрового размера с очень большой площадью поверхности и является микропористым, содержащим в порах Н2О и спирт. На поверхностях пористой структуры находятся концевые гидроксильные группы, ОН и некоторые непрореагировавшие функциональные группы OR. Путем соответствующего выбора концентраций, соотношений алкоксид/вода, температуры, спирта-растворителя и введения других химикатов, таких как кислоты или основания, можно сделать так, чтобы в спиртовом растворе происходили зародышеобразование и рост пористого оксидного материала-предшественника. Необходимо подобрать подходящие концентрации суспендированных частиц, выступающих в качестве центров роста материала покрытия.
Раствор алкоксида (алкоксидов) металла получают в безводном спирте и затем в течение нескольких часов его при непрерывном перемешивании медленно прибавляют к суспензии сверхтвердых частиц в аликвоте чистой воды обычно в том же спирте. Для стабилизации суспензии можно прибавить пептизирующий реагент, такой как кислоту или основание.
Альтернативно, если необходимо использовать особенно реакционноспособный алкоксидный реагент, лучшее регулирование образования покрытия можно обеспечить путем медленного прибавления аликвоты воды в спирте к суспензии сверхтвердых частиц в суспензии алкоксида в безводном спирте.
Побочные продукты реакции - воду и спирт - можно удалить путем сушки и термической обработки при низкой температуре 2(b). Аналогичным образом можно удалить поверхностные функциональные группы ОН. Обычно после фильтрования, центрифугирования или осаждения и декантирования суспензии с последующей промывкой свежим чистым спиртом и/или деионизированной водой обладающие покрытием частицы можно медленно высушить в течение примерно двух дней при температуре около 60°С в низком вакууме. Последующее удаление остаточной воды и спирта можно обеспечить путем нагревания примерно до 300°С на воздухе.
Многие элементы периодической системы могут образовывать алкоксиды. Алкоксиды, найденные пригодными для получения оксидных матриц способом, предлагаемым в настоящем изобретении, включают алкоксиды титана, алюминия, циркония, хрома, кремния, вольфрама, молибдена, тантала, ниобия, ванадия, а алкоксиды кальция, магния, гафния, иттрия иногда пригодны в качестве добавок, включая комбинации этих алкоксидов. Алкоксиды, найденные пригодными для получения нитридных покрытий способом, предлагаемым в настоящем изобретении, включают алкоксиды алюминия, титана, циркония, кремния, тантала, хрома, ниобия, гафния, ванадия, молибдена и вольфрама и их комбинации. Алкоксиды, найденные пригодными для получения карбидных покрытий способом, предлагаемым в настоящем изобретении, включают алкоксиды титана, циркония, кремния, тантала, хрома, ниобия, гафния, ванадия, молибдена и вольфрама и их комбинации.
Алкильные группы R в общей формуле алкоксидов металлов, M[OR]n, могут включать метил, этил, н-пропил, н-бутил и любую группу общей формулы -CxH2x+1. Кроме того, включаются алкильные группы, в которых содержатся боковые алкильные группы, такие как изопропильная группа, -СН(СН3)2, втор-бутильная группа, -СНСН2СН3СН3, трет-бутильная группа, -С(СН3)3 и др.
Скорость реакции гидролиза и время достижения точки гелеобразования для каждого алкоксида металла сильно зависят от длины цепи алкильной группы. Чем меньше длина цепи R, тем быстрее гидролиз и тем меньше время достижения точки гелеобразования оксидного материала-предшественника в покрытии сверхтвердых частиц. На характеристики покрытия для каждого типа требующегося гидратированного оксидного предшественника покрытия может сильно повлиять выбор R.
Спирты, применяющиеся в качестве растворителя для алкоксида и воды и в качестве суспендирующей жидкости для сверхтвердых частиц, можно выбрать из числа любых обычно имеющихся в продаже жидких растворителей. Предпочтительными спиртами являются этанол, метанол и изопропиловый спирт. Более предпочтительно, но не обязательно, использовать спирт, содержащий такую же группу, что и алкоксид.
В таблице 1 приведен примерный, но не полный перечень некоторых алкоксидов, наиболее подходящих для способа, предлагаемого в настоящем изобретении.
После сушки/предварительной термической обработки обладающие покрытием частицы можно исследовать с помощью сканирующего электронного микроскопа и/или трансмиссионного электронного микроскопа.
3. Термическая обработка с программированием температуры (ТПТ)
Затем обладающие покрытием частицы подвергают термической обработке с программированием температуры. Это выполняют в выбранных газовых средах, при выбранных скоростях нагрева, при выбранных максимальных температурах, в течение выбранных периодов времени для регулирования удаления остаточных летучих примесей, уплотнения и спекания, перехода в другие структурные фазы и проведения химической реакции покрытия с газами, приводящей к другим типам материалов и фаз. Предпочтительным подходом является использование проточных газовых систем при тщательно подобранной и регулируемой скорости потока. Нагревание обладающего покрытием измельченного материала можно проводить в трубчатой печи, вращающейся трубчатой печи, приспособленной для медленного перемешивания частиц, и тем самым предотвращения спекания или агломерации, или в любой конструкции печи, пригодной для регулируемого нагрева измельченных материалов в выбранных регулируемых газовых средах.
Как показано на схеме, приведенной на фиг.1, после предварительной сушки/термической обработки 2(b) существуют несколько возможных путей превращения обладающего покрытием материала в требующиеся материалы (сама предварительная сушка/термическая обработка 2(b) может представлять собой многостадийную процедуру, например, сушку в вакууме при температуре ниже 100°С для удаления большей части свободной воды из микропор покрытия с последующим нагреванием, например примерно до 300°С в вакууме или на воздухе для удаления остаточных спиртов и абсорбированных гидроксильных функциональных групп с поверхности).
Одним путем, путем А, является прокаливание обладающих покрытием частиц на воздухе или в кислороде, или в инертном газе для превращения покрытия в необходимый оксид. В зависимости от конкретного используемого пористого оксидного материала-предшественника будет происходить спекание и/или кристаллизация, включающая уплотнение. Также могут происходить фазовые превращения в оксиды различной кристаллической структуры и их можно осуществить для получения требующихся оксидов. По этой методике обычно получают нанометровый оксид(ы). Альтернативно, при спекании в стекла некоторые оксидные покрытия не кристаллизуются, а уплотняются с образованием стекол. В каждом случае необходимые условия термической обработки определяют с помощью методик мониторинга и исследования реакции, таких как термогравиметрический анализ (ТГА), дифференциальный термический анализ (ДТА), рентгеноструктурный анализ (РСА) и т.п. Нагревание можно проводить в любом обычном оборудовании, пригодном для обработки тонкоизмельченного материала, хотя предпочтительными являются вращающиеся печи и печи с псевдоожиженным слоем.
Путь В используют для нагревания высушенных обладающих покрытием частиц, полученных на стадии 2(b), в аммиаке или смеси аммиака с инертным газом для превращения пористого оксидного предшественника покрытия в нитрид(ы) или оксинитрид(ы). Аммиак разлагается с образованием высокоактивных азот- и водородсодержащих частиц, которые постепенно восстанавливают и азотируют оксидный предшественник покрытия. Путем подбора условий можно получить различные оксинитридные и нитридные структуры. И в этом случае необходимые условия термической обработки определяют с помощью методик мониторинга и исследования реакции, таких как термогравиметрический анализ (ТГА), дифференциальный термический анализ (ДТА), рентгеноструктурный анализ (РСА) и т.п. Обычно образуются обладающие зернами нанометрового размера покрытия.
Путь С используют для нагревания высушенных обладающих покрытием частиц, полученных на стадии 2(b), в смесях углеродсодержащих газов с водородом для превращения пористого оксидного предшественника покрытия в карбид(ы) или оксикарбид(ы). Углеродсодержащим газом, в принципе, может быть любой газообразный углеводород, но предпочтительно - метан или этан. Смеси углеродсодержащий газ/водород можно разбавить инертным газом-носителем, таким как, например, аргон. Если активные газы составляют не более 20% от инертного газа-носителя, то маловероятно, что при утечке образуется взрывоопасная смесь газов с воздухом, так что улучшается безопасность. Типичные значения отношений количества метана или этана к количеству водорода составляют от 1/5 до 1/20. Необходимые условия термической обработки определяют с помощью методик мониторинга и исследования реакции, таких как термогравиметрический анализ (ТГА), дифференциальный термический анализ (ДТА), рентгеноструктурный анализ (РСА) и т.п.
Альтернативой превращения покрытий в оксинитриды и нитриды является использование пути А для выбранного оксида с последующим использованием пути D путем проведения термической обработки в содержащей аммиак среде с получением нитридов. Кроме того, при последующем использовании пути Е путем проведения обработки полученных таким образом нитридных покрытий в системах углеродсодержащий газ/водород можно получить другие карбидные микроструктуры, не такие как для пути С.
Кроме того, после получения оксидных структур с использованием пути А можно использовать путь F для получения карбидных микроструктур непосредственно из оксидных фаз.
В случае, когда пористое оксидное покрытие в 2(b) легко восстанавливается водородом, можно использовать путь G и можно получить мелкозернистые металлические покрытия.
Альтернативные комбинации путей допускают внесение изменений в содержание углерода, азота и кислорода в каждом карбиде, нитриде и оксиде. Например, посредством выбора пути и условий ТПТ можно получить оксинитридные материалы, материалы MNOx, в которых М обозначает металл, с выбором х в диапазоне от 0,5 до 0,05. В другом примере посредством выбора пути и условий ТПТ можно получить карбонитридные материалы, материалы MCNy, в которых у может находиться в диапазоне от 0 до 1.
Температуры нагрева, необходимые для получения кристаллических систем требующегося состава и структуры для материалов покрытий, являются относительно низкими. Это может привести к образованию низкотемпературных кристаллических систем, которые не образуются по более часто применяющимся твердофазным реакциям, обычно проводимым при более высоких температурах. В большей части случаев необходимые температуры ниже 1200°С, часто ниже 1000°С и в некоторых случаях составляют лишь 550°С.
Настоящее изобретение будет более подробно описано с помощью приведенных ниже неограничивающих примеров.
Пример 1
50 г субмикрометрового кубического нитрида бора, обладающего средним размером частиц, равным 0,7 мкм, в диапазоне размеров от 0,5 до 1,0 мкм обрабатывали в дымящей концентрированной серной кислоте, к которой прибавлен нитрат калия. После промывания и сушки субмикрометровый cBN дополнительно нагревали на воздухе при 600°С в течение 30 мин. Эта процедура приводила к тому, что в составе поверхности cBN преобладали кислородсодержащие функциональные группы и поэтому она стала витреофильной.
Затем 15 г этого субмикрометрового cBN с подвергнутой обработке поверхностью суспендировали в 865 мл чистого этанола в стакане, в который прибавляли 7,3 мл деионизированной воды. Суспензию энергично перемешивали лопастной мешалкой примерно при 100 оборотов/мин. 15,3 г жидкого изопропоксида титана, Ti(ОС3Н7)4, растворяли в 100 мл безводного этанола. Затем этот раствор по каплям в течение 1 ч при комнатной температуре (примерно 25°С) медленно прибавляли к суспензии cBN/этанол/вода, продолжая перемешивание. Перемешивание продолжали в течение еще 2 ч и содержимое стакана выдерживали в течение ночи. Полученные обладающие покрытием частицы извлекали из суспензии путем вакуумного фильтрования, трижды промывали этанолом и трижды деионизованной водой и затем сушили при 60°С в течение 2 дней в вакуумном сушильном шкафу. С помощью исследования на сканирующем электронном микроскопе (СЭМ) обнаружено, что каждая частица cBN была полностью покрыта соединением оксида титана, предположительно представляющим собой микропористый аморфный диоксид титана, TiO2.
Затем 10 г частиц cBN, обладающих покрытием из TiO2, подвергали термической обработке в потоке воздуха при 700°С в течение 3 ч. Скорость нагревания и скорость охлаждения поддерживали равными 5°С/мин. С помощью исследования на рентгеновском дифрактометре обнаружено, что покрытие закристаллизовалось в виде преимущественно анатазной фазы диоксида титана, как это показано на фиг.2, на котором приведена рентгенограмма, показывающая, что этот материал состоит только из диоксида титана и cBN. С помощью исследования этого измельченного материала на трансмиссионном электронном микроскопе, ТЭМ, обнаружено, что покрытие из диоксида титана закристаллизовалось в форме нанометровых кристаллитов размером примерно 30 нм.
5 г нагретых на воздухе субмикрометровых частиц cBN, обладающих покрытием из диоксида титана, дополнительно нагревали при 1100°С в течение 5 ч в трубчатой печи при пропускании потока сухого газообразного аммиака, NH3. Использовали скорость нагревания, равную 10°С/мин. Эта термическая обработка в аммиаке приводила к превращению обладающего зернами нанометрового размера покрытия из диоксида титана в обладающий зернами нанометрового размера нитрид титана, TiN. Исследование этого материала с помощью ТЭМ показало, что теперь покрытие состоит из кристаллитов нитрида титана размером примерно 40 нм. На фиг.3 приведена рентгенограмма, показывающая, что полученное покрытие в действительности представляет собой нитрид титана, TiN, называющийся осборнитом.
Пример 2
30 г порошкообразного cBN со средним размером частиц, равным 2 мкм, суспендировали в смешанном растворе 15% пероксида водорода, Н2О2, и 15% гидроксида аммония, NH4OH, в воде, состава 1:1. Это приводило к гидролизу поверхностей частиц cBN и тем самым делало их витреофильными. Затем порошок cBN, обладающий частицами размером 2 мкм, извлекали из суспензии путем фильтрования и промывали деионизированной водой.
Затем 25,5 г полученного таким образом порошкообразного cBN суспендировали в 1440 мл этанола, в который прибавляли 13,1 мл деионизированной воды. Суспензию обрабатывали с помощью ультразвукового зонда в течение 15 мин для разрушения всех агломератов частиц cBN. 20,7 г изопропоксида титана растворяли в 100 мл безводного этанола. Затем этот раствор по каплям в течение 1 ч при комнатной температуре при энергичном перемешивании прибавляли к суспензии cBN в смеси этанол/вода. После прибавления суспензию перемешивали в течение еще 2 ч и затем выдерживали в течение ночи. Затем измельченный материал извлекали из суспензии путем фильтрования и трижды промывали чистым этанолом и затем трижды промывали деионизованной водой и затем сушили при 60°С в течение 2 дней в вакуумном сушильном шкафу. Исследование этого измельченного материала на электронном микроскопе с использованием устройства СДЭ показало, что на cBN имеется покрытие из соединения титана с кислородом. Все частицы были полностью покрыты в одинаковой степени.
Затем 20 г этого обладающего покрытием cBN прокаливали в трубчатой печи в потоке сухого воздуха при 450°С в течение 3 ч. Скорость нагревания и охлаждения поддерживали равными 5°С/мин. Исследование с помощью рентгеновского дифрактометра показало, что покрытие представляет собой диоксид титана, TiO2, со структурой анатаза.
8 г прокаленного cBN, обладающего покрытием из диоксида титана со структурой анатаза, нагревали в трубчатой печи при 1100°С в течение 5 ч в потоке сухого газообразного аммиака. С помощью исследования на рентгеновском дифрактометре показано, что покрытие из диоксида титана со структурой анатаза превратилось в нитрид титана.
Таким образом на образцы cBN, обладающие частицами со средним размером, равным 2 мкм, наносили покрытие из аморфного диоксида титана со структурой анатаза и нитрида титана соответственно.
Пример 3
105 г ограненного кристаллического cBN, пропущенного через сито США 120/140 меш (105-125 мкм), обрабатывали в кипящей 32 об.% хлористоводородной кислоте, промывали водой и сушили. Затем этот материал нагревали на воздухе при 650°С в течение 1 ч для небольшого окисления поверхностей частиц.
Затем частицы cBN суспендировали в 500 мл чистого этанола, к которому прибавляли 10,6 мл деионизированной воды. Суспензию образовывали и поддерживали с помощью механического перемешивания лопастной мешалкой примерно при 100 оборотов/мин. 20 г изопропоксида титана формулы Ti(ОСН(СН3)2)4 растворяли в 100 мл чистого безводного этилового спирта и этот раствор при перемешивании медленно прибавляли к суспензии, по каплям, в течение 2 ч. Затем суспензию перемешивали в течение еще 2 ч до завершения реакций гидролиза и поликонденсации. Затем измельченный материал cBN трижды промывали этиловым спиртом путем осаждения и декантации. После последней декантации материалу давали медленно высохнуть в течение двух дней при условиях окружающей среды, а затем в течение 24 ч в вакуумном сушильном шкафу при 60°С.
Затем обладающий покрытием cBN разделяли на два образца, один образец медленно нагревали на воздухе при 475°С и выдерживали при этой температуре в течение 3 ч, а второй образец аналогичным образом нагревали при температуре 800°С в течение 3 ч. На фиг.4 приведена рентгенограмма, показывающая, что (А), нагревавшийся при 475°С материал, представляет собой cBN, обладающий покрытием из очень мелкокристаллического диоксида титана, TiO2, со структурой анатаза, и что (В), нагревавшийся при 800°С материал представляет собой cBN, обладающий покрытием из мелкокристаллического диоксида титана, TiO2, преимущественно со структурой рутила при сохранении небольшого количества компонента со структурой анатаза.
Затем первый образец нагревали в сухом аммиаке при 1100°С в течение 5 ч и после этого покрытие из диоксида титана превращалось в нитрид титана. Изображения этого материала исследовали с помощью ТЭМ, и обнаружено, что все поверхности кристаллов cBN обладают хорошим покрытием, но в покрытии имеются небольшие трещины.
Пример 4
50 г алмаза, обладающего частицами микрометрового размера, полученного из синтетического алмаза с помощью дробления и сортировки, обладающего средним размером частиц, равным 1,0 мкм, в диапазоне размеров от 0,75 до 1,5 мкм обрабатывали в дымящей концентрированной серной кислоте, к которой прибавлен нитрат калия. Эта очистка показала, что на поверхности алмаза не содержится металлов и неорганических загрязнений. Затем алмаз нагревали в потоке 20% кислорода в аргоне при 480°С в течение 1 ч. Эта процедура приводила к доведению до максимума количества кислородсодержащих функциональных групп, присоединенных к поверхностям алмаза, и делала их витреофильными.
15 г этого алмаза, обладающего частицами размером 1 мкм, с подвергнутой обработке поверхностью затем суспендировали в 865 мл чистого этанола в стакане, в который прибавляли 7,3 мл деионизированной воды. Суспензию энергично перемешивали лопастной мешалкой примерно при 100 оборотов/мин. 15,6 г жидкого изопропоксида титана, Ti(ОС3Н7)4, растворяли в 100 мл безводного этанола. Затем этот раствор по каплям в течение 1 ч при комнатной температуре (примерно 25°С) медленно прибавляли к суспензии алмаз/этанол/вода, продолжая перемешивание. Перемешивание продолжали в течение еще 2 ч и содержимое стакана выдерживали в течение ночи. Полученные обладающие покрытием частицы извлекали из суспензии путем вакуумного фильтрования, трижды промывали этанолом и трижды деионизованной водой и затем сушили при 60°С в течение 2 дней в вакуумном сушильном шкафу.
Затем 12 г высушенного обладающего покрытием алмаза нагревали на воздухе в статических условиях при 450°С в течение 2 ч. Использовали скорость нагревания, равную 5°С/мин. Затем материал исследовали с помощью СЭМ рентгеновской дифракции и обнаружили, что теперь на алмазе имеется покрытие из кристаллического диоксида титана со структурой анатаза, а другие фазы и соединения не обнаружены.
Затем 5 г этого обладающего покрытием материала подвергали термической обработке в потоке сухого аммиака в течение 5 ч при 1100°С. Скорость потока аммиака составляла примерно 1 л/мин, и использовали скорость нагревания, равную примерно 10°С/мин. Исследование с помощью СЭМ и ДРИ (дифракции рентгеновского излучения) показало, что теперь на алмазе имеется покрытие из нитрида титана. На фиг.5 приведена рентгенограмма, показывающая наличие алмаза и нитрида титана, а другие фазы и соединения не обнаружены. Таким образом, этот алмаз, обладающий частицами размером 1 мкм, полностью покрыт нитридом титана.
Пример 5
Методику, описанную выше в примере 4, можно проводить до получения порошкообразного алмаза, обладающего покрытием из кристаллического анатаза. Предполагается, что, если этот порошок обрабатывать в потоке, состоящем из газовой смеси 10% метана в аргоне и 10% водорода в аргоне при соответствующем соотношении метан: водород (предположительно 1:4) и при температуре, равной примерно 1350°С, в течение нескольких часов (вероятно, более 5 ч), то покрытие из диоксида титана превратиться в карбид титана. Таким образом получен алмаз, обладающий частицами размером 1 мкм, обладающий покрытием из карбида титана.
Пример 6
20 г образца ограненного кристаллического синтетического алмаза со средним размером частиц в диапазоне от 105 до 125 мкм суспендировали в 1,25 л этилового спирта чистоты более 99%. Образец алмаза предварительно нагревали при 480°С в течение 10 мин в потоке 20% кислорода в аргоне для получения поверхностей с преимущественно кислородсодержащими функциональными группами. При энергичном перемешивании к этой суспензии прибавляли 250 мл деионизированной воды и 30 мл 25 об.% водного раствора гидроксида аммония.
40 г тетраэтоксисилана (Si(OC2H5)4) растворяли в 100 мл этилового спирта чистоты 99%. Этот раствор с постоянной скоростью при перемешивании медленно прибавляли к суспензии, поддерживаемой при комнатной температуре, в течение 8 ч. Перемешивание продолжали в течение еще 1 ч. Перемешивание прекращали и обладающим покрытием частицам алмаза давали осесть. Надосадочная жидкость над осевшим множеством частиц алмаза была в основном прозрачной, и ее декантировали. Затем обладающие покрытием частицы трижды промывали чистым этиловым спиртом. После отфильтровывания множество частиц алмаза сушили в вакуумном сушильном шкафу при 60°С в течение 24 ч.
Затем образец обладающих покрытием частиц исследовали на сканирующем электронном микроскопе (СЭМ), который показал, что покрытие полностью закрывает частицы, и с помощью энергодисперсионного анализа (ЭДА) показано, что покрытие состоит из кремния и кислорода. Толщина найдена равной примерно 0,4 мкм.
Затем половину образца нагревали в потоке чистого аргона в трубчатой печи до температуры, равной 670°С (образец А), и выдерживали при этой температуре в течение 3 ч. Скорость нагревания составляла 3°С/мин. При последующем исследовании с помощью СЭМ обнаружено, что произошло определенное слияние покрытия и определенная усадка покрытия.
Другую половину образца нагревали в потоке чистого аргона до температуры, равной 900°С (образец В) в течение 3 ч, также при скорости нагревания, равной 3°С/мин. При исследовании с помощью СЭМ обнаружено, что покрытие обладает стеклообразным видом и полностью закрывает все части ограненных поверхностей алмаза. Толщина найдена равной примерно от 0,2 до 0,3 мкм и в основном оно представляло не содержащее пор кварцевое стекло. При исследовании с помощью оптического микроскопа покрытие оказалось прозрачным. В покрытии не обнаружены трещины, и это показывает, что различие термического расширения измельченной алмазной подложки и покрытия из диоксида кремния являлось небольшим.
Образец, подвергнутый термической обработке при 670°С (образец А), и образец, подвергнутый термической обработке при 900°С (образец В), после этого сопоставляли с образцом такого же алмаза без покрытия в термогравиметрическом анализаторе в потоке воздуха при скорости нагревания, равной 20°С/мин. Окисление алмаза без покрытия начиналось при 781°С, а образцов А и В - при 791°С 893°С соответственно. Это показывало, что образец А содержал значительное количество открытых пор, слабо подавляющих окисление, тогда как в образце В, для которого начало окисления смещалось примерно на 110°С, обеспечивается значительная защита алмаза от окисления. Этот результат показывает, что образец, подвергнутый термической обработке при 900°С, образец В, был полностью покрыт преимущественно не содержащим пор стеклом SiO2, о чем свидетельствуют данные СЭМ и оптических изображений.
Пример 7
Множество частиц алмаза размером от 0,75 до 1,5 мкм, полученных с помощью хорошо известных технологий дробления и сортировки, обрабатывали в дымящей концентрированной серной кислоте, к которой прибавлен нитрат калия. Эта методика приводит к тому, что на поверхности алмаза преобладают кислородсодержащие функциональные группы и поэтому она является витреофильной, что позволяет находящимся на поверхности химическим соединениям участвовать в золь-гелевых реакциях. 20 г этого алмаза с помощью ультразвукового зонда диспергировали в 2,5 л этилового спирта чистоты 99%, к которому прибавляли 500 мл деионизированной воды и 60 мл 25 об.% водного раствора гидроксида аммония. Суспензию энергично перемешивали с помощью механической лопастной мешалки и выдерживали при комнатной температуре (25°С). 80 г тетраэтоксисиликата кремния (Si(OC2H5)4) растворяли в 100 мл этанола чистоты 99%. Этот раствор медленно прибавляли к суспензии в течение 12 ч. Перемешивание продолжали в течение еще 1 ч. Затем множество обладающих покрытием частиц алмаза извлекали из суспензии, промывали и сушили, как это описано в примере 6.
Исследование с помощью СЭМ показало, что каждая частица алмаза размером примерно 1 мкм полностью закрыта покрытием. Путем взвешивания до и после нанесения покрытия установлено, что масса покрытия составляет примерно 30 мас.% от полной массы.
Обладающий покрытием материал разделяли на 3 примерно одинаковые порции и образцы помечали как С, D и Е. Образец D нагревали в потоке чистого аргона до температуры, равной 670°С в течение 3 ч, скорость нагревания составляла 3°С/мин. Аналогичным образом образец Е подвергали термической обработке при максимальной температуре, равной 1000°С, также в течение 3 ч. Образец С сохраняли в высушенном состоянии и не подвергали дополнительной термической обработке. Исследование с помощью СЭМ показало, что частицы образца Е полностью закрыты не содержащим трещин покрытием, которое выглядело как плавленое стекло.
Удельную площадь поверхности не содержащего покрытия образца алмаза и образцов С, D и Е определяли с помощью хорошо известной методики адсорбции азота Брунауэра, Эметта и Теллера (БЭТ). Результаты приведены в таблице 2.
Из таблицы 2 следует, покрытие из диоксида кремния на образце С приводит к двадцатикратному увеличению удельной площади поверхности по сравнению с удельной площадью поверхности порошкообразного алмаза без покрытия. Это показывает, что покрытие действительно обладает сильно микропористой структурой с открытыми порами.
После термической обработки в аргоне при 670°С (образец D) происходило значительное вязкое течение диоксида кремния, находящегося в покрытии, так что открытые поры были у основном устранены, о чем свидетельствует значение удельной площади поверхности, которая уменьшилась до значения, близкого к значению для порошка без покрытия. Удельная площадь поверхности образца Е после термической обработки при 1000°С уменьшилась до значения, немного меньшего, чем значение для порошка без покрытия. Это указывает на немного более значительное закрывание пор и, возможно, не небольшое сглаживание поверхности частиц порошка, что согласуется с образованием не содержащего пор покрытия из кварцевого стекла, закрывающего грани и неровности частиц алмаза. Эти результаты показывают, что пористость и плотность покрытий из диоксида кремния можно регулировать путем выбора методики термической обработки, проводимой после нанесения покрытия по золь-гелевой методике.
Пример 8
20 г хорошо ограненных высококристаллических частиц зернистого материала cBN, обладающего частицами диаметром от 105 до 125 мкм, обрабатывали в кипящей 32 об.% хлористоводородной кислоте, промывали водой и сушили. Этот материал суспендировали путем энергичного перемешивания в смеси 1,8 л этанола чистоты 99%, 350 мл деионизированной воды и 40 мл 25 об.% водного раствора гидроксида аммония. Затем при перемешивании к суспензии в течение 10 ч медленно прибавляли 30 мас.% раствор тетраэтоксисилана (Si(OC2H5)4) в чистом сухом этаноле. Перемешивание продолжали в течение еще 1 ч. Материалу давали осесть, надосадочную жидкость удаляли, и обладающие покрытием частицы cBN промывали чистым сухим этанолом. Затем обладающий покрытием материал сушили при 60°С в вакуумном сушильном шкафу в течение 24 ч. Затем этот обладающий покрытием материал нагревали в сухом чистом аргоне со скоростью 3°С/мин до температуры, равной 800°С и выдерживали при этой температуре в течение 3 ч. Проведенное после этого исследование с помощью СЭМ показало, что частицы зернистого материала полностью закрыты не содержащим трещин покрытием из диоксида кремния, которое в основном выглядело гладким и бесструктурным.
Пример 9
Методику нанесения покрытия на алмаз, обладающий частицами размером от 0,75 до 1,5 мкм, описанную в примере 7, использовали для нанесения покрытия на микрометровый порошкообразный cBN, обладающий частицами со средним размером, равным 1,25 мкм. Мелкозернистый порошок, обладающий нанесенным по золь-гелевой методике покрытием, после сушки в течение 24 ч в вакууме при 60°С подвергали термической обработке в аргоне при 800°С в течение 3 ч. Исследование с помощью СЭМ показало, что каждая отдельная частица cBN полностью закрыта плотным покрытием из диоксида кремния.
Пример 10
110 г ограненного кристаллического cBN, пропущенного через сито США 120/140 меш (105-125 мкм), обрабатывали в кипящей 32 об.% хлористоводородной кислоте, промывали водой и сушили. Затем этот материал нагревали на воздухе при 650°С в течение 1 ч для небольшого окисления поверхностей частиц.
Затем эти частицы cBN суспендировали в 250 мл раствора н-пропоксида циркония формулы Zr(ОС3Н7)4 в безводном изопропаноле. Суспензию поддерживали с помощью механического перемешивания лопастной мешалкой примерно при 100 оборотах/мин. Масса н-пропоксида циркония в растворе составляла 15 г.
3,8 мл чистой деионизированной воды смешивали с 100 мл изопропанола и эту смесь в течение 90 мин медленно прибавляли к суспензии частиц cBN. Затем перемешивание продолжали в течение еще 3 ч до завершения реакций гидролиза и поликонденсации и на частицы cBN наносилось покрытие. Затем обладающим покрытием частицам cBN давали осесть в течение ночи и надосадочную жидкость сливали. Затем обладающие покрытием частицы cBN трижды промывали изопропанолом путем осаждения и декантации. Затем обладающие покрытием частицы cBN сушили в течение ночи путем медленного выпаривания оставшегося спирта при условиях окружающей среды. Затем материал сушили путем нагревания в вакуумном сушильном шкафу при 60°С в течение 24 ч. При исследовании с помощью сканирующего электронного микроскопа обнаружено, что все частицы cBN полностью закрыты в основном не содержащим трещин покрытием. Покрытие закрывало грани, ребра, ступени и входящие ребра. Как показано на фиг.6(А), исследование с помощью рентгеновского дифрактометра, обладающего покрытием высушенного cBN, обнаружило наличие только узких дифракционных полос, соответствующих кристаллическому cBN. Это показывает, что предполагаемое покрытие из диоксида циркония в основном является аморфным.
Затем 50 г обладающих покрытием и высушенных частиц cBN нагревали на воздухе при 475°С в течение 3 ч. Использовали очень низкую скорость нагревания, равную примерно 20°С/ч. Рентгенограмма этого материала, приведенная на фиг.6(В), показала, что покрытие из диоксида циркония закристаллизовалось с образованием микрокристаллического диоксида циркония, ZrO2, обладающего тетрагональной структурой.
Еще 50 г образца, обладающего покрытием, и высушенного cBN нагревали на воздухе при 800°С в течение 3 ч, также при сходной очень низкой скорости нагревания. Исследование с помощью рентгеновского дифрактометра показало, что, покрытие закристаллизовалось в моноклинную фазу диоксида циркония, ZrO2.
Предполагается, что cBN, обладающий покрытиями из такого кристаллического диоксида циркония, при изготовлении шлифовальных кругов и т.п. будет менее реакционноспособным по отношению к функциональным группам стекла.
Это является примером альтернативной методики нанесения покрытий, при которой спиртовый раствор воды медленно прибавляют к суспензии частиц, на которые наносят покрытие, в спиртовом растворе алкоксида.
Пример 11
Субмикрометровый кубический нитрид бора, обладающий размером частиц в диапазоне от 0,5 до 1 мкм (средний размер частиц равен 0,7 мкм), подвергали кислотной обработке, как это описано в примере 1. 34,04 г подвергнутого кислотной обработке порошкообразного cBN суспендировали в 2021 мл чистого этанола и 42 мл деионизированной воды. Эту суспензию cBN обрабатывали с помощью ультразвукового зонда в течение 20 мин для удаления агломератов, а затем энергично механически перемешивали лопастной мешалкой.
19,79 г н-пропоксида циркония(IV) (70 мас./мас.% в н-пропаноле), обладающего химической формулой Zr[O(СН2)2СН3]4, растворяли в 122 мл сухого этанола. Раствор алкоксида при перемешивании при комнатной температуре по каплям в течение 3 ч прибавляли к суспензии cBN и перемешивали в течение еще 1,5 ч после прибавления алкоксида. Суспензию обладающего покрытием cBN выдерживали при комнатной температуре в течение ночи. cBN, обладающий покрытием из оксида циркония, трижды промывали чистым этанолом и сушили на роторном испарителе в вакууме при давлении, равном от 600 до 390 мбар, и при температуре, равной от 70 до 80°С. Полученный порошок дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней. Высушенный порошок исследовали на сканирующем электронном микроскопе, и обнаружено, что частицы cBN обладают хорошим покрытием.
Затем этот высушенный порошок подвергали термической обработке на воздухе в статических условиях при 600°С в течение 3 ч. Использовали скорость нагревания, равную 5°С/мин. Рентгеноструктурный анализ подвергнутого термической обработке порошка показал, что покрытие представляет собой тетрагональный оксид циркония, ZrO2.
Микрофотографии, полученные с помощью ТЭМ, показали, что покрытие на поверхностях субмикрометровых частиц cBN состоит из нанометровых частиц диаметром примерно 5 нм.
Пример 12
Субмикрометровый кубический нитрид бора, обладающий размером частиц в диапазоне от 0,5 до 1 мкм (средний размер частиц равен 0,7 мкм), подвергали кислотной обработке, как это описано в примере 1. 25 г этого порошка суспендировали в 1,5 л чистого этанола и 30 мл деионизированной воды и в течение 25 мин подвергали ультразвуковой обработке для удаления агломератов. В отдельном стакане 0,48 г гексагидрата нитрата иттрия, Y(NO3)3·6H2O, растворяли в 50 мл чистого этанола, а затем прибавляли 13,9 г н-пропоксида циркония(IV), обладающего химической формулой Zr[O(СН2)2СН3]4, и еще 50 мл чистого этанола. Содержимое последнего стакана перемешивали стеклянной палочкой и дополнительно перемешивали путем встряхивания содержимого в делительной воронке. Раствор смеси гексагидрата нитрата иттрия с н-пропоксидом циркония(IV) при перемешивании по каплям при комнатной температуре в течение 2 ч прибавляли к суспензии cBN. После этого прибавления раствор дополнительно механически перемешивали в течение 1 ч 10 мин. Затем раствор выдерживали в течение ночи при комнатной температуре. Обнаружено, что после выдерживания в течение ночи полученное множество обладающих покрытием частиц образовало высоковязкий гель. После выдерживания всего в течение 48 ч золь-гель сушили на роторном испарителе в вакууме при давлении, равном 400 мбар, и при температуре, равной от 70 до 80°С.
Этот порошок дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней. Затем высушенный порошок cBN, обладающий покрытием из оксида циркония, подвергали термической обработке на воздухе в статических условиях при 600°С в течение 3 ч. Использовали скорость нагревания, равную 5°С/мин. Рентгеноструктурный анализ показал, что полученный порошок содержит фазы cBN и тетрагональную ZrO1,99. Полученные на ТЭМ микрофотографии показали, что зерна диоксида циркония обладают размером, равным от 4 до 5 нм.
Пример 13
12 г ограненного кристаллического cBN, пропущенного через сито США 120/140 меш (105-125 мкм) нагревали на воздухе при 650°С для окисления поверхностей. Затем этот материал перемешивали и суспендировали в 200 мл чистого изопропанола, к которому прибавляли 22 мл деионизированной воды. Суспензию нагревали и кипятили с обратным холодильником при 50°С.
К этой суспензии в течение 1 ч медленно прибавляли раствор 10 г втор-бутоксида алюминия формулы Al(ОС4Н9)3 в 50 безводного изопропанола. Суспензию перемешивали в течение еще 2 ч и затем прибавляли 1 мл 55% азотной кислоты и суспензию перемешивали в течение еще 1 ч. Затем измельченному материалу давали осесть и его промывали изопропанолом, а затем медленно сушили в течение ночи. В заключение обладающий покрытием cBN сушили в вакуумном сушильном шкафу в течение 24 ч при 60°С. Исследование с помощью СЭМ показало, что частицы cBN в основном закрыты тонким покрытием из соединения оксида алюминия.
Предполагается, что это покрытие можно превратить в ряд так называемых переходных структур оксида алюминия путем нагревания на воздухе при различных температурах в диапазоне от 400 до 1200°С, примерно вплоть до 1150°С и выше, и можно образовать так называемый 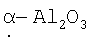 . Эти структуры оксида алюминия известны в области термической обработки оксидов алюминия, полученных по золь-гелевой методике.
. Эти структуры оксида алюминия известны в области термической обработки оксидов алюминия, полученных по золь-гелевой методике.
Пример 14
Синтетический порошкообразный алмаз, обладающий частицами размером 1 мкм с диапазоном размеров от 0,75 до 1,5 мкм, подвергали кислотной очистке, как это описано в примере 4. 20 г этого порошкообразного алмаза суспендировали в растворе, содержащем 258 мл чистого изопропанола и 175 мл деионизированной воды. Эту суспензию нагревали при 60°С в установке с обратным холодильником и механически перемешивали мешалкой лопастного типа примерно при 100 оборотов/мин. 24 г Втор-бутоксида алюминия, обладающего химической формулой AlO3C12H27, растворяли в 100 мл безводного изопропанола и по каплям в течение 1 ч 45 мин при нагревании и перемешивании прибавляли к суспензии алмаза суспензию. После прибавления алкоксида суспензию перемешивали в течение 1 ч 15 мин при 60°С. Затем к нагретой суспензии прибавляли примерно 1 мл хлористоводородной кислоты (32%) и затем нагревали до 80°С и перемешивали в течение еще 1 ч, поддерживая указанную температуру. Затем суспензии давали охладиться до комнатной температуры и ее выдерживали при комнатной температуре в течение ночи. Затем суспензию сушили на роторном испарителе при температуре, равной 80°С, и в вакууме при давлении, равном 400 мбар.
Алмаз, обладающий покрытием из соединения алюминия, дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней. Исследование с помощью СЭМ показало, что частицы алмаза обладают покрытием из соединения оксида алюминия.
Затем этот порошок подвергали термической обработке при 400°С на воздухе в статических условиях в течение 3 ч. Использовали скорость нагревания, равную 5°С/мин. Рентгеноструктурный анализ показал, что после этой термической обработки покрытие на алмазе являлось преимущественно аморфным. Это было подтверждено исследованием с помощью ТЭМ.
Пример 15
12 г хорошо ограненного чистого обладающего в основном октаэдрической морфологией синтетического алмаза, пропущенного через сито США 120/140 меш (105-125 мкм) нагревали на воздухе при 500°С в течение 1 ч для получения окисленных поверхностей. Этот алмаз суспендировали в 200 мл чистого этанола, к которому прибавляли 20 мл деионизированной воды. Суспензию поддерживали путем перемешивания.
Затем раствор 5,1 г этоксида вольфрама формулы W(OC2H5)5 в 50 мл чистого безводного этанола в течение примерно 1 ч при перемешивании по каплям медленно прибавляли к суспензии. Суспензию перемешивали в течение еще 1 ч до завершения реакций гидролиза и поликонденсации. После проводимых несколько раз осаждения, декантации и промывки чистым этанолом частицы алмаза сушили путем естественного испарения, а затем в вакуумном сушильном шкафу при 60°С. Исследование с помощью СЭМ и ЭДА показало, что каждый кристалл алмаза равномерно закрыт тонким покрытием из соединения вольфрама с кислородом, предположительно представляющего собой оксид вольфрама, WO3. Толщина найдена равной примерно 0,25 мкм.
Затем образец обладающего покрытием алмаза нагревали в атмосфере водорода при температуре 550°С в течение 1 ч. Исследование с помощью СЭМ показало, что покрытие из оксида вольфрама восстановилось в металлический вольфрам и образовало дисперсию очень мелкозернистых частиц или островков этого металла размером примерно 100 нм или менее. Сделан вывод о том, что покрытие из WO3 в этом случае было недостаточным для того, чтобы обеспечить покрытие всей поверхности после восстановления в металл.
Предполагается, что для того, чтобы полностью закрыть металлическим вольфрамом частицы алмаза такого размера, необходимо покрытие из WO3, обладающее толщиной, равной примерно 1 мкм или более. Это можно обеспечить путем соответствующего изменения методики, описанной в этом примере.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИКРИСТАЛЛИЧЕСКИЕ АБРАЗИВНЫЕ МАТЕРИАЛЫ И СПОСОБ ИХ ИЗГОТОВЛЕНИЯ | 2005 |
|
RU2404021C2 |
| СПОСОБ ПОЛУЧЕНИЯ СВЕРХТВЕРДОГО АБРАЗИВА С ПОКРЫТИЕМ | 2005 |
|
RU2378231C2 |
| АБРАЗИВЫ С ПОКРЫТИЕМ | 2005 |
|
RU2368489C2 |
| АБРАЗИВЫ С ПОКРЫТИЕМ | 2005 |
|
RU2372371C2 |
| Способ изготовления сверхтвердого композиционного материала для режущего инструмента | 2023 |
|
RU2829867C1 |
| СЛОЙ ИЛИ ПОКРЫТИЕ И КОМПОЗИЦИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2394798C2 |
| КОМПОЗИЦИОННЫЙ ПРИПОЙ ДЛЯ ПАЙКИ АБРАЗИВНЫХ ИНСТРУМЕНТОВ ИЗ СВЕРХТВЕРДЫХ МАТЕРИАЛОВ | 2014 |
|
RU2588928C1 |
| КОМПОЗИЦИОННЫЙ МАТЕРИАЛ НА ОСНОВЕ КАРБИДА БОРА | 2009 |
|
RU2515663C2 |
| Способ получения композиционного металл-дисперсного покрытия, дисперсная система для осаждения композиционного металл-дисперсного покрытия и способ ее получения | 2020 |
|
RU2746863C1 |
| Способ получения композиционного металл-дисперсного покрытия, дисперсная система для осаждения композиционного металл-дисперсного покрытия и способ ее получения | 2020 |
|
RU2746861C1 |
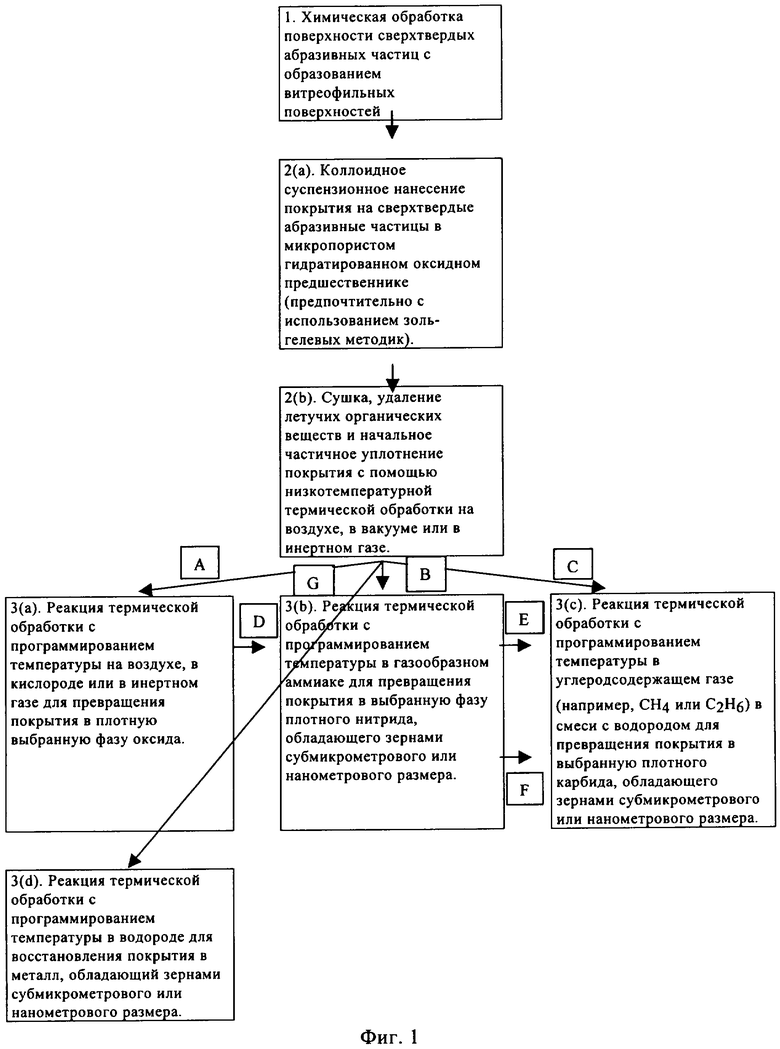
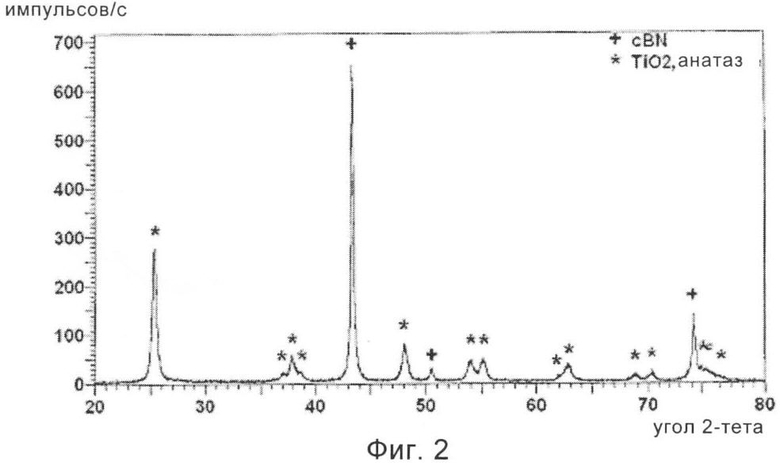
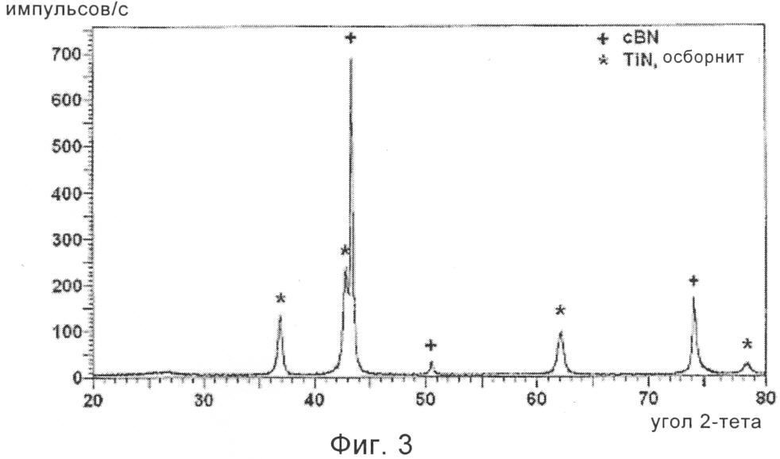
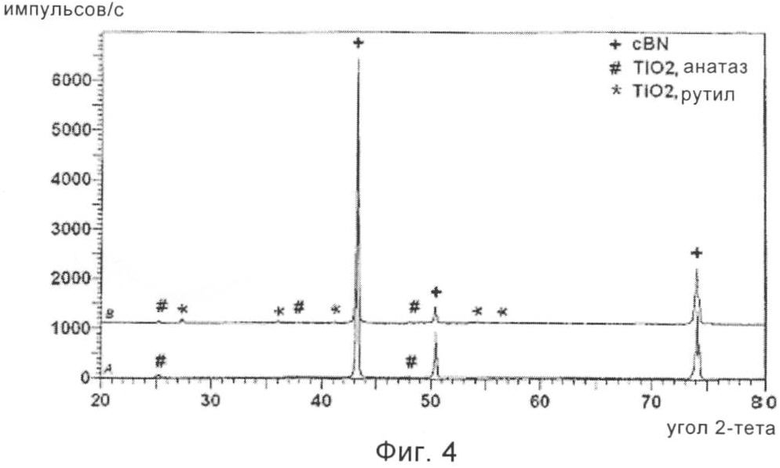
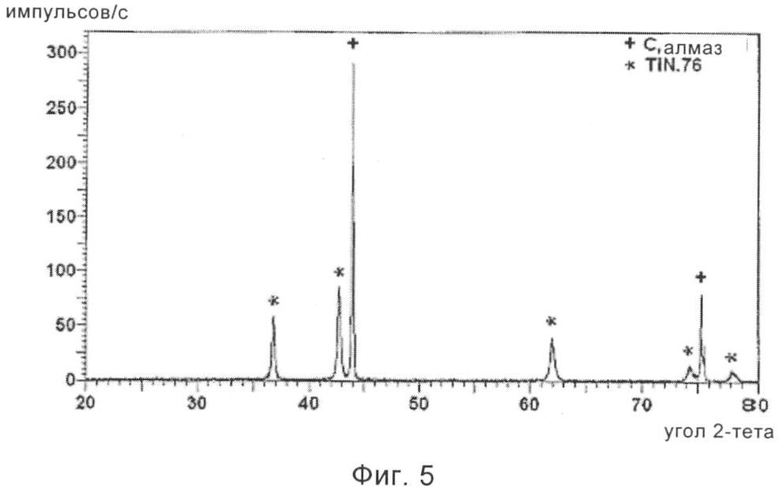
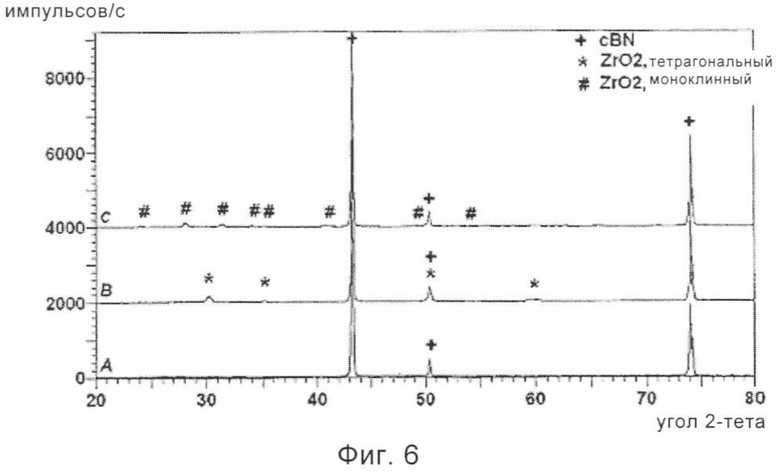
Изобретение может быть использовано при изготовлении абразивных инструментов. Используют множество сверхтвердых абразивных частиц субмикрометрового или нанометрового размера алмаза или кубического нитрида бора или комбинации этих материалов и обладающие витреофильными поверхностями, способными образовывать химические связи с оксидами. Наносят на частицы покрытия из оксидного материала-предшественника и затем термически обрабатывают для высушивания и очистки покрытий. Покрытия выбраны из группы, включающей нитриды титана, ванадия, ниобия, тантала, молибдена и вольфрама или карбиды ванадия, ниобия, тантала, молибдена и вольфрама, фазу анатаза диоксида титана, фазу рутила диоксида титана, тетрагональный диоксид циркония, моноклинный диоксид циркония, диоксид циркония, стабилизированный оксидом иттрия или оксидом магния, переходные структуры или альфа-фазу оксида алюминия и оксиды ванадия, ниобия, тантала, гафния, молибдена и вольфрама, кварцевое стекло. Изобретение позволяет наносить покрытия на абразивы субмикрометрового или нанометрового размера. 5 н. и 30 з.п. ф-лы, 6 ил., 2 табл.
1. Способ нанесения покрытия на сверхтвердые абразивные частицы субмикрометрового или нанометрового размера, выбранные из алмаза или кубического нитрида бора или комбинации этих материалов, включающий стадии использования множества сверхтвердых абразивных частиц, обладающих витреофильными поверхностями, способными образовывать химические связи с оксидами, нанесения на сверхтвердые абразивные частицы покрытия из оксидного материала-предшественника и термической обработки обладающих покрытием сверхтвердых абразивных частиц для высушивания и очистки покрытий.
2. Способ по п.1, в котором обладающие покрытием сверхтвердые абразивные частицы обрабатывают для превращения оксидного материала-предшественника в оксид, нитрид, карбид, оксинитрид, оксикарбид или карбонитрид оксидного материала-предшественника или в элементную форму оксидного материала-предшественника, или в их комбинации.
3. Способ по п.1, в котором оксидный материал-предшественник представляет собой аморфный или нанокристаллический оксид, гидроксид или оксогидроксид.
4. Способ по п.1, в котором частицы алмаза или кубического нитрида бора подвергнуты поверхностной обработке, чтобы сделать их поверхности витреофильными.
5. Способ по п.2, в котором подвергнутый превращению оксидный материал-предшественник выбран из группы, включающей обладающие зернами микрометрового, субмикрометрового или нанометрового размера оксиды, нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды оксидных материалов-предшественников или элементные материалы-предшественники, или их комбинации.
6. Способ по п.2, в котором подвергнутый превращению оксидный материал-предшественник выбран из группы, включающей оксиды, нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды алюминия, титана, кремния, ванадия, циркония, ниобия, гафния, тантала, хрома, молибдена и вольфрама и элементные формы молибдена и вольфрама, и любую подходящую комбинацию этих материалов.
7. Способ по п.2, в котором подвергнутый превращению оксидный материал-предшественник представляет собой обладающее зернами нанометрового размера соединение алюминия, титана, кремния, ванадия, циркония, ниобия, гафния, тантала, хрома, молибдена или вольфрама, или любую подходящую комбинацию этих материалов.
8. Способ по п.2, в котором подвергнутый превращению оксидный материал-предшественник представляет собой обладающую зернами нанометрового размера элементную форму вольфрама, молибдена или комбинацию, или сплав этих металлов.
9. Способ по п.1, в котором оксидный материал-предшественник в виде покрытия нанесен на сверхтвердые абразивные частицы с использованием так называемой золь-гелевой методики.
10. Способ по п.9, в котором сверхтвердые абразивные частицы суспендированы в жидкой среде и введен подходящий химический реагент для образования коллоидных частиц, которые связываются с поверхностями соответствующих частиц и образуют покрытия на частицах.
11. Способ по п.10, в котором подходящий химический реагент представляет собой по меньшей мере один алкоксид или раствор алкоксида (алкоксидов) в спирте.
12. Способ по п.10, в котором жидкой средой является аликвота воды и спирта.
13. Способ по п.10, в котором подходящим химическим реагентом является аликвота воды и спирта.
14. Способ по п.13, в котором жидкой средой является по меньшей мере один алкоксид или раствор алкоксида (алкоксидов) в спирте.
15. Способ по п.11 или 14, в котором алкоксид представляет собой алкоксид элемента, выбранного из группы, включающей алюминий, титан, кремний, цирконий, ванадий, ниобий, тантал, хром, молибден, вольфрам, гафний и иттрий.
16. Способ по п.11 или 12, в котором в жидкую среду введены 2 или большее количество алкоксидов, которые выбраны из группы, включающей алкоксиды элементов алюминия, титана, кремния, циркония, ванадия, ниобия, тантала, хрома, молибдена, вольфрама, гафния и иттрия.
17. Способ по п.13 или 14, в котором жидкая среда включает 2 или большее количество алкоксидов в спиртовом растворе, которые выбраны из группы, включающей алкоксиды элементов алюминия, титана, кремния, циркония, ванадия, ниобия, тантала, хрома, молибдена, вольфрама, гафния и иттрия.
18. Способ по любому из пп.10-12, в котором подходящий химический реагент представляет собой раствор смешанного алкоксидного соединения или комплекса, включающего два или большее количество элементов алюминия, титана, кремния, циркония, ванадия, ниобия, тантала, хрома, молибдена, вольфрама, гафния или иттрия.
19. Способ по любому из пп.10, 13 или 14, в котором жидкая среда представляет собой раствор смешанного алкоксидного соединения или комплекса, включающего два или большее количество элементов алюминия, титана, кремния, циркония, ванадия, ниобия, тантала, хрома, молибдена, вольфрама, гафния или иттрия.
20. Способ по любому из пп.11-14, в котором спирт содержит такую же алкильную группу, что и алкоксид (ы).
21. Способ по п.1, в котором оксидный материал-предшественник покрытия является в основном микропористым.
22. Способ по п.21, в котором обладающие покрытием сверхтвердые абразивные частицы подвергают нагреванию на воздухе, в вакууме или в инертном газе для удаления летучих и нежелательных химических веществ, присоединенных к большим участкам поверхности микропористых аморфных покрытий.
23. Способ по п.22, в котором обладающие покрытием сверхтвердые абразивные частицы подвергают дополнительной термической обработке или прокаливанию для кристаллизации покрытий в форме мелкозернистых или обладающих зернами нанометрового размера оксидных керамик.
24. Способ по п.22, в котором обладающие покрытием сверхтвердые абразивные частицы подвергают дополнительной термической обработке для витрификации покрытий с образованием стекол.
25. Способ по любому из пп.21-24, в котором обладающие покрытием сверхтвердые абразивные частицы нагревают в реакционноспособных газах для превращения материалов покрытий в неоксидные керамики или стекла.
26. Способ по п.25, в котором нитриды образуют по реакции покрытий с газообразным аммиаком.
27. Способ по п.26, в котором карбиды образуют по реакции покрытий в смесях углеродсодержащих газов с водородом.
28. Способ по п.27, в котором карбиды образуют по реакции покрытий в смеси метана или этана с водородом.
29. Способ по любому из пп.21-24, в котором обладающие покрытием сверхтвердые абразивные частицы нагревают в реакционноспособных газах для превращения материалов покрытий в оксинитридные или оксикарбидные керамики или стекла.
30. Способ по п.22 или 23, в котором оксидные покрытия восстанавливают водородом и превращают в обладающие зернами микрометрового или нанометрового размера элементы или металлы.
31. Обладающий покрытием сверхтвердый абразивный измельченный материал, включающий частицы алмаза или кубического нитрида бора субмикрометрового или нанометрового размера, обладающие покрытиями, выбранными из группы, включающей нитриды титана, ванадия, ниобия, тантала, молибдена и вольфрама или карбиды ванадия, ниобия, тантала, молибдена и вольфрама.
32. Обладающий покрытием сверхтвердый абразивный измельченный материал, включающий частицы алмаза или кубического нитрида бора субмикрометрового или нанометрового размера, обладающие покрытиями, выбранными из группы, включающей фазу анатаза диоксида титана, фазу рутила диоксида титана, тетрагональный диоксид циркония, моноклинный диоксид циркония, диоксид циркония, стабилизированный оксидом иттрия или оксидом магния, переходные структуры или альфа-фазу оксида алюминия и оксиды ванадия, ниобия, тантала, гафния, молибдена и вольфрама.
33. Обладающий покрытием сверхтвердый абразивный измельченный материал, включающий частицы алмаза или кубического нитрида бора субмикрометрового или нанометрового размера, обладающие покрытием из кварцевого стекла.
34. Обладающий покрытием сверхтвердый абразивный измельченный материал, включающий частицы алмаза или кубического нитрида бора субмикрометрового или нанометрового размера, обладающие покрытием из оксидов титана, ванадия, ниобия, тантала, алюминия, кремния, молибдена или вольфрама, или нитридов или карбидов титана, ванадия, ниобия, тантала или молибдена, или металлического молибдена или вольфрама.
35. Обладающий покрытием сверхтвердый абразивный измельченный материал по пп.31-33, в котором толщина покрытия составляет менее 2 мкм.
| US 5104422 A, 14.04.1992 | |||
| СПОСОБ ОЧИСТКИ СВЕРХТВЕРДЫХ МАТЕРИАЛОВ ОТ ПРИМЕСЕЙ | 1999 |
|
RU2163222C2 |
| Способ получения поликристаллического алмазсодержащего материала | 1980 |
|
SU961281A1 |
| US 4339281 A, 13.07.1982 | |||
| US 5344526 A, 06.09.1994 | |||
| Устройство для обнаружения юза колес транспортного средства | 1984 |
|
SU1318467A1 |
| Способ выращивания семян сахарной свеклы | 1984 |
|
SU1213999A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОГО ПОРОШКА МЕТАЛЛА | 1995 |
|
RU2126735C1 |
| TUROVA N.YA, TUREVSKAYA E.P, Chemistry of Metal Alkoxides, Nor-well, Kluwer Academic Publishers, 2002, p.107-125 | |||
| WO 9402560 A1, 03.02.1994 | |||
| Устройство для останова путевой машины | 1974 |
|
SU503974A1 |
| WO 03076337 A2, 18.09.2003. | |||

