Изобретение относится к способу получения новых производных хинолина, обладающих высокой бактерицидной активностью.
Цель изобретения - получение новых производных хинолина, которые по сравнению с известными структурными аналогами проявляют более высокую активность против грамположительных и грам- отрицательных бактерий.
Пример 1 (контрольный). Этил 2-пентафторбензоил-З-цикпопропил- аминоакрилат.
Смесь этилового эфира пентафтор- бензоилуксусной кислоты (25 г), этил- ортоформиата (20 г) и уксусного ангидрида (23 г) кипятят 2 ч. Реакционную смесь испаряют при пониженном давлении досуха. Остаток растворяют в диэ иловом эфире и вводят в реак-
ы
|Циюс циклопропиламином (5,1 г) с по -лучением этилового эфира 2-пентафтор- i бензоил-3 циклопропиламикоакриловой ; кислоты (28 г), т.пл. 89 с.
П р и м е р 2 (контрольный). ; Этиловый эфир 2-С4-(4-ацетил-3-метил-. : 1-пиперазинил)т-2,3,5,6-тетрафторбен- зоил -3-цикл 5пропиламиноакриловой
кислоты.
i а. Этиловый эфир пентафторбензоил- уксусной кислоты вводят в реакцию с 2-метилпиперазином, после .чего продукт реакции ацетилируют и получают этиловый эфир 4-(4-ацетил-3-метил-1- пиперазинил)-2,3,5,6-тетрафторбензо- илуксусной кислоты.
Ь. Полученное, соединение обрабатывают по методике примера 1 и получают этиловый эфир (4-ацетил-3- метил-1-пиперазинил)-2,3,5,6-тетра- фторбензоил j-3-циклопропиламиноакри- ловой кислоты.
Пример 3 (контрольный) . Используя методику примера 2, получают следующие соединения: этил (4- формил-1-пиперазинил)-2,3,5,6-тетра- фторбензоил }-3-циклопропиламиноакри- лат; этил (цис-3 ,5-димeтI-m-1-пи- пepaзинил)-2 ,3,5 ,6-тетрафторбензоил - 3-цикпопропиламиноакрилат; этил (3-фторметил-1 -пиперазинил)-2 ,3 ,5 ,6- тетрафторбензоил -3-циклопропиламино- акрилат этил (3-амино-3-метил- 1 -пирролидини.л )-2 ,3,5,6-тетрафторбен- зоил -3-циклопропиламиноакрилат; этил (цис-3-амино-4-метил-1-пирроли- динил)-2,3,5,6-тетрафторбензоилJ-3- циклопропиламиноакрилат; этил (транс-З-амино-4-метил-1-пирролиди- нил)-2,3,5,6-тетрафторбензош1 -3-циклолролиламиноакрилат; этил 2- 4-(цис 3-трифторацетиламино-4-фторметил-1- пирролидинил)-2,3,5,6-тетрафторбензо илЗ-3-циклопропиламиноакрилат; этил 2 4-(транс-З-амино-4-фторметил-З-ме тил-1-пирролидинил)-2,3,5,6-тетрафто бензоил -3-циклопропиламиноакрилат.
.Пример 1. 1-Циклопропил- 5,6,7,8-тетрафтор-1,4-дигидро-4-оксо- хинолин-3-карбоновая кислота.
а. Этил 2-пентафторбензоил.-3-цик- лопропиламиноакрйлат (28 г) рас1воря- ют в сухом тетрагидрофуране-и вводят в реакцию при комнатной температуре с 60%-ным гидридом натрия (3,85 г). Получают этиловый эфир 1-циклопропил- 5,6,7,8-тетрафтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (18,4 г), т.пл. 170-171 С.
Ь. Полученное соединение (10 г) гидролизуют кипячением 3Q мин в смеси ледяной уксусной кислоты (60 мл), воды (50U мл) и концентрированной серной кислоты (7 мл). Получают 1- циклопропил-5,6,7,8-тетрафтор-1,4-ди- гидро-4-оксохинолин-З-карбоновой кислоты (8,7 г), т.пл. 181-182 0.
П р и м е р 2. 1-Циклопропил- 5,6,8-трифтор-7-(З-метил-1-пиперазинил)- ,4-дигидро-4-оксохинолин-3-кар- боновая кислота.
а. Этиловый эфир (4-ацетил-3- метил-1-пиперазинил)-2,3,5 ,6-тетрафторбензоил -3-циклопропиламиноакри- лат (2,0 г) растворяют в сухом тетрагидрофуране (10 мл), добавляют 60%- ного .гидрида натрия (200 мг). Смесь перемешивают 10 мин при комнатной температуре.
После упаривания реакционной смеси под вакуумом добавляют воду до образования осадка и смесь экстрагируют хлороформом. 1,2 г., полученного соединения и 20 мл 20%-ной соляной кислоты подвергают дефлегмации в-течение 10 ч. После испарения реакционной смеси при пониженном давлении остаток растворяют в воде (20 мл), раствор доводят до рН 7-8. Образовавшиеся кристаллы отфильтровывают и получают 1-циклопропил-5,6,8-трифтор-7-(3-ме- тил-1-пиперазинил)-1,4-дйгидро-4-ок- сохинолин-3-карбоновой кислоты
(0,96 г), т.пл. 235-237 С.
I
Пример 3. 1-Циклопропил- 5,6,8-трифтор-7-(1-пиперазинил)-1,4- дигидро-4-оксохинолин-З-карбоновая кислота и ее хлоргидрат.
а. Смесь этил 2-{;4-(4-формил-1- пиперазинил)-2,3,5,6-тетрафторбензо- ил -3-циклопропиламиноакрилат (2,5 г) т-бутоксид натрия перемешивают при 0.°С в течение 1 ч. Затем реакционную смесь концентрируют досуха при пониженном давлении. Остаток смешивают с водой и экстрагируют хлороформом. . Экстракт сушат и хлороформ испаряют К остатку добавляют этанол, выпавшие кристаллы отфильтровывают. Перекристаллизацией из смеси хлороформ - этанол получают 1-ЦИКЛОПРОПИЛ-5,6,8-три фтор-7-(4-формил-1-пиперазинил)-1 ,4- дигидро-4-оксохинолин-З-карбоновую кислоту (1,58 г), т.пл. 290-297 С.
b.Смесь полученной карбоновой кислоты (0,5 г) и 15%-ной соляной кислоты перемешивают 1 ч при 90- . Реакционную смесь концентрируют досуха при пониженном давлении. Полученные кристаллы перекристаллизо вывают из воды с получением хлоргид- рата 1-циклопропил-5,6,8-трифтор-7(1-пиперазинил)-1,4-дигидро-4-оксохи нолин-3-карбоновой кислоты (0,25 г), ; т.пл. 270-280°С (с разложением).
c.Полученное соединение (170 мг) растворяют в воде (5 мл) и добавлением 10%-ного водного аммиака рН доводят до 7-8. Выпавшие кристаллы отфильтровывают, промывают водой и сушат с получением 1-циклопропил-5,6,8- трифтор-7-(1-пиперазинил)-1,4-дигид- ро-4-оксохинолин-З-карбоновой кислоты (140мг), т.пл. 208-213 С.
Пример 4. 7-(Цис-3-амино-4- фторметил-1-пирролидинил)-1-циклопро- пил-5,6,8-трифтор-1,4-дигидро-4-оксо- хинолин-3-карбоновая кислота.
a.По методике примера 2а этил (цис-3-трифторацетиламино-4- фт.орметил-1-пирролидинил)-2,3,5 ,6- тетрафторбензоил 3 3-циклопропиламино- акрилат (2,0 г) циклизуют, получая
1-цикло-пропил-5,6,8-трифтор-7-(цис- З-трифторацетиламино-4-фторметил-1- пирролидинил)-1,4-дигидро-4-оксохино- лин-3-карбоновую кислоту (1,25 г), т.пл. 283-284 С.
b.Смесь полученного соединения (1 г) и 10% -ного водного раствора гидроксида натрия (5 мл) перемешивают 1 ч при 80-90°С. Добавлением ледяной уксусной кислоты устанавливают в ре- акщюнной смеси рН 8-9, осадившиеся кристаллы отфильтровывают. Перекристаллизацией из диметилформамида получают 7-(цис-3-амино-4-фторметил-1- пирролидинил)-1-циклопропил-5,6,8- трифтор-1,4-дигидро-4-оксохинолин-3- карбоновую кислоту (0,52 г), т.пл. 252-253 С.
Пример 5. 1-Цикпопропил-5 6,8- трифтор-7(цис-3,5-диметил-1-пиперазинил)- ,4-дигидро-4-оксохинолингЗ- карбоновая кислота.
а. Смесь этил (цис-3 ,5-димe- тил-1-пипepaзинил)-2 ,3,5,6-тетрафтор- бензоил }-3-циклопропиламиноакрилата (2,0 г), этоксида натрия и этанола дефлегмируют 20 мин. Смесь обрабатывают- по методике примера 2а, получая этил 1-циклопропил-5,6,8-трифтор-710
15
25
(цис-3 ,5-диметил-1-пиперазинил)-1,4- дигидро-4-оксохинолин-З-карбоксилата (1,2 г).
Ь. По методике примера 2Ь полученное соединение гидролизуют с получением 1-циклопропил-$,6,8-трифтор-7- (цис-3 ,5-диметил-1-пиперазинил)-1,4- дигидро-4-оксохинолин-З-карбоновой кислоты, т.пл. 259-260 С (из смеси хлороформ - этанол).
Пример 6. 1-Циклопропил- 6,5,8-трифтор-7-(3-фторметил-1-пипе- разинил)-1 ,4-дигидро-4-оксохинолин- 3-карбоновая кислота.
a.Смесь этил (3-фторметил1-пиперазинил)-2,3,5,6-тетрафторбен- зоил J-3-циклопропиламиноакрилата (2,0 г), гидроксида натрия и диметил- 20 формамида перемешивают при 150 с в течение 10 мин. Реакционную смесь обрабатывают по методике примера 2а, получая этил-1-циклопропил-5,6,8-три- фтор-7-(3-фторметил-1-пиперазинил)1,4-дигидро-4-оксохинолин-3-карбокси- лат (1 ,25 г).
b.По методике примера 2Ь полученное соединение гидролизуют, получая
1-циклопропил-5 ,6,8-трифтор-7- (3- фторметил-1-пиперазинил)-1,4-дигидро- 4-оксохинолин-З-карбоковую кислоту, т.пл. 219-220 с.
Пример 7. 7-(3-Амино-3-ме- тил-1-пирролидинил)-1-циклопропил- 5,6,8-трифтор-1,4-дигидро-4-оксохино- лин-3-карбоновая кислота.
а. Смесь этил (3-aминo-3-мe- тшl-1-пиppoлидинил)-2 ,3 ,5 ,6-тетра- фторбензоил J-З-циклопропиламиноакри- лата, 60%-ного гидрида натрия и тет- рагидрофурана перемешивают при комнатной температуре в течение 10 мин. Реакционную смесь обрабатывают по методике примера 2а, получая этил-7- (З-амино-З-метил-1-пирролидинил) циклопропил-5,6,8-трифтор-1,4-дигид- ро-4-оксохинолин-З-карбоксилат.
Ь. Полученное соединение гидролизуют по методике примера 2Ь, названное соединение, т.пл.
Пример 8. 7-(цис-3-Амино-4- метил-1-пирролидинил)-1-циклопропил- 3,6,8-трифтор-1 ,4-дигидро-4-оксохино- лин-3-карбоновая кислота.
а. Смесь этил (цис-3-амино-4- метил-1-пирролидинил)-2,3,5,6-тетра- фторбензоилJ-3-циклопропиламиноакри- лата, триэтиламина и диоксана дефлег- мируют 20 мин. Реакционную смесь об30
5
0
5
0
получая 280-282 с.
5
рабатывают -по методике примера 2а с получением этил-7-(цис-З-амино-4-ме- тил-1-пирролидинил)-1-циклопропил- 5,6,8-трифтор-1,4-дигидрс-4-оксохино- лин-3-карбоксилата.
Ь, Полученный карбоксилат гидролизуют по методике примера 2Ь, получая 7-(цис-З-амино-4-метил-1-пирролидинил )-.1-Циклопропил-5, 6, 8-трифтор -1i дигидро-4-оксохинолин-З-карбоновук. кислоту, т.пл. 264-265 С.
Пример 9. 7-Странс-3-.Амино- 4-метил-1-пирролидинил)-1-циклопро- пил-5,6,8-трифтор-1,4-дигидро-4-оксо- хинолин-3-карбоновая кислота.
а. Смесь этил (транс-3-амино- 4-метил-1-пирролидинил)-2,3 ,5 ,6-тет- рафторбензоил 3-3-циклопропиламиноак- рилат, ДБУ и диоксана дефлегмируют 20 мин. Реакционную смесь обрабатывают по методике примера 2а, получая этил 7-(транс-3-амино-4-метил-1-пирролидинил )-1-циклопропил-5 ,6 ,8-трифтор-1 ,4-дигидро-4-оксохинолин-3-кар- боксилат.
Ь. Гидролизом по методике примера 2Ь получают 7-(транс-3-амино-4-метил- 1 -пирролидинил) -1 -циклопропил-5 ,6,8т- тр 1фтор-1 ,4-дигидро-4-оксохинолин-3- карбоновую кислоту, т.пл. 255-256 С.
Пример 10. 7-(транс-3-Амино 4-фторметил-3-метил-1-пирролидинил)- 1-циклопропил-5,6,8-трифтор-1,4-ди- гидро-4-оксохинолин-З-карбоновая кис лота и ее хлоргидрат.
а. Смесь этил (транс-3-ами- но-4-фторметил-З-метил-1-пирролиди- нил)-2,3,5,6-тетрафторбензоил -3- циклопропиламиноакрилата, карбоната калия и диметилсульфоксида перемешивают при 60°С в течение 2. ч. Реакционную смесь обрабатывают по методике примера 2а, получая этил 7-(транс-3- амино-4-фторметил-3-метил-1-пирроли-
динил)-1-циклопропил-5,6,8-дифтор- 1 ,4 -дигидро-4-оксохинолин-3-карбоксилат.
Ь. Полученное соединение гидроли зуют по методике примера 2Ь с получ нием 7-(транс-З-амино-4-фторметил-З метил-1-пирролидинил)-1-ЦИКлОпропил 5,6,8-трифтор-1,4-дигидро-4-оксохин лин-3-карбоновой кислоты, т.пл. 255 .256°С.
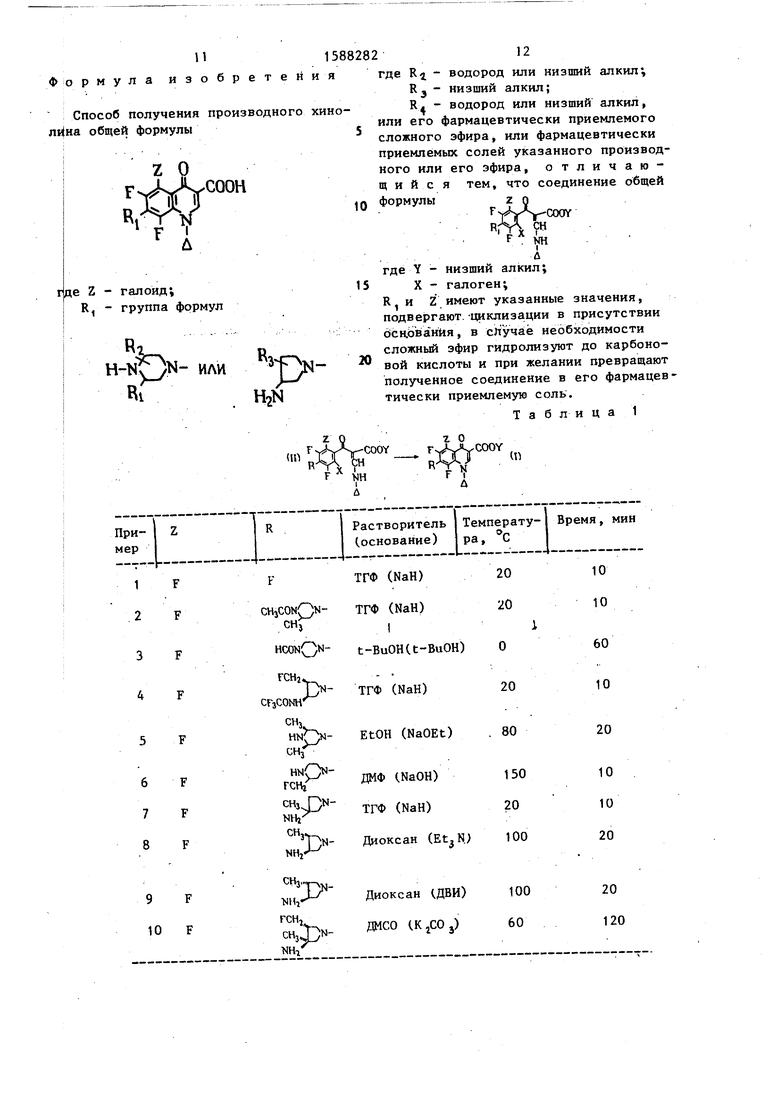
Условия получения предлагаемых соединений формулы U) представлены в табл. 1 .
Хемотерапевтическое действие соединений показано в примерах 11 - 13. Использованы следующие соединения: соединение 1: циклопропил-5,6 ,8-три- фтрр-7-(3-метил-1-пиперазинил)-1,4- дигидро-4-оксохинолин-З-карбоновая
кислота;
соединение 2: 1-циклопропил-5 ,6 ,8- трифтор-7-(цис-З-амино-4-метил-1-пир- ролидил)-1,4-дигидро-4-оксохинолин-3- карбоновая кислота; соединение 3: 1-циклопропил-5,6,8- трифтор-7-С1-пиперазинил)-1,4-дигид- ро-4-оксохинолин-З-карбоновая кислота;
соединение 4: 1-циклопропил- 5,6,8- тpифтop-7-(3,5-димeтил-1-пипepaзи- нил)-1,4-дигидро-4-оксохинолин-3-карбоновая кислота.
Со единение А: 5-амино-1-этил-6,8- дифтop-7-(1-пипepaзинил)-1 ,4-дигидро- 4-оксохинолин-З-карбоновая кислота
ННч
соои
формулы
Соединение В:.хлоргидрат 1-цикло- пропил-6-фтор-7-(1-пипераэинил)-1,4- , дигидро-4-оксохинолин-З-карбоновая кислота формулы
нсен
Г
-1
соон
I
л
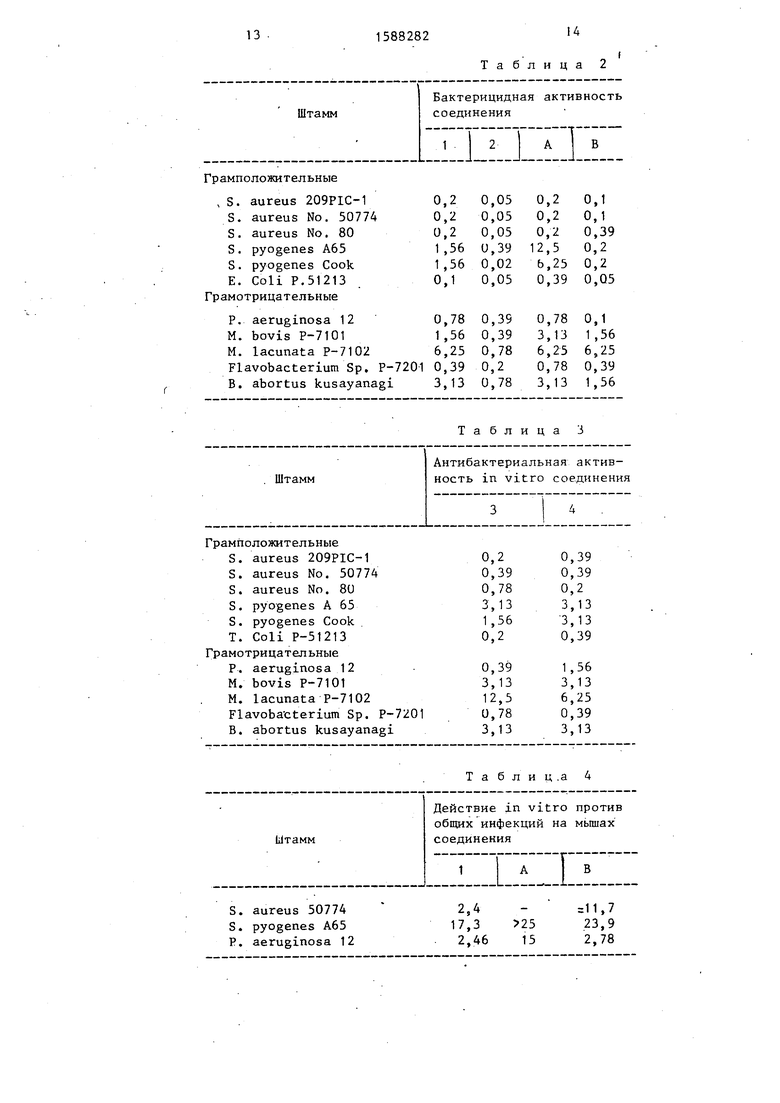
Пример 11. Бактерицидное действие in vitro показано в табл.2
и 3.
Указанные в табл. 2 значения соответствуют минимальной ингибирующей концентрации (г/мл в пересчете на свободное основание). Минимальная ин- гибирующая концентрация (МИК) определена методом двухкратного разбавления агара, рекомендованным хемотерапевти- .ческим обществом Японии с использованием агара Миллера-Хинтона. Одну петельку выращиваемой в течение суток культуры испытуемого организма в бульоне Мюллера-Хинтона наносят на содержащие лекарство слои в 10 мл агара в чашечках Петри. Бактериальная ино- кула содержит примерно 10 колонилоб- разующих единиц. Рост бактерий наблюдают через 20 ч инкубирования при . МИК определяют как самую низкую концентрацию лекарства, предотвращающую видимый рост ба.ктерий.
5158828210
Из приведенных в табл. 2 и 3 ре- 7 дней подсчитывают число мертвых зультатов видно, что предлагаемые сое- мьшей и в соответствии с методом динения 1 - 4 показывают очень высо- Бейренса-Кэрбера подсчитывают значекую бактерицидную активность против грамположительных и грамотрицательных бактерий.
Пример 12. Действие in vitro против общих инфекций на мьшах (табл. 4 и 5) .
Каждое соединение растворяют в деионизированной воде. Каждый раствор вводят перорально мышам, зараженным испытуемым организмом, в нижеприведенных условиях и на основе анализа проб подсчитывают значение средней эффективности дозы (ЭД), приведенные в таблицах цифры соответствуют значениям ЭЦ (.мг/кг) в пересчете на свободное основание.
Экспериментальные условия, Мыши - мужские особи () весом около
20 г.;
Заражение: Staphylococcus aureus 50774 (внутривенное заражение клеток на мьпиь, суспендированньк в солевом растворе); Streptococcus руо- genes А65 (заражение внутрибрюшинно 3 1.0 клеток на мышь, суспендированных в сердечно-мозговом бульоне для вливаний)V Pseudomonas aeruginosa 12 (заражение внутрибрюшинной 5 -10 клеток на мьшгь, суспендированных в трип- тосоевом бульоне с 4% муцина).
Медикация. Дважды, сразу же после заражения и спустя 6ч.
Наблюдения проводят в течение 14 дней для Staphylococcus aureus 50774 и в течение 7 дней для других организмов.
10
ние средней летальной дозы ДЦ гр (мг/кг) Полученные результаты приведены в табл. 6.
Таким образом, соединения 1 - 4 имеют низкую оральную токсичность.
Пример 14. Раствор соединения 1 растворяют в солевом или физиологическом растворе с эквимолярным количеством гидроокиси натрия или раствор соединения В в солевом или 15 физиологическом растворе дают орально или внутривенно самцу мышей (ddY) весом примерно 30 г в дозе до 5 мг/кг. Мочу и фекалии собирают в емкости для продуктов метаболизма в течение 24 ч.
Концентрацию соединений в отобранных образцах определяют тонкослойным чашечно-пластинчатым методом с использованием в качестве организма-индикатора Escherichia coli Кр.
В табл. 7 приведена уринарная и фекальная секреция мышей.
20
25
30
35
40
Таким образом, высокий общий выход соединения 1 указывает на его хорошую метаболическую устойчивость. Секреция в моче соединения 1 очень хорошая Это указывает на хорош то абсорбируе- мость соединения 1 при пероральном введении. Уровень в моче (34,3 г/мл) соединения 1 примерно в 22-2700 раз превышает значения МИК (0,0125 - 1,56 г/мл) против различных бактерий, приведенных в табл. 1.
Кроме того, соединение 1 превосходит соединение В по метаболической устойчивости и абсорбируемости при пероральном введении.
Таким образом, предлагаемые соединения 1, 3 и-4 оказывают сильное терапевтическое действие на общие инфекции, вызываемые грамположительными и грамотрицательными бактериями. В частности соединения 1 оказывают лучшее действие на общую инфекцию, вызываемую Р. aeruginosa по сравнению с соединением А, а соединение 1 показывает лучшее действие против общего заражения, вызываемого S .aureus 50774 по сравнению с соединением В.
Пример 13 (острая токсичность) . Мужским особям мышей (ddy-S) вводят перорально раствор, содержа- щий каждый в различной концентрации предлагаемое соединение в дозировке 0,1 мл на 10 г веса тела. Спустя
0
ние средней летальной дозы ДЦ гр (мг/кг) . Полученные результаты приведены в табл. 6.
Таким образом, соединения 1 - 4 имеют низкую оральную токсичность.
Пример 14. Раствор соединения 1 растворяют в солевом или физиологическом растворе с эквимолярным количеством гидроокиси натрия или раствор соединения В в солевом или 5 физиологическом растворе дают орально или внутривенно самцу мышей (ddY) весом примерно 30 г в дозе до 5 мг/кг. Мочу и фекалии собирают в емкости для продуктов метаболизма в течение 24 ч.
Концентрацию соединений в отобранных образцах определяют тонкослойным чашечно-пластинчатым методом с использованием в качестве организма-индикатора Escherichia coli Кр.
В табл. 7 приведена уринарная и фекальная секреция мышей.
0
5
0
5
0
с
0
5
Таким образом, высокий общий выход соединения 1 указывает на его хорошую, метаболическую устойчивость. Секреция в моче соединения 1 очень хорошая Это указывает на хорош то абсорбируе- мость соединения 1 при пероральном введении. Уровень в моче (34,3 г/мл) соединения 1 примерно в 22-2700 раз превышает значения МИК (0,0125 - 1,56 г/мл) против различных бактерий, приведенных в табл. 1.
Кроме того, соединение 1 превосходит соединение В по метаболической устойчивости и абсорбируемости при пероральном введении.
Как видно из табл. 2-7, предлагаемые соединения оказывают сильное терапевтическое де 1ствие на экспериментальные инфекции, вызываемые грамположительными и грамотрицательными бактериями, при низкой токсичности. Соединения также обладают хорошей аб- сорбируемостью и метаболической устойчивостью. Более того, соединения обладают низкой цитотоксичностью и- при парентеральном введении вызывают низкое местное раздражение. Соответственно, указанные соединения применимы в качестве бактерицидных средств для перорального введения или введения в виде инъекций.
Формула изобретения
Способ получения производного хино- пйна общей формулы
соон
где Z - галоид , i RI группа формул
или
PM
H2N
где RI - водород или низший алкил, Rj - низший алкил; R. - водород или низший алкил, или его фармацевтически приемлемого сложного эфира, или фармацевтически приемлемых солей указанного производного или его эфира, отличающий с я тем, что соединение общей формулы
;
F NH Л
где Y - низший алкил;
X - галоген;
R и Z имеют указанные значения, подве|ргают. -циклизации в присутствии 6сн.Ьва нйя, в случае необходимости сложный эфир гидролизуют до карбоновой кислоты и при желании превращают полученное соединение в его фармацев тически приемлемую соль.
Таблица 1
г О
Т а
лица





Изобретение относится к гетероциклическим соединениям , в частности к получению производного хинолина общей ф-лы CF = CR 1-CF = CZ-C = C-N(Δ)-CH=C(COOH)-C(O), где Z - атом галоида, R 1 - группа ф-л -N-CH 2-CHR 4-NH-CH 2-CH 2 или -N-CH 2-CH(NH 2)-CH 2-CH 2, где R 2 - H или низший алкил, R 3 - низший алкил, R 4 - H или низший алкил, или его фармацевтически приемлемого сложного эфира, или фармацевтически приемлемых солей указанного производного или его эфира, обладающих бактерицидной активностью. Получение ведут циклизацией соединения ф-лы @ где Y - низший алкил, X - атом галогена
R 1 и Z - указано выше, в присутствии основания. В случае необходимости сложный эфир гидролизуют до карбоновой кислоты и при желании превращают полученное соединение в его фармацевтически приемлемую соль. 7 табл.
Штамм
Действие in vitro против общих инфекций на мьшах соединения
Таблица 5
Действие ин виво против общих инфекций на мьппах Штаммсоединения
.ZZIIZII
genes А65-12,5
uginosa 12,243,05
Таблицаб
СоединениеДЦ , мг/кг
Таблица
--- ---- ----------...
Путь вве-| Обозна- Моча Фекалии Общее дения | чение
- «. .. .- i в.-.м--ей. в--в
К 24ТЗloi
В63,614. 77,6
П/о . 34,3144
В33,422,3 85,7
К24,3138
В35,48,96 44,4
П/о 05341
В6,3926,3 32,7
ечание. К- концентрация (г/мл); В - выход С%) .
| УСТРОЙСТВО для ЗАМЕНЫ НИЖНЕГО МАЛОГО КОНУСА С ЧАШЕЙ ГАЗОВЫМ ЗАТВОРОМ ЗАСЫПНОГО АППАРАТА ДОМЕННОЙ ПЕЧИ | 0 |
|
SU168737A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
