Область изобретения
Настоящее изобретение относится к области фармацевтического синтеза, в частности к синтезу противораковых препаратов.
Предшествующий уровень техники
В настоящее время рак является наиболее распространенным заболеванием в мире, а также имеет самый высокий уровень смертности. Он представляет собой серьезную угрозу жизни и здоровью людей, и во многом все еще остается загадкой для исследователей. В начале 1970-х годов ученые ничего не знали о причинах возникновения раковых опухолей. Искать лекарства от рака приходилось «вслепую». В 1966 году Национальный институт рака (США) начал спонсировать проект по скрининговому тестированию химических препаратов. Тысячи препаратов, которые могли оказаться эффективными в борьбе против рака, проверяли один за другим. В итоге были разработаны многие препараты, используемые при химиотерапии - амино-метил-фолиевая кислота, циклофосфамид, цисплатин, фторурацил, паклитаксел и другие. Хотя подобные цитотоксические препараты способны уменьшить или полностью устранить некоторые патологические проявления болезни, продолжительное лечение часто приводит к возникновению значительной мультилекарственной устойчивости, которая снижает эффективность лечения. Хуже всего, что со временем раковые клетки приобретают устойчивость к подобным лекарствам, и лечить рак таким способом уже может оказаться невозможным.
В 1971 году доктор Фолкман впервые высказал соображения теории ангиогенеза. Он выдвинул смелую гипотезу, которая характеризовалась следующими положениями: 1) рост опухоли опирается на ангиогенез; 2) опухоль может стимулировать формирование кровеносных сосудов; 3) опухоль способна выделять определенное химическое вещество, которое способствует прорастанию кровеносного сосуда к опухоли и образованию новых ответвлений. Рост опухоли зависит от количества неопластических и эндотелиальных клеток кровеносных сосудов опухоли. Жизнедеятельность этих двух типов клеток взаимозависима. Изменение числа клеток одного типа неизбежно приведет к соответствующему изменению числа клеток второго типа. Поэтому для лечения рака будет эффективным любое лекарство, которое подавляет либо неопластические, либо эндотелиальные клетки кровеносных сосудов опухоли. Для подавления неопластических клеток применяют химиотерапию, в основном с использованием цитотоксических препаратов, а для подавления эндотелиальных клеток применяется антиангиогенную терапию, которая в последнее время стала самым примечательным методом лечения. Согласно гипотезе доктора Фолкмана, рост и распространение опухоли опирается на реваскуляризацию. Следовательно, если подавить образование кровеносных сосудов опухоли, то это приведет к гибели клеток опухоли из-за недостаточного поступления крови и кислорода; таким образом, рост опухоли замедлится, а ее распространение будет сдерживаться. Доказано, что для роста опухоли необходим ангиогенез новых кровеносных сосудов. Если опухоль имеет объем менее 1-2 куб. мм, то она может выжить, получая питательные вещества из соседних тканей посредством осмоса. При этом рост опухоли будет происходить медленно, так как для дальнейшего ее развития требуется ангиогенез для предоставления необходимого питания. Ангиогенез опухоли - это динамичный многостадийный процесс, в который входят: отвод перицитов от наружной поверхности капилляров, выработка протеаз активированными эндотелиальными клетками, расщепление внеклеточного матрикса (ВКМ), который окружает существующие сосуды, миграция эндотелиальных клеток по направлению к стимулу ангиогенеза и их интенсивное размножение, образование трубкообразных структур, сращение образующихся сосудов и начало кровотока. Этот процесс регулируется как фактором внутренней секреции базального нерва, так и факторами роста, выражаемыми клетками опухоли и клетками матрикса опухоли.
Антиангиогенная терапия поражает эндотелиальные клетки сосудов, окружающих различные опухоли. Между сосудистыми эндотелиальными клетками опухоли и нормальными сосудистыми эндотелиальными клетками практически нет заметных отличий, за исключением более высокого темпа деления опухолевых эндотелиальных клеток. Нормальные сосудистые эндотелиальные клетки отличаются большим временем существования и более стабильным генотипом. Среди всех типов клеток, за исключением нервных, эндотелиальные клетки имеют наибольшую продолжительность жизни. Во взрослой стенке сосуда лишь 0,01% эндотелиальных клеток находится в состоянии деления. Сосудистые эндотелиальные клетки опухоли некоторым образом отличаются от нормальных эндотелиальных сосудистых клеток. Так, скорость размножения опухолевых эндотелиальных сосудистых клеток в среднем в 50 раз больше, чем у нормальных клеток, и, следовательно, опухолевые эндотелиальные клетки менее зрелые и развитые. Значит, факторы угнетения сосудов будут проявлять сравнительную специфичность в отношении кровеносных сосудов опухоли, тогда как на сосуды здоровых тканей заметного воздействия оказано не будет. Антиангиогенная терапия имеет несколько важных преимуществ по сравнению с традиционной химиотерапией, которая непосредственно атакует клетки опухоли, а именно: 1) антиангиогенные лекарства обладают хорошей специфичностью, так как ангиогенез возникает при образовании опухоли. Антиангиогенные лекарства напрямую атакуют неоваскулярные эндотелиальные клетки, что приводит к гибели тысяч клеток опухоли из-за недостатка кислорода, при условии, что будет уничтожен какой-либо сосуд, обеспечивающий зарастание. Исследования показали, что 99% клеток опухоли погибают в участке ишемии в течение двух часов после применения антиангиогенных лекарств. 2) Тот факт, что сосудистые эндотелиальные клетки непосредственно связаны с кровотоком, позволяет лекарствам воздействовать на эти клетки напрямую. Антиангиогенные лекарства не вызывают прямую гибель клеток опухоли, они лишь изменяют образование и темп роста этих клеток. Терапевтически эффективная доза антиангиогенных лекарств настолько мала, что составляет всего 1/10-1/100 от максимально переносимой дозы (МПД). Высокая терапевтическая эффективность малых доз препарата избавляет пациента от побочных эффектов, сопровождающих лечение радиотерапией и химиотерапией. 3) Экспрессия гена эндотелиальных клеток достаточно стабильна, поэтому развитие устойчивости клеток к лекарствам маловероятно. Скорость деления сосудистых эндотелиальных клеток опухоли в десятки раз выше, чем у клеток здоровых тканей. Ингибиторы ангиогенеза селективно действуют на сосудистые эндотелиальные клетки опухоли, которые быстро размножаются, тогда как для клеток здоровых тканей эффект оказывается минимальным. Таким образом, ингибиторы ангиогенеза имеют ряд значительных преимуществ.
Кустарники и деревья семейства комбретовых (combretaceae), произрастающие в тропическом и субтропическом поясе, широко используются в традиционной медицине. 25 видов рода комбретум используются в Африке и Индии для лечения болезни Хансена и рака. В конце 1970-х годов, после широкомасштабного скрининга, ученые Национального института рака (США) обнаружили, что растения рода комбретум (combretum) способны активно подавлять клетки лимфолейкоза Р-388. В начале 1980-х возник широкий интерес к изучению этих растений. В этот период доктор Дж. Роберт Петтит, директор Института по исследованию рака Аризонского государственного университета, и его коллеги смогли выделить комбретастатины из африканской ивы (Combretum caffrum), которую племена зулусов используют в качестве растительного лекарства, а также для окраски копий. В статье, опубликованной в журнале Journal of Canadian Chemistry, доктор Петтит заявил, что кора этого дерева обладает противораковым действием. Впоследствии из коры не только выделили и идентифицировали множество высокоактивных соединений, но и провели исследования по определению механизмов фармакологического действия и модификаций их структуры. Возглавляемые доктором Петтитом ученые первыми начали углубленные исследования в этой области. Эта группа ученых изучала растения рода комбретум, в результате чего удалось изолировать серию активных фенантренов, стильбенов и дибензилов. Особенно важным открытием стало обнаружение мощных ингибиторов роста клеток и тубулина - комбретастатинов А-1 и А-4 (далее обозначаются как КА-4 и КА-1, представлены формулой II). Оба препарата оказались чрезвычайно сильными ингибиторами полимеризации тубулина (US 5,561,122; WO 9935150).
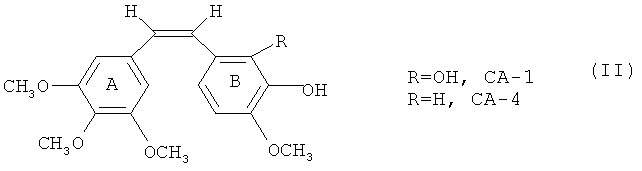
Изучение комбретастатинов вызывает большой интерес, хотя они были открыты сравнительно недавно. Дело не только в повышенной противораковой активности этих препаратов, важно то, что они представляют собой маленькие природные вещества, способные подавлять полимеризацию тубулина и ангиогенез. Изучение механизма действия КА-4 выявило, что А-кольцо и В-кольцо связываются с α-тубулином и β-тубулином, соответственно, что приводит к разрушению эндотелиальных клеток кровеносных сосудов опухоли. КА-4 ингибирует полимеризацию тубулина и, таким образом, атакует новые кровеносные сосуды, подавляя рост опухоли - ведь кровеносные сосуды доставляют кислород и питательные вещества, необходимые для роста опухоли.
КА-4 способен проникать в эндотелиальные клетки, которые выстилают кровеносные сосуды опухоли. Эндотелиальные клетки опухолей незрелые, и поэтому они особенно чувствительны к воздействию комбретастатинов, в отличие от эндотелиальных клеток здоровых тканей. Проникнув в эндотелиальные клетки, комбретастатин разрушает внутренний скелет клеток, меняя их форму от плоской к круглой, и эффективно закупоривает питающие опухоль капилляры, что в итоге создает обширный ишемический участок опухоли и приводит к ее регрессии. По данным предварительных экспериментов и клинических испытаний, ни один из классических противораковых препаратов на такое пока не способен. Эта теория уже подтвердилась во время I фазы клинических испытаний КА-4. У каждого ракового больного в течение 4-6 часов после введения КА-4 заметно уменьшился кровоток, и погибло более 95% клеток опухоли. КА-4 может использоваться не только в качестве системного средства для лечения рака, но и как препарат местного действия для лечения различных глазных болезней, в том числе возрастной дистрофии желтого пятна и пролиферативной диабетической ретинопатии. Возможно также, что КА-4 перспективен в борьбе с псориазом и артритом. Кроме того, КА-4 стимулирует иммунитет, что может пригодиться в лечении болезней, связанных со СПИДом (WO 02058535; US 6773702).
Недавно было сделано волнующее открытие - КА-4 показал свою эффективность в разрушении сосудистой сети опухоли, действуя в качестве целевого препарата васкулярной направленности (Thorpe РЕ. «Clin Cancer Res.», 15 января 2004 г., 10(2):415-27; West CM, Price P. «Anticancer Drugs», март 2004 г., 15(3):179-87; Young SL, Chaplin DJ. «Expert Opin Investig Drugs», сентябрь 2004 г., 13(9):1171-82). Так что разработка новых аналогов КА-4 становится все более значимой задачей. Компания Oxigene Inc., например, разработала серию функционализированных производных стильбена (US 6,919,324).
Хорошо известно, что введение фтора в биоактивную молекулу приведет к изменению ее биологической активности, однако неясно, будет ли это увеличение или снижение активности.
Так, синтезированный компанией Sigma-Tau Industrie Farmaceutiche Riunite S.P.A фторокомбретастатин, в который ввели один или два атома фтора в двойной связи, в своей активности не показал никаких отличий от КА-4.
Поэтому возникает актуальная задача нахождения новых производных комбретастатина с более высокой активностью.
Раскрытие изобретения
Целью настоящего изобретения является создание производных фторалкоксикомбретастатина по формуле I.
Еще одной целью изобретения является создание способа для получения соединений по формуле I.
Третьей целью изобретения является создание фармацевтической композиции, содержащей соединения по формуле I.
Четвертой целью изобретения является подготовка соединений формулы I к использованию в медицинских целях.
Согласно первой особенности изобретения, предлагаются соединения по формуле I:
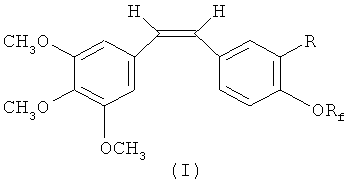
где:
Rf - алкильная группа, содержащая от 1 до 8 атомов углерода, в которой от 1 до 17 атомов водорода замещены 1-17 атомами фтора;
R - аминогруппа, замещенная аминогруппа, гидроксильная группа, нитрогруппа, галогеновая, алкоксильная, фосфатная группа либо боковая цепь аминокислоты или ее фармацевтически приемлемые соли.
Желательно, чтобы Rf=-CH2F, -CHF2, -CnF2n+1, -CH2CnF2n+1, -CHFCnF2n+1 или -CH2CHFCnF2n+1, где n - целое число от 1 до 3.
В предпочтительном варианте осуществления изобретения Rf и R представляют собой один из следующих вариантов:
(а) Rf - фторметил, R - гидроксильная группа;
(б) Rf - фторметил, R - аминогруппа или замещенная аминогруппа;
(в) Rf - фторметил, R - динатрийфосфат, фосфат аммония или внутренняя соль фосфорилхолина; либо
(г) Rf - фторметил, R - -NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, a m - целое число от 1 до 3.
В другом предпочтительном варианте осуществления изобретения Rf и R представляют собой один из следующих вариантов:
(а) Rf - фторэтил, R - гидроксильная группа;
(б) Rf - фторэтил, R - аминогруппа или замещенная аминогруппа;
(в) Rf - фторэтил, R - динатрийфосфат, фосфат аммония или внутренняя соль фосфорилхолина; либо
(г) Rf - фторэтил, R - -NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, а m - целое число от 1 до 3.
В еще одном предпочтительном варианте осуществления изобретения Rf и R представляют собой один из следующих вариантов:
(а) Rf представляет собой -CH2F, -CHF2, -CF3, -CH2CF3, -CH2CHF2 или -CF2CF3, а R представляет собой -ОН или -OPO3Na2; либо
(б) Rf представляет собой -CH2F, -CHF2, -CF3, -CH2CF3, -CH2CHF2 или -CF2CF3, а R представляет собой -NH2 или -NHCOCH(NH2)СН2ОН.
В другом предпочтительном варианте осуществления изобретения Rf=-CHF2, R=-OH.
В еще одном предпочтительном варианте осуществления изобретения Rf=CHF2, R=-OPO3Na2.
В еще одном предпочтительном варианте осуществления изобретения Rf=-CHF2, R=-NH2.
В еще одном предпочтительном варианте осуществления изобретения Rf=-CHF2, R=-NHCOCH(NH2)CH2OH.
В еще одном предпочтительном варианте осуществления изобретения Rf=-CH2CF3, R=-OH.
В еще одном предпочтительном варианте осуществления изобретения Rf=-CH2CF3, R=-OPO3Na2.
В еще одном предпочтительном варианте осуществления изобретения Rf=-CH2CF3, R=-NH2.
В еще одном предпочтительном варианте осуществления изобретения Rf=-CH2CF3, R=-NHCOCH(NH2)CH2OH.
Согласно второй особенности изобретения, предлагается способ подготовки соединений по формуле I, предусматривающий следующие шаги:
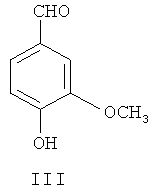
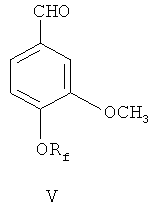
(1) 4-гидрокси-3-метоксибензальдегид III в присутствии катализатора межфазного переноса фторалкилируют посредством фторосодержащего агента, чтобы синтезировать 4-фторалкокси 3-метоксибензальдегид, представленный формулой V;
(2) С помощью дифенилфосфина лития деметилируют 4-фторалкокси-3-метоксибензальдегид V, чтобы синтезировать 4-фторалкокси-3-гидроксибензальдегид, представленный формулой VI;
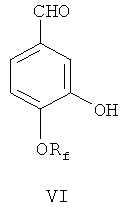
(3) Защищают гидроксильную группу 4-фторалкокси-3-гидроксибензальдегида VI, а затем 4-фторалкокси-3-гидроксибензальдегид VI с защищенной гидроксильной группой посредством реакции Виттига взаимодействует с илидом 3,4,5-триметоксибензилтрифенилфосфония, после чего из полученного соединения удаляют защиту для получения соединений по формуле I.
В другом предпочтительном варианте осуществления изобретения способ предусматривает следующие шаги:
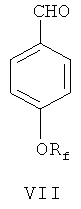
(а) 4-гидроксибензальдегид IV в присутствии катализатора межфазного переноса фторалкилируют посредством фторосодержащего агента, чтобы синтезировать 4-фторалкоксибензальдегид, представленный формулой VII;
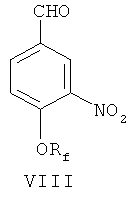
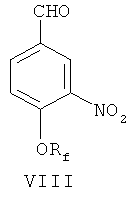
(б) 4-фторалкоксибензальдегид VII нитруют в 3-м положении фенильного кольца посредством азотной кислоты и уксусного ангидрида, чтобы синтезировать 4-фторалкокси-3-нитробензальдегид, представленный формулой VIII;
(в) 4-фторалкокси-3-нитробензальдегид VIII взаимодействует с илидом 3,4,5 - триметоксибензилтрифенилфосфония посредством реакции Виттига для получения соединений по формуле I.
В еще одном предпочтительном варианте осуществления изобретения упомянутый фторосодержащий агент представляет собой фторгалогенметан или фторалкилсульфонат.
Согласно третьей особенности изобретения, предлагается фармацевтическая композиция, содержащая эффективное количество соединений по формуле I в фармацевтически приемлемом носителе.
В другом предпочтительном варианте осуществления изобретения, упомянутые фармацевтические композиции могут вводиться перорально или внутривенно в виде одной из следующих лекарственных форм: лиофилизированный порошок, порошок, гранулы, таблетки, капсулы, сироп, суппозитории, препарат для инъекций, эмульсия, настойка, суспензия или раствор.
Согласно четвертой особенности изобретения, предусматривается использование соединений по формуле I для изготовления ингибитора, связывающегося с тубулином.
Согласно пятой особенности изобретения, предусматривается использование соединений по формуле I для изготовления препаратов для лечения заболеваний, вызванных нездоровым ангиогенезом.
В другом предпочтительном варианте осуществления изобретения, соединения по формуле I используются для борьбы с растущими опухолями и метастазированием, вызываемыми нездоровым ангиогенезом. В число упомянутых опухолей входят, в том числе, следующие: рак легкого, немелкоклеточный рак легкого, печеночно-клеточный рак, аденокарцинома поджелудочной железы, карцинома желудка, остеокарцинома, карцинома пищевода, рак груди, рак предстательной железы, рак яичек, карцинома толстой кишки, рак яичников, карцинома мочевого пузыря, рак шейки матки, меланокарцинома, плоскоклеточная карцинома, базально-клеточная карцинома, аденокарцинома, карцинома потовых желез, карцинома сальных желез, папиллярная карцинома, папиллярные аденокарциномы, цистаденокарцинома, цистокарцинома, медуллярный рак, бронхогенный рак, рак костных клеток, эпителиальная карцинома, карцинома желчных протоков, эмбриональный рак, хориокарцинома, семинома, опухоль Вильмса, олигодендроглиома, астроцитома, медуллобластома, краниофарингиома, эпендимома, пинеалома, гемангиобластома, акустическая невринома, менингиома, нейробластома, бластома зрительного нерва, ретинобластома, нейрофиброма, фибросаркома, фибробластома, фиброма, фиброаденома, хондрофиброма, фиброцистома, фибромиксома, фибро-строма, фибромиксосаркома, фибропапиллома, миксосаркома, опухоль синовиальной сумки, миксоэнходрома, миксохондросаркома, миксохондрофибросаркома, миксоаденома, миксобластома, липосаркома, липома, липоаденома, рак липобластов, липохондрома, липидная фиброма, липоангиома, миксолипома, хондросаркома, хондрома, хондромиома, хордома, хориоаденома, хориоэпителиома, хориобластома, остеосаркома, остеобластическая саркома, остеохондрофиброма, остеохондросаркома, остеохондрома, остеоцистома, остеодентинома, остеофиброма, фибросаркома костей, ангиосаркома, ангиома, ангиолипома, гематальная хондрома, ангиобластома, ангиокератома, ангиоглиома, гемангиоэндотелиома, гемангиофиброма, ангиомиома, ангиолипома, гематальная лимфангиома, ангиолиполейомиома, ангиомиолипома, гематальная мионеврома, гематальная миксома, ангиоретикулоэндотелиома, лимфангиосаркома, лимфогранулематоз, лимфангиома, лимфома, лимфомиксома, лимфосаркома, лимфангиофиброма, лимфоцитома, лимфоэпителиома, лимфобластома, эндотелиальная карцинома, эндобластома, синовиома, синовиосаркома, мезотелиома, мезоцитома, опухоль Юинга, лиомиома, лейомиосаркома, лейомиобластома, лиомиофиброма, рабдомиома, рабдомиосаркома, рабдомиомиксома, острый лимфоцитарный лейкоз, острый миелоцитарный лейкоз (миелобластный, промиелоцитарный, миеломоноцитарный, моноцитарный и эритро-лейкозы), хронический лейкоз (хронический миелоцитарный [гранулоцитарный] лейкоз и хронический лимфоцитарный лейкоз), истинная полицитемия, лимфома (болезнь Ходжкина и неходжкинские лимфомы), множественная миелома.
В другом предпочтительном варианте осуществления изобретения, соединения по формуле I используются для лечения других родственных болезней, вызываемых патологическим ангиогенезом, в число которых входят, в том числе, следующие заболевания: ревматоидный артрит, диабетическая ретинопатия, ретинопатия недоношенных, тромбоз ретинальных вен, псориаз, розацеа, саркома Капоши, аллергический кератит, эпидемический кератоконъюнктивит, неоваскулярная глаукома, бактериальные язвы, грибковые язвы, инфекции простого герпеса, инфекции опоясывающего лишая, простейшие инфекции, микобактериальные инфекции, полиартериит, саркоидоз, склерит, приливы крови, болезнь Шегрена, системная красная волчанка, синдром приобретенного иммунодефицита (СПИД), сифилис.
Таким образом, настоящее изобретение предлагает новые производные комбретастатинов, обладающие улучшенной биологической активностью.
Краткое описание чертежей
На фиг.1 изображен ход синтеза фторметоксикомбретастатина;
На фиг.2 изображен ход синтеза фторэтоксикомбретастатина;
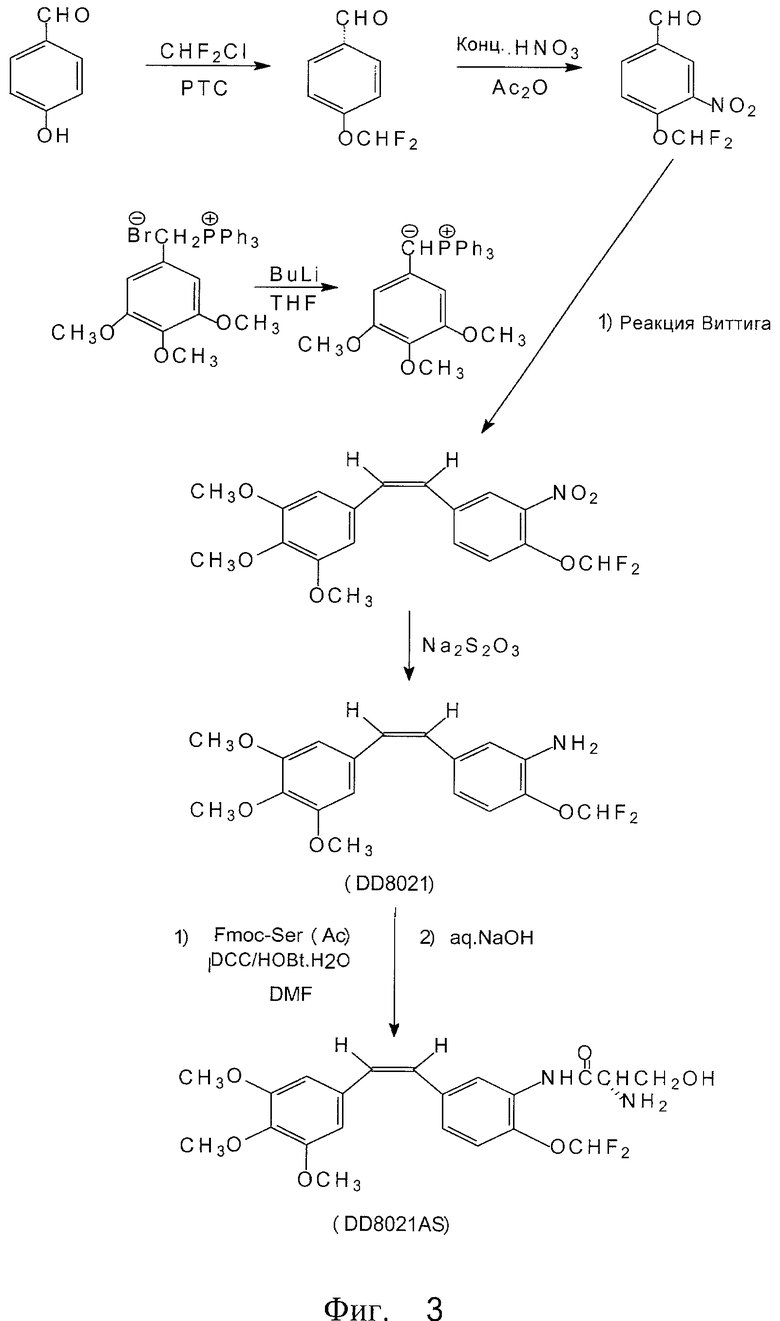
На фиг.3 изображен ход синтеза аминокислотных производных фторметоксикомбретастатина;
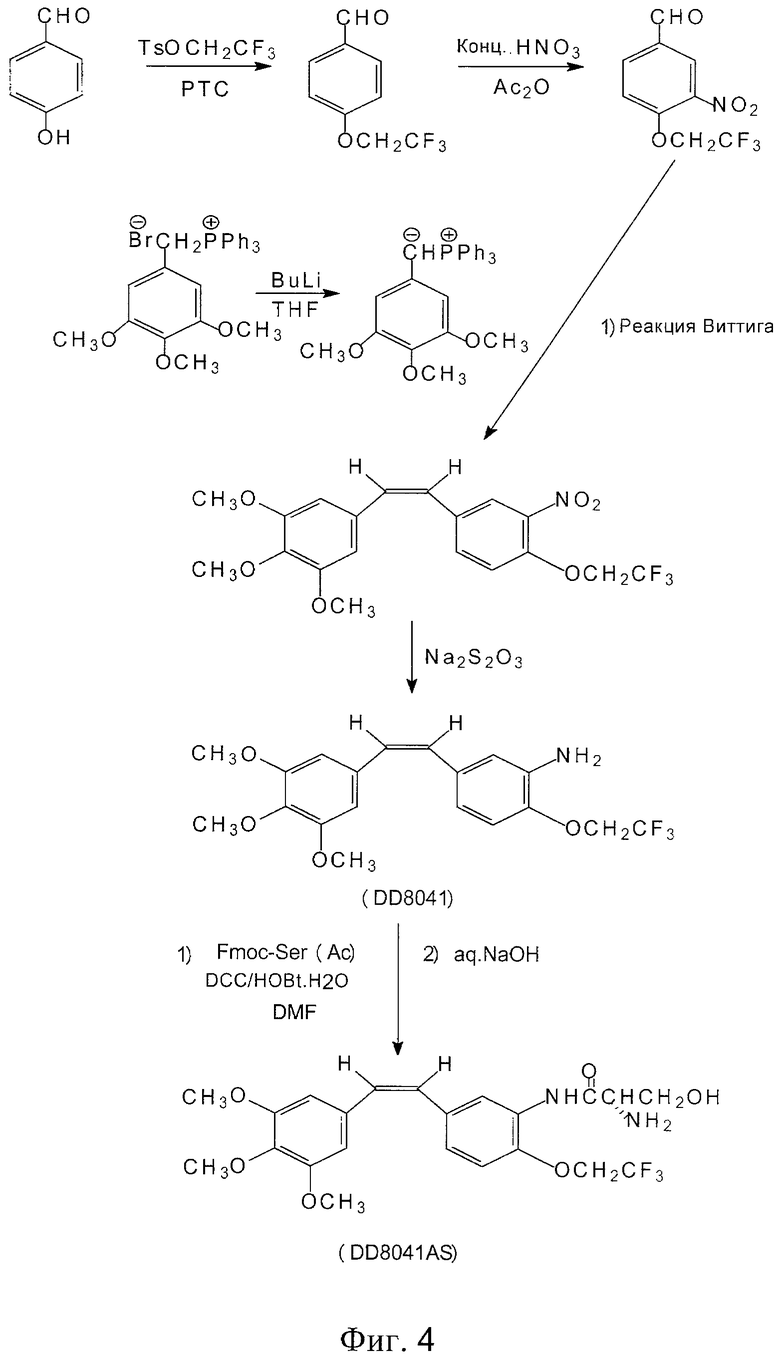
На фиг.4 изображен ход синтеза аминокислотных производных фторэтоксикомбретастатина.
Здесь:
РТС (КМП) - катализатор межфазного переноса;
Cat. (Кат.) - катализатор реакции Виттига;
Ph2PLi - дифенилфосфин лития;
THF (ТГФ) - тетрагидрофуран;
TFA (ТФУ) - трифторуксусная кислота;
iPr2EtN - диизопропилэтиламин;
(PhCH2)2P(O)H - дибензилфосфат;
TMBS (ТМБС) - триметилбромосилан;
Fmoc-Ser(Ac) - производное N-α-9-фторэнилметоксикарбонил серина;
DCC (ДЦК) - циклогексилкарбодиимид;
HOBt (ГОБТ) - 1-гидроксибензотриазол; DMF (ДМФ) - диметилформамид;
водн. HCl - слабый водный раствор соляной кислоты;
водн. NaOH - слабый водный раствор гидроксида натрия;
конц. HCl - концентрированная соляная кислота;
конц. HNO3 - концентрированная азотная кислота.
Лучший вариант осуществления изобретения
В ходе проведения интенсивных разносторонних исследований неожиданно обнаружилось, что 4-е положение В-кольца природного вещества комбретастатина является активным участком, и что в этот участок можно ввести фторалкоксильную группу, чтобы улучшить целевую активность в отношении сосудов опухоли.
В вышеупомянутое природное вещество комбретастатин, в 4-м положении ароматического В-кольца, успешно ввели фторалкоксильную группу посредством ключевой реакции деметилирования с селективным применением дифенилфосфина лития.
По сравнению с КА-4 новые соединения по формуле I усиливают подавление полимеризации тубулина, что может быть использовано для лечения патологического состояния, вызванного нездоровым ангиогенезом.
Под производными комбретастатина здесь понимаются соединения, представленные формулой II.
Соединения
Настоящее изобретение предлагает новые производные комбретастатина, которому в 4′ положении ароматического В-кольца ввели фторалкоксильную группу; эти производные представлены формулой I
где:
Rf - алкильная группа, содержащая от 1 до 8 атомов углерода, в которой от 1 до 17 атомов водорода замещены 1-17 атомами фтора;
R - аминогруппа, замещенная аминогруппа, гидроксильная группа, нитрогруппа, галогеновая, алкоксильная, фосфатная группа либо боковая цепь аминокислоты или ее фармацевтически приемлемые соли.
Желательно, чтобы Rf=-CH2F, -CHF2, -CnF2n+1, -CH2CnF2n+1, -CHFCnF2n+1 или -CH2CHFCnF2n+1, где n - целое число от 1 до 3.
Предпочтительными соединениями по формуле I в настоящем изобретении являются фторметоксикомбретастатин или его аминокислотные производные, представленные формулой I, в которой Rf=CH2F, -CHF2 или -CF3, a R=-ОН, -OPO3Na2, -NH2 или -NHCOCH(NH2)CH2OH;
Желательно, чтобы Rf=-CH2F, R=-OH, -OPO3Na2, -NH2 или -NHCOCH(NH2)CH2OH.
В другом предпочтительном варианте осуществления изобретения, предпочтительными соединениями по формуле I являются фторэтоксикомбретастатин или его аминокислотные производные, представленные формулой I, в которой Rf=-CH2CF3, -CH2CHF2 или -CF2CF3, a R=-ОН, -OPO3Na2, -NH2 или -NHCOCH(NH2)CH2OH.
Производные фторалкоксикомбретастатина настоящего изобретения способны образовывать фармацевтически приемлемые соли присоединения основания с неорганическими или органическими основаниями. В число упомянутых неорганических оснований входят, в том числе, гидроксид калия и гидроксид аммония, а в число упомянутых органических оснований, в том числе, алифатические амины (например, триэтиламин), гидроксиамины (например, этаноламин), аминокислоты (например, гистидин) и аминогликозиды (например, неоамин).
Производные фторалкоксикомбретастатина настоящего изобретения также способны образовывать фармацевтически приемлемые соли присоединения кислоты с неорганическими или органическими кислотами. В число упомянутых неорганических кислот входят, в том числе, соляная, серная и фосфорная кислота, а в число упомянутых органических кислот, в том числе, щавелевая, фумаровая, малеиновая, яблочная, лимонная, винная и глютаминовая кислоты.
Подготовка соединений
Настоящее изобретение предлагает способ подготовки соединений по формуле I, предусматривающий следующие шаги:
4-гидрокси-3-метоксибензальдегид фторалкилируют в присутствии катализатора межфазного переноса, а затем селективно деметилируют его посредством последовательного применения дифенилфосфина лития для получения ряда новаторских фторированных производных алкоксибензальдегида. Вышеупомянутое соединение нитруют, восстанавливают, вводят гидроксильную защиту, а затем подвергают реакции Виттига, удаляют защиту, фосфатируют, соединяют с аминокислотами и т.д., с целью получения ряда производных фторалкоксикомбретастатина.
Синтез производных фторалкоксибензальдегида
4-фторалкокси-3-метоксибензальдегид (V) или 4-фторалкоксибензальдегид (VII) получают из 4-гидрокси-3-метоксибензальдегида (III) или 4-гидроксибензальдегида (IV), применяя фторалкилирующий агент в присутствии неорганического основания и катализатора межфазного переноса.
В качестве упомянутого фторалкилирующего агента используют фторгалогеналкан или фторалкилсульфонат, желательно Фреон (F22) или фторалкил-п-толуолсульфонат. В качестве упомянутых неорганических оснований используют гидроксид или один и более карбонатов, желательно гидроксид калия и/или карбонат калия. В качестве упомянутого катализатора межфазного переноса используют четвертичные соли аммония, четвертичные соли фосфония, краун-эфир или полиэтиленгликоль (ПЭГ), желательно бензилтриэтиламмоний хлорид, тетрабутиламмоний бисульфат (ТБАБС), 18-краун-6, дифенил-18-краун-6, дициклогексил-18-краун-6 эфиры или ПЭГ-400. Формильную группу 4-фторалкокси-3-метоксибензальдегида (V) защищают с помощью соединений гликоля, после чего метоксил в 3-м положении селективно деметилируют посредством дифенилфосфина лития для получения 4-фторалкокси-3-гидроксибензальдегида VI. 4-фторалкоксибензальдегид VII нитруют в мета-положении с помощью концентрированной азотной кислоты в присутствии ацетил-ангидрида в качестве растворителя, для получения 3-нитро-4-фторалкоксибензальдегида VIII.
Синтез производных фторалкоксикомбретастатина
В присутствии катализатора органического основания 4-фторалкокси-3-гидроксибензальдегид VI взаимодействует с трифенилметил хлоридом для получения 3-трифенилметокси-4-фторалкоксибензальдегида.
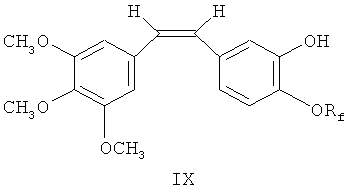
3,4,5-триметоксибензилтрифенилфосфин бромид превращают в соответствующий фосфорный илид с помощью н-бутиллития, а затем этот фосфорный илид взаимодействует с вышеупомянутым 3-трифенилметокси-4-фторалкоксибензальдегидом посредством реакции Виттига для получения фторалкоксистильбеновых производных. После этого удаляют защиту тритильной группы посредством совместного воздействия концентрированной соляной и трифторуксусной кислотами для получения производных 3′-гидроксифторалкоксикомбретастатина IX.
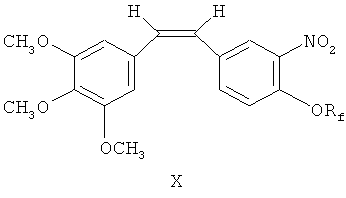
Аналогично, 4-фторалкокси-3-нитробензальдегид VIII взаимодействует с вышеупомянутым фосфорным илидом посредством реакции Виттига для получения производных 3′-нитрофторалкоксикомбретастатина X.
Синтез фосфатных или аминокислотных производных фторалкоксикомбретастатина
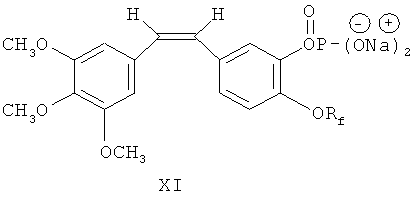
Как показано на фиг.1 и 2, гидроксильную группу в 3-м положении вышеупомянутого производного фторалкоксикомбретастатина IX превращают в фосфат натрия с использованием четыреххлористого углерода, ди(изопропил)этиламина, дибензилфосфоната, триметилсиланбромида и метилата натрия, в результате чего получают фторалкоксикомбретастатин фосфат XI.
Либо, как показано на фиг.3 и 4, нитрогруппу в 3-м положении вышеупомянутых производных фторалкоксикомбретастатина Х восстанавливают в аминогруппу с помощью восстанавливающих агентов. Желательно использовать в качестве восстанавливающих агентов хлорид олова (II), цинковую пыль/уксусную кислоту либо тиосульфат натрия. Затем восстановленный продукт обрабатывают аминокислотным производным N-α-9-фторэнилметоксикарбонила (FmocAA), циклогексилкарбодиимидом (DCC, ДЦК) и 1-гидроксибезнотриазолом (HOBt, ГОБТ), чтобы ввести в 3-м положении аминокислотную боковую цепь. Затем с продукта, имеющего в 3-м положении аминокислотную боковую цепь, удаляют защиту посредством гидроксида натрия до аминокарбоксамида, с целью получения аминокислотных производных фторалкоксикомбретастатина XII
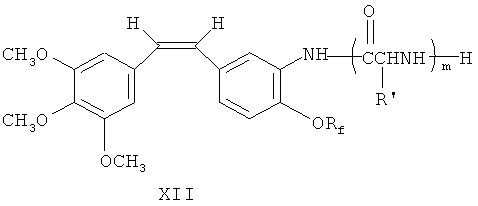
где R′=Н, фенильная группа или боковая цепь аминокислоты, m - целое число от 1 до 3.
Фармацевтические композиции
Предлагаемая фармацевтическая композиция содержит терапевтически эффективное количество соединений по формуле I в фармацевтически приемлемом носителе, при этом количество соединений по формуле I составляет 0,1 к 99% (вес/вес) композиций. Упомянутые фармацевтические композиции могут вводиться перорально или внутривенно в виде одной из следующих лекарственных форм: лиофилизированный порошок, порошок, гранулы, таблетки, капсулы, сироп, суппозитории, препарат для инъекций, эмульсия, настойка, суспензия или раствор.
Для внутривенного введения можно использовать композиции в виде лиофилизированного порошка, растворенного в физиологическом растворе или растворе глюкозы.
Для перорального приема можно использовать композиции в форме таблеток, настоек, капсул, суппозиториев, сиропа, гранул, эмульсий или растворов.
Дозировка активных ингредиентов зависит от способа введения, а также от стадии развития заболевания. При приеме предлагаемого соединения ежедневно, начиная с 0,5-500 мг/кг массы тела в день, наблюдался удовлетворительный терапевтический эффект. В одном предпочтительном варианте осуществления изобретения, предлагаемое соединение вводили пациенту в несколько приемов (по 2-4 раза в день), либо посредством лекарственной формы пролонгированного действия. Для большинства крупных млекопитающих суммарная дневная дозировка составляет от 1 до 100 мг. В эксперименте приемлемая форма препарата для перорального приема содержала 0,5-500 мг активного ингредиента, смешанного с твердым или жидким фармацевтически приемлемым носителем. Дозировку можно подобрать в соответствии с наиболее желательным результатом терапии. Например, в зависимости от конкретных условий лечения, предлагаемые композиции могут вводиться ежедневно в несколько приемов либо с равномерным уменьшением. Обычно клинически приемлемая пероральная доза для взрослого составляет от 1 до 1000 мг, а наиболее предпочтительным вариантом является интервал от 10 до 200 мг. Доза для применения иным способом для взрослого составляет от 0,1 до 100 мг, предпочтительно от 1 до 100 мг.
Предлагаемые настоящим изобретением фторалкоксикомбретастатины, подготовленные вышеописанными способами, при использовании в качестве агента васкулярной направленности могут вводиться как перорально, так и внутривенно. Дозировка активного ингредиента зависит от стадии развития заболевания. Ежедневная доза для взрослого человека составляет, как правило, от 1 до 3000 мг.
В предпочтительном варианте осуществления изобретения, предлагаемые соединения вводят перорально или внутривенно. В качестве твердых носителей можно использовать крахмал, лактозу, гидрофосфат кальция, кристаллическую целлюлозу, сахар или каолин; а в качестве жидких носителей - дистиллированную воду, полиэтиленгликоль, маннит, неионогенное поверхностно-активное вещество, пищевое масло (например, кукурузное, арахисовое или кунжутное), причем носители должны подходить для активного ингредиента и конкретной лекарственной формы. Также используются добавки, часто входящие в состав лекарств, например, ароматизаторы, красители, консерванты и антиоксиданты (например, витамин Е, витамин С, бутилгидрокситолуол, бутилгидроксианизол).
Как уже упоминалось в описании изобретения, при внутривенном введении препарата используют внутрибрюшные инъекции и капельные внутривенные влияния лиофилизированного порошка, растворенного в физиологическом растворе или растворе глюкозы. Приготовление лиофилизированного порошка производится общепринятым способом.
Предлагаемые композиции можно применять перорально, например, в виде таблеток или капсул. Для приготовления препаратов смешивают активный ингредиент с одной или несколькими фармацевтически приемлемыми добавками, такими как вспомогательные вещества, связывающие вещества, разрыхлители, скользящие вещества, красители, корригирующие вещества, антиоксиданты, консерванты и др., а затем формируют из получившейся смеси порошок, гранулы, таблетки, таблетки с оболочкой, пилюли, капсулы и т.д. В качестве подходящих вспомогательных веществ можно использовать любые из следующих: лактозу, кукурузный крахмал, сахарид, декстрозу, сорбитол, кристаллическую целлюлозу. В качестве подходящих связывающих веществ можно использовать любые из следующих: поливиниловый спирт, поливинилэфир, этилцеллюлозу, метилцеллюлозу, аравийскую камедь, трагант, желатин, шеллак, гидроксипропилцеллюлозу, оксипропилированный крахмал, поливинилпирролидон. В качестве подходящих разрыхлителей можно использовать любые из следующих: крахмал, агар, желатиновый порошок, кристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстран, пектин. В качестве подходящих скользящих веществ можно использовать любые из следующих: стеарат магния, тальк, полиэтиленгликоль, кремний, отвержденное растительное масло. В качестве подходящих красителей можно использовать любые красители, разрешенные к использованию в составе лекарств. В качестве подходящих корригирующих веществ можно использовать любые из следующих: порошок какао, ментол, масло перечной мяты, очищенный борнеол, корицу. Таблетки и гранулы можно при необходимости покрывать сахаром, желатином и т.д. Лекарственные препараты могут также содержать иные добавки, в том числе инертный разбавитель, консервант (например, сложные эфиры пара-оксибензойной кислоты, сорбиновую кислоту), антиоксидант (витамин Е, витамин С, бутилгидрокситолуол, бутилгидроксианизол), разлагающие вещества, клейкие вещества, газообразующие вещества, буфер, подслащивающие вещества, ароматизаторы, отдушки. Таблетки и пилюли также могут покрываться кишечнорастворимой оболочкой. В качестве антиоксидантов можно использовать витамин Е, витамин С, бутилгидрокситолуол, бутилгидроксианизол. В качестве консервантов можно использовать сложные эфиры пара-оксибензойной кислоты, сорбиновую кислоту. Таблетки и гранулы можно по желанию покрывать сахаром, желатином и т.п. В качестве жидких форм для перорального введения можно применять, например, эмульсии, сиропы, настойки, суспензии, растворы, содержащие часто используемый инертный разбавитель, например, воду.
Основным преимуществом настоящего изобретения является введение фторалкоксильной группы в 4-м положении ароматического В-кольца природного вещества комбретастатина, которое позволяет улучшить его целевую активность в отношении сосудов опухоли.
В дальнейшем изобретение поясняется подробным описанием примеров его осуществления. Эти примеры лишь поясняют изобретение, они не ограничивают его сущность приведенными конкретными вариантами. В экспериментальных способах, применяемых в нижеследующих примерах, все процедуры выполняются в стандартных условиях либо согласно указаниям производителя, а все части, проценты и доли указаны по массе, если прямо не указано иное.
Пример 1
Синтез 4-дифторметоксибензальдегида
В литровую колбу с четырьмя горловинами, оборудованную термометром, обратным холодильником, механической мешалкой и подводом газа, поместили 50 г (0,41 моль) 4-гидроксибензальдегида и 400 мл изопропилового спирта. Смесь перемешивали в течение 20 мин, после чего по капле добавили к ней 120 мл водного раствора, содержащего 5 г 18-краун-6 эфира и 106,3 г гидроксида натрия (2665 моль). Затем полученную смесь перемешивали в течение 30 мин, нагревали до 65°С, и в течение 5-6 часов через отверстие для подвода газа вводили хлордифторметан, отслеживая ход реакции посредством тонкослойной хроматографии. После завершения реакции смесь остудили до 15°С, а затем добавили 400 мл воды для заглушения реакции. Полученную смесь трижды экстрагировали эфиром (3×300 мл). Органический слой промыли водой с рН=7 и высушили безводным сульфатом магния. Эфир выпарили методом однократной перегонки, а остаток перегнали при пониженном давлении для получения 4-дифторалкоксибензальдегида (85-87°С/10 мм рт.ст.) с выходом 95%. Спектр ЯМР 1H (мд) δ: 9,87(1Н, с; -СНО); 7,70 (2Н, м; 2,6-ArH); 7,36 (1Н, т; J2 H-F=68 Гц; -CHF2); 6,96 (2Н, м; 3,5-ArH).
Пример 2
Синтез 4-дифторметокси-3-метоксибензальдегида
То же, что в Примере 1, только вместо 4-гидроксибензальдегида использовали 62,5 г (0,41 моль) 4-гидрокси-3-метокси-бензальдегида для получения 4-дифторметокси-3-метоксибензальдегида (117-120°С/10 мм рт.ст.) с выходом 93%. Спектр ЯМР 1Н (мд) δ: 9,85 (1Н, с; -СНО); 7,38 (1Н, т; J2 H-F=69 Гц; -CHF2); 7,27 (1Н, м; 6-ArH); 7,20 (1Н, м; 2-ArH); 6,83 (1Н, м; 5-ArH); 3,73 (3Н, с; -ОСН3).
Пример 3
Синтез 4-дифторметокси-3-гидроксибензальдегида
Шаг 1: в колбу с тремя горловинами в аргоновой атмосфере последовательно поместили 61 г (0,3 моль) 4-дифторметокси-3-метоксибензальдегида, 130 г (2,1 моль) этиленгликоля и 133 г (0,9 моль) триэтилортоформиата. Смесь нагревали с обратным холодильником, а затем в качестве катализатора добавили 1 мл эфирного раствора трехфтористого бора. Реакция смеси продолжалась в течение 24 часов, ход реакции отслеживали посредством тонкослойной хроматографии. Затем смесь остудили до комнатной температуры и добавили 200 мл 15% водного раствора гидроксида натрия. Полученную смесь экстрагировали 300 мл эфира. Экстракт промыли насыщенным рассолом, высушили безводным сульфатом магния и перегнали при пониженном давлении до получения желтого маслянистого продукта.
Шаг 2: в колбу поместили 200 мл 1,28 М тетрагидрофуранового раствора дифенилфосфина лития, а затем отдельными порциями добавили 50 г (0,2 моль) ранее подготовленного ацеталя. Затем полученную смесь в течение 3-4 часов перемешивали при комнатной температуре, отслеживая ход реакции посредством тонкослойной хроматографии. Для заглушения реакции добавили воду, а затем внесли 200 мл 30% водного раствора гидроксида натрия. Полученную смесь экстрагировали 300 мл эфира. Водный слой охладили и подкислили соляной кислотой до рН=3-4, а затем экстрагировали 500 мл эфира. Эфирные экстракты объединили, промыли насыщенным рассолом и высушили безводным сульфатом магния. Высушенный экстракт отфильтровали и при пониженном давлении удалили растворитель для получения желтого сухого остатка. Сырой продукт перекристаллизовали из бензола/петролейного эфира для получения 31,2 г желтоватого кристаллического сухого остатка (точка плавления 104-106°С) с выходом 83%. Спектр ЯМР 1H (мд) δ: 9,86 (1Н, с; -СНО); 7,37 (1H, т; J2 H-F=72 Гц; -CHF2; 7,26 (1Н, м; 6-ArH); 7,17 (1Н, м; 2-ArH); 6,79 (1Н, м; 5-ArH); 4,88 (1Н, с; -ОН). Спектр ЯМР 13С (мд) δ: 191,0 (СНО), 163,9 (т, CHF2), 157,2 (4-ArC), 146,2 (3-ArC), 130,6 (1-ArC), 123,5 (6-ArC), 116,7 (2-ArC), 116,2 (5-ArC).
Пример 4
Синтез 4-дифторметокси-3-нитробензальдегида
В литровую колбу с тремя горловинами, оборудованную капельной воронкой и механической мешалкой, поместили 72 г (0,42 моль) 4-дифторметоксибензальдегида и 400 мл уксусного ангидрида. Смесь охладили посредством льдосоляной бани. В течение 3-4 часов при температуре менее 5°С по каплям добавляли раствор 36 мл концентрированной азотной кислоты в 50 мл дихлорметана. За ходом реакции следили посредством тонкослойной хроматографии. Температуру увеличили до комнатной, после чего смесь перемешивали в течение 2 дней.
Реакционную смесь охладили до 0-5°С. Постоянно перемешивая, добавляли 20% водный раствор соляной кислоты до образования осадка, после чего смесь дополнительно охлаждали, пока осадок не исчез. Для получения желтых кристаллов смесь отфильтровали. Сырой продукт перекристаллизовали из 95% раствора этанола для получения 74 г (точка плавления 88-90°С) желтоватого кристаллического сухого остатка с выходом 81%. Спектр ЯМР 1H (мд) δ: 9,92 (1Н, с; -СНО); 7,87 (1H, т; J2 H-F=70 Гц; -CHF2); 7,68 (1Н, м; 6-ArH); 7,59 (1Н, м; 2-ArH); 7,22 (1Н, м; 5-ArH). Спектр 13С (мд) δ: 194,0 (СНО), 165,1 (т, CHF2), 160,2 (4-ArC), 157,4 (3-ArC), 137,3 (1-ArC), 130,2 (6-ArC), 122,5 (2-ArC), 120,2 (5-ArC).
Пример 5
Синтез 4-трифторэтокси-бензальдегида
В литровую колбу с четырьмя горловинами, оборудованную термометром, механической мешалкой и обратным холодильником, поместили 50 г (0,41 моль) 4-гидроксибензальдегида, 400 мл N,N-диметилформамида (ДМФА) и 5 г 18-краун-6 эфира, после чего смесь перемешивали в течение 20 минут. Затем отдельными порциями добавили 168 г (1,22 моль) порошка карбоната калия и перемешивали еще 30 минут. Реакционную смесь нагрели до 110°С, а затем в течение приблизительно 1 часа по каплям добавляли в нее раствор 115 г (0,45 моль) трифторэтильного толуол-4-сульфоната в 100 мл ДМФА. Затем смесь нагревали до 130°С в течение 3-4 часов. За ходом реакции следили посредством тонкослойной хроматографии. Смесь охладили до 0°С, вылили в охлажденные 600 мл 3N соляной кислоты и перемешали. Полученную смесь экстрагировали 1000 мл эфира. Водный слой отделили и экстрагировали эфиром (3×400 мл). Эфирные экстракты объединили и последовательно промыли 400 мл 3N соляной кислоты, дистиллированной водой, рассолом. Затем их высушили безводным сульфатом магния. Эфир выпарили методом однократной перегонки, а остаток перегнали в вакууме для получения 4-трифторэтоксибензальдегида (95-97°С/10 мм рт.ст.) с выходом 88%. Спектр ЯМР 1H (мд) δ: 9,80 (1Н, с; -СНО); 7,65 (2Н, м; 2,6-ArH); 6,83 (2Н, м; 3,5-ArH); 4,56 (2Н, кв; J3 H-F=7,2 Гц; -CH2CF3).
Пример 6
Синтез 4-трифторэтокси-3-метоксибензальдегида
То же, что в Примере 5, только вместо 4-гидроксибензальдегида использовали 4-гидрокси-3-метокси-бензальдегид для получения 4-трифторэтокси-3-метоксибензальдегида (126~129°С/10 мм рт.ст.) с выходом 83%. Спектр ЯМР 1H (мд) δ: 9,88 (1Н, с; -СНО); 7,27 (1Н, м; 6-ArH); 7,20 (1Н, м; 2-ArH); 6,83 (1Н, м; 5-ArH); 4,48 (2Н, кв; J3 H-F=7,2 Гц; -CH2CF3); 3,65 (3Н, с; -ОСН3).
Пример 7
Синтез 4-трифторэтокси-3-гидроксибензальдегида
То же, что в Примере 3, только вместо 4-дифторметокси-3-метоксибензальдегида использовали 70 г (0,3 моль) 4-трифторэтокси-3-метоксибензальдегида для получения 4-трифторэтокси-3-гидроксибензальдегида (точка плавления 133~135°С) с выходом 81%. Спектр ЯМР 1Н (мд) δ: 9,81 (1Н, с; -СНО); 7,26 (1Н, м; 6-ArH); 7,17 (1Н, м; 2-ArH); 6,79 (1Н, м; 5-ArH); 4,88 (1Н, с; -ОН); 4,45 (2Н, кв; J3 H-F=7,2 Гц; -CH2CF3). Спектр ЯМР 13С (мд) δ: 191,0 (СНО), 157,2 (4-ArC), 146,2 (3-ArC), 130,6 (1-ArC), 126 (кв, CF3), 123,5 (6-ArC), 116,7 (2-ArC), 116,2 (5-ArC), 87 (м, СН2).
Пример 8
Синтез 4-трифторэтокси-3-нитробензальдегида
То же, что в Примере 4, только вместо 4-дифторметоксибензальдегида использовали 86 г (0,42 моль) 4-трифторэтоксибензальдегида для получения 4-трифторэтокси-3-нитробензальдегида (точка плавления 126~127°С) с выходом 78%. Спектр ЯМР 1Н (мд) δ: 9,91 (1Н, с; -СНО); 7,28 (1Н, м; 6-ArH); 7,20 (1Н, м; 2-ArH); 6,77 (1Н, м; 5-ArH); 4,46 (2Н, кв; J3 H-F=7,2 Гц; -CH2CF3). Спектр ЯМР 13С (мд) δ: 191,0 (СНО), 157,2 (4-ArC), 146,2 (3-ArC), 130,6 (1-ArC), 127 (кв, CF3), 123,5 (6-ArC), 116,7 (2-ArC), 116,2 (5-ArC), 89 (м, СН2).
Пример 9
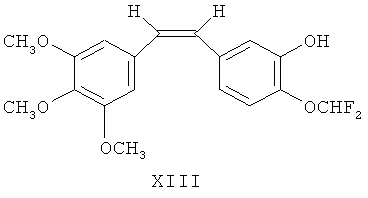
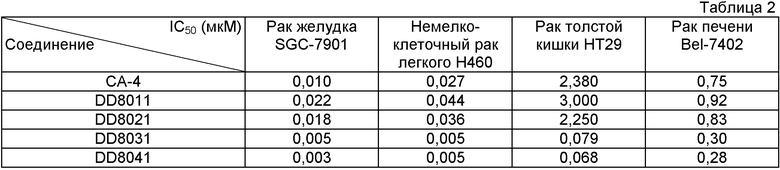
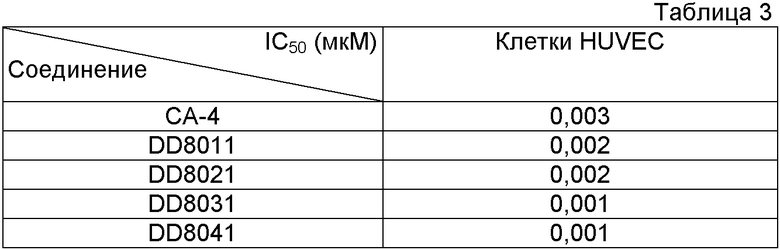
Синтез(Z)-1-(3,4,5-триметоксифенил)-2-(3′-гидрокси-4′-дифторметок-сифенил)-этена(DD8011, формула XIII)
Шаг 1: в поллитровой колбе с четырьмя горловинами в 42 мл сухого тетрагидрофурана растворили 12,5 г (0,066 моль) 4-дифторметокси-3-гидроксибензальдегида и 21,1 г (0,076 моль) трифенилметил-хлорида. Смесь равномерно перемешивали. Затем в смесь медленно по капле добавили 1,3 мл триэтиламина, после чего перемешивали в течение 1 часа. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции добавили 50 мл воды для заглушения реакции. Затем еще 30 минут перемешивали смесь, а потом добавили 100 мл этилацетата, чтобы растворить флоккулированный остаток. Для выделения бледно-желтого сухого осадка добавили 250 мл н-гептана. Осадок отфильтровали, дважды промыли водой, а затем промыли смесью 10 мл этилацетата и 20 мл петролейного эфира для получения бледно-желтого кристаллического сухого остатка. Затем кристаллический сухой остаток перекристаллизовали из этилацетата/петролейного эфира. Получили 25,8 г белого кристаллического сухого остатка с выходом 91%. Спектр ЯМР 1H (мд) δ: 9,87 (с, 1H, СНО), 7,37 (т, 1Н, J2 H-F=72 Гц, -CHF2), 7,26 (м, 2Н, Ar-Н), 7,19 (м, 15Н, Tr-Н), 6,85 (с, 1H, Ar-Н).
Шаг 2: в аргоновой атмосфере в 30 мл тетрагидрофурана взвесили 15 г (28,7 ммоль) триметоксифенилметилентрифенилфосфония бромида, полученную смесь охладили до ок. -15°С. Медленно по каплям добавили 22 мл раствора н-бутиллития в гексане (ок. 1,6 моль/л), а затем реакционную смесь перемешивали в течение 1 часа. Раствор 12,5 г (29 ммоль) ранее подготовленного альдегида из шага 1 медленно по каплям добавили в 24 мл тетрагидрофурана. Ночью смесь перемешивали, температура реакции медленно снизилась до комнатной. За ходом реакции следили посредством тонкослойной хроматографии. На следующий день реакционную смесь снова охладили до -5°С и добавили рассол для заглушения реакции. Отделили органический слой и удалили растворитель методом однократной перегонки. Сырой продукт очистили методом колоночной флеш-хроматографии для получения 15 г белого кристаллического сухого остатка с выходом 88%. Спектр ЯМР 1Н (мд) δ: 7,19 (м, 15Н, Tr-Н); 6,94 (д, 1Н, 2′-Н); 6,80 (дд, 1Н, 6′-Н); 6,74 (д, 1Н, 5′-Н); 6,55 (с, 2Н, 2,6-Н); 6,52 (т, 1H; J2 H-F=7,2 Гц; -CHF2) 6,47 (д, 1Н, 1а-Н); 6,41 (д, 1Н, 1а′-Н); 3,88 (с, 3Н, 4-ОСН3); 3,71 (с, 6Н, 3,5-ОСН3).
Шаг 3: при комнатной температуре растворили в 20 мг толуола 10 г (16,8 ммоль) ранее подготовленных соединений реакции Виттига шага 2. По каплям добавили 4 мл 37% водного раствора соляной кислоты. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции добавили воду для остановки реакции. Реакционную смесь охладили до 0-5°С для получения белого кристаллического сухого остатка, при помешивании. Сухой остаток отфильтровали и высушили для получения 5,6 г белого кристаллического остатка с выходом 95%. Спектр ЯМР 1Н (мд) δ: 7,02 (д, 1Н, 2′-Н); 6,94 (дд, 1Н, 6′-Н); 6,80 (д, 1Н, 5′-Н); 6,62 (с, 2Н, 2,6-Н); 6,53 (т, 1Н; J2 H-F=7,2 Гц; -CHF2) 6,46 (д, 1Н, 1а-Н); 6,40 (д, 1Н, 1а′-Н); 5,51 (шир, 1Н; ОН); 3,86 (с, 3Н, 4-ОСН3); 3,70 (с, 6Н, 3,5-ОСН3).
Пример 10
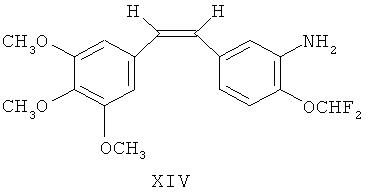
Синтез (Z)-1-(3,4,5-триметоксифенил)-2-(3′-амино-4′-дифторметоксифенил)-этена (DD8021, формула XIV)
Шаг 1: в аргоновой атмосфере в 30 мл тетрагидрофурана взвесили 15 г (28,7 ммоль) триметоксифенилметилентрифенилфосфония бромида, полученную смесь охладили до приблизительно -15°С. Медленно по каплям добавили 22 мл раствора н-бутиллития в гексане (ок. 1,6 моль/л), а затем реакционную смесь перемешивали в течение 1 часа. Медленно по каплям добавили раствор 6,3 г (29 ммоль) 4-дифторметокси-3-нитробензальдегида в 24 мл тетрагидрофурана. Ночью смесь перемешивали, температура реакции медленно снизилась до комнатной. За ходом реакции следили посредством тонкослойной хроматографии. На следующий день реакционную смесь снова охладили до -5°С и добавили рассол для заглушения реакции. Отделили органический слой и удалили растворитель методом однократной перегонки. Сырой продукт очистили методом колоночной флеш-хроматографии для получения 6,6 г бледно-желтого кристаллического сухого остатка с выходом 61%. Спектр ЯМР 1Н (мд) δ: 7,32 (д, 1H, 2′-Н), 7,16 (дд, 1Н, 6′-Н), 6,90 (д, 1Н, 5′-Н), 6,78 (т, 1H; J2 H-F=72 Гц; -CHF2), 6,64 (с, 2Н, 2,6-Н), 6,49 (д, 1Н, 1а-Н), 6,43 (д, 1Н, 1а′-Н), 3,86 (с, 3Н, 4-ОСН3), 3,70 (с, 6Н, 3,5-ОСН3).
Шаг 2: в 10 мл смеси ацетона с водой (объем/объем, 2:1) растворили 4,1 г (10,8 ммоль) (Z)-1-(3,4,5-триметоксифенил)-2-(3′-нитро-4′-дифторметокси-фенил) этена. Смесь нагрели до 50°С и перемешивали до полного растворения сухого остатка. Добавили 18,8 г тиосульфата натрия, а затем реакционную смесь нагревали с обратным холодильником в течение 6 часов. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции смесь охладили до комнатной температуры. Отделили органический слой, после чего экстрагировали водный слой 50 мл ×4 этилацетата. Органический слой объединили, промыли насыщенным рассолом, а затем высушили над безводным сульфатом магния. Растворитель частично удалили роторным испарителем, а затем смесь охладили для получения сырого продукта. Сырой продукт перекристаллизовали из петролейного эфира для получения 2,6 г желтоватого кристаллического сухого остатка с выходом 68,6%. Спектр 1H (мд) δ: 7,08 (д, 1Н, 2′-Н), 6,92 (дд, 1Н, 6′-Н), 6,76 (д, 1Н, 5′-Н), 6,62 (с, 2Н, 2,6-Н), 6,49 (д, 1Н, 1а-Н), 6,43 (д, 1Н, 1а′-Н), 6,28 (т, 1Н, J2 H-F=72 Гц; -CHF2), 5,13 (шир, 2Н, NH2), 3,86 (с, 3Н, 4-ОСН3), 3,70 (с, 6Н, 3,5-ОСН3).
Пример 11
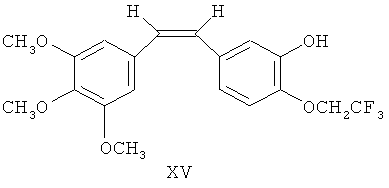
Синтез (Z)-1-(3,4,5-триметоксифенил)-2-(3′-гидрокси-4′-трифторэтоксифенил)-этен (DD8031, формула XV)
То же, что в Примере 9, только вместо 4-дифторметокси-3-гидроксибензальдегида использовали 14,5 г (66 ммоль) 4-трифторэтокси-3-гидроксибензальдегида. После трехступенчатой реакции получили (Z)-1-(3,4,5-триметоксифенил)-2-(3′-гидрокси-4′-трифторэтоксифенил)-этен с выходом 79,5%. Спектр ЯМР 1H (мд) δ: 6,93 (д, 1Н, 2′-Н), 6,84 (дд, 1Н, 6′-Н), 6,72 (д, 1Н, 5′-Н), 6,60 (с, 2Н, 2,6-Н), 6,45 (д, 1Н, 1а-Н), 6,38 (д, 1Н, 1а′-Н), 5,51 (шир, 1Н, ОН), 4,48 (2Н, кв, J3 H-F=7,2 Гц, -CH2CF3), 3,86 (с, 3Н, 4-ОСН3), 3,70 (с, 6Н, 3,5-ОСН3).
Пример 12
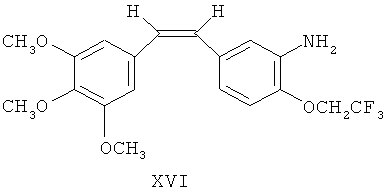
Синтез (Z)-1-(3,4,5-триметоксифенил)-2-(3′-амино-4′-трифторэтоксифенил)-этена (DD8041, формула XVI)
То же, что в Примере 10, только вместо 4-дифторметокси-3-нитробензальдегида использовали 14,5 г (66 ммоль) 4-трифторэтокси-3-нитробензальдегида. После двухступенчатой реакции получили (Z)-1-(3,4,5-триметокси-фенил)-2-(3′-амино-4′-трифторэтоксифенил)-этен с выходом 43,6%. Спектр ЯМР 1H (мд) δ: 7,08 (д, 1Н, 2′-Н), 6,92 (д, 1H, 6′-Н), 6,76 (д, 1Н, 5′-Н), 6,62 (с, 2Н, 2,6-Н), 6,49 (д, 1Н, 1а-Н), 6,43 (д, 1Н, 1а′-Н), 5,13 (шир, 2H, NH2), 4,40 (2Н, кв, J3 H-F=7,2 Гц, -CH3CF3), 3,86 (с, 3Н, 4-ОСН3), 3,70 (с, 6Н, 3,5-ОСН3).
Пример 13
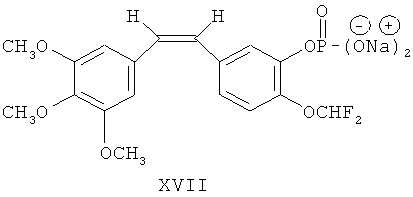
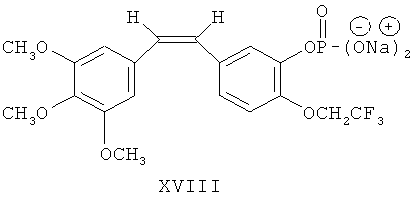
Синтез двунатриевой соли (Z)-1-(3,4,5-триметоксифенил)-2-(3′-гидрокси-4′-дифторметоксифенил)-этен-3′-O-фосфата (DD8011DP, формула XVII) и двунатриевой соли (Z)-1-(3,4,5-триметоксифенил)-2-(3′-гидрокси-4′-трифторэтоксифенил)-этен-3′-O-фосфата (DD8031DP, формула XVIII)
Процесс превращения гидроксильной группы комбретастатина А-4 в водорастворимое пролекарство (динатрий фосфат) описан в статье Дж. Роберта Петита и др. в журнале Anti-Cancer Drug Design, 1998 г., №13, стр.183-191 (см. фиг.1 и 2).
Пример 14
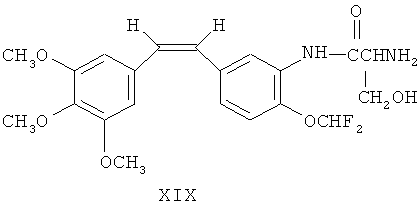
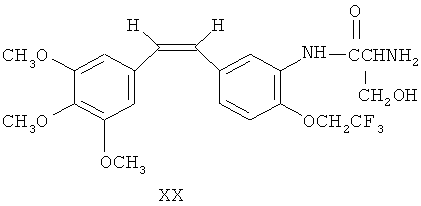
Синтез (Z)-1-(3,4,5-триметоксифенил)-2-(3′-амино-4′-дифторметоксифенил)-этен-3′-N-серинамида (DD8021AS, формула XIX) и (Z)-1-(3,4,5-триметоксифенил)-2-(3′-амино-4′-трифторэтоксифенил)-этен-3′-N-серинамида (DD8041AS, формула XX)
Аминозамещенные производные стильбена взаимодействовали с производным N-α-9-фторэнилметоксикарбонил серина (FmocAA) посредством реакции связывания, а затем из полученного продукта удалили защиту для получения аминокислотного пролекарства из соединений стильбена. Этот процесс описан в статье Дж. Роберта Петита и др. в журнале J.Med.Chem., 2002 г., №46, стр.525-531. См. фиг.3 и 4.
Пример 15
Оценка противораковой активности in vitro
Выращенную in vitro клетку опухоли подвергали воздействию фторалкоксикомбретастатина в течение 72 часов, оценивая подавление пролиферации опухоли посредством МТТ и SRB (сульфородамин В) тестов. В Таблице 2 приведены результаты, в сравнении с КА-4.
Группы клеток: Н460 - клетки рака легких человека, SGC7901 - клетки рака желудка человека, НТ-29 - клетки рака толстой кишки человека, Bel-7402 - клетки рака печени человека.
План эксперимента: в течение 72 часов клетки культивировали в теплой среде с различными концентрациями соединений (100, 10, 1, 0.1, 0.01, 0.001 мкМ). Для оценки степени подавления пролиферации клеток соединениями использовали SRB-тест (сульфородамин В). Вычисляли коэффициент подавления, по Логит модели вычисляли IC50 (ингибирующую концентрацию 50) согласно коэффициенту подавления. Также сравнивали противораковую активность соединений in vitro.
Для выражения коэффициента подавления использовали следующее равенство:
Коэффициент подавления (%)=[(среднее значение ОП контрольной группы-среднее значение ОП экспериментальной группы)/среднее значение ОП контрольной группы]×100%.
Результат показал, что фторметоксикомбретастатин обладает противораковой активностью in vitro, аналогичной активности природного комбретастатина А-4. А фторэтоксикомбретастатин продемонстрировал противоопухолевую активность в 3-30 раз выше, чем фторметоксикомбретастатин.
Пример 16
Оценка подавления реваскуляризации in vitro
Оценивали антиангиогенное действие фторалкоксикомбретастатинов на примере эндотелиальных клеток пупочной вены человека (HUVEC) тем же способом, что и в Примере 15.
Результаты продемонстрировали высокую подавляющую тубулин-связывающую активность фторалкоксикомбретастатинов, что говорит о том, что фторалкоксикомбретастатины представляют собой новый перспективный класс противораковых лекарств васкулярной направленности (Таблица 3).
Пример 17
Приготовление лиофилизированного порошка фторалкоксикомбретастатина
Материалы дозировали по массе в строгом соответствии с формулами (Таблица 4). Заданное количество маннита развели в 80% заданного количества воды для инъекций до получения прозрачного раствора, а затем добавили 0,1% (г/мл) инъекционного вещества. Смесь перемешали, осадили, профильтровали через мембрану с размером пор 0,45 мкм, после чего добавили оставшуюся воду для инъекций. Раствор снова отфильтровали через мембрану с размером пор 0,22 мкм. Для проверки качества препарата измерили значение рН и содержание продукта. Затем некоторое количество раствора залили во флакон, после чего произвели лиофилизацию. Ввели азот, остановили процесс, осуществили маркировку, упаковку и контрольные испытания для получения готового продукта (в связи с относительной чувствительностью производных комбретастатина к температуре и свету все процедуры проводили в темноте, а готовые препараты хранили в темном помещении при температуре 2-8°С).
Все упоминаемые в настоящем описании изобретения документы входят в состав изобретения в качестве источников информации, как если бы каждый документ был приложен отдельно. Также следует понимать, что специалисты могут вносить в вышеописанный вариант изобретения некоторые изменения, при этом полученные аналоги будут оставаться в пределах объема изобретения, описанного приложенной формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПРОИЗВОДСТВА И ИСПОЛЬЗОВАНИЯ ЭТОКСИКОМБРЕТАСТАТИНОВ И ПРОЛЕКАРСТВА НА ИХ ОСНОВЕ | 2007 |
|
RU2451664C2 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛО-СТИЛЬБЕНА (СОЕДИНЕНИЯ А-104815) | 2021 |
|
RU2786432C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2609018C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ФТАЛИДА | 2005 |
|
RU2394569C2 |
| ПРОИЗВОДНОЕ БЕНЗИЛФЕНИЛОВОГО ЭФИРА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2743165C2 |
| ФЕНИЛАТНОЕ ПРОИЗВОДНОЕ, ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2744975C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2762111C1 |
| ПРОСТОЕ ЭФИРНОЕ ПРОИЗВОДНОЕ НИКОТИНИЛОВОГО СПИРТА, ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЯ | 2017 |
|
RU2735541C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ СЕМЕЙСТВА ТАМ | 2016 |
|
RU2750727C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2009 |
|
RU2514427C2 |
Изобретение относится к новым производным комбретастатина формулы (I), обладающим свойствами ингибитора ангиогенеза, которые могут быть использованы в качестве противораковых и/или антиангиогенных средств. В формуле I
Rf - алкильная группа, содержащая от 1 до 2 атомов углерода, в которой от 1 до 5 атомов водорода замещены 1-5 атомами фтора, a R - аминогруппа, или группа, представляющая собой аминогруппу, замещенную остатком аминокислоты общей формулы NH(COCHR′NH)-H, где R′ - водород, боковая цепь природной аминокислоты, представляющая собой С1-С4алкил, возможно замещенный гидроксигруппой, или гидроксильная группа, или динатрий, или аммоний фосфатная группа. Изобретение также относится к вариантам способа получения соединений формулы I, предусматривающим следующие стадии: фторалкилирование 4-гидрокси-3-метоксибензальдегида или 4- гидроксибензальдегида в присутствии катализатора межфазного переноса, с получением соответственно 4-фторалкокси-3-метоксибензальдегида (V) или 4-фторалкоксибензальдегида (VII), последующее селективное деметилирование (V) с помощью дифенилфосфина лития и защитой гидроксильной группы, или нитрование (VII) в 3-е положение. Полученные при этом соединения подвергают реакции Виттига с использованием илида 3,4,5-триметоксибензилтрифенилфосфония и выделяют целевой продукт. 4 н. и 2 з.п. ф-лы, 4 ил., 4 табл.
1. Соединение формулы (I)
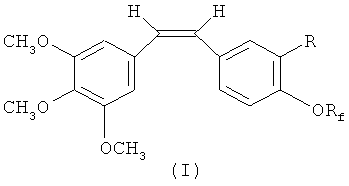
где Rf - алкильная группа, содержащая от 1 до 2 атомов углерода, в которой от 1 до 5 атомов водорода замещены 1-5 атомами фтора;
R - аминогруппа или группа, представляющая собой аминогруппу, замещенную остатком аминокислоты общей формулы NH(COCHR′NH)-H, где R′ - водород, боковая цепь природной аминокислоты, представляющая собой С1-С4алкил, возможно замещенный гидроксигруппой, или гидроксильная группа, или динатрий- или аммонийфосфатная группа.
2. Соединение по п.1, в котором Rf и R представляют собой один из следующих вариантов:
(а) Rf представляет собой -CH2F, -CHF2, -CF3, -CH2CF3, -CH2CHF3 или -CF2CF3, a
R=-OH или представляет собой -OPO3Na2; либо
(б) Rf представляет собой -CH2F, -CHF2, -CF3, -CH2CF3, -CH2CHF3 или -CF2CF3, a R представляет собой -NH2 или -NHCOCH(NH2)CH2OH.
3. Способ получения соединения по п.1, предусматривающий следующие стадии:
(1) 4-гидрокси-3-метоксибензальдегид III в присутствии катализатора межфазного переноса фторалкилируют посредством фторосодержащего агента для получения 4-фторалкокси-3-метоксибензальдегида, представленного формулой V
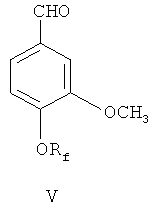
(2) деметилируют 4-фторалкокси-3-метоксибензальдегид дифенилфосфином лития для получения 4-фторалкокси-3-гидроксибензальдегида, представленного формулой VI
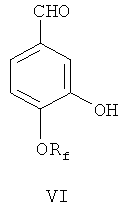
(3) защищают гидроксильную группу 4-фторалкокси-3-гидроксибензальдегида VI, а затем 4-фторалкокси-3-гидроксибензальдегид VI с защищенной гидроксильной группой в условиях реакции Виттига обрабатывают илидом 3,4,5-триметоксибензилтрифенилфосфония, после чего из полученного соединения удаляют защитную группу для получения соединений формулы I по п.1.
4. Способ получения соединений по п.1, предусматривающий следующие стадии:
(а) 4-гидроксибензальдегид IV в присутствии катализатора межфазного переноса фторалкилируют посредством фторосодержащего агента, чтобы синтезировать 4-фторалкоксибензальдегид, представленный формулой VII
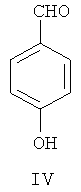
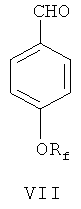
(б) полученный 4-фторалкоксибензальдегид VII нитруют в 3-м положении фенильного кольца посредством азотной кислоты и уксусного ангидрида для получения 4-фторалкокси-3-нитробензальдегида, представленного формулой VIII
(в) полученный 4-фторалкокси-3-нитробензальдегид VIII обрабатывают илидом 3,4,5-триметоксибензилтрифенилфосфония в условиях реакции Виттига для получения соединений формулы I по п.1.
5. Способ по п.3 или 4, в котором в качестве фторосодержащего агента используют фторгалогенметан или фторалкилсульфонат.
6. Фармацевтическая композиция, обладающая свойствами ингибитора ангиогенеза, содержащая терапевтически эффективное количество соединения по п.1 в фармацевтически приемлемом носителе.
| ПУЛЬСАЦИОННО-ЦИКЛИЧЕСКИЙ СПОСОБ ЭКСТРАКЦИОННОГО РАЗДЕЛЕНИЯ СМЕСИ КОМПОНЕНТОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2009 |
|
RU2403949C1 |
| WO 2006036743 А2, 06.04.2006 | |||
| NICHOLAS J | |||
| LAWRENCE et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ШЕППАРД У | |||
| и др | |||
| Органическая химия фтора | |||
| - М.: Мир, 1972, 374-375 | |||
| ЕА 200300516 А1, 28.08.2003 | |||
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО | 1999 |
|
RU2215525C2 |

