Область изобретения
Настоящее изобретение относится к области фармацевтического синтеза, в частности к синтезу противораковых препаратов.
Предшествующий уровень техники
Кустарники и деревья семейства комбретовых, произрастающие в тропическом и субтропическом поясе, широко используются в традиционной медицине. 25 видов рода комбретум семейства комбретовых используются в Африке и Индии для лечения болезни Хансена и рака. Однако изучены лишь немногие виды, в частности комбретум мелкоцветковый (combretum micranthum) и комбретум Зейера (combretum zeyheri). Южно-африканская ива (combretum caffrum) - вид рода комбретум - племена зулусов ЮАР называют «мдулу»; этот вид также известен как южно-африканская ива или южноафриканское дерево. В конце 1970-х годов, после широкомасштабного скрининга ученые Национального института рака (США) обнаружили, что растения рода комбретум способны активно подавлять клетки лимфолейкоза Р-388. В начале 1980-х возник широкий интерес к изучению этих растений. В этот период доктор Дж. Роберт Петтит, директор Института по исследованию рака Аризонского государственного университета, а также четверо его коллег смогли выделить комбретастатины из южно-африканской ивы (combretum caffrum), которую племена зулусов используют в качестве растительного лекарства, а также для окраски копий. В статье, опубликованной в журнале Journal of Canadian Chemistry, доктор Петтит заявил, что кора этого дерева обладает противораковым действием. Впоследствии из коры не только выделили и идентифицировали множество высокоактивных соединений, но и провели исследования по определению механизмов фармакологического действия и модификаций их структуры (Pettit, G.R. et al. 1) Can. J. Chem. 1987, 65, 2390-2396. 2) J. Nat. Prod. 1987, 50, 119-131. 3) Experieutia 1989, 45, 209-211). Комбретастатины - серия соединений, характеризующихся структурой 2-1,2-дифенилэтилен. Одно их таких соединений, комбретастатин А-4 [КА-4, комбретастатин, (Z)-1-(3,4,5-триметокси)фенил-2-(3'-гидрокси-4'-метокси)фенилэтилен], оказалось чрезвычайно сильным ингибитором полимеризации тубулина; оно представлено формулой XVII (Pettit, G. R., et al. J. Med. Chem. 1995, 38, 1666-1672).
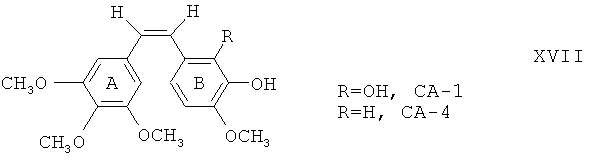
Недавно было сделано волнующее открытие - КА-4 показал свою эффективность в разрушении сосудистой сети опухоли, действуя в качестве целевого препарата васкулярной направленности (Thorpe, Р.Е. Clin. Cancer Res., 15 января 2004 г., 10(2):415-427; West, СМ.; Price, P. Anticancer Drugs, март 2004 г., 15(3):179-187; Young, S.L.; Chaplin D.J. Expert Opin. Investig. Drugs, сентябрь 2004 г., 13(9): 1171-1182). Разработанный компанией Oxigene Inc. (США) КА-4 сейчас находится в III стадии клинических испытаний в качестве лекарства от рака. В 1997 году Т. Хатанака и др. из компании Ajinomoto Со. (Япония) обнаружили, что если заменить гидроксильную группу КА-4 в 3-ем положении аминогруппой, а затем модифицировать до получения аминоамида, то получится водорастворимое пролекарство, обладающее заметно возросшими противораковыми свойствами и значительно меньшей токсичностью, по сравнению с КА-4 (USP5674906). В настоящее время 3'-амино КА-4 аминоамид (AVE8062), разрабатываемый компанией Aventis Pharma Со. (Франция), вошел во II стадию клинических испытаний.
Поэтому задача нахождения новых комбретастатинов с более высокой активностью является очень актуальной в данной области.
Раскрытие изобретения
Задачей настоящего изобретения является создание этоксикомбретастатинов формулы I. Еще одной задачей изобретения является создание способа для получения соединений формулы I. Также задачей изобретения является создание фармацевтической композиции, содержащей соединения формулы I. Еще одной задачей изобретения является подготовка соединений формулы I к использованию в медицинских целях.
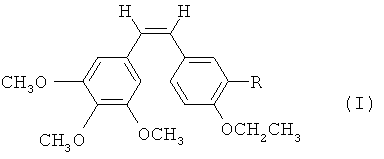
Согласно первой особенности настоящего изобретения предлагаются соединения формулы I
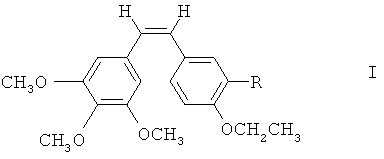
где R - гидроксильная группа, аминогруппа, фосфатная группа, выбранная из динатрийфосфата или фосфата аммония, или внутренняя соль фосфорилхолина, -NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, a m - целое число от 1 до 3.
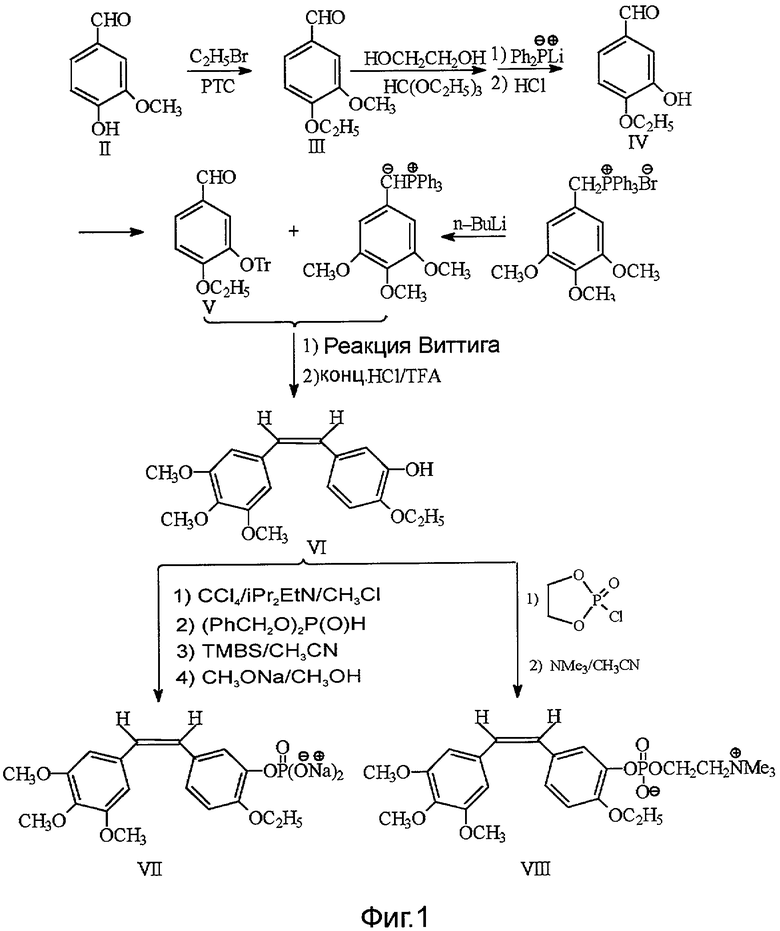
Согласно второй особенности изобретения предлагается способ получения соединения формулы I, в которой R представляет собой фосфатную группу или внутреннюю соль фосфорилхолина, предусматривающий следующие шаги:
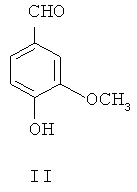
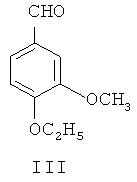
(1) 4-гидрокси-3-метоксибензальдегид II в присутствии катализатора межфазного переноса этилируют посредством этилбромида, чтобы синтезировать 4-этокси-3-метоксибензальдегид, представленный формулой III
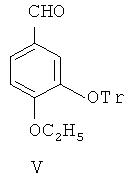
(2) с помощью дифенилфосфина лития селективно замещают m-метильную группу 4-этокси-3-метоксибензальдегида III гидроксильной группой, чтобы синтезировать 4-этокси-3-гидроксибензальдегид, представленный формулой IV
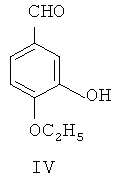
(3) гидроксильную группу защищают посредством трифенилхлорметана, затем 4-этокси-3-гидроксибензальдегид IV с защищенной гидроксильной группой посредством реакции Виттига взаимодействует с илидом 3,4,5-триметоксибензилтрифенилфосфония, после чего из полученного соединения удаляют защиту для получения этоксикомбретастатина формулы VI
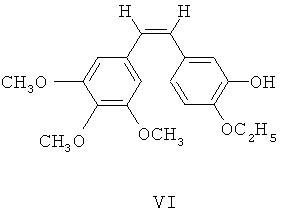
(4) этоксикомбретастатин VI фосфорилируют посредством фосфорилирующего агента, чтобы синтезировать его фосфатные производные;
(5) в щелочных условиях фосфатные производные этоксикомбретастатина превращают в фосфат этоксикомбретастатина или в соответствующую внутреннюю соль фосфорилхолина.
В предпочтительном варианте осуществления изобретения в качестве фосфорилирующего агента на шаге (4) используют дибензилфосфонат или 2-хлор-1,3,2-диоксафосфолан.
В предпочтительном варианте осуществления изобретения m-гидроксильную группу на шаге (3) защищают посредством трифенилхлорметана.
В предпочтительном варианте осуществления изобретения (Z)-l-(3,4,5-триметокси)фенил-2-(3'-тритилокси-4'-этокси)фенилэтилен на шаге (3) взаимодействует с концентрированной соляной и трифторуксусной кислотами для удаления тритила и получения этоксикомбретастатина VI.
В предпочтительном варианте осуществления изобретения фосфатное производное этоксикомбретастатина на шаге (5) превращают в фосфатное пролекарство этоксикомбретастатина посредством реакций крекинга в щелочных условиях, желательно при рН=8-10.
В предпочтительном варианте осуществления изобретения фосфатное производное этоксикомбретастатина на шаге (5) взаимодействует с третичным амином для получения пролекарства, внутренней соли фосфорилхолина.
Согласно третьей особенности изобретения предлагается способ получения соединения формулы I, в котором R - -NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, а m - целое число от 1 до 3, предусматривающий следующие шаги:
(а) 4-гидрокси-3-нитробензальдегид IX в присутствии катализатора межфазного переноса этилируют посредством этилбромида, чтобы синтезировать 4-этокси-3-нитробензальдегид, представленный формулой X
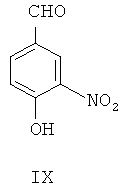
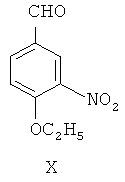
(б) при фотокатализе от ультрафиолетового излучения с длиной волны 254 нм, 4-этокси-3-нитробензальдегид Х посредством реакции Виттига взаимодействует с илидом 3,4,5-триметоксибензилтрифенилфосфина для получения (Z)-1-(3,4,5-триметокси)фенил-2-(3'-нитро-4'-этокси)фенилэтилена, представленного формулой XI
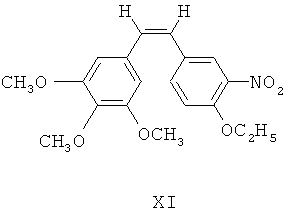
(в) (Z)-1-(3,4,5-триметокси)фенил-2-(3'-нитро-4'-этокси)фенилэтилен XI восстанавливают посредством восстанавливающих агентов для получения 3'-амино этоксикомбретастатина XII
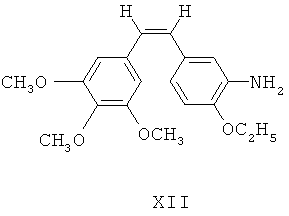
(г) 3'-амино этоксикомбретастатин XII взаимодействует с аминокислотными производными для получения соответствующих аминоамидных производных;
(д) в щелочных условиях вышеупомянутые аминоамидные производные превращают в аминоамиды 3'-аминоэтоксикомбретастатина.
В предпочтительном варианте осуществления изобретения в качестве восстанавливающих агентов на шаге (в) используют дихлорид олова, цинковую пыль/уксусную кислоту, тиосульфат натрия или хлорид никеля/борогидрид натрия.
В предпочтительном варианте осуществления изобретения на шаге (г) в присутствии катализаторов дициклогексилкарбодиимида (ДЦКД, DCC) и 1-гидроксибензотриазола (HOBt), или бензотриазол-1-илокси-трис(диметиламино)-фосфоний-гексафторфосфата (ВОР), (Z)-1-(3,4,5-триметокси)фенил-2-(3'-амино-4'-этокси)фенилэтилен взаимодействует с аминокислотными производными N-α-9-фторилметоксикарбонила (FmocAA), чтобы превратить 3'-аминогруппу в Fmoc-аминоамид.
В предпочтительном варианте осуществления изобретения на шаге (д) Fmoc-аминоамид (Z)-1-(3,4,5-триметокси)фенил-2-(3'-амино-4'-этокси)фенилэтилена превращают в водорастворимое пролекарство аминоамида 3'-аминоэтоксикомбретастатина посредством удаления Fmoc, при этом желательно для щелочной среды использовать водный раствор гидроксида натрия.
Согласно четвертой особенности изобретения предлагается фармацевтическая композиция, обладающая противораковой активностью, содержащая терапевтически эффективное количество соединения формулы 1 и фармацевтически приемлемый носитель.
В другом предпочтительном варианте осуществления изобретения упомянутые фармацевтические композиции могут вводиться перорально или внутривенно в виде одной из следующих лекарственных форм: лиофилизированный порошок, порошок, гранулы, таблетки, капсулы, сироп, суппозитории, препарат для инъекций, эмульсия, настойка, суспензия или раствор.
Согласно пятой особенности изобретения предусматривается использование соединений формулы I или их или его приемлемых солей для лечения опухоли путем введения соединений больным.
Согласно шестой особенности изобретения предусматривается использование соединений формулы I или их приемлемых солей для лечения заболеваний, вызванных нездоровым ангиогенезом.
В другом предпочтительном варианте осуществления изобретения соединения формулы I используются для борьбы с растущими опухолями и метастазированием, вызываемыми нездоровым ангиогенезом. В число упомянутых опухолей входят, в том числе, следующие: рак легкого, немелкоклеточный рак легкого, печеночно-клеточный рак, аденокарцинома поджелудочной железы, карцинома желудка, остеокарцинома, карцинома пищевода, рак груди, рак предстательной железы, рак яичек, карцинома толстой кишки, рак яичников, карцинома мочевого пузыря, рак шейки матки, меланокарцинома, плоскоклеточная карцинома, базально-клеточная карцинома, аденокарцинома, карцинома потовых желез, карцинома сальных желез, папиллярная карцинома, папиллярные аденокарциномы, цистаденокарцинома, цистокарцинома, медуллярный рак, бронхогенный рак, рак костных клеток, эпителиальная карцинома, карцинома желчных протоков, эмбриональный рак, хориокарцинома, семинома, опухоль Вильмса, спонгиобластома, астроцитома, медуллобластома, краниофарингиома, эпендимома, пинеалома, гемангиобластома, акустическая невринома, менингиома, нейробластома, бластома зрительного нерва, ретинобластома, нейрофиброма, фибросаркома, фибробластома, фиброма, фиброаденома, хондрофиброма, фиброцистома, фибромиксома, фибрострома, фибромиксосаркома, фибропапиллома, миксосаркома, миксоцистома, миксоэнходрома, миксохондросаркома, миксохондрофибросаркома, миксоаденома, миксобластома, липосаркома, липома, липоаденома, рак липобластов, липохондрома, липидная фиброма, липоангиома, миксолипома, хондросаркома, хондрома, хондромиома, хордома, хориоаденома, хориоэпителиома, хориобластома, остеосаркома, остеобластома, остеохондрофиброма, остеохондросаркома, остеохондрома, остеоцистома, остеодентинома, остеофиброма, фибросаркома костей, ангиосаркома, ангиома, ангиолипома, гематальная хондрома, ангиобластома, ангиокератома, ангиоглиома, гемангиоэндотелиома, гемангиофиброма, ангиомиома, ангиолипома, гематальная лимфангиома, ангиолиполейомиома, ангиомиолипома, гематальная мионеврома, гематальная миксома, ангиоретикулоэндотелиома, лимфангиосаркома, лимфогранулематоз, лимфангиома, лимфома, лимфомиксома, лимфосаркома, лимфангиофиброма, лимфоцитома, лимфоэпителиома, лимфобластома, эндотелиальная карцинома, эндобластома, синовиома, синовиосаркома, мезотелиома, мезоцитома, опухоль Юинга, лиомиома, лейомиосаркома, лейомиобластома, лиомиофиброма, рабдомиома, рабдомиосаркома, рабдомиомиксома, острый лимфоцитарный лейкоз, острый миелоцитарный лейкоз, хронический лейкоз, истинная полицитемия, лимфома, множественная миелома.
В другом предпочтительном варианте осуществления изобретения соединения формулы I используются для лечения других родственных болезней, вызываемых патологическим ангиогенезом, в число которых входят, в том числе, следующие заболевания: ревматоидный артрит, диабетическая ретинопатия, ретинопатия недоношенных, тромбоз ретинальных вен, псориаз, розацеа, саркома Капоши, аллергический кератит, эпидемический кератоконъюнктивит, неоваскулярная глаукома, бактериальные язвы, грибковые язвы, инфекции простого герпеса, инфекции опоясывающего лишая, простейшие инфекции, микобактериальные инфекции, полиартериит, саркоидоз, склерит, приливы крови, болезнь Шегрена, системная красная волчанка, синдром приобретенного иммунодефицита (СПИД), сифилис.
Таким образом, настоящее изобретение предлагает новые производные комбретастатина, обладающие улучшенной биологической активностью.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 изображен ход синтеза этоксикомбретастатина и его водорастворимых пролекарств;
На фиг.2 изображен ход синтеза 3'-аминоэтоксикомбретастатина и его аминоамидных производных.
Здесь:
РТС (КМП) - катализатор межфазного переноса;
Wittig reaction - реакция Виттига;
Ph2PLi - дифенилфосфин лития;
n-BuLi - нормальный бутиллитий;
TFA (ТФУ) - трифторуксусная кислота;
i-Pr2EtN - диизопропилэтиламин;
(PhCH2O)2P(O)H - дибензилфосфат;
TMBS (ТМБС) - триметилбромосилан;
Fmoc-L-Ser- производное N-α-9-фторэнилметоксикарбонил-L-серина;
Fmoc-Gly - производное N-α-9-фторэнилметоксикарбонил-глицина;
ВОР - бензотриазол-1-илокси-трис(диметиламино)-фосфоний-гексафторфосфат;
DCC (ДЦКД) - дициклогексилкарбодиимид;
HOBt - 1-гидроксибензотриазол;
DMF (ДМФ) - диметилформамид;
NMe3 - триметиламин;
aq. NaOH (водн. NaOH) - слабый водяной раствор гидроксида натрия;
conc. НСl (конц. НСl) - концентрированная соляная кислота.
Лучший вариант осуществления изобретения
В ходе проведения интенсивных разносторонних исследований неожиданно обнаружилось, что 4-е положение В-кольца природного вещества комбретастатина является активным участком и что исходную метоксильную группу в 4-м положении В-кольца комбретастатина можно заменить на этоксильную группу, чтобы улучшить целевую активность в отношении сосудов опухоли.
В вышеупомянутое природное вещество комбретастатин, в 4-м положении ароматического В-кольца, успешно ввели этоксильную группу посредством ключевой реакции деметилирования с селективным применением дифенилфосфина лития.
Также для синтеза упомянутых соединений посредством реакции Виттига использовали фотокатализ от ультрафиолетового излучения с длиной волны 254 нм, что позволило улучшить стереоселективность реакции и, таким образом, значительно увеличить выход продуктов в Z конфигурации.
Эти новые соединения обладают повышенной способностью к подавлению полимеризации тубулина, что может быть использовано для лечения опухолей и патологического состояния, вызванного нездоровым ангиогенезом.
Соединения
Настоящее изобретение предлагает новые производные комбретастатина, которому в 4-м положении ароматического В-кольца ввели этоксильную группу, а в 3-м положении находилась, в основном, гидроксильная группа с производным фосфатом или внутренней солью фосфорилхолина и аминогруппа с производными аминоамидными водорастворимыми пролекарствами, представленными формулой I
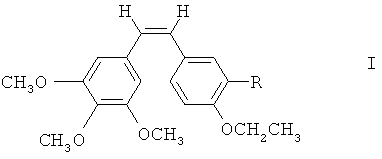
Здесь:
R - гидроксильная группа, аминогруппа, нитрогруппа, галогеновая, алкоксильная, фосфатная группа, внутренняя соль фосфорилхолина, боковая цепь аминокислоты или ее фармацевтически приемлемые соли.
Если R - фосфат, внутренняя соль фосфорилхолина, боковая цепь аминокислоты или ее фармацевтически приемлемые соли, то образуются соответствующие водорастворимые пролекарства.
Если R - гидроксильная группа, то производят водорастворимые пролекарства, фосфат или внутреннюю соль фосфорилхолина.
Если R - аминогруппа, то получают водорастворимые пролекарства, -NH(COCHR'NH)n-H (где R' - боковая цепь природной аминокислоты).
Желательно, чтобы в приоритете был этоксикомбретастатин с R-гидроксильной группой; 3'-аминоэтоксикомбретастатин с R-аминогруппой. Структура представлена формулой I, в которой R=-OH, -NH2, -OP(O)(ONa)2, -ОР(O)(O-)(OCH2CH2NMe3) или -NH(COCHR'NH)m-H (где R' - водород, боковая цепь природной аминокислоты или фенильная группа, a m - целое число от 1 до 3).
Этоксикомбретастатины настоящего изобретения способны образовывать фармацевтически приемлемые соли присоединения основания с неорганическими или органическими основаниями. В число упомянутых неорганических оснований входят, в том числе, гидроксид калия и гидроксид аммония, а в число упомянутых органических оснований входят, в том числе, алифатические амины (например, триэтиламин), гидроксиламины (например, этаноламин), аминокислоты (например, гистидин) и аминогликозиды (например, неоамин).
Этоксикомбретастатины настоящего изобретения также способны образовывать фармацевтически приемлемые соли присоединения кислоты с неорганическими или органическими кислотами. В число упомянутых неорганических кислот входят, в том числе, соляная, серная и фосфорная кислота, а в число упомянутых органических кислот входят, в том числе, щавелевая, фумаровая, малеиновая, яблочная, лимонная, винная и глютаминовая кислоты.
Подготовка соединений
Настоящее изобретение предлагает способ подготовки соединений формулы I, предусматривающий следующие шаги:
4-гидрокси-3-метоксибензальдегид этилируют в присутствии катализатора межфазного переноса, а затем селективно деметилируют его посредством применения дифенилфосфина лития для получения ряда новаторских производных p-этоксибензальдегида. Вышеупомянутые соединения производных p-этоксибензальдегида затем взаимодействуют посредством реакции Виттига в качестве исходных веществ с высокой стереоселективностью для получения ряда производных этоксикомбретастатинов, после чего фосфатируют или соединяют с аминокислотами и т.д. с целью получения ряда водорастворимых пролекарств этоксикомбретастатина.
Синтез производных p-этоксибензальдегида
4-этокси-3-метоксибензальдегид III или 4-этокси-3-нитробензальдегид Х получают из 4-гидрокси-3-метоксибензальдегида II (ванилина) или 4-гидрокси-3-нитробензальдегида IX, применяя этилбромид в присутствии неорганического основания и катализатора межфазного переноса.
В качестве вышеупомянутых неорганических оснований используют гидроксид или один и более карбонатов, желательно гидроксид калия и/или карбонат калия. В качестве упомянутого катализатора межфазного переноса используют четвертичные соли аммония, четвертичные соли фосфония, краун-эфир или полиэтиленгликоль (ПЭГ), желательно бензилтриэтиламмоний хлорид, тетрабутиламмоний бисульфат (ТБАБС), 18-краун-6, дифенил-18-краун-6, дициклогексил-18-краун-6 эфиры или ПЭГ-400.
Формильную группу 4-этокси-3-метоксибензальдегида III защищают с помощью соединений гликоля, после чего метоксил в 3-м положении селективно деметилируют посредством дифенилфосфина лития для получения 4-этокси-3-гидроксибензальдегида IV.
Синтез этоксикомбретастатина
В присутствии катализатора органического основания 4-этокси-3-гидроксибензальдегид IV взаимодействует с трифенилметилхлоридом для получения 3-трифенилметокси-4-этоксибензальдегида. 3,4,5-Триметокси-бензилтрифенилфосфин бромид превращают в соответствующий фосфониевый илид с помощью н-бутиллития, а затем этот фосфониевый илид взаимодействует с вышеупомянутым 3-трифенилметокси-4-этоксибензальдегидом посредством реакции Виттига для получения высокоэффективных цис-стильбеновых производных. После этого удаляют защиту тритильной группы посредством совместного воздействия концентрированной соляной и трифторуксусной кислотами для получения этоксикомбретастатина VI.
В качестве вышеупомянутых неорганических оснований использовали триэтиламин или диизопропилэтиламин.
Синтез 3'-аминоэтоксикомбретастатина
Под воздействием ультрафиолетового излучения с длиной волны 254 нм 4-этокси-3-нитробензальдегид Х посредством реакции Виттига взаимодействует с вышеупомянутым фосфониевым илидом для получения (Z)-3'-нитро этоксикомбретастатина XI с высокой селективностью. Затем 3'-нитро восстанавливают до аминогруппы посредством восстанавливающих агентов. Желательно использовать в качестве восстанавливающих агентов дихлорид олова, цинковую пыль/уксусную кислоту, тиосульфат натрия или хлорид никеля/борогидрид натрия. Таким образом, получают 3'-аминоэтоксикомбретастатин XII.
Синтез этоксикомбретастатин фосфата
Как показано на фиг.1, гидроксильную группу в 3-ем положении вышеупомянутого этоксикомбретастатина VI превращают в двунатриевую соль фосфата путем реакции с четыреххлористым углеродом, диизопропилэтиламином, дибензилфосфонатом, триметилсиланбромидом и метилатом натрия, в результате чего получают этоксикомбретастатин фосфат VII.
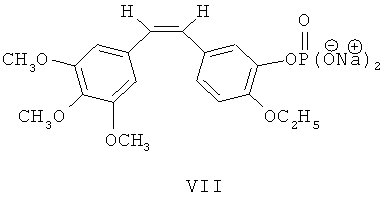
Синтез этоксикомбретастатин фосфорилхолина
Либо, что также изображено на фиг.1, гидроксильную группу в 3-м положении вышеупомянутого этоксикомбретастатина VI превращают в циклическое фосфатное производное этоксикомбретастатина путем реакции с 2-хлор-1,3,2-диоксафосфоланом (фосфорилирующим агентом). В присутствии триметиламина разрывают кольцо циклического фосфатного производного для получения внутренней соли этоксикомбретастатин фосфорилхолина VIII.
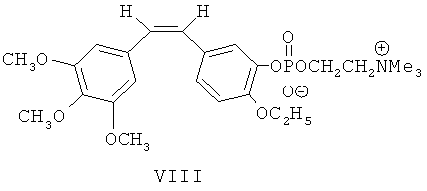
Синтез 3'-аминоэтоксикомбретастатин аминоамида
Как показано на фиг.2, 3'-аминоэтоксикомбретастатин XII обрабатывают аминокислотным производным N-α-9-фторэнилметоксикарбонила (FmocAA) и ВОР-агентом, либо циклогексилкарбодиимидом (DCC) и 1-гидроксибензотриазолом (HOBt), чтобы ввести в 3-м положении аминогруппы аминокислотную боковую цепь, в соответствии с XIII и XIV. После этого у вещества с аминокислотной боковой цепью в 3-м положении удаляют защиту посредством гидроксида натрия, а затем превращают в аминоамид для получения ряда аминоамидных производных 3'-аминоэтоксикомбретастатина в соответствии с XV и XVI.
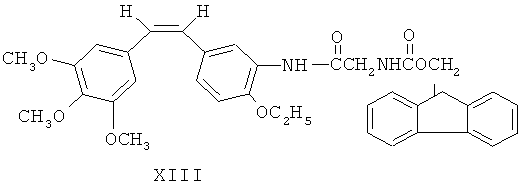
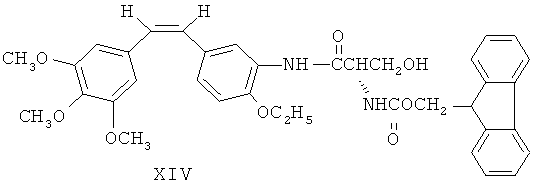
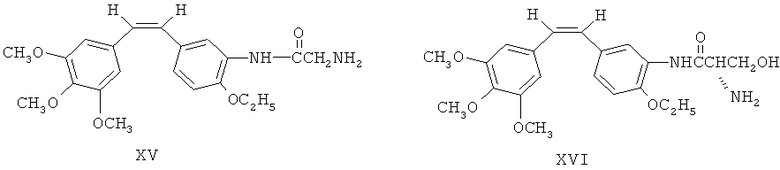
Фармацевтические композиции
Предлагаемая фармацевтическая композиция содержит терапевтически эффективное количество соединений формулы I в фармацевтически приемлемом носителе, при этом доля соединений формулы I может варьироваться от 0,1 до 99% (вес/вес) композиций. Упомянутые фармацевтические композиции могут использоваться в различных лекарственных формах. Эти фармацевтические композиции могут вводиться перорально или внутривенно в виде одной из следующих лекарственных форм: лиофилизированный порошок, гранулы, порошок, таблетки, капсулы, сироп, суппозитории, препарат для инъекций, эмульсия, настойка, суспензия или раствор.
Для внутривенного введения можно использовать композиции в виде лиофилизированного порошка, растворенного в физиологическом растворе или растворе глюкозы.
Дозировка активного ингредиента зависит от способа введения, а также от тяжести заболевания. В целом, при приеме предлагаемого соединения ежедневно в количестве от 0,5 до 500 мг/кг массы тела в день, наблюдается удовлетворительный терапевтический эффект. В одном предпочтительном варианте осуществления изобретения предлагаемое соединение вводят пациенту в несколько приемов (по 2-4 раза в день), либо посредством лекарственной формы пролонгированного действия. Для большинства крупных млекопитающих суммарная дневная дозировка составляет от 1 до 100 мг. В эксперименте приемлемая форма препарата для перорального приема содержала около 0,5-500 мг активного ингредиента, смешанного с твердым или жидким фармацевтически приемлемым носителем. Дозировку можно подобрать в соответствии с наиболее желательным результатом терапии. Например, в зависимости от конкретных условий лечения предлагаемые композиции могут вводиться ежедневно в несколько приемов либо с равномерным уменьшением. Обычно клинически приемлемая пероральная доза для взрослого составляет от 1 до 1000 мг, а наиболее предпочтительным вариантом является интервал от 10 до 200 мг. Доза для применения иным способом для взрослого составляет от 0,1 до 100 мг, предпочтительно от 1 до 100 мг.
Предлагаемые настоящим изобретением этоксикомбретастатины, подготовленные вышеописанными способами, при использовании в качестве агента васкулярной направленности могут вводиться как перорально, так и внутривенно. Дозировка активного ингредиента зависит от тяжести заболевания. Ежедневная доза для взрослого человека составляет, как правило, от 1 до 3000 мг.
В предпочтительном варианте осуществления изобретения предлагаемые соединения вводят перорально или внутривенно. В качестве твердых носителей можно использовать крахмал, лактозу, гидрофосфат кальция, кристаллическую целлюлозу, сахар или каолин, а в качестве жидких носителей - стерилизованную воду, полиэтиленгликоль, маннит, неионогенное поверхностно-активное вещество, пищевое масло (например, кукурузное, арахисовое или кунжутное), причем носители должны подходить для активного ингредиента и конкретной лекарственной формы. Также можно использовать добавки, часто входящие в состав фармацевтических композиций, например ароматизаторы, красители, консерванты и антиоксиданты (например, витамин Е, витамин С, бутилгидрокситолуол, бутилгидроксианизол).
Как уже упоминалось в описании изобретения, при внутривенном введении препарата также можно использовать внутрибрюшные инъекции и капельные внутривенные вливания лиофилизированного порошка, растворенного в физиологическом растворе или растворе глюкозы. Приготовление лиофилизированного порошка производится общепринятым способом.
Предлагаемые композиции можно применять перорально, например, в виде таблеток или капсул. Для приготовления препаратов можно смешивать активный ингредиент с одной или несколькими фармацевтически приемлемыми добавками, такими как вспомогательные вещества, связывающие вещества, разрыхлители, скользящие вещества, красители, корригирующие вещества и др., а затем формировать из получившейся смеси порошок, гранулы, таблетки, таблетки с оболочкой, драже, капсулы и т.д. В качестве подходящих вспомогательных веществ можно использовать любые из следующих: лактозу, кукурузный крахмал, сахарид, декстрозу, сорбитол, кристаллическую целлюлозу. В качестве подходящих связывающих веществ можно использовать любые из следующих: поливиниловый спирт, этилцеллюлозу, метилцеллюлозу, аравийскую камедь, трагант, желатин, шеллак, гидроксипропилцеллюлозу, гидроксипропилированный крахмал, поливинилпирролидон. В качестве подходящих разрыхлителей можно использовать любые из следующих: крахмал, агар, желатиновый порошок, кристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, циклодекстрин, пектин. В качестве подходящих скользящих веществ можно использовать любые из следующих: стеарат магния, тальк, полиэтиленгликоль, кремний, отвержденное растительное масло. В качестве подходящих красителей можно использовать любые красители, разрешенные к использованию в фармацевтике. В качестве подходящих корригирующих веществ можно использовать любые из следующих: порошок какао, ментол, масло перечной мяты, очищенный борнеол, корицу. Таблетки и гранулы можно при необходимости покрывать сахаром, желатином и т.д. Лекарственные препараты могут также содержать иные добавки, в том числе инертный разбавитель, консервант (например, сложные эфиры пара-оксибензойной кислоты, сорбиновую кислоту), антиоксидант (например, α-витамин Е, витамин С, цистеин), разлагающие вещества, клейкие вещества, загустители, буфер, подслащивающие вещества, ароматизаторы, отдушки. Таблетки и драже также можно покрывать кишечнорастворимой оболочкой. В качестве жидких форм для перорального введения можно применять, например, эмульсии, сиропы, настойки, суспензии, растворы, содержащие часто используемый инертный разбавитель, например, воду.
Основным преимуществом настоящего изобретения является введение этоксильной группы в 4-м положении ароматического В-кольца природного вещества комбретастатина, которое позволяет улучшить его целевую активность в отношении сосудов опухоли.
В дальнейшем изобретение поясняется подробным описанием примеров его осуществления. Эти примеры лишь поясняют изобретение, они не ограничивают его сущность приведенными конкретными вариантами. В экспериментальных способах, применяемых в нижеследующих примерах, все процедуры выполняются в стандартных условиях либо согласно указаниям производителя, если прямо не указано иное. Все части, проценты и доли указаны по массе, если прямо не указано иное.
Пример 1
Синтез 4-этокси-3-метоксибензальдегида
В литровую колбу с четырьмя горловинами, оборудованную термометром, механической мешалкой и обратным холодильником, поместили 62 г (0,41 моль) 4-гидрокси-3-метоксибензальдегида и 400 мл изопропилового спирта. Смесь перемешивали в течение 20 мин, после чего с помощью капельной воронки с постоянным давлением по капле добавили к ней 120 мл водного раствора, содержащего 5 г 18-краун-6 эфира и 106,3 г (2,66 моль) гидроксида натрия. Затем полученную смесь перемешивали в течение 30 мин и нагрели до 60°С. При этой температуре в течение 5-6 часов вводили 67,3 г (0,62 моль) этилбромида, отслеживая ход реакции посредством тонкослойной хроматографии. После завершения реакции смесь остудили до 15°С, а затем добавили 400 мл воды для заглушения реакции. Полученную смесь трижды экстрагировали эфиром (300 мл × 3). Органический слой промыли водой с рН=7 и высушили безводным сульфатом магния. Часть эфира выпарили методом однократной перегонки, а затем добавили большое количество петролейного эфира для получения осадка сырого продукта. Сырой продукт перекристаллизовали из диэтилового/петролейного эфира для получения 67 г 4-этокси-3-метоксибензальдегида с выходом 91%. Спектр ЯМР 1Н (м.д.), δ: 9,87 (1H, с; -СНО); 7,31(1H, м; 2-АrН); 7,26 (1Н, м; 6-АrН); 6,86 (1Н, м; 5-АrН); 3,98 (2Н, кв; -СН2); 3,73 (3Н, с; -ОСН3); 1,42 (3Н, т; -СН3). МС (m/z): 180 (М+).
Пример 2
Синтез 4-этокси-3-нитробензальдегида
То же, что в примере 1, только вместо 4-гидрокси-3-метоксибензальдегида использовали 68,5 г (0,41 моль) 4-гидрокси-3-нитробензальдегида для получения 68,7 г 4-этокси-3-нитробензальдегида с выходом 86%. Спектр ЯМР 1Н (м.д.), δ: 9,96 (1Н, с; -СНО); 7,73 (1Н, м; 2-АrН); 7,58 (1Н, м; 6-ArН); 7,33 (1Н, м; 5-ArН); 4,15 (2Н, кв; -СН2); 3,82 (3Н, с; -ОСН3); 1,53(3Н, т; -СН3). МС (m/z): 195 (М+).
Пример 3
Синтез 4-этокси-3-гидроксибензальдегида
Шаг 1: в колбу с тремя горловинами в аргоновой атмосфере последовательно поместили 54 г (0,3 моль) 4-этокси-3-метоксибензальдегида, 130 г (2,1 моль) этиленгликоля и 133 г (0,9 моль) триэтилортоформиата. Смесь нагрели с обратным холодильником при 100°С, а затем в качестве катализатора добавили 1 мл эфирного раствора трехфтористого бора. Реакция смеси продолжалась в течение 24 часов, ход реакции отслеживали посредством тонкослойной хроматографии. Затем смесь остудили до комнатной температуры и добавили 200 мл 15% водного раствора гидроксида натрия. Полученную смесь экстрагировали 300 мл диэтилового эфира. Экстракт промыли насыщенным рассолом, высушили безводным сульфатом магния и перегнали при пониженном давлении для удаления этиленгликоля и триэтилортоформиата до получения желтого маслянистого продукта.
Шаг 2: в колбу поместили 200 мл 1,28М тетрагидрофуранового раствора дифенилфосфина лития, затем отдельными порциями добавили 56 г (0,25 моль) ранее подготовленного ацеталя. Затем полученную смесь в течение 3-4 часов перемешивали при комнатной температуре, отслеживая ход реакции посредством тонкослойной хроматографии. Для заглушения реакции добавили воду, а затем внесли 200 мл 30% водного раствора гидроксида натрия. Полученную смесь экстрагировали 300 мл диэтилового эфира. Водный слой охладили и подкислили соляной кислотой до рН=3-4, а затем экстрагировали 500 мл диэтилового эфира. Эфирные экстракты объединили, промыли насыщенным рассолом и высушили безводным сульфатом магния. Высушенный экстракт отфильтровали и при пониженном давлении удалили растворитель для получения желтого сухого остатка. Сырой продукт перекристаллизовали из бензола/петролейного эфира для получения 35,3 г бледно-желтого кристаллического сухого остатка с выходом 85%. Спектр ЯМР 1Н (м.д.), δ: 9,90 (1Н, с; -СНО); 7,32 (1Н, м; 2-ArH); 7,27 (1Н, м; 6-ArH); 6,89 (1Н, м; 5-ArH); 4,88 (1Н, шир; -ОН); 4,17 (2Н, кв; -СН2); 1,53 (3Н, т; -СН3). Спектр ЯМР 13С (мл) д: 192,0 (СНО), 157,6 (4-ArC), 143,3 (3-ArC), 129,6 (1-ArC), 124,5 (6-ArC), 116,7 (2-ArC), 116,6 (5-ArC), 82,1 (-ОСН2), 23,5 (-СН3). МС (m/z): 166 (M+).
Пример 4
Синтез (Z)-1-(3,4,5-триметокси)фенил-2-(3'-гидрокси-4'-этокси)фенилэтилена (этоксикомбретастатина)
Шаг 1: в колбу объемом 500 мл с четырьмя горловинами в аргоновой атмосфере поместили 11,0 г (0,066 моль) 4-этокси-3-гидроксибензальдегида, 21,1 г (0,076 моль) трифенилметилхлорида и 42 мл сухого тетрагидрофурана. Смесь перемешивали до получения однородной смеси. Затем в смесь медленно по капле добавили 1,3 мл триэтиламина, после чего перемешивали в течение 1 часа. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции добавили 50 мл воды для заглушения реакции. Затем еще 30 минут перемешивали смесь, а потом добавили 100 мл этилацетата, чтобы растворить флоккулированный остаток. Для выделения бледно-желтого сухого осадка добавили 250 мл н-гептана. Осадок отфильтровали, дважды промыли водой, а затем промыли смесью 10 мл этилацетата и 20 мл петролейного эфира для получения мелочно-белого кристаллического сухого остатка. Затем сухой остаток перекристаллизовали из этилацетата/петролейного эфира для получения 25 г белого крупнокристаллического сухого остатка с выходом 93%. Спектр ЯМР 1Н (м.д.), δ: 9,91 (1Н, с; -СНО); 7,33 (1Н, м; 2-ArН); 7,26 (1Н, м; 6-АrН); 7,19 (15Н, м; Тr-Н); 6,89 (1Н, с; 5-ArН); 4,17 (2Н, кв; -СН2); 1,53 (3H, т; -СН3).
Шаг 2: в аргоновой атмосфере в 300 мл тетрагидрофурана взвесили 15 г (28,7 ммоль) триметоксифенилметилентрифенилфосфония бромида, полученную смесь охладили до -15°С. Медленно по каплям добавили 22 мл раствора н-бутиллития в гексане (ок. 1,6 моль/л), а затем реакционную смесь перемешивали в течение 1 часа. Медленно по каплям добавили раствор 11,8 г (29 ммоль) ранее подготовленного альдегида в 24 мл тетрагидрофурана. Ночью смесь перемешивали, следили за ходом реакции посредством тонкослойной хроматографии, при этом температура смеси медленно дошла до комнатной. Отделили органический слой и удалили растворитель. Сырой продукт очистили методом колоночной флеш-хроматографии (колонка с силикагелем, 4:1 н-гексан/этилацетат) для получения 13,7 г белого кристаллического сухого остатка с выходом 83,5%. Спектр ЯМР 1Н (м.д.), δ: 7,12 (15Н, м; Tr-Н); 6,97 (1Н, д; 2'-Н); 6,81 (1Н, дд; 6'-Н); 6,75 (1Н, д; 5'-Н); 6,59 (2Н, с; 2, 6-Н); 6,47 (1Н, д; 1а-Н); 6,41 (1Н, д; J=12 Гц; 1а'-Н); 4,13 (2Н, кв; -СН2); 3,88 (3H, с; 4-ОСН3); 3,71 (6Н, с; 3,5-ОСН3); 1,55 (3H, т; -СН3).
Шаг 3: при комнатной температуре растворили в 20 мл толуола 9,6 г (16,8 ммоль) вышеупомянутого продукта реакции Виттига шага 2. По каплям добавили 4 мл 37% водного раствора соляной кислоты (содержащего 0,2 мл трифторуксусной кислоты). За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции добавили воду для остановки реакции. Реакционную смесь охладили до 0-5°С для получения кристаллического остатка, при помешивании. Сухой остаток отфильтровали и высушили для получения 5,1 г белого кристаллического остатка с выходом 92%. Спектр ЯМР 1Н (м.д.), δ: 7,02 (1Н, д; 2'-Н); 6,94 (1Н, дд; 6'-Н); 6,80 (1Н, д; 5'-Н); 6,62(2Н, с; 2,6-Н); 6,46 (1Н, д; J=12 Гц; 1а-Н); 6,40 (1Н, д; J=12 Гц; 1а'-Н); 5,51 (1Н, шир; ОН); 4,16 (2Н, кв; -CH2); 3,86 (3Н, с; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 1,52 (3Н, т; -СН3). МС (m/z): 330 (M+). Масс-спектр высокого разрешения: рассчитано 330,15, найдено 330,16.
Пример 5
Синтез (Z)-1-(3,4,5-триметокси)фенил-2-(3'-амино-4'-этокси)фенилэтилена (3'-аминоэтоксикомбретастатина)
Шаг 1: в фотохимическом синтезаторе в аргоновой атмосфере взвесили в 300 мл тетрагидрофурана 15 г (28,7 ммоль) триметоксифенилметилентрифенилфосфония бромида, полученную смесь охладили до -15°С. Медленно по каплям во взвесь добавили 22 мл раствора н-бутиллития в гексане (ок. 1,6 моль/л), а затем реакционную смесь перемешивали в течение 1 часа. Затем включили ультрафиолетовое освещение с длиной волны 254 нм и медленно по каплям под воздействием УФ излучения в реакционную смесь добавили раствор 5,7 г (29 ммоль) 4-этокси-3-нитробензальдегида в 24 мл тетрагидрофурана. Ночью смесь перемешивали, следили за ходом реакции посредством тонкослойной хроматографии, при этом температура смеси медленно достигла комнатной. На следующий день выключили свет, реакционную смесь снова охладили до -5°С и добавили рассол для заглушения реакции. Отделили органический слой и удалили растворитель методом однократной перегонки. Сырой продукт очистили методом колоночной хроматографии обычного давления (колонка с силикагелем, 4:1 н-гексан/этилацетат) для получения 6,5 г бледно-желтого кристаллического сухого остатка с выходом 63%. Спектр ЯМР 1Н (м.д.), δ: 7,32 (1Н, д; 2'-Н); 7,16 (1Н, дд; 6'-Н); 6,90 (1Н, д; 5'-Н); 6,64 (2Н, с; 2,6-Н); 6,49 (1Н, д; J=12,2 Гц; 1a-H); 6,43 (1Н, д; J=12,2 Гц; 1а'-Н); 4,18 (2Н, кв; -СН2); 3,86 (3Н, с; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 1,55 (3Н, т; -СН3). МС (m/z): 359 (М+). Масс-спектр высокого разрешения: рассчитано 359,14, найдено 359,13.
Шаг 2: растворили 4,1 г (10,8 ммоль) (Z)-1-(3,4,5-триметокси)фенил-2-(3'-нитро-4'-этокси)фенилэтилена в 350 мл уксусной кислоты, а затем добавили 100 г цинковой пыли (<10 µм). Смесь перемешивали в течение 6 часов. После завершения реакции смесь отфильтровали посредством воронки Бюхнера, выстланной 1-сантиметровым слоем диатомита, после чего роторным испарителем конденсировали фильтрат. После этого сырой продукт очистили методом колоночной флеш-хроматографии (4:1 н-гексан/этилацетат), а затем перекристаллизовали из ок. 9:1 н-гексан/этилацетат для получения 2,7 г бесцветного кристаллического сухого остатка с выходом 77%. Спектр ЯМР 1Н (м.д.), δ: 7,08 (1Н, д; 2'-Н); 6,92 (1Н, дд; 6'-Н); 6,76 (1Н, д; 5'-Н); 6,62 (2Н, с; 2,6-Н); 6,49 (1Н, д; J=12,2 Гц; 1а-Н); 6,43 (1Н, д; J=12,2 Гц; 1а'-Н); 4,73-4,25 (2Н, шир; NH2); 4,18 (2Н, кв; -CH2); 3,86 (3H, c; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 1,55 (3Н, т; -СН3). МС (m/z): 329 (М+). Масс-спектр высокого разрешения: рассчитано 329,16, найдено 329,18.
Пример 6
Синтез двунатриевой соли фосфата этоксикомбретастатина
Шаг 1: в литровую колбу с четырьмя горловинами в аргоновой атмосфере поместили 41,6 г (126 ммоль) комбретастатина, растворенного в 400 мл сухого ацетонитрила. Смесь охладили до
-25°С, а затем добавили 61 мл четыреххлористого углерода. Смесь перемешивали в течение 5 минут, после чего добавили 47 мл диизопропилэтиламина и 1,5 г 4-диметиламинопиридина (DMAP). Через 1 минуту в реакционную смесь медленно добавили 41 мл дибензилфосфоната (80%), поддерживая температуру ниже -10°С в течение последующих 3,5 часов. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции добавили 100 мл 0,5М KH2PO4, после чего температура реакционной смеси естественным образом достигла комнатной температуры. Смесь экстрагировали этилацетатом, собрали органический слой, последовательно промыли его дистиллированной водой и рассолом, затем высушили безводным сульфатом магния и перегнали при пониженном давлении для удаления растворителей до получения непрозрачного маслянистого продукта. Сырой продукт очистили методом колоночной флеш-хроматографии (колонка с силикагелем, 3:2 петролейный эфир/этилацетат) для получения 75 г бледно-желтого маслянистого продукта, а затем перекристаллизовали из н-гексана/этилацетата для получения 68,4 г бесцветного игольчатого кристаллического сухого остатка с выходом 92%.
Шаг 2: в литровую колбу с четырьмя горловинами в аргоновой атмосфере поместили 65 г (110 ммоль) ранее высушенного бензилфосфата, растворенного в 250 мл сухого ацетонитрила при 15°С. Смесь перемешали и быстро добавили 45 мл триметилбромосилана (TMBS). Через 5-10 минут добавили 18 г метилата натрия, растворенного в 70 мл безводного метанола, после чего смесь моментально превратилась в молочно-белую взвесь. Через полчаса в реакционную смесь добавили 36 мл безводного метанола и 36 мл ацетона; ночью смесь перемешивали. Смесь отфильтровали для получения белого сухого остатка, который затем промыли безводным метанолом и ацетоном и высушили в вакууме. Сухой остаток перекристаллизовали из воды/метанола/ацетона для получения 43 г белого порошка с выходом 86%. Спектр ЯМР 1Н (м.д.), δ: 7,11 (1Н, д; 2'-Н); 6,98 (1Н, дд; 6'-Н); 6,87 (1Н, д; 5'-Н); 6,64 (2Н, с; 2,6-Н); 6,47 (1Н, д; J=12 Гц; 1а-Н); 6,42 (1Н, д; J=12 Гц; 1a'-H); 4,18 (2Н, кв; -СН2); 3,86 (3Н, с; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 1,52 (3Н, т; -СН3). МС (m/z): 454 (М+). Масс-спектр высокого разрешения: рассчитано 454,08, найдено 454,06.
Пример 7
Синтез внутренней соли этоксикомбретастатин фосфорилхолина
Шаг 1: в сухую колбу объемом 500 мл с тремя горловинами поместили раствор 68 г (0,5 моль) сухого трихлорида фосфора в 100 мл дихлорметана. Смесь охладили для поддержания температуры в 0°С, а затем по каплям добавили раствор 31 г (0,5 моль) сухого этиленгликоля в 100 мл дихлорметана. После этого реакционной смеси дали достичь комнатной температуры, после чего реакция продолжалась в течение еще 3 часов.
Затем удалили растворитель, а остаток перегнали при пониженном давлении для сбора фракции при 60°С/20 мм рт.ст., получив 41 г 2-хлор-1,3,2-диоксафосфолана с выходом 65%.
Спектр ЯМР 1Н (CDCl3, 500М), δ: 4,46 (2Н, м; а-СНСН-); 4,24 (2Н, м, е-СНСН-).
Шаг 2: в колбу объемом 100 мл с тремя горловинами поместили 50 мл безводного бензола и 25,3 г (0,2 моль) 2-хлор-1,3,2-диоксафосфолана, медленно вводя в раствор кислород. После ок. одного часа введения кислорода смесь нагрели с обратным холодильником и поддерживали температуру в течение 5-6 часов. Затем реакционную смесь охладили до комнатной температуры, удалили растворитель, а остаток перегнали при пониженном давлении для сбора фракции при 90°С/1 мм рт.ст., получив 12,3 г 2-хлор-1,3,2-диоксаоксидофосфолана с выходом 43%. Спектр ЯМР 1Н (CDCl3, 500М) д: 4,64 (2Н, м; а-СНСН-); 4,56 (2Н, м, е-СНСН-).
Шаг 3: в аргоновой атмосфере растворили в 20 мл сухого бензола 3,3 г (10 ммоль) этоксикомбретастатина и 1,0 г (10 ммоль) триэтиламина. Смесь охладили до -40°С, а затем по каплям, постоянно перемешивая, добавили раствор 1,42 г (10 ммоль) 2-хлор-1,3,2-диоксаоксидофосфолана в 20 мл бензола. После этого реакционную смесь оставили на полчаса так, затем вернули к комнатной температуре, после чего перемешивали в течение еще 10 часов. Затем смесь отфильтровали для удаления триэтиламингидрохлорида, после чего фильтрат перегнали для частичного удаления растворителя. Остаток промыли в 15 мл 15% карбоната натрия, экстрагировали 50 мл диэтилового эфира, после чего снова экстрагировали водный слой 50 мл диэтилового эфира. Собрали эфирный слой, промыли его 10% карбонатом натрия (20 мл × 2), высушили, отфильтровали, а затем перегнали для удаления диэтилового эфира, получив 3,5 г бледно-желтого фосфатного сухого остатка с выходом 81%. Вышеупомянутый фосфат затем растворили в 50 мл ацетонитрила, затем добавили 5 мл водного раствора триметиламина (28%), перемешивали в течение 20 часов при комнатной температуре, отслеживая ход реакции посредством тонкослойной хроматографии. После завершения реакции в смесь добавили 50 мл ацетона, охладили до -30°С, перемешивали до получения кристаллического сухого остатка, затем отфильтровали и высушили для получения белого сырого продукта. Сырой продукт перекристаллизовали из 95% этанола для получения 3 г белого кристаллического сухого остатка с выходом 76%. Спектр ЯМР 1H (м.д.), δ: 7,27 (1Н, д; 2'-Н); 6,63 (1Н, дд; 6'-Н); 6,51 (1Н, д; 5'-Н); 6,46 (1Н, д; 1а-Н); 6,380 (2Н, с; 2,6-Н); 6,20 (1Н, д; 1а'-Н); 4,62 (2Н, м; а-СНСН-); 4,53 (2Н, м, е-СНСН-); 4,18 (2Н, кв; -ОСН2); 3,86 (3Н, с; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 3,18 (9Н, с; NMe3); 1,52 (3Н, т; -СН3). МС (m/z): 495 (М+). Масс-спектр высокого разрешения: рассчитано 495,20, найдено 495,22.
Пример 8
Синтез 3'-аминоэтоксикомбретастатин глицинамида
Шаг 1: 4,28 г (13 ммоль) 3'-аминоэтоксикомбретастатина, 4,75 г (16 ммоль) Fmoc-глицина и 22,7 г (51,6 ммоль) ВОР-агента растворили в 100 мл ДМФ. Затем полученную смесь нагревали до 60°С, после чего реакция смеси продолжалась в течение 2 часов с постоянным перемешиванием. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции смесь остудили и тщательно смешали с 100 мл насыщенного раствора бикарбоната натрия. Затем смесь экстрагировали дихлорметаном (120 мл × 3), после чего высушили органический слой безводным сульфатом магния и сконденсировали при пониженном давлении. Полученный сырой продукт затем очистили методом колоночной флеш-хроматографии (колонка с силикагелем, 2:1 н-гексан/этилацетат) для получения 3,3 г белой пенящейся субстанции с выходом 42%. Спектр ЯМР 1Н (CDCl3, 500М), δ: 9,61 (1H, шир.с; -NH); 7,74 (2Н, м; Fmoc); 7,59 (2Н, д; J=6,2 Гц; Fmoc); 7,37(2Н, м; Fmoc); 7,29(2Н, м; Fmoc); 7,08 (1Н, д; 2'-Н); 6,92 (1Н, дд; 6'-Н); 6,76 (1Н,д; 5'-Н); 6,62 (2Н,с; 2,6-Н); 6,49 (1Н, д; J=12,2 Гц, 1а-Н); 6,43 (1Н, д; J=12,2 Гц; 1a'-Н); 5,79 (1Н, шир.с; Gly-NH); 4,38 (2Н, д; J=7,0 Гц; Fmoc); 4,22 (1Н, т; J=7,0 Гц; Fmoc); 4,18 (2Н, кв; -СН2); 4,04 (2Н, м; Gly-CH2); 3,86 (3H, с; 4-ОСНз); 3,70 (6Н, с; 3,5-ОСН3); 1,55 (3H, т; -СН3). МС (m/z): 608 (М+). Масс-спектр высокого разрешения: рассчитано 608,25, найдено 608,27.
Шаг 2: 2,2 г (3,6 ммоль) вышеупомянутого (Z)-1-(3,4,5-триметокси)фенил-2-(3'-амино-4' -этокси)фенилэтилен Fmoc-глицинамида растворили в 40 мл метанола. В раствор добавили 2 мл 2N раствора гидроксида натрия, после чего реакция продолжалась в течение 3 часов с постоянным помешиванием, при комнатной температуре. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции смесь остудили и тщательно смешали с 20 мл насыщенного раствора бикарбоната натрия. Затем смесь экстрагировали дихлорметаном (50 мл х 3), после чего высушили органический слой безводным сульфатом магния и сконденсировали при пониженном давлении. Полученный сырой продукт затем очистили методом колоночной хроматографии обычного давления (колонка с силикагелем, 9:1 дихлорметан/метанол) для получения 0,97 г бесцветной пенящейся субстанции с выходом 70%. Спектр ЯМР 1Н (CDCl3, 500М), δ: 9,61 (1Н, шир.с; -NH); 7,08 (1Н, д; 2'-Н); 6,92 (1Н, дд; 6'-Н); 6,76 (1Н, д; 5'-Н); 6,62 (2Н,с; 2,6-Н); 6,49 (1Н, д; J=12,2 Гц; 1а-Н); 6,43 (1Н, д; J=12,2 Гц; 1а'-Н); 4,81-4,32 (2Н, шир; Gly-NH2); 4,18 (2Н, кв; -СН2); 4,04 (2Н, шир. с; Gly-CH2); 3,86 (3H, с; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 1,55 (3H, т; -СН3). МС (m/z): 386 (М+). Масс-спектр высокого разрешения: рассчитано 386,18, найдено 386,20.
Пример 9
Синтез 3'-аминоэтоксикомбретастатин серинамида
Шаг 1: 4,28 г (13 ммоль) 3'-аминоэтоксикомбретастатина, 5,89 г (16 ммоль) Fmoc-серина, 3,37 г (16 ммоль) ДЦКД и 2,44 г HOBt растворили в 80 мл ДМФ. Реакция смеси продолжалась в течение 5 часов с постоянным перемешиванием при комнатной температуре, ход реакции отслеживали посредством тонкослойной хроматографии. После завершения реакции смесь остудили и тщательно смешали с 50 мл этилацетата, чтобы разбавить смесь. Затем смесь отфильтровали, высушили безводным сульфатом магния и сконденсировали при пониженном давлении. Полученный сырой продукт затем очистили методом колоночной флеш-хроматографии (колонка с силикагелем, 2:1 н-гексан/этилацетат) для получения 5,1 г белой пенящейся субстанции с выходом 61%. Спектр ЯМР 1Н (CDCl3, 500M), δ: 9,73 (1Н, шир.с; -NH); 7,73 (2Н, м; Fmoc); 7,56 (2Н, д; J=6,2 Гц; Fmoc); 7,35 (2Н, м; Fmoc); 7,22 (2Н, м; Fmoc); 7,05 (1Н, д; 2'-Н); 6,91 (1Н, дд; 6'-Н); 6,74 (1Н, д; 5'-Н); 6,60 (2Н, с; 2,6-Н); 6,51 (1Н, д; J=12,2 Гц; 1а-Н); 6,43 (1Н, д; J=12,2 Гц, 1a'-H); 5,82 (1Н, шир.с; Ser-NH); 4,63 (1Н, шир.с; Ser-OH); 4,38 (2Н, д; J=7,0 Гц; Fmoc); 4,22 (1Н, т; J=7,0 Гц; Fmoc); 4,18 (2Н, кв; -CH2); 3,91 (1Н, м; Ser-CH); 3,85 (3Н, с; 4-ОСН3); 3,71 (6Н, с; 3,5-ОСН3); 2,66 (2Н, м; Ser-СН2); 1,56 (3Н, т; -СН3). МС (m/z): 638 (М+). Масс-спектр высокого разрешения: рассчитано 638,26, найдено 638,27.
Шаг 2: 1,9 г (3,0 ммоль) вышеупомянутого (Z)-1-(3,4,5-триметокси)фенил-2-(3'-амино-4'-этокси)фенилэтилен Fmoc-серинамида растворили в смеси растворителей, состоящей из 20 мл метанола и 20 мл дихлорметана. В раствор добавили 3,4 мл 2N раствора гидроксида натрия, после чего реакция продолжалась в течение 24 часов с постоянным помешиванием при комнатной температуре. За ходом реакции следили посредством тонкослойной хроматографии. После завершения реакции смесь остудили и тщательно смешали с 20 мл насыщенного раствора хлорида натрия. Затем смесь экстрагировали дихлорметаном (50 мл × 3), после чего высушили органический слой безводным сульфатом магния и сконденсировали при пониженном давлении. Полученный сырой продукт затем очистили методом колоночной хроматографии обычного давления (колонка с силикагелем, 9:1 дихлорметан/метанол) для получения 0,62 г бесцветной пенящейся субстанции с выходом 50%. Спектр ЯМР 1Н (CDCl3, 500М), δ: 9,75 (1Н, шир.с; -NH); 7,08 (1Н, д; 2'-Н); 6,94 (1Н, дд; 6'-Н); 6,75 (1Н, д; 5'-Н); 6,63 (2Н, с; 2,6-Н); 6,53 (1Н, д; J=12,2 Гц; 1а-Н); 6,45 (1Н, д; J=12,2 Гц; 1a'-H); 5,51-4,72 (2Н, шир; Ser-NH2); 4,52 (1Н, шир.с; Ser-OH); 4,19 (2Н, кв; -СН2); 3,92 (1Н, м; Ser-CH); 3,86 (3H, c; 4-ОСН3); 3,72 (6Н, с; 3,5-ОСН3); 2,68 (2Н, м; Ser-СН2); 1,57 (3Н, т; -СН3). МС (m/z): 416 (M+). Масс-спектр высокого разрешения: рассчитано 416,19, найдено 416,20.
Сравнительный пример
Синтез пропоксикомбретастатина
То же, что в примере 1, только вместо этилбромида использовали пропилбромид для получения 4-пропокси-3-метоксибензальдегида. Затем согласно примерам 3 и 4 получали пропоксикомбретастатин. Спектр ЯМР 1Н (м.д.), δ: 7,02 (1Н, д; 2'-Н); 6,94 (1Н, дд; 6'-Н); 6,80 (1Н, д; 5'-Н); 6,62 (2Н, с; 2,6-Н); 6,46 (1Н, д; J=12 Гц; 1а-Н); 6,40 (1Н, д; J=12 Гц; 1a'-H); 5,51 (1Н, шир; ОН); 4,16 (2Н, кв; -CH2); 3,86 (3Н, с; 4-ОСН3); 3,70 (6Н, с; 3,5-ОСН3); 2,27 (2Н, м; -СН2); 1,18 (3Н, т; -СН3). МС (m/z): 344 (М+). Масс-спектр высокого разрешения: рассчитано 344,18, найдено 344,16.
Пример 10
Оценка противораковой активности в пробирке (in vitro) Выращенную в пробирке клетку опухоли подвергали воздействию производных этоксикомбретастатина в течение 72 часов, оценивая подавление пролиферации опухоли посредством МТТ и SRB (сульфородамин В) тестов. В таблице 1 приведены результаты в сравнении с КА-4.
Линия клеток: Н460 - клетки рака легких человека, SGC7901 - клетки рака желудка человека, НТ-29 - клетки рака толстой кишки человека, Bel-7402 - клетки рака печени человека.
План эксперимента: в течение 72 часов клетки инкубировали с различными концентрациями соединений (100, 10, 1, 0,1, 0,01, 0,001 µМ). Для оценки эффективности подавления пролиферации клеток соединениями использовали SRB-тест (сульфородамин В). Вычисляли коэффициент подавления, по Логит-модели вычисляли IC50 (ингибирующую концентрацию 50) как функцию коэффициента подавления. Также сравнивали противораковую активность соединений в пробирке (in vitro).
Для выражения коэффициента подавления использовали следующее равенство:
Коэффициент подавления (%)=[(среднее значение ОП контрольной группы - среднее значение ОП экспериментальной группы)/среднее значение ОП контрольной группы]×100%.
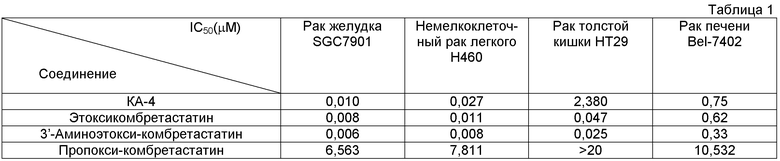
Результат показал, что все производные этоксикомбретастатина обладают более эффективной противораковой активностью в пробирке (in vitro), чем природный комбретастатин А-4, по различным линиям раковых клеток. В частности, в случае клеток рака толстой кишки, этоксикомбретастатины продемонстрировали противоопухолевую активность в 50-95 раз выше, нежели комбретастатин А-4, тогда как пропоксикомбретастатин (продукт, полученный в сравнительном примере) показал низкую противоопухолевую активность.
Пример 11
Оценка подавления реваскуляризации в пробирке (in vitro)
Оценивали антиангиогенное действие этоксикомбретастатинов на примере эндотелиальных клеток пупочной вены человека (HUVEC), тем же способом, что и в примере 10.
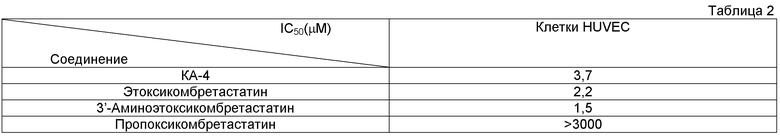
Результат продемонстрировал высокую подавляющую тубулин-связывающую активность этоксикомбретастатинов, что говорит о том, что этоксикомбретастатины представляют собой новый перспективный класс противораковых лекарств васкулярной направленности. У пропоксикомбретастатина же (продукта, полученного в сравнительном примере), напротив, такая активность практически полностью отсутствовала.
Пример 12
Приготовление лиофилизированного порошка этоксикомбретастатинов
Материалы дозировали по массе в строгом соответствии с формулами (таблица 3). Заданное количество маннита развели в 80% заданного общего количества воды для инъекций до получения прозрачного раствора, а затем добавили 0,1% (г/мл) инъекционного вещества - активированного угля. Смесь перемешали до достижения однородности, осаждали в течение 10 мин, а затем профильтровали через мембрану с размером пор 0,45 µм. После этого добавили оставшуюся воду для инъекций. Раствор снова отфильтровали через мембрану с размером пор 0,22 µм. Для проверки качества препарата измерили значение рН и содержание продукта. Затем некоторое количество раствора разлили по флаконам, после чего произвели лиофилизацию. Во флаконы ввели азот, закрыли флаконы крышками и осуществили маркировку. Затем флаконы упаковали по коробкам и провели контрольные испытания для получения готового продукта (в связи с относительной чувствительностью производных комбретастатина к температуре и свету, все процедуры проводили в темноте).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ФТОРАЛКОКСИКОМБРЕТАСТАТИНА, СПОСОБЫ ИХ ПРОИЗВОДСТВА И ИСПОЛЬЗОВАНИЯ | 2006 |
|
RU2417216C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4,5-ДИАРИЛАЗОЛОВ | 2022 |
|
RU2799312C2 |
| ПРОИЗВОДНЫЕ 4-АРИЛКУМАРИНОВ И ПРОТИВООПУХОЛЕВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2010 |
|
RU2440998C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛО-СТИЛЬБЕНА (СОЕДИНЕНИЯ А-104815) | 2021 |
|
RU2786432C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,6-7-ТРИМЕТОКСИ-3-ЭТИЛ-1,4-НАФТОХИНОНА | 2005 |
|
RU2277083C1 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| Производное 1'-бромо-2',3',4'-триметоксибензо[5',6':4,5]-(aR, 1S)-1-ацетамидо-6,7-дигидроциклогепта-[3,4-f]-1Н-индола и его применение | 2016 |
|
RU2630303C1 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ФТАЛИДА | 2005 |
|
RU2394569C2 |
| ПРОИЗВОДНОЕ 1',2',3'-ТРИМЕТОКСИБЕНЗО[4',5':4,5]-6,7-ДИГИДРОЦИКЛОГЕПТА-[3,2-f]-1H-1-МЕТИЛИНДОЛА И ЕГО ПРИМЕНЕНИЕ | 2012 |
|
RU2500671C1 |
| ПРОИЗВОДНЫЕ ГЛИЦЕРИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2040521C1 |
Изобретение относится к новым этоксикомбретастатинам формулы (I), обладающим противораковой активностью, к фармацевтической композиции, содержащей предлагаемые соединения, а также к способам получения некоторых из предлагаемых соединений.
 ,
,
где R - гидроксильная группа, аминогруппа, фосфатная группа, выбранная из динатрийфосфата или фосфата аммония, или внутренняя соль фосфорилхолина, -NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, a m - целое число от 1 до 3. 4 н. и 1 з.п. ф-лы, 3 табл., 2 ил., 13 пр.
1. Соединение по формуле (I)
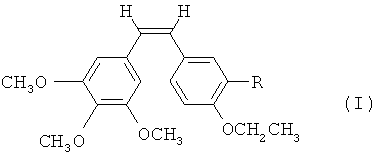
в котором R - гидроксильная группа, аминогруппа, фосфатная группа, выбранная из динатрийфосфата или фосфата аммония, или внутренняя соль фосфорилхолина, -NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, a m- целое число от 1 до 3.
2. Способ получения соединения по п.1, в котором R представляет собой фосфатную группу, предусматривающий следующие шаги:
(1) 4-гидрокси-3-метоксибензальдегид II в присутствии катализатора межфазного переноса этилируют посредством этилбромида, чтобы синтезировать 4-этокси-3-метоксибензальдегид, представленный формулой III;
(2) с помощью дифенилфосфина лития селективно замещают m-метильную группу 4-этокси-3-метоксибензальдегида III гидроксильной группой, чтобы синтезировать 4-этокси-3-гидроксибензальдегид, представленный формулой IV;
(3) гидроксильную группу защищают посредством трифенилхлорметана, затем 4-этокси-3-гидроксибензальдегид IV с защищенной гидроксильной группой посредством реакции Виттига взаимодействует с илидом 3,4,5-триметоксибензилтрифенилфосфония, после чего из полученного соединения удаляют защиту для получения этоксикомбретастатина, представленного формулой VI;
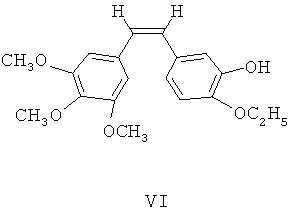
(4) этоксикомбретастатин VI фосфорилируют посредством фосфорилирующего агента, чтобы синтезировать его фосфатные производные;
(5) в щелочных условиях фосфатные производные этоксикомбретастатина превращают в фосфат этоксикомбретастатина.
3. Способ по п.2, в котором в качестве фосфорилирующего агента на шаге (4) используют дибензилфосфонат или 2-хлор-1,3,2-диоксафосфолан.
4. Способ получения соединения по п.1, в котором R - NH(COCHR'NH)m-H, где R' - водород, боковая цепь природной аминокислоты или фенильная группа, a m - целое число от 1 до 3, предусматривающий следующие шаги:
(а) 4-гидрокси-3-нитробензальдегид IX в присутствии катализатора межфазного переноса этилируют посредством этилбромида, чтобы синтезировать 4-этокси-3-нитробензальдегид, представленный формулой X;
(б) под воздействием ультрафиолетового излучения с длиной волны 254 нм, 4-этокси-3-нитробензальдегид X посредством реакции Виттига взаимодействует с илидом 3,4,5-триметоксибензилтрифенилфосфина для получения (Z)-1-(3,4,5-триметокси)фенил-2-(3'-нитро-4'-этокси)фенилэтилена, представленного формулой XI;
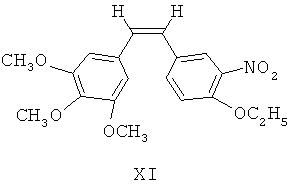
(в) (Z)-1-(3,4,5-триметокси)фенил-2-(3'-нитро-4'-этокси)фенилэтилен XI восстанавливают посредством восстанавливающих агентов для получения 3'-амино этоксикомбретастатина XII;
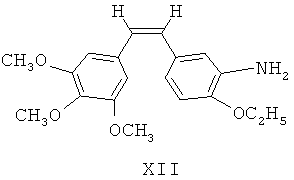
(г) 3'-амино этоксикомбретастатин XII взаимодействует с аминокислотными производными для получения соответствующих аминоамидных производных;
(д) в щелочных условиях вышеупомянутые аминоамидные производные превращают в аминоамиды 3'-амино этоксикомбретастатина.
5. Фармацевтическая композиция, обладающая противораковой активностью, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
| US 5430062А, 04.07.1995 | |||
| М | |||
| Cushman et al, Synthesis and Evaluation of Analogues of (Z)-l-(4-Methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethane as Potential Cytotoxic and Antimitotic Agents | |||
| J | |||
| Med | |||
| Chem., 1992, т.35, №14, с.2293-2306 | |||
| RU 2001106631 A, 20.03.2004. |

