Химические соединения
Область техники, к которой относится изобретение
Это изобретение касается некоторых производных ненасыщенных жирных кислот терапевтически активных 1,3-диоксолан нуклеозидных аналогов и фармацевтических препаратов, их содержащих. Упомянутые производные цитируются здесь как "Соединения с формулой I". Соединения с формулой I могут применяться при лечении раковых заболеваний. Также включается лечение как твердых опухолей, так и гематологических раков, таких как лейкемии, лимфомы и множественные миеломы.
Уровень техники
Нуклеозидные аналоги, производные природных нуклеозидов, известных как строительные блоки ДНК и РНК, являются эффективными для клинического лечения рака или вирусных заболеваний у человека, несмотря на то, что ранее такие соединения рассматривали в качестве антитуберкулезных средств. Такие соединения были зарегистрированы на рынке более чем 40 лет назад, и примерно 35 продуктов в настоящее время находятся в ежедневном употреблении. Природные нуклеозиды, показанные ниже, сконструированы из двух классов азотных оснований: пуринов, представленных аденином и гуанином, и пиримидинов, представленных тимином, урацилом и цитозином, и из моносахаридов рибозы или дезоксирибозы.
Пурины:
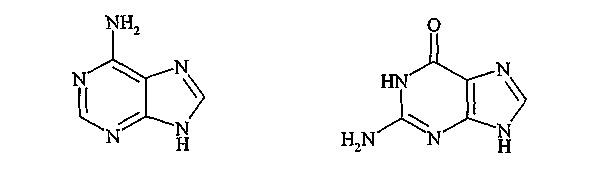
аденин гуанин
Пиримидины:
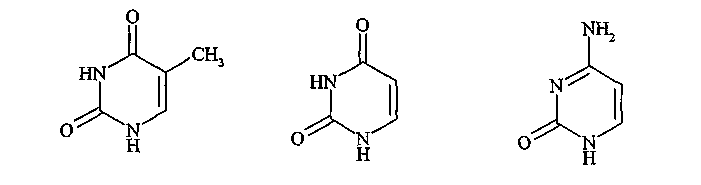
тимин урацил цитозин
Моносахариды:

рибоза 2-дезоксирибоза
Все природные нуклеозиды существуют в так называемой β-D конфигурации, как показано в формуле A ниже. И азотное основание, и гидроксиметильная боковая цепь кольца сахара расположены по одну сторону (цис-) плоскости кольца сахара.
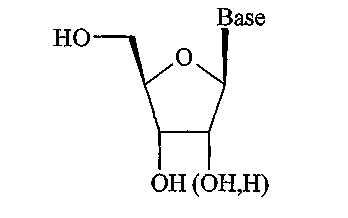
Формула А
Если две группы расположены по разные стороны (транс-), соединение цитируется как изомер. Для того чтобы получить нуклеозидные производные с антираковой или антивирусной активностью, были получены химические модификации и в азотном основании, и/или в моносахариде. Например, прибавление атомов галогена, замещение OH групп другими функциональными группами или стереохимическая замена рибозы на арабинозу может приводить к продуктам с потенциальным терапевтическим преимуществом. Во многих продуктах моносахаридное кольцо сохраняют, тогда как в других кольцо сахара превращают в цепь. Нуклеозидные аналоги представляют собой малые молекулы с растворимостью в воде от хорошей до отличной.
Всесторонние исследования и научные разработки, предпринятые в области нуклеозидных аналогов в рамках всемирной эпидемии СПИДа, содействовавшие получению основных знаний и пониманию механизма действия, видоизменения в профиле активности в зависимости от химических модификаций и т.д., также являются значимыми в области лечения рака.
Общим недостатком многих лекарств, включая нуклеозидные аналоги, является низкая активность и низкая специфичность при лечении рассматриваемого фактического заболевания. Некоторые из этих проблем могут быть связаны с присущей самому лекарственному средству активностью, некоторые могут быть связаны с определенными резистентными механизмами (или присущими пациенту, или приобретенными в процессе лечения, например, MDR при лечении рака). Некоторые проблемы могут быть связаны с низким транспортом или клеточным поглощением и активационными механизмами. Некоторые проблемы могут быть связаны с быстрой инактивацией и/или выделением лекарственного средства.
Эффективность нуклеозидных аналогов в большой степени зависит от их способности имитировать природные нуклеозиды, таким образом взаимодействуя с вирусными и/или клеточными белками и взаимодействуя или ингибируя критические процессы в метаболизме нуклеиновых кислот. Для того чтобы вызвать их антивирусную или антираковую активность, нуклеозидные аналоги должны быть трансформированы, через их моно- и дифосфаты в соответствующие трифосфаты под действием вирусных и/или клеточных киназ. В качестве общего правила трифосфат представляет собой активный агент, но для некоторых продуктов, например гемситабина, даже дифосфат может вызывать клинически значимый эффект.
Для того чтобы достичь зараженных, раковых или инфицированных вирусом клеток или тканей после энтерального или парентерального введения, нуклеозидные аналоги должны иметь благоприятные фармакокинетические характеристики. Дополнительно к быстрому выведению введенного лекарственного средства многие нуклеозидные аналоги могут дезактивироваться или в токе крови, или в тканях. Например, производные цитозина могут даже на уровне монофосфата быстро дезаминироваться под действием класса белков, называемых дезаминазами, в неактивный аналог урацила. Клеточное поглощение и, таким образом, хорошая терапевтическая эффективность многих нуклеозидных аналогов сильно зависит от мембрано-связанных нуклеозидных транспортных протеинов (называемых концентрирующими и эквилибрирующими нуклеозидными переносчиками). Поэтому существует потребность в соединениях, которые не зависят от таких специфичных механизмов поглощения. Кроме этого, другими факторами, лимитирующими активность, особенно в антираковой области, являются механизмы клеточного восстановления. Когда монофосфат антиракового нуклеозидного аналога связывается в клеточной ДНК, он не может быть удален из раковой клеточной ДНК благодаря экзонуклеазной активности, связанной с белком p53. Однако удаление нуклеозидного аналога из ДНК здоровой клетки является благоприятным для того, чтобы ограничить побочные эффекты лекарственного средства.
С годами были разработаны многочисленные нуклеозидные аналоги, которые в большой степени преодолевают некоторые или многие факторы, лимитирующие активность. Ацикловир (ACV), например, может применяться для иллюстрации соединения с отличной специфичностью. ACV-монофосфат может быть образован только вирусными киназами, то есть ACV не может быть активирован неинфицированными клетками. Несмотря на этот факт, ACV не является особенно активным продуктом. Для того чтобы обойти частую скорость, лимитирующую стадию активации нуклеозидного аналога, внутриклеточное образование монофосфата нуклеозидного аналога, были разработаны несколько фосфонатов, таких как сидофовир, или даже монофосфатных продуктов. Для того чтобы облегчить пероральное поглощение или сохранить благоприятное распределение лекарственного средства в организме, были разработаны конкретные пролекарства, такие как гепсера.
Дополнительно к структурным изменениям, произведенным в нуклеозидных аналогах для достижения улучшенной клинической полезности, были проведены дальнейшие модификации для улучшения активности. Заявитель настоящего изобретения (US 6153594, US 6548486 B1, US 6316425 B1, US 6384019 B1) и несколько других групп модифицировали нуклеозидные аналоги путем прибавления липидных компонентов (EP-A-56265, EP-A-393920, WO 99/26958). Это достигается связыванием жирных кислот, например, через сложноэфирную, амидную, карбонатную или карбаматную связь. Могут быть получены более сложные продукты, такие как фосфолипидные производные (Eur J Pharm Sci (2000) 1lb Suppl 2: p 15-27, EP 545966 B1, CA 2468099 A1, US 6372725 B1 и US 6670341 B1) нуклеозидных аналогов. Было описано, что такие аналоги имеют антивирусную активность, которая особенно подходит для терапии и профилактики инфекций, вызванных ДНК, РНК или ретровирусами. Они также подходят для лечения злокачественных опухолей. Липидные производные нуклеозидных аналогов могут служить для нескольких целей. Они могут рассматриваться как пролекарство, которое не является субстратом для дезаминаз, таким образом защищая нуклеозидный аналог от деактивации в процессе переноса в кровотоке. Липидные производные также могут более эффективно транспортироваться через клеточную мембрану, приводя к увеличению внутриклеточной концентрации нуклеозидного аналога. Липидные производные также могут быть более подходящими для применения в дермальных препаратах, пероральных продуктах (US 6576636 B2 и WO 01/18013 или специфических препаратах, таких как липосомы (US 5223263), разработанных конкретно для направленного воздействия на опухоли.
Ранее, некоторые авторы настоящего изобретения продемонстрировали, что для нуклеозидных аналогов с сохраненной β-D конфигурацией моносахаридного кольца или нуклеозидных аналогов с нециклической боковой цепью антивирусная или антираковая активность может быть наиболее эффективно улучшена путем образования липидных производных моно-незамещенных ω-9 C18 и C20 жирных кислот (Antimicrobial Agents and Chemotherapy, Jan. 1999, p.53-61, Cancer Research 59, 2944-2949, June 15, 1999, Gene Therapy (1998) 5, 419-426, Antiviral Research 45 (2000) 157-167, Biochemical Pharmacology 67 (2004) 503-511). Будучи не только более активными, чем поли-незамещенные аналоги, предпочтительные моно-незамещенные производные являются более кристаллическими и химически стабильными по отношению к окислению липидной цепи, и, следовательно, более предпочтительными с точек зрения химического и фармацевтического производства. Заявитель настоящего изобретения также продемонстрировал, что моно-незамещенные ω-9 C18 и C20 жирные кислоты подходят для улучшения терапевтической активности большого числа ненуклеозидных биологически активных соединений (EP 0977725 B1).
Относительно новой подгруппой нуклеозидных аналогов являются так называемые 1,3-диоксолановые производные. В этом классе соединений сохраняют пятичленное кольцо, аналогичное моносахаридному, присутствующему в природных нуклеозидах, но CH2 группу в положении 3 заменяют на атом O, как показано в формуле B ниже.
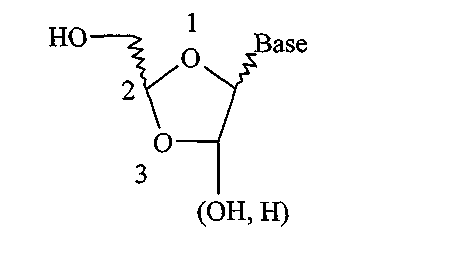
Формула B
Некоторые продукты 1,3-диоксоланового класса продемонстрировали многообещающую антивирусную и антираковую in-vitro и in-vivo активность. Для выбранных соединений были показаны улучшенные и независимые от нуклеозидного переносчика клеточное поглощение и уменьшенная скорость дезаминирования (= инактивации).
Известны некоторые типы липофильных производных таких 1,3-диоксолановых продуктов, включая сложные эфиры и амиды и поли-ненасыщенных, и моно-ненасыщенных ω-9 C18 и C20 жирных кислот (US 5817667, US 2001/0020026 A1 и US 2003/0013660 A1). Эти ссылки предоставляют и общие сведения о возможных достоинствах, полученных приготовлением таких липидных производных, и специфические преимущества таких производных, касающиеся активности на механистических моделях и моделях заболеваний. В частности, in-vitro антираковую активность N4-амидных производных (-)-β-L-диоксолан-цитидина (троксацитабина), приготовленных со следующими жирными кислотами: олеиновой кислотой (цис-9-октадеценовой кислотой), элаидиновой кислотой (транс-9-октадеценовой кислотой) и линоленовой кислотой (цис-9,12-октадекадиеновой кислотой), сравнивали на нескольких клеточных линиях. Три продукта показали вполне сопоставимую активность, амидное производное олеиновой кислоты было минимально лучше (2-5 раз), чем элаидиновый аналог.
Подробное описание изобретения
Неожиданно было найдено, что активность 1,3-диоксолановых нуклеозидных производных может быть значительно увеличена по сравнению с соединениями существующего уровня техники путем образования производных некоторых ненасыщенных длинноцепочечных жирных кислот с, по меньшей мере, одной двойной связью в положении 6, при подсчете от карбонильного атома углерода, обычно называемых Δ6 ненасыщенными жирными кислотами. В работе были продемонстрированы и сложноэфирные, и амидные производные моно- и поли- Δ6-ненасыщенных жирнокислотных производных 1,3-диоксолан нуклеозидных аналогов с конкретным продуктом троксацитабином, и было проведено их сравнение с соединениями троксацитабина из существующего уровня техники. Более конкретно, сложноэфирные и амидные производные петроселиновой кислоты (цис-6-октадеценовой кислоты), петроселаидиновой (транс-6-октадеценовой кислоты), гамма-линоленовой кислоты (цис-6,9,12-октадекатриеновой кислоты) и амид элаидиновой кислоты были протестированы в качестве системы сравнения с существующим уровнем техники. Производные петроселаидиновой кислоты имеют наибольший потенциал. Они являются идентичными по активности, минимально более активны, чем производные гамма-линоленовой кислоты, и в 20 раз более активны, чем продукт существующего уровня техники, амид троксацитабин-N4-элаидиновой кислоты.
В отдельном эксперименте сравнивали in-vitro активность амидного производного петроселиновой кислоты и продукта существующего уровня техники амида троксацитабина-N4-элаидиновой кислоты на клеточной линии опухоли шейки матки HeLa/mut и ее адриамицин резистентной сублинии HeLa/mut/Adr. Неожиданно было найдено, что хорошая активность троксацитабин-петроселинового амида на HeLa/mut линии остается неизменной на HeLa/mut/Adr сублинии, хотя для амида троксацитабин-N4-элаидиновой кислоты наблюдали фактор резистентности 6.
Соединение по изобретению может быть описано общей формулой I:
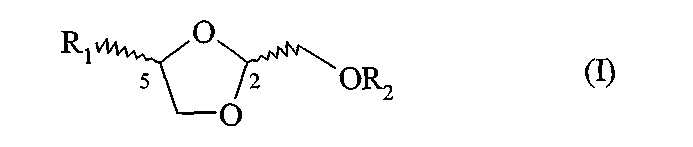
где заместители в положениях 2 и 5 1,3-диоксоланового кольца имеют возможность располагаться выше или ниже плоскости пятичленного кольца. Упомянутые заместители могут быть или в цис-, или в транс-конфигурации. R2 представляет собой атом водорода или ацильную группу ненасыщенной жирной кислоты R5C(О), R5CH2OC(О) или R5CH2NHC(О), где R5 представляет собой C7-23 алкенильный остаток формулы:
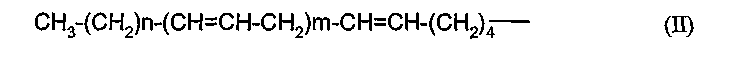
где m представляет собой число от 0 до 2, и n представляет собой число от 0 до 10.
R1 предсталяет собой, необязательно, замещенное азотное основание, необязательно, имеющее функциональную группу, такую как спиртовую или аминогруппу; такая группа, при необходимости, может быть ацилирована ненасыщенной жирной кислотой.
Более конкретно, R1 представляет собой:
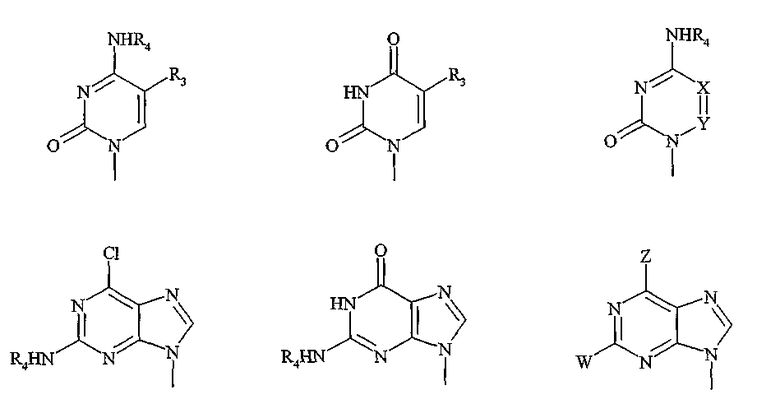
и R3 выбирают из группы, состоящей из водорода, метила, трифторметила, фтора, хлора, брома и иода; и каждый из Z и W независимо представляет собой: Br, Cl, I, F, OR4 или NHR4, и, по меньшей мере, один из Z и W представляет собой или OR4, или NHR4, и R4 представляет собой H или R5C(О), R5CH2OC(О) или R5CH2NHC(О), где R5 представляет собой C7-23 алкенил с общей формулой II. R2, R3 и R4 не могут одновременно представлять собой водород. X и Y могут представлять собой или CH, или N атом, при этом, по меньшей мере, один из X или Y представляет собой N.
Замечено, что Δ6 ненасыщенные жирные кислоты, рассмотренные в этом изобретении, могут иметь и цис-, и транс- стереохимию двойных связей.
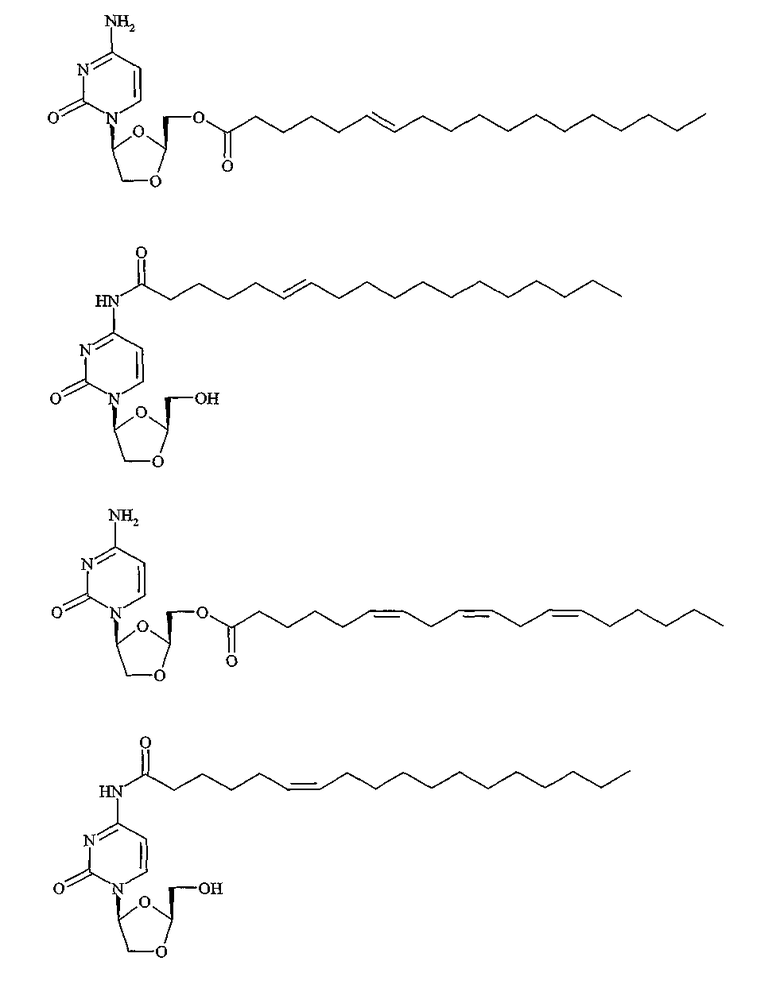
Особенно предпочтительные воплощения этого изобретения включают, но не ограничиваются сложным эфиром троксацитабин-2'-гидроксиметил-транс-6-октадеценовой кислоты, амидом троксацитабин-N4-транс-6-октадеценовой кислоты, сложным эфиром троксацитабин-2'-гидроксиметил-гамма-линоленовой кислоты и амидом троксацитабин-N4 -цис-6-октадеценовой кислоты.
Примеры
Пример 1
Клеточную линию рака шейки матки HeLa/mut высевают в 96-луночные планшеты по 5×103 клеток на лунку. Среда представляет собой RMPI 1640 с 2 мМ глутамина и 10% фетальной бычьей сыворотки. Через 24 часа прибавляют тестовые соединения в 6 концентрациях. Клетки инкубируют в течение 4 дней. В каждую лунку прибавляют раствор MTT и инкубируют в течение 4 часов. Образцы считывают с помощью ИФА ридера при 540 нм. Величины IC50 определяют из кривых роста. Тестовые соединения представляют собой троксацитабин элаидиновый амид, троксацитабин петроселаидоат, троксацитабин петроселаидиновый амид и троксацитабина γ-линоленоат. Неожиданным оказалось, что троксацитабин петроселаидоат и петроселаидиновый амид были в 20 раз более активны, чем троксацитабин элаидиновый амид.
Пример 2
Человеческую клеточную линию рака шейки матки HeLa/mut и адриамицин резистентную клеточную линию HeLa/mut/Adr высевают в 96-луночные планшеты по 5×l03 клеток на лунку. Среда представляет собой RMPI 1640 с 2 мМ глутамина и 10% фетальной бычьей сыворотки. Через 24 часа к клеткам прибавляют тестовые соединения в конечном объеме 20 мкл в шести различных концентрациях. Клетки инкубируют в течение 4 дней. В каждую лунку прибавляют раствор MTT и инкубируют в течение 4 часов. Образцы считывают с помощью ИФА ридера при 540 нм. Величины IC50 определяют из кривых роста. Фактор резистентности представляет собой отношение IC50 в HeLa/mut/Adr к IC50 в HeLa/mut. Тестовые соединения представляют собой троксацитабин элаидиновый амид и троксацитабин петроселиновый амид. Неожиданно было найдено, что троксацитабин петроселиновый амид не зависит от адриамициновой резистентности с фактором резистентности 1,0, по сравнению с 5,8 для троксацитабин элаидинового амида.
Пример 3
Клеточные линии U937 и THP-1 высевают в 96-луночные планшеты по 20 000 клеток на лунку. В каждую лунку прибавляют по 50 мкл среды (). В то же самое время прибавляют тестовые соединения в пяти различных концентрациях и инкубируют в течение 48 часов. Для изучения цитотоксичности теста для этих клеток проводят нерадиоактивный клеточный пролиферативный анализ CellTiter 96® (Promega). Этот анализ представляет собой колориметрическое определение числа жизнеспособных клеток при анализах пролиферации и хемочувствительности. Набор составлен из растворов нового соединения тетразолия (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2H-тетразолия, внутренней соли; MTS) и электрон связывающего реагента (феназин метосульфата; PMS). MTS биовосстанавливается клетками в продукт формазан, который растворяется в тканевой культуральной среде. Поглощение формазана при 490 нм может быть измерено непосредственно из анализируемых 96-луночных планшет без дополнительной обработки. Количество формазанового продукта, которое измеряют количеством поглощения 490 нм, является прямо пропорциональным количеству живых клеток в культуре (значения IC50). Тестовые соединения представляют собой сложный эфир троксацитабин 5'-элаидиновой кислоты, амид α-траксацитабин N4-элаидиновой кислоты, троксацитабин-5'-петроселаидоат, троксацитабин-5'-олеат, амид α-троксацитабин N4-петроселаидиновой кислоты, троксацитабин C5'-6,9,12-линоленоат, амид троксацитабин N4-6,9,12-линоленовой кислоты, троксацитабин-5'-петроселиноат и амид троксацитабин N4-петроселиновой кислоты. Все соединения продемонстрировали сильную клеточную цитотоксичность на обоих клеточных линиях, и троксацитабин-5'-петроселиноат, амид троксацитабин N4-6,9,12-линоленовой кислоты, амид троксацитабин N4-петроселиновой кислоты были даже более активны, чем другие соединения, которые также продемонстрировали высокую активность, за исключением амида α-троксацитабин N4-элаидиновой кислоты, как ожидалось.
IC50 мкМ ± SD
IC50 мкМ ± SD
nd = не определено
Пример 4
Амид троксацитабин N4-элаидиновой кислоты (соединение сравнения)
К троксацитабину (150 мг, 0,70 ммоль), TEA (0,1 мл, 0,74 ммоль) и ДМАП (90 мг, 0,74 ммоль) в (смеси) ДХМ/ДМФА (5 мл/2 мл) прибавляют элаидоил хлорид в дихлорметане (5 мл). Гидрохлорид кислоты приготавливают из элаидиновой кислоты (209 мг, 0,74 ммоль), оксалил хлорида (0,4 мл, 2,96 ммоль) и ДМФА (каталитическое количество) в толуоле (10 мл) при перемешивании при комнатной температуре в течение 2 часов и затем упаривают до сухого состояния. После перемешивания в течение 22 часов при комнатной температуре к смеси прибавляют насыщенный водный раствор NH4Cl и затем фазы разделяют. Водную фазу экстрагируют ДХМ (3x), комбинированные органические слои промывают насыщенным солевым раствором, высушивают (Na2SO4), фильтруют и упаривают в вакууме. Продукт очищают флэш-хроматографией на силикагеле и элюируют MeOH/ДХМ (25:975), затем MeOH/ДХМ (5:95) и получают 208 мг (62%) требуемого продукта в виде бесцветных кристаллов.
1H-ЯМР (200 МГц; CDCl3); δ 8,51 (1H, д), 7,43 (1H, д), 6,21 (1H, м), 5,39 (2H, м), 5,18 (м, 1H), 4,40-4,12 (м, 2H), 4,01 (м, 2H), 2,44 (т, 2H), 2,2-1,9 (м, 6H), 1,8-1,1 (м, 22H), 0,96 (т, 3H). МС (с электрораспылением); 500 [M+Na]+. ВЭЖХ:99,4%
Пример 5
Троксацитабин-2'-гидроксиметил-петроселаидоат
Троксацитабин (0,5 г, 2,4 ммоль) в сухом ДМФА (10 мл) прибавляют к раствору HCl в ДМФА (1,3 M, 2,2 мл, 2,8 ммоль), который приготавливают пропусканием газообразного HCl через ДМФА. Через несколько секунд из раствора выпадает бесцветный твердый осадок. Полученную смесь перемешивают в течение 30 минут и затем прибавляют петроселаидоил хлорид в ДМФА (5 мл). Гидрохлорид кислоты предварительно приготавливают из петроселаидиновой кислоты (1,0 г, 3,5 ммоль), оксалил хлорида (1,9 мл, 14 ммоль) и ДМФА (каталитическое количество) в ДХМ (30 мл) перемешиванием при комнатной температуре в течение 1 ч и затем упаривают до сухого состояния. После перемешивания в течение 64 часов при комнатной температуре смесь выливают в воду и экстрагируют ДХМ (3x). Объединенные органические экстракты промывают насыщенным водным раствором NaHCO3, соли, высушивают (Na2SO4), фильтруют и упаривают в вакууме. Продукт очищают флэш-хроматографией на силикагеле, элюируют MeOH/ДХМ (5:95) и получают 0,52 г (46%) требуемого продукта в виде бесцветных кристаллов.
1Н-ЯМР (200 МГц; CDCl3); δ 7,74 (д, 1H), 6,27 (м, 1H), 5,87 (д, 1H), 5,40 (м, 2H), 5,22 (м, 1H), 4,6-4,2 (м, 4H), 2,42 (т, 2H), 2,00 (м, 4H), 1,65 (м, 2H), 1,5-1,2 (м, 22H), 0,87 (т, 3H).
МС (с электрораспылением); 478 [M+H]+, 500 [M+Na]+. ВЭЖХ; 93,7%
Пример 6
Амид троксацитабин N4-петроселаидиновой кислоты
К петроселаидиновой кислоте (662 мг, 2,35 ммоль) в ДХМ (20 мл) прибавляют TEA (0,33 м, 2,35 ммоль), затем TBTU (753 мг, 2,35 ммоль) и перемешивают в течение 40 мин при комнатной температуре перед тем, как прибавляют троксацитабин (0,5 г, 2,35 ммоль) в ДМФА (2 мл). После перемешивания в течение 22 ч при комнатной температуре прибавляют насыщенный водный раствор NH4Cl и разделяют фазы. Водную фазу экстрагируют ДХМ (3x) и объединенные органические экстракты промывают насыщенным раствором NaHCO3, соли, высушивают (Na2SO4), фильтруют и упаривают до сухого состояния в вакууме. Продукт очищают флэш-хроматографией на силикагеле, элюируют MeOH/ДХМ (25:975) и получают 650 мг (58%) требуемого продукта в виде бесцветных кристаллов.
1H-ЯМР (200 МГц; CDCl3); δ 8,57 (д, 1H), 7,47 (д, 1H), 6,21 (д, 1H), 5,42 (м, 2H), 5,16 (с, 1H), 4,4-4,2 (м, 2H), 4,01 (м, 2H), 2,49 (т, 2H), 2,02 (м, 4H), 1,71 (м, 2H), 1,5-1,2 (м, 22H), 0,87 (т, 3H).
МС (с электрораспылением); 478 [M+H]+, 500 [M+Na]+. ВЭЖХ; 93,8%
Пример 7
Троксацитабин 2'-гидроксиметил-6,9,12-линоленоат
Троксацитабин (0,7 г, 3,3 ммоль) в сухом ДМФА (17 мл) прибавляют к раствору HCl в ДМФА (1,3 M, 3,0 мл, 3,9 ммоль), который приготавливают пропусканием газообразного HCl через ДМФА. Через несколько секунд из раствора выпадает бесцветный твердый осадок. Полученную смесь перемешивают в течение 30 минут перед прибавлением хлорангидрида кислоты в ДМФА (4 мл). Хлорангидрид кислоты предварительно приготавливают из GLA (2,19 г, 7,9 ммоль), оксалил хлорида (2,66 мл, 31,4 ммоль) и ДМФА (каталитическое количество) в ДХМ (50 мл) перемешиванием при комнатной температуре в течение 3 часов и затем упаривают до сухого состояния. После перемешивания в течение 48 часов при комнатной температуре смесь выливают в водный NaHCO3 и экстрагируют ДХМ (3x). Объединенные органические экстракты промывают раствором соли, высушивают (Na2SO4), фильтруют и упаривают в вакууме. Продукт очищают флэш-хроматографией на силикагеле, элюируют MeOH/ДХМ (3:100) и получают 0,85 г (54%) требуемого продукта в виде бледно-желтого твердого вещества.
1Н-ЯМР (200 МГц, CDCl3); δ 7,78 (д, 1H), 6,30 (м, 1H), 6,85 (д, 1H), 5,6-5,1 (м, 7H), 4,6-4,1 (м, 5H), 2,9-2,7 (м, 4H), 2,4 (т, 2H), 2,2-2,0 (м, 3H), 1,8-1,2 (м, 10H), 0,95 (т, 3H).
МС (с электрораспылением); 496 [M+Na+]. ВЭЖХ; 93,9%
Пример 8
Амид троксацитабин N4-петроселиновой кислоты
К петроселиновой кислоте (666 мг, 2,36 ммоль) в ДХМ (20 мл) прибавляют TEA (0,33 мл, 2,37 ммоль), затем TBTU (753 мг, 2,35 ммоль) и перемешивают в течение 40 мин при комнатной температуре перед прибавлением троксацитабина (0,501 г, 2,35 ммоль) в ДМФА (2 мл). После перемешивания в течение 22 часов при комнатной температуре прибавляют водный раствор NH4Cl (25 мл)и фазы разделяют. Водную фазу экстрагируют ДХМ (3×50 мл) и комбинированные органические экстракты промывают насыщенным NaHCO3 (25 мл), раствором соли (25 мл), высушивают (Na2SO4), фильтруют и упаривают в вакууме. Продукт очищают флэш-хроматографией на силикагеле, элюируют MeOH/ДХМ (25:975-50:950) и получают 599 мг (53%) требуемого продукта в виде бесцветнах кристаллов.
1H-ЯМР (200 МГц; CDCl3); δ 8,48 (уш.с, 1H), 8,42 (д, J=7,4 Гц, 1H), 7,39 (д, J=7,5 Гц, 1H), 6,16 (д, J=4,7 Гц, 1H), 5,31 (м, 2H), 5,09 (с, 1H), 4,18-4,33 (м, 2H), 3,94 (д, J=4,8 Гц, 2H), 2,40 (т, J=7,3 Гц, 2H), 2,02 (м, 4H), 1,63 (м, 2H), 1,23-1,44 (м, 21H), 0,84 (т, J=6,0 Гц, 3H).
МС (с электрораспылением, позитив.); 500 [M+Na]+.
МС (с электрораспылением, негатив.); 477 [M+]+. ВЭЖХ; 93%
Пример 9
Троксацитабин-2'-гидроксиметил-петроселиноат
К троксацитабину (0,503 г, 2,36 ммоль) в сухом ДМФА (7 мл) прибавляют раствор HCl в ДМФА (1,3 M, 2,2 мл, 2,86 ммоль), который приготавливают пропусканием газообразного HCl через ДМФА. Через несколько секунд из раствора выпадает бесцветный твердый осадок. Полученную смесь перемешивают в течение 30 мин и затем прибавляют гидрохлорид петроселиновой кислоты в ДМФА (12 мл). Хлорангидрид кислоты предварительно приготавливают из петроселиновой кислоты (1,59 г, 5,6 ммоль), оксалил хлорида (4,5 мл, 53 ммоль) и ДМФА (0,1 мл, каталитическое количество) в толуоле (35 мл) перемешиванием при комнатной температуре в течение 2 часов и затем упаривают до сухого состояния. После перемешивания в течение 43 часов при комнатной температуре смесь выливают в воду (50 мл) и экстрагируют ДХМ (3×100 мл). Объединенные органические экстракты промывают насыщенным водным раствором NaHCO3 (50 мл), раствором соли (50 мл), высушивают (Na2SO4), фильтруют и упаривают в вакууме. Продукт очищают флэш-хроматографией на силикагеле, элюируют MeOH/ДХМ (25:975-50:950) и получают 0,686 г (61%) требуемого продукта в виде бледно-желтых кристаллов.
1H-ЯМР (200 МГц; CDCl3); δ 7,69 (д, J=7,4 Гц, 1H), 6,21 (м, 1H), 5,70 (д, J=7,4 Гц, 1H), 5,30 (м, 2H), 5,16 (м, 1H), 4,45 (д.д., J=3,4 Гц, 12,3 Гц, 1H), 4,16-4,28 (м, 3H), 2,34 (т, J=7,4 Гц, 2H), 1,99 (м, 4H), 1,68 (м, 2H), 1,22-1,38 (м, 22H), 0,84 (т, J=6,4 Гц, 3H).
MC (с электрораспылением, позитив.); 500 [M+Na]+.
МС (с электрораспылением, негатив.); 477 [M+]+. ВЭЖХ; 94%
Пример 10
Амид троксацитабин N4-6,9,12-линоленовой кислоты
К раствору гамма-линоленовой кислоты (GLA) (0,38 г, 1,8 ммоль) в ДХМ (4 мл) прибавляют TEA (245 мкл, 1,8 ммоль) и TBTU (0,57 г, 1,8 ммоль). Раствор оставляют перемешиваться в течение 30 мин при комнатной температуре перед прибавлением троксацитабина (0,38 г, 1,8 ммоль) в ДХМ (2 мл). Реакционную смесь перемешивают в течение 70 часов при комнатной температуре, затем прибавляют насыщенный водный раствор NH4Cl и смесь экстрагируют ДХМ (3x). Объединенные органические фазы промывают раствором соли, высушивают (NaSO4), фильтруют и упаривают в вакууме. Требуемый продукт выделяют флэш-хроматографией на силикагеле, элюируют MeOH/ДХМ (3:200) и получают 0,30 г (35%) целевого продукта в виде бледно-желтого твердого вещества.
1H-ЯМР (200 МГц, CDCl3); δ 8,5 (д, 1H), 7,45 (д, 1H), 6,20 (д, 1H), 6,6-6,3 (м, 6H), 5,15 (с, 1H), 4,3 (м, 2H), 4,0 (с, 2H), 2,8 (м, 5H), 3,5 (м, 3H), 2,1 (м, 4H), 1,7 (м, 2H), 1,4 (м, 6H), 0,95 (м, 3H).
МС (с электрораспылением); 496 [M+Na+].
ВЭЖХ: 97,0%
ВЭЖХ параметры:
Система ВЭЖХ: Agilent 1100
Колонка: Cromolith Speedrod RP-C18e 50×4,6 мм
Время анализа:15 мин
Скорость элюирования:2 мл/мин
Подвижная фаза: A: 5% MeOH в воде с прибавленнием фосфатного буфера (pH 6)
B: MeOH с прибавленнием фосфатного буфера (pH 6)
Температура: 40°C
Детектор: UV 240-300 нм
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГЕМЦИТАБИНА | 1998 |
|
RU2194711C2 |
| АНАЛОГИ АЗАЦИТИДИНА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2488591C2 |
| АНАЛОГИ АЗАЦИТИДИНА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2487883C2 |
| СА-IX СПЕЦИФИЧЕСКИЕ РАДИОФАРМПРЕПАРАТЫ ДЛЯ ЛЕЧЕНИЯ И ВИЗУАЛАЗИЦИИ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 2009 |
|
RU2539565C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2005 |
|
RU2430088C2 |
| МОНОЭФИРНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2139884C1 |
| N-АЦИЛЬНЫЕ ПРОИЗВОДНЫЕ БИОГЕННЫХ АМИНОВ - МОДУЛЯТОРЫ ПЕРЕКИСНОГО ОКИСЛЕНИЯ ЛИПИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2093520C1 |
| 5-(ЦИТОЗИНИЛ-1-)-1,3-ОКСАТИОЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2142462C1 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНИЛАНТРАЦИКЛИНА | 2015 |
|
RU2715902C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИНА | 2005 |
|
RU2378257C2 |
Настоящее изобретение относится к соединениям формулы (I)
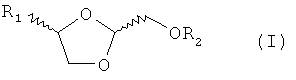
где R1 представляет собой 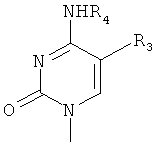
или
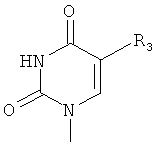
а значения радикалов R2, R3 и R4 раскрыты в формуле изобретения, а также к фармацевтическим композициям на основе таких соединений. Соединения формулы (I) и композиции на их основе могут применяться для лечения раковых заболеваний как твердых опухолей, так и гематологических раков, таких как лейкемии, лимфомы и множественные миеломы. 4 н. и 7 з.п. ф-лы, 2 табл.
1. Соединение формулы (I)
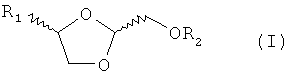
где R1 представляет собой
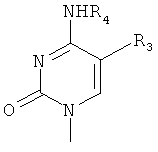
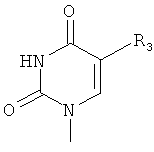
и R3 выбирают из группы, состоящей из водорода, метила, трифторметила, фтора, хлора, брома или иода; и
R4 представляет собой Н или R5CH2NHC(O), где R5 представляет собой C7-23 алкенил с общей формулой
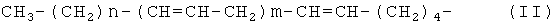
где m представляет собой число от 0 до 2, и n представляет собой число от 0 до 10,
R2 представляет собой Н или R5C(O), где R5 представляет собой C7-23 алкенил с общей формулой
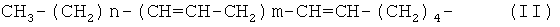
где m представляет собой число от 0 до 2, и n представляет собой число от 0 до 10, при условии, что R2, R3 и R4 одновременно не могут представлять собой водород, и при условии, что 2-гидроксиметил-4-(тимин-1'-ил)-1,3-диоксолан исключен, или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 представляет собой аналог цитозина
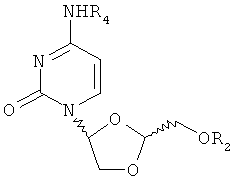
где R2 представляет собой Н или R5C(O), и R4 представляет собой Н или R5CH2NHC(O), где R5 представляет собой C7-23 алкенил с общей формулой
где m представляет собой число от 0 до 2, и n представляет собой число от 0 до 10, при условии, что R2 и R4 одновременно не могут представлять собой водород; или его фармацевтически приемлемая соль.
3. Соединение по п.1 или 2, определенное как
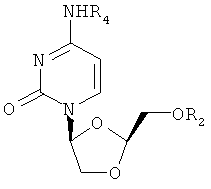
где R2 представляет собой Н или R5C(O), R4 представляет собой Н или R5CH2NHC(O), где R5 представляет собой С7-23 алкенил с общей формулой
где m представляет собой число от 0 до 2, и n представляет собой число от 0 до 10, при условии, что R2 и R4 одновременно не могут представлять собой водород; или его фармацевтически приемлемая соль.
4. Соединение по п.1 или 2, определенное как
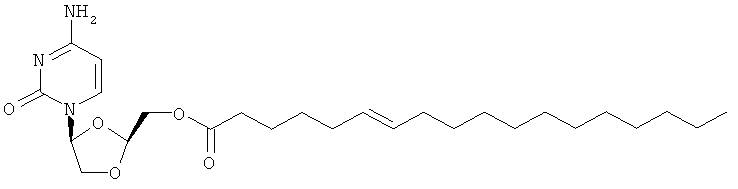
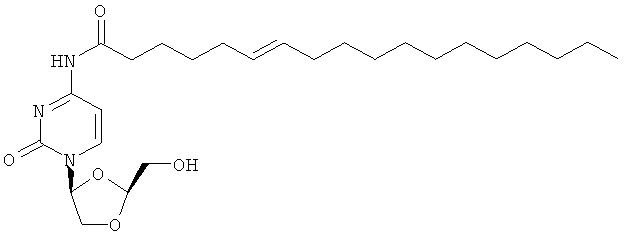
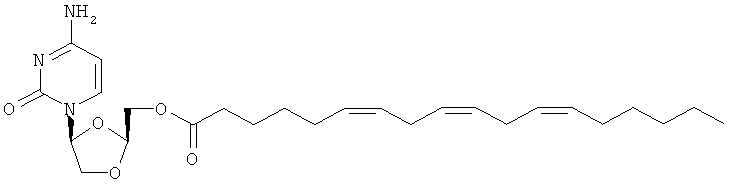
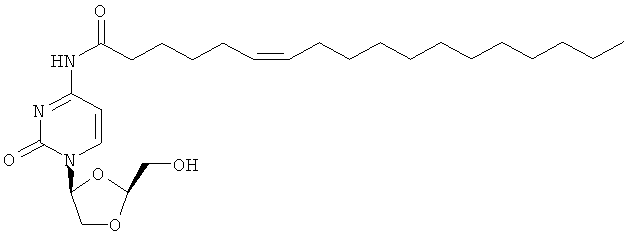
или его фармацевтически приемлемая соль.
5. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль, предназначенные для лечения рака.
6. Применение соединения формулы (I) по п.1 или его фармацевтически приемлемой соли для получения фармацевтического препарата для лечения рака.
7. Применение по п.6 для лечения твердых опухолей.
8. Применение по п.6 для лечения гематологических раков.
9. Применение по п.8, где гематологический рак выбран из группы, состоящей из лейкемий, лимфом и множественных миелом.
10. Способ лечения рака у субъекта, который нуждается в таком лечении, включающий введение такому субъекту терапевтически эффективного количества соединения формулы (I) по п.1; или его фармацевтически приемлемой соли.
11. Фармацевтическая композиция для лечения рака, содержащая соединение формулы (I) по п.1 и фармацевтически приемлемые эксципиенты, разбавители и/или носители.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Устройство для крепления осевого инструмента | 1987 |
|
SU1468687A1 |
| RU 97105381 A, 10.05.1999 | |||
| US 6525033 B1, 25.02.2003 | |||
| US 5270315 A, 14.12.1993 | |||
| US 6566365 B1, 20.03.2003. | |||

