Данное изобретение заявляет приоритет Предварительной Заявки на Патент США серийный № 60/975437, поданной 26 сентября 2007 года, которая во всей полноте включена в данное описание во в виде ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к аналогам азацитидина и их применению.
УРОВЕНЬ ТЕХНИКИ
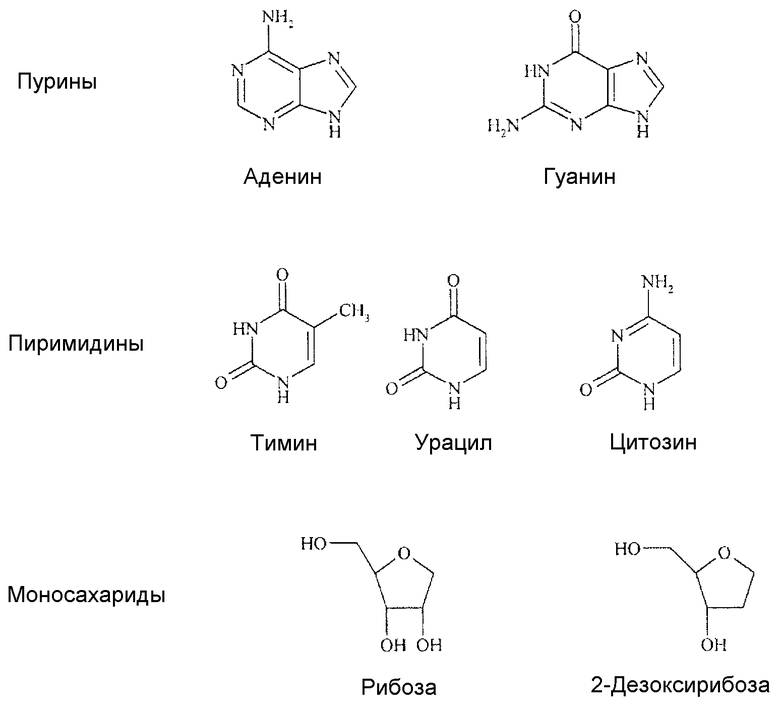
Аналоги нуклеозидов, производные природных нуклеозидов, рассматриваемых в качестве структурообразующих блоков ДНК и РНК, эффективны в клиническом лечении рака или вирусных заболеваний человека, хотя ранее такие соединения находили применение как противотуберкулезные средства. Такие соединения регистрировались на рынке в течение более 40 лет назад, и на сегодняшний день применяются 35 продуктов на их основе. Природные нуклеозиды, которые представлены в таблице 1 ниже, включают азотсодержащие соединения двух классов, т.е. пурины (примерами которых являются аденин и гуанин) и пиримидины (примерами которых являются тимин, урацил и цитозин), а также рибозу или дезоксирибозу, которые представляют собой моносахариды.
Таблица 1
Все природные нуклеозиды существуют в так называемой β-D-конфигурации, как показано в формуле А ниже. Азотсодержащее основание и гидроксиметильная боковая цепь на цикле сахара находятся по одну сторону (цис-положение) плоскости цикла сахара.
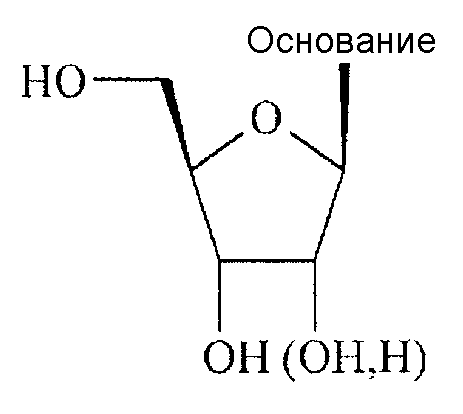
Формула А
Для получения нуклеозид-производных с противораковой или противовирусной активностью осуществлялись изменения химической структуры азотсодержащего основания и/или моносахарида. Например, введение атомов галогенов или других функциональных групп в азотсодержащее основание, вставка дополнительных атомов азота или стереохимические изменения в моносахаридном цикле с превращением рибозы в арабинозу или удаление гидроксильной группы с получением дезоксирибозы могут приводить к получению продуктов с возможным полезным терапевтическим действием. В ряде соединений моносахаридный цикл был сохранен, в то время как в других он превращен в цепь. Аналоги нуклеозидов представляют собой малые молекулы с от хорошей до прекрасной растворимостью в воде.
Широкие исследования и усилия, направленные на разработку нуклеозидных аналогов вследствие распространения СПИДа во всем мире и внесшие большой вклад в общую базу знаний и понимание механизма действия, изменения профиля активности при изменении химической структуры и т.д., также имеют большое значение для области техники, относящейся к лечению рака.
Общим недостатком применения многих лекарственных средств, включая нуклеозидные аналоги, является их низкая активность и низкая специфичность в отношении лечения целевого заболевания. Некоторые из этих проблем могут быть обусловлены активностью, свойственной действующему веществу самого лекарственного средства, другая часть может быть связана с определенными механизмами резистентности (свойственной пациенту или приобретенной им в процессе лечения рака, такой как, например, полирезистентность к лекарственным средствам (MDR)). Некоторые проблемы могут являться результатом недостаточного транспорта или клеточного поглощения, а также плохих механизмов активации. Некоторые другие проблемы могут быть связаны с быстрой инактивацией и/или экскрецией лекарственного средства.
Эффективность нуклеозидных аналогов в значительной степени зависит от их способности имитировать природные нуклеозиды, взаимодействуя с вирусными и/или клеточными ферментами и воздействуя на определяющие процессы в метаболизме нуклеиновых кислот или ингибируя эти процессы. Для того, чтобы привести в действие противовирусную или противораковую активность нуклеозидных аналогов, они должны быть превращены через их моно- и дифосфаты в соответствующие трифосфаты под действием вирусных и/или клеточных киназ. Как правило, активным соединением является трифосфат, но у некоторых производных, таких как, например, гемцитабин, даже дифосфат может проявлять клинически значимое действие.
Для того чтобы нуклеозидные аналоги достигали больных, раковых или вирусинфицированных клеток или тканей после энтерального или парентерального введения, нуклеозидные аналоги должны обладать благоприятными фармакокинетическими характеристиками. Помимо быстрой экскреции введенного лекарственного средства многие нуклеозидные аналоги могут дезактивироваться и в токе крови, и в тканях. Например, производные цитозина, даже на монофосфатном уровне, могут быстро дезаминироваться в результате действия ферментов класса дезаминаз с получением неактивного аналога урацила. Поэтому клеточное поглощение и, как следствие, хорошее терапевтическая эффективность многих нуклеозидных аналогов в значительной степени зависят от белков трансмембранного перемещения связанных нуклеозидов (называемых концентративными и разновесными переносчиками нуклеозидов). Следовательно, желательными являются соединения, действие которых не основывается на таких специфических механизмах поглощения. Еще одним фактором, ограничивающим активность, особенно в области борьбы с раковыми заболеваниями, являются клеточные механизмы восстановления. Когда противораковый монофосфатный нуклеозидный аналог вводится в клеточную ДНК, он не должен выводиться из ДНК раковой клетки вследствие экзонуклеазной активности, связанной с р53 белком. Однако удаление нуклеозидного аналога из ДНК здоровой клетки является благоприятным для ограничения побочных эффектов лекарственного средства.
В течение многих лет было разработано большое количество нуклеозидных аналогов, которые в значительной степени преодолевают некоторые или многие из факторов, неблагоприятно влияющих на их активность. Например, ацикловир (ACV) может служить иллюстрацией соединения с большой селективностью действия. ACV-монофостат может быть получен только под действием вирусных киназ, а это означает, что ACV не может активироваться в неинфицированных клетках. Несмотря на этот факт, ACV не является очень активным продуктом. Для того, чтобы обойти стадию, зачастую ограничивающую скорость активации нуклеозидного аналога, стадию внутриклеточного образования монофосфата нуклеозидного аналога, было разработано несколько фосфонатов, таких как цидофовир, и даже монофосфатов. Для облегчения перорального поглощения или гарантированно благоприятного использования лекарственного средства в организме, были разработаны специфические пролекарства, такие как гепсера.
Помимо структурных изменений, вводимых в нуклеозидные аналоги для способствования повышенной клинической применимости, для повышения их активности были осуществлены дополнительные модификации. Есть несколько примеров модифицированных нуклеозидных аналогов, полученных в результате добавления липидных фрагментов (Патенты США №№6153594, 6548486, 6316425 и 6384019; Европейские Заявки на Патент №№EP-A-56265 и EP-A-393920; WO 99/26958). Указанный синтез может осуществляться связыванием жирных кислот, например, через сложный эфир, амид, карбонат или карбаматную связь. Могут быть получены и более сложные продукты, такие как фосфолипид-производные нуклеозидных аналогов (см. Eur. J. Pharm. Sci. 11b Suppl 2: 15-27 (2000); EP No. 545966; Патент Канады №2468099; Патенты США №№6372725 и 6670341). Такие аналоги, как описано в литературе, обладают противовирусной активностью и особенно подходят для терапевтического лечения и профилактики инфекций, вызванных ДНК-, РНК- и ретровирусами. Они также подходят для лечения злокачественных опухолей. Липид-производные нуклеозидных аналогов могут служить нескольким целям. Они могут выступать в качестве пролекарства, которое не является субстратом для дезаминаз, в результате чего нуклеозидные аналоги защищаются от дезактивации в процессе транспорта в токе крови. Липидные производные также могут более эффективно транспортироваться через клеточную мембрану, что приводит к повышенной внутриклеточной концентрации нуклеозидного аналога. Липидные производные могут также более подходить для применения в дермальных препаратах, препаратах для перорального введения (см. Патент США №6576636 и WO 01/18013) или в особых препаратах, таких как липосомы (см. Патент США №5223263), разработанных для целенаправленного поражения опухолей.
Было показано, что противовирусная или противораковая активность нуклеозидных аналогов с сохраненной β-D-конфигурацией моносахаридного цикла или нуклеозидных аналогов с нециклической боковой цепью может наиболее эффективно улучшаться посредством образования липидных производных моно-замещенных ω-9 С18 и С20 жирных кислот (см. Antimicrobial Agents and Chemotherapy, Vol., 53-61 (1999); Cancer Research 59: 2944-2949 (1999); Gene Therapy, 5: 419-426 (1998); Antiviral Research, 45: 157-167 (2000); Biochemical Pharmacology, 67: 503-511 (2004). Предпочтительные мононенасыщенные производные являются не только более активными, чем полиненасыщенные аналоги, но и более кристалличными и химически стабильными в отношении окисления липидной цепи. Следовательно, они являются более благоприятными соединениями с химической точки зрения и с точки зрения производства лекарственного средства. Было также показано, что моно-ненасыщенные ω-9 С18 и С20 жирные кислоты подходят для повышения терапевтической активности в большом количестве ненуклеозидных биологических активных соединений (см. Европейский Патент №0977725).
Относительно новую подгруппу нуклеозидных аналогов составляют, так называемые, аза-С-производные. В данном классе соединений СН группа в 5 положении пиримидиновой основы заменена на атом азота, как показано в формуле В ниже.
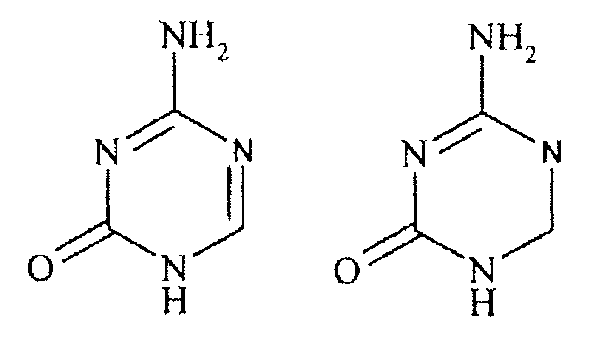
Формула В
Опухоль-подавляющие гены, которые были заглушены аберрантным ДНК метилированием, являются потенциальными мишенями для реактивации с помощью этих новых химиотерапевтических средств. Мощные ингибиторы ДНК метилирования и противолейкемические средства, аза-цитидин и 5-аза-2'-дезоксицитидиновые производные (5-аза-C, 5-аза-CdR, децитабин), могут повторно активировать заглушенные опухоль-подавляющие гены. При высоких концентрациях данные соединения являются цитотоксическими, но при более низких концентрациях гипометилирование приводит к дифференциации клеточных линий. Соединениям необходима метаболическая активация дезоксицитидинкиназой, и они обеспечивают ингибирование ДНК метилтрансферазы. Препятствием применения этих производных в качестве лекарственных средств является их быстрая in vivo инактивация цитидиндеаминазой (CD). Нестабильность в водных растворах, а также профили побочных эффектов также ограничили их клиническую активность.
Настоящее изобретение направлено на преодоление этих и других проблем предшествующего уровня.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
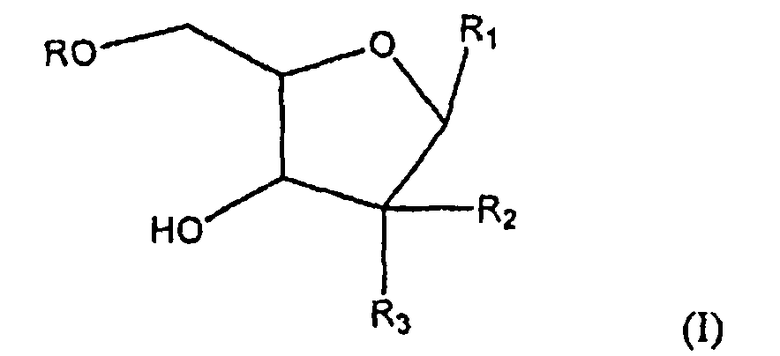
Один аспект настоящего изобретения относится к соединению формулы (I)
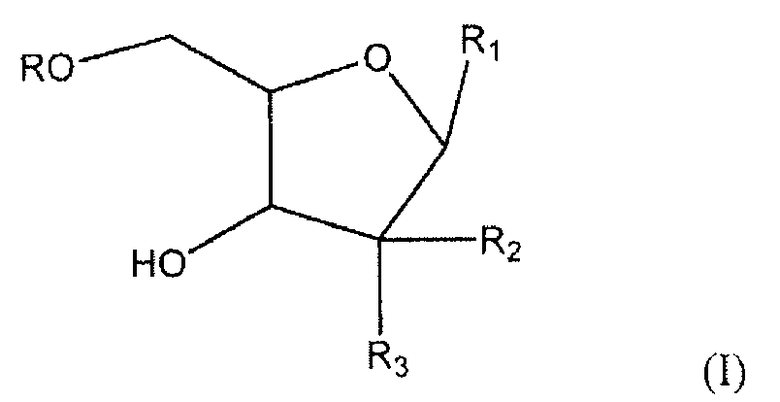
где R представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O), R1 представляет собой
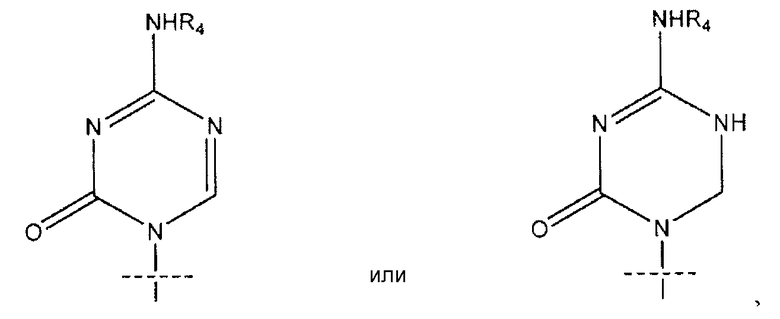
где перекрещивающаяся пунктирная линия показывает связь, образованную присоединением R1 к молекуле формулы (I), R2 и R3 независимо представляют собой OH или H при условии, что R2 и R3 одновременно не являются OH; R4 представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O) при условии, что R и R4 одновременно не являются H; R5 имеет общую формулу:
CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-;
k представляет целое число от 0 до 7; m представляет собой целое число от 0 до 2; и n представляет собой целое число от 0 до 10,
или его фармацевтически приемлемой соли.
Другой аспект настоящего изобретения относится к фармацевтической композиции, включающей соединение формулы (I) и фармацевтический наполнитель, разбавитель и/или носитель.
Еще один аспект настоящего изобретения относится к способу лечения у субъекта новообразования. Способ включает выбор субъекта с новообразованием и введение указанному субъекту соединения формулы (I), которая описана выше, или его фармацевтической соли в условиях, эффективных для лечения новообразования у субъекта.
Еще один аспект настоящего изобретения относится к способу лечения у субъекта воспалительного процесса. Способ включает выбор субъекта с воспалительным процессом и введение субъекту соединения формулы (I), которая описана выше, или его фармацевтически приемлемой соли в условиях, эффективных для лечения воспалительного состояния у субъекта.
Нестабильность аза-С в буфере и плазме хорошо известна (см. публикации Israili et al., Cancer Research 36, 1453-1461 (1976); Rudek et al., J. Clin. Oncol., 23:17, 3906-3911(2005); Rustum et al., J. Chromat, 421:12, 387-91 (1987); Zhao et al., J. Chromat B, 813, 81-88 (2004), которые во всей полноте введены в данное описание в виде ссылки). Средний период полураспада для аза-С в клинических образцах плазмы, как сообщалось, составляет 1,50±2,30 часа (см. публикацию Rudek et al., J. Clin. Oncol., 23:17, 3906-3911(2005), которая таким образом во всей полноте введена в данное описание в виде ссылки). В условиях in vitro при хранении aza-C в течение 4,5 дней даже при -60°С наблюдается потеря 20% соединения, а при хранении в течение 0,5 часа при комнатной температуре - потеря 10% (см. публикацию Zhao et al., J. Chromat В, 813, 81-88 (2004), которая во всей полноте введена в данное описание в виде ссылки). Считалось, что первичная нестабильность аза-C обусловлена быстрым (первая стадия является обратимой) раскрытием 5-азапиримидинового цикла с последующим отщеплением муравьиной кислоты (см. публикацию Chan et al., J. Pharma Sci., 68;7, 807-12 (1979), которая таким образом во всей полноте введена в данное описании в виде ссылки). Считается также, что другими путями разложения являются дезаминирование аминогруппы в положении 4 и гидролиз гликозидной связи с получение D-рибозы и 5-азацитозина. Неожиданно было обнаружено, что предпочтительные липидные производные аза-С обладают в значительной степени лучшим профилем стабильности в плазме, чем сам аза-С. Соединения являются стабильными (процент оставшегося соединения ≥94% от исходного содержания) в контрольной матрице плазмы человека при комнатной температуре в течение, по меньшей мере, 4 часов в условиях эксперимента, и не наблюдается значительного разложения продуктов в постэкстракционном супернатанте после осаждения белков плазмы. Стабильность в плазме предпочтительных липидных производных дополнительно исследовалась при хранении при 37°С. Показано, что раскрытие цикла аза-фрагмента или другое разложение соединения значительно снижается, когда липидная боковая цепь присоединена к аза-С.
Быстрое разложение аза-С является недостатком для его клинического применения. Повышенная стабильность в плазме липид-производных по сравнению с аза-С может обеспечивать высокий и длительный уровень содержания в плазме пациента липид-производного. Это может приводить в лучшему распределению в ткани/органе/опухоли, лучшей клеточной экспозиции и поглощению лекарственного средства по сравнению с аза-С, с одной стороны, и затем лучшую экспозицию опухолевой клеточной ДНК действию аза-С после внутриклеточного гидролиза аза-С-5'-сложноэфирной связи.
Вариантами осуществления настоящего изобретения являются новые соединения, молекулы которых получены через модификацию азацитидина и деоксицитидина (например, 5-аза-2'-деоксицитидина) и свойства которых неожиданно отличаются от свойств азацитидина и дезоксицитидина (например, 5-аза-2'-дезоксицитидина). В результате получен ряд соединений с активностью, которая расширяет уже достигнутую противораковую активность азатидина и деоксицитидина (например, 5-aza-2'-диоксицитидина), ограниченную гематобластозами. Эти новые соединения обладают противораковой эффективностью в отношении широкого спектра солидных опухолей, включая рак молочной железы и рак шейки матки. Соединения также неожиданно активны в отношении новообразований, резистентных к лечению и, таким образом, могут предоставить полезное терапевтическое действие в лечении солидных опухолей, для которых выбор терапевтических средств ограничен. Варианты осуществления настоящего изобретения могут применяться для лечения раковых заболеваний, для которых выбор и эффективность терапевтических средств остаются ограниченными, и удовлетворяют существующим потребностям.
Данные соединения показывают более быстрое проявление активности после ограниченной экспозиции и, следовательно, могут быть эффективными в течение короткого периода в патологическом процессе. Это должно привести к менее продолжительному лечению с более редким введением лекарственного средства и снижению токсичности, связанной с применением лекарственных средств, по сравнению с исходными лекарственными средствами. Это должно обеспечить более высокий терапевтический индекс.
Изменение структуры с добавлением липидного (включает сложные эфиры, амиды, карбаматы и карбонаты) компонента сохраняет азолцитидиновый цикл и, таким образом, обеспечивает воздействие молекулы на эпигенетические механизмы. Эпигенетическая модуляция предоставляет важный механизм для изменения экспрессии гена при раке и воспалении. Эти новые соединения обладают активностью при более низких концентрациях, чем азацитидин, и, следовательно, являются более эффективными. Указанные соединения с измененным спектром активности могут модулировать эпигенетические мишени солидных опухолей и воспалительных процессов.
Эпигенетические механизмы имеют большое значение для изменения протекания воспалительных процессов, которые включают, но не исключительно, воспаления легких, воспаления соединительных тканей, воспаления желудочно-кишечного тракта и воспаления сосудистой сети. Данные соединения посредством целевых эпигенетических механизмов могут снижать или обращать воспалительные процессы, ответственные за эти заболевания.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
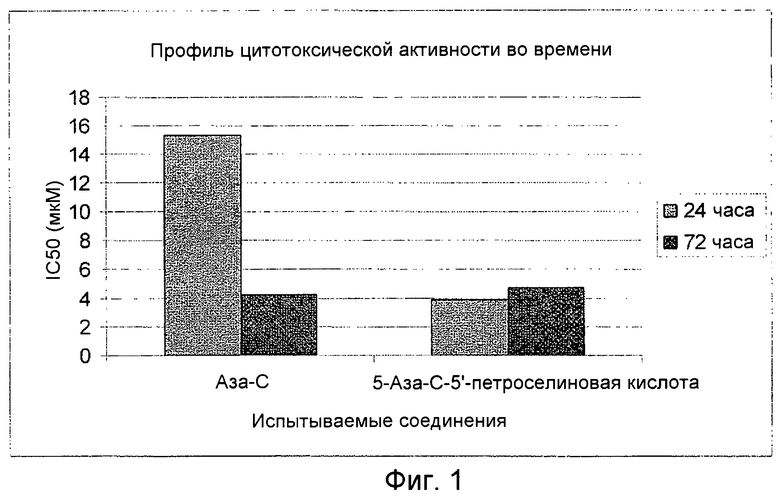
На фиг.1 представлен график, показывающий временной профиль цитотоксической активности аза-С и 5-аза-С-5'-петроселиновой кислоты.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Один аспект настоящего изобретения относится к соединению формулы (I)
где R представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O), R1 представляет собой
где перекрещивающаяся пунктирная линия показывает связь, образованную присоединением R1 к молекуле формулы (I); R2 и R3 независимо представляют собой OH или H при условии, что R2 и R3 одновременно не являются OH; R4 представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O) при условии, что R и R4 одновременно не являются H, R5 соответствует общей формуле:
CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-;
k представляет целое число от 0 до 7; m представляет собой целое число от 0 до 2; и n представляет собой целое число от 0 до 10,
или его фармацевтически приемлемой соли.
В предпочтительных вариантах осуществления изобретения k равно 4, n равно 10. В некоторых вариантах осуществления изобретения R1 представляет собой
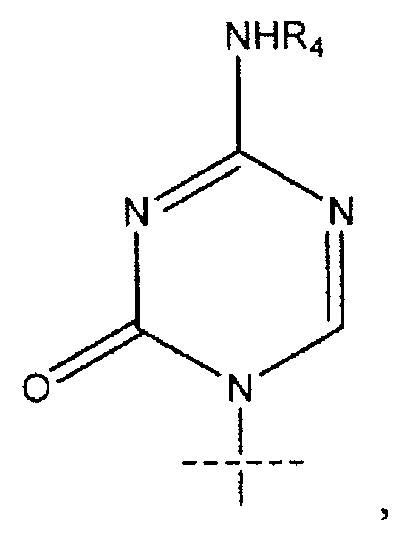
где перекрещивающаяся пунктирная лини показывает связь, образованную присоединением R1 к молекуле формулы (I). В некоторых вариантах осуществления изобретения R4 может представлять собой Н. В некоторых вариантах осуществления изобретения R представляет собой R5C(O), k равно 4, m равно 0, R2 представляет собой Н, R3 представляет собой ОН, R4 представляет собой Н.
В более широком аспекте настоящее изобретение относится к соединению формулы (I)'
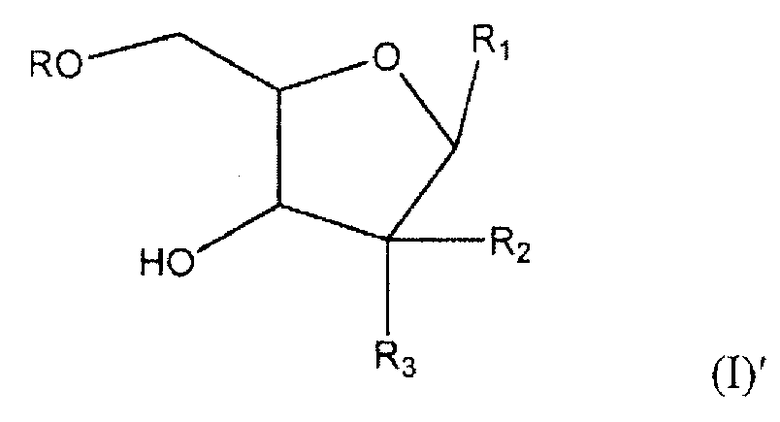
где R представляет собой Н, R5C(O), R5CH2OC(O) или R5CH2NHC(O), R1 представляет собой
где перекрещивающаяся пунктирная лини показывает связь, образованную присоединением R1 к молекуле формулы (I)'; R2 и R3 независимо представляют собой OH или H при условии, что R2 и R3 одновременно не являются OH; R4 представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O) при условии, что R и R4 одновременно не являются H; R5 представляет собой С3-С26 алкенил, или к его фармацевтически приемлемой соли.
В предпочтительном варианте осуществления соединения формулы (I)' k равно 4 и n равно 10. В некоторых вариантах осуществления изобретения R1 представляет собой
где перекрещивающаяся пунктирная лини показывает связь, образованную присоединением R1 к молекуле формулы (I)'. В некоторых вариантах осуществления изобретения R4 может представлять собой Н. В других вариантах осуществления R представляет собой R5C(O); k равно 4; m равно 0; n равно 10; R2 представляет собой Н; R3 представляет собой ОН. В некоторых вариантах осуществления R5 представляет собой С9-С26 алкенил.
Другой аспект настоящего изобретения относится к фармацевтической композиции, включающей соединение формулы (I) и фармацевтический наполнитель, разбавитель и/или носитель.
Лекарственные средства согласно настоящему изобретению могут вводиться перорально, парентерально, например, подкожно, внутривенно, внутримышечно, интраперитонеально, интраназальным вливанием или посредством нанесения на слизистые оболочки, такие как слизистая оболочка носа, горла или бронхиол. Они могут вводиться сами по себе или с подходящими фармацевтическими носителями, и могут быть в твердой или жидкой форме, такой как таблетки, капсулы, порошки, растворы, суспензии или эмульсии.
Активные ингредиенты согласно настоящему изобретению могут вводиться перорально, например, с инертным разбавителем или с ассимилируемым съедобным носителем, они могут заключаться в капсулы с твердыми или мягкими оболочками, они могут прессоваться в таблетки или они могут вводиться непосредственно с пищей рациона питания. Для перорального введения при терапевтическом лечении указанные активные ингредиенты могут вводиться с наполнителями и применяться в форме таблеток, капсул, эликсиров, суспензий, сиропов и т.п. Такие композиции и препараты должны содержать, по меньшей мере, 0,1% активного ингредиента. Процентное содержание активного ингредиента в указанных композициях, разумеется, может изменяться и традиционно может находиться в интервале от примерно 2% до примерно 60% массы стандартной лекарственной формы. Количество активного ингредиента в таких терапевтически применимых композициях является таким, чтобы была получена подходящая доза. Предпочтительные композиции согласно настоящему изобретению получают таким образом, что лекарственная форма стандартной дозы содержит от примерно 1 до 250 мг активного ингредиента.
Таблетки, капсулы и т.п. также могут содержать связующее вещество, такое как трагакантовая камедь, гуммиарабик, кукурузный крахмал или желатин; наполнители, такие как гидрофосфат кальция; дезинтегрирующее средство, такое как кукурузный крахмал, картофельный крахмал, альгиновая кислота; добавку, повышающую скольжение, такую как стеарат магния; и подслащивающую добавку, такую как сахароза, лактоза или сахарин. Когда лекарственная форма стандартной дозы представляет собой капсулу, она может содержать помимо добавок, указанных выше, жидкий носитель, такой как жирное масло.
Различные другие материалы могут присутствовать в виде покрытий или служить для модификации физического состояния лекарственной формы стандартной дозы. Например, таблетки могут быть покрыты щеллаком, сахаром или обоими этими веществами. Сироп может помимо активного ингредиента содержать сахарозу в качестве подслащивающего агента, метил и пропилпарабены в качестве консервантов, краситель и вкусовую добавку, такую как вишневая или апельсиновая вкусовая добавка.
Указанные активные ингредиенты также могут вводиться парентерально. Растворы или суспензии указанных активных ингредиентов могут быть получены в воде, подходящим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть получены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Типичными примерами масел являются масла нефтяного, животного, растительного происхождения или синтетические масла, например, арахисовое масло, соевое масло или минеральное масло. Предпочтительными жидкими носителями, в частности в растворах для инъекций, обычно являются вода, раствор соли, водная декстроза и раствор родственных сахаров, а также гликоли, такие как пропиленгликоль или полиэтиленгликоль. В стандартных условиях хранения и применения указанные препараты содержат консервант для предотвращения роста микроорганизмов.
Фармацевтические формы, подходящие для применения в виде инъекций, включают стерильные водные растворы или дисперсии и стерильные порошки для быстрого приготовления стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и должна быть жидкой в такой степени, чтобы можно было их применять с помощью шприца. Она должна быть стабильной в условиях производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, многоатомный спирт (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Активные ингредиенты согласно настоящему изобретению также могут вводиться непосредственно в дыхательные пути в форме аэрозоля. Для применения в виде аэрозолей активные ингредиенты согласно настоящему изобретению в растворе или суспензии могут расфасовываться в аэрозольный баллон под давлением вместе с подходящими пропеллентами, например углеводородными пропеллентами, такими как пропан, бутан или изобутан, с подходящими адъювантами. Соединения согласно настоящему изобретению также могут вводиться в форме без давления, такой как аэрозольный ингалятор или пульверизатор.
Дополнительный аспект настоящего изобретения относится к способу лечения у субъекта новообразования. Способ включает выбор субъекта с новообразованием и введение субъекту соединения формулы (I), которая описана выше, или его фармацевтически приемлемой соли в условиях, эффективных для лечения у субъекта новообразования.
В некоторых вариантах осуществления изобретения новообразование представляет собой раковое заболевание. Раковое заболевания может представлять собой солидную опухоль, гемобластоз или злокачественную опухоль кроветворной ткани. Раковое заболевание может представлять собой лейкемию, лимфому, множественную миелому или миелодиспластический синдром.
В некоторых вариантах осуществления изобретения солидная опухоль может представлять собой рак ткани, такой как ткань молочной железы, ткань яичника, ткань предстательной железы, ткань мозга, ткань мочевого пузыря и ткань легких.
Дополнительный аспект настоящего изобретения относится к способу лечения у субъекта воспалительного процесса. Способ включает выбор субъекта с воспалительным процессом и введение субъекту соединения формулы (I), которая описана выше, или его фармацевтически приемлемой соли в условиях, эффективных для лечения у субъекта воспалительного процесса.
В некоторых варианта осуществления изобретения воспалительное состояние представляет собой воспаление легких, воспаление соединительной ткани, воспаление желудочно-кишечного тракта или воспаление сосудистой сети.
За исключением особо оговоренных случаев, научные и технические термины, используемые в связи с настоящей заявкой, будут иметь значения, которые обычно вкладывает в них специалист данной области техники. Кроме того, за исключением случаев, когда того требует контекст, термины в единственном числе должны включать и термин в множественном числе, а термины в множественном числе должны включать термины в единственном числе.
ПРИМЕРЫ
Пример 1 - Реагенты, клеточные линии и клеточная культура
Реагент клеточной пролиферации WST-1 получают из Roche Applied Science (Manheim, Germany), PI и Annexin V - FITC (набор для определения процентной доли клеток, подвергшихся апоптозу) получают от BD Biosciences, Palo Alto, CA, 5-азатидин (5-аза-C), бромид этидия (EB), акридиновый оранжевый (АО), нитросиний тетразолий (NBT), форбол-12-миристат-13-ацетат (ТРА) получают от Sigma Chemical Co (St. Lous, MO).
Клеточные линии промиелоцитарной лейкемии человека HL60, гистиоцитной лимфомы человека U937, хронической миелогенной лейкемии человека К562, Т-клетки человека линии Jurkat, аденокарциномы молочной железы MCF-7, карциномы мочевого пузыря 5637, карциномы простаты DU-145 получают из Американской коллекции типов культур (American Type Culture Collection). Все клеточные линии, за исключением клеток линии Jurkat, выдерживают в RPMI 1640 среде (Gibco, Glasgow, UK), снабженной 10% термоинактивированной фетальной телячьей сывороткой (FCS), 100 U/мл пенициллина и 100 мг/мл стрептомицина в атмосфере 5% СО2 при 37°С. Клетки линии Jurkat выращивают в RPMI 1640 среде, снабженной 1,5 г/л гидрокарбоната натрия, 4,5 г/л глюкозы, 10 мМ пирувата натрия, 10% FCS, 100 U/мл пенициллина и 100 мг/мл стрептомицина.
Пример 2 - Определение цитотоксичности
Цитотоксичность 5-азацитидинового липида определяют колориметрическим анализом, который основан на расщеплении соли тетразолия WST-1 (дисульфонат 4-[3-(4-лодофенил)-2-(4-нитрофенил)-2Н-5-тетразолио]-1,3-бензола) посредством митохондриальных дегидрогеназ в жизнеспособных клетках. Клетки высевают в 96-луночные плоскодонные микропланшеты с начальной концентрацией 1×106/мл (HL60 клетки) или 1,25×105/мл (U937, K562 и Jurkat) в среде с добавлением или без добавления 5-азацитидинового липида различных концентраций и выращивают в течение от 24 до 72 часов. Клетки MCF-7, DU-145 и 5637 (1×104/мл) высевают, дают им возможность адгезировать и разрастаться в течение 24 часов. 5-азацитидиновый липид добавляют в различных концентрациях и культуры выдерживают в течение 24 или 72 часов. Культуры инкубируют с WST-1 реагентом в течение 1 часа.
Продуцирование формазана количественно определяют с помощью микропланшет-ридера (Bio-Tek Instruments, Elx 800) при 450 нм с эталонной длиной волны 650 нм. Ингибирование роста определяют при сравнении с необработанными клетками (%). Значения IC50 вычисляют с использованием программного обеспечения CalcuSyn (Biosoft).
Пример 3 - Подсчет апоптотических клеток
Апоптотические клетки идентифицируют с использованием морфологических критериев и флуоресцентного сортера клеток (fluorescence-activated cell sorting - FACS) после подкрашивания Annexin V - FITC. Для морфологического анализа 1 мкл исходного раствора, содержащего 100 мкг/мл АО и 100 мкг/мл ЕВ, добавляют к 25 мкг клеточной суспензии. Апоптотические клетки и апоптическую массу анализируют с помощью флуоресцентного микроскопа. Процент апоптотических клеток рассчитывают после подсчета всех 300 клеток. Для FACS анализируют 2×105 до 5×106 клеток промывают PBS и затем метят с помощью Annexin V-FICS и йодидом пропидиума (PI) в реагенте, связывающем среду, согласно инструкции набора Annexin V-FITC для определения процентной доли клеток, подвергшихся апоптозу, предоставленной производителем. Флуоресцентные сигналы FITC и PI обнаруживают, соответственно, при 518 нм и при 620 нм на FACSCAN (Becton Dickinson, San Jose, CA). Значения log Annexin V-FITC флуоресценции наносят на оси Х, значения log PI флуоресценции наносят на оси Y. Данные анализируют с помощью программного обеспечения CellQuest (Becton Dickinson). Для каждого анализа считывают результаты для 10000 клеток.
Пример 4 - Клеточный цикл
Клетки пеллетируют центрифугированием и дважды промывают PBS, фиксируют с помощью охлажденного 70% (об./об.) этанола (-20°С) и хранят при 4°С в течение, по меньшей мере, 24 часов. Клетки промывают в PBS. Клеточные пеллеты подкрашивают красящим раствором PI/RNase. Клеточную суспензию инкубируют в темноте при комнатной температуре в течение 30 минут. Содержание ДНК определяют с использованием аппарата проточного цитометрического анализа FACSCalibur (Becton Dickinson, Mount View, CA). Проценты клеток Sub-G1, G1, S и G2/M стадий клеточного цикла определяют с использованием программы, представляющей данные ДНК в виде гистограмм (Becton Dickinson). Для каждого образца считывают минимум 10000 результатов.
Пример 5 - Синтез сложного эфира аза-С-5'-петроселиновой кислоты
Петроселиновую кислоту (1,75 ммоль, 494 мг) растворяют в толуоле (3 мл). Добавляют сначала ДМФА (10 мкл), затем в течение 10 минут при комнатной температуре добавляют оксалилхлорид (3,6 ммоль, 457 мг). Спустя 3 часа толуол удаляют в вакууме.
Аза-С (1,57 ммоль, 427 мг) суспендируют в ДМА (6 мл), добавляют HCl (1М в Et2O, 2,0 ммоль, 2,0 мл), выдерживают в течении 5 минут при комнатной температуре, после чего Et2O удаляют в вакууме. Полученный мутный раствор охлаждают на водно-ледяной бане и в течение 40 минут добавляют хлорангидрид, растворенный в ДМА (2 мл). Реакционную смесь перемешивают в течение ночи, в процессе чего температура смеси медленно достигает комнатной температуры. Спустя 24 часа растворители удаляют при давлении 0,1 мбар. Остаток распределяют между насыщенным водным раствором NaHCO3 и EtOAc (25 мл каждого). Водную фазу экстрагируют EtOAc (3×25 мл). Органические фазы объединяют, промывают раствором соли и сушат (MgSO4). После удаления растворителей в вакууме сырой продукт (600 мг) очищают флэш-хроматографией (SiO2, CH2Cl2 с 2,5, 5 и 10% MeOH). И наконец, продукт сушат при ~0,25 мбар в течение ночи. Выход: 210 мг (24%).
Пример 6 - Синтез сложного эфира аза-С-5'-петроселаидиновой кислоты
Петроселаидиновую кислоту (1,77 ммоль, 500 мг) растворяют в толуоле (3 мл), добавляют сначала ДМФА (10 мкл), затем в течение 10 минут при комнатной температуре оксалилхлорид (3,6 ммоль, 457 мг). Спустя 3 часа толуол удаляют в вакууме.
Аза-С (1,75 ммоль, 427 мг) суспендируют в ДМА (6 мл), добавляют HCl (1М в Et2O, 2,0 ммоль, 2,0 мл), смесь выдерживают в течение 5 минут при комнатной температуре, после чего Et2O удаляют в вакууме. Полученный мутный раствор охлаждают на водно-ледяной бане и в течение 2 часов минут добавляют хлорангидрид, растворенный в ДМА (2 мл). Реакционную смесь перемешивают в течение ночи, в процессе чего температура смеси медленно достигает комнатной температуры, после этого смесь в течение 2 часов выдерживают при 30°С. После охлаждения до комнатной температуры остаток распределяют между насыщенным водным раствором NaHCO3 и EtOAc (по 25 мл каждого). Водную фазу экстрагируют EtOAc (3×25 мл). Органические фазы объединяют, промывают раствором соли и сушат (MgSO4). После удаления растворителей в вакууме получают сложный эфир в виде белого порошка. Выход: 500 мг.
Пример 7 - Метаболическая стабильность сложного эфира 5-аза-5'-петроселиновой кислоты в пуле плазмы человека
Сложный эфир 5-аза-С-5'-петроселиновой кислоты осторожно добавляют в пул плазмы человека в пяти концентрациях (0,1, 1, 3, 10 и 30 мкМ, соответственно). Смесь инкубируют на качающейся водяной бане при 37°С. Отбирают по три (n=3) аликвоты инкубированных растворов после истечения установленного периода инкубации (0, 15, 30, 60 и 120 минут) и сразу осаждают белок плазмы с использованием ацетонитрила, содержащего 0,1% муравьиной кислоты (300 мкл). Отрицательный контроль приготавливают с испытываемым соединением и аза-С в экспериментальном буфере (PBS, рН 7,4) при одной концентрации инкубации (1 мкМ). После центрифугирования супернатант непосредственно используют в ЖХ-МС-МС анализе. См. таблицу 1.
трация
(мкМ)
(среднее ± SD, n=3)
Пример 8 - Цитотоксичность аза-С и 5-аза-С-5'-петроселиновой кислоты
Цитотоксичность аза-С и 5-аза-С-5'-петроселиновой кислоты определяют в клеточной линии МТ-3 рака молочной железы и адриабластин-резистетной клеточной линии МТ-3/ADR. MT-3/ADR сверхэкспрессируют MDR-1/p-гликопротеин. Клетки высевают в 96-луночные планшеты с плотностью 5×103 клеток на лунку в среду RPMI 1640 c 2 мМ глютамина и 10% FBS. Клетки инкубируют в течение 24 часов. Испытываемые соединения растворяют в ДМСО и дополнительно разбавляют средой непосредственно перед применением. Для каждой опытной концентрации используют 6 лунок. Клетки инкубируют с испытываемым соединением в течение 24 часов. В каждую лунку добавляют 20 мкл свежеприготовленного МТТ раствора и инкубируют в течение 4 часов. Значения IC50 определяют из кривых роста, представленных графически для 8 различных концентраций в интервале от 0,01 мкМ до 100 мкМ. Результаты представлены в таблице 1. Аналогичную активность получают для аза-5 и 5-аза-С-5'-петролелиновой кислоты в клеточной линии карциномы молочной железы МТ-3, но в МТ-3/ADR резистентной клеточной линии активность аза-С теряется. Активность не наблюдается в области испытанных концентраций до 100 мкМ, в то время как 5-аза-С-5'-петроселиновая кислота остается активной со значением IC50 в резистентной клеточной линии, аналогичным значению с нерезистентной МТ-3 линии. См. таблицу 2.
IC50 (мкМ)
Пример 9 - Антипролиферативная активность Аза-С и 5-Аза-С-5'-петроселиновой кислоты
Антипролиферативную активность аза-С и 5-аза-С-5'-петроселиновой кислоты определяют в клеточной линии Hela рака шейки матки при 24- и 72-часовой экспозиции. Клетки высевают в 96-луночные микропланшеты при плотности 5×103 клеток на лунку в RPMI 1640 среде с 2 мМ глютамина и 10% FBS. Клетки инкубируют в течение 24 и 72 часов. Испытываемые соединения растворяют в ДМСО и дополнительно разбавляют средой непосредственно перед применением. Для каждой экспериментальной концентрации используют 6 лунок. Цитотоксичность определяют с использованием МТТ анализа, в каждую лунку добавляют 20 мкл свежеприготовленного МТТ раствора и инкубируют в течение 4 часов. Значения IC50 определяют из кривых роста, представленных графических, для 8 концентраций в интервале от 0,01 мкМ до 100 мкМ. Аналогичную цитотоксическую активность получают при пролонгированной экспозиции в течение 72 часов для двух указанных соединений, но неожиданно цитотоксический эффект 5-аза-С-5'-петроселиновой кислоты имел место уже после 24 часовой экспозиции. Различный временной профиль наблюдается для аза-С- и 5-аза-С-5'-петровелиновой кислоты с более быстрым началом фитотоксического действия 5-аза-С-5'-петроселиновой кислоты. См. фиг.1.
Пример 10 - Воздействие ингибирования нуклеозидного транспортера на цитотоксическую активность в раковых клетках для аза-С и 5-аза-С-5'-петроселиновой кислоты
Влияние ингибирования нуклеозидного транспортера на цитотоксическую активность оценивают в мутантных клетках Hela рака шейки матки для аза-С и 5-аза-С-5'-петроселиновой кислоты. В качестве ингибитора равновесных нуклеозидных транспортеров используют hENT1 и hENT2. Клетки высевают в 96-луночные планшеты при плотности 5×103 клеток на лунку в RPMI 1640 среду с 2 мМ глютамина и 10% FBS. Клетки предварительно инкубируют в течение 24 часов. В клетки за 30 минут до добавления испытываемых соединений добавляют дипиридамол (10 мкМ). Испытываемые соединения растворяют в ДМСО и дополнительно разбавляют средой непосредственно перед применением. Для каждой экспериментальной концентрации используют 6 лунок. Клетки инкубируют с испытываемым соединением в течение 72 часов. В каждую лунку добавляют 20 мкл свежеприготовленного МТТ раствора и инкубируют в течение 4 часов. Значения IC50 определяют из кривых роста, представленных графических, для 8 концентраций в интервале от 0,01 мкМ до 100 мкМ. Результаты представлены в таблице 3. Активность аза-С снижается в 3 раза в результате добавления ингибитора нуклеозидного транспорта дипиридамола, показывая, что приток и отток аза-С в Hela клетках в некоторой степени зависит от нуклеозидных транспортеров hENT1 и hENT2. Цитотоксическая активность 5-аза-С-5'-петроселиновой кислоты не только сохраняется, но и повышается в 10 раз при блокировании hENT1 и hENT2 нуклеозидных транспортеров посредством применения дипиридамола. Это может быть особенно важно для пациентов, у которых активность аза-С отсутствует вследствие потери экспрессии нуклеозидных транспортеров. См. таблицу 3.
IC50 (мкМ)
Пример 11 - Экспрессия гена эстрогенового рецептора β (ERβ) в линии клеток рака молочной железы после обработки азацитидином или 5-аза-С-5'-петроселиновой кислотой
Экспрессию гена (определенную на уровне РНК) эстрогенового рецептора бэта определяют методом ПЦР в реальном времени (TagMan). Клетки рака молочной железы MCF-7 выращивают в среде с дефицитом экстрогена (Phenol-Red-free RPMI с 2% глютамина и 10% фетальной телячьей сыворотки, обработанной смесью древесный уголь-декстран). Клетки высевают в чашки площадью 25 см2 и инкубируют их в течение 24 часов перед обработкой 1 мкМ азацитидина или 5-аза-С-5'-петроселиновой кислотой. В качестве контроля используют один необработанный контроль. Клетки собирают после 5 дневной экспозиции соединениями, их собирают трипсинацией, промывкой и быстрой заморозкой в жидком азоте.
Всю РНК экстрагируют из приблизительно 106 быстрозамороженных MCF-7 клеток, определяют концентрацию и чистоту РНК, РНК транскрибируют в кДНК с использованием TagMan Reverse Transcription реагента (N808-0234). Количественное определение в реальном времени осуществляют с использованием стандартных методик и предварительно смешанных ПЦР-реагентов. Исходные пробные смеси получают от Applied Biosystems, ER β (ID Hs00230957_m1) и гидроцилметил-билансинтазы гена «домашнего хозяйства» HMBS (ID Hs00609297_m1). Экспрессию гена вычисляют с использованием сравнительного дельта-дельта Ct способа. Индукция экспрессии ER β в 4,14 раза повышается после экспозиции 5-аза-С-5'-петроселиновой кислотой, в то время как после экспозиции в азацитидине она увеличивается только в 2,51 раза (см. таблицу 4). Это может иметь большое значение для лечения гормонально активных опухолей, когда может быть восстановлена гормональная чувствительность (см. таблицу 4).
Хотя предпочтительные варианты осуществления были представлены и описаны здесь подробно, специалисту данной области техники будет понятно, что различные модификации, дополнения, замещения и т.п. могут быть сделаны без выделения их из области данного изобретения и, следовательно, должны рассматриваться как часть области изобретения, которая определена в формуле изобретения, представленной далее.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДИОКСОЛАНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2418795C2 |
| АНАЛОГИ АЗАЦИТИДИНА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2487883C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2733393C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2805949C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ТРИЖДЫ НЕГАТИВНОГО РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ И РАКА ЯИЧНИКА | 2019 |
|
RU2776897C2 |
| НОВОЕ ПРИМЕНЕНИЕ АРИЛ-ХИНОЛИНОВЫХ ПРОИЗВОДНЫХ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСКУЛОГЕННОЙ МИМИКРИИ | 2016 |
|
RU2685731C1 |
| СПОСОБЫ ЛЕЧЕНИЯ | 2020 |
|
RU2822394C1 |
| БЕНЗО[b]ТИОФЕНОВЫЕ АГОНИСТЫ STING ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2771811C2 |
| СОЕДИНЕНИЯ, СВЯЗЫВАЮЩИЕСЯ С BIR ДОМЕНОМ IAP | 2007 |
|
RU2446170C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ТЕТРАЦИКЛИЧЕСКИМИ АНАЛОГАМИ ХИНОЛОНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2752506C2 |
Настоящее изобретение относится к новым соединениям формулы (I):
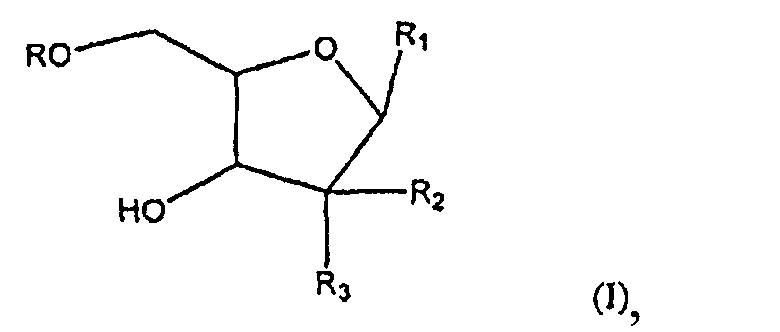
где R представляет собой Н, R5C(O), R5СН2ОС(О) или R5CH2NHC(О);
R1 представляет собой
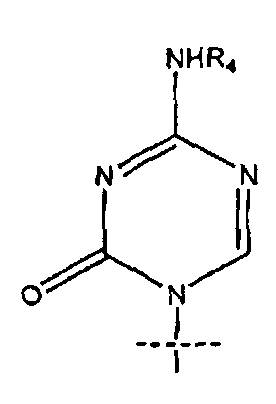 или
или 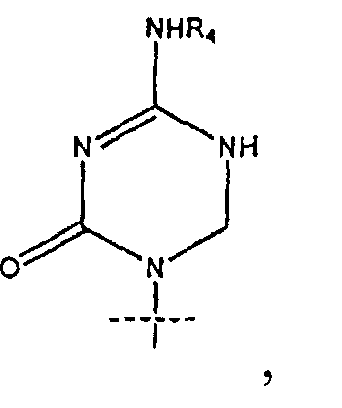
где перекрещивающаяся пунктирная линия показывает связь, образованную присоединением R1 к молекуле формулы (I);
R2 и R3 независимо представляют собой ОН или Н при условии, что R2 и R3 одновременно не являются ОН; R4 представляет собой Н; R5 имеет общую формулу: CH3-(CH2)n-(CH=CH-CH2)m-CH=CH-(CH2)k-; где k представляет целое число от 0 до 7; m представляет собой целое число от 0 до 2 и n представляет собой целое число от 0 до 10, или их фармацевтически приемлемым солям. Эти соединения обладают противораковой активностью. Изобретение также относится к фармацевтической композиции на основе этих соединений и применению их для изготовления лекарственного средства для лечения раковых заболеваний. 3 н. и 12 з.п. ф-лы, 4 табл., 11 пр., 1 ил.
1. Соединение формулы (I)
где R представляет собой Н, R5C(O), R5CH2OC(O) или R5CH2NHC(О);
R1 представляет собой
или
где перекрещивающая пунктирная линия показывает связь, образованную присоединением R1 к молекуле формулы (I);
R2 и R3 независимо представляют собой ОН или Н при условии, что R2 и R3 одновременно не являются ОН;
R4 представляет собой Н;
R5 имеет общую формулу:
СН3-(СН2)n-(СН=СН-СН2)m-СН=СН-(СН2)k;
k представляет целое число от 0 до 7;
m представляет собой целое число от 0 до 2; и
n представляет собой целое число от 0 до 10,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где k равно 4.
3. Соединение по п.2, где n равно 10.
4. Соединение по п.1, где R1 представляет собой
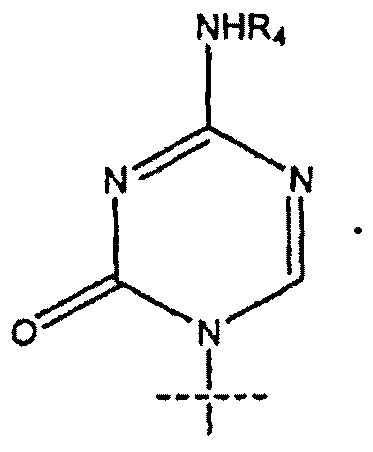
5. Соединение по п.1, где R представляет собой R5C(O); R1 представляет собой
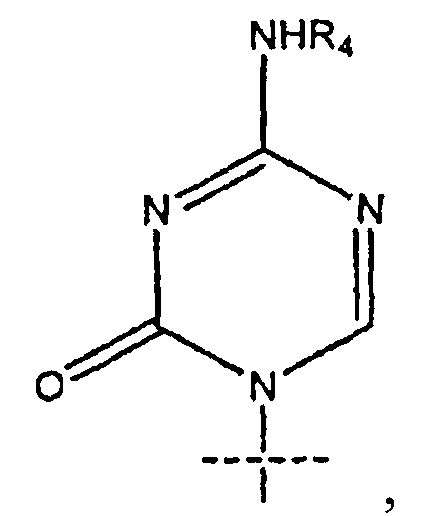
R2 представляет собой Н; R3 представляет собой ОН; R4 представляет собой Н; k равно 4; m равно 0; и n равно 10.
6. Фармацевтическая композиция для лечения рака, включающая:
соединение по п.1 и фармацевтический наполнитель, разбавитель и/или носитель.
7. Применение соединения формулы:
где R представляет собой Н, R5C(О), R5CH2OC(O) или
R5CH2NHC(O);
R1 представляет собой
где перекрещивающаяся пунктирная линия показывает связь, образованную присоединением R1 к молекуле формулы (I);
R2 и R3 независимо представляют собой ОН или Н, при условии, что R2 и R3 одновременно не являются ОН;
R4 представляет собой Н;
R5 имеет общую формулу
СН3-(СН2)n-(СН=СН-СН2)m-СН=СН-(СН2)]k-,
где k представляет целое число от 0 до 7;
m представляет собой целое число от 0 до 2; и n представляет собой целое число от 0 до 10;
или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения новообразования у субъекта.
8. Применение по п.7, где новообразование представляет собой раковое заболевание.
9. Применение по п.8, где раковое заболевание представляет собой солидную опухоль, гематологический рак или злокачественную опухоль.
10. Применение по п.8, где раковое заболевание представляет собой лейкемию, лимфому, множественную миелому или миелодиспластический синдром.
11. Применение по п.9, где солидная опухоль представляет собой рак ткани, выбранной из группы, состоящей из ткани молочной железы, ткани яичника, ткани предстательной железы, ткани мозга, ткани мочевого пузыря и ткани легкого.
12. Применение по п.7, где k равно 4.
13. Применение по п.12, где n равно 10.
14. Применение по п.7, где R1 представляет собой
15. Применение по п.7, где R представляет собой R5C(O); R1 представляет собой
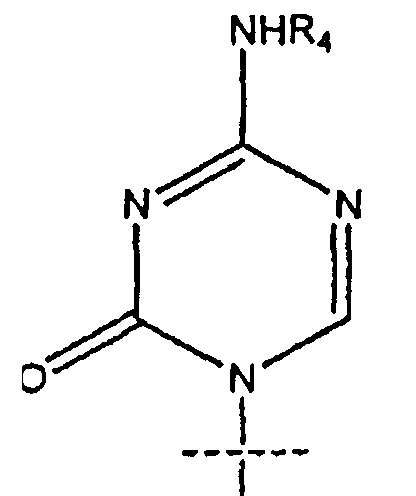
R2 представляет собой Н; R3 представляет собой ОН; R4 представляет собой Н; k равно 4; m равно 0 и n равно 10.
| WO 9705154 A1, 13.02.1997 | |||
| WO 2007035783 A2, 29.03.2007 | |||
| US 4172889 A, 30.10.1979 | |||
| RU 2000100373 A, 10.01.2002 | |||
| ПИРИМИДИН-НУКЛЕОЗИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116306C1 |
| 3-Фтор-2,3-дидезоксиаденозин,проявляющий цитостатическую активность | 1981 |
|
SU961354A1 |

