По настоящей заявке испрашивается приоритет на основании предварительной заявки на патент США №60/975437, поданной в патентное ведомство США 26 сентября 2007 г., содержание которой полностью включено в настоящее описание посредством ссылки.
Область техники
Настоящее изобретение относится к аналогам азацитидина и их применению.
Уровень техники
Аналоги нуклеозидов, производные природных нуклеозидов, которые встречаются в качестве строительных блоков ДНК и РНК, являются эффективными в клиническом лечении рака или вирусных заболеваний у человека, хотя раньше подобные соединения рассматривали в качестве противотуберкулезных средств. Такие соединения зарегистрированы на рынке уже в течение более 40 лет, и в настоящее время примерно 35 продуктов применяются ежедневно. Природные нуклеозиды, приведенные в таблице 1 ниже, состоят из азотистых оснований двух классов, а именно пуринов (представлены аденином и гуанином) и пиримидинов (представлены тимином, урацилом и цитозином), а также из моносахарида рибозы или дезоксирибозы.
Таблица 1
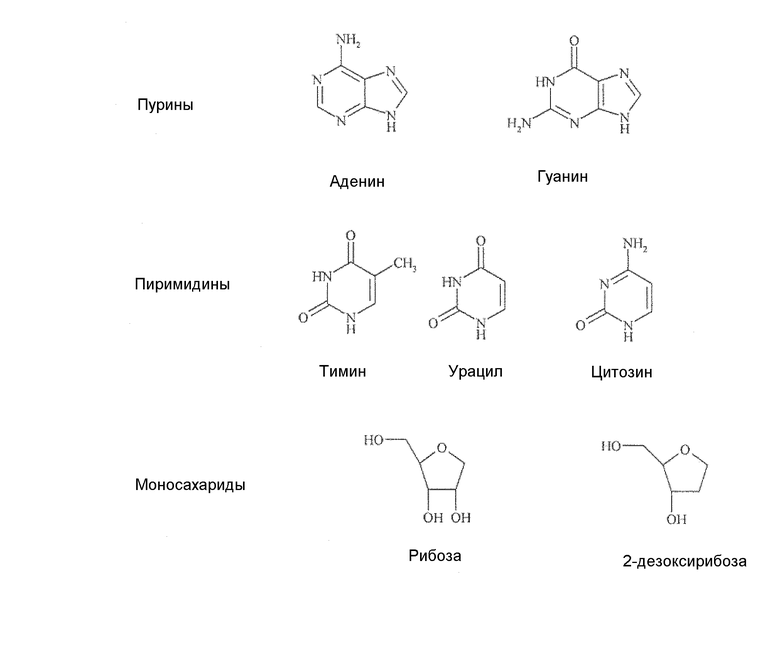
Все природные нуклеозиды существуют в так называемой β-D конфигурации, как показано ниже в формуле А. Азотистое основание и гидроксиметильная боковая цепь в цикле сахара находятся по одну и ту же сторону (цис) от плоскости кольца сахара.
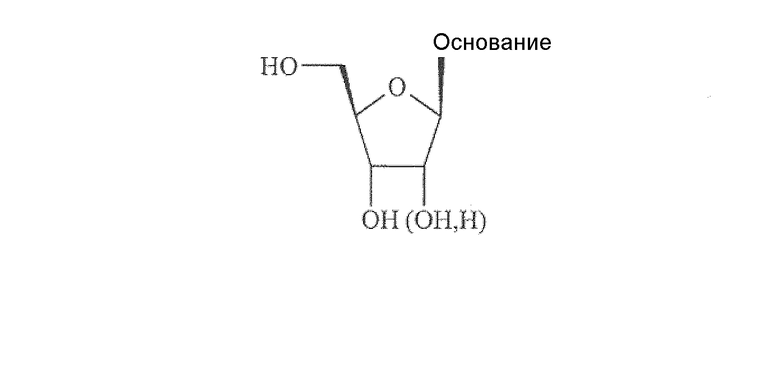
Формула A
С целью получения нуклеозидных производных с противораковой или противовирусной активностью были произведены химические модификации в азотистом основании и/или моносахариде. Например, введение в азотистое основание атомов галогена или других функциональных групп, введение дополнительных атомов азота, или же стереохимическая замена кольца моносахарида с рибозы на арабинозу или удаление гидроксильной группы с получением дезоксирибозы может приводить к образованию продуктов, обладающих терапевтическим потенциалом. Во многих продуктах моносахаридное кольцо сохраняется, тогда как в других кольцо сахара может быть трансформировано в цепь. Аналоги нуклеозидов представляют собой небольшие молекулы, имеющие превосходную растворимость в воде.
Многочисленные усилия, направленные на исследования и разработки в области аналогов нуклеозидов, обусловленные всемирной эпидемией СПИД, содействовали накоплению фундаментальной базы знаний и пониманию механизма действия, изменений в профиле активности благодаря химической модификации и т.п., относятся также и к области лечения рака.
Общим недостатком многих лекарственных средств, включая аналоги нуклеозидов, является низкая активность и низкая специфичность при лечении конкретного рассматриваемого заболевания. Некоторые из этих проблем можно отнести к активности, присущей самому лекарственному средству, некоторые можно отнести к определенным механизмам устойчивости (как врожденным, так и приобретенным в ходе лечения, например множественная лекарственная устойчивость (MDR) при лечении рака). Некоторые проблемы можно отнести к замедленному транспорту или клеточному захвату и механизмам активации. Некоторые проблемы можно отнести к быстрой инактивации и/или экскреции лекарственного средства.
Эффективность аналогов нуклеозидов зависит в немалой степени от их способности мимикрировать под природные нуклеозиды и, таким образом, взаимодействовать с вирусными и/или клеточными ферментами, а также препятствовать протеканию ключевых процессов в метаболизме нуклеиновых кислот (или ингибировать их). Для того чтобы привести в действие их противовирусную или противораковую активность, аналоги нуклеозидов следует трансформировать через их моно- и дифосфаты в их соответствующие трифосфаты под действием вирусных и/или клеточных киназ. В общем случае, активным средством является трифосфат, но для некоторых продуктов, например для гемцитабина, даже воздействие дифосфата может быть клинически значимым.
Для того чтобы достичь пораженных заболеванием, раковых или инфицированных вирусом клеток или тканей после энтерального или парентерального введения, аналоги нуклеозидов должны обладать подходящими фармакокинетическими характеристиками. Помимо быстрой экскреции введенного лекарства, многие аналоги нуклеозидов могут быть деактивированы как в кровотоке, так и в тканях. Например, производные цитозина, даже на уровне монофосфата, могут быть быстро деаминированы под действием класса ферментов, называемых деаминазами, до неактивного аналога урацила. Клеточный захват и, таким образом, высокая терапевтическая эффективность многих аналогов нуклеозидов сильно зависит от мембранных белков, осуществляющих транспорт нуклеозидов (называемых концентрирующим и уравновешивающим нуклеозидными переносчиками). Следовательно, объектом поиска являются соединения, которым не требуется такой специфический механизм захвата. Еще один фактор, ограничивающий активность, особенно в сфере лечения рака, представляет собой механизмы клеточного восстановления. Когда монофосфат противоракового нуклеозидного аналога встраивается в клеточную ДНК, он не удаляется из ДНК раковой клетки под действием эндонуклеазы, связанной с белком p53. Тем не менее удаление нуклеозидного аналога из ДНК здоровой клетки является желательным для того, чтобы ограничить побочные эффекты лекарственного средства.
В течение долгих лет были разработаны многие нуклеозидные аналоги, которые в значительной мере преодолевают некоторые или многие факторы, ограничивающие активность. В качестве примера, иллюстрирующего соединение, обладающее высокой специфичностью, можно привести ацикловир (ACV). ACV-монофосфат может образовываться только под действием вирусных киназ, и это означает, что ACV не может быть активирован в неинфицированных клетках. Несмотря на этот факт, ACV не является исключительно активным продуктом. Для того чтобы обойти распространенную скорость-лимитирующую стадию в активации нуклеозидного аналога, т.е. внутриклеточное образование монофосфата нуклеозидного аналога, были разработаны несколько фосфонатов, таких как цидофовир, или даже монофосфатные продукты. Для обеспечения возможности перорального приема или желаемой диспозиции лекарственного средства в организме, были получены конкретные пролекарства, такие как Hepsera.
В дополнение к структурным изменениям, произведенным в нуклеозидных аналогах для достижения повышенной клинической применимости, были осуществлены дальнейшие модификации для улучшения активности. Существует несколько примеров модифицированных нуклеозидных аналогов, получаемых в результате присоединения липидной части (патенты США №6153594, 6548486, 6316425 и 6384019; заявки на европейский патент № EP-A-56265 и EP-A-393920; и WO 99/26958). Присоединения липидной части можно достичь путем присоединения жирных кислот, например, посредством сложноэфирной, амидной, карбонатной или карбаматной связи. Можно получить более совершенные продукты, такие как фосфолипидные производные аналогов нуклеозидов. См. Eur. J. Pharm. Sci. 11b Suppl. 2: 15-27 (2000); европейский патент №545966; канадский патент №2468099; и патенты США №6372725 и 6670341. В указанных документах описывается, что такие аналоги имеют противовирусную активность, особенно подходящую для лечения и профилактики инфекций, вызванных ДНК, РНК или ретровирусами. Они также подходят для лечения злокачественных опухолей. Липидные производные аналогов нуклеозидов могут служить нескольким целям. Их можно рассматривать как пролекарство, которое не является субстратом для деаминаз, предохраняя таким образом нуклеозидные аналоги от деактивации во время транспорта в кровотоке. Липидные производные также могут быть более эффективно транспортированы через клеточную мембрану, что приведет к повышенной внутриклеточной концентрации нуклеозидного аналога. Липидные препараты могут быть также более подходящими для применения в дермальных препаратах, пероральных продуктах (см. патент США №6576636 и WO 01/18013), или конкретных композициях, таких как липосомы (см. патент США №5223263), разработанных для воздействия на опухоли.
Было показано, что в случае нуклеозидных аналогов с сохраненной β-D конфигурацией моносахаридного кольца или нуклеозидных аналогов с нециклической боковой цепью противовирусная или противораковая активность может быть наиболее эффективно улучшена путем получения производных мононенасыщенных ω-9 C18 и C20 жирных кислот. См. Antimicrobial Agents and Chemotherapy, Vol., 53-61 (1999); Cancer Research 59: 2944-2949 (1999); Gene Therapy, 5: 419-426 (1998); Antiviral Research, 45: 157-167 (2000); и Biochemical Pharmacology, 67: 503-511 (2004). Желательно, чтобы мононенасыщенные производные были не только более активными, чем полиненасыщенные аналоги, но и чтобы они легче подвергались кристаллизации и были более устойчивыми к окислению липидной цепи. Таким образом, указанные производные являются более предпочтительными соединениями с точки зрения химического и фармацевтического производства. Было также показано, что мононенасыщенные жирные кислоты ω-9 C18 и C20 подходят для улучшения терапевтической активности большого числа биологически активных веществ ненуклеозидной природы (см. европейский патент №0977725).
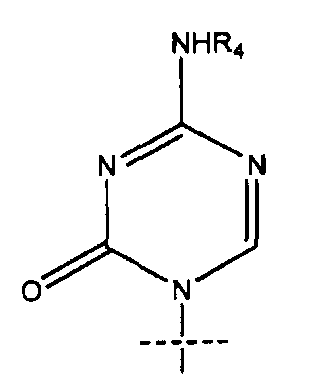
Сравнительно новой подгруппой нуклеозидных аналогов являются так называемые аза-C производные. У соединений этого класса CH-группа в положении 5 в пиримидиновом основании заменена атомом азота, как показано ниже в формуле B.
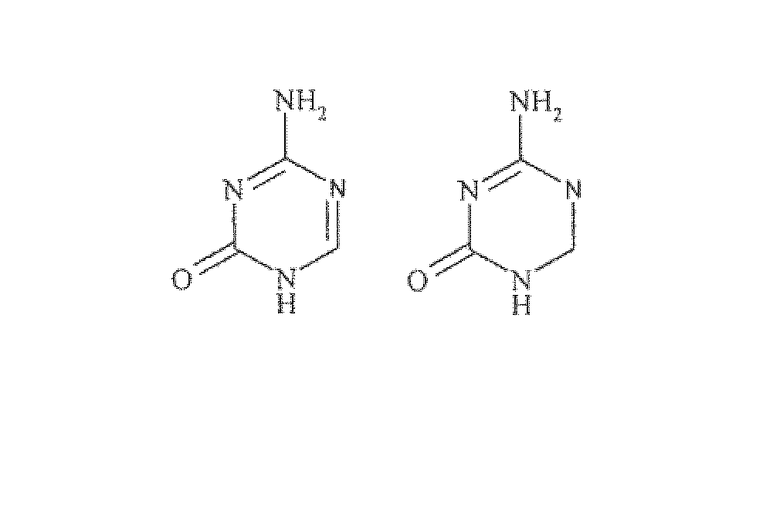
Формула B
Потенциальными мишенями для реактивации с помощью новых химиотерапевтических средств являются гены-супрессоры опухолей, которые были выключены вследствие абберантного метилирования ДНК. Производные аза-цитидина и 5-аза-2'-дезоксицитидина (5-аза-C, 5-аза-CdR, Децитабин), которые являются сильными ингибиторами метилирования ДНК и противолейкемическими средствами, могут реактивировать выключенный ген-супрессор опухолей. При высоких концентрациях эти соединения цитотоксичны, но при более низких концентрациях гипометилирование приводит к дифференцировке клеточных линий. Эти соединения требуют метаболической активации посредством дезоксицитидинкиназы и осуществляют ингибировние ДНК-метилтрансферазы. Одним из препятствий для их лечебного действия является их быстрая in vivo инактивация с участием цитидиндеаминазы (CD). Неустойчивость в водных растворах, а также совокупность побочных эффектов ограничивают клиническую активность указанных соединений.
Настоящее изобретение направлено на преодоление этих и других недостатков в данной области техники.
Краткое описание изобретения
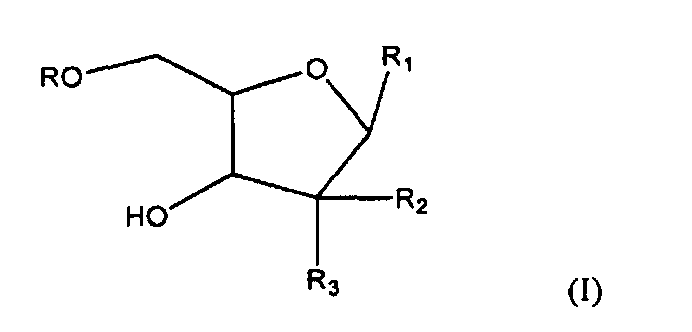
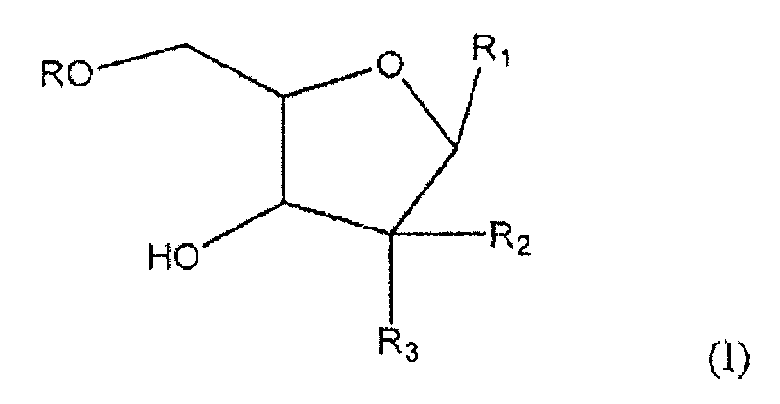
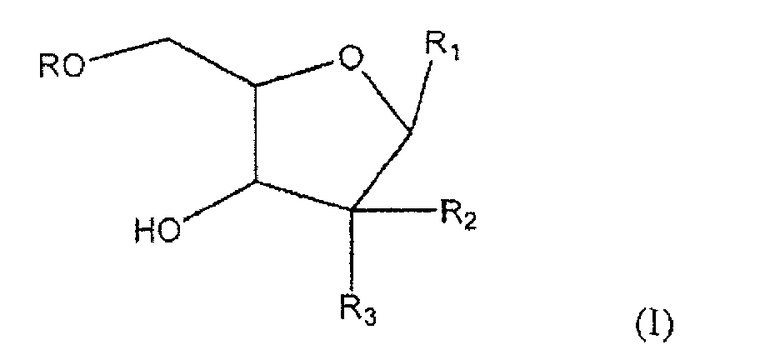
Один из аспектов настоящего изобретения направлен на соединение согласно формуле (I)
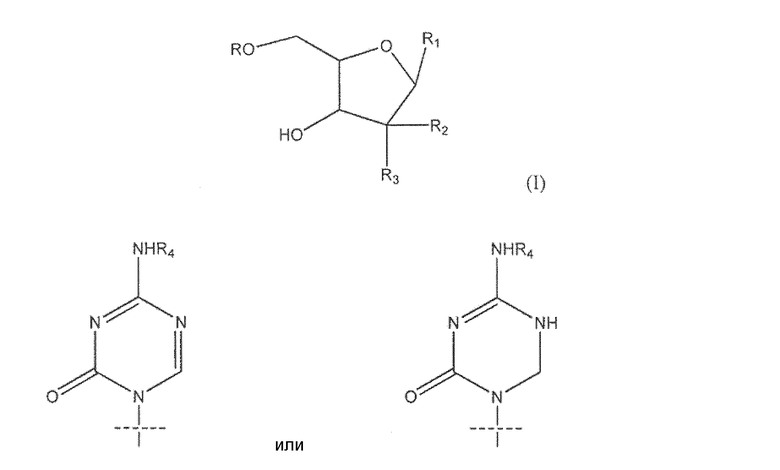
где R представляет собой H, R5C(O); R1 представляет собой
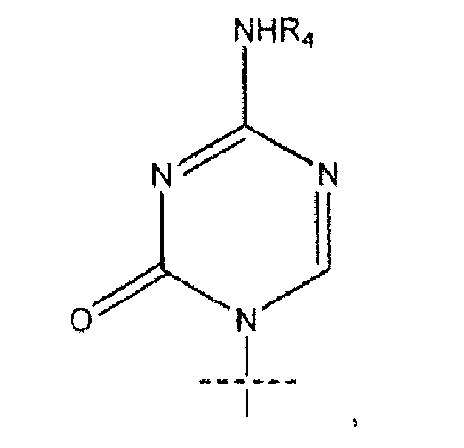
где пересекающая пунктирная линия изображает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I), R2 и R3 независимо представляют собой OH или H, при условии что R2 и R3 не являются одновременно OH, R4 представляет собой H или R5C(O), при условии, что R и R4 не являются одновременно H, R5 представляет собой C3-C26 алкенил; или фармацевтическую соль указанного соединения.
Другой аспект настоящего изобретения направлен на фармацевтическую композицию, содержащую соединение формулы (I) и фармацевтический эксципиент, разбавитель и/или носитель.
Еще один аспект настоящего изобретения направлен на способ лечения неопластического синдрома у пациентов. Данный способ включает отбор пациента с неопластическим синдромом и введение ему соединения формулы (I), как описано выше, или фармацевтической соли этого соединения, в условиях, эффективных для лечения неопластического синдрома у пациента.
Еще один аспект настоящего изобретения направлен на способ лечения воспалительного синдрома у пациентов. Данный способ включает отбор пациента с воспалительным синдромом и введение ему соединения формулы (I), как описано выше, или фармацевтической соли этого соединения, в условиях, эффективных для лечения воспалительного синдрома у пациента.
Неустойчивость Аза-C в буферном растворе и плазме крови хорошо известна (см. Israili et al., Cancer Research 36, 1453-1461 (1976); Rudek et al., J. Clin. Oncol., 23:17, 3906-3911 (2005); Rustum et al., J. Chromat., 421:12, 387-91 (1987); Zhao et al., J. Chromat B, 813, 81-88 (2004), полностью включенные в настоящую заявку посредством ссылки). Сообщалась средняя величина конечного периода полувыведения для Аза-C, составляющая 1,50±2,30 часа в клинических образцах плазмы крови (см. Rudek et al., J. Clin. Oncol., 23:17, 3906-3911 (2005), данный источник полностью включен в настоящую заявку посредством ссылки). В исследованиях in vitro была отмечена потеря 20% Аза-C даже при -60°C после 4,5 дней хранения, и потеря 10% в течение 0,5 часа при хранении при комнатной температуре (см. Zhao et al., J. Chromat B, 813, 81-88 (2004), данный источник полностью включен в настоящую заявку посредством ссылки). Предполагают, что первичная неустойчивость Аза-C обусловлена быстрым (первая стадия является обратимой) раскрытием цикла 5-Аза-пиримидинового кольца с последующим распадом муравьиной кислоты (см. Chan et al., J. Pharma Sci., 68;7, 807-12 (1979), данный источник полностью включен в настоящую заявку посредством ссылки). Предполагается, что прочие пути распада обусловлены деаминированием аминогруппы в 4 положении и гидролизом гликозидной связи с образованием D-рибозы и 5-азацитозина. Было неожиданно обнаружено, что предпочтительные липидные производные Аза-C имеют значительно лучший профиль устойчивости в плазме по сравнению с самим Аза-C. Соединения стабильны (доля остающегося от исходного ≥94%) в контрольной матрице человеческой плазмы при комнатной температуре в течение по меньшей мере 4 часов в экспериментальных условиях, и в постэкстракционном супернатанте после осаждения плазменных белков не было обнаружено значимых продуктов деградации. Устойчивость в плазме предпочтительных липидных соединений была изучена далее в условиях хранения при 37°C. Было показано, что возможность раскрытия кольца Аза-фрагмента или иного распада соединения существенно снижается, если к Аза-C присоединена липидная боковая цепь.
Быстрый распад Аза-C является препятствием для клинического применения Аза-C. Повышенная устойчивость в плазме крови липидных производных по сравнению с самим Аза-C может обеспечить высокий и устойчивый уровень липидного производного в плазме пациента. Это может привести к лучшему распределению в ткани/органе/опухоли и подверженности клетки действию лекарства, а также усвоению лекарственного средства, чем для самого Аза-C, с одной стороны, и, следовательно, к лучшей подверженности ДНК опухолевых клеток действию Аза-C после внутриклеточного гидролиза Аза-C-5'-сложноэфирной связи.
В вариантах реализации настоящего изобретения посредством модификации азацитидина и дезоксицитидина (например, 5-аза-2'-дезоксицитидина) предложены новые молекулы со свойствами, неожиданно отличающимися от свойств азацитидина и дезоксицитидина (например, 5-аза-2'-дезоксицитидина). Это обеспечивает серию соединений, имеющих активность значительно большую, чем противораковая активность азацитидина и дезоксицитидина (например, 5-аза-2'-дезоксицитидина), которая ограничена гематологическими злокачественными образованиями. Эти новые соединения обладают противораковым действием, направленным против широкого спектра солидных опухолей, включая рак молочной железы и шейки матки. Эти соединения проявляют также неожиданную активность против тех видов рака, которые устойчивы к лечению, и, таким образом, они могут быть полезны в терапии солидных опухолей в тех случаях, когда текущий выбор терапевтических средств ограничен. Варианты реализации настоящего изобретения терапевтически применимы при лечении рака в тех случаях, когда варианты и эффективность лечения ограничены, и указанные варианты реализации восполняют неудовлетворенную потребность.
Предложенные соединения демонстрируют более ранее проявление активности после ограниченного времени воздействия, и поэтому эффективно лишь краткосрочное лечение с их использованием в клинической ситуации. Это приводит к более короткому, менее частому терапевтическому воздействию и снижению связанных с лекарственным средством токсических эффектов, по сравнению с исходным лекарственным средством. Таким образом, обеспечивается повышенный терапевтический индекс.
При изменении структуры за счет введения липидного (включая как сложные эфиры, так и амиды) компонента сохраняется азольное цитидиновое кольцо и, следовательно, воздействие молекулы на эпигенетические механизмы. Эпигенетическая модуляция предоставляет важный механизм изменения экспрессии генов при раке и воспалении. Указанные новые соединения являются активными при меньших концентрациях, чем азацитидин и, таким образом, являются более активными. Эти соединения с измененным спектром активности могут модулировать эпигенетические мишени в солидных опухолях и при воспалительных заболеваниях.
Эпигенетические механизмы важны в провоспалительных состояниях, которые включают, но не ограничиваются ими, воспалительные состояния легких, соединительных тканей, желудочно-кишечного тракта и сосудистой системы. Данные соединения путем воздействия на эпигенетические механизмы могут снижать или обращать развитие воспалительных процессов, ответственных за эти заболевания.
Краткое описание чертежей
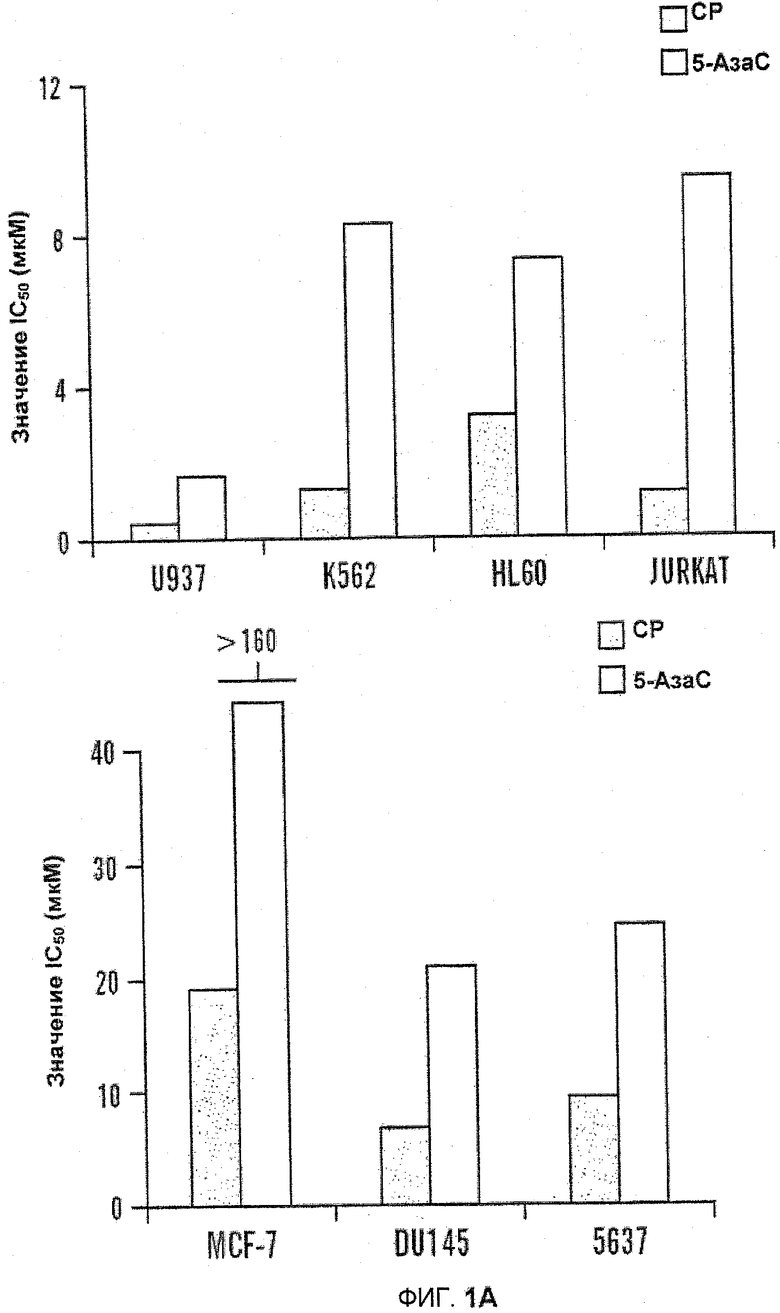
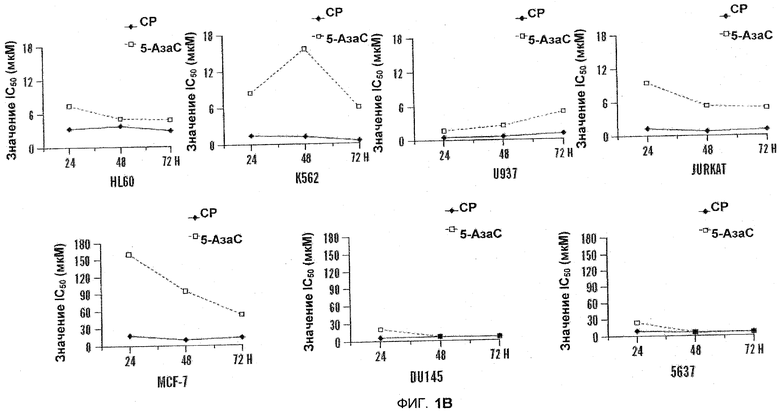
На фиг.1A-B изображены диаграммы сравнительной цитотоксической активности 5-азацитидин 5'-элаидат (CP) и 5-Аза-C на лейкемических и солидно-опухолевых клеточных линиях. Цитотоксичность определяли в ходе анализа WST-1. Рассчитывали IC50 в программной среде CalcySyn. Клетки обрабатывали в течение 24 часов (фиг.1A) или клетки обрабатывали в течение 24, 48 и 72 часов (фиг.1B).
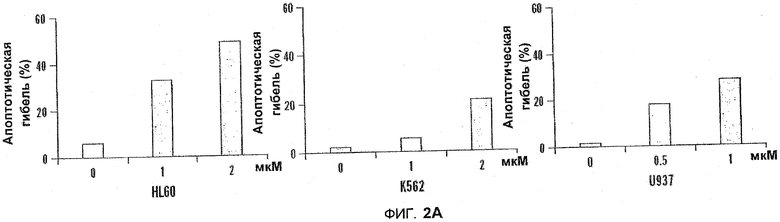
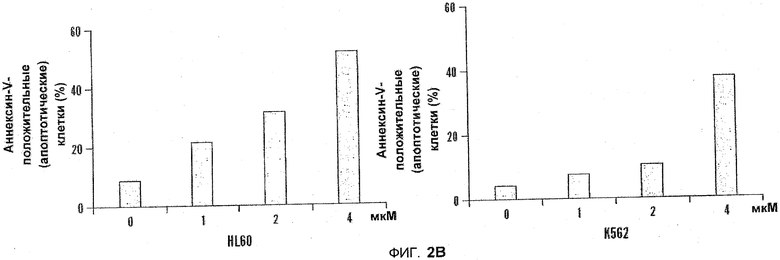
На фиг.2A-B показаны диаграммы индукции апоптоза под действием 5-азацитидин 5'-элаидат в лейкемических клетках. На фиг.2A клетки HL60, K562 и U937 не подвергались обработке или обрабатывались с помощью 5-азацитидин 5'-элаидат (CP) (0,5-4 мкм) в течение 24 часов. Процент апоптотических клеток определяли с помощью флуоресцентного микроскопа после окрашивания с помощью акридин оранжа и этидиумбромида. На фиг.2B как HL60, так и K562 не подвергались обработке или обрабатывались с помощью 5-азацитидин 5'-элаидат (1, 2 или 4 мкм) в течение 24 часов. Процент апоптотических клеток определяли с помощью проточной цитометрии с применением окрашивания Аннексином-V и PI.
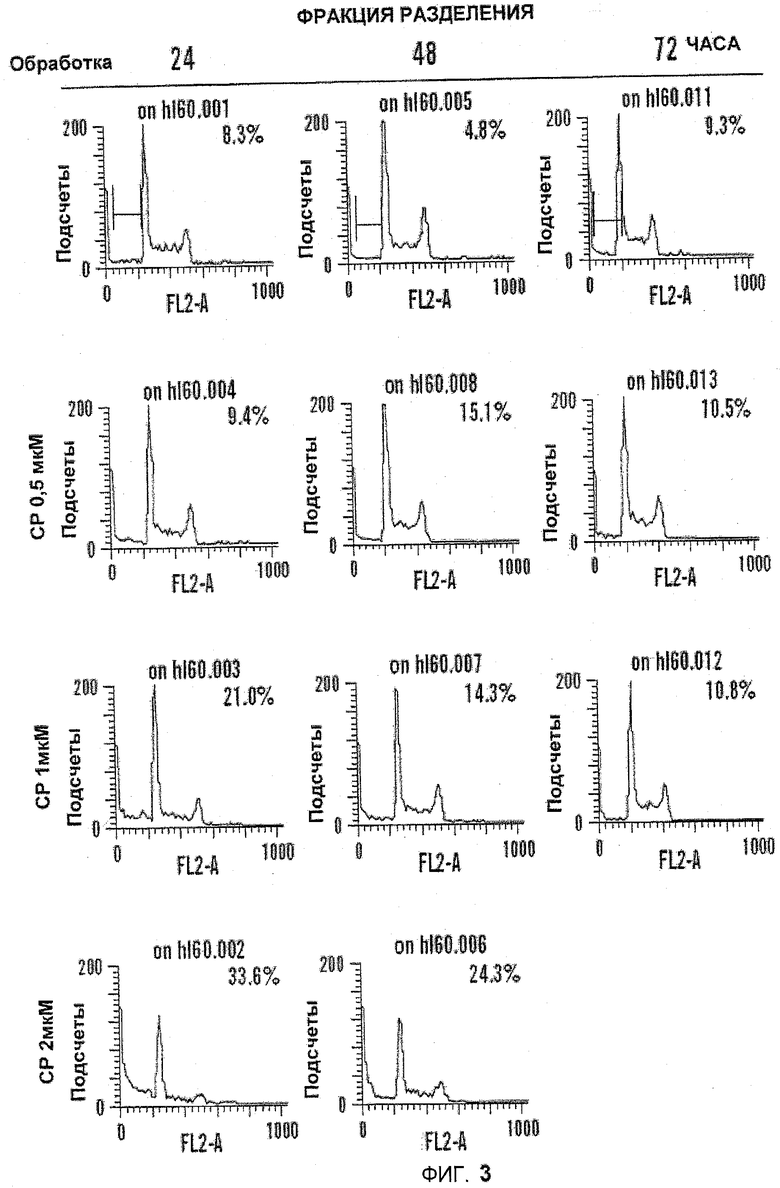
На фиг.3 показаны графики эффекта обработки 5-азацитидин 5'-элаидатом (CP) на клеточный цикл. Клетки культивировали в течение одинакового периода времени перед тем, как анализировать содержание ДНК с помощью проточной цитометрии. Численные значения на графике представляют популяцию регулируемых клеток, имеющих суб-G1 ДНК области.
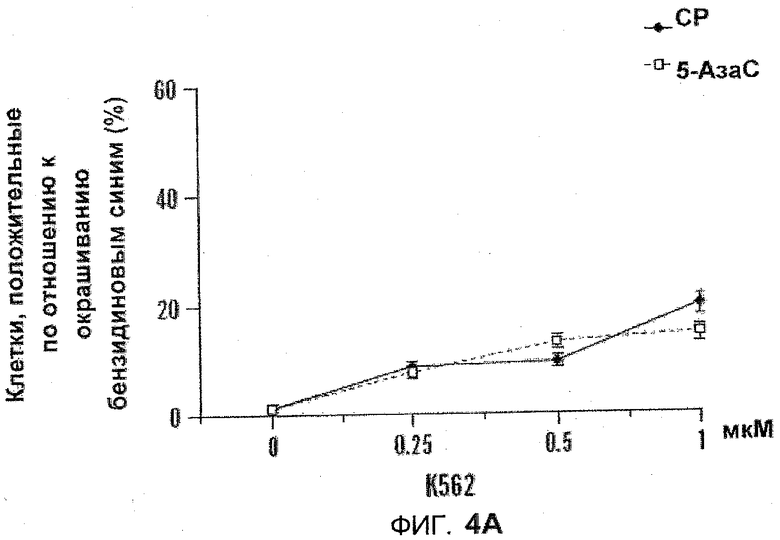
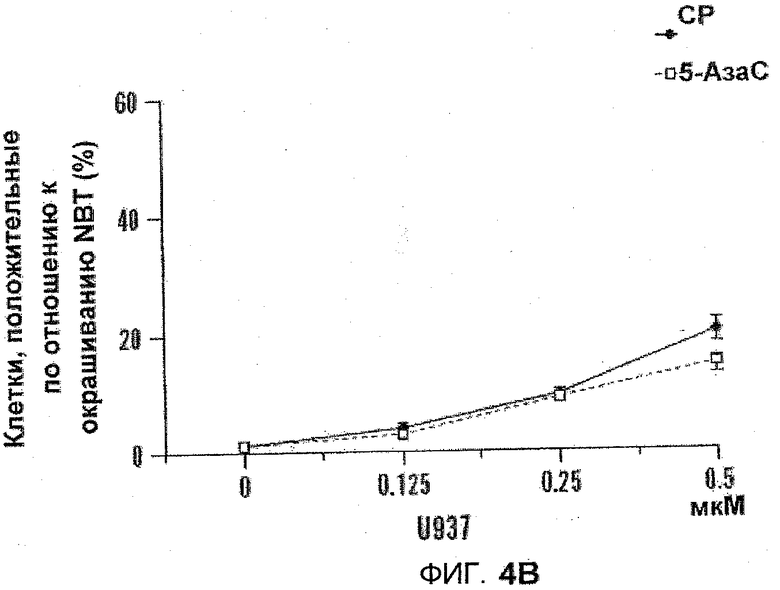
На фиг.4А-В показаны графики дифференцировки, включающие действие 5-азацитидин 5'-элаидат и 5-АзаС. K562 и U937 подвергали воздействию 5-азацитидин 5'-элаидат (CP) или 5-АзаС (0,125-1 мкм) в течение 14 дней. После начальной обработки в день 0 клетки подвергались воздействию новой порции лекарственных средств в день 4, день 7 и день 10. Дифференцировку оценивали с помощью окрашивания NBT (U937) (фиг.4A) или бензидином (K562) (фиг.4B).
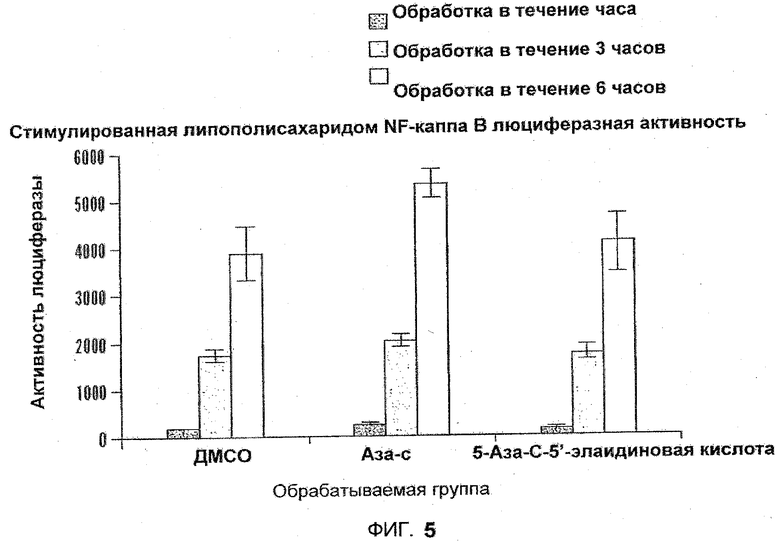
Фиг.5 представляет собой диаграмму, отображающую стимулированную липополисахаридом (LPS) NFκB люциферазную активность, измеренную в клетках U937, подвергавшихся воздействию сложного эфира кислоты и Аза-C или 5'-Аза-C-5'-элаидик.
Подробное описание изобретения
Один из аспектов настоящего изобретения направлен на соединение согласно формуле (I)
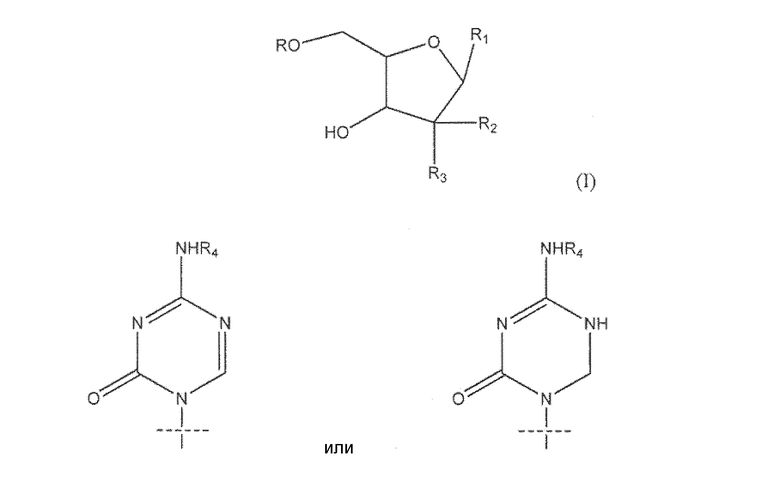
где R представляет собой H, R5C(O); R1 представляет собой
где пересекающая пунктирная линия изображает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I), R2 и R3 независимо представляют собой OH или H, при условии что R2 и R3 не являются одновременно OH, R4 представляет собой H или R5C(O), при условии что R и R4 не являются одновременно H, R5 представляет собой C3-C26 алкенил или его фармацевтическую соль.
В конкретном варианте реализации R5 может представлять C9-C26 алкенил.
В предпочтительном варианте реализации R представляет собой R5C(O), R1 представляет собой 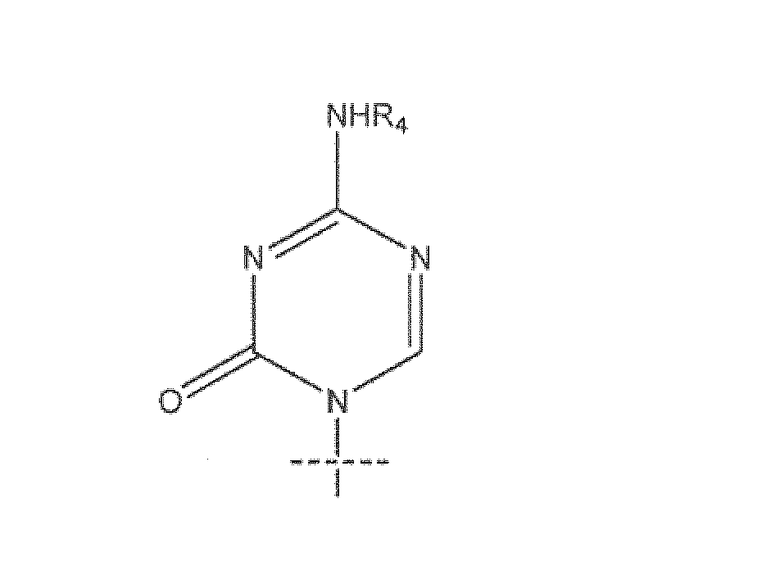 , R2 представляет собой H, R3 представляет собой OH, R4 представляет собой H и R5 представляет собой CH3-(CH2)7-CH=CH-(CH2)7-.
, R2 представляет собой H, R3 представляет собой OH, R4 представляет собой H и R5 представляет собой CH3-(CH2)7-CH=CH-(CH2)7-.
В более широком аспекте настоящее изобретение направлено на соединение согласно формуле (I)′
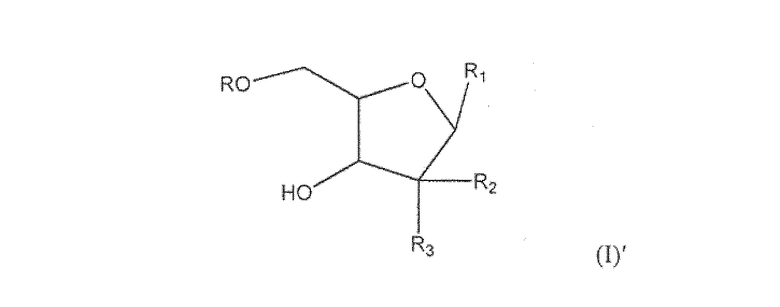
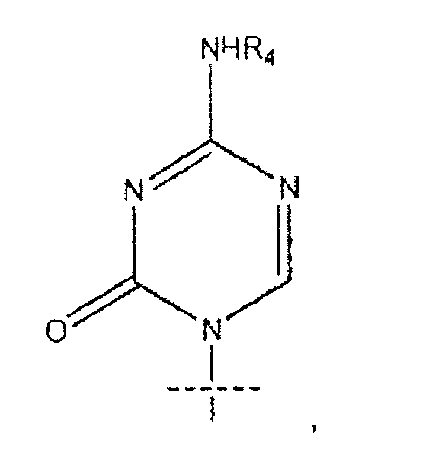
где R представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O), R1 представляет собой
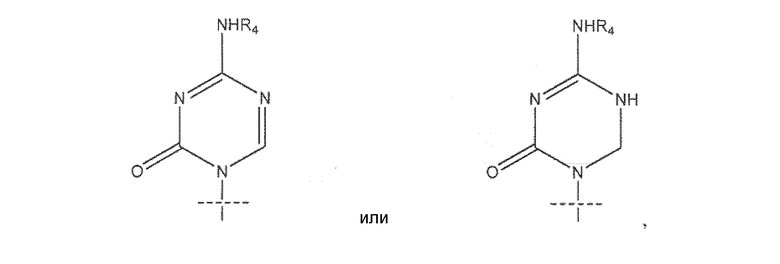
где пересекающая пунктирная линия изображает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I)', R2 и R3 независимо представляют собой OH или H, при условии что R2 и R3 не являются одновременно OH, R4 представляет собой H, R5C(O), R5CH2OC(O) или R5CH2NHC(O), при условии что R и R4 не являются одновременно H, и R5 представляет собой C3-C26 алкенил или его фармацевтическую соль.
В предпочтительном варианте реализации соединения согласно формуле (I)' k равно 7, а n равно 7. В некоторых вариантах реализации R1 представляет собой
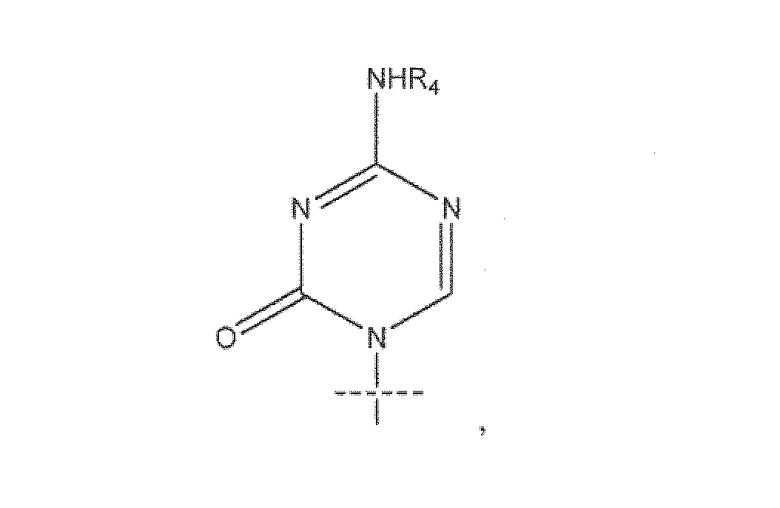
где пересекающая пунктирная линия изображает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I)'. В некоторых вариантах реализации R представляет собой R5C(O), k равно 7, m равно 0, n равно 7, R2 представляет собой H, а R3 представляет собой OH. В некоторых вариантах реализации R5 представляет собой C9-C26 алкенил.
Другой аспект настоящего изобретения направлен на фармацевтическую композицию, включающую соединение формулы (I), а также фармацевтический эксципиент, разбавитель и/или носитель.
Предложенные согласно настоящему изобретению средства можно вводить перорально, парентерально, например, подкожно, внутривенно, внутримышечно, интраперитонеально, путем закапывания в нос или посредством приложения к слизистым оболочкам, например, к слизистым носа, горла и бронхиол. Их можно вводить отдельно или же совместно с фармацевтическими носителями, и они могут быть в твердой или жидкой форме, например, в форме таблеток, капсул, порошков, растворов, суспензий или эмульсий.
Предложенные согласно настоящему изобретению активные средства можно вводить перорально, например, вместе с инертным разбавителем или вместе с усваиваемым съедобным носителем, или же их можно заключить в капсулы, имеющие твердую или мягкую оболочку, или их можно прессовать в таблетки, или их можно вводить непосредственно с пищей в соответствии с режимом питания. В случае перорального терапевтического введения эти активные вещества можно вводить вместе с эксципиентом и применять в форме таблеток, капсул, эликсиров, суспензий, сиропов и т.п. Подобные композиции и препараты должны содержать по меньшей мере 0,1% активного средства. Доля средства в этих композициях может, конечно, варьироваться и может предпочтительно находиться в пределах от примерно 2% до примерно 60% от веса дозированной формы. Количество действующего вещества в подобных терапевтически применимых композициях является таким, чтобы составлять подходящую дозу. Предпочтительные композиции согласно настоящему изобретению готовят таким образом, чтобы стандартная лекарственная форма для перорального приема содержала от примерно 1 до 250 мг действующего вещества.
Таблетки, капсулы и т.п. могут содержать также связующее вещество, например трагакантовую камедь, гуммиарабик, кукурузный крахмал или желатин; эксципиенты, такие как фосфат дикальция; разрыхлитель, например кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, например, стеарат магния; и подсластитель, например, сахарозу, лактозу или сахарин. Если стандартной лекарственной формой является капсула, то в дополнение к вышеуказанным типам материалов она может содержать жидкий носитель, например, жирные масла.
Могут присутствовать многие другие материалы в качестве оболочки или для модификации физической формы стандартной лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим. Сироп может содержать, в дополнение к активному ингредиенту, сахарозу в качестве подсластителя, метил и пропилпарабены в качестве консервантов, краситель и ароматизатор, например, со вкусом вишни или апельсина.
Указанные активные средства можно также вводить парентерально. Растворы или суспензии этих активных средств можно приготовить в воде, подходящим образом смешав с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть приготовлены в глицерине, жидких пропиленгликолях и также в их масляных смесях. Примерами масел могут служить масла нефтяного, животного, растительного, синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В общем случае, предпочтительными жидкими носителями, в частности, в случае растворов для инъекций, являются вода, солевой раствор, водный раствор декстрозы и родственных сахаров, а также гликоли, такие как пропиленгликоль или полиэтиленгликоль. При обычных условиях хранения и применения такие препараты содержат консервант для предотвращения роста микроорганизмов.
Фармацевтические формы, подходящие для применения для инъекций, включают стерильные водные растворы или суспензии, а также стерильные порошки для приготовления стерильных растворов или дисперсий для инъекций для немедленного использования. Во всех случаях форма должна быть стерильной и обладать текучестью в такой мере, чтобы ее легко можно было вводить шприцом. Она должна быть стабильной в условиях производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), подходящую смесь из этих компонентов или растительные масла.
Предложенные согласно настоящему изобретению средства можно также вводить непосредственно в дыхательные пути в форме аэрозоля. Для применения в виде аэрозоля предложенные согласно настоящему изобретению средства в виде раствора или суспензии могут быть упакованы в герметизированный аэрозольный контейнер вместе с подходящими газами-вытеснителями, например, углеводородными газами-вытеснителями, такими как пропан, бутан или изобутан, вместе с подходящими адъювантами. Вещества согласно настоящему изобретению можно также вводить в негерметизированной форме, например, с помощью распылителя или пульверизатора.
Следующий аспект настоящего изобретения направлен на способ лечения неопластического синдрома у пациента. Данный способ включает отбор пациента с неопластическим синдромом и введение указанному пациенту соединения формулы (I), как описано выше, или его фармацевтической соли в условиях, эффективных для лечения неопластического синдрома у пациента.
В некоторых вариантах реализации неопластический синдром представляет собой рак. Раковое заболевание может представлять собой солидную опухоль или гематологический рак или злокачественную опухоль. Раковое заболевание может представлять лейкемию, лимфому, множественную миелому или миелодиспластический синдром.
В некоторых вариантах реализации солидная опухоль может представлять рак ткани, например, тканей груди, яичника, простаты, мозга, мочевого пузыря и легких.
Следующий аспект настоящего изобретения направлен на способ лечения воспалительного синдрома у пациента. Данный способ включает отбор пациента с воспалительным синдромом и введение указанному пациенту соединения формулы (I), как описано выше, или его фармацевтической соли в условиях, эффективных для лечения воспалительного синдрома у пациента.
В некоторых вариантах реализации воспалительный синдром представляет воспалительное состояние легких, соединительной ткани, желудочно-кишечного тракта или сосудистой системы.
В материалах настоящей заявки, если не определено иное, научные и технические термины, используемые в связи с настоящей заявкой, имеют свои обычные значения, хорошо известные среднему специалисту в данной области техники. Далее, если в контексте не указано иное, термины в единственном числе включают их множественную форму, и термины во множественном числе включают их форму в единственном числе.
Примеры
Пример 1 - Реагенты, клеточные линии и клеточная культура
5-азацитидин 5'-элаидат (CP или CP-4200) был получен от компании Clavis Pharma AS, реагент для клеточной пролиферации WST-1 был получен от Roche Applied Science (Манхайм, Германия), PI и набор для определения апоптоза Annexin V-FITC apoptosis kit был заказан у BD Biosciences, Пало-Альто, CA, 5-азацитидин (5-АзаС), этидиумбромид (EB), акридиновый оранжевый (AO), нитросиний тетразолий (NBT), форбол 12-миристат 13-ацетат (TPA) были заказаны у Sigma Chemical Co (Сент-Луис, MO).
Линии человеческих промиелоцитных лейкемических клеток HL60, человеческой гистоцитной лимфомы U937, человеческой хронической миелогенной лейкемии K562, T-клетки линии Jurkat человека, аденокарциномы молочной железы MCF-7, карциномы мочевого пузыря 5637, карциномы простаты DU-145 были заказаны у American Type Culture Collection. Все клеточные линии, за исключением Jurkat, содержались в среде RPMI 1640 (Gibco, Глазго, Соединенное Королевство), включающей также 10% инактивированной нагреванием фетальной телячьей сыворотки (FCS), 100 Ед/мл пенициллина 100 мг/мл стрептомицина в атмосфере 5% CO2 при 37°C. Клетки линии Jurkat культивировали в среде RPMI 1640, включающей также 1,5 г/л бикарбоната натрия, 4,5 г/л глюкозы, 10 мМ пируват натрия и 10% FCS, 100 Ед/мл пенициллина и 100 мг/мл стрептомицина.
Пример 2 - Анализ цитотоксичности
Цитотоксичность липида 5-азацитидина определяли в ходе колориметрического анализа, основанного на расщеплении соли тетразолия WST-1 (4-[3-(4-лодофенил)-2-(4-нитрофенил)-2H-5-тетразолио]-1,3-бензолдисульфонат) митохондриальными дегидрогеназами в жизнеспособных клетках. Клетки высевали при исходной концентрации 1×106/мл (клетки HL60) или 1,25×105/мл (U937, K562 и Jurkat) в среде, не содержащей или содержащей различные концентрации липида 5-азацитидина в 96-луночных плоскодонных микропланшетах и культивировали в течение от 24 до 72 часов. Клетки MCF-7, DU-145 и 5637 (1×104/мл) высевали в чашки и оставляли для адгезии и роста на 24 часа. Добавляли различные концентрации липида 5-азацитидина, после чего культуры поддерживали еще в течение от 24 до 72 часов. Культуры инкубировали с реагентом WST-1 в течение 1 часа. Образование формазана измеряли с помощью ридера микропланшетов (Bio-Tek Instruments, Elx 800) при 450 нм с длиной волны сравнения 650 нм. Ингибирование роста определяли путем сравнения с необработанными клетками (%). Значения IC50 определяли с помощью программы CalcuSyn (Biosoft).
Пример 3 - Количественная оценка апоптотических клеток
Апоптотические клетки определяли с помощью морфологических критериев и флуоресцентно-активированного клеточного сортинга (FACS) после окрашивания Аннексином V-FITC. Для морфологического анализа добавляли 1 мкл готового раствора, содержащего 100 мкг/мл AO и 100 мкг/мл EB к 25 мкл клеточной суспензии. Апоптотические клетки и апоптотические тельца анализировали с помощью флуоресцентной микроскопии. Процент апоптотических клеток определяли после того, как насчитывали общее число всех клеток, равное 300. Для анализа FACS, от 2×105 до 5×106 клеток промывали с помощью PBS, а затем метили с помощью Аннексина V-FICS и пропидиумйодида (PI) в связывающем среду реагенте, согласно инструкциям, приложенным производителем к набору для определения апоптоза Annexin V-FITC apoptosis detection kit. Флуоресцентные сигналы FITC и PI детектировали, соответственно, при 518 нм и 620 нм на FACSCAN (Becton Dickinson, Сан-Хосе, Калифорния). Запись значений флуоресценции Аннексина V-FITC отображалась по оси X, а запись значений флуоресценции PI отображалась на оси Y. Данные обрабатывали с помощью программы CellQuest (Becton Dickinson). Для каждого анализа была произведена запись 10000 клеточных событий.
Пример 4 - Клеточный цикл
Клетки осаждали в ходе центрифугирования и промывали дважды с помощью PBS, фиксировали с помощью 70% (об./об.) холодного спирта (-20°C) и хранили при 4°C в течение по меньшей мере 24 часов. Клетки промывали с помощью PBS. Сгусток клеток окрашивали с помощью окрашивающего раствора PI/RNase. Клеточную суспензию инкубировали в темноте при комнатной температуре в течение 30 мин. Содержание ДНК определяли с помощью проточной цитометрии FACSCalibur (Becton Dickinson, Mount View, CA). Процентное содержание клеток в стадиях Sub-G1, G1, S и G2/M клеточного цикла определяли с помощью программы, вычерчивающей диаграммы ДНК (Becton Dickinson). Записывали минимум 10000 событий на образец.
Пример 5 - Синтез сложного эфира Аза-C-5'-олеиновой кислоты
Олеиновую кислоту (1,77 ммоль, 500 мг) растворяли в толуоле (3 мл). Добавляли сначала DMF (10 мкл), а затем оксалилхлорид (3,6 ммоль, 457 мг) в течение 10 мин при комнатной температуре. Через 3 часа удаляли толуол в вакууме.
Аза-C (1,75 ммоль, 427 мг) суспендировали в DMA (6 мл), добавляли HCl (1 M в Et2O, 2,0 ммоль, 2,0 мл) и через 5 мин при комнатной температуре удаляли Et2O в вакууме. Получаемый мутный раствор охлаждали на водно-ледяной бане и добавляли хлорид кислоты, растворенный в DMA (2 мл), в течение 2 ч. Реакционную смесь перемешивали в течение ночи, пока температура не достигала медленно комнатной температуры, а затем ее нагревали при 30°C в течение 2 ч. После охлаждения до комнатной температуры реакционную смесь делили между насыщенным водным NaHCO3 и EtOAc (25 мл каждого). Водную фазу экстрагировали с помощью дополнительных 3×25 мл EtOAc. Органические фазы объединяли, промывали с помощью соляного раствора и сушили (MgSO4). После удаления растворителей в вакууме неочищенный маслянистый продукт очищали с помощью флэш-хроматографии (SiO2, CH2Cl2 с 2,5, 5 и 10% MeOH). Выход: 110 мг (13%).
Пример 6 - Синтез сложного эфира 5-азацитидин 5'-элаидиновой кислоты
5-азацитидин (4,1 ммоль, 1,00 г) суспендировали в сухом DMA (15 мл) и медленно добавляли раствор HCl в диэтиловом эфире (4,9 ммоль, 1 M, 4,9 мл) при комнатной температуре с образованием прозрачного раствора. Эфир далее удаляли в вакууме, что приводило к образованию слегка мутного раствора. Раствор элаидилхлорида (4,8 ммоль, 1,44 г) в сухом DMA (8 мл) добавляли в течение 1 ч при комнатной температуре. Реакционную смесь далее нагревали в течение ночи при 30°C, охлаждали до комнатной температуры и гасили с помощью метанола (0,05 мл). По истечении по меньшей мере 1 ч реакционную смесь концентрировали при приблизительно 10-2 мбар, а остаток делили между насыщенным водным NaHCO3 и этилацетатом. Водную фазу экстрагировали этилацетатом. Органические фазы далее объединяли, промывали (солевым раствором), сушили (Na2SO4) и концентрировали в вакууме. После очистки с помощью флэш-хроматографии (SiO2, CH2Cl2 с 0-10% метанолом) было получено 0,9 г (43%) продукта.
Пример 7 - In vitro эффект 5-азацитидин 5'-элаидата на опухолевые клетки
Сравнивали цитотоксическую активность 5-азацитидин 5'-элаидата и 5-АзаС по отношению к линиям лейкемических клеток и клеток солидных опухолей. По сравнению с 5-АзаС, цитотоксический эффект 5-азацитидин 5'-элаидата был выше во всех изученных клеточных линиях. По сравнению с солидными опухолями, лейкемические клеточные линии были более чувствительны как к 5-азацитидин 5'-элаидату (CP), так и к 5-АзаС, см. таблицу 2 (ниже) и фиг.1A-B.
Поразительным результатом является раннее проявление активности в случае 5-азацитидин 5'-элаидата, чего не наблюдалось для азацитидина.
Пример 8 - Индукция апоптоза в лейкемических клетках
Клетки HL60, K562 и U937 не обрабатывали или обрабатывали 5-азацитидин 5'-элаидатом (0,5-4 мкМ) в течение 24 ч. Процентное содержание апоптотических клеток определяли с помощью флуоресцентного микроскопа после окрашивания с помощью акридинового оранжевого и этидиумбромида (см. фиг. 2A). В другой серии экспериментов HL60 и K562 не обрабатывали или обрабатывали 5-азацитидин 5'-элаидатом (1, 2 или 4 мкМ) в течение 24 часов. Процентное содержание апоптотических клеток определяли с помощью проточной цитометрии с применением окрашивания Аннексином-V и PI. Оба способа продемонстрировали, что 5-азацитидин 5'-элаидат индуцировал концентрационно-зависимое увеличение числа апоптотических клеток (см. фиг.2A и 2B).
Пример 9 - Протекание клеточного цикла
Изучали эффект от воздействия 5-азацитидин 5'-элаидата на клеточный цикл. Клетки культивировали в течение указанного периода времени, после чего их анализировали на состав ДНК с помощью проточной цитометрии. Численные значения на графике представляют популяцию клеток, дающих сигнал выше порогового значения с содержанием суб-G1 ДНК. Наиболее значительные изменения наблюдались через 24 часа обработки. По сравнению с контролем (8,3%) процентное содержание в суб-G1 фазы (апоптотические клетки) возрастало вплоть до 33,6% в клетках, обработанных 2 мкМ 5-азацитидин 5'-элаидатом, и уменьшалось в G1, S и G2/M фазе. Результаты обобщены на фиг.3 и в таблице 3, ниже. Эти результаты подтвердили индукцию апоптоза под действием 5-азацитидин 5'-элаидата и продемонстрировали, что действие 5-азацитидин 5'-элаидата на клеточный цикл имело концентрационно-зависимый характер.
Пример 10 - Индукция дифференциации
Изучали дифференциацию на основе морфологии. Поскольку одиночное воздействие (96 часов) не имело эффекта, клетки подвергались воздействию 5-азацитидин 5'-элаидата или 5-АзаС в течение 11 (клетки HL60) или 14 (клетки U937 и K562) дней. После начальной обработки в день 0 клетки подвергались воздействию новых порций лекарства в день 4, 7 и 10. Дифференциацию оценивали с помощью исследования изменения нитросинего тетразолия (U937 и HL60) или с помощью окрашивания бензидином (K562). Несмотря на то, что 5-азацитидин 5'-элаидат был несколько более эффективен, оба реагента не вызывали существенной дифференциации. См. фиг.4A-B.
Пример 11 - Метаболическая стабильность сложного эфира 5-аза-5'-элаидиновой кислоты в пуле человеческой плазмы
Сложный эфир 5-Аза-C-5'-элаидиновой кислоты помещали в пулированную человеческую плазму при пяти уровнях концентрации (0,1; 1; 3; 10 и 30 мкМ, соответственно). Смесь инкубировали на водяной бане с шейкером при 37°C. Отбирались тройные (n=3) аликвоты (100 мкл) инкубирующегося раствора через соответствующие периоды инкубации (0, 15, 30, 60 и 120 минут), и немедленно осаждали плазменный белок с помощью ацетонитрила, содержащего 0,1% муравьиную кислоту (300 мкл). Отрицательные контроли готовили с тестируемым соединением и Аза-C в буфере для анализа (PBS, pH 7,4) при одной концентрации инкубации (1 мкМ). После центрифугирования супернатант непосредственно вводили для анализа LC-MS-MS. См. таблицу 4.
Пример 12 - Цитотоксичность Аза-C и 5-Аза-C-5'-элаидиновой кислоты
Цитотоксичность Аза-C и 5-Аза-C-5'-элаидиновой кислоты определяли в линии раковых клеток молочной железы MT-3 в адриабластин-резистентной линии клеток MT-3/ADR. Для MT-3/ADR характерна избыточная экспрессия MDR-1/p-гликопротеина. Клетки высевали на 96-луночных планшетах по 5×103 клеток на лунку, в среде RPMI 1640 с 2 мМ глутамином и 10% FBS. Клетки инкубировали в течение 24 часов. Исследуемые соединения растворяли в ДМСО и далее разводили средой непосредственно перед использованием. Для анализа одной концентрации использовали шесть лунок. Клетки инкубировали с исследуемым соединением в течение 24 часов. Добавляли по 20 мкл свежеприготовленного раствора MTT в каждую лунку и инкубировали 4 часа. Величины IC50 определяли из кривых роста, представленных графически на основе 8 различных концентраций в диапазоне от 0,01 мкМ до 100 мкМ. Результаты представлены в таблице 5. Одинаковые активности были получены для Аза-C и 5-Аза-C-5'-элаидиновой кислоты на линии раковых клеток молочной железы MT-3, но в случае резистентной линии клеток MT-3/ADR активность Аза-C исчезала. Активность не наблюдалась в тестированном интервале концентраций вплоть до 100 мкМ, в то время как 5-Аза-C-5'-элаидиновая кислота была активна с одинаковым значением IC50 как в резистентной линии клеток, так и в нерезистентной линии MT-3. Это может быть очень важным в лечении резистентного рака. См. таблицу 5.
IC50 (мкМ)
IC50 (мкМ)
Пример 13 - Влияние ингибирования нуклеозидного переносчика Аза-C и 5-Аза-C-5'-элаидиновой кислоты
Оценивали влияние ингибирования нуклеозидного переносчика на цитотоксическую активность в мутантных клетках карциномы шейки матки Hela для Аза-С и 5-Аза-C-5'-элаидиновой кислоты. Использовали дипиридамол в качестве ингибитора уравновешивающих нуклеозидных переносчиков hENT1 и hENT2. Клетки высевали на 96-луночных планшетах по 5×103 клеток на лунку, в среде RPMI 1640 с 2 мМ глутамином и 10% FBS. Клетки преинкубировали в течение 24 часов. Добавляли дипиридамол (10 мкМ) к клеткам за 30 минут перед добавлением исследуемых соединений. Исследуемые соединения растворяли в ДМСО и далее разводили средой непосредственно перед использованием. Для анализа одной концентрации использовали шесть лунок. Клетки инкубировали с исследуемым соединением в течение 72 часов. Добавляли по 20 мкл свежеприготовленного раствора MTT в каждую лунку и инкубировали 4 часа. Величины IC50 определяли из кривых роста, представленных графически на основе 8 различных концентраций в диапазоне от 0,01 мкМ до 100 мкМ. Результаты представлены в таблице 6. Активность Аза-C снизилась в 3 раза в результате добавления ингибитора нуклеозидного транспорта дипиридамола, указывая на то, что приток и отток Аза-C частично зависят от нуклеозидных переносчиков hENT1 и hENT2. Цитотоксическая активность 5-Аза-C-5'-элаидиновой кислоты не только сохранилась, но возросла в 50 раз, когда нуклеозидные переносчики hENT1 и hENT2 были блокированы с помощью дипиридамола. Увеличение активности в клетках, где нуклеозидные переносчики блокированы с помощью дипиридамола, является неожиданным и может означать еще большую активность у пациентов с резистентностью к терапии, вызванной дефицитом нуклеозидных переносчиков. См. таблицу 6.
IC50 (мкМ)
Пример 14 - Активация ядерного фактора транскрипции -κB (NFκB)
Определяли активацию ядерного фактора транскрипции -κB (NFκB) для оценки влияния на воспаление. NFκB участвует в регуляции большого числа как иммунных, так и воспалительных реакций, процессов развития, клеточного роста и апоптоза.
Использовали гены-репортеры NFκB-люциферазы для определения влияния Аза-С и 5-Аза-C-5'-элаидиновой кислоты на NFκB-индуцированную люциферазную активность в линии клеток моноцитов человека U937, устойчиво трансфецированных репортером люциферазы, содержащим три NFκB-связывающих сайта. Индукцию NFκB проводили с применением липополисахарида (LPS) с концентрацией 1 мкг/мл в течение 30 минут перед добавлением к среде Аза-С или 5-Аза-C-5'-элаидиновой кислоты в концентрации 10 нМ. Клетки культивировали в среде RPMI-1640, содержащей 10% фетальную телячью сыворотку. Перед стимуляцией с помощью LPS клетки переносили в среду, содержащую только 2% фетальную сыворотку коровы. Люциферазную активность измеряли с помощью отображения в Системе отображения IVIS 100 (Xenogen Corp., USA) через 1,3 или 6 часов. LPS-стимуляция возрастала со временем в течение 6 часов, что наблюдалось для контрольных клеток, обработанных DMSO. Воздействие 10 нМ Аза-C еще более увеличивало LPS-индуцированную активацию NFκB, по сравнению с обработанным DMSO контролем. 5-азацитидин-5'-элаидат не отличался от контроля DMSO в плане LPS-стимулированной активности NFκB. Как правило, положительная стимуляция NFκB, наблюдаемая для азацитидина, не является позитивной, но хорошо, что стимуляция LPS не возрастает далее под воздействием 5-аза-C-5'-элаидиновой кислоты. См. фиг.5.
Пример 15 - Экспрессия гена рецептора эстрогена β (ERβ) в линии раковых клеток молочной железы после обработки с помощью азацитидина или 5-азацитидин 5'-элаидата
Экспрессия гена (определенная по уровню РНК) рецептора эстрогена бета определяли с помощью количественной ПЦР в реальном времени (TaqMan). Клетки карциномы молочной железы MCF-7 выращивали в среде с недостатком эстрогена (не содержащая феноловый красный среда RPMI с 2% глутамином и 10% фетальной сывороткой коровы, обработанной нагруженной декстраном угольной пылью). Клетки высевали в колбы 25 см2 и оставляли для адгезии на 24 часа перед обработкой с помощью 1 мкМ азацитидина или 5-азацитидин 5'-элаидата. Один необработанный образец был включен в качестве контроля. Клетки были собраны через 5 дней от начала воздействия соединений; их собирали путем трипсинизации, промывали и подвергали ударной заморозке в жидком азоте.
Общую РНК экстрагировали из примерно 106 клеток MCF-7, подвергавшихся ударной заморозке. Определяли концентрацию и чистоту РНК и транскрибировали РНК в кДНК с помощью реагентов обратной транскрипции (N808-0234). Проводили количественную оценку в реальном времени по стандартным протоколам с использованием стандартных заранее перемешанных реагентов для ПЦР. Смеси праймер-зонд, ER β (ID Hs00230957_m1) и гидроксиметилбилан-синтазу конститутивных генов HMBS (ID Hs00609297_m1) заказывали в Applied Biosystems. Генную экспрессию рассчитывали с помощью сравнительного дельта-дельта Ct метода. Индукция экспрессии ER β была 5,26-кратной после воздействия 5-азацитидин 5'-элаидата, по сравнению лишь с 2,51-кратной после воздействия азацитидина. См. таблицу 7. Это может быть очень существенно для гормон-рефракторной опухоли, при которой можно восстановить чувствительность к гормону. См. таблицу 7.
дельта
Несмотря на то, что в настоящей заявке приведены и подробно обсуждаются предпочтительные варианты реализации, для специалиста в данной области техники очевидно, что различные возможные модификации, дополнения, замены и т.п. также находятся в рамках настоящего изобретения, охарактеризованного в пунктах нижеследующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ АЗАЦИТИДИНА И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2488591C2 |
| ПРОИЗВОДНЫЕ ДИОКСОЛАНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2418795C2 |
| МОНОЭФИРНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2139884C1 |
| ПРОИЗВОДНЫЕ ГЕМЦИТАБИНА | 1998 |
|
RU2194711C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2462472C2 |
| ПРОИЗВОДНЫЕ АРАБИНОФУРАНОЗИЛ-ЦИТОЗИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2165260C2 |
| СЛОЖНЫЕ ЭФИРЫ НУКЛЕОЗИДОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2126417C1 |
| ПРИРОДНЫЕ ПРОТИВООПУХОЛЕВЫЕ ИЛИ ПРОТИВОВИРУСНЫЕ ВЕЩЕСТВА И ИХ ПРИМЕНЕНИЕ | 1998 |
|
RU2205010C2 |
| ФОСФОРАМИДАТНЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ | 2010 |
|
RU2553656C2 |
| ПУРИНОВЫЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЕ | 1997 |
|
RU2183639C2 |
Данное изобретение относится к новому соединению-аналогу азацитина следующей формулы (I):
где R представляет собой Н, R5C(O); R1 представляет собой
 или
или 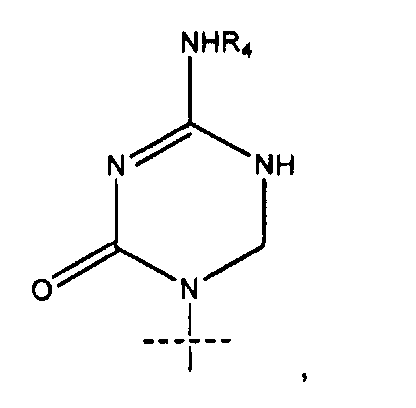
где пересекающая пунктирная линия обозначает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I); R2 и R3 независимо представляют собой ОН или Н при условии, что R2 и R3 не являются одновременно ОН; R4 представляет собой Н или R5C(O) при условии, что R и R4 не являются одновременно Н; и R5 представляет собой С3-С26 алкенил, или его фармацевтической соли. Эти соединения обладают противораковой и противовоспалительной активностью. Изобретение также относится к фармацевтической композиции на основе этих соединений и к способам лечения. 4 н. и 11 з.п. ф-лы, 6 табл., 8 ил., 15 пр.
1. Соединение формулы (I)
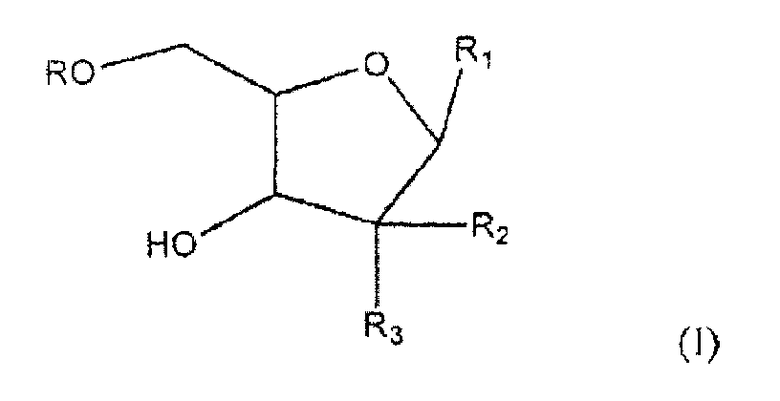
где
R представляет собой Н или R5C(О);
R1 представляет собой
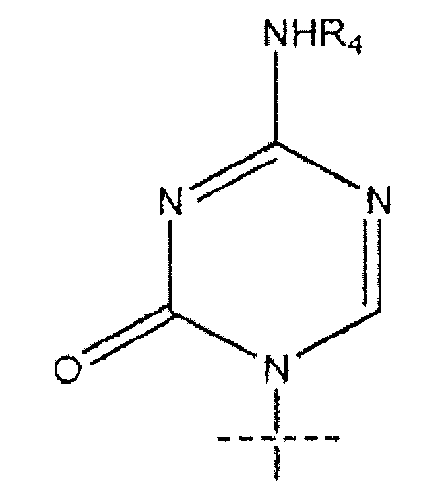
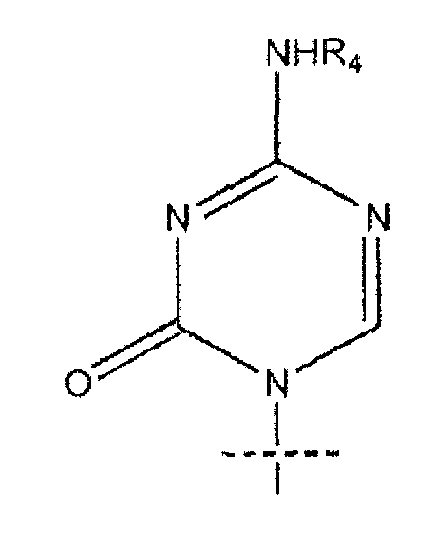 или
или 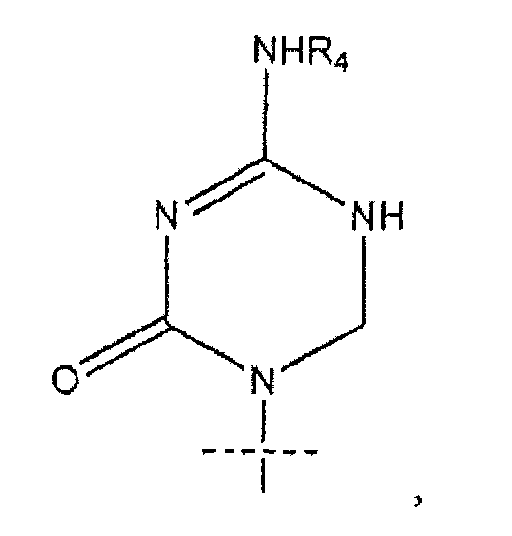
где пересекающая пунктирная линия обозначает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I);
R2 и R3 независимо представляют собой ОН или Н при условии, что R2 и R3 не являются одновременно ОН;
R4 представляет собой Н или R5C(O) при условии, что R и R4 не являются одновременно Н; и
R5 представляет собой С3-С26 алкенил,
или фармацевтическая соль указанного соединения.
2. Соединение по п.1, где R5 представляет собой С9-С26 алкенил.
3. Соединение по п.1, где R представляет собой R5C(O), R1 представляет собой
R2 представляет собой Н, R3 представляет собой ОН, R4 представляет собой Н и R5 представляет собой СН3-(СН2)7-СН=СН-(СН2)7-.
4. Фармацевтическая композиция, предназначенная для лечения состояния, выбранного из неопластического состояния и воспалительного состояния, содержащая соединение по п.1 и фармацевтический наполнитель, разбавитель и/или носитель.
5. Способ лечения неопластического состояния у пациента, включающий отбор пациента с неопластическим состоянием, введение указанному пациенту соединения формулы
где
R представляет собой Н или R5C(O);
R1 представляет собой
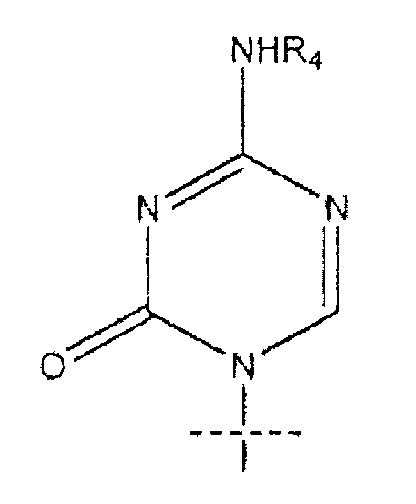 или
или 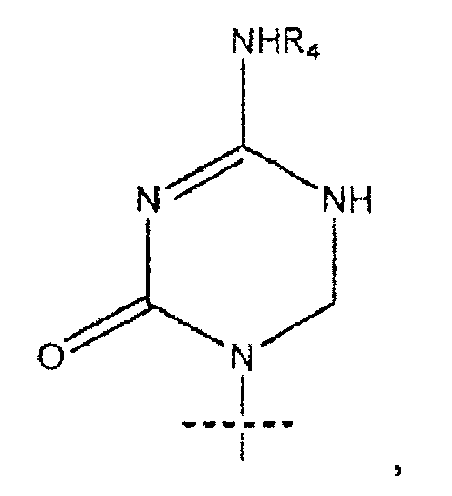
где пересекающая пунктирная линия обозначает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I);
R2 и R3 независимо представляют собой ОН или Н, при условии что R2 и R3 не являются одновременно ОН;
R4 представляет собой Н или R5C(O), при условии что R и R4 не являются одновременно Н; и
R5 представляет собой С3-С26 алкенил,
или фармацевтической соли указанного соединения в условиях, эффективных для лечения указанного неопластического состояния у пациента.
6. Способ по п.5, где R5 представляет собой С9-С26 алкенил.
7. Способ по п.5, где указанное неопластическое состояние представляет собой раковое заболевание.
8. Способ по п.7, где раковое заболевание представляет собой солидную опухоль, или гематологический рак, или злокачественное образование.
9. Способ по п.7, где указанное раковое заболевание представляет собой лейкемию, лимфому, множественную миелому или миелодиспластический синдром.
10. Способ по п.8, где солидная опухоль представляет собой рак ткани, выбранной из группы тканей, состоящей из ткани молочной железы, яичника, простаты, мозга, мочевого пузыря и легких.
11. Способ по п.7, где R представляет собой R5C(O), R1 представляет собой
R2 представляет собой Н, R3 представляет собой ОН, R4 представляет собой Н и R5 представляет собой СН2-(СН2)7-СН=СН-(СН2)7-.
12. Способ лечения воспалительного состояния у пациента, включающий отбор пациента с воспалительным состоянием и введение указанному пациенту соединения формулы
где
R представляет собой Н или R5C(O);
R1 представляет собой
 или
или 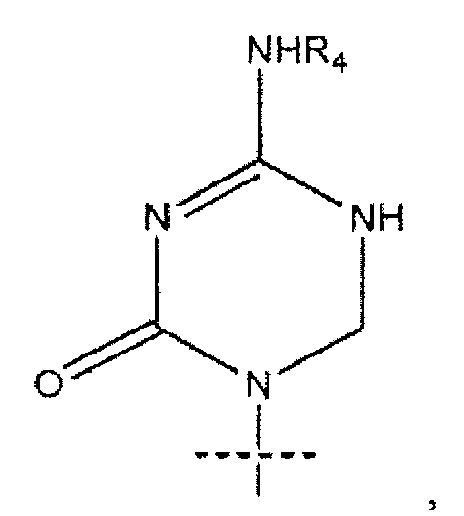
где пересекающая пунктирная линия обозначает образовавшуюся связь, присоединяющую R1 к молекуле формулы (I);
R2 и R3 независимо представляют собой ОН или Н при условии, что R2 и R3 не являются одновременно ОН;
R4 представляет собой Н или R5C(О) при условии, что R и R4 не являются одновременно Н; и
R5 представляет собой С3-С26 алкенил,
или фармацевтической соли указанного соединения в условиях, эффективных для лечения указанного воспалительного состояния.
13. Способ по п.12, где R5 представляет собой С9-С26 алкенил.
14. Способ по п.12, где указанное воспалительное состояние представляет воспалительное состояние легких, соединительной ткани, желудочно-кишечного тракта или сосудистой системы.
15. Способ по п.12, где R представляет собой R5C(O), R1 представляет собой
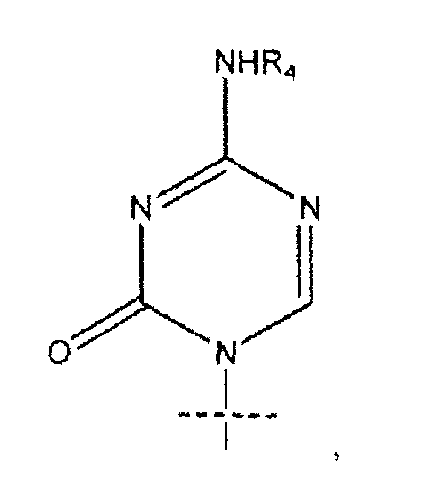
R2 представляет собой Н, R3 представляет собой ОН, R4 представляет собой Н и R5 представляет собой СН3-(СН2)7-СН=СН-(СН2)7-.
| WO 9705154 A1, 13.02.1997 | |||
| WO 2007035783 A2, 29.03.2007 | |||
| US 2006006735 A1, 23.03.2006 | |||
| RU 98104092 A, 27.01.2000. |

