Область техники, к которой относится изобретение
Настоящее изобретение относится к новому способу синтеза (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида из D-аланина и метоксибензола через реакцию Фриделя-Крафтса. Точнее, настоящее изобретение относится к новому способу получения тамсулозина.
Уровень техники
Тамсулозин является фармацевтически активным веществом из группы антагонистов α1-адренергических рецепторов, которые применяются для лечения функциональных нарушений предстательной железы. Химическое название тамсулозина - (R)-5-(2-((2-(2-этоксифенокси)этил)амино)пропил)-2-метоксибензолсульфонамид (формула 1) и он относится к группам бензолсульфонамидов и сульфамоилфенетиламинов.
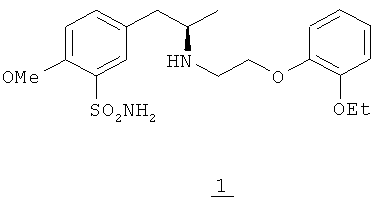
Тамсулозин имеется в продаже в форме гидрохлорида чистого (R)-энантиомера и применяется для лечения доброкачественной гиперплазии предстательной железы. В настоящем изобретении термин "тамсулозин" означает соединение формулы (1) в форме гидрохлорида, а соединение в несолевой форме называется основанием тамсулозина.
Постоянно необходимо получение фармацевтически активных веществ, обладающих качеством, пригодным для включения в готовое лекарственное средство возможно более технологически простым и экономичным способом. Тамсулозин является хиральной молекулой и его (R)-энантиомер применяется в качестве терапевтически активного вещества. Получение оптически чистого соединения, обладающего минимальным содержанием нежелательного (S)-изомера, является трудной технической задачей, которую относительно затруднительно решить, поскольку энантиомеры невозможно разделить с помощью обычных химических методик и способов разделения.
Тамсулозин можно получить в соответствии с основополагающим патентом ЕР 34432, в содержащихся в котором примерах раскрыто проводимое сначала получение рацемата, который затем очищается с помощью хроматографии на колонке на хиральной матрице с получением энантиомеров.
В заявках на патенты WO 03/37850 и WO 03/37851 раскрыто альтернативное получение тамсулозина путем синтеза кристаллического рацемического основания тамсулозина, которое необходимо очистить с помощью дорогостоящей камфор-5-сульфоновой кислоты. В WO 03/37850 явно описано такое выделение полученного рацемического основания тамсулозина, однако на последней стадии всегда существует опасность того, что хиральное разделение не будет полным и будет получен продукт с большим содержанием противоположного энантиомера.
В CZ 290708 раскрыт способ разделения рацемического тамсулозина с использованием кислоты.
Для исключения такого разделения необходимо получить оптически чистый (R)-5-(2-аминопропил)-2-метоксибензолсульфонамид (2).
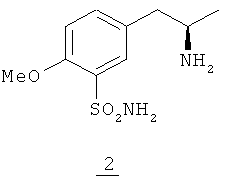
(R)-5-(2-Аминопропил)-2-метоксибензолсульфонамид (2) известен в данной области техники как ключевой промежуточный продукт при синтезе тамсулозина. Способы синтеза (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида (2) уже описаны в предшествующем уровне техники. Например:
В US 5447958 раскрыт синтез соединения (2). Исходным веществом является (-)-2-(п-метоксифенил)-1-метилэтиламин, но его синтез не раскрыт.
В ЕР 380144 раскрыт синтез соединения (2), начиная с восстановительного аминирования 5-ацетонил-2-метоксибензолсульфонамида с помощью R(+)α-метилбензиламина с последующим гидрогенезисом. Синтез 5-ацетонил-2-метоксибензолсульфонамида не раскрыт.
В СА 1282077 наряду с другими способами также раскрыт синтез соединения (2) с использованием (R)(-)-2-(п-метоксифенил)-1-метилэтиламина в качестве исходного вещества, но способ получения последнего не указан.
Поэтому необходимо разработать альтернативные способы более прямого получения (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида, которые не требуют оптического разделения промежуточных продуктов, позволяют упростить промышленное получение этого продукта и тем самым снизить производственные затраты.
Краткое содержание изобретения
Настоящее изобретение относится к новому и прямому способу получения (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида из D-аланина и метоксибензола через реакцию Фриделя-Крафтса.
Способ, предлагаемый в настоящем изобретении, исключает серьезные недостатки, связанные с указанными выше способами: сложный синтез, дорогие реагенты и исходные вещества, оптическое разделение промежуточных продуктов и/или выделение энантиомера тамсулозина после заключительной стадии способа.
Подробное описание изобретения
Реакция Фриделя-Крафтса известна в данной области техники. Например, Nordlander et al. показали, что хиральные N-трифторацетилзамещенные аминокислоты вступают в реакцию Фриделя-Крафтса с бензолом и метоксибензолом (J. Org. Chem., 50 (1985), 3481). Кроме того, сообщают, что реакция Фриделя-Крафтса ароматических соединений с защищенным по атому N аланилхлоридом является способом получения гомохиральных катинонов (М. Osorio-Olivares et al., Tetrahedron: Asymmetry, 14 (2003), 1473). Однако реакция Фриделя-Крафтса D-аланина с метоксибензолом не была раскрыта в предшествующем уровне техники.
Согласно изобретению неожиданно было обнаружено, что с использованием минимального количества стадий можно синтезировать (R)-5-(2-аминопропил)-2-метоксибензолсульфонамид из D-аланина и метоксибензола посредством реакций, при которых не происходит рацемизация асимметрического центра. Таким образом можно получить (R)-5-(2-аминопропил)-2-метоксибензолсульфонамид, обладающий оптической и химической чистотой, пригодными для получения тамсулозина, предназначенного для любого фармацевтического применения.
Первый вариант осуществления настоящего изобретения относится к способу получения (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида из D-аланина и метоксибензола через реакцию Фриделя-Крафтса.
Настоящее изобретение предпочтительно относится к способу получения (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида, включающему следующие стадии:
a) защиту аминогруппы D-аланина,
b) реакцию полученного защищенного по атому N D-аланина с метоксибензолом с получением соответствующего защищенного по аминогруппе 4'-метокси-2-аминопропиофенола,
c) полное восстановление оксогруппы образовавшегося защищенного по аминогруппе 4'-метокси-2-аминопропиофенола с получением соответствующего защищенного по аминогруппе 1-(4-метоксифенил)пропан-2-амина,
d) хлорсульфонирование полученного защищенного по аминогруппе 1-(4-метоксифенил)пропан-2-амина и последующий аммонолиз образовавшейся хлорсульфонильной группы и
e) удаление защитной группы аминогруппы.
D-Аланин является простым исходным веществом, имеющимся в продаже в больших количествах по умеренной цене и обладающим высокой оптической чистотой.
Способ, предлагаемый в настоящем изобретении, дает возможность выполнить синтез (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида (2) путем использования простого сырья, такого как D-аланин и метоксибензол, без рацемизации. Таким образом, способ, предлагаемый в настоящем изобретении, является прямым, не требует каких-либо специальных условий для обеспечения энантиоселективного синтеза и/или разделения энантиомеров, при котором имеется опасность того, что оно будет неполным, что приведет к продукту, обладающему слишком низким энантиомерным избытком и не являющемуся оптически чистым продуктом, соответственно. Поэтому необходимо выбрать реакции, при которых не происходит рацемизация по асимметрическому атому углерода, что приводит к оптически чистому продукту, который можно применять для синтеза фармацевтически активного вещества тамсулозина.
В соответствии со стадией (а) D-аланин защищают с помощью любой защитной группы аминогруппы (X), хорошо известной и обычно использующейся в химии пептидов, при условии, что указанная защитная группа не допускает рацемизации асимметрического центра D-аланина путем превращения в оксазолидинон на последующей стадии способа (схема 1). Например, подходящие защитные группы аминогруппы выбираются из группы, включающей трифторацетил; алкоксикарбонил, такой как трет-бутоксикарбонил, бензилоксикарбонил, флуоренилметилоксикарбонил; фталимид. Предпочтительной защитной группой аминогруппы является трифторацетильная группа.
Затем защищенный по аминогруппе D-аланин превращают в кислотно-активированный защищенный по аминогруппе D-аланин с помощью кислотно-активирующего реагента. Указанная кислотно-активирующая группа может представлять собой хлорангидрид, ангидрид кислоты или любую другую кислотно-активирующую группу, хорошо известную в синтезе пептидов. Например, подходящие кислотно-активирующие реагенты выбираются из группы, включающей хлорангидрид или ангидрид, предпочтительно - ангидриды кислот. Более предпочтительными кислотно-активирующими реагентами для получения (R)-(N-трифторацетил)пропионилхлорида являются оксалилхлорид и тионилхлорид. Наиболее предпочтительным является тионилхлорид.
Кислотно-активированный защищенный по аминогруппе D-аланин можно выделить и как таковой направить на следующую стадию реакции (b) или можно направить на следующую стадию реакции (b) в растворе, без выделения. Метиленхлорид является растворителем, подходящим и для образования кислотно-активированного защищенного по аминогруппе D-аланина, и для реакции Фриделя-Крафтса, так что образовавшийся кислотно-активированный защищенный по аминогруппе D-аланин можно направить непосредственно на следующую стадию (реакцию Фриделя-Крафтса).
Метоксибензол, также называющийся анизолом, имеется в продаже. Метоксибензол является достаточно активированным для электрофильного замещения типа Фриделя-Крафтса, чтобы обеспечить мягкую реакцию с кислотно-активированным защищенным по аминогруппе D-аланином. При проведении реакции Фриделя-Крафтса в соответствии со стадией (b) предпочтительно прибавлять кислоту Льюиса. Подходящая кислота Льюиса выбирается из группы, включающей соединения алюминия, железа(III), олова(IV), висмута, бора и соли других переходных металлов или лантанидов.
При реакции Фриделя-Крафтса между анизолом и кислотно-активированным защищенным по аминогруппе D-аланином замещение может протекать в пара-положении (в положении 4 относительно метоксигруппы) или в орто-положении (в положении 2 относительно метоксигруппы) с учетом активации для электрофильного замещения. Мы обнаружили, что в этой реакции более предпочтительным является пара-положение. Соотношение количеств орто- и пара-замещенных продуктов зависит от условий проведения реакции, растворителей и, прежде всего, от типа кислоты Льюиса. Доля орто-замещенного продукта во всем количестве замещенных продуктов составляет от 15 до 30%, если в качестве кислоты Льюиса используется АlСl3, FеСl3, TiCl4 или трифлат висмута при добавлении или без добавления вспомогательного лиганда, нитрометана. Реакцию проводят при температурах от примерно 0 до примерно 45°С, предпочтительно - примерно при 20-25°С. Оптимальный выход достигается при использовании хлорида алюминия при комнатной температуре, где термин "оптимальный" означает, что учитывается диапазон конверсии, доля пара-замещенного продукта и отсутствие рацемизации по асимметрическому атому углерода.
Оксогруппу пропионфенонового промежуточного продукта, полученного по реакции Фриделя-Крафтса, можно восстановить в соответствии со стадией (с) путем полного восстановления в метиленовую группу. Предпочтительными восстановительными реагентами являются гидриды кремния в виде полиалкилгидроксисиланов или алкилсиланов. Предпочтительно использовать триэтилсилан. Защищенный по атому N (R)-(4-метоксифенил)пропан-2-амин получают на стадии (с).
В соответствии со стадией (d) защищенный по атому N (R)-(4-метоксифенил)пропан-2-амин хлорсульфонируют хлорсульфоновой кислотой, при котором электрофильное замещение в основном протекает по положению 2 относительно метоксигруппы. Хлорсульфонирование в соответствии со стадией (d) можно провести в самом реагенте или его можно разбавить. Для указанной реакции предпочтительно использовать тионилхлорид. Полученный сульфонилхлорид после этого превращают в сульфонамид с помощью аммиака, предпочтительно - водного раствора аммиака.
Искомое соединение, (R)-5-(2-аминопропил)-2-метоксибензолсульфонамид, получают удалением защитной группы аминогруппы в соответствии со стадией (e). Это удаление защитной группы можно провести по любой обычной методике, хорошо известной в данной области техники, такой как описана в публикации T.W.Greene and P.G.Wuts in "Protective groups in organic synthesis", 2nd Edition. Например, удаление трифторацетильной группы можно провести или в кислой среде, например, разбавленной хлористо-водородной кислоте, или в щелочной среде, например, путем гидролиза карбонатом калия в спирте, предпочтительно - в метаноле, более предпочтительно - в водном растворе метанола.
(R)-5-(2-Аминопропил)-2-метоксибензолсульфонамид, полученный согласно настоящему изобретению, является подходящим сырьем для получения тамсулозина, поскольку с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) с использованием хиральной колонки можно обнаружить не более 0,3% нежелательного (S)-изомера. При использовании известного способа основание тамсулозина превращается в тамсулозина гидрохлорид, который пригоден для любого фармацевтического применения.
Предпочтительный способ, предлагаемый в настоящем изобретении, приводящий к превосходным результатам при синтезе (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида, представлен на схеме 2.
В предпочтительном варианте осуществления способ, предлагаемый в настоящем изобретении, включает дополнительную стадию, на которой тамсулозин получают после о-этоксифеноксиэтилирования аминогруппы (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида. Эта последующая стадия раскрыта в данной области техники, например, в ЕР 380144 и в US 5447958. Затем тамсулозина гидрохлорид можно получить путем обработки тамсулозина этанольным раствором НСl.
Настоящее изобретение также относится к (R)-5-(2-аминопропил)-2-метокси-бензолсульфонамиду, полученному способом получения, раскрытым ранее.
Настоящее изобретение также относится к тамсулозину или тамсулозину гидрохлориду, полученному из (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида, полученного способом получения, раскрытым ранее.
Еще один вариант осуществления настоящего изобретения относится к применению (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида для синтеза тамсулозина, характеризующемуся тем, что (R)-5-(2-аминопропил)-2-метоксибензолсульфонамид получают способом получения, раскрытым ранее.
Еще один вариант осуществления настоящего изобретения относится к следующим новым промежуточным продуктам:
(R)-1-(4-метокси-3-сульфамоилфенил)-2-трифторацетиламинопропану,
(R)-1-(4-метокси-3-сульфамоилфенил)-2-трифторацетиламино-1-пропанону.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, включающей тамсулозин или тамсулозина гидрохлорид, для которой тамсулозин получен из (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида, полученного способом получения, раскрытым ранее.
Примеры
Настоящее изобретение более подробно описано и разъяснено с помощью приведенных ниже примеров. Однако следует понимать, что настоящее изобретение не ограничивается этими примерами и что без отклонения от объема настоящего изобретения в него могут быть внесены различные изменения и модификации.
Пример 1
Синтез N-(трифторацетил)-D-аланина
К смеси D-аланина (20,00 г; 0,224 моль) и триэтиламина (31,3 мл; 0,224 моль) в абсолютном метаноле (100 мл) прибавляют этилтрифторацетат (33,4 мл; 0,280 моль) и перемешивают при комнатной температуре для гомогенизации смеси (примерно 1 день). Раствор концентрируют в роторном испарителе (35°С; 16 мм рт.ст.) и затем растворяют в смеси ТГФ (тетрагидрофуран)/вода (1:1; 140 мл). Прибавляют кислый ионообменник Dowex 50W-X8 (100 г), перемешивают в течение 10 мин, фильтруют и повторно концентрируют в роторном испарителе (35°С; 16 мм рт.ст.). Остаток возгоняют (80°С; 0,05 мм рт.ст.). Получают чистый продукт в виде бесцветных кристаллов (33,50 г; 80,8%).
Пример 2
Синтез (R)-N-(трифторацетил)-α-амино-4'-метоксипропиофенона
К смеси N-(трифторацетил)-D-аланина (10,00 г; 0,054 моль) и пиридина (100 мкл) в CH2Cl2 (100 мл) при комнатной температуре по каплям прибавляют тионилхлорид (4,1 мл; 0,057 моль) и затем перемешивают в течение 7 ч при 45°С. Прибавляют анизол (7,0 мл; 0,065 моль) и раствор охлаждают на бане со льдом. Порциями прибавляют АlСl3 (7,92 г; 0,059 моль) и перемешивают при комнатной температуре в течение 36 ч. Реакцию останавливают путем прибавления холодного 1 М НСl (150 мл) и льда (100 мл). Органическую фазу промывают с помощью 1 М НСl (2×100 мл), водой (2×100 мл), насыщенным раствором NаНСО3 (2×100 мл), сушат над безводным сульфатом натрия и концентрируют. К остатку при перемешивании прибавляют гептан (35 мл). Образовавшийся пара-продукт отфильтровывают и промывают гептаном (2×25 мл). Получают белые иглообразные кристаллы: 4,80 г (выход 32,3%); >99% энантиомерный избыток; 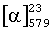 - 40,4;
- 40,4; 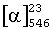 - 47,0 (с 1,0; МеОН), содержание орто-продукта 1-2%, температура плавления 110-114°С, 1Н-ЯМР (CDCl3): 1,52 (d, 3Н, СН3; J=7,2 Гц); 3,91 (s, 3Н, МеО); 5,47 (m, 1Н, CH); 7,00 (m, 2H, Н-2,6); 7,64 (широкий s, 1Н, NНСОСF3); 7,97 (m, 2H, Н-3,5).
- 47,0 (с 1,0; МеОН), содержание орто-продукта 1-2%, температура плавления 110-114°С, 1Н-ЯМР (CDCl3): 1,52 (d, 3Н, СН3; J=7,2 Гц); 3,91 (s, 3Н, МеО); 5,47 (m, 1Н, CH); 7,00 (m, 2H, Н-2,6); 7,64 (широкий s, 1Н, NНСОСF3); 7,97 (m, 2H, Н-3,5).
Дополнительная порция кристаллов образуется из фильтрата и их отфильтровывают и промывают петролейным эфиром (2×15 мл): 1,4 г; орто: пара = 1:1.
Пример 3
Синтез (R)-N-(трифторацетил)-α-амино-4'-метоксипропиофенона
К охлажденной (0°С) смеси D-N-(трифторацетил)аланина (1,00 г; 5,4 ммоль) и пиридина (1 капля) в СН2Сl2 (20 мл) по каплям прибавляют оксалилхлорид (0,50 мл; 5,7 ммоль) и затем перемешивают в течение 2 ч при комнатной температуре. Прибавляют нитрометан (330 мг; 5,4 ммоль) и анизол (0,7 мл; 6,5 ммоль) и раствор охлаждают на бане со льдом. Порциями прибавляют АlСl3 (0,79 г; 5,9 моль) и перемешивают при комнатной температуре в течение 36 ч. Реакцию останавливают путем прибавления холодного 1 M HCl (15 мл) и льда (10 мл). Органическую фазу промывают с помощью 1 M HCl (2×10 мл), водой (2×10 мл), насыщенным раствором NaHCO3 (2×10 мл), сушат над безводным сульфатом натрия и выпаривают. К остатку при перемешивании прибавляют петролейный эфир (2,5 мл). Образовавшийся пара-продукт отфильтровывают и промывают петролейным эфиром (2×2,5 мл). Получают белые иглообразные кристаллы: 330 мг, энантиомерный избыток >99%.
Пример 4
Синтез (R)-N-(трифторацетил)-α-амино-4'-метоксипропиофенона
К смеси D-N-(трифторацетил)аланина (1,0 г; 5,4 ммоль) и пиридина (5,0 мкл) в СН2Сl2 (10 мл) при комнатной температуре по каплям прибавляют тионилхлорид (0,41 мл; 5,7 ммоль) и затем перемешивают в течение 5 ч при 45°С. Прибавляют нитрометан (330 мг; 5,4 ммоль) и анизол (0,7 мл; 6,5 ммоль) и раствор охлаждают на бане со льдом. Порциями прибавляют FеСl3 (0,96 г; 5,9 моль) и перемешивают при комнатной температуре в течение 24 ч. Реакцию останавливают путем прибавления холодного 1 М HCl (15 мл) и льда (10 мл). Органическую фазу промывают с помощью 1 М HCl (2×10 мл), водой (2×10 мл), насыщенным раствором NаНСО3 (2×10 мл), сушат над безводным сульфатом натрия и выпаривают. К остатку при перемешивании прибавляют петролейный эфир (2,5 мл). Образовавшийся пара-продукт отфильтровывают и промывают петролейным эфиром (2×2,5 мл). Получают белые иглообразные кристаллы: 405 мг, энантиомерный избыток >99%.
Пример 5
Синтез (R)-N-(трифторацетил)-α-амино-4'-метоксипропиофенона
К смеси D-N-(трифторацетил)аланина (10,0 г; 0,054 ммоль) и пиридина (50 мкл) в CH2Cl2 (100 мл) при комнатной температуре по каплям прибавляют тионилхлорид (4,1 мл; 0,057 моль) и затем перемешивают в течение 5 ч при 45°С. Прибавляют анизол (7,0 мл; 0,065 моль) и раствор охлаждают на бане со льдом. Порциями прибавляют TiCl4 (32,1 г; 0,135 моль) и перемешивают при комнатной температуре в течение 36 ч. Реакцию останавливают путем прибавления холодного 1 М НСl (150 мл) и льда (100 мл). Органическую фазу промывают с помощью 1 М НСl (2×100 мл), водой (2×100 мл), насыщенным раствором NaHCO3 (2×100 мл), сушат над безводным сульфатом натрия и выпаривают. К остатку при перемешивании прибавляют петролейный эфир (25 мл). Образовавшийся пара-продукт отфильтровывают и промывают петролейным эфиром (2×25 мл). Получают белые иглообразные кристаллы: 0,6 г, энантиомерный избыток >99%.
Пример 6
Синтез (R)-2-(N-(трифторацетил)амино)-1-(4'-метоксифенил)пропана
К смеси (R)-2-N-(трифторацетил)-α-амино-4'-метоксипропиофенона (7,5 г; 27,2 ммоль) в СF3СО2Н (21 мл; 273 ммоль) по каплям прибавляют триэтилсилан (13,5 мл; 81,8 ммоль) и перемешивают при комнатной температуре в течение 1 дня. Затем смесь выливают на лед (40 мл) и нейтрализуют с помощью 4 н. NaOH. Продукт экстрагируют с помощью EtOAc (3×20 мл), сушат над MgSO4, фильтруют и выпаривают. Остаток промывают гептаном (3×30 мл) и сушат. Получают белые бесцветные кристаллы: 6,78 г; энантиомерный избыток >99%; 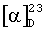 +15,0 (с 1,0; МеОН); температура плавления 100-102°С, 1Н-ЯМР (СDСl3): 1,21 (d, 3Н, СН3; J=6,6 Гц); 2,79 (m, 2H, CH2); 3,80 (s, 3Н, ОМе); 4,25 (m, 1H, СН); 6,05 (широкий s, 1H, NHCOCF3); 6,86 (m, 2H, H-3,5); 7,08 (m, 2H, H-2,6).
+15,0 (с 1,0; МеОН); температура плавления 100-102°С, 1Н-ЯМР (СDСl3): 1,21 (d, 3Н, СН3; J=6,6 Гц); 2,79 (m, 2H, CH2); 3,80 (s, 3Н, ОМе); 4,25 (m, 1H, СН); 6,05 (широкий s, 1H, NHCOCF3); 6,86 (m, 2H, H-3,5); 7,08 (m, 2H, H-2,6).
Пример 7
Синтез (R)-2-(N(трифторацетил)амино)-1-(4'-метокси-3'-сульфамоил)-фенилпропана
К охлажденному (-10°С) раствору (R)-2-(N-(трифторацетил)амино)-1-(4'-метокси)фенилпропана (5,00 г; 19,1 ммоль) в SOСl2 (4,2 мл; 57,4 ммоль) по каплям прибавляют СlSO3Н (2,5 мл; 38,2 ммоль). Смесь медленно нагревают до 40°С и перемешивают при такой же температуре в течение 3 ч. Получают вязкую смесь коричнево-красного цвета, охлаждают до комнатной температуры и прибавляют по каплям к охлажденному (0°С) 28% водному раствору аммиака (30 мл) и ацетона (15 мл). После завершения прибавления смесь перемешивают в течение 10 мин и затем ацетон выпаривают. Образовавшийся в виде белого осадка продукт отфильтровывают, промывают водой (2×20 мл), сушат и затем промывают с помощью i-Pr2O (40 мл): белый порошок; 6,13 г; выход 94%. 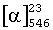 -5,0 (с 1,0; МеОН); температура плавления 171-173°С, 1Н-ЯМР (ДМСО(диметилсульфоксид)-d6): 1,14 (d, 3Н, СН3; J=6,6 Гц); 2,76 (m, 2H, СН2); 3,87 (s, 3Н, ОМе); 4,00 (m, 1Н, CH); 7,01 (br s, 2H, SO2NH2); 7,12 (d, 1H, H-5; J=8,4 Гц); 7,38 (dd, 1H, H-6; J=8,4 и 2,1 Гц); 7,58 (d, 1H, H-2; J=2,1 Гц); 9,34 (широкий d, 1H, NHCOCF3; J=8,1 Гц).
-5,0 (с 1,0; МеОН); температура плавления 171-173°С, 1Н-ЯМР (ДМСО(диметилсульфоксид)-d6): 1,14 (d, 3Н, СН3; J=6,6 Гц); 2,76 (m, 2H, СН2); 3,87 (s, 3Н, ОМе); 4,00 (m, 1Н, CH); 7,01 (br s, 2H, SO2NH2); 7,12 (d, 1H, H-5; J=8,4 Гц); 7,38 (dd, 1H, H-6; J=8,4 и 2,1 Гц); 7,58 (d, 1H, H-2; J=2,1 Гц); 9,34 (широкий d, 1H, NHCOCF3; J=8,1 Гц).
Пример 8
Синтез (R)-2-амино-1-(4'-метокси-3'-сульфамоил)фенилпропана
K2СО3 (13 г; 94 ммоль) и воду (5 мл) прибавляют к раствору R-2-(N-(трифторацетил)амино)-1-(4'-метокси-3'-сульфамоил)фенилпропана (4,00 г; 11,75 ммоль) в МеОН (80 мл). Смесь нагревают в течение 8 ч при температуре кипения и выпаривают. К остатку прибавляют воду (20 мл) и перемешивают в течение ночи. Образовавшийся в виде белого осадка продукт отфильтровывают, промывают водой (2×5 мл) и сушат; получают слабоокрашенный белый порошок, 2,65 г; 94% выход, по данным Н-ЯМР чистота составляет более 97%. Продукт перекристаллизовывают из i-PrOH (45 мл): получают слабоокрашенный белый порошок; 2,55 г; выход 89%, по данным 1Н-ЯМР чистота составляет >98%, 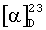 -17,8 (с 1,0, МеОН); температура плавления 168-170°С; 1Н-ЯМР (ДМСО-d6): 0,94 (d, 3Н, СН3; J=6,3 Гц); 2,51 (m, перекрывается с ДМСО, СН2); 2,94 (m, 1H, CH); 3,87 (s, 3H, ОМе); 7,11 (d, 1H, H-5; J=8,4 Гц); 7,36 (dd, 1H, H-6; J=8,4 и 2,0 Гц); 7,36 (d, 1H, H-2; J=2,0 Гц).
-17,8 (с 1,0, МеОН); температура плавления 168-170°С; 1Н-ЯМР (ДМСО-d6): 0,94 (d, 3Н, СН3; J=6,3 Гц); 2,51 (m, перекрывается с ДМСО, СН2); 2,94 (m, 1H, CH); 3,87 (s, 3H, ОМе); 7,11 (d, 1H, H-5; J=8,4 Гц); 7,36 (dd, 1H, H-6; J=8,4 и 2,0 Гц); 7,36 (d, 1H, H-2; J=2,0 Гц).
Пример 9
Синтез 5-(2-(2-(2-этоксифенокси)-этиламино)-пропил)-2-метоксибензол-сульфонамидгидрохлорида (тамсулозина)
(R)-2-(N-(Трифторацетил)амино)-1-(4'-метокси-3'-сульфамоил)фенилпропан (10 г), 2-(о-этоксифенокси)этилбромид (19 г) и МеОН (170 мл) кипятят с обратным холодильником в течение 43 ч. МеОН выпаривают в вакууме в роторном испарителе при 60°С. К остатку прибавляют 170 мл воды, 130 мл этилацетата и при охлаждении и перемешивании 16 г 50% NaOH. Если обе фазы непрозрачны, то прибавляют NaOH до осветления. После разделения фаз водную фазу экстрагируют с помощью 2×100 мл этилацетата. Объединенные экстракты промывают с помощью 2×130 мл воды и выпаривают в вакууме в роторном испарителе при 60°С. Полученное неочищенное основание тамсулозина все еще содержит большой избыток 2-(2-этоксифенокси)этилбромида. Его растворяют в 100 мл EtOH и при охлаждении и перемешивании прибавляют 7 мл этанольного раствора НСl (примерно 300 мг НСl/мл). При охлаждении (0°С) перемешивают в течение 4 ч и образовавшийся неочищенный тамсулозин в виде гидрохлорида удаляют центрифугированием, промывают с помощью 20 мл холодного EtOH и сушат в вакууме при 40°С и получают 7,0 г продукта.
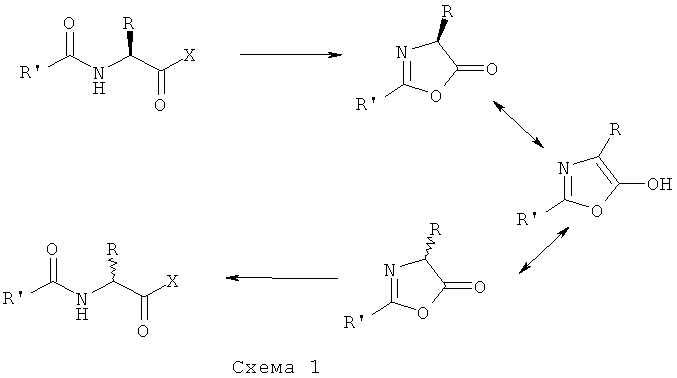
Х = защитная группа аминогруппы, описанная выше.
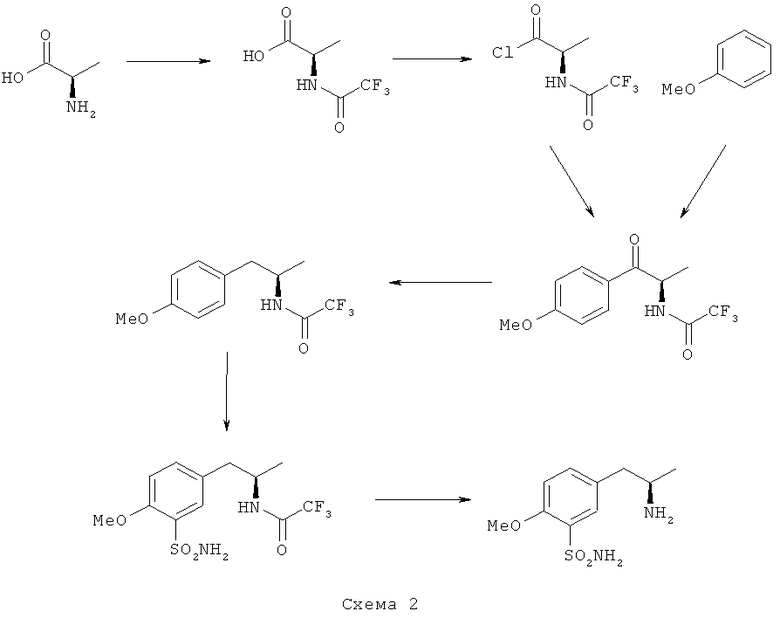
Изобретение относится к способу получения (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида. Способ осуществляют из D-аланина и метоксибензола через реакцию Фриделя-Крафтса, и через (R)-1-(4-метокси-3-сульфамоилфенил)-2-трифторацетиламинопропан в качестве промежуточного продукта. Способ включает следующие стадии: (a) защиту аминогруппы D-аланина, (b) реакцию полученного защищенного по атому N D-аланина с метоксибензолом с получением соответствующего защищенного по аминогруппе 4'-метокси-2-аминопропиофенола, (c) полное восстановление оксогруппы образовавшегося защищенного по аминогруппе 4'-метокси-2-аминопропиофенола с получением соответствующего защищенного по аминогруппе 1-(4-метоксифенил)пропан-2-амина, (d) хлорсульфонирование полученного защищенного по аминогруппе 1-(4-метоксифенил)пропан-2-амина и последующий аммонолиз образовавшейся хлорсульфонильной группы, и (е) удаление защитной группы аминогруппы. Изобретение также относится к способу получения тамсулозина или тамсулозин гидрохлорида, включающий стадии от (а) до (е), стадию о-этоксифеноксиэтилирования аминогруппы (R)-5-(2-аминопропил)-2-метоксибензола и, при необходимости, стадию обработки полученного тамсулозина раствором HCl. Технический результата - повышение оптической чистоты (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида. 3 н. и 9 з.п. ф-лы.
1. Способ получения (R)-5-(2-аминопропил)-2-метоксибензолсульфонамида, характеризующийся тем, что он осуществляется из D-аланина и метоксибензола через реакцию Фриделя-Крафтса, и через (R)-1-(4-метокси-3-сульфамоилфенил)-2-трифторацетиламинопропан в качестве промежуточного продукта и включает следующие стадии:
a) защиту аминогруппы D-аланина,
b) реакцию полученного защищенного по атому N D-аланина с метоксибензолом с получением соответствующего защищенного по аминогруппе 4'-метокси-2-аминопропиофенола,
c) полное восстановление оксогруппы образовавшегося защищенного по аминогруппе 4'-метокси-2-аминопропиофенола с получением соответствующего защищенного по аминогруппе 1-(4-метоксифенил)пропан-2-амина,
d) хлорсульфонирование полученного защищенного по аминогруппе 1-(4-метоксифенил)пропан-2-амина и последующий аммонолиз образовавшейся хлорсульфонильной группы, и
e) удаление защитной группы аминогруппы.
2. Способ по п.1, в котором указанная защита на стадии (a) проводится с помощью этилтрифторацетата.
3. Способ по п.1, в котором на стадии (b) прибавляют кислоту Льюиса.
4. Способ по п.3, в котором указанная кислота Льюиса представляет собой соль висмута, титана, железа (III) или алюминия.
5. Способ по п.4, в котором указанная кислота Льюиса представляет собой хлорид железа (III).
6. Способ по п.4, в котором указанная кислота Льюиса представляет собой хлорид алюминия.
7. Способ по п.1, в котором стадия (с) проводится с использованием триэтилсилана в качестве восстановительного реагента.
8. Способ по п.1, в котором стадия (d) проводится с использованием хлорсульфоновой кислоты в качестве хлорсульфонирующего реагента.
9. Способ по п.1, в котором реагентом для аммонолиза хлорсульфонильной группы является водный раствор аммиака.
10. Способ по п.1, в котором удаление защитной группы на стадии (е) проводится с помощью карбоната калия.
11. Способ получения тамсулозина или тамсулозин гидрохлорида, включающий стадии от (а) до (е) по пп.1-10 и стадию, на которой тамсулозин получают после о-этоксифеноксиэтилирования аминогруппы (R)-5-(2-аминопропил)-2-метоксибензола и, при необходимости, путем обработки полученного тамсулозина, раствором HCl, получают тамсулозин гидрохлорид.
12. (R)-1-(4-Метокси-3-сульфамоилфенил)-2-трифторацетиламинопропан.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Устройство для управления исполнительным механизмом манипулятора | 1983 |
|
SU1282077A1 |
| Пневмогидравлический прибор для испытания автосвечей на герметичность | 1932 |
|
SU34432A1 |
| US 5447958 A, 05.09.1995 | |||
| MEILING QI et al.: "Determination of the enantiomers of tamsulosin hydrochloride and its synthetic intermediates by chiral liquid chromatography", CHROMATOGRAPHY, 2004, vol.59, no.3/4, pp.251-254 | |||
| СПОСОБ ОПРЕДЕЛЕНИЯ ВИДА ЛЕЧЕНИЯ БОЛЬНЫХ С ДОБРОКАЧЕСТВЕННОЙ ГИПЕРПЛАЗИЕЙ ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ | 2001 |
|
RU2205001C2 |

