Изобретение относится к новому антивирусному лекарственному средству, в частности к эфиру аминокислоты пуринового производного, а наиболее конкретно к эфиру, полученному из ганцикловира (ganciclovir) и L-валина, и его фармацевтически приемлемым солям. Кроме того, настоящее изобретение относится к промежуточным соединениям, способам синтеза для получения антивирусного лекарственного средства, фармацевтическим композициям для него и его применению при лечении вирусных и связанных с ними заболеваний.
Более конкретно изобретение относится к L-моновалиновому эфиру, полученному из 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)- метокси-1,3-пропандиола, и к его фармацевтически приемлемым солям.
В описании к патенту Великобритании 1523865 предлагаются антивирусные пуриновые производные с ациклической цепью в 9-ом положении. Установлено, что среди их производных хорошим действием против герпетических вирусов, в частности против простого герпеса, обладает 2-(2-амино-1,6- дигидро-6-оксо-1,6-дигидропурин-9-ил)-метоксиэтанол с наименованием ацикловир (acyclovir) по INN-номенклатуре. Несмотря на то, что, как установлено, ацикловир очень эффективен при введении в организм локальным или парентеральным путем, при пероральном применении он абсорбируется лишь умеренно.
В описании к американскому патенту 4355032 представлено соединение 9-[(2-гидрокси-1-оксиметилэтокси)- метил]-гуанин или 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси- 1,3-пропандиол с наименованием ганцикловир по INN-номенклатуре. Ганцикловир высокоэффективен против вирусов семейства герпесов, например против простого герпеса и цитомегаловируса. При пероральном введении в организм степень его усвоения относительно низка, поэтому при употреблении таким путем его дозы должны быть большими. Чаще всего ганцикловир вводят в организм внутривенным вливанием. Недостаток такого метода применения состоит в очень большом неудобстве для пациента, частой потребности в услугах врача, медицинской сестры или другого медицинского персонала. Существует также определенная опасность инфицирования, которая особенно проблематична для пациентов с подверженной риску иммунной системой, которых лечат ганцикловиром и которые могут обладать слабой сопротивляемостью против инфекций. Таким образом, весьма желательно создать ганцикловир с улучшенным профилем поглощения при пероральном употреблении.
В описании к заявке 2122618 на патент Великобритании предлагаются производные 9-(2- гидроксиэтоксиметил)-гуанина общей формулы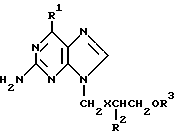
где X обозначает атом кислорода или серы, R1 обозначает гидроксил или аминогруппу, R2 обозначает атом водорода или группу формулы - CH2ORa 3, а каждый из R3 и Ra 3, которые могут быть одинаковыми или различными, обозначает ацильный радикал аминокислоты, и их физиологически приемлемые соли. Такие соединения могут быть использованы для лечения при вирусном инфицировании и обладают высокой водорастворимостью, которая обуславливает их ценность при приготовлении водных фармацевтических препаратов. Хотя общая формула, приведенная в описании к заявке на патент Великобритании, охватывает соединения, в которых R2 обозначает группу -CH2ORa 3, конкретные соединения этой группы не описаны. В описании к этой заявке на патент говорится также, что композиции, в которых использованы такие соединения с улучшенной водорастворимостью, охватывают пероральные, ректальные, назальные, топические, вагинальные или парентеральные препараты.
В заявке 2104070 на патент Великобритании предлагаются антивирусные соединения формулы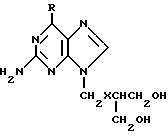
где R обозначает гидроксильную или аминогруппу, а X обозначает атом кислорода или серы, и физиологически приемлемые соли и сложные эфиры. Общая формула охватывает ганцикловир и физиологические приемлемые соли и сложные эфиры. Сложные эфиры включают в себя те, что содержат формилоксигруппу, C1-C16-алканоилокси (в частности, C1-C6), например, ацетокси или пропионилокси, необязательно замещенную аралканоилокси (например, фенил-C1-C4-алканоилокси, как фенилацетокси) или необязательно замещенную ароилокси (например, бензоилокси или нафтоилокси) эфирную группу в одном или обоих концевых положениях 9 - боковой цепи соединений общей формулы. Вышеуказанные аралканоилокси- или ароилоксиэфирные группы могут быть замещенными, например, одним или несколькими атомами галогена (например, атомами хлора или брома) или аминовыми, нитрильными или сульфамидными группами, причем предпочтительный арильный остаток такой группы содержит от 6 до 10 углеродных атомов.
В заявке на Европейский патент, публикация 375329, предлагаются пролекарственные формы соединений, отвечающие нижеследующей формуле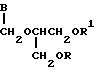
где значения R и R1 независимо друг от друга выбирают из атома водорода и аминоацильного остатка при условии, что по меньшей мере одним из R и R1 обозначен аминокислотный ацильный остаток, а В обозначает группу формул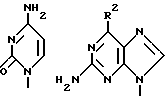
где R обозначает C1-C6прямую цепь, C3-C6-разветвленную цепь или C3-C6-циклическую алкоксигруппу, гидрокси- или аминогруппу, или водородный атом, и их физиологически приемлемые соли. Эти пролекарственные формы соединений описаны как характеризующиеся выгодной биологической доступностью при введении в организм пероральным путем, благодаря чему в организме достигается высокая концентрация исходного соединения.
В примере 3b) описания к заявке на Европейский патент, публикация 375329, описано получение в виде белой пеноподобной массы бис-L-изолейцинатного эфира ганцикловира. В примере 4b) представлено получение бис-глицинатного эфира ганцикловира в виде белого твердого вещества. В примере 5b) показано получение в виде твердого вещества бис-L-валинатного эфира ганцикловира. В примере 6b) описано получение бис-L-аланинатного эфира ганцикловира в виде сиропа, содержащего 90% диэфира и 10% моноэфира. Описанные диэфиры представляют собой некристаллические материалы, которые трудно обрабатывать при приготовлении пероральных фармацевтических дозированных препаратов.
В описании к заявке 8829571 на патент Великобритании предлагаются аминокислотные эфиры соединений формулы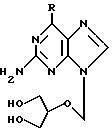
(где R обозначает гидроксильную или аминогруппу или водородный атом) и их физиологически приемлемые соли. Примеры предпочтительных аминокислот охватывают алифатические кислоты, например, содержащие до 6 углеродных атомов, в частности глицин, аланин, валин и изолейцин. Такие аминокислотные эфиры включают в себя как моно-, так и диэфиры. Однако в описаниях к этой заявке на патент, а также к публикации 375329 заявки на Европейский патент и к патенту США 5043339 получение сложных моноэфиров не проиллюстрировано и не содержатся какие-либо данные, позволяющие предположить их полезность.
Jensen и др. в Acta Pharm. Nord. 3(4) 243-247 (1991) описывают синтез, ферментативный гидролиз и физико-химические свойства N-замещенных 4-(аминометил) - бензоатдиэфирных пролекарственных форм ганцикловира формулы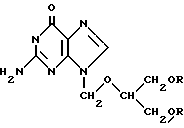
где R может обозначать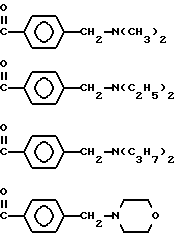
Эти сложные эфиры синтезировали и оценивали с целью улучшения способности выделять ганцикловир. Такие сложные эфиры подвергались ферментативному гидролизу плазмой человека до исходного лекарственного соединения, причем этот гидролиз протекал с промежуточным образованием соответствующего сложного моноэфира. Авторы оценивали эти сложные эфиры по скорости их ферментативного гидролиза, липофильности и пришли к выводу, что свойства таких сложных эфиров обусловливают улучшение сложными диэфирами (обещающим пролекарством типа ганцикловира) своих характеристик лекарственного снабжения, например, при введении в организм парентеральным путем.
Martin и др. в J. Pharm. Sci. 76(2), 180-184 (1987) описывают моно- и диацильные сложные эфиры ганцикловира, которые подвергали испытаниям для изучения их биологической доступности после перорального употребления. Авторы указывают на то, что дипропионатный эфир примерно на 42% биологически доступнее, чем сам ганцикловир.
В описании к заявке на Европейский патент, публикация 158847, говорится, помимо прочего, о том, что 6-дезоксиацикловир и 6- дезоксиганцикловир под действием ферментов способны in vivo легко конвертироваться соответственно в ацикловир и ганцикловир. В ходе экспериментов на крысах авторы установили, что пероральное введение в организм этих 6-дезокси-пролекарств приводит к их эффективному всасыванию в желудочно-кишечном тракте и созданию в плазме высокой концентрации исходных лекарственных веществ.
Maudgal и др. в Arch. Ophthalmol. 102, 140-142 (1984) показали эффективность глицинового эфира ацикловира при локальном лечении эпителиального и стромного кератита простого герпеса и связанного с ним ирита у кроликов с использованием 1%-ных глазных капель. Авторы представляют глициновый, аланиновый, β- аланиновый и сукциниловый эфиры ацикловира и указывают на то, что растворимость глицинового эфира примерно в 30 раз превышает растворимость самого ацикловира, что позволяет использовать этот глициновый эфир для глазных капель с концентрацией до 6%, в то время как сам ацикловир применяют в виде мази, которая малоэффективна при заболевании стромы или ирите.
Colla и др. в J. Med. Chem.,9S, 602- 604 (1983) описывают несколько водорастворимых сложноэфирных производных ацикловира и их солей как пролекарственных форм ацикловира. Авторы указывают на то, что из-за ограниченной растворимости в воде ацикловир нельзя употреблять в форме глазных капель или внутримышечных инъекций, поэтому ими синтезированы производные ацикловира, которые более растворимы в воде, чем исходное соединение. Авторы дают описание гидрохлоридной соли глицилового эфира, гидрохлоридной соли аланилового эфира, гидрохлоридной соли β- аланилового эфира, натриевой соли сукцинилового эфира и азидоацетатного эфира. Согласно мнению авторов, в процессе испытаний на культурах первичных клеток почек кроликов против различных вирусов простого герпеса штаммов типа 1 и типа 2 первые четыре эфира почти столь же активны, что и сам ацикловир. Авторы полагают, что эти ацикловирные эфиры должны быть более практичными для клинического применения, чем исходное соединение при локальной обработке в форме глазных капель и для системного лечения при инфицировании герпетическими вирусами, что хорошо согласуется с внутривенной обработкой ацикловиром. В противоположность ацикловиру такие сложные эфиры можно было бы вводить в существенно меньших объемах, что позволяет применять внутримышечные инъекции.
Beauchamp и др. в Antiviral Chemistry & Chemotherapy 3, 157-164 (1992) описывают восемнадцать аминокислотных эфиров антигерпетического медикаментозного ацикловира и их эффективность в качестве пролекарственных форм ацикловира, которую оценивали на крысах путем измерения выделения ацикловира с мочой. При использовании десяти перечисленных ниже пролекарств в моче содержались более значительные количества исходного лекарственного вещества, чем в случае применения ацикловира как такового: глициловый, D,L-аланиловый, L-аланиловый, L-2-аминобутиратный, D, L-валиловый, L-валиловый, DL-изолейциловый, L-изолейциловый, L-метиониловый и L-пролиловый эфиры. L-аминокислотные эфиры оказались более эффективными пролекарствами, чем соответствующие D- или D,L-изомеры, что, как полагают, объясняется наличием стереоселективного переносчика. Из таблицы 1 этой публикации, в которую сведены химические данные и данные о пероральной биологической доступности таких восемнадцати аминокислотных эфиров, следует, что D- аминокислотные эфиры обладают более низкой биологической доступностью при пероральном введении, чем сам ацикловир. Таким образом, поскольку D-аминокислотные эфиры не обладают никакими преимуществами перед ацикловиром, они не могут быть использованы в качестве пролекарственных форм ацикловира. Однако пероральная биологическая доступность ахирального глицилового эфира ацикловира выше, чем у ацикловира (в ходе проведения испытаний на выделение с мочой зафиксировали выделение 30% ацикловира, дозированного в форме глицилового эфира, в то время как в случае дозирования ацикловира выделили 19% ацикловира). По мнению авторов, из исследованных сложных эфиров лучшим пролекарством оказался L-валиловый эфир ацикловира.
В описании к заявке на Европейский патент, публикация 308065, предлагаются валиновые и изолейциновые эфиры ацикловира, предпочтительно в L-форме, как проявляющие более высокую всасываемость кишечником после перорального введения в организм в сравнении с другими сложными эфирами и ацикловиром.
В настоящее время лидирующим лекарственным средством для лечения при цитомегаловирусной инфекции является ганцикловир. Однако его очень ограниченная пероральная биологическая доступность и потребность в медленном ежедневном внутривенном вливании этого лекарственного средства (или в интравитреальных инъекциях или имплантатах) указывают на крайнюю необходимость создания пероральных дозированных форм с улучшенной биологической доступностью.
По настоящему изобретению предлагается стойкая пролекарственная композиция ганцикловира с улучшенным пероральным поглощением и пониженной токсичностью. Такие характеристики особенно ценны для подавления герпетических инфекций у пациентов с подверженной риску иммунной системой, когда предпочтительно терапевтическое пероральное введение в организм. Кроме того, активнодействующие компоненты проявляют фармацевтические свойства, которые обусловливают их улучшенные характеристики и фармацевтическую технологию. Установлено, что такие желательные характеристики проявляют L-моновалиновый эфир ганцикловира и его фармацевтически приемлемые соли.
Первым предметом настоящего изобретения является соединение формулы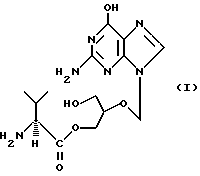
и его фармацевтически приемлемые соли. Это соединение в дальнейшем называется 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)- метокси-3-гидрокси-1-пропанил-L-валинат или моно-L-валинганцикловир.
Вторым предметом настоящего изобретения является фармацевтическая композиция, которая содержит моно-L-валиновый эфир ганцикловира или его фармацевтически приемлемую соль или его диастереомер, предпочтительно в смеси с одним или несколькими приемлемыми эксципиентами или носителями, для использования при лечении вирусных и связанных с ними заболеваний.
Третьим предметом настоящего изобретения является способ лечения или профилактики вирусных инфекций или связанных с ними заболеваний, предусматривающий введение моно-L-валинового эфира ганцикловира, его фармацевтически приемлемой соли или содержащей его композиции в организм животного, нуждающегося в таком лечении или профилактике.
Четвертым предметом настоящего изобретения являются соединения, которые служат полезными промежуточными продуктами для получения моно-L-валинганцикловира или его фармацевтически приемлемых солей формулы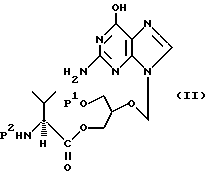
где P1 является гидроксизащитной группой, а P2 является аминозащитной группой.
Пятый предмет настоящего изобретения состоит в способе получения пролекарственной формы соединения по изобретению и его фармацевтически приемлемых солей. Этот способ предусматривает этерификацию ганцикловира и его производных, удаление защитных групп из ганцикловира, этерифицированного L-валином, частичный гидролиз бис-L-валинового эфира ганцикловира до моно-L-валинового эфира формулы I, конденсацию гуанина с замещенным глицерином, оптическое выделение соединения формулы 1 и получение солей пролекарства формулы I. Способ изложен подробно ниже.
Если не оговорено иное, следующие термины, используемые в описании и формуле настоящего изобретения, имеют приведенные ниже значения.
Термин "алкил" обозначает насыщенный углеводородный радикал с прямой или разветвленной цепью, содержащий от одного до указанного числа углеродных атомов. Так, например, C1-C7-алкил представляет собой алкил, содержащий по меньшей мере один, но не более семи углеродных атомов, например метил, этил, изопропил, н-пропил, н-бутил, н-пентил, н-гептил и тому подобное.
Термин "низший алкил" обозначает алкил, содержащий от одного до шести углеродных атомов.
Термин "арил" обозначает органический радикал, полученный из ароматического углеводорода удалением одного водородного атома. Предпочтительные арильные радикалы содержат от шести до двенадцати углеродных атомов в виде кольцевых углеродных атомов в ароматическом углеводороде.
Термин "аралкил" обозначает органический радикал, полученный из аралкана, в котором алкильный водородный атом замещен определенной выше арильной группой.
Термин "ацил" обозначает органический радикал, полученный из органической кислоты удалением гидроксильной группы. Так, например, CH3CO представляет собой ацильный радикал CH3COOH или ацетил. Другими примерами таких ацильных групп являются пропионил, бензоил и тому подобное. Значения термина "ацил" включают "алканоил", который является органическим радикалом RCO-, где R обозначает алкильную группу, определенную выше.
Термин "низший алкокси", "(низший алкил)амино", "ди(низший алкил)амино", "(низший алканоил) амино" и аналогичные термины обозначают алкокси-, алкиламино-, диалкиламино-, алканоиламиногруппы и тому подобное, в которых каждый из алкильных радикалов представляет собой "низший алкил", как определено выше.
Термин "галоген" обозначает фтор, хлор, бром или иод.
В соответствии с Hackh в Chemical Dictionary, McGraw-Hill Book Company, 1969, "производным" соединения является соединение, которое может быть получено из исходного соединения проведением простого химического процесса.
Термин "активированное производное" соединения обозначает реакционноспособную форму исходного соединения, которая придает соединению активность в ходе проведения желаемой химической реакции, где такое исходное соединение проявляет лишь умеренную реакционную способность или не является реакционноспособным. Активирование достигается получением производного или химической группировки внутри молекулы с более высоким содержанием свободной энергии, чем у исходного соединения, благодаря чему придается активированная форма, более чувствительная к реакции с другим реагентом. В отношении настоящего изобретения активирование карбоксильной группы имеет особое значение; соответствующие активирующие агенты или группировки, которые активируют карбоксильную группу, более подробно описаны ниже. Примером активированного производного L-валина служит соединение формулы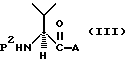
где P1 является аминозащитной группой и A является карбоксиактивирующей группой, например галоидом или низший ацилоксигруппой. Другим примером служит ангидрид аминокислоты, который является активированной формой аминокислоты, придающей аминокислоте (прежде всего L-валину) чувствительность к этерификации. Другим примером служат UNCA, более подробно описанные ниже.
Термин "защитная группа" обозначает химическую группу, которая (а) защищает реакционноспособную группу от участия в нежелательной химической реакции и (б) может быть легко удалена после того, как защита реакционноспособной группы уже не требуется. Так, например, для первичной гидроксильной функциональной группы защитной группой служит бензильная группа.
Термин "аминозащитная группа" служит для обозначения защитной группы, защищающей реакционноспособную аминогруппу, которая в ином случае, в ходе протекания определенных химических реакций могла бы подвергаться модификации. Это определение охватывает формильную группу или низшие алканоильные группы с 2-4 углеродными атомами, в частности ацетильную или пропионильную группу, тритильные или замещенные тритильные группы, в частности монометокситритильные группу, диметокситритильные группы, в частности 4,4'-диметокситритильную или 4,4'-диметокситрифенилметильную группу, трифторацетильную и N-9- фторфенилметоксикарбонильную или "ФМОК"-группу, аллилоксикарбонильную группу или другие защитные группы, являющиеся производными галоидкарбонатов, в частности C6-C12арил - низший алкил-карбонаты (в частности, N-бензилоксикарбонильная группа, являющаяся производной бензилхлоркарбоната), или производными дифенилалкилгалоидкарбонатов, или третичных алкилгалоидкарбонатов, таких, как трет-бутилгалоидкарбонаты, в частности трет-бутилхлоркарбонат, или ди- низший алкил-дикарбонаты, в частности ди(трет-бутил)ди-карбонат, и фталильную группу.
Термин "гидроксизащитная группа" служит для обозначения защитной группы, защищающей гидроксильную группу, которая в противном случае, при некоторых химических реакциях может быть модифицирована. Приемлемые гидроксизащитные группы включают в себя эфиробразующие группы, которые можно легко удалять после завершения всех других реакционных стадий, в частности бензильную или третильную группу, возможно замещенную в ее фенильном кольце. Другие приемлемые гидроксизащитные группы охватывают алкилэфирные группы, тетрагидропиранильную, силильную, триалкилсилилэфирные группы и аллильную группу.
Термин "отщепляемая группа" обозначает подвижную группу, которая в химической реакции замещается другой группой. Примерами отщепляемых групп служат атом галогена, необязательно замещенная бензилоксигруппа, изопропилоксигруппа, мезилоксигруппа, тозилоксигруппа или ацилоксигруппа.
Все активирующие и защитные агенты, используемые при получении соединения формулы I, должны отвечать нижеследующим требованиям: (1) их введение должно происходить количественно и без рацемизации L-валинового компонента; (2) защитная группа, присутствующая во время желаемой реакции, должна быть стойкой в создаваемых реакционных условиях и (3) эта группа должна быть легко удаляемой в условиях, в которых сложноэфирная связь обладает стойкостью и в которых не происходит рацемизация L- валинового компонента сложного эфира.
Термин "хиральность" означает свойство молекулы, которое показывает наличие симметрии элементов молекулы или ее отсутствие элементов. Молекулы с недостаточной симметрией элементов молекулы являются "хиральными". Хиральная молекула, у которой отсутствует симметрия всех элементов молекулы, даже простой оси, носит название "асимметричной".
Термин "ахиральный" означает наличие в молекуле по меньшей мере одного элемента симметрии, в частности простой оси.
Термин "изомерия" относится к соединениям, которые одинаковы по атомной массе и атомному числу, но различаются одним или несколькими физическими или химическими свойствами. Известны изомеры следующих различных типов:
"Cтереоизомер" обозначает химическое соединение, которое по молекулярной массе, химическому составу и строению идентично другому соединению, но атомы в нем сгруппированы по-другому, то есть некоторые идентичные химические остатки сориентированы в пространстве по-разному, и, следовательно, в чистом виде они способны вращать плоскость поляризованного света. Однако некоторые чистые стереоизомеры могут обладать настолько слабой способностью вращать плоскость поляризации света, что с помощью имеющихся приборов ее определение невозможно.
"Оптический изомер" относится к одному типу стереоизомерии, которая проявляется во вращении, сообщаемом изомером в чистом виде или в растворе плоскости поляризации света. Во многих случаях это вызвано присоединением четырех различных химических атомов или групп по крайней мере к одному углеродному атому в молекуле или иначе вышеописанной хиральностью молекулы.
Стереоизомеры или оптические изомеры, которые являются зеркальным отражением друг друга, называются "энантиомерами", то есть о них можно сказать, что они энантиомерны. Хиральные группы, которые являются зеркальным отражением друг друга, называются энантиомерными группами. Энантиомеры, чьи абсолютные конфигурации не известны, в зависимости от направления вращения ими плоскости поляризации света в определенных экспериментальных условиях можно различать как правовращающие (со знаком + впереди) или левовращающие (со знаком - впереди).
В том случае, когда продукт состоит из равных количеств совместно присутствующих энантиомерных молекул, его называют рацемическим независимо от того, является ли он кристаллическим, жидким или газообразным. Гомогенную твердую фазу, состоящую из эквимолярных количеств энантиомерных молекул, называют рацемическим соединением. Смесь эквимолярных количеств энантиомерных молекул, присутствующих в виде отдельных твердых фаз, называют рацемической смесью. Любую гомогенную фазу, содержащую эквимолярные количества энантиомерных молекул, называют рацематом.
Соединения с двумя асимметричными углеродными атомами (хиральными центрами) имеют четыре стереоизомера, которые образуют две пары энантиомеров. В то время как энантиомеры пары являются зеркальными отражениями друг друга, энантиомеры двух отдельных пар не являются зеркальными отражениями друг друга и называются "диастереомерами". Диастереомеры проявляют похожие, но не идентичные химические свойства, и обладают различными физическими свойствами, например температурой плавления, растворимостью и т.п.
Оптически активные соединения по настоящему изобретению могут быть определены рядом правил, то есть по правилам R- и S-последовательности Кана и Прелога; как эритро- и трео-изомеры, D- и L-изомеры; d- и l-изомеры; (+)-и (-)-изомеры, что указывает на направление вращения плоскости поляризации света благодаря химическому строению соединения либо в чистом виде, либо в растворе. Эти правила хорошо известны в данной области техники и подробно описаны в работе E.L. Eliel "Stereochemistry of Carbon Compounds", опубликованной в издательстве McGraw Hill Book Company, Inc. of New York в 1962 г. и упомянутой в настоящем описании в качестве ссылки. Таким образом, в зависимости от используемой номенклатурной системы эти изомеры могут быть обозначены как d-, l- или d.l-пара; или D-, L- или D,L-пара; или R-, S-или R, S-пара. В настоящем описании, как правило, использованы обозначения (D), (L) и (D,L) для аминокислоты (валина) и обозначения (R), (S) и (R,S) для асимметричного углерода в остатке ганцикловира, что позволяет подчеркнуть различие между ними.
Соединение формулы I и соединения формулы II содержат по два асимметричных центра (по 2 углеродных атома), один из которых находится в валиновом компоненте, а другой - в алифатической боковой цепи ганцикловирного компонента. Этим последним является 2-й углеродный атом пропанильного радикала. Таким образом, соединение формулы I и соединения формулы II существуют в виде диастереомеров и в виде смесей диастереомеров. Что касается соединений настоящего изобретения, то можно использовать любой диастереомер или смесь диастереомеров, и, если отсутствуют другие ограничения, рамки формулы изобретения охватывают и индивидуальные диастереомеры и их смеси, предпочтительно эквимолярные смеси. Формула 1 охватывает оба диастереомера формулы I, а также их смеси, предпочтительно эквимолярные смеси (R)- и (S)-диастереомеров.
Термины "необязательный" и "необязательно" означают, что описываемое обстоятельство может иметь место или может отсутствовать и что данное описание охватывает примеры, когда упомянутое обстоятельство имеет место, и примеры, когда этого нет. Так, например, фраза "необязательно замещенный фенил" означает, что фенил может быть замещенным или незамещенным и что данное описание охватывает как незамещенный фенил, так и фенил, у которого имеется заместитель; фраза "с необязательной последующей конверсией свободного основания в кислотно-аддитивную соль означает, что для того, чтобы описываемая процедура охватывалась рамками изобретения, указанную конверсию можно проводить или не проводить, и рамками изобретения охватываются те процедуры, в которых предусмотрена конверсия свободного основания в кислотно-аддитивную соль, и те процедуры, в которых она не предусмотрена.
Термин "фармацевтически приемлемый" относится к тому, что может быть применимо при приготовлении фармацевтической композиции, которая обычно безвредна и нетоксична, и охватывает то, что допустимо для использования в ветеринарии, а также для фармацевтического использования в отношении человека.
Термин "фармацевтически приемлемые соли" обозначает соли, которые обладают целевым фармакологическим действием и которые ни с биологической, ни с какой-либо другой точки зрения не являются нежелательными. Такие соли охватывают кислотно-аддитивные соли, полученные с использованием минеральных кислот, в частности соляной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты и тому подобного, или органических кислот, в частности уксусной кислоты, пропионовой кислоты, гексановой кислоты, гептановой кислоты, циклогептанпропионовой кислоты, гликолевой кислоты, пировиноградной кислоты, молочной кислоты, малоновой кислоты, янтарной кислоты, яблочной кислоты, малеиновой кислоты, фумаровой кислоты, винной кислоты, лимонной кислоты, бензойной кислоты, о-(4-гидрокси- бензоил)-бензойной кислоты, коричной кислоты, миндальной кислоты, метансульфокислоты, этансульфокислоты, 1,2-этанди-сульфокислоты, 2- гидроксиэтансульфокислоты, бензолсульфокислоты, п-хлорбензолсульфокислоты, 2 - нафталинсульфокислоты, п-толуолсульфокислоты, камфорсульфоновой кислоты, 4-метилдицикло [2,2,2] окт-2-ен-1- карбоновой кислоты, глюкогептоновой кислоты, 4,4'-метилен-бис-(3- гидрокси-2-нафтойной кислоты, 3-фенилпропионовой кислоты, триметилуксусной кислоты, третичной бутилуксусной кислоты, лаурилсульфокислоты, глюконовой кислоты, глутаминовой кислоты, гидроксинафтойных кислот, салициловой кислоты, стеариновой кислоты, муконовой кислоты и т.п. Предпочтительными фармацевтически приемлемыми солями являются те, что получены с использованием соляной, серной, фосфорной кислот, уксусной или метансульфокислоты, этансульфокислоты, 1,2-этандисульфокислоты, 2-гидроксиэтансульфокислоты, бензолсульфокислоты, п-хлорбензолсульфокислоты, 2-нафталинсульфокислоты, п-толуолсульфокислоты и камфорсульфоновой кислоты.
Термин "животное" включает людей, других млекопитающих (например, собак, кошек, кроликов, крупный рогатый скот, лошадей, овец, коз, свиней и оленей), а также таких немлекопитающих, как птицы, рыбы и т.п.
Термин "заболевание" включает конкретно любое нездоровое состояние животного или части его тела. Таким образом, в рамках данного описания термин "заболевание" охватывает любое вирусное или связанное с ним заболевание, которое поддается лечению моно-L-валинганцикловиром или его фармацевтически приемлемыми солями.
Термин "лечение" обозначает любое лечение заболевания у животных и включает в себя:
(1) профилактику заболевания у животного, которое предрасположено к заболеванию, но все еще не испытывает или не проявляет симптомов болезни, например профилактику начала клинических симптомов;
(2) подавление заболевания, например остановку его развития или
(3) ослабление заболевания, например путем регрессии симптомов заболевания.
Термин "эффективное количество" для лечения заболевания служит для обозначения того количества, которое при введении в организм нуждающегося в нем животного достаточно для лечения, как это определено выше, такого заболевания.
Если не оговорено иное, реакции по настоящему изобретению протекают при атмосферном давлении в температурном интервале от 5 до 170oC (предпочтительно от 10 до 50oC, а наиболее предпочтительно при "комнатной" температуре или температуре "окружающей среды", например при 20-30oC). Однако очевидна возможность проведения некоторых реакций в интервале температур, создаваемых для химической реакции, который выше или ниже таких температурных интервалов. Далее во всех случаях, за исключением специально оговоренных, параметры продолжительности реакции и условий ее проведения принимаются как приближенные. Так, в частности, ее проводят примерно при атмосферном давлении и в температурном интервале от примерно 5 до примерно 100oC (предпочтительно от примерно 10 до примерно 50oC, наиболее предпочтительно при примерно 20oC) в течение от примерно 1 до примерно 100 ч (предпочтительно от примерно 5 до 60 ч). Параметры, приведенные в примерах, следует считать конкретными, а не приближенными.
Выделение и очистку соединений и промежуточных продуктов по настоящему описанию при желании можно проводить осуществлением любой пригодной процедуры разделения или очистки, как фильтрование, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография, толстослойная хроматография или сочетание этих методов. Конкретные пригодные методы разделения и выделения могут быть проиллюстрированы со ссылкой на нижеприведенные примеры. Однако совершенно очевидно, что можно также прибегать к применению других эквивалентных методов разделения или выделения.
Соединение формулы I или его фармацевтически приемлемые соли получают различными методами. В нижеследующей формуле I участки доступа для операций синтеза обозначены пунктирными линиями [от (а) до (f)]. Эти пунктирные линии схематически указывают на соответствующие реакционные участки, а в нижеприведенной таблице представлено краткое описание различных методов, которые более подробно описаны ниже. Буквенные символы в скобках использованы для ссылки на соответствующую стадию описываемого способа/пункта (пунктов) формулы изобретения.
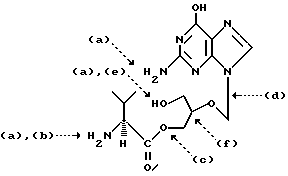
Участки доступа для операций - Метод
(а) - Удаление защитной группы
(b) - Получение соли
(с) - Этерификация
(d) - Конденсация
(е) - Частичный гидролиз
(f) - Оптическое разделение/разделение диастереомеров
Таким образом, способ получения соединения формулы I или его фармацевтически приемлемой соли включает в себя одну или несколько нижеследующих стадий:
(а) удаления амино- и/или гидроксизащитной группы из соединения формулы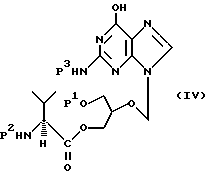
где P1 обозначает гидроксизащитную группу или водородный атом, P2 обозначает аминозащитную группу, а P3 обозначает водородный атом или P2, с получением соединения формулы I;
(b) конверсии соединения формулы I в его фармацевтически приемлемую соль или
c) этерификации 2-(2-амино-1,6-дигидро-6-оксопурин-9ил)-метокси-1,3-пропандиола (ганцикловира) или его соли активированным производным L-валина, или
(d) конденсации необязательно замещенного гуанина формулы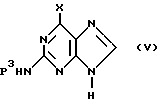
необязательно в персилилированной форме, где P3 обозначает водородный атом или аминозащитную группу, с 2-замещенным глицерином формулы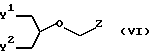
где Y1 и Y2 независимо друг от друга обозначают атомы галогена, низший ацилокси-, низший алкилокси- или аралкилоксигруппы, a Z обозначает отщепляемую группу, выбранную из низший ацилокси-, метокси-, изопропилокси-, бензилоксирадикалов, атомов галогена, мезилокси-, тозилоксирадикалов и т.п., необязательно в присутствии кислоты Льюиса в качестве катализатора с получением соединения формулы I, или
(е) частичного гидролиза бис-(L-валината) бис-эфира 2-(2-амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-1,3-пропандиила или его соли с получением моноэфира формулы I, или
f) оптического разделения или разделения диастереомеров соединения формулы (I).
Соединение формулы I и его фармацевтически приемлемые соли проявляют фармацевтическое действие и прежде всего антивирусное действие. Как таковые соединение и его фармацевтически приемлемые соли могут быть использованы для лечения широкого диапазона болезненных состояний животных, в частности людей.
Примеры болезней, которые можно лечить с использованием соединения и солей по настоящему изобретению, охватывают герпетические инфекции, в частности герпес типов 1, 2 и 6, ветряную оспу Зостера, вирус Эппштейна-Барра и прежде всего цитомегаловирус, а также гепатит Б и родственные вирусы у человека или других животных, прежде всего у человека. К примерам клинических состояний, вызываемых этими вирусами, относятся герпетический кератит, герпетический энцефалит, лихорадки, инфицирование гениталий (вызванное простым герпесом), ветряная оспа, опоясывающий лишай (вызванный оспой Зостера), центральная мигрирующая вирусная пневмония и центральный мигрирующий вирусный ретинит, в особенности у пациентов с подверженной риску иммунной системой, включая реципиентов трансплантатов (например, сердечных, почечных и костномозговых трансплантатов), и пациентов с синдромом приобретенного иммунного дефицита (СПИДа), инфекционный мононуклеоз, вызванный вирусом Эппштейна-Барра. Соединение по настоящему изобретению может быть также использовано для лечения некоторых карцином или лимфом, вызванных вирусными инфекциями или связанных с ними, в частности рака носоглотки, иммунобластовой лимфомы, лимфомы Баркитта (Burkitt) и волосяной лейкоплакии.
Еще одним предметом настоящего изобретения является способ лечения животного (предпочтительно человека), проявляющего состояние, при котором отражается действие вышеописанных вирусных инфекций, или профилактической обработки животного, когда такое вирусное инфицирование ожидается, осуществляемый с помощью врача или ветеринара. Способ предусматривает введение в организм такого животного терапевтически эффективного количества моно-L-валин- ганцикловира или его фармацевтически приемлемых солей. Терапевтически эффективным количеством соединения или его фармацевтически приемлемых солей является такое количество, которое оказывается эффективным для лечения болезненного состояния, то есть болезни. Вводимое в организм точное количество может варьироваться в широком интервале в зависимости от серьезности конкретного болезненного состояния, когда проводят лечение, возраста и веса, относительного состояния здоровья субъекта и других факторов (в частности, от формы препарата). Для пероральной композиции терапевтически эффективное количество может варьироваться от примерно 1 до 250 мг/кг веса тела в день, предпочтительно от примерно 7 до 100 мг/кг веса тела в день. Наиболее предпочтительно терапевтически эффективное количество составляет от примерно 10 до 50 мг/кг/день, прежде всего при лечении центрального мигрирующего вирусного ринита и центральной мигрирующей вирусной пневмонии. Таким образом, для человека весом 70 кг терапевтически эффективное количество составляет от примерно 70 мг/день до примерно 7 г/день, предпочтительно от примерно 500 мг/день до примерно 5 г/день, наиболее предпочтительно от 700 мг/день до 3,5 г/день. Однако для интравитреального (intravitreal) имплантата доза пролекарства находится в интервале от 0,5 до 25 мг, предпочтительно от 5 до 10 мг/имплантат. Для любого специалиста в данной области техники очевидно, что различные дозировочные интервалы определяются различием дозированных форм пролекарств по настоящему изобретению.
Ганцикловир является испытанным антивирусным лекарственным средством. Полезность пролекарственной формы ганцикловира по настоящему изобретению установлена определением концентрации ганцикловира в крови подопытных животных (крыс и обезьян) после перорального введения в их организм пролекарства. Концентрацию в плазме крови определяли в соответствии с методами, изложенными в примерах 9 и 10, являющихся процедурами, которые представляют собой модифицированные процедуры, описанные Sommadossi и др. в REVIEWS OF INFECTIOUS DISEASES 10(3), S507 (1988) и в Journal of Chromatography, Biomedical Applications 414, 429-433 (1987).
Соединение или его фармацевтически приемлемые соли по настоящему изобретению можно вводить в организм любым обычным и приемлемым способом, который известен в данной области техники, либо отдельно, либо в сочетании с другим терапевтическим средством. Обычно соединение и соли по настоящему изобретению вводят в форме фармацевтической композиции с фармацевтически приемлемым носителем в организм перорально, системно (например, через кожу или посредством суппозитория) или парентерально (например, внутримышечно [вм], внутривенно [вв] , подкожно [пк] или интравитреально с помощью имплантата. Таким образом, соединение по изобретению можно вводить в составе композиции, которая представляет собой полутвердую, порошкообразную, аэрозольную, растворную, суспензионную или другую приемлемую композицию, как описано ниже. Предпочтительными являются пероральные фармацевтические композиции.
Фармацевтическая композиция включает в себя соединение формулы I или его фармацевтически приемлемые соли, предпочтительно в сочетании с фармацевтически приемлемым носителем. Таким носителем служит нетоксичный материал. Носителем может быть любой твердый, жидкий, полутвердый, газообразный (в аэрозольных препаратах) носитель, который обычно доступен для любого специалиста в данной области техники и не оказывает нежелательного влияния на эффективность активнодействующего вещества.
Обычно фармацевтическая композиция по настоящему изобретению содержит терапевтически эффективное количество соединения или его фармацевтически приемлемых солей в сочетании по меньшей мере с одним носителем. В зависимости от типа композиции, размера единичной дозы, типа носителей и других факторов, известных любому специалисту в области фармацевтики, количество соединения по настоящему изобретению может варьироваться в композиции в широком интервале. Обычно готовая композиция включает в себя от примерно 1 до примерно 99,5 вес.% соединения по изобретению, а остальное приходится на долю носителя или носителей. В предпочтительном варианте содержание активнодействующего соединения составляет от примерно 10,0 до примерно 99 вес.% и наиболее предпочтительно от примерно 50 до примерно 99 вес. %, причем остальная часть приходится на долю носителя или носителей. Пригодные фармацевтические носители при приготовлении фармацевтических композиций, о которых идет речь, могут быть твердыми, полутвердыми, жидкими или газообразными. Таким образом, эти композиции могут иметь форму таблеток, пилюль, капсул, порошков, суппозиториев, накладок для введения через кожу, препаратов длительного выделения, интравитреальных имплантатов, растворов, в частности внутривенных растворов, суспензий, эликсиров, аэрозолей и т.п. К твердым фармацевтическим носителям относятся крахмалы, в частности кукурузный крахмал, целлюлоза, тальк, глюкоза, лактоза, сахароза, желатин, солод, рис, мука, мел, силикагель, стеарат магния, стеарат натрия, стеариновая кислота, моностеарат глицерина, хлористый натрий, сухое сепарированное молоко и т.п. Жидкие и полутвердые носители могут быть выбраны из воды, этанола, глицерина, пропиленгликоля, различных масел, включая получаемые из нефти, животного, растительного или синтетического происхождения, например арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Предпочтительными жидкими носителями являются вода, рассол, водная декстроза и гликоли, в частности при приготовлении растворов для инъекций. Другие приемлемые фармацевтические эксципиенты и носители и их композиции описаны E.W. Martin в работе "Remington's Pharmaceutical Sciences", Mack Publishing Company, Easton, Pennsylvania (1980), которая упомянута в качестве ссылки.
Предпочтительную фармацевтическую композицию вводят в организм в единичной дозированной форме, более предпочтительно в пероральной дозированной форме для непрерывного лечения или в единичной дозированной форме по желанию, когда конкретно требуется ослабление симптомов.
Хотя в самом широком аспекте объект настоящего изобретения определен как соединение формулы I и его фармацевтически приемлемые соли, предпочтительными являются (R,S)-смесь и некоторые соли.
Для получения фармацевтически приемлемых солей соединения формулы I предпочтительны следующие кислоты: соляная, серная, фосфорная кислоты, уксусная, метансульфо-, этансульфо-, 1,2-этандисульфо-, 2- гидроксиэтансульфо-, бензолсульфо-, п-хлор-бензолсульфо-, 2- нафталинсульфо-, п-толуолсульфокислоты и камфорсульфоновая кислота.
Наиболее предпочтительны такие сильные минеральные кислоты, как соляная, серная и фосфорная кислоты. Наиболее предпочтительными соединениями являются гидрохлорид и ацетат 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)метокси- 3-гидрокси-1-пропанил-L-валината. Эти соединения могут быть получены в виде кристаллических материалов и, следовательно, с их использованием можно легко приготовить стабильные пероральные композиции. Предпочтительными являются пероральные и внутривенные композиции. Достоинством пероральных композиций является высокая биологическая доступность; достоинство внутривенных композиций состоит в том, что пролекарство по изобретению в отличие от внутривенных ганцикловирных композиций, готовят в условиях физиологически более приемлемой величины pH (4-6). Внутривенная композиция ганцикловира требует величины pH 11, что вызывает раздражающее действие.
Очевидно, что предлагаемые соединения особенно пригодны в фармацевтических композициях и при осуществлении способов лечения по настоящему изобретению.
В описании процессов при осуществлении любых заключительных стадий ссылка на формулу I, II, III, IV, V или VI относится к таким формулам, у которых значения символов P1, P2 и P3, A, Y1, Y2 и Z в их самых широких аспектах определены выше, причем эти процессы, в частности, применимы к настоящим предпочтительным вариантам выполнения.
Предпочтительные фармацевтические композиции по настоящему изобретению содержат фармацевтически приемлемую соль пролекарства формулы I. Таким образом, если процесс приготовления фармацевтических композиций включает в себя гомогенное смешение фармацевтических носителей с активнодействующим компонентом в его солевой форме, предпочтительно использовать фармацевтические носители, которые по своей природе не обладают основными свойствами, то есть являются либо кислыми, либо нейтральными.
Предпочтительный в настоящее время способ получения соединения формулы I включает в себя стадию (а), предпочтительно осуществляемую с сопутствующим образованием соли соединения формулы I, или стадию (с), или же сочетание стадий (а) и (с) (см. приведенное ниже описание стадий III и IV). Получение моноэфира в соответствии со стадией (а) требует селективной защиты одной из двух первичных гидроксильных функциональных групп ганцикловира или его производного. Это обычно может включать в себя или не включать защиту аминогруппы во 2-м положении гуанинового основания (см. приведенное ниже подробное описание стадий I-III для случая, когда процесс проводят с защищенной аминогруппой). Кроме того, перед проведением этерификации (стадия III) аминогруппу аминокислотного реагента необходимо защитить с целью избежать ее влияния (образования амида) в ходе реакции этерификации. Защита аминогруппы описана ниже.
При осуществлении способа по настоящему изобретению те амино-, гидроксигруппы или карбоксильные группы, которые не принимают участия в реакции синтеза, обычно должны быть защищены до тех пор, пока (1) либо в результате удаления защитной группы не будет получен конечный продукт, либо (2) конкретная защищенная группа не примет участия в последующей стадии синтеза, либо (3) наличие незащищенной группы на последующих стадиях реакции, ведущих к образованию конечного продукта, не изменит намеченной последовательности реакций. Примером, удовлетворяющим требованию (1) при получении моноэфиров по настоящему изобретению, является бензильная группа, которая защищает первичную гидроксильную функциональную группу ганцикловира до тех пор, пока ее не удаляют на стадии удаления защитной группы. Примером, удовлетворяющим требованию (2), служит вторая бензильная группа, защищающая вторую первичную гидроксильную функциональную группу ганцикловира, которую удаляют непосредственно перед стадией этерификации. Примером, удовлетворяющим требованию (3), служит ацетильная группа, тритильная или монометокситритильная группа, защищающая аминогруппу гуаниновой циклической системы ганцикловира, поскольку незащищенная аминогруппа не влияет на этерификацию (стадия III).
В основном характеристики потенциальных блокирующих агентов, которые определяют их пригодность для использования при получении соединения формулы I, включают следующее:
(1) их введение должно происходить количественно и равномерно, без рацемизации L-валина;
(2) блокированный промежуточный продукт должен быть стойким в условиях проводимых реакций до момента необходимости удаления защитной группы;
(3) блокирующая группа должна быть способной легко удаляться в условиях, в которых не происходит изменения химической природы оставшейся части молекулы или рацемизации L-валинового компонента.
Все исходные материалы (ганцикловир и L-валин), а также реагенты, защищающие и активирующие карбоксильные группы, используемые для получения соединения формулы I, известны. Известны также различные L-валиновые производные с защищенным амином, в частности N-бензилоксикарбонил-L-валин, БОС-L-валин, ФМОК-L-валин, М-формил-L-валин и N- бензилоксикарбонил-N-карбокси-L-валиновый ангидрид, которые являются коммерчески доступными полупродуктами или описаны в литературе, в частности N-аллилоксикарбонил-L-валин.
Предпочтительным защищенным ганцикловировым исходным материалом для получения предпочтительного соединения по изобретению служит N2-ацетил-бис-O-бензилганцикловир (N2-ацетил-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1,3-бис (бензилокси)-пропан), который представлен в описании к патенту США 4355032. Другими предпочтительными защищенными ганцикловировыми исходными материалами служат N2- тритил-9-[(3 -гидрокси-2-пропокси-1-тритилокси)-метил] -гуанин-[N2-тритил-2--(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1 тритилоксипропан -3-ол] и N2-монометокситритил-9-[(3-гидрокси-2- (пропокси-1-монометокситритилокси)-метил] -гуанин, получение которого описано в J. Pharm. Sci. 76(2), 180-184 (1987), который упомянут в настоящем описании в качестве ссылки. Бис-(L-валинат) 2-(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1,3-пропандиила, который служит исходным материалом для осуществления стадии частичного гидролиза, представлен в описании к заявке на Европейский патент, публикация 375329.
Перед осуществлением стадии III (стадии этерификации) с целью избежать помех аминогруппы L-валина протеканию этерификации из-за образования нежелательного амида ее необходимо блокировать. С этой целью можно использовать нижеследующие аминозащитные группы: галоидкарбонаты, как C6C12-арил-низший алкилкарбонаты (в частности, карбобензилокси-группа, являющаяся производной бензилхлоркарбоната), дифенилалкилгалоидкарбонаты, трет-алкилгалоидкарбонаты, такие, как трет-бутилгалоидкарбонаты, например, трет-бутилхлоркарбонат, или ди-низший алкил-дикарбонаты, в частности ди-трет-бутилдикарбонат, трифенилметилгалогениды, например трифенилметилхлорид, и трифторуксусный ангидрид. Стадию блокирования осуществляют растворением или суспендированием L-валина в щелочном водном растворе, который может включать в себя низший алканол. Реакционную смесь охлаждают и одновременно небольшими порциями добавляют в нее блокирующий агент, в частности галоидкарбонат, предпочтительно в форме раствора в воде или низшем алканоле. В процессе такого добавления температуру реакционной смеси поддерживают в интервале от 0 до 30oC, предпочтительно 0-5oC, в течение нескольких часов, пока она не достигнет комнатной температуры. Реакционную смесь концентрируют досуха и остаток разделяют между органической фазой и водой. Водный слой подкисляют и экстрагируют органическим растворителем для блокированной аминокислоты. Органическую фазу промывают водой, а затем рассолом и сушат над сульфатом магния с последующим выпариванием досуха. N-защищенную аминокислоту выделяют и очищают с помощью обычных способов выделения и очистки.
Получение моно-L-валинганцикловира
Стадия I: ганцикловир с необязательно защищенной 2-аминогруппой и обеими защищенными первичными гидроксильными функциональными группами частично деблокируют, например, гидрогенизацией до ганцикловира с 2- аминогруппой, которая сохраняется в защищенной форме, и одной защищенной первичной гидроксильной функциональной группой. Подходящие аминозащитные группы представляют собой низшие алканоильные группы с 2-4 углеродными атомами, в частности ацетильную или пропионильную группу. Другими пригодными аминозащитными группами являются тритильные или замещенные тритильные группы, в частности монометокситритильная группа, и 4,4'-диметокситритильная группа.
Пригодными гидроксизащитными группами служат группы, образующие простые эфиры, которые можно легко удалять после завершения всех других реакционных стадий. Эти гидроксизащитные группы простых эфиров охватывают бензильную и тритильную группу. Такие группы могут быть замещенными в фенильном кольце. К другим подходящим гидроксилзащитным группам относятся остаток аллилового простого эфира, тетрагидропиранил, силил, остатки простых триалкилсилиловых эфиров, которые могут быть удалены фтористым водородом проведением процедуры, которая хорошо известна любому специалисту в данной области техники.
Гидрогенизацию для удаления одной гидроксизащитной группы предпочтительно проводить растворением защищенного ганцикловира в растворительной системе, которая выделяет водород в присутствии катализатора, такого, как палладиевое соединение, в частности гидроксид палладия. Это достигается обменной гидрогенизацией или другими обычными процедурами гидрогенизации. К другим приемлемым катализаторам гидрогенизации относятся гидрогенизационные катализаторы вообще, в частности Pd, Pd на угле и гомогенные гидрогенизационные катализаторы. Растворительная система включает в себя такой низший алканол, как метанол или этанол, и циклогексен. Обычно реакцию следует проводить при температурах от комнатной до точки кипения растворительной системы с обратным холодильником, например, в кипящем с обратным холодильником этаноле и циклогексене в инертной атмосфере и при исключении доступа кислорода или воздуха, предпочтительно в атмосфере азота. Катализатор выделяют фильтрованием. Объем фильтрата можно уменьшить выпариванием избыточного растворителя. Полученная сырая реакционная смесь в качестве основных продуктов, как правило, содержит не подвергшийся изменению исходный материал и 2- аминозащищенный ганцикловир с одной защищенной алифатической гидроксильной группой. Разделение этих двух продуктов обычно проводят осуществлением процедур выделения, известных в данной области техники, часто по методам хроматографии, предпочтительно на силикагеле, с последующим элюированием соответствующими элюентами, такими, как смеси низшего спирта с галоидированным низшим алканом (предпочтительно с этанолом и дихлорметаном), в результате чего образуется 2- аминозащищенный ганцикловир с одной защищенной алифатической гидроксильной группой.
Стадия II: ганцикловир с защищенной 2- аминогруппой и одной защищенной алифатической гидроксильной группой подвергают деблокированию аминогруппы. Если на этой стадии аминозащитной группой служит низшая алканоильная группа, для удаления защитной группы создают основные условия (величина pH между 9 и 14). Так, например, М2-ацетилмоно-O-бензилганцикловир обрабатывают щелочным реагентом, таким, как гидроксид аммония, карбонат натрия или калия или гидроксид натрия или калия, до завершения процесса удаления ацетильной группы. Обычно эту реакцию проводят в присутствии пригодного растворителя, такого, как низший алканол. В предпочтительном варианте исходный материал растворяют в метаноле и добавляют в раствор стехиометрический избыток гидроксида аммония. Реакционную температуру поддерживают между 0 и 50oC, предпочтительно на уровне комнатной температуры. После завершения реакции (определяется по данным тонкослойной хроматографии) для упрощения выделения деблокированного продукта можно добавлять другой растворитель, в частности диэтиловый эфир, что приводит к осаждению деацилированного продукта, который можно отфильтровать и выделить с использованием обычных методов разделения.
Стадия III: на этой стадии активированное производное аминозащищенного L-валина формулы III этерифицируют защищенным производным ганцикловира, полученным на стадии II. Пригодными аминозащитными группами для L-валина служат N- бензилоксикарбонильная группа, фталильная группа, трет-бутил- оксикарбонильная группа и N-9-фторенилметоксикарбонильная или "ФМОК"- группа.
В начале необходимо использовать по меньшей мере 1 экв. защищенной аминокислоты и 1 экв. приемлемого связующего вещества или дегидратационного агента, например, 1,3-дициклогексилкарбодиимида, или солей таких диимидов с основными группами. Можно также применять другие карбодиимиды, в частности N, N'-карбонилдиимидазол. Другими пригодными дегидратационными агентами служат трифторуксусный ангидрид, смешанные ангидриды, хлорангидриды кислот, 1- бензотриазолилокситрис(диметиламино)-фосфонийгексафторфосфат, PYBOP, 1-гидроксибензотриазол, 1-гидрокси-4-азабензотриазол, 1-гидрокси-7- азабензотриазол, N-этил-N'-[3-(диметиламино)-пропил] - карбодиимидгидрохлорид, 3-гидрокси-3,4-дигидро-4-оксо-1,2,3- бензотриазин, O-(бензотриазол-1-ил)-1,1,3,3-тетраметилуроний- гексафторфосфат, O-(7-аза-бензотриазол-1-ил)-1,1,3,3- тетраметилуроний-гексафторфосфат, O-(7-азабензотриазол-1-ил)-1,1,3,3- тетраметилуроний-тетрафторборат, O-(1H-бензотриазол-1-ил)-1,1,3,3-бис- (тетраметилен)-уронийгексафторфосфат или O-(7-азабензотриазол-1-ил)- 1,1,3,3-бис-(тетраметилен)-уронийгексафторфосфат. Описание таких связующих веществ можно найти в J. Am. Chem. Soc. 115, 4397-4398 (1993). Кроме того, с этой целью полезными являются уретанзащищенные N-карбоновые ангидриды аминокислот (UNKA), описанные Fuller и др. в журнале J. Am. Chem. Soc. 112, 7414-7416 (1990), который упомянут в настоящем описании в качестве ссылки. Вообще в качестве связующего вещества можно использовать любой другой реагент, который в мягких условиях образует ангидрид или другое активированное производное защищенной аминокислоты.
Аминозащищенную аминокислоту растворяют в инертном растворителе таком, как галоидированный низший алкан, предпочтительно дихлорметан, в инертной атмосфере, например в токе азота, и добавляют связующее вещество (предпочтительно 1,3-дициклогексилкарбодиимид). Реакционную смесь перемешивают при температурах между 0 и 50oC, предпочтительно приблизительно при комнатной температуре. Реакционную смесь фильтруют и выделяют продукт реакции (ангидрид защищенной аминокислоты). Конечный продукт растворяют в сухом инертном растворителе, как сухой диметилформамид (ДМФ), и создают азотную атмосферу. В вышеуказанный раствор ангидрида добавляют раствор эквивалентного количества продукта стадии II в инертном растворителе. Реакцию проводят при температуре между 0 и 50oC, предпочтительно примерно при комнатной температуре, в течение от 5 до 90 ч. Реакционный продукт можно выделять и очищать по обычным методам, в частности хроматографией. Обычно продукт содержит непрореагировавшую N-защищенную аминокислоту, которую можно удалять обработкой не смешивающегося с водой раствора (органической фазы) продукта водной щелочью, как бикарбонат натрия, карбонат натрия, рассолом и их смесями. Из органической фазы L-валиновый эфир ганцикловира с защищенной алифатической гидроксильной группой и N- защищенную аминокислоту можно выделять и очищать с использованием обычной техники выделения и очистки.
Стадия IV (конечное деблокирование с получением продукта формулы I)
Обе защитные группы продукта стадии III удаляют реакциями деблокирования, предпочтительно в кислой среде или растворителе, наиболее предпочтительно гидрогенизацией. Деблокирование в кислых условиях предпочтительно, поскольку при этом обеспечивается протонирование аминогруппы, высвобождающейся в ходе реакции деблокирования, то есть обеспечивается нейтрализация основания формулы I по мере его образования в ходе реакции деблокирования по меньшей мере стехиометрическим количеством присутствующей кислоты. Выделение соединения формулы I в форме его кислотно-аддитивной соли позволяет защищать желаемую стереоконфигурацию соединения формулы I. Таким образом, примеры, приведенные ниже, в которых описывается стадия (а) деблокирования, иллюстрируют также сопутствующую стадию (b) солеобразования.
Реакцию деблокирования проводят растворением продукта стадии этерификации в инертном растворителе, предпочтительно в кислом растворителе, с использованием катализатора гидрогенизации, в частности палладия на угле, платины, с созданием повышенного давления водорода, находящегося между 1 и 2000 фунтов/кв. дюйм (0,070-1406 кг/см2), предпочтительно от 20 до 200 фунтов/кв. дюйм (1,40-14,1 кг/см2. Завершение реакции можно определить слежением по данным обычного тонкослойного хроматографического (ТСХ) анализа. Гидрогенизацию проводят до завершения конверсии с добавлением, если требуется, дополнительного катализатора гидрогенизации. Катализатор выделяют и промывают. Объединенные фильтраты операции фильтрования и промывные жидкости концентрируют и лиофилизуют для выделения L-валинового эфира ганцикловира. Очистку продукта и выделение кристаллического сложного эфира проводят перекристаллизацией или с помощью другой техники очистки, в частности по методу жидкостной хроматографии.
Если в качестве аминозащитной группы используют третичную бутилоксикарбонильную группу, ее удаление осуществляют с помощью кислоты, в частности HCl, и изопропанола в качестве растворителя или же только трифторуксусной кислоты.
Если же стадию этерификации осуществляют с использованием производного ганцикловира с защищенными тритилами или защищенными замещенными тритилами, такие защитные группы можно удалять обработкой водной алкановой кислотой, трифторуксусной или соляной кислотой при температуре между -20 и 100oC, например водной уксусной кислотой.
Аллиловые группы удаляют изомеризацией до простых виниловых эфиров с помощью родиевых или палладиевых катализаторов с последующим кислым водным гидролизом.
Другие методы получения [стадии (b), (d) и (е)].
Специалисту в данной области техники известно также, что соединение формулы I может быть получено в форме кислотно- аддитивной соли или соответствующего свободного основания. Если оно получено в форме кислотно-аддитивной соли, соединение можно подвергать конверсии в свободное основание обработкой пригодным основанием, в частности раствором гидроксида аммония, гидроксида натрия, гидроксида калия или т.п. Однако важно указать на то, что свободное основание формулы I труднее охарактеризовать, чем кислый аддукт. В процессе конверсии свободного основания в кислотно-аддитивную соль проводят реакцию соединения с пригодной органической или минеральной кислотой (описанной выше). Эти реакции проводят обработкой по меньшей мере стехиометрическим количеством соответствующей кислоты (в случае получения кислотно-аддитивной соли) или основания (в случае высвобождения свободного соединения формулы I). На стадии солеобразования по настоящему изобретению свободное основание обычно растворяют в полярном растворителе таком, как вода или низший алканол (предпочтительно изопропанол), и их смеси, и в воду или низший алканол добавляют в требуемом количестве кислоту. Реакционную температуру обычно поддерживают в интервале примерно от 0 до 50oC, предпочтительно приблизительно на уровне комнатной. Соответствующая соль выпадает в осадок самопроизвольно или же ее можно выделить из раствора добавлением менее полярного растворителя, удалением растворителя выпариванием или в вакууме или же охлаждением раствора.
Реакционные условия стадии конденсации (d) представлены в публикации описания к заявке на Европейский патент, публикация 187297. Эту стадию конденсации осуществляют по одному из предпочтительных методов получения диастереомеров моноэфира. На этой стадии конденсации проводят реакцию гуанина, предпочтительно с защищенной 2-аминогруппой, с производным глицерина. Это производное глицерина, в частности 1-галоид-3-бензилокси-2- ацилоксиметоксиглицерин, вводят в реакцию с гуанином или замещенным гуаниновым производным в апротонном углеводородном растворителе (в частности, в бензоле, толуоле или ксилолах) или ДМФ совместно с гекса(низший)алкилсилазаном, например с гексаметилсилазаном, гексаэтилсилазаном или т.п., и катализатором при температурах между 30oC и температурой кипения с обратным холодильником. Катализатором служит соль кислоты Льюиса, в частности, триалкилсилиловая соль, например, сульфат или трифторалкилсульфонат, хлорсилан или сульфат аммония и пиридин. Более подробное описание реакционных условий на стадии конденсации (d) представлено в описании к заявке на Европейский патент, публикация 187297, которая включена в настоящее описание в качестве ссылки. Как правило, значения Y1 и Y2 необходимо выбирать таким образом, чтобы обеспечивалась возможность получения моно-L-валинового эфира формулы I. Y1 может обозначать аминозащищенную L-валиниловую группу или группу, которую можно трансформировать в L-валиниловую группу.
Соединение по настоящему изобретению можно также получать из бис-(L-валината) 2-(2-амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-1,3-пропандиила, который приведен в описании к заявке на Европейский патент, публикация 375329. Конверсию в 2-(2-амино-1,6-дигидро-6- оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L-валинат проводят частичным гидролизом [Стадия (е)] одной из групп L-валинового эфира в регулируемых условиях, которые приводят к предпочтительному отщеплению только одного ацильного остатка аминокислоты. Бис-L-валинат 2-(2- амино-1,6-дигидро-6-оксо-9H-пурин-9-ил)-метокси-1,3-пропандиила, предпочтительно бис-ацетатную соль, растворяют в деионизированной воде и частично нейтрализуют слабым основанием, в частности разбавленным раствором гидроокиси аммония. Эту смесь выдерживают при комнатной температуре в течение от одного до нескольких дней, предпочтительно от 48 до 72 ч.
По другому варианту для осуществления частичного гидролиза можно прибегнуть также к ферментативному гидролизу, в частности, с использованием эстеразы, например порцинэстеразы или пептидазы, такой, как карбоксипептидаза.
Моноэфир можно отделить от бис-эфира препаративной хроматографией в слабокислых условиях (pH 3-5, предпочтительно pH 4). Используемый для хроматографического разделения растворитель удаляют и L-валинат 2-(2-амино-1,6-дигидро-6-оксо-9H- пурин-9-ил)-метокси-3-гидрокси-1-пропанила выделяют в виде смеси двух диастереомеров.
Выделение стереоизомеров
Из формулы (I) следует, что, помимо асимметричного углеродного атома в молекуле L-валина, у соединения по изобретению имеется один асимметричный углеродный атом (хиральный центр) в пропанильной цепи. Таким образом, существуют две диастереомерные формы, а именно (R)- и (S)-формы, как это определяется правилами Кана и др.
При разделении диастереомеров можно применять ряд приемлемых для этого методов, но предпочтительные методы основаны на технике использования преимуществ различных физических свойств этих двух диастереомеров. Обычно диастереомеры разделяют хроматографическим путем, но предпочтительной является техника разделения/выделения в зависимости от различий растворимости, в частности техника фракционной кристаллизации.
Особенности техники разделения, приемлемой для получения диастереомеров формулы I, изложены Jean Jacques, Andre Collet, Samuel H.Wilen в работе Enantiomers, Racemates and Resolutions, John Wiley & Sons, Inc. (1981), которая включена в настоящее описание в качестве ссылки.
По другому варианту соединение по настоящему изобретению может быть получено с помощью оптически активных реагентов. Когда получают чистые диастереомеры моно-L- валинганцикловира, предпочтительным техническим приемом в процессе синтеза является стадия конденсации (d). Однако в случае использования оптически активных реагентов важной становится необходимость избегать величин pH выше 6, поскольку в интервале более высоких величин pH происходит взаимопревращение свободного соединения формулы I. Так, например, при величине pH 7 и 40oC период полупревращения диастереомерных смесей формулы I составляет менее одного часа.
Стереоконфигурацию при втором хиральном центре соединения формулы I можно определять по круговому дихроизму, предпочтительно рентгеновским анализом по единственному кристаллу производного с тяжелым атомом или корреляцией с материалом, полученным полным синтезом из единственного энантиомера глицерина известной конфигурации.
Получение кристаллического 2-(2-амино-1,6-дигидро-6-оксопурин- 9-ил)-метокси-3-гидрокси-1-пропанил-L-валината
Соединение по изобретению может быть получено и его получают в кристаллической форме. В этом состоит его решающее преимущество перед соединениями, известными ранее и описанными как некристаллические материалы. Преимущество состоит в том, что использование кристаллического материала упрощает приготовление фармацевтических композиций. Кристаллический материал более эффективно поддается переработке и обладает более воспроизводимыми свойствами, чем некристаллический; в качестве кристаллических материалов изобретения можно удостовериться намного легче, чем в качестве некристаллических материалов.
С целью получить кристаллический материал предпочтительнее использовать соль 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1- пропанил-L-валината. Предпочтительными кристаллическими солями являются ацетат и гидрохлоридная соль. Кристаллизацию такой соли предпочтительно инициировать растворением гидрохлоридной или ацетатной соли в воде и добавлением органического растворителя, смешивающегося с водой, в частности метанола, этанола, изопропанола, тетрагидрофурана или ацетонитрила. По другому варианту гидрохлоридную соль можно кристаллизовать из безводного раствора в низшем алканоле, в частности в метаноле, этаноле, добавлением других органических растворителей, например этилацетата, изопропанола, тетрагидрофурана или толуола.
Существо и возможность практического выполнения настоящего изобретения для любого специалиста в данной области техники становятся более ясными из нижеприведенных описаний получения и примеров. Их следует рассматривать не как ограничивающие рамки изобретения, а только как иллюстрирующие его сущность и типичные примеры его выполнения.
Пример 1. Получение (5)-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)- метокси-3-бензилоксипропан-1-ола
А. (R)-(1-Хлор-2-ацетоксиметокси-3-бензилокси)-пропан
Пузырьки газообразной HCl (высушенной пропусканием через концентрированную H2SO4) пропускали при 0oC через перемешиваемую смесь 500 мг (3,06 ммоля) (S)-(+)-бензилокси-метилоксирана и 201 мг (6,71 ммоля) параформальдегида в 8 мл дихлорметана до полного растворения твердого материала (примерно 45 мин). Образовавшийся раствор хранили при 0oC в течение 16 ч. После сушки над сульфатом магния растворитель выпаривали с получением (R)-(1-хлор-2-хлорметокси-З-бензилокси) -пропана. Этот простой хлорметилэфирный промежуточный продукт растворяли в 3 мл ацетона и по каплям добавляли в смесь 2,1 г (21,4 ммоля) ацетата калия в 7 мл ацетона. Смесь перемешивали при комнатной температуре в течение 16 ч. Твердый материал отфильтровывали и концентрировали фильтрат. Остаток растворяли в 20 мл толуола и промывали 10 мл насыщенного раствора бикарбоната натрия и 2 порциями по 20 мл воды. Органический слой сушили над сульфатом натрия. После фильтрования фильтрат концентрировали и остаток очищали высокоскоростной хроматографией на силикагеле (гексан/этилацетат = 7/1) с получением в виде бесцветного маслоподобного продукта 810 мг (2,97 ммоля, 97%-ный выход) (R)-(1- хлор-2-ацетоксиметокси-3-бензилокси)-пропана (соотношение между изомерами составляло 12:1).
Б. (R)-2-(2-амино-1,6-дигидро-6- оксопурин-9-ил)-метокси-1-хлор-3-бензилоксипропан
Раствор 1,09 г (2,95 ммоля) персилилированного гуанина в 3,2 мл ДМФ добавляли в 810 мг (R)-(1-хлор-2-ацетоксиметокси-3-бензилокси)-пропана. Раствор перемешивали при 130oC 1 ч с последующим введением триметилсилилтрифторметансульфоната. Перемешивание продолжали при той же температуре 4 ч. Смесь охлаждали до комнатной температуры и разделяли между водой и этилацетатом. Водный слой тщательно экстрагировали этилацетатом. Объединенный органический слой сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали хроматографией над силикагелем, получая (R)-2-(2-амино-1,6-дигидро-6-оксопурин-9- ил)-метокси-1-хлор-3-бензилоксипропан совместно с его N-7-изомером. Соотношение между N-9-и N-7-изомерами составляло приблизительно 2,3:1.
В. (R)-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1-ацетокси-3- бензилоксипропан
Смесь продукта из предыдущей стадии с ацетатом калия (в большом избытке) и ДМФ кипятили с обратным холодильником 5 ч. Образовавшуюся коричневую смесь охлаждали до комнатной температуры и фильтровали через слой целита. Фильтровальный слой промывали метанолом. Фильтрат выпаривали и остаток ДМФ удаляли в вакууме. Сырой продукт очищали высокоскоростной хроматографией на силикагеле (CH2Cl2: метанол = 10:1), получая в виде бледно-желтого твердого продукта (R) 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1- ацетокси-3-бензилоксипропан.
Г. (R)-2-(2-амино-1,6-дигидро-6- оксопурин-9-ил)-метокси-1-бензилоксипропан-3-ол
Смесь (R)-2-(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1-ацетокси-3- бензилоксипропана с 30%-ным аммиачным раствором в метаноле (1:2) перемешивали при комнатной температуре в течение 18 ч. Выпаривали растворитель и остаток растирали в небольшом количестве метанола. Собирали бледно-желтый твердый продукт, получая (S)-2-(2-амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-1-бензилоксипропан-3-ол. С целью получить вторую порцию продукта маточный раствор концентрировали и остаток перекристаллизовывали из горячего метанола.
Пример 2. Получение 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил) -метокси-1 - бензилоксипропан -3-ола
А. 54,2 г (114 ммоля) N2-ацетил-2-(2-амино- 1,6-дигидро-6-оксопурин-9-ил)-метокси-1,3-бис-(бензилокси)-пропана растворяли в 815 мл кипящего с обратным холодильником этанола и в атмосфере азота добавляли 610 мл циклогексена. В реакционную смесь добавляли шлам 16 г гидроксида палладия в 50 мл этанола и смесь кипятили с обратным холодильником в атмосфере азота 1,5 ч. Горячую смесь фильтровали через целит и фильтрат концентрировали в роторном испарителе. Конечную сырую реакционную смесь хроматографировали на силикагеле. В результате элюирования смесью 8 % метанола и 92% дихлорметана и затем смесью 10% метанола с 90% дихлорметана получали 18,6 г (16%) N2-ацетил-2-(2-амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-1,3-бис-(бензилокси)-пропана (исходный материал) и 17,6 г (40%) N2-ацетил-2-(2-амино-1,6-дигидро-6- оксопурин-9-ил)-метокси -1 -бензилоксипропан-3-ола.
Б. 21,9г (56,5 ммоля) N2-Ацетил-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1- бензилоксипропан-3-ола растворяли в 200 мл метанола и добавляли 101 мл гидроксида аммония. Смесь перемешивали в течение ночи при комнатной температуре. В белый шлам добавляли 400 мл диэтилового эфира и смесь фильтровали. Осадок последовательно промывали 100 мл диэтилового эфира, 100 мл воды и 100 мл диэтилового эфира и сушили в глубоком вакууме в течение ночи, получая 15,9 г (46,13 ммоля, 82%) 2-(2-амино- 1,6-дигидро-6-оксопурин-9-ил)-метокси-1-бензилоксипропан-3-ола. В результате выпаривания фильтрата и суспензии полученного осадка в 200 мл диэтилового эфира с последующими фильтрованием и сушкой в глубоком вакууме дополнительно получали 2,3 г (6,7 ммоля, 12%) продукта.
Данные элементарного анализа для C16H19N50O4 (345,36): Рассчитано: C 55,65; H 5,55; N 20,28; найдено: C 55,25; H 5,60; N 20,12.
Пример 3. Получение 2-(2-Амино-1,6-дигидро- 6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L-валината
А. 2-[(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси] -З-бензилокси-1-пропанил-N-(бензилоксикарбонил)-L-валинат
43,66 г (0,174 моля, 3 экв.) N-бензилоксикарбонил-L-валина суспендировали в 72 мл дихлорметана и добавляли 14,34 г (69,5 ммол., 1,2 экв.) 1,3- дициклогексилкарбодиимида. Смесь перемешивали в токе азота в течение 48 ч. Эту смесь фильтровали через стеклянную фритту и белый твердый остаток промывали 75 мл дихлорметана. Объединенный фильтрат перемешивали в токе азота и добавляли суспензию 20 г (57,91 ммоля, 1 экв.) 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- бензилоксипропан-1-ола в 90 мл диметилформамида, а затем 1,77 г (14,4 ммоля, 0,25 экв.) 4- диметиламинопиридина. Смесь перемешивали в токе азота 18 ч, выливали в 1200 мл воды и экстрагировали смесью 350 мл этилацетата с 350 мл толуола. Отделяли водный слой, а органический слой промывали 600 мл полунасыщенного раствора бикарбоната натрия, а затем 200 мл воды. Органический слой сушили над сульфатом магния и концентрировали под пониженным давлением. Остаток осаждали из смеси этилацетата с циклогексаном, получая в виде аморфного твердого вещества 2-[(2-амино- 1,6-дигидро-6-оксопурин-9-ил) метокси]-3-бензилокси-1- пропанил-N-(бензилоксикарбонил)-L-валинат.
Б. Гидрохлорид 2-(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L- валината
224,8 г (0,39 моля) 2-[(2-амино-1,6-дигидро-6-оксопурин-9- ил)-метокси] -3-бензилокси-1-пропанил-N-(бензилоксикарбонил)-L-валината растворяли в 1,2 л метанола и по каплям добавляли 32,4 мл (0,39 моля) концентрированной соляной кислоты. Над смесью создавали азотную атмосферу и добавляли 67,4 г палладия на угле. Смесь гидрогенизовали в сосуде высокого давления Парра под давлением водорода (40-100 фунтов/кв.дюйм, 2,8-7,0 кг/см2 в среднем 80 фунтов/кв. дюйм, 6,3 кг/см2) в течение 48 ч. Добавляли дополнительно 5 г палладия на угле и смесь гидрогенизовали под давлением 100 фунтов/кв.дюйм (7,0 кг/см2) 24 ч. Эту смесь фильтровали через слой целита и остаток промывали 1 л метанола. Фильтрат выпаривали под пониженным давлением досуха. Остаток растворяли в 150 мл воды и нагревали до 60oC. Осторожно по каплям при перемешивании добавляли 830 мл изопропанола, поддерживая температуру 60-70oC. Раствор медленно охлаждали до комнатной температуры в течение 16 ч. Образовавшийся кристаллический раствор нагревали до 30oC и добавляли дополнительно 220 мл изопропанола. Смесь медленно охлаждали до конечной температуры-11oC в течение 4 ч. Кристаллы выделяли фильтрованием и промывали 200 мл холодной смеси 2% воды с изопропанолом, получая 120,5 г (79%-ный выход) гидрохлорида 2-(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L- валината. Это соединение подвергается фазовому превращению при 142oC и при 175oC разлагается.
Пример 4. Получение кристаллического 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- гидрокси-1-пропанил-L-валината
150 г Гидрохлорида 2-(2-амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L-валината растворяли в 150 мл воды и нагревали до 50-60oC. Осторожно по каплям при перемешивании добавляли 830 мл изопропанола, тогда как температуру медленно повышали до 60-70oC. Раствор медленно, в течение 20 ч охлаждали до 25oC. Образовавшийся кристаллический раствор нагревали до 30oC и добавляли дополнительно 220 мл изопропанола. Смесь медленно, в течение 6 ч охлаждали до конечной температуры-11oC. Кристаллы отделяли фильтрованием и промывали 200 мл холодной смеси 2% воды с изопропанолом, получая 135 г (90%-ный выход) кристаллов гидрохлорида 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- гидрокси-1-пропанил-L-валината. При 142oC это соединение подвергается фазовому превращению и разлагается при температуре выше 175oC.
Аналогичным путем в кристаллической форме может быть получен ацетат 2- (2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил- L-валината.
Пример 5. Получение гидрохлорида (S)-2-(2-мино-1,6- дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L- валината.
А. (S)-2-(2-Амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- бензилокси-1-пропанил-N-(бензилоксикарбонил)-L-валинат
437 мг (1,74 ммоля, 3 экв.) N-Бензилоксикарбонил-L-валина суспендируют в 1 мл дихлорметана и добавляют 143 мг (0,7 ммоля, 1,2 экв.) 1,3- дициклогексилкарбодиимида. Смесь перемешивают в токе азота 48 ч. Эту смесь фильтруют через стеклянную фритту и белый твердый остаток промывают 1 мл дихлорметана. Объединенный фильтрат перемешивают в токе азота и добавляют суспензию 200 мг (0,58 ммоля, 1 экв.) (R)-2-(2- амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-3-бензилоксипропан-1-ола в 1,5 мл диметилформамида, а затем 18 мг (14,4 ммоля, 0,25 экв.) 4- диметилами-нопиридина. Смесь перемешивают в токе азота 18 ч, выливают в 12 мл воды и экстрагируют смесью 3,5 мл этилацетата с 3,5 мл толуола. Водный слой отделяют и органический слой промывают 6 мл полунасыщенного раствора бикарбоната натрия, а затем 2 мл воды. Органический слой сушат над сульфатом магния и концентрируют под пониженным давлением. Остаток осаждают из смеси этилацетата с циклогексаном, получая в виде твердого вещества (S)-2-(2-амино-1,6- дигидро-6-оксопурин-9-ил)-метокси-3-бензилокси-1-пропанил -N-(бензилоксикарбонил)-L-валинат.
Б. Гидрохлорид (S)-2-(2-амино- 1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L- валината
225 мг (3,9 моля) (S)-2-(2-Амино-1,6-дигидро-6-оксопурин-9- ил)-метокси-3-бензилокси-1-пропанил-N-(бензилоксикарбонил)-L- валината растворяют в 12 мл метанола и добавляют 0,3 мл (3,9 ммоль) концентрированной соляной кислоты. Над смесью создают азотную атмосферу и добавляют 674 мг палладия на угле. Эту смесь гидрогенизуют в сосуде высокого давления Парра под давлением водорода (40-100 фунтов/кв. дюйм, 2,8-7,0 кг/см2, в среднем 80 фунтов/кв. дюйм, 6,3 кг/см2 в течение 48 ч. Добавляют дополнительно 50 мг палладия на угле и эту смесь гидрогенизуют под давление 100 фунтов/кв. дюйм 24 ч. Смесь фильтруют через слой целита и остаток промывают 10 мл метанола. Фильтрат выпаривают под пониженным давлением досуха. Остаток растворяют в 1,5 мл воды и нагревают до 60oC. Осторожно при перемешивании добавляют 8 мл изопропанола, поддерживая температуру 60- 70oC. Раствор медленно охлаждают до комнатной температуры в течение 16 ч. Образовавшийся раствор нагревают до 30oC и добавляют дополнительно 2 мл изопропанола. Смесь медленно охлаждают до конечной температуры -11oC в течение 4 ч. Кристаллы выделяют фильтрованием и промывают 2 мл холодной смеси 2% воды с изопропанолом, получая гидрохлорид (S)-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- гидрокси-1-пропанил-L-валината.
Пример 6. Получение ацетата 2-(2-амино-1,6-дигидро-6- оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L-валината из бис- ацетата бис-L-валината 2-(2-амино-1,6-дигидро-6-оксопурин-9- ил)-метокси-1,3-пропандиила
100 мг бис-ацетатной соли бис-L- валината 2-[(2-амино-1,6-дигидро-6-оксо-9H-пурин-9-ил)-метокси] -1,3- пропандиила [лиофилизованный образец, содержавший 0,6-эквивалентный избыток уксусной кислоты, 0,164 ммоля (= в общем 0,426 ммоля уксусной кислоты)] растворяли в 0,4 мл деионизированной воды и частично нейтрализовывали добавлением 24 мл 0,015М раствора гидроксида аммония (=0,36 ммоля). Смесь выдерживали при комнатной температуре 67 ч. Образец вводили в виде двух равных порций в колонку для высокоэффективной жидкостной препаративной хроматографии с обращенной фазой (YMC-Pack, ODS-AM DM-33-5, 2 х 250 мм; YMC Inc. ). Разделения достигали с использованием растворительной системы из 10% метанола и 90% 0,1М раствора ацетата аммония, содержавшей в качестве буфера, обеспечивавшего величину pH 4, уксусную кислоту, подаваемой с расходом потока 9,5 мл/мин; детектор настраивали на длину волны 256 нм. Собирали моноэфирный продукт, двум стереомерам которого соответствовали оба пика. В глубоком вакууме до остаточного объема около 2 мл удаляли растворитель и остаток дважды лиофилизировали из воды, содержавшей 0,1% уксусной кислоты, для удаления буфера.
В виде эквимолярной смеси двух диастереомеров получали 45 мг (0,112 ммоля, 68%) ацетатной соли 2-[(2-амино-1,6-дигидро-6-оксо-9H-пурин-9-ил)- метокси]-3-гидрокси-1-пропанил-L-валината с нижеследующими характеристическими пиками на ЯМР-спектрограмме
(1H-ЯМР, 300 МГц, ДМСО-d6 раствор: δ ): 7,78 (1H, с., H C-8), 6,48 и 6,45 (2 ш.с., 2H, NH2), 5,44 (м.AB, J=ll Гц) и 5,43 (с.) (все 2H, CH2); 1,91 (с., 3H, CH3COO-), 0,83+0,82 (2д., J=7 Гц, 3H, CH3), 0,75+0,76 (2д., J=7 Гц, 3H, CH3).
Пример 7. Разделение (R,S)-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1 -пропанил-L-валината
Раствор 1,66 г (R,S)-2-(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L- валината в 9,60 мл смеси 90% 0,1М ацетата аммония, подкисленного до величины pH 4 уксусной кислотой, с 10% метанола пропускали через колонку для высокоэффективной жидкостной хроматографии YMC-Pack, ODS-AM [каталожный N DM-33-5, размеры: 250 х 20 мм (внутренний диаметр); с размерами частиц 5 мм, 120А] в виде 48 инъекций по 200 мкл, элюируя с расходом потока подвижной фазы 9,5 мл/ мин, которой служила смесь 90% 0,1М ацетата аммония, подкисленного до величины pH 4 соляной кислотой, с 10% метанола. Пики определяли с помощью монитора переменной длины волны Кнауэра, настроенного на длину волны 256 им, собирая фракции вручную. Собирали три комплекта фракций, соответствующих пику 1 (время удержания 24,4 мин), перекрывающей зоне между пиками и пику 2 (время удержания 27,8 мин). Фракции каждого из комплектов объединяли, выпаривали под пониженным давлением для удаления метанола, а затем лиофилизировали для удаления остаточных летучих компонентов. Остаток растворяли в воде, подкисляли раствор до величины pH 4 уксусной кислотой и вновь лиофилизировали, получая пик 1 (1,57 г) и пик 2 (0,91 г). Высокоэффективный жидкостной хроматографический анализ продуктов с использованием колонки YMCPack, ODS-AM [каталожный N RM-33-5, размеры: 250 х 4,6 мм (внутренний диаметр); с размерами частиц 5 мм, 120А] и элюированием с расходом потока подвижной фазы 0,5 мл/мин, которой служила смесь 90% 0,1М ацетата аммония, подкисленного до величины pH 4 соляной кислотой, с 10% метанола, показывал, что пику 1 (время удержания в препаративной колонке 24,4 мин) соответствовала смесь 70,8% продукта пика 1 (время удержания 21,1 мин), 26,4% продукта пика 2 (время удержания 24,6 мин) и 2,8% полностью гидролизованного продукта (время удержания 12,4 мин); а пику 2 (время удержания в препаративной колонке 27,8 мин) соответствовала смесь 68,5% продукта пика 2 (время удержания 22,0 мин), 27,5% продукта пика 1 (время удержания 20,2 мин) и 4% полностью гидролизованного продукта (время удержания 11,6 мин). Продукты каждого из пиков, а именно: 0,91 г пика 2 и 1,57 г пика 1, растворяли в 3,60 мл смеси 90% 0,1М ацетата аммония, подкисленного до величины pH 4 уксусной кислотой, с 10% метанола и вновь очищали с применением вышеописанной системы в виде 18 инъекций по 200 мл. Два комплекта фракций, которые соответствовали пикам 1 и 2, собирали, объединяли, частично выпаривали под пониженным давлением для удаления метанола и остаток лиофилизировали для удаления остаточных летучих компонентов. Остаток от каждого из комплектов фракций растворяли в воде, осторожно подкисляли до величины pH 4 уксусной кислотой и еще раз лиофилизировали. Из фракций, которые соответствовали пику 1, получали 0,70 г белого "пушистого" твердого материала, который при стоянии на воздухе проявлял гигроскопичность. Высокоэффективный жидкостной анализ (проведенный по вышеизложенному) показывал, что материал представлял собой смесь, которая содержала 94,4% продукта пика 1 (время удержания 21,1 мин), 4,6% продукта пика 2 (время удержания 26,7 мин) и 0,5% полностью гидролизованного продукта (время удержания 11,8 мин). 1H-ЯМР-спектральный анализ (DMSO-d6, величины δ брали относительно тетраметилсилана в качестве внутреннего стандарта) давал характеристические пики δ 5,43 (м. AB, 2H, JAB=11,1 Гц, д.A 5,44, д.B 5,43), 3,02 (д., 1H, J=5,2 Гц), 0,82 (д., 3H, J=6,8 Гц), 0,75 (д., 3H, J=6,8 Гц). Из фракций, которые соответствовали пику 2, получали 0,81 г белого "пушистого" твердого материала, который при стоянии на воздухе проявлял гигроскопичность. Высокоэффективный жидкостной хроматографический анализ (проведенный по вышеизложенному) показывал, что материал представлял собой смесь, которая включала в себя 91,0% продукта пика 2 (время удержания 29,8 мин), 8,4% продукта пика 1 (время удержания 28,4 мин) и 0,6% полностью гидролизованного продукта (время удержания 14,4 мин). 1H-ЯМР - спектральный анализ (DMSO-d6, величины δ брали относительно тетраметилсилана в качестве внутреннего стандарта) давал характеристические пики δ 5,43 (с., 2H), 2,99 (д., 1H, J=5,2 Гц), 0,83 (д., 3H, J=6,8 Гц), 0,76 (д., 3H, J=6,8 Гц).
Пример 8. А. Получение (R)-2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- бензилокси-1-пропанил-(N-бензилоксикарбонил)-L-валината
В раствор 327 мг (1,30 ммоля, 3 экв.) N-бензилоксикарбонил-L-валина в 25 мл дихлорметана в токе азота добавляли 134 мг (0,65 ммоля, 1,5 экв.) 1,3- дициклогексилкарбодиимида и реакционную смесь перемешивали при комнатной температуре 13,5 ч. Образовавшуюся смесь фильтровали для удаления нерастворимого материала и под пониженным давлением в роторном испарителе выпаривали растворитель. Полученный белый пеноподобный материал растворяли в 10 мл сухого ДМФ и непосредственно после этого добавляли в раствор 150 мг (0,43 ммоля, 1 экв.) (S)-2-(2- амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-бензилокси-1-пропан-1- ола также в 10 мл сухого ДМФ. В этот ДМФ-раствор добавляли 13 мг (0,11 ммоля, 0,25 экв.) 4,4-диметиламинопиридина и реакционную смесь оставляли перемешиваться при комнатной температуре 27 ч, после чего тонкослойный хроматографический анализ указывал на полный расход исходных материалов. Реакционную смесь выпаривали под пониженным давлением и сырой продукт очищали высокоскоростной хроматографией с использованием в качестве подвижной фазы 95% хлористого метилена с 5% метанола, получив в виде аморфного твердого продукта 158 мг (63%) соединения, указанного в заголовке.
Б. Получение гидрохлорида (R)-2- (2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил- L-валината
158 мг (0,27 ммоль) (R)-2-(2-амино-1,6-дигидро-6- оксопурин-9-ил)-метокси-3-бензилокси-1-пропанил-(N- бензилоксикарбонил)-L-валината растворяли в 15 мл метанола и добавляли 24 мл (0,27 ммоля) концентрированной соляной кислоты. Над этой смесью создавали азотную атмосферу и добавляли в нее 48 мг 10%- ного палладия на угле. Смесь гидрировали в течение 5 ч в шейкере Парра под избыточным давлением водорода 50 фунтов/кв.дюйм (3,5 кг/см2). Смесь фильтровали через слой целита и остаток промывали 25 мл метанола. В результате выпаривания объединенных фильтрата и промывных жидкостей под пониженным давлением получали 107 мг (95%) соединения, указанного в заголовке. Высокоэффективный жидкостной хроматографический анализ продукта с использованием колонки YMC-Pack, ODS-AM [каталожный N RM-33-5, размеры: 250 х 4,6 мм (внутренний диаметр); с размерами частиц 5 мм, 120A] и элюированием с расходом потока подвижной фазы 0,5 мл/мин, которой служила смесь 90% 0,1М ацетата аммония, подкисленного до величины pH 4 уксусной кислотой, с 10% метанола, показывал, что он представлял собой смесь диастереомеров (R) и (S) в соотношении 85:15.
Пример 9. Определение перорального поглощения (биологической доступности) на крысах (см. в конце описания).
В сравнительных целях для определения перорального поглощения (пероральной биологической доступности) соединения формулы I (L-моновалинового эфира ганцикловира), а также других ганцикловировых эфиров аминокислот, других ганцикловировых сложных и простых эфиров проводили нижеследующее испытание. Для измерения пероральной биологической доступности соединения содержание этого соединения в плазме самцов крыс вначале определяли после одинарной пероральной (по) дозы соединения.
Для измерения пероральной биологической доступности пролекарства у самцов крыс вначале концентрацию в плазме активнодействующего вещества (в данном случае ганцикловира) определяли после однократной пероральной дозы этого пролекарства. Затем концентрацию активнодействующего вещества, ганцикловира, в плазме у самцов крыс определяют после однократной внутривенной (вв) дозы такого соединения. Для ганцикловира однократная по- и вв-доза в каждом случае составляет 10 мг/кг; для сложноэфирного пролекарства (включая L-моновалиновый эфир ганцикловира) однократная пероральная и вв-доза в каждом случае равна дозе, которая эквимолярна 10 мг/кг ганцикловира. Из двух измерений после по- и вв-введения в организм пероральную биологическую доступность соединения рассчитывали делением всей площади участка под графиком зависимости концентрации от времени после введения в организм на всю площадь участка под графиком зависимости концентрации от времени после вв-введения в организм с соответствующей корректировкой на дозу согласно уравнению:
F(по)(%) = [ППГ(по)/ППГ(вв)] • [Доза(вв)/Доза(по)] • 100
Величины ППГ (общей площади участка под графиком) рассчитывали во всем интервале времени анализа 0-24 ч.
Лекарственная среда дозируемого вещества для пероральной и внутривенной дозы представляла собой нормальный солевой раствор, содержавший 2% уксусной кислоты. В обоих случаях концентрация соединения была эквивалентной 4,0 мг/мл ганцикловира, а доза - эквивалентной 10 мг/кг (2,5 мл/кг) ганцикловира. Животному весом 200 г давали по 0,5 мл раствора перорального лекарства путем принудительного кормления или его вводили инъекцией в хвостовую вену.
Крыс акклиматизировали в лабораторных условиях в течение трех дней и до начала испытаний им не давали корма в течение ночи и до истечения 4 ч после введения дозы. У 4 особей брали кровь в каждый из следующих моментов: 0 мин (перед введением дозы), 5 мин (только вв), 15 мин, 30 мин, 1 ч, 2 ч, 3 ч, 5 ч, 7 ч, 10 ч и 24 ч. Кровь немедленно центрифугировали для выделения плазмы и плазму замораживали до анализа при температуре-20oC.
Испытание с ганцикловиром в плазме
0,50-Миллилитровые аликвоты плазмы смешивали с 0,020 мл внутреннего стандарта (ацикловир, 15 мкг/мл в 10%-ном растворе метанола в воде) и 3,0 мл ацетонитрила. Смесь подвергали вихревому перемешиванию и образовавшийся осадок удаляли центрифугированием (4000 г, 10 мин). Верхний слой выпаривали досуха в токе азота и воссоздавали в 200 мкл подвижной фазы для высокоэффективной жидкостной хроматографии. 0,05-Миллилитровые аликвоты анализировали высокоэффективной жидкостной хроматографией с использованием колонки Кейстона размерами 250 х 4,6 мм, C 18 с продуктом Hypersil BDS. Подвижная фаза включала в себя 2% ацетонитрила в 30 мМ натрийфосфатном буфере, содержавшем 5 мМ гептансульфокислоты, с величиной pH 2,0; ее прокачивали с расходом потока 1,0 мл/мин. Ганцикловир и внутренний стандарт определяли и измеряли по УФ- поглощению при длине волны 254 нм.
Пример 10. Определение перорального поглощения (биологической доступности) у обезьян Cynomolgus.
Для определения перорального поглощения (пероральной биологической доступности) соединения формулы I на обезьянах Cynomolgus проводили нижеследующее испытание.
Животные, дозирование и сбор проб
Использовали самцов обезьян Cynomolgus весом от 5 до 7 кг. Животным давали корм для обезьян, фрукты и воду и соблюдали 12-часовой световой цикл. Из испытываемых соединений готовили композиции, концентрация которых была эквимолярной 10 мг/мл солевого раствора ганцикловира. Пероральную композицию в случае конечной дозы, эквимолярной дозе ганцикловира 10 мг/кг, вводили в организм принудительным кормлением из расчета 1,0 мл/кг. Вв-композицию ганцикловира готовили с использованием солевого раствора, содержавшего 0,2% HCI, концентрацией 20 мг/мл и вводили в организм из расчета 0,5 мл/кг.
Животным не давали корма, начиная с вечера перед дозированием соединения, и до истечения 4 ч после дозирования. У каждой обезьяны брали кровь в момент 0 (перед введением дозы), 5 мин (только вв), 15 мин, 30 мин, 1 ч, 2 ч, 3 ч, 5 ч, 7 ч, 10 ч и 24 ч после дозирования. Пробы крови собирали в гепаринизированных шприцах и плазму немедленно после этого выделяли центрифугированием и замораживали до анализа при температуре-20oC.
Испытание с ганцикловиром в плазме
0,5-Миллилитровые аликвоты плазмы смешивали с 0,020 мл внутреннего стандарта (ацикловир, 15 мкг/мл в 10%-ном растворе метанола в воде) и 3,0 мл ацетонитрила. Смесь подвергали вихревому перемешиванию и образовавшийся осадок удаляли центрифугированием (4000 г, 10 мин). Верхний слой выпаривали досуха в токе азота и воссоздавали в 200 мкл подвижной фазы для высокоэффективной жидкостной хроматографии. 0,05-Миллилитровые аликвоты анализировали высокоэффективной жидкостной хроматографией с использованием колонки Кейстона размерами 250 х 4,6 мм, C 18 с продуктом Hypersil BDS. Подвижная фаза включала в себя 2% ацетонитрила в 30 мМ натрийфосфатном буфере, содержавшем 5 мМ гептансульфокислоты, с величиной pH 2,0; ее прокачивали с расходом потока 1,0 мл/мин. Ганцикловир и внутренний стандарт определяли и измеряли по УФ- поглощению при длине волны 254 нм.
Биологическую доступность (F) рассчитывали в соответствии с уравнением, приведенным в примере 9.
Пероральная биологическая доступность пролекарства, 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3- гидрокси-1-пропанил-L-валината составляла 35,7%. Пероральная биологическая доступность пролекарства, бис-L-валината 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-1,3-пропандиила, составляла 23,5%. Биологическая доступность ганцикловира была равной 9,9%. При пероральном введении в организм тех же самых обезьян того же пролекарства и вв-введении ганцикловира средняя пероральная биологическая доступность пролекарства достигала 41,6%.
Пример 11. В нижеследующих примерах предлагаемые капсулы для L-валинового моноэфира ганцикловира в качестве эксципиентов включают в себя повидон, связующее вещество; кукурузный крахмал, агент, придающий рассыпчатость, и стеариновую кислоту, смазочная добавка и вещество, понижающее трение, которые помещают в состоящую из двух частей капсульную оболочку из твердого желатина. В процессе приготовления практически полностью удаляют воду, которая служит жидкостью для гранулирования (см. в конце описания).
Порошкообразной смесью наполняют оболочки твердых желатиновых капсул, состоящих из двух частей.
1Количество стеариновой кислоты можно варьировать от 0,1 до 5,0 вес.%.
2Количество воды для достижения приемлемого эффекта гранулирования можно варьировать с последующей сушкой.
3Общий полный вес (теоретический) не включает в себя остаточной влажности, которая будет присутствовать в готовом продукте.
Порошкообразной смесью наполняют оболочки твердых желатиновых капсул, состоящих из двух частей.
1Количество стеариновой кислоты можно варьировать от 0,1 до 5,0 вес.%.
2Количество воды для достижения приемлемого эффекта гранулирования можно варьировать с последующей сушкой.
3Общий полный вес (теоретический) не включает в себя остаточной влажности, которая будет присутствовать в готовом продукте.
Пример процедуры приготовления капсул с L-валиновым моноэфиром ганцикловира.
1. L-валиновый моноэфир ганцикловира в пригодном смесителе смешивают с частью кукурузного крахмала.
2. В воде с перемешиванием растворяют повидон.
3. Компонент п.(2) добавляют к компоненту п.(1), продолжая перемешивать до образования гранулята.
4. При необходимости мокрый гранулят измельчают.
5. Мокрый гранулят сушат в сушилке.
6. Через мельницу пропускают сухой гранулят, остаток кукурузного крахмала и стеариновую кислоту.
7. Массу п.(6) перемешивают в подходящем смесителе.
8. Соответствующее количество продукта по п.(7) инкапсулируют в твердые оболочки желатиновых капсул, состоящие из 2 частей.

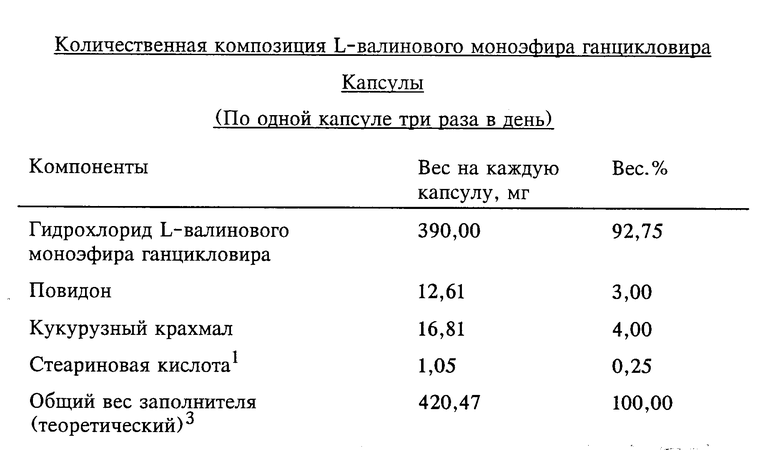
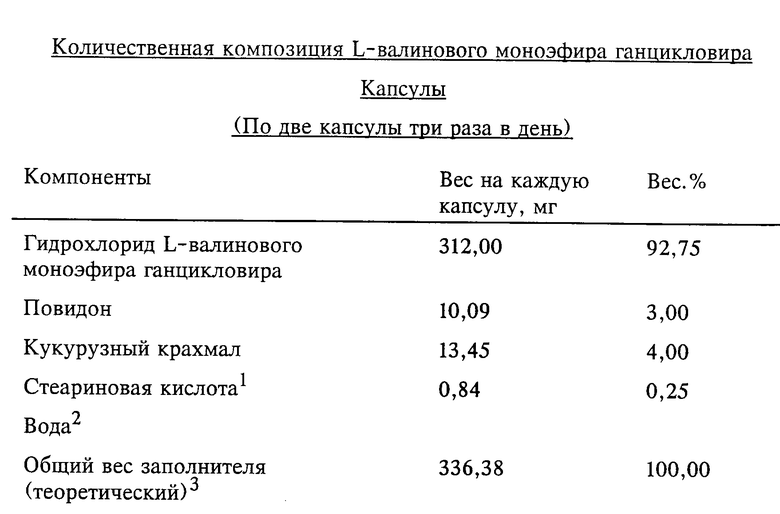
Описываются новые соединения -2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L-валинат или его фармацевтически приемлемая соль в форме его (R)- или (S)-диастереомеров или в форме смесей обоих диастремеров. Они являются ценными антивирусными агентами с улучшенной всасываемостью. Описывается также фармацевтическая композиция на базе соединений формулы (I), гидрокси- и/или аминозащищенный 2-(2-амино-1,6-дигидро-6-оксопурин-9-ил)-метокси-3-гидрокси-1-пропанил-L-валинат. 3 с. и 1 з.п.ф-лы.
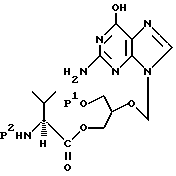
где Р1 - атом водорода или гидроксизащитная группа;
Р2 - аминозащитная группа.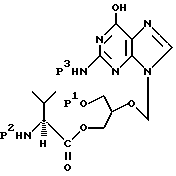
где Р1 - гидроксизащитная группа или атом водорода;
Р2 - аминозащитная группа;
Р3 - атом водорода или группа Р2,
в кислой среде или в условиях гидрогенолиза с последующим в случае необходимости разделением полученного соединения на (R)- и (S)-диастереомеры и/или превращением полученного соединения в его фармацевтически приемлемую соль.
| Способ получения производных гуанина или их кислотно-аддитивных фармацевтически приемлемых солей | 1988 |
|
SU1634138A3 |
| УСТРОЙСТВО для ЗАРЯДКИ ШПУЛЬ ЧЕЛНОКОВ НА МНОГОЗЕВНЫХ ТКАЦКИХ СТАНКАХ | 0 |
|
SU375329A1 |
| Способ посева озимых зерновых культур | 2019 |
|
RU2723093C1 |
| US 4897479 A, 30.06.90 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, т.2, с.380. | |||
