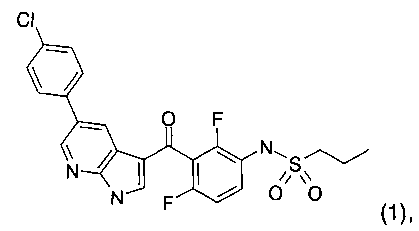
Настоящее изобретение относится к альтернативным путям синтеза для получения соединения пропан-1-сульфоновой кислоты {3-[5-(4-хлор-фенил)-1H-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифтор-фенил}-амида (формула 1).
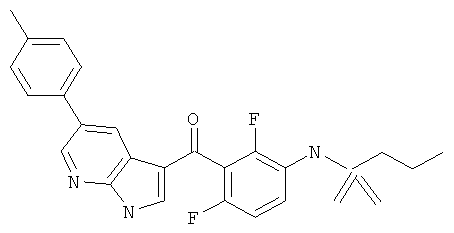
Синтез соединения формулы 1 был описан ранее в WO 2007002433 и WO 2007002325.
В настоящем изобретении раскрыты альтернативные реакции для получения соединения 1. В раскрытых здесь реакциях и способах используется мало реакционных стадий, и они приводят к хорошему общему выходу соединения 1, среди прочего, из-за малого числа стадий разделения и процедур обработки. Кроме того, раскрытые здесь способы можно проводить с относительно малыми количествами исходного вещества, и, следовательно, они могут быть более безопасными для применения в крупномасштабном производстве, интересными с точки зрения затрат и экологически безопасными.
Кроме того, в настоящем изобретении предложены новые способы синтеза для получения ключевых промежуточных соединений, используемых при получении соединения формулы 1. Одна группа указанных ключевых промежуточных соединений включает 5-замещенные-7-азаиндолы. Другая группа ключевых промежуточных соединений включает пинаконвинилборонаты.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном воплощении настоящего изобретения предложен способ получения соединения формулы 1,
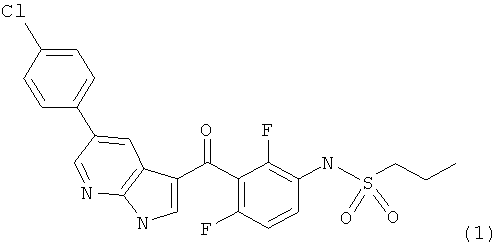 ,
,
включающий по меньшей мере одну реакцию Сузуки-Мияура с последующим ацилированием Фриделя-Крафтса.
В другом воплощении настоящего изобретения предложен способ получения соединения формулы 1, включающий две реакции Сузуки-Мияура с последующим ацилированием Фриделя-Крафтса.
В другом воплощении настоящего изобретения предложен способ получения соединения формулы 1, включающий одну реакцию Сузуки-Мияура с последующей реакцией Соногашира, кроме того, с последующим ацилированием Фриделя-Крафтса.
В предпочтительном воплощении согласно настоящему изобретению после первой реакции Сузуки-Мияура в каждом из способов, как описано выше, следует реакция галогенирования, в частности реакционная стадия для введения в соответствующее промежуточное соединение атома галогена, предпочтительно йодо или бромо.
Более конкретно, предложен способ получения соединения формулы 1, где
a) соединение формулы 2
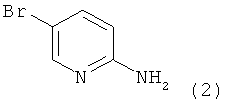
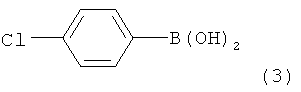
подвергают взаимодействию в присутствии палладиевого катализатора, основания и соединения формулы 3 (1-я реакция Сузуки-Мияура)
с получением соединения формулы 4
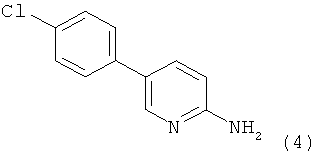 ;
;
б) указанное соединение формулы 4 далее подвергают взаимодействию в присутствии галогенирующего реактива с получением соединения формулы 5
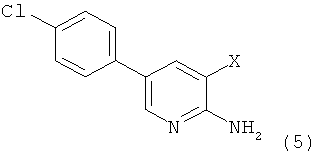 , где X представляет собой I (5а) или Br (5б); и
, где X представляет собой I (5а) или Br (5б); и
указанное соединение формулы 5 далее подвергают взаимодействию в присутствии одного из двух
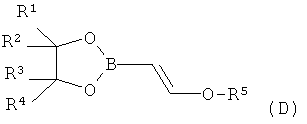
в-1) соединения формулы (D) (2-я реакция Мияура); или
в-2) соединения формулы 7 (реакция Соногашира)
 или
или 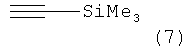 ;
;
с получением соединения формулы 8
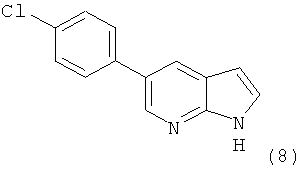 ; и
; и
г) указанное соединение формулы 8 далее подвергают взаимодействию в присутствии соединения формулы 9 и при условиях ацилирования Фриделя-Крафтса
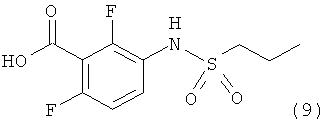
с получением соединения формулы 1, где
все R1, R2, R3 и R4 представляют собой метил, или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо; и R5 представляет собой -(C1-С6)алкил или бензил.
В другом предпочтительном воплощении предложен описанный выше способ, где все R1-R4 представляют собой метил; и R5 представляет собой этил.
В еще одном другом воплощении согласно настоящему изобретению предложен описанный выше способ а)-г) получения соединения формулы 1, где
стадии а)-б) являются такими же, как описано выше; и
в-1) указанное соединение формулы 5 согласно стадии б) далее подвергают взаимодействию в присутствии палладиевого катализатора и основания, которые оба могут быть такими же или другими, чем на стадии a), и соединения формулы D
с последующей обработкой кислотой с получением соединения формулы 8
; и
г) указанное соединение формулы 8 далее подвергают взаимодействию с соединением формулы 9
в присутствии (COCl)2 и AlCl3 с получением соединения формулы 1, и R1-R5 являются такими же, как определено выше.
В еще одном другом предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединения формулы 1 согласно стадиям а)-г), приведенным выше, где
а) соединение формулы 2
подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, Pd(OAc)2, Na2CO3 и соединения формулы 3
с получением соединения формулы 4
б) указанное соединение формулы 4 далее подвергают взаимодействию в присутствии N-йодсукцинимида (NIS) и CF3COOH или N-бромсукцинимида (NBS) с получением соединения формулы 5
, где X представляет собой -I (5a) или -Br (5б);
в-1) указанное соединение формулы 5 далее подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, LiOH и соединения формулы 6,
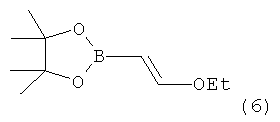
и затем обрабатывают HCl с получением соединения формулы 8
; и
г) указанное соединение формулы 8 далее подвергают взаимодействию с соединением формулы 9
в присутствии (COCl)2 и AlCl3 с получением соединения формулы 1.
В особенно предпочтительном воплощении согласно настоящему изобретению реакционную стадию б) перед реакционной стадией в-1), как упомянуто выше, проводят в присутствии NBS с получением соединения формулы 5, где X представляет собой бромо (5б).
В еще одном другом воплощении согласно настоящему изобретению предложен способ получения соединения формулы 1, где реакционные стадии а) и б) являются такими же, как описано здесь выше; и
в-2) соединение формулы 5 с реакционной стадии б) далее подвергают
взаимодействию в присутствии палладиевого катализатора и основания, которые
оба могут быть такими же или другими, чем на стадии a), Cul, тетраметилгуанидина (TMG) и соединения формулы 7
с последующей реакцией с сильным основанием с получением соединения формулы 8
; и
г) указанное соединение формулы 8 далее подвергают взаимодействию с соединением формулы 9
в присутствии (СОСд)2 и АдСд3 с получением соединения формулы 1.
В еще одном предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединения формулы 1 согласно стадиям а)-г), приведенным выше, где
ф) соединение формулы 2
подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, Pd(OAc)2, Na2Cl3 и соединения формулы 3
с получением соединения формулы 4
б) указанное соединение формулы 4 далее подвергают взаимодействию в присутствии N-йодсукцинимида (NIS) и CF3COOH или N-бромсукцинимида (NBS) с получением соединения формулы 5
, где X представляет собой -I (5a) или -Br (5б);
в-2) указанное соединение формулы 5 далее подвергают взаимодействию в присутствии Pd(PPh3)2Cl2, Cul, тетраметилгуанидина (TMG) и соединения формулы 7,
с последующей реакцией с KOtBu в NMP, с получением соединения формулы 8
; и
г) указанное соединение формулы 8 далее подвергают взаимодействию с соединением формулы 9
,
в присутствии (COCl)2 и AlCl3 с получением соединения формулы 1.
В еще одном другом предпочтительном воплощении предложен способ, как описано выше, где реакционная стадия б) перед реакционной стадией в-2) проводится в присутствии NBS с получением соединения формулы 5, где X представляет собой бромо (5б).
Основной путь синтеза
Участники реакции и условия вышеупомянутых реакций Сузуки-Мияура, Соногашира и Фриделя-Крафтса обычно известны специалисту в области синтетической органической химии и, среди прочего, описаны или на них дается ссылка в обычных руководствах по органической химии. Если прямо не указано иное, вышеописанные предпочтительные условия реакции для получения соединения формулы 1 можно обобщить, но не ограничить, согласно следующей общей схеме реакции 1. Ключевую реакционную стадию согласно настоящему изобретению, а именно реакции от соединения 5 до 8, можно осуществлять либо через реакцию с соединением D, предпочтительно с соединением 6 (путь Сузуки-Мияура), либо через реакцию с соединением 7 (путь Соногашира):
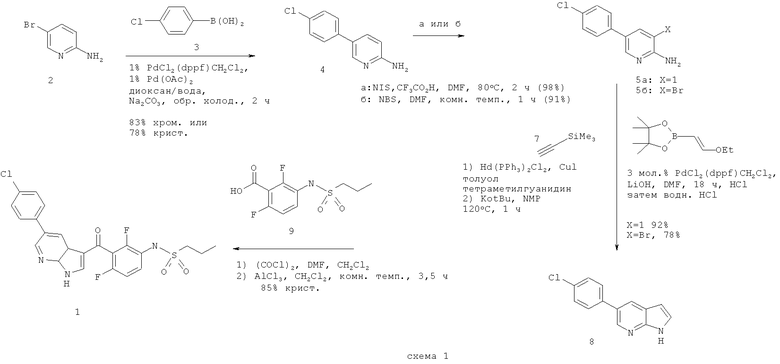
Согласно настоящему изобретению участником реакции первой реакции Сузуки-Мияура является 4-хлорфенилборйновая кислота (3). Участником реакции второй реакции Сузуки-Мияура является винилэфирборонат формулы (D), такой как катехолвинилборонат или пинаконвинилборонат (сложный пинаконовый эфир 1-этоксиэтен-2-бориновой кислоты, б), причем пинаконвинилборонат является особенно предпочтительным. Если прямо не указано иное, соединения (D) или (6) присутствуют в виде смесей их E/Z изомеров. Реакция проводится в присутствии палладиевых (Pd) катализаторов, более предпочтительно PdCl2(dppf)CH2Cl2, Pd(OAc)2 и тому подобных. Кроме того, обе реакции Сузуки-Мияура идут при основных условиях (значения pH выше 7) в органических растворителях или смесях органических растворителей с водой. Предпочтительными основаниями согласно настоящему изобретению являются органические основания или основания щелочных металлов, более конкретно N(CH2CH3)3, Na2CO3, LiOH и тому подобное. Предпочтительными органическими растворителями являются толуол, диметилформамид (DMF) или смеси диоксана и воды.
После второй реакции Сузуки-Мияура соединение формулы (8) получают реакцией циклизации в присутствии кислоты. Подходящие кислоты хорошо известны специалисту в данной области и охватывают органические и неорганические или минеральные кислоты. Предпочтительными кислотами согласно настоящему изобретению являются HCl, H2SO4, HNO3, CF3COOH и тому подобные; причем HCL является особенно предпочтительной.
Согласно настоящему изобретению предпочтительным участником реакции в реакции Соногашира является этинилтриметилсилан (7). Реакция предпочтительно проводится в присутствии Pd(PPh3)2Cl2 и Cul в толуоле. Сильные основания в 1М-метил-2-пирролидоне (NMP), используемые затем в реакции для получения соединения формулы (8), представляют собой основания с большей силой, чем основания, используемые в реакциях Сузуки-Мияура вместе с палладиевыми катализаторами, как описано ранее. Такие сильные основания предпочтительно представляют собой алкоголяты щелочных металлов и тому подобное. Особенно предпочтительным согласно настоящему изобретению является KOtBu, используемый в N-метил-2-пирролидоне (NMP).
Реакции Сузуки-Мияура и Соногашира предпочтительно проводятся в интервале температуры от 70 до 120°C или с обратным холодильником, и используется растворитель или смесь растворителей.
Конечная реакция Фриделя-Крафтса, также именуемая ацилированием Фриделя-Крафтса, предпочтительно идет в присутствии (COCl)2 и AlCl3 в DMF и CH2Cl2 при комнатной температуре (комн. темп.).
Термин «комнатная температура» (комн. темп.) в том виде, как он здесь используется, означает температуру окружающей среды места, где проводится реакция, без какого-либо дополнительного нагревания или охлаждения. Согласно настоящему изобретению комнатная температура предпочтительно составляет от 18 до 26°C, более предпочтительно - от 20 до 24°C.
Термин -(C1-С6)алкил в том виде, как он здесь используется, означает линейный или разветвленный насыщенный углеводород, содержащий от одного до шести атомов углерода, предпочтительно от 2 до 4 атомов углерода. Наиболее предпочтительная -(C1-С6)алкильная группа - согласно настоящему изобретению представляет собой этил.
Термин «реакция галогенирования» в том виде, в котором она упоминается выше, представляет собой реакцию продукта первой реакции Сузуки-Мияура с «галогенирующим реактивом», выбранным либо из N-йодсукцинимида (NIS) для введения одного атома йода или других (например, I2); либо из N-бромсукцинимида (NBS) для введения одного атома брома в указанный продукт первой реакции Сузуки-Мияура. Реакция с NIS предпочтительно проводится в присутствии трифторуксусной кислоты (TFA) и DMF. Реакция с NBS предпочтительно проводится в DMF.
Промежуточные соединения
I) 5-замещенные-7-азаиндолы
Одной из ключевых характеристик в пути синтеза к соединению формулы 1 согласно настоящему изобретению является новый и улучшенный способ получения соединения формулы 8. Из-за данных улучшений, вышеописанные пути синтеза впервые дают способ получения соединения формулы 1, который быстрее, дешевле и безопаснее, чем способы, описанные ранее.
На основе этого, другим воплощением настоящего изобретения является предложение способа получения соединения формулы A,
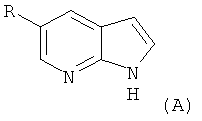 ,
,
причем указанный способ характеризуется тем, что
а) соединение формулы B
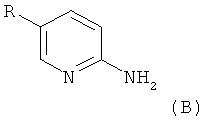
подвергают взаимодействию в присутствии галогенирующего реактива, возможно с последующим введением групп, защищающих аминогруппы, с получением соединения формулы C
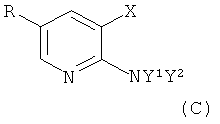 ;
;
б) указанное соединение формулы C далее подвергают взаимодействию в присутствии палладиевого катализатора, основания и соединения формулы D или 7
или
с получением соединения формулы E или F соответственно
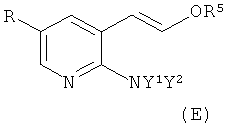 , или
, или 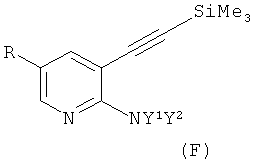
в-1) соединение формулы E далее подвергают взаимодействию с кислотой; или
в-2) соединение формулы F далее подвергают взаимодействию с сильным основанием с получением соединения формулы A;
где R представляет собой фенил, который является незамещенным или один или несколько раз замещенным галогеном, или
-Br.
Все R1, R2, R3 и R4 представляют собой метил или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо;
R5 представляет собой -(C1-С6)алкил или бензил;
X представляет собой -Br или -I; и
Y1 и Y2 независимо выбраны из бензила, трифторацетила, ацетила и водорода.
Соединения формулы E и F, как определено выше, являются новыми и образуют другое воплощение настоящего изобретения.
Если прямо не указано иное, соединения формулы (D) и (E) присутствуют в виде смесей их E/Z изомеров.
Термин «галогенирующий реактив» в том виде, как он здесь используется, означает N-бромсукцинимид (NBS), N-йодсукцинимид (NIS) или перйодат натрия в комбинации с йодом (I2/NalO4). Для йодирования соединения формулы B особенно предпочтительным является применение NIS в присутствии трифторуксусной кислоты (TFA).
Термин -(C1-С6)алкил в том виде, как он здесь используется, означает линейный или разветвленный насыщенный углеводород, содержащий от одного до шести атомов углерода, предпочтительно от 2 до 4 атомов углерода. Наиболее предпочтительная -(C1-С6)алкильная группа согласно настоящему изобретению представляет собой этил.
Термин «группы, защищающие аминогруппы» в том виде, как он здесь используется, означает любую защитную группу, известную специалисту в области органической химии, для защиты аминогруппы от реаций. Предпочтительными группами, защищающими аминогруппы, согласно настоящему изобретению являются бензил, трифторацетил и ацетил.
Предпочтительные палладиевые катализаторы и основания в том виде, в котором они используются в способе для получения соединений формулы A, являются такими же, как описано выше в связи с реакциями согласно схеме 1. В частности, реакционная стадия б), которая приводит к соединению формулы E, как описано выше, предпочтительно проводится в присутствии PdCl2(dppf)CH2Cl2 в качестве палладиевого катализатора и LiOH в качестве основания. Реакционная стадия б), которая приводит к соединению формулы F, как описано выше, предпочтительно проводится в присутствии Pd(PPh3)2Cl2, Cul и тетраметилгуанидина (TMG).
Термин «сильные основания» означает основания с большей силой, чем основания, используемые в реакциях Сузуки-Мияура вместе с палладиевыми катализаторами, как описано выше. Предпочтительно термин «сильные основания» означает алкоголяты щелочных металлов и тому подобное. Особенно предпочтительным сильным основанием согласно настоящему изобретению является KOtBu, который предпочтительно используется в N-метил-2-пирролидоне (NMP).
Термин «кислота» в том виде, как он здесь используется, означает органические и неорганические или минеральные кислоты. Предпочтительными кислотами согласно настоящему изобретению являются HCl, H2SO4, HNO3, CF3COOH и тому подобное; причем HCl является особенно предпочтительной.
В предпочтительном воплощении настоящего изобретения предложен способ получения соединения формулы A, как описано выше,
.
причем указанный способ характеризуется тем, что
а) соединение формулы B
подвергают взаимодействию в присутствии N-бромсукцинимида (NBS) или N-йодсукцинимида (NIS) или перйодата натрия в комбинации с йодом (I2/NalO4), возможно с последующим введением групп, защищающих аминогруппы, с получением соединения формулы C
.
б) указанное соединение формулы C далее подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, LiOH и соедиения формулы D
,
с получением соединения формулы E
и
в-1) соединение формулы E далее обрабатывают HCl с получением соединения формулы A;
где R представляет собой фенил, который является незамещенным или один или несколько раз замещенным галогеном, или
-Br;
все R1, R2, R3 и R4 представляют собой метил или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо; R5 представляет собой -(C1-С6)алкил или бензил; X представляет собой -Br или -I; и
Y1 и Y2 независимо выбраны из бензила, трифторацетила, ацетила и водорода.
В предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединения формулы A через соединение формулы E и реакционную стадию в-1), как описано выше,
где R представляет собой -Br, и
все остальные заместители являются такими же, как определено выше.
В другом предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединения формулы A через соединение формулы E и реакционную стадию в-1), как описано выше,
где R представляет собой -Br;
все R1-R4 представляют собой метил;
R5 представляет собой этил;
X представляет собой -I; и
оба Y1 и Y2 представляют собой водород.
В другом предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединения формулы A через соединение формулы E и реакционную стадию в-1), как описано выше,
где R представляет собой 4-C1-фенил, и
все остальные заместители являются такими же, как определено выше.
В другом предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединения формулы А, через соединение формулы Е и реакционную стадию в-1), как описано выше, где
R представляет собой 4-С1-фенил;
все R1-R4 представляют собой метил;
R5 представляет собой этил;
X представляет собой -Br; и
оба Y1 и Y2 представляют собой водород.
В другом воплощении настоящего изобретения предложен способ получения соединения формулы A через соединение формулы F и реакционную стадию в-2), как описано выше,
,
причем указанный способ характеризуется тем, что
а) соединение формулы B
подвергают взаимодействию в присутствии N-бромсукцинимида (NBS) или N-йодсукцинимида (NIS), или перйодата натрия в комбинации с йодом (I2/NalO4), возможно с последующим введением групп, защищающих аминогруппы, с получением соединения формулы C
б) указанное соединение формулы C далее подвергают взаимодействию в присутствии Pd(PPh3)2Cl2, Cul, тетраметилгуанидина и соединения формулы 7
;
с получением соединения формулы F
, и
в-2) указанное соединение формулы F далее подвергают взаимодействию в присутствии KOtBu с получением соединения формулы A; где
R представляет собой фенил, который является незамещенным или один или несколько раз замещенным галогеном, или
-Br;
X представляет собой -Br или -I; и
Y1 и Y2 независимо выбраны из бензила, трифторацетила, ацетила и водорода.
В предпочтительном воплощении согласно настоящему изобретению предложен способ получения соединений формулы (А) через соединения формулы (F), как описано выше, где
реакционная стадия а) проводится в присутствии N-йодсукцинимида (NIS) и трифторуксусной кислоты;
реакционная стадия б) проводится в присутствии Pd(PPh3)2Cl2, Cul, тетраметилгуанидина и соединения формулы 7 в толуоле;
реакционная стадия в-2) проводится в присутствии KOtBu в N-метил-2-пирролидоне; и
R представляет собой -Br;
X представляет собой -I; и
оба Y1 и Y2 представляют собой водород.
II) Пинаконвинилборонаты
Согласно настоящему изобретению описанный выше улучшенный синтез соединений формулы A и затем также соединения формулы 1 основан, в частности, на применении винилборонатов в соответствующих реакциях Сузуки-Мияура, которые приводят к соединениям формулы A, в частности 5-Br-7-азаиндолу и 5-(4-С1-фенил)-7-азаиндолу. Применение винилборонатов и их получение обычно известно в данной области.
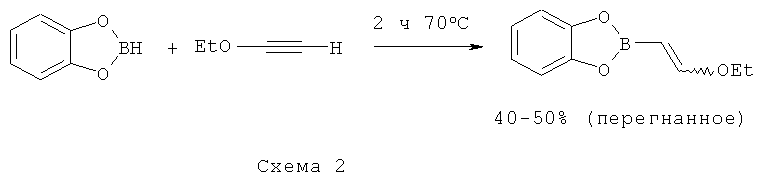
Катехолвинилэфирборонат (соединение формулы D, где R1-R4 совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо) можно получить согласно методике, описанной в Satoh, М.; Miyaura, N,; Suzuki, А.; Synthesis 1987, 373 и согласно следующей схеме реакции 2:
Пинаконвинилэфирборонат (соединение формулы D, где все R1-R4 представляют собой метил) можно получить согласно схеме реакции 3, приведенной ниже.
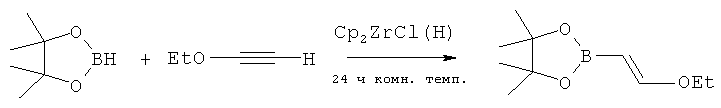
Однако в обоих путях синтеза используется чрезвычайно легковоспламеняющийся этоксиацетилен, что приводит к огромным опасениям относительно безопасности при использовании в промышленном масштабе.
Винилэфирборонаты также были получены с использованием дегидрирующего борилирования алкенов. Данная реакция состоит из каталитического гидроборирования алкена моногидридбораном с последующим отщеплением водорода. В данном типе реакции часто используются родиевые, титановые и рутениевые катализаторы. Главной проблемой с данными катализаторами является то, что многие из них также являются эффективными катализаторами гидрирования и, следовательно, при условиях реакции могут происходить конкурирующие гидрирование и гидроборирование.
Сохраняется потребность в нахождении катализаторов, способных к проведению дегидрирующего борилирования без конкурирующего гидроборирования или гидрирования винилбороната, и в преодолении вышеописанных проблем безопасности.
Теперь неожиданно обнаружили, что с определенными родиевыми, рутениевыми или палладиевыми катализаторами из списка обычно используемых катализаторов образовался практически исключительно желательный пинаконвинилэфирборонат. Можно было детектировать лишь небольшие количества (примерно 3%) продукта гидроборирования.
Следовательно, в другом воплощении настоящего изобретения предложен способ получения соединений формулы D
,
где соединение формулы G
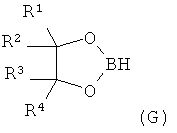
реагирует с соединением формулы H
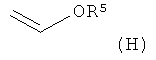
в присутствии палладиевого, родиевого или рутениевого катализатора с получением соединения формулы D,
где все R1, R2, R3 и R4 представляют собой метил или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо; и
R5 представляет собой -(C1-С6)алкил или бензил.
В предпочтительном воплощении предложен вышеописанный способ получения соединений формулы D,
где все R1, R2, R3 и R4 представляют собой метил; и R5 представляет собой этил.
В другом предпочтительном воплощении вышеописанный способ получения соединений формулы D проводится в присутствии Pd(OAc)2, Pd2(dba)3, Pd(NO3)2, Pd/C (5%), PdCl2, Rh(OAc)2 или RuCl3.
В другом предпочтительном воплощении вышеописанный способ получения соединений формулы D проводится в присутствии от 0,05 до 0,5 мол. % Pd(OAc)2 при комнатной температуре.
Термин «комнатная температура» (комн. темп.) в том виде, как он здесь используется, означает температуру среды в месте, где проводится реакция, без какого-либо дополнительного нагревания или охлаждения.
Согласно настоящему изобретению комнатная температура предпочтительно составляет от 18 до 26°C, более предпочтительно от 20 до 24°C.
Изобретение теперь проиллюстрировано следующими сопровождающими, неограничивающими примерами.
ПРИМЕРЫ
Пример 1
2-амино-5-(4-хлорфенил)-пиридин (4, первая реакция Сузуки-Мияура)
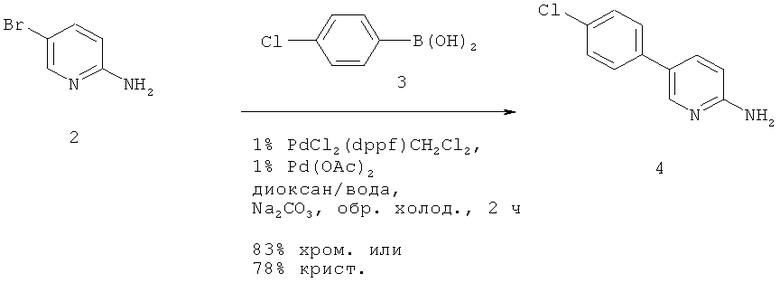
2-Амино-5-бромпиридин (2) подвергают взаимодействию с 4-хлорфенилборйновой кислотой (3) в диоксане/воде в присутствии 2,2 эквивалентов Na2CO3 и 1 мол.% Pd(OAc)2 плюс 1 мол.% PdCl2(dppf)CH2Cl2 при 90°С в течение 1,5 ч. После охлаждения до комнатной температуры продукт (4) осаждался в виде HCL соли путем добвления HCl (25%, 6 экв.) с последующим удалением диоксана под вакуумом. Соль фильтровали, расщепляли в диэтиловом эфире, фильтровали и затем превращали до свободного амина путем обработки водным NaOH. После фильтрования продукт выделяли с 78%-ным выходом. В качестве альтернативы продукт был выделен хроматографией с 83%-ным выходом. МС (масс-спектрометрия) (Turbo Spray): 207 (52%), 205 (М+Н\ 100%), 170 (9%).
Пример 2
Галогенирование 2-амино-5-(4-хлорфенил)-пиридина (4)
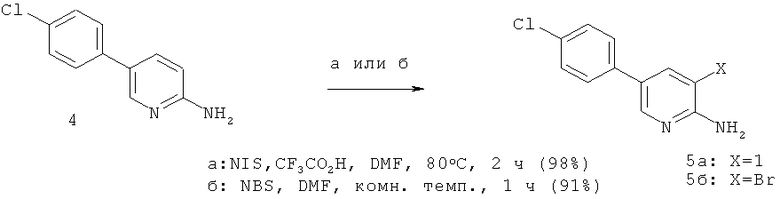
2-Амино-5-(4-хлорфенил)-пиридин (4) превращали до аминопиридинйодида (5а) и аминопиридинбромида (56). Йодирование (4) с использованием йода и AgSO4 давало только 75%-ное превращение через 3 суток при комнатной температуре. Лучшие результаты были получены с NIS/TFA. Пиридинбромид мог быть получен с использованием NBS.
а) Йодирование (4)
К раствору (4) и трифторуксусной кислоты (1,2 экв.) в DMF добавляли N-йодсукцинимид (NIS, 1,1 экв.) в DMF. После перемешивания в течение 2,5 ч при 80°C реакция была завершена. После обработки водой продукт 5а выделяли с 98%-ным выходом. МС (Turbo Spray): 331 (100%), 205 (42%), 122 (19%).
(б) Бромирование (4)
К раствору (4) в DMF добавляли N-бромсукцинимид, и образующийся реакционный раствор перемешивали при комнатной температуре в течение 1 ч. После добавления раствора в воду продукт осаждался и затем был отфильтрован. Минорные примеси удаляли путем расщепления продукта в гексанах. Аминопиридинбромид (5б) выделяли с выходом 91%. МС (GC-Split): 284 (100%), 282 (77%, М), 168 (34%), 151 (14%), 140 (18%), 113 (10%).
Пример 3
5-(4-С1-фенил)азаиндол (8, через вторую реакцию Сузуки-Мияура)
I. Связывание пинаконвинилбороната
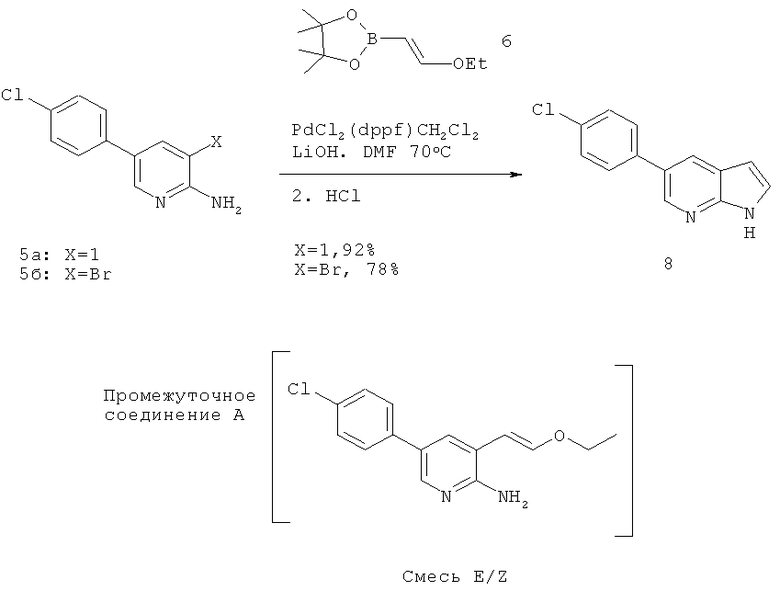
1-a) Циклизация йодида (5a):
К смеси йодида (5a) и пинаконвинилбороната (6, 1,3 экв.) в DMF добавляли LiOH (3 экв.) с последующим добавлением PdCL2(dppf).CH2CL2 (3 мол. %) в инертной атмосфере (Аг). Реакционную смесь нагревали до 70°C и перемешивали в течение 18 ч. Анализ HPLC (высокоэффективная жидкостная хроматография) показал полное превращение до винилэфира «Промежуточное соединение-А» (МС: (Turbo Spray) 277 (33%), 275 (М+Н+, 100%), 231 (22%), 229 (84%), 205 (11%)).
После охлаждения до 50°C, добавляли 25% HCL (15 экв.). Смесь выдерживали при данной температуре в течение 1 ч. После обработки выделяли азаиндол 8 в виде кристаллического твердого вещества с 92%-ным выходом. МС (Turbo Spray): 231 (26%), 229 (М+Н+, 100%).
1-б) Циклизация бромида:
К смеси бромида (56)и пинаконвинилбороната (6, 1,2 экв.) в DMF добавляли LiOH (3 экв.) с последующим добавлением PdCl2(dppf).CH2Cl2 (3 мол. %). Реакционную смесь нагревали до 70°C и перемешивали в течение 18 ч. Анализ ВЭЖХ показал полное превращение до винилэфирного промежуточного соединения. После охлаждения реакционной смеси до 50°С добавляли HCL (25%), и смесь перемешивали при данной температуре в течение 1 ч. В этот момент времени реакция была завершена. После обработки выделяли соединение 8 в виде кристаллического твердого вещества с 78%-ным выходом. МС (Turbo Spray): 231 (35%), 229 (М+Н+, 100%).
II. Связывание катехолвинилбороната
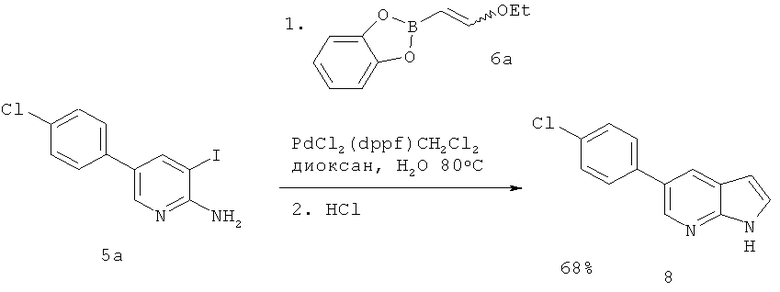
К смеси йодида (5a) и катехолвинилбороната (6a, 1,1 экв.) в диоксане/воде (80:20) добавляли LiOH (3 экв.) с последующим добавлением PdCl2(dppf).CH2Cl2 (3 мол. %). Реакционную смесь нагревали до 80°C и перемешивали в течение 24 ч. Анализ ВЭЖХ показал полное превращение до винилэфирного промежуточного соединения. После охлаждения до 50°C добавляли 25% HCL (15 экв.), и смесь выдерживали при данной температуре в течение 1 ч. После обработки выделяли азаиндол 8 в виде кристаллического твердого вещества с 68%-ным выходом. МС (Turbo Spray): 231 (26%), 229 (М+Н+, 100%).
Пример 4
Пропан-1-сульфоновой кислоты (3-[5-(4-хлорфенил)-1Н-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}-амид (1)
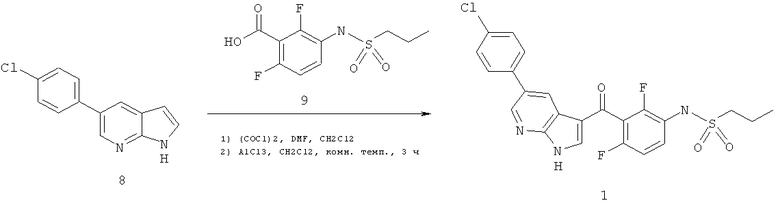
Суспензию сульфонамидной кислоты (9) (1,2 экв.) в CH2Cl2 обрабатывали при комнатной температуре кат.количеством DMF (0,11 экв.). За 30 мин добавляли раствор оксалилхлорида (1,30 экв.) в CH2Cl2, и реакционную смесь перемешивали в течение 2 ч, посредством чего образовался соответствующий хлорангидрид. Суспензию хлорида алюминия (AlCl3, 4 экв.) в CH2Cl2 обрабатывали при 0°C раствором Cl-фенилазаиндола (8) в CH2Cl2. К реакционной смеси при комнатной температуре затем добавляли свежеприготовленный (вышеописанный) хлорангидрид. Перемешивание при комнатной температуре в течение 3 ч, обработка водой и кристаллизация из THF/гептана давала соединение, указанное в заголовке (1), в виде беловатого порошка с 85%-ным выходом. МС (Turbo Spray): 509 (48%), 507 (M+NH4 +, 100%), 492 (40%), 490 (М+Н+, 84%).
Пример 5
5-(4-С1-фенил)азаиндол (8, через реакцию Соногашира)
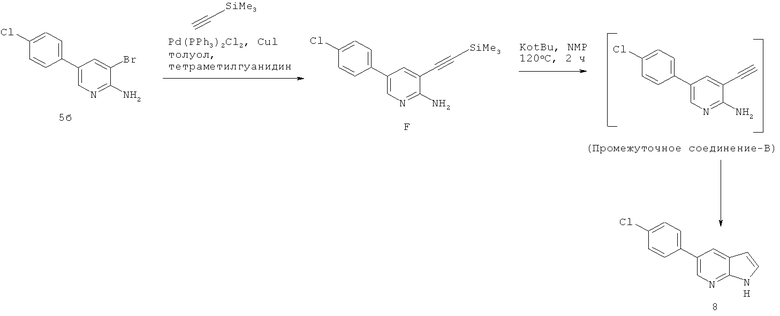
Раствор бромида 5б в толуоле дегазировали аргоном и затем обрабатывали PdCl2(PPh3)2Cl2 (0,17 экв.) и Cul (0,17 экв.) и вновь дегазировали аргоном. К суспензии добавляли тетраметилгуанидин (1,4 экв.) и этилтриметилсилан (2 экв.) и перемешивали при 100°C в течение 2 ч. После охлаждения до комнатной температуры, экстрактивного выделения и хроматографической очистки промежуточное соединение F собирали с 93%-ным выходом. МС (Turbo Spray): 303 (49%), 301 (М+Н+, 100%).
Раствор промежуточного соединения F в 1-метил-2-пирролидиноне (NMP) обрабатывали при комнатной температуре трет-бутилатом калия (2 экв.). Смесь нагревали до 120°C и перемешивали в течение 2 ч. После охлаждения до комнатной температуры, экстрактивного выделения и кристаллизации из ЕtOН/воды собирали соединение, указанное в заголовке (8), с 80%-ным выходом. МС (Turbo Spray): 231 (26%), 229 (М+Н+, 100%).
Промежуточное соединение X: MC (Turbo Spray): 231 (41%), 229 (М+Н+, 100%).
Пример 6
2-амино-5-бром-3-йодпиридин (11)
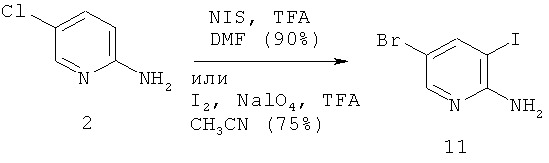
а) Методика с NIS:
К раствору 2-амино-5-бромпиридина (2) в DMF добавляли трифторуксусную кислоту (1,2 экв.). При комнатной температуре добавляли N-йодсукцинимид (1,1 экв.), и реакционную смесь нагревали при 50°С в течение 3 ч. ВЭЖХ показала полное превращение. После охлаждения до комнатной температуры продукт осаждали добавлением реакционной смеси в воду. После нейтрализации тиосульфатом натрия и 1 н. NaOH соединение, указанное в заголовке (11), собирали посредством фильтрования в виде коричневого твердого вещества с 90%-ным выходом.
б) Методика с I2/NaIO4:
К раствору 2-амино-5-бромпиридина (2) в ацетонитриле добавляли перйодат натрия (0,4 экв.) и йод (0,65 экв.). Добавляли трифторуксусную кислоту (0,65 экв.) в течение 15 мин, и реакционную смесь нагревали при 80°C в течение ночи. ВЭЖХ показала 96%-ное превращение в данный момент времени. Добавляли водный раствор сульфита натрия с последующим добавлением дополнительного количества воды для осаждения продукта, который отфильтровывали и промывали водой. Соединение, указанное в заголовке (11), выделяли в виде коричневого кристаллического твердого вещества с 75%-ным выходом. МС (Turbo Spray): 298 (М+Н+, 100%).
Пример 7
5-бром-7-азаиндол (12, через реакцию Сузуки-Мияура)
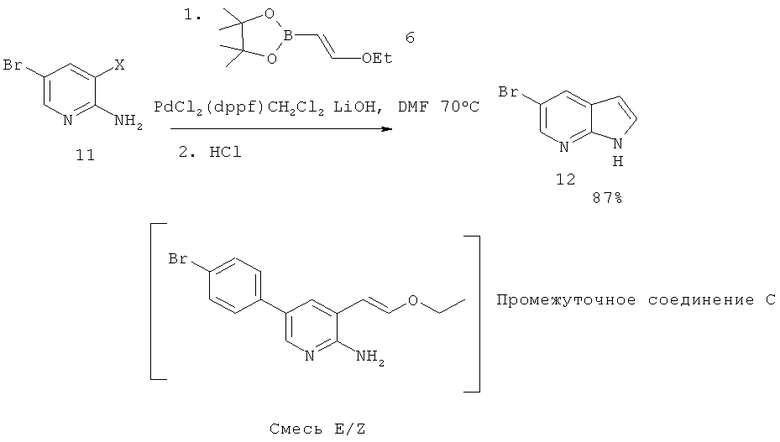
2-Амино-3-йод-5-бромпиридин (11) подвергали взаимодействию с пинаконвинилэфирборонатом (6, 1,3 экв.) в присутствии LiOH (3 экв.) и PdCl2(dppf).CH2Cl2 (1 мол. %). Через 18 ч при 70°C наблюдали полное превращение до винилового эфира аминопиридина (Промежуточное соединение-C). Виниловый эфир немедленно гидролизовали добавлением 25% HCl и перемешиванием реакционной смеси при 50°C в течение 1 ч. Выделение и кристаллизация из толуола/гептана давало соединение, указанное в заголовке (12), с 87%-ным выходом. МС (Turbo Spray): 199 (М+Н+, 100%). Промежуточное соединение: MS (Turbo Spray): 243 (М+Н+), 199.
Пример 8
5-бром-7-азаиндол (12, через реакцию Соногашира)
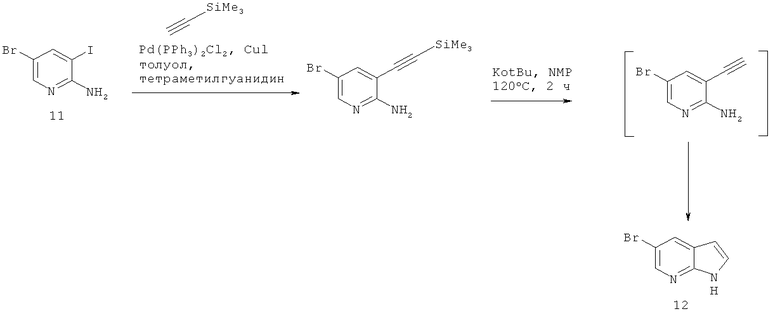
Раствор 2-амино-3-йод-5-бромпиридина (11) в толуоле дегазировали аргоном и затем обрабатывали PdCl2(PPh3)2Cl2 (0,17 экв.) и Cul (0,17 экв.) и вновь дегазировали аргоном. К суспензии добавляли тетраметилгуанидин (1,4 экв.) и этилтриметилсилан (2 экв.) и перемешивали при 80°С в течение 1 ч. После охлаждения до комнатной температуры, экстрактивного выделения промежуточное соединение собирали с 83%-ным выходом в виде коричневого твердого вещества.
Раствор промежуточного соединения в 1-метил-2-пирролидиноне (NMP) обрабатывали при комнатной температуре трет-бутилатом калия (2 экв.). Смесь нагревали до 80°С и перемешивали в течение 1 ч. После охлаждения до комнатной температуры, экстрактивного выделения и хроматографической очистки собирали соединение, указанное в заголовке (12), с неоптимизированным 53%-ным выходом.
Пример 9
Пинаконвинилборонат (6)
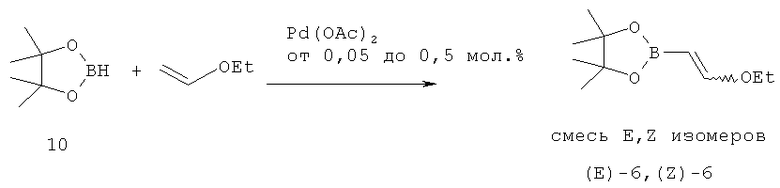
Пинаконборан (10) и 4,1 эквивалентов этил винилового эфира перемешивают при комнатной температуре в присутствии 0,05 мол. % Pd(OAc)2 до завершения реакции (20 ч). Затем концентрируют смесь, и перегоняют продукт под вакуумом с получением пинаконвинилбороната (6) в виде бесцветной жидкости с 83%-ным выходом. Продукт состоит из смеси E/Z изомеров (соотношение примерно 2:1). МС (Turbo Spray): 199 (М+Н+, 100%), 216 (M+NH4 +).
Изобретение относится к способу получения соединения формулы 1,
который включает по меньшей мере одну реакцию Сузуки-Мияура с последующим ацилированием Фриделя-Крафтса, где соединение формулы 2 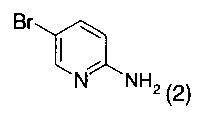 подвергают взаимодействию в присутствии палладиевого катализатора, основания и соединения формулы 3 (1-я реакция Сузуки-Мияура)
подвергают взаимодействию в присутствии палладиевого катализатора, основания и соединения формулы 3 (1-я реакция Сузуки-Мияура) 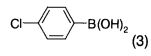 с получением соединения формулы 4
с получением соединения формулы 4 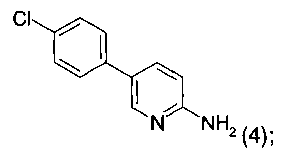 соединение формулы 4 далее подвергают взаимодействию в присутствии галогенирующего реактива с получением соединения формулы 5
соединение формулы 4 далее подвергают взаимодействию в присутствии галогенирующего реактива с получением соединения формулы 5 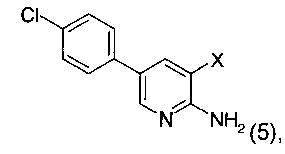 где X представляет собой I (5а) или Br (5б); и соединение формулы 5 далее подвергают взаимодействию в присутствии либо соединения формулы (D)
где X представляет собой I (5а) или Br (5б); и соединение формулы 5 далее подвергают взаимодействию в присутствии либо соединения формулы (D) 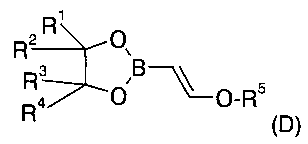 , либо соединения формулы 7
, либо соединения формулы 7 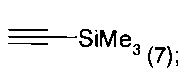 (реакция Соногашира), с получением соединения формулы 8
(реакция Соногашира), с получением соединения формулы 8 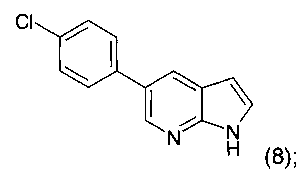 и которое далее подвергают взаимодействию в присутствии соединения формулы 9
и которое далее подвергают взаимодействию в присутствии соединения формулы 9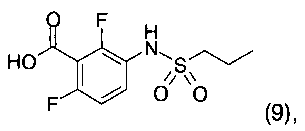 и при условиях ацилирования Фриделя-Крафтса с получением соединения формулы 1, где все R1, R2, R3 и R4 представляют собой метил, или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо; и R5 представляет собой -(С1-С6)алкил. Технический результат: предложен новый способ получения соединения формулы (1) с высоким выходом. 3 з. п. ф-лы, 9 пр.
и при условиях ацилирования Фриделя-Крафтса с получением соединения формулы 1, где все R1, R2, R3 и R4 представляют собой метил, или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо; и R5 представляет собой -(С1-С6)алкил. Технический результат: предложен новый способ получения соединения формулы (1) с высоким выходом. 3 з. п. ф-лы, 9 пр.
1. Способ получения соединения формулы 1,
включающий по меньшей мере одну реакцию Сузуки-Мияура с последующим ацилированием Фриделя-Крафтса, где
а) соединение формулы 2
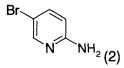
подвергают взаимодействию в присутствии палладиевого катализатора, основания и соединения формулы 3 (1-ая реакция Сузуки-Мияура)
с получением соединения формулы 4
б) указанное соединение формулы 4 далее подвергают взаимодействию в присутствии галогенирующего реактива с получением соединения формулы 5
где X представляет собой I (5а) или Br (5б); и
указанное соединение формулы 5 далее подвергают взаимодействию в присутствии либо
в-1) соединения формулы (D) (2-я реакция Мияура); либо
в-2) соединения формулы 7 (реакция Соногашира)
или
с получением соединения формулы 8
и
г) указанное соединение формулы 8 далее подвергают взаимодействию в присутствии соединения формулы 9 и при условиях ацилирования Фриделя-Крафтса
с получением соединения формулы 1, где
все R1, R2, R3 и R4 представляют собой метил, или совместно с атомами углерода, к которым они присоединены, образуют фенильное кольцо; и
R5 представляет собой -(С1-С6)алкил.
2. Способ по п. 1, где
а) соединение формулы 2
подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, Pd(OAc)2, Na2CO3 и соединения формулы 3
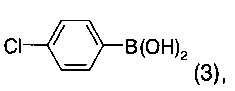
с получением соединения формулы 4
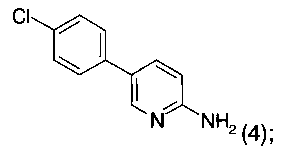
б) указанное соединение формулы 4 далее подвергают взаимодействию в присутствии N-йодсукцинимида (NIS) и CF3COOH или N-бромсукцинимида (NBS) с получением соединения формулы 5
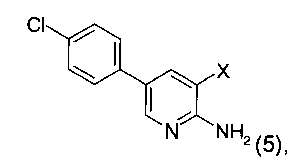
где X представляет собой -I (5а) или -Br (5б);
в-1) указанное соединение формулы 5 далее подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, LiOH и соединения формулы 6,
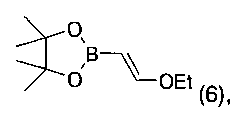
с получением соединения формулы 8
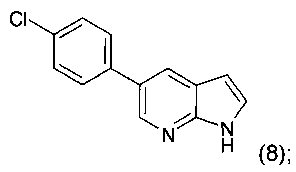 и
и
г) указанное соединение формулы 8 далее подвергают взаимодействию с соединением формулы 9
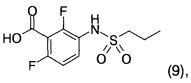
в присутствии (COCl)2 и AlCl3 с получением соединения формулы 1.
3. Способ по п. 2, где стадия реакции б) проводится в присутствии NBS с получением соединения формулы 5, где X представляет собой бром (5б).
4. Способ по п. 1, где
а) соединение формулы 2
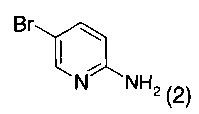
подвергают взаимодействию в присутствии PdCl2(dppf)CH2Cl2, Pd(OAc)2, Na2CO3 и соединения формулы 3
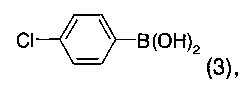
с получением соединения формулы 4
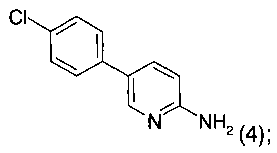
б) указанное соединение формулы 4 далее подвергают взаимодействию в присутствии N-йодсукцинимида (NIS) и CF3COOH или N-бромсукцинимида (NBS) с получением соединения формулы 5
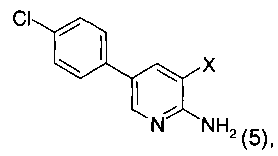
где X представляет собой -I (5а) или -Br (5б);
в-2) указанное соединение формулы 5 далее подвергают взаимодействию в присутствии Pd(PPh3)2Cl2, CuI, тетраметилгуанидина (TMG) и соединения формулы 7,
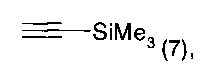
с последующей реакцией с KOtBu с получением соединения формулы 8
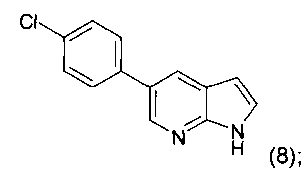 и
и
г) указанное соединение формулы 8 далее подвергают взаимодействию с соединением формулы 9
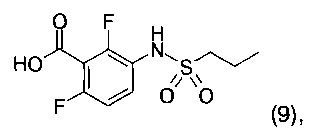
в присутствии (COCl)2 и AlCl3 с получением соединения формулы 1.
| WO 2007002325 A1, 04.01.2007 | |||
| Shu-Bin Zhao et al | |||
| "New Bimetallic Reactivity in Pt /Pt Transformation Mediated by a Benzene Ring" ORGANOMETALLICS, 2010, Vol.29, N4, 998-1002 | |||
| МОДУЛЯТОРЫ КИНАЗЫ НА ОСНОВЕ ПРОИЗВОДНЫХ ПИРРОЛОПИРИДИНА | 2005 |
|
RU2389728C2 |
| WO 2005095400 A1, 13.10.2005. | |||

