ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способам получения олефина, позволяющим эффективно конвертировать стартовые олефины в другие виды олефинов с помощью реакции метатезиса в присутствии водорода.
СВЕДЕНИЯ О ПРЕДШЕСТВУЮЩЕМ УРОВНЕ ТЕХНИКИ
В процессе крекинга углеводородного сырья получают различные углеводородные соединения, такие как этилен, пропилен и бутен, с установленным процентным выходом целевых олефинов. Пропорции, в которых получают эти олефины, не всегда находятся в согласии с требованиями, предъявляемыми к олефинам. Поэтому необходимо, чтобы олефины, получаемые в процессе крекинга углеводородного сырья, были преобразованы так, чтобы они удовлетворяли требованиям потребителей. Реакция используется для конверсии олефинов. В реакции метатезиса олефины одинаковых или различных видов вступают в реакцию друг с другом, для получения олефинов, имеющих различные структуры. Реакция метатезиса имеет важное значение, поскольку она допускает конверсию олефинов при крекинге углеводородного сырья в ответ на изменения требований к олефинам.
В 1931 г. было открыто, что реакция метатезиса олефинов протекает при высокой температуре 725°С без катализаторов.
Однако промышленная ценность реакции метатезиса была признана только после создания катализатора, имеющего окись металла, такого как молибден, вольфрам или рений, нанесенного на несущий элемент с большой площадью поверхности. Первая реакция метатезиса с такими катализаторами была разработана компанией Phillips Petroleum в 1964 году, в которой этилен и 2-бутен были синтезированы из пропилена с помощью катализа окиси молибдена, нанесенного на оксид алюминия (γ-глинозем).
Реакция метатезиса обратима, и, таким образом, в ней существует равновесный состав. Например, в реакции метатезиса этилена и 2-бутена в пропилен равновесие изменяется таким образом, чтобы получить больше пропилена при более низких температурах. После этого были выполнены исследования с целью понижения температуры реакции за счет улучшения свойств катализаторов. Компания Phillips Petroleum разработала способ, в котором катализатор, содержащий окись вольфрама, нанесенный на кварц, и сокатализатор окиси магния используются в комбинации. Этот метод был основан как процесс производства пропилена компанией Lummus Global, Inc.
В частности, в патенте США 4,575,575 (Патентный документ 1) и в Журнале Молекулярного Катализа, том 28, стр.117 (1985) (Непатентный документ 1) сообщается, что, когда реакция метатезиса между этиленом и 2-бутеном происходит при температуре 330°С в присутствии катализатора окиси вольфрама, нанесенного на кварц, конверсия бутена составляет только 31%, в то время как в комбинации с сокатализатором из окиси магния конверсия повышается до 67%.
Кроме того, в патенте США 4,754,098 (Патентный документ 2) сообщается, что в той же самой реакции метатезиса при температуре 330°С использование сокатализатора, в котором окись магния нанесена на оксид алюминия (γ-глинозем), повышает конверсию бутена до 75%. Об этом также сообщается в патенте США 4,684,760 (Патентный документ 3), в котором используется сокатализатор, включающий окись магния и гидроксид лития, нанесенный на оксид алюминия (γ-глинозем), конверсия бутена поддерживается на уровне 74% даже при более низкой температуре в 270°С.
Однако температуры реакции, приведенные в вышеупомянутых документах (например, 270°С в Патентном документе 3), достаточно высоки для практических производственных процессов, и для достижения таких температур реакции, например 270°С, требуется использование оборудования типа нагревающих печей. Поэтому температура в реакции метатезиса должна быть понижена до уровня приблизительно в 200°С, для чего наиболее простым решением служит паровое нагревание.
Для решения данной проблемы заявитель ранее установил, что катализатор окиси вольфрама, нанесенный на кварц, в комбинации с сокатализатором в виде окиси магния или системы, в которой гидроокись натрия нанесена на оксид алюминия (γ-глинозем), позволяет достигать радикального повышения каталитической активности в реакции в присутствии небольшого количества водорода (Патентный документ 4). Согласно этому способу температура реакции понижается, и пропилен может быть получен с намного более высокой селективностью. Кроме того, при условии, что температура реакции является постоянной, пропилен можно устойчиво получать в течение более длительного периода времени.
Однако вышеупомянутый способ требует присутствия водорода, хотя его количество небольшое. Водород вступает в нежеланные реакции с этиленом или пропиленом, что приводит к получению побочного этана или пропана.
Пропан, который в вышеупомянутой реакции является побочным продуктом, уменьшает чистоту получаемого пропилена. Побочное получение этана вызывает технологические проблемы, а именно когда при возвращении в реактор непрореагировавшего этилена происходит концентрация и накопление этана в системе.
JP-B-S48-16482 (Патентный документ 5) и JP-B-S57-13532 (Патентный документ 6) описывают, что в реакции метатезиса используется водород в присутствии катализатора, в котором окись молибдена и окись рения нанесены на оксид алюминия (γ-глинозем). В указанных патентных документах нет информации о формировании побочных продуктов за счет гидрирования, и отсутствуют данные о приведенной скорости.
[Патентный документ 1] Патент США 4,575,575.
[Патентный документ 2] Патент США 4,754,098.
[Патентный документ 3] US Patent No. 4,684,760.
[Патентный документ 4] WO 2006/093058.
[Патентный документ 5] JP-B-S48-16482.
[Патентный документ 6] JP-B-S57-13532 5.
[Непатентный документ 1] Журнал Молекулярного Катализа, том. 28, стр.117 (1985).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является повышение эффективности процесса реакции метатезиса по получению олефинов в присутствии водорода наряду с подавлением побочного получения парафинов.
В процессе получения олефинов с помощью реакции метатезиса, включающей подачу газообразного олефина, для прохождения через слой катализатора в присутствии водородного газа, для конверсии олефина в другой вид олефина, слой катализатора, содержащий катализатор, включающий, по крайней мере, один металл, выбранный из группы, состоящей из вольфрама, молибдена, рения, ниобия, тантала и ванадия, и сокатализатора, включающего основной состав, содержащий, по крайней мере, один металл из группы, состоящей из Группы Ia (щелочные металлы), Группы IIa (щелочноземельные металлы), Группы IIb и Группы IIIa Периодической таблицы, усовершенствование включает управление приведенной скоростью газа, проходящего через слой катализатора, в диапазоне от 0.01 до 2.0 м/сек.
В общей реакции метатезиса приведенная скорость прохождения через слой катализатора обычно составляет приблизительно 0.001 м/сек, для того чтобы гарантировать достаточное время контакта с катализатором. Однако было установлено, что при такой низкой скорости олефины реагируют с водородом при катализе с катализатором и получением парафинов. Заявителем было обнаружено, что процесс конверсии олефинов с помощью реакции метатезиса не требует длительного контакта с катализатором. В настоящем изобретении газ пропускают через слой катализатора с очень высокой приведенной скоростью, в диапазоне от 0.01 до 2.0 м/сек, сводя, таким образом, олефины с катализатором добиваются повышения производства целевых олефинов, подавляя формирование побочных продуктов, таких как парафины. Подробно, в реакции метатезиса этилена и 2-бутена, управление приведенной скоростью пропускания газа через слой катализатора в диапазоне от 0.01-2.0 м/сек позволяет эффективно формировать целевой пропилен и уменьшает количество побочных парафинов, таких как этан и пропан.
ПРЕИМУЩЕСТВА ИЗОБРЕТЕНИЯ
Согласно процессу получения олефина по настоящему изобретению газ пропускают с высокой приведенной скоростью, и поэтому стартовые олефины и водородный газ находятся в контакте с катализатором реакции метатезиса в течение короткого промежутка времени.
Время контакта таково, что олефины преобразуются в другие виды олефинов, но при этом подавляется гидрирование олефинов. Таким образом, предотвращается нежеланное формирование парафинов в реакции.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На чертеже представлен график, показывающий связь между приведенной скоростью в реакторе и количеством водорода, потребляемого при формировании парафинов, согласно Примерам 1 и 2 и Сравнительным Примерам 3 и 5.
ПРЕДПОЧТИТЕЛЬНЫЙ ВАРИАНТ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Получение олефинов с помощью реакции метатезиса согласно настоящему изобретению будет описано подробно.
В настоящем изобретении, в процессе получения олефинов реакцией метатезиса катализатор и сокатализатор используются в комбинации.
Катализатор, используемый в изобретении, содержит, по крайней мере, один металл, отобранный из вольфрама, молибдена, рения, ниобия, тантала и ванадия. Из них вольфрам, молибден и рений обеспечивают относительно высокую каталитическую активность. В частности, катализатор, включающий вольфрам, показывает более высокую активность.
Металлический катализатор может быть окисью, сульфидом или гидроокисью металла. Они могут использоваться отдельно, или по два или более видов могут использоваться в комбинации. Металлический состав может быть нанесен на несущий элемент. Реакция метатезиса, катализируемая вышеупомянутым катализатором, предпочтительно является проточной реакцией с неподвижным слоем. В проточной реакции с неподвижным слоем катализатором предпочтительно является окись металла, потому что дезактивированный катализатор может быть восстановлен прокаливанием.
Несущий элемент для катализатора предпочтительно имеет большую площадь поверхности, обычно не меньше чем 10 м2/г. Несущий элемент предпочтительно выполнен из некислотного состава, потому что кислый несущий элемент вызывает олигомеризацию олефинов.
Примеры несущих элементов включают кварц, оксид алюминия (γ-глинозем) и диоксид титана, причем кварц является предпочтительным из-за большой площади поверхности. При использовании кварца в качестве несущего элемента количество наносимого металла, с точки зрения окиси, предпочтительно находится в диапазоне от 0.01 до 50% веса, а более предпочтительно - в диапазоне от 0.1 до 20% веса относительно несущего элемента.
Металлический состав (катализатора) может быть нанесен на несущий элемент различными способами. Описан образцовый метод в случае нитрата или гидроокиси металла, или вольфрама, молибдена или рения: поликислота или изополикислота металлического состава или металла, или соли аммония поликислоты, или соли аммония изополикислоты используются как стартовый материал. Стартовый материал формируется в виде водного раствора, а несущий элемент пропитывается. После этого раствор выпаривается насухо, а остаток обжигается при температуре 300°С или выше в кислородной атмосфере.
Поскольку несущий элемент, использованный в данном случае, является коммерчески доступным, то он может использоваться без модификации. Также возможно использование несущего элемента, который получен в результате реакции соли соответствующего металла с основой и сжиганием получающейся гидроокиси в окись.
Когда несущий элемент выполнен из соответствующей металлической соли, может использоваться способ совместного осаждения, в котором металлическая соль, формирующая катализатор, добавляется вместе с металлической солью для несущего элемента и одновременно осуществляется синтез несущего элемента и наносимого металлического составного катализатора.
В общем случае несущий элемент имеет различные формы, такие как сферические формы, цилиндрические формы, экструзионные формы и дробленые формы. Размер разноформенных частиц не ограничен до тех пор, пока частицы могут заполнять реактор метатезиса, и, в общем случае, находится в диапазоне от 0.01 до 100 мм.
В настоящем изобретении вышеупомянутый катализатор используется в комбинации с сокатализатором. Примеры сокатализаторов, используемых в изобретении, включают составы металлических элементов, принадлежащих Группе Ia (щелочные металлы), Группе IIa (щелочноземельные металлы), Группе IIb и Группе IIIa Периодической таблицы. Эти составы могут использоваться как отдельно, так и по два или более видов в комбинации. Предпочтително использование следующих металлов из Групп Периодической таблицы: литий, натрий, калий, цезий, магний, кальций, стронций, барий, цинк и иттрий.
Патенты США 4,575,575, 4,754,098 и 4,684,760 (Компании Phillips Petroleum) описывают, что окись магния по существу используется как сокатализатор. В настоящем изобретении окись магния не является существенным компонентом и может быть заменена или может использоваться в комбинации с другими металлами, типа лития, натрия и калия ввиду их каталитической активности в реакции метатезиса. Эти составы могут использоваться отдельно или по два или более видов в комбинации.
Сокатализатор может быть в твердом состоянии типа окиси, гидроокиси, нитрата или ацетата. Такие металлические составы могут также содержать другие металлы. Примерами таких сокатализаторов с другими металлами могут быть гидроталцит, который является слоистой двойной гидроокисью алюминия и магния, и твердый раствор окиси алюминия и окиси магния, полученного путем сжигания гидроталцита. Окиси, смешанные окиси, гидроокиси, двойные гидроокиси, нитраты и ацетаты металлов могут быть нанесены на несущие элементы, имеющие большую площадь поверхности.
Несущий элемент для сокатализатора предпочтительно является составом, который не проявляет кислотность даже после нанесения сокатализатора, потому что кислый несущий элемент может вызвать олигомеризацию олефинов.
Предпочтительные примеры несущих элементов для сокатализаторов включают оксид алюминия (γ-глинозем), двуокись циркония и диоксид титана. Они могут использоваться отдельно или по два или более видов в комбинации. Площадь поверхности несущего элемента предпочтительно не должна быть меньше чем 10 м2/г.
В настоящем изобретении окись магния может использоваться как несущий элемент для сокатализатора из-за его большой площади поверхности. Возможно также использование окиси магния и вышеописанных металлических окисей в комбинации. В частности, комбинация оксида алюминия (γ-глинозем) и магния предпочтительна из-за высокой химической стабильности оксида алюминия (γ-глинозем). Смесь оксида алюминия и магния также пригодна к употреблению.
При использовании сокатализатора, нанесенного на несущий элемент, количество металла сокатализатора в части окиси относительно несущего элемента находится, в общем случае, в диапазоне от 0.01 до 50% веса, а в предпочтительном случае находится в диапазоне от 0.1 до 20% веса.
Поскольку несущий элемент для сокатализатора является коммерчески доступным, то он может использоваться без модификации. Возможно также использование несущего элемента, который получают в результате реакции соли соответствующего металла с основой известным методом и сжигания получающейся гидроокиси в окись.
Оксиды могут наноситься на несущий элемент известными способами, например, из металлических составов в виде сокатализаторов. В типичном способе нитрат или гидроокись металла преобразуют в водный раствор или суспензию, которыми пропитывают несущий элемент. После этого водный раствор или суспензию насухо выпаривают, а остаток сжигают при температуре 300°С или выше в воздушной атмосфере.
Если несущий элемент изготовлен из соответствующих металлических солей, то в этом случае может быть использован способ совместного осаждения, в который металлическая соль, формирующая сокатализатор, добавляется вместе с металлической солью для несущего элемента, а синтез несущего элемента и нанесение металлического состава сокатализатора выполняется одновременно.
Формы сокатализатора и несущего элемента для сокатализатора ничем особенно не ограничены. В общем случае они могут быть различной формы: сферической, цилиндрической, экструзионной и дробленой формы. Размер частиц не ограничен до тех пор, пока частицы могут быть загружены в реактор метатезиса, и, в общем случае, находится в диапазоне от 0.01 до 100 мм.
В настоящем изобретении реакция метатезиса может катализироваться системой катализатора, в которой катализатор реакции метатезиса (металл(ы), выбранные из вольфрама, молибдена, рения, ниобия, тантала и ванадия) и сокатализатор (металл(ы), отобранные из Группы Ia (щелочные металлы), Группы IIа (щелочноземельные металлы), Группы IIb и Группы IIIa Периодической таблицы) совместно наносятся на несущий элемент.
В реакции метатезиса количество сокатализатора относительно катализатора (отношение веса сокатализатор/катализатор) в общем случае находится в диапазоне от 0.1 до 20. Когда отношение будет меньше нижней границы диапазона, водородный газ, который добавляется в небольшом количестве в реакцию метатезиса, не будет оказывать достаточного влияния. Когда отношение превышает верхнюю границу диапазона, количество катализатора является слишком маленьким относительно общего количества катализатора и сокатализатора, и каталитическая активность заметно снижается.
Активность катализатора метатезиса значительно снижает влага, содержащаяся в катализаторе, углекислый газ, угарный газ, составы диенов, меркапто составы, спирты и карбоксильные составы. Соответственно, олефины, используемые в качестве стартовых материалов предпочтительно, не должны содержать указанные компоненты, которые снижают каталитическую активность. Такие примеси предпочтительно должны быть удалены достаточной очисткой стартовых олефинов с помощью дистилляции, адсорбции, выделения или промывки перед контактом с катализатором реакции метатезиса. Чрезмерная очистка увеличивает затраты, и поэтому очистку стартовых олефинов предпочтительно выполнять так, чтобы оптимизировать устойчивость активности катализатора метатезиса и затраты на очистку сырья.
Другие материалы, такие как газы азота и водорода, которые вводятся в реактор, предпочтительно должны быть настолько чистыми, насколько это возможно осуществить.
Катализатор и сокатализатор, в общем случае, помещаются в цилиндрический реактор. Верхние и более низкие части реактора заполняются шариками α-глинозема и т.п., для того чтобы удерживать катализатор и сокатализатор в середине реактора.
Достаточно часто катализатор и сокатализатор, помещаемые в реактор, содержат небольшое количество воды. Такая вода существенно снижает каталитическую активность в реакции метатезиса, и поэтому предпочтительно, чтобы она была удалена перед подачей стартовых олефинов. Например, катализатор и сокатализатор, помещенные в реактор, могут быть подвергнуты пропусканию инертного газа, типа гелия, азота, аргона или ксенона, нагретого до 300°С или выше в течение, по крайней мере, 10 минут, посредством чего удаляют влагу, адсорбируемую катализатором, сокатализатором, несущим элементом и шариками α-глинозема.
Течение горячего инертного газа, в общем случае, сопровождается восстановительной обработкой для активации катализатора, за счет пропускания нагретого восстановительного газа. Используемые здесь восстановительные газы включают угарный газ и водород. Восстановительный газ, в общем случае, нагревается не менее чем до 300°С и пропускается в течение, по крайней мере, 10 минут. В способе получения олефинов с помощью реакции метатезиса в настоящем изобретении конверсия олефинов протекает в присутствии небольшого количества водородного газа. Поэтому, даже если небольшое количество водорода, используемого в качестве восстановительного газа, остается в реакторе метатезиса, такой остаточный водородный газ существенно не уменьшает выход в реакции метатезиса.
В настоящем изобретении олефины вступают в контакт с катализатором метатезиса и сокатализатором, которые подвергаются рассмотренной выше предварительной обработке, посредством чего олефины преобразуются в другие виды олефинов.
Стартовые олефины, используемые в реакции метатезиса, могут быть одного или отличного друг от друга состава. В способе получения олефина с помощью реакции метатезиса по настоящему изобретению может использоваться смесь олефинов, получаемая в результате крекинга сырой нефти.
Олефины, используемые в реакции метатезиса по настоящему изобретению, являются низшими олефинами. Примеры олефинов включают этилен, пропилен, 1-бутен, 2-бутен, 2-пентен, 2-гексен, 4-метил-2-пентен и 3-метил-1-бутен. Они могут использоваться отдельно или по два или более вида в комбинации.
Например, пропилен может быть получен из этилена и 2-бутена; пропилен и 1-бутен из этилена и 2-пентена; пропилен и 1-пентен из этилена и 2-гексена; пропилен и изобутен из этилена и 2-метил-2-бутена; и пропилен и 3-метил-1-бутен из этилена и 4-метил-2-пентена. Так как реакция метатезиса обратима, то выбор условий реакции позволяет получать стартовые олефины из олефинов, полученных в вышеупомянутых реакциях.
Когда в реакции метатезиса для получения другого олефина используются два или более вида олефинов, молярное отношение олефинов в стартовых материалах особенно не ограничено. Когда этилен включен в два или более вида олефинов, количество этилена находится предпочтительно в избытке по отношению к другим олефинам. В случае реакции этилена и 2-бутена для получения пропилена молярное отношение этилена к n-бутену (общее количество 1-бутена и 2-бутена) (этилен/n-бутен), в общем случае, будет составлять 1-50, а предпочтительно примерно 1-5. Когда отношение будет слишком малым, реакция, предпочтительно, будет иметь место между молекулами бутена. Когда отношение слишком большое, то в этом случае потребуется крупногабаритное оборудование и большое количество энергии для восстановления непрореагировавшего этилена.
При получении олефина реакцией метатезиса стартовые олефины могут вступать в контакт с катализатором метатезиса и сокатализатором, которые формируют неподвижный слой, псевдоожиженный слой, взвешенный слой или ступенчатый неподвижный слой.
Когда проточный реактор с неподвижным слоем упакован катализаторами, как описано в Журнале Молекулярного Катализа, том 28, стр.117 (1985), катализатор и сокатализатор могут быть физически смешаны и помещены в реактор, или сокатализатор и катализатор могут быть помещены в таком же порядке вдоль направления, в котором подают материальный газ. Эти способы упаковки могут использоваться в комбинации.
В производстве олефина с помощью реакции метатезиса газ проходит через слой катализатора с приведенной скоростью от 0.01 до 2.0 м/сек. Предпочтительно, чтобы нижний предел приведенной скорости составлял 0.014 м/сек, верхний предел - 1.5 м/сек, а реакция метатезиса выполнялась с приведенной скоростью в этом диапазоне.
Приведенная скорость будет объяснена на основе выполнения реакции метатезиса в промышленном масштабе, когда используется проточный реактор с неподвижным слоем, позволяющий легко разделять катализаторы и создавать пробковый режим потока. Приведенная скорость (Uavg) описывается следующим уравнением (1):
 ,
,
в котором - Di - внутренний диаметр (м) реактора, а Fv - скорость загрузки сырья (м3/сек).
Более точно, приведенная скорость (Uavg) является скоростью газа (сырья), проходящего через катализатор метатезиса и сокатализатор.
В обычной реакции метатезиса приведенная скорость низка и обеспечивает вступление стартовых олефинов в контакт с катализаторами в достаточной степени. Поэтому стартовые олефины находятся в контакте с катализатором метатезиса и сокатализатором в течение относительно долгого времени. Когда водородный газ присутствует, олефины гидрогенизируются в течение такого длительного времени контакта. Это вероятно и есть причина образования парафинов в значительном количестве.
Конверсия олефинов произойдет, даже если сильно сократить время контакта между стартовыми олефинами и катализатором метатезиса и сокатализатором. Любой более длительный контакт с катализаторами приведет только к гидрированию олефина с образованием парафинов. Учитывая это, нижний предел приведенной скорости в изобретении установлен на очень высоком уровне по сравнению с традиционными способами. Верхний предел приведенной скорости гарантирует минимальное время, требуемое для завершения реакции метатезиса. Если приведенная скорость больше, чем верхний предел, контакт между стартовыми олефинами и катализаторами недостаточен, и реакция метатезиса не будет протекать в течение достаточного времени.
Приведенная скорость эффективна, когда скорость загрузки сырья в час относительно общего веса катализатора (общая масса катализатора метатезиса и сокатализатора) (WHSV: 1/час) находится в диапазоне от 0.1 до 50 час-1, а предпочтительно в диапазоне от 0.5 до 30 час-1. Если скорость загрузки сырья (WHSV) меньше нижнего предела, время контакта с катализатором метатезиса и сокатализатором длительное, и при этом получают большее количество парафинов. Если WHSV выше верхнего предела, реакция метатезиса не будет протекать в течение достаточного времени.
В настоящем изобретении стартовые олефины предпочтительно загружаются в реактор метатезиса с небольшим количеством водородного газа. Водородный газ может быть добавлен в количестве от 0.05 до 10% объема, а предпочтительно в диапазоне от 0.08 до 5% объема, в зависимости от суммарного объема газа (суммарный объем газа стартовых олефинов и водородного газа). Подавая водородный газ в таком небольшом количестве, катализаторы метатезиса поддерживают каталитическую активность в течение длительного времени. Если количество водорода меньше, чем вышеупомянутый диапазон, каталитическая активность не может поддерживаться. Если количество больше, чем описано выше, выделение непрореагировавшего водорода увеличивает трудности. Даже при том что водородный газ подается в вышеупомянутом количестве, это не приводит к существенному увеличению количества побочно получаемых парафинов, а катализаторы метатезиса поддерживают каталитическую активность в течение долгого времени. Такая длительная активность катализаторов метатезиса вероятно способствует эффективному течению реакции метатезиса с приведенной скоростью в вышеупомянутом диапазоне.
В общем случае водород подается непрерывно в газовом состоянии. В варианте изобретения, однако, водородный газ может подаваться периодически, то есть водородный газ может быть добавлен в начале реакции метатезиса, затем его подача может быть прекращена во время реакции и может быть возобновлена после предопределенного времени.
В изобретении стартовые олефины и водородный газ вступают в контакт с катализатором метатезиса и сокатализатором. Водородный газ может быть восстановлен фракциями с низкой точкой кипения в верхней части колонны и может быть переработан.
Как описано выше, водород используется в восстановительной обработке катализаторов. Если такой водород остается даже после того, как реактор очищен азотом, остаточный водород поможет катализаторам метатезиса поддерживать активность, по крайней мере, в самом начале реакции. Однако каталитическая активность будет постепенно уменьшаться, если водородный газ не будет заново добавлен, а результаты реакции будут, в конечном счете, равны результатам, полученным без добавления водородного газа. В производстве олефина с помощью реакции метатезиса согласно настоящему изобретению водородный газ непрерывно подается с определенной скоростью в систему реакции, и, таким образом, катализатор метатезиса и сокатализатор могут устойчиво использоваться в течение длительного времени.
Температура реакции обычно находится в диапазоне от 100 до 500°С, предпочтительно в диапазоне от 130 до 350°С. Когда температура реакции чрезвычайно низка, скорость реакции понижена, и производительность продуктов реакции уменьшается. Когда температура реакции чрезвычайно высока, протекают побочные реакции с образованием большого количества побочных продуктов, таких как парафины, и при этом ускоряется дезактивация катализатора.
В изобретении стартовые олефины и водород предварительно подогреваются приблизительно до температуры от 100 до 300°С и обычно загружаются в газовом состоянии в реактор, заполненный катализатором метатезиса и сокатализатором.
Давление реакции при вышеупомянутой температуре может быть пониженным давлением, избыточным давлением или атмосферным давлением. С точки зрения эффективности реакции (эффективность реакции на единицу объема) давление находится обычно в диапазоне от 0.01 до 20 МПа, и предпочтительно в диапазоне 0.05 к 10 МПа. При этом условии давления реакция метатезиса пройдет равномерно. Чрезмерно высокое давление невыгодно с точки зрения оборудования, т.е. необходимости использования реакторов, способных выдерживать высокое давление.
В настоящем изобретении получение олефина с помощью реакции метатезиса не обязательно включает реакционные растворители или газы растворения. Однако система реакции может включать растворители или газы, которые являются инертными к катализаторам и стартовым материалам. Определенные примеры включают алканы типа метана, этана, пропана и бутана, и инертные газы растворения, типа азота и гелия. Такие компоненты могут присутствовать в системе реакции или могут быть добавлены к системе реакции для управления реакцией.
После реакции метатезиса продукты реакции отделяют и восстанавливают от катализаторов, стартовых олефинов, водородного газа и т.п. Более детально, целевые олефины отделяют от смеси реакции известным методом, типа дистилляции, выделения или адсорбции. Непрореагировавшее сырье может быть восстановлено и многократно использоваться в системе реакции. Реакция метатезиса получения олефина может быть выполнена в любой жидкой фазе, газовой фазе и фазе смеси газ-жидкость. С точки зрения эффективности реакции предпочтительно выполнять реакцию в газовой фазе.
В производстве олефина по настоящему изобретению требуемая конечная продукция олефина может быть получена альтернативным способом, используя два или более реакторов, параллельно расположенных так, что катализатор метатезиса и сокатализатор в одном реакторе восстанавливаются с горячим воздухом или воздухом с растворенным азотом, в то время как олефины получают реакцией метатезиса в другом реакторе (реакторах). В общем случае этот способ называют способом карусели. Используя способ карусели, олефины получают более эффективно. Реакция может использовать три реактора, два из которых связаны последовательно, чтобы стабилизировать конечную продукцию. Когда реакция выполняется в режиме проточной реакции с псевдоожиженным слоем или в режиме реакции с движущимся слоем, все или часть катализаторов могут извлекаться из реактора непрерывно или периодически, с добавлением соответствующего количества новых катализаторов для поддержания активности на определенном уровне.
При получении олефина реакцией метатезиса катализатор метатезиса и сокатализатор со временем уменьшают свою каталитическую активность. Дезактивированные катализаторы могут быть регенерированы для восстановления каталитической активности. В общем случае олефины, адсорбируемые на катализаторах, очищают газом азота, а катализаторы окисляют воздухом или воздухом с растворенным азотом при температуре 300°С или выше. При использовании металла - вольфрама или молибдена, катализаторы затем подвергаются сокращению с помощью восстановительного газа, типа водорода или угарного газа. Таким образом, катализаторы реактивируют.
ПРИМЕРЫ
Настоящее изобретение ниже будет описано Примерами, не ограничивающими объем изобретения.
[Пример 1]
8.3 г метавольфрамата аммония (Олдрич) было растворено в 1 литре дистиллированной воды, и 50 г силикагеля Q-10, производимого компанией Fuji Silysia Chemical Ltd. (поверхностная площадь: 300 м2/г, объем поры: 1 мл/г, 150-500 мкм), находится в растворе во взвешенном состоянии. Суспензия размешивалась при комнатной температуре в течение 30 минут. Впоследствии вода была выпарена в испарителе.
Получающийся белый сухой остаток был сожжен при атмосферном давлении при температуре 550°С в течение 6 часов. Сожженный продукт упоминался как катализатор WQ-10.
Окись магния (Kyowamag 150 (зарегистрированная торговая марка), производимая Kyowa Chemical Industry Co., Ltd.), в виде тонкоизмельченного порошка, была спрессована гидравлическим прессом с давлением 200 кг/см2 в течение 3 минут. Затем прессовку дробили, а частицы классифицировали. Использовались частицы, имеющие диаметры 150-500 мкм.
12 г катализатора WQ-10 и 48 г окиси магния были физически смешаны и упакованы в центре трубы из нержавеющей стали SUS, имеющей внешний диаметр 18 мм, внутренний диаметр (Di) 16 мм и длину 1000 мм. Вершина и основание трубы были заполнены шариками α-глинозема. Таким образом, реактор метатезиса был готов.
Слой катализатора был высотой 0,5 м.
Перед реакцией метатезиса катализаторы подвергались предварительной обработке следующим образом. Газ азота при нормальном давлении подавался из верхней части реактора со скоростью 100 мл/мин, при этом температура была поднята до 500°С и оставалась постоянной в течение 1 часа. Впоследствии газ водорода при нормальном давлении и газ азота подавались со скоростью 20 мл/мин и 80 мл/мин соответственно, при температуре 500°С в течение 30 минут. После этого газ азота снова подавался со скоростью 100 мл/мин при температуре 500°С в течение 2 часов. Затем температура была снижена до 300°С, при подаче азотного газа.
Сжиженный 1-бутен (чистота: 99%, производимый Mitsui Chemicals, Inc.) был очищен адсорбционной обработкой оксидом алюминия (γ-глиноземом) (NKHD-24, производимый Sumitomo Chemical Co., Ltd.).
После предобработки этилен и водород подавались со скоростью 1.87 л/мин и 31.6 мл/мин соответственно, при температуре 25°С и нормальном давлении. Давление в реакторе было установлено в размере 3.5 МПа через обратный клапан.
Очищенный сжиженный 1-бутен подавался со скоростью 2.88 г/мин на подогреватель (200°С), расположенный вверх по течению от реактора метатезиса. Газ 1-бутен от подогревателя вместе с этиленом и водородом подавался в реактор для начала реакции.
В реакторе (300°С, 3.5 МПа) скорость подачи (Fv) сырья была 2.87×10-6 м3/сек. Другими словами, объемная скорость (WHSV) веса сырья (г/час) к общему весу катализатора (г) была равна 5 час-1. Концентрация водорода в сырье была равна 1% по объему. Приведенная скорость (Uavg) в реакторе была равна 0.014 м/сек из следующего уравнения (1):
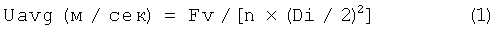 ,
,
в котором - Di обозначает внутренний диаметр реактора метатезиса, а Fv - скорость загрузки сырья.
Давление соответствовало нормальному давлению и устанавливалось с помощью обратного клапана, а образцы продуктов реакции автоматически выбирались. Образец анализировался в режиме онлайн с помощью газовой хроматографии.
На основе состава, выбранного спустя 3 часа после начала реакции, подсчитанный объем конвертированного бутена был равен 70% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от количества подаваемого 1-бутена.
Селективность пропилена, основанная на бутене, была 88%. Также были получены небольшие количества пентена и гексена.
Из поданного водорода 20% было израсходовано на побочное получение этана и пропана. Реакция продолжалась в течение еще 40 часов, но конверсия бутена не уменьшалась.
В вышеупомянутой реакции, выполненной с приведенной скоростью 0.014 м/сек с дополнением по объему 1% водорода, каталитическая активность поддерживалась в течение существенно увеличенного промежутка времени, а побочное получение парафинов из-за присутствия водорода было существенно снижено.
[Сравнительный Пример 1]
Реактор метатезиса был подготовлен так же, как и в Примере 1, за исключением того, что 0.6 г и 2.4 г катализатора WQ-10 и окиси магния, используемых в Примере 1, были физически смешаны и упакованы в трубе из нержавеющей стали SUS, имеющей внешний диаметр 18 мм, внутренний диаметр (Di) 16 мм и длину 400 мм. Катализаторы предварительно обрабатывались тем же способом, как и в Примере 1.
Сжиженный 1-бутен, обработанный с помощью адсорбции глинозема, загружался со скоростью 0.145 г/мин, а этилен загружался со скоростью 87 мл/мин при температуре 25°С и нормальном давлении. Водород не подавался. Газы сырья проходили через слой катализатора при давлении 3.5 МПа и температуре 300°С.
Объемная скорость (WHSV) веса сырья (г/час) к общему весу катализатора (г) была 5 час-1. Концентрация водорода в сырье была 0% по объему. Приведенная скорость (Uavg) из вышеупомянутого уравнения (1) была 0.0007 м/сек.
Давление соответствовало нормальному давлению и устанавливалось с помощью обратного клапана, а образцы продуктов реакции автоматически выбирались. Образец анализировался в режиме онлайн с помощью газовой хроматографии.
На основе состава, выбранного спустя 1 час после начала реакции, подсчитанный объем конвертированного бутена был равен 70% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от количества подаваемого 1-бутена.
Селективность пропилена, основанная на бутене, была 88%. Также были получены небольшие количества пентена и гексена.
Хотя этан или пропан не были побочно получены, конверсия бутена уменьшилась до 0% через 10 часов после начала реакции.
[Сравнительный Пример 2]
Процедуры Сравнительного Примера 1 были повторены за исключением того, что устройство очистки сырья было помещено вверх по направлению от подогревателя. Устройство очистки было трубой из нержавеющей стали SUS (внешний диаметр: 18 мм, внутренний диаметр (Di): 16 мм, длина: 700 мм), заполненной 6 г и 24 г катализатора WQ-10 и окисью магния соответственно. До реакции катализаторы нагревались в устройстве очистки в присутствии азота, затем их количество уменьшалось за счет водорода, затем они снова нагревались в присутствии азота, а температуру снижали до окружающей. Сырьевой газ подавали через устройство очистки, и таким образом примеси в 1-бутене, типа бутадиена, полностью удалялись адсорбцией.
Давление соответствовало нормальному давлению и устанавливалось с помощью обратного клапана, а образцы продуктов реакции автоматически выбирались. Образец анализировался в режиме онлайн с помощью газовой хроматографии.
На основе состава, выбранного спустя 1 час после начала реакции, подсчитанный объем конвертированного бутена был равен 70% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от количества подаваемого 1-бутена. Селективность пропилена, основанная на бутене, была равна 88%. Также были получены небольшие количества пентена и гексена. Этан или пропан не были побочно получены, а конверсия бутена не уменьшилась в течение 20 часов после начала реакции.
Результаты Сравнительного Примера 2 показали, что при полном удалении веществ отравления из сырья бутена катализаторы оставались активными в течение длительного периода времени. Однако такая полная очистка слишком дорога для реализации в промышленном масштабе.
В настоящем изобретении сырье 1-бутен очищают, используя оксид алюминия (γ-глинозем), и очищенный 1-бутен используют в реакции метатезиса. Если сырье 1-бутен будет очищено до высокого уровня, то катализаторы метатезиса останутся активными в течение дальнейшего длительного периода времени. Однако очищение сырья увеличивает затраты. Очистка, такая как очистка 1-бутена с помощью оксида алюминия (γ-глинозема) в Примере 1, может позволить сделать реакцию метатезиса промышленно рентабельной.
[Сравнительный Пример 3]
Процедуры Сравнительного Примера 1 были повторены за исключением того, что водородный газ подавался в реактор метатезиса со скоростью 1.5 мл/мин при температуре 25°С и нормальном давлении, то есть концентрация водорода составляла 1% по объему относительно сырья.
На основе состава, выбранного спустя 3 часа после начала реакции, подсчитанный объем конвертированного бутена был равен 70% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от количества подаваемого 1-бутена. Селективность пропилена, основанная на бутене, была 88%. Также были получены небольшие количества пентена и гексена. Из поданного водорода 90% было израсходовано на побочное получение этана и пропана. Реакция продолжалась в течение еще 20 часов, но конверсия бутена не уменьшалась.
[Сравнительный Пример 4]
Процедуры Сравнительного Примера 3 были повторены за исключением того, что водородный газ подавался в реактор метатезиса со скоростью 0.7 мл/мин, то есть концентрация водорода составляла 0.5% по объему относительно сырья.
На основе состава, выбранного спустя 3 часа после начала реакции, подсчитанный объем конвертированного бутена был равен 70% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от количества подаваемого 1-бутена. Селективность пропилена, основанная на бутене, была 88%. Также были получены небольшие количества пентена и гексена. Из поданного водорода 90% было израсходовано на побочное получение этана и пропана. Реакция продолжалась в течение еще 20 часов, но конверсия бутена не уменьшалась.
Вышеупомянутые результаты свидетельствуют, что даже при том, что бутадиен не был полностью удален из сырья, катализаторы поддерживали активность в течение длительного периода времени за счет добавления водорода. Однако добавление водорода также вызвало побочное получение парафинов, которого нельзя избежать просто с помощью управления подаваемым количеством водорода.
[Сравнительный Пример 5]
Реакция была выполнена при давлении в 3.5 МПа и температуре 300°С таким же образом, как и в Сравнительном Примере 1, за исключением того, что катализатор, WQ-10 и окись магния использовались в количестве 1.2 г и 4.8 г соответственно, сжиженный 1-бутен подавался со скоростью 0.29 г/мин, а этилен и водород подавались со скоростью 180 мл/мин и 3 мл/мин соответственно, при температуре 25°С и нормальном давлении.
Объемная скорость (WHSV) веса сырья (г/час) к общему весу катализатора (г) была 5 час-1. Концентрация водорода в сырье была 1% по объему. Приведенная скорость (Uavg) из вышеупомянутого уравнения (1) была 0.0014 м/сек.
На основе состава, замеренного спустя 3 часа после начала реакции, подсчитанный объем конвертированного бутена был равен 70% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от количества подаваемого 1-бутена. Селективность пропилена, основанная на бутене, была 88%. Также были получены небольшие количества пентена и гексена. Из поданного водорода 88% было израсходовано на побочное получение этана и пропана. Реакция продолжалась в течение еще 20 часов, но конверсия бутена не уменьшалась. Вышеупомянутые результаты свидетельствуют о том, что при увеличении (по сравнению со Сравнительным Примером 1) приведенной скорости до 0.0014 м/сек каталитическая активность и поддержание каталитической активности были эквивалентны значениям, достигнутым согласно настоящему изобретению; однако побочное получение парафинов из-за использования водорода не было устранено в достаточной степени.
[Пример 2]
WO3/SiO2 и окись магния были подготовлены согласно процессам, описанным в Примере 1, затем подвергались формованию. 180 г формованного WO3/SiO2 и 550 г формованной окиси магния физически смешивались и упаковывались в трубчатом реакторе из нержавеющей стали SUS, имеющем внешний диаметр 48.6 мм, внутренний диаметр (Di) 41.2 мм и длину 2 м. Затем 240 г формованной окиси магния были размещены на верхней поверхности смеси. Вершина и основание реактора были заполнены шариками α-глинозема, и, таким образом, слой катализатора был зафиксирован.
Слой катализатора был высотой 1 м.
Газ азота при нормальном давлении подавался из верхней части реактора со скоростью 10.2 л/мин, при этом температура была поднята до 550°С и оставалась постоянной в течение 10 часов. Впоследствии газ водорода и газ азота подавались со скоростью 1.5 мл/мин и 14.7 л/мин, соответственно, при температуре 550°С в течение 3 часов для уменьшения катализаторов. После этого газ азота снова подавался со скоростью 10.2 л/мин при температуре 550°С в течение 24 часов. Затем температура была снижена до 300°С (температура реакции), при подаче азотного газа.
Затем газ азота переключали на химически активный газ, который подавался при внутреннем давлении в 2.7 МПа через обратный клапан. Таким образом, реакция инициировалась.
Химически активный газ был составлен из этилена, смеси соединений с 4 углеродистыми атомами (в дальнейшем называемой как смесь С4) и водорода. Смесь С4 содержала 2-бутены (включая цис-изомер и транс-изомер), 1-бутен, изобутен, изобутан и n-бутаны. У смеси С4 был типичный состав, 50-60% веса которого составляли n-бутены, то есть 2-бутен и 1-бутен.
Этилен и смесь С4 подавались со скоростью 2.1 кг/час и 4.7 кг/час соответственно, а водород подавался со скоростью 40 л/час при температуре 25°С и нормальном давлении. В реакторе (300°С, 2.7 МПа) скорость подачи сырья (Fv) была 7.6×10-5 м3/сек. Другими словами, объемная скорость (WHSV) веса сырья (г/час) к общему весу катализатора (г) была 7 час-1. Концентрация водорода в сырье была 1% по объему. Приведенная скорость (Uavg) из вышеупомянутого уравнения (1) была 0.055 м/сек.
Произведенный газ отбирался через базовую линию осуществления выборки, расположенную вверх по направлению от обратного клапана, и периодически анализировался с помощью газовой хроматографии.
На основе состава, выбранного спустя 24 часа после начала реакции, подсчитанный объем конвертированного бутена был равен 68.5% за вычетом общего количества транс-бутена-2, цис-бутена-2 и 1-бутена в выходном газе от общего количества подаваемого 1-бутена и 2-бутена в смеси С4. Из поданного водорода 20% было израсходовано на побочное получение этана и пропана. Реакция была продолжена, и конверсия бутена составила 58% через 500 часов после начала реакции.
[Пример 3]
Процедуры Примера 2 были повторены за исключением того, что водородный газ подавался в реактор метатезиса со скоростью 12 л/мин при температуре 25°С и нормальном давлении, то есть концентрация водорода составляла 0.3% по объему относительно сырья.
Через 24 часа после начала реакции конверсия бутена составляла 68.6%. Из поданного водорода 20% было израсходовано на побочное получение этана и пропана. Реакция была продолжена, и конверсия бутена составила 53% через 500 часов после начала реакции.
[Пример 4]
Процедуры Примера 2 были повторены за исключением того, что формованная окись магния была заменена формованным продуктом в виде твердого раствора оксид магния/оксид алюминия (MgO·Al2O3), полученным прокаливанием гидроталцита (формованный продукт, производимый компанией Mitsui Chemicals, Inc.).
Через 24 часа после начала реакции конверсия бутена составляла 71%. Из поданного водорода 20% было израсходовано на побочное получение этана и пропана. Реакция была продолжена, и конверсия бутена составила 65% через 500 часов после начала реакции.
[Сравнительный Пример 6]
Процедуры Примера 2 были повторены за исключением того, что водород не подавался.
Через 24 часа после начала реакции конверсия бутена составляла 70%. Конверсия бутена уменьшилась до 40% в течение 500 часов после начала реакции.
Результаты Примеров 2-4 и Сравнительного Примера 6 свидетельствуют о том, что добавление водорода резко увеличивает длительность каталитической активности, а побочное получение парафинов, из-за присутствия водорода, существенно снижается при приведенной скорости, равной 0.055 м/сек.
ПРИМЕНИМОСТЬ В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
В процессе получения олефинов с помощью реакции метатезиса, включающей подачу газообразного олефина, для прохождения через слой катализатора в присутствии водородного газа, для конверсии олефина в другой вид олефина, слой катализатора, содержащий катализатор, включающий, по крайней мере, один металл, выбранный из группы, состоящей из вольфрама, молибдена, рения, ниобия, тантала и ванадия, и сокатализатор, включающий основной состав, содержащий, по крайней мере, один металл из группы, состоящей из Группы Ia (щелочные металлы), Группы IIa (щелочноземельные металлы), Группы IIb и Группы IIIa Периодической таблицы, усовершенствование включает управление приведенной скоростью газа, проходящего через слой катализатора, в диапазоне от 0.01 до 2.0 м/сек. Согласно изобретению достигается высокая выработка целевых олефинов, в то время как побочные реакции, формирующие парафины, подавляются.
При получении пропилена согласно настоящему изобретению, в качестве примера, формирование пропана, которое уменьшает чистоту пропилена, подавляется. Кроме того, формирование этана также подавляется, и таким образом рециркуляция непрореагировавшего этилена приводит к незначительной концентрации и накоплению этана в системе. Другими словами, настоящее изобретение не требует большого количества энергии для отделения побочно получаемых парафинов, таких как этан, а процесс получения олефинов обладает существенными преимуществами с технологической и экономической точек зрения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2006 |
|
RU2367644C2 |
| ИЗОМЕРИЗАЦИЯ В ЖИДКОЙ ФАЗЕ ДЛЯ ПРОЦЕССА МЕТАТЕЗИСА | 2018 |
|
RU2783161C2 |
| ИНТЕГРАЦИЯ ОКИСЛИТЕЛЬНОГО СОЧЕТАНИЯ В МЕТАНОВЫЕ УСТАНОВКИ | 2018 |
|
RU2764097C2 |
| СПОСОБ И ИНТЕГРИРОВАННАЯ СИСТЕМА ДЛЯ ПРИГОТОВЛЕНИЯ НИЗШЕГО ОЛЕФИНОВОГО ПРОДУКТА | 2010 |
|
RU2560185C2 |
| СПОСОБЫ ОЧИСТКИ И ПРОИЗВОДСТВА ТОПЛИВА ИЗ НАТУРАЛЬНОГО МАСЛЯНОГО ИСХОДНОГО СЫРЬЯ | 2010 |
|
RU2565057C2 |
| БОЛЕЕ ЭНЕРГОЭФФЕКТИВНЫЙ СПОСОБ ГИДРОГЕНИЗАЦИИ С5 | 2013 |
|
RU2627657C2 |
| РЕГЕНЕРАЦИЯ КАТАЛИЗАТОРА | 2009 |
|
RU2503499C2 |
| СПОСОБ ГИДРОИЗОМЕРИЗАЦИИ ДВОЙНОЙ СВЯЗИ | 2006 |
|
RU2376272C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2010 |
|
RU2554511C2 |
| ТРЕХСТАДИЙНЫЙ СПОСОБ ПОЛУЧЕНИЯ ЛЕГКИХ ОЛЕФИНОВ ИЗ МЕТАНА И/ИЛИ ЭТАНА | 1998 |
|
RU2165955C2 |
Способ получения олефинов с помощью реакции метатезиса, включающей подачу газообразного олефина, для прохождения через слой катализатора в присутствии водородного газа, для конверсии олефина в другой вид олефина, слой катализатора, содержащий катализатор, включающий, по крайней мере, один металл, выбранный из группы, состоящей из вольфрама, молибдена, рения, ниобия, тантала и ванадия, и сокатализатор, включающий основной состав, содержащий, по крайней мере, один металл из группы, состоящей из Группы Iа (щелочные металлы), Группы IIа (щелочноземельные металлы), Группы IIb и Группы IIIa Периодической таблицы, усовершенствование включает управление приведенной скоростью газа, проходящего через слой катализатора, в диапазоне от 0.01 до 2.0 м/сек, при этом давление реакции находится в диапазоне от 0.01 до 20 МПа и количество сокатализатора относительно катализатора находится в диапазоне от 0.1 до 20 по весу. Применение настоящего способа позволяет повысить эффективность процесса реакции метатезиса по получению олефинов в присутствии водорода наряду с подавлением побочного получения парафинов. 2 з.п. ф-лы, 1 ил.
1. Способ получения олефинов с помощью реакции метатезиса, включающий подачу газообразного олефина для прохождения через слой катализатора в присутствии водородного газа, для конверсии олефина в другой вид олефина, слой катализатора, содержащий катализатор, включающий, по крайней мере, один металл, выбранный из группы, состоящей из вольфрама, молибдена, рения, ниобия, тантала и ванадия, и сокатализатор, включающий основной состав, содержащий, по крайней мере, один металл из группы, состоящей из Группы Iа (щелочные металлы), Группы IIа (щелочноземельные металлы), Группы IIb и Группы IIIa Периодической таблицы, усовершенствование включает управление приведенной скоростью газа, проходящего через слой катализатора, в диапазоне от 0,01 до 2,0 м/с, при этом давление реакции находится в диапазоне от 0,01 до 20 МПа и количество сокатализатора относительно катализатора находится в диапазоне от 0,1 до 20 по весу.
2. Способ по п.1, в котором газ, подвергнутый реакции метатезиса, содержит водородный газ, и водородный газ составляет от 0,05 до 10,0% по объему от суммарного объема (100% по объему) газа.
3. Способ по п.1, в котором реакция метатезиса протекает при температуре в диапазоне от 100 до 500°С.
| ЕР 1854776 А1, 24.02.2006 | |||
| GB 1338429 A, 21.11.1973 | |||
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| GB 1117968 A, 26.06.1968 | |||
| КАТАЛИЗАТОР, СОСТОЯЩИЙ ИЗ ПЕРЕХОДНОГО МЕТАЛЛА, НАНЕСЕННОГО НА ДИОКСИД КРЕМНИЯ ВЫСОКОЙ ЧИСТОТЫ, ДЛЯ МЕТАТЕЗИСА ОЛЕФИНА (ОЛЕФИНОВ) | 2002 |
|
RU2291743C2 |
| Способ получения олефиновых углеводородов | 1971 |
|
SU448635A3 |

