Область техники, к которой относится изобретение
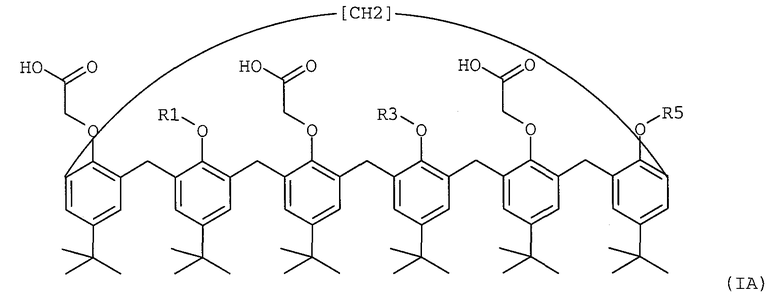
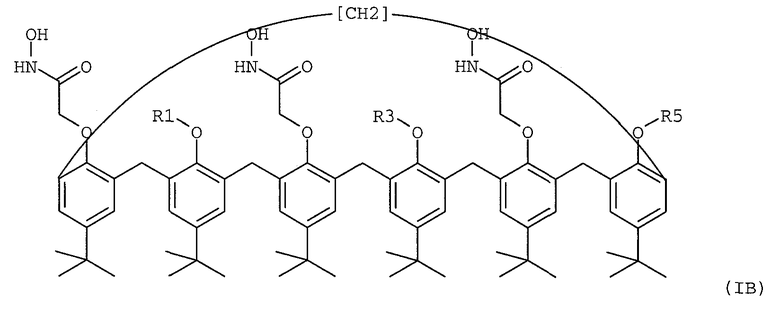
Настоящее изобретение относится к новым пара-трет-бутилкаликс[6]аренам формулы (IA) и (IB), содержащим три кислотные карбоксильные функциональные группы или три кислотные гидроксаминовые функциональные группы в положениях 1, 4 и 6 и содержащим другие функциональные группы в положениях 1, 3 и 5, к нанесенным на подложку жидким мембранам и к содержащим их материалам-подложкам, а также к их применению.
Уровень техники
В топливном цикле, начиная от уранового рудника и заканчивая заводом по переработке, радиоактивные соединения присутствуют в самых разных физических и химических формах с разной степенью токсичности и с разной степенью опасности радиоактивного поражения персонала.
Для обеспечения охраны здоровья работников необходимо осуществлять медицинский контроль. Работники проходят через различные исследования, среди которых можно указать анализ актинидов, являющихся источниками альфа-излучений и удаляемых из организма естественным путем (моча и кал).
В настоящее время в качестве известной технологии анализа используют α-спектрометрию. Из-за небольшой длины пробега α-частиц в веществе невозможно измерить напрямую уран, америций и плутоний в моче. Поэтому необходимо изготовить тонкослойный источник для каждого из актинидов. Это предполагает осуществление первого этапа минерализации образца с последующей химической очисткой, которая позволяет изолировать актиниды из мочи и отделить их друг от друга, чтобы ограничить спектральные интерференции.
В протоколах, используемых на сегодняшний день всеми лабораториями радиологического токсилогического анализа, эта очистка основана на последовательных разделениях актинидов при помощи хроматографических колонок (Harduin et coll., Radioprotection, 31, No 2, 229-245, 1996). Эти протоколы являются продолжительными (6 дней для получения конечного результата), порядка 3 дней для химической обработки и 3 дней для подсчета путем α-спектрометрии, чтобы достичь уровней активности для каждого изотопа менее 1 мБк · 1-1 мочи.
В контексте постоянного технологического усовершенствования в ядерной промышленности и изменения регламентных норм средства, позволяющие осуществлять постоянное наблюдение за работниками и оценивать внутреннее облучение каждого человека в случае аварии, должны быть тоже усовершенствованы.
Поэтому в рамках охраны здоровья существует реальная и важнейшая необходимость получать в кратчайшие сроки результаты измерений в отношении урана, америция и плутония, чтобы иметь возможность предупредить гибельные последствия α-излучений. Получение более быстрых и более эффективных технологий позволило бы также отслеживать α-излучения в окружающей среде, что отвечает проблематике долгосрочного развития.
Для решения этой проблемы были проведены многочисленные исследования, однако ни одно из них до сегодняшнего дня не привело к желаемому результату. Исследования были направлены на изучение селективных комплексообразующих соединений для трех актинидов с целью их выделения из сложных комплексных форм, таких как биологические среды.
Большинство работ проводилось на специальных комплексообразующих соединениях: каликсаренах. Например, в 1993 году Araki et coll. выявили комплексообразующие свойства при жидкофазной экстракции 1,3,5-O-триметил-2,4,5-O-трикислотного карбоксил-пара-трет-бутил-каликс[6]-арена (молекула показана ниже в виде формулы IA) в отношении урана (Chem. Lett., 829-832, 1993). В 1994 году Van Dutnoven et coll. использовали эту же молекулу для изучения конформационных балансов (J. Av. Chem. Soc., 116, 5814-5822). В 1997 году С.Dinse et coll. (Radioprotection, 32 No 5, 659-671) показали селективность при жидкофазной экстракции молекулы формулы А в отношении урана и натрия.
Сравнительно недавно для образования комплексов с катионами Bennoura et coll. в Journal of Inclusion Phenomena and Macrocyclic Chemistry, 40, 95-98, 2001 предложили новый экстрагент, являющийся синтетическим производным молекулы формулы А: 1,2,5-O-триметил1-2,4,6-O-трикислотный гидроксамин-пара-трет-бутил-каликс[6]-арен (молекула показана ниже в виде формулы В).
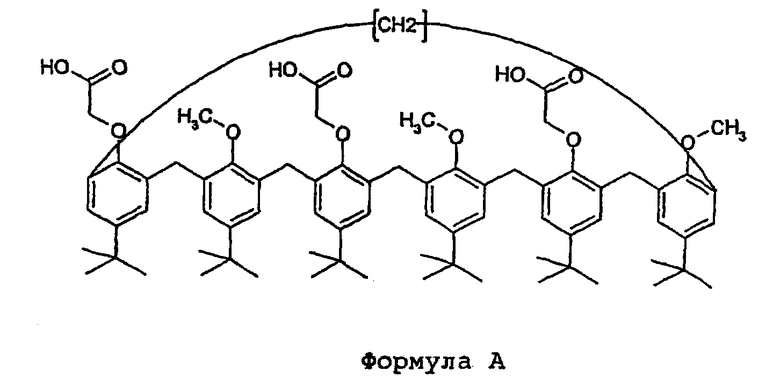
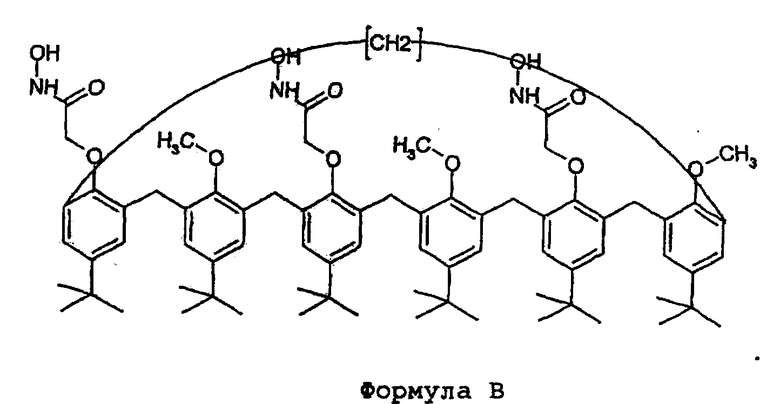
Насколько нам известно, молекулы формул А и В никогда не исследовались в рамках нанесенных на подложку мембран или привитых подложек.
Иммобилизация каликсаренов при помощи ковалентной связи на материалах подложки уже была описана S.P. Alexandratos et coll. в Macromolecules, 2001, 34, 206-210, затем в 2002 году Trivedi et coll. в Reactive and Functional Polymers, 50, 205-216. Кроме того, в книге Z. Asfari, изданной в 2001 году издательством Kluwer Academic Press (Нидерланды) и озаглавленной «Каликсарены» в главе, написанной R.Milbradt, V. Bohmer, стр.663-676, представлен обзор технологий иммобилизации каликсаренов и полученные продукты. Однако иммобилизация каликсаренов формул А и В никогда не была описана.
Ни один из описанных продуктов и ни одна из известных технологий не позволяет осуществить анализ катионов урана, и/или америция, и/или плутония с содержанием порядка 1 мБк на литр меньше, чем за 6 дней.
Раскрытие изобретения
Изобретатели неожиданно выявили, что замещение метоксигруппы в формулах А и В, описанных в литературе, другой группой и предпочтительно гидроксильной группой не снижает способностей комплексообразования по отношению к катионам урана, америция и плутония.
Это открытие было использовано для получения нанесенных на подложку жидких мембран, содержащих соединения общей формулы IA и/или IB, и новых материалов-подложек. Эти мембраны и эти материалы-подложки имеют свойство селективного комплексообразования с ураном, и/или америцием, и/или плутонием. Применение этих нанесенных мембран в соответствующих колонках для анализа при помощи экстракционной хроматографии позволяет произвести анализ вышеупомянутых элементов при содержании порядка 1 мБк/л.
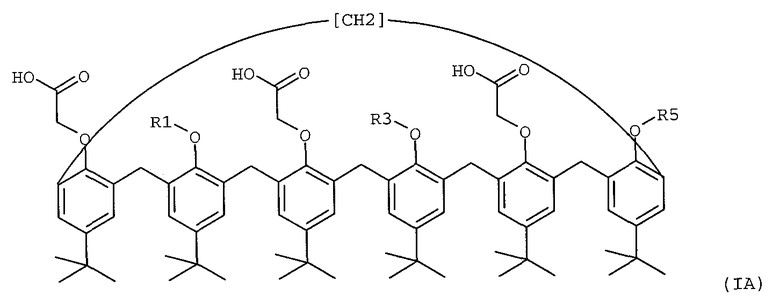
Таким образом, настоящее изобретение касается нового семейства соединений пара-трет-бутил-каликс[6]-аренов, имеющих формулу (IA) или (IB)
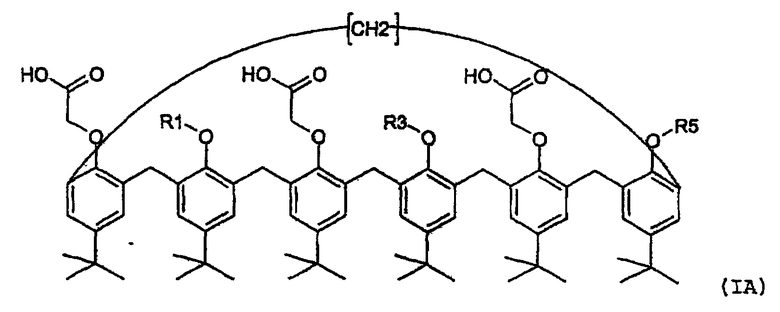
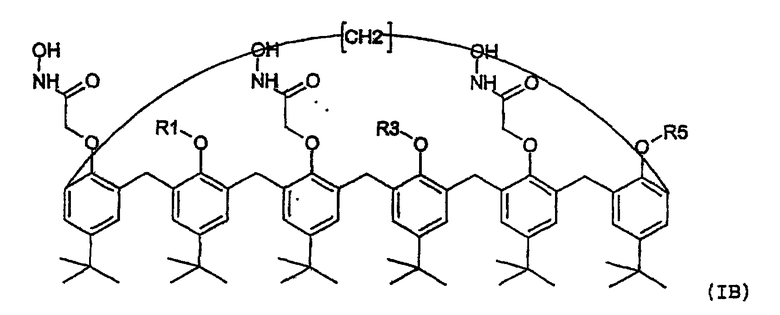
(где R1, R3 и R5, идентичные или разные, представляют собой, каждый отдельно:
(i) атом водорода или галогена,
(ii) ацетил, амино, фосфат, нитро, сульфат, карбокси, карбоксил, тиокарбокси, карбамат, тиокарбамат,
(iii) линейный или разветвленный алкил, содержащий от 1 до 60, предпочтительно от 1 до 30 атомов углерода, при необходимости, замещенный, и имеющий, при необходимости, по меньшей мере, одну двойную или тройную связь,
(iv) циклоалкил, содержащий от 3 до 12 атомов углерода, при необходимости, замещенный, и имеющий, при необходимости, по меньшей мере, одну двойную или тройную связь,
(v) арил, нафтил, арил-(С1-С30 алкил), (С1-С30 алкил)-арил, при необходимости, замещенные;
при этом радикалы (ii) - (v) могут замещаться атомами галогена, металлоорганическими соединениями, спиртовыми группами, аминогруппами, карбоксильными, сульфоновыми, сернокислыми, фосфорнокислыми, фосфоновыми или гидроксамино-кислотными или эфирными группами, карбаматами, тиокарбаматами, группами простых эфиров, тиоловыми группами, эпоксидными группами, тиоэпоксидными группами, изоцианатными, тиоизоцианатными группами, или один углерод этих радикалов может быть замещен гетероатомом азота, серы, фосфора, кислорода, бора, мышьяка;
(vi) полимер, выбранный из группы, в которую входят полистиролы, сополимеры хлор- и/или бромметилстирола и дивинилбензола, полиэфиры, полиакриламиды, полиглицидил метакрилаты, декстраны и агарозы;
при следующих условиях:
R1, R2 и R3 не представляют собой одновременно СН3 в (IA) и (IB),
R1, R2 и R3 не представляют собой одновременно CF2COOH в (IA) и (IB),
R1, R2 и R3 не представляют собой одновременно CH2CONHOH в (IA) и (IB).
Согласно предпочтительному варианту выполнения в формуле (IA) или (IB) два из R1, R2 и R3 представляют водород или метил, при этом третий является выбранным из (vi) полимером, выбранным из группы, в которую входят полистиролы, сополимеры хлор- и/или бромметилстирола и дивинилбензола, полиэфиры, полиакриламиды, полиглицидил метакрилаты, декстраны и агарозы.
Согласно другому предпочтительному варианту выполнения пара-трет-бутил-каликс[6]-арены в соответствии с настоящим изобретением являются соединениями формулы (IA) или (IB), в которой R1, R2 и R3 являются идентичными и предпочтительно представляют водород.
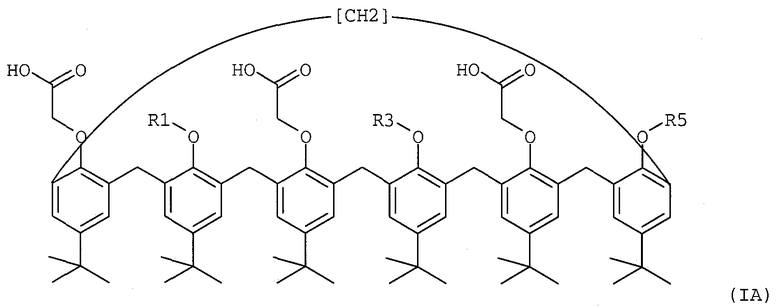
Объектом настоящего изобретения являются также нанесенные на подложку жидкие мембраны, содержащие пара-трети-бутил-каликс[6]-арен формулы (IA) или (IB)
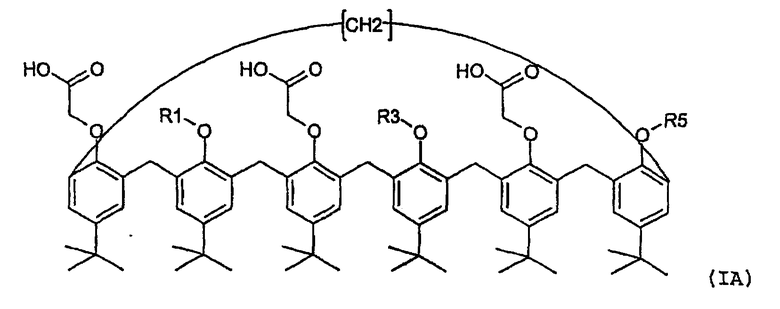
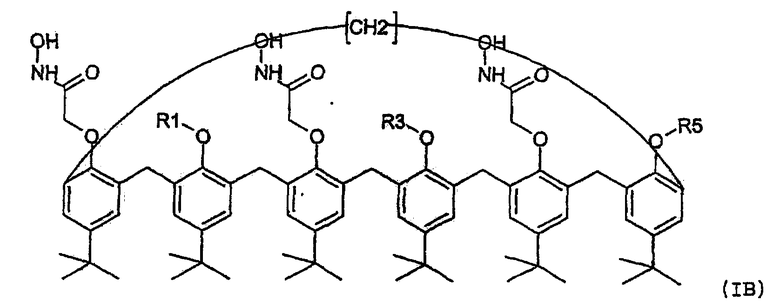
где R1, R3 и R5, идентичные или разные, представляют собой, каждый отдельно:
(i) атом водорода или галогена,
(ii) ацетил, амино, фосфат, нитро, сульфат, карбокси, карбоксил, тиокарбокси, карбамат, тиокарбамат,
(iii) линейный или разветвленный алкил, содержащий от 1 до 60, предпочтительно от 1 до 30 атомов углерода, при необходимости, замещенный, и имеющий, при необходимости, по меньшей мере, одну двойную или тройную связь,
(iv) циклоалкил, содержащий от 3 до 12 атомов углерода, при необходимости, замещенный, и имеющий, при необходимости, по меньшей мере, одну двойную или тройную связь,
(v) арил, нафтил, арил-(С1-С30 алкил), (С1-С30 алкил)-арил, при необходимости, замещенные;
при этом радикалы (ii) - (v) могут замещаться атомами галогена, металлоорганическими соединениями, спиртовыми группами, аминогруппами, карбоксильными, сульфоновыми, сернокислыми, фосфорнокислыми, фосфоновыми или гидроксамино-кислотными или эфирными группами, карбаматами, тиокарбаматами, группами простых эфиров, тиоловыми группами, эпоксидными группами, тиоэпоксидными группами, изоцианатными, изотиоцианатными группами, или один углерод этих радикалов может быть замещен гетероатомом азота, серы, фосфора, кислорода, бора, мышьяка;
(vi) полимер, выбранный из группы, в которую входят полистиролы, сополимеры хлор- и/или бромметилстирола и дивинилбензола, полиэфиры, полиакриламиды, полиглицидил метакрилаты, декстраны и агарозы;
при этом указанный продукт формулы (IA) или (IB) растворен в органическом растворителе и абсорбирован на подложке.
Органический растворитель в соответствии с настоящим изобретением имеет точку кипения, превышающую 60°С, что ограничивает, таким образом, испарение указанного растворителя во время хранения. Кроме того, он должен обладать хорошими свойствами растворения по отношению к каликсаренам в соответствии с настоящим изобретением. Среди наиболее предпочтительных растворителей можно указать толуол, ксилол, хлорбензол, орто-дихлорбензол, нитробензол, 1,4-диизопропил бензол, гексилбензол, керосин, тетрагидропиран, 1,2,3,4-тетрагидронафталин, пентанол и высшие спирты-гомологи, гликоли и их простые эфиры, например, такие как диэтиленгликоль дибутилэфир, сложные эфиры, такие как метилбензоат, простые эфиры, такие как ортонитрофенил пентилэфир или нитрофенил октилэфир.
В качестве примера можно указать следующие значения растворимости в моль/л соединения (IA) в различных растворителях: при 25°С 1,2-дихлорбензол: 3,13·10-3 М, хлорбутан: 1,6·10-3 М, изооктан: 1,25·10-3 М, изобутилацетат: 1,75·10-3 М, трет-бутилацетат: 5,25·10-3 М, изоамилбензоат: 2,40·10-3 М, бензилацетат: 3,71·10-3 М, метилбензоат: 6,41·10-3 М, бензонитрил: 1,81·10-3 М, 1-гексанол: 17,65·10-3 М, 1-гептанол: 14,28·10-3 М, диметиловый эфир диэтиленгликоля: 23,53·10-3 М, трет-бутил этиловый эфир диэтиленгликоля: 11,34·10-3 М, дибутиловый эфир диэтиленгликоля: 19,33·10-3 М, дипентиловый эфир: 2,32·10-3 М, изоамиловый эфир: 2,02·10-3 М, изобутиловый эфир: 2,32·10-3 М, 1,1,2-трихлор-трифторэтан: 1,57·10-3 М, 1,2,3,4-тетрагидронафталин: 6,4·10-3 М.
Материалы-подложки, образующие жидкие мембраны, имеют минеральное происхождение и выбраны из группы, в которую входят кремнеземные гели, оксиды, такие как глинозем, циркон или оксид титана, или имеют органическое происхождение и выбраны из группы, в которую входят полистирол-дивинилбензол, полиэфиры, полиакриламиды, полиглицидил метакрилаты, или имеют минералоорганическое происхождение и выбраны из группы, в которую входят композиты кремнезем/декстран или гидроксиапатит/агароза, и их смеси.
Предпочтительно подложка имеет форму частиц, при этом размер частиц колеблется от 10 нм до 10 мм, предпочтительно от 10 до 50 микрон, и диаметр пор колеблется от 10 до 5000 Å, предпочтительно от 100 до 500 Å.
Объектом настоящего изобретения являются также жидкие мембраны, содержащиеся в нанесенных на подложку жидких мембранах, то есть пара-трет-бутил-каликс[6]-арены формул (IA) и (IB), растворенные в растворителе, при этом растворитель является водонерастворимьм органическим растворителем, указанным выше.
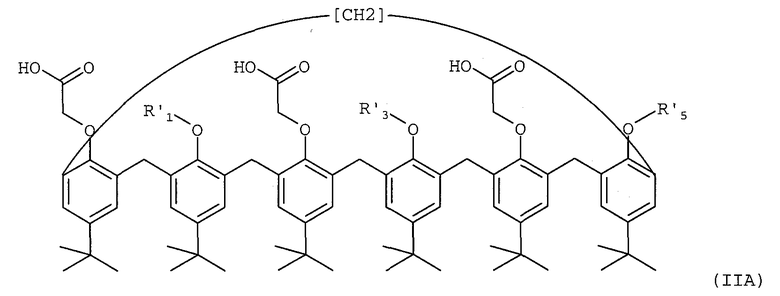
Объектом настоящего изобретения является также материал-подложка, который является пара-трет-бутил-каликс[6]ареном формулы (IIА) или (IIB)
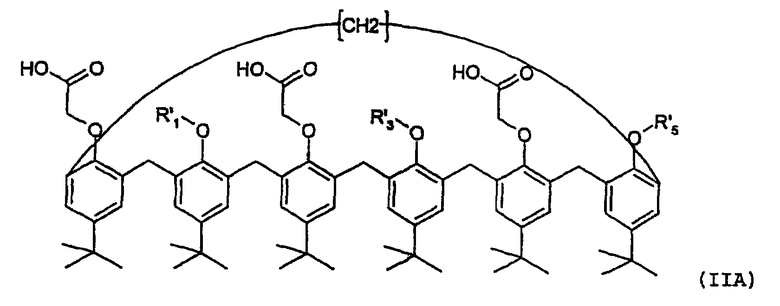
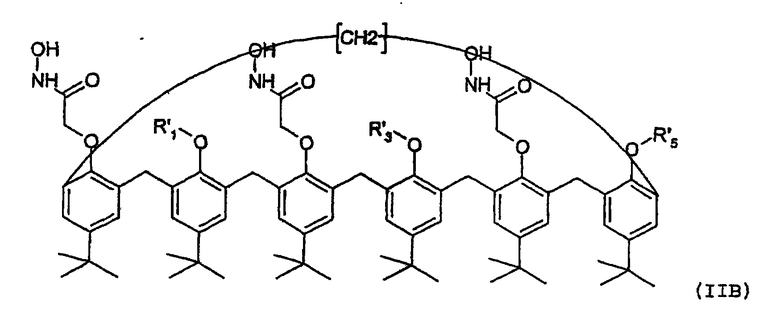
где R'1, R'3 и R'5, идентичные или разные, представляют собой, каждый отдельно:
(i) атом водорода или галогена,
(ii) ацетил, амино, фосфат, нитро, сульфат, карбокси, карбоксил, тиокарбокси, карбамат, тиокарбамат,
(iii) линейный или разветвленный алкил, содержащий от 1 до 60, предпочтительно от 1 до 30 атомов углерода, при необходимости, замещенный, и имеющий, при необходимости, по меньшей мере, одну двойную или тройную связь,
(iv) циклоалкил, содержащий от 3 до 12 атомов углерода, при необходимости, замещенный, и имеющий, при необходимости, по меньшей мере, одну двойную или тройную связь,
(v) арил, нафтил, арил-(С1-С30 алкил), (С1-С30 алкил)-арил, при необходимости, замещенные;
при этом радикалы (ii) - (v) могут замещаться атомами галогена, металлоорганическими соединениями, спиртовыми группами, аминогруппами, карбоксильными, сульфоновыми, сернокислыми, фосфорнокислыми, фосфоновыми или гидроксамино-кислотными или эфирными группами, карбаматами, тиокарбаматами, группами простых эфиров, тиоловыми группами, эпоксидными группами, тиоэпоксидными группами, изоцианатными, изотиоцианатными группами, или один углерод этих радикалов может быть замещен гетероатомом азота, серы, фосфора, кислорода, бора, мышьяка;
(vi) полимер, выбранный из группы, в которую входят полистиролы, сополимеры хлор- и/или бромметилстирола и дивинилбензола, полиэфиры, полиакриламиды, полиглицидил метакрилаты, декстраны и агарозы;
(vii) спейсер - подложка,
при этом спейсер является двухвалентным радикалом, выбранным из группы, в которую входят С1-С60 алкилены, предпочтительно С1-С30 алкилены, (С1-С60 алкил)-арилены, арил(С1-С60 алкилены), арил(С1-С60 алкил)-арилы, при этом двухвалентный радикал может быть замещен атомами галогена, металлоорганическими соединениями, спиртовыми группами, аминогруппами, кислотными группами, группами сложных эфиров, карбаматами, тиокарбаматами, группами простых эфиров, тиоловыми группами, эпоксидными группами, тиоэпоксидными группами, изоцианатными, изотиоцианатными группами, или один углерод этого двухвалентного радикала может быть замещен гетероатомом азота, серы, фосфора, кислорода, бора, мышьяка;
при этом спейсер выбирают из подложек минерального, органического или минералоорганического происхождения, предпочтительно в виде частиц, в которых размер частиц колеблется от 10 нм до 10 мм, предпочтительно от 10 до 50 микрон, и диаметр пор колеблется от 10 до 5000 Å, предпочтительно от 100 до 500 Å;
при условии, что, по меньшей мере, один из R'1, R'3 или R'5 является группой (vi) или (vii).
Материал-подложка входит в состав стационарной фазы хроматографической колонки.
В материале-подложке в соответствии с настоящим изобретением подложка имеет минеральное происхождение, и ее выбирают из группы, в которую входят кремнеземные гели, оксиды, такие как глинозем, циркон или оксид титана, или имеет органическое происхождение и выбрана из группы, в которую входят полистирол-дивинилбензол, полиэфиры, полиакриламиды, полиглицидил метакрилаты, или имеет минералоорганическое происхождение и выбрана из группы, в которую входят композиты кремнезем/декстран или гидроксиапатит/агароза.
Эту подложку получают из подложки, содержащей реактивные химические функциональные группы, при этом указанные химические функциональные группы могут быть органическими или минеральными, такими, например, как хлориды карбоновых кислот, аминокислоты, альдегиды, тиолы, сульфохлорид, изоцианат, металлический галогенид, причем этот перечень может быть продолжен.
Например, функционализированные подложки из частиц являются сополимером [5-(4-хлорметил)фенил]пентил-стирола и дивинилбензола, а также сополимером [5-(4-бромметил)фенил]пентил-стирола и дивинилбензола и сополимером 4-хлорметил-стирола и дивинилбензола.
Они могут иметь вид твердых частиц сферической или неправильной формы, гранулометрический состав и пористость которых могут меняться. На рынке имеются различные виды, такие как смолы Stratospheres®, продаваемые компанией Aldrich.
Для синтеза новых соединений общей формулы (IA) и (IB) осуществляют следующие операции:
синтезируют соединение 1,3,5-триметокси-пара-трет-бутилкаликс[6]арен формулы (а)
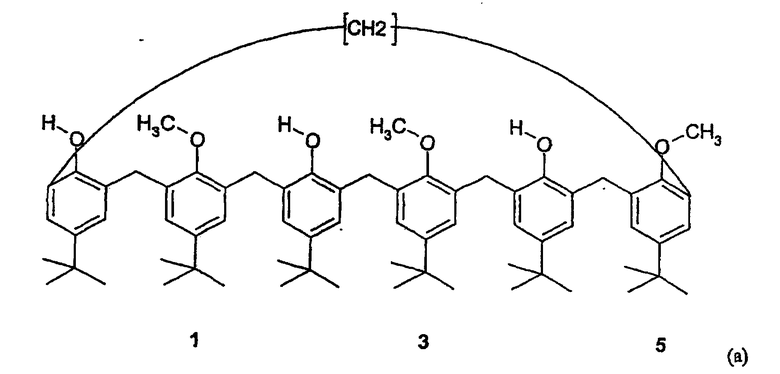
затем исходное соединение 2,4,6-триэтилэфир-1,3,5-триметокси-пара-трет-бутилкаликс[6]арен формулы (b)
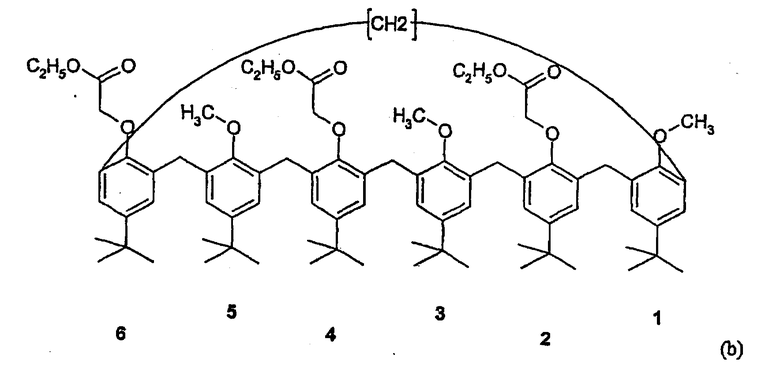
После этого, в случае необходимости, соединение формулы (b) модифицируют путем омыления или путем замещения функциональных групп этилового эфира карбоновой кислоты для получения соединений формул А или В.
Соединение формулы (b) можно также частично или полностью деметилировать, чтобы получить соединения, имеющие общие формулы (с1), (с2) или (с3).

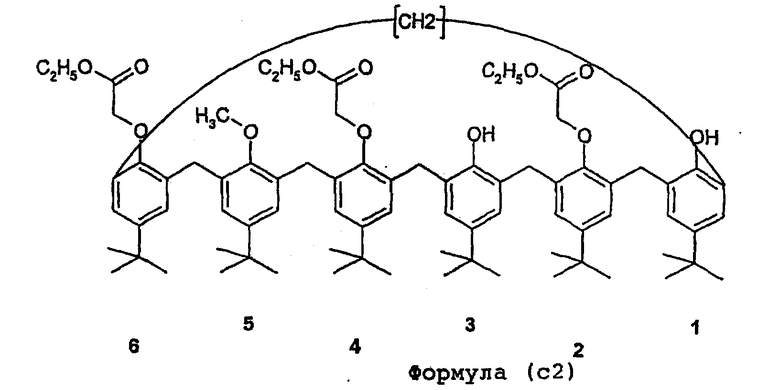
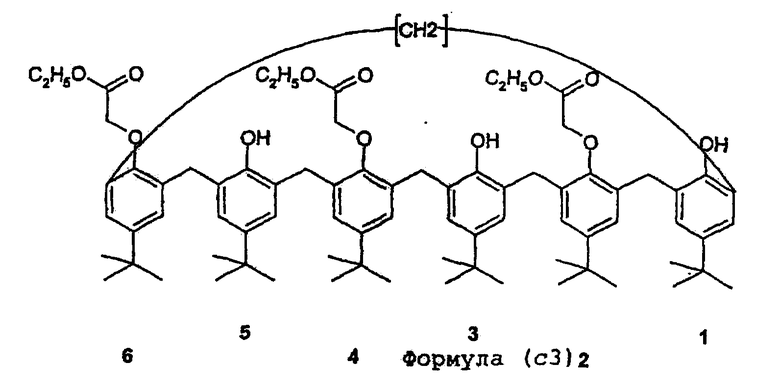
После этого соединения формул (с1), или (с2), или (с3) модифицируют путем химической модификации гидроксилов для получения, с одной стороны, соединений общей формулы (IA) путем омыления этиловых эфиров или, с другой стороны, соединений общей формулы (IB) путем удаления гидроксаминовых кислотных функциональных групп из этиловых эфиров.
Согласно варианту способа соединения формул (с1), или (с2), или (с3) могут затем вступать в прямую реакцию с соответственно функционализированными подложками для осуществления реакций ковалентной привитой сополимеризации через активирование их свободных фенольных гидроксилов, чтобы получить, с одной стороны, материалы-подложки общей формулы (IIA) путем омыления этиловых эфиров или, с другой стороны, материалы-подложки общей формулы (IIB) путем удаления гидроксаминовых кислотных функциональных групп из этиловых эфиров.
Таким образом, материал-подложку в соответствии с настоящим изобретением можно представить в следующем виде:
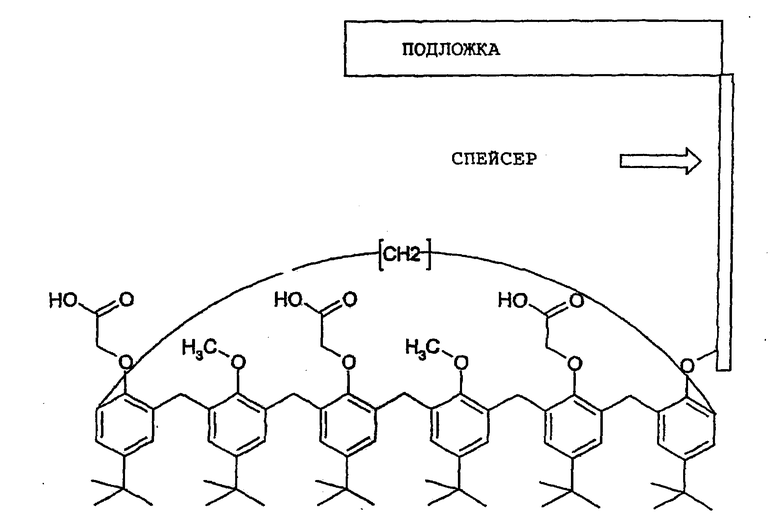
Далее детально представлены этапы этого синтеза:
А) синтез 1,3,5-триметокси-пара-тирет-бутилкаликс[6]арена (Mr=1014)
Согласно способу, прежде всего, синтезируют общее исходное вещество, содержащее функциональные группы сложного эфира карбоновой кислоты в положении 2, 4 и 6 и три метоксигруппы в положении 1, 3 и 5.
Для этого сначала необходимо синтезировать симметричное соединение 1,3,5-триметокси-n-трет-бутилкаликс[6]арена (Mr=1014) вышеуказанной формулы (а).
Пара-трет-бутилкаликс[6]арен, продаваемый на рынке, растворяют в безводном адетоне. Добавляют карбонат калия и взвесь взбалтывают в течение 3 часов в атмосфере азота. После этого добавляют избыточный метилиодид и реакционную суспензию постепенно доводят до состояния флегмы в течение 24 часов при взбалтывании.
После этого ацетон выпаривают насухо в вакууме при помощи ламинарного насоса, при этом температура водяной бани равна 60°С. Полученный сухой остаток растворяют в хлороформе. После растворения добавляют воду и двухфазную среду подвергают активному перемешиванию, затем подкисляют концентрированной до 12 М соляной кислотой. Затем нижнюю органическую фазу выделяют путем отстаивания, после чего промывают водой до нейтрализации промывочной воды, содержащейся в верхней фазе. После этого органическую фазу сушат на безводном сульфате натрия и фильтруют. Прозрачную органическую фазу концентрируют до сухого состояния при помощи вакуумного ламинарного насоса при температуре водяной бани 60°С. Сухой остаток очищают при помощи хроматографии низкого давления на кремнеземном геле хроматографического качества. Используемым элюентом является чистый синтетический метиленхлорид, стабилизированный амиленом. Степень чистоты различных фракций контролируют в метилен/этанол хлориде 95/5 при помощи тонкослойной хроматографии на пластинах самородного кремнезема.
Фракции, содержащие симметричное соединение 1,3,5-триметокси-n-трет-бутилкаликс[6]арена, проявляющееся в виде монохромного пятна при тонкослойной хроматографии (проявление паром йода), объединяют, затем концентрируют до сухого состояния.
Остаток используют в полученном виде на следующем этапе.
В) синтез 2,4,6-триэтилового эфира-1,3,5-триметокси-пара-трет-бутилкаликс[6]арена (Mr=1272,5) (формула (b))
На втором этапе способа синтезируют общее исходное вещество, содержащее функциональные группы эфира карбоновой кислоты в положении 2, 4 и 6 и три метоксигруппы в положении 1, 3 и 5, модифицируя все фенольные гидроксильные группы соединения, полученного на предыдущем этапе, в положениях 2, 4 и 6, при этом положения 1, 3 и 5 защищены метоксигруппами.
Продукт, полученный в предыдущем примере (формула (а)), растворяют в безводном (высушенном на гидриде натрия) ДМФ (диметилформамид) в атмосфере азота. Добавляют карбонат цезия в очень большом избытке и полученную суспензию взбалтывают в течение 4 часов в атмосфере азота. После этого в течение 5 минут на реакционную суспензию выливают этилбромацетат в большом избытке и среду при сильном взбалтывании постепенно доводят до состояния флегмы в течение 24 часов в атмосфере азота.
ДМФ выпаривают насухо в вакууме при помощи ламинарного насоса, температура водяной ванны составляет 60°С. Полученный твердый сухой остаток растворяют в хлороформе. После растворения добавляют воду и двухфазную среду подвергают активному перемешиванию, затем подкисляют концентрированной до 12М соляной кислотой. Затем нижнюю органическую фазу несколько раз промывают водой, после чего сушат на безводном сульфате натрия. После фильтрования органическую фазу концентрируют до сухого состояния, при этом температура водяной ванны составляет 60°С, и используют вакуум ламинарного насоса. Сухой остаток помещают в этанол. Получают белую суспензию. Белое твердое вещество выделяют путем фильтрования. Его промывают этанолом несколько раз на фильтре, затем сушат при температуре 40°С в вакуумном сушильном шкафу.
С) синтез 2,4,6-триэтилового эфира-1-гидрокси, 3,5-диметокси-пара-трет-бутилкаликс[6]арена (соединение (с1))
1,3-дигидрокси,5-монометокси-пара-трет-бутилкаликс[6]арен (соединение (с2))
1,3,5-тригидрокси-пара-трет-бутилкаликс[6]арен (соединение (с3))
На третьем этапе способа частично или полностью деметилируют общее исходное вещество формулы (b), содержащее функциональные группы эфира карбоксильной кислоты в положениях 2, 4 и 6 и одновременно фенольные гидроксильные группы и метоксигруппы в положениях 1, 3 и 5 (соединения (с1) или (с2)) или только фенольные гидроксильные группы в положениях 1, 3 и 5 (соединение (с3)).
Для этого ранее полученное соединение формулы (b) растворяют в безводном хлороформе, предварительно высушенном на гидриде натрия. Среду перемешивают в атмосфере азота, затем добавляют триметилсилил иодид (агент деметоксилирования каликсаренов) в стехиометрии или в недостатке, в зависимости от того, отдается ли предпочтение моно-, ди- или тридеметилированию, и реакционную среду в течение 2 часов доводят до состояния флегмы в атмосфере азота. Реакцию осуществляют под контролем тонкослойной хроматографии в толуоле/этилацетате 90/10 на пластинах кремнезем/полиэфир. В случае необходимости снова однократно добавляют триметилсилил иодид после охлаждения в реакционной среде, в зависимости от кинетики образований необходимых видов (молекулы формулы (с1), или (с2), или (с3)). Реакционную среду снова доводят до состояния флегмы в течение 2 или более часов.
Реакцию прекращают путем добавления воды. Реакционную среду подкисляют НСl 1М и нижнюю органическую фазу красно-кирпичного цвета выделяют путем отстаивания. Ее промывают водой до достижения нейтрального рН промывочной воды (верхняя фаза).
Нижнюю хлороформную фазу выпаривают насухо в вакууме при помощи ламинарного насоса, при этом температура водяной ванны составляет 60°С, после сушки на безводном сульфате натрия. Полученный сухой остаток затем очищают при помощи хроматографии низкого давления на кремнеземном геле хроматографического качества. Используемым элюентом является смесь толуол/этилацетат 90/10. Степень чистоты различных фракций проверяют в смеси толуол/этилацетат 90/10 при помощи тонкослойной хроматографии на пластинах из самородного кремнезема, нанесенного на полиэфирную подложку.
Фракции, содержащие соединения (с1), или (с2), или (с3), объединяют и отдельно сушат в описанных выше условиях выпаривания.
Каждый сухой остаток используют в полученном виде для возможных дальнейших химических модификаций.
D) синтез 2,4,6-триэтилового эфира-1-R1,3,5-диметокси-пара-трет-бутилкаликс[6]арена (соединение (d1))
1,3-R1-R3,5-монометокси-пара-трет-бутилкаликс[6]арен (соединение (d2))
1,3,5-R1-R5-пара-трет-бутилкаликс[6]арен (соединение d3))
На другом этапе способа синтезируют соединение общей формулы (d1), или (d2), или (d3) соответственно из полученных ранее соединений (с1), или (с2), или (с3). Фенольные гидроксильные функциональные группы в положениях 1, и/или 3, и/или 5 модифицируют и вводят группы R1, и/или R3, и/или R5, учитывая, что они не могут представлять собой гидроксилы или метилы.
Для этого полученное ранее соединение формулы (с1), или (с2), или (с3) растворяют в безводном (высушенном на гидриде натрия) ДМФ (диметил формамид) в атмосфере азота. Добавляют карбонат цезия в очень большом избытке и полученную суспензию взбалтывают в течение 4 часов в атмосфере азота. После этого в течение 5 минут на реакционную суспензию выливают галогенид в большом избытке, органический остаток которого представляет группу R1, или R3, или R5, и среду при сильном взбалтывании постепенно доводят до состояния флегмы в течение 24 часов в атмосфере азота.
ДМФ выпаривают насухо в вакууме при помощи ламинарного насоса, температура водяной ванны составляет 80°С. Полученный твердый сухой остаток растворяют в хлороформе. После растворения добавляют воду и двухфазную среду подвергают активному перемешиванию, затем подкисляют концентрированной до 12 М соляной кислотой. Затем нижнюю органическую фазу несколько раз промывают водой, после чего сушат на безводном сульфате натрия. После фильтрования органическую фазу концентрируют до сухого состояния, при этом температура водяной ванны составляет 60°С, и используют вакуум ламинарного насоса. Сухой остаток помещают в хлороформ.
Нижнюю хлороформную фазу выпаривают насухо в вакууме при помощи ламинарного насоса, при этом температура водяной ванны составляет 60°С, после сушки на безводном сульфате натрия. Полученный сухой остаток затем очищают при помощи хроматографии низкого давления на кремнеземном геле хроматографического качества. Используемым элюентом является смесь толуол/этилацетат 90/10. Степень чистоты различных фракций проверяют в смеси толуол/этилацетат 90/10 при помощи тонкослойной хроматографии на пластинах из самородного кремнезема, нанесенного на полиэфирную подложку.
Фракции, содержащие соединения (d1), или (d2), или (d3) объединяют и отдельно сушат в описанных выше условиях выпаривания.
Каждый сухой остаток используют в полученном виде для возможных дальнейших химических модификаций.
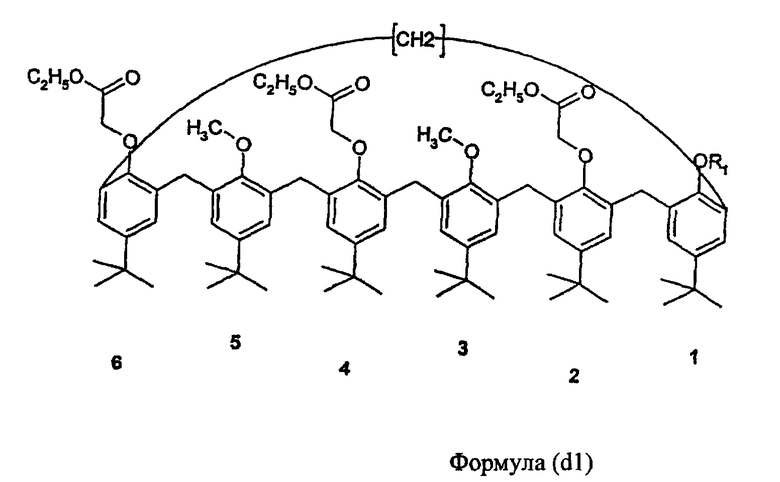
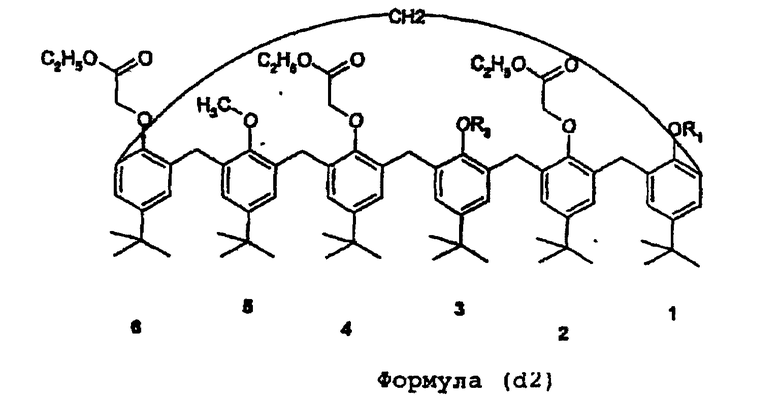
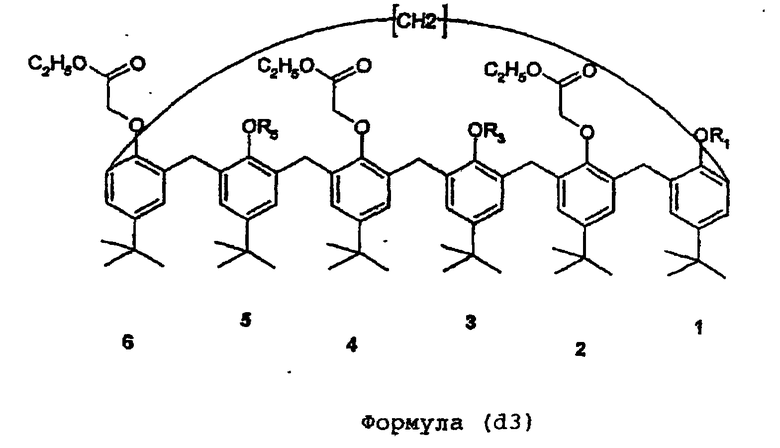
Е) привитая сополимеризация на подложке одного из соединений (с1), (с2), (с3), (d1), (d2), (d3)
На другом этапе способа на подложке ковалентно осуществляют привитую сополимеризацию соединения формулы (с1), или (с2), или (с3), или (d1), или (d2), или (d3), содержащего функциональные группы эфира карбоновой кислоты в положениях 2, 4 и 6 и одновременно фенольные гидроксильные функциональные группы и метоксигруппы в положениях 1 и/или 3 (соединения (с1) или (с3)), или только фенольные гидроксильные группы в положениях 1, 3 и 5 (соединение (с3)), или группы R1, и/или R3, и/или R5 (где R1, R3, R5 являются отличными от водорода и от метила) в положениях 1, 3 или 5 (соединения (d1), (d2), (d3)).
Соединение формулы (с1), или (с2), или (с3), или (d1), или (d2), или (d3) растворяют в безводном диметилформамиде, предварительно высушенном на гидриде натрия. Среду перемешивают в атмосфере азота до полного растворения. Добавляют карбонат цезия в очень большом избытке, затем имеющуюся на рынке смолу с известными и количественно определенными функциональными группами, при этом указанные группы выбирают таким образом, чтобы иметь возможность реакции на фенольном гидроксиле или на группе R1, и/или R3, и/или R5, и реакционную среду выдерживают в течение 72 часов при температуре 60°С в атмосфере азота.
Реакционную суспензию фильтруют и твердую фазу промывают при помощи ДМФ, затем ацетоном, затем НСl 1М до состояния кислого рН промывочной воды. После этого смолу промывают водой до нейтрального состояния, затем этанолом.
Е) Синтез соединения общей формулы IIA
В факультативном варианте способа используют соединение общей формулы (а), или (с1), или (с2), или (с3), или (d1), или (d2), или (d3) или соединение, полученное при помощи способа, описанного в разделе Е.
Указанное соединение растворяют или суспендируют в этаноле. Добавляют водный раствор поташа в очень большом избытке относительно стехиометрии, вычисленной по предназначенному для омыления количеству групп этилового эфира, и реакционную среду (раствор или суспензия) доводят до состояния флегмы в течение 4 часов.
Реакционную массу охлаждают и среду подкисляют НСl 12М.
Суспензию фильтруют и осадок (или подложку) промывают водой до нейтрального рН фильтрационных вод, затем этанолом.
Твердую фазу сушат при температуре 40°С в вакууме до постоянного веса.
G) Синтез соединения общей формулы IIB
В факультативном варианте способа используют соединение общей формулы (а), или (с1), или (с2), или (с3), или (d1), или (d2), или (d3) или соединение, полученное при помощи способа, описанного в разделе Е.
Указанное соединение растворяют или суспендируют в тетрагидрофуране (ТГФ). Добавляют метанольный раствор гидроксиламинхлоргидрата в избытке по отношению к стехиометрии, вычисленной по количеству групп этилового эфира. При этом добавляют второй раствор, содержащий расслоенный поташ, предварительно растворенный в смеси метанол/ТГФ, и выдерживают при температуре +5°С. Среду (раствор или суспензия) перемешивают в течение 7 дней при окружающей температуре в атмосфере азота.
Реакционную среду выпаривают до сухого состояния при температуре водяной бани 60°С в вакууме.
Остаток помещают в метиленхлорид, затем добавляют уксусную кислоту. Среду перемешивают в течение 4 часов при 20-25°С, затем снова сушат.
Реакционную среду выпаривают до сухого состояния при водной бане температурой 60°С в вакууме.
Объектом настоящего изобретения является также применение нанесенной на подложку жидкой мембраны, описанной выше, и/или материала-подложки в соответствии с настоящим изобретением для селективного комплексообразования и анализа элементов урана, америция и плутония и других радиоактивных элементов в их катионной форме.
Объектом настоящего изобретения является также применение нанесенной на подложку жидкой мембраны и/или материала-подложки в соответствии с настоящим изобретением для извлечения из смеси, по меньшей мере, двух компонентов, выбранных из группы, в которую входят органические, минеральные или минералоорганические молекулы, по меньшей мере, части одного из этих компонентов или для разделения указанных компонентов хроматографическим методом.
Нанесенные на подложку жидкие мембраны и материалы-подложки в соответствии с настоящим изобретением предпочтительно предназначены для эксклюзионной хроматографии. Они обеспечивают детектирование и выделение ничтожных количеств продукта, в частности выделение урана, и/или америция, и/или плутония с содержанием порядка 1 мБк/л.
Далее следует более подробное описание настоящего изобретения на следующих примерах, представленных в качестве иллюстраций и не являющихся ограничительными.
ПРИМЕРЫ
Пример 1:
Материал-подложка, содержащий 3,5-диметокси-2,4,6-карбоновая кислота-n-трет-бутилкаликс[6]арен
1-1: синтез 1,3,5-триметокси-p-трет-бутилкаликс[6]арена (Mr=1014)
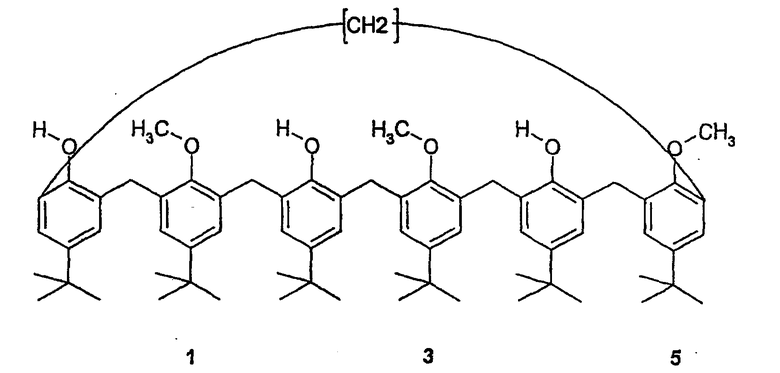
В стеклянный 20-литровый реактор, оборудованный конденсатором, вводят 194,7 г n-трет-бутилкаликс[6]арена (0,2 моль - Mr=972) и 15 л безводного ацетона. Среду взбалтывают в атмосфере азота до полного растворения. Добавляют 82,9 г карбоната калия (0,6 моль) и суспензию взбалтывают в атмосфере азота в течение 3 часов. В течение 5 минут выливают 113,6 г метилиодида (0,8 моль) и реакционную среду постепенно доводят до состояния флегмы в течение 24 часов. Ацетон выпаривают до сухого состояния в вакууме ламинарного насоса, установив температуру водяной ванны в 60°С. Полученный твердый сухой остаток растворяют в 5,0 л хлороформа. После растворения добавляют 1,0 л воды и двухфазную среду подвергают сильному взбалтыванию. Медленно, с учетом выделения углекислого газа, добавляют 0,1 л концентрированной до 12 М соляной кислоты. После этого органическую фазу промывают 5 раз при помощи 1,0 л воды, затем сушат на 200 г безводного сульфата натрия. После фильтрования органическую фазу концентрируют до сухого состояния при водяной ванне температурой 60°С в ламинарном вакууме. Получают 250 г сухого остатка, который затем очищают при помощи хроматографии низкого давления на 15 кг кремнеземного геля 40-200 мкм (размеры поры 60 Å). Используемым элюентом является метиленхлорид (40 л).
Степень чистоты различных фракций проверяют в метиленхлориде/этаноле 95/5 при помощи тонкослойной хроматографии на пластинах самородного кремнезема.
Из фракций, содержащих чистый 1,3,5-триметокси-каликсарен, получают 40 г остатка (39,45 ммоль: выход = 19,7%).
Н2 ЯМР-спектр в CDCl3 показывает следующие химические сдвиги:
7,00 частей на миллион (s, 6H, ArH мета ОН) - 6,90 частей на миллион (s, 6H, ArH мета ОСН3) - 6,75 частей на миллион (s, 3Н, ОН) - 3,89 частей на миллион (s, 9H, ОСН3) -3,47 частей на миллион (s, 12H, ArCH2CH), 1,20 частей на миллион (s, 27H, трет-бутил пара ОН) - 1,00 частей на миллион (s, 27H, трет-бутил пара ОСН3).
1-2: синтез 2,4,6-триэтилового эфира-1,2,5-триметокси-р-трет-бутилкаликс[6]арена (Mr=1272,5)
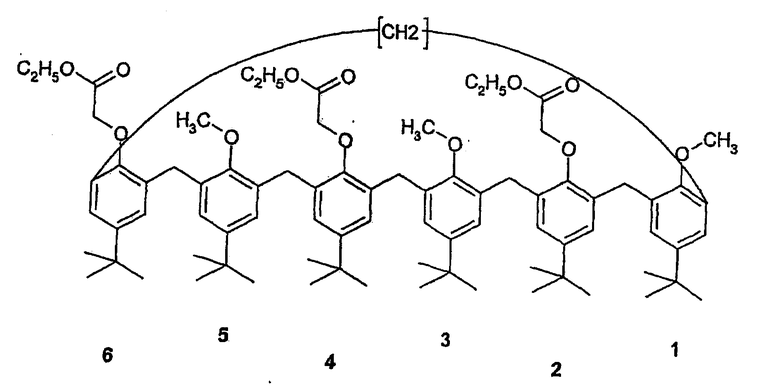
В стеклянный 10-литровый реактор, оборудованный конденсатором, вводят 40,0 г продукта, полученного в 1-1 (39,45 ммоль), и 4 л безводного диметилформамида (ДМФ) (высушенного на гидриде натрия). Среду взбалтывают в атмосфере азота до полного растворения. Добавляют 81,6 г карбоната цезия (0,25 моль) и суспензию взбалтывают в атмосфере азота в течение 4 часов. В течение 5 минут выливают 52,7 г этилбромацетата (0,31 моль) и реакционную среду постепенно доводят до состояния флегмы в течение 24 часов в атмосфере азота.
ДМФ выпаривают до сухого состояния в вакууме ламинарного насоса, установив температуру водяной ванны в 80°С. Полученный твердый сухой остаток растворяют в 2,0 л хлороформа. После растворения добавляют 0,5 л воды и двухфазную среду подвергают сильному взбалтыванию. Медленно, с учетом выделения углекислого газа, добавляют 20 мл концентрированной до 12 М соляной кислоты. После этого органическую фазу промывают 5 раз при помощи 0,5 л воды, затем сушат на 20 г безводного сульфата натрия. После фильтрования органическую фазу концентрируют до сухого состояния при водяной ванне температурой 60°С в ламинарном вакууме. Остаток помещают в 300 мл этанола. Получают белую суспензию. Твердое вещество извлекают при помощи фильтрования и промывают 3 раза при помощи 40 мл этанола, затем сушат при температуре 40°С в вакуумном сушильном шкафу.
После сушки при постоянном весе получают 43,2 г белого порошка (33,95 ммоль:выход = 86%).
Н+-ЯМР-спектр в CDCl3 показывает следующие химические сдвиги:
6,71 частей на миллион (s широкий, 12Н, ArH мета) - 4,55 частей на миллион (s, 6Н, ArCH2CO2R) - 4,29 частей на миллион (qt, 6H, О-СН2-метил J=7 Гц) - 3,47 частей на миллион (s, 21H, ArCH2Ar + метил ОСН3), 1,38 частей на миллион (s, 54P, трет-бутил) - 1,33 частей на миллион (t, 9H, метил ОСН2СН3 J=7 Гц).
1-3: синтез 1-гидрокси-3,5-диметокси-2,4,6-триэтилового эфира-n-трет-бутилкаликс[6]арена (Mr=1258,5)
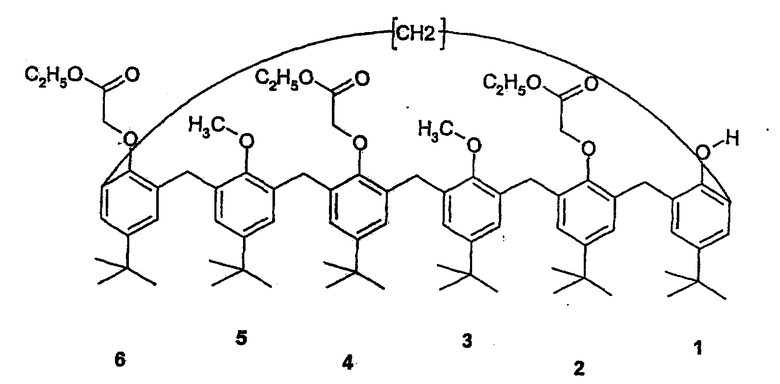
В 5-литровый стеклянный реактор, оборудованный конденсатором, вводят 27,8 г продукта (белый порошок), полученного ранее на 1-2 (21,85 ммоль - Mr=1272,5), и 1,5 л безводного хлороформа, предварительно высушенного на гидриде натрия. Среду взбалтывают в атмосфере азота до полного растворения. Добавляют 3,1 мл (4,37 г) триметилсилилиодида (21,85 ммоль - Mr=200,1) и реакционную среду в течение 2 часов доводят до состояния флегмы в атмосфере азота. Реакцию осуществляют под контролем тонкослойной хроматографией в толуоле/этилацетате 90/10 на пластине кремнезем/полиэфир. После охлаждения реакционной среды опять однократно добавляют 3,1 мл (4,37 г) триметилсилилиодида. Среду снова постепенно в течение 2 часов доводят до состояния флегмы.
Реакцию останавливают добавлением 2 л воды. Добавляют 100 мл HCl 1М и извлекают органическую фазу цвета красного кирпича. Ее 2 раза промывают 1 л воды.
Хлороформную фазу выпаривают до сухого состояния в вакууме ламинарного насоса, при этом температура водяной ванны составляет 60°С, после сушки при помощи 200 г безводного сульфата натрия. Получают 27,2 г сухого остатка, который затем очищают при помощи хроматографии низкого давления на 3 кг кремнеземного геля 40-200 мкм (размер поры 60 Å). Используемым элюентом является смесь толуол/этилацетат 90/10 (20 л). Чистоту различных фракций контролируют в смеси толуола/этилацетата 90/10 при помощи тонкослойной хроматографии на пластинах самородного кремнезема, нанесенного на полиэфирную подложку.
Получают 8,5 г остатка (6,75 ммоль: выход=30,7%) после сушки при водяной ванне 60°С в вакууме фракций, содержащих чистый 1-гидрокси-3,5-диметокси-2,4,6-триэтиловый эфир-n-трет-бутилкаликс[6]арен.
Масс-спектр, полученный в режиме FAB при помощи положительной химической ионизации, подтверждает наличие ожидаемого продукта (МН+ 1259 дальтон).
H+ - ЯМР-спектр в CDCl3 показывает следующие химические сдвиги:
6,75 частей на миллион (s, 13H, ArH мета + ОН фенол) - 4,55 частей на миллион (s, 6Н, ArCH2CO2R) - 4,29 частей на миллион (qt, 6H, О-СН2-метил J=7 Гц) - 3,47 частей на миллион (s, 20H, ArCH2Ar + метил ОСН3), 1,38 частей на миллион (s, 54H, трет-бутил)-1,33 частей на миллион (t, 9H, метил ОСН2СН3).
1-4: Материал-подложка, содержащий 1е-3,5-диметокси-2,4,6-триэтиловый эфир-n-трет-бутилкаликс[6]арен
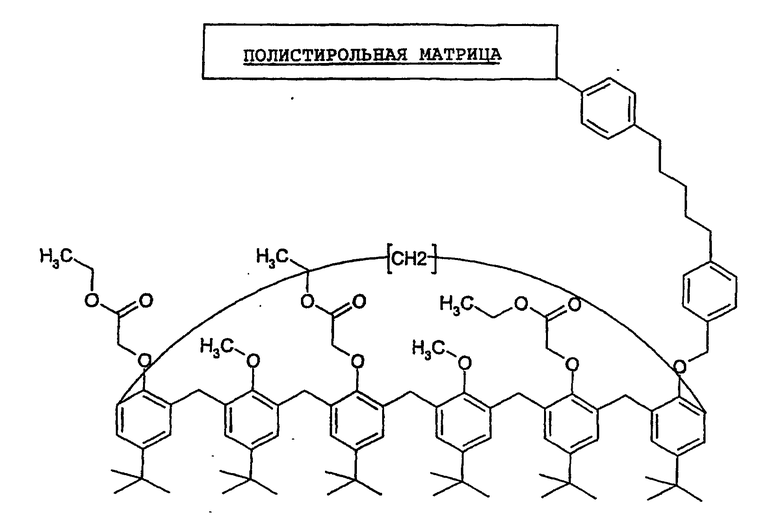
В 250-миллилитровый стеклянный реактор, оборудованный конденсатором, вводят 8,5 г продукта, полученного ранее на 1-3 (6,75 ммоль - Mr=1258,5), и 150 мл безводного ДМФ, предварительно высушенного на гидриде натрия. Среду взбалтывают в атмосфере азота до полного растворения. Добавляют 20 г карбоната цезия (61 ммоль), затем 10 г выпускаемой в продажу смолы полистирола, модифицированной хлорметилфенил-пентилом (закупаемая у Aldric смола ХМФП: [5-[4-(хлорметил)фенил]пентил]стирол, связанный полимер номер 513776) и реакционную среду доводят в течение 72 часов при 60°С в атмосфере азота.
Реакционную суспензию фильтруют и твердую фазу промывают в ДМФ (2 раза по 50 мл), затем в ацетоне (3 раза по 50 мл), затем в HCl 1М (4 раза по 100 мл), затем в воде (5 раз по 100 мл), затем в этаноле (3 раза по 50 мл).
Получают 16,15 г сухой смолы после сушки в вакууме при 60°С до постоянного веса.
Расчетная степень сшивания после микроанализа составляет 0,2 ммоль каликса/г смолы.
Микроанализ является следующим:
С%: 80,80 Н%: 7,29 Cl: 0,79
Микроанализ исходной смолы хлорметилфенил-пентила является следующим:
С%: 86,52 Н%: 7,87 Cl: 3,91
1-5: Материал-подложка, содержащий 1-3,5-диметокси-2,4,6-трикарбоновая кислота-n-трет-бутилкаликс[6]арен
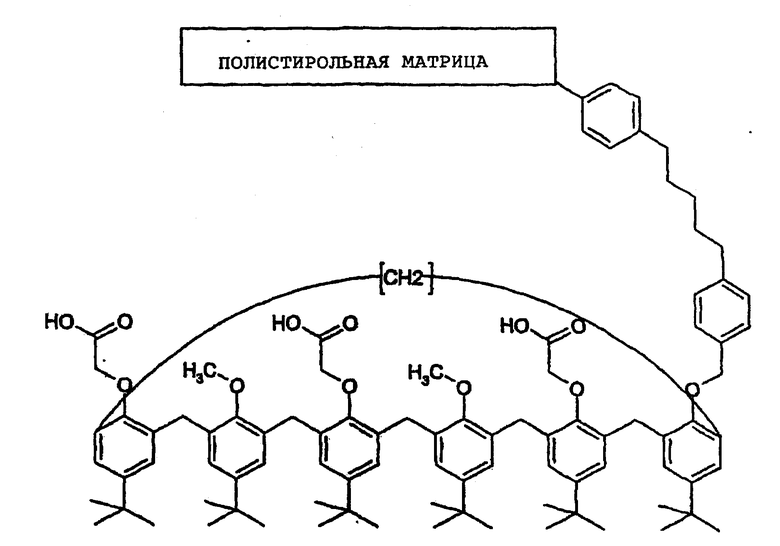
В 250-миллилитровый стеклянный реактор, оборудованный конденсатором, вводят 10 г сухой смолы, полученной ранее на 1-4 и 100 мл этанола. 6,1 г Поташа в таблетках 85% растворяют в 100 мл воды и полученный раствор добавляют однократно в реактор. Реакционную среду доводят до состояния флегмы в атмосфере азота в течение 4 часов.
Реакционную суспензию охлаждают, затем добавляют 12 мл 12М HCl. рН Суспензии составляет 1. После 1 часа взбалтывания суспензию фильтруют, затем промывают водой 8 раз по 100 мл, затем этанолом (3 раза по 50 мл).
Получают 9,7 г смолы после сутки при 60°С в вакууме до постоянного веса.
Смолу используют в полученном виде без дополнительной аналитической характеристики.
ПРИМЕР 2
Нанесенная на подложку жидкая мембрана, содержащая 1-3,5-триметокси-2,4,6-тригидроксаминовая кислота-n-трет-бутилкаликс[6]арен (Mr=1234,6)
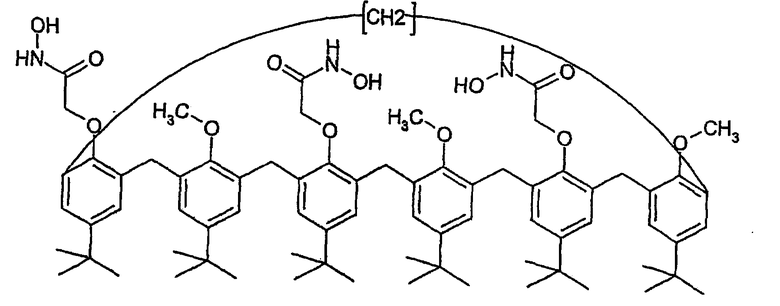
В 250-миллилитровый стеклянный реактор, оборудованный конденсатором, вводят 1,8 г продукта, полученного в примере 1-2 (1,4 ммоль), и 50 мл тетрагидрофурана (ТТФ). 2,02 г гидроксиламинхлоргидрата (29 ммоль) растворяют в 80 мл метанола и 40 мл ТГФ, затем полученный раствор добавляют к раствору, полученному в реакторе. После этого другой предварительно полученный (и выдержанный при +5°С) раствор расслоенного поташа (2,04 г 100%, то есть 36 моль) в 24 мл метанола и 12 мл ТГФ добавляют однократно в реактор. Реакционную среду перемешивают в течение 7 дней при окружающей температуре в атмосфере азота.
Реакционную суспензию выпаривают до сухого состояния при температуре водяной ванны 60°С в вакууме. Остаток помещают в смесь метиленхлорид/уксусная кислота 50 мл/10 мл и взбалтывают в течение 4 часов. Реакционную массу сушат в вакууме при температуре водяной ванны 60°С.
Сухой остаток помещают в 20 мл дихлорметана. Ожидаемый продукт осаждается в виде белой твердой фазы.
Получают 1,5 г белой твердой фазы после сушки при 60°С в вакууме до постоянного веса (выход=85,9%).
Масс-спектр, полученный при электроспрей-ионизации ESI, подтверждает наличие ожидаемого продукта (M/z 1234 дальтон).
H+ - ЯМР-спектр при 300 МГц в DMSO дает следующие химические сдвиги:
10,8 частей на миллион (s, 3Н, -NH) - 9,08 частей на миллион (s, 3H, -ОН гидроксаминовой группы) - 7,23 частей на миллион (s, 6H, ArH мета OCH2COOEt) - 6,57 частей на миллион (s, 6H, ArH мета ОСН3) - 4,44-4,33 частей на миллион (q, 18H, ArCH2CO2R+ArCH2Ar) - 2,50 частей на миллион (s широкий, 9Н, метокси) - 1,36 частей на миллион (s, 27H, трет-бутил пара OCH2CONHOH), 0,73 частей на миллион (s, 27H, трет-бутил пара ОСН3).
2 г эпоксидной смолы Macrocrep от компании BIORAD с гранулометрией 70-100 мкм, серия 11/99, добавляют в предварительно полученный раствор 15,6 мг полученной белой твердой фазы, отобранной ранее из 1,5 г полученной твердой фазы, ранее растворенной в 20 мл дихлорметана, и 1,12 мл 1,2,3,4-тетрагидронафталина. Суспензию медленно выпаривают при окружающей температуре до постоянного веса: получение белой твердой фазы.
Полученный вес: 2,2 г.
ПРИМЕР 3
Нанесенная на подложку жидкая мембрана, содержащая 1-3,5-триметокси-2,4,6-трикарбоновая кислота-n-трет-бутилкаликс[6]арен (Mr=1174,4)
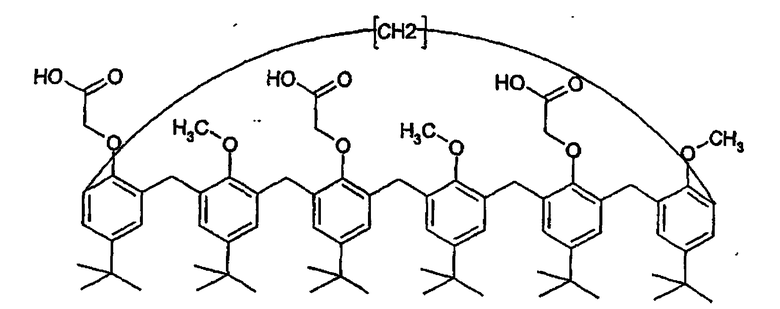
В 250-миллилитровый стеклянный реактор, оборудованный конденсатором, вводят 5,9 г продукта (белый порошок), полученного в примере 1-2 (4,00 ммоль), и 150 мл этанола. Предварительно приготовленный (и выдержанный при +5°С) раствор расслоенного поташа (12 г 100%, то есть 214 ммоль) в 150 мл воды добавляют однократно в реактор. Реакционную среду доводят до состояния флегмы в течение 4 часов в атмосфере азота.
25 мл HCl 12М медленно добавляют после охлаждения реакционной массы до 20°С. Ожидаемый продукт осаждается в виде белой твердой фазы. После этого суспензию фильтруют и твердое вещество промывают водой 8 раз по 50 мл и этанолом 2 раза по 50 мл. Затем твердую фазу сушат при 40°С в вакууме до постоянного веса,
Получают 4,2 г твердого вещества после сушки при 40°С в вакууме до постоянного веса (выход = 99%).
H+ - ЯМР-спектр при 300 МГц в CDCl3 дает следующие химические сдвиги:
6,97 частей на миллион (s, 6H, ArH мета ОСН2СООН) - 6,94 частей на миллион (s, 6Н, ArH мета ОСН3) - 3,84 частей на миллион (s, 6H, ArCH2COOH) - 3,73 частей на миллион (s широкий, 9Н, метокси) - 1,12 частей на миллион (s, 27H + трет-бутил пара OCH2COOH), 1,09 частей на миллион (s, 27H, трет-бутил пара ОСН3).
2 г эпоксидной смолы Macrocrep от компании BIORAD с гранулометрией 70-100 мкм, серия 11/99, добавляют в предварительно полученный раствор 12,5 мг полученной белой твердой фазы, отобранной ранее из 4,2 г полученной твердой фазы, ранее растворенной в 20 мл дихлорметана, и 1,12 мл 1,2,3,4-тетрагидронафталина. Суспензию медленно выпаривают при окружающей температуре до постоянного веса. Получение белой твердой фазы.
Полученный вес: 2,2 г.
ПРИМЕР 4
Комплексообразование и селективная экстракция америция
В этом примере при помощи каликсаренов в соответствии с настоящим изобретением фиксируют америций, присутствующий в концентрации 10-11 моль·л-1 в водном растворе NaNO3, 0,04 моль·л-1, имитирующем среду мочи и доведенном до рН=4.
Для этого опыта используют 100 мг материала-подложки из примера 1-5, доведенного до кондиции в колонке.
Раствор NaNO3, 0,04 моль·л-1, при рН=4 пропускают через колонку, чтобы создать для него оптимальные условия экстракции (этап доведения до кондиции). Затем через колонку пропускают водный раствор, содержащий америций (этап фиксации). Раствор NaNO3, 0,04 моль·л-1, при рН=4 снова пропускают через колонку, чтобы удалить америций, не экстрагированный каликсареном (этап промывки). Наконец, зафиксированный америций вымывают раствором HNO3 2M (этап вымывания). Растворы вытекают за счет естественной силы тяжести. Америций измеряют при помощи альфа-спектрометрии в каждом растворе в нижней части колонки. Результаты измерений позволяют рассчитать степень фиксации и степень вымывания америция.
Результаты этого опыта приведены в таблице I ниже.
ПРИМЕР 5
Комплексообразование и селективная экстракция урана
В этом примере при помощи каликсаренов в соответствии с настоящим изобретением фиксируют уран, присутствующий в концентрации 10-8 моль·л-1 в водном растворе NaNO3, 0,04 моль·л-1, имитирующем среду мочи и доведенном до рН=4.
Для этого опыта используют 1 г нанесенной на подложку жидкой мембраны из примера 2, доведенной до кондиции в колонке.
Этапы доведения до кондиции, фиксации и промывки идентичны этапам, описанным в примере 4. Зафиксированный уран вымывают раствором HNO3 1М. Уран измеряют при помощи альфа-спектрометрии или масс-спектрометрии (ICP-MS) в каждом растворе в нижней части колонки. Результаты измерений позволяют рассчитать степень фиксации и степень вымывания урана.
Полученные результаты приведены в таблице I ниже.
ПРИМЕР 6
Комплексообразование и селективная экстракция урана из мочи
В этом примере при помощи каликсаренов в соответствии с настоящим изобретением фиксируют уран, присутствующий в концентрации 5·10-6 г/л в моче.
Для этого опыта используют 1 г нанесенной на подложку жидкой мембраны из примера 3, доведенной до кондиции в колонке.
Путем нагревания микроволнами осуществляют предварительный этап минерализации мочи. Минерализационный остаток помещают в раствор HNO3 2M, затем рН раствора доводят до 4 перед пропусканием через колонку.
Этапы доведения до кондиции, фиксации, промывки и вымывания идентичны этапам, описанным в примере 4. Уран измеряют и результаты выражают так же, как и в предыдущих примерах.
Полученные результаты приведены в таблице I ниже.
ПРИМЕР 7
Комплексообразование и селективная экстракция плутония
В этом примере при помощи каликсаренов в соответствии с настоящим изобретением фиксируют плутоний, присутствующий в концентрации 10-10 моль·л-1 в водном растворе NaNO3, 0,04 моль·л-1, имитирующем среду мочи и доведенном до рН=4.
Для этого опыта используют 100 г материала-подложки из примера 1-5, доведенного до кондиции в колонке.
Этапы доведения до кондиции, фиксации, промывки и вымывания идентичны этапам, описанным в примере 4. Плутоний измеряют при помощи альфа-спектрометрии или масс-спектрометрии (ICP-MS) в каждом растворе в нижней части колонки. Результаты измерений позволяют рассчитать степень фиксации и степень вымывания урана.
Полученные результаты приведены в таблице I ниже.
ПРИМЕР 8
Нанесенная на подложку жидкая мембрана, содержащая 1-гидрокси-3,5-диметокси-2,4,6-трикарбоновая кислота-n-трет-бутилкаликс[6]арен (Mr=1174,4)
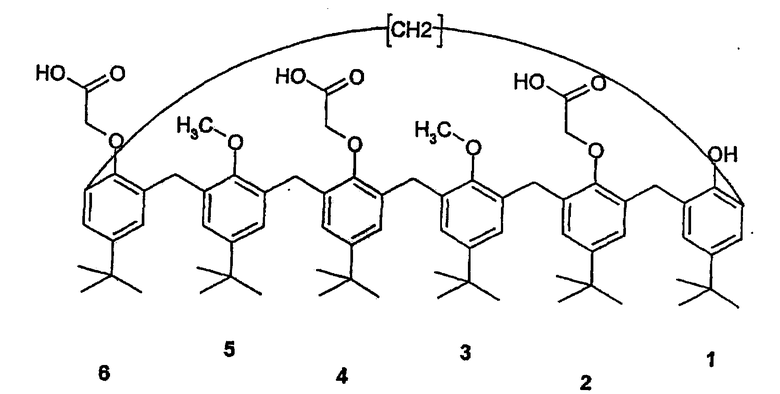
В 250-миллилитровый стеклянный реактор, оборудованный конденсатором, вводят 5,03 г продукта (белый порошок), полученного в примере 1-3, во время применения второго опыта, идентичного предыдущему (4,00 ммоль), и 150 мл этанола. Предварительно приготовленный (и выдержанный при +5°С) раствор расслоенного поташа (12 г 100%, то есть 214 ммоль) в 150 мл воды добавляют однократно в реактор. Реакционную среду доводят до состояния флегмы в течение 4 часов в атмосфере азота.
25 мл HCl 12М медленно добавляют после охлаждения реакционной массы до 20°С. Ожидаемый продукт осаждается в виде белой твердой фазы. После этого суспензию фильтруют и твердое вещество промывают водой 8 раз по 50 мл, затем этанолом 2 раза по 50 мл. Затем твердую фазу сушат при 40°С в вакууме до постоянного веса.
Получают 4,65 г твердого вещества после сушки при 40°С в вакууме до постоянного веса (выход=99%).
Н+ - ЯМР-спектр протона при 300 МГц в CDCl3 дает следующие химические сдвиги:
6,86 частей на миллион (s, 13H, ArH мета + ОН фенол) - 3,95 частей на миллион (s, 6Н, ArCH2COOH) - 3,73 частей на миллион (s широкий, 6Н, метокси) - 1,12 частей на миллион (s, 27H+трет-бутил пара ОСН2СООН), 1,09 частей на миллион (s, 27H, трет-бутил пара ОСН3).
2 г Эпоксидной смолы Macrocrep от компании BIORAD с гранулометрией 70-100 мкм, серия 11/99, добавляют в предварительно полученный раствор 12,5 мг полученной белой твердой фазы, отобранной ранее из 4,65 г полученной твердой фазы, ранее растворенной в 20 мл дихлорметана, и 1,12 мл 1-гептанола. Суспензию медленно выпаривают при окружающей температуре до постоянного веса. Получение белой твердой фазы.
Полученный вес: 2,2 г.
Комплексообразование и селективная экстракция урана
В этом примере при помощи каликсаренов в соответствии с настоящим изобретением фиксируют уран, присутствующий в концентрации 10-8 моль·л-1 в водном растворе NaNO3, 0,04 моль·л-1, имитирующем среду мочи и доведенном до рН=4.
Для этого опыта используют 1 г нанесенной на подложку жидкой мембраны, полученной ранее и доведенной до кондиции в колонке.
Этапы доведения до кондиции, фиксации, промывки и вымывания идентичны этапам, описанным в примере 4. Уран измеряют и результаты выражают так же, как и в предыдущих примерах.
Полученные результаты приведены в таблице II ниже.
Комплексообразование и селективная экстракция тория
В этом примере при помощи каликсаренов в соответствии с настоящим изобретением фиксируют торий, присутствующий в концентрации 10-8 моль·л-1 в водном растворе NaNO3, 0,04 моль·л-1, имитирующем среду мочи и доведенном до рН=3.
Для этого опыта используют 1 г нанесенной на подложку жидкой мембраны, полученной выше и доведенной до кондиции в колонке.
Этапы доведения до кондиции, фиксации и промывки идентичны этапам, описанным в примере 4, при этом растворы для доведения до кондиции и промывки доводят до рН=3. Торий измеряют при помощи альфа-спектрометрии или масс-спектрометрии (ICP-MS) в каждом растворе в нижней части колонки и результаты выражают так же, как и в предыдущих примерах.
Полученные результаты приведены в таблице II ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОСМЕТИЧЕСКИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ МОЛЕКУЛ КАЛИКСАРЕНА | 2009 |
|
RU2493818C2 |
| ГИДРИРОВАННЫЕ ПОЛИМЕРЫ С РАДИАЛЬНОЙ СТРУКТУРОЙ, ИМЕЮЩИЕ ЯДРО НА ОСНОВЕ КАЛИКСАРЕНОВ, И ИХ ПРИМЕНЕНИЕ В СМАЗОЧНЫХ КОМПОЗИЦИЯХ | 2015 |
|
RU2672421C2 |
| ПРОИЗВОДНОЕ ТЕТРАДОДЕЦИЛОКСИФЕНИЛКАЛИКС[4]АРЕНА ДЛЯ СОРБЦИИ АЗО-КРАСИТЕЛЕЙ ИЗ ВОДНЫХ РАСТВОРОВ | 2010 |
|
RU2428411C1 |
| ВОДНАЯ СИСТЕМА, СОДЕРЖАЩАЯ СУПРАМОЛЕКУЛЯРНЫЕ КАЛИКСАРЕНОВЫЕ НАНОКОНТЕЙНЕРЫ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2362761C1 |
| СПОСОБ ОБНАРУЖЕНИЯ РЕАГЕНТА, ПОЛИМЕР, ЭЛЕКТРОДНАЯ ОСНОВА, ЭЛЕКТРОД И КАЛИКСАРЕН | 1994 |
|
RU2133463C1 |
| ПРОИЗВОДНОЕ ТЕТРАМЕТИЛОКСИФЕНИЛКАЛИКС[4]АРЕНА ДЛЯ СОРБЦИИ АЗО-КРАСИТЕЛЕЙ ИЗ ВОДНЫХ РАСТВОРОВ | 2012 |
|
RU2480451C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ИОНОВ МЕТАЛЛА | 1989 |
|
RU2091311C1 |
| КОМПОЗИЦИЯ КАЛИКС[4]АРЕНОВ ДЛЯ СОРБЦИИ АЗО-КРАСИТЕЛЕЙ ИЗ ВОДНЫХ РАСТВОРОВ | 2012 |
|
RU2489205C1 |
| Способ получения масляного связующего для лаков и эмалей | 1990 |
|
SU1819901A1 |
| СПОСОБ ПОЛУЧЕНИЯ СВЕТО- И ТЕРМОСТАБИЛИЗАТОРА ПОГОДОСТОЙКОГО ПОЛИЭТИЛЕНА | 2004 |
|
RU2265008C2 |
Настоящее изобретение относится к материалу-подложке для комплексообразования и селективной экстракции америция, плутония, урана или тория в их катионной форме, который является пара-трет-бутил-каликс[6]ареном формулы (IIA), где R'1, R'3 и R'5, идентичные или разные, представляют собой, каждый отдельно: (i) линейный или разветвленный C1-6 алкил, нанесенный на подложку, при этом одна из групп R'1, R'3 и R'5 в соединении формулы (IIA) является группой (ii); (ii) спейсер-подложка, где спейсер является двухвалентным радикалом, выбранным из группы, в которую входят арил(С1-6алкил)арилы; и подложку выбирают из подложки, являющейся сополимером хлор- или бром-метилстирола и дивинилбензола. Изобретение также относится к нанесенной на подложку жидкой мембране для комплексообразования и селективной экстракции америция, плутония, урана или тория в их катионной форме, содержащей пара-трет-бутил-каликс[6]-арен формулы (IA) или (IB), который растворен в органическом растворителе, имеющем точку кипения, превышающую 60°С, и абсорбирован на подложке, представляющей собой эпоксидную смолу, где R1, R3 и R5, идентичные или разные, представляют собой, каждый отдельно: (i) атом водорода, (ii) линейный или разветвленный C1-6 алкил.

6 н. и 1 з.п. ф-лы, 2 табл.
1. Материал-подложка для комплексообразования и селективной экстракции америция, плутония, урана или тория в их катионной форме, который является пара-трет-бутил-каликс[6]ареном формулы (IIA)
где R'1, R'3 и R'5, идентичные или разные, представляют собой, каждый отдельно:
(i) линейный или разветвленный C1-6 алкил, нанесенный на подложку, при этом одна из групп R'1, R'3 и R'5 в соединении формулы (IIA) является группой (ii);
(ii) спейсер-подложка, где спейсер является двухвалентным радикалом, выбранным из группы, в которую входят арил(С1-6алкил)арилы; и подложку выбирают из подложки являющейся сополимером хлор- или бром-метилстирола и дивинилбензола.
2. Нанесенная на подложку жидкая мембрана для комплексообразования и селективной экстракции америция, плутония, урана или тория в их катионной форме, содержащая пара-трет-бутил-каликс[6]-арен формулы (IA) или (IB), который растворен в органическом растворителе, имеющем точку кипения, превышающую 60°С, и абсорбирован на подложке, представляющей собой эпоксидную смолу,
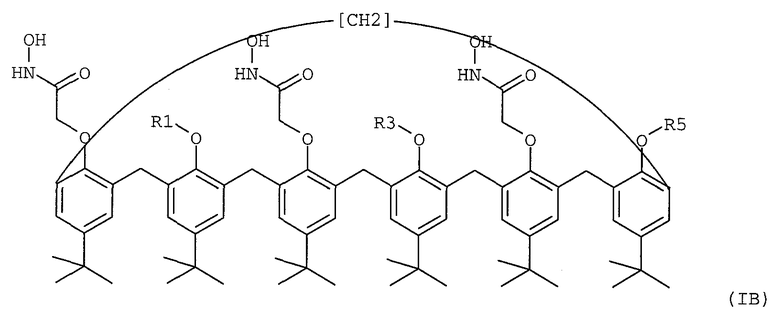
где R1, R3 и R5, идентичные или разные, представляют собой, каждый отдельно:
(i) атом водорода,
(ii) линейный или разветвленный C1-6 алкил.
3. Нанесенная на подложку жидкая мембрана для комплексообразования и селективной экстракции америция, плутония, урана или тория в их катионной форме, содержащая пара-трет-бутил-каликс[6]-арен формулы (IA) или (IB), который растворен в органическом растворителе, имеющем точку кипения, превышающую 60°С, и абсорбирован на подложке, представляющей собой эпоксидную смолу,

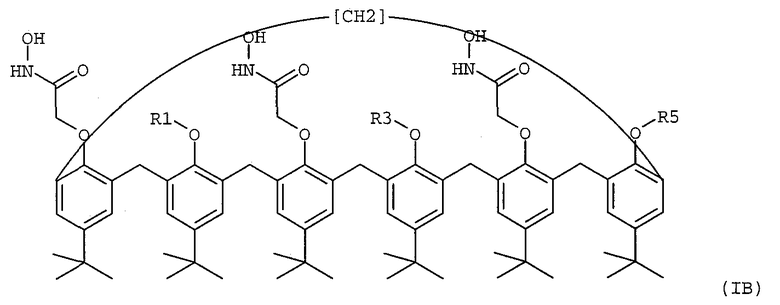
где R1, R3 и R5 являются одинаковыми и представляют собой, каждый отдельно:
(i) атом водорода,
(ii) линейный или разветвленный C1-6 алкил.
4. Нанесенная на подложку жидкая мембрана для комплексообразования и селективной экстракции америция, плутония, урана или тория в их катионной форме, содержащая пара-трет-бутил-каликс[6]-арен формулы (IA) или (IB), который растворен в органическом растворителе, имеющем точку кипения, превышающую 60°С, и абсорбирован на подложке, представляющей собой эпоксидную смолу,
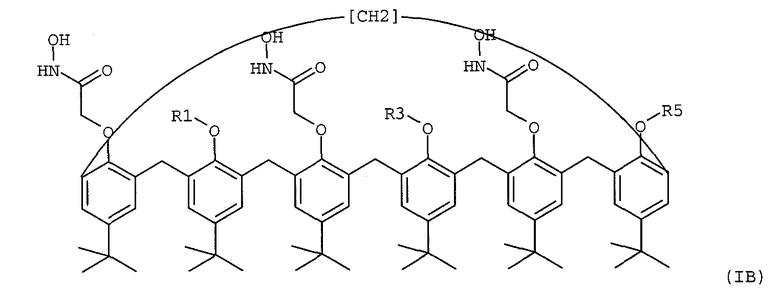
где R1, R3 и R5 представляют собой атом водорода.
5. Нанесенная на подложку жидкая мембрана по пп.2-4, отличающаяся тем, что органический растворитель имеющий точку кипения, превышающую 60°С, выбран из группы, в которую входят толуол, ксилол, хлорбензол, орто-дихлорбензол, нитробензол, 1,4-диизопропил бензол, гексилбензол, керосин, тетрагидропиран, 1,2,3,4-тетрагидронафталин, пентанол и высшие спирты-гомологи, гликоли и их простые эфиры, такие как дибутиловый эфир диэтиленгликоля, сложные эфиры, такие как метилбензоат, простые эфиры, такие как пентиловый эфир ортонитрофенила или нитрофенил октиловый эфир, и их смеси.
6. Применение нанесенной на подложку жидкой мембраны по любому из пп.2-5 для комплексообразования и селективной экстракции урана, америция, плутония или тория в их катионной форме.
7. Применение материала-подложки по п.1, для комплексообразования и селективной экстракции урана, америция, плутония или тория в их катионной форме.
| ЕР 1295861 А2, 26.03.2003 | |||
| DE 10210115 А1, 18.09.2003 | |||
| КАСКАДНЫЙ ПРЕОБРАЗОВАТЕЛЬ ЧАСТОТЫ С УВЕЛИЧЕННЫМ ЧИСЛОМ УРОВНЕЙ ВЫХОДНОГО НАПРЯЖЕНИЯ | 2020 |
|
RU2767491C1 |
| DINSE С ЕТ AL: "New purification protocol for actinide measurement in excreta based on calixarene chemistry" APPLIED RADIATION AND ISOTOPES, vol | |||
| Веникодробильный станок | 1921 |
|
SU53A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| К | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |

