Изобретение относится к области органической химии, а именно к способу получения N-нитрометильных азолов формулы
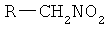
где R= 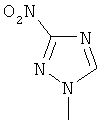 ,
, 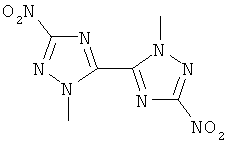 ,
,
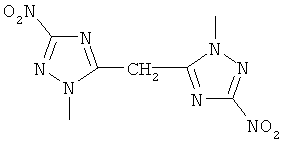 ,
, 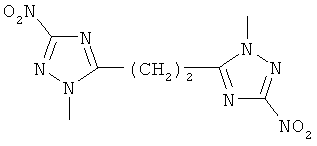 ,
,
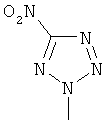 ,
,
которые могут найти применение в качестве биологически активных веществ, интермедиатов при синтезе других производных азолов, энергонасыщенных материалов для пиротехнических составов систем безопасности.
В настоящее время существует несколько способов получения нитрометильных соединений.
Известен синтез соединений с нитрометильной группой через производные индандиона-1,3: производное индандиона-1,3 с необходимым заместителем обрабатывают азотной кислотой в среде уксусной, а затем при подщелачивании выделяют продукт в виде его соли [Залукаев Л.П., Ванаг Э. // ЖОХ. Т.26. 1956. С.607-609; Залукаев Л.П. Синтезы и реакции алъфа-нитрокетонов. Рига: Изд-во АН ЛатССР. 1958. С.111].
Недостатком этого способа является невозможность получения N-нитрометильных производных азолов.
Наиболее близким способом того же назначения к заявленному изобретению является способ получения на основе кетонов. В этом способе N-ацетонильное производное 5-нитро-1,2,3,4-тетразола растворяют в серно-азотной смеси. Соотношение (об.) азотная кислота:серная:вода=5:12:5 на 1 г исходного соединения. Реакционную массу выдерживают 2 часа при комнатной температуре и выливают на лед. Отфильтровывают полученный 2-нитрометил-5-нитро-1,2,3,4-тетразол [Семенов В.В., Шевелев С.А., Брускин А.Б., Канищев М.И., Барышников А.Т. // Изв. АН. Сер. хим. 2009. №10. С.2014-2033].
Недостатками этого способа являются: 1) очень низкий выход целевого продукта (14%); 2) данный способ не приемлем для получения N-нитрометильных производных ряда 1,2,4-триазола.
Известными способами нельзя получить N-нитрометильные производные данного ряда 1,2,4-триазола. Поэтому существует необходимость разработки способа получения соединений такого ряда.
Изобретение решает задачу создания нового способа получения N-нитрометильных производных азолов.
Техническим результатом предлагаемого способа получения является создание нового способа N-нитрометильных производных азолов, синтез новых N-нитрометильных производных 3-нитро-1,2,4- триазола.
Технический результат предлагаемого способа получения достигается за счет использования нитрующей смеси, состоящей из слабой азотной и уксусной кислот, повышения температуры до 50-60°С, увеличения времени выдержки 3-5 часов, добавления нитрита натрия.
Указанный технический результат достигается тем, что способ получения N-нитрометильных замещенных азолов формулы
где R= , ,
, ,
,
заключается в нитровании кетонов формулы
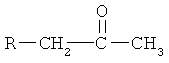
где R= , ,
, ,
,
смесью разбавленной азотной кислоты концентрации 58-70% и ледяной уксусной кислоты при их объемном соотношении 1:6 с добавлением 0,05-0,25 молей нитрита натрия на 1 моль исходного кетона, при этом процесс нитрования проводят при температуре 50-60°С в течение 3-5 часов с последующим охлаждением до комнатной температуры и выливанием на лед с дальнейшим выделением конечного продукта.
Проведение процесса при температуре менее 50°С значительно снижает выход целевых продуктов, в то время как при температурах выше 60°С происходит более глубокая деструкция, которая также ведет к снижению выхода.
В случае выдержки реакционной массы менее 3 часов наблюдается низкий выход конечного продукта. Увеличение времени более 5 часов нецелесообразно, так как это затягивает продолжительность процесса и не приводит к повышению выхода продукта.
Использование в качестве нитрующей системы азотной и уксусной кислоты обусловлено тем, что исходные кетоны лучше всего растворимы в уксусной кислоте.
Использование азотной кислоты концентрации 58-70%, объемного соотношения азотной кислоты к уксусной равного 1:6 и 0,05-0,25 молей нитрита натрия на 1 моль исходного кетона получено опытным путем и приводит к оптимальному выходу целевого продукта.
Способ получения N-нитрометильных производных азолов осуществляется следующим образом: кетон соответствующего азола растворяют в уксусно-азотной кислотной смеси при 50-60°С. Соотношение (об.) азотная кислота (концентрации 58-70%) к уксусной 1:6. Выдерживают при этой температуре в течение 3-5 часов. Во время выдержки в три приема через равные промежутки времени в реакционную массу вводят 0,05-0,25 молей нитрита натрия на 1 моль исходного кетона. Выделяют продукты реакции, перекристаллизовывают из подходящих растворителей.
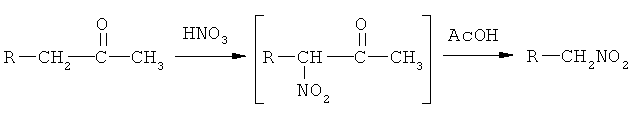
где R= , ,
, ,
Этим способом получены неописанные ранее вещества:
1. Бис-(1 -нитрометил-3-нитро-1,2,4-триазол-5-ил)
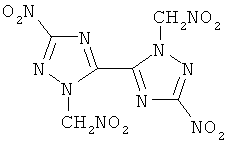
Кристаллы желтого цвета растворимы в ацетоне, этилацетате. Тпл(разл.) 245°С. Найдено, %: С 20,81; Н 1,21; N 40,83. C6H4N10O8. Вычислено, %: С 20,93; Н 1,16; N 40,70. ИК-спектр, см-1: 1562, 1580, 1310 (NO2-группы); 2993, 3057 (СН2-группа).
2. Бис-(1-нитрометил-3-нитро-1,2,4-триазол-5-ил)метан
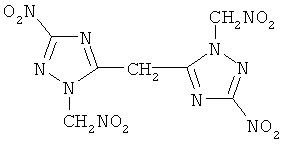
Кристаллы желтого цвета растворимы в ацетоне, этилацетате. Тпл(разл.) 258°С. Найдено, %: С 23,72; Н 1,63; N 39,32. C7H6N10O8. Вычислено, %: С 23,46; Н 1,68; N 39,11. ИК-спектр, см-1: 1561, 1579, 1310 (NO2-группы); 2993, 3056 (СН2-группа).
3. Бис-(1-нитрометил-3-нитро-1,2,4-триазол-5-ил)этан
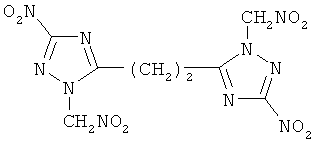
Кристаллы желтого цвета растворимы в ацетоне, этилацетате. Тпл(разл.) 157°С. Найдено, %: С 25,62; Н 2,17; N 37,83. C8H8N10O8. Вычислено, %: С 25,81; Н 2,15; N 37,63. ИК-спектр, см-1: 1561, 1595, 1304 (CH2-группы); 2979 (СН2-группа)
Пример 1. Получение 1-нитрометил-3-нитро-1,2,4-триазола
В кислотной смеси, состоящей из 20 мл 58%-ной азотной кислоты и 120 мл ледяной уксусной кислоты, растворяли 0,058 моль 1-ацетонил-3-нитро-1,2,4-триазола. Реакционную массу нагревали до 50°С и при этой температуре выдерживали в течение 4 часов. Во время выдержки в три приема через равные промежутки времени в реакционную массу вводят 0,0043 моль нитрита натрия. По истечении выдержки содержимое колбы при перемешивании выливали на 50 г льда и доводили водой реакционный объем до 240 мл. Полученную смесь выдерживали при -15°С в течение 24 часов. Осадок отфильтровывали, перекристаллизовывали из смеси (этанол-вода=1:1). Выход 40%. Тпл 181°С. рКа 7,8. Электронный спектр, λmax, нм, вода: 204,5. Найдено, %: С 20,78; Н 1,86; N 40,41. C3H3N5O4. Вычислено, %: С 20,82; Н 1,75; N 40,46. ИК-спектр, см-1: 1552, 1579, 1307 (NO2-группы); 2992, 3049 (СН2-группа); 3137 (С-Н в 5 положении кольца).
Пример 2. Получение бис(1-нитрометил-3-нитро-1,2,4-триазол-5-ил)
В кислотной смеси, состоящей из 20 мл 65%-ной азотной кислоты и 120 мл ледяной уксусной кислоты, растворяли 0,029 моль бис(1-ацетонил-3-нитро-1,2,4-триазол-5-ил). Реакционную массу нагревали до 60°С и при этой температуре выдерживали в течение 5 часов. Во время выдержки в три приема через равные промежутки времени в реакционную массу вводят 0,0072 моль нитрита натрия. По истечении выдержки содержимое колбы при перемешивании выливали на 50 г льда и доводили водой реакционный объем до 240 мл. Продукт извлекали из реакционной смеси этилацетатом. После отгонки растворителя остаток перекристаллизовывали из смеси (этанол-вода=1:1). Выход 30%. Тпл(разл.) 245°С. Найдено, %: С 20,81; Н 1,21; N 40,83. C6H4N10O8. Вычислено, %: С 20,93; Н 1,16; N 40,70. ИК-спектр, см-1: 1562, 1580, 1310 (NO2-группы); 2993, 3057 (СН2-группа).
Пример 3. Получение 2-нитрометил-5-нитро-1,2,3,4-тетразола.
В кислотной смеси, состоящей из 8 мл 70%-ной азотной кислоты и 48 мл ледяной уксусной кислоты, растворяли 0,057 моль 2-ацетонил-5-нитро-1,2,3,4-тетразола. Реакционную массу нагревали до 55°С и при этой температуре выдерживали в течение 3 часов. Во время выдержки в три приема через равные промежутки времени в реакционную массу вводят 0,0029 моль нитрита натрия. По истечении выдержки содержимое колбы охлаждали и испаряли на воздухе при комнатной температуре досуха. Затем сухой остаток перекристаллизовывали из смеси (этанол-вода=1:1). Выход 80%. Тпл 132°С. рКа 7,5. Электронный спектр, λmax, нм, вода: 206. Найдено, %: С 13,71; Н 1,08; N 48,17. C2H2N6O4. Вычислено, %: С 13,80; Н 1,16; N 48,28. ИК-спектр, см-1: 1304, 1561, 1591 (NO2-группы); 2995, 3056 (СН2-группа).
Пример 4. Получение бис-(1-нитрометил-3-нитро-1,2,4-триазол-5-ил)метана
В кислотной смеси, состоящей из 20 мл 70%-ной азотной кислоты и 120 мл ледяной уксусной кислоты, растворяли 0,028 моль бис(1-ацетонил-3-нитро-1,2,4-триазол-5-ил)-метана. Реакционную массу нагревали до 60°С и при этой температуре выдерживали в течение 5 часов. Во время выдержки в три приема через равные промежутки времени в реакционную массу вводят 0,0072 моль нитрита натрия. По истечении выдержки содержимое колбы при перемешивании выливали на 50 г льда и доводили водой реакционный объем до 240 мл. Продукт извлекали из реакционной смеси этилацетатом. Выход 25%. Тпл(разл.) 258°С. Найдено, %: С 23,72; Н 1,63; N 39,32. C7H6N10O8. Вычислено, %: С 23,46; Н 1,68; N 39,11. ИК-спектр, см-1: 1561, 1579, 1310 (NO2-группы); 2993, 3056 (СН2-группа).
Пример 5. Получение бис-(1-нитрометил-3-нитро-1,2,4-триазол-5-ил)этана
В кислотной смеси, состоящей из 20 мл 66%-ной азотной кислоты и 120 мл ледяной уксусной кислоты, растворяли 0,027 моль бис(1-ацетонил-3-нитро-1,2,4-триазол-5-ил)-этана. Реакционную массу нагревали до 60°С и при этой температуре выдерживали в течение 5 часов. Во время выдержки в три приема через равные промежутки времени в реакционную массу вводят 0,0072 моль нитрита натрия. По истечении выдержки содержимое колбы при перемешивании выливали на 50 г льда и доводили водой реакционный объем до 240 мл. Продукт извлекали из реакционной смеси этилацетатом. Выход 34%. Тпл(разл.) 157°С. Найдено, %: С 25,62; Н 2,17; N 37,83. C8H8N10O8. Вычислено, %: С 25,81; Н 2,15; N 37,63. ИК-спектр, см-1: 1561, 1595, 1304 (NO2-группы); 2979 (СН2-группа).
Полученные авторами N-нитрометильные замещенные азолы могут найти применение в качестве биологически активных веществ, интермедиатов при синтезе других производных азолов, энергонасыщенных материалов для пиротехнических составов систем безопасности.
| название | год | авторы | номер документа |
|---|---|---|---|
| Замещенные [(3-нитро-1Н-1,2,4-триазол-1-ил)-NNO-азокси]фуразаны и способ их получения | 2020 |
|
RU2747110C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИНИТРОМЕТИЛ-ONN-АЗОКСИСОЕДИНЕНИЙ | 2014 |
|
RU2558138C1 |
| Способ получения 3-азолилпропанолов | 2022 |
|
RU2786670C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ 2-МЕТИЛТИО-6-НИТРО-1,2,4-ТРИАЗОЛО[5,1-c]-1,2,4-ТРИАЗИН-7-ОНА, ДИГИДРАТА | 2007 |
|
RU2343154C2 |
| СПОСОБ ПОЛУЧЕНИЯ R-МЕТИЛПРОИЗВОДНЫХ 3,5-ДИАМИНО-1,2,4-ТРИАЗОЛА | 2005 |
|
RU2292340C1 |
| 1-ТРИНИТРОМЕТИЛ-3-НИТРО-5-R -1,2,4-ТРИАЗОЛ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1976 |
|
SU1840302A1 |
| 2-(5-НИТРОНИЛФУРАН-2-ИЛ)-5-МЕТИЛ-6-НИТРО-1,2,4-ТРИАЗОЛО[1,5-А]ПИРИМИДИН-7(4Н)-ОН И ЕГО СОЛИ | 2018 |
|
RU2716715C2 |
| Замещенные [(3,4-динитро-1H-пиразол-1-ил)-NNO-азокси]фуразаны и способ их получения | 2020 |
|
RU2756321C1 |
| Способ получения первичных алифатических нитраминов | 2016 |
|
RU2610282C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДРАЗОНОВ 4-R-1,2,5-ОКСАДИАЗОЛ-3-КАРБОНОВОЙ КИСЛОТЫ | 2014 |
|
RU2557659C1 |
Изобретение относится к области органической химии, а именно к способу получения N-нитрометильных азолов формулы
где R=
, , ,
, ,
которые могут найти применение в качестве биологически активных веществ, интермедиатов при синтезе других производных азолов. Техническим результатом описываемого способа получения является создание нового способа получения N-нитрометильных производных азолов и синтез новых N-нитрометильных производных 3-нитро-1,2,4-триазола. Способ получения N-нитрометильных производных азолов осуществляется путем обработки кетона соответствующего азола уксусно-азотной кислотной смесью при 50-60°С, в присутствии 0,05-0,25 молей нитрита натрия на 1 моль исходного кетона. Соотношение (об.) азотная кислота (концентрации 58-70%) к ледяной уксусной 1:6.
Способ получения N-нитрометильных замещенных азолов формулы
где R=
, ,
,
заключающийся в том, что кетон формулы
,
где R=
, ,
,
подвергают нитрованию нитрующей смесью разбавленной азотной кислоты концентрации 58-70% и ледяной уксусной кислоты при соотношении 1:6, при этом процесс нитрования проводят при температуре 50-60°С в течение 3-5 ч, с добавлением 0,05-0,25 молей нитрита натрия на 1 моль исходного кетона, с последующим охлаждением до комнатной температуры и выделением конечного продукта.
| Семёнов В.В | |||
| и др | |||
| Известия АН Сер | |||
| Хим | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Разборное приспособление для накатки на рельсы сошедших с них колес подвижного состава | 1920 |
|
SU65A1 |
| Голубцова О.А | |||
| и др | |||
| Материалы докладов Всероссийской научно-технической конференции | |||
| - Казань, РФ, сентябрь 26-28, 2002, т.2 с.23-26 Chemistry of Heterocyclic Compounds, NY, 39(5), 604-607, 2003. | |||

