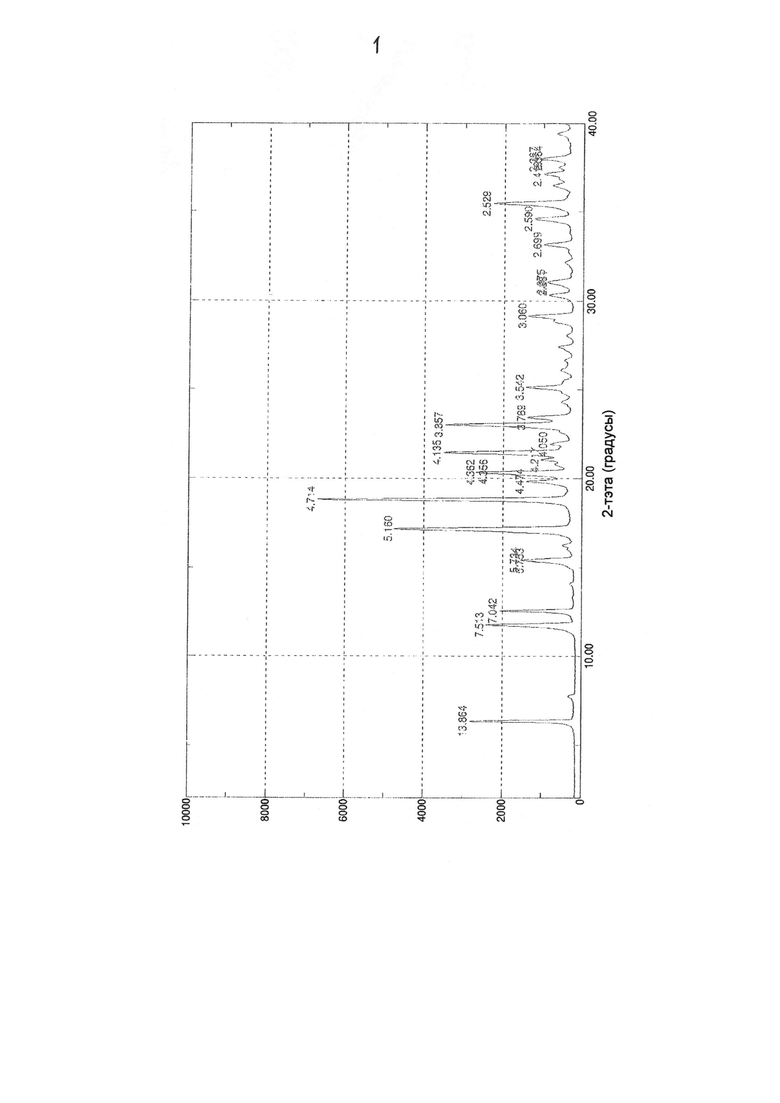
Настоящее изобретение относится к солям соединений формулы
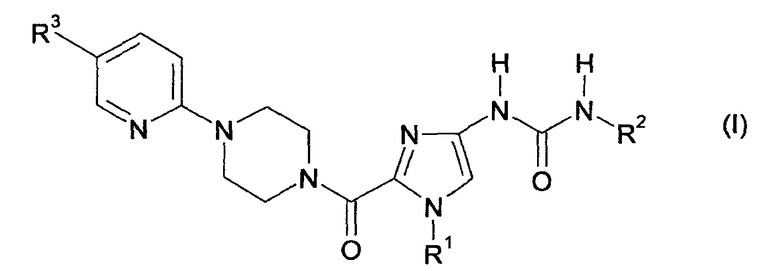
в которой
R1 обозначает метил, этил, бутил или циклопропилметил,
R2 обозначает фенил, где фенил содержит заместитель, который выбран из группы, включающей трифторметоксигруппу и дифторметоксигруппу, и
R3 обозначает водород, метил, хлор, метоксигруппу или трифторметил.
Настоящее изобретение также относится к способу их получения, их применению для лечения и/или предупреждения заболеваний, а также к их применению для приготовления фармацевтических средств, предназначенных для лечения и/или предупреждения заболеваний, предпочтительно предназначенных для применения в качестве противовирусных средств, предпочтительно для борьбы с цитомегаловирусами.
Соединения формулы (I) известны, например, из WO 2006/089664 и разработаны авторами настоящего изобретения, как перспективные противовирусно эффективные вещества, в частности, предназначенные для борьбы с инфекцией цитомегаловирусом человека (ЦМВЧ). Однако в ходе разработки показано, что эти вещества обладают не отвечающей требованиям растворимостью в водных растворителях, а также в сильно полярных растворителях. Затруднения, связанные с растворимостью, усиливаются еще и тем, что соединения также обладают недостаточной растворимостью при условиях, существующих в желудке человека (примерно 0,1 М раствор HCl, рН~1), и в этом случае может начаться образование соли с HCl in situ.
Таким образом, задачей настоящего изобретения является описание солей, которые обладают значительно лучшей растворимостью, чем свободное основание соединений формулы (I). Кроме того, необходимо, чтобы эти соли оставались стабильными в течение длительного периода времени при обычных условиях хранения. В частности, необходимо, чтобы соединения не обладали повышенной гигроскопичностью. Также необходимо, чтобы соли в присутствии разбавленного раствора HCl превращались в соль с HCl лишь медленно, чтобы обеспечить максимально быстрое и равномерное высвобождение даже при условиях, существующих в желудке человека.
Согласно изобретению весьма неожиданно обнаружено, что соли соединений формулы (I) с органическими сульфоновыми кислотами обладают лучшей растворимостью, чем свободное основание, а также широкий спектр других солей соединений формулы (I). Кроме того, эти соли также обладают длительной стабильностью, которая необходима при их применении в лекарственных средствах. Кроме того, показано, что соли, предлагаемые в настоящем изобретении, также обладают высокой и постоянной растворимостью при условиях, существующих в желудке человека.
Объектами настоящего изобретения являются соли соединений формулы (I) с органической сульфоновой кислотой или их сольваты или гидраты.
В объеме настоящего изобретения соли органических сульфоновых кислот представляют собой аддукты, полученные по реакции соединения формулы (I) с органической сульфоновой кислотой. При этом соединения формулы (I) и органические сульфоновые кислоты могут содержаться в любом соотношении. В этом случае соотношение предпочтительно описывается целыми числами (например, 1:1, 1:2, 1:3, 3:1, 2:1). В этом случае это соли можно получить путем прямой реакции соединений формулы (I) и органической сульфоновой кислоты или путем получения солей соединений формулы (I) с другими кислотами с последующим обменом противоиона.
В объеме настоящего изобретения сольваты означают такие формы соединений, предлагаемых в настоящем изобретении, которые образуют комплекс путем координации с молекулами растворителя. Гидраты являются особой формой сольватов, в которых координация происходит с водой.
В объеме настоящего изобретения предпочтительными являются соли, в которых органической сульфоновой кислотой является метансульфоновая кислота.
В объеме настоящего изобретения особенно предпочтительными являются димезилаты.
В объеме настоящего изобретения предпочтительной является соль, обладающая следующей формулой:
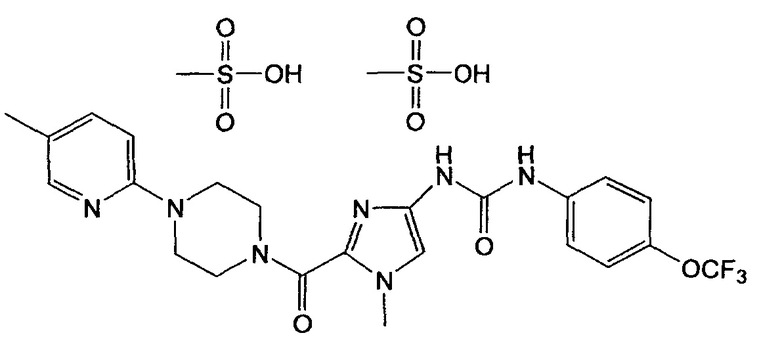
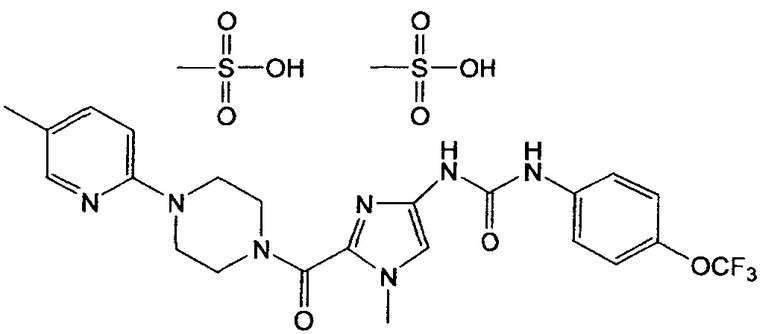
В объеме настоящего изобретения особенно предпочтительным является кристаллический димезилат N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины, обладающий порошковой рентгенограммой, содержащей характеристические пики примерно при 6,37, 11,77, 12,56, 17,17, 18,81, 20,34, 21,47, 23,04, 35,46 градуса 2-тэта.
Кроме того, в объеме настоящего изобретения предпочтительным является кристаллический димезилат N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины, обладающий порошковой рентгенограммой, в общем виде представленной на фиг. 1.
Соли, предлагаемые в настоящем изобретении, обычно получают по реакции соединения формулы (I) с органической сульфоновой кислотой в растворителе.
Кроме того, соли, предлагаемые в настоящем изобретении, можно получить по реакции соли соединений формулы (I) с кислотой, которая не является солью с органической сульфоновой кислотой, с источником сульфонат-анионов органической сульфоновой кислоты в растворителе.
В последнем случае источником сульфонат-анионов может быть органическая сульфоновая кислота или соль органической сульфоновой кислоты.
Таким образом, объектом настоящего изобретения также является способ получения солей соединений формулы (I) с органической сульфоновой кислоты, который включает реакцию соединений формулы (I) или солей соединений формулы (I), которые не является солями с органической сульфоновой кислотой, с органической сульфоновой кислотой или с источником сульфонат-анионов в растворителе.
Растворитель предпочтительно выбирать таким образом, чтобы он обеспечивал хороший баланс растворимости соединений формулы (I) или солей соединения формулы (I), которые не является солями с сульфоновой кислотой, и растворимости органической сульфоновой кислоты или источника сульфонат-анионов. Предпочтительно, чтобы соли, предлагаемые в настоящем изобретении, были как можно хуже растворимы в использующемся растворителе. Однако соли, предлагаемые в настоящем изобретении, также можно необязательно осадить путем добавления антирастворителя.
Примеры растворителей, которые используют для получения солей, предлагаемых в настоящем изобретении, включают следующие: а именно, спирты, такие как метанол, этанол, н-пропанол, изопропанол и бутанол; простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан или тетрагидрофуран; углеводороды, такие как бензол или толуол; или другие растворители, такие как ацетон, этилацетат, метилэтилкетон, метилизобутилкетон, ацетонитрил, гептан, диметилсульфоксид или диметилформамид.
Для осаждения солей, предлагаемых в настоящем изобретении, необязательно добавляют антирастворитель. Примеры таких антирастворителей включают следующие: а именно, воду и спирты, такие как метанол, этанол или пропанол.
Соли, таким образом полученные в соответствии с настоящим изобретением, можно необязательно дополнительно обработать, например, перекристаллизовать или микронизировать, чтобы дополнительно изменить их физические характеристики для конкретного случая применения.
Гетероциклиламидзамещенные имидазолы, использующиеся для получения солей, предлагаемых в настоящем изобретении, известны и их можно получить, например, по методике, описанной в WO 2006/089664.
В частности, получение использующихся гетероциклиламидзамещенных имидазолов осуществляют путем реакции соединений формулы
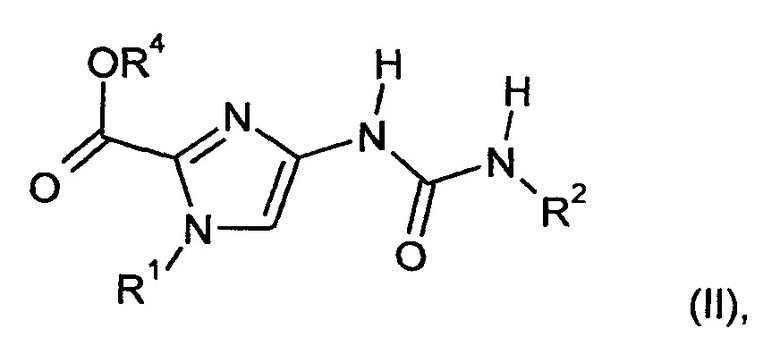
в которой
R1 и R2 являются такими, как определено выше, и
R4 обозначает метил или этил,
на первой стадии с основанием и на второй стадии с соединениями формулы
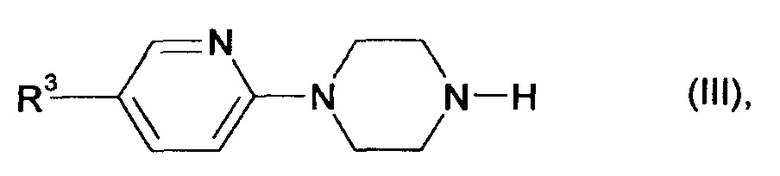
в которой
R3 является таким, как определено выше,
в присутствии дегидратирующих реагентов.
Первую стадию реакции обычно проводят в инертных растворителях, предпочтительно при температуре, находящейся в диапазоне от 0°C до температуры кипения растворителя, при нормальном давлении.
Основаниями являются, например, гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид лития или гидроксид калия, или карбонаты щелочных металлов, такие как карбонат цезия, карбонат натрия или карбонат калия. В этом случае предпочтительным является гидроксид натрия.
Инертными растворителями являются, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен; простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол; углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или неочищенные фракции нефти; или другие растворители, такие как диметилформамид, диметилацетамид, диметилсульфоксид, ацетонитрил или пиридин, или смеси растворителей с водой. Предпочтительным растворителем является смесь этанола и воды.
Вторую стадию реакции обычно проводят в инертных растворителях, необязательно в присутствии основания, предпочтительно при температуре, находящейся в диапазоне от -70 до 40°C, при нормальном давлении.
При этом подходящими дегидратирующими реагентами являются, например, карбодиимиды, такие как, например, N,N'-диэтил-, N,N'-дипропил-, N,N'-диизопропил-, N,N'-дициклогексилкарбодиимид, N-(3-диметиламиноизопропил)-N'-этилкарбодиимидгидрохлорид (EDC), N-циклогексилкарбодиимид-N'-пропилоксиметил-полистирол (ПС-карбодиимид); или карбонильные соединения, такие как карбонилдиимидазол; или соединения 1,2-оксазолия, такие как 2-этил-5-фенил-1,2-оксазолий-3-сульфат или 2-трет-бутил-5-метилизоксазолийперхлорат; ациламиносоединения, такие как 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, или ангидрид пропанфосфоновой кислоты, или изобутилхлорформиат, или бис-(2-оксо-3-оксазолидинил)фосфорилхлорид, или бензотриазолилокси-три(диметиламино)фосфонийгексафторфосфат, или O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (HBTU), 2-(2-оксо-1-(2Н)-пиридил)-1,1,3,3-тетраметилуронийтетрафторборат (TPTU) или O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (ГАТУ), или 1-гидроксибензотриазол (HOBt), или бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфат (ВОР) или смеси последних с основаниями.
Основаниями являются, например, карбонаты щелочных металлов, такие как, например, карбонат натрия или карбонат калия, или бикарбонат калия; или органические основания, такие как триалкиламины, например, триэтйламин, N-метилморфолин, N-метилпиперидин, 4-диметиламинопиридин или диизопропилэтиламин, или ДБУ (1,8-диазабицикло[5,4,0]ундец-7-ен), ДБН (1,5-диазабицикло[4,3,0]нон-5-ен), или пиридин; предпочтительным является N-метилморфолин.
Реакцию конденсации с ангидридом пропанфосфоновой кислоты (ТЗР) предпочтительно проводят в присутствии N-метилморфолина (NMM).
Инертными растворителями являются, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен; простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля; углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или неочищенные фракции нефти; или другие растворители, такие как этилацетат, ацетон, диметилформамид, диметилацетамид, 2-бутанон, диметилсульфоксид, ацетонитрил или пиридин, в случае смешиваемых с водой растворителей также их смеси с водой; предпочтительным является диметилформамид.
Соединения формулы (II) являются известными или их можно получить путем введения в реакцию соединений формулы (IV)
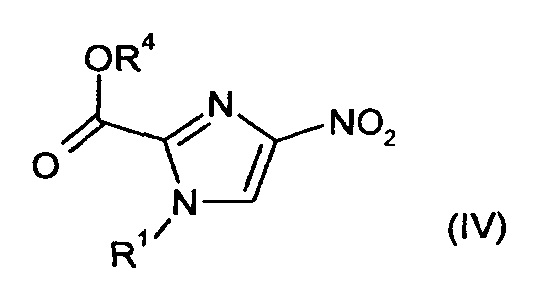
в которой
R1 и R4 являются такими, как определено выше,
на первой стадии с восстановительным реагентом и на второй стадии в присутствии производного угольной кислоты с соединениями формулы
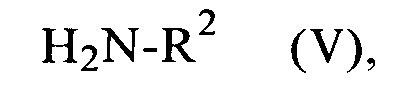
в которой
R2 является таким, как определено выше,
или на второй стадии с соединениями формулы
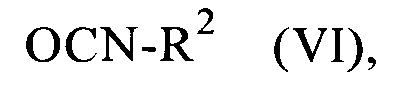
в которой
R2 является таким, как определено выше.
В этом случае реакцию первую стадию реакции обычно проводят в инертных растворителях, предпочтительно при температуре, находящейся в диапазоне от 0°C до температуры кипения растворителя, при давлении от нормального до равного не более 3 бар.
Восстановительными реагентами являются, например, палладий на активированном угле и водород, муравьиная кислота/триэтиламин/палладий на активированном угле, цинк, цинк/хлористоводородная кислота, железо, железо/хлористоводородная кислота, сульфат железа(II)/хлористоводородная кислота, сульфид натрия, дисульфид натрия, дитионит натрия, полисульфид аммония, борогидрид натрия/хлорид никеля, дихлорид олова, трихлорид титана или никель Ренея и водный раствор гидразина; предпочтительными являются никель Ренея и водный раствор гидразина, палладий на активированном угле и водород или муравьиная кислота/триэтиламин/палладий на активированном угле.
Инертными растворителями являются, например, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол; углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или неочищенные фракции нефти; или другие растворители, такие как диметилформамид, диметилацетамид, ацетонитрил или пиридин, в случае смешиваемых с водой растворителей также их смеси с водой. Предпочтительными растворителями являются метанол, этанол, изопропанол или, в случае использования никеля Ренея и водного раствора гидразина, предпочтительным является тетрагидрофуран.
В первом варианте вторую стадию реакции обычно проводят в инертных растворителях, предпочтительно при температуре, находящейся в диапазоне от комнатной температуры до 40°C, при нормальном давлении.
Производными угольной кислоты являются, например, N,N-карбонилдиимидазол, фосген, дифосген, трифосген, фенилхлорформиат или 4-нитрофениловый эфир хлормуравьиной кислоты; предпочтительным является N,N-карбонилдиимидазол.
Инертными растворителями являются, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен; простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля; углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или неочищенные фракции нефти; или другие растворители, такие как этилацетат, ацетон, диметилформамид, диметилацетамид, 2-бутанон, диметилсульфоксид, ацетонитрил или пиридин, и в случае смешиваемых с водой растворителей также их смеси с водой; предпочтительным является диметилсульфоксид.
Во втором варианте вторую стадию реакции обычно проводят в инертных растворителях, необязательно в присутствии основания, предпочтительно при температуре, находящейся в диапазоне от комнатной температуры до температуры кипения растворителя, при нормальном давлении.
Инертными растворителями являются, например, галогенированные углеводороды, такие как метиленхлорид, трихлорметан, тетрахлорметан, трихлорэтан, тетрахлорэтан, 1,2-дихлорэтан или трихлорэтилен; простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, 1,2-диметоксиэтан, диоксан, тетрагидрофуран, диметиловый эфир гликоля или диметиловый эфир диэтиленгликоля; углеводороды, такие как бензол, ксилол, толуол, гексан, циклогексан или неочищенные фракции нефти; или другие растворители, такие как этилацетат, ацетон, диметилформамид, диметилацетамид, 2-бутанон, диметилсульфоксид, ацетонитрил или пиридин; предпочтительными являются тетрагидрофуран или метиленхлорид.
Основаниями являются, например, карбонаты щелочных металлов, такие как карбонат цезия, карбонат натрия или карбонат калия, или трет-бутанолят калия, или другие основания, такие как гидрид натрия, ДБУ, триэтиламин или диизопропилэтиламин, предпочтительно триэтиламин.
Соединения формулы (IV) являются известными или их можно получить путем введения в реакцию соединений формулы
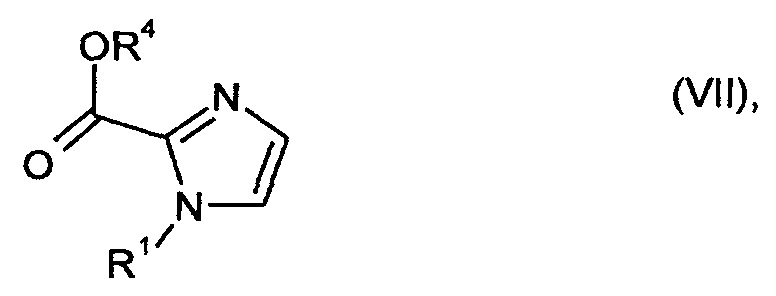
в которой
R1 и R4 являются такими, как определено выше,
с дымящей азотной кислотой, концентрированной азотной кислотой, нитрующей кислотой или смесями серной и азотной кислоты других составов, необязательно в уксусном ангидриде в качестве растворителя, предпочтительно при температуре, находящейся в диапазоне от комнатной температуры до 60°C, при нормальном давлении.
Соединения формул (III), (IV), (V), (VII) являются известными или их можно синтезировать из соответствующих эдуктов по известным методикам.
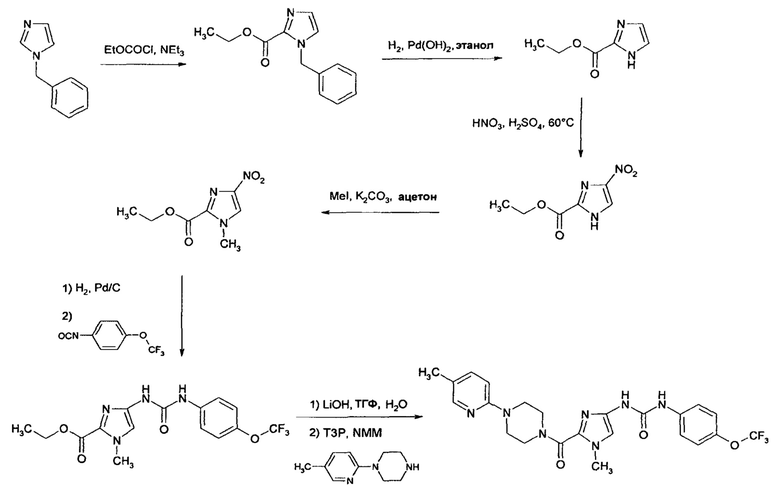
Получение гетероциклиламидзамещенных имидазолов, использующихся для получения солей, предлагаемых в настоящем изобретении, более подробно в качестве примера представлено на приведенной ниже схеме синтеза. В этом контексте схема синтеза приведена только в качестве примера и никоим образом не является ограничивающей.
Схема синтеза:
Соли, предлагаемые в настоящем изобретении, обладают противовирусным воздействием по отношению к представителям группы Herpesviridae (вирусы герпеса), главным образом по отношению к цитомегаловирусам (ЦМВ), в особенности по отношению к цитомегаловирусу человека (ЦМВЧ). Таким образом, они пригодны для лечения и/или предупреждения заболеваний, главным образом инфекций вирусами, предпочтительно вирусами, указанными в настоящем изобретении, и возникающих при этом инфекционных заболеваний. В настоящем изобретении вирусная инфекция означает инфицирование вирусом, а также заболевание, вызванное инфицированием вирусом.
С учетом их особых характеристик соли, предлагаемые в настоящем изобретении, можно использовать для приготовления фармацевтических средств, которые являются подходящими для предупреждения и/или лечения заболеваний, предпочтительно вирусных инфекций.
В качестве типов показаний можно отметить следующие:
1) Лечение или предупреждение инфекций ЦМВЧ у пациентов, страдающих СПИД (синдром приобретенного иммунодефицита) (ретинит, пневмонит, желудочно-кишечные инфекционные заболевания).
2) Лечение или предупреждение инфекций цитомегаловирусом у пациентов, которым трансплантирован костный мозг или орган и у которых развивается часто опасный для жизни вызванный ЦМВЧ пневмонит, вызванный ЦМВЧ энцефалит, а также желудочно-кишечные и системные инфекции ЦМВЧ.
3) Лечение или предупреждение инфекций ЦМВЧ у новорожденных и детей ясельного возраста.
4) Лечение острой инфекции ЦМВЧ у беременных женщин.
5) Лечение инфекции ЦМВЧ у пациентов с ослабленным иммунитетом, вызванным раком и противораковой терапией.
Лечение инфекции ЦМВЧ у страдающих раком пациентов с целью ослабления опосредуемого ЦМВЧ прогрессирования опухоли (см. J. Cinati, et al., FEMS Microbiology Reviews 2004, 28, 59-77).
Соли, предлагаемые в настоящем изобретении, предпочтительно используют для приготовления фармацевтических средств, которые являются подходящими для предупреждения и/или лечения инфекций представителями группы Herpesviridae, в особенности цитомегаловирусом, предпочтительно цитомегаловирусом человека.
С учетом их фармакологических характеристик по отдельности и при необходимости также в комбинации с другими активными ингредиентами, предпочтительно противовирусными активными ингредиентами, такими как, например, валганцикловир, ганцикловир, валацикловир, ацикловир, фоскарнет, цидофовир и родственные производные, соли, предлагаемые в настоящем изобретении, можно использовать для лечения и/или предупреждения вирусных инфекций, предпочтительно инфекций ЦМВЧ.
Другим объектом настоящего изобретения является применение солей, предлагаемых в настоящем изобретении, в способе лечения и/или предупреждения заболеваний, предпочтительно вирусных инфекций, предпочтительно, инфекций цитомегаловирусом человека (ЦМВЧ) или другим представителем группы Herpesviridae.
Другим объектом настоящего изобретения является применение солей, предлагаемых в настоящем изобретении, для лечения и/или предупреждения заболеваний, предпочтительно указанных выше заболеваний.
Другим объектом настоящего изобретения является применение солей, предлагаемых в настоящем изобретении, для приготовления фармацевтического средства, предназначенного для лечения и/или предупреждения заболеваний, предпочтительно указанных выше заболеваний.
Другим объектом настоящего изобретения является способ лечения и/или предупреждения заболеваний, предпочтительно указанных выше заболеваний, с использованием солей, предлагаемых в настоящем изобретении, в противовирусно эффективном количестве.
Соли, предлагаемые в настоящем изобретении, могут действовать системно и/или местно. Для обеспечения такого воздействия их можно вводить подходящим путем, таким как, например, пероральный, парентеральный, пульмональный, назальный, сублингвальный, лингвальный, трансбуккальный, ректальный, кожный, чрескожный, конъюнктивальный, ушной или в виде имплантата или стента.
При этих путях введения соли, предлагаемые в настоящем изобретении, можно вводить в подходящих препаративных формах.
В соответствии с предшествующим уровнем техники подходящими для перорального введения являются препаративные формы, которые высвобождают соли, предлагаемые в настоящем изобретении, быстро и/или модифицированным образом и которые содержат соединения, предлагаемые в настоящем изобретении, в кристаллической и/или аморфизированной и/или растворенной форме, такие как, например, таблетки (таблетки без покрытия или с покрытием, например, обладающие покрытиями, которые являются стойкими к воздействию желудочного сока, или медленно растворимы или нерастворимы, которые регулируют высвобождение соединений, предлагаемых в настоящем изобретении), таблетки или пленки/облатки, которые быстро растворяются в полости рта, пленки/лиофилизаты, капсулы (например, капсулы из твердого или мягкого желатина), таблетки с покрытием, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное ведение можно проводить с исключением стадии всасывания (например, внутривенно, внутриартериально, внутрикардиально, внутрипозвоночно или внутрипояснично) или с включением стадии всасывания (например, внутримышечно, подкожно, внутрикожно, накожно или внутрибрюшинно). Препаративными формами, пригодными для парентерального ведения, в частности, являются, препараты для инъекции и вливания в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Подходящими для других путей введения являются, например, формы лекарственных средств для ингаляции (в частности, для порошковых ингаляторов, распылителей), капли для носа, растворы для носа, спреи для носа; таблетки, которые предназначены для лингвального, сублингвального или трансбуккального введения; пленки/облатки или капсулы, суппозитории, препараты для глаз и ушей, вагинальные капсулы, водные суспензии (лосьоны, взбалтываемые смеси), липофильные суспензии, мази, кремы, чрескожные терапевтические системы (такие как, например, пластыри), молочко, пасты, пенки, присыпки, имплантаты или стенты.
Соли, предлагаемые в настоящем изобретении, можно превратить в указанные препаративные формы. Это можно выполнить известным в данной области техники образом путем смешивания с инертными, нетоксичными фармацевтически приемлемыми инертными наполнителями. Эти инертные наполнители, в частности, включают наполнители (например, микрокристаллическую целлюлозу, лактозу, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергирующие агенты или смачивающие агенты (например, додецилсульфат натрия, полиоксисорбитанолеат), связующие (например, поливинилпирролидон), синтетические и натуральные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, такие как, например, аскорбиновая кислота), красители (например, неорганические пигменты, такие как, например, оксиды железа) и вещества, исправляющие вкус и/или запах.
В объеме настоящего изобретения предпочтительным является фармацевтическое средство, которое содержит от 5 до 12,5 мг/мл соли, предлагаемой в настоящем изобретении, от 50 до 150 мг/мл гидроксипропил-β-циклодекстрина, от 0,5 до 2,0 мг/мл ацетата натрия, а также воду и необязательно другие фармацевтически безопасные вспомогательные вещества.
Другими объектами настоящего изобретения являются фармацевтические средства, которые содержат по меньшей мере одну соль, предлагаемую в настоящем изобретении, обычно вместе с одним или большим количеством инертных, нетоксичных, фармацевтически приемлемых вспомогательных веществ, а также их применение для указанных выше целей.
Установлено, что для обеспечения эффективного результата при внутривенном введении обычно предпочтительно вводить количества, составляющие примерно от 0,001 до 10 мг/(кг массы тела), предпочтительно примерно от 0,01 до 5 мг/(кг массы тела) в пересчете на чистый активный ингредиент. При пероральном введении доза обычно составляет примерно от 0,01 до 25 мг/(кг массы тела), предпочтительно от 0,1 до 10 мг/(кг массы тела).
Тем не менее необязательно может оказаться необходимым отклонение от указанных количеств, в частности, в зависимости от массы тела, пути введения, индивидуальной реакции на активный ингредиент, природы препарата и времени или интервала между введениями. Таким образом, в некоторых случаях будет достаточно использовать количество, меньшее указанного минимального, а в других случаях необходимо превысить указанный верхний предел. В случае введения более значительных количеств может оказаться целесообразным их разделение на несколько отдельных доз, вводимых в течение суток.
Настоящее изобретение будет более подробно описано ниже с помощью примеров, а также со ссылкой на прилагающийся чертеж.
На фиг. 1 приведена порошковая рентгенограмма соли соединения примера 1.
В приведенных ниже исследованиях и примерах, если не указано иное, выраженные в процентах значения являются массовыми; выраженные в частях значения также являются массовыми. Соотношения количеств растворителей, соотношения разбавления и значения концентрации в растворах жидкость/жидкость во всех случаях являются объемными.
Примеры
Использующиеся аббревиатуры:
Методики ВЭЖХ и ЖХ-МС:
Методика 1 (ЖХ-МС): Прибор: Micromass Quattro LCZ с HPLC Agilent Series 1100; колонка: Phenomenex Synergi 2 мкм Hydro-RP Mercury 20 мм×4 мм; элюент А: 1 л воды +0,5 мл 50% муравьиной кислоты, элюент В: 1 л ацетонитрила +0,5 мл 50% муравьиной кислоты; градиентный режим: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи: 50°C; детектирование в УФ-области: 208-400 нм.
Методика 2 (ЖХ-МС): Прибор: Micromass Platform LCZ с HPLC Agilent Series 1100; колонка: Phenomenex Synergi 2 мкм Hydro-RP Mercury 20 мм×4 мм; элюент А: 1 л воды +0,5 мл 50% муравьиной кислоты, элюент В: 1 л ацетонитрила +0,5 мл 50% муравьиной кислоты; градиентный режим: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи: 50°C; детектирование в УФ-области: 210 нм.
Методика 3 (ЖХ-МС): Тип прибора для МС: Micromass ZQ; тип прибора для ВЭЖХ: Waters Alliance 2795; колонка: Phenomenex Synergi 2 мкм Hydro-RP Mercury 20 мм×4 мм; элюент А: 1 л воды +0,5 мл 50% муравьиной кислоты, элюент В: 1 л ацетонитрила +0,5 мл 50% муравьиной кислоты; градиентный режим: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи: 50°C; детектирование в УФ-области: 210 нм.
Методика 4 (ЖХ-МС): Тип прибора для МС: Micromass ZQ; тип прибора для ВЭЖХ: HP 1100 Series; УФ ДДМ (детектор на диодной матрице); колонка: Phenomenex Synergi 2 мкм Hydro-RP Mercury 20 мм×4 мм; элюент А: 1 л воды +0,5 мл 50% муравьиной кислоты, элюент В: 1 л ацетонитрила +0,5 мл 50% муравьиной кислоты; градиентный режим: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; скорость потока: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи: 50°C; детектирование в УФ-области: 210 нм.
Методика 5 (аналитическая ВЭЖХ): колонка: Kromasil 100 RP-18, 60 мм×2,1 мм, 3,5 мкм; элюент А: вода +0,5% хлорной кислоты (70%), элюент В: ацетонитрил; градиентный режим: 0 мин 2% В, 0,5 мин 2% В, 4,5 мин 90% В, 9 мин 90% В, 9,2 мин 2% В, 10 мин 2% В; скорость потока: 0,75 мл/мин; температура колонки: 30°C; детектирование: в УФ-области при 210 нм.
Исходные соединения
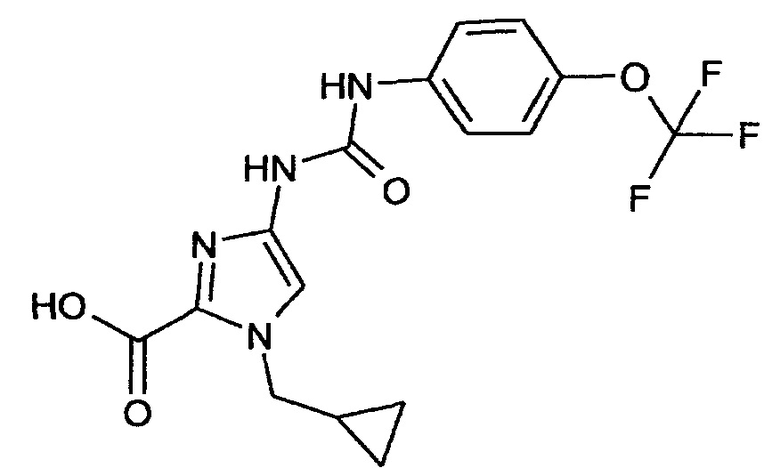
Пример 1А
1-(Циклопропилметил)-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1Н-имидазол-2-карбоновая кислота
Стадия 1
Этиловый эфир 1-(циклопропилметил)-4-нитро-1Н-имидазол-2-карбоновой кислоты
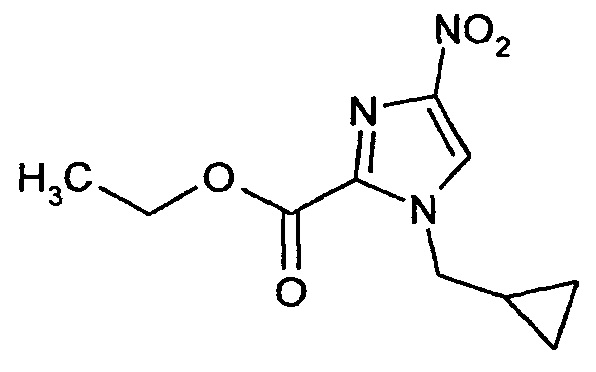
15 г (81 ммоль) Этилового эфира 4-нитро-! Н-имидазол-2-карбоновой кислоты, 13,13 г (97,2 ммоля) циклопропилметилбромида и 22,4 г (162 ммоля) карбоната калия в 165 мл ДМФ перемешивают в атмосфере аргона при 80°C в течение 1 ч. После охлаждения реакционную смесь разбавляют водой и четырежды экстрагируют этилацетатом. Объединенные органические фазы один раз промывают водой и трижды насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют путем выпаривания в вакууме. Кристаллический остаток сразу используют в следующей реакции.
Выход: 17,59 г (70% от теоретического значения)
ЖХ-МС (методика I): Rt=2,02 мин.
МС (ИЭР+): m/z=240 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,2 (s, 1Н), 4,4 (q, 2H), 4,3 (d, 2H), 1,4 (m, 4H), 0,55 (q, 2H), 0,45 (q, 2H) част./млн.
Стадия 2
Этиловый эфир 4-амино-1-(циклопропилметил)-1Н-имидазол-2-карбоновой кислоты
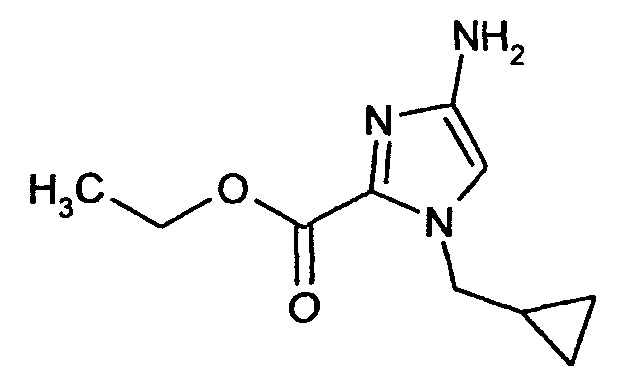
3,89 г (16,26 ммоля) Этилового эфира 1-(циклопропилметил)-4-нитро-1Н-имидазол-2-карбоновой кислоты растворяют в 50 мл ТГФ и смешивают с никелем Ренея (полный кончик шпателя). Реакционную смесь гидрируют водородом в аппарате для гидрирования при комнатной температуре. Катализатор отфильтровывают и фильтрат концентрируют путем выпаривания в вакууме. Полученный после выпаривания остаток сразу используют в следующей реакции.
Выход: 3,46 г (100% от теоретического значения)
ЖХ-МС (методика 2): Rt=1,21 мин.
МС (ИЭР+): m/z=210 [M+H]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=6,55 (s, 1Н), 4,55 (s, 2H), 4,2 (q, 2H), 4,1 (d, 2H), 1,25 (tr, 3Н), 1,2 (m, 1Н), 0,5 (q, 2H), 0,3 (q, 2H) част./млн.
Стадия 3
Этиловый эфир 4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1-(циклопропилметил)-1 Н-имидазол-2-карбоновой кислоты
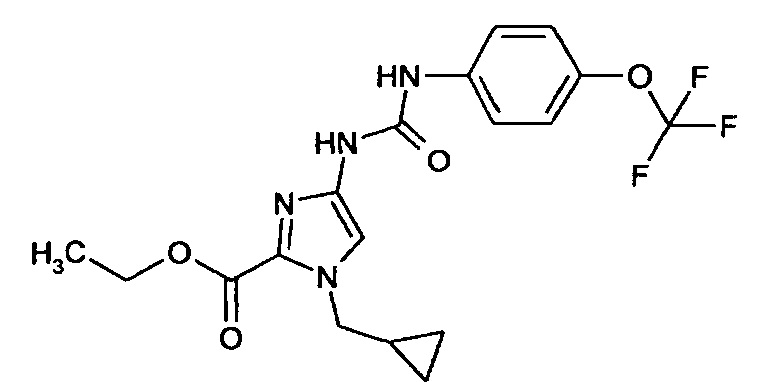
7,49 г (35,8 ммоля) Этилового эфира 4-амино-1-(циклопропилметил)-1Н-имидазол-2-карбоновой кислоты в 18 мл ТГФ в атмосфере аргона смешивают с 6 г (35,8 ммоля) 4-(трифторметокси)фенилизоцианата и перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь концентрируют путем выпаривания в вакууме и продукт, который при этом кристаллизуется, перемешивают в 40 мл этилацетата и отсасывают.
Выход: 11,1 г (82% от теоретического значения)
ЖХ-МС (методика I): Rt=2,66 мин.
МС (ИЭР+): m/z=376 [M+H]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=9,45 (s, 1Н), 8,0 (d, 1Н), 7,35 (s, 1Н), 7,3 (d, 1Н), 7,2 (dd, 1Н), 4,3 (q, 2H), 4,25 (d, 2H), 2,25 (s, ЗН), 1,3 (tr, 3Н), 1,25 (m, 1Н), 0,55 (q, 2H), 0,35 (q, 2H) част./млн.
Стадия 4
4-[({[4-(Трифторметокси)фенил]амино}карбонил)амино]-1-(циклопропилметил)-1 Н-имидазол-2-карбоновая кислота
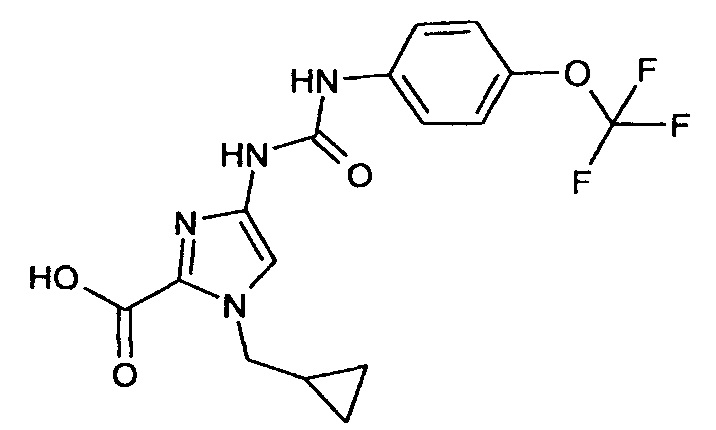
10,6 г (28,1 ммоля) Этилового эфира 4-[({[4-(трифторметокси)фенил]амино} -карбонил)амино]-1-(циклопропилметил)-1 Н-имидазол-2-карбоновой кислоты суспендируют в 158 мл этанола. При охлаждении льдом добавляют 16,4 мл воды и 6 мл (112 ммолей) 50% водного раствора гидроксида натрия. Реакционную смесь перемешивают в течение 1 ч при комнатной температуре и затем концентрируют путем выпаривания в вакууме. Остаток переносят в 100 мл изопропанола и при охлаждении льдом смешивают с 100 мл 1 н. раствора хлористоводородной кислоты. Кристаллы отсасывают и сушат в вакууме при 40°C.
Выход: 9,85 г (100% от теоретического значения)
ЖХ-МС (методика 3): Rt=1,74 мин.
МС (ИЭР+): m/z=349 [М+Н]+
1Н-ЯМР (400 МГц, ДМСО-d6): δ=9,4 (s, 1Н), 8,0 (d, 1H), 7,3 (s, 1H), 7,25 (d, 1H), 7,2 (dd, 1H), 4,25 (d, 2H), 2,25 (s, 3H), 1,25 (m, 1H), 0,55 (q, 2H), 0,35 (q, 2H) част./млн.
Пример 2А
1-Бутил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1Н-имидазол-2-карбоновая кислота
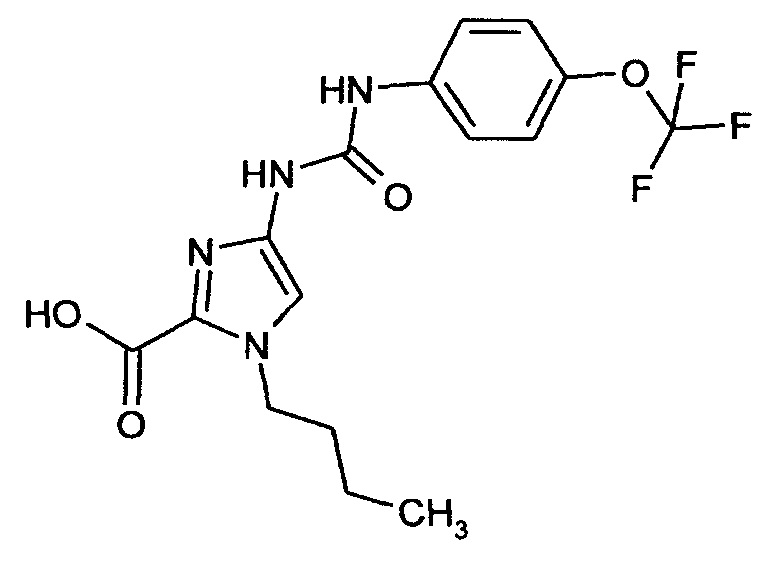
Получают аналогично получению соединения примера 1А.
Выход: 2,05 г (96% от теоретического значения)
ЖХ-МС (методика 3): Rt=1,96 мин.
МС (ИЭР+): m/z=387 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=9,0 (s, 1Н), 8,9 (s, 1H), 7,55 (d, 2H), 7,3 (s, 1H), 7,25 (d, 1H), 4,35 (tr, 2H), 1,7 (квинтет, 2H), 1,25 (секстет, 2H), 0,9 (tr, 3H) част./млн.
Пример 3А
Этиловый эфир 1-метил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1Н-имидазол-2-карбоновой кислоты
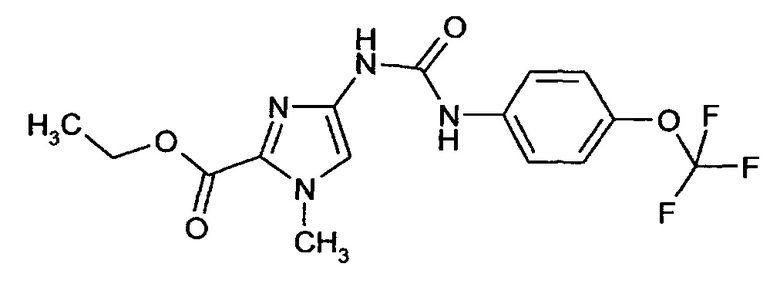
1,22 г (3,61 ммоля) Этилового эфира 4-амино-! -метил- 1Н-имидазол-2-карбоновой кислоты (синтез проводят аналогично описанному в примере 1А, стадия 3, или в соответствии с публикацией Tetrahedron Lett. 2003, 44, 1607 и цитированной в ней литературе) в 50 мл ТГФ в атмосфере аргона смешивают с 1,46 г (7,21 ммоля) 4-(трифторметокси)фенилизоцианата и перемешивают при комнатной температуре в течение ночи. Реакционную смесь фильтруют, фильтрат концентрируют путем выпаривания в вакууме и очищают с помощью хроматографии.
Выход: 860 мг (62% от теоретического значения)
ЖХ-МС (методика 4): Rt=2,41 мин.
МС (ИЭР+): m/z=373 [M+H]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,98 (bs, 2H), 7,55 (m, 2H), 7,36 (s, 1H), 7,29 (m, 2H), 4,28 (q, 2H), 3,91 (s, 3H), 1,30 (t, 3H).
Пример 4А
1-Метил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1Н-имидазол-2-карбоновая кислота
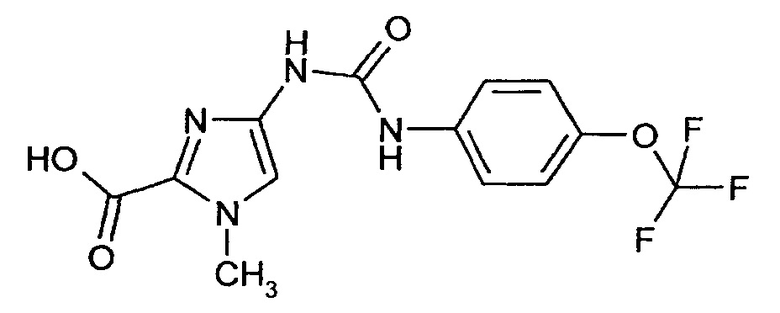
835 мг (2,13 ммоля) Этилового эфира 1-метил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1Н-имидазол-2-карбоновой кислоты (соединение примера 3А) суспендируют в 5 мл этанола и 12 мл тетрагидрофурана. При охлаждении льдом добавляют 2 мл (25 ммолей) 50% водного раствора гидроксида натрия. Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем при охлаждении льдом подкисляют 1 н. раствором хлористоводородной кислоты. Раствор экстрагируют дихлорметаном. Органическую фазу концентрируют путем выпаривания в вакууме. Остаток очищают с помощью препаративной ВЭЖХ. Выход: 346 мг (44% от теоретического значения)
ЖХ-МС (методика 3): Rt=1,62 мин.
МС (ИЭР+): m/z=345 [М+Н]+
1Н-ЯМР (400 МГц, ДМСО-d6): 5=9,33 (bs, 1Н), 8,98 (bs, 1H), 7,55 (m, 2H), 7,30 (s, 1H), 7,28 (m, 2H), 3,90 (s, 3H).
Пример 5А
1-Этил-4-[({[4-(трифторметокси)фенил]амино}карбонил)амино]-1Н-имидазол-2-карбоновая кислота
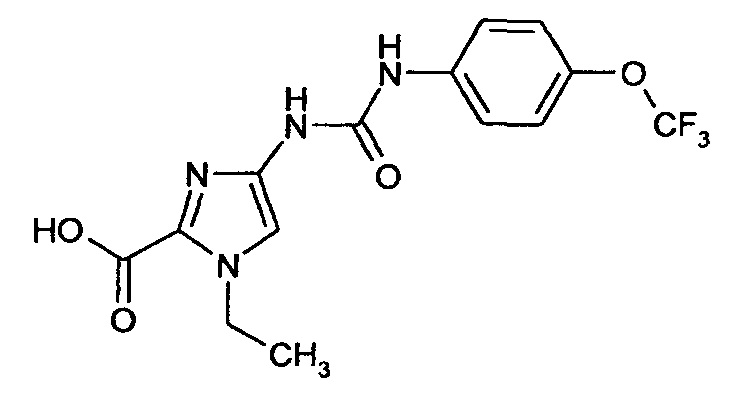
Получают аналогично получению соединения примера 4А.
Выход: 425 мг (91% от теоретического значения)
ЖХ-МС (методика 4): Rt=1,94 мин.
МС (ИЭР+): m/z=359 [M+H]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=10,3 (bs, 1Н), 7,67 (m, 2H), 7,24 (s, 1H), 7,20 (m, 2H), 4,45 (q, 2H), 1,33 (t, 3Н).
Пример 6А
4-[({[4-(Дифторметокси)фенил]амино}карбонил)амино]-1-метил-1Н-имидазол-2-карбоновая кислота
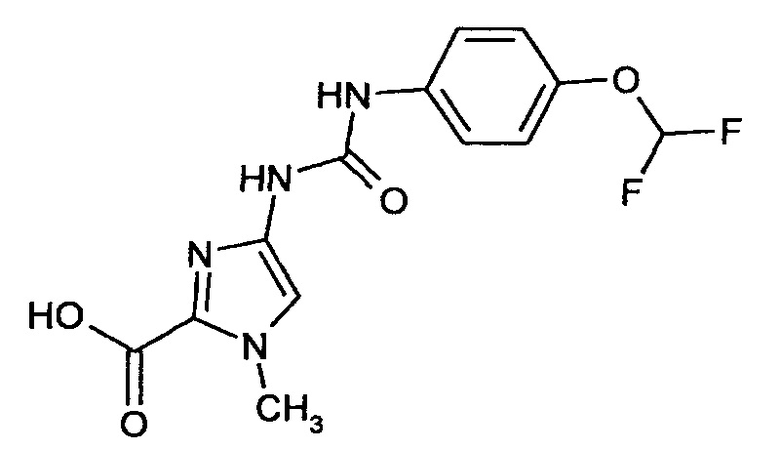
Получают аналогично получению соединения примера 4А.
Выход: 964 мг (81% от теоретического значения)
HPLC (методика 5): Rt=3,57 мин.
МС (ИЭР+): m/z=327 [М+Н]+
1Н-ЯМР (400 МГц, CDCl3): δ=8,9 (s, 1H), 8,8 (s, 1H), 7,5 (d, 2H), 7,3 (s, 2H), 7,1 (t, 1 H), 7,09 (d, 2H), 3,9 (s, 3Н).
Пример 7А
1-(5-Метилпиридин-2-ил)пиперазин
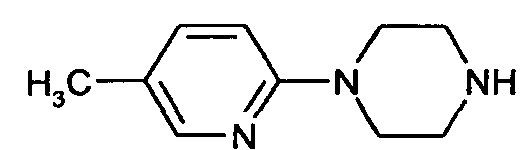
Стадия 1 1-(трет-Бутилоксикарбонил)-4-(5-метилпиридин-2-ил)пиперазин
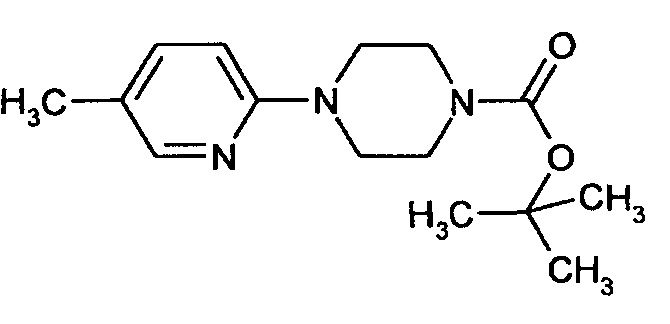
В атмосфере аргона 2,50 г (19,6 ммоля) 2-метил-5-хлорпиридина и 4,38 г (23,5 ммоля) N-(трет-бутилоксикарбонил)пиперазина растворяют в 50 мл абсолютного толуола. Затем добавляют 2,26 г (23,5 ммоля) трет-бутилата натрия, 0,37 г (0,59 ммоля) БИНАФ (2,2'-бис(дифенилфосфино)-1,1'-бинафтил) и 0,36 г (0,39 ммоля) трис(дибензилиденацетон)дипалладия и смесь нагревают при 70°C в течение 12 ч. После охлаждения реакционную смесь смешивают с диэтиловым эфиром, трижды промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель удаляют в вакууме. Остаток очищают с помощью флэш-хроматографии (циклогексан/этилацетат 9:1).
Альтернативно, реакцию сочетания также можно провести с использованием в качестве катализатора ацетата палладия(II).
Выход: 5,27 г (97% от теоретического значения)
ЖХ-МС (методика 3): Rt=1,26 мин.
МС (ИЭР+): m/z=278 [М+Н]+
1Н-ЯМР (300 МГц, CDCl3): δ=8,02 (d, 1Н), 7,34 (dd, 1H), 6,59 (d, 1H), 3,55 (m, 4H), 3,45 (m, 4H), 2,21 (s, 3H), 1,49 (s, 9H).
Стадия 2
1-(5-Метилпиридин-2-ил)пиперазин
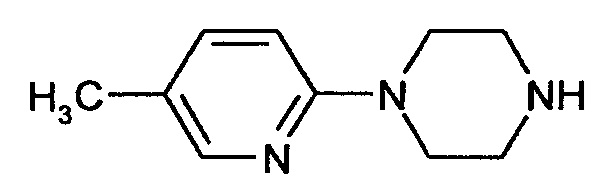
3,47 г (12,5 ммоля) 1-(трет-Бутилоксикарбонил)-4-(5-метилпиридин-2-ил)пиперазина растворяют в 10 мл диоксана и смешивают с 31 мл (125 ммолей) раствора хлорида водорода в диоксане (4 моля). Смесь перемешивают при комнатной температуре в течение 2 ч. Затем ее концентрируют путем выпаривания, остаток подщелачивают 1 М раствором гидроксида натрия и смесь несколько раз экстрагируют дихлорметаном. Объединенные органические фазы сушат над сульфатом натрия, концентрируют путем выпаривания и сушат в вакууме.
Альтернативно, соединение примера 7А также можно выделить в форме его гидрохлорида.
Выход: 2,18 г (98% от теоретического значения)
ЖХ-МС (методика 4): Rt=0,38 мин.
МС (ИЭР+): m/z=177 [М+Н]+
1Н-ЯМР (300 МГц, CDCl3): δ=8,02 (d, 1H), 7,32 (dd, 1H), 6,59 (d, 1H), 3,45 (m, 4H), 3,00 (m, 4H), 2,20 (s, 3H).
Пример 8А
N-{1-Метил-2-[(4-пиридин-2-илпиперазин-1-ил)карбонил]-1Н-имидазол-4-ил}-N'-[4-(трифторметокси)фенил]мочевина
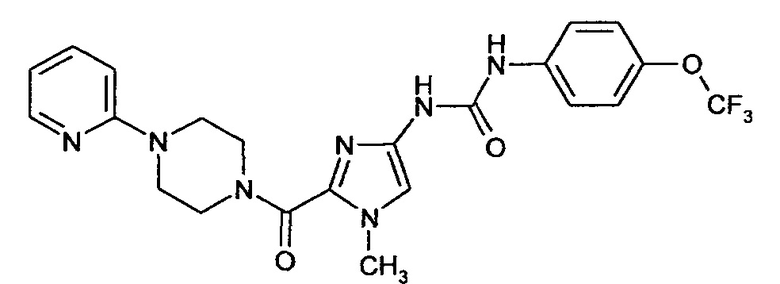
1,50 г (4,36 ммоля) Соединения примера 4А растворяют в 30 мл ДМФ и смешивают с 1,82 г (5,66 ммоля) 0-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуронийтетрафторбората (TBTU) и 266 мг (2,18 ммоля) 4-диметиламинопиридина. После добавления 925 мг (5,66 ммоля) 1-(пиридин-2-ил)пиперазина смесь перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь очищают с помощью ОФ-ВЭЖХ.
Выход: 1,79 г (83% от теоретического значения)
ЖХ-МС (методика I): Rt=1,83 мин.
МС (ИЭР+): m/z=490 [M+H]+
1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,89 (bs, 2Н), 8,12 (d, 1H), 7,55 (m, 3H), 7,29 (m, 2Н), 7,20 (s, 1H), 6,88 (d, 1H), 6,68 (dd, 1H), 4,02 (bs, 2Н), 3,77 (s, 3H), 3,71 (bs, 2Н), 3,58 (bs, 4H).
Пример 9А
N-(1-Метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-(трифторметокси)фенил]мочевина
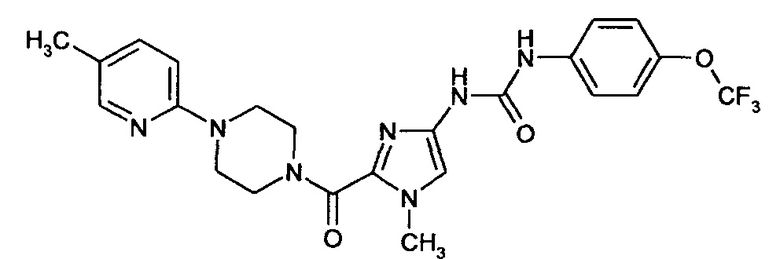
5,6 г (26,14 ммоля) Соединения примера 7А и 13,22 г (130,7 ммоля) N-метилморфолина добавляют к раствору 9,0 г (26,14 ммоля) соединения примера 4А в 110 мл этилацетата и реакционную смесь охлаждают до 0°C. К реакционной смеси в течение 90 мин добавляют 16,63 г (52,26 ммоля) раствора ангидрида пропанфосфоновой кислоты (ТЗР) и полученную суспензию перемешивают при этой температуре в течение еще 10 мин. Затем реакционную смесь в течение 60 мин нагревают до 20°C и перемешивают при этой температуре в течение ночи. Непрореагировавший ТЗР нейтрализуют путем добавления 45 мл воды и реакционную смесь перемешивают в течение еще 10 мин. Затем фазы разделяют и органическую фазу несколько раз промывают водой (3×45 мл), значение рН которой установлено равным 5. Объединенные водные фазы один раз промывают дополнительным количеством этилацетата и объединенные органические фазы дважды промывают с помощью 45 мл водного раствора бикарбоната натрия, сушат над сульфатом натрия и концентрируют путем выпаривания. Полученный неочищенный продукт перекристаллизовывают из этанола и после этого получают конечный продукт в виде бледно-желтого твердого вещества.
Выход: 8,42 г (64% от теоретического значения)
ЖХ-МС (методика 4): Rt=2,01 мин.
МС (ИЭР4"): m/z=504 [М+Н]+
1H-ЯМР (300 МГц, ДМСО-d6): δ=8,92 (bs, 2Н), 7,99 (d, 1H), 7,54 (m, 2H), 7,42 (dd, 1H), 7,28 (m, 2H), 7,20 (s, 1H), 6,80 (d, 1H), 4,00 (bs, 2H), 3,77 (s, 3H), 3,72 (bs, 2H), 3,51 (bs, 4H), 2,16 (s, 3H).
Пример 10А
N-(2-{[4-(5-Хлорпиридин-2-ил)пиперазин-1-ил]карбонил}-1-этил-1Н-имидазол-4-ил)-N'-[4-(трифторметокси)фенил]мочевина
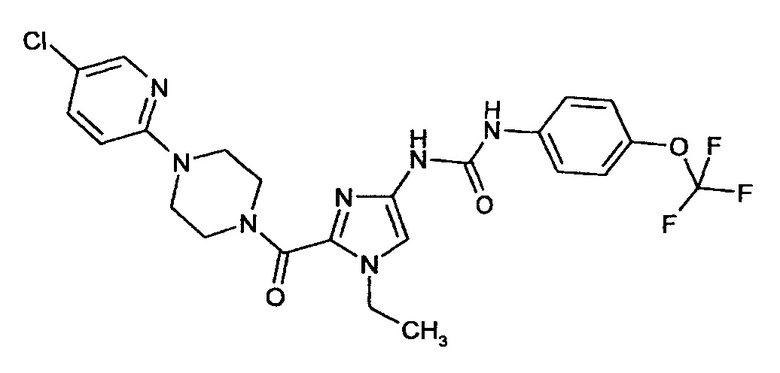
Получают аналогично получению соединения примера 9А из соединения примера 5А.
Выход: 55 мг (68% от теоретического значения)
ЖХ-МС (методика 4): Rt=2,76 мин.
МС (ИЭР+): m/z=538 [М+Н]+
1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,97 (bs, 1H), 8,92 (bs, 1H), 8,14 (d, 1H), 7,65 (dd, 1H), 7,54 (m, 2H), 7,28 (m, 2H), 7,24 (s, 1H), 6,92 (d, 1H), 4,16 (q, 2H), 3,97 (bs, 2H), 3,72 (bs, 2H), 3,59 (bs, 4H), 1,32 (t, 3H).
Пример 11A
N-(2-{[4-(4-Метоксифенил)пиперазин-1-ил]карбонил}-1-метил-1Н-имидазол-4-ил)-N'-[4-(трифторметокси)фенил]мочевина
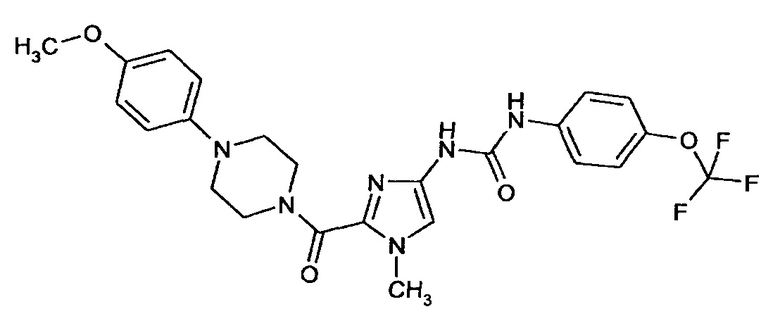
Получают аналогично получению соединения примера 9А из соединения примера 4А.
Выход: 35 мг (58% от теоретического значения)
ЖХ-МС (методика 3): Rt=2,24 мин.
МС (ИЭР+): m/z=519 [М+Н]+
1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,89 (bs, 2H), 7,53 (m, 2H), 7,28 (m, 2H), 7,19 (s, 1H), 6,92 (m, 2H), 6,84 (m, 2H), 4,05 (bs, 2H), 3,75 (m, 5H), 3,69 (s, 3H), 3,08 (bs, 4H).
Пример 12А
N-[4-(Дифторметокси)фенил]-N'-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)мочевина
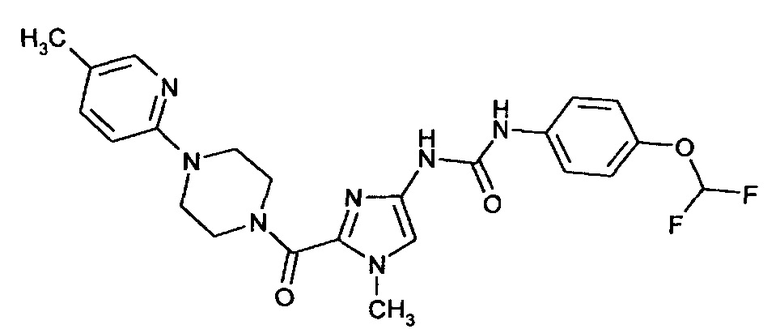
Получают аналогично получению соединения примера 9А из соединения примера 6А.
Выход: 17 мг (29% от теоретического значения)
ЖХ-МС (методика 4): Rt=1,70 мин.
МС (ИЭР+): m/z=486 [M+H]+
1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,84 (bs, 1Н), 8,77 (bs, 1H), 7,98 (d, 1H), 7,47 (m, 2H), 7,42 (dd, 1H), 7,18 (s, 1H), 7,11 (t, 1H), 7,10 (m, 2H), 6,80 (d, 1H), 4,01 (bs, 2H), 3,77 (s, 3H), 3,71 (bs, 2H), 3,50 (bs, 4H), 2,16 (s, 3H).
Соединения примеров, приведенные в таблице 1, получают аналогично получению соединения примера 8А.
Таблица 1
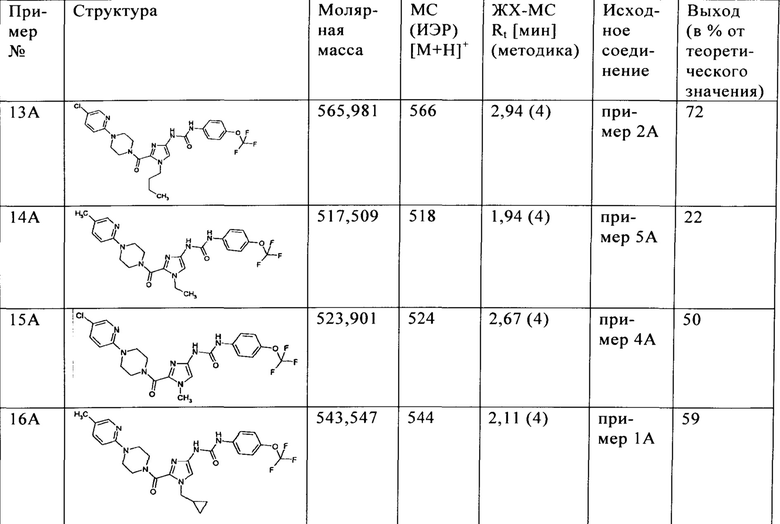
Варианты осуществления Пример 1
Димезилат N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины
Все операции проводят в защитной атмосфере азота. В сосуде для проведения реакции 3,202 г соединения примера 9А (6,36 моля, 1 экв.) смешивают со смесью, содержащей 15 л ТГФ и 1 л воды. Полученную суспензию медленно нагревают до 60°C и затем перемешивают при этой температуре в течение 30 мин. К образовавшемуся желтоватому раствору добавляют 1,252 г метансульфоновой кислоты (13,03 моля, 2,05 экв.) и затем вносят затравку димезилата N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины. К суспензии, полученной из кристаллического димезилата N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины, в течение 2 ч добавляют еще 30 л ТГФ. Суспензию медленно охлаждают до 20°C и затем перемешивают при этой температуре в течение еще 12 ч. Образовавшиеся кристаллы собирают с помощью вакуумного фильтрования и реактор промывают с помощью ТГФ и затем н-гептаном, причем эти органические фазы затем используют для промывки кристаллов. В заключение кристаллы сушат на фильтре в вакууме и в потоке азота. Получают 4,262 г (выход: 96,4%, чистота >99%) искомого димезилата.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=9,07 (s, 1Н), 8,98 (s, 1H), 7,99 (s, 1H), 7,92 (d, 1H), 7,56 (d, 2H), 7,41 (d, 1H), 7,33-7,24 (m, 3H), 4,18 (s, br., 2H), 3,92-3,69 (m, 9H), 2,43-2,39 (s, 6H), 2,25 (s, 3H).
Рентгенограмму, представленную на фиг. 1, снимали с использованием порошкового дифрактометра Rigaku MiniFlex.
Соединение примера 8А, а также соединения примеров 10А-15А можно превратить в димезилаты аналогичным образом.
Исследование растворимости
20 мг Соединения примера 1, а также, для сравнения, цитрата, малеата, сульфата и тартрата соединения примера 9А, а также хлоридов соединения примера 9А, полученных с использованием 1, 2 и 4 экв. хлористоводородной кислоты, а также соединения примера 9А в виде свободного основания, отвешивают в стеклянные сосуды для ВЭЖХ, снабженные стержнями для магнитных мешалок. В каждый сосуд добавляют 1 мл Н2О и стеклянные сосуды для ВЭЖХ герметизируют. Полученные суспензии перемешивают при 25°C в течение ночи. Для определения количества растворившегося вещества суспензии фильтруют через пипетки с микрофильтрами и полученные фильтраты разбавляют в соотношении 1:4 и анализируют с помощью ВЭЖХ. Анализ с помощью ВЭЖХ проводят с использованием колонки Dionex Luna RP18 (100А), обладающей следующими размерами: 5 мкм, 50×4,6 мм, и с использованием смеси ацетонитрила и Н2О+0,1% ТФК (трифторуксусная кислота) состава 3:7 в изократическом режиме.
Значения, полученные при исследовании растворимости, приведены ниже в таблице 2.
Эти значения ясно указывают на то, что растворимость соли соединения примера 1 в водной среде существенно выше, чем растворимость других солей соединения примера 9А.
Растворимость при искусственно созданных условиях, соответствующих условиях в желудке человека
Для определения растворимости при искусственно созданных условиях, соответствующих условиях в желудке человека, суспензии соли соединения 1, а также цитрата и тартрата соединения примера 9А и соответствующего свободного основания в водном растворе хлорида натрия (0,2 мас. %), значение рН которого устанавливают равным 1,2 хлористоводородной кислотой, перемешивают в течение 5 ч. Затем образцы обрабатывают так, как описано выше, и с помощью ВЭЖХ определяют количество свободного основания в растворе. Соответствующие значения при искусственно созданных условиях, соответствующих условиях в желудке человека, приведены ниже в таблице 3.
Приведенные в таблице данные ясно указывают на значительно лучшую растворимость солей, предлагаемых в настоящем изобретении, при искусственно созданных условиях, соответствующих условиях в желудке человека. В связи с этим следует отметить, что после выдерживания раствора в течение длительного периода времени, можно наблюдать повторное образование суспензии. Можно полагать, что это происходит вследствие образования плохо растворимых хлоридов соединения примера 9А. Это образование нерастворимых хлоридов не считается существенной проблемой, однако, поскольку последнее, главным образом при использовании соединения примера 1, происходит с задержкой, то, таким образом, изначально существует метастабильный перенасыщенный раствор. Это является дополнительным подтверждением преимуществ, которые можно обеспечить при использовании солей, предлагаемых в настоящем изобретении, для приготовления лекарственных средств.
Гигроскопичность
Гигроскопичность солей, предлагаемых в настоящем изобретении, исследуют при хранении соли соединения примера 1 в чистом виде в атмосфере с относительной влажностью, равной примерно 46%, и при 24°C. В этом случае после хранения в течение примерно 2 дней масса соли соединения примера 1 увеличивается менее, чем на 0,11%, что означает, что соединение обладает гигроскопичностью, приемлемой для его использования в лекарственных средствах.
В. Оценка физиологической эффективности
Воздействие соединений, предлагаемых в настоящем изобретении, in vitro на репликацию ЦМВЧ (цитомегаловирус человека) может быть продемонстрировано с помощью приведенного ниже исследования противовирусной активности:
Исследование уменьшения интенсивности флуоресценции ЦМВЧ
Исследуемые соединения используют в виде 50 мМ растворов в диметилсульфоксиде (ДМСО). В качестве соединений для сравнения можно использовать, например, ганцикловир®, фоскарнет®, цидофовир® или соединение примера 9А. За 1 день до начала проведения исследования, 1,5×10 фибробластных клеток крайней плоти полового члена мужчины (клетки NHDF)/лунка в 200 мкл культуральной среды для клеток высевают в лунки В2-G11 96-луночных планшетов (черные с прозрачным дном). Для устранения краевых эффектов в лунки, расположенные по краям помещают только 200 мкл среды. В день проведения исследования из лунок В2 - G11 каждого 96-луночного планшета отсасывают культуральную среду для клеток и ее заменяют на 100 мкл суспензии вируса (множественность заражения (МНЗ): 0,1-0,2). Использующимся вирусом является рекомбинантный ЦМВЧ, который содержит в геноме вируса кассету экспрессии белка, обладающего зеленой флуоресценцией (БЗФ) (ЦМВЧ AD 169 RV-HG, E.M. Borst, К. Wagner, A. Binz, В. Sodeik, и M. Messerle, 2008, J. Virol. 82: 2065-2078). После инкубации при 37°C и 5% СО2 в течение 2 ч инокулированный вирус отсасывают и во все лунки, за исключением лунок столбца 3, помещают 200 мкл культуральной среды для клеток. Лунки столбца 2 больше не обрабатывают и используют в качестве вирусного контроля. В лунки столбца 3 помещают по 300 мкл исследуемого соединения (разводят в культуральной среде для клеток), в каждом случае проводят двукратные исследования. Концентрация соответствующего противовирусного соединения в лунках столбца 3 составляет ~27х значение концентрации ЕС50, предполагаемое в каждом случае. Исследуемое соединение в лунках столбца 3 96-луночного планшета разводят 8 раз в соотношении 1:3 путем переноса в каждом случае по 100 мкл из лунок столбца в лунки находящегося справа столбца и смешивания с находящимися в них 200 мкл культуральной среды для клеток. Таким образом, три противовирусных соединения исследуют двукратно. Планшеты инкубируют при 37°C/5% СО2 в течение 7 дней. Затем все лунки планшета 3 раза промывают с помощью ЗФФ (забуференный фосфатом физиологический раствор) и в них помещают по 50 мкл ЗФФ. Затем измеряют интенсивность флуоресценции БЗФ в каждой лунке 96-луночного планшета с помощью устройства для считывания флуоресценции (FluoBox; Bayer Technology Services GmbH; параметры фильтра: БЗФ, длина волны возбуждения: 480 нм, длина волны испускания: 520 нм). Из полученных таким образом значений можно определить значения ЕС50 для соединения, обладающего активностью по отношению к ЦМВЧ: ЕС50 (БЗФ-RA)= концентрация соединения в мкм, которая приводит к снижению интенсивности флуоресценции БЗФ в инфицированных клетках на 50% по сравнению с не подвергнутым воздействию вирусным контролем.
Полученные in vitro типичные значения активности соединений, предлагаемых в настоящем изобретении, приведены в таблице 4:
Варианты осуществления фармацевтических композиций
Соединения, предлагаемые в настоящем изобретении, можно переработать в фармацевтические препараты следующим образом:
Таблетка:
Состав:
100 мг Соединения примера 1, 50 мг лактозы (моногидрат), 50 мг кукурузного крахмала (натурального), 10 мг поливинилпирролидона (ПВП 25) (выпускается фирмой BASF, Ludwigshafen, Germany) и 2 мг стеарата магния.
Масса таблетки 212 мг, диаметр 8 мм, радиус кривизны 12 мм.
Приготовление:
Смесь активного ингредиента, лактозы и крахмала гранулируют вместе с 5% (мас./мас.) водным раствором ПВП. После сушки гранулы перемешивают со стеаратом магния в течение 5 мин. Эту смесь прессуют с помощью обычного таблетирующего пресса (размеры таблетки указаны выше). Рекомендуемая прессующая сила при прессовании равна 15 кН.
Суспензия, которую можно вводить перорально:
Состав:
1,000 мг Соединения примера 1, 1,000 мг этанола (96%), 400 мг Rhodigel (ксантановая камедь, выпускается фирмой FMC, Pennsylvania, USA) и 99 г воды.
10 мл Суспензии для перорального введения эквивалентны равной 100 мг разовой дозе соединения, предлагаемого в настоящем изобретении.
Приготовление:
Rhodigel суспендируют в этаноле и к суспензии прибавляют активный ингредиент. При перемешивании прибавляют воду. Смесь перемешивают в течение примерно 6 ч до завершения набухания Rhodige.
Раствор, который можно вводить внутривенно:
Состав:
5,53 г Соединения примера 1, 1,000 г воды для инъекций, которая содержит 10% (мас./об.) гидроксипропил-Р-циклодекстрина (Aldrich) и 985 мг ацетата натрия.
Приготовление:
Соединение, предлагаемое в настоящем изобретении, при перемешивании растворяют в воде и значение рН доводят до равного примерно 3,94 ацетатом натрия. Раствор стерилизуют фильтрованием (диаметр пор 0,22 мкм) и в асептических условиях переливают в термически стерилизованные флаконы для вливания. Последние закрывают пробками для вливания и фланцевыми колпачками.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДЫ КАК ПРОТИВОВИРУСНЫЕ СРЕДСТВА | 2007 |
|
RU2423352C2 |
| ГЕТЕРОЦИКЛИЛАМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 2006 |
|
RU2415851C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЛАМИДОМ ТИАЗОЛЫ, ПИРРОЛЫ И ТИОФЕНЫ | 2006 |
|
RU2425829C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ EGFR | 2021 |
|
RU2815022C1 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| Пан-геномные ингибиторы белка NS5A вируса гепатита С, фармацевтические композиции, промежуточные продукты для синтеза ингибиторов и способы их получения и применения | 2016 |
|
RU2669919C1 |
| АЦИЛИРОВАННЫЕ НОНАДЕПСИПЕПТИДЫ В КАЧЕСТВЕ ПРОИЗВОДНЫХ ЛИЗОБАКТИНА | 2005 |
|
RU2414477C2 |
| Производные N-пиперидин-3-илбензамида для лечения сердечно-сосудистых заболеваний | 2014 |
|
RU2618628C1 |
| ОКСОХИНАЗОЛИНИЛБУТАНАМИДНЫЕ ПРОИЗВОДНЫЕ | 2014 |
|
RU2669393C2 |
Изобретение относится к соли соединения формулы
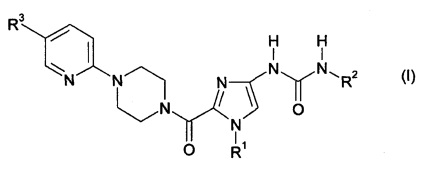
в которой R1 обозначает метил, этил, бутил или циклопропилметил, R2 обозначает фенил, где фенил содержит заместитель, который выбран из группы, включающей трифторметоксигруппу и дифторметоксигруппу, и R3 обозначает водород, метил, хлор, метоксигруппу или трифторметил, с органической сульфоновой кислотой или ее сольват, или гидрат, их сольватам и гидратам и их применению в качестве противовирусных средств. 8 н. и 6 з.п. ф-лы., 1 ил., 4 табл., 1 пр.
1. Соль соединения формулы
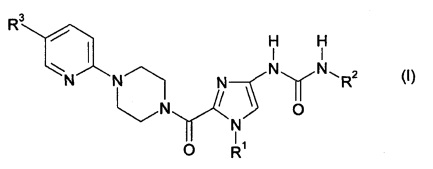
в которой
R1 обозначает метил, этил, бутил или циклопропилметил,
R2 обозначает фенил, где фенил содержит заместитель, который выбран из группы, включающей трифторметоксигруппу и дифторметоксигруппу, и
R3 обозначает водород, метил, хлор, метоксигруппу или трифторметил с органической сульфоновой кислотой или ее сольват, или гидрат.
2. Соль по п. 1, отличающаяся тем, что органической сульфоновой кислотой является метансульфоновая кислота.
3. Соль по п. 2, где соль является димезилатом.
4. Соль по п. 3, описывающаяся следующей формулой:
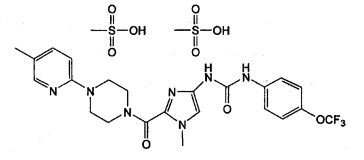
5. Кристаллический димезилат N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины, на порошковой рентгенограмме которого содержатся характеристические пики примерно при 6,37, 11,77, 12,56, 17,17, 18,81, 20,34, 21,47, 23,04 и 35,46 градуса 2-тэта.
6. Кристаллический димезилат N-(1-метил-2-{[4-(5-метилпиридин-2-ил)пиперазин-1-ил]карбонил}-1Н-имидазол-4-ил)-N'-[4-трифторметоксифенил]мочевины, обладающий порошковой рентгенограммой, в общем виде представленной на фиг. 1.
7. Способ получения соединений формулы (I) по п. 1, который включает реакцию соединения формулы (I) или соли соединений формулы (I) с кислотой, которая не является солью с органической сульфоновой кислотой, с органической сульфоновой кислотой или с источником сульфонат-анионов в растворителе.
8. Соль по одному из пп. 1-6, предназначенная для применения в способе лечения и/или предупреждения заболеваний, предпочтительно вирусных инфекций, предпочтительно инфекций ЦМВЧ или другим представителем группы Herpesviridae.
9. Фармацевтическое средство для лечения и/или предупреждения вирусных инфекций, предпочтительно инфекций ЦМВЧ или другим представителем группы Herpesviridae, которое содержит эффективное количество соли по одному из пп. 1-6 в комбинации по меньшей мере с одним инертным, нетоксичным, фармацевтически приемлемым вспомогательным веществом.
10. Фармацевтическое средство по п. 9, которое содержит от 5 до 12,5 мг/мл соли по одному из пп. 1-6, от 50 до 150 мг/мл гидроксипропил-β-циклодекстрина, от 0,5 до 2,0 мг/мл ацетата натрия, а также воду и необязательно другие фармацевтически безопасные вспомогательные вещества.
11. Применение соли по одному из пп. 1-6 для приготовления фармацевтического средства, предназначенного для лечения и/или предупреждения вирусных инфекций, предпочтительно инфекций ЦМВЧ или другим представителем группы Herpesviridae.
12. Применение соли по одному из пп. 1-6 в способе лечения и/или предупреждения заболеваний, предпочтительно вирусных инфекций, предпочтительно инфекций ЦМВЧ или другим представителем группы Herpesviridae.
13. Фармацевтическое средство по п. 9 или 10, предназначенное для применения в способе лечения и/или предупреждения вирусных инфекций, предпочтительно инфекций ЦМВЧ или другим представителем группы Herpesviridae.
14. Способ борьбы с вирусными инфекциями, предпочтительно с инфекциями ЦМВЧ или другим представителем группы Herpesviridae, у людей и животных, который включает введение по меньшей мере одной соли по одному из пп. 1-6, фармацевтического средства по п. 9 или 10 или фармацевтического средства, полученного в п. 11 или 12, в противовирусно эффективном количестве человеку или животному, которое нуждается в таком лечении.
| WO 2006089664 A2, 31.08.2006 | |||
| WO 2004052852 A1, 24.06.2004 | |||
| НОВЫЕ ПРОИЗВОДНЫЕ АРИЛИМИДАЗОЛА, ИХ ПРЕПАРАТЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2003 |
|
RU2323216C2 |

