Область техники, к которой относится изобретение
Настоящее изобретение относится в целом к композициям хинолиноновых производных. Более конкретно представленное в настоящем описании изобретение относится к композициям в виде твердых лекарственных форм, содержащим фармацевтически приемлемые соли, такие как лактаты 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она, и способам получения и применению таких композиций.
Предпосылки создания изобретения
Известно большое количество разнообразных химических соединений и композиций, обладающих активностью в отношении одной или нескольких рецепторных тирозинкиназ сосудистого эндотелиального фактора роста (VEGF-RTK). Их примерами являются хинолиновые производные, такие как соединения, описанные в WO 98/13350, аминоникотинамидные производные (см, например, WO 01/55114), антисмысловые соединения (см., например, WO 01/52904), пептидомиметики (см., например, WO 01/52875), хиназолиновые производные (см., например, US 6258951), моноклональные антитела (см., например, ЕР 1086705 А1), различные 5,10,15,20-тетраарилпорфирины и 5,10,15-триарилкорролы (см., например, WO 00/27379), гетероциклические производные алкансульфоновой и алканкарбоновой кислот (см., например, DE19841985), оксиндолилхиназолиновые производные (см., например, WO 99/10349), 1,4-диазаантрациновые производные (см., например US 5763441) и циннолиновые производные (см., например, WO 97/34876) и различные индазольные производные (см., например, WO 01/02369 и WO 01/53268).
Синтез 4-гидроксихинолоновых и 4-гидроксихинолиновых производных описан в многочисленных публикациях. Например, Ukrainets с соавторами описал синтез 3-(бензимидазол-2-ил)-4-гидрокси-2-оксо-1,2-дигидрохинолина - (Ukrainets I. и др. Tetrahedron Lett. 42, 1995, cc.7747-7748; Ukrainets I. и др. - Khimiya Geterotsiklicheskikh Soedinii, 2, 1992, сс.239-241). Ukrainets описал также синтез, противосудорожную и антитиреоидную активность других 4-гидроксихинолонов и тиоаналогов, таких как 1Н-2-оксо-3-(2-бензимидазолил)-4-гидроксихинолин (Ukrainets I. и др. Khimiya Geterotsiklicheskikh Soedinii, 1, 1995, сс.105-108; Ukrainets I. и др. Khimiya Geterotsiklicheskikh Soedinii, 8, 1993, сс.1105-1108; Ukrainets I. и др. Chem. Heterocyclic Comp. 33, 1997, сс.600-604. Синтез различных хинолиновых производных описан в WO 97/48694. Описано, что эти соединения обладают способностью связываться с рецепторами ядерных гормонов и что их можно применять для стимулирования пролиферации остеобластов и роста кости. Указано также, что эти соединения можно применять для лечения или предупреждения заболеваний, ассоциированных с семействами рецепторов ядерных гормонов.
Различные хинолиновые производные, в которых бензольное кольцо хинолина замещено группой, представляющей собой серу, описаны в WO 92/18483. Указано, что эти соединения можно применять в фармацевтических композициях и в качестве лекарственных средств.
Хинолоновые и кумариновые производные описаны в качестве соединений, которые находят применение в различных областях, не связанных с медициной и фармацевтическими композициями. Получение хинолоновых производных, предназначенных для применения в фотополимеризующихся композициях или в областях, связанных с люминисцентными свойствами, описано, в частности, в US 5801212 на имя Okamoto и др.; JP 8-29973; JP 7-43896; JP 6-9952; JP 63-258903; EP 797376 и DE 2363459.
Многочисленные замещенные хинолиноновые производные, включая хинолинонбензимидазолильные производные и 4-аминозамещенные хинолинонбензимидазолильные производные, такие как 4-амино-5-фтор-3-[5-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]хинолин-2(1Н)-он, описаны в последние годы, например, в WO 02/22598, WO 2004/043389, WO 2005/047244, US 2004/0220196, US 2005/0137399, WO 2005/046590 и WO 2005/046589. Описано, что такие соединения обладают способностью ингибировать VEGF-RTK. Такие соединения описаны также в опубликованных заявках US 2002/0107392 и US 2003/0028018 и в патентах US 6605617, 6774237, 6762194 и 6800760. Другие такие соединения, а также новые применения таких соединений для ингибирования серин/треониновых киназ и тирозиновых киназ описаны в WO 2004/018419 и US 2004/0092535, зарегистрированной 19 августа 2003 г. и претендующей на приоритет каждой из следующих предварительных заявок на патент: предварительной заявки на патент США №60/405729, зарегистрированной 23 августа 2002 г.; предварительной заявки на патент США №60/426107, зарегистрированной 13 ноября 2002 г.; предварительной заявки на патент США №60/426226, зарегистрированной 13 ноября 2002 г.; предварительной заявки на патент США №60/426282, зарегистрированной 13 ноября 2002 г.; предварительной заявки на патент США №60/428210, зарегистрированной 21 ноября 2002 г.; предварительной заявки на патент США №60/460327, зарегистрированной 3 апреля 2003 г.; предварительной заявки на патент США №, зарегистрированной 3 апреля 2003 г.; предварительной заявки на патент США №60/460493, зарегистрированной 3 апреля 2003 г.; предварительной заявки на патент США №60/478916, зарегистрированной 16 июня 2003 г., и предварительной заявки на патент США №60/484048, зарегистрированной 1 июля 2003 г. Дополнительная информация, касающаяся хинолиноновых производных и их применений, приведена в предварительной заявке на патент США №60/680722, зарегистрированной 13 мая 2005 г.; предварительной заявке на патент США №60/681893, зарегистрированной 17 мая 2005 г.; предварительной заявке на патент США №60/546395, зарегистрированной 20 февраля 2004 г.; предварительной заявке на патент США №60/547103, зарегистрированной 23 февраля 2004 г.; предварительной заявке на патент США №60/554771, зарегистрированной 19 мая 2004 г.; предварительной заявке на патент США №60/647568, зарегистрированной 27 января 2005 г.; предварительной заявке на патент США №60/669245, зарегистрированной 6 апреля 2005 г.; предварительной заявке на патент США №60/538594, зарегистрированной 23 января 2004; предварительной заявке на патент США №60/683999, зарегистрированной 23 мая 2005 г. заявке на патент США №11/061386, зарегистрированной 18 февраля 2005 г.; заявке на патент США №11/041191, зарегистрированной 21 января 2005 г., и заявке РСТ №PCT/US2005/05316, зарегистрированной 18 февраля 2005 г. Гетероциклические соединения, родственные бензимидазолилхинолинонам, описаны в последние годы в WO 02/18383, US 2002/0103230 и US 6756383. Каждый из перечисленных в данном параграфе документов включен в настоящее описание в качестве ссылки во всей полноте и для всех целей, которые полностью изложены в настоящем описании.
Хотя в литературе уже описаны различные хинолиноновые производные, существует необходимость в новых стабильных композициях, лекарственных средствах и способах введения таких соединений, поскольку эти соединения находят важное фармацевтическое применение при ингибировании ангиогенеза и лечении рака.
Краткое изложение сущности изобретения
Настоящее изобретение относится к фармацевтическим композициям хинолиноновых производных, таким как композиции в виде капсулы или таблетки, которые содержат лактаты 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она, и к способам получения и применения таких композиций. Композиции можно получать с помощью методов сухого смешения или мокрой грануляции.
Одним из объектов настоящего изобретения является фармацевтическая композиция, которая содержит соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь
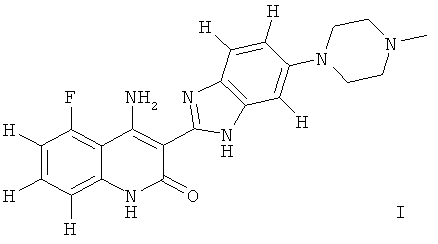 ;
;
и по меньшей мере один ингредиент, выбранный из группы, включающей (I) целлюлозу; (II) лактозу, крахмал или их смесь; (III) повидон; (IV) диоксид кремния или тальк; (V) фармацевтически приемлемый замасливатель и (VI) ингредиент, выбранный из кросповидона, кроскармеллозы натрия и натрийгликолят крахмала.
Другой объект настоящего изобретения относится к фармацевтической композиции, которая содержит соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь; по меньшей мере один ингредиент, выбранный из группы, включающей целлюлозу, повидон, диоксид кремния, тальк и фармацевтически приемлемый замасливатель, и по меньшей мере один ингредиент, выбранный из группы, включающей лактозу, крахмал, кросповидон, кроскармеллозу натрия и натрийгликолят крахмала.
В определенных вариантах осуществления изобретения композиция содержит: (I) целлюлозу; (II) диоксид кремния; (III) стеариновую кислоту или соль стеариновой кислоты и (IV) по меньшей мере один ингредиент, выбранный из кросповидона, крахмала, лактозы, кроскармеллозы натрия и натрийгликолята крахмала. В некоторых из таких вариантов осуществления изобретения композиция содержит кросповидон. В некоторых других вариантах осуществления изобретения композиция содержит крахмал, такой как частично предварительно желированный крахмал. В других вариантах осуществления изобретения композиция содержит лактозу.
В определенных вариантах осуществления изобретения композиция содержит лактат соединения формулы I.
В других вариантах осуществления изобретения композиция включена в состав капсулы или таблетки. В некоторых из таких вариантов осуществления изобретения общая масса соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в капсуле составляет от 25 до 500 мг.
В следующих вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 10 до 50 мас.% в пересчете на общую массу композиции. В некоторых из таких вариантов осуществления изобретения композиция содержит лактат соединения в количестве от 20 до 45 мас.% в пересчете на общую массу композиции. В некоторых других из таких вариантов осуществления изобретения композиция содержит лактат соединения в количестве от 30 до 40 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения целлюлоза, применяемая в композиции, представляет собой микрокристаллическую целлюлозу.
В определенных вариантах осуществления изобретения композиция содержит целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу композиции. В некоторых из таких вариантов осуществления изобретения композиция содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу композиции и композиция содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения композиция содержит крахмал в количестве от 10 до 40 мас.% в пересчете на общую массу композиции, и крахмал представляет собой частично предварительно желированный крахмал.
В определенных вариантах осуществления изобретения композиция содержит диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения композиция содержит стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 30 до 40 мас.% в пересчете на общую массу композиции; диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции, целлюлозу в количестве от 25 до 40 мас.% в пересчете на общую массу композиции, стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции и кросповидон в количестве от 2 до 4 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 50 до 80 мас.% в пересчете на общую массу композиции; диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции, целлюлозу в количестве от 0 до 50 мас.% в пересчете на общую массу композиции, стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции и крахмал в количестве от 10 до 40 мас.% в пересчете на общую массу композиции.
Объектом изобретения являются также контейнеры для упаковки фармацевтических продуктов. В одном из вариантов осуществления изобретения контейнер для упаковки представляет собой сосуд для хранения, содержащий две или более капсул или таблеток, где капсулы или таблетки содержат фармацевтическую композицию, предлагаемую в одном из вариантов осуществления изобретения. В некоторых из таких вариантов осуществления изобретения сосуд для хранения сделан из полиэтилена высокой плотности (ПЭВП). В некоторых из таких вариантов осуществления изобретения сосуд для хранения содержит прокладку из хлопка или искусственного волокна и в определенных вариантах осуществления изобретения включает термокрышку. Другой вариант осуществления изобретения относится к контейнеру для упаковки фармацевтических продуктов, который представляет собой блистерную упаковку, где блистерная упаковка содержит по меньшей мере одну капсулу или таблетку, которая включает фармацевтическую композицию, предлагаемую в одном из вариантов изобретения.
Изобретение относится также к нанесению на таблетку, предлагаемую в настоящем изобретении, покрытия из вещества, выбранного из группы, включающей сахар, полимер на основе целлюлозы и полиметакрилатный полимер. В некоторых вариантах осуществления изобретения на таблетку можно наносить также желатиновое покрытие или капсулировать таблетку в желатиновую оболочку.
Изобретение относится также к окрашиванию таблетки или капсулы, предлагаемой в настоящем изобретении, фармацевтически приемлемым красителем или замутнителем.
Одним из объектов изобретения является способ получения фармацевтической композиции. Способ заключается в том, что (а) смешивают первую смесь с получением первой перемешанной смеси, где первая смесь содержит: (I) соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь и (II) по меньшей мере один ингредиент, выбранный из группы, включающей целлюлозу; лактозу, крахмал или их смесь; повидон; диоксид кремния или тальк; фармацевтически приемлемый замасливатель; и ингредиент, выбранный из кросповидона, кроскармелозы натрия и натрийгликолят крахмала. В некоторых из таких вариантов осуществления изобретения соединение формулы I смешивают с (I) целлюлозой; (II) диоксидом кремния и (III) ингредиентом, выбранным из кросповидона, крахмала и лактозы. Кроме того, способ заключается в том, что (б) смешивают стеариновую кислоту, соль стеариновой кислоты или их смесь с первой перемешанной смесью с получением второй перемешанной смеси и/или (в) изготавливают по меньшей мере одну капсулу или по меньшей мере одну таблетку из второй перемешанной смеси.
Другой объект изобретения относится к способу получения фармацевтической композиции. Способ заключается в том, что (а) смешивают смесь ингредиентов с получением первой перемешанной смеси. Первая перемешанная смесь содержит: (I) соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь, (II) по меньшей мере один ингредиент, выбранный из группы, включающей целлюлозу; крахмал; лактозу; и повидон; (III) по меньшей мере один ингредиент, выбранный из группы, включающей кросповидон, кроскармелозу натрия и натрийгликолят крахмала; жидкость для грануляции, выбранную из группы, включающей водную кислоту, спирт, водный спирт или смесь любых двух или большего количества из этих компонентов. Способ заключается также в том, что (б) удаляют жидкость для грануляции. Кроме того, способ заключается в том, что (в) получают вторую перемешанную смесь путем смешения первой перемешанной смеси с по меньшей мере одним дополнительным ингредиентом, выбранным из группы, включающей (I) кросповидон, кроскармеллозу натрия или натрийгликолят крахмала; (II) стеариновую кислоту или соль стеариновой кислоты и (III) диоксид кремния или тальк. Способ может заключаться также в том, что (г) изготавливают по меньшей мере одну капсулу или по меньшей мере одну таблетку из второй перемешанной смеси.
Изобретение относится также к способам получения фармацевтической композиции, в которых фармацевтическую композицию приготавливают с использованием по меньшей мере одного аппарата, выбранного из группы, включающей (I) гранулятор с псевдоожиженным слоем, снабженный механизмом нижнего распыления, верхнего распыления или тангенциального распыления; (II) гранулятор с высокими сдвиговыми усилиями; (III) гранулятор с низкими сдвиговыми усилиями; (IV) роллерный уплотнитель и (V) таблеточный пресс.
В определенных вариантах осуществления изобретения общая масса соединения формулы I, таутомера соединения, фармацевтически приемлемой соли соединения, фармацевтически приемлемой соли таутомера или их смеси в капсуле или таблетке составляет от 25 до 500 мг.
В определенных вариантах осуществления способа вторая перемешанная смесь содержит лактат соединения формулы I. В других вариантах осуществления изобретения вторая перемешанная смесь содержит лактат соединения в количестве от 10 до 50 мас.% в пересчете на общую массу второй перемешанной смеси.
В некоторых вариантах способа получения фармацевтической композиции целлюлоза представляет собой микрокристаллическую целлюлозу. В определенных вариантах осуществления изобретения крахмал представляет собой предварительно желированный крахмал.
В определенных вариантах способа вторая перемешанная смесь содержит целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу второй перемешанной смеси. В некоторых из таких вариантов осуществления изобретения вторая перемешанная смесь содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и вторая перемешанная смесь содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу второй перемешанной смеси.
В определенных вариантах способа вторая перемешанная смесь содержит крахмал в количестве от 20 до 40 мас.% в пересчете на общую массу второй перемешанной смеси, и крахмал представляет собой частично предварительно желированный крахмал.
В конкретных вариантах способа вторая перемешанная смесь содержит диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу второй перемешанной смеси.
В конкретных вариантах способа получения фармацевтической композиции вторая перемешанная смесь содержит стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу второй перемешанной смеси.
В конкретных вариантах способа вторая перемешанная смесь содержит лактат соединения в количестве от 50 до 80 мас.% в пересчете на общую массу второй перемешанной смеси, в количестве от 55 до 75 мас.% в пересчете на общую массу второй перемешанной смеси или в количестве от 60 до 70 мас.% в пересчете на общую массу второй перемешанной смеси.
В некоторых из вариантов способа диоксид кремния присутствует в количестве от 0,3 до 2 мас.% в пересчете на общую массу второй перемешанной смеси. В других вариантах этого способа целлюлоза присутствует в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси. В следующих вариантах этого способа стеарат магния присутствует в количестве от 0,1 до 2 мас.% в пересчете на общую массу второй перемешанной смеси. В других вариантах этого способа вторая перемешанная смесь дополнительно содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу второй перемешанной смеси. В следующих вариантах этого способа вторая перемешанная смесь содержит диоксид кремния в количестве от 0,5 до 2 мас.% в пересчете на общую массу второй перемешанной смеси, целлюлозу в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси, стеарат магния в количестве от 0,5 до 2 мас.% в пересчете на общую массу второй перемешанной смеси и кросповидон в количестве от 2 до 4 мас.% в пересчете на общую массу второй перемешанной смеси.
Конкретным объектом изобретения является способ лечения рака и/или ингибирования ангиогенеза у индивидуума. Способ заключается в том, что индивидууму вводят композицию, предлагаемую в одном из представленных в настоящем описании вариантов осуществления изобретения. В некоторых из вариантов осуществления изобретения композиция представляет собой капсулу. В других вариантах осуществления этого способа композиция представляет собой таблетку.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума композицию вводят в количестве, достаточном для обеспечения концентрации Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 20 до 4000 нг/мл, или концентрации Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 40 до 8000 нг/мл.
В конкретных вариантах осуществления способа лечения рака и/или ингибирования ангиогенеза у индивидуума композицию вводят в количестве, достаточном для обеспечения концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 10 до 2000 нг/мл через 24 ч после введения, или концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 20 до 4000 нг/мл через 24 ч после введения.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума композицию вводят в количестве, достаточном для обеспечения величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 500 до 60000 нг·ч/мл, или величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 750 до 120000 нг·ч/мл.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума композицию вводят один, два, три или четыре раза в день.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума количество соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси, вводимое индивидууму, составляет от 0,25 до 30 мг/кг веса тела индивидуума.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума рак, подлежащий лечению, выбирают из группы, включающей рак предстательной железы, колоректальный рак, рак молочной железы, множественную миелому, рак поджелудочной железы, мелкоклеточный рак, острый миелогенный лейкоз, хронический миелогенный лейкоз, миелопролиферативное заболевание, немелкоклеточный рак легкого, мелкоклеточный рак легкого, хронический лимфолейкоз, саркому, меланому, лимфому, рак щитовидной железы, нейроэндокринный рак, почечно-клеточный рак, рак желудка, желудочно-кишечный стромальный рак, глиому, рак головного мозга или рак мочевого пузыря. В определенных вариантах осуществления изобретения рак является метастазирующим.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума способ дополнительно заключается в том, что композицию вводят в качестве элемента цикла лечения, где цикл лечения предусматривает ежедневное введение композиции в течение 7, 14, 21 или 28 дней, после чего следует 7- или 14-дневный период без введения композиции. В конкретных вариантах осуществления изобретения цикл лечения предусматривает ежедневное введение определенного количества соединения в течение 7 дней, после чего следует 7-дневный период без введения соединения. В некоторых вариантах осуществления изобретения цикл лечения повторяют один или несколько раз.
Другие объекты, отличительные признаки и преимущества изобретения станут очевидными после ознакомления с чертежами и прилагаемым подробным описанием.
Краткое описание чертежей
На фиг.1 показана картина дифракции рентгеновских лучей на порошке (XRPD) для формы А,
на фиг.2 - схема, изображающая различные стадии процесса приготовления капсул, содержащих композицию.
Подробное описание изобретения
Настоящее изобретение относится к композициям хинолиноновых производных. Такие композиции можно применять в качестве антагонистов рецепторных тирозинкиназ и более конкретно для ингибирования функции PDGFRα и PDGFRβ, bFGF и/или VEGF-RTK. Такие композиции можно применять также для ингибирования других тирозинкиназ и различных сериновых/треониновых киназ. Композиции можно применять, например, для лечения пациентов, страдающих раком и/или нуждающихся в ингибировании VEGF-RTK. Композиции можно применять также для лечения индивидуума, нуждающегося в ингибировании ангиогенеза.
В настоящем описании используются следующие сокращения и определения.
Сокращение «AUC» относится к площади под кривой на графике зависимости концентрации соединения в плазме крови от времени.
Сокращение «API» обозначает фармацевтическое действующее вещество.
Сокращение «bFGF» обозначает основной фактор роста фибробластов.
Сокращение «bFGFR», синонимом которого является «FGFR1», относится к тирозинкиназе, которая взаимодействует с фактором роста фибробластов FGF.
Сокращение «Cmax» обозначает максимальную концентрацию соединения в плазме, ткани или крови индивидуума, которому ввели соединение. Как правило, Cmax достигается в течение нескольких часов после введения соединения индивидууму.
Сокращение «DVS» обозначает динамическую сорбцию паров.
Сокращение «ПЭВП» обозначает полиэтилен высокой плотности.
Сокращение «LLOQ» обозначает нижний предел количественной оценки.
Сокращение «PDGF» обозначает тромбоцитарный фактор роста. PDGF взаимодействует с тирозинкиназами PDGFRα и PDGFRβ.
Сокращение «PIB» обозначает композицию типа «порошок в пузырьке».
Сокращение «RH» обозначает относительную влажность.
Сокращение «RTK» обозначает рецепторную тирозинкиназу.
Сокращение «VEGF» обозначает сосудистый эндотелиальный фактор роста.
Сокращение «VEGF-RTK» обозначает рецепторную тирозинкиназу сосудистого эндотелиального фактора роста.
Сокращение «XRPD» обозначает дифракцию рентгеновских лучей на порошке.
Понятие «фармацевтически приемлемая соль» обозначает соль неорганического основания, органического основания, неорганической кислоты, органической кислоты или оснòвной или кислотной аминокислоты. В контексте изобретения к солям неорганических оснований относятся, например, соли щелочных металлов, таких как натрий и калий, щелочно-земельных металлов, таких как кальций и магний или алюминий, и соли аммония. В контексте изобретения к солям органических оснований относятся, например, соли триметиламина, триэтиламина, пиридина, пиколина, этаноламина, диэтаноламина и триэтаноламина. В контексте настоящего изобретения к солям неорганических кислот относятся, например, соли таких кислот, как соляная кислота, бромистоводородная кислота, азотная кислота, серная кислота и фосфорная кислота. В контексте настоящего изобретения к солям органических кислот относятся, например, соли таких кислот, как муравьиная кислота, уксусная кислота, фумаровая кислота, щавелевая кислота, винная кислота, малеиновая кислота, молочная кислота, лимонная кислота, янтарная кислота, яблочная кислота, метансульфоновая кислота, бензолсульфоновая кислота и пара-толуолсульфоновая кислота. В контексте изобретения к солям оснòвных аминокислот относятся, например, соли таких кислот, как аргинин, лизин и орнитин. К кислотным аминокислотам относятся, например, аспарагиновая кислота и глутаминовая кислота.
Понятие «индивидуум» в контексте настоящего описания относится к любому животному, на которое можно оказывать благоприятное воздействие с помощью способов, предлагаемых в изобретении. Так, соединение формулы I, его фармацевтически приемлемые соли, таутомеры или фармацевтически приемлемую соль таутомера можно вводить любому животному, на которое может оказывать благоприятное воздействие соединение, предлагаемое в изобретении, при его применении с помощью способов лечения рака, предлагаемых в изобретении. Предпочтительно животное представляет собой млекопитающее и прежде всего человека, хотя объем изобретения не ограничен только им. Примерами других пригодных животных являются (но не ограничиваясь только ими) крысы, мыши, обезьяны, собаки, кошки, крупный рогатый скот, лошади, свиньи, овцы и т.п.
Понятие «лечение» в контексте настоящего изобретения обозначает ослабление симптомов, ассоциированных с нарушением или заболеванием, или прекращение дальнейшего развития или ухудшения этих симптомов, или предупреждение или профилактику заболевания или нарушения. Например, касательно рака успешное лечение может заключаться в ослаблении симптомов или прекращении развития заболевания, что оценивают по уменьшению скорости роста опухоли, прекращению роста опухоли, уменьшению размера опухоли, частичной или полной ремиссии рака, или по увеличению коэффициента выживаемости или клиническому благоприятному действию.
Одним из объектов настоящего изобретения является фармацевтическая композиция, которая содержит соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь
и по меньшей мере один ингредиент, выбранный из группы, включающей (I) целлюлозу; (II) лактозу, крахмал или их смесь; (III) повидон; (IV) диоксид кремния или тальк; (V) фармацевтически приемлемый замасливатель и (VI) ингредиент, выбранный из кросповидона, кроскармеллозы натрия и натрийгликолята крахмала. В других вариантах осуществления изобретения фармацевтическая композиция включает по меньшей мере два, три или четыре ингредиента, выбранных из (I) целлюлозы; (II) лактозы, крахмала или их смеси; (III) повидона; (IV) диоксида кремния или талька; (V) фармацевтически приемлемого замасливателя и (VI) ингредиента, выбранного из кросповидона, кроскармеллозы натрия и натрийгликолята крахмала.
Другим объектом настоящего изобретения является фармацевтическая композиция, которая содержит соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь и по меньшей мере один ингредиент, выбранный из группы, включающей целлюлозу, повидон, диоксид кремния, тальк, фармацевтически приемлемый замасливатель; и по меньшей мере один ингредиент, выбранный из группы, включающей лактозу, крахмал, кросповидон, кроскармеллозу натрия и натрийгликолят крахмала.
Композиция может содержать фармацевтически приемлемый замасливатель, который уменьшает способность порошков прилипать к металлическим частям машин для заполнения капсул или таблетировочных машин. Такие замасливатели хорошо известны в данной области и к ним относятся жирная С16-С22-кислота, соль жирной С16-С22-кислоты, эфир жирной С16-С22-кислоты; полиэтиленгликоль со средней молекулярной массой от 6000 до 10000 и смесь любых двух или большего количества из указанных веществ. В конкретных вариантах осуществления изобретения фармацевтически приемлемый замасливатель представляет собой стеариновую кислоту, ее соли, ее эфиры, соли эфиров или их смеси. Например, композиция может содержать стеарат магния, стеарат натрия, стеарат кальция, стеарат цинка, моностеарат глицерила, пальмитостеарил глицерила, бегенат глицерила или стеарилфумарат натрия. Как должно быть очевидно специалистам в данной области, стеариновая кислота, ее соли, эфиры и соли эфиров, включая смеси жирных C16- и C18-кислот, подпадают под объем изобретения.
Композиция может содержать, практически состоять или состоять из соединения формулы I, таутомера соединения, фармацевтически приемлемой соли соединения, фармацевтически приемлемой соли таутомера или их смеси и (I) целлюлозы; (II) диоксида кремния; (III) стеариновой кислоты, соли стеариновой кислоты или их смеси и (IV) по меньшей мере одного ингредиента, выбранного из кросповидона, крахмала, лактозы, кроскармеллозы натрия и натрийгликолята крахмала. В конкретных вариантах осуществления изобретения композиции содержат (I) микрокристаллическую целлюлозу; (II) диоксид кремния; (III) стеарат магния; (IV) по меньшей мере один ингредиент, выбранный из кросповидона, частично предварительно желированного крахмала и лактозы.
Композиция может содержать лактат соединения формулы I. В конкретных вариантах осуществления изобретения лактат может находиться в безводной кристаллической форме, такой как форма А, которая более подробно описана и охарактеризована в разделе «Примеры» настоящего документа.
Композиция может содержаться в капсуле или таблетке. В некоторых из таких вариантов осуществления изобретения общая масса соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в капсуле или таблетке составляет от 25 до 500 мг. В качестве капсул можно использовать, например, белые непрозрачные желатиновые капсулы размера №0, такие как CS-капсулы, поступающие от фирмы Capsugel, или ГПМЦ-капсулы, поступающие от фирм Quali-V и Shinogi.
В конкретных вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 10 до 50 мас.% в пересчете на общую массу композиции. В некоторых из таких вариантов осуществления изобретения композиция содержит лактат соединения в количестве от 20 до 45 мас.% в пересчете на общую массу композиции. В других вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 30 до 40 мас.% в пересчете на общую массу композиции.
В конкретных вариантах осуществления изобретения целлюлоза, применяемая в композиции, представляет собой микрокристаллическую целлюлозу. В других вариантах осуществления изобретения применяемая целлюлоза представляет собой силикатированную микрокристаллическую целлюлозу, натрийкарбоксиметилцеллюлозу или гидроксипропилцеллюлозу.
В конкретных вариантах осуществления изобретения композиция содержит целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу композиции. В некоторых из таких вариантов осуществления изобретения композиция содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу композиции и композиция содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу композиции. В определенных вариантах осуществления изобретения композиция содержит целлюлозу в количестве от 20 до 45 мас.% в пересчете на общую массу композиции и композиция содержит крахмал или лактозу в количестве от 10 до 40 мас.% в пересчете на общую массу композиции.
В конкретных вариантах осуществления изобретения композиция содержит крахмал в количестве от 10 до 40 мас.% в пересчете на общую массу композиции, и крахмал представляет собой частично предварительно желированный крахмал.
Композиции могут содержать диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции. В конкретных вариантах осуществления изобретения диоксид кремния присутствует в количествах от 0,2 до 5 мас.%, от 0,4 до 4 мас.%, от 0,5 до 2 мас.%, от 0,75 до 1,5 мас.% или от 0,8 до 1,2 мас.% в пересчете на общую массу композиции. В определенных вариантах осуществления изобретения диоксид кремния присутствует в количестве, составляющем примерно 1 мас.% в пересчете на общую массу композиции. В других вариантах осуществления изобретения диоксид кремния можно заменять на коллоидный диоксид кремния, силикат магния, трисиликат магния или тальк в таком же или близком процентном содержании (мас.%).
Композиции могут содержать стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции. В конкретных вариантах осуществления изобретения стеарат присутствует в количествах от 0,2 до 5 мас.%, от 0,4 до 4 мас.%, от 0,5 до 2 мас.%, от 0,75 до 1,5 мас.% или от 0,8 до 1,2 мас.% в пересчете на общую массу композиции. В определенных вариантах осуществления изобретения стеарат присутствует в количестве, составляющем примерно 1 мас.% в пересчете на общую массу композиции. В других вариантах осуществления изобретения стеарат магния можно заменять на стеариновую кислоту, ее соли, их смеси и/или другие фармацевтически приемлемые замасливатели в таком же или близком процентном содержании (мас.%).
В некоторых вариантах осуществления изобретения композиция содержит, практически состоит или состоит из лактата соединения в количестве от 30 до 40 мас.% в пересчете на общую массу композиции, диоксида кремния в количестве от 0,3 до 2% мас.% в пересчете на общую массу композиции, целлюлозы в количестве от 25 до 40 мас.% в пересчете на общую массу композиции, стеарата магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции и кросповидона в количестве от 2 до 4 мас.% в пересчете на общую массу композиции.
В других композициях, таких как композиции с высокой дозой (например, 200-500 мг API или более), композиция содержит лактат соединения формулы I в количестве от 50 до 80 мас.% в пересчете на общую массу композиции, от 55 до 75 мас.% в пересчете на общую массу композиции или от 60 до 70 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения композиция содержит лактат соединения формулы I в количестве от 50 до 80 мас.% в пересчете на общую массу композиции, диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции, целлюлозу в количестве от 0 до 50 мас.% в пересчете на общую массу композиции, стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции и крахмал в количестве от 10 до 40 мас.% в пересчете на общую массу композиции. В некоторых из таких вариантов осуществления изобретения композиция содержит лактат соединения в количестве от 55 до 75 мас.% в пересчете на общую массу композиции, целлюлозу в количестве от 5 до 40 мас.% в пересчете на общую массу композиции и крахмал в количестве от 15 до 30 мас.% в пересчете на общую массу композиции. В других вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 60 до 70 мас.% в пересчете на общую массу композиции и целлюлозу в количестве от 5 до 25 мас.% в пересчете на общую массу композиции.
В определенных вариантах осуществления изобретения композиция содержит, практически состоит или состоит из лактата соединения в количестве от 50 до 80 мас.% в пересчете на общую массу композиции, диоксида кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции, целлюлозы в количестве от 0 до 50 мас.% в пересчете на общую массу композиции, стеарата магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции и лактозы в количестве от 10 до 40 мас.% в пересчете на общую массу композиции. В некоторых из таких вариантов осуществления изобретения композиция содержит лактат соединения в количестве от 55 до 75 мас.% в пересчете на общую массу композиции и целлюлозу в количестве от 5 до 40 мас.% в пересчете на общую массу композиции. В других вариантах осуществления изобретения композиция содержит лактат соединения в количестве от 60 до 70 мас.% в пересчете на общую массу композиции и целлюлозу в количестве от 5 до 40 мас.% в пересчете на общую массу композиции.
В конкретных вариантах осуществления изобретения композиция дополнительно содержит антиоксидант, хелатирующий агент, аскорбиновую кислоту, редуцирующий сахар или смесь любых двух или большего количества из указанных веществ. К пригодным антиоксидантам для оральных и других композиций относятся, например, аскорбиновая кислота, например, в количестве от 0,01 до 0,1 мас.%, бисульфит натрия, например, в количестве вплоть до 0,65 мг/стандартную дозу, гидрохлорид цистеина, например, в количестве вплоть до 16 мг/стандартную дозу, метионин и метабисульфит натрия, например, в количестве от 0,01 до 0,1 мас.%. Другими пригодными антиоксидантами для оральных и других композиций являются редуцирующие сахара, содержащие кетоновые или альдегидные группы, такие как фруктоза, глюкоза, арабиноза и мальтоза, применяемые, например, в количестве от 1 до 55 мас.%. К пригодным хелатирующим агентам относятся, например, этилендиаминтетрауксусная кислота (ЭДТК) и ее соли, такие как кальцийдвунатриевая соль этилендиаминтетрауксусной кислоты (кальцийдинатрийэдетат) и тетранатриевая соль этилендиаминтетрауксусной кислоты (тетранатрийэдетат), применяемые, например, в количестве от 0,005 до 0,1 мас.%, и цитрат натрия, применяемый, например, в количестве от 0,3 до 2 мас.%.
Представленные в настоящем описании фармацевтические композиции являются стабильными. Например, продукты расщепления соединения формулы I в композициях, предлагаемых в изобретении, как правило, присутствуют в количестве менее 10 мас.% в пересчете на общую массу композиции после хранения композиции в течение трех месяцев при 40°С и 75%-ной комнатной влажности. В конкретных вариантах осуществления изобретения количество продуктов расщепления составляет менее 8, менее 5, менее 4, менее 3, менее 2 или даже менее 1 мас.% в пересчете на общую массу композиции после хранения композиции в течение трех месяцев при 40°С и 75%-ной комнатной влажности.
Изобретение относится также к контейнерам для упаковки фармацевтических продуктов. В одном из вариантов осуществления изобретения контейнер для упаковки представляет собой сосуд для хранения, содержащий две или более капсул или таблеток, где капсулы или таблетки содержат фармацевтическую композицию, предлагаемую в одном из вариантов осуществления изобретения. В некоторых из таких вариантов осуществления изобретения фармацевтическая композиция, предлагаемая в одном из вариантов осуществления изобретения, содержится в нескольких капсулах или таблетках. В некоторых из таких вариантов осуществления изобретения сосуд для хранения сделан из полиэтилена высокой плотности (ПЭВП). В некоторых из таких вариантов осуществления изобретения сосуд для хранения содержит прокладку из хлопка или искусственного волокна, а в определенных вариантах осуществления изобретения включает термокрышку. В других вариантах осуществления изобретения сосуд для хранения сделан из полиэтилена высокой плотности без прокладки из хлопка, но с термокрышкой. Другие варианты осуществления изобретения относятся к контейнеру для упаковки фармацевтических продуктов, который представляет собой блистерную упаковку, такую как Al-Al-блистерную упаковку или упаковку из поливинилхлорида (ПВХ), или упаковку из поливинилиденхлорида (ПВДХ), или упаковку Aclar®. Блистерная упаковка содержит по меньшей мере одну капсулу или таблетку, которые включают фармацевтическую композицию, предлагаемую в одном из вариантов осуществления изобретения.
В других вариантах осуществления изобретения на таблетку или капсулу, предлагаемую в настоящем изобретении, можно наносить покрытие из материала для нанесения покрытия, такого как сахар, полимер на основе целлюлозы, полиметакрилатный полимер. Примерами агентов для нанесения покрытия из полимеров на основе целлюлозы являются (но не ограничиваясь только ими) метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксиэтилметилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза и этилцеллюлоза. К пригодным агентам для нанесения покрытия, представляющим собой полиметакрилатные полимеры, относятся (но не ограничиваясь только ими) сополимеры метакриловой кислоты, такие как поли(метакриловая кислота-метилметакрилат) и поли(метакриловая кислота-этилакрилат); аммонийметакрилатные сополимеры, такие как поли(этилакрилат-метилметакрилат-триметиламмонийэтилметакрилатхлорид), и поли(этилакрилат-метилметакрилат). К другим материалам, которые можно применять для нанесения покрытия, относятся продукты, поступающие на рынок под товарными знаками Opadry®, Surelease®, Aquacoat® и Eudragit®. В других вариантах осуществления изобретения на таблетку можно наносить желатиновое покрытие или капсулировать таблетку в желатиновую оболочку.
Другие объекты изобретения относятся к материалу для нанесения покрытия, содержащему фармацевтически приемлемый краситель. Еще в одном варианте осуществления изобретения материал для нанесения покрытия может содержать фармацевтически приемлемый замутнитель. В качестве пригодных замутнителей можно применять диоксид титана или тальк.
Один из объектов изобретения относится к способу получения фармацевтической композиции. Способ заключается в том, что (а) смешивают первую смесь с получением первой перемешанной смеси, где первая смесь содержит: (I) соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь и (II) по меньшей мере один ингредиент, выбранный из группы, включающей целлюлозу; лактозу, крахмал или их смесь; повидон; диоксид кремния или тальк; фармацевтически приемлемый замасливатель и ингредиент, выбранный из кросповидона, кроскармеллозы натрия и натрийгликолята крахмала. В некоторых из этих вариантов осуществления изобретения соединение формулы I смешивают с (I) целлюлозой; (II) диоксидом кремния и (III) ингредиентом, выбранным из кросповидона, крахмала и лактозы. Кроме того, способ может заключаться в том, что (б) смешивают стеариновую кислоту, соль стеариновой кислоты или их смесь с первой перемешанной смесью с получением второй перемешанной смеси и/или (в) формируют по меньшей мере одну капсулу или по меньшей мере одну таблетку из второй перемешанной смеси.
Следующий объект изобретения относится к способу получения фармацевтической композиции. Способ заключается в том, что (а) смешивают смесь ингредиентов с получением первой перемешанной смеси. Первая перемешанная смесь содержит: I) соединение формулы I, таутомер соединения, фармацевтически приемлемую соль соединения, фармацевтически приемлемую соль таутомера или их смесь, (II) по меньшей мере один ингредиент, выбранный из группы, включающей целлюлозу, крахмал, лактозу и повидон, (III) по меньшей мере один ингредиент, выбранный из группы, включающей кросповидон; кроскармеллозу натрия и натрийгликолят крахмала; жидкость для грануляции, выбранную из группы, включающей водную кислоту, спирт, водный спирт или смесь любых двух или большего количества из указанных веществ. Например, жидкость для грануляции может представлять собой воду или водный раствор соляной кислоты. Способ заключается также в том, что (б) удаляют жидкость для грануляции, например, путем сушки. Способ заключается также в том, что (в) получают вторую перемешанную смесь путем смешения первой перемешанной смеси с по меньшей мере одним дополнительным ингредиентом, выбранным из группы, включающей (I) кросповидон, кроскармеллозу натрия или натрийгликолят крахмала; (II) стеариновую кислоту или соль стеариновой кислоты и (III) диоксид кремния или тальк. Стадии (а), (б) и (в) можно осуществлять последовательно или одновременно, или стадию (в) можно осуществлять перед осуществлением стадии (б). Способ может заключаться также в том, что (г) формируют по меньшей мере одну капсулу или по меньшей мере одну таблетку из второй перемешанной смеси.
В методах получения фармацевтических композиций, представленных в настоящем описании, можно применять различное оборудование, хорошо известное специалистам в данной области.
Пригодное оборудование включает гранулятор с псевдоожиженным слоем, снабженный механизмом нижнего распыления, верхнего распыления или тангенциального распыления; гранулятор с высокими сдвиговыми усилиями; гранулятор с низкими сдвиговыми усилиями; роллерный уплотнитель; калибровочную машину; машину для заполнения капсул и/или таблеточный пресс. Так, например, можно применять грануляторы с псевдоожиженным слоем, выпускаемые фирмой Niro Pharma Systems, такие как Sirocco®, Multiprocessor®, MP-Micro®, STREA-1®, MP-1 Multi-processor®, а также гранулятор с псевдоожиженным слоем/сушилку/машину для нанесения покрытия, выпускаемую фирмой Glatt; грануляты с высокими сдвиговыми усилиями, выпускаемые фирмой Niro Pharma Systems, такие как Collette Gral®, UltimaGral®, PMA Pharma Matrix®, фирмой Bohle, такой как минигранулятор Bohle, и фирмой Glatt Air Techniques, такой как вертикальный гранулятор Glatt-Powrex; грануляторы с низкими сдвиговыми усилиями, такие как V-Blender и Hobart миксер/гранулятор; роллерные уплотнители, выпускаемые фирмой Fitzpatrick Chilsonators, микро-, мини- и макроуплотнители фирмы Gerteis, и роллерный уплотнитель Vector TFC; калибровочное оборудование типа Quadro, выпускаемое фирмой Comil, молотковую мельницу, выпускаемую фирмой Fitzpatrick Chilsonators, вибраторы, поступающие в продажу от нескольких фирм; машины для заполнения капсул, выпускаемые фирмами MG2 (MG), Bosch (GKF) и IMA (Zanasi); и/или таблеточный пресс, такой как пресс, выпускаемый фирмами Manesty, Fette и Courtoy.
В конкретных вариантах способа общая масса соединения формулы I, таутомера соединения, фармацевтически приемлемой соли соединения, фармацевтически приемлемой соли таутомера или их смеси в капсуле или таблетке составляет от 25 до 500 мг.
В определенных вариантах осуществления изобретения вторая перемешанная смесь содержит лактат соединения формулы I. В других вариантах осуществления изобретения вторая перемешанная смесь содержит лактат соединения в количестве от 10 до 50 мас.% в пересчете на общую массу второй перемешанной смеси, в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси или в количестве от 30 до 40 мас.% в пересчете на общую массу второй перемешанной смеси.
В конкретных вариантах способа получения фармацевтической композиции целлюлоза представляет собой микрокристаллическую целлюлозу. В определенных вариантах осуществления изобретения крахмал представляет собой предварительно желированный крахмал.
В конкретных вариантах способа вторая перемешанная смесь содержит целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу второй перемешанной смеси. В некоторых вариантах осуществления изобретения вторая перемешанная смесь содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и вторая перемешанная смесь содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу второй перемешанной смеси. В некоторых вариантах осуществления изобретения вторая перемешанная смесь содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и вторая перемешанная смесь содержит крахмал или лактозу в количестве от 10 до 40 мас.% в пересчете на общую массу второй перемешанной смеси.
В конкретных вариантах способа вторая перемешанная смесь содержит крахмал в количестве от 20 до 40 мас.% в пересчете на общую массу второй перемешанной смеси, и крахмал представляет собой частично предварительно желированный крахмал.
В конкретных вариантах способа вторая перемешанная смесь содержит диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу второй перемешанной смеси. В других вариантах осуществления изобретения диоксид кремния присутствует в количествах от 0,2 до 5 мас.%, от 0,4 до 4 мас.%, от 0,5 до 2 мас.%, от 0,75 до 1,25 мас.% или от 0,8 до 1,2 мас.% в пересчете на общую массу второй перемешанной смеси. В некоторых вариантах осуществления изобретения диоксид кремния присутствует в количестве, составляющем примерно 1 мас.% в пересчете на общую массу второй перемешанной смеси.
В конкретных вариантах способа вторая перемешанная смесь содержит соль стеариновой кислоты, такую как стеарат магния. Например, в некоторых вариантах способа стеарат магния присутствует в количестве от 0,1 до 2 мас.% в пересчете на общую массу второй перемешанной смеси. В других вариантах способа стеарат присутствует в количествах от 0,2 до 5 мас.%, от 0,4 до 4 мас.%, от 0,5 до 1,5 мас.%, от 0,75 до 1,25 мас.% или от 0,8 до 1,2 мас.% в пересчете на общую массу второй перемешанной смеси. В некоторых вариантах осуществления способа стеарат присутствует в количестве, составляющем примерно 1 мас.% или 1 мас.% в пересчете на общую массу второй перемешанной смеси.
В конкретных вариантах способа вторая перемешанная смесь содержит лактат соединения в количестве от 50 до 80 мас.% в пересчете на общую массу второй перемешанной смеси, в количестве от 55 до 75 мас.% в пересчете на общую массу второй перемешанной смеси или в количестве от 60 до 70 мас.% в пересчете на общую массу второй перемешанной смеси.
В некоторых из таких вариантов способа диоксид кремния присутствует в количестве от 0,3 до 2 мас.% в пересчете на общую массу второй перемешанной смеси. В других вариантах способа целлюлоза присутствует в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси. В следующих вариантах способа стеарат магния присутствует в количестве от 0,1 до 2 мас.% в пересчете на общую массу второй перемешанной смеси. В других вариантах способа вторая перемешанная смесь дополнительно содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу второй перемешанной смеси. В следующих вариантах способа вторая перемешанная смесь содержит диоксид кремния в количестве от 0,5 до 2 мас.% в пересчете на общую массу второй перемешанной смеси, целлюлозу в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси, стеарат магния в количестве от 0,5 до 2 мас.% в пересчете на общую массу второй перемешанной смеси и кросповидон в количестве от 2 до 4 мас.% в пересчете на общую массу второй перемешанной смеси.
Изобретение относится также к способу лечения рака и/или ингибирования ангиогенеза у индивидуума. Способ заключается в том, что индивидууму вводят композицию, предлагаемую в одном из вариантов осуществления изобретения. В конкретных вариантах осуществления изобретения композиция представляет собой капсулу или таблетку. К индивидуумам, для которых можно применять способ, относятся млекопитающие, такие как крысы, мыши, обезьяны и другие приматы, собаки, кошки, крупный рогатый скот, лошади, свиньи, овцы и т.п. В конкретных вариантах осуществления изобретения индивидуум представляет собой человека, а в некоторых из этих вариантов осуществления изобретения он представляет собой страдающего раком пациента. В определенных вариантах осуществления изобретения композицию вводят орально в виде капсулы или таблетки пациенту, такому как страдающий раком пациент.
Композицию можно вводить в количестве, достаточном для обеспечения концентрации Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 20 до 4000 нг/мл, или концентрации Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 40 до 8000 нг/мл. В конкретных вариантах осуществления изобретения вводимое количество является достаточным для обеспечения величины Cmax в плазме индивидуума, составляющей примерно от 35 до 2000 нг/мл, или величины Cmax в крови индивидуума, составляющей примерно от 70 до 4000 нг/мл, величины Cmax в плазме индивидуума, составляющей примерно от 50 до 500 нг/мл, или величины Cmax в крови индивидуума, составляющей примерно от 100 до 1000 нг/мл, величины Cmax в плазме индивидуума, составляющей примерно от 50 до 250 нг/мл, или величины Cmax в крови индивидуума, составляющей примерно от 100 до 500 нг/мл, величины Cmax в плазме индивидуума, составляющей примерно от 75 до 150 нг/мл, или величины Cmax в крови индивидуума, составляющей примерно от 150 до 300 нг/мл, величины Cmax в плазме индивидуума, составляющей примерно от 100 до 2000 нг/мл, или величины Cmax в крови индивидуума, составляющей примерно от 200 до 4000 нг/мл, или величины Cmax в плазме индивидуума, составляющей примерно от 100 до 1000 нг/мл, или величины Cmax в крови индивидуума, составляющей примерно от 200 до 2000 нг/мл.
Композицию можно вводить также в количестве, достаточном для обеспечения концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 10 до 2000 нг/мл через 24 ч после введения, или концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно 20 до 4000 нг/мл через 24 ч после введения. В конкретных вариантах осуществления изобретения вводимое количество достаточно для обеспечения концентрации в плазме пациента, составляющей примерно от 20 до 1000 нг/мл через 24 ч после введения, или концентрации в крови пациента, составляющей примерно от 40 до 2000 нг/мл через 24 ч после введения, концентрации в плазме пациента, составляющей примерно от 40 до 500 нг/мл через 24 ч после введения, или концентрации в крови пациента, составляющей примерно от 80 до 1000 нг/мл через 24 ч после введения, или концентрации в плазме пациента, составляющей примерно от 40 до 250 нг/мл через 24 ч после введения, или концентрации в крови пациента, составляющей примерно от 80 до 500 нг/мл через 24 ч после введения.
Композицию можно вводить также в количестве, достаточном для обеспечения величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 500 до 60000 нг·ч/мл, или величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 750 до 120000 нг·ч/мл. В других вариантах осуществления изобретения вводимое количество является достаточным для обеспечения величины AUC, составляющей примерно от 1000 до 30000 нг·ч/мл в плазме индивидуума или примерно от 1500 до 60000 нг·ч/мл в крови индивидуума. В других вариантах осуществления изобретения величина AUC составляет примерно от 2000 до 15000 нг·ч/мл в плазме индивидуума или примерно от 3000 до 30000 нг·ч/мл в крови индивидуума.
Композиции, предлагаемые в изобретении, могут представлять собой капсулу или таблетку, достаточную для обеспечения по меньшей мере одного из следующих показателей:
(а) величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 20 до 4000 нг/мл, или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 40 до 8000 нг/мл, после введения индивидууму,
(б) концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 10 до 2000 нг/мл через 24 ч после введения, или концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 20 до 4000 нг/мл через 24 ч после введения индивидууму, или
(в) величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 500 до 60000 нг·ч/мл, или величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 750 до 120000 нг·ч/мл, после введения индивидууму.
Композиции, предлагаемые в изобретении, могут представлять собой также капсулу или таблетку, достаточную для обеспечения по меньшей мере одного из следующих показателей:
(а) величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 50 до 500 нг/мл, или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 100 до 1000 нг/мл, после введения индивидууму,
(б) концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 20 до 1000 нг/мл через 24 ч после введения, или концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 40 до 2000 нг/мл через 24 ч после введения индивидууму, или
(в) величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 1000 до 30000 нг·ч/мл, или величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 1500 до 60000 нг·ч/мл, после введения индивидууму.
Кроме того, композиции, предлагаемые в изобретении, могут представлять собой капсулу или таблетку, достаточную для обеспечения по меньшей мере одного из следующих показателей:
(а) величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 50 до 250 нг/мл, или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 100 до 500 нг/мл, после введения индивидууму,
(б) концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 40 до 500 нг/мл через 24 ч после введения, или концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 80 до 1000 нг/мл через 24 ч после введения, или
(в) величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 2000 до 15000 нг·ч/мл, или величины AUC соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 3000 до 30000 нг·ч/мл, после введения.
Кроме того, композиции, предлагаемые в изобретении, могут представлять собой также капсулу или таблетку, достаточную для обеспечения по меньшей мере одного из следующих показателей:
(а) величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 75 до 150 нг/мл, или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 150 до 300 нг/мл, после введения, или
(б) концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 40 до 250 нг/мл через 24 ч после введения, или концентрации соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 80 до 500 нг/мл через 24 ч после введения.
В конкретных вариантах осуществления изобретения каждая стандартная доза композиции достаточна для обеспечения величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 100 до 2000 нг/мл, или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 200 до 4000 нг/мл; или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в плазме индивидуума, составляющей примерно от 100 до 1000 нг/мл, или величины Cmax соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в крови индивидуума, составляющей примерно от 200 до 2000 нг/мл, после введения.
В определенных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума композицию вводят один раз, два раза, три раза или четыре раза в день.
Количество соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси, вводимое индивидууму, может составлять от примерно 0,25 до 30 мг/кг веса тела индивидуума. В других вариантах осуществления изобретения количество, вводимое индивидууму, может составлять от примерно 25 до 1500 мг/индивидуума в день, от примерно 100 до 1000 мг/индивидуума в день или от примерно 200 до 500 мг/индивидуума в день.
В конкретных вариантах способа лечения рака и/или ингибирования ангиогенеза у индивидуума подлежащий лечению рак выбирают из группы, включающей рак предстательной железы, колоректальный рак, рак молочной железы, множественную миелому, рак поджелудочной железы, мелкоклеточный рак, острый миелогенный лейкоз, хронический миелогенный лейкоз, миелопролиферативное заболевание, немелкоклеточный рак легкого, мелкоклеточный рак легкого, хронический лимфолейкоз, саркому, меланому, лимфому, рак щитовидной железы, нейроэндокринный рак, почечно-клеточный рак, рак желудка, желудочно-кишечный стромальный рак, глиому, рак головного мозга, рефракторную множественную миелому или рак мочевого пузыря. В определенных вариантах осуществления изобретения рак является метастазирующим.
В конкретных вариантах осуществления изобретения способ лечения рака и/или ингибирования ангиогенеза у индивидуума дополнительно заключается в том, что композицию вводят в качестве элемента цикла лечения, где цикл лечения предусматривает ежедневное введение композиции в течение 7, 14, 21 или 28 дней, после чего следует 7- или 14-дневный период без введения композиции. В конкретных вариантах осуществления этого способа цикл лечения предусматривает ежедневное введение определенного количества соединения в течение 7 дней, после чего следует 7-дневный период без введения соединения. В некоторых вариантах осуществления этого способа цикл лечения повторяют один или несколько раз.
В композиции, предлагаемые в настоящем изобретении, можно включать другие ингредиенты в дополнение к указанным в настоящем описании. Такие дополнительные или альтернативные ингредиенты описаны, например, в справочнике «Remington's Pharmaceutical Sciences», изд-во Mack Pub. Co., New Jersey, 1991, который включен в настоящее описание в качестве ссылки. К таким дополнительным или альтернативным ингредиентам относятся (но не ограничиваясь только ими) метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксиэтилметилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, этилцеллюлоза, лаурилсульфат натрия, cab-o-sil, Avicel РН, поли(этилакрилат-метилметакрилат), сополимеры метакриловой кислоты, такие как (но не ограничиваясь только ими) поли(метакриловая кислота-метилметакрилат) и поли(метакриловая кислота-этилметакрилат), и сополимеры аминометилметакрилата, такие как (но не ограничиваясь только ими) поли(этилакрилат-метилметакрилат-хлорид триметиламмонийэтилметакрилата).
Композиции, предлагаемые в изобретении, могут быть предназначены для кратковременного действия, быстрого высвобождения, продолжительного действия и пролонгированного высвобождения. Так, фармацевтические композиции можно приготавливать в форме, предназначенной для контролируемого высвобождения или медленного высвобождения.
Терапевтически эффективная доза обозначает количество соединения, которое приводит к ослаблению симптомов. Можно регулировать конкретные дозы в зависимости от состояния заболевания, возраста, веса тела, общего состояния здоровья, пола, диеты индивидуума, интервала между дозами, путей введения, скорости экскреции и комбинации лекарственных средств. Эффективные количества любой из указанных выше форм лекарственного средства можно определять с помощью стандартных экспериментов и, следовательно, они подпадают под объем настоящего изобретения. Терапевтически эффективная доза может варьироваться в зависимости от пути введения и формы лекарственного средства. Предпочтительное соединение или соединения, предлагаемое(ые) в настоящем изобретении, представляют собой композицию, которая имеет высокий терапевтический индекс. Терапевтический индекс представляет собой отношение доз, вызывающих токсическое и терапевтическое действия, которое можно выражать в виде отношения величин LD50 и ED50. LD50 представляет собой дозу, летальную для 50% популяции, а ED50 представляет собой дозу, обладающую терапевтической эффективностью для 50% популяции. Величины LD50 и ED50 определяют с помощью стандартных фармацевтических процедур на культурах клеток животных или на подопытных животных.
К ассоциированным с RTK нарушениям или опосредованным RTK заболеваниям, которые можно лечить с помощью способов, предлагаемых в изобретении, относится любое биологическое нарушение или заболевание, в котором участвует RTK, или при котором ингибирование RTK усиливает биохимические пути, нарушенные при этом нарушенном или болезненном состоянии. Примерами таких заболеваний являются различные виды рака, такие как рак предстательной железы, колоректальный рак, рак молочной железы, множественная миелома, рак поджелудочной железы, мелкоклеточный рак, острый миелогенный лейкоз, хронический миелогенный лейкоз или миелопролиферативное заболевание.
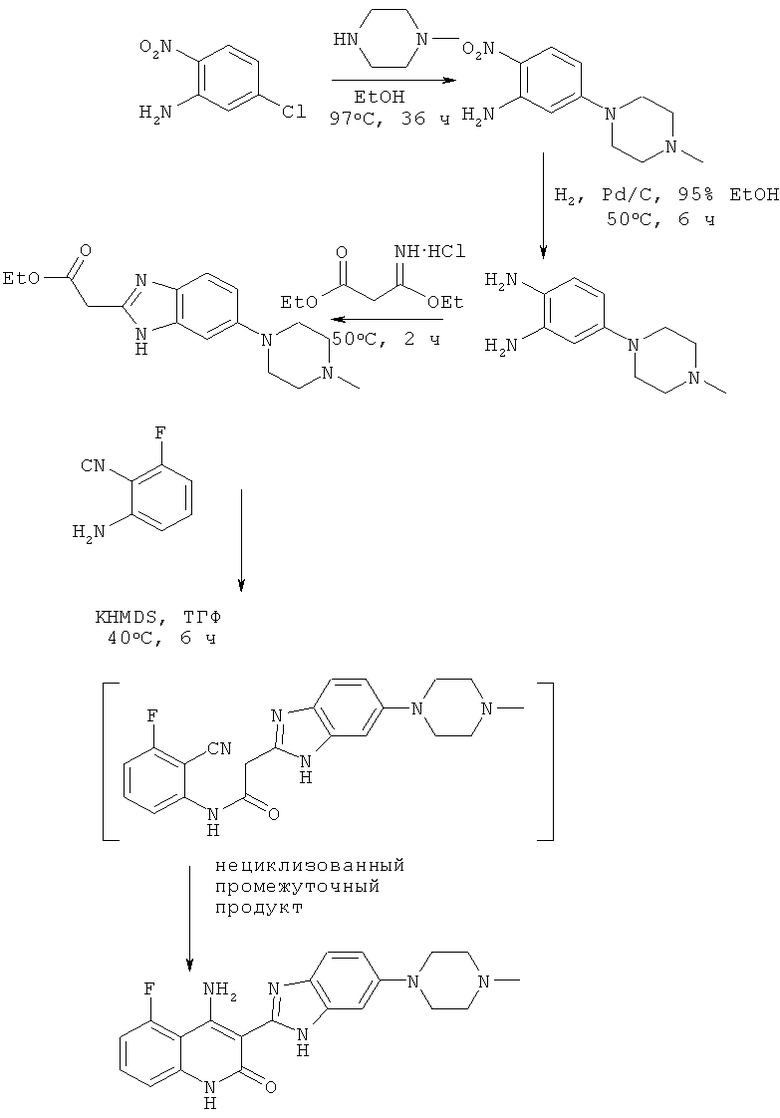
На схеме 1 представлен один из путей синтеза соединения, применяемого в композициях, предлагаемых в настоящем изобретении, и его никоим образом не следует рассматривать в качестве ограничивающего объем изобретения.
Касательно любой композиции, способа или упаковки, предлагаемых в настоящем изобретении, подразумевается, что, когда рассматривается применение капсул, то можно применять также таблетки, и когда рассматривается применение таблеток, то можно применять также капсулы.
Схема 1
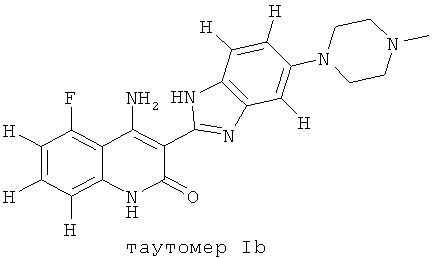
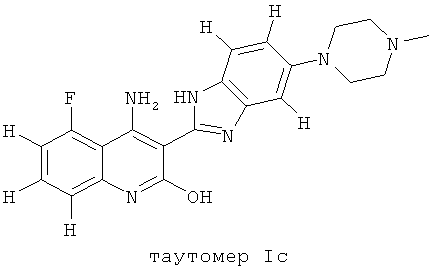
Следует иметь ввиду, что органические соединения, предлагаемые в изобретении, могут обладать таутомерией. В настоящем описании в качестве химической структуры можно представить каждый раз только одну из возможных таутомерных форм, при этом следует иметь ввиду, что под объем изобретения подпадают все таутомерные формы представленной структуры. Например, соединение, имеющее формулу I, представлено ниже одним таутомером, а именно таутомером Ia:
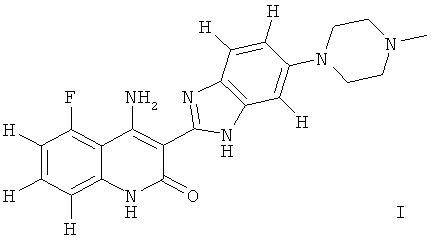
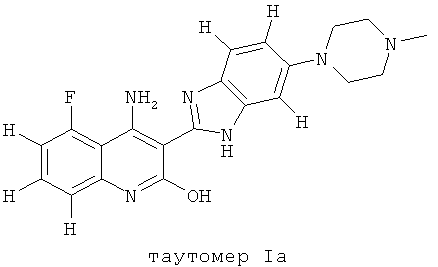
Ниже представлены другие таутомеры соединения, имеющего формулу I, a именно таутомер Ib и таутомер Ic
Настоящее изобретение, в целом представленное в описании, должно стать более понятным после ознакомления с приведенными примерами, которые даны для целей иллюстрации и не направлены на ограничение объема настоящего изобретения.
Примеры
В примерах используются следующие сокращения:
EtOH: этанол
H2O: вода
HCl: соляная кислота
ЖХВР: жидкостная хроматография высокого разрешения
KHMDS: бис(триметилсилил)амид калия
LiHMDS: бис(триметилсилил)амид лития
NaHMDS: бис(триметилсилил)амид натрия
NaOH: гидроксид натрия
N2: азот
ТБМЭ: метил-трет-бутиловый эфир
ТГФ: тетрагидрофуран
Номенклатура указанных в примерах соединений дана с использованием программы ACD Name, версия 5.07 (14 ноября 2001 г.), которую можно получить от фирмы Advanced Chemistry Development, Inc., программы с запатентованным названием Chemlnnovation NamExpert + Nomenclator™, которую можно получить от фирмы Chemlnnovation Software, Inc., и программы AutoNom, версия 2.2, входящей в пакет программ ChemOffice® Ultra, версия 7.0, который можно получить от фирмы CambridgeSoft Corporation (Кэмбридж, шт.Массачусетс). Некоторые из соединений и исходных продуктов обозначены с использованием стандартной номенклатуры ИЮПАК.
Различные исходные продукты можно приобретать на рынке от различных поставщиков или получать с помощью методов, известных специалисту в данной области.
Пример 1
Синтез 5-(4-метилпиперазин-1-ил)-2-нитроанилина
Процедура А
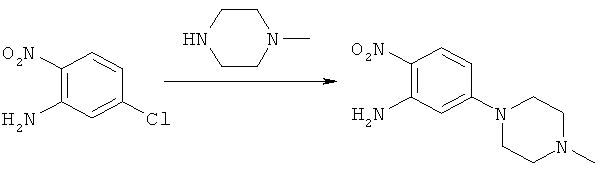
5-Хлор-2-нитроанилин (500 г, 2,898 моля) и 1-метилпиперазин (871 г, 8,693 моля) вносили в колбу вместимостью 2000 мл, снабженную конденсатором, и продували N2. Колбу помещали в масляную баню при 100°С и нагревали до тех пор, пока 5-хлор-2-нитроанилин по данным ЖХВР не прореагирует полностью (как правило, в течение ночи).
После подтверждения с помощью ЖХВР исчезновения 5-хлор-2-нитроанилина реакционную смесь сливали (еще теплую) при механическом перемешивании непосредственно в 2500 мл воды, находящейся при комнатной температуре. Образовавшуюся смесь перемешивали до тех пор, пока ее температура не достигала комнатной, и затем фильтровали. Полученный таким путем твердый продукт желтого цвета вносили в 1000 мл воды и перемешивали в течение 30 мин. Полученную смесь фильтровали и образовавшийся твердый продукт промывали с помощью ТБМЭ (500 мл, 2Х), затем сушили в вакууме в течение 1 ч с использованием резиновой перегородки. Образовавшийся твердый продукт переносили на сушильный поддон и сушили в вакуумной печи при 50°С до постоянной массы, получая 670 г (выход 97,8%) указанного в заголовке соединения в виде порошка желтого цвета.
Процедура Б
5-хлор-2-нитроанилин (308,2 г, 1,79 моля) вносили в 4-горлую круглодонную колбу вместимостью 5000 мл, снабженную верхней мешалкой, конденсатором, впускным газовым клапаном, капельной воронкой и датчиком температуры. Затем колбу продували N2. В реакционную колбу добавляли при перемешивании 1-метилпиперазин (758,1 г, 840 мл, 7,57 моля) и 200 Proof этанола (508 мл). Колбу снова продували N2 и реакционную смесь выдерживали в атмосфере N2. Колбу нагревали с помощью нагревательного кожуха до внутренней температуры 97°С (±5°С) и поддерживали при этой температуре до завершения реакции (как правило, в течение 40 ч) по данным ЖХВР. После завершения реакции нагревание прекращали и реакционную смесь охлаждали при перемешивании до внутренней температуры, составляющей примерно от 20 до 25°С, и реакционную смесь перемешивали в течение 2-3 ч. Если при этом не происходило осаждение, то в реакционную смесь вносили затравочные кристаллы (0,20 г, 0,85 ммоля) 5-(4-метилпиперазин-1-ил)-2-нитроанилина. К реакционной смеси добавляли при перемешивании воду (2450 мл) в течение промежутка времени, составлявшего примерно один час, при этом внутреннюю температуру поддерживали на уровне примерно 20-30°С. После завершения добавления воды образовавшуюся смесь перемешивали в течение примерно 1 ч при температуре от 20 до 30°С. Затем образовавшуюся смесь фильтровали и колбу и осадок на фильтре промывали водой (3×2,56 л). Твердый продукт золотисто-желтого цвета сушили в вакууме в вакуумной печи при 50°С до постоянной массы, получая 416 г продукта (выход 98,6%).
Процедура В
5-Хлор-2-нитроанилин (401 г, 2,32 моля) вносили в 4-горлую круглодонную колбу вместимостью 12 л, снабженную верхней мешалкой, конденсатором, впускным газовым клапаном, капельной воронкой и датчиком температуры. Затем колбу продували N2. В реакционную колбу добавляли при перемешивании 1-метилпиперазин (977 г, 1,08 л, 9,75 моля) и 100%-ный этанол (650 мл). Колбу снова продували N2 и реакционную смесь выдерживали в атмосфере N2. Колбу нагревали в нагревательном кожухе до достижения внутренней температуры 97°С (±5°С) и поддерживали при этой температуре до завершения реакции (как правило, в течение 40 ч) по данным ЖХВР. После завершения реакции нагревание прекращали и реакционную смесь охлаждали при перемешивании до внутренней температуры, составляющей примерно 80°С, и к смеси добавляли через капельную воронку воду (3,15 л) в течение промежутка времени, составляющего 1 ч, при этом внутреннюю температуру поддерживали на уровне 82°С (±3°С). После завершения добавления воды нагревание прекращали и реакционной смеси давали охладиться в течение периода времени не менее 4 ч до внутренней температуры, составляющей 20-30°С. Затем реакционную смесь перемешивали еще в течение 1 ч при внутренней температуре 20-30°С. Затем образовавшуюся смесь фильтровали и колбу и осадок на фильтре промывали водой (1×1 л), 50%-ным этанолом (1×1 л) и 95%-ным этанолом (1×1 л). Твердый продукт золотисто-желтого цвета сушили в вакууме в вакуумной печи при 50°С до постоянной массы, получая 546 г продукта (выход 99%).
Пример 2
Синтез этилового эфира [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты
Процедура А
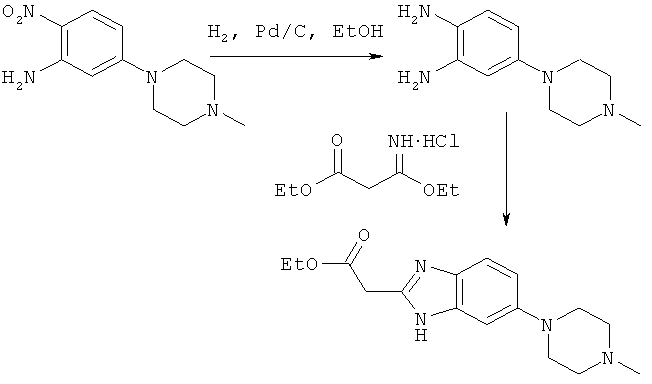
4-горлую колбу вместимостью 5000 мл снабжали мешалкой, термометром, конденсатором и впускным/выпускным газовым клапаном. В снабженную этими устройствами колбу вносили 265,7 г (1,12 моля, 1,0 экв.) 5-(4-метилпиперазин-1-ил)-2-нитроанилина и 2125 мл 200 Proof EtOH. Образовавшийся раствор продували N2 в течение 15 мин. Затем добавляли 20,0 г 5%-ного Pd/C (50 мас.% H2O). Реакционную смесь интенсивно перемешивали при 40-50°С (внутренняя температура), при этом смесь барботировали Н2. Через каждый час осуществляли мониторинг реакции, оценивая с помощью ЖХВР исчезновение 5-(4-метилпиперазин-1-ил)-2-нитроанилина. Как правило, время реакции составляло 6 ч.
После полного исчезновения 5-(4-метилпиперазин-1-ил)-2-нитроанилина из реакционной смеси раствор продували N2 в течение 15 мин. Затем добавляли 440,0 г (2,25 моля) гидрохлорида этил-3-этокси-3-иминопропаноата в виде твердого вещества. Реакционную смесь перемешивали при 40-50°С (внутренняя температура) до завершения реакции. Осуществляли мониторинг реакции, оценивая с помощью ЖХВР исчезновение диаминового производного. Как правило, время реакции составляло 1-2 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры и фильтровали через подушку из фильтрующего материала целита (Celite). Фильтрующий материал целит промывали абсолютным EtOH (2×250 мл) и фильтрат концентрировали при пониженном давлении, получая густое масло коричневого/оранжевого цвета. Образовавшееся масло растворяли в 850 мл 0,37%-ного раствора HCl. Затем добавляли твердый NaOH (25 г) в виде одной порции, после чего происходило образование осадка. Образовавшуюся смесь перемешивали в течение 1 ч и затем фильтровали. Твердый продукт промывали Н2О (2×400 мл) и сушили при 50°С в вакуумной печи, получая 251,7 г (выход 74,1%) этилового эфира [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты в виде порошка светло-желтого цвета.
Процедура Б
4-горлую колбу вместимостью 5000 мл, помещенную в кожух, снабжали механической мешалкой, конденсатором, датчиком температуры, впускным газовым клапаном и масляным барботером. В колбу, снабженную этими устройствами, вносили 300 г (1,27 моля) 5-(4-метилпиперазин-1-ил)-2-нитроанилина и 2400 мл 200 Proof EtOH (для этой реакции не является необходимым применение 200 Proof этанола, ее можно осуществлять и ее осуществляли с использованием 95%-ного этанола). Образовавшийся раствор перемешивали и продували N2 в течение 15 мин. Затем в реакционную колбу добавляли 22,7 г 5%-ного Pd/C (50 мас.% Н2О). Реакционный сосуд продували N2 в течение 15 мин. После продувания N2 реакционный сосуд продували Н2, поддерживая слабый, но постоянный поток Н2 через колбу. Реакционную смесь перемешивали при 45-55°С (внутренняя температура), при этом смесь барботировали с помощью Н2 до полного поглощения 5-(4-метилпиперазин-1-ил)-2-нитроанилина по данным ЖХВР. Как правило, время реакции составляло 6 ч.
После полного исчезновения 5-(4-метилпиперазин-1-ил)-2-нитроанилина из реакционной смеси раствор продували N2 в течение 15 мин. Промежуточный продукт, представляющий собой диаминовое производное, является чувствительным к действию воздуха, поэтому принимали меры предосторожности для того, чтобы избегать контакта с воздухом. В реакционную смесь добавляли 500 г (2,56 моля) гидрохлорида этил-3-этокси-3-иминопропаноата в течение примерно 30-минутного промежутка времени. Реакционную смесь перемешивали в атмосфере N2 при 45-55°С (внутренняя температура) до полного поглощения диамина по данным ЖХВР. Как правило, время реакции составляло примерно 2 ч. После завершения реакции еще теплую реакционную смесь фильтровали через подушку из целита. Затем реакционную колбу и целит промывали 200 Proof EtOH (3×285 мл). Фильтраты объединяли в колбе вместимостью 5000 мл и отгоняли примерно 3300 мл этанола в вакууме, получая масло оранжевого цвета. К образовавшемуся маслу добавляли воду (530 мл) и затем 1 М HCl (350 мл) и перемешивали образовавшуюся смесь. Образовавшийся раствор интенсивно перемешивали, при этом в течение примерно 20-минутного периода времени добавляли 30%-ный NaOH (200 мл), поддерживая внутреннюю температуру на уровне примерно 25-30°С, в результате чего доводили значение рН до 9-10. Образовавшуюся суспензию перемешивали в течение примерно 4 ч, поддерживая внутреннюю температуру на уровне примерно 20-25°С. Образовавшуюся смесь фильтровали и осадок на фильтре промывали H2O (3×300 мл). Собранный твердый продукт сушили в вакууме при 50°С в вакуумной печи до постоянной массы, получая 345,9 г (выход 90,1%) этилового эфира [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты в виде порошка светло-желтого цвета. При использовании альтернативной процедуры обработки фильтраты объединяли и этанол отгоняли в вакууме до удаления по меньшей мере примерно 90%. Затем к образовавшемся маслу добавляли воду при нейтральном значении рН и раствор охлаждали до примерно 0°С. Затем медленно добавляли при интенсивном перемешивании водный 20%-ный раствор NaOH, доводя значение рН до 9,2 (что определяли с помощью рН-метра). Затем образовавшуюся смесь фильтровали и сушили, как описано выше. Альтернативная процедура обработки позволяла получать продукт от светло-рыжевато-коричневого до светло-желтого цвета с выходом до 97%.
Пример 3
Метод уменьшения содержания воды в этиловом эфире [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты
Этиловый эфир [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты (120,7 г), который предварительно обрабатывали и сушили до содержания H2O примерно 8-9%, вносили в круглодонную колбу вместимостью 2000 мл и растворяли в абсолютном этаноле (500 мл). Раствор янтарного цвета концентрировали с использованием роторного испарителя при нагревании до полного удаления растворителя с получением густого масла. Процедуру повторяли еще дважды. Полученное таким образом густое масло оставляли в колбе и помещали на ночь в вакуумную печь, нагретую до 50°С. По результатам анализа методом Карла Фишера содержание воды составляло 5,25%. Уменьшение содержание воды, достигаемое с помощью этого метода, позволяло получать более высокие выходы продукта при использовании процедуры, описанной в примере 4. Вместо этанола в данном процессе сушки можно использовать другие растворители, такие как толуол и ТГФ.
Пример 4
Синтез 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она
Процедура А
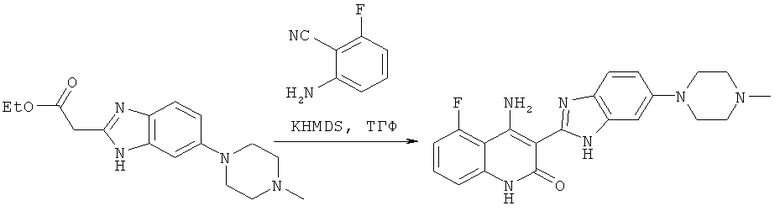
Этиловый эфир [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты (250 г, 820 ммолей) (высушенный с использованием этанола описанным выше методом) растворяли в ТГФ (3800 мл) в колбе вместимостью 5000 мл, снабженной конденсатором, механической мешалкой, датчиком температуры, и продували аргоном. К раствору добавляли 2-амино-6-фторбензонитрил (95,3 г, 700 ммолей) и внутренняя температура повышалась до 40°С. После того, как происходило полное растворение твердых частиц и внутренняя температура достигала 40°С, добавляли твердый KHMDS (376,2 г, 1890 ммолей) в течение 5 мин. После завершения добавления калиевого основания получали гетерогенный раствор желтого цвета и внутренняя температура повышалась до 62°С. Через 60 мин внутренняя температура снова понижалась до 40°С, и реакция завершалась по данным ЖХВР (отсутствовал исходный продукт или нециклизованный промежуточный продукт). Затем реакцию прекращали, сливая густую реакционную смесь на H2O (6000 мл) и перемешивали образовавшуюся смесь до достижения комнатной температуры. Затем смесь фильтровали и осадок на фильтре промывали водой (1000 мл, 2Х). Твердый продукт ярко-желтого цвета помещали в сушильный поддон и сушили в течение ночи в вакуумной печи при 50°С, получая 155,3 г (выход 47,9%) требуемого 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она.
Процедура Б
4-горлую колбу вместимостью 5000 мл, помещенную в кожух, снабжали дистиллятором, датчиком температуры, газовым клапаном для впуска N2, капельной воронкой и механической мешалкой. В реактор загружали этиловый эфир [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты (173,0 г, 570 ммолей) и реактор продували N2 в течение 15 мин. Затем в колбу загружали при перемешивании безводный ТГФ (2600 мл). После полного растворения твердого продукта растворитель удаляли дистилляцией (в вакууме или при атмосферном давлении), применяя при необходимости нагревание (более высокая температура способствует удалению воды). После удаления 1000 мл растворителя дистилляцию прекращали и реакционную смесь продували N2. Затем в реакционный сосуд добавляли 1000 мл безводного ТГФ и после полного растворения твердого продукта снова осуществляли дистилляцию (в вакууме или при атмосферном давлении) до удаления еще 1000 мл растворителя. Этот процесс добавления безводного ТГФ и удаления растворителя повторяли по меньшей мере 4 раза (в ходе 4-го цикла дистилляции удаляли 60% растворителя, а не почти 40%, как в ходе первых трех циклов дистилляции), после чего отбирали образец объемом 1 мл для определения содержания воды методом Карла Фишера. Если результаты анализа показывали, что содержание воды в образце составляло менее 0,20%, то реакцию продолжали согласно процедуре, описанной в следующем параграфе. Однако, если результаты анализа показывали, что содержание воды составляло более 0,20%, то продолжали описанный выше процесс обезвоживания до тех пор, пока содержание воды не становилось меньше 0,20%.
После того, как в результате осуществления описанной в предыдущем параграфе процедуры содержание воды достигало уровня менее 0,20% или становилось примерно равным ему, дистиллятор заменяли на парциальный конденсатор горячего орошения и в реакционную смесь вносили 2-амино-6-фторбензнитрил (66,2 г, 470 ммолей) (в некоторых вариантах осуществления этой процедуры использовали 0,95 экв.). Затем реакционную смесь нагревали до внутренней температуры 38-42°С. Когда внутренняя температура достигала 38-42°С, к реакционной смеси в течение 5-минутного периода добавляли через капельную воронку раствор KHMDS (1313 г, 1,32 моля, 20% KHMDS в ТГФ), поддерживая в процессе добавления внутреннюю температуру на уровне примерно 38-50°С. После завершения добавления калиевого основания реакционную смесь перемешивали в течение 3,5-4,5 ч (в некоторых таких примерах перемешивание осуществляли в течение 30-60 мин, и реакция могла завершаться в течение этого периода времени), поддерживая внутреннюю температуру на уровне 38-42°С. Затем отбирали образец реакционной смеси и анализировали с помощью ЖХВР. Если реакция еще не была завершена, то в колбу добавляли дополнительную порцию раствора KHMDS в течение 5-минутного промежутка времени и реакционную смесь перемешивали при 38-42°С в течение 45-60 мин (количество добавляемого раствора KHMDS определяли следующим образом: если коэффициент IPC<3,50, то добавляли 125 мл; если 10,0>IPC>3,50, то добавляли 56 мл; если 20,0>IPC>10, то добавляли 30 мл, где коэффициент IPC определяли как отношение площади, соответствующей 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-ону, к площади, соответствующей нециклизованному промежуточному продукту). После завершения реакции (коэффициент IPC>20) реактор охлаждали до внутренней температуры 25-30°С и в реактор вносили воду (350 мл) в течение промежутка времени, составлявшего 15 мин, поддерживая внутреннюю температуру на уровне 25-35°С (в одном из альтернативных вариантов реакцию осуществляли при 40°С и добавляли воду в течение 5 мин. Более быстрое прекращение реакции снижает количество примесей, которые образуются с течением времени). Затем парциальный конденсатор горячего орошения заменяли на дистиллятор и удаляли растворитель путем дистилляции (в вакууме или при атмосферном давлении), применяя при необходимости нагревание. После удаления 1500 мл растворителя дистилляцию прекращали и реакционную смесь продували N2. После этого в реакционную колбу добавляли воду (1660 мл), поддерживая внутреннюю температуру на уровне 20-30°С. Затем реакционную смесь перемешивали при 20-30°С в течение 30 мин, после чего охлаждали до внутренней температуры 5-10°С и затем перемешивали в течение 1 ч. Образовавшуюся суспензию фильтровали и колбу и осадок на фильтре промывали водой (3×650 мл). Полученный таким образом твердый продукт подвергали вакуумной сушке в вакуумной печи при 50°С до постоянной массы, получая 103,9 г (выход 42,6%) 4-амино-5-фтор-3-[6-(4-метилпиперазин-1 -ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она в виде порошка желтого цвета.
Процедура В
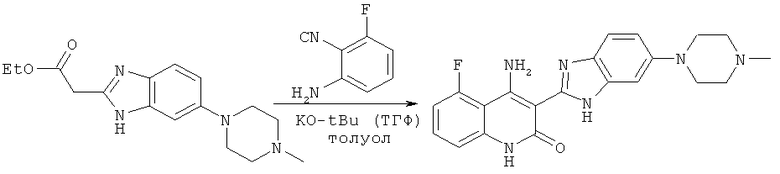
Этиловый эфир [6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]уксусной кислоты (608 г, 2,01 моля) (безводный) и 2-амино-6-фторбензнитрил (274 г, 2,01 моля) вносили в 4-горлую колбу вместимостью 12 л, помещенную в нагревательный кожух и снабженную конденсатором, механической мешалкой, впускным газовым клапаном и датчиком температуры. Реакционный сосуд продували N2 и в реакционную смесь вносили при перемешивании толуол (7,7 л). Реакционный сосуд снова продували N2 и выдерживали в атмосфере N2. Внутреннюю температуру смеси повышали до достижения температуры 63°С (±3°С). Внутреннюю температуру смеси поддерживали на уровне 63°С (±3°С), при этом из колбы удаляли 2,6 л толуола путем дистилляции при пониженном давлении (380±10 торр, температура дистилляционной насадки t=40°С (±10°С) (для проверки содержания воды в смеси применяли анализ методом Карла Фишера. Если содержание воды превышало 0,03%, то добавляли еще 2,6 л толуола и повторяли процедуру дистилляции. Этот процесс повторяли до тех пор, пока содержание воды не становилось менее 0,03%). После того, как содержание воды становилось менее 0,03%, нагревание прекращали и реакционную смесь охлаждали в атмосфере N2 до внутренней температуры 17-19°С. Затем к реакционной смеси в атмосфере N2 добавляли трет-бутоксид калия в ТГФ (20% в ТГФ; 3,39 кг, 6,04 моля трет-бутоксида калия) с такой скоростью, чтобы внутренняя температура реакционной смеси поддерживалась на уровне ниже 20°С. После завершения добавления трет-бутоксида калия реакционную смесь перемешивали при внутренней температуре ниже 20°С в течение 30 мин. После этого температуру повышали до 25°С и реакционную смесь перемешивали в течение по меньшей мере 1 ч. Затем температуру повышали до 30°С и реакционную смесь перемешивали в течение по меньшей мере 30 мин. Затем осуществляли мониторинг завершения реакции с помощью ЖХВР, проверяя поглощение исходных продуктов (как правило, в течение 2-3 ч происходило поглощение обоих исходных продуктов (соответствующая им площадь составляла менее 0,5% по данным ЖХВР). Если реакция не завершалась по истечении 2 ч, то в этот момент времени добавляли еще 0,05 экв. трет-бутоксида калия и осуществляли процесс до тех пор, пока по данным ЖХВР реакция не завершалась. После завершения реакции к реакционной смеси добавляли при перемешивании 650 мл воды. Затем реакционную смесь нагревали до внутренней температуры 50°С и удаляли ТГФ из реакционной смеси путем дистилляции при пониженном давлении (в объеме, составляющем примерно 3 л). Затем к реакционной смеси добавляли по каплям с помощью капельной воронки воду (2,6 л). После этого смесь охлаждали до комнатной температуры и перемешивали в течение по меньшей мере 1 ч. Затем смесь фильтровали и осадок на фильтре промывали водой (1,2 л), 70%-ным этанолом (1,2 л) и 95%-ным этанолом (1,2 л). Твердый продукт ярко-желтого цвета помещали в сушильный поддон и сушили в вакуумной печи при 50°С до достижения постоянной массы, получая 674 г (выход 85,4%) требуемого 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она.
Пример 5
Очистка 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она
4-горлую колбу вместимостью 3000 мл, снабженную конденсатором, датчиком температуры, впускным газовым клапаном для N2 и механической мешалкой, помещали в нагревательный кожух. Затем колбу загружали 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-оном (101,0 г, 0,26 моля), суспендировали твердый продукт желтого цвета в 95%-ном этаноле (1000 мл) и перемешивали. В некоторых случаях использовали соотношение 8:1 (продукт: растворитель). Затем суспензию выдерживали при осторожном кипячении с обратным холодильником (температура примерно 76°С) при перемешивании в течение примерно 1 ч.
После этого реакционную смесь перемешивали при кипячении с обратным холодильником в течение 45-75 мин. По истечении этого времени нагревание колбы прекращали и суспензии давали охладиться до температуры 25-30°С. Затем суспензию фильтровали и осадок на фильтре промывали водой (2×500 мл). После этого твердый продукт желтого цвета помещали на сушильный поддон и сушили в вакуумной печи при 50°С до достижения постоянной массы (как правило, в течение 16 ч), в результате получали 97,2 г (выход 96,2%) очищенного продукта в виде порошка желтого цвета.
Пример 6
Получение лактата 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она
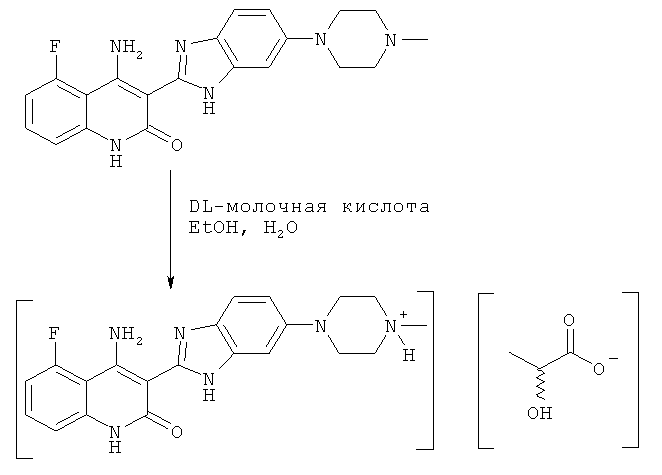
4-горлую колбу вместимостью 3000 мл, помещенную в кожух, снабжали конденсатором, датчиком температуры, впускным газовым клапаном для N2 и механической мешалкой. Реакционный сосуд продували N2 в течение по меньшей мере 15 мин и затем в него вносили 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-он (484 г, 1,23 моля). Получали раствор, содержащий D,L-молочную кислоту (243,3 г, 1,72 моля мономера, воду (339 мл) и этанол (1211 мл), и затем вносили в реакционную колбу. Начинали перемешивание со средней скоростью и реакционную смесь нагревали до внутренней температуры 68-72°С. Внутреннюю температуру реакционной смеси поддерживали на уровне 68-72°С в течение 15-45 мин и затем прекращали нагревание. Образовавшуюся смесь фильтровали через фритту с размерами отверстий 10-20 мкм, собирая фильтрат в колбу вместимостью 12 л. Колбу вместимостью 12 л снабжали датчиком внутренней температуры, парциальным конденсатором горячего орошения, капельной воронкой, газовым впускным/выпускным клапаном и верхней мешалкой. Затем фильтрат перемешивали со средней скоростью и нагревали до температуры дефлегмации (внутренняя температура примерно 78°С). При осторожном кипячении с обратным холодильником в колбу вносили этанол (3596 мл) в течение промежутка времени, составлявшего примерно 20 мин. Затем реакционную колбу охлаждали в течение 15-25 мин до внутренней температуры, составлявшей примерно 64-70°С, и поддерживали эту температуру в течение примерно 30 мин. Реактор обследовали в отношении присутствия кристаллов. Если кристаллы отсутствовали, то в колбу вносили кристаллы лактата 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1H-хинолин-2-она (484 мг, 0,1 мол.%) и перемешивали реакционную смесь при 64-70°С в течение 30 мин, после чего колбу снова обследовали в отношении присутствия кристаллов. Если кристаллы присутствовали, то скорость перемешивания снижали до низкого уровня и перемешивали реакционную смесь при 64-70°С еще в течение 90 мин. Затем реакционную смесь охлаждали примерно до 0°С в течение примерно 2 ч и образовавшуюся смесь фильтровали через фильтр из фритты с размерами отверстий 25-50 мкм. Реактор промывали этанолом (484 мл) и перемешивали до тех пор, пока внутренняя температура не достигала примерно 0°С. Для промывки осадка на фильтре применяли холодный этанол и повторяли эту процедуру еще два раза. Собранный твердый продукт подвергали вакуумной сушке в вакуумной печи при 50°С до достижения постоянной массы, в результате чего получали 510,7 г (выход 85,7%) кристаллического лактата 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она. Эта процедура позволяет получать форму А лактата соединения. В процессе фильтрации, как правило, применяли резиновую перегородку или инертные условия. Хотя безводный твердый продукт, по-видимому, не является высоко гигроскопичным, влажный осадок на фильтре имеет тенденцию впитывать влагу и становиться липким. Предпринимали меры для того, чтобы избегать продолжительного воздействия атмосферы на влажный осадок на фильтре.
Имеющаяся на рынке молочная кислота, как правило, содержит примерно 8-12 мас.% воды и помимо мономерной молочной кислоты содержит димеры и тримеры. Молярное соотношение димеров и мономеров молочной кислоты, как правило, составляет примерно 1,0:4,7. В процессе, описанном ранее, можно использовать молочную кислоту с чистотой, соответствующей поступающему на рынок продукту, поскольку из реакционной смеси происходит осаждение преимущественно монолактата.
Пример 7
Рентгеноструктурный анализ лактата формы А
Предварительные кристаллографические исследования
Предварительные анализы методом XRPD (дифракция рентгеновских лучей на порошке) проводили с помощью рентгеновского порошкового дифрактометра типа Shimadzu XRD-6000 с использованием Кα-излучения от медного источника (Cu-анод). Прибор оборудован рентгеновской трубкой с тонкой фокусировкой. Напряжение и силу тока в трубке устанавливали на уровне 40 кВ и 40 мА соответственно. Устанавливали щели для углов расходимости и рассеяния, равные 1°, а размер входной щели устанавливали равным 0,15 мм. Дифрагированное излучение регистрировали с помощью сцинтилляционного NaI-детектора. Применяли непрерывное тета-два тета-(θ-2θ)-сканирование со скоростью 3°/мин (с шагом 0,4 с/0,02°) в диапазоне от 2,5 до 40°С. Было установлено, что лактат 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она имеет высокую степень кристалличности и характеризуется четкой картиной дифракции рентгеновских лучей на порошке.
Дополнительная характеризация с помощью XRPD лактата 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она, форма А
XRPD осуществляли с помощью порошкового дифрактометра типа Philips X'Pert (Кα-излучение от медного источника). Использовали металлические держатели образца глубиной 0,4 или 0,8 мм (ТТК-типа). Вследствие высокой активности исследуемой лекарственной субстанции держатели образца покрывали тонкой каптоновой пленкой после получения на рабочем столе с ламинарным воздухопотоком. Длина волны CuKα1-излучения была равна 1,54060 Å. Рентгеновская трубка работала при напряжении 40 кВ и силе тока 40 мА. Использовали размер шага 0,02° и время счета от 2,0 до 2,4 с на шаг. Вследствие вариабельности плотности упаковки в держателе образца регистрируемая интенсивность может варьироваться и слабый аморфный фон, обусловленный каптоновой пленкой, трудно отличать от любой аморфной лекарственной субстанции, которая может присутствовать в образце, полученном в эксперименте по кристаллизации.
XRPD-картина для формы А представлена на фиг.1. Относительно выраженные 2θ-пики были выявлены при примерно 5,7, примерно 11,3, примерно 12,4, примерно 15,3, примерно 15,9, примерно 17,0, примерно 19,1, примерно 19,7, примерно 20,5, примерно 20,9, примерно 22,8, примерно 23,4, примерно 23,7, примерно 24,7, примерно 25,0, примерно 25,9, примерно 26,9 и примерно 31,2 градусах.
Пример 8
Гигроскопичность формы А
Исследование лактата 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она, форма А, в DVS-эксперименте позволило установить, что исследуемая форма А при относительной влажности (ОВ) ниже примерно 80% является негигроскопичной (см. таблицу 1). Все DVS-измерения осуществляли при изменении относительной влажности на 2,5% в час. Однако выдерживание в условиях, когда ОВ превышала 90%, приводило к существенному поглощению воды, которое не было полностью обратимым в течение принятого времени измерений. Кроме того, поглощение воды не заканчивалось, когда по истечении 4500 мин относительную влажность возвращали от 95% к исходной величине 50%. Результаты DVS-измерений представлены на фиг.2 и 3.
Пример 9
Композиции 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она
Композиции в форме капсул получали согласно общему методу, представленному на фиг.2. Лактат 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она получали согласно описанному выше методу и для приготовления композиций, представленных в настоящем описании, предпочтительно использовали безводную кристаллическую форму (форма А). Применяемые в композиции эксципиенты представляли собой моногидрат лактозы (например, FAST FLO №316, поставляемый фирмами Foremost Whey Products и DMV Corp.), микрокристаллическую целлюлозу (например, AV1CEL 101, поставляемую фирмой FMC Corp.), частично предварительно желированный крахмал (например, STARCH 1500, поставляемый фирмой Colorcon, Inc.), повидон (поставляемый фирмой ISP или фирмой Base), кросповидон (например, POLYPLASDONE XL, поставляемый фирмой ISP), диоксид кремния (например, SYLOID 244FP, поставляемый фирмой Grace Davison или фирмой Cabot) и стеарат магния (например, поставляемый фирмой Mallinckrodt).
Получали двенадцать композиций (препаративных форм) в форме капсул (композиции 1-12), которые имели процентные содержания ингредиентов, указанные в таблице 2. Капсулы приготавливали таким образом, чтобы они содержали по 15 мг API (соединение формулы I) каждая. Приготавливали три дополнительные композиции (количество продукта (масштаб производства) составляло примерно 1,5 кг) с двумя различными содержаниями действующего вещества (25 мг и 100 мг (композиция 13) и 30 мг и 100 мг (композиции 14 и 15)). Ингредиенты и их количества, которые использовали для приготовления композиций, представлены в таблицах 3-5.
В целом метод заключался в следующем. Все ингредиенты за исключением стеарата магния объединяли и приготавливали премикс перед осуществлением измельчения. После измельчения и смешения добавляли стеарат магния и смесь перемешивали второй раз. После смешения с добавленным стеаратом магния композициями заполняли капсулы таким образом, чтобы они содержали по 25, 30 и 100 мг композиции. Когда количества композиций составляли 25 и 30 мг, использовали шведские непрозрачные оранжевые капсулы размера 2, а когда количество композиции составляло 100 мг, применяли непрозрачные серые желатиновые (CS, фирма Capsugel) или ГПМЦ-капсулы (QUALI-V, фирма Shanogi) размера 0. Такую же или аналогичную процедуру можно использовать для приготовления капсул других размеров, отличных от тех, которые представлены в таблицах 2-5. Например, такую же процедуру можно применять для приготовления капсул, содержащих по 25, 30, 50, 100, 150, 200, 250, 300, 350, 400, 450 и 500 мг, путем простого регулирования размера капсул и количества ингредиентов в композиции, как должно быть очевидно специалисту в данной области.
Стабильность каждой композиции, представленной в таблице 2, оценивали после хранения в течение 3 месяцев при 40°С/75% комнатной влажности. Было установлено, что в течение этого периода времени количество примесей и продуктов разложения составляло для всех композиций менее 0,6% и для большинства композиций менее 0,4%.
С учетом этих результатов приготавливали согласно описанному выше методу три дополнительные композиции (композиции 13-15). Было установлено, что каждая из композиций обладала требуемой стабильностью и характеристиками растворимости, как показано в таблицах 6-15. Таким образом, любые из представленных в настоящем описании композиций представляют собой различные варианты осуществления изобретения. Ни в одной из композиций не было обнаружено продуктов расщепления. Каждая из композиций 13-15 обладала высокой растворимостью. Например, 80% композиции 1 растворялось в течение 10 мин, 85% композиции 2 растворялось в течение 10 мин и 85% композиции 3 растворялось в течение 20 мин. Все эти результаты полностью удовлетворяют стандартам, установленным Департаментом по контролю за качеством пищевых продуктов, медикаментов и косметических средств, согласно которым 85% вещества должно растворяться в течение 45 мин. В конкретных вариантах осуществления изобретения предпочтительной композицией является композиция 14. В других вариантах осуществления изобретения предпочтительной композицией является композиция 13 или композиция 15.
Осуществляли тестирование в ходе процесса для оценки однородности смесей, распределения размеров частиц (PSD) смеси и объемной/насыпной плотности для всех композиций. Для хранения капсул после изготовления использовали три варианта упаковки. В одном варианте капсулы хранили в бутылке из полиэтилена высокой плотности (ПЭВП), снабженной прокладкой из искусственного волокна и термокрышкой. Во втором варианте капсулы хранили в ПЭВП-бутылке без прокладки из искусственного волокна, но с термокрышкой. В третьем варианте капсулы хранили в блистерной Al-Al-упаковке. Проводили тестирование стабильности с позиций однородности содержимого, внешнего вида и характеристик растворимости. Для оценки стабильности композиций в форме капсул использовали также ЖХВР-анализ.
Вариант упаковки представлял собой ПЭВП-бутылку без вкладыша из искусственного волокна и термокрышки.
Вариант упаковки представлял собой ПЭВП-бутылку с вкладышем из искусственного волокна и термокрышкой.
Вариант упаковки представлял собой ПЭВП-бутылку без вкладыша из искусственного волокна и термокрышки.
Вариант упаковки представлял собой ПЭВП-бутылку с вкладышем из искусственного волокна и термокрышкой.
Стабильность растворимости
Стабильность растворимости фармацевтических композиций 13, 14 и 15 оценивали в искусственной желудочной жидкости с помощью устройства USP Apparatus II при 50 об/мин. Тестировали композиции, содержащие 100 и 25 или 30 мг API. Было установлено, что через 60 мин все композиции полностью растворялись в искусственной желудочной жидкости. Результаты исследований стабильности представлены в таблицах 10-15.
Пример 10
Исследование характеристик композиций 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил1-1Н-хинолин-2-она в виде порошка
Проводили изучение влияния лекарственной загрузки при содержании API 200 мг. Приготавливали композиции, обозначенные как композиции 16, 17, 18 и 19 (таблицы 16-19), с загрузками 70, 60, 50 и 60% соответственно с использованием метода смешения в полиэтиленовом мешке. API и эксципиенты за исключением стеарата магния смешивали в мешке с использованием ПЭ-мешка в течение 3 мин. Затем смесь пропускали через ручное сито с размером отверстий 30 меш и вносили в ПЭ-мешок для дополнительного смешения в течение 3 мин. Затем пропускали стеарат магния через ручное сито с размером отверстий 30 меш, добавляли к смеси и перемешивали еще в течение 3 мин или до тех пор, пока смесь не становилась однородной при визуальном осмотре.
Все композиции оценивали в отношении приемлемых характеристик текучести при содержании API 200 мг. Все оценки текучести проводили путем определения индекса Карра. Индекс Карра рассчитывали по следующей формуле: (насыпная плотность - объемная плотность)/(насыпная плотность). В таблице 20 представлены результаты оценки, включая объемную плотность, насыпную плотность, индекс Карра и угол естественного откоса. Было установлено, что все композиции, представленные в таблицах 16-19, обладают приемлемыми характеристиками текучести. Таким образом, любые из указанных в настоящем описании композиций представляют собой различные варианты осуществления изобретения.
Пример 11
Композиции 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-6ензимидазол-2-ил]-1Н-хинолин-2-она, полученные методом мокрой грануляции
Композиции в форме капсул получали с помощью процедуры, аналогичной общему методу, описанному в примере 10, с использованием двух дополнительных стадий: (1) первое смешивание осуществляли в присутствии жидкости для грануляции, такой как водная, спиртовая или водно-спиртовая жидкость, и (2) применяли стадию сушки для удаления жидкости для грануляции. Лактат 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она получали согласно описанной выше процедуре и для приготовления описанных выше композиций предпочтительно использовали безводную кристаллическую форму (форма А). Различные композиции получали в масштабе производства примерно 15 г, и каждая капсула была рассчитана на то, чтобы содержать примерно 15 мг API. Помимо лактата 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она добавляли в качестве разбавителей моногидрат лактозы, микрокристаллическую целлюлозу и частично предварительно желированный крахмал; в качестве связующего вещества добавляли повидон; в качестве разрыхлителя добавляли кросповидон; в качестве агента, повышающего текучесть, добавляли диоксид кремния; в качестве смачивающего агента добавляли лаурилсульфат натрия и в качестве растворителя или жидкости для грануляции добавляли воду или 0,5 н. HCl.
Процедура заключалась в следующем. Все ингредиенты за исключением лаурилсульфата натрия и половины кросповидона объединяли и предварительно смешивали в сухом состоянии (2 мин с помощью лопастной мешалки при 650 об/мин и измельчителя при 2500 об/мин). Лаурилсульфат натрия, если он присутствовал, растворяли в жидкости для грануляции. В процессе смешения при скорости вращения лопастной мешалки 650 об/мин (при отключенном измельчителе) к смеси добавляли с помощью пипетки жидкость для грануляции. После завершения добавления жидкости для грануляции включали измельчитель (2500 об/мин) и смесь перемешивали еще в течение 2 мин. Мокрую смесь для грануляции пропускали через сито с размером отверстий 20 меш и смесь сушили в печи примерно при 50°С до тех пор, пока потеря при сушке не становилась менее 1%. Продукт для грануляции снова калибровали с помощью сита с размером отверстий 20 меш и затем смешивали с оставшейся частью кросповидона в течение 10 мин.
Композиции капсулировали таким образом, чтобы в каждой капсуле присутствовало по 15 мг API. Применяемые капсулы представляли собой либо белые непрозрачные размера 0 желатиновые капсулы фирмы Capsugel, либо ГПМЦ-капсулы фирмы Shanogi. Эту же или аналогичную процедуру можно применять для приготовления капсул, имеющих размеры, отличные от описанного. Например, эту же процедуру можно применять для приготовления капсул, содержащих по 25, 30, 50, 100, 150, 200, 250, 300, 350, 400, 450 и 500 мг, путем простого регулирования размера капсул и количества ингредиентов в композиции, как должно быть очевидно специалисту в данной области.
В таблице 21 представлены 12 композиций (20-31), полученных описанными выше методами мокрой грануляции.
Пример 12
Оценка композиций на собаках
Четыре композиции оценивали на собаках. Композиции представляли собой композицию 13, композицию 14, композицию 15 и композицию типа «порошок в бутылке» (PIB). Вводимые собакам дозы составляли 100 мг соединения на собаку на период времени (100 мг/собаку/период). Исследование проводили на четырех собаках-самцах и четырех собаках-самках (N=4/пол). При оценке использовали четырехсторонний рандомизированный перекрестный план исследования. Между обработками предусматривали 1-недельный промежуток времени для вымывания лекарственного средства. Нижний предел количественной оценки (LLOQ) составлял примерно 1 нг/мл. Оценивали величину AUC (нг·ч/мл) для каждой композиции, и было установлено, что она составляет от 100 до 350 нг·ч/мл. Для PIB-композиции величины AUC составляли 150-450 нг·ч/мл. Не было выявлено заметного влияния пола животного на результаты. Не было выявлено влияния вида обработки или периода времени на результаты. Аналогичные данные были получены касательно величины Cmax. Результаты этих исследований свидетельствуют о том, что каждая из композиций и, прежде всего композиция 14, обладают свойствами, требуемыми для фармацевтической композиции.
Содержание каждого из патентов, заявок на патент и журнальных статей, процитированных выше, включено в настоящее описание в качестве ссылки и для всех целей, как если бы оно было полностью приведено как единое целое.
Подразумевается, что объем изобретения не ограничивается вариантами осуществления изобретения, приведенными в настоящем описании для иллюстрации, но он включает все такие его формы, которые определяются объемом представленного описания и прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2008 |
|
RU2485951C2 |
| СОЛИ ЭФИРОВ БЕНЗИМИДАЗОЛИЛПИРИДИЛА И ИХ СОДЕРЖАЩИЕ СОСТАВЫ | 2007 |
|
RU2457206C2 |
| СОЛИ 4-((2-(6-(4-МЕТИЛПИПЕРАЗИН-1-КАРБОНИЛ)НАФТАЛИН-2-ИЛ)ЭТИЛ)АМИНО)ХИНАЗОЛИН-6-КАРБОНИТРИЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2017 |
|
RU2641001C1 |
| НОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2779429C2 |
| СОСТАВЫ БЕНЗИМИДАЗОЛИЛПИРИДИЛЭФИРОВ | 2007 |
|
RU2452469C2 |
| СПОСОБ ГРАНУЛЯЦИИ ИЗ РАСПЛАВА | 2009 |
|
RU2491918C2 |
| Дихлорацетаты замещенных N4-[2-(диметилфосфорил)фенил]-N2-(2-метокси-4-пиперидин-1-илфенил)-5-хлорпиримидин-2,4-диаминов в качестве модуляторов ALK и EGFR, предназначенных для лечения рака | 2015 |
|
RU2606951C1 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 2020 |
|
RU2822063C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЧАСТИЧНЫЙ АГОНИСТ 5HT | 2002 |
|
RU2322978C2 |
| СТАБИЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ВОДОРАСТВОРИМУЮ СОЛЬ ВИНФЛУНИНА | 2008 |
|
RU2462248C2 |
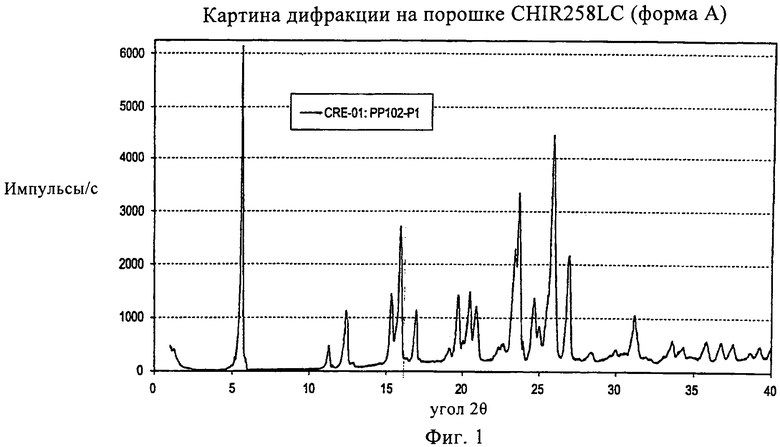
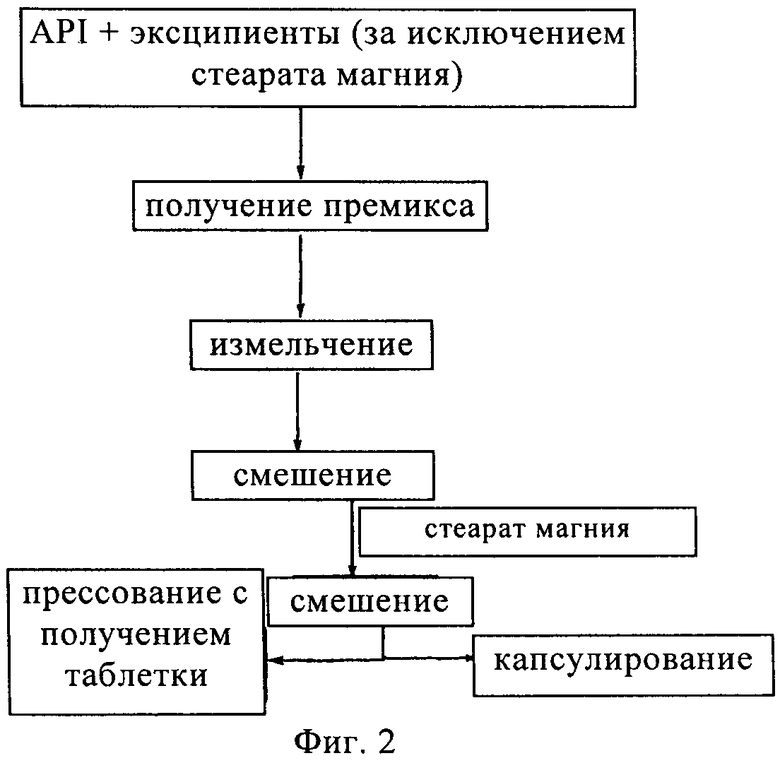
Изобретение относится к композициям хинолиноновых производных, способам получения и применению таких композиций. Фармацевтическая композиция содержит лактат соединения 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1Н-бензимидазол-2-ил]-1Н-хинолин-2-она, или его таутомер, или их смесь в количестве от 10 до 80 мас.% и по меньшей мере один ингредиент, выбранный из группы, которая включает (I) целлюлозу; (II) диоксид кремния; (III) стеарат магния и (IV) ингредиент, выбранный из кросповидона, крахмала и лактозы. Композиция включена в состав капсулы или таблетки. Композиции по изобретению проявляют удовлетворительные свойства в отношении скоростей растворения и стабильности. 8 н. и 41 з.п. ф-лы, 2 ил., 21 табл.
1. Фармацевтическая композиция, включающая соль лактат соединения формулы I, или его таутомер, или их смесь, в количестве от 10 до 50 мас.% в пересчете на общую массу композиции
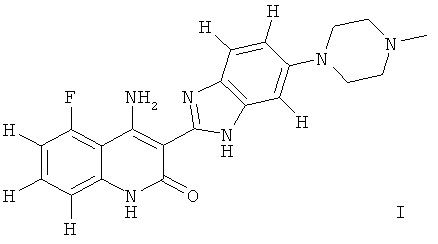
и целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу композиции.
2. Фармацевтическая композиция по п.1, содержащая фармацевтически приемлемый замасливатель, выбранный из группы, включающей жирную С16-С22-кислоту, соль жирной С16-С22-кислоты, эфир жирной C16-C22-кислоты, соль эфира жирной С16-С22-кислоты; полиэтиленгликоль со средней молекулярной массой от 6000 до 10000.
3. Фармацевтическая композиция по п.1, где композиция дополнительно содержит:
(I) диоксид кремния;
(II) стеариновую кислоту или соль стеариновой кислоты; и
(III) по меньшей мере один ингредиент, выбранный из кросповидона, крахмала, лактозы, кроскармеллозы натрия и натрийгликоляткрахмала.
4. Фармацевтическая композиция по п.1, в которой лактат соединения формулы I представляет собой безводную кристаллическую форму А.
5. Фармацевтическая композиция по п.1, где композиция содержит:
(I) микрокристаллическую целлюлозу;
(II) диоксид кремния;
(III) стеарат магния;
(IV) по меньшей мере один ингредиент, выбранный из кросповидона, частично предварительно желированного крахмала и лактозы.
6. Фармацевтическая композиция по п.5, где композиция содержит лактат соединения в количестве от 20 до 45 мас.% в пересчете на общую массу композиции.
7. Фармацевтическая композиция по п.5, где композиция содержит лактат соединения в количестве от 30 до 40 мас.% в пересчете на общую массу композиции.
8. Фармацевтическая композиция по одному из пп.1-7, в которой целлюлоза представляет собой микрокристаллическую целлюлозу.
9. Фармацевтическая композиция по п.1, где композиция содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу композиции и где композиция содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу композиции.
10. Фармацевтическая композиция по п.1, где композиция содержит целлюлозу в количестве от 20 до 45 мас.% в пересчете на общую массу композиции и где композиция содержит крахмал или лактозу в количестве от 10 до 40 мас.% в пересчете на общую массу композиции.
11. Фармацевтическая композиция по п.1, где композиция содержит крахмал в количестве от 10 до 40 мас.% в пересчете на общую массу композиции, причем крахмал представляет собой частично предварительно желированный крахмал.
12. Фармацевтическая композиция по одному из пп.1-7, где композиция содержит диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции.
13. Фармацевтическая композиция по одному из пп.1-7, где композиция содержит стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции.
14. Фармацевтическая композиция по одному из пп.1-5, где композиция содержит лактат соединения в количестве от 30 до 40 мас.% в пересчете на общую массу композиции; диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции; целлюлозу в количестве от 25 до 40 мас.% в пересчете на общую массу композиции; стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции; и кросповидон в количестве от 2 до 4 мас.% в пересчете на общую массу композиции.
15. Фармацевтическая композиция, включающая
соль лактат соединения формулы I, его таутомер или их смесь в количестве от 50 до 80 мас.% в пересчете на общую массу композиции
и по меньшей мере один ингредиент, выбранный из группы, состоящей из (i) целлюлозы, (ii) лактозы, крахмала или их смеси; (iii) повидона, (iv) диоксида кремния или талька; (v) фармацевтически приемлемого замасливателя; и (vi) ингредиента, выбранного из кросповидона, кроскармеллозы натрия и натрийгликоляткрахмала.
16. Фармацевтическая композиция по п.15, содержащая лактат соединения формулы I, или его таутомер, или их смесь в количестве от 50 до 80 мас.% в пересчете на общую массу композиции; диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции; целлюлозу в количестве от 0 до 50 мас.% в пересчете на общую массу композиции; стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции; и крахмал в количестве от 10 до 40 мас.% в пересчете на общую массу композиции.
17. Фармацевтическая композиция по п.16, где композиция содержит лактат соединения в количестве от 55 до 75 мас.% в пересчете на общую массу композиции; целлюлозу в количестве от 5 до 40 мас.% в пересчете на общую массу композиции; и крахмал в количестве от 15 до 30 мас.% в пересчете на общую массу композиции.
18. Фармацевтическая композиция по п.16, где композиция содержит лактат соединения в количестве от 60 до 70 мас.% в пересчете на общую массу композиции; и целлюлозу в количестве от 5 до 25 мас.% в пересчете на общую массу композиции.
19. Фармацевтическая композиция по п.15, где композиция содержит лактат соединения формулы I, или его таутомер, или их смесь в количестве от 50 до 80 мас.% в пересчете на общую массу композиции; диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу композиции; целлюлозу в количестве от 0 до 50 мас.% в пересчете на общую массу композиции; стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу композиции; и лактозу в количестве от 10 до 40 мас.% в пересчете на общую массу композиции.
20. Фармацевтическая композиция по п.19, где композиция содержит лактат соединения в количестве от 55 до 75 мас.% в пересчете на общую массу композиции; и целлюлозу в количестве от 5 до 40 мас.% в пересчете на общую массу композиции.
21. Фармацевтическая композиция по п.19, где композиция содержит лактат соединения в количестве от 60 до 70 мас.% в пересчете на общую массу композиции; и целлюлозу в количестве от 5 до 40 мас.% в пересчете на общую массу композиции.
22. Фармацевтическая композиция, включающая соль лактат соединения формулы I, его таутомер или их смесь в количестве от 20 до 45 мас.% в пересчете на общую массу композиции
и по меньшей мере один ингредиент, выбранный из группы, состоящей из целлюлозы, повидона, диоксида кремния, талька и фармацевтически приемлемого замасливателя; и
по меньшей мере один ингредиент, выбранный из группы, состоящей из лактозы, крахмала, кросповидона, кроскармеллозы натрия и натрийгликоляткрахмала.
23. Фармацевтическая композиция по одному из пп.1, 15 или 22, которая дополнительно содержит антиоксидант, хелатирующий агент, аскорбиновую кислоту, редуцирующий сахар или смесь двух или большего количества из указанных ингредиентов.
24. Фармацевтическая композиция по одному из пп.1, 15 или 22, где композиция включена в состав капсулы или таблетки.
25. Фармацевтическая композиция по п.24, где общая масса соединения формулы I, таутомера соединения, лактата соединения, лактата таутомера или их смеси в капсуле или таблетке составляет от 25 до 500 мг.
26. Фармацевтическая композиция по одному из пп.1, 15 или 22, в которой количество продуктов расщепления соли лактата соединения формулы I после хранения композиции в течение трех месяцев при 40°С и 57%-й комнатной влажности составляет менее 10 мас.% в пересчете на общую массу композиции.
27. Способ получения фармацевтической композиции, включающий:
(а) смешение первой смеси с получением первой перемешанной смеси, где первая смесь содержит:
(I) соль лактат соединения формулы I, его таутомер или их смесь
и (II) по меньшей мере один ингредиент, выбранный из группы, состоящей из целлюлозы; лактозы, крахмала или их смеси; повидона; диоксида кремния или талька; фармацевтически приемлемого замасливателя; и ингредиента, выбранного из кросповидона, кроскармеллозы натрия и натрийгликолята крахмала; и
б) смешение стеариновой кислоты, соли стеариновой кислоты или их смеси с первой перемешанной смесью с получением второй перемешанной смеси, где вторая перемешанная смесь включает соль лактат соединения в количестве от 10 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и включает целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу второй перемешанной смеси.
28. Способ по п.27, в котором соль лактат соединения формулы I смешивают с (I) целлюлозой; (II) диоксидом кремния и (III) ингредиентом, выбранным из кросповидона, крахмала и лактозы.
29. Способ по п.27, который дополнительно включает (в) формирование по меньшей мере одной капсулы или по меньшей мере одной таблетки из второй перемешанной смеси.
30. Способ получения фармацевтической композиции, включающий:
(а) смешивание смеси ингредиентов с получением первой перемешанной смеси, где перемешанная смесь содержит:
(I) соль лактат соединения формулы I, его таутомер или их смесь,
(II) по меньшей мере один ингредиент, выбранный из группы, состоящей из целлюлозы, крахмала, лактозы и повидона,
(III) по меньшей мере один ингредиент, выбранный из группы, состоящей из кросповидона; кроскармеллозы натрия и натрийгликолят крахмала;
(IV) жидкость для грануляции, выбранную из группы, состоящей из водной кислоты, спирта, водного спирта или смеси любых двух или большего количества из указанных веществ;
(б) удаление жидкости для грануляции;
(в) получение второй перемешанной смеси путем смешения первой перемешанной смеси по меньшей мере с одним дополнительным ингредиентом, выбранным из группы, состоящей из:
(I) кросповидона, кроскармеллозы натрия или натрийгликолят крахмала;
(II) стеариновой кислоты или соли стеариновой кислоты и
(III) диоксида кремния или талька,
причем вторая перемешанная смесь содержит соль лактат соединения в количестве от 10 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и включает целлюлозу в количестве от 10 до 70 мас.% в пересчете на общую массу второй перемешанной смеси.
31. Способ по п.30, который дополнительно включает (г) формирование по меньшей мере одной капсулы или по меньшей мере одной таблетки из второй перемешанной смеси.
32. Способ по п.31, где общая масса соли лактата соединения формулы I, его таутомера или их смеси в капсуле или таблетке составляет от 25 до 500 мг.
33. Способ по п.30, в котором вторая перемешанная смесь содержит лактат соединения в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси.
34. Способ по п.30, в котором вторая перемешанная смесь содержит лактат соединения в количестве от 30 до 40 мас.% в пересчете на общую массу второй перемешанной смеси.
35. Способ по п.30, в котором вторая перемешанная смесь содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и в котором вторая перемешанная смесь содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу второй перемешанной смеси.
36. Способ по п.28, в котором вторая перемешанная смесь содержит целлюлозу в количестве от 20 до 50 мас.% в пересчете на общую массу второй перемешанной смеси и в котором вторая перемешанная смесь содержит крахмал или лактозу в количестве от 10 до 40 мас.% в пересчете на общую массу второй перемешанной смеси.
37. Способ по одному из пп.27-34, в котором целлюлоза представляет собой микрокристаллическую целлюлозу.
38. Способ по одному из пп.27-34, в котором вторая перемешанная смесь содержит крахмал в количестве от 20 до 40 мас.% в пересчете на общую массу второй перемешанной смеси и крахмал представляет собой частично предварительно желированный крахмал.
39. Способ получения фармацевтической композиции, включающий:
(а) смешивание первой смеси с получением первой перемешанной смеси, где первая перемешанная смесь содержит:
(I) соль лактат соединения формулы (I), его таутомер или их смесь,
и (II) по меньшей мере один ингредиент, выбранный из группы, состоящей из целлюлозы, лактозы, крахмала или их смеси, повидона, диоксида кремния или талька, фармацевтически приемлемого замасливателя и ингредиента, выбранного из кросповидона; кроскармеллозы натрия и натрийгликолят крахмала; и
(б) смешивание стеариновой кислоты, соли стеариновой кислоты или их смеси с первой перемешанной смесью с получением второй перемешанной смеси, где вторая перемешанная смесь содержит соль лактат соединения в количестве от 50 до 80 мас.% в пересчете на общую массу второй перемешанной смеси.
40. Способ по п.39, в котором вторая перемешанная смесь содержит лактат соединения в количестве от 55 до 75 мас.% в пересчете на общую массу второй перемешанной смеси.
41. Способ по одному из пп.39-40, в котором вторая перемешанная смесь дополнительно содержит диоксид кремния в количестве от 0,3 до 2 мас.% в пересчете на общую массу второй перемешанной смеси.
42. Способ по одному из пп.39-40, в котором вторая перемешанная смесь дополнительно содержит целлюлозу в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси.
43. Способ по одному из пп.39-40, в котором вторая перемешанная смесь дополнительно содержит стеарат магния в количестве от 0,1 до 2 мас.% в пересчете на общую массу второй перемешанной смеси.
44. Способ по одному из пп.39-40, в котором вторая перемешанная смесь дополнительно содержит кросповидон в количестве от 2 до 6 мас.% в пересчете на общую массу второй перемешанной смеси.
45. Способ по одному из пп.39-40, в котором вторая перемешанная смесь содержит диоксид кремния в количестве от 0,5 до 2 мас.% в пересчете на общую массу второй перемешанной смеси, целлюлозу в количестве от 20 до 45 мас.% в пересчете на общую массу второй перемешанной смеси, стеарат магния в количестве от 0,5 до 2 мас.% в пересчете на общую массу второй перемешанной смеси и кросповидон в количестве от 2 до 4 мас.% в пересчете на общую массу второй перемешанной смеси.
46. Способ по одному из пп.28-35 или 41-42, в котором фармацевтическую композицию получают с помощью по меньшей мере одного устройства, выбранного из группы, состоящей из (I) гранулятора с псевдоожиженным слоем, снабженного механизмом нижнего распыления, верхнего распыления или тангенциального распыления; (II) гранулятора с высокими сдвиговыми усилиями; (III) гранулятора с низкими сдвиговыми усилиями; (IV) роллерного уплотнителя и (V) таблеточного пресса.
47. Применение композиции по любому из пп.1, 15 или 22 при изготовлении лекарственного средства для лечения рака.
48. Применение по п.47, в котором рак выбран из группы, включающей рак предстательной железы, колоректальный рак, рак молочной железы, множественную миелому, рак поджелудочной железы, мелкоклеточный рак, острый миелогенный лейкоз, хронический миелогенный лейкоз, миелопролиферативное заболевание, немелкоклеточный рак легкого, мелкоклеточный рак легкого, хронический лимфолейкоз, саркому, меланому, лимфому, рак щитовидной железы, нейроэндокринный рак, почечно-клеточный рак, рак желудка, желудочно-кишечный стромальный рак, глиому, рак головного мозга или рак мочевого пузыря.
49. Применение композиции по любому из пп.1, 15 или 22 при изготовлении лекарственного средства для ингибирования ангиогенеза.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

