Настоящее изобретение относится к улучшенному способу получения периндоприла и его фармацевтически приемлемых солей, которые могут быть использованы для производства фармацевтических препаратов.
Периндоприл и его фармацевтически приемлемые соли, предпочтительно его трет-бутиламиновая соль, проявляют ценные фармакологические свойства при лечении сердечно-сосудистых заболеваний, нормализуя артериальное давление
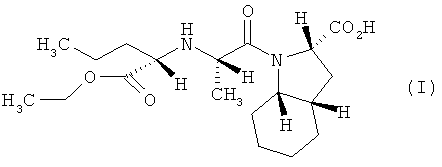
В патенте ЕР 1279665 (и его модификации ЕР 1362864, WO 03010142, WO 2005003153) описан способ получения периндоприла и его фармацевтически приемлемых солей, согласно которому получение периндоприла осуществляют путем конденсации N-[(S)-1-карбэтоксибутил]-(5)-аланина, за счет активации карбоксильной группы фосгеном или его производными, и октагидроиндол-2-карбоновой кислоты.
Недостатком способа является применение высокотоксичного фосгена и его производных, что приводит к определенным трудностям в процессе реализации в промышленности данного изобретения.
Известен способ получения периндоприла и его фармацевтически приемлемой соли периндоприл эрбумина (WO 2005/068425), заключающийся в защите атома азота и карбоксильной функции N-[(S)-1-карбэтоксибутил]-(S)-аланина триметилсилильными группами с последующей активацией карбоксильной группы путем перевода ее в соответствующий хлор- или бромангидрид. Дальнейшее взаимодействие ангидрида с защищенной (бензиловый, триметилсилиловый, трет-бутиловый эфир) октагидроиндол-2-карбоновой кислотой приводит к эфиру периндоприла. Удаление защитной группы и получение соли с трет-бутиламином дают периндоприл эрбумин.
Данный способ также имеет ряд существенных недостатков: большое количество стадий и необходимость очистки конечного продукта от кремнийорганических производных.
В патенте US 4914214 описан способ получения периндоприла и его трет-бутиламиновой соли, согласно которому N-[(S)-1-карбэтоксибутил]-(S)-аланин конденсируют с бензиловым эфиром октагидроиндол-2-карбоновой кислоты с последующим удалением защитной группы (бензильной) путем гидрирования в присутствии катализатора на основе благородного металла (палладия) на угле. Также в качестве защитной группы, помимо бензильной, могут быть использованы: моно-, ди-, триарилзамещенные (алкил, алкокси, гало, нитро) бензильные группы; дифенилметильная и трифенилметильная или их производные. В синтезе периндоприла могут быть использованы соответствующие гексагидроиндол-2-карбоновая кислота и дигидроиндол-2-карбоновая кислота (патенты WO 2004078708, WO 2004078107, ЕР 1362864, WO 2005003153, ЕР 1367062, ЕР 1256590, WO 2005023842, WO 2005066199).
Недостатком способа является использование в процессе гидрирования водорода, который обладает способностью к образованию взрывоопасной смеси с воздухом.
В патенте RU 2339645 этот способ был улучшен, устранив некоторые из перечисленных ранее недостатков, многостадийность технологического процесса и образование нежелательных примесей. При этом удаление защитной группы осуществляют в присутствии основания, которое добавляют непосредственно в реакционную смесь, получаемую в процессе удаления защитной группы, что приводит к сокращению стадий технологического процесса.
Недостатком данного способа также является использование водорода, так как удаление защитной группы включает процесс гидрирования в присутствии катализатора на основе благородного металла, предпочтительно палладия на угле.
Получаемую вышеописанными способами трет-бутиламиновую соль периндоприла используют в производстве лекарственных препаратов в виде различных кристаллических форм или в виде моногидрата. Известны следующие формы трет-бутиламиновой соли периндоприла и способы их получения, которые защищены патентами: α-кристаллическая форма (РСТ WO 01/87835), β-кристаллическая форма (РСТ WO 01/87836), γ-кристаллическая форма (РСТ WO 01/87835), моногидрат периндоприла (RU 2339645).
Основной задачей изобретения является разработка простого и безопасного способа получения периндоприла и его фармацевтически приемлемых солей, легко и быстро осуществимого в промышленных условиях.
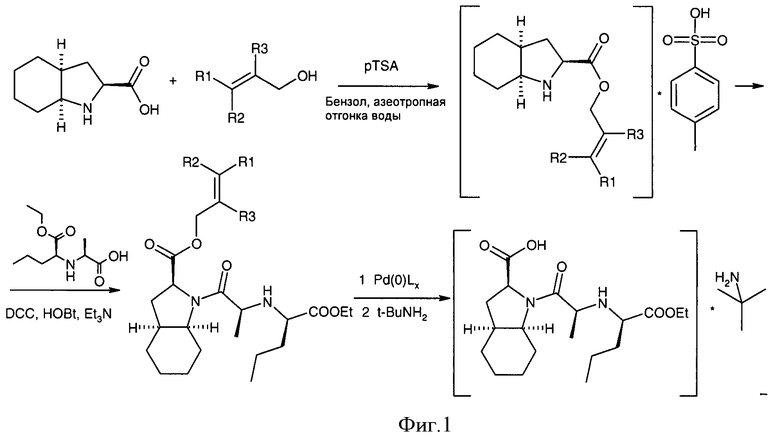
Авторы настоящего изобретения разработали экологически чистый и взрывобезопасный способ получения периндоприла и его фармацевтически приемлемых солей, позволяющий исключить использование высокотоксичных и взрывоопасных реагентов, который включает стадии этерификации (А), конденсации (Б), удаления защитной группы (В) и солеобразования (Г), при этом две последние стадии могут быть объединены в одну (фиг.1).
Изобретение относится, прежде всего, к способу получения периндоприла (I) или его фармацевтически приемлемых солей из защищенного соединения (IV)
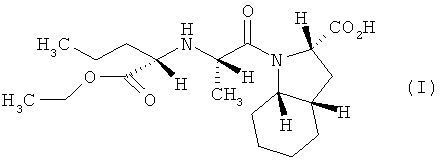
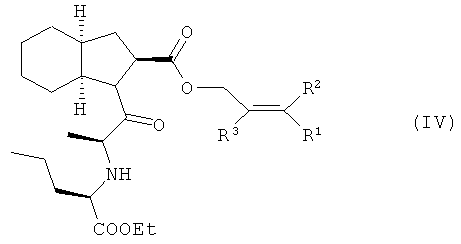
характеризующемуся тем, что удаление защитной группы из защищенного соединения (IV), в котором R1, R2 и R3 представляют собой предпочтительно водород, алкил, арил или хлор в любой комбинации, а защитная группа представляет собой алкенильную группу, осуществляют путем обработки защищенного соединения (IV) производными палладия с арилфосфинами или бифосфинами, при этом получают периндоприл (I), который, при необходимости, обрабатывают основанием, предпочтительно трет-бутиламином, и получают соль, предпочтительно трет-бутиламиновую соль периндоприла.
В качестве катализатора используют производные палладия с арилфосфином или бифосфином, при этом могут быть использованы производные палладия, как получаемые in situ, так и вносимые в реакционную смесь с нулевой степенью окисления металла, предпочтительно Рd(0)/РРh3, Рd(0)/Р(о-толлил)3, Рd(0)/(2-бифенил)дитрет-бутилфосфина, Pd(0)/BINAP и др.
Удаление защитной группы проводят в органическом растворителе, при этом могут быть использованы как протонные растворители, предпочтительно алифатические линейные или разветвленные спирты C1-C4, так и апротонные растворители, предпочтительно 1,4-диоксан, тетрагидрофуран, этилацетат, дихлорметан, хлороформ.
С целью сокращения количества стадий технологического процесса соль, предпочтительно трет-бутиламиновую соль периндоприла, можно получить непосредственно из соединения (IV), при этом удаление защитной группы осуществляют в присутствии основания, предпочтительно трет-бутиламина, непосредственно в реакционной смеси, получаемой во время процесса обработки соединения (IV) производными палладия.
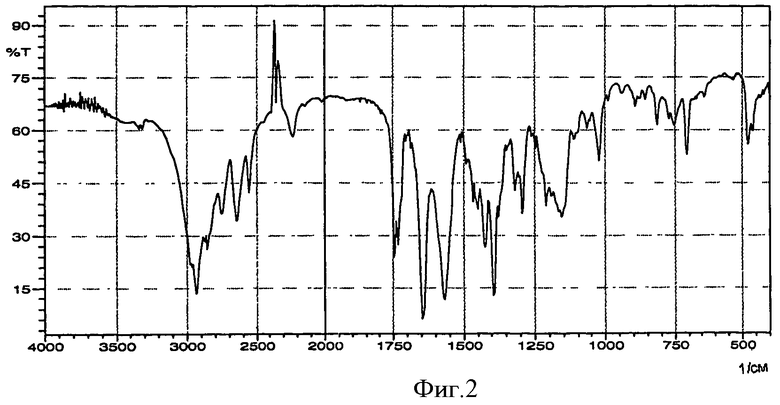
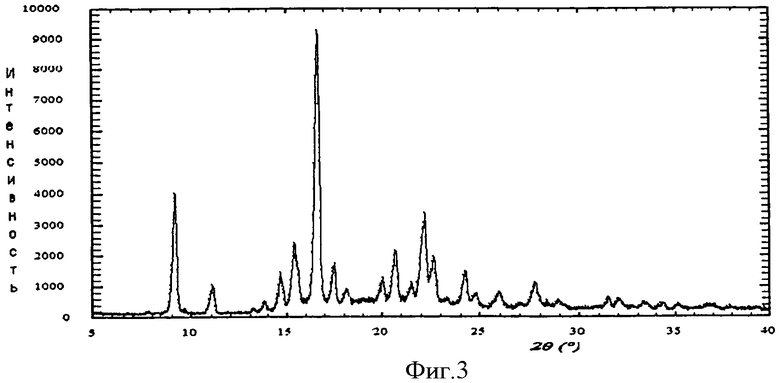
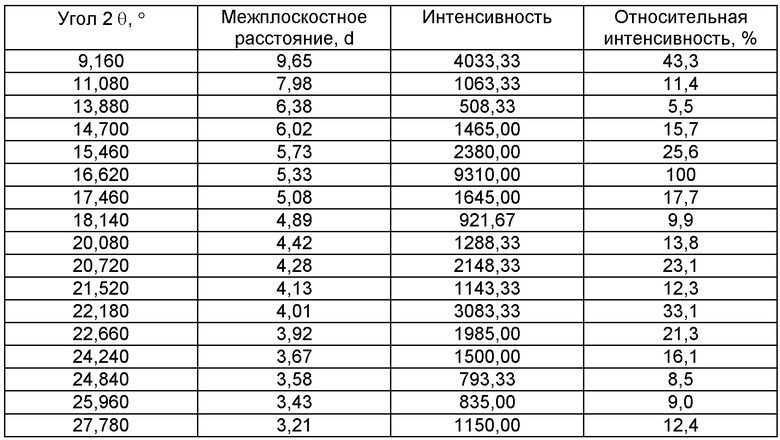
Авторы установили, что конкретную соль периндоприла - трет-бутиламиновую соль можно получать в хорошо обозначенной, отлично воспроизводимой кристаллической форме, характеризующейся ИК-спектром (фиг.2) и диаграммой рентгеновской дифракции порошка (фиг.3), выражаемой в терминах: межплоскостное расстояние d, угол 2 θ Брэгга (Bragg's), интенсивность и относительная интенсивность (выражаемая в процентах от наиболее интенсивного луча), представленных в таблице.
Спектр рентгеновской дифракции порошка измеряли при следующих условиях: дифрактометр Thermo ARL X'TRA, полупроводниковый детектор, излучение СuKα, напряжение 45 кВ, ток 35 мА, непрерывная съемка со скоростью 2°/мин, шаг 0.02°, ширина щелей трубки 2 и 4 мм, детектора - 0.5 и 0.2 мм. Данные эксперимента обрабатывались при помощи программного обеспечения WinXRD 2.0.
Данную кристаллическую форму можно получить в процессе перекристаллизации, предпочтительно, трет-бутиламиновой соли периндоприла, полученной любым известным способом, а также при перекристаллизации других кристаллических форм (например α-, β-, γ-формы) этой соли из ацетонитрила, предпочтительно из смеси ацетонитрила и этанола, при этом содержание этанола в смеси не должно превышать 30% объемных. Добавление этанола позволяет уменьшить общий объем растворителя, необходимого для перекристаллизации, при этом увеличение содержания этанола свыше 30% объемных приводит к резкому сокращению выхода целевого продукта.
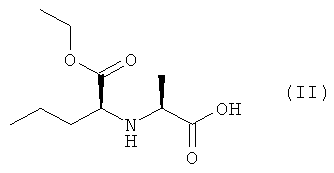
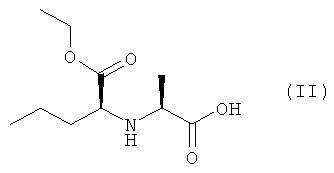
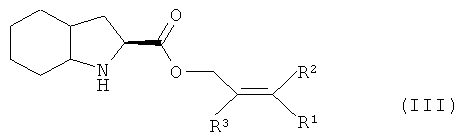
Изобретение также относится к соединению (IV), которое можно получить конденсацией кислоты (II) и эфира (III), который, в свою очередь, получают этерефикацией октагидроиндол-2-карбоновой кислоты спиртом, в котором R1, R2 и R3 представляют собой предпочтительно водород, а также алкил, арил или хлор в любой комбинации
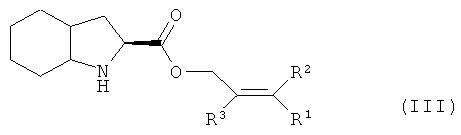
Приведенные ниже примеры иллюстрируют данное изобретение, но не ограничивают его.
Пример 1
А. Получение паратолуолсульфоната аллилового эфира (2S, 3aS, 7aS)-октагидроиндол-2-карбоновой кислоты.
4,23 г (0,025 моль) (2S, 3aS, 7aS)-пepгидpoиндoл-2-кapбoнoвoй кислоты, 16,9 мл (0,25 моль, 14,5 г) аллилового спирта и 5,71 г (0,03 моль) паратолуолсульфокислоты (в виде моногидрата) в 200 мл бензола кипятят с ловушкой Дина-Старка до прекращения отделения воды (~24 часа). Полученную смесь охлаждают и упаривают досуха, разбавляют 20 мл гексана и выпавший осадок отфильтровывают. Полученный продукт перекристаллизовывают из метилтрет-бутилового эфира. Выход - количественный. Т. пл. 110°С. Спектр ЯМР 1Н, δ, м.д. (DMSO-d6):1,23 м (1Н), 1,36 м (3Н), 1,61 м (1Н), 1,86 м (1Н), 2,01 м (1Н), 2,04 с (3Н, СН3), 2,32 м (1Н), 3,61 м (1Н), 4,50 м (1Н), 4,70 м (2Н, СH2-O), 5,27 д (1Н, J 10,8 Гц, СH2=СН), 5,36 д (1Н, J 16,8 Гц, СH2=СН), 5,93 м (1Н, СН2=СH), 7,11 д (1Н, J 7.9 Гц, Ar), 7,49 д (1Н, J 7,9 Гц, Ar), 8.51 с (1Н), 9,78 с (1Н).
Б. Получение аллилового эфира (2S, 3aS, 7аS)-1-{2-[1(этоксикарбонил)-(S)-бутиламин]-S-пропионил}-октагидроиндол-2-карбоновой кислоты.
К суспензии 2,39 г (0,011 моль) N-[(S)-1-карбэтоксибутил]-(S)-аланина и 1,485 г (0,011 моль) 1-гидроксибензтриазола в 20 мл этилацетата при комнатной температуре добавляют 4,2 мл (3,05 г, 0,03 моль) триэтиламина, 2,27 г (0,011 моль) N,N'-дициклогексилкарбодиимида и через 10 мин 3,81 г (0,01 моль) паратолуолсульфоната аллилового эфира (2S, 3aS, 7aS)-oктaгидpoиндoл-2-кapбoнoвoй кислоты. Полученную смесь перемешивают 12 часов, затем выпавший осадок мочевины отфильтровывают и фильтрат промывают содой, водой и насыщенным раствором соли. Сушат сульфатом натрия и упаривают досуха. Полученный остаток растворяют в холодном гексане и отфильтровывают от остатков мочевины, упаривают. Получают 4,01 г (98%) продукта (аллилового эфира периндоприла) в виде желтоватого масла. Продукт без дополнительной очистки используют на следующей стадии.
В. 0,511 г (1,25 ммоль) аллилового эфира периндоприла и 8 мг (0.03 ммоль) трифенилфосфина растворяют в 10 мл 1,4-диоксана, продувают азотом и добавляют 9 мг (0,0078 ммоль) тетракис(трифенилфосфин)палладия. Затем через 24 часа реакционную смесь упаривают досуха, получая технический периндоприл, который растворяют в смеси вода/циклогексан, органический слой отделяют, а водный упаривают, получая периндоприл. Выход: 285 мг (62%).
Г. 285 мг (0,77 ммоль) периндоприла растворяют в этаноле (2 мл) и добавляют трет-бутиламин (0,2 мл), полученную смесь упаривают и остаток перекристаллизовывают из ацетонитрила. Выход: 272 мг (80%).
Пример 2
0,511 г (1,25 ммоль) аллилового эфира периндоприла (полученного как описано в примере 1 стадии А-Б) и 8 мг (0,03 ммоль) трифенилфосфина растворяют в 10 мл 1,4-диоксана, продувают азотом и добавляют 9 мг (0,0078 ммоль) тетракис(трифенилфосфин)палладия. Затем прибавляют 0,26 мл (0,183 г, 2,5 ммоль) трет-бутиламина. Через 2 часа наблюдается выпадение осадка, реакционную смесь перемешивают еще 22 часа и упаривают досуха. Полученный продукт разбавляют 10 мл этилацетата, доводят до кипения, охлаждают и выпавший осадок отфильтровывают. Перекристаллизовывают из смеси ацетонитрил/этанол (9/1). Выход: 0,424 г (77%).
Пример 3
0,606 г (1,25 ммоль) 3-фенилаллилового эфира периндоприла (полученного по аналогии с примером 1 стадии А-Б) и 19 мг (0,03 ммоль) 1,1-бинафталин-2,2-диил-бис-дифенилфосфина (BINAP) растворяют в 10 мл тетрагидрофурана, продувают азотом и добавляют 5,5 мг (0,0078 ммоль) бис(трифенилфосфин)палладия дихлорид. Через 24 часа реакционную смесь упаривают досуха. Полученный продукт разбавляют 10 мл этилацетата, добавляют 0,26 мл (0,183 г, 2,5 ммоль) трет-бутиламина, доводят до кипения, охлаждают и выпавший осадок отфильтровывают. Перекристаллизовывают из смеси ацетонитрил/этанол (9/1). Выход: 0,292 г (53%).
Пример 4
528 мг (1,25 ммоль) бут-2-енилового эфира периндоприла (полученного по аналогии с примером 1 стадии А-Б) и 9 мг (0,03 ммоль) трис(о-метилфенил)фосфина растворяют в 10 мл этанола, продувают азотом и добавляют 5,5 мг (0,0078 ммоль) бис(трифенилфосфин)палладия дихлорид. Затем нагревают до кипения и выдерживают реакционную смесь при данной температуре 10 часов, охлаждают и упаривают досуха. Полученный продукт разбавляют 10 мл этилацетата, добавляют 0,26 мл (0,183 г, 2,5 ммоль) трет-бутиламина, доводят до кипения, охлаждают и выпавший осадок отфильтровывают. Перекристаллизовывают из ацетонитрила как в примере 1. Выход: 0,397 г (72%).
Пример 5
546 мг (1,25 ммоль) пент-2-енилового эфира периндоприла (полученного по аналогии с примером 1 стадии А-Б) и 9 мг (0,03 ммоль) трифенилфосфина растворяют в 10 мл изопропанола, продувают азотом и добавляют 5,5 мг (0,0078 ммоль) бис(трифенилфосфин)палладия дихлорид. Затем нагревают до 70°С и выдерживают реакционную смесь при данной температуре 10 часов, охлаждают и упаривают досуха. Полученный продукт разбавляют 10 мл этилацетата, добавляют 0,26 мл (0,183 г, 2,5 ммоль) трет-бутиламина, доводят до кипения, охлаждают и выпавший осадок отфильтровывают. Перекристаллизовывают из смеси ацетонитрил/этанол (7/1). Выход: 0,376 г (68%).
Пример 6
554 мг (1,25 ммоль) 3-хлораллилового эфира периндоприла (полученного по аналогии с примером 1 стадии А-Б) и 9 мг (0,03 ммоль) (2-бифенил)дитрет-бутилфосфина растворяют в 10 мл 1,4-диоксана, продувают азотом и добавляют 5,5 мг (0,0078 ммоль) бис(трифенилфосфин)палладия дихлорид. Затем нагревают до 70°С и выдерживают реакционную смесь при данной температуре 10 часов, охлаждают и упаривают досуха. Полученный продукт разбавляют 10 мл этилацетата, добавляют 0,26 мл (0,183 г, 2,5 ммоль) трет-бутиламина, доводят до кипения, охлаждают и выпавший осадок отфильтровывают. Перекристаллизовывают из смеси ацетонитрил/этанол (15/1). Выход: 0,353 г (64%).
Пример 7
623 мг (1,25 ммоль) 3-(4-метилфенил)аллилового эфира периндоприла (полученного по аналогии с примером 1 стадии, А-Б) и 9 мг (0,03 ммоль) (2-бифенил)дитрет-бутилфосфина растворяют в 10 мл этилацетата, продувают азотом и добавляют 5,5 мг (0,0078 ммоль) бис(трифенилфосфин)палладия дихлорид. Затем выдерживают реакционную смесь 24 часа и упаривают досуха. Полученный продукт разбавляют 10 мл этилацетата, добавляют 0,26 мл (0,183 г, 2,5 ммоль) трет-бутиламина, доводят до кипения, охлаждают и выпавший осадок отфильтровывают. Перекристаллизовывают из смеси ацетонитрил/этанол (12/1). Выход: 0,259 г (47%).
Изобретение относится к улучшенному способу получения периндоприла формулы (I) и его фармацевтически приемлемых солей из защищенного соединения
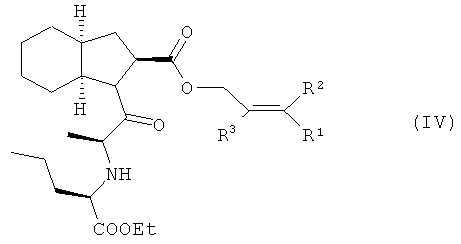
Способ заключается в том, что с защищенного соединения формулы (IV), где R1, R2 и R3 независимо означают Н, алкил, арил, Сl, снимают защитные группы путем его обработки производными палладия с арилфосфинами или бифосфинами в протонных или апротонных растворителях с получением периндоприла (I), который, при необходимости, обрабатывают основанием для получения его фармацевтически приемлемой соли, в последующем, в случае необходимости, ее перекристаллизовывают из ацетонитрила или смеси ацетонитрила с этанолом. 22 з.п. ф-лы, 1 табл., 3 ил.
1. Способ получения периндоприла (I) и его фармацевтически приемлемых солей из защищенного соединения
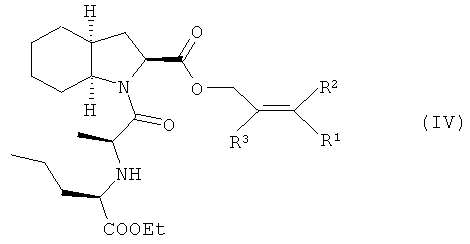
отличающийся тем, что удаление защитной группы из защищенного соединения (IV), в котором R1, R2 и R3 представляют собой водород, алкил, арил или хлор в любой комбинации, а защитная группа представляет собой алкенильную группу, осуществляют путем обработки защищенного соединения (IV) производными палладия с арилфосфинами или бифосфинами в протонных или апротонных растворителях, при этом получают периндоприл (I), который при необходимости обрабатывают основанием, и получают фармацевтически приемлемую соль периндоприла, которую при необходимости перекристаллизовывают.
2. Способ по п.1, отличающийся тем, что защищенное соединение (IV) получают конденсацией кислоты (II) с эфиром (III), который, в свою очередь, получают этерификацией октагидроиндол-2-карбоновой кислоты спиртом, в котором R1, R2 и R3 представляют собой водород, а также алкил, арил или хлор в любой комбинации
3. Способ по п.1, отличающийся тем, что производные палладия представляют собой комплекс палладия с трифенилфосфином.
4. Способ по п.1, отличающийся тем, что производные палладия представляют собой комплекс палладия с три(о-метилфенил)фосфином.
5. Способ по п.1, отличающийся тем, что производные палладия представляют собой комплекс палладия с 1,1-бинафталин-2,2-диил-бис-дифенилфосфином.
6. Способ по п.1, отличающийся тем, что производные палладия представляют собой комплекс палладия с (2-бифенил)-дитрет-бутилфосфином.
7. Способ по п.1, отличающийся тем, что R1, R2, R3 представляют собой водород.
8. Способ по п.1, отличающийся тем, что R1 представляет собой фенил, а R2, R3 - водород.
9. Способ по п.1, отличающийся тем, что R1 представляет собой 4-метилфенил, а R2, R3 - водород.
10. Способ по п.1, отличающийся тем, что R1 представляет собой метил, a R2, R3 - водород.
11. Способ по п.1, отличающийся тем, что R1 представляет собой этил, а R2, R3 - водород.
12. Способ по п.1, отличающийся тем, что R1 представляет собой хлор, а R2, R3 - водород.
13. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии удаления защитной группы используют спирты C1-C4.
14. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии удаления защитной группы используют этанол.
15. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии удаления защитной группы используют изопропанол.
16. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии удаления защитной группы используют апротонные растворители.
17. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии удаления защитной группы используют 1,4-диоксан.
18. Способ по п.1, отличающийся тем, что в качестве растворителя на стадии удаления защитной группы используют тетрагидрофуран.
19. Способ по п.1, отличающийся тем, что стадию удаления защитной группы проводят в присутствии основания в количестве большем или равном 1 экв., получая соль.
20. Способ по любому из пп.1, 19, отличающийся тем, что для получения фармацевтически приемлемой соли периндоприла в качестве основания используют трет-бутиламин и получают трет-бутиламиновую соль периндоприла.
21. Способ по п.20, отличающийся тем, что трет-бутиламиновую соль периндоприла перекристаллизовывают из ацетонитрила.
22. Способ по п.20, отличающийся тем, что трет-бутиламиновую соль периндоприла перекристаллизовывают из смеси ацетонитрила и этанола при содержании этанола не выше 30 об.%.
23. Способ по любому из пп.21, 22, отличающийся тем, что трет-бутиламиновую соль периндоприла переводят в кристаллическую форму, характеризующуюся следующей диаграммой рентгеновской дифракции порошка, измеряемой с применением дифрактометра Thermo ARL X'TRA и выражаемой в терминах: межплоскостное расстояние d, угол 2 θ Брэгга, интенсивность и относительная интенсивность.
| Кнопочный переключатель | 1986 |
|
SU1367061A1 |
| ЕР 1256590 А1, 23.07.2002 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| J | |||
| of Organic Chemistry, 74(23), 9010-9026, 2009 | |||
| ПЕРИНДОПРИЛ | 2003 |
|
RU2339645C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРИНДОПРИЛА С ИСПОЛЬЗОВАНИЕМ ТЕТРАМЕТИЛУРОНИЕВЫХ СОЛЕЙ В КАЧЕСТВЕ РЕАГЕНТОВ РЕАКЦИИ СОЧЕТАНИЯ | 2004 |
|
RU2363695C2 |

