Область изобретения
Настоящее изобретение относится к области синтетической органической химии и касается способа получения соединений, обладающих АПФ (ангиотензинпревращающий фермент) ингибиторной активностью.
В частности, настоящее изобретение относится к надежному, селективному и простому способу получения соединений, обладающих АПФ ингибиторной активностью.
Известный уровень техники
Молекулы большинства АПФ I ингибиторов химически состоят из аминокислотной и неаминокислотной частей. Конденсация этих различных частей в конечную структуру протекает при помощи, главным образом, общих реагентов, применяемых для образования пептидной связи, таких как:
фосген, дифосген или трифосген;
карбонилдиимидазол, хлортионилимидазол или тионилдиимидазол;
дихлоргексилкарбодиимид/1-гидроксибензотриазол.
Различные предшественники АПФ ингибиторов нуждаются в различных конденсирующих агентах для наиболее подходящего получения, хороших выходов и соответствующего фармацевтического качества. Например, в SI 9400290 описан синтез эналаприла с тионилимидазолом в качестве конденсирующего агента для образования пептидной связи. Эта реакция дает хороший результат, но требует очень строгого контроля над реакционными условиями и абсолютно безводной среды, иначе сопровождается побочными реакциями, например образованием дикетопиперазидов и т.п., и снижением выхода.
В другом случае, описанном в патенте US 4914214, синтез между (2S,3aS,7aS)-2-кapбoкcипepгидpoиндoлoм, где карбоксильная группа защищена бензильной группой, и N-((S)-1-карбэтокси-изо-бутил)-(S)-аланином с дициклогексилкарбодиимид/1-гидроксибензотриазолом в качестве конденсирующего агента применяется в процессе получения другого АПФ ингибитора, периндоприла. Этот процесс дает хорошие результаты, но требует тщательного выделения из-за образования дициклогексилмочевины в качестве побочного продукта, которая с трудом выделяется из реакционной смеси. В более поздней заявке ЕР 1279665 описан способ получения предшественников того же периндоприла из хлорангидридов кислот с помощью трифосгена. Этот процесс дает хорошие результаты, но требует особого внимания из-за работы с фосгеном.
Целью настоящего изобретения является получение соединений с АПФ ингибирующей активностью новым и простым способом, в котором АПФ ингибиторы получают с высоким выходом и высокой чистотой.
Детальное описание изобретения
При поиске эффективного способа получения АПФ I ингибиторов неожиданно было обнаружено, что конденсация между карбоновой кислотой и аминогруппой соответствующей аминокислоты с использованием солей урония протекает быстро, гладко и чисто. Реакция сама по себе очень проста и не требует никаких специальных условий. Очень важно, что она может очень быстро заканчиваться, например через 15 мин, по сравнению с другими процессами получения АПФ ингибиторов. Кроме того, способ по настоящему изобретению высокоселективен и дает в высокой степени чистый конечный продукт без каких-либо побочных продуктов.
Первым вариантом по настоящему изобретению является способ получения соединений с АПФ ингибирующей активностью формулы I:
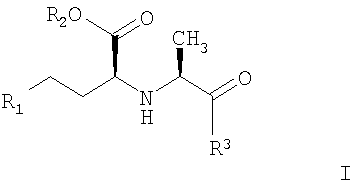
где R1 обозначает Н, алкил, фенил;
R2 обозначает Н, алкил;
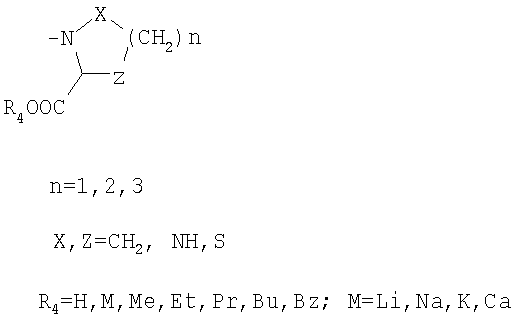
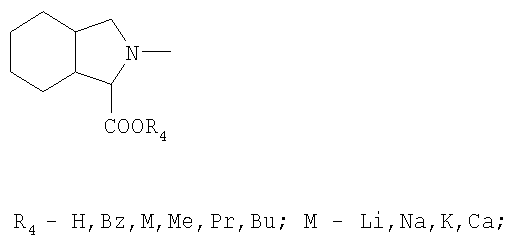
R3 обозначает
или
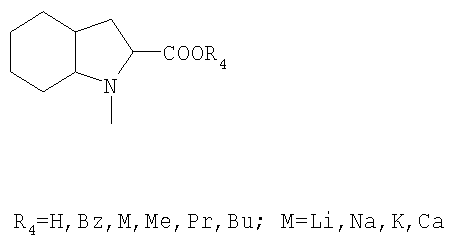
или
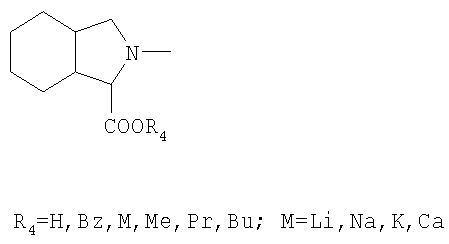
или их фармацевтически акцепторные соли,
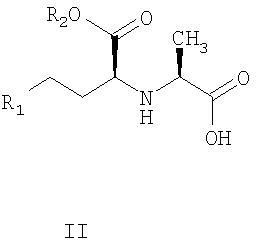
отличающиеся тем, что карбоксильную группу стереоспецифической аминокислоты II:
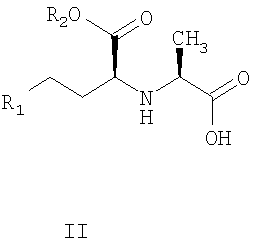
R2, R2 обозначены выше,
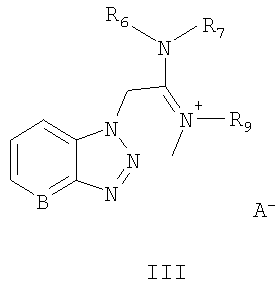
активируют солью урония формулы III:
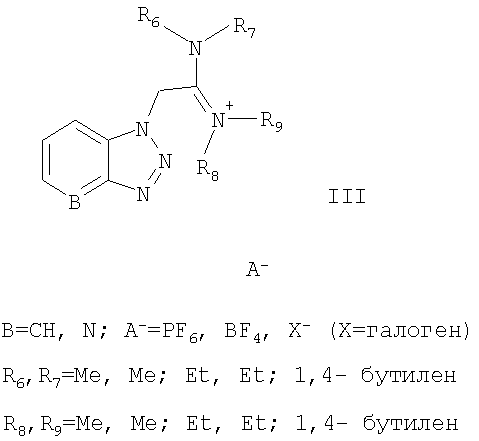
в присутствии апротонного растворителя, а затем активированная кислота превращается в пептид с помощью соответствующего амина из ряда НR3, где R3 обозначает:
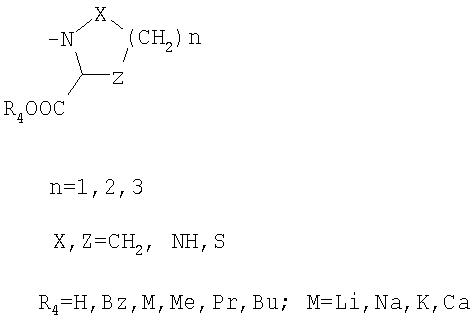
или
или
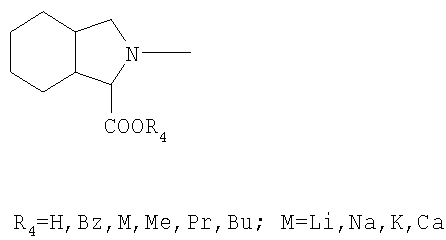
Полученный таким образом продукт может быть переведен в активную фармацевтическую форму превращениями полученных промежуточных продуктов посредством реакций удаления защитных групп, расщепления соответствующих сложных эфиров, нейтрализации, подщелачивания, подкисления и, при необходимости, получения фармацевтически приемлемой соли.
Соль урония формулы
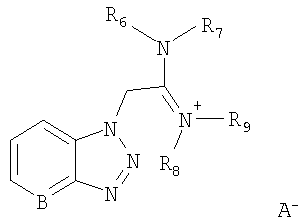
может быть выбрана из группы, состоящей из:
O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата,
O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний тетрафторфосфата,
O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата.
Названные выше реагенты известны из литературы (G.A.Grant, Synthetic Peptides, Oxford University Press, 1992, 119) как исключительно подходящие реагенты для синтеза пептидов без побочных реакций изомеризации на хиральных центрах. Эти реагенты особенно применимы для конденсации различных аминокислот, но не были еще использованы для получения АПФ ингибиторов, которые представляют собой непептидный тип молекулы.
В другом варианте по настоящему изобретению конденсация кислоты с реагентом урония может протекать при добавлении органического основания, такого как третичный амин, в от 1,5 до 3-молярном избытке по отношению к реагенту, предпочтительно в 2-молярном избытке. Третичный амин может быть выбран из группы, состоящей из триэтиламина, N,N-диизопропиламина, пиридина, лутидина.
Растворители для этого типа реакций могут быть выбраны из группы, состоящей из хлорированных углеводородов, например хлористого метилена, хлороформа, циклических или нециклических углеводородов, сложных эфиров органических кислот, например, этилацетата, амидных растворителей, например, N,N-диметилформамида, N,N-диметилацетамида, N-метилпирролидона или других апротонных растворителей, таких как ацетонитрил.
Реакция образования пептидной связи описывается следующим образом.
Кислоту и амин вводят в реакцию в стехиометрических количествах или в количественных соотношениях от 1,1:1 до 1:1,1 вместе с названными выше реагентами пептидной конденсации O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфатом или тетрафторборатом в от 1 до 1,5-молярном избытке в присутствии третичного амина в соответствующем растворителе. Реакцию проводят в течение 10-120 мин, предпочтительно от 10 до 45 мин и наиболее предпочтительно от 15 до 30 мин, при температурах от 0 до 40°С, предпочтительно при комнатной температуре, например при 20-25°С. В реакционную смесь добавляют воду, и продукт выделяют добавлением соответствующего растворителя, в котором продукт не растворяется. После промывания экстрактов водой и выпаривания растворителя продукт, имеющий хорошую химическую и оптическую чистоту, выделяют с высоким выходом (87-95%).
Реакционная схема:
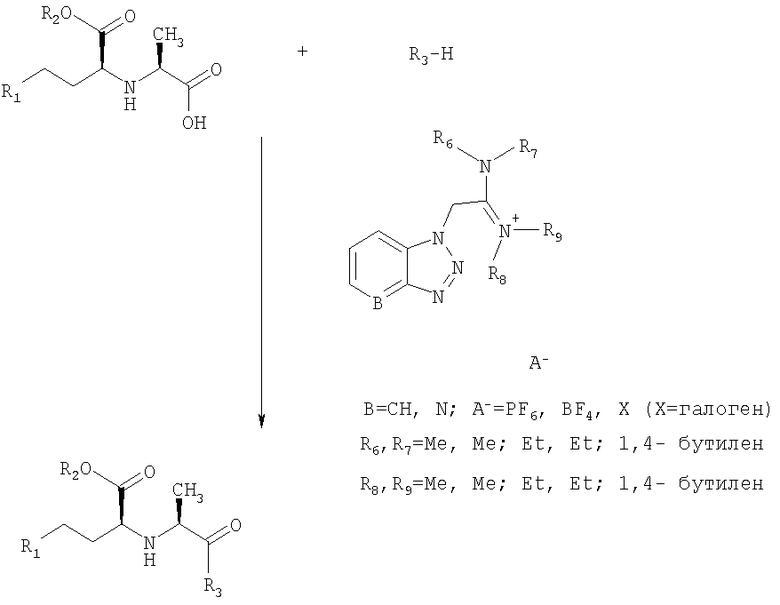
Настоящее изобретение иллюстрируется следующими примерами, которые не лимитируют объем изобретения:
Пример 1: Получение эналаприлмалеата
К смеси 2,79 г 1-((S)-N-(1-(этоксикарбонил)-3-фенилпропил)-L-аланина и 1,15 г L-пролина в 100 мл ацетонитрила и 5 мл ДМФ добавляют 2,9 г триэтиламина и при перемешивании добавляют 3,8 г O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата. Реакционную смесь перемешивают затем в течение 30 мин при комнатной температуре, добавляют 300 мл насыщенного раствора NaCl и дважды экстрагируют смесь 100 мл этилацетата. Объединенные экстракты промывают смесью 70 мл вода/1 мл конц. НСl и затем 130 мл воды, высушивают над Na2SO4 и выпаривают в вакууме при 40°С, получая 3,55 г эналаприла.
Промежуточный продукт растворяют в 100 мл этилацетата, затем добавляют 1,00 г малеиновой кислоты в 50 мл этилацетата. После перемешивания в течение 30 мин эналаприлмалеат отфильтровывают и высушивают в вакууме при 40°С, получая 4, 2 г продукта (85,4%).
Пример 2: Получение бензил (2S,3aS,7aS)-((2-(1-(этоксикабонил)-(S)-бутиламино)-(S)-пропионил)октагидроиндол-2-карбоксилата (бензиловый эфир периндоприла)
Смесь 855 мг бензилового эфира (2S,3аS,7аS)-2-карбоксипергидроиндола, 651 мг N-((S)-1-карбэтоксибутил)-(S)-аланина, 1137 мг O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 0,84 мл триэтиламина перемешивают в 20 мл ацетониторила в течение 30 мин. Добавляют 50 мл насыщенного раствора NaCl и дважды экстрагируют 35 мл этилацетата. Объединенные экстракты промывают смесью 70 мл вода/1 мл конц. НСl и затем 130 мл воды, высушивают над Na2SO4 и выпаривают в вакууме при 40°С, получая 1210 мг (87,7%) бензил (2S,3аS,7аS)-((2-(1-(этоксикабонил)-(S)-бутиламино)-(S)-пропионил)октагидроиндол-2-карбоксилата (бензиловый эфир периндоприла).
Полученный продукт является предшественником для получения периндоприла или периндоприл эрбумина, которые могут быть получены известными из литературы методами, например превращением в свободную кислоту и затем в фармацевтически приемлемую соль известными методами, описанными в патенте US 4914214.
Пример 3: Получение трандолаприла
К 2,79 г (S)-1-(N-(1-(этоксикарбонил)-3-фенилпропил)-L-аланина и 1,75 г (2S,3аR,7аS)-октагидро-1Н-индол-2-карбоновой кислоты в 100 мл ацетонитрила и 5 мл ДМФ сначала добавляют 2,9 мл триэтиламина, а затем при перемешивании 3,8 г O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и продолжают перемешивание смеси в течение 30 мин при комнатной температуре. Наконец, добавляют 300 мл насыщенного раствора NaCl и дважды экстрагируют по 100 мл этилацетата. После выделения продукта, которое осуществляют аналогично примеру 1, получают 3,96 г (92) трандолаприла.
Настоящее изобретение, относится к способу получения соединений формулы (I), обладающих АПФ ингибирующей активностью, и их фармацевтически приемлемых солей, в котором карбоксильную группу аминокислоты формулы (II) активируют солью урония формулы (III) в присутствии апротонного растворителя, и активированную аминокислоту вводят в реакцию с соответствующим амином из ряда НR3. Заместители R1-R3, R6-R9, В указаны в формуле изобретения. Предложенный способ позволяет получать целевой продукт с высоким выходом и высокой чистотой. 16 з.п. ф-лы.
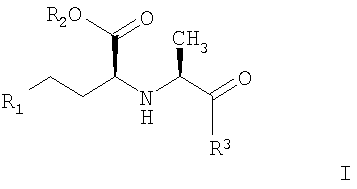
1. Способ получения соединений с АПФ ингибирующей активностью, представленных формулой I
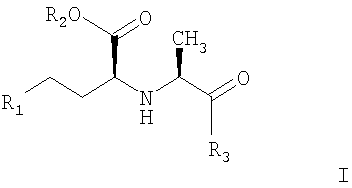
и их фармацевтически приемлемых солей, где R1-R3 обозначают
R1 - H, алкил, фенил;
R2 - Н, алкил и
R3:
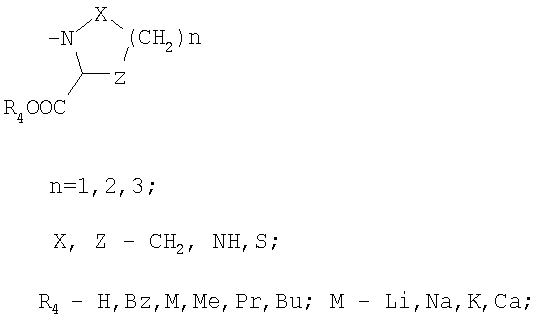
или
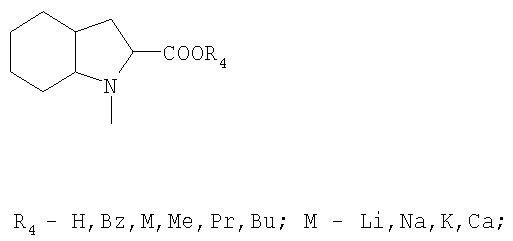
или
отличающийся тем, что карбоксильную группу аминокислоты формулы II
активируют солью урония формулы III
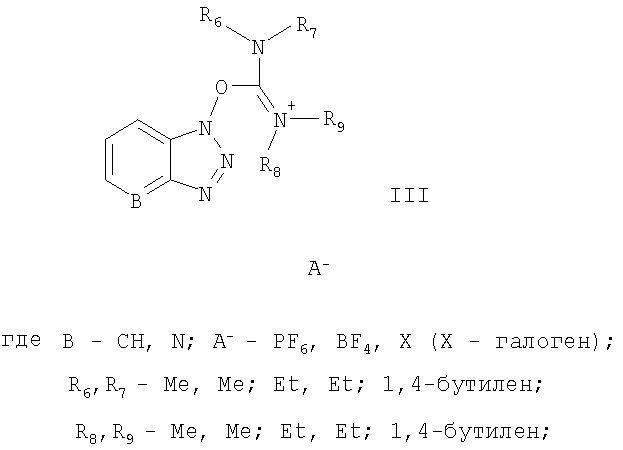
в присутствии апротонного растворителя и активированную аминокислоту вводят в реакцию с соответствующим амином из ряда НR3, где R3
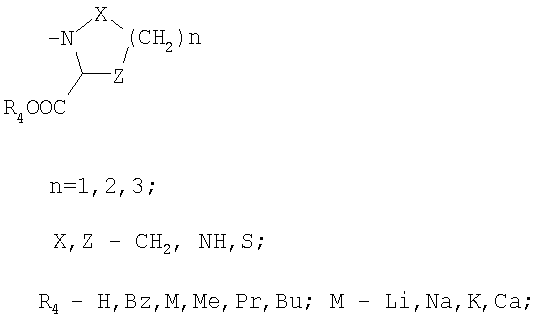
или
обозначает
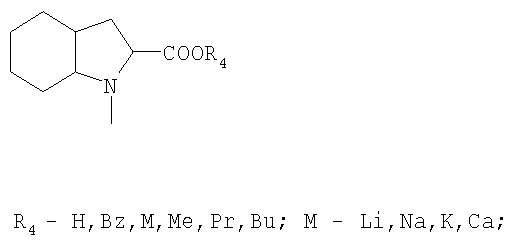
или
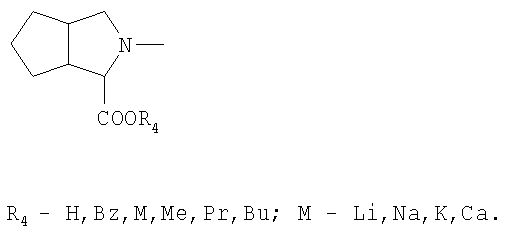
2. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют N-((S)-1-карбэтоксибутил)-(S)-аланин.
3. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют 1-((S)-N-(1-(этоксикарбонил)-3-фенилпропил)-L-аланин.
4. Способ по п.1, отличающийся тем, что в качестве амина используют L-пролин.
5. Способ по п.1, отличающийся тем, что в качестве амина используют (2S,3аR,7аS)-октагидро-1Н-индол-2-карбоновую кислоту или ее эфир.
6. Способ по п.1, отличающийся тем, что в качестве соли урония используют О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат.
7. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют 1-((S)-N-(1-(этоксикарбонил)-3-фенилпропил)-L-аланин, в качестве амина используют L-пролин, а в качестве соли урония - O-(бензотриалол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат.
8. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют N-((S)-1-карбэтоксибутил)-(S)-аланин, в качестве амина используют (2S,3аR,7аS)-октагидро-1Н-индол-2-карбоновую кислоту или ее эфир, а в качестве соли урония - O-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуроний гексафторфосфат.
9. Способ по одному из пп.1-5, отличающийся тем, что конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления органического основания.
10. Способ по одному из пп.1-5, отличающийся тем, что в качестве соли урония используют O-(бензотриазол-1-ил)-N,N,N'N'-тетраметилуроний гексафторфосфат, а конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления органического основания.
11. Способ по одному из пп.1-5, отличающийся тем, что конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления триэтиламина.
12. Способ по одному из пп.1-5, отличающийся тем, что в качестве соли урония используют O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат, а конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления триэтиламина.
13. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют 1-((S)-N-(1-(этоксикарбонил)-3-фенилпропил)-L-аланин, в качестве амина используют L-пролин, а в качестве соли урония - O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат, а конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления органического основания.
14. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют 1-((S)-N-(1-(этоксикарбонил)-3-фенилпропил)-L-аланин, в качестве амина используют L-пролин, а в качестве соли урония - O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат, а конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления триэтиламина.
15. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют N-((S)-1-карбэтоксибутил)-(S)-аланин, в качестве амина используют (2S,3аR,7аS)-октагидро-1Н-индол-2-карбоновую кислоту или ее эфир, а в качестве соли урония - O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат, а конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления органического основания.
16. Способ по п.1, отличающийся тем, что в качестве аминокислоты формулы II используют N-((S)-1-карбэтоксибутил)-(S)-аланин, в качестве амина используют (2S,3аR,7аS)-октагидро-1Н-индол-2-карбоновую кислоту или ее эфир, а в качестве соли урония - O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат, а конденсацию аминокислоты формулы II и соли урония на стадии активации аминокислоты осуществляют посредством добавления триэтиламина.
17. Способ по пп.8, 15 или 16, отличающийся тем, что количество N-((S)-1-карбэтоксибутил)-(S)-аланина и (2S,3аR,7аS)-октагидро-1Н-индол-2-карбоновой кислоты или ее эфира находится в соотношении от 1,1:1 до 1:1,1.
| DE 4334936 C1, 22.06.1995 | |||
| Устройство для измерения электрических величин | 1925 |
|
SU2968A1 |
| ПРОИЗВОДНЫЕ ДИПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, АНТИГИПЕРТОНИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2098424C1 |
| Способ получения катализатора на основе нитрата алюминия | 1985 |
|
SU1279665A1 |
| СПОСОБ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ИЗЪЯЗВЛЕНИЙ РОГОВИЦЫ | 1996 |
|
RU2119314C1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

