Изобретение относится к способу получения фармацевтически приемлемой соли периндоприла и к ее новым полиморфным формам.
Периндоприл - это международное незапатентованное название (2S,3aS,7aS)-1-{2-[1-(этоксикарбонил)-(S)-бутиламино]-(S)-пропионил}октагидроиндол-2-карбоновой кислоты. Периндоприл известен своим терапевтическим использованием в качестве ингибитора ангиотензин-преобразующего фермента (АСЕ). АСЕ представляет собой пептидил дипептитазу, которая катализирует преобразование ангиотензина I в ангиотензин II, а также вызывает разложение брадикинина. Ангиотензин II является вазоконстриктором, который также стимулирует секрецию альдостерона с помощью коркового вещества надпочечника. Таким образом, было показано, что ингибирование АСЕ может быть использовано в терапии пациентов, страдающих от болезненных состояний, таких как гипертензия и застойная сердечная недостаточность. Кроме того, было обнаружено, что ингибиторы АСЕ могут использоваться при лечении расстройств познавательной способности.
Периндоприл имеет следующую структурную формулу (I)
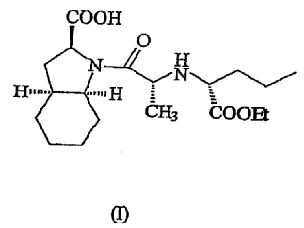
Периндоприл описан в патенте США № 4508729. Способы его получения, описанные в этом патенте США, осуществляют в спиртовой среде, в присутствии нейтрального дегидратирующего агента и органического или неорганического цианоборогидрида. Могут быть проведены, если они необходимы, процессы удаления защитных групп, например, имеется указание на гидролиз и/или гидрирование.
В патенте США № 4914214 описан способ получения периндоприла и его трет-бутиламиновой соли. Способ включает конденсацию сложного эфира (2S,3aS,7aS)-2-карбоксипергидроиндола с (S,S)-диастереомером N-[(S)-1-карбэтоксибутил]-(S)-аланина с последующим использованием активированного угля, содержащего 5% палладия, и воды. Затем добавляют трет-бутиламин для получения трет-бутиламиновой соли периндоприла.
В патентной заявке РСТ WO 01/87835 описана новая кристаллическая форма, а именно, α-кристаллическая форма, трет-бутиламиновой соли периндоприла, способ ее получения и содержащие ее фармацевтические препараты.
В патентной заявке РСТ WO 01/87836 описана новая кристаллическая форма, а именно, β-кристаллическая форма, трет-бутиламиновой соли периндоприла, способ ее получения и содержащие ее фармацевтические препараты.
В патентной заявке РСТ WO 01/87835 описана новая кристаллическая форма, а именно, γ-кристаллическая форма, трет-бутиламиновой соли периндоприла, способ ее получения и содержащие ее фармацевтические препараты.
В патентной заявке РСТ WO 01/58868 описан способ получения периндоприла или его фармацевтически приемлемой соли, который позволяет получить периндоприл или его соль с улучшенной чистотой. Более конкретно, описано, что содержание известных примесей, имеющихся в периндоприле или его соли, полученных в соответствии с патентной заявкой РСТ WO 01/58868, составляет менее 0,2 или 0,1 мас.%. Промежуточные стадии процесса проводят в присутствии 1-гидроксибензотриазола, дициклогексилкарбодиимида и, необязательно, триэтиламина, при температуре в интервале от 20 до 77°С, с последующим удалением защитных групп и, если необходимо, преобразованием солей.
Способы предшествующего уровня техники получения периндоприла или его фармацевтически приемлемой соли обычно продолжительные и часто приводят к нежелательным примесям, таким как аналоги дикетопиперазина. Поэтому существует потребность в улучшенном способе получения периндоприла или его фармацевтически приемлемых солей, которые позволять решить указанные выше проблемы.
Авторы настоящего изобретения разработали способ получения фармацевтически приемлемой соли периндоприла, который превосходит известные способы получения фармацевтически приемлемой соли периндоприла тем, что время проведения реакции сокращается, а также тем, что позволяет избежать образования нежелательных примесей и, таким образом, получить продукт высокой степени чистоты.
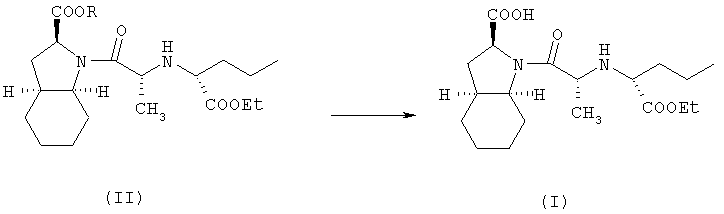
В соответствии с одним аспектом изобретения предложен способ получения фармацевтически приемлемой соли периндоприла формулы формулы (I) из защищенного соединения-предшественника формулы (II)
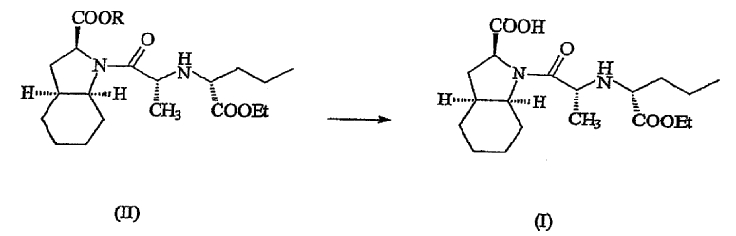
где R представляет собой карбоксизащитную группу,
заключающийся в том, что соединение формулы (II) подвергают удалению защиты с карбоксильной группы COOR, присоединенной к гетероциклическому кольцу, с получением, таким образом, соответствующей свободной кислоты, это удаление защиты осуществляют в присутствии основания, что приводит к получению фармацевтически приемлемой соли указанной свободной кислоты, образующейся при таком удалении защиты.
Обычно, R может представлять собой любую подходящую карбоксизащитную группу, которая может быть избирательно удалена способом в соответствии с настоящим изобретением. Предпочтительно, R может представлять собой необязательно замещенный аралкил, в частности, необязательно замещенный бензил. Следовательно, обычно R может представлять собой незамещенный бензил; альтернативно, может быть использован замещенный бензил, такой как 4-галогензамещенный или 4-C1-4алкоксизамещенный бензил, в частности 4-Cl бензил или 4-метокси бензил.
Удобным является, когда удаление защиты, используемое в способе по настоящему изобретению, может включать гидрирование в присутствии катализатора на основе благородного металла, предпочтительно, палладия на угле.
Способ по настоящему изобретению имеет преимущество в том, что позволяет получить продукт высокой степени чистоты. Фармацевтически приемлемая соль периндоприла, полученная способом в соответствии с настоящим изобретением, имеет степень чистоты более 99% масс./масс., и, более предпочтительно, более 99,5% масс./масс. Чистота фармацевтически приемлемой соли периндоприла, полученная способом в соответствии с настоящим изобретением, может быть и далее повышена с помощью необязательной стадии кристаллизации в подходящем растворителе, таком как этилацетат, изопропанол или тому подобное, что позволяет получить фармацевтически приемлемую соль периндоприла, которая предпочтительно имеет степень чистоты около 99,8% масс./масс.
Предпочтительно, основание, используемое в способе по настоящему изобретению, выбирают таким образом, чтобы оно приводило к образованию фармацевтически приемлемой соли свободной кислоты, образующейся при удалении защиты, как указано выше, поскольку можно получать фармацевтически приемлемую соль периндоприла непосредственно в реакционной смеси в процессе обработки. В частном предпочтительном варианте осуществления в соответствии с настоящим изобретением можно получить трет-бутиламиновую соль периндоприла высокой степени чистоты непосредственно в реакционном процессе.
В соответствии с вышеуказанным предпочтительным вариантом осуществления предложен способ получения периндоприла трет-бутиламина (который хорошо известен специалистам в данной области как периндоприл эрбумин) из защищенного соединения-предшественника формулы (II), как по существу описано выше (предпочтительно, бензилзащищенное соединение-предшественник формулы (II), где R представляет собой бензил), который заключается в том, что соединение формулы (II) подвергают удалению защиты (предпочтительно, гидрированием в присутствии катализатора на основе благородного металла, такого как палладий на угле) с карбоксильной группы COOR, присоединенной к гетероциклическому кольцу, с получением таким образом соответствующей свободной кислоты, это удаление защиты осуществляют в присутствии трет-бутиламина, что приводит к получению трет-бутиламиновой соли периндоприла.
Удобно, когда соединение-предшественник формулы (II) сначала растворяют в спиртовом растворителе, таком как изопропанол или тому подобное, и затем добавляют к нему основание. Далее подвергают удалению защиты с карбоксильной группы COOR, предпочтительно, добавлением палладия-на-угле и гидрированием в течение нескольких часов. Спиртовой растворитель предпочтительно концентрируют в вакууме и остаток помещают в несмешивающийся с водой растворитель, такой как этилацетат или тому подобное. Образовавшийся твердый осадок может быть охлажден и отфильтрован с получением фармацевтически приемлемой соли периндоприла.
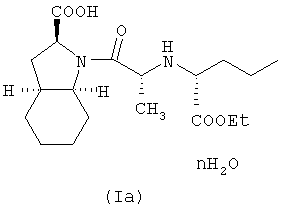
Способ по настоящему изобретению, по существу описанный выше, может далее включать гидратирование фармацевтически приемлемой соли периндоприла, полученной по данному способу, с получением, таким образом, фармацевтически приемлемой соли гидратированного периндоприла формулы (Ia)
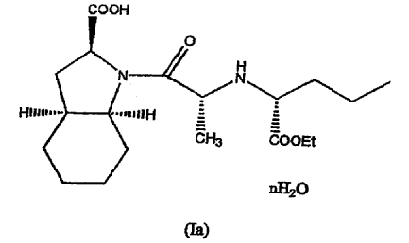
где n равно целому числу от 1 до 5, или обратному целому числу от 2 до 5. Гидратирование может быть осуществлено путем добавления воды или при высушивании на воздухе.
Предпочтительно, n равно 1, поэтому по способу в соответствии с настоящим изобретением образуется моногидрат фармацевтически приемлемой соли периндоприла.
Настоящее изобретение относится также к способу получения моногидрата фармацевтически приемлемой соли периндоприла, который включает гидратирование фармацевтически приемлемой соли периндоприла с получением, таким образом, указанного моногидрата. Гидратирование может быть осуществлено путем добавления воды или при высушивании на воздухе, и, предпочтительно, периндоприл трет-бутиламин гидратируют с получением моногидрата периндоприл трет-бутиламина.
Настоящее изобретение относится далее к фармацевтически приемлемой соли периндоприла, необязательно, в гидратированном виде, полученная по способу, по существу описанному выше. В частности, предложена фармацевтически приемлемая соль гидратированного периндоприла формулы (Ia)
где n равно целому числу от 1 до 5, или обратному целому числу от 2 до 5. Предпочтительно, n равно 1. Предпочтительной фармацевтически приемлемой солью гидратированного периндоприла формулы (Ia) является трет-бутиламиновая соль. В частном случае предпочтительного осуществления настоящее изобретение относится к моногидрату периндоприл трет-бутиламина (или эрбумина).
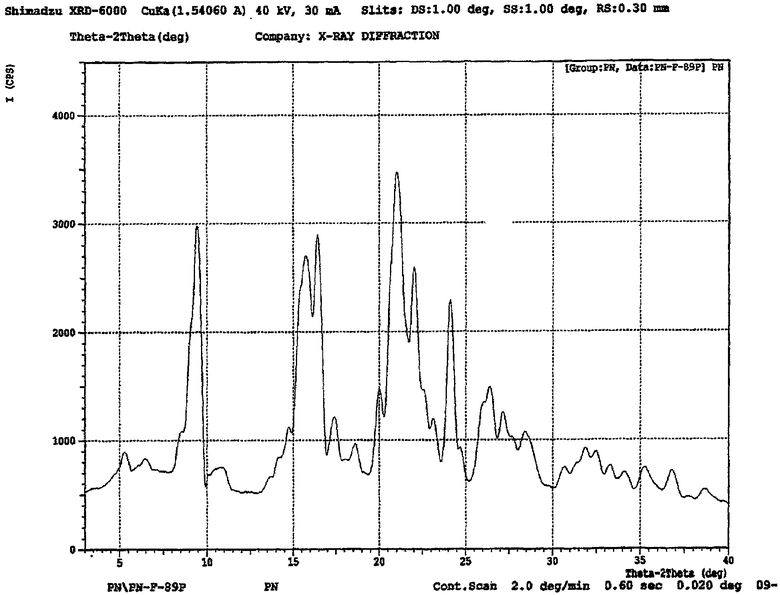
Настоящее изобретение относится также к моногидрату периндоприла трет-бутиламина, имеющему рентгеновскую дифрактограмму или по существу такую же рентгеновскую дифрактограмму, как показано на чертеже. Более предпочтительно, периндоприл трет-бутиламин моногидрат в соответствии с настоящим изобретением может быть охарактеризован пиками рентгеновской порошковой рентгенограммы (2 θ): 9,5504, 14,8600, 15,7486, 16,5400, 20,0400, 21,0499, 22,0600, 24,1744, 26,3300 и 27,1600.
Другие характеристические данные для моногидрата периндоприл трет-бутиламина по настоящему изобретению, полученные при рентгенографии показаны в следующей таблице I.
(град)
(A)
(град)
I (рассчит.)
(рассчит.)
Периндоприл, предложенный по настоящему изобретению, может быть использован в терапии в качестве ингибитора АСЕ.
Кроме того, настоящее изобретение относится к способу ингибирования АСЕ у пациента, при необходимости такого ингибирования, включающему введение указанному пациенту эффективного количества фармацевтически приемлемой соли периндоприла для ингибирования (предпочтительно перинодоприла трет-бутиламин моногидрата) по настоящему изобретению.
Настоящее изобретение также относится к применению периндоприла по настоящему изобретению (предпочтительно перинодоприла трет-бутиламин моногидрата) для производства лекарственного средства для ингибирования АСЕ.
Ингибирование АСЕ для лечения пациента необходимо тогда, когда, например, пациент страдает гипертонией, хронической застойной сердечной недостаточностью или тому подобным. Ингибирование АСЕ снижает уровни ангиотензина II и, таким образом, ингибирует эффекты, вызывающие спазм сосудов, гипертензию и гиперальдостеронию. Ингибирование АСЕ также может повышать эндогенные уровни брадикинина. Эффективным количеством приндоприла для ингибирования АСЕ по настоящему изобретению является количество, которое эффективно для ингибирования АСЕ у пациента, при необходимости такого ингибирования, что приводит, например, к гипотензивному эффекту.
При эффективном лечении пациента, периндоприл по настоящему изобретению может вводиться в любой форме, которая позволяет соединению быть биоактивным в эффективных количествах, включая пероральные и парентеральные пути. Например, периндоприл по настоящему изобретению может быть введен перорально, подкожно, внутримышечно, внутривенно, чрескожно, интраназально, ректально и тому подобное. Как правило, предпочтительным является пероральное введение. Специалист в области изготовления лекарственных препаратов может легко выбрать подходящую форму и способ введения в зависимости от тяжести заболевания, на которое направлено лечение, и стадии заболевания.
Периндоприл по настоящему изобретению может быть введен в форме фармацевтических композиций или лекарственных препаратов, которые получают объединением периндоприла по настоящему изобретению с фармацевтически приемлемыми носителями, разбавителями или эксципиентами, количественное соотношение и природа которого определяется выбранным подходящим путем введения и стандартной фармацевтической методикой.
В еще одном варианте осуществления, настоящее изобретение относится к фармацевтическим композициям, содержащим эффективное количество периндоприла для ингибирования ACE по настоящему изобретению (предпочтительно моногидрата периндоприл трет-бутиламина) вместе с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Под «фармацевтически приемлемым» понимают, что носитель, разбавитель и эксципиент должны быть совместимы с периндоприлом по настоящему изобретению, и не оказывать отрицательное действие на пациента.
Фармацевтические композиции или лекарственные средства получают способом, хорошо известным в фармацевтической области. Носитель, разбавитель или эксципиент может быть твердым, полутвердым или жидким веществом, которое может служить в качестве переносчика или среды для активного ингредиента. Подходящие носители, разбавители или эксципиенты хорошо известны в данной области. Фармацевтические композиции по настоящему изобретению могут применяться перорально или парентерально и могут вводиться пациенту в форме таблеток, капсул, суппозиториев, растворов, суспензий и тому подобного.
Фармацевтические композиции могут вводиться перорально, например, с инертным растворителем или с пищевым носителем. Они могут быть включены в желатиновые капсулы или спрессованы в таблетки. При использовании для перорального терапевтического введения, моногидрат по настоящему изобретению может быть объединен с эксципиентами и использоваться в форме таблеток, капсул, эликсиров, суспензий, сиропов и тому подобного.
Таблетки, пилюли, капсулы и тому подобное могут также содержать один или несколько нижеследующих адъювантов: связывающие агенты, такие как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиенты, такие как крахмал или лактоза; дезинтигрирующие агенты, такие как альгиновая кислота, кукурузный крахмал и тому подобное; смазывающие агенты, такие как стеарат магния; агенты, придающие скольжение, такие как коллоидный диоксид силикона; и подсластители, такие как сахароза или сахарин. Если лекарственной формой является капсула, то она может содержать, кроме веществ, указанных выше, жидкий носитель, такой как полиэтиленгликоль или жирная кислота. Другая лекарственная форма может содержать другие вещества, которые модифицируют физическую форму лекарственной формы, например, агенты покрытия. Таким образом, таблетки или пилюли могут быть покрыты сахаром, шеллаком или другим энтеросолюбильным агентом покрытия. Сироп может содержать, кроме активного ингредиента, сахарозу, в качестве подсластителя и некоторые консерванты. Вещества, используемые для получения таких разнообразных композиций, должны быть фармацевтически чистыми и нетоксичными в используемом количестве.
Для парентерального введения периндоприл по настоящему изобретению может быть получен в виде раствора или суспензии. Растворы или суспензии также могут содержать один или несколько дополнительных адъювантов: стерильные разбавители, такие как вода для инъекции, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфат натрия; хелатирующие агенты, такие как этилен диаминтетрауксусная кислота; и буферы, такие как ацетаты, цитраты или фосфаты. Парентеральные препараты могут быть заключены в ампулы, одноразовые шприцы или пузырьки для многократного приема, сделанные из стекла или пластика.
Настоящее изобретение далее будет проиллюстрировано нижеследующими примерами и чертежом, которые никоим образом не ограничивают область настоящего изобретения.
На чертеже представлена дифракционная рентгенограмма моногидрата периндоприл эрбумина по настоящему изобретению. Образец анализировали с помощью рентгеновского диффрактометра Shimadzu-6000. Используемым источником была Kα монохроматическая радиация Cu с длиной волны 1,5406 Å. Значение использующейся выходной щели составляло 1°. Значение входной щели составляло 0,30 мм. В качестве детектора использовали сцинциляционный счетчик с диапазоном от 3° до 40° (2θ) со скоростью сканирования 2° в минуту.
Пример 1
Бензиловый эфир (2S,3aS,7aS)-1-{2-[1-(этоксикарбонил)-(S)-бутиламино]-(S)-пропионил}октагидроиндол-2-карбоновой кислоты, называемый бензил периндоприл, (10 г) растворяли в изопропаноле (100 мл). К прозрачному раствору добавляли трет-бутиламин (2,5 г) и 10% мас./мас. палладия на угле (2 г). Реакционную смесь гидрировали при давлении 1 кг/см2 в течение 2 часов.
Реакционную смесь фильтровали и удаляли катализатор. Растворитель концентрировали в вакууме и изопропанол заменяли, постепенно добавляя этилацетат. Полученные твердые продукты охлаждали до 0°C и фильтровали с получением периндоприла эрбумина (7,8 г).
Пример 2
Периндоприл эрбумин (10 г) суспенидровали в ацетоне (80 мл). К этому составу добавляли воду (0,4 мл) и нагревали до растворения твердых продуктов и охлаждали до комнатной температуры. Полученный твердый продукт фильтровали с получением моногидрата периндоприл эрбумина (9,4 г).
Пример 3
Периндоприл эрбумин (20 г) суспендировали в этилацетате (300 мл). К этому составу добавляли воду (1,5 мл) и нагревали до растворения твердых продуктов и охлаждали до 10°C. Полученную взвесь фильтровали с получением моногидрата периндоприл эрбумина (17 г).
Пример 4
Периндоприл эрбумин (5 г) суспендировали в ацетонитриле (75 мл). К этому составу добавляли воду (0,4 мл) и нагревали до растворения твердых продуктов и охлаждали до 0°C. Полученную взвесь фильтровали с получением моногидрата периндоприл эрбумина (2,9 г).
Пример 5
Периндоприл эрбумин (20 г) суспендировали в этилацетате (300 мл). Состав нагревали до растворения твердых продуктов и охлаждали до 10°C. Полученную взвесь фильтровали и сушили на воздухе с относительной влажностью по меньшей мере 75% с получением моногидрата периндоприл эрбумина (17 г).
Пример 6
Получение моногидрата периндоприл эрбумина
Исходные продукты:
1. Безводный периндоприл эрбумин = 10 г.
2. Изопропиловый спирт = 70 мл.
3. Вода = 2 мл.
4. Этилацетат = 85 мл.
Процедура:
1. В круглодонную колбу помещали 10 г периндоприла эрбумина (безводного). Добавляли 70 мл изопропилового спирта. Перемешивали в течение 1/2 часа (растворялось приблизительно 95% продукта).
2. Добавляли 2 мл воды. Перемешивали в течение 15 минут (получался прозрачный раствор).
3. Перемешивали реакционную смесь при 38-40°C в течение 2 часов.
4. Полностью отгоняли изопропиловый спирт под вакуумом (ниже 600 мм) при температуре ниже 40°C (наблюдали образование гелеобразного вещества).
5. Добавляли 30 мл этилацетата. Перемешивали в течение 15 минут ниже 40°C (наблюдали образование прозрачного раствора). Отгоняли под вакуумом при температуре ниже 40°C (наблюдали образование полутвердого продукта).
6. Добавляли 40 мл этилацетата при 36-38°C. Перемешивали в течение 15 минут. (Наблюдали образование свободных кристаллов).
7. Перемешивали при комнатной температуре (25-30°C). (Наблюдали образование свободных кристаллов).
8. Охлаждали до 10°С. Перемешивали в течение 2 часов.
9. Твердый продукт отфильтровывали и промывали 15 мл этилацетата. Отсасывали досуха в течение 2 часов.
10. Сушили под вакуумом при температуре ниже 40°C в течение 12 часов.
Содержание воды = 3,2-3,8%.
Т.пл. = 145-150°C.
Пример 7
Получали нижеследующие таблетки:
(a) Композиция I:
Способ: Вышеуказанные ингредиенты просеивали через соответствующие сита. Смешивали ингредиенты в подходящем блендере. Прессовали в таблетки в подходящем аппарате.
(b) Композиция II:
Способ:
1. Моногидрат периндоприл эрбумин растворяли в этаноле.
2. Гранулировали вышеуказанные ингредиенты, кроме гидрированного касторового масла, с вышеуказанным раствором. Гранулы сушили и калибровали.
3. Увлажняли гидрированным касторовым маслом в подходящем блендере. Прессовали гранулы в подходящем аппарате.
Изобретение относится к способу получения фармацевтически приемлемой соли периндоприла формулы (I), которую получают из защищенного соединения-предшественника формулы (II), где R представляет собой карбоксизащитную группу, и заключается в том, что соединение формулы (II) подвергают удалению защиты с карбоксильной группы COOR, присоединенной к гетероциклическому кольцу, с получением, таким образом, соответствующей свободной кислоты, это удаление защиты осуществляют в присутствии основания, которое образует фармацевтически приемлемую соль с указанной свободной кислотой, и, при необходимости, с последующим гидратированием фармацевтически приемлемой соли периндоприла. 3 н. и 12 з.п. ф-лы, 1 ил., 1 табл.
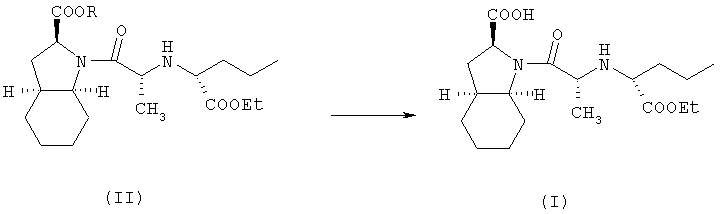
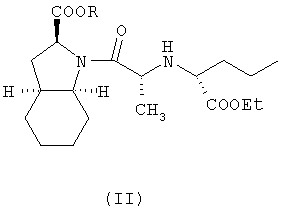
где R представляет собой карбоксизащитную группу,
заключающийся в том, что соединение формулы (II) подвергают удалению защиты с карбоксильной группы COOR, присоединенной к гетероциклическому кольцу, с получением, таким образом, соответствующей свободной кислоты, это удаление защиты осуществляют в присутствии основания, которое образует фармацевтически приемлемую соль с указанной образующейся свободной кислотой, и, при необходимости, с последующим гидратированием фармацевтически приемлемой соли периндоприла.
где R представляет собой карбоксизащитную группу,
заключающийся в том, что соединение формулы (II) подвергают удалению защиты с карбоксильной группы COOR, присоединенной к гетероциклическому кольцу, с получением таким образом соответствующей свободной кислоты, это удаление защиты осуществляют в присутствии трет-бутиламина, что приводит к получению соли трет-бутиламина периндоприла.
где n равно целому числу от 1 до 5 или обратному целому числу от 2 до 5.
| Способ получения замещенных аминодикислот,их рацематов или оптических изомеров,или их фармацевтически приемлемых солей | 1981 |
|
SU1153827A3 |
| US 4914214 А, 03.04.1990. | |||

