Область техники
Настоящее изобретение относится к промежуточным соединениям, которые полезны при получении антибактериальных соединений, в частности антибиотика левофлоксацина, обладающего высокой антимикробной активностью; и способам их получения.
Предшествующий уровень техники
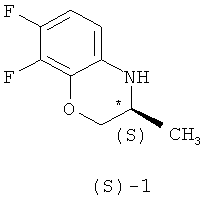
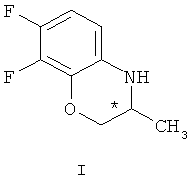
Известные методы получения (S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина ((S)-1) можно разделить на две основные группы.
Первая группа методов включает методы асимметрического синтеза, основанные на 1) асимметрическом восстановлении иминов под действием хиральных восстанавливающих агентов [ЕР 273399. Preparation of optically active 2,3-dihydrobenzoxazine derivatives as intermediates for ofloxacin and related antibacterials / Hayakawa J., Atarashi S. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. 06.07.88 JP 86/314095, 25.12.86. 12 pp.; Atarashi S., Tsurumi H., Fujiwara Т., Hayakawa J. Asymmetric reduction of 7,8-difluoro-3-methyl-2H[l,4]benzoxazine. Synthesis of a key intermediate of (S)-(-)-ofloxacin (DR-3355) // J. Heterocyclic Chem. - 1991. - V.28. - P.329-331] и хиральных катализаторов на основе комплексов Ir(I) [JP 05246995. Preparation of optically active benzo-fused cyclic amine / Yukimoto J., Kanai K., hianaga M., Achinami K. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. 02.03.92 JP 92-44308, 24.09.93. 6 pp.; Satoh К., Inenaga M., Kanai K. Synthesis of a key intermediate of Levofloxacin via enantioselective hydrogenation catalyzed by iridium(I) complexes // Tetrahedron: Asymmetry. - 1998. - V.9(15). - P.2657-2662; PCT Int. Appl. (2009) WO 2009005024. Method for producing optically active amines by asymmetric reduction of imines / Maeda S. (Hamari Chemicals, Ltd., Japan) // Appl. 30.06.2008 JP 61809]; 2) асимметрическом синтезе с использованием хиральных синтонов - диолов [ЕР 322815. Propoxybenzene derivatives, their preparation and use in the preparation of benzoxazine derivatives, especially antibacterials such as ofloxacin / Fujiwara Т., Ebata T. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. 23.12.88 JP 87-336489, 22 pp.; EP 368410. Optically active benzoxazines and benzothiazines and a process for their stereospecific preparation / Van Zoest W.J., Marx A.F., Koger H.S., Booy J. (Gist-Brocades N.V., Neth.) // 16.05.90 EP Appl. 88/202484, 07.11.88; 56 pp.; JP 08269024. Preparation of benzoxazine derivative as intermediate for bactericides / (Korea institute of Science and Technology, S. Korea) // 15.10.96 Appl. JP 96-54702, 12.03.96; 5 pp.; Kang S.B., Ahn E.J., Kim Y., Kim Y.H. A facile synthesis of (S)-(-)-7,8-difluoro-3,4-dihydro-3-methyl-2H-1,4-benzoxazme by zinc chloride assisted Mitsunobu cyclization reaction // Tetrahedron Lett. - 1996. - V.37(52). - P.9317-9320], производных аланинола [Bower J.F., Szeto P., Gallagher T. Enantiopure 1,4-benzoxazines via 1,2-cyclic sulfamidates. Synthesis of Levofloxacin // Org. Lett. - 2007. - V.9(17). - P.3283-3286; Parai M.K., Panda G. A convenient synthesis of chiral amino acid derived 3,4-dihydro-2H-benzo[b][1,4]thiazines and antibiotic Levofloxacin // Tetrahedron Lett. - 2009. - V.50(33). - P.4703-4705] и молочной кислоты [Патент РФ 2258069. Способ получения производного бензоксазина, способы получения его промежуточного соединения и промежуточные соединения / Сато К., Такаянаги Ё., Окано К., Накаяма К., Имура А., Итох М., Яги Ц., Кобаяси Ю., Нагаи Т. (ДАЙИТИ ФАРМАСЬЮТИКАЛ КО., ЛТД., Japan) // Заявл. 2002105997/04, 07.09.2000; 121 с. (БИ: 36/2007)].
Все эти методы многостадийны, требуют труднодоступного исходного сырья, не обеспечивают высокий выход целевого продукта и, кроме того, не дают возможности получения бензоксазина (-S)-1 достаточно высокой степени оптической чистоты. Другую группу методов составляют методы разделения рацемического бензоксазина формулы (I) и его производных.
Описаны методы микробиологического и ферментативного разделения в результате гидролиза амидов рацемического бензоксазина [Sakano К., Yokohama S., Hayakawa I., Atarashi S., Kadoya S. Optical resolution of (R,S)-3-(acetoxymethyl)-7,8-difluoro-2,3-dihydro-4H-[1,4]benzoxazine // Agricult. Biol. Chem. - 1987. - V.51(5). - P.1265-1270; JP 02031695. Optical separation of (-)-3-alkyl-3,4-dihydro-2H-[1,4]-benzooxazines with bacteria / Kuroda H., Miyadera A.. Imura A. (Daiichi Seiyaku Co.. Ltd., Japan) // Appl. JP 88-179616, 19880719; 4 pp.; Miyadera A., Imura A. Enantioselective synthesis of a key intermediate of Levofloxacin using microbial resolution // Tetrahedron: Asymmetry. - 1999. - V.10(1). - P.119-123; JP 2003180390. Manufacture of optically-active phenoxypropanol derivatives and preparation of levofloxacin from the derivatives / Sakamoto K., Tsusaki K. (Daiichi Fine Chemical Co., Ltd., Japan) // Appl.: JP 2002-298544 20021011, 13 pp.].
В патенте Японии [JP 01261380. Resolution of 7,8-dihalo-3-alkyl-3,4-dihydro-2H-[1,4]-benzoxazines by HPLC / Kuroda H., Miyadera A., Tojo Y. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. JP 88-90952, 19880413; 4 pp.] описано препаративное хроматографическое разделение рацемического бензоксазина (I) методом ВЭЖХ на хиральной неподвижной фазе Chiralcel OD, в результате которого получают (S)-1 с энантиомерным избытком 99,8%.
Известен также способ оптического разделения соответствующего рацемического соединения (I) путем образования соли с (R)-(-)-камфор-10-сульфокислотой, отделения менее растворимой соли бензоксазина (S)-1, с последующим выделением целевого продукта (S)-1 из соли [ЕР 304684. Process for preparation of benzoxazine derivatives / Fujiwara Т., Tsurumi H., Sato Y. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. 01.03.89 JP Appl. 87/194017, 03.08.87; 14 pp.]. Недостатком указанного способа является весьма низкая (49,2%) оптическая чистота получаемого (S)-1.
Одними из методов получения стереоизомера (S)-1 являются методы, основанные на оптическом разделении соответствующего рацемического соединения (соединение (I)) путем образования диастереомерных амидов с производными оптически активных кислот. Так, например, известен способ разделения соответствующего рацемического соединения (I) путем образования диастереомерных амидов с оптически активными производными циклических аминокислот, отделением амида, производного (S)-1, методами кристаллизации и/или жидкостной хроматографии на силикагеле и щелочного гидролиза последнего [ЕР 0206283. Optically active pyridobenzoxazine derivatives and intermediates thereof / Hayakawa J., Atarashi S., Yokohama S. et al. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. 86108442.4. 20.06.86; 57 pp.]. Недостатком этого метода является недостаточно высокий выход целевого соединения и его сложность, связанная с необходимостью использовать хроматографию для получения целевого соединения (S)-1 достаточно высокой степени оптической чистоты.
Другой способ получения бензоксазина (S)-1 основан на оптическом расщеплении рацемата с помощью N-(сульфонилзамещенного)-(R)-пролина [JP 01175975. Preparation of (S)-3-alkyl-3,4-dihydro-4H[1,4]benzoxazine derivatives by optical resolution with N-(substituted-sulfonyl)-(R)-proline / Fujiwara Т., Yokota T. (Daiichi Seiyaku Co., Ltd., Japan) // Appl. JP 87/333340, 29.12.87; 6 pp.]. Сущность метода состоит во взаимодействии оптически активного (R)-пролина (2) с арилсульфохлоридом и последующей обработке тионилхлоридом с образованием хлорангидрида Н-(сульфонил-замещенного)-(R)-пролина (3), реакции соединения (3) с рацемическим соединением (I) в дихлорэтане в присутствии пиридина с последующим выделением из смеси диастереомерных амидов (4) производного Н-(R)-пролинил-(S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина (5) фракционной кристаллизацией, его щелочным гидролизом и выделением целевого продукта ((S)-1) (Схема 1). Оптическая чистота (энантиомерный избыток, э.и.) 99%.
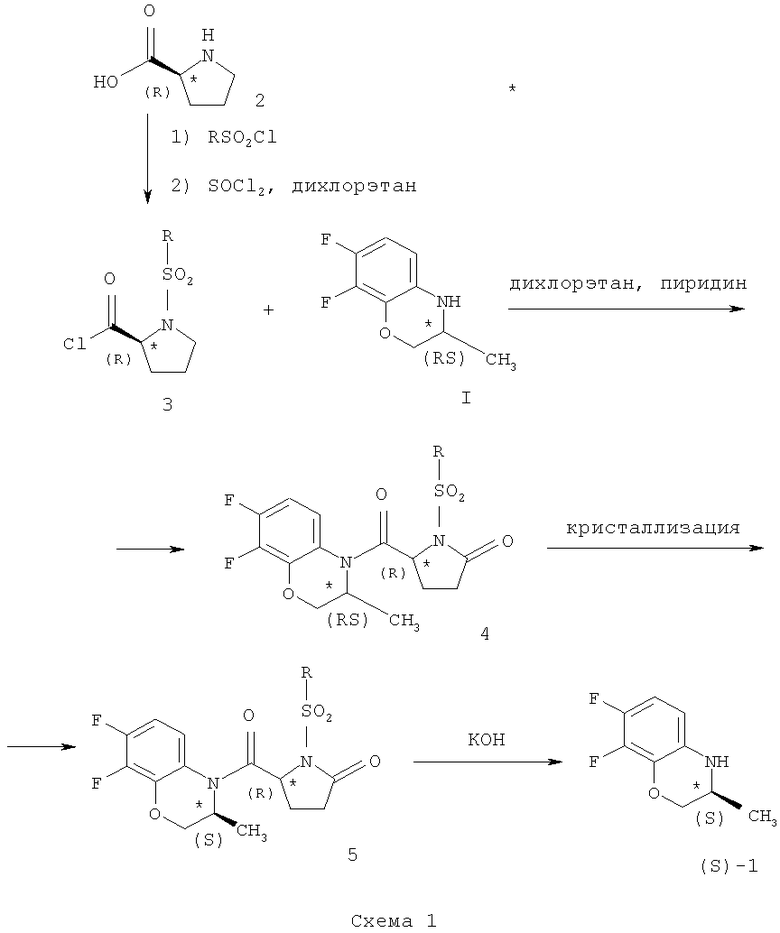
Недостатками данного способа являются: 1) малая доступность исходного асимметрического расщепляющего реагента, N-(сульфонилзамещенного)-(R)-пролина, вследствие чего необходим его предварительный синтез из (R)-пролина, являющегося непротеиногенной и, следовательно, относительно дорогой аминокислотой; 2) большой расход оптически активного расщепляющего реагента: 1,2 моль на 1 моль исходного рацемического бензоксазина (I); 3) недостаточный общий выход бензоксазина (S)-1, 30,3%, в расчете на взятый в реакцию бензоксазин (I).
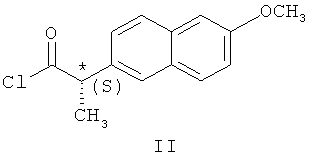
Наиболее близким по технической сущности к заявляемому методу является способ получения бензоксазина (S)-1 путем кинетического расщепления рацемата с помощью хлорангидрида (2S)-2-(6-метоксинафт-2-ил)пропионовой кислоты ((2S)-напроксена) - соединения (II) [JP 2000178265. Production of (S)-benzoxazme derivative and racemization of (R)-benzoxazine derivative / Chupakhin O.N., Krasnov V.P., Levit G.L., Charushin V.N., Korolyova M.A., Tzoi E.V., Lee H.S., Park Y.J., Kim M.H., Kim K.C. (Samsung General Chem. Co, Ltd., S. Korea) // Appl. 16.12.1998, priority 16.12.1998, 10 pp.] - прототип.
Сущность метода состоит во взаимодействии в органическом растворителе соединения (II) с рацематом (соединение (I)) при мольном соотношении соединение (II) - соединение (I) 0,5-0,6:1,0, соответственно, которое приводит к образованию смеси диастереомерных амидов - N-(2S)-2-(6-метоксинафт-2-ил)пропионил]-(3S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]-бензоксазина (соединение (III)) и N-[2S)-2-(6-метоксинафт-2-ил)пропионил]-(3R)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4] бензоксазина (соединение (IV)).
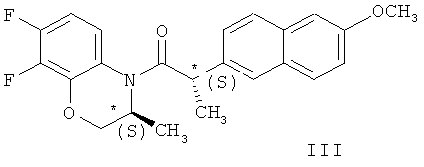
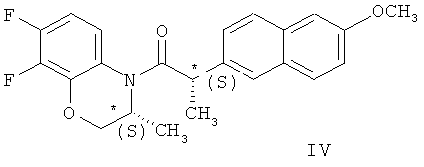
В формуле данного изобретения указано, что основной химический процесс (ацилирование рацемического бензоксазина (I) хлорангидридом II) проводят в органическом растворителе, а в примерах реализации изобретения уточняется, что имеются в виду хлорсодержащие растворители - дихлорметан, 1,2-дихлорэтан и хлороформ. Указано, что желательно проведение реакции в температурном интервале 10-40°С.
Очевидно, что реакцию ацилирования нельзя проводить в любом органическом растворителе, как указано в формуле изобретения. Так, например, этиловый спирт, используемый в качестве растворителя, будет подвергаться ацилированию хлорангидридом II, что приведет к значительному снижению выхода образующегося амида. По нашим данным, использование продажных хлорсодержащих растворителей требует специальной предварительной очистки от содержащихся в них стабилизаторов, чаще всего спиртов.
Смесь амидов обрабатывают гексаном для их разделения, амид IV подвергают кислотному гидролизу при нагревании, продукт гидролиза амида IV объединяют с непрореагировавшим (R)-1 и подвергают рацемизации при нагревании с серной кислотой с получением I, а амид III подвергают кислотному гидролизу при нагревании в смеси уксусной и соляной кислот (температура реакции не указана ни в формуле изобретения, ни в примерах реализации) с получением целевого продукта (Схема 2).
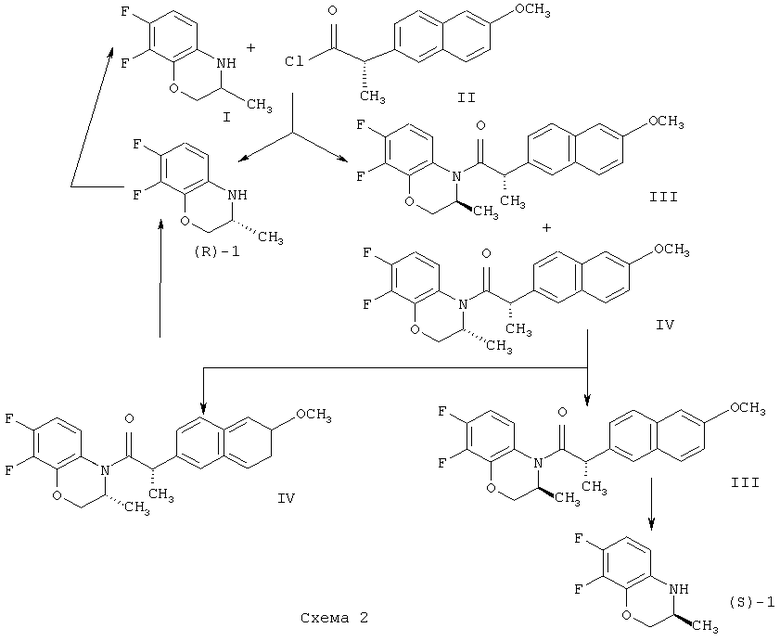
Следует отметить, что по результатам наших исследований однократная обработка смеси амидов III-IV гексаном не позволяет получать (S,S)-амид (III) достаточной степени оптической чистоты (диастереомерный избыток не более 99,0%), что в дальнейшем приводит к получению целевого соединения, (S)-бензоксазина ((S)-1) с не достаточно высоким энантиомерным избытком (не более 99,0%).
Задачей предлагаемого изобретения является повышение оптической чистоты (энантиомерного избытка) и химической чистоты целевого продукта при одновременном сохранении высокого выхода и обеспечение возможности получения его в промышленных масштабах.
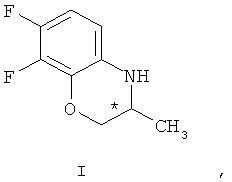
Поставленная задача решается тем, что в способе получения (S)-7,8-дифтор-2,3-дигидро-3метил-4Н-[1,4]бензоксазина формулы (S)-1
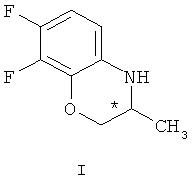
взаимодействием рацемического 7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина формулы (I)
,
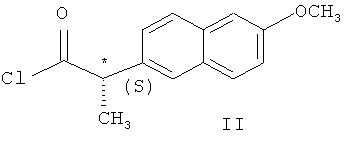
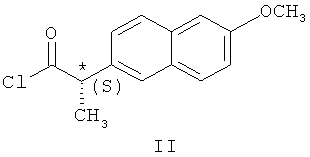
с хлорангидридом (S)-напроксена формулы (II)
в среде органического растворителя, последующей обработкой полученной смеси амидов формул (III) и (IV)
гексаном и гидролизом выделенного соединения (III) смесью уксусной и соляной кислот при нагревании, в качестве органического растворителя используют толуол или дихлорметан, взаимодействие осуществляют при температуре 0-(-40)°С при мольном соотношении соединение (I) - соединение (II)=1:0,5, а после обработки гексаном осадок дополнительно перекристаллизовывают из этилового спирта, затем выделенное соединение формулы (III) гидролизуют при температуре 92-96°С в течение 9-10 часов. Получают целевой продукт с содержанием основного вещества не менее 99,9% и энантиомерным избытком (э.и.) не менее 99,8% (Схема 3).
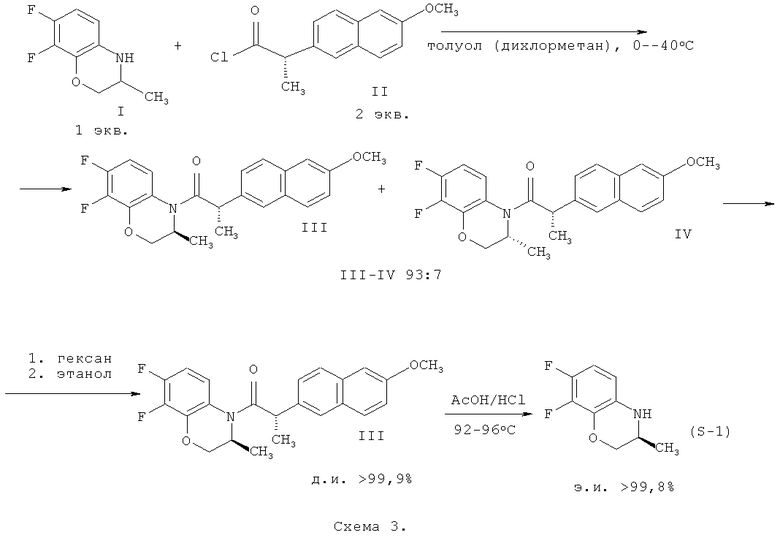
(S)-Напроксен используется в качестве субстанции для получения многих противовоспалительных и обезболивающих препаратов и доступен в значительных количествах. Установлено, что хлорангидрид (S)-напроксена (II) значительно быстрее реагирует с (S)-формой, чем с (R)-формой бензоксазина (1) [Charushin V.N., Krasnov V.P., Levit G.L., Korolyova M.A., Kodess M.I., Chupakhin O.N., Kim M.H., Lee H.S., Park Y.J., Kim K. -C. Kinetic resolution of (±)-2,3-dihydro-3-methyl-4H-1,4-benzoxazines with (S)-naprохеn // Tetrahedron: Asymmetry. - 1999. - V.10 (14). - P.2691-2702], поэтому в предлагаемом способе при взаимодействии хлорангидрида (S)-напроксена (II) с рацематом (I) при мольном соотношении (I)-(II) 1,0:0,5 образуется преимущественно (S,S)-амид (III), причем согласно результатам наших исследований условия реакции (природа растворителя и температура реакции) существенным образом влияют на величину диастереомерного избытка (д.и.) и выход образующегося амида III (табл.1). Реакцию ацилирования следует проводить при мольном соотношении (I)-(II) 1,0:0,5, поскольку изменение соотношения реагентов в сторону уменьшения количества хлорангидрида II (менее 0,5) приводит к уменьшению выхода образующегося амида, а увеличение количества хлорангидрида (более 0,5) приводит к снижению содержания (S,S)-амида (III) и увеличению содержания (R,S)-амида (IV) в смеси образующихся амидов.
Диастереомерный избыток (д.и., %) образующегося (S,S)-амида III определяют методом ВЭЖХ на хроматографе Agilent-1100 с диодно-матричным детектором и автосамплером (колонка Phenomenex Luna С 18(2) 250×4.6 мм, размер частиц сорбента 5 мкм; подвижная фаза ацетонитрил:вода 70:30; скорость потока 0.8 мл/мин; детектирование при 230 нм; времена удерживания (S,S)- и (R,S)-амидов: 14.5-15.7 и 17.7-19.3 мин, соответственно). После соответствующей обработки и выделения, методом ВЭЖХ на хиральной фазе (хроматограф Knauer Smartline; колонка 4,6×250 мм Chiralcel-OD-H, элюент - гексан - пропанол-2 100:1, скорость элюирования 1 мл/мин, УФ-детектирование при 230 нм; времена удерживания (R)- и (S)-энантиомеров бензоксазина: 8,3-8,6 и 10,3-10,8 мин, соответственно), определяют энантиомерный избыток (э.и., %) непрореагировавшего (R)-1. Это позволяет рассчитывать значения степени превращения исходного рацемата I (С, %):
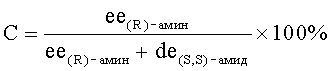
и фактора селективности (s), представляющего собой отношение констант скорости реакции отдельных энантиомеров: 
[Kagan H.B., Fiaud J.C. Kinetic resolution // Topic Stereochem. - 1988. - V.18. - P.249-330]. Показатели С и s характеризуют эффективность процесса кинетического разделения.
Таблица 1. Влияние условий ацилирования на стереохимические результаты кинетического разделения рацемического бензоксазина (I) хлорангидридом II.
мид
Как видно из приведенных в табл.1 данных, диастереоселективность ацилирования рацемического бензоксазина (I) хлорангидридом II существенным образом зависит от природы растворителя. Наибольшие значения диастереоселективного избытка (S,S)-амида III наблюдаются в бензоле, толуоле и дихлорметане. Снижение температуры реакции приводит к увеличению диастереоселективности ацилирования (фактор селективности достигает 57 в толуоле при минус 40°С) при некотором снижении степени превращения исходного рацемата. Поскольку бензол имеет температуру замерзания +5,5°С, в настоящей заявке нами предлагается использовать толуол или дихлорметан в качестве растворителей, а реакцию ацилирования проводить при температуре от 0 до минус 40°С, чтобы обеспечить высокую оптическую чистоту (диастереомерный избыток) и выход (S,S)-амида III.
Образующуюся в результате кинетического разделения смесь амидов (III) и (IV) в соотношении от 93:7 до 90:10 обрабатывают гексаном и получают (S,S)-амид III с содержанием основного вещества около 98,0% и диастереомерным избытком около 99,0%. Дополнительная стадия перекристаллизации полученного соединения из этилового спирта приводит к получению чистого (S,S)-амида III (содержание основного вещества не менее 99,9% и диастереомерный избыток не менее 99,9%), который подвергают кислотному гидролизу при нагревании в смеси соляной и уксусной кислот. Общий выход (S,S)-амида III составляет от 72 до 77%, считая на хлорангидрид II.
Гексановые маточные растворы, полученные после обработки смеси амидов и содержащие, в основном, (R,S)-амид (соединение IV) и (R)-бензоксазин ((R)-1), упаривают, остаток подвергают кислотному гидролизу с целью выделения хирального «балласта» - (R)-бензоксазина ((R)-1), который может быть возвращен в процесс путем рацемизации при нагревании в серной кислоте.
Обычно гидролиз амидов протекает с разрывом N-ацильной связи, при этом регенерируется исходная кислота и амин (или его соль). Поскольку первой стадией гидролиза является нуклеофильное присоединение по карбонильной группе, а амины являются плохой уходящей группой, эти реакции часто протекают медленно. Гидролиз протекает легче как в щелочных условиях, где имеется сильный нуклеофил НО-, так и в кислых условиях, где протонирование субстрата способствует нуклеофильной атаке водой и отщеплению амина, причем в том и другом случае гидролиз проводится при нагревании. Следует отметить, что в случае хиральных соединений при проведении гидролиза в достаточно жестких условиях (щелочная или кислая среда, нагревание) существует опасность рацемизации целевых соединений.
Проведенные нами исследования (особое внимание уделялось как оптической чистоте (энантиомерный избыток) целевого соединения - (S)-бензоксазина (S)-1, так и его химической чистоте (содержание основного вещества)), показали, что оптимальным является проведение кислотного гидролиза при температуре от +92 до +96°С в течение 9-10 ч. Выходы целевого соединения высокой оптической (э.и. более 99,8%) и химической чистоты (содержание основного вещества более 99,9%) на этой стадии составляют от 85 до 87%.
Увеличение продолжительности гидролиза и повышение температуры реакции приводит к заметной рацемизации и снижению оптической чистоты целевого соединения, а снижение температуры гидролиза и уменьшение времени реакции приводит к снижению выхода целевого соединения вследствие неполноты прохождения процесса.
Полученный по разработанной схеме (S)-бензоксазин (S)-1 не требует дополнительной очистки.
Поскольку хлорангидрид (S)-напроксена, (S)-2-(6-метоксинафтил-2)пропионил хлорид (соединение II) является соединением не устойчивым при хранении, его получают непосредственно перед проведением стадии ацилирования следующим образом: к суспензии (S)-напроксена в бензоле добавляют при комнатной температуре и перемешивании оксалил хлорид; реакционную массу перемешивают 3-4 часа при комнатной температуре до образования прозрачного желтого раствора, затем упаривают досуха при пониженном давлении; к полученному остатку добавляют гексан, суспензию охлаждают до минус 18°С и оставляют при этой температуре на 2-3 часа; полученный осадок отфильтровывают и промывают гексаном; после высушивании получают соединение II в виде бесцветных кристаллов (температура плавления 96°С). 1Н ЯМР (СDCl3) δ, м.д.: 7,78-7,10 м (6Н, аром.); 4,25 кв (1H, СН, J=6,9 Гц); 3,92 с (3Н, ОСН3); 1,67 д (3Н, СН3, J=6,9 Гц).
Пример 1:
Получение N-[(2S)-2-(6-метоксинафт-2-ил)пропионил1-7,8-дифтор-2.3-дигидро-3(S)-метил-4Н-[1,4]бензоксазина (III).
К раствору 30,0 г (0,162 моль) (RS)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина в 410 мл толуола при перемешивании и температуре 0°С добавляют по каплям раствор 20,1 г (0,081 моль) соединения II в 400 мл дихлорметана. Реакционную массу перемешивают в течение 6 часов при 0°С, затем промывают водой (3 раза по 150 мл) и сушат над безводным сульфатом магния. После упаривания растворителя и высушивания в вакууме полученный остаток перемешивают в 500 мл гексана при комнатной температуре 2 часа. Полученную суспензию охлаждают до минус 18°С и оставляют при этой температуре на 16 часов. Осадок отфильтровывают в вакууме, промывают на фильтре 2 раза 50 мл охлажденного гексана. После высушивания на воздухе при комнатной температуре получают 24,2 г (75%) соединения III с диастереоизомерным избытком (д.и.) 99,0% (ВЭЖХ: хроматограф Agilent 1100, колонка Phenomenex Luna С 18(2) 250×4.6 мм; подвижная фаза ацетонитрил:вода 70:30; скорость потока 0.8 мл/мин; детектирование при 230 нм; времена удерживания (S,S)- и (R,S)-амидов: 14.5-15.7 и 17.7-19.3 мин). Полученное соединение перекристаллизовывают из 220 мл этанола и получают 24,1 г (75%) соединения III в виде бесцветных кристаллов с диастереоизомерным избытком (д.и.) 99,9%.
Температура плавления от 114 до 116°С.
[α]D от +72,5 до +73,5 (С 1,4; СНСl3).
1Н ЯМР (400 МГц, DMSO-d6, 100°С) δ, м.д.: 7,77-6,84 м (8Н, аром.); 4,85 квдд (1Н, С3H-бензоксазина, J=6,8; 3,0 и 1,6 Гц); 4,52 кв (1Н, СН-напроксена, J=6,8 Гц); 4,22 дд (1Н, С2HА-бензоксазина, J=11,0 и 1,2 Гц); 4,02 дд (1Н, С2HB-бензоксазина, J=11,0 и 2,7 Гц); 3,86 с (3Н, ОСН3); 1,47 д (3Н, СН3-напроксена, J=6,8 Гц); 0,70 д (3Н, СН3-бензоксазина, J=6,8 Гц).
19F ЯМР (DMSO-d6) δ, м.д.: 20,6 м (F8); 1.75 ддд (F7, J=20,8; 8,2 и 2,4 Гц).
Элементный анализ для C23H21F2NO3.
Вычислено, %: С 69,54; Н 5,33; N 3,52; F 9,56.
Найдено, %: С 69,70; Н 5,33; N 3,53; F 9,54.
ГЖХ (хроматограф Shimadzu GC-2010 с пламенно-ионизационным детектором (кварцевая капиллярная колонка ZB-5 30 м×0.25 мм×0.25 мкм, скорость потока газа-носителя (азот) 30 мл/мин, скорость расхода через колонку 1.0 мл/мин): содержание основного вещества 99,9% (время удерживания (S,S)-амида III 34.5 мин).
Получение (S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина ((S)-1).
80 г (0,201 моль) соединения III (д.и. 99,9%) растворяют в 500 мл ледяной уксусной кислоты, к полученному раствору добавляют 500 мл концентрированной соляной кислоты, затем нагревают при температуре 92°С в течение 10 часов. Затем реакционную массу упаривают в вакууме до объема 500 мл и выливают в 4000 мл дистиллированной воды. Выпавший осадок отфильтровывают и промывают на фильтре 2 раза по 100 мл воды. Фильтрат и промывные растворы нейтрализовывают карбонатом натрия до рН 8-9. Выделяющееся масло экстрагируют бензолом (3 раза по 300 мл). Органический слой промывают насыщенным раствором NaCl (300 мл) и сушат над безводным сульфатом магния. После упаривания растворителя и высушивания в вакууме получают 32,4 г (87%) соединения (S)-1 в виде бесцветных легкоплавких кристаллов с энантиомерным избытком (э.и.) 99,95% (ВЭЖХ: хроматограф Knauer SmartLine, колонка Chiralcel OD Н, элюент гексан-изопропанол 40:1; время удерживания τS 14-15 мин; (R)-изомер τR 11-12 мин).
Температура плавления от 28 до 29°С.
[α]D от -7,3 до -7,7 (С2; хлороформ).
1Н ЯМР (400 МГц, CDCl3) δ, м.д.: 6,55 ддд (1Н, C6H, J=10,0; 9,0 и 8,0 Гц); 6,27 ддд (1Н, С5H, J=9,0; 4,6 и 2,3 Гц); 4,28 дд (1Н, С2HA, J=10,5 и 2,7 Гц); 3,79 дд (1Н, С2HB, J=10,5 и 8,1 Гц); 3,51 дквд (1Н, С3H J=8,1; 6,5 и 2,7 Гц); 1,20 д (3Н, Me, J-6,5 Гц).
19F ЯМР (CDCl3) δ, м.д.: 11,9 ддд (F7, J=21,0; 10.0 и 4,6 Гц); 0,92 ддд (F7, J=21,0; 8,1 и 2,3 Гц).
Элементный анализ для C9H9F2NO.
Вычислено, %: С 58,38; Н 4,90; N 7,57; F 20,52.
Найдено, %: С 58,45; Н 4,78; N 7,72; F 20,30.
ГЖХ (хроматограф Shimadzu GC-2010 с пламенно-ионизационным детектором (кварцевая капиллярная колонка ZB-5 30 м×0.25 мм×0.25 мкм, скорость потока газа-носителя (азот) 30 мл/мин, скорость расхода через колонку 1.0 мл/мин): содержание основного вещества 99,9% (время удерживания (S)-бензоксазина (S)-1 18.3 мин).
Пример 2:
Получение N-[(2S)-2-(6-метоксинафт-2-ил)пропионил]-7,8-дифтор-2.3-дигидро-3(S)-метил-4Н-[1,4]бензоксазина (III).
К раствору 30,0 г (0,162 моль) (R)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина в 410 мл толуола при перемешивании и температуре минус 40°С добавляют по каплям раствор 20,1 г (0,081 моль) соединения II в 400 мл дихлорметана. Реакционную массу перемешивают в течение 6 часов при минус 40°С, затем нагревают до 0°С, промывают водой (3 раза по 150 мл) и сушат над безводным сульфатом магния. После упаривания растворителя и высушивания в вакууме полученный остаток перемешивают в 500 мл гексана при комнатной температуре 2 часа. Полученную суспензию охлаждают до минус 18°С и оставляют при этой температуре на 16 часов. Осадок отфильтровывают в вакууме, промывают на фильтре 2 раза по 50 мл охлажденного гексана. После высушивания на воздухе при комнатной температуре получают 23,2 г (72%) соединения III с диастереоизомерным избытком (д.и.) 99,1% (ВЭЖХ: хроматограф Agilent 1100, колонка Phenomenex Luna С 18(2) 250×4.6 мм; подвижная фаза ацетонитрил:вода 70:30; скорость потока 0.8 мл/мин; детектирование при 230 нм; времена удерживания (S,S)- и (R,S)-амидов: 14.5-15.7 и 17.7-19.3 мин). Полученное соединение перекристаллизовывают из 220 мл этанола и получают 23,1 г (72%) соединения III в виде бесцветных кристаллов с диастереоизомерным избытком (д.и.) 99,9%.
Получение (S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина ((S)-1).
80 г (0,201 моль) соединения III (д.и. 99,9%), растворяют в 500 мл ледяной уксусной кислоты, к полученному раствору добавляют 500 мл концентрированной соляной кислоты, затем нагревают при температуре 96°С в течение 9 часов. Затем реакционную массу упаривают в вакууме до объема 500 мл и выливают в 4000 мл дистиллированной воды. Выпавший осадок отфильтровывают и промывают на фильтре 2 раза по 100 мл воды. Фильтрат и промывные растворы нейтрализовывают карбонатом натрия до рН 8-9. Выделяющееся масло экстрагируют бензолом (3 раза по 300 мл). Органический слой промывают насыщенным раствором NaCl (300 мл) и сушат над безводным сульфатом магния. После упаривания растворителя и высушивания в вакууме получают 31,7 г (85%) (S)-1 в виде бесцветных легкоплавких кристаллов с энантиомерным избытком (э.и.) 99,95% (ВЭЖХ: хроматограф Knauer SmartLine, колонка Chiralcel OD Н, элюент гексан-изопропанол 40:1; время удерживания τS 14-15 мин; (R)-изомер τR 11-12 мин).
Пример 3:
Получение N-[(2S)-2-(6-метоксинафт-2-ил)пропионил]-7,8-дифтор-2.3-дигидро-3(S)-метил-4Н-[1,4]бензоксазина (III).
К раствору 400 г (2,16 моль) (R,S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина в 4200 мл дихлорметана при перемешивании и температуре минус 20°С добавляют по каплям раствор 268,6 г (1,08 моль) соединения II в 3000 мл дихлорметана. Реакционную массу перемешивают в течение 6 часов при минус 20°С, нагревают до 0°С, затем промывают водой (3 раза по 2000 мл) и сушат над безводным сульфатом магния. После упаривания растворителя и высушивания в вакууме полученный остаток перемешивают в 7200 мл гексана при комнатной температуре 2 часа. Полученную суспензию охлаждают до минус 18°С и оставляют при этой температуре на 16 часов. Осадок отфильтровывают в вакууме, промывают на фильтре 2 раза по 200 мл охлажденного гексана. После высушивания на воздухе при комнатной температуре получают 334 г (78%) соединения III с диастереоизомерным избытком (д.и.) 99,2% (ВЭЖХ: Phenomenex Luna С 18(2) 250×4.6 мм; подвижная фаза ацетонитрил:вода 70:30; скорость потока 0.8 мл/мин; детектирование при 230 нм; времена удерживания (S,S)- и (R,S)-амидов: 14.5-15.7 и 17.7-19.3 мин). Полученное соединение перекристаллизовывают из 3000 мл этанола и получают 330 г (77%) соединения III в виде бесцветных кристаллов с диастереоизомерным избытком (д.и.) 99,9%.
Получение (S)-7,8-дифтор-2.3-дигидро-3-метил-4Н-[1,4]бензоксазина ((S)-1).
80 г (0,201 моль) соединения III (д.и. 99,9%) растворяют в 500 мл ледяной уксусной кислоты, к полученному раствору добавляют 500 мл концентрированной соляной кислоты, затем нагревают при температуре 92°С в течение 10 часов. Затем реакционную массу упаривают в вакууме до объема 500 мл и выливают в 4000 мл дистиллированной воды. Выпавший осадок отфильтровывают и промывают на фильтре 2 раза по 100 мл воды. Фильтрат и промывные растворы нейтрализовывают карбонатом натрия до рН 8-9. Выделяющееся масло экстрагируют бензолом (3 раза по 300 мл). Органический слой промывают насыщенным раствором NaCl (300 мл) и сушат над безводным сульфатом магния. После упаривания растворителя и высушивания в вакууме получают 32,4 г (87%) (S)-1 в виде бесцветных легкоплавких кристаллов с энантиомерным избытком (э.и.) 99,95% (ВЭЖХ: хроматограф Knauer SmartLine, колонка Chiralcel OD Н, элюент гексан-изопропанол 40:1; время удерживания τS 14-15 мин; (R)-изомер τR 11-12 мин).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 7,8-ДИФТОР-2,3-ДИГИДРО-3-МЕТИЛ-4Н-1,4-БЕНЗОКСАЗИНА | 2010 |
|
RU2434005C1 |
| (3S)-4-[6-(Пурин-6-иламино)гексаноил]-3,4-дигидро-3-метил-7,8-дифтор-2Н-[1,4]бензоксазин и (3R)-4-[6-(Пурин-6-иламино)гексаноил]-3,4-дигидро-3-метил-7,8-дифтор-2Н-[1,4]бензоксазин, обладающие противовирусной активностью | 2016 |
|
RU2644351C1 |
| АМИДЫ N-(2-АМИНОПУРИН-6-ИЛ)-6-АМИНОКАПРОНОВОЙ КИСЛОТЫ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2015 |
|
RU2599577C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО БЕНЗОКСАЗИНА, СПОСОБЫ ПОЛУЧЕНИЯ ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2258069C2 |
| 4-[(Пурин-6-ил)аминополиметиленкарбонил]-производные 3,4-дигидро-3-метил-7,8-дифтор-2H-[1,4]бензоксазина, обладающие противоопухолевой активностью | 2021 |
|
RU2760305C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2044735C1 |
| СПИРОСОЕДИНЕНИЕ ИЛИ ЕГО СОЛИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ОБЛАДАЮЩАЯ ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2094432C1 |
| 2-АМИНО-5,5-ДИФТОР-5,6-ДИГИДРО-4Н-ОКСАЗИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ 1 И(ИЛИ) ВАСЕ 2 | 2010 |
|
RU2515221C2 |
| АМИДЫ АКРИЛОВОЙ И МЕТАКРИЛОВОЙ КИСЛОТ С ОЛИГОПИПЕРИДИНАМИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2015 |
|
RU2617694C1 |
| БИЦИКЛИЧЕСКИЕ АРОМАТИЧЕСКИЕ АМИНОКИСЛОТЫ | 1998 |
|
RU2187506C2 |
Изобретение относится к способу получения (S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина формулы (S)-1. Способ осуществляют взаимодействием рацемического 7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина формулы (I) с хлорангидридом (S)-напроксена формулы (II) в толуоле или дихлорметане при температуре от 0 до -40°С и при мольном соотношении соединение (I) - соединение (II) 1:0,5. Полученную смесь обрабатывают гексаном и перекристаллизовывают из этилового спирта с получением соединения формулы (III). Проводят кислотный гидролиз соединения (III) в смеси уксусной и соляной кислот при температуре 92-96°С в течение 8-10 часов с получением соединения (S)-1. Технический результат - усовершенствованный способ получения (S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина, имеющего содержание основного вещества не менее 99,9% и энантиомерный избыток не менее 99,8%. 1 табл.
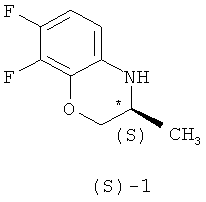

Способ получения (S)-7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина формулы (S)-1

взаимодействием рацемического 7,8-дифтор-2,3-дигидро-3-метил-4Н-[1,4]бензоксазина формулы (I)
с хлорангидридом (S)-напроксена формулы (II)
в среде органического растворителя, последующей обработкой полученной смеси амидов формул (III) и (IV)
,
гексаном и гидролизом выделенного соединения (III) смесью уксусной и соляной кислот при нагревании, отличающийся тем, что в качестве органического растворителя используют толуол или дихлорметан, взаимодействие осуществляют при температуре 0-(-40)°С при мольном соотношении соединение (I) - соединение (II)=1:0,5, а после обработки гексаном осадок дополнительно перекристаллизовывают из этилового спирта, затем выделенное соединение формулы (III) гидролизуют при температуре 92-96°С в течение 9-10 ч.
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| ЛЕВИТ Г.Л | |||
| Автореферат диссертации на соискание ученой степени доктора химических наук: Аминокислоты в регио- и стереонаправленном синтезе физиологически активных соединений | |||
| - Екатеринбург, 19.10.2009 | |||
| КРАСНОВ В.П., ЛЕВИТ Г.Л | |||
| и др | |||
| Кинетическое разделение гетероциклических аминов реакцией с хлорангидридами оптически | |||

