Изобретение относится к каталитическим системам, более конкретно - к катализатору на основе смешанных оксидов для гидрирования органических соединений, способу его получения и способу гидрирования.
Для катализа реакций гидрирования используют кобальтовые катализаторы в виде губчатого кобальта (кобальта Ренея), которые получают, как правило, кальцинированием и восстановлением соответствующих исходных соединений, таких как гидроксид кобальта, нитрат кобальта и оксид кобальта.
Гидрирование органических нитрилов с использованием катализаторов Ренея часто осуществляют в присутствии обладающих основным характером соединений щелочных или щелочноземельных металлов, например, таких как указаны в патентах США US 3821305, US 5874625, US 5151543, US 4375003, европейских заявках на патент ЕР-А-0316761, ЕР-А-0913388 и патенте США US 6660887.
Кроме того, содержащие кобальт катализаторы могут быть получены восстановлением оксида, гидроксида или карбоната кобальта. В опубликованной немецкой заявке на патент DE 3403377 описаны содержащие частицы металлического кобальта и/или металлического никеля катализаторы, которые могут быть получены благодаря реализации контакта частиц оксида кобальта и/или оксида никеля с водородом. Согласно указанной заявке содержание щелочного и/или щелочноземельного металла предпочтительно составляет менее 0,1% мас. В европейской заявке на патент ЕР-В-0742045 описаны кобальтовые катализаторы, получаемые кальцинированием оксидов кобальта (от 55 до 98% мас.), фосфора (от 0,2 до 15% мас.), марганца (от 0,2 до 15% мас.) и щелочного металла (от 0,05 до 5% мас.) и восстановлением продуктов кальцинирования в токе водорода. Кобальтовые катализаторы, которые могут быть получены осаждением карбоната кобальта из водного раствора соли кобальта и последующим восстановлением водородом, приведены в европейской заявке на патент ЕР-А-0322760. Указанные катализаторы дополнительно могут содержать от 0,25 до 15% мас. (в пересчете на общую массу катализатора) SiO2, MnO2, ZrO2, Аl2O3 и МgО в виде оксидов, гидроксидов или гидратированных оксидов. Катализаторы гидрирования, состоящие из одного или нескольких оксидов железа, никеля, марганца, хрома, молибдена, вольфрама или фосфора и одного или нескольких оксидов щелочных металлов, щелочноземельных металлов или представителей группы редкоземельных элементов, описаны в европейской заявке на патент ЕР-В-0445589. Согласно указанной заявке часть оксидов после восстановления находится в виде соответствующих металлов.
В основу настоящего изобретения была положена задача разработать катализатор для гидрирования органических соединений, который обладает преимуществами по сравнению с уровнем техники. В частности, из катализатора должны выщелачиваться как можно меньшие количества металлов, например таких как алюминий в случае скелетных катализаторов, или щелочных промоторов, таких как литий, поскольку подобный процесс обусловливает снижение стабильности и деактивирование катализатора. Твердые фракции алюминатов, образующихся в щелочных условиях из выделившегося в результате выщелачивания алюминия, могут вызывать закупоривание технологического оборудования и формирование отложений, а также деструкцию ценных продуктов. Кроме того, новый катализатор должен был бы позволить осуществление гидрирования органических соединений в менее сложных реакционных условиях. В частности, следовало найти катализаторы, позволяющие осуществлять реакцию гидрирования при низких давлениях. Кроме того, должны быть доступны способы гидрирования, которые можно было бы реализовать в отсутствие воды, аммиака и водного основания.
Кроме того, настоящее изобретение направлено на разработку способа гидрирования, обеспечивающего возможность с высокой селективностью гидрировать нитрилы до первичных аминов.
Поставленная задача решается предлагаемым катализатором для гидрирования органических соединений, получаемым восстановлением предварительного катализатора, состоящего из а) кобальта и b) одного или нескольких элементов группы щелочных или щелочноземельных металлов, причем элементы а) и b) по меньшей мере частично находятся в виде их смешанных оксидов.
Смешанный оксид отличается тем, что его кристаллическая решетка наряду с кобальтом и кислородом содержит также по меньшей мере один другой элемент b) группы щелочных металлов или щелочноземельных металлов. Так, например, элементом b) может являться литий, натрий, калий, рубидий, цезий, бериллий, магний, кальций, стронций, барий, радий, предпочтительно литий, натрий, калий, магний, кальций или смесь, состоящая из двух или более указанных элементов.
В соответствии с массовым отношением кобальта к элементу b)
1) элемент b) может занимать место кобальта в узле кристаллической решетки (твердый раствор замещения) или ее междоузлии (твердый раствор внедрения),
2) кобальт может занимать место элемента b) в узле кристаллической решетки или ее междоузлии или
3) кобальт и элемент b) совместно с кислородом могут образовывать общую кристаллическую решетку, не похожую на кристаллическую решетку ни одного из базовых соединений.
Смешанными оксидами в соответствии с настоящим изобретением называют также так называемые твердые растворы, то есть непрерывные последовательности смешанных кристаллов.
Смесь оксидов или оксидная смесь отличается от предлагаемого в настоящем изобретении смешанного оксида тем, что кристаллические структуры оксида кобальта и оксидов элементов b) одновременно находятся в смеси оксидов или оксидной смеси в более или менее тонко распределенном состоянии. Присутствие предлагаемого в изобретении смешанного оксида аналитически можно обнаружить, например, благодаря измерению дифракции рентгеновских лучей. Соответствующие сравнительные спектры приведены в банке кристаллографических данных [ICSD (Inorganic Crystal Structure Database), Bergerhoff и другие, Universität Bonn (D), или в Powder Diffraction File, Berry и другие, International Centre for Diffraction Data (ICDD), Swarthmore, США)].
Как указано выше, предварительные катализаторы, используемые для получения предлагаемых в изобретении катализаторов, частично находятся в виде смешанного оксида, содержащего кобальт и по меньшей мере один из указанных выше элементов b). Предварительные катализаторы частично предпочтительно находятся в виде смешанных оксидов кобальта и лития, кобальта и натрия, кобальта и калия, кобальта и рубидия, кобальта и цезия, кобальта и бериллия, кобальта и магния, кобальта и кальция, кобальта и стронция, кобальта или бария. Предварительные катализаторы частично особенно предпочтительно находятся в виде смешанных оксидов кобальта и лития, кобальта и магния. Предварительные катализаторы частично еще более предпочтительно находятся в виде смешанных оксидов кобальта и лития или кобальта и магния.
В другом предпочтительном варианте осуществления изобретения исходные соединения (предварительные катализаторы), используемые для получения предлагаемых в изобретении катализаторов, частично находятся в виде смешанных оксидов лития, натрия и кобальта, лития, калия и кобальта, лития, магния и кобальта, лития, кальция и кобальта, натрия, магния и кобальта, калия, магния и кобальта, натрия, кальция и кобальта или калия, кальция и кобальта.
В предпочтительном варианте осуществления изобретения предварительные катализаторы могут быть восстановлены до одного или нескольких соединений, обладающих суммарной формулой MI xMII yCozO(x/2+y+z*1,5), в которой x означает 0 или число от 0,1 до 1, у означает 0 или число от 0,1 до 1, z означает число от 0,1 до 1, причем х и у одновременно не могут означать 0, МI означает по меньшей мере один элемент группы щелочных металлов и МII означает по меньшей мере один элемент группы щелочноземельных металлов.
Прежде всего предпочтительным является предварительный катализатор, который обладает суммарной формулой LiСоО2 (кобальтит лития). Кобальтит лития может находиться в виде низкотемпературной фазы (LT-LiCoO2), высокотемпературной фазы (HT-LiCoO2) или смеси указанных фаз.
В другом предпочтительном варианте осуществления изобретения в качестве предварительного катализатора используют кобальтит лития, который получают при вторичной переработке батарей.
Кроме того, для использования в качестве предварительных катализаторов пригодны непрерывные последовательности смешанных кристаллов оксида кобальта и оксида магния формулы MgaCObO1, в которой 0<а<1, 0<b<1 и сумма а+b означает 1.
Согласно изобретению предварительные катализаторы частично находятся в виде смешанных оксидов. Однако предварительные катализаторы могут также полностью состоять из смешанных оксидов. В предпочтительном варианте количество кобальта, находящегося в предварительном катализаторе в виде смешанных оксидов, составляет по меньшей мере 10% мол., предпочтительно по меньшей мере 20% мол. и особенно предпочтительно по меньшей мере 30% мол. (соответственно в пересчете на общий кобальт, содержащийся в предварительном катализаторе), предварительный катализатор наряду с одним или несколькими смешанными оксидами может содержать также один или несколько дополнительных компонентов. Подобными дополнительными компонентами могут являться оксиды элементов. Пригодными оксидами элементов могут быть оксиды элементов от первой до пятой главных групп или от третьей до восьмой побочных групп периодической системы элементов, прежде всего оксиды кобальта, никеля, меди, марганца, фосфора, хрома, серебра, железа, циркония, алюминия, титана, лития, натрия, калия, магния, кальция, лантана или иттрия.
Предварительный катализатор может содержать один или несколько легирующих элементов. Пригодными легирующими элементами являются элементы от третьей до восьмой побочных групп периодической системы элементов (согласно редакции ЮПАК от 03.10.2005 (http://www.iupac.org/reports/periodic_table/IUPAC_Periodic_Table-3Oct05.pdf)), a также элементы третьей, четвертой и пятой главных групп периодической системы. Предпочтительными легирующими элементами являются железо, никель, хром, марганец, фосфор, титан, ниобий, ванадий, медь, серебро, палладий, платина, родий, иридий, рутений и золото. Содержание легирующих элементов предпочтительно не превышает 10% мас., составляя, например, от 0,1 до 10% мас., особенно предпочтительно от 1 до 5% мас. соответственно в пересчете на используемый предварительный катализатор.
Предварительные катализаторы в общем случае могут быть получены термической обработкой соответствующих соединений кобальта и одного или нескольких соединений группы щелочных металлов или соединений группы щелочноземельных металлов, например, нитратов, карбонатов, гидроксидов, оксидов, ацетатов, оксалатов или цитратов. Под термической обработкой подразумевают, например, сплавление или кальцинирование указанных соединений. При этом термическая обработка указанных соединений, таких как нитраты, карбонаты, гидроксиды и оксиды, может быть выполнена на воздухе.
В предпочтительном варианте термическую обработку, в особенности карбонатов, осуществляют в атмосфере инертного газа. Пригодными инертными газами являются, например, азот, диоксид углерода, гелий, неон, аргон, ксенон, криптон или смеси указанных инертных газов. В качестве предпочтительного инертного газа используют азот. Преимущество получения предварительного катализатора термической обработкой указанных соединений в атмосфере инертного газа состоит в том, что восстановление предварительного катализатора может быть выполнено непосредственно после подобной обработки. При получении предварительного катализатора не в атмосфере инертного газа перед восстановлением должна быть осуществлена дополнительная операция инертизации. На стадии инертизации мешающие соединения, такие как кислород воздуха, которые способны взаимодействовать с восстанавливающим агентом по реакции восстановления, могут быть удалены, например, насыщением исходного соединения инертным газом или многократной эвакуацией и продувкой инертным газом.
Другим способом получения предварительных катализаторов является осаждение водорастворимых соединений кобальта и по меньшей мере одного или нескольких элементов, выбранных из группы, включающей водорастворимые соединения щелочных металлов, водорастворимые соединения щелочноземельных металлов, водорастворимые соединения редкоземельных элементов и водорастворимые соединения цинка, добавлением щелочного раствора и последующим осуществлением сушки и кальцинирования.
Способ получения LiCoO2 описан, например, в Е. Antolini, Solid State lonics, 159-171 (2004), а также в W.M. Fenton, P.A. Huppert, Sheet Metal Industries, 25(1948), 2255-2259.
В частности, LiCoO2 может быть получен термической обработкой соответствующих соединений лития и кобальта, таких как нитраты, карбонаты, гидроксиды, оксиды, ацетаты, цитраты или оксалаты.
Кроме того, LiCoO2 может быть получен осаждением водорастворимых солей лития и кобальта, реализуемым благодаря добавлению щелочного раствора и последующему кальцинированию.
Кроме того, LiCoO2 может быть получен золь-гель процессом.
LiCoO2 может быть получен также в соответствии с S.W. Song, K.S. Han, M. Yoshimura, Y. Sata, A. Tatsuhiro, Mat. Res. Soc. Symp.Proc, 606, 205-210 (2000) гидротермальной обработкой металлического кобальта водными растворами гидроксида лития.
Согласно изобретению в качестве исходного соединения для катализатора (предварительного катализатора) можно использовать также LiCoO2, выделенный при регенерации батарей. Метод извлечения кобальтита лития из отработанных батарей, соответственно его регенерации, приведен, например, в заявке CN 1594109. В результате механического вскрытия батарей и выщелачивания содержащих алюминий компонентов посредством концентрированного раствора едкого натра может быть получен обогащенный LiCoO2 фильтровальный осадок.
Непосредственно после синтеза оксидного предварительного катализатора перед его восстановлением может быть реализована его промывка или промывка в сочетании с последующей сушкой. Промывкой можно удалить примеси, побочные продукты или непревращенные эдукты.
Как указано выше, предварительный катализатор может содержать один или несколько легирующих элементов.
Подобные легирующие добавки могут быть введены при получении предварительного катализатора добавлением комплексов и солей металлов, таких как карбонаты и оксиды металлов, или самих металлов, которое осуществляют сплавлением соответствующих оксидов, карбонатов или их смесей. Кроме того, легирующие добавки можно ввести при получении предварительного катализатора по реакции осаждения в виде водорастворимых солей и комплексов в смеси с осаждающим реактивом. Наряду с этим возможный вариант введения легирующих добавок предусматривает реализуемое до восстановления поверхностное легирование оксидного предварительного катализатора солями металлов, в соответствии с которым находящийся, например, в водном растворе предварительный катализатор в течение определенного времени подвергают контактированию со смешанным оксидом. Подобным образом можно легировать также уже полученный восстановлением предварительный катализатор после восстановления и даже во время осуществления реакции гидрирования. При этом в предварительный катализатор и/или также в катализатор уже могут быть введены легирующие элементы.
Получаемый, как правило в порошкообразном состоянии, предварительный катализатор перед восстановлением можно подвергать формованию или он может абсорбироваться пористыми и поверхностно-активными материалами (носителями). Общепринятые методы формования и нанесения на носители приведены, например, в Ullmann's Encyclopedia Electronic Release, 2000, глава Catalysis and Catalysts, страницы 28-32. Кроме того, пригодные вещества могут быть нанесены на носитель и подвергнуты на нем реакции, в результате которой образуется предварительный катализатор.
Восстановление предварительного катализатора можно осуществлять в органическом растворителе, в котором его суспендируют, например в автоклаве с мешалкой, насадочной барботажной колонне, циркуляционном реакторе или реакторе со стационарным слоем.
Восстановлению можно подвергать также предварительный катализатор в виде сухого порошка, причем соответствующий процесс осуществляют в подвижных или неподвижных восстановительных печах, а также в стационарном или псевдоожиженном слое.
В предпочтительном варианте восстановление предварительного катализатора осуществляют в органическом растворителе, в котором его подвергают суспендированию.
В качестве органических растворителей можно назвать, например, простые эфиры, такие как метил-трет-бутиловый эфир, этил-трет-бутиловый эфир и тетрагидрофуран, спирты, такие как метанол, этанол и изопропанол, углеводороды, такие как гексан, гептан и фракции рафината, ароматические соединения, такие как толуол, амиды, такие как диметилформамид и диметилацетамид, или лактамы, такие как N-метилпирролидон, N-этмлпирролидон, N-метилкапролактам и N-этилкапролактам. Пригодны также смеси указанных растворителей.
Предпочтительные органические растворители, используемые для суспендирования предварительного катализатора, содержат продукты подлежащего гидрированию. Особенно предпочтительные органические растворители состоят только из продуктов подлежащего гидрированию.
Температуре реализуемого в суспензии восстановления предварительного катализатора в общем случае соответствует интервал от 50 до 300°С, прежде всего интервал от 100 до 250°С, особенно предпочтительно от 120 до 200°С.
Восстановление в суспензии, как правило, осуществляют под давлением, составляющим от 1 до 300 бар, предпочтительно от 10 до 250 бар, особенно предпочтительно от 30 до 200 бар, причем в данном случае и ниже речь идет об абсолютном давлении.
Пригодным восстанавливающим агентом является водород, содержащий водород газ или источник гидрид-ионов.
В качестве водорода в общем случае используют технически чистый водород. Водород можно использовать также в виде содержащего водород газа, то есть в виде смеси водорода с инертными газами, такими как азот, гелий, неон, аргон или диоксид углерода. Водород можно также возвращать на стадию восстановления в виде рециркулируемого газа, при необходимости предварительно смешав его со свежим водородом и при необходимости удалив из него воду конденсацией.
Восстановление сухого, как правило порошкообразного, предварительного катализатора можно осуществлять при повышенной температуре в подвижных или стационарных восстановительных печах. При этом температура восстановления предварительного как правило составляет от 50 до 600°С, прежде всего от 100 до 500°С, особенно предпочтительно от 150 до 400°С.
При этом рабочее давление, как правило, составляет от 1 до 300 бар, прежде всего от 1 до 200 бар, особенно предпочтительно от 1 до 10 бар, причем поток водорода или содержащий водород поток, полученный указанным выше смешиванием водорода с инертными газами, можно пропускать через слой катализатора или над слоем катализатора. В соответствии с данным вариантом осуществления изобретения поток водорода также можно возвращать на стадию восстановления в виде рециркулируемого газа, при необходимости предварительно смешав его со свежим водородом и при необходимости удалив из него воду конденсацией.
Восстановление предварительного катализатора предпочтительно осуществляют таким образом, чтобы степень его восстановления составляла по меньшей мере 50%. Методом измерения степени восстановления является сравнение потерь массы сухого предварительного катализатора с потерями массы сухого восстановленного катализатора, причем образцы восстанавливают при температуре от комнатной до 900°С в потоке содержащего водород газа и регистрируют интегральное снижение массы. Степень восстановления рассчитывают по отношению потерь массы, используя следующую формулу:
степень восстановления (%)=100·[1 - (потеря массы восстановленного катализатора / потеря массы оксидного предварительного катализатора)].
В процессе восстановления с целью выведения образующейся реакционной воды в реакционную систему можно подавать растворитель. При этом можно подавать также растворитель в надкритическом состоянии.
Пригодными являются растворители, указанные выше в качестве пригодных для суспендирования катализатора. Предпочтительными растворителями являются простые эфиры, такие как метил-трет-бутиловый эфир, этил-трет-бутиловый эфир и тетрагидрофуран, спирты, такие как метанол, этанол и изопропанол, углеводороды, такие как гексан, гептан и фракции рафината, ароматические соединения, такие как толуол, амиды, такие как диметилформамид и диметилацетамид, или лактамы, такие как N-метилпирролидон, N-этилпирролидон, N-метилкапролактам и N-этилкапролактам. Особенно предпочтительными растворителями являются метанол и тетрагидрофуран. Пригодными являются также смеси указанных растворителей.
Указанные выше реакционные условия в общем случае относятся к восстановлению предварительного катализатора, например, в автоклаве с мешалкой, а также в псевдоожиженном или стационарном слое.
Предлагаемый в изобретении катализатор может быть получен также восстановлением предварительного катализатора источником гидрид-ионов в растворителе. Пригодными источниками гидрид-ионов являются комплексные гидриды, такие как LiAlH4 или NaBH4. Пригодными растворителями являются простые эфиры, такие как метил-трет-бутиловый эфир, этил-трет-бутиловый эфир и тетрагидрофуран, углеводороды, такие как гексан, гептан и фракции рафината, или ароматические соединения, такие как толуол. Особенно предпочтительным растворителем является тетрагидрофуран. Пригодны также смеси указанных растворителей.
В случае использования источника гидрид-ионов восстановление предпочтительно осуществляют в температурном интервале от 10 до 200°С при соответствующем собственном давлении в системе.
Восстановление предварительного катализатора можно осуществлять до степени восстановления, предпочтительно составляющей от 50 до 100%.
С подвергнутым восстановлению катализатором можно обращаться и хранить его в атмосфере инертного газа, такого как азот, или под слоем инертной жидкости, например спирта, воды или продукта той реакции, для осуществления которой используют данный катализатор. Катализатор после восстановления можно также подвергать пассивированию (то есть снабдить его защитным оксидным слоем) посредством содержащего кислород газового потока, такого как воздух или смесь воздуха с азотом.
Термин «катализатор» в нижеследующем описании используют для обозначения катализатора, который получен осуществляемым согласно изобретению восстановлением соответствующего предварительного катализатора, или катализатора, который после активирования подвергнут указанному выше пассивированию содержащим кислород газовым потоком.
Хранение катализатора под слоем инертного вещества или пассивирование катализатора позволяет обращаться с ним и хранить его несложным и безопасным образом. Катализатор перед началом непосредственной реакции при необходимости должен быть освобожден от инертной жидкости, соответственно от пассивирующего слоя, например, благодаря обработке водородом или содержащим водород газом.
Предлагаемые в изобретении катализаторы можно использовать для осуществления способа гидрирования соединений, содержащих по меньшей мере одну ненасыщенную углерод-углеродную, углерод-азотную или углерод-кислородную связь, или частичного или полного гидрирования ядра ароматических соединений.
Для гидрирования, как правило, пригодны соединения, содержащие по меньшей мере одну или несколько гидрируемых до аминов групп амидов карбоновых кислот, нитрильных групп, иминогрупп, енаминовых, или азиновых групп, или групп оксима.
Кроме того, предлагаемый способ можно использовать для гидрирования до спиртов соединений, содержащих по меньшей мере одну или несколько групп сложных эфиров карбоновой кислоты, групп карбоновой кислоты, альдегидных или кетогрупп.
Пригодными являются также ароматические соединения, которые могут быть превращены в ненасыщенные или насыщенные карбоциклы или гетероциклы.
Особенно пригодными соединениями, которые можно использовать для гидрирования предлагаемым в изобретении способом, являются органические нитрильные соединения. Подобные соединения можно гидрировать до первичных аминов.
Пригодными являются следующие нитрилы: для получения этиламина ацетонитрил, для получения пропиламина пропионитрил, для получения бутиламина бутиронитрил, для получения лауриламина лауронитрил, для получения стеариламина стеарилнитрил, для получения N,N-диметил-аминопропиламина N,N-диметиламинопропионитрил и для получения бензиламина бензонитрил. Пригодными являются следующие динитрилы: для получения гескаметилендиамина и/или аминокапронитрила адиподинитрил, для получения 2-метилглутародиамина 2-метилглутародинитрил, для получения 1,4-бутандиамина сукцинонитрил и для получения октаметилендиамина динитрил корковой кислоты. Кроме того, пригодными являются циклические нитрилы; для получения изофорондиамина такие как изофороннитрилимин (изофороннитрил), а для получения м-ксилилендиамина изофталодинитрил. Кроме того, пригодными являются α-аминонитрилы и β-аминонитрилы: для получения 1,3-диаминопропана или ω-аминонитрила такие как аминопропионитрил, а для получения гескаметилендиамина такие как аминокапронитрил. Другими пригодными нитрильными соединениями являются так называемые нитрилы Штреккера, такие как используемый для получения диэтилентриамина иминодиацетонитрил. Для получения толуидиндиамина пригоден динитротолуол. Другими пригодными нитрилами являются β-аминонитрилы, например, продукты присоединения алкиламинов, алкилдиаминов или алканоламинов к акрилонитрилу. Так, например, продукты присоединения этилендиамина к акрилонитрилу можно превратить в соответствующие диамины. В частности, 3-[2-аминоэтил)амино]пропионитрил может быть превращен в 3-(2-амино-этил)аминопропиламин, а 3,3'-(этилендиимино)биспропионитрил, соответственно 3-[2-(3-аминопропиламино)этиламино]пропионитрил, в N,N'-бис(3-аминопропил)этилендиамин.
Предлагаемый в изобретении способ особенно предпочтительно используют для получения N,N-диметиламинопропиламина из N,N-диметиламинопропионитрила и для получения гескаметилендиамина из адиподинитрила.
В качестве восстанавливающего агента можно использовать водород, содержащий водород газ или источник гидрид-ионов.
В общем случае водород используют для гидрирования в значительном стехиометрическом избытке, составляющем от 1 до 25, предпочтительно от 2 до 10, или в стехиометрических количествах. Водород можно возвращать в реакцию в качестве рециркулируемого газа. В общем случае для гидрирования пригоден технически чистый водород. Водород можно использовать для гидрирования также в виде содержащего водород газа, то есть в смеси с инертными газами, такими как азот, гелий, неон, аргон или диоксид углерода.
Гидрирование можно осуществлять также посредством источника гидрид-ионов. Пригодным источником гидрид-ионов являются комплексные гидриды, такие как LiAlH4 или NaBH4.
В случае осуществления способа получения аминов восстановлением нитрилов гидрирование можно осуществлять при добавлении аммиака. При этом молярное отношение добавляемого аммиака к нитрильным группам, как правило, составляет от 0,5:1 до 100:1, предпочтительно от 2:1 до 20:1. Предпочтительным является осуществление способа без добавления аммиака.
Гидрирование можно осуществлять в присутствии жидкости.
Используемой для гидрирования жидкостью может быть жидкость, аналогичная той, в которой осуществляют рассмотренное выше восстановление или суспендирование предварительного катализатора.
Пригодными жидкостями являются, например, спирты с 1-4 атомами углерода, простые алкиловые эфиры с 4-12 атомами углерода или циклические простые эфиры с 4-12 атомами углерода, такие как тетрагидрофуран или трет-бутилметиловый эфир. Пригодными являются также смеси указанных жидкостей. Жидкость может представлять собой также продукт гидрирования.
В предпочтительном варианте гидрирование осуществляют в безводной жидкости.
Перед началом гидрирования катализатор может быть освобожден от инертной жидкости, соответственно от пассивирующего слоя. Процесс подобного освобождения реализуют, например, обработкой катализатора водородом или содержащим водород газом. Предпочтительным является осуществление гидрирования непосредственно перед восстановлением предварительного катализатора в том же реакторе, в котором было выполнено восстановление.
Гидрирование, как правило, осуществляют под давлением, составляющим от 1 до 300 бар, прежде всего от 5 до 200 бар, предпочтительно от 8 до 85 бар и особенно предпочтительно от 10 до 65 бар. Гидрирование предпочтительно осуществляют в соответствии с техникой низкого давления, которое не превышает 65 бар.
Температура гидрирования, как правило, составляет от 40 до 250°С, прежде всего от 60 до 160°С, предпочтительно от 70 до 150°С, особенно предпочтительно от 80 до 130°С.
Гидрирование можно осуществлять, например, в жидкой фазе в автоклаве с мешалкой, барботажной колонне, циркуляционном реакторе, например, таком как струйный петлевой реактор, или в реакторе с неподвижным слоем.
Катализатор можно выделять известными специалистам методами, например, фильтрованием или седиментацией.
Гидрирование можно осуществлять также в газовой фазе в реакторе с неподвижным слоем или реакторе с псевдоожиженным слоем. Общеупотребительные реакторы, используемые для осуществления реакции гидрирования, приведены, например, в Ullmann's Encyclopedia Electronic Release, 2000, раздел «Hydrogenation and Dehydrogenation», страницы 2-3.
Предпочтительным является осуществление гидрирования в суспензии.
В соответствии с особым вариантом осуществления изобретения чаще всего по причине технологической простоты гидрирование осуществляют в том же реакционном сосуде, что и восстановление предварительного катализатора.
Гидрирование можно осуществлять в периодическом, полунепрерывном или непрерывном режиме. Предпочтительным является полунепрерывный или непрерывный режим гидрирования.
Активность и/или селективность предлагаемых в изобретении катализаторов могут снижаться по мере увеличения срока их службы. В связи с этим найден способ регенерации предлагаемых в изобретении катализаторов, в соответствии с которым катализатор подвергают обработке жидкостью. Обработка катализатора жидкостью должна обеспечивать растворение возможно адгезированных соединений, которые блокируют активные центры катализатора. Обработка катализатора жидкостью может состоять в его перемешивании в жидкости или его промывке жидкостью, которая после подобной обработки может быть отделена от катализатора вместе с растворившимися в ней примесями фильтрованием или декантированием.
Пригодной жидкостью, как правило является продукт гидрирования, вода или органический растворитель, предпочтительно выбранный из группы, включающей простые эфиры, спирты или амиды.
В соответствии с другим вариантом осуществления изобретения обработку катализатора жидкостью можно выполнять в присутствии водорода или содержащего водород газа.
Подобную регенерацию можно осуществлять при повышенной температуре, как правило составляющей от 20 до 250°С. Кроме того, использованный катализатор можно подвергнуть сушке и оксидировать адегизированные органические соединения в атмосфере воздуха, превратив их в летучие соединения, такие как диоксид углерода. Перед дальнейшим использованием подвергнутого окислению катализатора для гидрирования его как правило следует активировать указанным выше методом.
В процессе регенерации катализатор можно дополнительно легировать тем или иным соединением элемента b). Подобная операция может быть выполнена таким образом, чтобы произошли пропитка и смачивание катализатора водорастворимым основанием элемента b).
Преимущество настоящего изобретения состоит в том, что благодаря использованию предлагаемого в изобретении катализатора уменьшается потребность в оборудовании и капиталовложениях, а также затраты на эксплуатацию установок гидрирования. Капитальные затраты обычно возрастают прежде всего по мере повышения рабочего давления и вследствие использования растворителей и добавок. Согласно настоящему изобретению гидрирование можно осуществлять также в отсутствие воды и аммиака, в связи с чем необходимость в использовании технологической операции отделения воды и аммиака от продуктов реакции (дистилляция) отсутствует или подобная операция упрощается. Кроме того, отсутствие воды и аммиака позволяет более эффективно использовать существующий объем реактора, поскольку освободившийся объем может служить в качестве дополнительного реакционного объема.
Благодаря осуществлению стадии восстановления предлагаемого в изобретении предварительного катализатора в среде жидкости можно получать катализаторы с меньшими размерами частиц и большей площадью поверхности.
Приведенные ниже примеры служат для пояснения изобретения. Термины
Нагрузку на катализатор указывают в виде частного от деления количества продукта на произведение массы катализатора и времени:
нагрузка на катализатор = количество продукта / (масса катализатора·время реакции).
Нагрузку на катализатор выражают в следующих единицах: [КГпродукт / (КГкатализатор·Ч)] или [Гпродукт / (Гкатализатор·Ч)].
Указанные ниже значения селективности определяли методом газовой хроматографии и рассчитывали по выраженным в процентах площадям хроматографических пиков.
Степень превращения эдукта U(E) вычисляли по формуле:
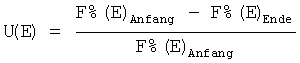 ,
,
в которой (E)Anfang означает площадь хроматографического пика эдукта в начале реакции, %, и (E)Ende - его площадь в конце реакции, %.
Выход продукта А(Р) вычисляли по выраженной в процентах площади соответствующего хроматографического пика:
A(P)=F%(P),
причем выраженная в процентах площадь хроматографического пика F%(i)) эдукта (F%(E)), продукта (F%(P)), побочного продукта (F%(N)) или в общем случае i-го вещества (F%(i)) рассчитывали в виде умноженного на 100 частного от деления площади F(i) хроматографического пика i-го вещества на суммарную площадь Foesamt, то есть сумму площадей хроматографических пиков всех веществ i:
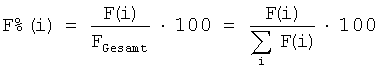
Селективность образования эдукта S(E) рассчитывали в виде частного от деления выхода продукта А(Р) на степень превращения эдукта U(Е):
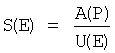 .
.
В случае смешивания диметиламинопропионитрила с диметиламином (DMA) процентные данные относятся к общей площади за вычетом площади хроматографического пика DMA:
 .
.
Это справедливо при условии, что обнаруженный в продукте диметиламин не образовался вследствие расщепления эдукта, а присутствует в нем исключительно благодаря добавлению на более ранней стадии.
Используемые сокращения:
g грамм,
% мас. массовые проценты,
h час(-ы),
кг килограмм,
min минута,
ml миллилитр,
млн-1 части на миллион частей,
% об. объемные проценты,
XRD дифракция рентгеновских лучей,
ADN адиподинитрил,
ACN аминокапронитрил,
DMA диметиламин,
DMAPA N,N-диметиламинопропиламин,
DMAPN диметиламинопропионитрил,
HMD гескаметилендиамин,
ТГФ тетрагидрофуран.
Пример 1
A) Получение предлагаемого в изобретении катализатора
В автоклав высокого давления загружали 80 г ТГФ и 3,0 г LiCoO2. Автоклав герметично закрывали, смесь инертизировали, и автоклав заполняли водородом под давлением 10 бар. Смесь при перемешивании нагревали до 150°С под собственным давлением. По достижении указанной температуры автоклав заполняли водородом под давлением 100 бар. В течение последующих 12 часов осуществляли восстановление предварительного катализатора. После охлаждения давление в автоклаве снижали примерно до 36 бар.
B) Гидрирование диметиламинопропионитрила
Непосредственно после получения катализатора (1А) в реактор при температуре 100°С и давлении 36 бар подавали насосом 0,44 мл/мин сырого диметиламинопропионитрила, содержащего 2,5% мас. диметиламина, поддерживая примерно постоянное давление подачей сжатого водорода. Нагрузка диметиламинопропионитрила на катализатор составляла 7,5 г/г LiCoO2·ч. Селективность образования сырого диметиламинпропиламина в течение периода от 8 до 20 часов составляла от 98,7 до 99,6%. По истечении 20 часов нагрузку на катализатор удваивали. Увеличение нагрузки сопровождалось снижением степени превращения диметиламинопропионитрила до 95% и селективности образования диметиламинпропиламина до 96,1%. Повышение температуры до 140°С и давления до 60 бар обеспечивало восстановление полноты превращения и увеличение селективности до 98,8% (53 часа). Анализ продуктов реакции на литий и кобальт по истечении периода гидрирования, составляющего от 62 до 74 часов, приводил к отрицательному результату (было обнаружено менее 1 млн-1 указанных элементов).
Пример 2
A) Получение предлагаемого в изобретении катализатора
В автоклав с мешалкой загружали 1,5 г LiCoO2 в 35 г ТГФ, и исходное вещество в течение 24 часов активировали при температуре 150°С, давлении водорода 100 бар и энергичном перемешивании. По истечении указанного времени автоклав охлаждали и давление в нем снижали до 10 бар.
B) Гидрирование диметиламинопропионитрила
Непосредственно после получения катализатора (2А) в автоклаве устанавливали температуру 100°С. По достижении указанной температуры автоклав заполняли водородом под давлением 36 бар. Затем в течение 2 часов при перемешивании дозировали 24 г чистого диметиламинопропионитрила при нагрузке на LiCoO2, составляющей 7 г/г·ч, поддерживая примерно постоянное давление подачей сжатого водорода. Спустя два часа дозирование диметиламинопропионитрила прекращали и после минутной выдержки из реактора выгружали 17 г его содержимого. Указанную процедуру повторяли дважды, отбирая соответственно 22 г и 27 г содержимого реактора. В результате анализа в каждом случае было обнаружено полное превращение диметиламинопропионитрила, причем селективность образования диметиламинопропиламина составляла 99,7%. Содержание лития и кобальта в последней выгруженной из реактора порции составляло менее 1 млн-1.
Примеры 1 и 2 подтверждают высокую эффективность предлагаемых в изобретении катализаторов, получаемых из используемого в качестве предварительного катализатора LiCoO2, которая сохранялась в течение длительного времени. Кроме того, удалось показать, что содержащийся в предварительном катализаторе литий в результате восстановления не переходит в растворимую форму и непрерывно выводится из системы. Другое вытекающее из приведенных выше примеров преимущество состоит в том, что катализатор может быть активирован в стандартной аппаратуре в мягких условиях. Присутствие воды с целью повышения активности предлагаемых в изобретении катализаторов на начальном этапе эксперимента не требуется, поскольку несмотря на непрерывное удаление воды катализатор сохранял свою активность.
Пример 3
A) Получение предлагаемого в изобретении катализатора
В автоклав высокого давления загружали 100 г ТГФ и 12 г LiCoO2. Автоклав герметично закрывали, смесь инертизировали, и автоклав заполняли водородом под давлением 10 бар. Смесь нагревали до 200°С при перемешивании под собственным давлением. По достижении указанной температуры в автоклав вводили водород под давлением 100 бар. В течение последующих 24 часов осуществляли восстановление предварительного катализатора катализатора. После охлаждения автоклав заполняли азотом. Затем полученный катализатор (3А) фильтровали под избыточным давлением азота и промывали тетрагидрофураном. Содержание сухого вещества в образовавшейся черной пасте составляло около 37% (33,8 г).
B) Гидрирование ненасыщенных соединений
Катализатор (3А) использовали для выполнения приведенных в таблице 1 опытов 3.1-3.5.
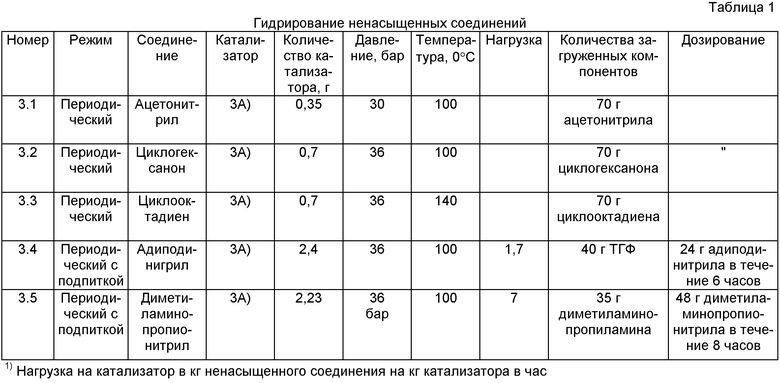
Указанные в таблице 1 количества полученного согласно примеру 3А) катализатора загружали в автоклав с мешалкой и добавляли указанные в таблице 1 количества компонентов. Затем в реакторе устанавливали указанную в таблице 1 температуру. По достижении указанной температуры в автоклав вводили водород, создавая указанное в таблице 1 давление.
После этого в периодических экспериментах (3.1-3.3) включали мешалку и осуществляли гидрирование, поддерживая давление в автоклаве на примерно постоянном уровне подачей сжатого водорода. Длительность гидрирования приведена в соответствующей графе таблицы 2. В таблице 2 приведена также степень превращения и селективность образования продуктов гидрирования.

В периодических экспериментах с подпиткой (3.4-3.6) после активирования предварительного катализатора при перемешивании дозировали указанные количества эдукта, причем дозирование осуществляли в соответствии с последней графой таблицы 1, поддерживая давление в автоклаве на примерно постоянном уровне подачей сжатого водорода. В таблице 2 приведены также результаты выполненного по истечении указанного времени анализа.
В примере 3 показана возможность осуществляемого с чрезвычайно высокой селективностью гидрирования весьма разных соединений с ненасыщенными углерод-углеродными, углерод-азотными или углерод-кислородными связями.
Пример 4
A) Получение предлагаемого в изобретении катализатора
1) Легирование LiCoO2 никелем
12 г LiCoO2 и 1,2 г тетрагидрата ацетата никеля (II) в 50 мл деминерализованной воды в течение 10 часов интенсивно перемешивали в герметичном стеклянном сосуде. Полученный черный порошок (4А-1) выделяли фильтрованием и промывали водой и тетрагидрофураном.
2) Получение катализатора
13,2 г полученного в примере 4А-1 предварительного катализатора в 100 г ТГФ, загруженного в автоклав объемом 300 мл, восстанавливали в течение 24 часов при температуре 200°С и давлении 100 бар. В результате фильтрования продуктов реакции получали 17,8 г восстановленного катализатора, увлажненного тетрагидрофураном. Содержание сухого вещества в восстановленном катализаторе (4А-2) составляло, например, 57%.
B) Гидрирование диметиламинопропионитрила
2,2 г катализатора (4А-2) загружали в автоклав с мешалкой и повышали температуру до 100°С. По достижении указанной температуры автоклав заполняли водородом под давлением 36 бар. Затем в течение 8 часов при перемешивании дозировали 48 г чистого диметиламинопропионитрила при нагрузке на катализатор 4,1 г/г·ч, поддерживая примерно постоянное давление подачей сжатого водорода. Степень превращения исходного диметиламинопропионитрила в диметиламинопропиламин по истечении 8 часов составляла 99,0% при селективности 99,7%.
Пример 4 показывает, что легированный никелем катализатор обладает низкой активностью, однако обеспечивает более высокую селективность гидрирования диметиламинопропионитрила по сравнению с нелегированным катализатором из примера 1А).
Пример 5
А) Использование предлагаемого в изобретении катализатора для гидрирования адиподинитрила
6 г LiCoO2 в 80 г ТГФ восстанавливали аналогично примеру 2А. Затем в течение 6 часов при давлении 36 бар и температуре 100°С дозировали 60 г адиподинитрила. Давление поддерживали на постоянном уровне непрерывной подачей сжатого водорода. Через 6 часов дозирование адиподинитрила прекращали и продолжали гидрирование еще в течение 6 часов. Согласно результатам анализа образца продуктов реакции, выполненного методом газовой хроматографии спустя 6 часов, степень превращения адиподинитрила в гексаметилендиамин и аминокапронитрил составляла 99,8% при селективности 97,6%. При этом было обнаружено образование 97,0% гексаметилендиамина и 0,5% аминокапронитрила. Сравнительный пример 1
A) Получение сравнительного катализатора
В автоклав высокого давления загружали 6 г Со3O4 в 80 г ТГФ и в течение 12 часов при температуре 200°С, давлении 100 бар и энергичном перемешивании предварительный катализатор активировали водородом. По истечении указанного периода автоклав охлаждали до 100°С и снижали давление до 36 бар.
B) Гидрирование адиподинитрила
Непосредственно после получения сравнительного катализатора (V1-A) в течение 6 часов при температуре 100°С, давлении 36 бар и перемешивании дозировали 60 г чистого адиподинитрила при нагрузке на катализатор 1,7 г/г·ч, поддерживая давление на уровне 36 бар подачей сжатого водорода. Через 6 часов дозирование адиподинитрила прекращали и продолжали гидрирование в указанных условиях еще в течение 6 часов. Согласно результатам анализа образца продуктов реакции, выполненного методом газовой хроматографии спустя 6 часов, степень превращения адиподинитрила в гексаметилендиамин и аминокапронитрил составляла 57% при селективности 87,7%. При этом было обнаружено образование 30,5% гексаметилекдиамина и 19,4% аминокапронитрила. Согласно результатам анализа образца продуктов реакции, выполненного методом газовой хроматографии по истечении 12 часов, степень превращения адиподинитрила в гексаметилендиамин и аминокапронитрил составляла 81% при селективности 88,5%. При этом было обнаружено образование 44,4% гексаметилендиамина и 27,2% аминокапронитрила.
Пример 5 и сравнительный пример 1 показывают, что катализатор, полученный восстановлением предварительного катализатора, содержащего предлагаемый в изобретении смешанный оксид, обладает преимуществами по сравнению с катализатором, полученным восстановлением предварительного катализатора, состоящего из чистого оксида кобальта. При одинаковой нагрузке на катализатор производительность предлагаемого в изобретении катализатора значительно превышала производительность катализатора, полученного из чистого оксида кобальта в качестве предварительного катализатора. Несмотря на повышение температуры восстановления на 50°С обеспечиваемая последним степень превращения, в том числе и по истечении дополнительных 6 часов гидрирования, не достигала степени превращения, достигаемой на LiCoO2 уже после 6 часов.
Пример 6
A) Получение предварительного катализатора
Порошкообразный карбонат магния интенсивно перемешивали с гидратированным карбонатом кобальта(II) (регистрационный номер в Chemical Abstracts 513-79-1) при молярном соотношении Мg:Со, составляющем 0,5:1, и полученную смесь подвергали кальцинированию в печи в атмосфере воздуха. Кальцинирование осуществляли, в течение 2 часов повышая температуру до 400°С и выдерживая указанную температуру в течение последующих 2 часов. На рентгеновской дифрактограмме полученного оксидного предварительного катализатора обнаружены рефлексы смешанных кристаллов СоО/МgО и структуры шпинели.
B) Получение предлагаемого в изобретении катализатора
Полученный в результате кальцинирования порошок (пример 6А) в течение двух часов нагревали в обогреваемой, инертизированной азотом восстановительной печи до 300°С, пропуская состоящий из 90% об. азота и 10% об. водорода газовый поток, в течение 16 часов восстанавливали при указанной температуре, а затем охлаждали. После охлаждения содержащую водород атмосферу заменяли на азот. Согласно данным рентгеновской дифракции полученный восстановленный катализатор преимущественно содержал кубический и гексагональный кобальт, а также СоО/МgО.
Полученный восстановленный катализатор (6В) использовали для гидрирования диметиламинопропионитрила.
С) Гидрирование диметиламинопропионитрила
В автоклав с мешалкой загружали 3 г катализатора (6В) и 35 г диметиламинопропиламина. Автоклав заполняли водородом под давлением 10 бар и при слабом перемешивании нагревали до 100°С. По достижении указанной температуры давление водорода в автоклаве повышали до 36 бар и приступали к дозированию диметиламинопропионитрила (6 г/ч). Давление в автоклаве поддерживали на примерно постоянном уровне непрерывной подачей сжатого водорода. Через 8 часов дозирование диметиламинопропионитрила прекращали и осуществляли его гидрирование еще в течение 3 часов. Измеренная через 8 часов степень превращения диметиламинопропионитрила составляла 99,8% при селективности 99,3%. Через 11 часов степень превращения составляла 99,95%, селективность 99,2%.
Пример 7
А) Получение предварительного катализатора
Порошкообразный карбонат лития (регистрационный номер в Chemical Abstracts 554-13-2) интенсивно перемешивали с гидратированным карбонатом кобальта(II) (регистрационный номер в Chemical Abstracts 513-79-1) при молярном соотношении Li:Со, составляющем 1:1, и полученную смесь подвергали кальцинированию в печи в атмосфере воздуха. Кальцинирование осуществляли повышением температуры до 400°С в течение 2 часов и выдерживанием указанной температуры в течение последующих 2 часов. Согласно результатам элементарного анализа молярное отношение Li:Со в полученном предварительном катализаторе составляло 1:1, а измеренная методом БЭТ площадь поверхности составляла 34 м2/г. На рентгеновской дифрактограмме порошкообразного предварительного катализатора (излучение Сu-K-альфа) были обнаружены рефлексы, согласующиеся с тем, что основным кристаллическим компонентом указанного предварительного катализатора является смешанный оксид LiCoO2.
В) Получение предлагаемого в изобретении катализатора
Полученный в результате кальцинирования порошок (пример 7А) в течение двух часов нагревали в обогреваемой, инертизированной азотом восстановительной печи до 300°С, пропуская состоящий из 90% об. азота и 10% об. водорода газовый поток, в течение 16 часов восстанавливали при указанной температуре, а затем охлаждали. После охлаждения содержащую водород атмосферу заменяли на азот.
Полученный восстановленный катализатор (7В) использовали для выполнения примера (7С).
С целью пассивирования катализатора в азотную атмосферу медленно вводили воздух, пока азот не оказывался полностью заменен на воздух.
Полученный пассивированный катализатор использовали для выполнения примеров (7D) и (7Е).
C) Полунепрерывное гидрирование диметиламинопропионитрила Полунепрерывное гидрирование диметиламинопропионитрила осуществляли, используя 3,0 г катализатора из примера (7 В). В автоклав с мешалкой загружали 35 г диметиламинопропиламина, и повышали температуру до 100°С. По достижении указанной температуры автоклав заполняли водородом до давления 36 бар. Затем в течение 8 часов при перемешивании дозировали 35 г диметиламинопропионитрила, нагрузка которого на катализатор составляла около 2 г/г·ч, при этом давление в автоклаве поддерживали на примерно постоянном уровне подачей сжатого водорода. Степень превращения диметиламинопропионитрила после 8-часового дозирования и гидрирования составляла 99,9% при селективности образования диметиламинопропиламина 99,6%.
D) Полунепрерывное гидрирование диметиламинопропионитрила
Для осуществления полунепрерывного гидрирования диметиламинопропионитрила использовали 3,0 г пассивированного катализатора из примера (7В). В автоклав с мешалкой загружали 35 г диметиламинопропиламина, и температуру повышали до 100°С. По достижении указанной температуры автоклав заполняли водородом до давления 36 бар. Затем в течение 8 часов при перемешивании дозировали 35 г диметиламинопропионитрила, нагрузка которого на катализатор составляла около 2 г/г·ч, при этом давление в автоклаве поддерживали на примерно постоянном уровне подачей сжатого водорода. Степень превращения диметиламинопропионитрила после 8-часового дозирования и гидрирования составляла 99,9% при селективности образования диметиламинопропиламина 99,7%.
Е) Непрерывное гидрирование диметиламинопропионитрила
Пассивированный катализатор из примера (7В) использовали для непрерывного гидрирования диметиламинопропионитрила в суспензии без предварительного активирования. При давлении водорода 40 бар, температуре 120°С, содержании катализатора 2,5% мас. и нагрузке диметиламинопропионитрила на катализатор 1,2 кг/кг·ч степень превращения диметиламинопропионитрила после 400 часов гидрирования оставалась на постоянно высоком уровне, составляющем более 99,9%, при одновременном сохранении высокой селективности, составляющей 99,5%, что свидетельствует об отсутствии деактивирования катализатора.
В примере 7 показана возможность использования полностью восстановленного или пассивированного катализатора, причем специальное активирование пассивированного катализатора перед гидрированием не является обязательным.
Пример 7 показывает также, что катализатор пригоден и для осуществления гидрирования в непрерывном режиме.
Пример 8
A) Получение предварительного катализатора
Порошкообразный карбонат лития (регистрационный номер в Chemical Abstracts 554-13-2) интенсивно перемешивали с гидратированным карбонатом кобальта(II) (регистрационный номер в Chemical Abstracts 513-79-1) при молярном соотношении Li:Со, составляющем 0,8:1, и полученную смесь подвергали кальцинированию в печи в атмосфере воздуха. Кальцинирование осуществляли повышением температуры до 400°С в течение 2 часов и выдерживанием указанной температуры в течение последующих 2 часов. На рентгеновской дифрактограмме порошка предварительного катализатора (излучение Сu-K-альфа) обнаружены рефлексы, согласующиеся с тем, что наряду с основным кристаллическим компонентом, представляющим собой смешанный оксид LixCO(1+x/3)O2 нестехиометрического состава, присутствует также, например, Со3O4.
B) Получение предлагаемого в изобретении катализатора
Полученное в результате кальцинирования предварительного катализатора (пример 8А) в течение двух часов нагревали в обогреваемой, инертизированной азотом восстановительной печи до 300°С, пропуская состоящий из 90% об. азота и 10% об. водорода газовый поток, в течение 16 часов восстанавливали при указанной температуре, а затем охлаждали. После охлаждения содержащую водород атмосферу заменяли на азот.
Восстановленный указанным образом катализатор (8В) использовали для гидрирования диметиламинопропионитрила.
С) Гидрирование диметиламинопропионитрила
Полунепрерывное гидрирование диметиламинопропионитрила осуществляли, используя 3,0 г катализатора из примера (8В). В автоклав с мешалкой загружали 35 г диметиламинопропиламина, и температуру повышали до 100°С. По достижении указанной температуры автоклав заполняли водородом под давлением 36 бар. Затем в течение 8 часов при перемешивании дозировали 35 г диметиламинопропионитрила, нагрузка которого на катализатор составляла около 2 г/г·ч, при этом давление в автоклаве поддерживали подачей сжатого водорода на примерно постоянном уровне. Степень превращения диметиламинопропионитрила после 8-часового дозирования и гидрирования составляла 99,8% при селективности образования диметиламинопропиламина 99,8%.
В примере 8 показано, что согласно изобретению пригодными являются также составы предварительного катализатора, которые не состоят только из смешанного оксида.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ АЛЬФА, ОМЕГА-АМИНОНИТРИЛОВ | 1995 |
|
RU2154630C2 |
| СПОСОБ ПОЛУЧЕНИЯ N, N-ЗАМЕЩЕННЫХ 3-АМИНОПРОПАН-1-ОЛОВ | 2009 |
|
RU2522761C2 |
| СПОСОБ ПРОИЗВОДСТВА 3-ДИМЕТИЛАМИНОПРОПИЛАМИНА (ДМАПА) ПРИ НИЗКОМ ДАВЛЕНИИ | 2003 |
|
RU2326108C2 |
| СПОСОБ ОДНОВРЕМЕННОГО ПОЛУЧЕНИЯ КАПРОЛАКТАМА И ГЕКСАМЕТИЛЕНДИАМИНА | 1995 |
|
RU2153493C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ГИДРИРОВАНИЯ НА ОСНОВЕ СПЛАВА АЛЮМИНИЯ И ПЕРЕХОДНОГО МЕТАЛЛА И СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ α, ω-АМИНОНИТРИЛОВ С ЕГО ПОМОЩЬЮ | 1995 |
|
RU2174047C2 |
| СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА ГИДРИРОВАНИЯ, СПОСОБ ГИДРИРОВАНИЯ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ НИТРИЛЬНЫЕ ГРУППЫ | 1998 |
|
RU2190469C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ АЛЬФА, ОМЕГА-АМИНОНИТРИЛОВ | 1995 |
|
RU2158254C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОНИТРИЛА, УЛУЧШЕНИЯ ВЫХОДА И/ИЛИ СЕЛЕКТИВНОСТИ ПО АМИНОНИТРИЛУ И КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2284989C2 |
| ПОЛУЧЕНИЕ АМИНОНИТРИЛОВ (ВАРИАНТЫ) | 1999 |
|
RU2220132C2 |
| СПОСОБ ГИДРИРОВАНИЯ ДИНИТРИЛОВ | 1998 |
|
RU2181716C2 |
Изобретение относится к катализатору на основе смешанных оксидов для гидрирования органических соединений, способу его получения и способу гидрирования. Описан катализатор для гидрирования органических соединений, получаемый восстановлением предварительного катализатора, состоящего из а) кобальта и b) одного или нескольких элементов группы щелочных металлов или щелочноземельных металлов, причем элементы а) и b) по меньшей мере частично находятся в виде их смешанных оксидов. Описан способ получения катализатора для гидрирования органических соединений путем восстановления указанного выше предварительного катализатора. Описан способ гидрирования соединений, содержащих по меньшей мере одну ненасыщенную углерод-углеродную, углерод-азотную или углерод-кислородную связь или частичного или полного гидрирования ядра ароматических соединений с использованием описанного выше катализатора. Технический эффект - повышение стабильности катализатора и упрощение технологии гидрирования органических соединений. 3 н. и 12 з.п. ф-лы, 2 табл.
1. Катализатор для гидрирования органических соединений, получаемый восстановлением предварительного катализатора, состоящего из а) кобальта и b) одного или нескольких элементов группы щелочных или щелочноземельных металлов, причем элементы а) и b) по меньшей мере частично находятся в виде их смешанных оксидов.
2. Катализатор по п.1, получаемый с использованием LiCoO2 в качестве предварительного катализатора.
3. Катализатор по п.1 или 2, получаемый с использованием в качестве предварительного катализатора LiCoO2, выделенного при регенерации батарей.
4. Катализатор по п.1 или 2, получаемый восстановлением предварительного катализатора в органическом растворителе.
5. Катализатор по п.1 или 2, отличающийся тем, что предварительный катализатор получен термообработкой соединений кобальта и одного или нескольких соединений группы щелочных или щелочноземельных металлов.
6. Катализатор по п.5, отличающийся тем, что предварительный катализатор получен термообработкой карбонатов кобальта и одного или нескольких соединений группы щелочных или щелочноземельных металлов.
7. Способ получения катализатора для гидрирования органических соединений, отличающийся тем, что восстанавливают предварительный катализатор, состоящий из а) кобальта и b) одного или нескольких элементов группы щелочных или щелочноземельных металлов, причем элементы а) и b) по меньшей мере частично находятся в виде их смешанных оксидов.
8. Способ по п.7, отличающийся тем, что в качестве предварительного катализатора используют LiCoO2.
9. Способ по п.7 или 8, отличающийся тем, что предварительный катализатор получают термообработкой соединений кобальта и одного или нескольких соединений группы щелочных или щелочноземельных металлов.
10. Способ по п.9, отличающийся тем, что предварительный катализатор получают термообработкой карбонатов кобальта и одного или нескольких соединений группы щелочных или щелочноземельных металлов.
11. Способ гидрирования соединений, содержащих по меньшей мере одну ненасыщенную углерод-углеродную, углерод-азотную или углерод-кислородную связь, или частичного или полного гидрирования ядра ароматических соединений, отличающийся тем, что используют катализатор, получаемый восстановлением предварительного катализатора, состоящего из а) кобальта и b) одного или нескольких элементов группы щелочных или щелочноземельных металлов, причем элементы а) и b) по меньшей мере частично находятся в виде их смешанных оксидов.
12. Способ по п.11, используемый для получения первичных аминов из соединений, содержащих по меньшей мере одну нитрильную группу.
13. Способ по п.11, отличающийся тем, что гидрирование осуществляют при низком давлении.
14. Способ по п.11, отличающийся тем, что используемый катализатор регенерируют путем его обработки жидкостью.
15. Способ по одному из пп.11-14, отличающийся тем, что в качестве предварительного катализатора используют LiCoO2.
| DE 4325847 A1, 02.02.1995 | |||
| Устройство для вращения колесных пар экипажа | 1973 |
|
SU445589A1 |
| DE 19630788 C1, 11.09.1997 | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| JP 2004031165 A, 29.01.2004 | |||
| RU 99104182 A, 27.12.2000 | |||
| СПОСОБЫ ПОЛУЧЕНИЯ АМИНА ИЗ НИТРИЛА ГИДРИРОВАНИЕМ (ВАРИАНТЫ) | 1999 |
|
RU2233266C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОНИТРИЛА И ДИАМИНА | 1999 |
|
RU2210564C2 |

