Настоящее изобретение относится к способу получения N,N-замещенных 3-аминопропан-1-олов.
Получение N,N-замещенных 3-аминопропан-1-олов описано, например, в европейской заявке на патент EP-A1-0673918. Согласно этой публикации N,N-замещенные 3-аминопропан-1-олы получают путем превращения этиленциангидрина со вторичными аминами на палладиевом катализаторе при повышенных температурах и давлениях.
В европейской заявке на патент EP-A2-0869113 описано получение N,N-замещенных 3-аминопропан-1-олов из этиленциангидрина и вторичных аминов, таких как диметиламин, причем реакцию осуществляют в присутствии содержащего палладий катализатора при температурах от 50 до 250°C и давлении от 5 до 350 бар. Используемый катализатор на носителе содержит от 0,1 до 10% масс. палладия в пересчете на общую массу катализатора и по меньшей мере один другой металл, выбранный из элементов групп IB и VIII периодической системы, церия и лантана.
В основу настоящего изобретения была положена задача предложить способ получения N,N-замещенных 3-аминопропан-1-олов из акролеина. Цель изобретения прежде всего состояла в том, чтобы предложить новую технологию синтеза технически важных N,N-замещенных 3-аминопропан-1-олов, таких как N,N-диметил-3-аминопропан-1-ол, в соответствии с которой используют исходные вещества, получаемые из возобновляемого сырья. Акролеин может быть получен, например, путем дегидратации глицерина, который, в свою очередь, можно выделять в качестве побочного продукта из продуктов омыления жиров или из продуктов синтеза биодизельного топлива. Использование возобновляемых ресурсов для получения N,N-замещенных 3-аминопропан-1-олов может способствовать сбережению обычно используемых для этой цели нефтехимических ресурсов.
Кроме того, задача настоящего изобретения состояла в том, чтобы предложить способ получения N,N-замещенных 3-аминопропан-1-олов, позволяющий синтезировать их с высоким выходом и высокой селективностью, причем способ должен отличаться технической простотой и высокой экономичностью.
Превращение акролеина со вторичными аминами описано в немецкой заявке на патент DE-A-4232424, а также в публикации H.D.Finch, E.A.Peterson, S.A.Ballard, J. Am. Chem. Soc., 74, 2016 (1952).
Согласно заявке DE-A-4232424 из 2-алкеналей и вторичных аминов получают соответствующие N,N,N',N'-замещенные ненасыщенные амины, которые на последующей стадии можно подвергать гидрированию до насыщенных аминов. Однако в цитируемой публикации отсутствуют данные относительно возможности высокоселективного получения N,N-замещенных 3-аминопропан-1-олов путем превращения акролеина со вторичными аминами и последующего гидрирования продуктов этого превращения.
В публикации H.D.Finch, E.A.Peterson, S.A.Ballard, J. Am. Chem. Soc., 74, 2016 (1952) описано взаимодействие акролеина, соответственно метакролеина, с первичными или вторичными аминами. Образующиеся при этом N,N'-замещенные 1,3-пропилендиамины, соответственно N,N,N',N'-замещенные 1,3-пропилендиамины, на последующей стадии гидрируют до соответствующих насыщенных N,N'-замещенных 1,3-пропандиаминов, соответственно N,N,N',N'-замещенных 1,3-пропан-диаминов, или нагревают вместе с другими аминами с целью осуществления аминного обмена. В цитируемой публикации сообщается, что в присутствии используемого в качестве катализатора никеля Ренея® аминный обмен и гидрирование можно осуществлять также одновременно. Так, например, смесь изопропилпропиламина и N-изопропил-1,3-пропандиамина получают в две стадии, причем на первой стадии осуществляют взаимодействие акролеина с изопропиламином, в то время как на второй стадии полученную реакционную смесь после удаления избыточного амина и растворителя гидрируют аммиаком в присутствии никеля Ренея® при температуре 105°C. В цитируемой публикации отсутствуют сведения относительно возможности высокоселективного образования N,N-замещенных 3-аминопропан-1-олов.
Неожиданно было обнаружено, что благодаря целенаправленному выбору и комбинированию технологических параметров N,N-замещенные 3-аминопропан-1-олы можно получать из акролеина и вторичных аминов с высокой селективностью.
Таким образом, указанная выше задача согласно изобретению решается благодаря способу получения N,N-замещенных 3-аминопропан-1-олов путем:
a) превращения вторичного амина с акролеином при температуре от -50 до 100°C и давлении от 0,01 до 300 бар, и
b) превращения полученной на стадии a) реакционной смеси с водородом и аммиаком в присутствии катализатора гидрирования при давлении от 1 до 400 бар,
отличающемуся тем, что молярное отношение вторичного амина к акролеину на стадии a) составляет 1:1 или более и температура на стадии b) находится в интервале от -50 до 70°C.
На первой стадии a) предлагаемого в изобретении способа осуществляют взаимодействие акролеина со вторичным амином.
Используемый при этом акролеин обычно получают путем окисления пропилена или путем дегидратации глицерина.
Обычно акролеин получают путем окисления пропилена. Получение акролеина путем окисления пропилена подробно описано, например, в энциклопедии Ульмана (Ullmann's Encyclopedia of Industrial Chemistry, Acrolein and Methacrolein, раздел 3.1 "Acrolein by Propene Oxidation", издательство Wiley-VCH-Verlag, электронная версия, 2007).
Однако в предпочтительном варианте осуществления предлагаемого в настоящем изобретении способа используют акролеин, получаемый путем дегидратации глицерина. Получение акролеина путем дегидратации глицерина описано, например, в международной заявке на патент WO-A2-2006087083, европейском патенте EP-B1-598228, международной заявке на патент WO-A1-2007090990, патентах США US 5079266 и US 2558520, а также в публикации S.H.Chai, H.P.Wang, Y.Lang, В.Q.Xu, Journal of Catalysis, 250 (2), 342-349 (2007).
Глицерин обычно образуется в качестве побочного продукта при превращении жиров и масел в жирные кислоты (омылении жиров) или сложные метиловые эфиры жирных кислот (синтезе биодизельного топлива). Получение глицерина из жиров и масел описано, например, в энциклопедии Ульмана (Ullmann's Encyclopedia of Industrial Chemistry, Glycerol, раздел 4.1 "Glycerol from Fat and Oils", издательство Wiley-VCH-Verlag, электронная версия, 2007).
Глицерин можно получать также из пропилена в качестве исходного нефтехимического продукта. Технология синтеза глицерина из пропилена также описана в энциклопедии Ульмана (Ullmann's Encyclopedia of Industrial Chemistry, Glycerol, раздел 4.1 "Synthesis from Propene", издательство Wiley-VCH-Verlag, электронная версия, 2007).
Для осуществления предлагаемого в изобретении способа как правило не имеет значения, каким методом получен глицерин. Для осуществления предлагаемого в изобретении способа в качестве исходного вещества можно использовать глицерин на растительной, животной или нефтехимической основе.
В еще более предпочтительном варианте осуществления предлагаемого в изобретении способа в качестве исходного продукта для получения акролеина используют глицерин на основе возобновляемого сырья, например глицерин, который образуется в качестве побочного продукта при омылении жиров, соответственно синтезе биодизельного топлива. Преимущество указанного особого варианта состоит в том, что технически важные аминоспирты, такие как N,N-диметил-3-аминопропан-1-ол, можно получать из возобновляемых сырьевых ресурсов. Использование возобновляемых ресурсов для получения подобных продуктов может способствовать сбережению обычно используемых для этой цели нефтехимических ресурсов.
В соответствии с предлагаемым в изобретении способом предпочтительно используют акролеин с содержанием основного вещества, составляющим по меньшей мере 95%, предпочтительно по меньшей мере 98%, особенно предпочтительно по меньшей мере 99%.
В качестве другого исходного вещества в соответствии с предлагаемым в изобретении способом используют вторичный амин.
В качестве вторичного амина можно использовать алифатические, циклоалифатические или циклические вторичные амины.
В качестве циклического вторичного амина можно использовать, например, пирролидин, имидазол, пиперидин, морфолин или пиперазин.
Согласно изобретению предпочтительно используют вторичные амины формулы (I):
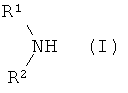 ,
,
в которой остатки R1 и R2 одновременно или независимо друг от друга соответственно означают неразветвленный, разветвленный или циклический углеводородный остаток с 1-20 атомами углерода, соответственно 3-20 атомами углерода, который может быть однократно или многократно ненасыщенным.
Остатки R1 и/или R2, например, могут означать:
алкил с 1-6 атомами углерода, например метил, этил, н-пропил, 1-метилэтил, н-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил-н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 2,2-диметилпропил, 1-этилпропил, н-гексил, 1,1-диметилпропил, 1,2-диметилпропил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,2-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, 1-этилбутил, 2-этилбутил, 1,1,2-триметилпропил, 1-этил-1-метилпропил или 1-этил-3-метилпропил,
алкил с 1-12 атомами углерода, например, указанный выше алкил с 1-6 атомами углерода, а также гептил, 2-метилгексил, 3-метилгексил, 2,2-диметилпентил, 2,3-диметилпентил, 2,4-диметилпентил, 3,3-диметил-пентил, 2,2-диметил-3-метилбутил, октил, 2-метилгептил, 3-метил-гептил, 4-метилгептил, 2,2-диметилгексил, 2,3-диметилгексил, 2,4-диметилгексил, 3,3-диметилгексил, 2,2-диметил-3-метилпентил, 2-метил-3,3-диметилпентил, 2,3,4-триметилпентил, 2,2,3,3-тетраметил-бутил, 1-нонил, 1-децил, 1-ундецил или 1-додецил,
алкил с 1-20 атомами углерода, например, указанный выше алкил с 1-12 атомами углерода, а также 1-тридецил, 1-тетрадецил, 1-пентадецил, 1-гексадецил, 1-гептадецил, 1-октадецил, нонадецил или эйкозил,
циклоалкил с 3-8 атомами углерода, например циклопропил, циклобутил, циклопентил, циклогексил или циклооктил,
циклоалкил с 3-12 атомами углерода, например, указанный выше циклоалкил с 3-8 атомами углерода, а также циклододецил,
алкенил с 2-6 атомами углерода, например этенил, пропиленил, бутенил, пентенил или гексенил,
алкенил с 2-20 атомами углерода, например указанный выше алкенил с 2-6 атомами углерода, а также гептенил, октенил, ноненил или деценил,
циклоалкенил с 3-6 атомами углерода, например циклопропенил, циклобутенил, циклопентенил или циклогексенил,
арил, например одноядерный, двухядерный или трехядерный ароматический карбоцикл с 6-14 кольцевыми членами, такой как фенил, нафтил или антраценил,
гетероарил, например, тиенил, фурил, пиразолил, имидазолил, тиазолил или оксазолил; или аралкил с 7-12 атомами углерода, например, такой как фенилметил, 1-фенилэтил 2-фенилэтил, 1-фенилпропил, 2-фенилпропил или 3-фенилпропил.
Остатки R1 и R2 при необходимости могут содержать заместители, тип которых можно варьировать в широких пределах.
Остатки R1 и R2 одновременно или независимо друг от друга предпочтительно означают неразветвленный или разветвленный насыщенный углеводородный остаток с 1-20, соответственно 3-20 атомами углерода.
В случае использования вторичных аминов формулы (I) с содержащими ненасыщенные связи заместителями R1 и/или R2 последние также могут участвовать в реакции гидрирования.
В соответствии с предлагаемым в изобретении способом в качестве вторичных аминов предпочтительно используют диметиламин, диэтиламин, ди-н-пропиламин, диизопропиламин, изопропилэтиламин, ди-н-бутиламин, ди-втор-бутиламин или дициклогексиламин.
При этом особенно предпочтительно используют диметиламин или диэтиламин, прежде всего предпочтительно диметиламин.
Молярное отношение акролеина к вторичному амину согласно изобретению составляет 1:1 или более. Молярное отношение вторичного амина к акролеину как правило находится в интервале от 1,4:1 до 200:1, предпочтительно от 1,8:1 до 100:1, особенно предпочтительно от 2:1 до 50:1, прежде всего предпочтительно от 2:1 до 10:1 и еще более предпочтительно от 2:1 до 5:1.
Превращение акролеина со вторичным амином можно осуществлять без использования катализатора или в присутствии катализатора.
В качестве катализаторов можно использовать, например, твердые кислоты Бренстэда или Льюиса, описанные, в частности, в европейской заявке на патент EP-A1-449089 (страница 2, колонка 2, строки 11-20), а также в статье K.Tanabe, Studies in Surface Science and Catalysis, том 51, 1989, с.1 и следующие. Соответствующими примерами являются кислые катализаторы на основе оксидов металлов, таких как оксид алюминия, диоксид титана, диоксид циркония и диоксид кремния. Кроме того, в качестве катализатора можно использовать содержащие ионы аммония неорганические или органические ионообменные вещества, такие как цеолиты, сульфированные сополимеры стирола с дивинилбензолом (например, продукты Lewatit® фирмы Lanxess или Amberlite® фирмы Rohm & Haas) или ионообменные вещества на основе силоксана (например, продукт Deloксан® фирмы Degussa).
Превращение акролеина со вторичным амином можно осуществлять в присутствии растворителя, например простого эфира, такого как метил-трет-бутиловый эфир, этил-трет-бутиловый эфир или тетрагидрофуран; спиртов, таких как метанол, этанол или изопропанол; углеводородов, таких как гексан, гептан или фракции рафината; ароматические соединения, такие как толуол; амиды, такие как диметилформамид или диметилацетамид, а также лактамы, такие как N-метилпирролидон, N-этилпирролидон, N-метилкапролактам или N-этилкапролактам. Кроме того, в качестве растворителя можно использовать соответствующие смеси указанных выше растворителей. Предпочтительным растворителем является тетрагидрофуран. Содержание растворителя может составлять от 5 до 95% масс., предпочтительно от 20 до 70% масс., особенно предпочтительно от 30 до 60% масс. соответственно в пересчете на общую массу реакционной смеси (то есть сумму массы используемых исходных веществ и массы растворителя).
Превращение акролеина со вторичным амином предпочтительно осуществляют без добавления растворителя.
Превращение акролеина со вторичным амином осуществляют при температуре от -50 до 100°C, предпочтительно от -20 до 70°C, особенно предпочтительно от -10 до 40°C, и давлении от 0,01 до 300 бар, предпочтительно от 0,1 до 200 бар, особенно предпочтительно от 1 до 200 бар, еще более предпочтительно при нормальном (атмосферном) давлении. В случае газообразных вторичных аминов, таких как диметиламин, превращение предпочтительно осуществляют при давлении от 5 до 400 бар, особенно предпочтительно от 10 до 300 бар, прежде всего от 15 до 200 бар.
Превращение акролеина со вторичным амином можно осуществлять как в периодическом, так и в непрерывном режиме.
Превращение акролеина со вторичным амином в периодическом режиме можно осуществлять, например, в автоклаве с мешалкой, барботажной колонне или циркуляционном реакторе, таком как струйный реактор с внутренним контуром циркуляции.
В случае периодического превращения акролеина со вторичным амином вторичный амин, соответственно суспензию вторичного амина и катализатора, а также при необходимости используемый растворитель обычно загружают в реактор. С целью обеспечения высокой степени превращения и высокой селективности суспензию вторичного амина и катализатора обычно тщательно смешивают с акролеином в автоклаве, например, посредством турбинной мешалки.
Суспендированный катализатор можно вводить в реакционную смесь и вновь отделять обычными методами (седиментацией, центрифугированием, обычным фильтрованием, фильтрованием в поперечном потоке). Катализатор можно использовать один или несколько раз.
В случае превращения акролеина со вторичным амином, осуществляемого в присутствии катализатора, концентрация последнего преимущественно составляет от 0,1 до 50% масс., предпочтительно от 0,5 до 40% масс., особенно предпочтительно от 1 до 30% масс., прежде всего от 5 до 20% масс. соответственно в пересчете на общую массу состоящей из вторичного амина и катализатора суспензии.
Средний размер частиц катализатора предпочтительно находится в интервале от 0,001 до 1 мм, предпочтительно от 0,005 до 0,5 мм, прежде всего от 0,01 до 0,25 мм.
Превращение акролеина со вторичным амином предпочтительно осуществляют в непрерывном режиме, обычно используя резервуары высокого давления или каскады из резервуаров высокого давления.
Акролеин и вторичный амин предпочтительно пропускают через трубчатый реактор, катализатор в котором находится в виде стационарного слоя.
Акролеин и вторичный амин как правило тщательно перемешивают перед подачей в резервуар высокого давления или в самом резервуаре высокого давления. Перемешивание акролеина со вторичным амином перед подачей в резервуар можно осуществлять, например, посредством статических смесителей. В резервуаре высокого давления могут быть смонтированы также внутренние устройства или смесительные элементы, повышающие эффективность перемешивания акролеина со вторичным амином. Перемешивание указанных реагентов при необходимости можно осуществлять также посредством встроенных мешалок или путем перекачивания реакционной смеси.
Нагрузка на катализатор при непрерывном превращении акролеина со вторичным амином предпочтительно составляет от 0,01 до 10, предпочтительно от 0,05 до 7, особенно предпочтительно от 0,1 до 5 кг акролеина на кг катализатора в час.
Полученная на стадии a) реакционная смесь обычно содержит N,N,N',N'-замещенный 1,3-пропендиамин.
С целью концентрирования N,N,N',N'-замещенного 1,3-пропендиамина полученную на стадии a) реакционную смесь перед использованием на стадии b) подвергают переработке, осуществляемой, например, путем дистилляции или ректификации.
Однако полученную на стадии a) реакционную смесь предпочтительно используют на стадии b) без дополнительной очистки или переработки.
Полученную на стадии a) реакционную смесь на стадии b) подвергают превращению с водородом и аммиаком, осуществляемому в присутствии катализатора гидрирования.
В соответствии с предлагаемым в изобретении способом для превращения на стадии b) используют водород.
В общем случае для гидрирования можно использовать технически чистый водород. Водород можно использовать также в виде водородсодержащего газа, то есть в виде смеси водорода с инертными газами, такими как азот, гелий, неон, аргон или диоксид углерода. В качестве водородсодержащих газов можно использовать, например, отходящие газы печи для риформинга, отходящие газы системы очистки нефтепродуктов и так далее при условии, что указанные газы не содержат отравляющих катализаторы гидрирования веществ, например, таких как монооксид углерода. Однако для осуществления предлагаемого в изобретении способа предпочтительно, соответственно преимущественно, используют чистый водород, например водород, степень чистоты которого составляет более 99% масс., предпочтительно более 99,9% масс., особенно предпочтительно более 99,99% масс., прежде всего более 99,999% масс.
Кроме того, в соответствии с предлагаемым в изобретении способом используют аммиак.
В качестве аммиака можно использовать обычный коммерчески доступный продукт, степень чистоты которого составляет, например, более 98% масс., предпочтительно более 99% масс., предпочтительно более 99,5% масс., прежде всего более 99,9% масс.
Молярное отношение используемого на стадии b) аммиака к используемому на стадии a) акролеину предпочтительно составляет от 1:1 до 1000:1, предпочтительно от 2:1 до 100:1, особенно предпочтительно от 4:1 до 50:1.
Превращение на стадии b) можно осуществлять также в присутствии воды.
Количество воды предпочтительно выбирают таким образом, чтобы молярное отношение воды к используемому на стадии a) акролеину находилось в интервале от 0,01:1 до 2:1, предпочтительно от 0,1:1 до 1,8:1, особенно предпочтительно от 0,3:1 до 1,7:1, прежде всего предпочтительно от 0,4: 1 до 1,6:1.
Воду и полученную на стадии a) реакционную смесь можно подавать на стадию b) совместно, например, в виде предварительно перемешанного потока реагентов, или по отдельности. В случае раздельного введения воду и полученную на стадии a) реакционную смесь можно подавать на стадию b) одновременно, со смещением во времени или последовательно. Возможен также вариант, в соответствии с которым подачу воды осуществляют до реализации стадии a), а, следовательно, вода присутствует при реализации стадии a), не оказывая на нее существенного влияния. Однако воду предпочтительно добавляют лишь перед началом осуществления стадии b).
Предлагаемый в изобретении способ реализуют в присутствии катализатора гидрирования.
Используемые в соответствии с предлагаемым в изобретении способом катализаторы гидрирования содержат один или несколько металлов групп 8, 9, 10 и/или 11 периодической системы элементов (в редакции IUPAC от 22.06.2007, смотри http://www.iupac.org/reports/periodic_table/IUPAC_Periodic_Table-22Jun07b.pdf). Примерами подобных металлов являются медь, кобальт, никель и/или железо, а также благородные металлы, такие как родий, иридий, рутений, платина, палладий и рений.
Для осуществления предлагаемого в изобретении способа катализатор гидрирования можно использовать в металлической форме, например, в виде сеток или решеток, или в виде губчатых или скелетных катализаторов Ренея.
В предпочтительном варианте осуществления предлагаемого в изобретении способа металлические катализаторы используют в виде губчатых или скелетных катализаторов Ренея. При этом особенно предпочтительно используют никелевые и/или кобальтовые катализаторы Ренея.
Никелевые и кобальтовые катализаторы Ренея обычно получают путем обработки сплава алюминия с никелем, соответственно сплава алюминия с кобальтом, концентрированным едким натром, в процессе которой происходит выщелачивание алюминия и образование губчатого никеля, соответственно кобальта. Получение катализаторов Ренея описано, например, в справочнике по гетерогенному катализу (статья М.S.Wainright в G.Ertl, H.Knözinger, J.Weitkamp (издатели), Handbook of Heterogeneous Catalysis, том 1, издательство Wiley-VCH, Вейнгейм, Германия, 1997, с.64 и следующие). Указанные катализаторы поставляет, например, фирма Grace (катализаторы Ренея®) или фирма Johnson Matthey (катализаторы Sponge Metal®).
В предпочтительном варианте осуществления предлагаемого в изобретении способа используют кобальтовые катализаторы Ренея.
Молярное содержание атомов кобальта в катализаторе гидрирования предпочтительно составляет 50% мол. и более, особенно предпочтительно 75% мол., еще более предпочтительно 90% мол. и более, прежде всего предпочтительно 99% мол. и более (в пересчете на сумму всех атомов металлов используемого для гидрирования металлического катализатора).
Состав используемого в металлической форме катализатора гидрирования может быть установлен методом атомной абсорбционной спектрофотометрии, атомной эмиссионной спектрометрии, рентгенофлуоресцентного анализа или оптической эмиссионной спектрометрии с использованием индуктивно связанной плазмы.
Катализаторы гидрирования, используемые в соответствии с предлагаемым в изобретении способом, могут быть получены также путем восстановления соответствующих предварительных веществ.
Предварительное вещество катализатора содержит активную массу, которая состоит из одного или нескольких каталитически активных компонентов и при необходимости материала носителя.
Под каталитически активными компонентами подразумевают кислородсодержащие соединения металлов групп 8, 9, 10 и/или 11 периодической системы элементов (в редакции IUPAC от 22.06.2007). Примерами подобных металлов являются медь, кобальт, никель и/или железо, а также благородные металлы, такие как родий, иридий, рутений, платина, палладий или рений, которые находятся в форме кислородсодержащих соединений, например оксидов, соответственно гидроксидов металлов, таких как CoO, NiO, CuO или RuO(OH)X.
Предпочтительными металлами являются медь, кобальт, никель и/или железо, а также благородные металлы, такие как родий, иридий, рутений, платина и палладий. Еще более предпочтительными металлами являются медь, никель и/или кобальт.
В особенно предпочтительном варианте осуществления предлагаемого в изобретении способа предварительное вещество катализатора гидрирования в качестве каталитически активного компонента содержит кислородсодержащие соединения кобальта, например CoO и/или смешанный оксид кобальта, такой как LiCoO2. Предварительное вещество катализатора гидрирования в качестве каталитически активного компонента предпочтительно содержит CoO. В соответствии с указанным предпочтительным вариантом предварительное вещество катализатора гидрирования помимо кислородсодержащего соединения кобальта может содержать другие каталитически активные компоненты. В соответствии с указанным предпочтительным вариантом молярное содержание атомов кобальта в пересчете на сумму всех атомов металлов, присутствующих в используемых каталитически активных компонентах, предпочтительно составляет 10% мол. и более, особенно предпочтительно 30% мол. и более, еще более предпочтительно 50% мол. и более, прежде всего 90% мол. и более. Атомарный состав каталитически активных компонентов может быть установлен методом атомной абсорбционной спектрофотометрии, атомной эмиссионной спектрометрии, рентгенофлуоресцентного анализа или оптической эмиссионной спектрометрии с использованием индуктивно связанной плазмы.
В соответствии с настоящим изобретением под каталитически активными компонентами подразумевают указанные выше кислородсодержащие соединения металлов, которые сами по себе не обладают каталитической активностью. Каталитическую активность, необходимую для реализации предлагаемого в изобретении превращения, они как правило приобретают (то есть становятся каталитически активными компонентами) только после восстановления.
В соответствии с настоящим изобретением под массой активной массы подразумевают суммарную массу материала носителя и каталитически активных компонентов.
Используемые для осуществления способа предварительные вещества катализатора помимо активной массы могут содержать также вспомогательные средства для формования, такие как графит, стеариновая кислота и фосфорная кислота, или другие технологические добавки.
Кроме того, используемые для осуществления способа предварительные вещества катализатора могут содержать один или несколько легирующих элементов (степень окисления 0), выбранных из групп 1-14 периодической системы элементов, или их неорганические или органические соединения. Примерами подобных элементов, соответственно их соединений, являются переходные металлы, такие как марганец, соответственно оксиды марганца, рений, соответственно оксиды рения, хром, соответственно оксиды хрома, молибден, соответственно оксиды молибдена, вольфрам, соответственно оксиды вольфрама, тантал, соответственно оксиды тантала, ниобий, соответственно оксиды ниобия или оксалат ниобия, ванадий, соответственно оксиды ванадия или ванадилпирофосфат, цинк, соответственно оксиды цинка, серебро, соответственно оксиды серебра, лантаниды, такие как цезий, соответственно CeO2, или празеодим, соответственно Pr2O3, оксиды щелочных металлов, такие как K2O, карбонаты щелочных металлов, такие как Na2CO3 и K2CO3, оксиды щелочноземельных металлов, такие как SrO, карбонаты щелочноземельных металлов, такие как MgCO3, CaCO3, BaCO3, олово, соответственно оксиды олова, ангидриды фосфорной кислоты и оксид бора (B2O3).
В соответствии с предлагаемым в изобретении способом предварительные вещества катализатора предпочтительно используют в виде веществ, состоящих только из каталитически активной массы, при необходимости вспомогательного средства для формования, например, такого как графит или стеариновая кислота (оно присутствует в том случае, если катализатор используют в виде формованного изделия), и при необходимости одного или нескольких легирующих элементов, причем помимо указанных компонентов предварительные вещества не содержат никаких других каталитически активных сопутствующих веществ. В этой связи материал носителя рассматривают в качестве компонента каталитически активной массы.
Указанные ниже составы относятся к составу предварительного вещества катализатора после его последней термической обработки (в общем случае заключающейся в прокаливании), выполняемой перед восстановлением водородом.
Содержание активной массы в пересчете на общую массу предварительного вещества катализатора обычно составляет 50% масс. или более, предпочтительно 70% масс. или более, особенно предпочтительно от 80 до 100% масс., еще более предпочтительно от 90 до 99% масс., прежде всего от 92 до 98% масс.
Предварительные вещества катализатора можно получать известными методами, например, в соответствии с осадительными реакциями (в частности, путем смешанного осаждения или последующего осаждения) или путем пропитки.
В предпочтительном варианте осуществления предлагаемого в изобретении способа используют предварительные вещества катализатора, которые получают путем пропитки материалов носителя (пропитанные предварительные вещества катализатора).
Используемые для пропитки материалы носителя могут находиться, например, в виде порошков или формованных изделий, таких как стерженьки, таблетки, шарики или кольца. Материал носителя, пригодный для использования в реакторах с псевдоожиженным слоем катализатора, предпочтительно получают путем распылительной сушки.
В качестве материалов носителя можно использовать, например, углерод, такой как графит, сажа и/или активированный уголь, оксид алюминия (гамма, дельта, тета, альфа, каппа, хи или их смеси), диоксид кремния, диоксид циркония, цеолиты, алюмосиликаты или их смеси.
Пропитку указанных выше материалов носителя можно выполнять обычными методами (А.В.Stiles, Catalyst Manufacture-Laboratory and Commercial Preperations, издательство Marcel Dekker, Нью-Йорк, 1983), например, путем поверхностного нанесения раствора соли металла, реализуемого в одну или несколько стадий. В качестве соли металла как правило следует использовать водорастворимые соли, такие как нитраты, ацетаты или хлориды каталитически активных компонентов, соответственно легирующих элементов. Пропитку можно осуществлять также другими пригодными растворимыми соединениями соответствующих элементов.
Непосредственно после пропитки материал носителя как правило подвергают сушке и прокаливанию.
Сушку материала носителя обычно осуществляют при температурах от 80 до 200°C, предпочтительно от 100 до 150°C. Прокаливание материала носителя в общем случае осуществляют при температурах от 300 до 800°C, предпочтительно от 400 до 600°C, особенно предпочтительно от 450 до 550°C.
Пропитку можно осуществлять также так называемым методом начального смачивания, в соответствии с которым материал носителя подвергают увлажнению пропиточным раствором до тех пор, пока водопоглощение не достигнет максимального значения (насыщения). Кроме того, пропитку можно осуществлять отстоявшимся раствором.
В случае многостадийной технологии пропитки целесообразным является осуществление сушки и при необходимости прокаливания между отдельными операциями пропитки. Многостадийную пропитку материала носителя предпочтительно следует выполнять в том случае, если его необходимо снабдить максимальным количеством солей металлов.
Для введения в материал носителя нескольких компонентов пропитку можно осуществлять, например, как одновременно всеми солями металлов, так и последовательно отдельными солями металлов, причем последовательность пропитки в последнем случае может быть произвольной.
Получаемые в результате пропитки предварительные вещества катализатора содержат каталитически активные компоненты в виде смеси соответствующих кислородсодержащих соединений, то есть прежде всего оксидов, смешанных оксидов и/или гидроксидов. Получаемые при этом предварительные вещества катализатора можно хранить как таковые.
В другом предпочтительном варианте осуществления изобретения предварительные вещества катализатора получают путем совместного осаждения всех компонентов. При этом как правило растворимую соль каталитически активных компонентов, соответственно легирующих элементов, и при необходимости растворимое соединение материала носителя перемешивают с осадителем при нагревании в жидкости до тех пор, пока не наступит полное осаждение.
В качестве жидкости как правило используют воду.
В качестве растворимых солей соответствующих каталитически активных компонентов обычно используют соответствующие нитраты, сульфаты, ацетаты или хлориды металлов групп 8, и/или 9, и/или 10, и/или 11 периодической системы элементов (в редакции IUPAC от 22.06.2007). Примерами подобных металлов являются медь, кобальт, никель и/или железо, а также благородные металлы, такие как родий, иридий, рутений, платина и палладий. Кроме того, пригодными растворимыми солями являются также соответствующие соединения легирующих элементов.
В качестве водорастворимых соединений материала носителя как правило используют водорастворимые соединения алюминия, циркония, кремния и других элементов, например соответствующие водорастворимые нитраты, сульфаты, ацетаты или хлориды.
Кроме того, предварительные вещества катализатора можно получать путем последующего осаждения.
Под последующим осаждением подразумевают метод получения предварительных веществ катализатора, в соответствии с которым трудно растворимый или нерастворимый материал носителя суспендируют в жидкости, после чего к соответствующим оксидам металлов добавляют растворимые соли, которые путем последующего добавления осадителя осаждают на суспендированной подложке (смотри, например, европейскую заявку на патент EP-A2-1106600, с.4, а также А.В.Stiles, Catalyst Manufacture, издательство Marcel Dekker, Inc., 1983, с 15).
В качестве трудно растворимых или нерастворимых материалов носителя можно использовать, например, углеродные соединения, такие как графит, сажа и/или активированный уголь, оксид алюминия (гамма, дельта, тета, альфа, каппа, хи или их смеси), диоксид кремния, диоксид циркония, цеолиты, алюмосиликаты или их смеси.
Материал носителя как правило находится в виде порошка или мелких частиц.
В качестве жидкости, в которой суспендируют материал носителя, обычно используют воду.
В качестве растворимых солей соответствующих каталитически активных компонентов как правило используют соответствующие нитраты, сульфаты, ацетаты или хлориды металлов групп 8, и/или 9, и/или 10, и/или 11 периодической системы элементов (в редакции IUPAC от 22.06.2007). Примерами подобных металлов являются медь, кобальт, никель, железо и/или олово, а также благородные металлы, такие как родий, иридий, рутений, платина и палладий. Кроме того, в качестве растворимых солей можно использовать также соответствующие соединения легирующих элементов.
Тип растворимых солей металлов, используемых при осадительных реакциях (совместном осаждении, соответственно последующем осаждении) в общем случае не является критическим. Поскольку основное значение в данном случае имеет растворимость солей в воде, критерием при их выборе является высокая растворимость в воде, которая необходима для получения растворов, обладающих сравнительно высокой концентрацией. Очевидным является то обстоятельство, что при выборе солей отдельных компонентов безусловно выбирают только соли с такими анионами, которые не могут привести к проблемам, связанным с нежелательными осадительными реакциями, или осложнениям или нарушениям процесса осаждения, обусловленным комплексообразованием.
При осуществлении осадительных реакций растворимые соединения обычно осаждают в виде труднорастворимых или нерастворимых основных солей путем добавления осадителя.
В качестве осадителя предпочтительно используют щелочи, прежде всего минеральные основания, такие как основания щелочных металлов. Примером пригодного осадителя являются карбонат натрия, гидроксид натрия, карбонат калия или гидроксид калия.
В качестве осадителя можно использовать также соли аммония, например галогениды аммония, карбонат аммония, гидроксид аммония или карбоксилаты аммония.
Осадительные реакции можно осуществлять, например, при температурах от 20 до 100°C, в особенности от 30 до 90°C, прежде всего от 50 до 70°C.
Осадки, получаемые в результате реализации осадительных реакций, в общем случае характеризуются отсутствием химической чувствительности и как правило содержат смеси оксидов, гидратов оксидов, гидроксидов, карбонатов и/или гидрокарбонатов используемых металлов. Пригодность осадков для фильтрования может быть повышена благодаря их выдержке, то есть хранению в течение определенного промежутка времени после осаждения, при необходимости реализуемому при одновременном воздействии тепла или пропускании воздуха.
Осадки, получаемые указанными выше методами осаждения, обычно подвергают обработке, состоящей в промывке, сушке, прокаливании и доведении до кондиции.
После промывки осадки в общем случае сушат при температуре от 80 до 200°C, предпочтительно от 100 до 150°C, а затем прокаливают.
Прокаливание в общем случае осуществляют при температурах от 300 до 800°C, предпочтительно от 400 до 600°C, особенно предпочтительно от 450 до 550°C.
Полученные в соответствии с реакциями осаждения предварительные вещества катализатора после прокаливания обычно доводят до кондиции.
Доведение до кондиции может заключаться, например, в измельчении полученного в результате осаждения катализатора до частиц определенного размера.
Полученные в результате измельчения предварительные вещества катализатора можно смешивать со средствами для облегчения извлечения изделий из формы, такими как графит или стеариновая кислота, и подвергать дальнейшей переработке в формованные изделия.
Общеупотребительные методы формования приведены, например, в энциклопедии Ульмана (Ullmann's Encyclopedia Electronic Release 2000, глава "Catalysis and Catalysts", c. 28-32), а также в справочнике Ertl, Knözinger, Weitkamp, Handbook of Heterogenoeous Catalysis, издательство VCH, Вейнгейм, 1997, с.98 и следующие.
Согласно указанным выше литературным источникам путем формования могут быть изготовлены формованные изделия в любом пространственном исполнении, например круглые, многогранные или продолговатые формованные изделия, в частности формованные изделия в виде стержней, таблеток, гранул, шариков, цилиндров или зерен. Общеупотребительными методами формования являются, например, экструзия, таблетирование, то есть механическое прессование, или гранулирование (прессование с использованием круговых и/или вращательных движений).
После доведения до кондиции, соответственно формования, как правило выполняют термическую обработку полученных формованных изделий. Температура термической обработки обычно аналогична температуре прокаливания.
Предварительные вещества катализатора, полученные в результате осуществления осадительных реакций, содержат каталитически активные компоненты в виде смеси соответствующих кислородсодержащих соединений, то есть прежде всего в виде оксидов, смешанных оксидов и/или гидроксидов. Указанные вещества могут быть подвергнуты выдержке как таковые.
В особом предпочтительном варианте осуществления предлагаемого в изобретении способа активная масса предварительного вещества катализатора не содержит материала носителя.
Катализаторы, активная масса которых не содержат материала носителя, как правило получают путем совместного осаждения.
В состав активной массы предварительных веществ катализатора, которые не содержат материала носителя, предпочтительно входит один или несколько активных компонентов, выбранных из группы, включающей CoO, NiO, CuO, RuO(OH)X и LiCoO2.
Активная масса предварительных веществ, не содержащих материала носителя, особенно предпочтительно включает NiO и/или CoO, прежде всего CoO.
Указанными предварительными веществами являются, например, описанные в заявке на патент РСТ/ЕР 2007/052013 катализаторы, в которых перед восстановлением водородом присутствуют a) кобальт и b) один или несколько элементов из группы щелочных металлов, группы щелочноземельных металлов, группы редкоземельных элементов, цинк или смеси указанных элементов, причем по меньшей мере часть элементов A) и b) находится в виде соответствующих смешанных оксидов (например, LiCoO2); описанные в европейской заявке на патент EP-A-0636409 особенно предпочтительные катализаторы, каталитически активная масса которых перед восстановлением водородом содержит от 55 до 98% масс. кобальта (в расчете на CoO), от 0,2 до 15% масс. фосфора (в расчете на H3PO4), от 0,2 до 15% масс. марганца (в расчете на MnO2) и от 0,2 до 15% масс. щелочного металла М (в расчете на M2O), или описанные в европейской заявке на патент EP-A-0742045 катализаторы, каталитически активная масса которых перед восстановлением водородом содержит от 55 до 98% масс. кобальта (в расчете на CoO), от 0,2 до 15% масс. фосфора (в расчете на Н3РО4), от 0,2 до 15% масс. марганца (в расчете на MnO2) и от 0,05 до 5% масс. щелочного металла М (в расчете на M2O).
Предварительные вещества катализатора, которые содержат смешанные оксиды кобальта (такие как LiCoO2), но предпочтительно не содержат материала носителя, в общем случае можно получать путем термической обработки соответствующих соединений кобальта и одного или нескольких соединений элемента из группы щелочных металлов, элемента из группы щелочноземельных металлов, элемента из группы редкоземельных элементов или цинка, например нитратов, карбонатов, гидроксидов, оксидов, ацетатов, оксалатов или цитратов. При этом под термической обработкой подразумевают, например, совместное плавление или прокаливание указанных соединений. Термическую обработку указанных соединений, таких как нитраты, карбонаты, гидроксиды и оксиды, можно осуществлять в атмосфере воздуха. В предпочтительном варианте термическую обработку осуществляют в атмосфере инертного газа, что прежде всего относится к карбонатам. В качестве инертного газа можно использовать, например, азот, диоксид углерода, гелий, неон, аргон, ксенон, криптон или смеси указанных инертных газов. Предпочтительно используют азот.
Способ получения LiCoO2 описан, например, в Е.Antolini, Solid State lonics, 159-171 (2004), а также в W.M.Fenton, P.A.Huppert, Sheet Metal Industries, 25 (1948), 2255-2259.
В качестве предварительного вещества катализатора, которое предпочтительно не содержит материала носителя, можно использовать также LiCoO2, выделенный путем регенерации аккумуляторных батарей. Повторное использование, соответственно регенерацию кобальтита лития из бывших в употреблении аккумуляторных батарей можно осуществлять, например, в соответствии с заявкой Канады на патент CN-A-1594109. Путем механического вскрытия аккумуляторной батареи и выщелачивания содержащих алюминий компонентов концентрированным раствором едкого натра можно получать обогащенный LiCoO2 фильтровальный осадок.
В другом предпочтительном варианте осуществления предлагаемого в изобретении способа активная масса помимо каталитически активных компонентов содержит материал носителя.
Катализаторы, в активной массе которых присутствует материал носителя, как правило получают путем последующего осаждения или пропитки.
В предварительных веществах, которые содержат материал носителя, могут присутствовать один или несколько каталитически активных компонентов, предпочтительно CoO, NiO, CuO и/или кислородсодержащие соединения родия, рутения, платины, палладия и/или иридия.
Активная масса содержащих материал носителя предварительных веществ особенно предпочтительно содержит CuO, NiO и/или CoO, особенно CoO.
В качестве материала носителя предпочтительно используют углерод, такой как графит, сажа и/или активированный уголь, оксид алюминия (гамма, дельта, тета, альфа, каппа, хи или их смеси), диоксид кремния, диоксид циркония, цеолиты, алюмосиликаты и другие, а также смеси указанных материалов.
Содержание материала носителя в активной массе можно варьировать в широких пределах, зависящих от метода получения предварительных веществ катализатора.
В случае предварительных веществ катализатора, получаемых путем пропитки, содержание материала носителя в активной массе как правило составляет более 50% масс., предпочтительно более 75% масс., особенно предпочтительно более 85% масс.
В случае предварительных веществ катализатора, получаемых в соответствии с осадительными реакциями, такими как совместное осаждение или последующее осаждение, содержание материала носителя в активной массе как правило находится в интервале от 10 до 90% масс., предпочтительно от 15 до 80% масс., особенно предпочтительно от 20 до 70% масс.
К подобным предварительным веществам, которые получают в соответствии с осадительными реакциями, относятся, например:
описанные в европейской заявке на патент EP-A-696572 катализаторы, каталитически активная масса которых перед восстановлением водородом содержит от 20 до 85% масс. ZrO2, от 1 до 30% масс. кислородсодержащих соединений меди (в расчете на CuO), от 30 до 70% масс. кислородсодержащих соединений никеля (в расчете на NiO), от 0,1 до 5% масс. кислородсодержащих соединений молибдена (в расчете на MoO3) и от 0 до 10% масс. кислородсодержащих соединений алюминия и/или марганца (в расчете на Al2O3, соответственно MnO2) (смотри, например, приведенный на с.8 цитируемой заявки катализатор, содержащий 31,5% масс. ZrO2, 50% масс. NiO, 17% масс. CuO и 1,5% масс. МоО3),
описанные в европейской заявке на патент EP-A-963975 катализаторы, каталитически активная масса которых перед восстановлением водородом содержит от 22 до 40% масс. ZrO2, от 1 до 30% масс. кислородсодержащих соединений меди (в расчете на CuO), от 15 до 50% масс. кислородсодержащих соединений никеля (в расчете на NiO), причем молярное отношения никеля к меди превышает 1:1, от 15 до 50% масс. кислородсодержащих соединений кобальта (в расчете на CoO), от 0 до 10% масс. кислородсодержащих соединений алюминия и/или марганца (в расчете Al2O3, соответственно MnO2) и не содержит кислородсодержащих соединений молибдена [смотри, например, приведенный на с.17 цитируемой заявки катализатор, содержащий 33% масс. циркония (в расчете на ZrO2), 28% масс. никеля (в расчете на NiO), 11% масс. меди (в расчете на CuO) и 28% масс. кобальта (в расчете на CoO)],
описанные в немецкой заявке на патент DE-A-2445303 медьсодержащие катализаторы (например, указанный в примере 1 медьсодержащий катализатор осаждения, который получают путем обработки раствора нитрата меди и нитрата алюминия бикарбонатом натрия и последующей промывки, сушки и термической обработки осадка, и который содержит около 53% масс. CuO и около 47% масс. Al2O3),
описанные в международной заявке WO 96/36589 катализаторы, прежде всего содержащие иридий, рутений и/или родий, а также активированный уголь в качестве материала носителя,
описанные в европейской заявке на патент EP-А2-1106600 катализаторы, каталитически активная масса которых перед восстановление водородом содержит от 22 до 45% масс. кислородсодержащих соединений циркония, (в расчете на ZrO2), от 1 до 30% масс. кислородсодержащих соединений меди (в расчете на CuO), от 5 до 50% масс. кислородсодержащих соединений никеля (в расчете на NiO), причем молярное отношение никеля к меди превышает 1:1, от 5 до 50% масс. кислородсодержащих соединений кобальта (в расчете на CoO), от 0 до 5% масс. кислородсодержащих соединений молибдена (в расчете на MoO3) и от 0 до 10% масс. кислородсодержащих соединений алюминия и/или марганца (в расчете на Al2O3, соответственно MnO2), описанные в европейской заявке на патент EP-A-1852182 катализаторы, которые содержат кобальт на подложке из оксида цинка и обладают следующим распределением частиц по размерам: <10% частиц размером менее 1 мкм, от 70 до 99% частиц размером от 1 до 5 мкм и <20% частиц размером более 5 мкм,
или описанные в международных заявках WO 2004085356, WO 2006005505 и WO 2006005506 катализаторы, каталитически активная масса которых содержит оксидный материал, состоящий из оксида меди в количестве в интервале от 50≤x≤80% масс., предпочтительно 55≤x≤75% масс., оксида алюминия в количестве в интервале 15≤y≤35% масс., предпочтительно 20≤y≤30% масс., и оксида лантана в количестве в интервале 1≤z≤30% масс., предпочтительно от 2≤z≤25% масс., соответственно в пересчете на общую массу оксидного материала после прокаливания, причем справедливо соотношение 80≤(x+y+z)≤100, особенно 95≤(x+y+z)≤100, а также от 1 до 40% масс. (в пересчете на общую массу оксидного материала) порошкообразной металлической меди, медных чешуек, цементного порошка или их смеси, и от 0,5 до 5% масс. (в пересчете на общую массу оксидного материала) графита, причем суммарное содержание оксидного материала, порошкообразной металлической меди, медных чешуек, цементного порошка или их смеси и графита в изготавливаемом из указанного материала формованном изделии составляет по меньшей мере 95% масс.
Согласно предпочтительному варианту осуществления предлагаемого в изобретении способа предварительные вещества, полученные путем описанной выше пропитки или осаждения, перед использованием в качестве катализаторов подвергают предварительному восстановлению путем обработки водородом, которое как правило выполняют после прокаливания, соответственно доведения до кондиции.
В общем случае предварительные вещества с целью предварительного восстановления сначала подвергают воздействию смеси азота с водородом при температуре от 150 до 200°C в течение промежутка времени от 12 до 20 часов, а затем дополнительной обработке в атмосфере водорода при температуре от 200 до 400°C, длительность которой составляет около 24 часов. При подобном предварительном восстановлении происходит восстановление части присутствующих в предварительных веществах кислородсодержащих соединений металлов до соответствующих металлов, вследствие которого последние присутствуют в активной форме катализатора вместе с разнотипными кислородсодержащими соединениями.
В соответствии с особенно предпочтительным вариантом осуществления изобретения предварительное восстановление предварительного вещества катализатора выполняют в том же реакторе, в котором впоследствии реализуют предлагаемый в изобретении способ.
Манипуляции с полученным в результате предварительного восстановления катализатором, в том числе его хранение, можно осуществлять в атмосфере инертного газа, такого как азот, или в инертной жидкости, например спирте, воде или продукте, для синтеза которого предназначен катализатор. Катализатор после предварительного восстановления может быть подвергнут также пассивированию в содержащем кислород газовом потоке, таком как воздух или смесь воздуха с азотом, обеспечивающему формирование защитного оксидного слоя.
Хранение катализаторов, получаемых путем предварительного восстановления предварительных веществ, в инертной среде или пассивирование указанных катализаторов упрощает обращение с ними и повышает его безопасность. Перед началом непосредственного последующего синтеза катализатор при необходимости следует освобождать от инертной жидкости, соответственно следует удалять пассивирующий слой, например, путем обработки водородом или содержащим водород газом.
Перед началом гидроаминирования катализатор можно не освобождать от инертной жидкости или пассивирующего слоя. Подобное освобождение осуществляют, например, путем обработки катализатора водородом или содержащим водород газом. Гидроаминирование предпочтительно осуществляют непосредственно после восстановления предварительного вещества катализатора в том же реакторе, в котором было выполнено его восстановление.
Однако предварительные вещества катализатора можно использовать также для осуществления синтеза без предварительного восстановления: в подобном случае восстановление происходит в условиях гидрирующего аминирования посредством присутствующего в реакторе водорода, причем катализатор как правило образуется in situ.
Перед использованием на стадии b) катализатор гидрирования можно освобождать от инертной жидкости или пассивирующего слоя. Подобное освобождение осуществляют, например, путем обработки катализатора водородом или содержащим водород газом. Стадию b) предпочтительно реализуют непосредственно после обработки катализатора гидрирования в том же реакторе, в котором была выполнена также его обработка водородом или содержащим водород газом.
Однако предварительные вещества катализатора можно использовать также для реализации стадии b) без предварительного восстановления: в подобном случае восстановление происходит в условиях выполняемого на стадии b) гидрирования посредством присутствующего в реакторе водорода, причем катализатор гидрирования как правило образуется in situ.
Стадию b) (гидрирование) можно выполнять в периодическом или предпочтительно непрерывном режиме.
Стадию b) можно осуществлять как в жидкой, так и в газовой фазе. Стадию b) предпочтительно осуществляют в жидкой фазе.
Превращение полученной на стадии a) реакционной смеси с водородом и аммиаком можно осуществлять в присутствии растворителя, причем предпочтительно используют растворитель, аналогичный используемому на стадии a), например простые эфиры, такие как метил-трет-бутиловый эфир, этил-трет-бутиловый эфир или тетрагидрофуран; спирты, такие как метанол, этанол или изопропанол; углеводороды, такие как гексан, гептан или фракции рафината; ароматические соединения, такие как толуол; амиды, такие как диметилформамид или диметилацетамид, а также лактамы, такие как N-метилпирролидон, N-этилпирролидон, N-метилкапролактам или N-этилкапролактам. В качестве растворителя можно использовать также смеси указанных выше соединений. Растворитель можно использовать в количестве от 5 до 95% % масс., предпочтительно от 20 до 70%, особенно предпочтительно от 30 до 60%, соответственно в пересчете на суммарную массу реакционной смеси стадии a) и растворителя.
Превращение акролеина со вторичным амином предпочтительно осуществляют в отсутствие растворителя.
Гидрирование (стадию b)) в периодическом режиме можно осуществлять, например, в автоклаве с мешалкой, барботажной колонне или циркуляционном реакторе, таком как струйный реактор с внутренним контуром циркуляции.
В случае периодического осуществления гидрирования в реактор обычно загружают суспензию реакционной смеси со стадии a), катализатор гидрирования и при необходимости используемый растворитель. Для достижения высокой степени превращения и высокой селективности суспензию реакционной смеси со стадии a) и катализатора обычно тщательно перемешивают с аммиаком в автоклаве, например, посредством турбинной мешалки. Суспендированный катализатор можно вводить в реакционную смесь и отделять от нее обычными методами (седиментацией, центрифугированием, обычным фильтрованием, фильтрованием в поперечном потоке). Катализатор можно использовать один или несколько раз.
Содержание катализатора преимущественно составляет от 0,1 до 50% масс., предпочтительно от 0,5 до 40% масс., особенно предпочтительно от 1 до 30% масс., прежде всего от 5 до 20% масс. соответственно в пересчете на суммарную массу суспензии, состоящей из реакционной смеси со стадии a) и катализатора.
Средний размер частиц катализатора преимущественно находится в интервале от 0,001 до 1 мм, предпочтительно от 0,005 до 0,5 мм, прежде всего от 0,01 до 0,25 мм.
При периодическом осуществлении стадии b) давление в общем случае составляет от 1 до 400 бар, предпочтительно от 5 до 300 бар, особенно предпочтительно от 10 до 250 бар, прежде всего предпочтительно от 30 до 100 бар.
При этом температура согласно изобретению составляет от -50 до 70°C, предпочтительно от 0 до 70°C, особенно предпочтительно от 20 до 70°C, прежде всего от 35 до 65°C. В другом варианте осуществления способа температура составляет от -50 до 39°C, предпочтительно от 0 до 39°C, особенно предпочтительно от 10 до 39°C.
Стадию гидрирования предпочтительно осуществляют в непрерывном режиме, обычно используя резервуары высокого давления или каскады из резервуаров высокого давления.
В случае непрерывного осуществления гидрирования на стадии b) в жидкой фазе реакционную смесь со стадии a), включая водород и аммиак, предпочтительно пропускают через катализатор гидрирования, который предпочтительно находится в реакторе со стационарным слоем катализатора. Пропускание указанных реагентов можно осуществлять как в режиме орошения, так и в режиме кубовой подачи.
При непрерывном осуществлении стадии b) в жидкой фазе давление в общем случае составляет от 1 до 400 бар, предпочтительно от 5 до 300 бар, особенно предпочтительно от 10 до 250 бар, более предпочтительно от 30 до 100 бар.
При этом температура согласно изобретению составляет от -50 до 70°C, предпочтительно от 0 до 70°C, особенно предпочтительно от 20 до 70°C, прежде всего от 35 до 65°C. В другом варианте осуществления способа температура составляет от -50 до 39°C, предпочтительно от 0 до 39°C, особенно предпочтительно от 10 до 39°C.
В случае непрерывного осуществления гидрирования на стадии b) в газовой фазе реакционную смесь со стадии a) совместно с аммиаком пропускают через катализатор в виде газового потока достаточной для испарения интенсивности в присутствии водорода.
При осуществлении стадии b) в газовой фазе давление в общем случае составляет от 0,1 до 400 бар, предпочтительно от 1 до 100 бар, особенно предпочтительно от 1 до 50 бар. При этом температура согласно изобретению составляет от -50 до 70°C, предпочтительно от 0 до 70°C, особенно предпочтительно от 20 до 70°C, прежде всего от 35 до 65°C. В другом варианте осуществления способа температура составляет от -50 до 39°C, предпочтительно от 0 до 39°C, особенно предпочтительно от 10 до 39°C.
Нагрузка на катализатор при непрерывном осуществлении стадии b) в общем случае находится в интервале от 0,05 до 20, предпочтительно от 0,1 до 15, особенно предпочтительно от 0,2 до 10 кг реакционной смеси со стадии a) на литр катализатора (насыпной объем) в час.
Полученная на стадии b) реакционная смесь содержит N,N-замещенные 3-пропан-1-олы.
Для повышения концентрации N,N-замещенного 3-аминопропан-1-ола полученную на стадии b) реакционную смесь перед дальнейшим использованием, соответственно дальнейшей переработкой можно подвергать переработке, например, путем дистилляции или ректификации.
Непревращенные эдукты, такие как вторичные амины, водород или аммиак, можно возвращать в технологический процесс.
N,N-диметил-3-аминопропан-1-ол можно использовать в качестве катализатора синтеза полиуретанов или в качестве промывочной жидкости для абсорбционной очистки газов. N,N-диметил-3-аминопропан-1-ол можно использовать также в качестве химиката в сфере электроники и гальванотехники.
Кроме того, N,N-диметил-3-аминопропан-1-ол является важным исходным продуктом органического синтеза, а также его можно использовать, например, в качестве промежуточного продукта при получении фармацевтических препаратов и средств защиты растений.
В соответствии с этим объектом настоящего изобретения является также применение N,N-диметил-3-аминопропан-1-ола, который может быть получен предлагаемым в изобретении способом, в указанных выше сферах.
Преимущество настоящего изобретения состоит в том, что предложен способ получения N,N-замещенных 3-аминопропан-1-олов из акролеина, в соответствии с которым достигают высокой селективности превращения исходного акролеина. Благодаря отсутствию необходимости выделения, соответственно очистки образующихся на первой стадии N,N,N',N'-замещенных 1,3-пропилендиаминов перед их превращением в N,N-замещенный 3-аминопропан-1-ол предлагаемый в изобретении способ отличается простотой управления и оптимальной пригодностью для технической реализации. Настоящее изобретение позволяет получать N,N-диметил-3-аминопропан-1-ол из исходных веществ на основе возобновляемых сырьевых ресурсов. Наряду с этим N,N-замещенные 3-аминопропан-1-олы в качестве целевых продуктов образуются с высокой селективностью. Другим преимуществом предлагаемого в изобретении способа является возможность использования на стадии b) катализатора гидрирования, который отличается более высокой экономичностью по сравнению с используемыми согласно уровню техники катализаторами, содержащими платину.
Приведенные ниже примеры служат для более подробного пояснения настоящего изобретения.
Пример 1
В автоклав объемом 270 мл загружают 33,8 г диметиламина (0,75 моль) и 30 г тетрагидрофурана, после чего в течение 60 минут при охлаждении до 4°C насосом дозируют 16,8 г акролеина (0,3 моль) в 30 г тетрагидрофурана. Реагенты перемешивают в течение последующих 15 минут. После этого отбирают образец, который анализируют методом газовой хроматографии. Затем содержимое автоклава передавливают водородом по соединительной линии в автоклав высокого давления объемом 270 мл, в котором уже находится 1,8 г кобальта Ренея (промытого тетрагидрофураном) в 25,5 г аммиака (1,5 моль). Второй автоклав нагревают до 40°C и заполняют находящимся под давлением 60 бар водородом. В течение последующих 3 часов осуществляют гидрирование, поддерживая давление на указанном уровне путем дополнительной подачи водорода. Через 3 часа отбирают образец реакционной смеси, который анализируют методом газовой хроматографии. Площади хроматографического пика N,N-диметил-3-аминопропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 91,5%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропан-диамин (4,6%).
Пример 2
В автоклав объемом 270 мл загружают 67,6 г диметиламина (1,5 моль), после чего в течение 60 минут при охлаждении до 4°C насосом дозируют 16,8 г акролеина (0,3 моль). Реагенты перемешивают в течение последующих 15 минут. После этого отбирают образец, который анализируют методом газовой хроматографии. Затем содержимое автоклава передавливают водородом по соединительной линии в автоклав высокого давления объемом 270 мл, в котором уже находится 1,8 г кобальта Ренея (промытого тетрагидрофураном) в 51,0 г аммиака (1,5 моль). Второй автоклав нагревают до 40°C и заполняют находящимся под давлением 60 бар водородом. В течение последующих 4 часов осуществляют гидрирование, поддерживая давление на указанном уровне путем дополнительной подачи водорода. Через 4 часа отбирают образец реакционной смеси, который анализируют методом газовой хроматографии. Площади хроматографического пика N,N-диметил-3-амино-пропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 91,4%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропандиамин (4,5%).
Пример 3
В автоклав объемом 270 мл загружают 67,6 г диметиламина (1,5 моль), после чего в течение 60 минут при охлаждении до 4°C насосом дозируют 16,8 г акролеина (0,3 моль). Реагенты перемешивают в течение последующих 15 минут. После этого отбирают образец, который анализируют методом газовой хроматографии. Затем содержимое автоклава передавливают водородом по соединительной линии в автоклав высокого давления объемом 270 мл, в котором уже находится 1,8 г кобальта Ренея (промытого тетрагидрофураном) в 51,0 г аммиака (1,5 моль) и 10,8 г (0,6 моль) воды. Второй автоклав нагревают до 23°C и заполняют находящимся под давлением 60 бар водородом. В течение последующих 6 часов осуществляют гидрирование, поддерживая давление на указанном уровне путем дополнительной подачи водорода. Через 6 часов отбирают образец реакционной смеси, который анализируют методом газовой хроматографии. Площади хроматографического пика N,N-диметил-3-аминопропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 58,3%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропандиамин (4,0%).
Пример 4
В автоклав объемом 270 мл загружают 67,6 г диметиламина (1,5 моль), после чего в течение 60 минут при охлаждении до 4°C насосом дозируют 16,8 г акролеина (0,3 моль). Реагенты перемешивают в течение последующих 15 минут. После этого отбирают образец, который анализируют методом газовой хроматографии. Затем содержимое автоклава передавливают водородом по соединительной линии в автоклав высокого давления объемом 270 мл, в котором уже находится 1,8 г кобальта Ренея (промытого тетрагидрофураном) в 51,0 г аммиака (1,5 моль) и 10,8 г (0,6 моль) воды. Второй автоклав нагревают до 60°C и заполняют находящимся под давлением 60 бар водородом. В течение последующих 2 часов осуществляют гидрирование, поддерживая давление на указанном уровне путем дополнительной подачи водорода. Через 2 часа отбирают образец реакционной смеси, который анализируют методом газовой хроматографии. Площади хроматографического пика N,N-диметил-3-аминопропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 33,8%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропандиамин (58,2%).
Пример 5
В автоклав объемом 270 мл загружают 67,6 г диметиламина (1,5 моль), после чего в течение 60 минут при охлаждении до 4°C насосом дозируют 16,8 г акролеина (0,3 моль). Реагенты перемешивают в течение последующих 15 минут. После этого отбирают образец, который анализируют методом газовой хроматографии. Затем содержимое автоклава передавливают водородом по соединительной линии в автоклав высокого давления объемом 270 мл, в котором уже находится 1,8 г кобальта Ренея (промытого тетрагидрофураном) в 51,0 г аммиака (1,5 моль) и 10,8 г (0,6 моль) воды. Второй автоклав нагревают до 40°C и заполняют находящимся под давлением 60 бар водородом. В течение последующих 3 часов осуществляют гидрирование, поддерживая давление на указанном уровне путем дополнительной подачи водорода. Через 3 часа отбирают образец реакционной смеси, который анализируют методом газовой хроматографии. Площади хроматографического пика N,N-диметил-3-аминопропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 57,5%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропандиамин (29,4%).
Сравнительный пример 1
Данный пример выполняют аналогично примеру 5. Однако стадию b) реализуют при температуре 100°C. Площади хроматографического пика N,N-диметил-3-аминопропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 0,4%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропандиамин (91,4%).
Сравнительный пример 2
Данный пример выполняют аналогично примеру 5. Однако стадию b) реализуют при температуре 80°C. Площади хроматографического пика N,N-диметил-3-аминопропан-1-ола, образовавшегося в качестве целевого продукта, соответствует 13,9%. Наряду с указанным целевым продуктом обнаружен N,N-диметилпропандиамин (80,3%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АМИНОВ ИЗ ГЛИЦЕРИНА | 2008 |
|
RU2480449C2 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ ЭТИЛЕНАМИНОВ | 2008 |
|
RU2473537C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКИХ α-АМИНОКИСЛОТ | 2012 |
|
RU2622402C2 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСЕЙ ЭТИЛЕНАМИНОВ | 2008 |
|
RU2478092C2 |
| ПРЕВРАЩЕНИЕ ГЛИКОЛЕВОГО АЛЬДЕГИДА СО СРЕДСТВОМ АМИНИРОВАНИЯ | 2010 |
|
RU2573570C2 |
| Способ каталитического N-полифторалкилирования полиэтиленполиамина полифторированным спиртом | 2024 |
|
RU2830371C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИЭТИЛЕНТЕТРААМИНА | 2008 |
|
RU2470009C2 |
| СПОСОБ АКТИВАЦИИ КАТАЛИЗАТОРА ГИДРООЧИСТКИ | 2004 |
|
RU2351634C2 |
| КАТАЛИЗАТОР НА ОСНОВЕ СМЕШАННЫХ ОКСИДОВ ДЛЯ ГИДРИРОВАНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ГИДРИРОВАНИЯ | 2007 |
|
RU2434676C9 |
| СПОСОБ ПРОИЗВОДСТВА КАТАЛИЗАТОРА ДЛЯ СИНТЕЗИРОВАНИЯ НЕНАСЫЩЕННОЙ КАРБОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2806328C2 |
Изобретение относится к способу получения N,N-замещенных 3-аминопропан-1-олов путем: a) взаимодействия вторичного алифатического амина с акролеином при температуре от -50 до 100°C и давлении от 0,01 до 300 бар и b) взаимодействия полученной на стадии а) реакционной смеси с водородом и аммиаком в присутствии катализатора гидрирования при давлении от 1 до 400 бар. При этом молярное отношение вторичного алифатического амина к акролеину на стадии a) составляет 1:1 до 8:1, температура на стадии b) находится в интервале от 20 до 40°C и молярное отношение используемого на стадии b) аммиака к используемому на стадии a) акролеину составляет от 1:1 до 10:1. Способ позволяет получать целевой продукт с высокой селективностью. 12 з.п. ф-лы, 6 пр.
1. Способ получения N,N-замещенных 3-аминопропан-1-олов путем:
a) взаимодействия вторичного алифатического амина с акролеином при температуре от -50 до 100°C и давлении от 0,01 до 300 бар, и
b) Взаимодействия полученной на стадии а) реакционной смеси с водородом и аммиаком в присутствии катализатора гидрирования при давлении от 1 до 400 бар,
отличающийся тем, что молярное отношение вторичного алифатического амина к акролеину на стадии a) составляет от 1:1 до 8:1, температура на стадии b) находится в интервале от 20 до 40°C и молярное отношение используемого на стадии b) аммиака к используемому на стадии a) акролеину составляет от 1:1 до 10:1.
2. Способ по п.1, отличающийся тем, что молярное отношение вторичного алифатического амина к акролеину составляет от 2:1 до 5:1.
3. Способ по п.1, отличающийся тем, что превращение на стадии b) осуществляют при давлении от 20 до 250 бар.
4. Способ по п.1, отличающийся тем, что катализатор гидрирования находится в металлической форме.
5. Способ по п.1, отличающийся тем, что молярное количество атомов кобальта в пересчете на сумму всех атомов металлов в катализаторе гидрирования, используемом для осуществления способа в металлической форме, составляет 50% мол. или более.
6. Способ по п.4, отличающийся тем, что катализатором гидрирования в металлической форме является губчатый или скелетный катализатор Ренея.
7. Способ по п.1, отличающийся тем, что катализатор гидрирования получают путем восстановления предварительных веществ, которые содержат один или несколько каталитически активных компонентов в виде кислородсодержащих соединений элементов групп 8, и/или 9, и/или 10, и/или 11 периодической системы.
8. Способ по п.7, отличающийся тем, что молярное количество атомов кобальта в пересчете на сумму всех атомов металлов, содержащихся в используемых каталитически активных компонентах, составляет 30% мол. или более.
9. Способ по п.1, отличающийся тем, что полученную на стадии a) реакционную смесь используют на стадии b) без предшествующей дополнительной очистки или переработки.
10. Способ по п.1, отличающийся тем, что используют вторичный алифатический амин, выбранный из группы, включающей диметиламин, диэтиламин, ди-н-пропиламин, диизопропиламин, изопропилэтиламин, ди-н-бутиламин, ди-втор-бутиламин и дициклогексиламин.
11. Способ по п.10, отличающийся тем, что в качестве вторичного алифатического амина используют диметиламин.
12. Способ по одному из пп.1-11, отличающийся тем, что используют акролеин, полученный из глицерина.
13. Способ по п.12, отличающийся тем, что глицерин является глицерином на основе воспроизводимого сырья.
| A.CHESNEY ET AL., “Synthetic approaches towards manzamine | |||
| An easy preparation of β-amino aldehydes”, Synthetic Communications, 20(20),1990, pp.3167-3180 | |||
| УСТРОЙСТВО ДЛЯ РАБОЧЕГО ПЕРЕМЕЩЕНИЯ ГЕОФИЗИЧЕСКИХ АНТЕНН | 2001 |
|
RU2194294C1 |
| Способ дуговой сварки в среде зашитных газов тонкостенного торцевого соединения | 1977 |
|
SU653056A1 |
| RU2006117985 A, 20.01.2008 | |||

