Предметом настоящей заявки являются новые производные имидазола. Изобретение также относится к фармацевтическим композициям, содержащим эти производные, и их применению для получения лекарственных средств.
Производные имидазола по настоящему изобретению имеют противоопухолевую активность и, в частности, являются ингибиторами полимеризации тубулина.
Тубулин, являющийся мишенью ряда противоопухолевых препаратов, представляет собой небольшого размера белок, при полимеризации образующий микротрубочки митотического веретена, которое делает возможным деление клетки во время митоза. Винка-алкалоиды ингибируют полимеризацию тубулина, в то время как паклитаксел и доцетаксел чрезмерно его стабилизируют. В обоих случаях нарушается нормальное протекание митоза, что препятствует пролиферации клеток.
Благодаря своей противоопухолевой активности соединения по настоящему изобретению могут быть использованы для лечения опухолей или рака, включая различные виды рака пищевода, желудка, кишечника, прямой кишки, полости рта, глотки, гортани, легкого, толстой кишки, молочной железы, шейки матки, эндометрия матки, яичников, простаты, яичков, мочевого пузыря, почек, печени, поджелудочной железы, костей, соединительных тканей, кожи, глаз, мозга и центральной нервной системы, а также различных видов рака щитовидной железы, лейкоза, болезни Ходжкина, не-Ходжкинских лимфом, множественных миелом и других. Кроме того, эти соединения могут быть использованы для лечения некоторых вирусных инфекций, таких как синдром приобретенного иммунодефицита, гепатит С, аутоиммунные заболевания и некоторые дегенеративные болезни.
Соединения по изобретению могут быть также использованы для лечения заболеваний, таких как подагра, ревматические заболевания, к примеру наследственная средиземноморская лихорадка, и всех других воспалительных заболеваний (Ben-Chetrit E.; Bergmann S.; Sood R.; Rheumatology 2005; 1-9).
Предметом настоящего изобретения является соединение общей формулы (I)
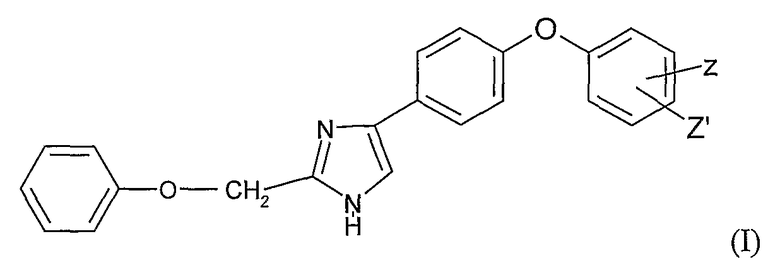
в рацемической, энантиомерной форме или любой комбинации этих форм, в которой
Z' представляет собой атом водорода или галоидный радикал;
Z представляет собой радикал формулы -NH-X-NR1R2;
R1 и R2 независимо представляют собой атом водорода, (С1-С6)алкильный радикал, фенил, необязательно замещенный (С1-С6)алкильным радикалом, или бензил, необязательно замещенный по кольцу (С1-С6)алкильным радикалом;
Х представляет собой -SO2- или
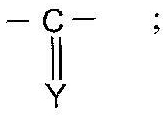
Y представляет собой S, O, CH-R или N-R;
R представляет собой -CN или -NO2;
или его фармацевтически приемлемая соль.
Очевидно, что изобретение охватывает все таутомерные формы соединений формулы (I), как определено выше.
В определениях, указанных выше, выражение галоидный (галогено) означает фторо-, хлоро-, бромо- или йодо-, предпочтительно фторо-, хлоро- или бромо-, радикал. Выражение (С1-С6)алкил означает алкильный радикал, имеющий от 1 до 6 атомов углерода, линейный или разветвленный, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил, пентил или амил, изопентил, неопентил, 2,2-диметилпропил, гексил, изогексил или 1,2,2-триметилпропил.
Предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что R1 и R2 независимо представляют собой атом водорода или (С1-С6)алкильный радикал. Наиболее предпочтительно, R1 представляет собой атом водорода, и R2 представляет собой атом водорода или (С1-С6)алкильный радикал.
Также, предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Х представляет собой -С(Y)-, и Y представляет собой S, O, CH-R или N-R. Наиболее предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Х представляет собой -С(Y)-, и Y представляет собой S, O или N-R. Также, наиболее предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Х представляет собой -С(Y)-, и Y представляет собой S или N-R, и R представляет собой -CN.
Также, предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Х представляет -SO2.
Предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Z находится в параположении.
Предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Z' находится в метаположении.
Предпочтительно, изобретение относится к соединению формулы (I), определенной выше и характеризующейся тем, что Z' представляет собой галоидный радикал, предпочтительно фтор.
Предметом изобретения являются также соединения, приведенные в качестве примеров в экспериментальной части и соответствующие одной из следующих формул:
- N-(4-метилфенил)-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- трифторацетат N-бутил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
- N-этил-N'-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
- N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил] фенокси}фенил)тиомочевина;
- N-изопропил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N'-фенилмочевина;
- N-(4-метилбензил)-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- трифторацетат N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид гидрохлорид;
- N”-циано-N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)гуанидин;
- N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N”-нитрогуанидин;
и, в особенности, имеющие одну из следующих формул:
- N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
- N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
- N-этил-N'-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
- N-(2-фтор-4-{4-[2-(феноксиметил)-1H-имидазол-4-ил]фенокси}фенил)тиомочевина;
- N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид гидрохлорид;
- N”-циано-N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)гуанидин;
- N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N”-нитрогуанидин.
В зависимости от значений варьирующихся групп Z и Z' соединения по изобретению могут быть получены согласно процедурам A-Е, описанным ниже.
Как представлено ниже на Диаграмме А, соединения общей формулы (I), в которой R2 представляет собой атом водорода, R1 представляет собой атом водорода, (С1-С6)алкильный радикал, необязательно замещенный фенильный радикал или необязательно замещенный бензильный радикал, X представляет собой радикал -C(Y), Y представляет собой атом кислорода или атом серы, могут быть получены из коммерчески доступных изоцианатных или изотиоцианатных соединений общей формулы (II) с использованием стандартных методов органического синтеза, известных из уровня техники
Анилиновые производные общей формулы (I') могут быть получены по методу, описанному в WO 2004/106307.
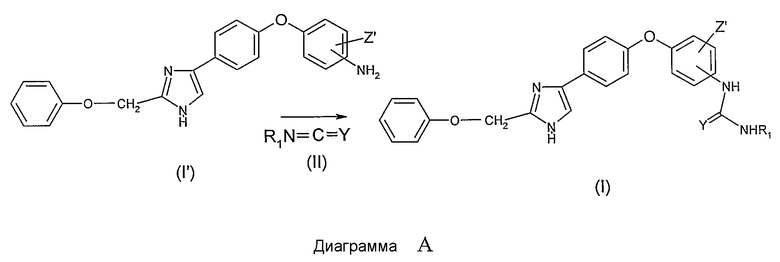
Как представлено ниже на Диаграмме B, соединения общей формулы (I), в которой R1 и R2 представляют собой атомы водорода, X представляет собой радикал -C(Y), Y представляет собой атом кислорода или атом серы, могут быть получены, к примеру, реакцией (тио)фосгена формулы Cl2C=Y в инертном растворителе, таком как тетрагидрофуран или диоксан, с анилиновым производным (I') с получением изо(тио)цианатного производного общей формулы (III) и последующей конденсацией с газообразным аммиаком в растворителе, таком как тетрагидрофуран или диоксан, с получением соединения общей формулы (I)
Как представлено ниже на Диаграмме C, соединения общей формулы (I), в которой R1 представляет собой атом водорода, R2 представляет собой атом водорода или алкильный радикал, X представляет собой -SO2- радикал, могут быть легко получены удалением защиты из промежуточных соединений общей формулы (V), в которой Gp представляет собой защитную группу для аминофункции (например, защитная группа карбаматного типа или любая другая защитная группа, известная из уровня техники, в особенности защитные группы, упомянутые в Protective groups in organic synthesis, 2-е издание, (John Wiley&Sons Inc, 1991)). Производные общей формулы (V) могут быть получены реакцией сульфонилгалоидных соединений, защищенных защитной группой Gp, определенной выше, общей формулы (IV), которые получают по методу, описанному в Bioorganic&Medicinal Chemistry Letters, 13 (2003), 837-840, с анилиновым производным общей формулы (I')
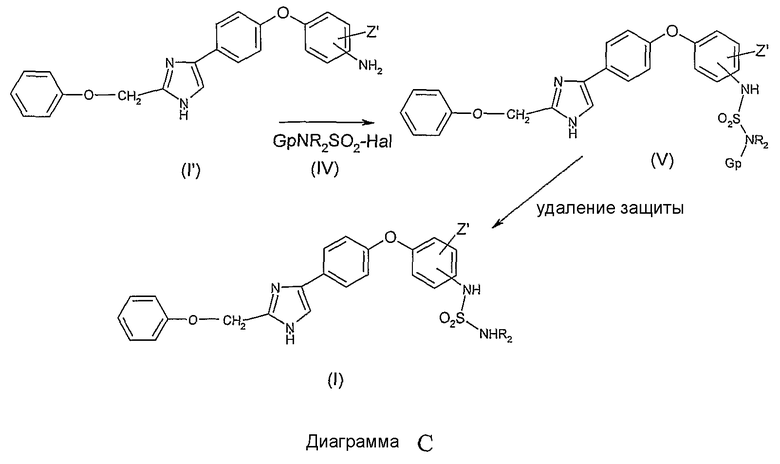
Как представлено ниже на Диаграмме D, соединения общей формулы (I), в которой R1 и R2 представляют собой атомы водорода, X представляет собой радикал -C(Y)-, Y представляет собой радикал N-NO2, могут быть легко получены кипячением с обратным холодильником в растворителе, таком как ацетонитрил, нитрированного производного общей формулы (VI), в которой G'p представляет собой уходящую группу, такую как диметилпиразольную или пиразольную группы, с анилиновым производным общей формулы (I)
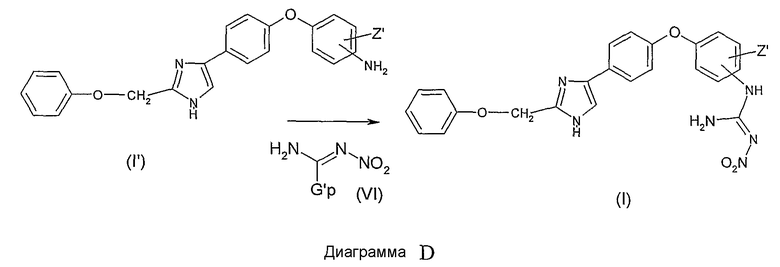
Производные общей формулы (VI) могут быть получены, к примеру, реакцией нитрогуанидина и гидразингидрата в полярном растворителе, таком как вода, и последующей конденсацией получившегося продукта с 2,4-пентадионом в полярном растворителе, таком как, например, вода.
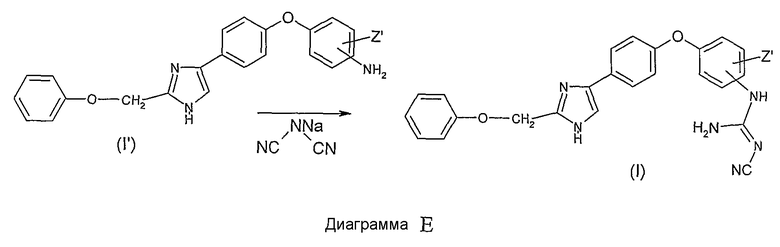
Как представлено ниже на Диаграмме E, соединения общей формулы (I), в которой R1 и R2 представляют собой атомы водорода, X представляет собой радикал -C(Y)-, Y представляет собой радикал -N-CN, могут быть получены способами, известными из уровня техники, к примеру конденсацией анилинового соединения общей формулы (I') с натриевой солью дицианоамида при кипячении с обратным холодильником в растворителе, таком как диметилформамид.
Предметом настоящего изобретения также является способ получения соединения формулы (I) по изобретению, в которой R2 представляет собой атом водорода, R1 представляет собой атом водорода, (С1-С6)алкильный радикал, необязательно замещенный фенильный радикал или необязательно замещенный бензильный радикал, X представляет собой радикал -C(Y), Y представляет собой атом кислорода или атом серы, отличающийся тем, что соединение общей формулы (I')
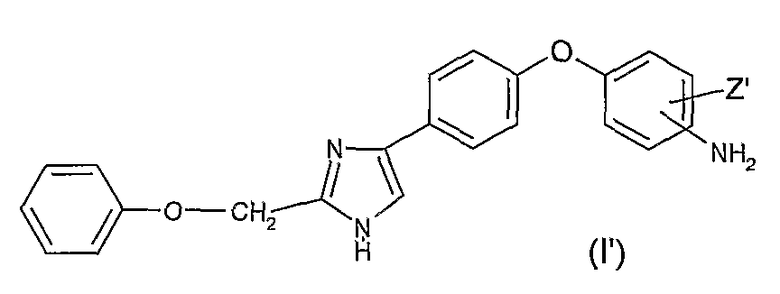
в которой Z' имеет значения, определенные выше, вводят в реакцию с изоцианатным или изотиоцианатным соединением формулы (II) R1N=C=Y, в которой R1 и Y имеют значения, определенные выше.
Предметом изобретения также является способ получения соединения формулы (I) по изобретению, в которой R1 и R2 представляют собой атомы водорода, X представляет собой радикал -C(Y), Y представляет собой атом кислорода или атом серы, отличающийся тем, что (тио)фосген формулы Cl2C=Y вводят в реакцию с соединением формулы (I'), определенной выше, в инертном растворителе с получением изо(тио)цианатного производного общей формулы (III)
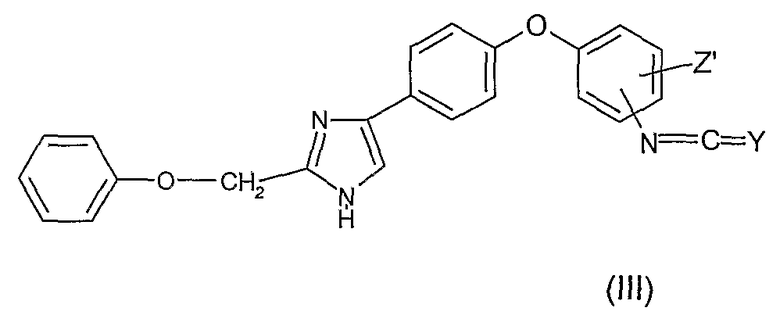
которое на следующей стадии конденсируют с газообразным аммиаком в растворителе с получением соединения общей формулы (I).
Предметом изобретения также является способ получения соединения формулы (I) по изобретению, в которой R1 представляет собой атом водорода, R2 представляет собой атом водорода или алкильный радикал, X представляет собой -SO2- радикал, отличающийся тем, что
- соединение формулы (I'), определенной выше, вводят в реакцию с сульфонилгалоидом формулы GpNR2-SO2-Hal, в которой Gp представляет собой защитную группу для аминофункции, Hal представляет собой галоидный радикал с получением соединения (V)
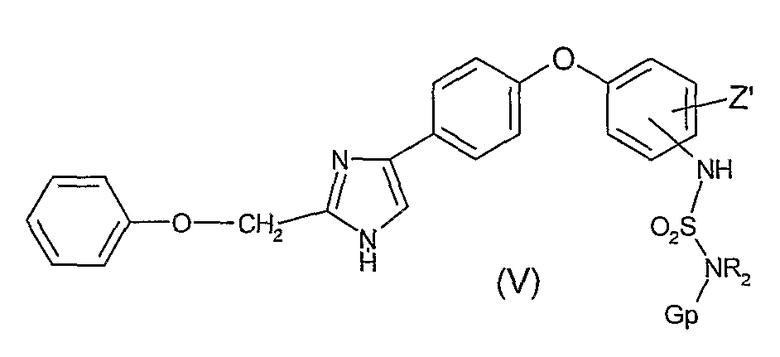
в которой Z' имеет значения, определенные в п. 1,
- из полученного таким образом соединения (V) удаляют защитную группу, получая соединение (I).
Предметом изобретения также является способ получения соединения формулы (I) по изобретению, в которой R1 и R2 представляют собой атомы водорода, X представляет собой радикал -C(Y)-, Y представляет собой радикал N-NO2, отличающийся тем, что нитрированное производное общей формулы (VI)
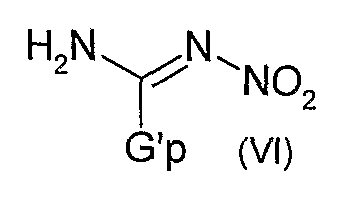
в которой G'p представляет собой уходящую группу, кипятят с обратным холодильником в растворителе с соединением общей формулы (I'), определенной выше.
Предметом изобретения также является способ получения соединения формулы (I) по изобретению, в которой R1 и R2 представляют собой атомы водорода, X представляет собой радикал -C(Y)-, Y представляет собой радикал -N-CN, отличающийся тем, что натриевую соль дицианоамида конденсируют с соединением формулы (I'), определенной выше, при кипячении с обратным холодильником в растворителе.
Соединения формулы (I) по настоящему изобретению обладают полезными фармакологическими свойствами. Было обнаружено, что соединения формулы (I) по изобретению имеют противоопухолевую (противораковую) активность и, в частности, являются ингибиторами полимеризации тубулина. Они также обладают антиревматической активностью. Кроме того, они могут иметь противовоспалительную активность.
Таким образом, соединения по настоящему изобретению могут иметь различные терапевтические применения. Они, преимущественно, могут быть использованы для лечения рака, как определено выше, предпочтительно для лечения рака молочной железы, толстой кишки, простаты, поджелудочной железы и меланом. Они также могут быть использованы для лечения ревматоидных или воспалительных заболеваний, таких как подагра. Иллюстрация фармакологических свойств соединений по изобретению будет приведена ниже в экспериментальной части.
Предметом настоящей заявки на изобретение также являются фармацевтические композиции, содержащие в качестве активного ингредиента, по крайней мере, одно соединение формулы (I), определенной выше, а также дополнительные соли указанных соединений формулы (I) с фармацевтически приемлемыми минеральными или органическими кислотами в комбинации с фармацевтически приемлемой основой.
Под фармацевтически приемлемыми солями подразумеваются, в частности, дополнительные соли с неорганическими кислотами, такие как гидрохлорид, гидробромид, гидройодид, сульфат, фосфат, дифосфат и нитрат, или с органическими кислотами, такие как ацетат, малеат, фумарат, тартрат, сукцинат, лактат, метансульфонат, бензолсульфонат, п-толуолсульфонат, памоат и стеарат. Также в область настоящего изобретения входят, когда они могут быть использованы, соли с основаниями, такими как гидроксид натрия или калия. Другие примеры фармацевтически приемлемых солей могут быть найдены в «Salts selection for basic drugs», Int. J. Pharm (1986), 33, 201-217.
Предметом настоящей заявки на изобретение также является применение соединения формулы (I) по настоящему изобретению для получения противоопухолевого лекарственного средства.
Предметом настоящей заявки на изобретение также является применение соединения формулы (I) по настоящему изобретению для получения лекарственного средства, предназначенного для ингибирования полимеризации тубулина.
Предметом настоящей заявки на изобретение также является применение соединения формулы (I) по настоящему изобретению для получения лекарственного средства, предназначенного для лечения ревматоидных или воспалительных заболеваний.
Предметом настоящей заявки на изобретение также является применение соединения формулы (I) по настоящему изобретению для получения лекарственного средства, предназначенного для лечения подагры.
Соединения по настоящему изобретению могут вводиться в одиночку, или одновременно, или последовательно в комбинации с другими средствами, обладающими противоопухолевой активностью. Среди средств, обладающих противоопухолевой активностью, могут быть упомянуты ингибиторы топоизомеразы I, такие как дифломотекан, иринотекан или топотекан; ингибиторы топозомеразы II; ингибиторы полимеризации тубулина, такие как навельбин; ингибиторы деполимеризации микротрубочек, такие как таксол; алкилирующие агенты, такие как циклофосфамид; фосфамиды или мелфалан; производные платины, такие как цисплатин, карбоплатин или оксалилплатин; антибиотики, такие как блеомицин или митомицин; антиметаболиты, такие как 5-фторурацил; антигормональные агенты и агенты, направленные против факторов роста.
Введение композиций по изобретению также может сочетаться с радиотерапией.
Фармацевтические композиции могут быть в твердой форме, например в виде порошков, гранул, таблеток, желатиновых капсул. Подходящими твердыми основами могут быть, к примеру, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, поливинилпирролидин и воск.
Фармацевтические композиции, содержащие соединение по изобретению, могут быть также представлены в жидком виде, например в виде растворов, эмульсий, суспензий или сиропов. Подходящими жидкими основами могут быть, к примеру, вода, органические растворители, такие как глицерин или гликоли, а также их смеси в различных пропорциях с водой, с добавлением фармацевтически приемлемых масел или жиров. Стерильные жидкие композиции могут быть использованы для внутримышечных, внутрибрюшинных или подкожных инъекций, стерильные композиции также могут быть введены внутривенно.
Все технические и научные термины, использованные в настоящем тексте, имеют значения, известные из уровня техники. Кроме этого, в виде ссылок включены все патенты (или заявки на изобретения), а также библиографические ссылки.
Экспериментальная часть
В зависимости от значений варьирующихся групп Z и Z' соединения по изобретению могут быть получены согласно различным процедурам A-Е, описанным выше.
Следующие примеры приведены в качестве иллюстраций к описанным выше процедурам и не должны рассматриваться как ограничивающие область изобретения.
Пример 1
N-(4-метилфенил)-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и параметилфенилизоцианат (0,089 г, 0,67 ммоль) в 3 мл тетрагидрофурана, перемешивают 24 часа при умеренной температуре. Образовавшийся осадок отфильтровывают, осадок перемешивают в смеси растворителей, таких как изопропиловый эфир/этилацетат/изопропанол (5/4/1). После фильтрации и высушивания получают целевое вещество в виде бежевого порошка с выходом 73%.
1Н-ЯМР (δ мд, ДМСО): 2,24 (с, 3Н); 5,07 (с, 2Н); 6,94-7,94 (м, 18 Н); 8,51 (с, 1Н); 8,60 (с, 1Н); 12,4 (уш.с, 1Н).
МН+ экспериментальное = 491,20; М теоретическое = 491,56; точка плавления: 192-194°С.
Пример 2
Трифторацетат N-бутил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и бутилизоцианат (0,094 г, 0,84 ммоль) в 3 мл тетрагидрофурана, перемешивают в течение дня при умеренной температуре. Добавляют аминометилполистирольную смолу EHL (Novabiochem, 200-400 меш, 2% DVB) (200 мг, приблизительно 1 ммоль) и перемешивают в течение 3 часов. После фильтрования через фритту фильтрат концентрируют на роторном испарителе, полученный остаток делят методом колоночной хроматографии на колонке с обращенно-фазовым силикагелем RP18 (элюент:ацетонитрил - 0,1 N трифторуксусная кислота: 5-5), получая целевое вещество в виде бежевого порошка с выходом 43%.
1Н-ЯМР (δ мд, ДМСО): 0,83-0,91 (м, 5Н); 1,39-1,43 (м, 2Н); 3,07-3,08 (м, 2Н); 5,3 (с, 1Н); 6,11 (м, 1Н); 6,96-7,44 (м, 13Н); 7,91 (м, 1Н); 8,44 (м, 1Н).
МН+ экспериментальное = 457,30; М теоретическое = 456,54; точка плавления: 85-87°С.
Пример 3
N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и триметилсилилизоцианат (0,11 мл, 0,84 ммоль) в 3 мл тетрагидрофурана, перемешивают в течение 24 часов при умеренной температуре. Добавляют аминометилполистирольную смолу EHL (Novabiochem, 200-400 меш, 2% DVB) (200 мг, приблизительно 1 ммоль) и перемешивают в течение 3 часов. После фильтрования через фритту фильтрат концентрируют на роторном испарителе. К полученному остатку добавляют изопропилацетат и следы изопропанола. Полученную смесь нагревают до растворения, отфильтровывают через фритту и оставляют кристаллизоваться в течение 24 часов. После фильтрации и высушивании в вакуумном колпаке получают светло-бежевый порошок.
1Н-ЯМР (δ мд, ДМСО): 5,06 (с, 2Н); 5,79 (с, 2Н); 6,90-7,74 (м, 14Н); 8,50 (с, 1Н); 12,35 (уш.с, 1Н).
МН+ экспериментальное = 401,20; М теоретическое = 400,44; точка плавления: 200-202°С.
Пример 4
N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,5 г, 0,140 ммоль) и этилизотиоцианат (0,162 г, 1,70 ммоль) в 10 мл этанола, кипятят с обратным холодильником в течение 4 часов. К полученному остатку добавляют 5 мл дихлорометана и следы этилацетата и этанола. Полученную смесь перемешивают в течение 30 минут, отфильтровывают, полученный осадок высушивают. Целевое вещество получают в виде порошка кремового цвета с выходом 45%.
1Н-ЯМР (δ мд, ДМСО): 1,1 (м, 3Н); 3,4 (м, 2Н); 5,07 (с, 2Н); 6,94-7,78 (м, 15Н); 9,35 (уш.с, 1Н); 12.40 (уш.с, 1Н).
МН+ экспериментальное = 445,20; М теоретическое = 444,57; точка плавления: 178-180°С.
Пример 5
N-этил-N'-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина
Смесь, содержащую 2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (1 г, 2,66 ммоль) и этилизотиоцианат (0,31 г, 2,32 ммоль) в 20 мл этанола, кипятят с обратным холодильником в течение 5 часов. Реакционную смесь концентрируют на роторном испарителе. Остаток очищают методом колоночной хроматографии на колонке с силикагелем (элюент:дихлорометан/этанол/жидкий аммиак 96/2/2), получая целевое вещество в виде желтого порошка с выходом 11%.
1Н-ЯМР (δ мд, ДМСО): 1,06 (т, 3Н); 3.46 (уш.м, 2Н); 5,08 (с, 2Н); 6,73-7,82 (м, 15Н); 9,03 (уш.с, 1Н).
МН+ экспериментальное = 463,20; М теоретическое = 462,55; точка плавления: 105-107°С.
Пример 6
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина
1) 4-[4-(3-Фтор-4-изотиоцианатофенокси)фенил]-2-(феноксиметил)-1Н-имидазол
К раствору 2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилина (3 г, 0,008 моль) в тетрагидрофуране (120 мл) при 23°С в атмосфере аргона добавляют триэтиламин (3,34 г, 0,024 моль). Реакционную смесь охлаждают до 0°С и по каплям прибавляют тиофосгеновое соединение (1,01 г, 0,0088 моль). Продолжают перемешивание при той же температуре в течение 30 минут, затем повышают температуру реакционной смеси до 23°С и перемешивают в течение 20 минут. К реакционной смеси добавляют 60 мл воды и 150 мл этилового эфира. Разделяют фазы, органическую фракцию промывают насыщенным раствором хлорида натрия. Органическую фракцию высушивают над сульфатом натрия, упаривают на роторном испарителе досуха, остаток растирают с изопентаном, содержащим следы дихлорометана. Полученный осадок отфильтровывают и высушивают, получая целевое вещество в виде желтого порошка с выходом 78%.
МН+ экспериментальное = 418,20; М теоретическое = 417,462.
2) N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина
4-[4-(3-Фтор-4-изотиоцианатофенокси)фенил]-2-(феноксиметил)-1Н-имидазол, полученный на предыдущей стадии (2,58 г, 0,0062 моль), солюбилизируют при 23°С в 50 мл смеси растворителей дихлорометан/метанол/тетрагидрофуран 6/3/1. Реакционную смесь охлаждают до 0°С и барботируют газообразный аммиак до насыщения раствора. Продолжают перемешивание при той же температуре в течение 30 минут, затем повышают температуру реакционной смеси до умеренной и перемешивают в течение 30 минут. Реакционную смесь упаривают на роторном испарителе, полученный твердый остаток растирают со смесью изопропиловый эфир - изопропанол (5:5). Полученный осадок отфильтровывают, затем перекристаллизовывают из минимального количества изопропанола при нагревании. Смесь оставляют охлаждаться, затем отфильтровывают. Целевое вещество получают в виде светло-коричневого порошка с выходом 42%.
1Н-ЯМР (δ мд, ДМСО): 5,08 (с, 2Н); 6,79-7,82 (м, 15Н); 9,26 (с, 1Н); 12,4 (уш.с, 1Н).
МН+ экспериментальное = 435,20; М теоретическое = 434,493; точка плавления: 171-173°С.
Пример 7
N-изопропил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и изопропилизоцианат (0,155 г, 1,8 ммоль) в 3 мл диметилформамида, нагревают до 100°С в течение 2 часов, затем перемешивают при умеренной температуре в течение 2 часов. К реакционной смеси добавляют 20 мл воды и 30 мл этилацетата. Проводят экстракцию, органическую фракцию промывают насыщенным раствором хлорида натрия. Органическую фракцию высушивают над сульфатом натрия, упаривают на роторном испарителе досуха, полученное масло растирают с изопентаном. После фильтрации и высушивания твердый осадок очищают хроматографией на колонке с силикагелем (элюент:дихлорометан-этанол-аммиак 96/2/2), целевое вещество получают в виде розово-кремового порошка с выходом 20%.
1Н-ЯМР (δ мд, ДМСО): 1,07 (д, 6Н); 3,74 (м, 1Н); 5,06 (с, 2Н); 5,93 (м, 1Н); 6,91-7,74 (м, 14Н); 10,19 (с, 1Н); 12,4 (уш.с, 1Н).
МН+ экспериментальное = 443,20; М теоретическое = 442,516; точка плавления: 184-186°С.
Пример 8
N- (4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N'-фенилмочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и фенилизоцианат (0,136 г, 1,12 ммоль) в 3 мл диметилформамида, нагревают до 100°С в течение 2 часов. Смесь охлаждают до умеренной температуры и добавляют 20 мл воды и 30 мл этилацетата. Проводят экстракцию, органическую фракцию промывают насыщенным раствором хлорида натрия. Органическую фракцию высушивают над сульфатом натрия, отфильтровывают, концентрируют досуха в вакууме. Полученный твердый остаток очищают хроматографией на колонке с силикагелем (элюент:дихлорометан-этанол-аммиак 96/2/2), целевое вещество получают в виде белого порошка с выходом 21%.
1Н-ЯМР (δ мд, ДМСО): 5,08 (с, 2Н); 6,94-7,77 (м, 19Н); 8,52 (м, 2Н); 12,4 (уш.с, 1Н).
МН+ экспериментальное = 477,20; М теоретическое = 476,534; точка плавления: 146-148°С.
Пример 9
N-(4-метилбензил)-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и 4-метилбензилизоцианат (0,17 г, 1,12 ммоль) в 3 мл диметилформамида, нагревают до 100°С в течение 2 часов. Смесь охлаждают до умеренной температуры и добавляют 20 мл воды и 30 мл этилацетата. Проводят экстракцию, органическую фракцию промывают насыщенным раствором хлорида натрия. Органическую фракцию высушивают над сульфатом натрия, отфильтровывают, концентрируют досуха в вакууме. Полученный твердый остаток перемешивают в 5 мл смеси растворителей этиловый эфир/этилацетат/изопропанол 6/3/1. Полученный осадок отфильтровывают и высушивают. Целевое вещество получают в виде кремового порошка с выходом 21%.
1Н-ЯМР (δ мд, ДМСО): 4,16-4,25 (м, 2Н); 5,07 (с, 2Н); 6,31-6,52 (м, 1Н); 6,91-7,75 (м, 20Н); 8,52 (м, 1Н); 10,18 (с, 1Н); 12,4 (уш.с, 1Н).
МН+ экспериментальное = 477,20; М теоретическое = 476,534; точка плавления: 146-148°С.
Пример 10
Трифторацетат N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина
Смесь, содержащую 4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,2 г, 0,56 ммоль) и этилизоцианат (0,066 мл, 0,84 ммоль) в 3 мл тетрагидрофурана, перемешивают в течение дня при умеренной температуре. Добавляют аминометилполистирольную смолу EHL (Novabiochem, 200-400 меш, 2% DVB) (200 мг, приблизительно 1 ммоль) и перемешивают в течение 3 часов. После фильтрования через фритту, фильтрат концентрируют на роторном испарителе, полученный остаток делят методом колоночной хроматографии на колонке с обращенно-фазовым силикагелем RP18 (элюент:ацетонитрил - 0,1 N трифторуксусная кислота: 5-5), получая целевое вещество в виде коричневой смолы с выходом 16%.
1Н-ЯМР (δ мд, ДМСО): 0,96-1,03 (м, 3Н); 3,2-4,2 (уш.м, 1Н); 3,60 (м, 2Н); 5,29 (с, 2Н); 6,11 (уш.с, 1Н); 6,95-7,89 (м, 14Н); 8,48 (с, 1Н); 12-15 (уш.с, 1Н).
МН+ экспериментальное = 429,20; М теоретическое = 428,90.
Пример 11
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид гидрохлорид
1) Трет-бутил {[(2-фтор-4-{4-[2-(феноксилметил)-1Н-имидазол-4-ил]фенокси}фенил)амино]сульфонил}карбамат
2-Фтор-4-{4-[2-(феноксилметил)-1Н-имидазол-4-ил]фенокси}анилин (1,5 г, 4 ммоль) и триэтиламин (0,63 мл, 4,52 ммоль) солюбилизируют при 0°С в 10 мл дихлорометана. К этому раствору при 0°С добавляют смесь сульфонилизоцианат хлорида (0,623 г, 4,40 ммоль) и трет-бутанол (0,325 г, 4,40 ммоль) в 10 мл дихлорометана. Повышают температуру реакционной смеси до умеренной и перемешивают в течение 2 часов. После упаривания растворителя, полученный остаток очищают хроматографией на колонке с силикагелем (элюент:дихлорометан/метанол 95/5). Целевое вещество получают после промывания дихлорометаном и фильтрации в виде желтого порошка с выходом 75%.
2) N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид гидрохлорид
Через смесь, содержащую трет-бутил {[(2-фтор-4-{4-[2-(феноксилметил)-1Н-имидазол-4-ил]фенокси}фенил)амино]сульфонил}карбамат (1,66 г, 2,9 ммоль), полученный на предыдущей стадии, в смеси растворителей этилацетат/этанол (3/1), при умеренной температуре барботируют газообразный хлороводород до насыщения. Осадок отфильтровывают, промывают изопропиловым эфиром и изопропанолом. После высушивания целевое вещество получают в виде белого порошка с выходом 80%.
1Н-ЯМР (δ мд, ДМСО): 5,43 (с, 2Н); 6,87-8,09 (м, 15Н); 9,01 (с, 1Н); 12,4 (уш.с, 2Н).
МН+ экспериментальное = 455,10; М теоретическое = 454,48, точка плавления: 185-187°С.
Пример 12
N”-циано-N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)гуанидин
Смесь 2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилина (0,1 г, 0,27 ммоль), дицианамида натрия (37 мг, 0,4 ммоль), 2,5 мл диметилформамида, 0,5 мл 1N соляной кислоты нагревают до 75°С в микроволновой печи (Biotage, Emrys Optimiser) в течение 900 секунд в герметичной стеклянной пробирке, пригодной для микроволнового нагревания. Реакционную смесь упаривают досуха. Остаток очищают хроматографией на колонке с силикагелем (элюент:дихлорометан/этанол 93/7), получая целевое вещество в виде белого порошка с выходом 31%.
1Н-ЯМР (δ мд, ДМСО): 5,07 (с, 2Н); 6,82-7,81 (м, 15Н); 8,72 (с, 1Н); 12,40 (уш.с, 1Н).
МН+ экспериментальное = 443,20; М теоретическое = 442,452, точка плавления: 139-141°С.
Пример 13
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N”-нитрогуанидин
1) N'-нитрогидразинкарбоксимидамид
К раствору нитрогуанидина (2 г, 19,2 ммоль) в 25 мл воды при 55°С добавляют по каплям гидразингидрат (1,05 мл, 21,6 ммоль). Перемешивание продолжают в течение 15 минут до появления четкого оранжево-желтого окрашивания реакционной смеси. После охлаждения добавляют 2 мл 37% соляной кислоты. Реакционную смесь отфильтровывают, полученный осадок промывают ледяной водой. Осадок перекристаллизовывают из минимального количества воды, после фильтрации получают целевое вещество в виде белого порошка с выходом 35%. Точка плавления: 187-189°С.
2) 3,5-диметил-N'-нитро-1Н-пиразол-1-карбоксимидамид
К смеси N'-нитрогидразинкарбоксимидамида, полученного на предыдущей стадии, (0,5 г, 4,20 ммоль) в 15 мл при кипячении с обратным холодильником добавляют 2.7 мл уксусной кислоты и 2,4-пентадион (0,785 мг, 7,83 ммоль). Реакционную смесь охлаждают до умеренной температуры и отфильтровывают. Полученный осадок промывают дихлорометаном. Целевое вещество получают в виде белого порошка с выходом 89%.
МН+ экспериментальное = 184,10; М теоретическое = 183,17.
3) N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N”-нитрогуанидин
2-Фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}анилин (0,6 г, 1,6 ммоль), 5-диметил-N'-нитро-1Н-пиразол-1-карбоксимидамид (0,296 г, 1,6 ммоль) в 5 мл метанола нагревают до 150°С в микроволновой печи (Biotage, Emrys Optimiser) в течение 2700 секунд в герметичной стеклянной пробирке, пригодной для микроволнового нагревания. Реакционную смесь упаривают досуха. Остаток очищают хроматографией на колонке с силикагелем (элюент:дихлорометан/этанол 98/2), получая целевое вещество в виде белого порошка.
1Н-ЯМР (δ мд, ДМСО): 5,08 (с, 2Н); 6,86-7,83 (м, 13Н); 8,2-9,5 (м, 3Н); 12,40 (уш.с, 1Н).
МН+ экспериментальное = 463,20; М теоретическое = 422,439, точка плавления: 193-195°С.
Фармакологические исследования
Антипролиферативную активность соединений по изобретению определяли, использую следующую экспериментальную процедуру.
Различные клеточные линии инкубируют при 37°С в атмосфере, содержащей 5% СО2 (Format Scientific incubators), в среде DMEM (среда Dulbecco, модифицированная средой Eagle), содержащей 4,5 г/л глюкозы, к которой добавляют инактивированную нагревом 10% сыворотку теленка, 50 U/мл пенициллина, 50 мкг/мл стрептомицина и 2 мМ глютамина (Gibco).
Ингибирование пролиферации клеток наблюдают по WST колоромитрическому тесту (соль тетразолия, Boehringer Mannhiem, Мейлан, Франция). Клетки рассевают в 96-луночный планшет (TPP) в 95 мкл среды со скоростью 1300 клеток на лунку в случае DU145 и скоростью 1200 клеток на лунку в случае MIA-Pa-Ca-2. Через 24 часа добавляют 5 мкл раствора препаратов в различных концентрациях (продукт растворяют в DMA в концентрации 10-3М и затем разводят культуральной средой). Конечные концентрации варьируются в диапазоне от 500 нМ до 97 нМ. После инкубации в течение 72 часов в каждую лунку добавляют 10 мкл WST и через два часа определяют абсорбцию при длине волны 450 нм (Victor, Perkin Elmer).
Каждый эксперимент, являющийся результатом измерения абсорбции в 8 лунках, повторяют дважды. Для каждого соединения значение IC50, соответствующее концентрации вещества, приводящей к 50% замедлению роста клеток, определяют методом линейной регрессии (линейное отклонение, линейность отклонения и разница между экспериментами, вычислительная программа TSAR) на линейной части сигмоиды.
Полученные значения IC50 были порядка 20 нМ и менее; для определенных соединений они варьируются от 1 нМ до 10 нМ.
Настоящее изобретение относится к новым производным имидазола общей формулы (I), в которой Z' представляет собой атом водорода или галоидный радикал; Z представляет собой радикал формулы -NH-X-NR1R2; R1 и R2 независимо представляют собой атом водорода, (С1-С6)алкильный радикал, фенил, необязательно замещенный (С1-С6)алкильным радикалом, или бензил, необязательно замещенный по кольцу (С1-С6)алкильным радикалом; Х представляет собой -SO2- или 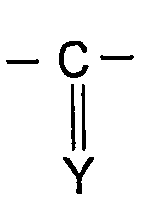 ; Y представляет собой S, О или N-R; R представляет собой -CN или -NO2; или к его фармацевтически приемлемой соли. Также изобретение относится к способам получения соединения формулы (I), к фармацевтической композиции на основе соединения формулы (I), к применению соединения формулы (I). Технический результат: получены новые производные имидазола - ингибиторы полимеризации тубулина, полезные при лечении опухолей и подагры. 8 н. и 12 з.п. ф-лы.
; Y представляет собой S, О или N-R; R представляет собой -CN или -NO2; или к его фармацевтически приемлемой соли. Также изобретение относится к способам получения соединения формулы (I), к фармацевтической композиции на основе соединения формулы (I), к применению соединения формулы (I). Технический результат: получены новые производные имидазола - ингибиторы полимеризации тубулина, полезные при лечении опухолей и подагры. 8 н. и 12 з.п. ф-лы.
1. Соединение общей формулы (I)
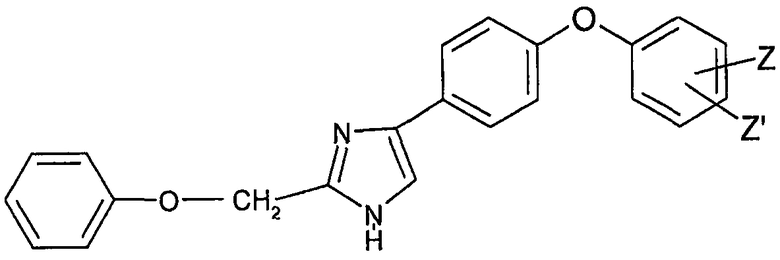
в которой Z' представляет собой атом водорода или галоидный радикал;
Z представляет собой радикал формулы -NH-X-NR1R2;
R1 и R2 независимо представляют собой атом водорода, (С1-С6)алкильный радикал, фенил, необязательно замещенный (С1-С6)алкильным радикалом, или бензил, необязательно замещенный по кольцу (С1-С6)алкильным радикалом;
Х представляет собой -SO2- или
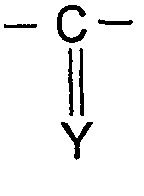 ;
;
Y представляет собой S, О или N-R;
R представляет собой -CN или -NO2; или его фармацевтически приемлемую соль.
2. Соединение по п.1, отличающееся тем, что R1 и R2 независимо представляют собой атом водорода или (С1-С6)алкильный радикал.
3. Соединение по одному из предыдущих пунктов, отличающееся тем, что R1 представляет собой атом водорода, а R2 представляет собой атом водорода или (С1-С6)алкильный радикал.
4. Соединение по п.1, отличающееся тем, что Х представляет собой -C(Y)-, и Y представляет собой S, О или N-R.
5. Соединение по п.1, отличающееся тем, что Х представляет собой -C(Y)-, и Y представляет собой S, О или N-R.
6. Соединение по п.1, отличающееся тем, что Х представляет собой -C(Y)-, и Y представляет собой S или N-R, и R представляет собой -CN.
7. Соединение по п.1, отличающееся тем, что Х представляет собой -SO2.
8. Соединение по п.1, отличающееся тем, что Z находится в параположении.
9. Соединение по п.1, отличающееся тем, что Z' находится в метаположении.
10. Соединение по п.9, отличающееся тем, что Z' представляет собой галоидный радикал, предпочтительно фтор.
11. Соединение по п.1, отличающееся тем, что оно соответствует одной из следующих формул:
N-(4-метилфенил)-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
трифторацетат N-бутил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевины;
N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
N-этил-N'-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
N-изопропил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N'-фенилмочевина;
N-(4-метилбензил)-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
трифторацетат N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевины;
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид гидрохлорид;
N-циано-1N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)гуанидин;
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N"-нитрогуанидин.
12. Соединение по п.1, отличающееся тем, что оно соответствует одной из следующих формул:
N-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)мочевина;
N-этил-N'-(4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
N-этил-N'-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)тиомочевина;
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид гидрохлорид;
N"-циано-N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)гуанидин;
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)-N"-нитрогуанидин.
13. Соединение по п.1, отличающееся тем, что оно соответствует следующей формуле:
N-(2-фтор-4-{4-[2-(феноксиметил)-1Н-имидазол-4-ил]фенокси}фенил)сульфамид, или его фармацевтически приемлемая соль.
14. Способ получения соединения формулы (I) по одному из предыдущих пунктов, в которой R2 представляет собой атом водорода, R1 представляет собой атом водорода, (С1-С6)алкильный радикал, необязательно замещенный фенильный радикал или необязательно замещенный бензильный радикал, Х представляет собой радикал -C(Y), Y представляет собой атом кислорода или атом серы, отличающийся тем, что соединение общей формулы (I')
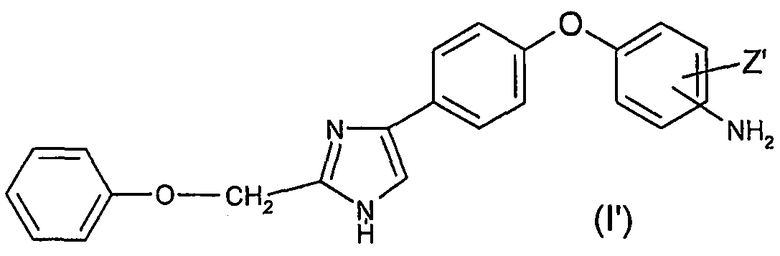
в которой Z' имеет-значения, определенные в п.1, вводят в реакцию с изоцианатным или изотиоцианатным соединением формулы R1N=C=Y, в которой R1 и Y имеют значения, определенные выше.
15. Способ получения соединения формулы (I) по одному из пп.1-12, в которой R1 и R2 представляют собой атомы водорода, Х представляет собой радикал -C(Y), Y представляет собой атом кислорода или атом серы, отличающийся тем, что (тио)фосген формулы Cl2C=Y в инертном растворителе вводят в реакцию с соединением формулы (I')
в которой Z' имеет значения, определенные в п.1, с получением изо(тио)цианатного производного общей формулы (III)
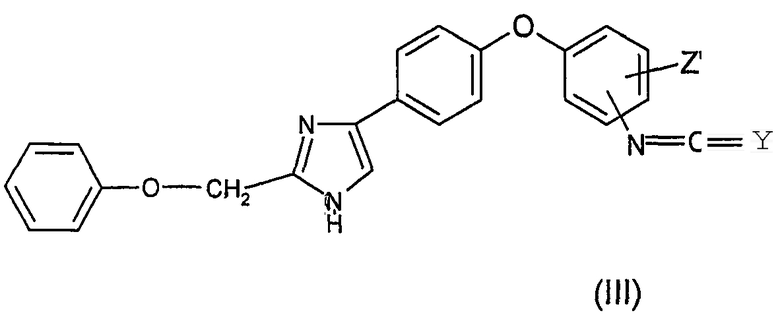 ,
,
которое на следующей стадии конденсируют с газообразным аммиаком в растворителе с получением соединения общей формулы (I).
16. Способ получения соединения формулы (I) по одному из пп.1-12, в которой R1 представляет собой атом водорода, R2 представляет собой атом водорода, или алкильный радикал, Х представляет собой -SO2- радикал, отличающийся тем, что
соединение формулы (I'), как определено в п.14, вводят в реакцию с сульфонил галоидом формулы GpNR2-SO2-Hal, в которой Gp представляет собой защитную группу для амино функции, Hal представляет собой галоидный радикал с получением соединения (V)
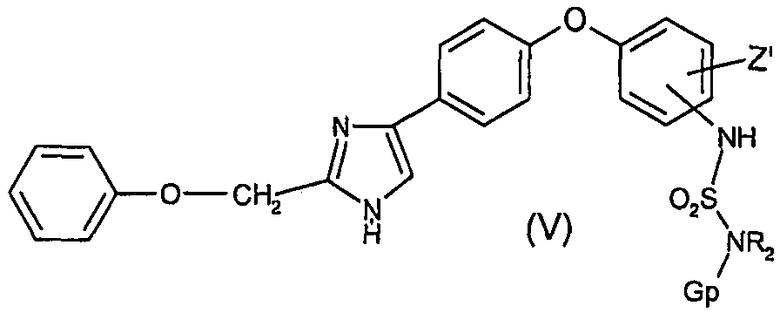
в которой Z' имеет значения, определенные в п.1,
из полученного таким образом соединения (V)удаляют защитную группу, получая соединение (I).
17. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая в качестве активного ингредиента, по крайней мере, одно соединение по одному из пп.1-13, в комбинации с фармацевтически приемлемой основой.
18. Применение соединения по одному из пп.1-13 для получения противоопухолевого лекарственного средства.
19. Применение соединения по одному из пп.1-13 для получения лекарственного средства, предназначенного для ингибирования полимеризации тубулина.
20. Применение соединения по одному из пп.1-13 для получения лекарственного средства, предназначенного для лечения подагры.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ получения азолил(1)-метанов или их солей | 1975 |
|
SU624574A3 |
| RU 2001120701 A, 10.12.2003. | |||

