Все процитированные документы включены в настоящее описание посредством ссылки.
Область изобретения
Настоящее изобретение относится к области комбинированных вакцин, в частности вакцин для защиты от дифтерии, столбняка, коклюша и Н.influenzae типа b ("Hib").
Предшествующий уровень техники
Известны комбинированные вакцины, содержащие антигены для иммунизации против дифтерии, столбняка, коклюша и Hib (вакцины "DTP-Hib"). Три вакцины этого типа доступны на рынке под названиями TETRAMUNE™ и QUATTVAXEM™ (содержащие клеточные антигены коклюша "DTwP-Hib") и INFANRDC-Hib™ (содержащие бесклеточные антигены коклюша "DTaP-Hib").
Включение Hib-конъюгатов в вакцины DTwP-Hib было связано со снижением ответа анти-Hib [1, 2]. Кроме того, Hib-конъюгаты нестабильны в водной среде и не выдерживают долгого хранения в такой форме [3]. По этой причине обычной практикой является предоставление Hib компонента в виде лиофилизированного порошка, который перед применением смешивают с жидкой композицией, содержащей другие антигены.
Hib-конъюгатные антигены недешевы в производстве, и существует опасение, что их стоимость будет сдерживать широкое распространение в развивающихся странах, поэтому были разработаны альтернативные стратегии для применения Hib-конъюгатов [4-6]. Один подход для расширения дальнейшего использования этих конъюгатов заключается во введении двух доз (например, в возрасте 3 и 5 месяцев [5] или 4 и 6 месяцев [6]) по сравнению с обычными тремя дозами (в 2, 4 и 6 месяцев [7]). В другом подходе использовали более низкие дозы (обычно дробные дозы 1/2, 1/3, 1/4 и т.д.) [4, 6], в то время как обычно Hib-конъюгаты используют в дозе 10 мкг/дозу. Например, в работе [6] Hib-конъюгаты вводили в дозе 5 мкг/дозу или 3,33 мкг/дозу.
Такой же подход распространяется и на применение Hib-конъюгатов в составе вакцин DTP-Hib. Например, в работе [8] сравнивают применение Hib-конъюгата в полной дозе, половине и одной трети дозы в комбинации с вакциной DTwP, и, хотя средние геометрические концентрации антител анти-PRP были ниже у пациентов, получавших комбинированные вакцины DTP-Hib по сравнению с пациентами, получавшими отдельно DTP и Hib, во всех случаях наблюдались приемлемые протективные анти-Hib иммунные ответы. В работе [9] использовали 10-кратное разведение дозы Hib-конъюгата при восстановлении одной дозы Hib в 10-дозовом флаконе с DTwP. В работе [10] описано восстановление лиофилизированного Hib-конъюгата в полной дозе, половине или четверти дозы при использовании вакцины TRTTANRIX™ DTwP-HBsAg.
Однако в каждом из этих случаев Hib-конъюгат находился в лиофилизированной форме, и его нужно было восстанавливать в водном растворе антигенов DTP перед применением. Таким образом, эти вакцины должны поставляться в двух отдельных контейнерах (в одном - водный раствор DTP, в другом - лиофилизированный Hib), что влечет за собой дополнительные расходы и логистические требования на стадии упаковки, транспортировки, хранения и применения. Поскольку вакцины со сниженной дозой предназначены для сокращения расходов и поощрения применения в развивающихся странах, эти дополнительные требования являются значительным недостатком. Необходимость этапа восстановления также означает наличие риска ошибки конечного пользователя, тенденции к нестандартным дозировкам, риска загрязнения смешанного продукта и необходимости обучения персонала процедуре восстановления. Все эти проблемы препятствуют продвижению на целевой рынок, т.е. развивающийся мир.
Таким образом, остается потребность в комбинированной вакцине, содержащей низкую дозу Hib-конъюгатного антигена и не требующей отдельной упаковки Hib антигена.
Раскрытие изобретения
Изобретение обеспечивает комбинированную вакцину, включающую антигены для защиты субъекта от, по меньшей мере, дифтерии (′D′), столбняка (′Т′), коклюша (′Р′) и H.influenzae типа b (′Hib′), в которой: (а) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (b) концентрация конъюгата Hib в вакцине <15 мкг/мл; и (с) конъюгат Hib никогда не лиофилизируют. Было обнаружено, что вакцины по изобретению безопасны и высокоиммуногенны по сравнению с иммунными ответами, наблюдаемыми в ссылках 6, 8 и 9.
Изобретение также обеспечивает комбинированную вакцину, включающую антигены для защиты от, по меньшей мере, дифтерии (′D′), столбняка (′Т′), коклюша (′Р′) и H.influenzae типа b (′Hib′), в которой: (а) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (b) концентрация конъюгата Hib в вакцине <15 мкг/мл; и (с) вакцина (i) не содержит гидроксида алюминия в качестве адъюванта и/или (ii) не содержит сульфата алюминия калия в качестве адъюванта. Считается, что гидроксид алюминия участвует в деградации конъюгатов сахаридов Hib, поэтому в качестве адъюванта вакцина предпочтительно включает фосфат алюминия. Если присутствует алюминиевый адъювант (например, фосфат алюминия в качестве адъюванта), предпочтительно, чтобы Hib-конъюгат не адсорбировался на нем.
Изобретение также обеспечивает флакон с прокалываемой герметичной крышкой и содержащий комбинированную вакцину, причем эта комбинированная вакцина содержит антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H.influenzae типа b (′Hib′), в которой антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; и в которой: (а) концентрация конъюгата Hib в вакцине <15 мкг/мл, и (b) прокалываемая герметичная крышка флакона не была проколота.
Изобретение также обеспечивает герметично закрытый контейнер, содержащий комбинированную вакцину, включающую антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H.influenzae типа b (′Hib′), причем антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib, и концентрация конъюгата Hib в вакцине <15 мкг/мл.
Изобретение также обеспечивает способ получения комбинированной вакцины, включающей антигены для защиты субъекта от, по меньшей мере, дифтерии (′D′), столбняка (′Т′), коклюша (′Р′) и H.influenzae типа b (′Hib′), причем антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib, и концентрация конъюгата Hib в вакцине <15 мкг/мл, и где: (а) способ включает этап смешивания указанных антигенов для защиты от D, Т, Р и Hib, и (b) процесс (i) не включает этап лиофилизации Hib-конъюгатного антигена, и/или (ii) не включает этап упаковки дифтерийных, столбнячных и коклюшных антигенов в смешанной форме отдельно от антигена - Hib-конъюгата.
Изобретение также обеспечивает способ помещения комбинированной вакцины в контейнер, причем: (а) вакцина включает антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H.influenzae типа b (′Hib,); (b) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; и (с) концентрация конъюгата Hib в вакцине <15 мкг/мл.
Изобретение также обеспечивает способ прикрепления этикетки на контейнер, причем:
(а) контейнер содержит комбинированную вакцину, включающую антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H.influenzae типа b (THib,); (b) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; и (с) концентрация конъюгата Hib в вакцине <15 мкг/мл. Этикетка может указывать на то, что в контейнере содержится вакцина.
Изобретение также обеспечивает способ помещения комбинированной вакцины в контейнер и последующего извлечения вакцины из контейнера, причем: (а) вакцина включает антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H.influenzae типа b ('Hib,); (b) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; и (с) концентрация конъюгата Hib в вакцине <15 мкг/мл.
Компоненты DTP
Антиген дифтерии предпочтительно представляет собой дифтерийный токсоид. Получение дифтерийных токсоидов хорошо описано [например, глава 13 ссылки 11]. Можно использовать любой подходящий дифтерийный токсоид. Концентрация дифтерийного токсоида обычно находится в пределах от 5 до 100 Lf/мл (флоккулирующих единиц/мл). Предпочтительная концентрация находится в пределах от 10 до 50 Lf/мл. Более предпочтительная концентрация находится в пределах от 20 до 40 Lf/мл, наиболее предпочтительная концентрация составляет приблизительно 30 Lf/мл. Как вариант, предпочтительная концентрация находится в пределах от 5 до 25 Lf/мл, более предпочтительная концентрация находится в пределах от 10 до 20 Lf/мл, и наиболее предпочтительная концентрация составляет около 15 Lf/мл. Однако если используется бесклеточный антиген коклюша, то предпочтительная концентрация дифтерийного токсоида составляет около 50 Lf/мл.
Антиген столбняка предпочтительно представляет собой столбнячный токсоид. Получение токсоидов столбняка хорошо описано [например, глава 27 ссылки 11]. Можно использовать любой подходящий столбнячный токсоид. Концентрация токсоида столбняка обычно находится в пределах от 1 до 50 Lf/мл. Предпочтительная концентрация находится в пределах от 2 до 9 Lf/мл. Более предпочтительная концентрация находится в пределах от 5 до 8 Lf/мл. Наиболее предпочтительная концентрация составляет около 6,5 Lf/мл. Однако если используется внеклеточный антиген коклюша, то предпочтительная концентрация столбнячного токсоида составляет около 20 Lf/мл.
Антиген коклюша, используемый согласно изобретению, может быть клеточным (например, цельноклеточным) или бесклеточным.
Получение обоих типов антигена хорошо описано [например, см. главу 21 ссылки 11; см. ссылку 12]. Для клеточных антигенов коклюша концентрация антигенов коклюша обычно составляет от 5 до 50 ОЕ/мл. Предпочтительная концентрация составляет от 10 до 40 ОЕ/мл. Более предпочтительная концентрация составляет от 25 до 35 ОЕ/мл. Наиболее предпочтительная концентрация составляет приблизительно 30 ОЕ/мл. Если используются внеклеточные антигены, то предпочтительно использовать голотоксин коклюша (РТ) и филаментный гемагглютинин (FHA), более предпочтительно в комбинации с пертактином (также известным как PRN или 69кДа антиген) и, при необходимости, с агглютиногенами (также известными как фимбрии) 2 и 3 [13]. Типичные уровни антигенов коклюша в дозе вакцины (например, в 0,5 мл) составляют: 10 мкг РТ, 5 мкг FHA, 3 мкг или 5 мкг PRN, 5 мкг объединенных фимбрий. РТ представляет собой токсический белок, и, если он присутствует в антигене коклюша, его предпочтительно детоксицируют. Детоксикация может быть осуществлена химическими и/или генетическими методами. Предпочтительным детоксицированным мутантом является двойной мутант 9K/129G [14].
Конъюгат Hib
Антиген H.influenzae типа b, используемый в вакцинах изобретения, включает антиген капсульного сахарида Hib. Антигены сахарида H.influenzae b хорошо известны [например, глава 14 ссылки 11]. Hib-сахарид конъюгирован с белком-носителем для усиления его иммуногенности, особенно у детей. Получение капсульного сахарида Hib хорошо описано [например, ссылки с 15 по 24]. В изобретении можно использовать любой подходящий Hib-конъюгат. Подходящие белки-носители описаны выше, предпочтительными носителями для сахаридов Hib являются CRM197 (′HbOC′), токсоид столбняка (′PRP-T′) и комплекс наружной мембраны N.meningitidis (TRP-OMP').
Сахаридный фрагмент конъюгата может представлять собой полисахарид (например, полноразмерный полирибозилрибитол фосфат (PRP)), но предпочтительно использовать олигосахариды (например, с молекулярной массой от ~1 до ~5 кДа). Их удобно получать фрагментацией очищенного PRP (например, гидролизом), за которой обычно следует выделение фрагментов желательного размера. Если композиция по изобретению включает конъюгированный олигосахарид, получение олигосахарида должно предшествовать конъюгированию.
Предпочтительные белки-носители для ковалентного конъюгирования представляют собой бактериальные токсины или токсоиды, такие как токсоид дифтерии или токсоид столбняка. Особенно предпочтителен мутант токсина дифтерии CRM197 [25-27]. К другим подходящим белкам-носителям относятся белок наружной мембраны N.meningitidis [28], синтетические пептиды [29, 30], белки теплового шока [31, 32], белки коклюша [33, 34], цитокины [35], лимфокины [35], гормоны [35], факторы роста [35], искусственные белки, включающие множественные эпитопы CD4+ Т-клеток человека от различных антигенов, производных от патогенов [36], белок D H.influenzae [37, 38], пневмококковый поверхностный белок PspA [39], белки поглощения железа [40], токсин А или В С.difficile [41], и т.д. Предпочтительными носителями являются токсоид дифтерии, токсоид столбняка и CRM197.
Можно использовать конъюгаты с соотношением сахарид:белок (по массе) от 1:5 (то есть избыток белка) до 5:1 (то есть избыток сахарида), например соотношения от 1:2 до 5:1 и соотношения от 1:1,25 до 1:2,5.
Конъюгаты могут использоваться в сочетании со свободным белком-носителем [42]. Если данный белок-носитель присутствует в композиции изобретения как в свободной, так и в конъюгированной форме, то неконъюгированная форма предпочтительно составляет не более 5% общего количества белка-носителя в композиции в целом и более предпочтительно составляет не более 2 мас.%.
Можно использовать любую подходящую реакцию конъюгирования, при необходимости - с любым подходящим линкером.
Перед конъюгированием сахарид обычно активизируют или функционализируют. Активация может включать, например, цианилирующие реагенты, такие как CDAP (например, 1-циано-4-диметиламино пиридиния тетрафторборат [43, 44 и т.д.]). В других подходящих методах используют цианамиды, гидразиды, активные сложные эфиры, норборан, п-нитробензойную кислоту, N-гидроксисукцинимид, S-NHS, EDC, TSTU; см. также введение к ссылке 22).
Связи через линкерную группу могут быть образованы при использовании любой известной методики, например, методами, описанными в ссылках 45 и 46. Один тип связи включает восстановительное аминирование полисахарида, соединение получившейся аминогруппы с одним концом линкерной группы, представляющей собой адипиновую кислоту, и затем соединение белка с другим концом линкерной группы, представляющей собой адипиновую кислоту [20, 47, 48]. К другим линкерам относятся В-пропионамидо [49], нитрофенил-этиламин [50], галоацил галиды [51], гликозидные связи [52], 6-аминокапроновая кислота [53], ADH [54], C4-C12 фрагменты [55] и т.д. Как альтернатива использованию линкера, можно использовать прямое связывание. Прямое связывание с белком может включать окисление полисахарида с последующим восстановительным аминированием белком, как описано, например, в ссылках 56 и 57.
Предпочтительным является процесс, включающий введение аминогрупп в сахарид (например, путем замены концевых =O групп на -NH2), с последующей дериватизацией адипиновым диэфиром (например, N-гидроксисукцинимидный диэфир адипиновой кислоты) и реакцией с белком-носителем. Другая предпочтительная реакция включает активацию CDAP с белком-носителем D.
После конъюгирования свободные и конъюгированные сахариды можно разделить. Для такого разделения подходит множество методов, включая гидрофобную хроматографию, тангенциальную ультрафильтрацию, диафильтрацию, и т.д. [см. также ссылки 58 и 59 и т.д.]. Если вакцина включает данный сахарид как в свободной, так и в конъюгированной форме, неконъюгированная форма предпочтительно составляет не более 20 мас.% от общего количества сахарида в композиции в целом (например, ≤15%, ≤10%, ≤5%, ≤2%, ≤1%).
Предпочтительный конъюгат содержит олигосахарид Hib, ковалентно связанный с CRM197 через линкер, представляющий собой адипиновую кислоту [60, 61]. Токсоид столбняка также является предпочтительным носителем.
Введение Hib антигена предпочтительно приводит к концентрации антител анти-PRP≥0,15 мкг/мл и более предпочтительно ≥1 мкг/мл. Это стандартные приемлемые пороги реакции.
Концентрация конъюгата Hib в вакцинах изобретения <15 мкг/мл, например ≤14 мкг/мл, ≤12 мкг/мл, ≤10 мкг/мл, ≤7,5 мкг/мл, ≤5 мкг/мл, ≤4 мкг/мл, ≤3 мкг/мл, ≤2 мкг/мл, ≤1 мкг/мл и т.д. Если белок-носитель не представляет собой ОМРС, то можно использовать немного более высокие дозы, например <20 мкг/мл, ≤19 мкг/мл, ≤18 мкг/мл, ≤17 мкг/мл, ≤16 мкг/мл и т.д. Концентрация конъюгата Hib в вакцинах по изобретению обычно составляет, по меньшей мере, 0,1 мкг/мл, например ≥0,2 мкг/мл, ≥0,3 мкг/мл, ≥0,4 мкг/мл, ≥0,5 мкг/мл, ≥0,6 мкг/мл, ≥0,7 мкг/мл, ≥0,8 мкг/мл, ≥0,9 мкг/мл, ≥1,0 мкг/мл, ≥1,25 мкг/мл, ≥1,5 мкг/мл, ≥2,0 мкг/мл, ≥3,5 мкг/мл и.т.д. Таким образом, предпочтительные пределы концентраций Hib конъюгата в вакцинах составляют от d1 до d2 мкг/мл, где: (i) d1<d2, (ii) d1 выбирают из группы, включающей 0,1, 02, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,0, 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2,0, 2,25, 2,5, 2,75, 3,0, 3,5 и 4,0; и (iii) d2 выбирают из группы, включающей 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 и 15.
Концентрации Hib конъюгатов определены в описании и формуле изобретения в терминах массы сахарида (т.е. доза конъюгата (носителя+сахарида) в целом выше указанной дозы) во избежание вариаций из-за выбора носителя.
Антигены Hib конъюгата по изобретению не являются и никогда не были лиофилизированными.
Адъюванты
Считается, что гидроксид алюминия участвует в деградации конъюгатов Hib сахарида [62], поэтому предпочтительно, чтобы вакцина содержала в качестве адъюванта фосфат алюминия.
Если в качестве адъюванта присутствует фосфат алюминия, то предпочтительно, чтобы конъюгат Hib не адсорбировался на нем, в противоположность ссылке [63]. Отсутствие адсорбции может быть достигнуто при получении путем подбора соответствующего порядка смешивания, подбора соответствующего уровня рН при смешивании антигена с адъювантом и/или подбора адъюванта с соответствующей точкой нулевого заряда (PZC) [64] (см. ниже).
Если присутствует фосфат алюминия, то токсоид дифтерии обычно адсорбирован на фосфат алюминия. Предпочтительно, чтобы адсорбция являлась частичной, например, из общего количества токсоида дифтерии в композици около 30-80 мас.% адсорбировано (например, около 40%-70%, около 50%-60% и т.д.). Адсорбция токсоида дифтерии повышается с течением времени при хранении приблизительно при 37°С. Токсоид столбняка обычно адсорбирован на фосфат алюминия. Предпочтительно, чтобы адсорбция являлась частичной, например, из общего количества токсоида столбняка в композици не более 40 мас.% адсорбировано (например, не более 30%, не более 20%, не более 10% и т.д.). Уровни адсорбции токсоида столбняка могут составлять около 0%. Hib-конъюгат остается неадсорбированным на алюминиевом адъюванте. Предпочтительно, чтобы не более 15 мас.% Hib-конъюгата в композиции было адсорбировано на фосфате алюминия (например, не больше 10%, не больше 5%, не больше 4%, не больше 3%, не больше 2% или не больше 1%).
Термин "фосфат алюминия" в настоящем описании включает фосфат алюминия, гидроксифосфат алюминия и гидроксифосфат сульфат алюминия. Предпочтительной формой фосфата алюминия для использования по настоящему изобретению является гидроксифосфатная соль.
Молярное соотношение РO4/Аl3+ в фосфате алюминия обычно составляет от 0,3 до 1,2, предпочтительно от 0,8 до 1,2 и более предпочтительно 0,95±0,1. Типичный адъювант представляет собой аморфный гидроксифосфат алюминия с молярным соотношением РO4/Аl между 0,84 и 0,92, включая 0,6 мг Al3+/мл. Обычно фосфат алюминия аморфен, особенно гидроксифосфатные соли. Обычно фосфат алюминия находится в форме частиц. Обычные диаметры частиц находятся в пределах 0,5-20 мкм (например, около 5-10 мкм) после адсорбции любого антигена.
Точка нулевого заряда (PZC) фосфата алюминия обратно связана со степенью замещения фосфата на гидроксил, и эта степень замещения может изменяться в зависимости от условий реакции и концентрации реагентов, использованных для получения соли преципитацией. Точка PZC также изменяется при изменении концентрации свободных фосфатных ионов в растворе (чем больше фосфата, тем более кислая точка PZC) или при добавлении буфера, такого как гистидиновый буфер (делает точку PZC более основной). Фосфаты алюминия, используемые по изобретению, обычно имеют точку PZC между 5,0 и 7,0, более предпочтительно между 5,5 и 6,0, например приблизительно 5,7.
Фосфат алюминия предпочтительно используют в форме водного раствора, к которому добавлены антигены (примечание: принято обозначать водный фосфат алюминия как "раствор", хотя, в строгом физико-химическом представлении, соль нерастворима и образует суспензию). Предпочтительно разбавлять фосфат алюминия до заданной концентрации и обеспечивать гомогенность раствора перед добавлением антигенных компонентов.
Концентрация Al3+ перед добавлением антигенов обычно составляет от 0 до 10 мг/мл. Предпочтительная концентрация составляет от 2 до 6 мг/мл. Более предпочтительная концентрация составляет от 4 до 5 мг/мл, например 4,4 мг/мл (соответствующая концентрации фосфата алюминия 20 мг/мл). Концентрация Al3+ в конечных вакцинах по изобретению обычно составляет от 0,1 до 2,0 мг/мл. Предпочтительная концентрация составляет от 0,2 до 1,5 мг/мл. Более предпочтительная концентрация составляет от 0,3 до 1,0 мг/мл. Наиболее предпочтительная концентрация около 0,6 мг/мл.
Раствор фосфата алюминия, используемый для получения вакцин по изобретению, может содержать буфер (например, фосфатный или гистидиновый буфер), но это не является необходимым. Раствор фосфата алюминия предпочтительно является стерильным и апирогенным. Раствор фосфата алюминия может содержать свободные водные ионы фосфата, например, в концентрации от 1,0 до 20 мМ, предпочтительно от 5 до 15 мМ и более предпочтительно около 10 мМ. Раствор фосфата алюминия может также содержать хлорид натрия. Концентрация хлорида натрия находится предпочтительно в пределах от 0,1 до 100 мг/мл (например, 0,5-50 мг/мл, 1-20 мг/мл, 2-10 мг/мл) и более предпочтительно составляет около 3±1 мг/мл. Наличие NaCl облегчает правильное измерение рН перед адсорбцией антигенов.
Хотя использование алюминиевых солей в качестве единственного адъюванта нормально, к другим адъювантам, которые могут быть включены в вакцины по настоящему изобретению, относятся, без ограничения:
А. Композиции, содержащие минерал
К композициям, содержащим минерал, подходящим для использования в качестве адъювантов по изобретению, относятся минеральные соли, такие как алюминиевые соли и кальциевые соли. Изобретение включает минеральные соли, такие как гидроксиды (например, оксигидроксиды), фосфаты (например, гидроксифосфаты, ортофосфаты), сульфаты и т.д. [например, см. главы 8 и 9 ссылки 65], или смеси различных минеральных соединений, причем соединения могут быть в любой подходящей форме (например, гель, кристалл, аморфное вещество и т.д.), причем адсорбция является предпочтительной. Композиции, содержащие минералы, также могут быть приведены в форму частиц соли металла [66].
Предпочтительно, чтобы вакцинные композиции по изобретению были по существу свободны от гидроксидов алюминия (например, оксигидроксидов алюминия). Концентрация гидроксидов алюминия в композиции обычно составляет менее 100 мкг/мл, предпочтительно менее 50 мкг/мл, более предпочтительно менее 10 мкг/мл и наиболее предпочтительно менее 1 мкг/мл. В частности, антиген НIb-конъюгата предпочтительно не адсорбирован на гидроксид алюминия.
В качестве адъюванта можно использовать фосфат кальция.
В. Масляные эмульсии
К масляным эмульсиям, подходящим для использования в качестве адъювантов по изобретению, относятся сквален-водные эмульсии, такие как MF59 [глава 10 ссылки 65; см. также ссылку 67] (5% сквалена, 0,5% Tween 80 и 0,5% Span 85, приведенные к виду субмикронных частиц при использовании микрофлюидайзера). Также можно использовать полный адъювант Фрейнда (CFA) и неполный адъювант Фрейнда (IFA).
С. Сапониновые составы [глава 22 ссылки 65]
В качестве адъюванта по изобретению также можно использовать сапониновые составы. Сапонины представляют собой гетерологичную группу стериновых гликозидов и тритерпеноидных гликозидов, которые обнаружены в коре, листьях, стволах, корнях и даже цветах широкого спектра растений. Сапонин из коры мыльного дерева Quillaia saponaria Molina широко изучался в качестве адъюванта. Сапонин также можно получать из Smilax ornata (сарсапарели), Gypsophilla paniculata (невестиной вуали) и Saponaria officianalis (мыльного корня). Составы с сапониновыми адъювантами включают как очищенные составы, такие как QS21, так и липидные составы, такие как ISCOM (иммуностимулирующие комплексы). QS21 представлен на рынке как Stimulon™.
Сапониновые композиции очищают при помощи ВЭЖХ (высокоэффективной жидкостной хроматографии) и обращено-фазовой ВЭЖХ. При использовании этих методов были идентифицированы специфические фракции, включая QS7, QS17, QS18, QS21, QH-А, QH-B и QH-C. Предпочтительно сапонин представляет собой QS21. Метод получения QS21 описан в ссылке [68]. Сапониновые составы также могут включать стерин, например холестерин [69].
Комбинации сапонинов и холестеринов можно использовать для образования уникальных частиц, называемых иммуностимулирующими комплексами (ISCOM, "ИСКОМ") [глава 23 ссылки 65]. Иммуностимулирующие комплексы обычно включают фосфолипид, такой как фосфатидилэтаноламин или фосфатидилхолин. В таких комплексах можно использовать любой известный сапонин. Предпочтительно ISCOM включает один или более из следующих сапонинов: QuilA, QHA и QHC. Более подробно эти комплексы описаны в ссылках [69-71]. При необходимости, комплексы ISCOM могут не содержать дополнительного детергента [72].
Обзор разработки адъювантов на основе сапонина можно найти в ссылках [73] и [74].
D. Виросомы и вирусоподобные частицы
В качестве адъювантов по изобретению также можно использовать виросомы и вирусоподобные частицы (VLP, ВПЧ). Эти структуры обычно содержат один или более белков вируса, необязательно в комбинации или в составе с фосфолипидом. Они обычно не патогенны, не реплицируются и обычно не содержат нативного вирусного генома. Вирусные белки могут быть получены рекомбинантным методом или выделены из целых вирусов. Эти вирусные белки, подходящие для использования в виросомах или ВПЧ, включают белки, полученные из вируса гриппа (такие как НА или NA), вируса гепатита В вирус (такие как белки сердцевины или капсида), вируса гепатита Е, вируса кори, вируса Sindbis, ротавируса, вируса ящура, ретровируса, Норуолкского вируса, вируса папилломы человека, ВИЧ, РНК-содержащих фагов, QB-фага (такие как белки оболочки), GA-фага, fr-фага, фага АР205 и Ту (такие как ретротранспозон белка р1 Ту). ВПЧ более подробно обсуждаются в ссылках [75-80]. Виросомы более подробно обсуждаются, например, в ссылке [81].
Е. Бактериальные или микробные производные
К адъювантам, подходящим для использования по изобретению, относятся бактериальные или микробные производные, такие как нетоксичные производные липополисахарида энтеробактерий (ЛПС), производные липида А, иммуностимулирующие олигонуклеотиды и ADP-рибозилирующие токсины и детоксицированные производные этих соединений.
Нетоксичные производные ЛПС включают монофосфорил липид A (MPL) и 3-O-деацилированный MPL (3dMPL). 3dMPL представляет собой смесь 3 де-O-ацилированных монофосфорил липидов А с 4, 5 или 6 ацилируемыми цепями. Предпочтительная форма 3 де-O-ацилированного монофосфорил липида А в виде "мелких частиц" описана в ссылке [82]. Такие "мелкие частицы" 3dMPL являются достаточно малыми, чтобы стерильно фильтроваться через 0,22 мкм мембрану [82]. Другие нетоксичные производные ЛПС включают миметики монофосфорил липида А, такие как производные аминоалкил глюкозаминид фосфата, например, RC-529 [83, 84].
К производным липида А относятся производные липида A Escherichia coli, такие как ОМ-174. Соединение ОМ-174 описано, например, в работах [85 и 86].
К иммуностимулирующим олигонуклеотидам, подходящим для использования в качестве адъювантов по изобретению, относятся нуклеотидные последовательности, содержащие CpG мотив (динуклеотидная последовательность, содержащая неметилированный цитозин, связанный фосфатной связью с гуанозином). Иммуностимулирующая активность показана также для двухцепочечных РНК и олигонуклеотидов, содержащих палиндромные или поли(dG) последовательности.
Фрагменты CpG могут включать модификации/аналоги нуклеотидов, такие как фосфоротиоатные модификации, и могут быть двухцепочечными или одноцепочечными. В работах [87, 88 и 89] описаны возможные аналоговые замены, например замена гуанозина на 2'-дезокси-7-деазагуанозин. Адъювантный эффект олигонуклеотидов CpG более подробно описан в ссылках [90-95].
Последовательность CpG может быть направлена на TLR9, такая как мотив GTCGTT или TTCGTT [96]. Последовательность CpG может быть специфической для индукции иммунной реакции Th1, такой как CpG-A ODN (олигодезоксинуклеотид), или может быть более специфичной для индукции В-клеточного ответа, такой как CpG-B ODN. Олигонуклеотиды CpG-A и CpG-B ODNs обсуждены в работах [97-99]. Предпочтительно CpG представляет собой CpG-A ODN.
Предпочтительно, чтобы олигонуклеотид CpG был сконструирован таким образом, чтобы 5′-конец был доступен для распознавания рецептором. При необходимости, две олигонуклеотидные последовательности CpG могут быть соединены 3′-концами, образуя "иммуномеры". См., например, ссылки [96] и [100-102].
В качестве адъювантов по изобретению могут быть использованы бактериальные ADP-рибозилирующие токсины и их детоксицированные производные. Предпочтительно, чтобы белок был получен из E.coli (термолабильный энтеротоксин E.coli "LT"), холеры ("СТ") или коклюша ("РТ"). Использование детоксицированных ADP-рибозилирующих токсинов в качестве адъюванта для слизистой оболочки описано в ссылке [103], а в качестве парентерального адъюванта - в ссылке [104]. Токсин или токсоид предпочтительно находится в форме голотоксина, включающего как А, так и В субъедницы. Предпочтительно субъединица А содержит детоксицирующую мутацию; предпочтительно субъединица В не мутирована. Предпочтительно адъювант представляет собой детоксицированный мутант LT, такой как LT-K63, LT-R72 и LT-G192. Использование ADP-рибозилирующих токсинов и их детоксицированных производных, в частности LT-K63 и LT-R72, в качестве адъювантов описано в ссылках [105-112]. Многочисленные ссылки на аминокислотные замены предпочтительно основаны на выравнивании субъединиц А и В ADP-рибозилирующих токсинов, приведенном в ссылке [113], специфически включенной в настоящее описание в своей полноте посредством ссылки.
F. Иммуномодуляторы человека
Иммуномодуляторы человека, подходящие для использования в качестве адъювантов по изобретению, включают цитокины, такие как интерлейкины (например, IL-1, IL-2, IL-4, IL-5, IL-6, IL-7, IL-12 [114] и т.д.) [115], интерфероны (например, интерферон-γ), колониестимулирующий фактор макрофага и фактор некроза опухоли.
G. Биоадгезивные и мукоадгезивные соединения
Биоадгезивные и мукоадгезивные соединения также могут использоваться в качестве адъювантов по изобретению. К подходящим биоадгезивным агентам относятся микросферы этерифицированной гиалуроновой кислоты [116] или мукоадгезивные агенты, такие как поперечно сшитые производные поли(акриловой кислоты), поливиниловый спирт, поливинилпирролидон, полисахариды и карбоксиметилцеллюлоза. Хитозан и его производные также могут использоваться в качестве адъювантов в изобретении [117].
Н. Микрочастицы
Микрочастицы могут также использоваться в качестве адъювантов в изобретении. Предпочтительными являются микрочастицы (то есть частицы диаметром от ~100 нм до ~150 мкм, более предпочтительно от ~200 нм до ~30 мкм и наиболее предпочтительно от ~500 нм до ~10 мкм), полученные из материалов, являющихся биоразлагаемыми и нетоксичными (например, поли(α-оксикислота), полигидроксимасляная кислота, полиортоэфир, полиангидрид, поликапролактон и т.д.), причем предпочтительным является поли(лактид-ко-гликолид), при необходимости обработанный для получения негативно-заряженной поверхности (например, додецилсульфатом натрия, SDS) или положительно-заряженной поверхности (например, катионным детергентом, таким как цетилтриметиламмония бромид, СТАВ).
I. Липосомы (Главы 13 и 14 ссылки 65)
Примеры липосомных составов, подходящих для использования в качестве адъювантов, описаны в ссылках [118-120].
J. Полиоксиэтиленовый эфир и составы с полиоксиэтиленовым сложным эфиром
Адъюванты, подходящие для использования в изобретении, включают полиоксиэтиленовые эфиры и полиоксиэтиленовые сложные эфиры [121]. Такие составы далее включают поверхностно-активные вещества, представляющие собой полиоксиэтиленовый эфир сорбита, в комбинации с октоксинолом [122], а также поверхностно-активные вещества, представляющие собой полиоксиэтиленовые алкильные простые эфиры или сложные эфиры, в комбинации с, по меньшей мере, одним дополнительным неионным поверхностно-активным веществом, таким как октоксинол [123]. Предпочтительные полиоксиэтиленовые эфиры выбирают из группы, включающей: полиоксиэтилен-9-лауриловый эфир (лаурет-9), полиоксиэтилен-9-стеариловый эфир, полиоксиэтилен-8-стеариловый эфир, полиоксиэтилен-4-лауриловый эфир, полиоксиэтилен-35-лауриловый эфир и полиоксиэтилен-23-лауриловый эфир.
К. Полифосфазен(РСРР)
Составы с РСРР описаны, например, в ссылках [124 и 125].
L. Мурамиловые пептиды
Примерами мурамиловых пептидов, подходящих для использования в качестве адъювантов в изобретении, являются N-ацетил-мурамил-L-треонил-D-изоглутамин (thr-MDP), N-ацетил-нормурамил-L-аланил-D-изоглутамин (нор-MDP) и N-ацетилмурамил-L-аланил-D-изоглутаминил-L-аланин-2-(1′-2′-дипальмитоил-sn-глицеро-3-гидроксифосфорилокси)-этиламин МТР-РЕ).
М. Имидазохинолоновые соединения.
Примеры имидазохинолоновых соединений, подходящих для использования в качестве адъювантов в изобретении, включают Имиквимод (Imiquamod) и его гомологи (например, "Resiquimod 3М"), описанные подробнее в ссылках [126 и 127].
Изобретение может также включать комбинации аспектов одного или более адъювантов, указанных выше. Например, в изобретении можно использовать следующие композиции адьюванта: (1) сапонин и эмульсия типа "масло в воде" [128]; (2) сапонин (например, QS21) + нетоксичное производное ЛПС (например, 3dMPL) [129]; (3) сапонин (например, QS21) + нетоксичное производное ЛПС (например, 3dMPL) + холестерин; (4) сапонин (например, QS21) + 3dMPL+IL-12 (при необходимости + стерин) [130]; (5) комбинации 3dMPL с, например, QS21 и/или эмульсиями типа "масло в воде" [131]; (6) SAF (Syntex adjuvant formulation), содержащий 10% сквалена, 0,4% Tween 80™, 5% блок-полимера плуроник LI 21, и thr-MDP, либо микрофлюидизированного до субмикронной эмульсии, или смешанного до эмульсии с большим размером частиц; (7) адъювантная система Ribi™ (RAS), (Ribi Immunochem), содержащая 2% сквалена, 0,2% Tween 80 и один или более компонентов клеточной стенки бактерий из группы, включающей монофосфориллипид A (MPL), димиколат трегалозы (TDM), и клеточный цитоскелет (cell wall skeleton - CWS), предпочтительно MPL+CWS (Detox™); и (8) одна или более минеральных солей (таких как соль алюминия)+нетоксичное производное ЛПС (такое как 3dMPL).
Другие вещества, действующие в качестве иммуностимулирующих агентов, описаны в главе 7 ссылки 65.
Дополнительные антигены
Композиции по изобретению содержат антигены D, Т, Р и Hib. Они могут также включать дополнительные антигены, такие как:
- антиген сахарида N.meningitidis серологической группы А, С, W135 и/или Y, такой как олигосахарид, описанный в ссылке [132], из серологической группы С или олигосахариды, описанные в ссылке [133]. Вакцина предпочтительно содержит конъюгаты из 2, 3 или 4 из серологических групп А, С, W135 и Y,
- антиген сахарида Streptococcus pneumoniae [например, ссылки 134-136],
- антиген вируса гепатита А, такой как инактивированный вирус [например, 137, 138],
- антиген вируса гепатита В, такой как поверхностные антигены и/или антигены сердцевины [например, 138, 139],
- везикула наружной мембраны (OMV) или препарат пузырька N.meningitidis серологической группы В, такие как описаны в ссылках [140, 141, 142, 143 и т.д],
- белковый антиген N.meningitidis серологической группы В, такой как описанные в ссылках [144-150], причем предпочтительными являются белок ′287′ (см. ниже) и производные (например, ′ΔG287′),
- антиген(ы) полиомиелита [например, 151, 152], такие как IPV.
Композиция может включать один или более из этих дополнительных антигенов. Антигены обычно присутствуют в концентрации, по меньшей мере, 1 мкг/мл каждого. Как правило, концентрация любого данного антигена достаточна для индукции иммунной реакции против этого антигена. Предпочтительно, чтобы протективная эффективность индивидуальных сахаридных антигенов не исчезала при их комбинировании, хотя фактическая иммуногенность (например, титры ELISA) может быть снижена.
Если используется сахаридный антиген, предпочтительно, чтобы он был конъюгирован с белком-носителем для усиления иммуногенности.
В качестве альтернативы использованию белковых антигенов в композиции по изобретению может быть использована нуклеиновая кислота, кодирующая антиген [например, ссылки 153-161]. Таким образом, белковые компоненты композиций по изобретению могут быть заменены нуклеиновой кислотой (предпочтительно ДНК, например, в виде плазмиды), кодирующей белок. Аналогично, композиции по изобретению могут включать белки, имитирующие сахаридные антигены, например мимотопы [162] или анти-идиотипические антитела. Они могут заменять отдельные сахаридные компоненты или могут дополнять их. Например, вакцина может включать пептидомиметик капсульного полисахарида МеnС [163] или МеnА [164] вместо самого сахарида.
Если вакцина по изобретению включает поверхностный антиген гепатита В (′HBsAg′), этот антиген может быть получен двумя способами. Первый метод включает выделение антигена в виде частиц из плазмы носителей хронического гепатита В, поскольку большие количества HBsAg синтезируются в печени и высвобождаются в кровоток в течение инфекции HBV. Второй метод является предпочтительным и включает экспрессию белка ДНК-рекомбинантными методами. Предпочтительно, чтобы HBsAg был получен экспрессией в дрожжах Saccharomyces cerevisiae. Ген HBsAg может быть вставлен в плазмиду, и его экспрессию из плазмиды можно контролировать при помощи промотора, такого как промотор 'GAPDH' (из гена глицеральдегид-3-фосфат-дегидрогеназы). Дрожжи могут культивироваться в синтетической среде. HBsAg затем можно очистить на этапах осаждения, ионообменной хроматографии и ультрафильтрации. После очистки HBsAg может быть подвергнут диализу (например, с цистеином). HBsAg может использоваться в форме частиц.
Если вакцина по изобретению включает антиген полиомиелита, предпочтительно использовать три антигена полиовируса - полиовируса типа 1 (например, штамм Mahoney), полиовируса типа 2 (например, штамм MEF-1) и полиовируса типа 3 (например, штамм Saukett). Полиовирусы могут быть выращены в культуре клеток. Предпочтительно использовать клетки линии VERO, которая является непрерывной линией клеток, полученных из почки обезьяны. Клетки VERO удобно культивировать на микроносителях. Культивирование клеток VERO до и в течение вирусной инфекции может включить использование материала, полученного от крупного рогатого скота, такого как сыворотка теленка, и этот материал должен быть получен из источников, свободных от бычьего губчатого энцефалита (BSE). Культивирование может также включать материалы типа гидролизата лактальбумина. После роста вирионы можно очистить с использованием методов типа ультрафильтрации, диафильтрации и хроматографии. Перед введением пациентам вирусы должны быть инактивированы, это может быть достигнуто путем обработки формальдегидом.
Предпочтительно вирусы выращивают, очищают и инактивируют по отдельности, а затем объединяют с образованием смеси для добавления к адсорбированным антигенам дифтерии и столбняка.
Антигены в вакцинах по изобретению присутствуют в "иммунологически эффективных количествах", то есть введение такого количества субъекту, в единичной дозе или как часть серии доз, эффективно для лечения или профилактики заболевания. Это количество изменяется в зависимости от здоровья и физического состояния субъекта, требующего лечения, возраста, таксономической группы субъекта, требующего лечения (например, человек, примат и т.д.), способности иммунной системы субъекта к синтезу антител, степени требуемой защиты, состава вакцины, оценки медицинской ситуации лечащим врачом и других связанных факторов. Ожидается, что количество попадает в относительно широкие пределы, которые можно определить в ходе рутинных исследований. Можно использовать режим единственной дозы или нескольких доз (например, включая бустерные дозы).
Неиммунологические компоненты вакцин по изобретению
Вакцины по изобретению в дополнение к антигенным и адъювантным компонентам, упомянутым выше, обычно включают один или более "фармацевтически приемлемых носителей", которые включают любой носитель, который самостоятельно не вызывает продукцию антител, вредных для субъекта, получающего композицию. Подходящие носители обычно представляют собой большие, медленно метаболизируемые макромолекулы, такие как белки, полисахариды, полимолочные кислоты, полигликолевые кислоты, полимерные аминокислоты, сополимеры аминокислот, сахарозу [165], трегалозу [166], лактозу и липидные агрегаты (такие как масляные капельки или липосомы). Такие носители известны специалистам в данной области техники. Вакцины могут также содержать разбавители, такие как вода, солевой раствор, глицерин и т.д. Дополнительно могут присутствовать вспомогательные вещества, такие как смачивающие или эмульгирующие агенты, буферные агенты и подобные. Типичным носителем является стерильный апирогенный фосфатно-забуференный физиологический солевой раствор. Подробное обсуждение фармацевтически приемлемых наполнителей приведено в работе [167].
Композиции изобретения находятся в водной форме, то есть в растворах или суспензиях. Жидкий состав такого типа позволяет вводить композиции непосредственно из упакованной формы, без необходимости восстановления в водной среде, и, таким образом, они идеальны для инъекций. Композиции могут быть представлены во флаконах или предварительно заполненных шприцах. Шприцы могут поставляться с иглами или без них. В шприце содержится одна доза, в то время как флакон может содержать одну или несколько доз.
Жидкие вакцины по изобретению также подходят для восстановления других вакцин из лиофилизированной формы. Если вакцина предназначена для такого восстановления непосредственно перед применением, изобретение обеспечивает набор, который может содержать два флакона, или может содержать один предварительно заполненный шприц и один флакон, причем содержимое шприца используют для реактивации содержимого флакона перед инъекцией.
Вакцины по изобретению могут быть упакованы в форме единичных доз или в форме нескольких доз. Для форм в виде нескольких доз предпочтительно использовать флаконы, а не предварительно заполненные шприцы. Эффективные объемы доз могут быть установлены обычными способами, но типичная доза композиции для инъекции человеку имеет объем 0,5 мл.
Уровень рН вакцин по изобретению обычно составляет от 6,0 до 8,0, более предпочтительно от 6,3 до 6,9, например 6,6±0,2. Вакцины предпочтительно забуферены при этом значении рН. Стабильный уровень рН может поддерживаться при помощи буфера. Если композиция включает соль гидроксида алюминия, предпочтительно использовать гистидиновый буфер [168]. Композиция может быть стерильной и/или апирогенной. Фосфат алюминия и цельноклеточные антигены коклюша несовместимы со стерилизацией фильтрованием, поэтому, если композиция по изобретению включает один из этих компонентов, предпочтительно стерилизовать композицию автоклавированием и/или использовать при получении композиции стерильные компоненты.
Композиции по изобретению могут быть изотоническими по отношению к людям.
Вакцины по изобретению могут включать антимикробный агент, особенно если упаковка содержит несколько доз. Многие антимикробные агенты основаны на ртути (например, тиомерсал), хотя ртутных консервантов предпочтительно избегать, например, можно использовать 2-феноксиэтанол. Любой консервант предпочтительно присутствует в низких концентрациях (например, 0,01% по объему). Консервант можно добавлять экзогенно, и/или он может быть компонентом исходной смеси антигенов, которые смешивают для получения композиции (например, он может присутствовать в качестве консерванта в антигенах коклюша).
Вакцины по изобретению могут включать детергент, например Tween (полисорбат), такой как Tween 80. Детергенты обычно присутствуют в низких концентрациях, например <0,01%.
Вакцины по изобретению могут содержать соли натрия (например, хлорид натрия) для придания тоничности. Композиция может включать хлорид натрия. Концентрация хлорида натрия в композиции находится предпочтительно в пределах от 0,1 до 100 мг/мл (например, 1-50 мг/мл, 2-20 мг/мл, 5-15 мг/мл) и более предпочтительно 10±2 мг/мл NaCl, например около 9 мг/мл.
Вакцины по изобретению обычно содержат буфер. Типичными являются фосфатный или гистидиновый буферы.
Вакцины по изобретению могут включать свободные фосфатные ионы в растворе (например, при использовании фосфатного буфера) для способствования отсутствию адсорбции антигенов. Концентрация свободных фосфатных ионов в композиции по изобретению обычно составляет от 0,1 до 10,0 мМ, предпочтительно от 1 до 5 мМ и более предпочтительно около 2,5 мМ.
Упаковка вакцин по изобретению
Вакцины по изобретению могут быть упакованы в различные типы емкостей, например во флаконы, шприцы и т.д.
При использовании вакцин DTP-Hib предшествующего уровня техники, которые требуют восстановления лиофилизированного компонента Hib, водный раствор антигенов DTP набирают из герметично закрытого флакона в шприц и затем вводят во второй герметично закрытый флакон, содержащий лиофилизированный материал. Затем ресуспендированную вакцину снова набирают в тот же самый шприц для введения пациенту. Напротив, Hib-конъюгаты в вакцинах по изобретению стабильны в водных растворах и не требуют лиофилизации. Изобретение таким образом может обеспечить флакон с прокалываемой герметичной крышкой и содержащий вакцину DTP-Hib, причем прокалываемая герметичная крышка не проколота. Аналогично, изобретение может обеспечить герметично закрытую емкость, содержащую вакцину по изобретению.
Вакцины DTP-Hib предшествующего уровня техники, требующие восстановления лиофилизированного Hib компонента, должны быть упакованы в две отдельные емкости. Напротив, изобретение позволяет обеспечить способ помещения вакцины по изобретению в контейнер в форме, уже включающей антигены DTP-Hib. Вакцину помещают в емкость предпочтительно не через герметичную крышку емкости.
Аналогично, поскольку упаковка вакцины маркируется при производстве, вакцины DTP-Hib предшествующего уровня техники маркируются в то время, как они находятся отдельно в виде DTP и Hib, тогда как изобретение позволяет маркировку вакцины в ее конечном виде DTP-Hib.
Кроме того, восстанавливаемые вакцины DTP-Hib предшествующего уровня техники извлекают из их емкостей в виде DTP-Hib, но они не были помещены в емкости в этой форме. Согласно изобретению такие вакцины DTP-Hib могут как помещаться, так и извлекаться в предварительно смешанной форме DTP-Hib. Временной интервал между помещением в емкость и извлечением может составлять, по меньшей мере, n недель, где n выбирают из группы, включающей 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25 или больше. Извлечение обычно производят через иглу (например, иглу стерильного средства доставки, такого как шприц), хотя помещение в емкость обычно осуществляется из производственной линии, а не из средства доставки.
Получение вакцин по изобретению
Вакцины по изобретению содержат, по меньшей мере, антигены DTP-Hib, и таким образом, их получение включает смешивание этих четырех антигенов. В отличие от получения восстанавливаемых вакцин DTP-Hib предшествующего уровня техники изобретение может обеспечить процесс получения, не включающий этапа лиофилизации конъюгата Hib. Аналогично, изобретение может обеспечить способ получения, не включающий этапа упаковки смешанных антигенов DTP отдельно от Hib антигена.
В процессе получения добавляемые антигены обычно не адсорбированы на соль алюминия (то есть они не являются "предадсорбированными"). Таким образом предпочтительно, чтобы для каждого добавляемого антигена не более 5 мас.% (предпочтительно нисколько) уже адсорбировано на соли алюминия (например, не более 4%, не более 3% или не более 2%). Однако в некоторых ситуациях можно добавлять предадсорбированные антигены.
Типичный способ получения вакцины по изобретению включает добавление Hib компонента к смеси D, Т и Р компонентов, то есть компоненты DTP смешаны до добавления компонента Hib. Этот порядок смешивания позволяет регулировать ионную силу и/или рН композиции (например, рН<7) до добавления компонента Hib, чтобы предотвратить адсорбцию на любой алюминиевый адъювант, который может присутствовать.
Вакцины по изобретению предпочтительно получают при температуре от 15°С до 30°С (например, от 19°С до 27°С или при 23±4°С).
Введение вакцин по изобретению
Изобретение обеспечивает способ индуцирования ответного образования антител у млекопитающего, включающий введение млекопитающему вакцины по изобретению. Вакцины могут вводится профилактически (то есть для предотвращения инфекции) или терапевтически (то есть для лечения болезни после инфицирования).
Изобретение обеспечивает способ возбуждения иммунного ответа у млекопитающего, включающий этап введения эффективного количества вакцины по изобретению. Иммунный ответ предпочтительно является протективным и предпочтительно включает антитела. Метод может индуцировать бустерный ответ.
Млекопитающее предпочтительно представляет собой человека. Если вакцина применяется профилактически, человек предпочтительно представляет собой ребенка (например, младшего возраста) или подростка; если вакцина применяется терапевтически, человек предпочтительно представляет собой взрослого. Вакцина, предназначенная для детей, может также вводиться взрослым, например, для оценки безопасности, дозировки, иммуногенности и т.д.
Изобретение также обеспечивает композиции по изобретению для использования в качестве медикамента. Медикамент предпочтительно способен вызвать иммунную реакцию у млекопитающего (то есть представляет собой иммуногенную композицию) и более предпочтительно представляет собой вакцину.
Изобретение также обеспечивает применение, по меньшей мере, антигенов дифтерии, столбняка, коклюша и H.influenzae типа b ('Hib') в производстве комбинированной вакцины для иммунизации пациента, причем (а) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (b) концентрация конъюгата Hib в вакцине <15 мкг/мл; и (с) производство не включает лиофилизации конъюгата Hib.
Эти применения и способы предпочтительно предназначены для профилактики и/или лечения инфекций C.diphtheriae, C.tetani, В.pertussis и H.influenzae и заболеваний, вызванных этими инфекциями, например, для профилактики дифтерии, столбняка, коклюша, бактериального менингита и т.д.
Один из способов проверки эффективности терапевтического применения включает контроль бактериальной инфекции после введения композиции по изобретению. Один из способов проверки эффективности профилактического применения включает контроль иммунного ответа на антигены после введения композиции. Иммуногенность композиций по изобретению можно определить при введении их исследуемым субъектам (например, детям возраста 12-16 месяцев или животным моделям [169]) и последующем определении стандартных иммунологических параметров. Эти иммунные реакции обычно определяют приблизительно через 4 недели после введения композиции и сравнивают с величинами, определенными перед введением композиции. Вместо оценки фактической протективной эффективности у пациентов для оценки эффективности вакцин Hib и DTP можно использовать известные стандартные животные модели и модели in vitro и соотношение с защитным эффектом.
Композиции по изобретению обычно вводят непосредственно пациенту. Прямую доставку можно осуществлять либо парентеральной инъекцией (например, подкожно, внутрибрюшинно, внутривенно, внутримышечно, или в интерстициальное пространство ткани) или ректальным, пероральным, вагинальным, местным, трансдермальным, интраназальным, пульмонарным способом, через глаза, через уши, или другим способом доставки через слизистые оболочки. Предпочтительным является внутримышечное введение в бедро или плечо. Инъекцию можно осуществлять через иглу (например, иглу для подкожных инъекций), но как вариант можно применять инъекцию без использования иглы. Типичная доза для внутримышечного введения составляет 0,5 мл.
Изобретение можно применять, чтобы вызвать системный иммунный ответ или ответ слизистых оболочек.
Режим введения может включать единственную дозу или несколько доз. Режим множественных доз можно использовать для первичной иммунизации и/или бустерной иммунизации. После режима первичной дозы можно применять режим бустерной дозы. Подходящий временной интервал между дозами примирования (например, от 4 до 16 недель) или между примированием и повторной иммунизацией можно определить рутинными методами.
Бактериальные инфекции поражают различные системы организма, поэтому композицию по изобретению можно получать в различных формах. Например, композиции можно получить в форме для инъекций, либо в виде жидких растворов, либо в виде суспензий. Композицию можно получить в форме для пульмонарного применения, например, в виде ингаляций с использованием тонкого порошка или спрея. Композицию можно получить в форме суппозитория или маточного кольца. Композицию можно получить в форме для назального применения, применения через глаза и уши, например, в виде спрея, капель, геля или порошка [например, ссылки 170 и 171]. Сообщалось об успешном интраназальном применении сахаридов Hib [172] и вакцин DTP [173, 174].
Общие понятия
Термин "содержащий" охватывает как понятие "включающий", так и "состоящий из", например, композиция, "содержащая" X, может состоять только из Х или может включать что-либо дополнительно, например, X+Y.
Термин "около" по отношению к численной величине х означает, например, х±10%.
Термин "по существу" не исключает понятия "полностью", например, композиция, "по существу свободная от" Y, может быть полностью свободна от Y. Где необходимо, слово "по существу" может быть опущено в определении изобретения.
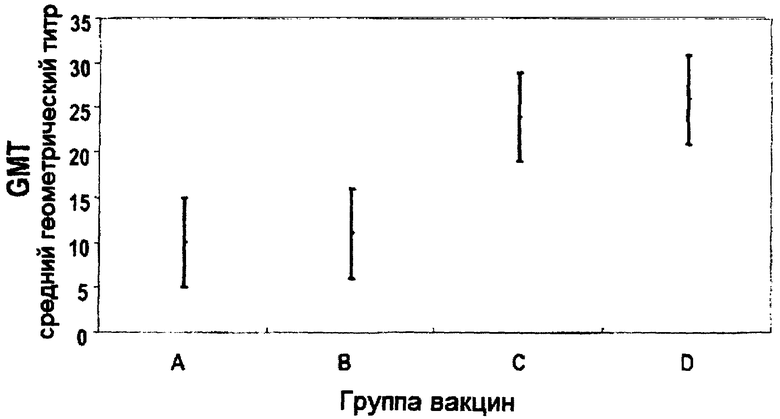
Краткое описание чертежа
На чертеже приведены GMT (средние геометрические титры) анти-PRP и 95% доверительные интервалы в группах вакцин А, В, С и D.
Варианты осуществления настоящего изобретения
Были получены четыре состава вакцины DTwP-Hib, отличающиеся только дозой конъюгата Hib-CRM197. Вакцины получены в виде доз по 0,5 мл, со следующим антигенным составом:
Процесс производства по сути был следующим: к воде для инъекций добавляли адъювант фосфат алюминия; добавляли компонент D; добавляли компонент Т; добавляли компонент wP; добавляли NaCl; проверяли и доводили рН; и добавляли компонент Hib. В отличие от утверждения в ссылке 175 компонент Hib не адсорбировался на адъюванте.
Стабильность
Проводили два исследования стабильности: одно при нормальных условиях хранения при 2-8°С в течение 2 лет и другое при ускоренных условиях при 37°С в течение 14 дней. Вакцины тестировали после хранения в прямом положении и перевернутом положении. Стабильность оценивали, определяя уровень рН и свободного сахарида.
Результаты первого исследования были следующими:
Результаты исследования при ускоренных условиях были следующими:
Таким образом, вакцины стабильны в течение продолжительного времени. В тех же временных интервалах анализировали адсорбцию, конъюгат Hib оставался неадсорбированным в течение времени хранения.
Клиническое исследование
В исследование был включен в общем 261 ребенок, дети были рандомизированы на получение одной из четырех вакцин в двойном слепом исследовании для оценки безопасности и иммуногенности (для сравнения, исследования в ссылках 6, 8 и 9 были либо частично слепыми, либо открытыми). В исследование включались здоровые дети трехмесячного возраста, родившиеся на или позже 37 гестационной недели с минимальным весом при рождении 2500 г и подходящие согласно местному календарю прививок (EPI Expanded Programme on Immunization), их рандомизировали в соотношении 66:65:65:65 A:B:C:D на получение трех единичных внутримышечных доз одной из вакцин в возрасте 3, 4 и 5 месяцев. Согласно местному календарю прививок субъекты получали пероральную вакцину против полиомиелита (OPV) параллельно в возрасте 2, 4 и 6 месяцев.
Средний возраст на момент включения составлял 94 дня (от 69 до 108), и среди четырех групп не было разницы по распределению пола, расовой принадлежности, веса и роста. Отслеживались постинъекционные реакции пациентов и нежелательные явления, возраст в месяцах. В анализ безопасности были включены 260 субъектов (65 на каждую из четырех вакцинных групп), и 251 субъект был включен в анализ иммуногенности (61 из группы А, по 64 в группах В и С и 62 в группе D). Девять субъектов не завершили исследования, так как их родители/законные представители не завершили исследования, так как их родители/законные представители отозвали согласие до завершения исследования (5 субъектов в группе А, 1 субъект в группе В и 3 субъекта в группе D).
Образцы крови собирали в начале исследования и через месяц после третьей дозы. Измеряли содержание антител к PRP, коклюшу, дифтерии и столбняку. Содержание антител IgG анти-PRP исследовали модифицированным методом ELISA, адаптированным из метода FDA ELISA [176]. Содержание антител IgG против дифтерийного токсина и IgG против столбнячного токсина исследовали методом ELISA. Содержание серологических маркеров к В.pertussis (анти-перактин и aimi-Agg2-3) также исследовали методом ELISA.
Безопасность и реактогенность
Исследователи отслеживали состояние субъектов в течение 30 минут после введения каждой дозы вакцины. Родителей просили заносить в дневник ежедневную ректальную температуру и локальные и системные реакции в течение семи дней после каждой инъекции. Кроме того, исследователи активно контактировали с родителями/законными представителями по телефону на второй и седьмой день после каждой вакцинации для получения сведений о каких-либо нежелательных явлениях. Случаи нежелательных явлений или серьезных нежелательных явлений и явлений, требующих визита врача и/или медикаментозного лечения, тщательно исследовались и регистрировались в течение всего исследования. Субъекты, получившие, по меньшей мере, одну вакцинацию, были включены в анализ безопасности.
Локальные и системные реакции в большинстве были умеренными и кратковременными. Наиболее частыми местными реакциями были чувствительность, эритема и уплотнение, а наиболее частыми системными реакциями были раздражительность, сонливость и необычный плач:
Между четырьмя группами вакцин не наблюдалось значительной разницы, все реакции после вакцинации прошли без последствий. Не сообщалось о серьезных нежелательных явлениях, связанных с вакциной исследования.
Иммуногенность
Уровни сероконверсии анти-PRP (% пациентов с сероконверсией) были следующими:

Уровни сероконверсии против коклюша оценивали по (а) увеличению ответа анти-пертактин и анти-Agg2-3 и (b) GMT. Результаты (% пациентов) были следующими:
GMT антител против дифтерии и столбняка были следующими:
Часть субъектов с титрами анти-PRP≥0,15 мкг/мл была сходна в четырех группах, но часть субъектов с титрами анти-PRP≥1,0 мкг/мл была выше в группах С и D (97% и 95% соответственно), чем в группах А и В (90% и 88% соответственно). GMT анти-PRP показывает четкий эффект зависимости от дозы, будучи сходным в группах А (6,94 мкг/мл) и В (7,82 мкг/мл) и в группах С (17 мкг/мл) и D (18 мкг/мл), но значительно ниже в группах А и В по сравнению с группами С и D (чертеж).
Спустя один месяц после третьей вакцинации у всех субъектов в каждой из четырех групп наблюдалась сероконверсия к дифтерии и столбняку (уровень антител ≥0,1 МЕ/мл). Между четырьмя группами не наблюдалось значительной разницы по GMT.
Между четырьмя группами не наблюдалось никаких различий по доле пациентов с 2- или 4-кратным увеличением антител анти-пертактин и анти-Agg2-3 по сравнению с исходными данными. Титры GMT среди четырех групп также были сходны.
Выводы
Составы вакцины DTwPHib с дробными дозами 5, 2,5 или 1,25 мкг Hib-конъюгата на дозу были столь же иммуногенны, как и известный в литературе состав с 10 мкг, в части, касающейся сероконверсии. Составы с 10 и 5 мкг конъюгата вызывали образование антител анти-PRP с высоким и эквивалентным уровнем GMT. Хотя составы с 2,5 мкг и 1,25 мкг конъюгата на дозу вызывали образование антител анти-PRP с более низкими уровнями GMT, они были адекватно иммуногенны.
Таким образом, работа подтверждает, что количество Hib антигена может быть снижено до 1,25 мкг на дозу в соответствующем составе, содержащем вакцины DTP, без влияния на протективную эффективность других компонентов вакцины.
Все четыре состава вакцины DTP-Hib были безопасны и иммуногенны по всем компонентам вакцины. В терминах уровней серологической защиты анти-PRP эти четыре состава были одинаково иммуногенны как в короткие сроки (титры ≥0,15 мкг/мл), так и в течение длительного промежутка времени (титры ≥1 мкг/мл). Состав, содержащий 5 мкг конъюгата, был столь же иммуногенен, как и известный в литературе состав, содержащий 10 мкг, и для обоих составов уровни GMT анти-PRP были особенно высоки (17 и 18 мкг/мл). Составы, содержащие 1,25 и 2,5 конъюгата, были высокоиммуногенны по сравнению с иммунными ответами, наблюдавшимися в других исследованиях с использованием дробных доз вакцин Hib [6, 8, 9].
Дальнейшую информацию можно найти в ссылке [177].
Следует понимать, что изобретение было описано только посредством примера, и могут существовать модификации, не выходящие за рамки объема и сущности изобретения.






Изобретение относится к области медицины и касается комбинированных вакцин с низкой дозой конъюгата HIB. Сущность изобретения включает комбинированные вакцины, содержащие антигены для иммунизации против дифтерии, столбняка, коклюша и HIB (вакцины "DTP-HIB), в которых: (а) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (b) концентрация конъюгата Hib в вакцине составляет от 1,25 до менее 15 мкг/мл; и (с) конъюгат Hib никогда не лиофилизируют. Преимущество изобретения заключается в создании безопасных и высокоиммуногенных комбинированных вакцин. 7 н. и 10 з.п. ф-лы, 1 ил.
1. Жидкая комбинированная вакцина, включающая антигены для защиты субъекта от, по меньшей мере, дифтерии (′D′), столбняка (′T′), коклюша (′Р′) и Haemophilus influenzae типа b (′Hib′), в которой: (а) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (b) концентрация конъюгата Hib в вакцине составляет от 1,25 до менее 15 мкг/мл; (с) вакцина включает в качестве адъюванта фосфат алюминия; и (d) не более 5 мас.% Hib-конъюгата в вакцине адсорбируется на фосфате алюминия.
2. Жидкая комбинированная вакцина по п.1, в которой вакцина не содержит в качестве адъюванта сульфат алюминия калия.
3. Вакцина по п.1, в которой дифтерийный антиген содержит токсоид дифтерии, столбнячный антиген содержит токсоид столбняка, а коклюшный антиген содержит клеточный компонент коклюша.
4. Вакцина по п.1, в которой конъюгат содержит в качестве носителя CRM197, токсоид столбняка или комплекс наружной мембраны N. meningitidis.
5. Вакцина по п.1, в которой конъюгат содержит олигосахаридный фрагмент полирибозилрибитол фосфата Hib.
6. Вакцина по п.1, в которой комбинированная вакцина дополнительно содержит поверхностный антиген вируса гепатита В.
7. Вакцина по п.1, в которой комбинированная вакцина дополнительно содержит антиген полиомиелита.
8. Вакцина по п.1, в которой комбинированная вакцина дополнительно содержит сахаридный антиген из N. meningitidis серогруппы А, С, W135 и/или Y.
9. Вакцина по п.1, в которой конъюгат имеет соотношение сахарид: белок (мас./мас.) от 1:5 до 5:1.
10. Вакцина по п.1, в которой введение вакцины приводит к концентрации антитела анти-PRP≥0,15 мкг/мл.
11. Вакцина по п.3, в которой токсоид дифтерии и токсоид столбняка адсорбируются на фосфате алюминия.
12. Флакон с прокалываемой герметичной крышкой, содержащий комбинированную вакцину по пп.1-11, причем эта комбинированная вакцина содержит антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и Н. influenzae типа b (′Hib′), где антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib, и где: (а) концентрация конъюгата Hib в вакцине составляет от 1,25 до менее 15 мкг/мл; (b) прокалываемая герметичная крышка флакона не была проколота; (с) вакцина включает в качестве адъюванта фосфат алюминия; и (d) не более 5 мас.% Hib-конъюгата в вакцине адсорбируется на фосфате алюминия.
13. Герметично закрытая емкость, содержащая комбинированную вакцину по пп.1-11, включающую антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H. influenzae типа b (′Hib′), в которой: (а) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (b) концентрация конъюгата Hib в вакцине составляет от 1,25 до менее 15 мкг/мл; (с) вакцина включает в качестве адъюванта фосфат алюминия; и (d) не более 5 мас.% Hib-конъюгата в вакцине адсорбируется на фосфате алюминия.
14. Способ получения жидкой комбинированной вакцины по пп.1-11, характеризующийся тем, что процесс включает следующие стадии: добавление к воде для инъекций фосфата алюминия в качестве адъюванта; добавление антигена для защиты субъекта от дифтерии (′D′); добавление антигена для защиты субъекта от столбняка (′Т′); добавление антигена для защиты субъекта от коклюша (′Р′); добавление NaCl; проверку и доведение рН; и добавление антигена для защиты субъекта от Haemophilus influenzae типа b (′Hib′).
15. Способ помещения комбинированной вакцины по пп.1-11 в контейнер, причем: (а) вакцина включает антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H. influenzae типа b (′Hib′); (b) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (с) концентрация конъюгата Hib в вакцине составляет от 1,25 до менее 15 мкг/мл; (d) вакцина включает в качестве адъюванта фосфат алюминия; и (е) не более 5 мас.% Hib-конъюгата в вакцине адсорбируется на фосфате алюминия.
16. Способ помещения комбинированной вакцины по пп.1-11 в контейнер и последующего извлечения вакцины из контейнера, причем: (а) вакцина включает антигены для защиты субъекта от, по меньшей мере, дифтерии, столбняка, коклюша и H. influenzae типа b (′Hib′); (b) антиген для защиты от Hib представляет собой конъюгат капсульного сахарида Hib; (с) концентрация конъюгата Hib в вакцине составляет от 1,25 до менее 15 мкг/мл; (d) вакцина включает в качестве адъюванта фосфат алюминия; и (е) не более 5 мас.% Hib-конъюгата в вакцине адсорбируется на фосфате алюминия.
17. Способ индуцирования ответного образования антител у млекопитающего, включающий введение млекопитающему вакцины по пп.1-11.
| AMIR J | |||
| et al | |||
| Immunogenicity and safety of a liquid combination | |||
| Of DT-PRP-T vs lyophilized PRP-T recontituted with DTP, VACCINE, BUTTERWORTH SCIENTIFIC, GUILDFORD, GB, 1997, v.15, №2, pp.149-154 | |||
| NICOL M., et al | |||
| Haemophilus influenzae type В conjugate vaccine diluted tenfold in diphtheria-tetanus-whole cell pertussis vaccine: A randomized |

