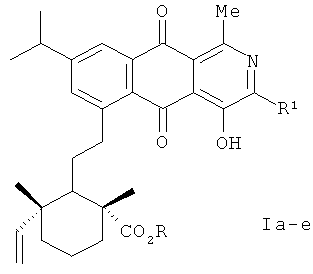
Изобретение относится к органической химии, а именно к новым оптически активным производным 4-гидрокси-2-аза-9,10-антрахинона формулы (Iа-е)
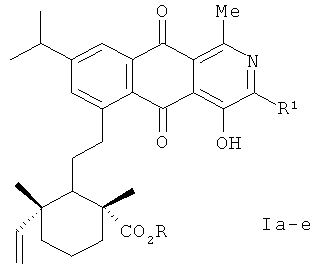
R=H, R1=Me (Ia); R=H, R1=Et (Iб); R=H, R1=i-Рr (Iв); R=R1=Me (Iг); R=Me, R1=Еt (Iд); R=Me, R1=i-Рr (Iе),
обладающим противовоспалительной активностью.
Указанное свойство позволяет предполагать возможность использования указанных соединений в медицине в качестве фармацевтических препаратов.
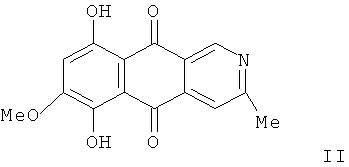
Группа замещенных 1-, 2-, 3- или 4-аза-9,10-антрахинонов перспективна для создания новых лекарственных препаратов в связи с тем, что многие из них обладают выраженной противоопухолевой активностью, широким спектром антибиотических свойств [R.H. Thomson. Naturally Occurring Quinones. IV. Recent Advances. // London: Chapman & Hall, 1997. P.583-648]. Заявляемые соединения являются структурными аналогами природных 2-азаантрахинонов, например бострикоидина (II), проявляющего высокую активность in vitro против Mycobacterium tuberculosis [2. Cameron D.W., Dentscher K.R., Feutrill G.I., Tetrahedron Lett., 1980, 21, 5089-5090; D. Parisot, M. Deoys, M. Barbier, J. Antibiotics, 1989, 42, 1189-1190].
Задачей изобретения является создание новых средств, обладающих противовоспалительной активностью с улучшенными свойствами.
Поставленная задача достигается новыми химическими соединениями указанной формулы (Iа-е), обладающими выраженной противовоспалительной активностью.
Следует подчеркнуть, что природные 2-аза-9,10-антрахиноны являются редкими малодоступными соединениями, и разработка методов синтеза их аналогов представляет существенный интерес. Ценность предлагаемого способа получения оптически активных 2-азаантрахинонов состоит в том, что на основе таких доступных продуктов, как сосновая живица, 1,4-бензохинон и α-аминокислоты (используются для получения необходимых 2-метил-4-алкил-5-метоксиоксазолов [Кондратьева Г.Я., Чжи-Хэн Л. Журнал общей химии, 1962, 32, 2348-2356; Doctorova N.D., Somova L.V., Karpeisky M.Ya., Padyukova N.Sh., Turchin K.F., Florentiev V.L. Tetrahedron, 1969, 25, 3527-3553]) с помощью простых технологических операций получаются оптически активные соединения уникальной структуры. Способ осуществляется по показанной на схеме 1 последовательности превращений из левопимаровой кислоты (III). Левопимаровая кислота является основным компонентом суммы кислот живицы сосны обыкновенной Pinus silvestris L. и отличается высокой реакционной способностью [Пентегова В.А., Дубовенко Ж.В., Ралдугин В.А., Шмидт Э.Н. Терпеноиды хвойных растений // Новосибирск: Наука, 1987].
На первой стадии проводится реакция 1,4-бензохинона с левопимаровой кислотой (III), которая присутствует в сосновой живице и не требует выделения. Выход дикетона (IVa), выделяющегося в виде кристаллов, не ниже 80%. Далее следует растворение в 5%-ном растворе гидроокиси натрия и обработка кислотой, что дает гидрохинон (Va) (выход 87%). Окисление последнего с помощью церий аммоний нитрата или броматом калия дает кислоту (VIa) с выходом 84% или 96% соответственно. Термолиз проходит с выходом до 97% нафтохинона (VIIa). Диеновый синтез между хиноном (VIa) и 4-алкил-2-метил-5-метоксиоксазолами (VIIIa-в) проходит при катализе эфиратом трехфтористого бора в хлористом метилене, давая трициклические эпоксиаддукты (IXa-в) (выход 62-68%). Заключительной стадией синтеза является термолиз эпоксисоединений (IХа-в) в растворе 1,4-диоксана в присутствии трифторуксусной кислоты, что приводит к 3-замещенным-4-гидрокси-2-аза-9,10-антрахинонам (Iа-в) (выход 72-81%).
Метилирование аддукта (IVa) диметилсульфатом дает метиловый эфир (IVб) (выход 83%), для которого осуществлена аналогичная последовательность превращений. Обработка соединения (IVб) гидроокисью натрия приводит к гидрохинону (Vб) (выход 90%). Окислением гидрохинона (Vб) получают метиловый эфир хризенхинона (VIб) (выход 89%). Термолизом полициклического хинона (VIб) получают замещенный 1,4-нафтохинон (VIIб) (выход 98%). Взаимодействие хинона (VIIб) с оксазолами (VIIIa-в) при катализе эфиратом трехфтористого бора в хлористом метилене приводит к смеси региоизомерных циклоаддуктов, из которой выделяют основные региоизомеры - эпоксиаддукты (IХг-е) (выход 62-64%). Термолиз соединений (IХг-е) в растворе диоксана в присутствии трифторуксусной кислотой приводит к 3-замещенным-4-гидрокси-2-аза-9,10-антрахинонам (Iг-е) (выход 76-85%). Проведение реакции циклоприсоединения хинонов (VIIa,б) с оксазолами при катализе эфиратом трехфтористого бора отличается региоселективностью и в конечном итоге [после кислотной обработки окса-аддуктов (IХа-в)] (схема 1) приводит к образованию 4-гидрокси-2-азаантрахинонов (Iа-в) с общим выходом 28-34% в расчете на вводимую в реакцию левопимаровую кислоту (III). Общий выход соединений (Iг-е) по схеме 1 в расчете на левопимаровую кислоту (III) составляет 26-28%. Физико-химические константы новых, впервые полученных соединений, приведены в примерах 1-6.
Достоинством изобретения является способ получения соединений (Iа-е) путем химической модификации доступного растительного метаболита сосны обыкновенной - левопимаровой кислоты (III) и 1,4-бензохинона. Благодаря высокой активности обоих компонентов в реакции диенового синтеза, не требуется выделения левопимаровой кислоты из живицы, а 1,4-бензохинон непосредственно добавляется к живице, как это описано в работе [Толстиков Г.А., Шульц Э.Э., Мухаметзянова Т.Ш., Байкова И.П., Спирихин Л.В. ЖОрХ, 1993, 29, 698-716]. Отличительной особенностью изобретения является получение оптически активных замещенных 1,4-нафтохинонов (VIIa, VIIб) (общий выход 53-62% в расчете на содержащуюся в сосновой живице левопимаровую кислоту) и введение их в реакцию диенового синтеза с оксазолами (VIIIa,б). Региоселективность реакции диенового синтеза обеспечивается использованием катализатора, в качестве которого выступает доступная и удобная в использовании кислота Льюиса - эфират трехфтористого бора.
В работе [Толстиков Г.А., Шульц Э.Э., Мухаметзянова Т.Ш., Султанова B.C., Спирихин Л.В. ЖОрХ, 1992, 28, 1310-1313] описывается способ получения изомерного 1-гидрокси-2-аза-9,10-антрахинона (X), основанный на термической реакции циклоприсоединения полициклического хинона (VIa) к 5-метоксиоксазолу (VIIIб) (кипячение в ацетонитриле) и последующем ретро-диеновом расщеплении полициклического аддукта (XI) (схема 2). Как видно из схемы 2, в результате реакции диенового синтеза образуются смесь региоизомеров - первичных продуктов реакции (ХI, ХII) [выход соединения (XI) составляет 58%]. Термическое расщепление (XI) путем нагревания в ДМФА и последующая колоночная хроматография приводит к соединению (X) (выход 66%). Соединение (XII) (минорный продукт) в индивидуальном виде не выделяли, его образование фиксировали по данным спектра ЯМР 1Н смеси соединений (XI, XII).
Таким образом, способ получения 4-гидрокси-2-азаантрахинонов (Iа-е) заключается в реакции оптически активных 1,4-нафтохинонов (VIIa,б) с 4-алкил-5-метоксиоксазолами (VIIIa-в), катализируемой кислотой Льюиса, и последующем кислотно-катализируемом термическом расщеплении региоселективно образующихся эпоксиаддуктов (IХа-е). Оптически активные нафтохиноны (VIIa,б) синтезируются из левопимаровой кислоты (III), содержащейся в сосновой живице, и 1,4-бензохинона.
Широкая распространенность воспалительных заболеваний, а также несовершенство известных противовоспалительных средств, заключающееся не столько в недостаточной эффективности, сколько в существовании побочных эффектов практически всех существующих групп соединений этого типа, стали стимулом для поиска новых нестероидных противовоспалительных средств (НПВС).
Известно использование в качестве противовоспалительного средства ортофена (вольтарен, диклофенак-натрий), имеющего формулу (XIII)
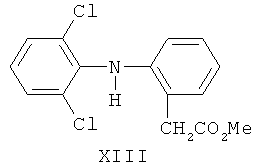
Соединение обладает выраженным ульцерогенным эффектом [М.Д. Машковский. Лекарственные средства, Торсинг, Харьков, 1998, т.1, с.198].
Анализ литературных данных показывает, что синтез и фармакологические испытания новых химических соединений, отличающихся от существующих НПВС способностью и избирательностью действия, пролонгированным эффектом и отсутствием побочных явлений, является актуальной задачей.
Эффективными направлениями противовоспалительной терапии является поиск средств, стабилизирующих лизосомные мембраны [H.G. Vogel, V. Vogel, Drug Discovery and Evaluation, Springer Verlag, Heidelberg, 1996, 236 p.], применение антиоксидантов [А.С. Саратиков, Т.П. Прищеп. Современные концепции анифлогистического действия НПВС. // Фармакол. и токсикол., 1982, v.45, №2, с.133-138], а также веществ, оказывающих тормозящее влияние на выработку антител лимфоидными и плазматическими клетками [A.M. Чернух. Воспаление, Медицина, М., 1999, 448 с.].
Биологическая активность заявляемых соединений (Iа-е) изучалась путем определения токсичности и противовоспалительной активности на модели каррагенинового и формалинового воспалений.
Острую токсичность 4-гидрокси-2-азаантрахинонов (Iа-е) определяли на белых беспородных мышах обоего пола при однократном введении в желудок. Параметры токсичности вычисляли по Литчфильду и Уилкоксону методом пробит-анализа.
LD50 новых соединений приведены в таблице 1. Как видно из данных таблицы 1, широко используемые в медицине противовоспалительные вещества токсичнее исследуемых соединений. Все соединения относятся к III классу умеренно опасных веществ.
Противовоспалительное действие наименее токсичных 4-гидрокси-2-азаантрахинонов (Iа,б) изучено на двух моделях воспаления, вызванных 1%-ным раствором каррагенина и 3%-ным раствором формалина под апоневроз одной из задних лапок мыши в дозе 0.05 мл. О противовоспалительном действии соединения судили по проценту увеличения массы воспаленной лапки по сравнению со здоровой. Препаратом сравнения служил ортофен, взятый в дозе ED50.
На модели воспаления, вызванного каррагенином, на мышах и крысах вычислена ED50 соединений (Iа,б), которая составила:
для соединения (Iа) - 30 (24-38) мг/кг;
для соединения (Iб) - 33 (25-49.4) мг/кг.
Результаты исследования противовоспалительной активности соединений (Iа,б) в дозе ED50 приведены в таблицах 2, 3.
На модели воспаления, вызванного введением формалина, соединения (Iа,б) статистически значимо задерживают развитие воспалительного отека (Р<0.001) (таблица 2). Противовоспалительное действие соединений в дозах 30 мг/кг (1/83 от LD50) для (Iа) и 33 мг/кг (1/60 от LD50) для (Iб) аналогично эффекту ортофена.
Оба исследуемых соединения в эффективных дозах достоверно задерживают развитие воспалительного отека, вызванного карагенином (таблица 3). Оба соединения в 1.7-1.8 раза уменьшают отек лапок животных по сравнению с контрольной группой.
К отличительным признакам способа следует отнести:
- получение оптически активных 4-гидрокси-2-азаантрахинонов из доступного отечественного сырья - сосновой живицы и 1,4-бензохинона;
- получение 4-гидрокси-2-азаантрахинонов с использованием на ключевой стадии катализируемой кислотой Льюиса реакцией диенового синтеза оптически активных полифункциональных 1,4-нафтохинонов с 4-алкил-2-метил-5-метоксиоксазолами;
- получение новых малотоксичных 4-гидрокси-2-азаантрахинонов, обладающих противовоспалительной активностью.
Данное изобретение иллюстрируется примерами.
Пример 1. Синтез (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-азаантрахинона (Iа).
1. Получение 7,10а-диметил-13-изопропил-7-карбокси-4,4а,5,6,6а,7,8,9,10,10а,10b,11, 1212а-тетрадекагидро-4b,12-(этено)-1Н-хризен-1,4-диона (IVa). К раствору 110 г сосновой живицы в 100 мл ацетонитрила приливают раствор 5.9 г бензохинона в 50 мл ацетонитрила и оставляют при комнатной температуре в темном месте на 7 дней. Растворитель отгоняют, к остатку приливают 50 мл этанола, к образовавшемуся раствору добавляют 30 мл диэтилового эфира. Выпавший осадок отфильтровывают, промывают холодным эфиром, получают 13.6 г аддукта (IVa) [выход 82.4% в расчете на левопимаровую кислоту (III)], в виде желтого кристаллического осадка. Т. пл. 208-210°С,  - 168° (с 10.1, хлф) (т.пл. 212-214°С [Herz W., Blackstone R.C., Nair M.G. J.Org. Chem. 1967, 32, 2992]; 192°С [Ruzicka L., Kaufmann S.Helv. Chim. Acta. 1941, 24, 1425]).
- 168° (с 10.1, хлф) (т.пл. 212-214°С [Herz W., Blackstone R.C., Nair M.G. J.Org. Chem. 1967, 32, 2992]; 192°С [Ruzicka L., Kaufmann S.Helv. Chim. Acta. 1941, 24, 1425]).
2. Получение 7,10а-диметил-1,4-дигидрокси-13-изопропил-7-карбокси-4b,5,6,6а,7,8, 9,10,10а,10b,11,12-додекагидро-4b,12-этено-хризена (Va). К 5 мл 5% раствора NaOH, предварительно продутого аргоном, при перемешивании прибавляют 8.52 г (20.4 ммоль) ендиона (IVa) и выдерживают 30 мин, затем реакционную массу подкисляют 3%-ным раствором соляной кислоты (30 мл, рН 6). Выпавший осадок отфильтровывают, тщательно промывают водой остаток и сушат на воздухе, а затем 2 ч при 90°С. Получают 8.0 г (87%) гидрохинона (Va), т. пл. 208-210°С.
3. Получение 7,10а-Диметил-7-карбокси-13-изопропил-4b,5,6,6а,7,8,9,10,10а,10b,11,12-додекагидро-4b,12-(этено)хризен-1,4-диона(VIа).
а). К перемешиваемому раствору 16.76 г (40.9 ммоль) гидрохинона (Va) в 70 мл диоксана прибавляют нагретый до 60°С раствор 3.41 г (20.4 ммоль) KBrO3 в 34 мл воды и 3.4 мл 1 н. Н2SO4. Через 10 мин к смеси добавляют 100 мл воды, продукт извлекают хлороформом. Хлороформный слой промывают водой, упаривают в вакууме, остаток растворяют в 100 мл эфира и добавляют 100 мл гексана. Отфильтровывают 6.44 г (39%) хризенхинона (VIa), по охлаждении дополнительно отфильтровывают 9.57 г (57%) (общий выход 96%) соединения (VIa).
б). К раствору 0.94 г (2.3 ммоль) гидрохинона (Va) в 40 мл безводного ацетонитрила при перемешивании добавляют 2.3 ммоль (NH4)2Ce(NO3)6. Перемешивание продолжают 5-6 ч (контроль ТСХ). Реакционную смесь выливают в 70 мл воды, продукт экстрагируют хлороформом (3×20 мл). Экстракт промывают водой, насыщенным раствором соли (2×20 мл), сушат MgSO4, упаривают в вакууме. После очистки с помощью колоночной хроматографии (силикагель, элюент-хлороформ) получают 0.78 г (выход 84%) полициклического хинона (VIa). Т. пл. 140-145°С (из смеси эфир-гексан), - 13.2° (с 5.2, EtOH). Найдено, %: С 76.65; Н 7.80. С26Н32O4. Вычислено, %: С 76.44; Н 7.90. Спектральные данные аналогичны приведенным в работе [Шульц Э.Э., Олейников Д.С., Нечепуренко И.В., Шакиров М.М., Толстиков Г.А. Журнал органической химии, 2009, 45, 108].
4. Получение 5-[2-(6-винил-2,6-диметил-2-карбоксициклогексил)этил]7-изопропил-1,4-нафтохинона (VIIa). Раствор 0.94 г (2.3 ммоль) хинона (VIa) в 15 мл диоксана нагревают в запаянной ампуле в атмосфере аргона 10 ч при 150°С. По охлаждении ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме, остаток хроматографируют на силикагеле (элюент-хлороформ). Получают 0.91 г (выход 97%) замещенного 1,4-нафтохинона (VIIa). Т. пл. 86-100°С. [α]580 - 22.9° (с 1.8, EtOH). Масс-спектр, m/z (Iотн, %): 408 (33), 362 (100), 214 (38), 43 (36). Найдено: [М]+ 408.23001. С26Н32O4. Вычислено: М 408.23004. Данные спектров ЯМР 1Н и 13С аналогичны приведенным в работе [Шульц Э.Э., Олейников Д.С., Нечепуренко И.В., Шакиров М.М., Толстиков Г.А. Журнал органической химии, 2009, 45, 108].
5. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-метокси-7-изопропил-1,3-диметил-1,4-эпокси-2-азаантрацен-9,10-диона (IXa).
Раствор 1.04 г (2.55 ммоль) соединения (VIIa) в 20 мл хлористого метилена загружают в стеклянную ампулу, прибавляют 0.032 мл (0.26 ммоль) ВF3·Et2О, затем 0.42 г (3.31 ммоль) оксазола (VIIIa). Ампулу продувают аргоном, охлаждают и запаивают. Реакционную смесь выдерживают при 20°С 48 ч. По прошествии указанного времени ампулу вскрывают, реакционную массу выливают в 100 мл воды, продукт экстрагируют хлористым метиленом (20×3 мл), органические слои объединяют, промывают водой, затем сушат над прокаленным сульфатом магния. Осушитель отфильтровывают, маточный раствор упаривают, остаток разделяют колоночной хроматографией на силикагеле (элюенты: хлороформ, хлороформ - этанол, в соотношении 10:1). Собирают 3 фракции. Последнюю фракцию (1.26 г) дополнительно очищают колоночной хроматографией на силикагеле (элюент: бензол-этилацетат). Получают 2 фракции. Последующей хроматографией фракции 2 выделяют 0.90 г (выход 68%) аддукта (IXa) в виде смеси стереоизомеров (эндо- и экзорасположение эпоксимостика). Маслообразное вещество. Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: 1.00 с, 1.03 с (3Н, СН3 при С6'), 1.18 с, 1.20 с (3Н, СН3 при С1), 1.26 д, 1.28 д [6Н, СН(СН 3)2 при С7, J=6.9], 1.28 с (3Н, СН3 при С2'), 1.30-1.52 м (5Н, H4',4',5',12',12'), 1.65 с, 1.70 с (3Н, СН3 при С3), 1.80-2.02 м (3Н, Н1',3',5'), 2.22 м [2Н, 1Н, СН(СН3)2 и 1Н, Н3'], 2.46-2.60 м (2Н, Н13',13'), 3.59 с, 3.65 с (3Н, СН3), 3.85 м (1Н, Н9a), 4.12 м (1Н, Н4a), 4.82-4.91 м (2Н, Н8'), 5.60-5.68 м (1Н, Н7'), 7.27 д (1Н, Н6, J=1.8), 7.84 д (1Н, Н8, J=1.8), 10.01 с (1Н, ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 16.2 к, 16.6 к (СН3 при С6'), 17.68 т (С4'), 18.01 к (СН3 при С1), 18.43 к, 18.63 к (СН(СН3)2), 19.4 к, 19.7 к (СН3 при С2'), 22.79 к (СН3 при С3), 28.76 т (С12'), 33.37 д (СН-i-Pr), 35.01 т (С3'), 36.46 т (С5'), 38.05 т (С13'), 41.43 с (С6'), 44.25 д (С1'), 45.97 с (С2'), 46.05 д (С4a), 50.32 д (С9a), 52.42 к (ОСН3), 73.01 с (С1), 88,54 с (С4), 110.46 т (С8'), 123.73 д (С8), 130.68 с (С10a), 134.17 с (С8a), 135.16 д (С6), 140.08 с (С5), 149.35 с (С7), 150.24 д (С7'), 160.35 с (С3), 187.21 с (С9), 189.81 с (С10), 184.61 с (С=O). Найдено, %: С 71.55; Н 7.45; N 2.44. С32Н41НО6. Вычислено, %: С 71.75; Н 7.71; N 2.61.
6. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-азаантрахинона (Iа). В стеклянную ампулу, в токе аргона, загружают 2.14 г (4 ммоль) эпоксисоединения (IXa) в 10 мл диоксана, 0.08 мл трифторуксусной кислоты, молекулярные сита 3А, охлаждают до 0°С и запаивают. Ампулу осторожно нагревают и выдерживают 15 ч при 125°С (баня). После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме. Колоночной хроматографией (силикагель, хлороформ, хлороформ-спирт, 10:1) выделяют 1.77 г (2R,6R)-5-[2(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Iа) в виде желтого маслообразного вещества. При перекристаллизации из петролейного эфира получают 1.48 г (выход 74%) (2R,6R)-5-[2(6-винил-2-карбокси-2,6-диметилциклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Iа). Т. пл. 102-104°С. +31.8° (с 1.2, хлф.). Спектр ЯМР 1Н (CDCl3), δ, м.д., J, Гц: 1.04 с (3Н, СН3 при С2'), 1.18 д, 1.24 д (6Н, СН(СН
3)2 при С7), 1.32 с (3Н, СН3 при С6'), 1.40-1.79 м (8Н, H1',3',4',4',5',5',12',12'), 1.88 с (3Н, СН3), 2.08 м (2Н, 1Н, CH-i-Pr и 1Н, Н3'), 2.42-2.56 м (2Н, Н13',13'), 2.18 с (3Н, СН3), 4.91 д, 4.98 д (2Н, Н8', Jгем 2.2, Jвиц 10.8 и 15.0). 5.74 м (1Н, Н7', J 10.8 и 15.0), 7.08 д (1Н, Н6, J 1.8), 7.82 д (1Н, Н8, J 1.8 Гц), 8.05 с (1Н, ОН), 9.90 с (1Н, ОН) Спектр ЯМР 13С (СDСl3), δ, м.д.: 17.46 к (СН3 при С6'), 17.8 т (С4'), 18.66 к (2xСН3-i-Рr), 21.88 к, 22.92 к (СН
3 при С1 и С3), 25.61 к (СН3 при С2'), 29.30 т (С12'), 32.10 д (CH-i-Pr), 35.10 т (С13'), 36.90 т (С5'), 38.00 т (С3'), 39.80 с (С6'), 43.10 с (С2'), 46.10 д (С1'), 110.50 т (С8'), 123.90 д (С8), 125.80 с (С8a), 126.10 с (С10a), 129.05 с (С9a), 130.80 д (С6), 133.18 с (С4a), 140.67 с (С5), 146.91 с (С4), 151.10 д (С7'), 156.10 с (С1), 158.20 с (С3), 161.10 с (С7), 179.86 с (С10), 183.02 с (С9), 184.62 с (СООН). Найдено, %: С 73.61; Н 7.22; N 2.59. С31Н37NO5. Вычислено, %: С 73.93; Н 7.41; N 2.78. Общий выход соединения (Iа) в расчете на исходную левопимаровую кислоту (III) составляет 31%.
Пример 2. Синтез (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-7-изопропил-1-метил-3-этил-2-азаантрахинонона (1б).
Стадии 1-4 выполняются по методикам, приведенным в описании примера 1.
5. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-метокси-7-изопропил-1-метил-3-этил-1,4-эпокси-2-азаантрацен-9,10-диона (IХб).
Раствор 1.04 г (2.55 ммоль) соединения (VIIa) в 20 мл хлористого метилена загружают в стеклянную ампулу, прибавляют 0.032 мл (0.26 ммоль) ВF3 ·Et2О, затем 0.46 г (3.26 ммоль) оксазола (VIIIб). Ампулу продувают аргоном, охлаждают и запаивают. Реакционную смесь выдерживают при 20°С 52 ч. По прошествии указанного времени ампулу вскрывают, реакционную массу выливают в 100 мл воды, продукт экстрагируют хлористым метиленом (20×3 мл), органические слои объединяют, промывают водой, затем сушат над прокаленным сульфатом магния. Осушитель отфильтровывают, маточный раствор упаривают, остаток разделяют колоночной хроматографией на силикагеле (элюенты: хлороформ, хлороформ - этанол, в соотношении 10:1). Собирают 3 фракции. Последнюю фракцию (1.18 г) дополнительно очищают колоночной хроматографией на силикагеле (элюент: бензол-этилацетат). Получают 2 фракции. Последующей хроматографией фракции 2 выделяют 0.95 г (выход 67%) аддукта (IХб) в виде смеси стереоизомеров (эндо- и экзорасположение эпоксимостика). Маслообразное вещество. Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: 1.06 с, 1.08 с (3Н, СН3 при С2'), 1.18 с, 1.20 с (3Н, СН3 при С1), 1.20 т (3Н, СН3, J=7), 1.29 д [6Н, СН(СН 3)2 при С7, J=6.9], 1.32 с (3Н, СН3 при С 6'), 1.38-1.81 м (8Н, Н1',3',4',4',5',5',12',12'), 2.02-2.12 м [2Н, 1Н, CH-i-Pr и 1Н, Н3'], 2.46-2.65 м (2Н, Н13',13'), 3.58 с, 3.61 с (3Н, СН3), 3.72 м (1Н, Н9a), 4.02 к (2Н, СН2, J=7), 4.14 м (1Н, Н4a), 4.85-4.94 м (2Н, Н8'), 5.60-5.66 м (1Н, Н7'), 7.26 д (1Н, Н6, J=1.8), 7.82 д (1Н, Н8, J=1.8), 10.11 с (1Н, ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 13.24 к (СН3), 16.4 к, 16.6 к (СН3 при С2'), 18.01 т (С4'), 18.12 к (СН3 при С1), 18.68 к, 18.73 к (СН(СН3)2), 22.41 к, 22.61 к (СН3 при С6'), 28.85 т (С12'), 33.37 д (СН-i-Рr), 36.12 т (С3'), 37.05 т (С13'), 37.96 т (С5'), 41.02 с (С6'), 43.97 с (С2'), 45.05 д (С1'), 45.85 д (С4a), 49.83 д (С9a), 52.42 к (ОСН3), 57.46 т (CH2), 74.11 с (С1), 89.08 с (С4), 111.26 т (С8'), 122.96 д (С8), 131.01 с (С10a), 134.21 с (С8a), 133.01 д (С6), 139.35 с (С5), 150.08 с (С7), 150.41 д (С7'), 161.17 с (С3), 188.04 с (С9), 189.75 с (С10), 184.80 с (С=О). Найдено, %: С 71.92; Н 7.68; N 2.41. С33Н43NO6. Вычислено, %: С 72.10; Н 7.88; N 2.55.
6. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-7-изопропил-1-метил-3-этил-2-азаантрахинонона (Iб). В стеклянную ампулу, в токе аргона, загружают 1.85 г (1.7 ммоль) эпокси-соединений (IXб) в 10 мл диоксана, 0.08 мл трифторуксусной кислоты, молекулярные сита 3А, охлаждают до 0°С и запаивают. Ампулу осторожно нагревают и выдерживают 15 ч при 125°С. После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме. Колоночной хроматографией остатка (1.93 г) (силикагель, хлороформ, хлороформ-спирт, 10:1) выделяют 1.44 г (выход 81%) (2R,6R)-5-[2(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Iб) в виде желтого маслообразного вещества, которое кристаллизуется при стоянии. Т. пл. 116-122°С (из эфира). +45.1° (с 3.1, СНСl3). Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: 0.99 с (3Н, CH3 пpи C2'), 1.18 д, 1.21 д (6H, CH(CH
3)2 при C7, J=6.8), 1.23 к (3H, CH3, J=7.0),1.32 с (3Н, СН3 при С6'), 1.35-1.78 м (8Н,Н1',3',4',4',5',5',12',12'), 2.15 м(2Н, 1Н, CH-i-Рr и 1Н, Н3'), 2.06 с (3Н, СН3), 2.41-2.59 м (2Н, Н13',13'), 4.18 к (2Н, СН2, J=7.0), 4.88-4.98 м (2Н, Н8'), 5.75 м (1Н, Н7', J 10.8 и 15.0), 7.20 д (1Н, Н6, J=1.8), 7.85 д (1Н, Н8, J=1.8), 8.09 с (1Н, ОН), 10.0 с (1Н, ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 12.42 к (СН3), 17.46 т (С4'), 17.88 к (СН3 при С6'), 18.66 к, 18.83 к (2 х СН3-i-Рr), 21.88 к (СН
3 при С1), 25.46 к (СН3 при С2'), 29.30 т (С12'), 32.10 д (CH-i-Pr), 34.58 т (С3'), 36.02 т (С13'), 37.90 т (С5'), 39.80 с (С6'), 40.26 т (СН2), 42.12 с (С2'), 46.73 д (С1'), 110.50 т (С8'), 124.08 д (С8), 125.88 с (С8a), 126.65 с (С10a), 129.45 с (С9a), 130.14 д (С6), 133.18 с (С4a), 139.68 с (С5), 147.18 с (С4), 151.18 д (С7'), 156.18 (С1), 158.27 с (С3), 161.34 с (С7), 179.80 с (С10), 183.34 с (С9). 184.46 с (СООН). Найдено, %: С 74.43; Н 7.01; N 2.46. С32Н39NO5. Вычислено, %: С 74.25; Н 7.59; N 2.71. Общий выход соединения (Iб) в расчете на исходную левопимаровую кислоту (III) составляет 34%.
Пример 3. Синтез (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-3,7-диизопропил-1-метил-2-азаантрацен-9,10-диона (Iв).
Стадии 1-4 аналогичны приведенным в описании примера 1.
5. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-метокси-3,7-диизопропил-1-метил-1,4-эпокси-2-азаантрацен-9,10-диона (IХв).
Раствор 1.58 г (3.87 ммоль) соединения (VIIa) в 20 мл хлористого метилена загружают в стеклянную ампулу, прибавляют 0.05 мл (0.4 ммоль) ВF3·Et2О, затем 0.67 г (4.32 ммоль) оксазола (VIIIa). Ампулу продувают аргоном, охлаждают и запаивают. Реакционную смесь выдерживают при 20°С 48 ч. По прошествии указанного времени ампулу вскрывают, реакционную массу выливают в 100 мл воды, продукт экстрагируют хлористым метиленом (25×3 мл), органические слои объединяют, промывают водой, затем сушат над прокаленным сульфатом магния. Осушитель отфильтровывают, маточный раствор упаривают, остаток разделяют колоночной хроматографией на силикагеле (элюенты: хлороформ, хлороформ - этанол, в соотношении 10:1). Собирают 3 фракции. Последнюю фракцию (1.66 г) дополнительно очищают колоночной хроматографией на силикагеле (элюент: бензол-этилацетат).
Получают 2 фракции. Последующей хроматографией фракции 2 выделяют 1.31 г (выход 60%) аддукта (IХв) в виде смеси стереоизомеров (эндо- и экзорасположение эпоксимостика). Маслообразное вещество. Спектр ЯМР 1Н (CDCl3), δ, м.д., J, Гц: 1.05 с, 1.08 с (3Н, СН3 при С6'), 1.18 д, 1.20 д, 1.26 д, 1.28 д (12Н, СН(СН 3)2 при С3 и СН(СН 3)2 при С6), 1.30 с, 1.33 с (3Н, СН3 при С2'), 1.38-1.79 м (8Н, H1',3',4',4',5',5',12',12'), 2.09 м (2Н, 1Н, CH-i-Pr и 1Н, Н3'), 2.22 м (1Н, СH-i-Pr), 2.42-2.60 м (2Н, Н13',13'), 2.62 с, 2.67 с (3Н, СН3), 3.59 с, 3.64 с (3Н, СН3), 3.78 м (1Н, Н4a), 4.06 м (1Н, Н4a), 4.81-4.91 м (2Н, Н8'), 5.60-5.68 м (1Н, Н7), 7.28 д (1Н, Н6, J 1.8 Гц), 7.81 д (1Н, Н8, J 1.8 Гц), 10.01 с (ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 16.2 к, 16.8 к (СН3 при С2'), 17.71 т (С4'), 18.61 к (СН3 при С1), 18.91 к, 19.03 к (СН(СН3)2), 21.55 к, 21.78 к (СН(СН3)2), 23.22 к (СН3 при С6'), 28.74 т (С12'), 32.18 д. 32.66 д (CH-i-Pr), 35.12 т (С13'), 36.19 т (С3'), 38.08 т (С5'), 40.66 с (С6'), 43.73 с (С2'), 45.38 д (С1'), 45.32 д (С4a), 49.51 д (С9a), 52.46 к (ОСН3), 75.01 с (С4), 89.54 с (С1), 110.46 т (С8'), 123.03 д (С8), 131.21 с (С10a), 134.17 с (С8a), 132.16 д (С6), 140.08 с (С5), 149.65 с (С7), 150.24 д (С7'), 160.35 с (С3), 184.53 с (С=O), 188.01 с (С9), 189.95 с (С10). Найдено, %: С 72.18; Н 7.77; N 2.04. C34H45NO6. Вычислено, %: С 72.44; Н 8.05; N 2.48.
6. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-3,7-диизопропил-1-метил-2-азаантрацен-9,10-диона (Iв). В стеклянную ампулу, в токе аргона, загружают 1.07 г (2 ммоль) эпокси-соединений (IХв) в 15 мл диоксана, 0,1 мл трифторуксусной кислоты, молекулярные сита 3А, охлаждают до 0°С и запаивают. Ампулу осторожно нагревают и выдерживают 15 ч при 105°С. После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме. Колоночной хроматографией (силикагель, хлороформ, хлороформ-спирт, 10:1) выделяют (2R,6R)-5-[2(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинон (Iв) в виде желтого маслообразного вещества (0.88 г). Перекристаллизацией из петролейного эфира получают 0.77 г (выход 72%) кристаллического продукта (Iв). Т. пл. 133-136°С. +28.5° (с 2.3, СНСl3). Спектр ЯМР 1H (СDСl3), δ, м.д., J, Гц: 1.12 с (3Н, СН3 при С6'), 1.18 д, 1.21 д (6Н, СН(СН
3)2 при С7, J=6.8), 1.25 с (3Н, СН
3 при С2'), 1.28 д, 1.32 д (6Н, СН(СН
3)2 при C3, J=7.0), 1.40-1.79 м (8Н, H1',3',4',4',5',5',12',12'), 2.08-2.22 м (2Н, 1Н, CH-i-Pr и 1Н, Н3'), 2.28 м (1Н, CH-i-Pr), 2.38 с (3Н, СН
3 при С1), 2.48-2.58 м (2Н, Н13',13'), 4.91 д, 4.96 д (2Н, Н8', Jгем 2.2, Jвиц 10.8 и 15.0), 5.70 м (1Н, Н7', J 10.8 и 15.0), 7.18 д (1Н, H6, J=1.8), 7.81 д (1Н, H8, J=1.8), 8.05 с (1Н, ОН), 9.90 с (1Н, ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 17.21 к (СН3 при С6'), 17.68 т (С4'), 18.66 к, 18.73 к (СН3-i-Рr), 20.08 к, 20.39 к (СН3-i-Рr), 21.92 к (СН
3 при С1), 25.81 д (CH-i-Pr), 26.08 к (СН3 при С2'), 29.68 т (С12'), 32.19 д (CH-i-Pr), 35.88 т (С3'). 36.10 т (С13'), 37.67 т (С5'), 39.43 с (С6'), 42.31 с (С2'), 46.10 д (C1'), 111.65 т (С8'), 123.90 д (С8), 125.80 с (С8a), 126.10 с (С10a), 130.05 с (С9a), 130.80 д (С6), 132.18 с (С4a), 140.07 с (С5), 148.16 с (С4), 151.10 д (С7'), 155.42 (С1), 158.04 с (С3), 161.10 с (С7), 179.86 с (С10), 183.02 с (С9), 184.62 с (СООН). Найдено, %: С 74.32; Н 7.43; N 2.51. С33Н41NO5. Вычислено, %: С 74.55; Н 7.77; N 2.63. Общий выход соединения (Iв) в расчете на исходную левопимаровую кислоту (III) составляет 28%.
Пример 4. Синтез (2R,6R)-5-[2-(6-винил-2,6-диметил-2-метоксикарбонилцикло-гексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-азаантрацен-9,10-диона (Iг).
1. Получение 7,10а-диметил-13-изопропил-7-метоксикарбонил-4,4а,5,6,6а,7,8,9,10,10а,10b,11,12,12а-тетрадекагидро-4b,12-этено-1Н-хризен-1,4-диона (IVб). К раствору 10.25 г (25 ммоль) ендиона (IVa) в 150 мл ацетона прибавляют 4.14 г (30 ммоль) K2СО3 и 2.84 мл (30 ммоль) диметилсульфата. Реакционную смесь кипятят 20 ч, после охлаждения отфильтровывают осадок, промывают хлороформом, органические слои упаривают. К остатку добавляют 50 мл воды, продукт экстрагируют хлороформом, экстракт промывают водой, сушат над MgSO4 и упаривают. Остаток (9.62 г) растворяют в 120 мл этанола, добавляют 1.5 г активированного угля и кипятят 20 мин, уголь отфильтровывают, осветленный этанольный раствор упаривают. Остаток перекристаллизовывают из смеси этанол-петролейный эфир (1:3), получают 8.82 г (83%) метилового эфира хинопимаровой кислоты (IVб), т. пл. 167-168°С, - 118° (с 4.5, СНСl3) (литературные данные 163°С [Herz W., Blackstone R.C., Nair M.G. J. Org. Chem. 1967, 32, 2992]).
2. Получение 7,10а-диметил-1,4-дигидрокси-13-изопропил-7-метоксикарбонил-5b,6a,8,9,10,10b,11,12-додекагидро-4b,12-этенохризена (Vб). К раствору 4.21 г (10 ммоль) метилхинопимарата (IVб) в 30 мл ЕtOН добавляют при перемешивании 25 мл 5% раствора NaOH, смесь выдерживают 40 мин, затем подкисляют 3%-ным раствором НС1 до рН 5 (≈ 24 мл). Выпавший осадок отфильтровывают, промывают водой, сушат и перекристаллизовывают из этилацетата. Отфильтровывают 3.84 г (90%) гидрохинона (Vб) т. пл. 185-187°С, - 106° (с 1.5, СНСl3). Спектр ЯМР 1Н (CDCl3), δ, м.д.: 0.67 с (3Н, СН3 при С10a), 0.76-0.86 м (3Н, Н11a,e,10b), 0.97 д, 1.0 д (6Н, СН(СН
3)2, J 6.9 Гц), 1.15 с (3Н, СН3 при С7), 1.18-1.32 м (4Н, H6a,e,9a,10a), 1.62.1.83 м (6Н, Н8a,e,5a,9e,6а,10е), 2.30 м (1Н, СН(СН3)2, J 6.9 Гц), 2.80 м (1Н, Н5e), 3.66 с (3Н, ОСН3), 4.08 с (1Н, Н12) 5.62 с (1Н, Н14), 6.30 д (1Н, H2, J 8.0 Гц), 6.38 д (1Н, Н3, J 8.0 Гц), 8.0 с (2Н, ОН).
3. Получение 7,10а-диметил-13-изопропил-7-метоксикарбонил-4,4а,5,6,7,8,9,10,10а,10b,11,12-додекагидро-4b,12-этено-1Н-хризен-1,4-диона (VIб). К раствору 3.85 г (8.2 ммоль) гидрохинона (Vб) в 40 мл безводного ацетонитрила при перемешивании постепенно порциями прибавляют 4.55 г (8.3 ммоль) (NH4)2Ce(NO3)6. Перемешивание продолжают 6 ч (контроль ТСХ). Реакционную смесь выливают в 150 мл воды, продукт экстрагируют хлороформом (3×60 мл). Экстракт промывают водой, насыщенным раствором соли (2×20 мл), сушат MgSO4, упаривают в вакууме. После очистки с помощью колоночной хроматографии (силикагель, элюент-хлороформ) получают 3.43 г (выход 89%) полициклического хинона (VIб). Спектральные данные соединения (VIб) аналогичны приведенным в работе [Шульц Э.Э., Олейников Д.С., Нечепуренко И.В., Шакиров М.М., Толстиков Г.А. Журнал органической химии, 2009, 45, 108].
4. Получение 5-[2-(6-винил-2,6-диметил-2-метоксикарбонилциклогексил)этил]-7-изопропил-1,4-нафтохинона (VIIб). Раствор 0.96 г (2.3 ммоль) хинона (VIб) в 15 мл диоксана нагревают в запаянной ампуле в атмосфере аргона 10 ч при 150°С. После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме, остаток хроматографируют на силикагеле (элюент-хлороформ). Получают 0.91 г (выход 98%) замещенного 1,4-нафтохинона (VIIб). Т. пл. 116-118°С. [α]D - 8.6° (с 4.0, СНСl3). Найдено, %: С 76.3; Н 8.2. С27Н34O4. Вычислено, %: С 76.74; Н 8.11. Данные спектров ЯМР 1Н и 13С аналогичны приведенным в работе [Шульц Э.Э., Олейников Д.С., Нечепуренко И.В., Шакиров М.М., Толстиков Г.А. Журнал органической химии, 2009, 45, 108]. Найдено, %: С 76.3; Н 8.2. С27Н34O4. Вычислено, %: С 76.74; Н 8.11.
5. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-метокси-7-изопропил-1,3-диметил-1,4-эпокси-2-азаантрацен-9,10-диона (IХг).
Раствор 2.16 г (5.12 ммоль) соединения (VIIб) в 20 мл хлористого метилена загружают в стеклянную ампулу, прибавляют 0.066 мл (0.52 ммоль) ВF3·Et2О, затем 0.70 г (5.51 ммоль) оксазола (VIIIa). Ампулу продувают аргоном, охлаждают и запаивают. Реакционную смесь выдерживают при 20°С 48 ч. По прошествии указанного времени ампулу вскрывают, реакционную массу выливают в 100 мл воды, продукт экстрагируют хлористым метиленом (20×3 мл), органические слои объединяют, промывают водой, затем сушат над прокаленным сульфатом магния. Осушитель отфильтровывают, маточный раствор упаривают, остаток разделяют колоночной хроматографией на силикагеле (элюенты: хлороформ, хлороформ - этанол, в соотношении 10:1). Собирают 3 фракции. Последнюю фракцию (2.11 г) дополнительно очищают колоночной хроматографией на силикагеле (элюент: бензол-этилацетат). Получают 2 фракции. Последующей хроматографией фракции 2 выделяют 1.77 г (выход 63%) аддукта (IХг) в виде смеси стереоизомеров (эндо- и экзорасположение эпоксимостика). Маслообразное вещество. Спектр ЯМР 1H (CDCl3), δ, м.д., J, Гц: 1.16 с, 1.18 с (3Н, СН3 при С1), 1.22 с (3Н, СН3 при С6'), 1.26 д, 1.28 д [6Н, СН(СН 3)2 при С7, J=6.9], 1.30 с, 1.33 с (3Н, СН3 при С2'), 1.35-1.56 м (5Н, H4',4',5',12',12'), 1.65 с, 1.67 с (3Н, СН3 при С3), 1.77-2.04 м (3Н, Н1',3',5'), 2.22 м [2Н, 1Н, СН(СН3)2 и 1Н, Н3'], 2.48-2.60 м (2Н, Н13',13'), 3.59 с (3Н, ОСН3), 3.65 с (3Н, ОСН3), 3.82 м (1Н, Н9a), 4.12 м (1Н, Н4a), 4.86-4.91 м (2Н, Н8'), 5.62-5.71 м (1Н, Н7'), 7.22 д (1Н, Н6, J=1.8), 7.80 д (1Н, Н8, J=1.8). Спектр ЯМР 13С (СDСl3), δ, м.д.: 16.8 к, 17.0 к (СН3 при С6'), 17.88 т (С4'), 18.01 к (СН3 при С1), 18.43 к, 18.63 к (СН(СН3)2), 20.4 к, 20.7 к (СН3 при С2'), 22.79 к (СН3 при С3), 29.17 т (С12'), 32.45 д (СН-i-Рr). 34.97 т (С13), 36.12 т (С3'), 38.66 т (С5'), 42.08 с (С2'), 41.48 с (С6'), 44.89 д (С1'), 44.31 д (С4a), 50.65 д (С9a), 52.89 к (ОСН3), 56,11 к (ОСН3), 74.12 с (С1), 91.41 с (С4), 110.46 т (С8'), 123.41 д (С8), 131.11 с (С10a), 133.51 с (С8a), 134.09 д (С6), 139.25 с (С5), 150.08 с (С7), 150.55 д (С7'), 161.17 с (С3), 177.61 с (С=О), 187.21 с (С9), 189.81 с (С10). Найдено, %: С 71.86; Н 7.75; N 2.32. С33Н43NO6. Вычислено, %: С 72.10; Н 7.88; N 2.55.
6. Получение (1R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-азаантрахинона (Iг). В стеклянную ампулу, в токе аргона, загружают 1.55 г (2.82 ммоль) эпоксисоединения (IХг) в 10 мл диоксана, 0.03 мл трифторуксусной кислоты, молекулярные сита 3А, охлаждают до 0°С и запаивают. Ампулу осторожно нагревают и выдерживают 15 ч при 130°С. После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме. Остаток (1.65 г) хроматографируют на колонке (силикагель, хлороформ, хлороформ-спирт, 10:1) получают 1.39 г (2R,6R)-5-[2(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Iг) в виде желтого маслообразного вещества, которое при растирании с петролейным эфиром кристаллизуется. Отфильтровывают 1.19 г (выход 82%) (2R,6R)-5-[2(6-винил-2-карбокси-2,6-диметилциклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Iг), т. пл. 142-144°С. [α]D + 18.2° (с 4.5, СНСl3). Спектр ЯМР 1Н (CDCl3), δ, м.д., J, Гц: 1.12 с (3Н, СН3 при С2'), 1.18 д, 1.24 д (6Н, СН(СН 3)2 при С7), 1.32 с (3Н, СН3 при С6'), 1.45-1.77 м (8Н, H1',3',4',4',5',5',12',12'), 1.86 с (3Н, СН3), 1.99-2.18 м (2Н. 1Н, CH-i-Pr и 1Н, Н3'), 2.44-2.61 м (2Н, Н13',13'), 2.18 с (3Н, СН3), 3.68 (3Н, ОСН3), 4.92 д, 4.98 д (2Н, Н8', Jгем 2.2, Jвиц 10.8 и 15.0), 5.74 м (1Н, Н7', J 10.8 и 15.0), 7.08 д (1Н, Н6, J 1.8), 7.82 д (1Н, Н8, J 1.8 Гц), 9.90 с (1Н, ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 17.66 к (СН3 при С6'), 17.45 т (С4'), 18.66 к, 19.03 к (2 х СН3-i-Рr), 22.54 к, 23.08 к (СН3 при С1 и С3), 25.88 к (СН3 при С2'), 29.30 т (С12'), 32.10 д (CH-i-Pr), 35.10 т (С13'), 37.08 т (С5'), 38.65 с (С3'), 40.05 с (С6'), 43.29 т (С2'), 46.28 д (С1'), 55.09 к (ОСН3), 111.06 т (С8'), 123.90 д (С8), 125.38 с (С10a), 126.12 с (С8a), 129.45 с (С9a), 131.11 д (С6), 133.15 с (С4a), 132.67 с (С5), 139.98 с (С3), 146.18 с (С4), 151.68 д (С7'), 155.11 с (С7), 156.48 (С1), 177.68 с (С=O), 179.86 с (С10), 183.02 с (С9). Найдено, %: С 74.14; Н 7.28; N 2.81. С32Н39NO5. Вычислено, %: С 74.25; Н 7.59; N 2.71. Общий выход соединения (Iг) в расчете на исходную левопимаровую кислоту (III) составляет 27,6%.
Пример 5. Синтез (2R,6R)-5-[2-(6-винил-2,6-диметил-2-метоксикарбонилцикло-гексил)этил]-4-гидрокси-7-изопропил-1-метил-3-этил-2-азаантрацен-9,10-диона (Iд).
Стадии 1-4 аналогичны приведенным в описании примера 4.
5. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-метокси-7-изопропил-1-метил-3-этил-1,4-эпокси-2-азаантрацен-9,10-диона (IХд).
Раствор 1.52 г (3.6 ммоль) соединения (VIIб) в 20 мл хлористого метилена загружают в стеклянную ампулу, прибавляют 0.045 мл (0.36 ммоль) ВF3·Et2О, затем 0.62 г (4.4 ммоль) оксазола (VIIIб). Ампулу продувают аргоном, охлаждают и запаивают. Реакционную смесь выдерживают при 20°С 52 ч. По прошествии указанного времени ампулу вскрывают, реакционную массу выливают в 100 мл воды, продукт экстрагируют хлористым метиленом (30×3 мл), органические слои объединяют, промывают водой, затем сушат над прокаленным сульфатом магния. Осушитель отфильтровывают, маточный раствор упаривают, остаток разделяют колоночной хроматографией на силикагеле (элюенты: хлороформ, хлороформ - этанол, в соотношении 10:1). Собирают 3 фракции. Последнюю фракцию (1.58 г) дополнительно очищают колоночной хроматографией на силикагеле (элюент: бензол-этилацетат). Получают 2 фракции. Последующей хроматографией фракции 2 выделяют 1.29 г (выход 64%) аддукта (IХд) в виде смеси стереоизомеров (эндо- и экзорасположение эпоксимостика). Маслообразное вещество. Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: 1.06 с, 1.08 с (3Н, СН3 при С2'), 1.18 с, 1.20 с (3Н, СН3 при С1), 1.20 т (3Н, СН3, J=7), 1.29 д [6Н, СН(СН 3)2 при С7, J=6.9], 1.32 с (3Н, СН3 при С6'), 1.38-1.81 м (8Н, Н1',3',4',4',5',5',12',12'), 2.02-2.12 м [2Н, 1Н, CH-i-Pr и 1Н, Н3'], 2.46-2.65 м (2Н, Н13',13'), 3.58 с (3Н, ОСН3), 3.61 с (3Н, ОСН3), 3.72 м (1Н, Н9a), 4.02 к (2Н, СН2, J=7), 4.14 м (1Н, Н4a), 4.85-4.94 м (2Н, Н8'), 5.60-5.66 м (1Н, Н7'), 7.26 д (1Н, Н6, J=1.8), 7.82 д (1Н, Н8, J=1.8). Спектр ЯМР 13С (СDСl3), δ, м.д.: 16.2 к, 16.6 к (СН3 при С2'), 17.4 к, 17.7 к (СН3 при С6'), 17.68 т (С4'), 18.01 к (СН3 при С1), 18.43 к, 18.63 к (СН(СН3)2), 22.79 к (СН3 при С1), 28.76 т (С12'), 33.37 д (СН-i-Рr), 35,01 т (С3'), 36.46 т (С5'), 38.05 с (С2'), 40.25 д (С1'), 41.43 с (С6'), 45.97 т (С13'), 46.05 д (С9a), 52.32 д (С4a), 52.42 к (ОСН3), 54.88 к (ОСН3), 70.01 с (С1), 83.54 с (С4), 110.46 т (С8'), 123.73 д (С8), 127.21 с (С8a), 134.17 с (С3), 135.16 д (С6), 142.08 с (С7), 145.35 с (С12'), 150.24 д (С7'). 160.35 с (С5), 177.78 с (С=O), 187.21 с (С9), 189.81 с (С10). Найдено, %: С 71.92; Н 7.68; N 2.41. С33Н43NO6. Вычислено, %: С 72.10; Н 7.88; N.55.
6. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-7-изопропил-1-метил-3-этил-2-азаантрахинонона (Iд). В стеклянную ампулу, в токе аргона, загружают 1.15 г (1.7 ммоль) эпокси-соединений (IХд) в 8 мл диоксана, 0.02 мл трифторуксусной кислоты, молекулярные сита 3А, охлаждают до 0°С и запаивают. Ампулу осторожно нагревают и выдерживают 15 ч при 130°С. После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме. Колоночной хроматографией (силикагель, хлороформ, хлороформ-спирт, 10:1) выделяют 0.83 г (выход 76%) (2R,6R)-5-[2(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Iд) в виде желтого маслообразного вещества, которое кристаллизуется при стоянии. Т. пл. 148-152°С (из эфира). [α]D + 25.5° (с 3.6, СНСl3). Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: 1.09 с (3Н, СН3 при С6'), 1.21 д, 1.25 д (6Н, СН(СН 3)2 при С7, J=6.8), 1.25 к (3Н, СН3, J=7.0), 1.30 с (3Н, СН3 при С2'), 1.38-1.78 м (8Н, H1',3',4',4',5',5',12',12'), 2.10 с (3Н, СН3). 2.16 м (2Н, 1Н, CH-i-Pr и 1Н, Н3'), 2.40-2.61 м (2Н, Н13',13'), 3.55 с (3Н, ОСН3), 4.18 к (2Н, СН2, J=7.0), 4.86-4.94 м (2Н, Н8'), 5.75 м (1Н, Н7', J 10.8 и 15.0), 7.21 д (1Н, Н6, J=1.8), 7.85 д (1Н, Н8, J=1.8), 10.08 с (1Н, ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 12.48 к (СН3), 17.69 т (С4'), 18.66 к, 18.75 к (2 х СН3-i-Рr), 19.05 к (СН3 при С6'), 22.84 к (СН 3 при С1), 25.81 к (СН3 при С2'), 29.30 т (С12'), 32.16 д (CH-i-Pr), 34.72 т (С13'), 36.10 т (С3'), 38.18 т (С5'), 40.02 с (С6'), 41.85 с (С2'), 48.26 т (СН2), 46.81 д (С1'), 55.19 к (ОСН3), 110.50 т (С8'), 124.61 д (С8), 126.05 с (С10a), 125.75 с (С8a), 129.33 с (С9a), 130.91 д (С6), 133.29 с (С4a), 140.06 с (С5), 147.76 с (С4), 151.04 д (С7'), 153.84 с (С7), 155.65 (С1), 160.02 с (С3), 177.86 с (С=О), 179.80 с (С10), 183.34 с (С9). Найдено, %: С 74.31; Н 7.43; N 2.41. С33Н41NO5. Вычислено, %: С 74.55; Н 7.77; N 2.63. Общий выход соединения (Iд) в расчете на исходную левопимаровую кислоту (III) составляет 26%.
Пример 6. Синтез (2R,6R)-5-[2-(6-винил-2,6-диметил-2-метоксикарбонилцикло-гексил)этил]-4-гидрокси-3,7-диизопропил-1-метил-2-азаантрацен-9,10-диона (Ie).
Стадии 1-4 выполняли по описанию в примере 4.
5. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-метокси-3,7-диизопропил-1-метил-1,4-эпокси-2-азаантрацен-9,10-диона (IХе).
Раствор 2.07 г (4.90 ммоль) соединения (VIIб) в 20 мл хлористого метилена загружают в стеклянную ампулу, прибавляют 0.062 мл (0.49 ммоль) BF3·Eе2O, затем 0.84 г (5.42 ммоль) оксазола (VIIIв). Ампулу продувают аргоном, охлаждают и запаивают. Реакционную смесь выдерживают при 20°С 48 ч. По прошествии указанного времени ампулу вскрывают, реакционную массу выливают в 100 мл воды, продукт экстрагируют хлористым метиленом (40×3 мл), органические слои объединяют, промывают водой, затем сушат над прокаленным сульфатом магния. Осушитель отфильтровывают, маточный раствор упаривают, остаток разделяют колоночной хроматографией на силикагеле (элюенты: хлороформ, хлороформ - этанол, в соотношении 10:1). Собирают 3 фракции. Последнюю фракцию (2.02 г) дополнительно очищают колоночной хроматографией на силикагеле (элюент: бензол-этилацетат). Получают 2 фракции. Последующей хроматографией фракции 2 выделяют 1.76 г (выход 62%) аддукта (IХе) в виде смеси стереоизомеров (эндо- и экзорасположение эпоксимостика). Маслообразное вещество. Спектр ЯМР 1Н (CDCд3), δ, м.д., J, Гц: 1.08 д, 1.10 д [6Н, СН(СН 3)2 при С3, J 6.8], 1.18 д, 1.23 д (6Н, и СН(СН 3)2 при С6, J 6.8), 1.25 с, 1.27 с (3Н, СН3 при С1), 1.25 с, 1.28 с (3Н, СН3 при С6'), 1.30 с, 1.33 с (3Н, СН3 при С2'), 1.38-1.79 м (8Н, H1',3',4',4',5',5',12',12'), 1.80-2.08 м (2Н, 1Н, CH-i-Pr и 1Н, Н3'), 2.22 м (1Н, СH-i-Pr), 2.42-2.60 м (2Н, Н13',13'), 3.59 с (3Н, ОСН3), 3.64 с (3Н, ОСН3), 3.85 д (1Н, Н4a, J 6.0), 4.26 д (1Н, Н4a, J 6.0), 4.82-4.90 м (2Н, Н8'), 5.60-5.68 м (1Н, Н7), 7.28 д (1Н, Н6, J 1.8), 7.81 д (1Н, Н8, J 1.8). Спектр ЯМР 13С (СDСl3), δ, м.д.: 17.71 к, 17.86 к (СН3 при С6'), 18.01 к (СН3 при С1), 17.71 т (С4'), 18.61 к, 18.63 к [СН(СН3)2], 22.79 к, 22.80 к [СН(СН3)2], 23.22 к, 24.21 к, (СН3 при С2'). 30.04 т (С12'), 32.12 д (CH-i-Pr), 32.48 д (CH-i-Pr), 34.32 т (С13'), 35.15 т (С3'), 37.98 т (С5'), 40.73 с (С6'), 43.28 с (С2'), 46.65 д (С1'), 44.51 д (С4a), 50.38 д (С9a), 52.42 к (ОСН3), 55.42 к (ОСН3), 76.13 с (С1), 90.09 с (С4), 110.88 т (С3'), 123.35 д (С8), 130.68 д (С6), 132.35 с (С10a), 133.87 с (С8a), 140.73 с (С5), 149.87 д (С7'), 151.66 с (С7), 163.17 с (С3), 177.83 с (С=О), 188.01 с (С9), 189.95 с (С10). Найдено, %: С 72.33; Н 7.93; N 2.41. С35Н47NO6. Вычислено, %: С 72.76; Н 8.20; N 2.42.
6. Получение (2R,6R)-5-[2-(6-винил-2,6-диметил-2-карбоксицикло-гексил)этил]-4-гидрокси-3,7-диизопропил-1-метил-2-азаантрацен-9,10-диона (Ie). В стеклянную ампулу, в токе аргона, загружают 1.62 г (2 ммоль) эпокси-соединений (IХе) в 10 мл диоксана, 0.01 мл трифторуксусной кислоты, молекулярные сита 3А, охлаждают до 0°С и запаивают. Ампулу осторожно нагревают и выдерживают 12 ч при 133°С. После охлаждения ампулу вскрывают, к остатку добавляют хлороформ, отфильтровывают механические примеси, раствор упаривают в вакууме. Колоночной хроматографией (силикагель, хлороформ, хлороформ-спирт, 10:1) выделяют 1.30 г (выход 85%) (2R,6R)-5-[2(6-винил-2,6-диметил-2-карбоксициклогексил)этил]-4-гидрокси-7-изопропил-1,3-диметил-2-аза-9,10-антрахинона (Ie) в виде желтого маслообразного вещества. При стоянии вещество кристаллизуется, т. пл. 157-160°С (из петролейного эфира). [α]D + 26.4° (с 4.1, СНСl3). Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: Найдено, %: С 73.61; Н 7.22; N 2.59. С31Н37NO5. Вычислено, %: С 73.93; Н 7.41; N 2.78. Спектр ЯМР 1Н (СDСl3), δ, м.д., J, Гц: 0.98 д, 1.08 д [6Н, СН(СН 3)2 при С3, J=6.8], 1.18 д, 1.20 д [6Н, СН(СН 3)2 при С7, J=7.0], 1.22 с (3Н, СН 3 при С6'), 1.28 с (3Н, СН3 при С2'), 1.42-1.80 м (8Н, H1',3',4',4',5',5',12',12'), 2.00-2.20 м (2Н, 1Н, CH-i-Pr и 1Н, Н3'), 2.35 м (1Н, СH-i-Pr), 2.40 с (3Н, СН 3 при С1), 2.48-2.58 м (2Н, Н13',13'), 3.61 с (3Н, ОСН3), 4.91 д, 4.98 д (2Н, Н8', Jгем 2.2, Jвиц 10.8 и 15.0), 5.62-5.68 м (1Н, Н7', J 10.8 и 15.0), 7.20 д (1Н, Н6, J=1.8), 7.81 д (1Н, Н8, J=1.8), 10.15 с (ОН). Спектр ЯМР 13С (СDСl3), δ, м.д.: 17.36 к (СН3 при С6'), 17.88 т (С4'), 18.58 к, 18.70 к (СН3-i-Рr), 20.18 к, 20.41 к (СН3-i-Рr), 22.92 к (СН 3 при С1), 25.18 к (СН3 при С2'), 26.48 д (CH-i-Pr), 29.30 т (С12'), 32.19 д (CH-i-Pr), 35.49 т (С13'), 36.34 т (С5'), 37.87 т (С3'), 39.80 с (С6'), 42.00 с (С2'), 46.66 д (С1'), 55.11 к (ОСН3), 110.92 т (С8'), 124.08 д (С8), 126.12 с (С8a), 126.47 с (С10a), 129.68 с (С9a), 131.19 д (С6), 132.42 с (С4a), 140.22 с (С5), 147.56 с (С4), 151.22 д (С7'), 151.78 с (С7), 156.10 (С1), 158.20 с (С3), 177.92 с (С=O), 179.86 с (С10), 183.02 с (С9). Найдено, %: С 74.52; Н 7.88; N 2.36. С34Н43NO5. Вычислено, %: С 74.83; Н 7.94; N 2.57. Общий выход соединения (Ie) в расчете на исходную левопимаровую кислоту (III) составляет 28%.
Пример 7. Влияние соединений на формалиновый отек изучено на мышах в дозе 1/280 и (или) 1/100 от LD50. Воспаление вызывалось введением под плантарный апоневроз правой задней лапки 0,05 мл 3% раствора формалина. Исследуемые соединения вводили внутрь за 2 ч до введения формалина, непосредственно перед введением и через 3 и 6 ч после флогогенного воздействия. Результаты исследований приведены в таблице 2.
Пример 8. Влияние соединений на карагениновый отек изучено на белых мышах. Воспаление вызывалось введением под плантарный апоневроз правой задней лапки 0,05 мл 1% раствора карагенина. Соединения вводили внутрь за 2 часа до введения карагенина, через 3 и 6 ч после введения. Результаты исследований приведены в таблице 3.
Таким образом, заявляемое изобретение обладает следующими преимуществами, а именно:
- получение новых малотоксичных оптически активных 4-гидрокси-2-азаантрахинонов, обладающих противовоспалительной активностью;
- предлагаемый способ получения оптически активных производных 4-гидрокси-2-аза-9,10-антрахинона основан на использовании доступного сырья - сосновой живицы, аминокислот и 1,4-бензохинона;
- применение в заявляемом способе получения новых веществ на ключевой стадии реакции диенового синтеза оптически активных полифункциональных 1,4-нафтохинонов с 4-алкил-2-метил-5-метоксиоксазолами, катализируемой кислотой Льюиса.

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2697090C1 |
| КОНДЕНСИРОВАННЫЕ БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2685234C1 |
| МЕТАЛЛОЦЕНОВОЕ СОЕДИНЕНИЕ, ЛИГАНД, СПОСОБЫ ПОЛУЧЕНИЯ ЛИГАНДА, КАТАЛИЗАТОР, СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРА АЛЬФА-ОЛЕФИНОВ, ГОМОПОЛИМЕР ПРОПИЛЕНА, СОПОЛИМЕР ПРОПИЛЕНА | 2000 |
|
RU2243229C2 |
| МЕТАЛЛООРГАНИЧЕСКОЕ СОЕДИНЕНИЕ, ПРИГОДНОЕ В КАЧЕСТВЕ СОКАТАЛИЗАТОРА ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 2001 |
|
RU2248980C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2684641C1 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679609C1 |
| (3aR,4aS,8aR,9aR,E)-3-АРИЛИДЕН-8a-МЕТИЛ-5-МЕТИЛЕН-ДЕКАГИДРОНАФТО[2,3-b]ФУРАН-2(3Н)-ОНЫ, ОБЛАДАЮЩИЕ ПРОТИВОЯЗВЕННОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2413724C1 |
Настоящее изобретение относится к органической химии, конкретно к новым оптически активным 3-алкил-4-гидрокси-2-аза-9,10-антрахинонам формулы (Ia-e): R=H, R1=Me (Ia); R=H, R1=Et (Iб); R=H, R1=i-Pr (Iв); R=R1=Me (Iг); R=Me, R1=Et (Iд); R=Me, R1=i-Pr (Ie). Также изобретение относится к способу получения соединения формулы (Ia-e). Технический результат: получены новые малотоксичные оптически активные производные 3-алкил-4-гидрокси-2-аза-9,10-антрахинона, обладающие противовоспалительной активностью. 2 н.п. ф-лы, 3 табл.
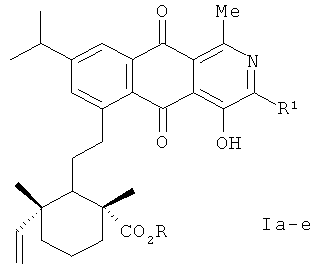
1. Оптически активные производные 4-гидрокси-2-аза-9,10-антрахинона формулы (Ia-e):
R=H, R1=Me (Ia); R=H, R1-Et (Iб); R=H, R1=i-Pr (Iв); R=R1=Me (Iг); R=Me, R1=Et (Iд); R=Me, R1=i-Pr (Ie)
обладающие противовоспалительной активностью.
2. Способ получения оптически активных производных 4-гидрокси-2-аза-9,10-антрахинона формулы (Ia-e):
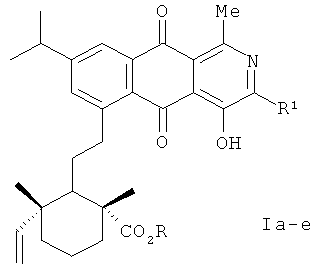
R=H, R1=Me (Ia); R=H, R1=Et (Iб); R=H, R1=i-Pr (Iв); R=R1=Me (Iг); R=Me, R1=Et (Iд); R=Me, R1=i-Pr (Ie)
из содержащейся в живице хвойных растений левопимаровой кислоты, которая под действием 1,4-бензохинона дает 7,10а-диметил-13-изопропил-7-карбокси-4,4а,5,6,6а,7,8,9,10,10а,10b,11,12,12а-тетрадекагидро-4b,12-этенохризен-1Н-1,4-дион, указанное соединение или его эфир обрабатывают щелочью, изомеризуя в соответствующие гидрохиноны, окисляемые далее в 7,10а-диметил-7-карбокси- или 7,10а-диметил-7-метоксикарбонил-13-изопропил-4b,5,6,6а,7,8,9,10,10а,10b,11,12-додекагидро-4b,12-этенохризен-1,4-дионы, последующее термическое расщепление которых дает 5-[2-(6-винил-2,6-диметил-2-карбоксициклогексил)этил]- или 5-[2-(6-винил-2,6-диметил-2-метоксикарбонилциклогексил)этил]-7-изопропил-1,4-нафтохинона, которые во взаимодействии с 4-алкил-2-метил-5-метоксиоксазолом в условиях катализа эфиратом трехфтористого бора и последующей кислотной обработки полученных аддуктов приводят к целевым продуктам.
| Koyama J | |||
| et al | |||
| Structure-activity relations of azafluorenone and azaanthraquinone as antimicrobial compounds | |||
| Bioorganic and Medicinal Chemistry Letters, v | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| Artamonov A.A | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Chemistry of Heterocyclic Compounds, v.16(4), 1980, p.397-401 | |||
| Clark A.M | |||
| et al | |||
| In vitro | |||

