Настоящее изобретение относится к новому классу металлоценовых соединений, к содержащим их катализаторам и к проводимому в присутствии указанных катализаторов способу получения полимеров альфа-олефинов, в особенности пропиленовых полимеров, имеющих низкую степень кристалличности. Настоящее изобретение также относится к лигандам для этих металлоценов и удобному способу их получения.
Продукты гомополимеризации пропилена могут иметь различные степени кристалличности. Тип и величина кристалличности в значительной степени зависят от микроструктуры полипропилена. Полипропилен, имеющий преимущественно изотактическую или синдиотактическую структуру, является частично кристаллическим, тогда как полипропилен, имеющий преимущественно атактическую структуру, является аморфным. Также известны пропиленовые полимеры, которые имеют пониженную степень кристалличности и обладают эластомерными свойствами. Например, в патенте США №4335225 раскрывается разделяемый на фракции эластичный полипропилен, имеющий содержание изотактической фракции 55% или менее, который содержит растворимую в диэтиловом эфире фракцию с содержанием изотактического кристаллического полимера приблизительно 0,5-5 мас.%. Этот полипропилен получен с помощью катализатора на основе тетраалкилциркония, нанесенного на оксид металла. Однако эластомерные полипропилены этого типа вследствие того, что каталитические системы, которые используются для их получения, имеют различные каталитические участки, характеризуются широким молекулярно-массовым распределением, что оказывает отрицательное влияние на их свойства.
С недавнего времени в реакции полимеризации олефинов стали использоваться металлоценовые катализаторы. При работе в присутствии этих катализаторов получают полимеры, отличающиеся узким молекулярно-массовым распределением и имеющие желаемые структурные характеристики. При полимеризации пропилена в присутствии металлоценовых катализаторов в зависимости от используемого металлоцена могут быть получены аморфные или высококристаллические полипропилены.
Также известны некоторые металлоценовые катализаторы, которые могут давать частично кристаллический эластомерный полипропилен. В международной заявке WO 95/25757, например, описаны не соединенные мостиковой связью металлоценовые катализаторы, которые дают изотактические/атактические стереоблокполипропилены, имеющие эластомерные термопластичные свойства. Несмотря на однородность молекулярно-массового распределения, распределение тактичности молекулярной структуры этих полимеров не является однородным. Более того, активность является низкой. В публикации U. Dietrich и др. в J. Am. Chem. Soc., 1999, 121, 4348-4355 описаны металлоценовые катализаторы, которые могут давать термопластичные эластичные полипропилены.
С совсем недавнего времени гетероциклические металлоценовые соединения стали использовать при полимеризации альфа-олефинов. В международной заявке WO 98/22486 раскрывается класс металлоценов, содержащих циклопентадиенильный радикал, непосредственно координированный с центральным атомом металла, который конденсирован с одним или более кольцами, содержащими, по меньшей мере, один гетероатом. Такие металлоцены в сочетании с подходящим сокатализатором используют при полимеризации олефинов, таких как пропилен. Рабочие примеры относятся к получению в высокой степени стереорегулярного полипропилена.
Было бы желательно создание нового класса металлоценов, которые при использовании в катализаторах для полимеризации олефинов, в частности пропилена, способны давать полимеры, характеризующиеся высокими молекулярными массами, узким молекулярно-массовым распределением и пониженной степенью кристалличности. Наиболее желательно получение металлоценовых катализаторов, которые могут давать такие полимеры, имеющих высокую активность, так что количество катализатора, остающегося в образованном полимере, будет минимальным.
Неожиданно найден новый класс металлоценовых соединений, которые обеспечивают названные выше и другие результаты.
В соответствии с первым аспектом настоящее изобретение предлагает металлоценовое соединение общей формулы (I):

где L представляет собой двухвалентную группу, соединяющую мостиковой связью остатки G и Z, выбранную из CR1R2, SiR1R2 и (CR1R2)2, R1 и R2, которые могут быть одними и теми же или отличаться друг от друга, выбраны из атома водорода, C1-C20-алкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, и R1 и R2 также могут образовывать кольцо, содержащее от 3 до 8 атомов, которое может иметь заместители;
Z представляет собой остаток формулы (II):
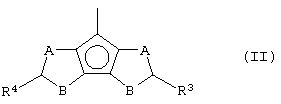
где R3 и R4, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из атома водорода, C1-C20-алкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, причем предпочтительно, чтобы, по меньшей мере, один из заместителей R3 и R4 отличался от атома водорода;
А и В выбраны из атома серы (S), кислорода (О) или CR5, где заместитель R5 выбран из атома водорода, C1-C20-алкильного, C3-C20-циклоалкильного, C2-C20-алкенильного, C6-C20-арильного, C7-C20-алкиларильного, C7-C20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, при условии, что если А представляет собой атомы S или О, то В представляет собой CR5, или, если В представляет собой атомы S или О, то А представляет собой CR5, то есть или А или В отличается от CR5, и где кольца, содержащие А и В, имеют двойную связь в допустимом положении, являясь двумя ароматическими кольцами;
G представляет собой фрагмент формулы (III):
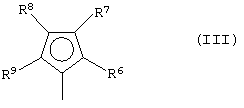
где R6, R7, R8 и R9, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из группы, включающей атом водорода, C1-C20-алкильный, C3-C20-циклоалкильный, C2-C20-алкенильный, C6-C20-арильный, C7-C20-алкиларильный, C7-C20-арилалкильный радикал, необязательно содержащий гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, при этом заместители R6 и R7 и/или R8 и R9 могут образовывать кольцо, содержащее от 3 до 8 атомов, которое может иметь заместители; при условии, что заместитель R7 отличается от заместителя R8 и, когда R7 представляет собой трет-бутильный радикал, заместитель R8 не является атомом водорода;
М представляет собой атом переходного металла, выбранный из атомов металлов, принадлежащих 3, 4, 5, 6 группам или группам лантанидов и актинидов Периодической таблицы элементов (новая версия IUPAC),
X, одинаковые или разные, выбраны из атома водорода, атома галогена, групп R10, OR10, ОSО2СF3, OCOR10, SR10, NR
р представляет собой целое число от 0 до 3, предпочтительно от 1 до 3, равное формальной степени окисления металла М минус 2;
при этом исключены:
изопропилиден(3-триметилсилилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
диметилсиландиил(3-триметилсилилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
изопропилиден(3-этилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
диметилсиландиил(3-этилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
изопропилиден(3-н-бутилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
диметилсиландиил(3-н-бутилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
изопропилиден(3-метилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
диметилсиландиил(3-метилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид;
изопропилиден(3-изопропилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид и
диметилсиландиил(3-изопропилциклопентадиенил)(7-циклопентадитиофен)цирконийдихлорид.
Переходный металл М предпочтительно выбран из титана, циркония и гафния, предпочтительно имеющих формальную степень окисления +4. Заместители Х предпочтительно представляют собой атомы хлора, бензильные или метильные группы. Предпочтительно мостиковая группа L представляет собой группы СМе2 или SiMe2. Предпочтительно один из А или В представляет собой атом серы, а другой остаток представляет собой группу СН, более предпочтительно А представляет собой атом серы, а В представляет собой группу СН. Предпочтительно заместители R3 и R4 являются одинаковыми и выбраны из С1-С20-алкильных групп, которые могут содержать атом кремния. Более предпочтительно заместители R3 и R4 представляют собой метильный, этильный, фенильный или триметилсилильный радикал.
Не ограничивающими примерами металлоценовых соединений в соответствии с настоящим изобретением являются:
метилен(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(3-этилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(3-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(2,4-диэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(2,4-диизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(2,3,5-триметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(2,3,5-триэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(2,3,5-триизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(3-циклогексилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(инденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(инденил)-7-(2,5-дитриметилсилилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(3-изопропилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(3-трет-бутилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(2,3-диметилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(3-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
метилен(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-этилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2,4-диэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2-метил-4-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2,4-диизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2,3,5-триметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2,3,5-триэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(2,3,5-триизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-циклогексилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен-1-(инденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен-1-(2-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен-1-(2,3-диметилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-метилциклопентадиенил)-4-(2,6-диметилциклопентадиенил)-[2,1-b;4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-изопропилциклопентадиенил)-7-(2,5-дитриметил-силилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
метилен(3-метилциклопентадиенил)-7-(2,5-дитриметил-силилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)гафнийдихлорид и диметил;
изопропилиден(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-этилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,4-диметилциклопентадиенил)-7-(2,5-дитриметил-силилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,4-диэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,4-диизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2-метил-4-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2-метил-4-фенилциклопентадиенил)-7-(2,5-дитри-метилсилилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2-метил-4-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,3,5-триметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,3,5-триэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(2,3,5-триизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-циклогексилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-дитриметил-силилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-изопропилциклопентадиенил)-4-(2,6-диметилциклопентадиенил)-[2,1-b:3,4-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден-1-(инденил)-1-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден-1-(3-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден-1-(2,3-диметилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден-1-(3-изопропилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден-1-(3-трет-бутилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-метилциклопентадиенил)-4-(2,6-диметилциклопентадиенил)-[2,1-b:3,4-b’]дитиофен)гафнийдихлорид и диметил;
диметилсиландиил(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(3-этилциклопентадиенил)-7-(2,5-диметилцикло-пентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(3-фенилциклопентадиенил)-7-(2,5-диметилцикло-пентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(2,4-диэтилциклопентадиенил)-7-(2,5-диметил-циклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(2,4-диизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(2,3,5-триметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(2,3,5-триэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(2,3,5-триизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(3-циклогексилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил(3-триметилсилилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(инденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(2,3-диметилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-этилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-изопропилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-изопропилинденил)-4-(2,6-диметилциклопентадиенил)-[2,1-b:3,4-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-изопропилинденил)-7-(2,5-дитриметил-силилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)гафнийдихлорид и диметил;
диметилсиландиил-1-(3-трет-бутилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
диметилсиландиил-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и диметил;
изопропилиден(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’)диоксазол)цирконийдихлорид и диметил;
изопропилиден(3-этилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(3-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(2,4-диэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(2,4-диизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(2,3,5-триметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(2,3,5-триэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(2,3,5-триизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(3-циклогексилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(инденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(3-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(3-этилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(3-изопропилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(3-трет-бутилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)гафнийдихлорид и диметил;
диметилсиландиил(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(3-этилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(3-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(2,4-диэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(2,4-диизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(2,3,5-триметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(2,3,5-триэтилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(2,3,5-триизопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(3-циклогексилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил-1-(инденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил-1-(2,3-диметилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил-1-(3-изопропилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
диметилсиландиил-1-(тетрагидроинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]диоксазол)цирконийдихлорид и диметил;
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b;4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)гафнийдихлорид и диметил.
Особенно интересный класс металлоценов с мостиковой связью формулы (I) в соответствии с настоящим изобретением составляют соединения, где G представляет собой остаток формулы (IIIa):
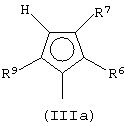
где R6 и R9, одинаковые или отличающиеся друг от друга, выбраны из атома водорода, С1-С20-алкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, и R6 предпочтительно представляет собой атом водорода, а R9 отличается от атома водорода;
R7 выбран из С6-С20-арильной, С7-С20-алкиларильной группы или группы QR11R12R13, где Q выбран из атомов С, Si, Ge;
R11, R12 и R13, которые могут быть одинаковыми или могут отличаться друг от друга, представляют собой атом водорода, C1-C20-алкильный, С3-С20-циклоалкильный, С2-С20-алкенильный, С6-С20-арильный, С7-С20-алкиларильный, С7-С20-арилалкильный радикал, необязательно содержащий гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, при условии, что, когда Q представляет собой атом углерода, по меньшей мере, один из заместителей R11, R12 и R13 представляет собой атом водорода.
Особенно предпочтительными металлоценами названного выше класса являются металлоцены, где заместитель R7 выбран из фенильной группы, группы CHR11R12 и группы SiR11R12R13, причем R11, R12 и R13 представляют собой атом водорода или С1-С20-алкильные группы.
Наиболее предпочтительными являются металлоцены, где QR11R12R13 представляет собой изопропильную или триметилсилильную группу.
Не ограничивающими примерами этого класса металлоценов являются:
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
диметилсиландиил(3-триметилсилилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(2-метил-4-изопропилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(3-изопропилциклопентадиенил)-7-(2,5-дитриметилсилилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(3-изопропилциклопентадиенил)-4-(2,6-диметилциклопентадиенил)-[2,1-b:3,4-b’]дитиофен)цирконийдихлорид;
изопропилиден(2-метил-4-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(3-фенилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(2-метил-4-фенилциклопентадиенил)-7-(2,5-дитриметилсилилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
изопропилиден(2,4-диметилциклопентадиенил)-7-(2,5-дитриметилсилилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
диметилсиландиил(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид и изопропилиден(3-метилциклопентадиенил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)гафнийдихлорид.
Другой предпочтительный класс металлоценов с мостиковой связью формулы (I) составляют металлоцены, где G представляет собой остаток формулы (IV):
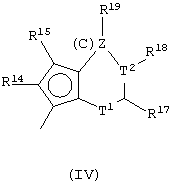
где Т1 представляет собой атом серы или группу CR16;
Т2 представляет собой атом углерода или атом азота;
z равно 1 или 0;
кольцо, содержащее Т1 и Т2, имеет двойные связи в допустимом положении;
при условии, что, если z равно 1, Т1 представляет собой группу CR16 и Т2 представляет собой атом углерода, а образованное кольцо является бензольным кольцом; и, если z равно 0, Т2 связан непосредственно с циклопентадиенильным кольцом, образованное 5-членное кольцо имеет двойные связи в любом разрешенном положении, имея ароматический характер, и Т1 и Т2 не являются одновременно атомом серы и атомом азота;
R14, R15, R16, R17, R18 и R19, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из атома водорода, С1-С20-алкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, и любые два соседних заместителя R14, R15, R16, R17, R18 и R19 могут образовывать кольцо, содержащее от 4 до 8 атомов, которое может иметь заместители.
Предпочтительный подкласс соединений, принадлежащих к описанному выше классу, составляют соединения, где G представляет собой остаток формулы (IVa):
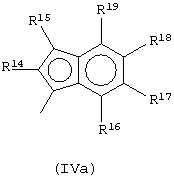
где R14, R15, R16, R17, R18 и R19, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из атома водорода, С1-С20-алкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, и любые два соседних заместителя R14, R15, R16, R17, R18 и R19 могут образовывать кольцо, содержащее от 4 до 8 атомов, которое может иметь заместители, причем бензольное кольцо может быть пергидрированным.
Не ограничивающими примерами металлоценов, принадлежащих этому классу, являются:
диметилсиландиил-1-(инденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид;
диметилсиландиил-1-(2-метилинденил)-7-(2,5-диметилциклопентадиенил)-[1,2-b:4,3-b’]дитиофен)цирконийдихлорид.
Предпочтительная структура соединений формулы (IVa) имеет формулу (IVb):
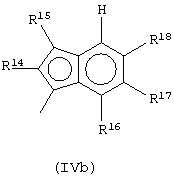
где R15, R16, R17 и R18 выбраны из атома водорода, C1-C20-aлкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, а любые два соседних заместителя R14, R16, R17 и R18 могут образовывать кольцо, содержащее от 4 до 8 атомов углерода, которое может иметь заместители; R14 выбран из группы, включающей C1-C20-алкильную или С6-С20-арильную группу, например метильную, этильную или фенильную группу.
Предпочтительно, когда G представляет собой остаток формулы (IVb), L представляет собой группу формулы SiR1R2, где R1 и R2 имеют значения, указанные выше, более предпочтительно L представляет собой SiMe2.
Еще одна предпочтительная структура соединений формулы (IVa) имеет формулу (IVc):
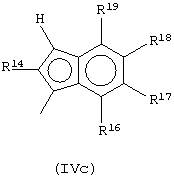
где R14, R16, R17 и R18 выбраны из атома водорода, C1-C20-aлкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, и необязательно любые два соседних заместителя R14, R16, R17, R18 и R19 могут образовывать кольцо, содержащее от 4 до 8 атомов углерода, которое может иметь заместители;
R19 выбран из группы, включающей C1-C20-алкильную или С6-С20-арильную группы, такие как метильная, этильная или фенильная группа, или образует с заместителем R18 бензольное кольцо, которое может иметь заместители.
Предпочтительно заместитель R4 выбран из группы, включающей C1-C20-алкильную или С6-С20-арильную группы, такие как метильная, этильная или фенильная группы; предпочтительно заместитель R16 выбран из группы, включающей C1-C20-алкильную или С6-С20-арильную группы, такие как метильная, этильная или фенильная группы.
Предпочтительно, когда G представляет собой остаток формулы (IVc), L представляет собой группу SiR1R2, где заместители R1 и R2 имеют описанные выше значения, более предпочтительно L представляет собой SiMe2.
Другой предпочтительный подкласс соединений, где G представляет собой остаток формулы (IV), составляют соединения, где G представляет собой остаток формулы (IVd):
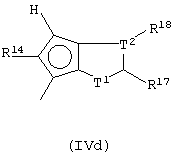
где Т1 представляет собой атом серы или группу CR16;
Т2 представляет собой атом углерода или атом азота;
5-членное кольцо, образованное Т1 и Т2, имеет двойные связи в любом допустимом положении, имея ароматический характер;
при условии, что, если Т1 представляет собой атом серы, Т2 не является атомом азота;
R14, R17 и R18, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из атома водорода, С1-С20-алкильного, С3-С20-циклоалкильного, С2-С20-алкенильного, С6-С20-арильного, С7-С20-алкиларильного, С7-С20-арилалкильного радикала, необязательно содержащего гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, и заместители R17 и R18 могут образовывать кольцо, содержащее от 4 до 8 атомов, которое может иметь заместители.
Особенно предпочтительными являются соединения, в которых Т2 представляет собой атом углерода; Т1 представляет собой атом серы, а заместители R14, R17 и R18, одинаковые или отличающиеся друг от друга, представляют собой С1-С20-алкил, С6-С20-арил; предпочтительно R14, R17 и R18 представляют собой метильную или фенильную группы.
Предпочтительно, когда G представляет собой остаток формулы (IVd), L представляет собой группу SiR1R2, где заместители R1 и R2 имеют описанные выше значения, более предпочтительно L представляет собой SiMe2.
В соответствии с другим аспектом настоящего изобретения предлагается класс лигандов формулы (V):

где L принимает значения, описанные выше;
Z’ представляет собой остаток формулы (VI):
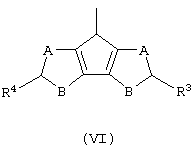
и его изомеры по двойной связи;
где А, В, R3 и R4 принимают значения, описанные выше, а двойные связи находятся в любом из разрешенных положений;
G’ представляет собой остаток формулы (VII):
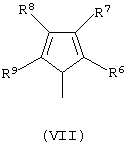
и его изомеры по двойной связи;
где R6, R7, R8 и R9 принимают описанные выше значения. Лиганд формулы (V) может быть получен в соответствии с методикой, известной в данной области техники, в частности, когда оба заместителя R4 и R5 представляют собой атом водорода, лиганд формулы (V) может быть получен так, как описано в публикации WO 98/22486.
В соответствии с еще одним аспектом настоящее изобретение предлагает способ получения лиганда формулы (V), который включает следующие стадии:
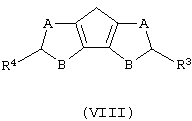
а) контактирование соединения формулы (VIII) с основанием, выбранным из группы, включающей металлический натрий и калий, гидроксид натрия и калия и литийорганическое соединение, где мольное соотношение между соединением формулы (VIII) и указанным основанием составляет, по меньшей мере, 1:1;
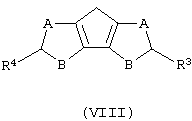
где А, В, R3 и R4 описаны выше;
б) контактирование полученных анионных соединений формулы (VIII) с соединением формулы (IX):
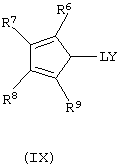
где L, R6, R7, R8 и R9 имеют значения, определенные выше, a Y представляет собой галогеновый радикал, выбранный из группы, состоящей из хлора, брома и йода, предпочтительно хлора и брома.
Когда L представляет собой CR1R2, лиганд формулы (V) может быть получен с помощью альтернативного способа, включающего следующие стадии:
а) контактирование соединения формулы (VIII) с основанием, выбранным из группы, включающей металлический натрий и калий, гидроксид натрия и калия и литийорганическое соединение, где мольное соотношение между соединением формулы (VIII) и указанным основанием составляет, по меньшей мере, 1:1;
б) контактирование полученного анионного остатка формулы (VI) с соединением формулы (X):
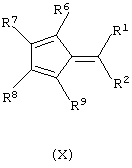
где R1, R2, R6, R7, R8 и R9 имеют значения, определенные выше; а затем обработку полученного продукта протонирующим агентом.
Основание, используемое на стадии а) обоих способов, предпочтительно представляет собой метиллитий или н-бутиллитий.
Предпочтительно протонирующий агент, используемый в описанном выше способе, представляет собой четвертичную аммонийную соль и наиболее предпочтительно протонирующий агент представляет собой хлорид аммония.
Не ограничивающие примеры соединения формулы (X) выбирают из 6,6-диметилфульвена и 3-изопропил-6,6-диметилфульвена.
Не ограничивающими примерами соединений формулы (IX) являются (3-метилциклопентадиенил)диметилхлорсилан, (3-изопропилциклопентадиенил)диметилхлорсилан, 1-(3-метилциклопентадиенил)-1,1-диметил-2,2-диметил-2-хлорэтан и 1-(3-изопропилциклопентадиенил)-1,1-диметил-2,2-диметил-2-хлорэтан.
Соединения формулы (VIII), когда В представляет собой CR5, могут быть получены способом, который включает следующие стадии:
i) обработку соединения формулы (XI):
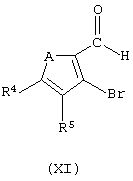
где А представляет собой атом серы или кислорода, соединением формулы (XII):
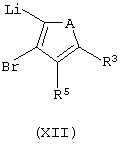
где А представляет собой атом серы или кислорода,
ii) контактирование полученного таким способом продукта с восстанавливающим агентом при мольном отношении между указанным восстанавливающим агентом и продуктом, полученным на стадии i), по меньшей мере, 1;
iii) контактирование продукта, полученного на стадии ii), с соединением, выбранным из литийорганического соединения, натрия и калия, при мольном отношении между указанным соединением и продутом, полученным на стадии ii), равном или большем чем 2;
iv) обработку полученного таким способом продукта агентом, выбранным из группы, включающей хлорид меди, йодид меди и Mg/Pd, для получения соединения общей формулы (XIII):
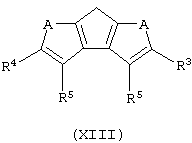
Когда В представляет собой атом серы или кислорода, а А представляет собой группу CR5, соединение формулы (VIII) может быть получено в соответствии со способом, который включает следующие стадии:
i) контактирование соединения формулы (XIV):
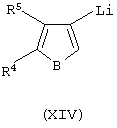
где В представляет собой атом серы или кислорода, с соединением формулы (XV):
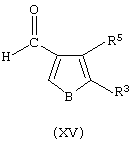
где В представляет собой атом серы или кислорода, и последующую обработку нейтрализующим агентом;
ii) обработку полученного таким способом продукта восстанавливающим агентом при мольном отношении между указанным восстанавливающим агентом и соединением, полученным на стадии i), составляющем, по меньшей мере, 1;
iii) контактирование полученного таким способом продукта со смесью литийорганического соединения и тетраметилэтилендиамина (ТМЭДА) при мольном отношении между указанной смесью и продуктом, полученным на стадии ii), составляющем, по меньшей мере, 2;
iv) контактирование полученного таким образом продукта с агентом, выбранным из группы, включающей хлорид меди, йодид меди и Mg/Pd, с получением соединения формулы (XVI):
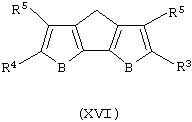
Альтернативный способ получения соединения формулы (VIII), когда А представляет собой S или О, включает следующие стадии:
i) контактирование эквимолярной смеси соединений формул (XVII) и (XVIII):
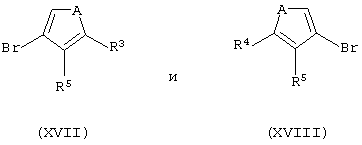
где А представляет собой атом серы или кислорода,
с кислотой Льюиса или смесью кислоты Льюиса и протонсодержащей кислоты;
ii) обработку полученного таким образом продукта СН2О при мольном соотношении между указанной смесью и СН2О в пределах интервала от 10:1 до 1:10;
iii) контактирование полученного таким образом продукта с соединением, выбранным из литийорганического соединения, натрия и калия;
iv) контактирование полученного таким образом продукта с агентом, выбранным из группы, включающей хлорид меди, йодид меди и Mg/Pd, с получением соединения общей формулы (XIII).
Кислоту Льюиса, используемую в описанном выше способе, предпочтительно выбирают из дихлорида цинка, дихлорида кадмия, дихлорида ртути, тетрахлорида олова, трифторборана, тетрахлорида циркония и тетрахлорида титана. Наиболее предпочтительно, когда кислота Льюиса представляет собой дихлорид цинка.
Агент, используемый в описанных выше способах изобретения, предпочтительно представляет собой хлорид меди.
Предпочтительно восстанавливающий агент представляет собой смесь АlСl3/LiАlН4.
Органическое соединение лития, используемое выше, предпочтительно представляет собой бутиллитий.
Другой альтернативный способ получения соединения формулы (VIII), когда А представляет собой S или О, включает следующие стадии:
i) контактирование соединения формулы (XIX):
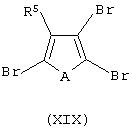
с основанием, выбранным из литийорганического соединения, натрия или калия; обработку полученного продукта эфиром муравьиной кислоты, где мольное соотношение между указанным эфиром и соединением формулы (XIX) составляет, по меньшей мере, 1:2, и затем обработку полученного продукта восстанавливающим агентом с получением соединения формулы (XX):
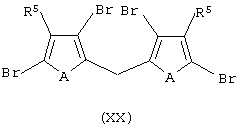
ii) контактирование соединения формулы (XX) с основанием, выбранным из литийорганического соединения, натрия или калия, затем обработку диметаллированного соединения алкилирующим агентом с получением соединения формулы (XXI):
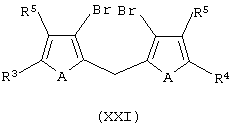
или, в соответствии с альтернативной методикой, обработку диметаллированного соединения эфиром борной кислоты и протонирующим агентом с получением соединения формулы (XXII):
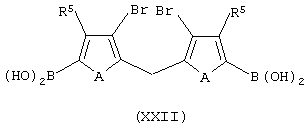
и последующее контактирование со смесью алкилирующего агента в присутствии комплексного соединения переходного металла с получением соединения формулы (XXI);
iii) контактирование алкилированного соединения, полученного на стадии b), с сочетающим агентом с получением соединения формулы (XIII).
Алкилирующий агент предпочтительно выбирают из диметилсульфата (Me2SO4), триметилхлорсилана (Me3SiCl) и смеси соединений формул R3Y’ и R4Y’, где R3 и R4 определены выше, a Y’ выбран из хлора, брома и йода, предпочтительно Y’ представляет собой хлор. Предпочтительно комплексное соединение переходного металла представляет собой PdCl2(dppf).
В описанных выше способах восстановитель предпочтительно представляет собой смесь АlСl3/LiАlН4 или смесь триэтилсилана (Et3SiH) и СF3СООН. Предпочтительным основанием является бутиллитий.
Предпочтительным эфиром органической кислоты является эфир муравьиной кислоты. Предпочтительный сочетающий агент выбирают из группы, включающей хлорид меди, йодид меди и Mg/Pd.
Все реакции проводят в апротонных растворителях. Не ограничивающими примерами апротонных растворителей, подходящих для описанных выше процессов, являются тетрагидрофуран, диметоксиэтан, диэтиловый эфир, толуол, дихлорметан, пентан, гексан и бензол.
В течение всего процесса температуру обычно поддерживают между -100 и 80°С, предпочтительно между -20 и 40° С.
Соединения формулы (V) могут быть использованы в качестве промежуточных соединений для получения металлоценов формулы (I).
Таким образом, еще одним объектом настоящего изобретения является способ получения металлоценового соединения формулы (I), которое может быть получено путем контактирования лиганда общей формулы (V) с соединением, способным образовывать соответствующее дианионное соединение, и затем с соединением общей формулы МХр+2, где М, Х и р определены выше.
Соединение, способное образовывать указанное соответствующее дианионное соединение, выбрано из группы, включающей гидроксиды щелочных и щелочноземельных металлов, металлический натрий и калий и металлорганические литиевые соли.
Предпочтительно соединение, способное образовывать указанное соответствующее дианионное соединение, представляет собой бутиллитий.
Не ограничивающими примерами соединений формулы МХр+2 являются тетрахлориды титана, циркония и гафния.
Более конкретно, лиганд формулы (V) растворяют в полярном апротонном растворителе и к полученному раствору добавляют раствор литийорганического соединения в неполярном растворителе. Полученное таким образом анионное соединение необязательно выделяют, растворяют или суспендируют в полярном апротонном растворителе и затем добавляют к суспензии соединения МХр+2 в полярном апротонном растворителе. По окончании реакции полученный твердый продукт отделяют от реакционной смеси с помощью методик, обычно используемых в данной области техники, таких как фильтрование или перекристаллизация. Не ограничивающими примерами полярных апротонных растворителей, подходящих для описанных выше способов, являются тетрагидрофуран, диметоксиэтан, диэтиловый эфир и дихлорметан. Не ограничивающими примерами неполярных растворителей, подходящих для использования в описанных выше способах, являются пентан, гексан, бензол и толуол.
В течение всего процесса температуру обычно поддерживают между -100 и 80° С, предпочтительно между -20 и 40° С.
В случае, когда, по меньшей мере, один заместитель Х в металлоценовом соединении формулы (I) отличается от атома галогена, альтернативный способ его получения состоит в приготовлении дигалогенированного производного, то есть комплекса, где оба заместителя Х являются галогеном, и в последующем замещении атомов галогена соответствующими группами Х с помощью обычно используемых методов. Например, если желаемыми заместителями Х являются алкильные группы, металлоцен может быть получен реакцией с алкилмагнийгалогенидами (реактивами Гриньяра) или алкиллитиевыми соединениями. Общие методы замещения Х заместителями, отличными от атома галогена, такими как сера, фосфор, кислород и др., описаны в публикации Chem. Rev., 1994, 94, 1661-1717, а также в цитируемых в ней ссылках.
В соответствии с еще одним аспектом настоящего изобретения предлагается катализатор для полимеризации альфа-олефинов, получаемый путем контактирования:
(А) металлоценового соединения формулы (I)

где L, Z, М, Х и р имеют значения, определенные выше, и G представляет собой остаток формулы (III):
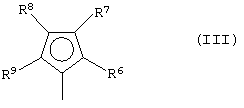
где R6, R7, R8 и R9, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из группы, включающей атом водорода, С1-С20-алкильный, С3-С20-циклоалкильный, С2-С20-алкенильный, С6-С20-арильный, С7-С20-алкиларильный, С7-С20-арилалкильный радикал, необязательно содержащий гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, заместители R6 и R7 и/или R8 и R9 могут образовывать кольцо, содержащее от 3 до 8 атомов, которое может иметь заместители; при условии, что R7 отличается от R8 и, когда R7 представляет собой трет-бутильный радикал, R8 не является атомом водорода; и
(В) алюмоксана и/или соединения, способного образовывать алкилметаллоценовый катион.
Предпочтительно в металлоценовом соединении формулы (I) G представляет собой остаток формулы (IIIa) или (IV), более предпочтительно G представляет собой остаток, выбранный из соединений формул (IIIa), (IVb), (IVc) или (IVd).
Алюмоксан, используемый в качестве компонента (В), может быть получен при взаимодействии воды с алюминийорганическим соединением формулы HjAlR
Мольное соотношение между алюминием и металлом металлоценового соединения составляет от около 10:1 до около 20000:1 и более предпочтительно от около 100:1 до около 5000:1.
Алюмоксаны, используемые в катализаторе в соответствии с изобретением, представляют собой, как считают, линейные, разветвленные или циклические соединения, содержащие, по меньшей мере, одну группу типа
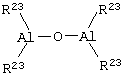
где заместители R23, одинаковые или разные, определены выше.
В частности, алюмоксаны формулы
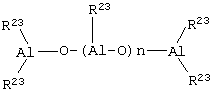
могут быть использованы в случае линейных соединений, где u равно 0 или целому числу от 1 до 40, а заместители R23 определены выше,
или алюмоксаны формулы:
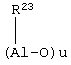
могут быть использованы в случае циклических соединений, где u представляет собой целое число от 2 до 40, а заместители R23 определены выше.
Примерами алюмоксанов, подходящих для использования в настоящем изобретении, являются метилалюмоксан (МАО), тетра(изобутил)алюмоксан (TIBAO), тетра-(2,4,4-триметилпентил)-алюмоксан (TIOAO), тетра-(2,3-диметилбутил)алюмоксан (TDMBAO) и тетра-(2,3,3-триметилбутил)алюмоксан (ТТМВАО).
Особенно интересными сокатализаторами являются сокатализаторы, описанные в публикациях WO 99/21899 и РСТ/ЕР 00/09111, в которых алкильные и арильные группы имеют определенные модели разветвления.
Не ограничивающими примерами алюминиевых соединений в соответствии с указанными заявками РСТ являются: трис(2,3,3-триметилбутил)алюминий, трис(2,3-диметилгексил)алюминий, трис(2,3-диметилбутил)алюминий, трис(2,3-диметилпентил)алюминий, трис(2,3-диметилгептил)алюминий, трис(2-метил-3-этилпентил)алюминий, трис(2-метил-3-этилгексил)алюминий, трис(2-метил-3-этилгептил)алюминий, трис(2-метил-3-пропилгексил)алюминий, трис(2-этил-3-метилбутил)алюминий, трис(2-этил-3-метилпентил)алюминий, трис(2,3-диэтилпентил)алюминий, трис(2-пропил-3-метилбутил)алюминий, трис(2-изопропил-3-метилбутил)алюминий, трис(2-изобутил-3-метилпентил)алюминий, трис(2,3,3-триметилпентил)алюминий, трис(2,3,3-триметилгексил)алюминий, трис(2-этил-3,3-диметилбутил)алюминий, трис(2-этил-3,3-диметилпентил)алюминий, трис(2-изопропил-3,3-диметилбутил)алюминий, трис(2-триметилсилилпропил)алюминий, трис(2-метил-3-фенилбутил)алюминий, трис(2-этил-3-фенилбутил)алюминий, трис-(2,3-диметил-3-фенилбутил)алюминий, трис(2-фенилпропил)алюминий, трис[2-(4-фторфенил)пропил]алюминий, трис[2-(4-хлорфенил)пропил]алюминий, трис[2-(3-изопропилфенил)пропил]алюминий, трис(2-фенилбутил)алюминий, трис(3-метил-2-фенилбутил)алюминий, трис(2-фенилпентил)алюминий, трис[2-(пентафторфенил)пропил]алюминий, трис[2,2-дифенилэтил]алюминий и трис[2-фенил-2-метилпропил]алюминий, а также соответствующие соединения, где одна из углеводородных групп замещена атомом водорода, и соединения, где одна или две из углеводородных групп замещены изобутильной группой.
Среди названных выше алюминиевых соединений наиболее предпочтительными являются триметилалюминий (ТМА), триизобутилалюминий (TIBAL) трис(2,4,4-триметилпентил)алюминий (TIOA), трис(2,3-диметилбутил)алюминий (TDMBA), и трис (2,3,3-триметилбутил)алюминий (ТТМВА).
Не ограничивающими примерами соединений, способных образовывать алкилметаллоценовый катион, являются соединения формулы D+Е-, где D+ представляет собой кислоту Брэнстеда, способную быть донором протона и необратимо реагировать с заместителем Х металлоцена формулы (I), и Е- представляет собой совместимый анион, который способен стабилизировать активные каталитические молекулы, образованные в реакции двух соединений, и который является достаточно подвижным, чтобы он мог быть удален олефиновым мономером. Предпочтительно анион Е- состоит из одного или двух атомов бора. Более предпочтительно анион Е- представляет собой анион формулы BAr
Катализаторы настоящего изобретения также могут быть нанесены на инертный носитель. Это достигается путем осаждения металлоценового соединения (А) или продукта его реакции с компонентом (В), или компонента (В), а затем металлоценового соединения (А) на носитель, такой как, например, двуокись кремния, окись алюминия, галогениды магния, сополимеры стирол/дивинилбензол, полиэтилен или полипропилен. Процесс нанесения проводят в инертном растворителе, таком как углеводород, например толуол, гексан, пентан или пропан, и при температуре в интервале от 0 до 100° С, предпочтительно процесс осуществляют при комнатной температуре.
Подходящим классом носителей, которые могут быть использованы, является класс, составленный пористыми органическими носителями с функциональными группами, имеющими активные атомы водорода. Особенно подходящими являются те органические носители, в которых органический носитель представляет собой частично поперечно сшитый стирольный полимер. Носители этого типа описаны в европейской заявке ЕР 633272.
Другой класс инертных носителей, особенно подходящих для применения в настоящем изобретении, составляют олефиновые, в особенности пропиленовые, пористые форполимеры, описанные в международной заявке WO 95/26369.
Еще один подходящий класс инертных носителей для применения в настоящем изобретении составляют пористые галогениды магния, такие как описанные в международной заявке WO 95/32995.
Твердое соединение, полученное таким образом, в сочетании с добавлением алкилалюминиевого соединения или в том виде, как оно есть, или предварительно прореагировавшего с водой, если это необходимо, может быть использовано при газофазной полимеризации.
В соответствии с еще одним аспектом настоящего изобретения предлагается способ получения полимеров альфа-олефинов, включающий контактирование одного или нескольких альфа-олефинов в условиях реакции полимеризации с катализатором, содержащим продукт, получаемый контактированием:
(А) металлоценового соединения формулы (I):

где L, Z, М, Х и р имеют значения, определенные выше, и G представляет собой остаток формулы (III):
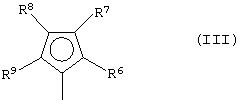
где R6, R7, R8 и R9, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из группы, включающей атом водорода, С1-С20-алкильный, С3-С20-циклоалкильный, С2-С20-алкенильный, С6-С20-арильный, С7-С20-алкиларильный, С7-С20-арилалкильный радикал, необязательно содержащий гетероатомы, принадлежащие группам 13-17 Периодической таблицы элементов, заместители R6 и R7 и/или R8 и R9 могут образовывать кольцо, содержащее от 3 до 8 атомов, которое может иметь заместители; при условии, что R7 отличается от R8, и, когда R7 представляет собой трет-бутильный радикал, R8 не является атомом водорода; и
(В) алюмоксана и/или соединения, способного образовывать алкилметаллоценовый катион.
Предпочтительно в металлоценовом соединении формулы (I) G представляет собой остаток формулы (IIIa) или (IV), более предпочтительно G представляет собой остаток, выбранный из соединений формулы (IIIa), (IVb), (IVc) или (IVd).
Процесс полимеризации олефинов в соответствии с настоящим изобретением может быть проведен в жидкой фазе в присутствии или в отсутствие инертного углеводородного растворителя или в газовой фазе. Углеводородный растворитель может быть либо ароматическим, таким как толуол, либо алифатическим, таким как пропан, гексан, гептан, изобутан или циклогексан.
Температура полимеризации обычно находится между -100 и +100° С и в особенности между 10 и +90° С. Давление при полимеризации обычно составляет от 0,5 до 100 бар.
Чем ниже температура полимеризации, тем выше конечные молекулярные массы полученных полимеров.
Выход реакции полимеризации зависит от чистоты металлоценового соединения, образующего катализатор. Металлоценовые соединения, полученные способом настоящего изобретения, могут быть использованы как они есть или могут быть подвергнуты очистке.
Компоненты катализатора могут быть приведены в контакт друг с другом до полимеризации. При предварительном контактировании концентрации металлоценового соединения (А) обычно находятся между 0,1 и 10-8 моль/л, тогда как концентрации компонента (В) находятся между 2 и 10-8 моль/л. Предварительное контактирование проводят обычно в присутствии углеводородного растворителя и, если необходимо, небольших количеств мономера. При предварительном контактировании также можно использовать неполимеризиуемые олефины, такие как изобутен, 2-бутен и другие подобные олефины.
Кроме того, молекулярные массы полученного полимера, в частности гомо- или сополимеров пропилена, полимеров 1-бутена или гомо- или сополимеров этилена, распределены в пределах относительно ограниченных интервалов. Молекулярно-массовое распределение может быть представлено отношением Мw/Мn, которое для данных полимеров обычно составляет меньше, чем 4, предпочтительно меньше, чем 3,5 и более предпочтительно меньше, чем 3.
Молекулярно-массовое распределение можно менять за счет использования смесей различных металлоценовых соединений или путем проведения полимеризации в несколько стадий, которые отличаются температурой полимеризации и/или концентрациями регуляторов молекулярной массы.
Одним из предпочтительных альфа-олефинов, используемых в процессе полимеризации настоящего изобретения, является пропилен. Когда полимеризации подвергают пропилен, a G представляет собой остаток, выбранный из соединений формул (IIIa) и (IVb), обычно может быть получен полимер пропилена, имеющий энтальпию плавления <70 Дж/г; и триады (mm), удовлетворяющие отношению: 30<mm<85. Когда G представляет собой фрагмент, выбранный из соединений формулы (IVc), (IVd), полученный полимер обычно имеет каталитическую активность и/или характеристическую вязкость выше, чем эти характеристики, достигаемые с помощью аналогичного катализатора, используемого в предшествующем уровне техники. Например, в публикации в J. Am. Chem. Soc. 1998, 120, 10786-10787 изопропилиден{(3-трет-бутилциклопентадиенил)-7-(2,5-дитриметилсилилциклопента[1,2-b:4,3-b’]-дитиофен)}цирконийдихлорид используется при полимеризации пропилена с каталитической активностью только 13 кг/ммоль кат· час. Полученные полимеры обычно имеют триады (mm), удовлетворяющие соотношению: 70<mm<95, предпочтительно 85<mm<95, и характеристическую вязкость (Х.В.), измеренную в тетрагидронафталине (ТГН), выше,чем 0,7, предпочтительно 0,8, более предпочтительно выше, чем 1 и еще более предпочтительно выше, чем 2.
Более интересными пропиленовыми полимерами, получаемыми способом, описанным выше, являются пропиленовые полимеры, имеющие следующие характеристики:
- триады (mm), удовлетворяющие соотношению: 30<mm<85, предпочтительно 55<mm<85 и более предпочтительно 65<mm<85;
- энтальпию плавления (Δ Н) <70 Дж/г, предпочтительно находящуюся в интервале между 5 и 70 Дж/г, более предпочтительно между 20 и 70 Дж/г.
Молекулярные массы указанных выше пропиленовых полимеров могут быть достаточно высокими. Следовательно, характеристическая вязкость может достигать значений больше, чем 0,7 децилитр/г (дл/г), предпочтительно больше, чем 1 дл/г, более предпочтительно больше, чем 2 дл/г.
Пропиленовые полимеры, описанные выше, обладают хорошим соотношением между оптическими свойствами, являясь полностью прозрачными, и эластичными свойствами. Таким образом, полипропилен настоящего изобретения имеет следующие свойства:
- Мутность (ASTM 2457) от 15 до 30%, предпочтительно от 20 до 30%;
- Блеск (60° С) (ASTM 2457) от 60 до 95%, предпочтительно от 70 до 85%;
Модуль упругости при растяжении (ASTM D4065) от 1000 до 200 мПа, предпочтительно от 700 до 400 мПа;
Относительное удлинение при разрыве (ASTM D4065) от 300 до 900%, предпочтительно от 500 до 700%;
Предел прочности при разрыве (ASTM D638) от 10 до 40%, предпочтительно от 10 до 30%.
Микроструктуры полипропилена, полученного способом настоящего изобретения, охватывают интервал коммерческих сополимеров, таких как эластомерный, эластичный и похожий на статистический полипропилен, но с тем отличием, что температура плавления полипропилена настоящего изобретения всегда выше, чем указанных известных сополимеров. Следовательно, полипропилен настоящего изобретения может легко заменять эти более дорогие сополимеры.
Реакция полимеризации пропилена в соответствии с настоящим изобретением может быть проведена в присутствии этилена или С4-С10-альфа-олефинового сомономера. Следовательно, еще одним объектом настоящего изобретения является пропиленовый сополимер, содержащий от 0,1 до 30 мол.%, предпочтительно от 0,1 до 20 мол.%, более предпочтительно от 0,1 до 10 мол.%, и еще более предпочтительно от 0,1 до 5 мол.% звеньев, полученных из олефина формулы CH2=CHR’, где R’ представляет собой атом водорода, С2-С20-алкильную или С6-С12-арильную группу, причем указанный пропиленовый сополимер имеет следующие характеристики:
- энтальпию плавления <70 Дж/г, предпочтительно <50 Дж/г;
- триады (mm) полипропиленовой гомопоследовательности, удовлетворяющие соотношению: 30<mm<85, предпочтительно 55<mm<85.
Не ограничивающими примерами альфа-олефинов, которые могут быть использованы в качестве сомономеров в сополимерах в соответствии с настоящим изобретением, являются этилен, 1-бутен, 1-пентен, 1-гексен, 4-метил-1-пентен, 1-октен, 1-децен, 1-додецен, стирол, 1,5-гексадиен и 1,7-октадиен. Предпочтительным сополимером является этилен.
Способ в соответствии с настоящим изобретением также подходит для получения гомо- и сополимеров этилена, где олефиновыми сомономерами могут быть альфа-олефины, циклоолефины или полиены. Могут быть получены этиленовые гомополимеры, имеющие замечательно высокую молекулярную массу. Действительно, с помощью способа настоящего изобретения можно получать этиленовые полимеры, имеющие значение характеристической вязкости (Х.В.) до 5,0 дл/г и даже выше.
В сополимерах, получаемых способом изобретения, молярное содержание этиленовых звеньев обычно выше, чем 40%, и предпочтительно оно находится между 50 и 99%, и наиболее предпочтительно оно находится между 80 и 98%.
Молярное содержание звеньев, полученных из альфа-олефина, предпочтительно составляет от 0 до 60%, более предпочтительно от 1 до 50% и наиболее предпочтительно от 2 до 20%.
Не ограничивающими примерами альфа-олефинов, которые могут быть использованы в способе изобретения, являются пропилен, 1-бутен, 1-пентен, 4-метил-1-пентен, 1-гексен, 1-октен, 4,5-диметил-1-гептен, 1-децен, 1-додецен, 1-тетрадецен, 1-гексадецен, 1-октадецен, 1-эйкозен и аллилциклогексан.
Не ограничивающими примерами циклоолефинов, которые могут быть использованы в качестве сомономеров в способе настоящего изобретения, являются циклопентен, циклогексен и норборнен.
Сополимеры в соответствии с настоящим изобретением также могут содержать звенья, полученные из полиенов. Содержание звеньев, полученных из полиенов, если они присутствуют, находится между 0 и 30 мол.%, более предпочтительно между 0 и 20 моль.%.
Полиены, которые могут быть использованы в качестве сомономеров в сополимерах настоящего изобретения, входят в следующие классы;
- несопряженные диолефины, способные к циклополимеризации, такие как, например, 1,5-гексадиен, 1,6-гептадиен, 2-метил-1,5-гексадиен;
- диены, способные давать ненасыщенные мономерные звенья, в частности сопряженные диены, такие как, например, бутадиен и изопрен, и линейные несопряженные диены, такие как, например, транс-1,4-гексадиен, цис-1,4-гексадиен, 6-метил-1,5-гептадиен, 3,7-диметил-1,6-октадиен, 11-метил-1,10-додекадиен, и циклические несопряженные диены, такие как 5-этилиден-2-норборнен.
Металлоцены настоящего изобретения также могут быть использованы для полимеризации в газовой фазе этилена с альфа-олефинами, такими как пропилен, 1-бутен, 1-пентен, 4-метил-1-пентен, 1-гексен, 1-октен, 4,6-диметил-1-гептен, 1-децен, 1-додецен, 1-тетрадецен, 1-гексадецен, 1-октадецен, 1-эйкозен и аллилциклогексан.
В случае этилен/пропиленовых сополимеров произведение констант сополимеризации r1·r2, где r1 представляет собой константу сополимеризации пропилена и r2 представляет собой константу сополимеризации этилена, рассчитывают в соответствии со следующей формулой:
r1·r2=1+f(χ +1)-(f+1)(χ +1)1/2
где f - отношение между числом молей этиленовых звеньев и числом молей пропиленовых звеньев в сополимере и
χ =(ППП+ППЭ)/ЭПЭ.
Молекулярные массы полимеров могут быть изменены за счет изменения типа или концентрации компонентов катализатора или за счет использования регуляторов молекулярной массы, таких как, например, водород.
Тактичность молекулярной структуры, то есть распределение относительной конфигурации третичных атомов углерода, определяют с помощью ЯМР, как это описано в публикации Resconi et al., Chem. Rev., 2000, 100, 1253-1345 и цитируемых в ней ссылках.
Полимеры настоящего изобретения могут быть преобразованы в формованные изделия обычной переработкой материалов, такой как формование, экструзия, литье и др. Полимеры настоящего изобретения могут быть использованы для получения синтетической кожи, кровельных смесей, геомембран, прозрачных изделий, вспененных слоев, в качестве добавок к битуму или в качестве полимерного носителя для пигментов и/или красителей в концентрированных красителях.
ПРИМЕРЫ
Общие методики
Все операции проводят в атмосфере азота с использованием обычных методик по схеме Шленка (Schlenk-line techniques). Растворители очищают обезгаживанием с помощью N2 и пропусканием над активированной (8 час, продувка N2, 300° C) Аl2O3 и хранят в атмосфере азота. н-BuLi (Aldrich) используют в том виде, в каком он поставляется.
Спектры лигандов и металлоценов на протонах и атомах углерода получены с использованием спектрометра Bruker DPX 200, работающего с преобразованием Фурье, при комнатной температуре и при 200,13 МГц и 50,32 МГц соответственно. Образцы растворяют в CDCl3, CD2Cl2 или C6D6. В качестве стандарта используют остаточный сигнал СНСl3, CHDCl2 или С6НD5 в спектрах на ядрах 1H (7,25, 5,35 и 7,15 м.д. соответственно) и сигнал растворителя в спектрах на ядрах 13С (77,00 м.д. для CDCl3). Спектр ПМР получают с импульсом 15° и задержкой 2 сек между импульсами; для каждого спектра накапливают 32 скана. Спектры на атомах углерода получены с импульсом 45° и задержкой между импульсами 6 секунд; для каждого спектра накапливают приблизительно 512 сканов. СDСl3 (Aldrich, 99,8% атом. D) и C6D6 (Aldrich, 99,6% атом. D) хранят над молекулярными ситами (4-5  ), тогда как CD2Cl2 (Aldrich, 99,8% атом. D) используют в том виде, в каком он получен.
), тогда как CD2Cl2 (Aldrich, 99,8% атом. D) используют в том виде, в каком он получен.
Приготовление образцов проводят в атмосфере азота с использованием стандартных методик с инертной атмосферой.
Спектры полимеров на протонах и атомах углерода получают с использованием спектрометра Bruker DPX 400, работающего с преобразованием Фурье, при температуре 120° С и при 400,13 МГц и 100,61 МГц соответственно. Образцы растворяют в C2D2Cl4. В качестве стандарта используют остаточный сигнал C2DHCl4 (5,95 м.д.) в спектрах на ядрах 1Н и сигнал mmmm-пентад в спектрах на ядрах 13С (21,8 м.д.). Спектры ПМР получают с импульсом 45° и задержкой 5 сек между импульсами; для каждого спектра накапливают 256 сканов. Спектры на атомах углерода получены с импульсом 90° и задержкой между импульсами 12 секунд (15 секунд для полимеров на основе этилена) и развязкой CPD (16 waltz) для подавления взаимодействия 1H-13С. Для каждого спектра накапливают приблизительно 3000 сканов.
Анализ ГХ-МС проводят на газовом хроматографе HP 5890 - серия 2 и квадрупольном масс-спектрометре HP 5989B.
Характеристическую вязкость (Х.В.) измеряют в тетрагидронафталине (ТГН) при 135° С.
Температуры плавления полимеров (Тпл) измеряют с помощью дифференциальной сканирующей калориметрии (ДСК) на приборе Perkin Elmer DSC-7 в соответствии со стандартным методом. Взвешенный образец (5-10 мг), полученный при полимеризации, герметично закрывают на алюминиевых поддонах и нагревают до 200° С, поддерживая скорость, соответствующую 20° С/мин. Образец выдерживают при 200° С в течение 5 мин, позволяя полностью расплавиться всем кристаллам. Затем после охлаждения до 0° С при скорости сканирования, соответствующей 20° С/мин, температурный пик принимают за температуру кристаллизации (Тк). После стояния 5 мин при 0° С образец нагревают второй раз до 200°С со скоростью, соответствующей 20° С/мин. При этом втором нагревании температурный пик принимают за температуру плавления (Тпл), а площадь - за суммарную энтальпию плавления (Δ Hf).
Молекулярно-массовое распределение определяют с помощью вытеснительной хроматографии (SEC) на приборе WATERS 200 в трихлорбензоле при 135° С.
Используются следующие сокращения:
водн. - водный
ТГФ - тетрагидрофуран
Et2O - диэтиловый эфир
CH2Cl2 - дихлорметан
ДМФА - N,N-диметилформамид
Me2SiCl2 - дихлордиметилсилан
Me3SiCl - хлортриметилсилан
CuCl2 - хлорид меди (II)
РОСl3 - оксихлорид фосфора
В(ОМе)3 - триметилборат
АlСl3 - трихлорид алюминия
н-BuLi - нормальный бутиллитий
Dppf, Дффф - дифенилфостинферроцен
ТМЭДА - N,N,N’,N’-тетраметилэтилендиамин
ZrCl4 - тетрахлорид циркония
HfCl4 - хлорид гафния
Th2Cp - 7Н-циклопента[1,2-b:4,3-b’]дитиофен или 7Н-тиено-[3’,2’:3,4]циклопента[b]тиофен
MeTh2Cp - 2,5-диметил-7Н-циклопента[1,2-b:4,3-b’]дитиофен
EtTh2Cp - 2,5-диэтил-7Н-циклопента[1,2-b:4,3-b’]дитиофен
PhTh2Cp - 2,5-дифенил-7Н-циклопента[1,2-b:4,3-b’]дитиофен
АсОН - уксусная кислота
(МеО)2СН2 - диметоксиметан
ПОЛУЧЕНИЕ ПРЕДШЕСТВЕННИКОВ ЛИГАНДОВ
Синтез 3,3’-дибром-2,2’-дитиенилметанола
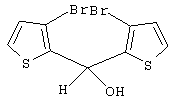
Раствор н-BuLi в гексане (2,5 М, 24,30 мл, 60,76 ммоль) добавляют по каплям при -20° С к раствору 15,00 г 2,3-дибромтиофена (Aldrich, 98%, Мм=241,94, d=2,137, 60,76 ммоль, н-BuLi:2,3-Вr2-тиофен 1:1) в 90 мл эфира. Раствор меняет цвет от бледно-желтого до желтого. После перемешивания в течение 1 час при -20° С добавляют по каплям 2,53 мл этилформиата (Aldrich, 97%, Мм=74,08, d=0,917, 30,38 ммоль, HCOOEt:2,3-Вr2-тиофен 0,5:1) в 30 мл эфира. При добавлении цвет раствора изменяется от желтого до темно-желтого. Реакционную смесь выдерживают при -20° С в течение 15 мин, затем дают нагреться до комнатной температуры и перемешивают 20 часов. Полученную бледно-оранжевую суспензию выливают при 0° С в подкисленную воду (1,65 г NH4Cl в 75 мл воды), органический слой отделяют и водный слой экстрагируют эфиром (3× 25 мл). Органические слои собирают, сушат над Na2SO4, и растворители удаляют под вакуумом при 30-35° С, получают оранжевое масло (9,52 г), которое характеризуют с помощью анализа ГХ-МС и спектроскопии 1H-ЯМР.
Чистота (данные ГХ-МС) 96,0%. Выход чистого продукта 85,0%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): 7,28 (д, 2Н, J=5,29 Гц, СН); 6,95 (д, 2Н, J=5,29 Гц, СН); 6,41 (с, 1Н, СН); 2,86 (уш.с, 1Н, СН).
m/z (%): 356 (23) [M++4], 354 (42) [M++2], 352 (22) [М+], 339 (10), 337 (18), 275 (10), 273 (10), 194 (11), 193 (23), 192 (11), 191 (100), 177 (32), 166 (10), 164 (10), 121 (17), 111 (14), 84 (33), 83 (15), 82 (26), 81 (14), 69 (11), 45 (33), 39 (15).
Синтез 3,3’-дибром-2,2’-дитиенилметана
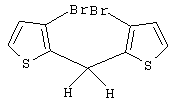
В трехгорлой колбе объемом 250 мл в атмосфере азота растворяют 9,45 г 3,3’-дибром-2,2’-дитиенилметанола (Мм=354,09, 26,69 ммоль, соответствует исходному материалу чистотой 100%), полученного, как описано выше, в 85 мл дихлорметана и добавляют при 0° С 4,26 мл триэтилсилана (Aldrich, Мм=116,28, d=0,728, 26,69 ммоль). Затем к смеси при перемешивании и при 0° С добавляют по каплям 2,06 мл СF3СООН (Aldrich, Мм=114,02, d=1,48, 26,69 ммоль). При добавлении цвет реакционной смеси меняется от темно-оранжевого до темно-красного. Реакционную смесь выдерживают при 0° С в течение 15-20 мин, затем дают нагреться до комнатной температуры и перемешивают 3 час и 30 мин при той же температуре. После охлаждения до 0° С к темно-красному раствору добавляют карбонат калия (Fluka, 3,69 г, Мм=138,21, 26,69 ммоль), полученную смесь перемешивают 30 мин при комнатной температуре и затем фильтруют на фильтре Шотта G4. Остаток на фильтре промывают дважды CH2Cl2 (2× 20 мл) до неокрашенного продукта, фильтрат сушат в вакууме при 45°С в течение 3 час, получают темно-красное масло (9,07 г), которое анализируют с помощью ГХ-МС и спектроскопии 1H ЯМР. Чистота (ГХ-МС) 79,9%. Выход чистого продукта 80,3%. В качестве побочных продуктов присутствуют 3-бром-2,2’-дитиенилметан (9,9 мас.%) и гексаэтилдисилоксан (6,2 мас.%). Продукт используют в том виде, как он есть, без дополнительной очистки.
Спектр 1Н ЯМР (CDCl3, δ , м.д.): 7,16 (д, 2Н, J=5,38 Гц, СН); 6,94 (д, 2Н, J=5,38 Гц, СН); 4,27 (с, 2Н, CH2).
m/z (%): 340 (28) [M++4], 338 (51) [M++2], 336 (26) [М+], 259 (55), 257 (51), 179 (15), 178 (100), 177 (43), 89 (16), 45 (10).
Синтез 7Н-циклопента[1,2-b:4,3-b’]дитиофена
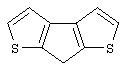
В колбе объемом 250 мл в атмосфере азота при -50° С к раствору 8,99 г 3,3’-дибром-2,2’-дитиенилметана (Мм=338,09, 26,59 ммоль), полученного, как описано выше, в 75 мл эфира добавляют по каплям раствор н-BuLi в гексане (2,5 М, 21,30 мл, 53,25 ммоль). После перемешивания в течение 1 час при -50° С темно-коричневую суспензию дилитиевой соли медленно добавляют к суспензии 7,26 г CuCl2 (Aldrich, 98%, Мм=134,45, 52,92 ммоль) в 50 мл Et2O. Реакционную смесь выдерживают при -50° С 30 мин, дают нагреться до -20° С за 2 час 30 мин и затем до 0° С в течение нескольких минут. Для наблюдения за ходом реакции с помощью анализа ГХ-МС отбирают аликвоты через 30 мин при -50° С, при -20° С и через 1 час при 0° С. Оказалось, что индуцированная CuCl2 реакция сочетания начинается при -50° С, но протекает медленно до температуры 0° С. Только 10 мас.%. 7Н-циклопента[1,2-b:4,3-b’]дитиофена образуется через 1 час при 0° С. После выдерживания при 0° С 1 час 30 мин реакционную смесь перемешивают в течение ночи при комнатной температуре и затем выливают при 0° С в 100 мл 2 М водного раствора НСl. Полученную смесь перемешивают 15 мин при комнатной температуре, фильтруют для удаления сероватого осадка Cu2Cl2, эфирный слой отделяют и водную фазу экстрагируют эфиром. Объединенные эфирные экстракты промывают 2 М НСl (100 мл), дважды водным NаНСО3 и затем эфиром. Полученную органическую фазу (конечный объем 300 мл) сушат с помощью Na2SO4, растворители удаляют в вакууме, получают 3,16 г темно-красного масла, которое анализируют с помощью ГХ-МС и спектроскопии 1H ЯМР. Анализ показывает наличие целевого продукта вместе с димерами, тримерами и смолами. К сырому продукту добавляют 40 мл этанола и перемешивают в течение 1 часа при комнатной температуре. Желто-оранжевый экстракт концентрируют в вакууме при 55° С в течение 4 час, получают темно-оранжевое масло (1,92 г), которое кристаллизуется при стоянии при 0° С в течение ночи.
Чистота (ГХ-МС) ~ 50%. Выход чистого продукта 20,2%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,30 (д, 2Н, J=4,93 Гц, СН); 7,13 (д, 2Н, J=4,93 Гц, СН); 3,80 (с, 2Н, CH2).
m/z (%): 180 (9) [M++2], 179 (16) [M++1], 178 (100) [М+], 177 (92), 134 (13), 89 (7), 69 (6), 45 (6).
Синтез бис(3,5-дибром-2-тиенил)метанола (или 3,3’,5,5’-тетрабром-2,2’-дитиенилкарбинола)
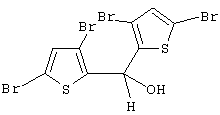
Раствор 31,35 г 2,3,5-трибромтиофена (Lancaster, 98%, Мм=320,84, 95,75 ммоль) в 70 мл эфира охлаждают до -78° С и добавляют по каплям 38,3 мл 2,5 М раствора н-BuLi в гексане (95,75 ммоль). Полученной смеси дают нагреться до комнатной температуры, перемешивают еще 1 час и затем добавляют при температуре 0--10° С к раствору 3,86 мл этилформиата (Aldrich, 97%, Мм=74,08, d=0,917, 46,35 ммоль) в 20 мл гексана, предварительно охлажденного до 0--10° С. По окончании добавления (~ 20 мин) реакционной массе дают нагреться до комнатной температуры и затем кипятят с обратным холодильником 1 час. Полученную смесь гасят 7,5 мл воды, органический слой отделяют, сушат над сульфатом магния, растворители упаривают, получают 23,2 г бледно-коричневого твердого вещества, которое анализируют с помощью спектров 1H и 13С ЯМР и ГХ-МС.
Чистота продукта 93,0%. Выход относительно этилформиата 90,9%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): 6,92 (с, 2Н, СН); 6,26 (д, 1Н, J=3,2 Гц, мостиковый СН); 2,73 (д, 1Н, J=3,2 Гц, ОН).
Спектр 13С ЯМР (СDСl3, δ , м.д.): 67,38 (СНОН), 108,60, 113,58, 132,18 (СН), 141,10.
m/z (%): 512 (67) [М+], 494 (50), 433 (54), 352 (53), 335 (35), 285 (43), 269 (100), 242(19), 162(33), 81 (27), 39 (13).
Синтез 3,3’,5,5’-тетрабром-2,2’-дитиенилметана
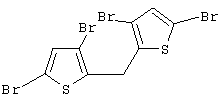
При комнатной температуре добавляют трифторуксусную кислоту (0,25 мл, Aldrich, 99%, Мм=114,02, d=1,48, 3,24 ммоль) к раствору 1,75 г бис(3,5-дибром-2-тиенил)метанола (93,0%, Мм=511,90, 3,18 ммоль) в 15 мл метиленхлорида, содержащего 0,50 мл триэтилсилана (Aldrich, 99%, Мм=116,28, d=0,728, 3,13 ммоль). Полученный красный раствор перемешивают в течение 1 часа при комнатной температуре, нейтрализуют твердым карбонатом калия (0,4 г, Мм=138,21, 2,89 ммоль), фильтруют и упаривают, получают бледно-красное твердое вещество. Выход сырого продукта 100%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 6,94 (с, 2Н, СН); 4,17 (с, 2Н, CH2).
Спектр 13С ЯМР (СDСl3, δ , м.д.): 29,30 (СН2), 109,07, 111,38, 131,98 (СН), 137,22.
m/z (%): 496 (71) [М++4], 417 (76) [М+], 336 (91), 255 (100), 176 (41), 125 (46), 95 (30), 69 (40), 45 (22).
Синтез 3,3’-дибром-5,5’-диметил-2,2’-дитиенилметана
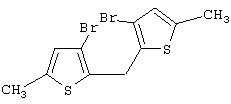
Предварительно охлажденный (-20° С) 2,5 М раствор н-BuLi в гексане (41,1 мл, 102,75 ммоль) добавляют при -20° С к раствору 25,48 г 3,3’,5,5’-тетрабром-2,2’-дитиенилметана (Мм=495,90, 51,38 ммоль) в 100 мл Et2O. После 30 мин перемешивания при -20° С добавляют предварительно охлажденный (-20° С) эфирный (10 мл) раствор диметилсульфатa (Aldrich, 9,72 мл. Мм=126,13, d=1,333, 102,75 ммоль). Полученную черную суспензию перемешивают 45 мин при -20° С; охлаждающую баню затем убирают, и останавливают ток азота. Добавляют 4 н. раствор гидроксида натрия (2,5 мл, 10 ммоль), и смесь интенсивно перемешивают 2 час при комнатной температуре. Полученную реакционную массу сушат сульфатом магния, фильтруют, остаток на фильтре промывают дважды эфиром (для извлечения всего продукта), и фильтрат концентрируют при пониженном давлении при 40° С в течение 2 час, получают 17,8 г коричневого твердого вещества. Чистота 87,8% (ГХ-МС). Выход чистого продукта 83,1% (выход сырого продукта 94,6%).
Спектр 1H ЯМР (СDСl3, δ , м.д.): 6,58 (кв, 2Н, J=1,0 Гц, СН); 4,11 (с, 2Н, СН2); 2,39 (д, 6Н, J=1,0 Гц, СН3).
Спектр 13С ЯМР (СDСl3, δ , м.д.): 15,41 (СН3), 28,88 (CH2), 108,20, 127,57 (СН), 134,10, 138,70.
m/z (%): 366 (43) [М+], 287 (47), 206 (100), 191 (21), 173 (14), 103 (10), 59 (20).
Синтез 2,5-диметил-7Н-циклопента[1,2-b:4,3-b’]дитиофена (или 2,5-диметил-7Н-тиено[3’,2’:3,4]циклопента[b]тиофена)
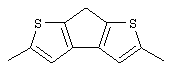
Предварительно охлажденный (-70° С) 2,5 М раствор н-BuLi в гексане (27,1 мл, 67,75 ммоль) добавляют по каплям при -70° С к раствору 10,77 г 3,3’-дибром-5,5’-диметил-2,2’-дитиенилметана (Мм=366,15, 29,41 ммоль) в 60 мл эфира. По окончании добавления коричневую суспензию перемешивают еще 30 мин при той же температуре. Затем быстро добавляют предварительно охлажденную (-70° С) суспензию 10,28 г безводного CuCl2 (высушенного при 130° С в течение 1 час, Мм=134,45, 76,46 ммоль) в 35 мл эфира. Полученную черную суспензию выдерживают при -70° С 10 мин, при -50° С 1 час, при -20° С 1 час и при 0° С 1 час. Затем смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. При повышении температуры цвет реакционной смеси меняется от черного до бледно-коричневого. При проведении реакции для анализа с помощью ГХ-МС отбирают аликвоты: при -50° С титр целевого соединения равен 8,6%, при -20° С титр целевого соединения равен 20,9%, при 0° С титр целевого соединения равен 68,8%. Полученную суспензию выливают в 160 мл насыщенного водного раствора хлорида аммония, органический слой отделяют, водный слой промывают эфиром, органические слои собирают и сушат. Получают 4,79 г черного твердого вещества. Чистота (ГХ-МС) 75,9%. Выход чистого продукта 60,0% (выход сырого продукта 79,0%).
Спектр 1H ЯМР (СDСl3, δ , м.д.): 6,78 (с, 2Н, СН); 3,69 (с, 2Н, СН2); 2,54 (с, 6Н, СН3).
Спектр 13С ЯМР (СDСl3, δ , м.д.): 15,96 (СН3), 33,13 (СН2), 116,43 (СН), 140,16, 142,16, 143,67.
m/z (%): 206 (100) [М+], 191 (54), 173 (29), 158 (6), 147 (8).
Синтез 3,3’-дибром-5,5’-дитриметилсилил-2,2’-дитиенилметана
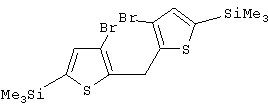
2,18 М раствор н-BuLi (65 мл, 141,7 ммоль) добавляют при -70° С к раствору 34,8 г 3,3’,5,5’-тетрабром-2,2’-дитиенилметана (70,2 ммоль) в 150 мл эфира. Смесь перемешивают в течение 30 мин при той же температуре и затем добавляют 35,5 мл Me2SiCi (280 ммоль) в 65 мл эфира. Полученной смеси дают нагреться до комнатной температуры, отфильтровывают LiCl, маточный раствор упаривают, получают масло, которое представляет собой целевое соединение чистотой, по меньшей мере, 95%. К этому маслу добавляют 50 мл гексана, и полученный раствор выдерживают при -30° С 10 часов. Большие кристаллы отделяют, промывают охлажденным гексаном и сушат. Выход перекристаллизованного продукта составляет 60%. Названное соединение характеризуют с помощью спектроскопии 1H и 13С ЯМР.
Синтез 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофена (или 2,5-диметилтриметилсилил-7Н-тиено[3’,2’:3,4]циклопента[b]тиофена)
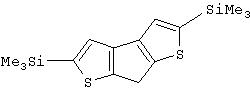
Раствор 0,1 моль 3,3’-дибром-5,5’-дитриметилсилил-2,2’-дитиенилметана в 200 мл эфира обрабатывают при -70°С 0,23 моль н-BuLi. По окончании добавления реакционную смесь перемешивают еще 30 мин при той же температуре. Затем быстро добавляют 0,265 моль CuCl2. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную суспензию выливают в воду, органическую фазу отделяют и концентрируют. Остаток пропускают через колонку, заполненную SiO2, с использованием в качестве элюента гексана или смеси гексан/эфир. Полученный раствор упаривают, получают кристаллы или маслянисто-кристаллическое твердое вещество, которые представляют собой целевой продукт. Выход 50-60%. Сырой продукт может быть дополнительно очищен фильтрованием в эфире при 0° С или перекристаллизацией из пентана. Названное соединение характеризуют с помощью спектроскопии 1H и 13С ЯМР.
Синтез 3,3’-дибром-5,5’-дигидроксиборил-2,2’-дитиенилметана
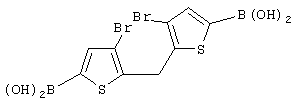
При -70° С добавляют 1,6 н. раствор н-BuLi (100 мл, 160 ммоль) к раствору 39,6 г 3,3’,5,5’-тетрабром-2,2’-дитиенилметана (79,8 ммоль) в 150 мл эфира. Смесь перемешивают в течение 30 мин при той же температуре и затем добавляют 23,3 г В(ОМе)3 (220 ммоль) в 100 мл эфира. Реакционной смеси дают нагреться до комнатной температуры. Полученную суспензию обрабатывают 100 мл 10%-ного водного раствора НСl, органический слой отделяют, промывают дважды 50 мл 10%-ного водного раствора Nа2СО3, упаривают и сушат. Полученное твердое вещество, которое представляет собой сырую дибороновую кислоту, используют на следующей стадии без дополнительной очистки. Названное соединение характеризуют с помощью спектроскопии 1H и 13С ЯМР.
Синтез 3,3’-дибром-5,5’-дифенил-2,2’-дитиенилметана
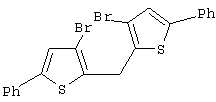
В реакционную колбу помещают 1,81 г 3,3’-дибром-5,5’-дигидроксиборил-2,2’-дитиенилметана (3,76 ммоль), 1,40 г PhI (6,84 ммоль), 0,15 г PdCl2 (dppf)2 (0,21 ммоль), 120 мл ДМФА и 8 мл Еt3N и смесь перемешивают при 80° С в течение 2 часов. Полученную смесь выливают в двухфазную систему СН2Сl2/вода. Органической слой собирают, промывают дважды 30 мл 10%-ной фосфорной кислоты, затем водой и упаривают. Остаток пропускают через колонку, наполненную SiO2, используя в качестве элюента смесь гексан/СН2Сl2 1/1. Полученный раствор упаривают, остаток промывают гексаном и сушат, получают 0,6 г дифенильного производного. Выход 32%. Названное соединение характеризуют с помощью спектроскопии 1H и 13С ЯМР.
Синтез 2,5-дифенил-7Н-циклопента[1,2-b:4,3-b’]дитиофена (или 2,5-дифенил-7Н-тиено[3’,2’:3,4]циклопента[b]тиофена)
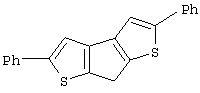
При -70° С добавляют 1,6 н. раствор н-BuLi (11,9 мл, 19 ммоль) к раствору 4,24 г 3,3’-дибром-5,5’-дифенил-2,2’-дитиенилметана (8,65 ммоль) в 50 мл эфира. По окончании добавления реакционную смесь перемешивают в течение еще 30 мин при той же температуре. Затем быстро добавляют 5,6 г (41,8 ммоль) CuCl2. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную суспензию выливают в воду, органическую фазу отделяют и растворитель упаривают. Полученный остаток пропускают через колонку, заполненную SiO2, с использованием в качестве элюента смеси гексан/СН2Сl2 4/1. Полученный раствор упаривают, получают остаток, который промывают гексаном и сушат, получают 1,1 г кристаллического твердого вещества. Выход 38%. Названное соединение характеризуют с помощью спектроскопии 1H и 13С ЯМР.
Синтез 2-метил-4-бромтиофена
К 2,5 моль измельченного в порошок АlСl3 при перемешивании и при поддержании температуры ниже 40°С добавляют 1 моль 2-тиофенкарбоксальдегида. По окончании добавления жидкий комплекс становится твердым; затем осторожно по каплям при перемешивании добавляют 1,2 моль брома. По окончании добавления перемешивание становится невозможным, так как смесь полностью затвердевает. Полученное твердое вещество выгружают в смесь льда (0,5 кг) и соляной кислоты (100 мл, 32%), затем добавляют 300 мл CH2Cl2. Органическую фазу отделяют и растворитель упаривают. Полученное вещество (4-бром-2-тиофенкарбоксальдегид) растворяют в 700 мл диэтиленгликоля и приготовленный таким образом раствор обрабатывают 5,5 моль гидразингидрата. Полученную смесь кипятят с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры добавляют 2,75 моль гидроксида калия. После окончания выделения газа начинают перегонку и собирают фракцию до 150° С. Эта фракция представляет собой смесь воды и целевого продукта; органический слой отделяют и перегоняют при 60° С/10 мм рт.ст. Выход 52%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): 6,99 (д, 1Н, Нα ); 6,69 (кв, 1Н, Нβ ); 2,48 (д, 3Н, СН3).
Синтез 2-метил-4-формилтиофена
К раствору 44,26 г 2-метил-4-бромтиофена (0,25 ммоль) в 300 мл эфира при перемешивании и при -70° С добавляют 1,6 М раствор н-BuLi (164 мл, 0,26 моль). Полученный раствор перемешивают при -60--70° С 30 мин и затем обрабатывают 27,4 г диметилформамида (0,37 моль) в 100 мл эфира. Смеси дают нагреться до комнатной температуры, затем нейтрализуют 10% водным раствором NH4Cl, промывают 10% водным раствором Н3РO4 и затем водой до нейтрального значения рН. Органическую фазу собирают, упаривают и перегоняют при 110° С/10 мм рт.ст. Выход 22,3 г (71%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез 2,2’-диметил-4,4’-дитиенилметана
К раствору 31,3 г 2-метил-4-бромтиофена (0,177 моль) в 150 мл эфира при -70° С и перемешивании добавляют 113 мл 1,6 М раствора (0,18 моль) н-BuLi. Полученный раствор перемешивают при -60--70° С в течение 30 мин и затем добавляют 22,3 г 2-метил-4-формилтиофена (0,177 моль) в 100 мл эфира. Смеси дают нагреться до комнатной температуры, затем нейтрализуют 10% водным раствором NH4Cl и промывают водой. Органическую фазу отделяют и упаривают. Получают сырой бис(2-метил-4-тиенил)метанол (или 2,2’-диметил-4,4’-дитиенилкарбинол).
Суспензию 35,5 г АlСl3 (0,266 моль) в 100 мл эфира медленно добавляют к суспензии 10 г LiAlH4 (0,266 моль) в 100 мл эфира. Полученную смесь обрабатывают раствором карбинола (полученного как описано выше) в 100 мл эфира. Реакционную смесь кипятят с обратным холодильником еще 1 час, охлаждают до комнатной температуры и затем добавляют 100 мл этилацетата. Затем смесь обрабатывают 300 мл воды и 300 мл эфира. Органическую фазу собирают, промывают водой, сушат MgSO4 и упаривают. Остаток перегоняют при 90-110° С/0,5 мм рт.ст. Выход 23,2 г (63%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез 2,6-диметил-4Н-циклопента[2,1-b:3,4-b’]дитиофена (или 2,6-диметил-4Н-тиено[3’,2’:2,3]циклопента[b]тиофена)
В 30 мл эфира растворяют 1,04 г 2,2’-диметил-4,4’-дитиенилметана (5 ммоль) и при -70° С при перемешивании добавляют 1,6 М раствор н-BuLi (14,4 ммоль) и 1,74 г ТМЭДА (15 ммоль). Полученной смеси дают нагреться до комнатной температуры, перемешивают 1 час, затем снова охлаждают до -70° С и обрабатывают 2,7 г CuCl2 (20 ммоль). Полученной реакционной смеси дают нагреться до комнатной температуры и добавляют 30 мл воды. Органическую фазу собирают и пропускают через колонку, заполненную силикагелем. Раствор упаривают, получают 0,34 г продукта. Выход 34%. Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез 2-этил-4-бромтиофена
К суспензии 2,5 моль АlСl3 в 1000 мл СНСl3 при перемешивании и, поддерживая температуру ниже 40° С, медленно добавляют 1 моль ацетилтиофена, растворенного в 250 мл СНСl3. По окончании добавления осторожно по каплям при перемешивании добавляют 1,2 моль Вr2. Полученную смесь перемешивают в течение ночи и затем выливают в смесь льда (0,5 кг) и соляной кислоты (100 мл, 32%). Органическую фазу отделяют, и растворитель удаляют. Полученное вещество растворяют в 700 мл диэтиленгликоля, и приготовленный таким образом раствор обрабатывают 5,5 моль 100%-ного гидразингидрата. Полученную смесь кипятят с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры добавляют 2,75 моль КОН. После окончания выделения газа начинают перегонку. Собирают фракцию при температуре 150° С. Эта фракция представляет собой смесь воды и целевого продукта; органический слой отделяют и перегоняют при 80° С/10 мм рт.ст. Выход 45%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): 7,05 (д, 1Н, Н5); 6,76 (кв, 1Н, Н3); 2,86 (кв, 2H, CH2), 1,33 (т, 3Н, СН3).
Синтез 3,3’-дибром-5,5’-диэтил-2,2’-дитиенилметана
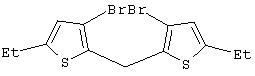
2-Этил-4-бромтиофен, полученный на предыдущей стадии, растворяют в 120 мл АсОН и обрабатывают смесью 6,1 мл H2SO4 и 9,1 мл (МеО)2СН2. Реакционную смесь перемешивают в течение ночи, затем промывают 300 мл воды и экстрагируют CH2Cl2. Органическую фазу отделяют и сушат при пониженном давлении. Остаток пропускают через колонку, наполненную Al2O3, используя в качестве элюента гексан. Растворитель удаляют и получают целевой продукт в виде желтого масла. Выход 90%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): 6,68 (м, 2Н, СН); 4,20 (с, 2Н, мостиковые CH2); 2,80 (кв, 4Н, СН2), 1,30 (т, 6Н, СН3).
Синтез 2,5-диэтил-7Н-циклопента[1,2-b:4,3-b’]дитиофена
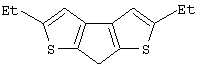
Раствор 0,1 моль 3,3’-дибром-5,5’-диэтил-2,2’-дитиенилметана в 200 мл эфира обрабатывают при -70° С 0,23 моль н-BuLi. По окончании добавления смесь перемешивают еще 30 мин при той же температуре. Образуется белый осадок дилитиевой соли. Затем быстро при -70° С добавляют 0,265 моль CuCl2. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 12 часов. Полученную суспензию выливают в воду, органический слой отделяют и концентрируют. Остаток перекристаллизовывают из эфира. Выход 25%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): 6,86 (м, 2Н, СН); 3,74 (с, 2Н, CH2); 2,98 (кв, 4Н, СН2); 1,38 (т, 6Н, СН3).
ПРИМЕР 1
Синтез 2,2-(3-метилциклопентадиенил)-7-(2,5-диметилциклопента-[1,2-b:4,3-b’]дитиофен)пропана
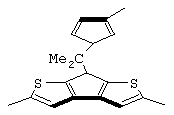
При -70° С добавляют 3,13 мл 1,6 М раствора н-BuLi (5 ммоль) к раствору 1,03 г (5 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 20 мл эфира. Реакционную смесь перемешивают еще 30 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают 0,6 г (5 ммоль) 3,6,6-триметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу выделяют, сушат MgSO4 и концентрируют. Остаток перекристаллизовывают из гексана. Выход 1,0 г (62%). Названное соединение характеризуют с помощью спектроскопии 1Н ЯМР.
Синтез изопропилиден{(3-метилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-1
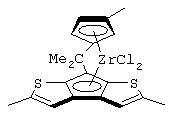
При -70° С добавляют 2,3 мл 1,6 М раствора н-BuLi (3,7 ммоль) к суспензии 0,6 г (1,85 ммоль) 2,2-(3-метилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)пропана в 20 мл эфира. Смеси дают нагреться до 0° С и затем обрабатывают 0,43 г (1,85 ммоль) ZrCl4. Реакционную смесь кипятят с обратным холодильником при перемешивании 3 час, затем отфильтровывают желтый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из СН2Сl2. Выход 0,72 г (80%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 2
Синтез изопропилиден{(3-метилциклопентадиенил)-7-(2,5-диметил-циклопента[1,2-b:4,3-b’]дитиофен)}гафнийдихлорида СН-1
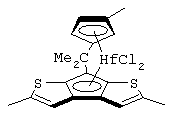
При -70° С добавляют 2,5 мл 1,6 М раствора н-BuLi (4,0 ммоль) к суспензии 0,65 г (2,0 ммоль) 2,2-(3-метилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)пропана в 20 мл эфира. Смеси дают нагреться до 0° С и затем обрабатывают 0,64 г (2,0 ммоль) HfCl4. Реакционную смесь кипятят с обратным холодильником при перемешивании 3 час, затем отфильтровывают желтый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 0,48 г (42%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 3
Синтез 2,2-(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопента [1,2-b:4,3-b’]дитиофен)пропана
Используют ту же методику, которая описана для синтеза 2,2-(3-метилциклопентадиенил)-7-(2,5-диметилциклопента-[1,2-b:4,3-b’]дитиофен)пропана (см. ниже).
Синтез изопропилиден{(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-12
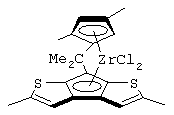
При -70° С добавляют 3,13 мл 1,6 М раствора н-BuLi (5 ммоль) к раствору 1,03 г (5,0 ммоль) 2,5-диметил-7Н-циклопента[1,2-b:4,3-b’]дитиофена в 20 мл эфира. Реакционную смесь перемешивают еще 30 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают 0,67 г (5,0 ммоль) 1,3,6,6-тетраметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 8 часов. Затем реакционную смесь охлаждают до -30° С и добавляют 3,13 мл 1,6 М раствора н-BuLi (5 ммоль). Смеси дают нагреться до 0° С и обрабатывают 1,16 г (5,0 ммоль) ZrCl4. Реакционную смесь кипятят с обратным холодильником при перемешивании 3 час и затем при комнатной температуре добавляют 10 мл CH2Cl2. Раствор выделяют, концентрируют, остаток перекристаллизовывают из смеси CH2Cl2/гексан. Выход 0,58 г (23% из расчета на 2,5-диметил-7Н-циклопента-[1,2-b:3,4-b’]дитиофен).
ПРИМЕР 4
Синтез 2,2-(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)пропана
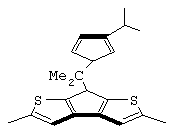
При -70° С добавляют 3,13 мл 1,6 М раствора н-BuLi (5 ммоль) к раствору 1,03 г (5 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 20 мл эфира. Реакционную смесь перемешивают еще 30 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают 0,74 г (5 ммоль) 3-изопропил-6,6-диметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу выделяют, сушат MgSO4 и концентрируют. Остаток перекристаллизовывают из гексана. Выход 0,85 г (48%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез изопропилиден{(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-2
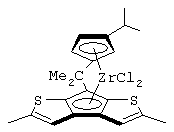
При -70° С добавляют 3,75 мл 1,6 М раствора н-BuLi (6,0 ммоль) к суспензии 1,06 г (3,0 ммоль) 2,2-(3-изопропилциклопентадиенил) -7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)-пропана в 20 мл эфира. Смеси дают нагреться до 0° С и затем обрабатывают 0,7 г (3,0 ммоль) ZrCl4. Реакционную смесь кипятят с обратным холодильником при перемешивании в течение 3 часов, затем отфильтровывают желтый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,24 г (80%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 5
Синтез 2,2-(3-изопропилциклопентадиенил)-7-(2,5-дитриметилсилилциклопента[1,2-b:4,3-b’]дитиофен)пропана
Используют ту же методику, которая описана в примере 4 для синтеза 2,2-(3-изопропилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)пропана, за исключением того, что используют 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофен (или 2,5-диметилтриметилсилил-7Н-тиено-[3’,2’:3,4]-циклопента[b]тиофен) (см. ниже).
Синтез изопропилиден{(3-изопропилциклопентадиенил)-7-(2,5-дитриметилсилилциклопента[1,2-b:4,3-b’]дитиофен)}
цирконийдихлорида С-7
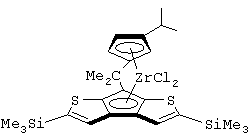
При -70° С добавляют 3,05 мл 1,6 М раствора н-BuLi (4,9 ммоль) к раствору 1,57 г (4,86 ммоль) 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофена в 20 мл эфира. Полученную смесь перемешивают еще 30 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают 0,72 г (4,9 ммоль) 3-изопропил-6,6-диметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 4 часа. Затем ее охлаждают до -30° С и добавляют 3,05 мл 1,6 М раствора н-BuLi (4,9 ммоль). Смеси дают нагреться до 0° С и обрабатывают 1,14 г (4,9 ммоль) ZrCl4. Полученную реакционную массу кипятят с обратным холодильником при перемешивании 3 час, затем раствор выделяют и концентрируют. Остаток перекристаллизовывают из пентана. Выход 0,23 г (7,4% из расчета на 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофен). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 6
Синтез 2,2-(3-изопропилциклопентадиенил)-4-(2,6-диметилциклопента[2,1-b:3,4-b’]дитиофен)пропана
Осуществляют ту же методику, которая описана в примере 4, за исключением того, что используют 2,6-диметил-4Н-циклопента-[2,1-b:3,4-b’]дитиофен (см. ниже).
Синтез изопропилиден{(3-изопропилциклопентадиенил)-4-(2,6-диметилциклопента[2,1-b:3,4-b’]дитиофен)}цирконийдихлорида С-8
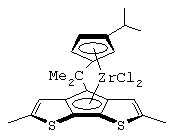
При -70° С добавляют 3,13 мл 1,6 М раствора н-BuLi (5,0 ммоль) к раствору 1,03 г (5,0 ммоль) 2,6-диметил-4Н-циклопента[2,1-b:3,4-b’]дитиофена в 20 мл эфира. Полученную смесь перемешивают еще 30 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают 0,74 г (5,0 ммоль) 3-изопропил-6,6-диметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 4 часа. Затем ее охлаждают до -30° С и добавляют 3,13 мл 1,6 М раствора н-BuLi (5,0 ммоль). Реакционной смеси дают нагреться до 0° С и обрабатывают 1,16 г (5,0 ммоль) ZrCl4. Полученную реакционную смесь кипятят с обратным холодильником при перемешивании 3 час и потом при комнатной температуре добавляют 30 мл CH2Cl2. Затем раствор выделяют и концентрируют. Остаток перекристаллизовывают из смеси CH2Cl2/гексан. Выход 0,87 г (34% из расчета на 2,6-диметил-4Н-циклопента[2,1-b:3,4-b’]дитиофен). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 7
Синтез 2,2-(3-трет-бутилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)пропана
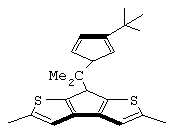
При -70° С добавляют 3,13 мл 1,6 М раствора н-BuLi (5 ммоль) к раствору 1,03 г (5 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 20 мл эфира. Полученную смесь перемешивают еще 30 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают 0,81 г (5 ммоль) 3-трет-бутил-6,6-диметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу выделяют, сушат над МgSO4 и концентрируют. Остаток перекристаллизовывают из гексана. Выход 0,94 г (51%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез изопропилиден{(3-трет-бутилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-3
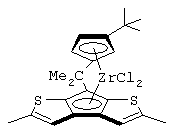
При -70° С добавляют 3,75 мл 1,6 М раствора н-BuLi (6,0 ммоль) к суспензии 1,11 г (3,0 ммоль) 2,2-(3-трет-бутилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)-пропана в 20 мл эфира. Смеси дают нагреться до 0° С и затем обрабатывают 0,7 г (3,0 ммоль) ZrCl4. Реакционную смесь кипятят с обратным холодильником при перемешивании 3 час, затем отфильтровывают желтый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,27 г (80%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 8
Синтез 2,2-(3-изопропилциклопентадиенил)-7-(циклопента-[1,2-b:4,3-b’]дитиофен)пропана
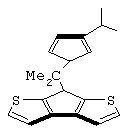
При -70° С добавляют 3,13 мл 1,6 М раствора н-BuLi (5 ммоль) к раствору 0,89 г (5 ммоль) 7Н-циклопента[1,2-b:4,3-b’]дитиофена в 20 мл ТГФ. Полученную смесь перемешивают еще 30 мин при 0° С, затем охлаждают снова до -70° С и обрабатывают 0,74 г (5 ммоль) 3-изопропил-6,6-диметилфульвена в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу выделяют, сушат с помощью МgSO4 и концентрируют. Остаток пропускают через колонку, наполненную силикагелем, с использованием в качестве элюента гексана (Rf=0,8). Выход 1,05 г (64%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез изопропилиден{(3-изопропилциклопентадиенил)-7-(циклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-16
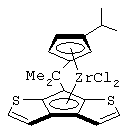
Раствор 1,05 г (3,22 ммоль) 2,2-(3-изопропилциклопентадиенил)-7-(циклопента[1,2-b:4,3-b’]дитиофен)пропана в смеси 10 мл эфира и 60 мл гексана обрабатывают при -70° С 4,1 мл (6,6 ммоль) 1,6 М раствора н-BuLi. Смеси дают нагреться до 0° С и обрабатывают 0,75 г (3,2 ммоль) ZrCl4. Полученную смесь кипятят с обратным холодильником при перемешивании 3 час, затем отфильтровывают желтый осадок, промывают дважды гексаном, сушат и, наконец, перекристаллизовывают из смеси CH2Cl2/гексан. Выход 0,32 г (21%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 9
Синтез изопропилиден{(циклопентадиенил)-7-(циклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-0
Синтез осуществляют в соответствии с описанием примера 6 публикации WO 98/22486.
ПРИМЕР 10
Синтез хлор(1-инденил)диметилсилана
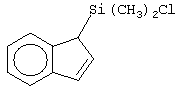
К раствору индена (чистота 90%, 11 г, 85,23 ммоль) в 60 мл Et2O, предварительно охлажденному до -78° С, добавляют по каплям 37,5 мл 2,5 М раствора н-BuLi в гексане (93,75 ммоль, н-ВuLi:инден 1,1:1). По окончании добавления желтой суспензии дают нагреться до комнатной температуры и перемешивают 4 час, получают оранжевый раствор. Растворители упаривают при пониженном давлении, получают желтое твердое вещество, которое поглощают в 75 мл гексана; молочную суспензию перемешивают в течение нескольких минут, отфильтровывают литиевую соль индена (белый осадок) и промывают гексаном (3× 20 мл). Твердое вещество снова суспендируют в гексане (40 мл) и добавляют при перемешивании к раствору Ме2SiСl2 (15,5 мл, 127,84 ммоль, Me2SiCl2:IndLi 5:1) в 50 мл гексана, предварительно охлажденному до -78° С. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Суспензию затем фильтруют, и фильтрат упаривают досуха в вакууме, получают бледно-желтое масло (16,5 г) (1-Ind) SiMe2Cl без его винильного изомера (выход 89%).
Спектр 1H ЯМР (СDСl3, δ , м.д.): 0,21 (с, 3Н, Si-СН3); 0,26 (с, 3Н, Si-СН3); 3,77 (уш.с, 1Н, Ср-Н); 6,68 (дд, 1Н, Ср-Н); 7,03 (дд, 1Н, Ср-Н); 7,19-7,36 (м, 2Н, Аr); 7,48-7,52 (м, 1Н, Аr); 7,57-7,61 (м, 1Н, Аr).
Синтез (1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
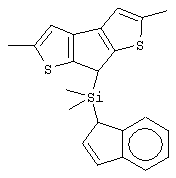
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (4,80 мл, 12,00 ммоль) к суспензии 2,25 г 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена (Мм=206,32, 10,90 ммоль, н-BuLi:MeTh2Cp 1,1:1) в 50 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С с образованием в конечном итоге темно-коричневой суспензии. Эту суспензию снова охлаждают до -20° С и добавляют раствор 2,20 г хлор(1-инденил)диметилсилана (Мм=208,76, 10,54 ммоль, IndSiMe2Cl:MeTh2Cp 1:1) в 10 мл эфира. Реакционной смеси затем дают нагреться до комнатной температуры и перемешивают 2 часа. Полученную темную (почти черную) суспензию концентрируют в вакууме, и остаток экстрагируют 50 мл толуола. Экстракт сушат в вакууме, получают 4,06 г коричневого продукта, который характеризуют с помощью спектроскопии 1H ЯМР. Спектр 1H ЯМР показывает присутствие желаемого лиганда (78,5 мас.%) вместе с 15,1 мас%. исходного IndSiMe2Cl и 6,4 мас.% толуола. Лиганд используют в том виде, как он есть, на следующей стадии без дополнительной очистки.
Выход чистого продукта составляет 79,9%.
Спектр 1H ЯМР (СDСl3, δ , м.д.): -0,39 (с, 3Н, Si-СН3); -0,20 (с, 3Н, Si-СН3); 2,57 (с, 6Н, СН3); 3,82 (т, 1Н, J=1,85 Гц, СН); 3,89 (с, 1Н, СН); 6,45 (дд, 1Н, J=5,33 Гц, J=1,85 Гц, СН); 6,77-7,52 (м, 7Н, Ar).
Синтез диметилсилил{(1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-10
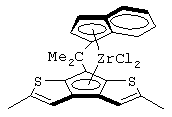
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (9,00 мл, 22,50 ммоль) к раствору 4,06 г (1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана (Мм=378,64, 10,72 ммоль, н-BuLi:лиганд 2:1, исходя из лиганда чистотой 100%) в 50 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С с образованием темно-коричневой суспензии. Полученную суспензию снова охлаждают до -20° С и добавляют суспензию 2,5 г ZrCl4 (Мм=233,03, 10,72 ммоль, ZrCl4:лиганд 1:1, исходя из лиганда чистотой 100%) в 50 мл пентана, предварительно охлажденную до -20° С. Реакционную смесь выдерживают при -20° С 20 мин, затем дают ей нагреться до комнатной температуры и перемешивают в течение 2 часов. Полученную оранжево-коричневую суспензию упаривают в вакууме, и остаток экстрагируют 50 мл толуола. Экстракт выбрасывают, а нерастворимый в толуоле остаток промывают эфиром, получают оранжевый порошок (4,10 г), который, как показывают данные спектра 1Н ЯМР, является целевым катализатором. Аликвоту полученного порошка (1,5 г) очень быстро промывают EtOH (10 мл) и затем Et2O. После сушки получают 0,9 г чистого катализатора в виде оранжевого порошка. Выход сырого продукта (с LiCl) 71,0%.
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 1,01 (с, 3Н, Si-СН3); 1,29 (с, 3Н, Si-СН3); 2,46 (д, 3Н, J=1,17 Гц, СН3); 2,58 (д, 3Н, J=1,17 Гц, СН3); 6,07 (д, 1Н, J=3,28 Гц, СН); 6,70 (кв, 1Н, J=1,17 Гц, СН); 6,85 (кв, 1Н, J=1,17 Гц, СН); 6,90-7,64 (м, 5Н, Ar).
ПРИМЕР 11
Синтез хлор(2-метил-1-инденил)диметилсилана
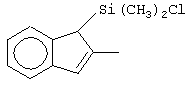
Добавляют по каплям 2,5 М раствор н-BuLi в гексане (22,1 мл, 55,25 ммоль, н-BuLi:2-Ме-инден 1,1:1) к раствору 6,54 г 2-метилиндена (Boulder Scientific Company 419-0128, Мм=130,19, 50,23 ммоль) в 70 мл Et2O, предварительно охлажденного до -20° С. По окончании добавления смесь выдерживают при -20° С 15 мин, затем смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Растворители упаривают при пониженном давлении, получают светло-оранжевый твердый продукт, который поглощают в 50 мл гексана; суспензию перемешивают 10 мин при комнатной температуре и фильтруют. Литиевую соль 2-метилиндена промывают на фильтре гексаном (2× 10 мл) и сушат. Твердый остаток снова суспендируют в 70 мл гексана и добавляют при перемешивании к раствору Me2SiCl2 (9,1 мл, d=1,064, Мм=129,06, 75,02 ммоль, Me2SiCl2:2-Me-IndLi 1,5:1) в 60 мл гексана, предварительно охлажденного до -20° С. По окончании добавления бледно-оранжевую суспензию выдерживают при -20° С в течение 15 мин, затем дают нагреться до комнатной температуры и перемешивают в течение ночи. Конечную бело-бледно-желтую суспензию фильтруют, фильтрат сушат досуха в вакууме при 40° С, получают продукт в виде желто-оранжевого масла (8,40 г). Выход 75,1%. Чистота 89,1%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 0,22 (с, 3Н, Si-СН3); 0,47 (с, 3Н, Si-СН3); 2,36 (м, 3Н, СН3); 3,65 (уш.с, 1Н, СН); 6,70 (м, 1Н, Ср-Н); 7,18-7,56 (м, 4Н, Ar).
Присутствует приблизительно 6% (ГХ-МС) бис(2-метил-1-инденил)диметилсилана (рац/мезо=1,3:1).
m/z (%): 224 (28) [M++2], 222 (74) [М+], 129 (20), 128 (67), 127 (17), 95 (35), 93 (100).
Альтернативный способ без выделения соли 2-Me-1-Ind-Li+
Добавляют по каплям 2,5 М раствор н-BuLi в гексане (23,6 мл, 59,00 ммоль, н-ВuLi:2-Ме-инден 1:1) к раствору 7,87 г 2-метилиндена (Boulder Scientific Company 419-0128, Мм=130,19, 97,6%, 59,00 ммоль) в 50 мл Et2O, предварительно охлажденного до 0° С. По окончании добавления смесь выдерживают при 0° С 15 мин, затем дают нагреться до комнатной температуры и перемешивают 2 час до образования бледно-желтой суспензии. Суспензию снова охлаждают до 0° С и добавляют по каплям Me2SiCl2 (7,86 мл, d=1,064, Мм=129,06, 64,80 ммоль, Me2SiCl2:2-Me-IndLi 1,1:1). По окончании добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную бело-бледно-желтую суспензию концентрируют в вакууме, и остаток экстрагируют 30 мл толуола. Экстракт сушат досуха в вакууме при 40° С, получают продукт в виде оранжевого масла (10,41 г). Выход 79,2%. Чистота 83,6%. Также присутствуют следы исходного 2-метилиндена и 9,8% бис(2-метил-1-инденил)диметилсилана (данные ГХ-МС).
m/z для бис(2-метил-1-инденил)диметилсилана (%): 316 (21) [М+], 187 (100), 159 (24), 128 (18), 59 (57).
Синтез (2-метил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
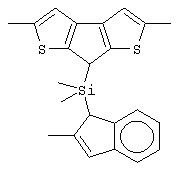
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (4,15 мл, 10,37 ммоль) к раствору 2,13 г 2,5-диметил-7Н-циклопента[1,2-b:4,3-b’]дитиофена (Мм=206,32, 87,9% (ГХ-МС), 9,07 ммоль, н-BuLi:MeTh2Cp 1,1:1) в 20 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С с образованием в конечном итоге темно-коричневого раствора. Полученный раствор снова охлаждают до -20° С и добавляют раствор 2,03 г хлор(2-метил-1-инденил)-диметилсилана (9,10 ммоль, (2-Me-l-Ind) SiMe2Cl:MeTh2Cp 1:1) в 3 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 2 часа. Полученный темный раствор (почти черный) концентрируют в вакууме, и липкий остаток экстрагируют 50 мл толуола. Экстракт сушат в вакууме, получают 3,93 г коричневого липкого продукта, который характеризуют с помощью ГХ-МС и спектроскопии 1H ЯМР. Данные 1H ЯМР показывают присутствие желаемого лиганда вместе с 10 мас.% толуола.
Чистота (данные ГХ-МС) 90,4%. Выход чистого продукта 89,9%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): -0,37 (с, 6Н, Si-СН3); 2,26 (д, 3Н, J=0,8 Гц, СН3); 2,56 (дд, 3Н, J=1,1 Гц, J=0,6 Гц, СН3); 2,58 (дд, 3Н, J=1,1 Гц, J=0, 6 Гц, СН3); 3,88 (уш.с, 1Н, СН); 4,04 (с, 1Н, СН); 6,65-6,66 (м, 1Н, СН); 6,87 (кв, 1H, J=1,1 Гц, СН), 6,89 (кв, 1H, J=1,1 Гц, СН), 7,10-7,50 (м, 4Н, Ar).
m/z (%): 393 (13) [M++1], 392 (40) [М+], 263 (100), 235 (18), 187 (44), 159 (15), 59 (13).
Синтез диметилсилил{(2-метил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-20
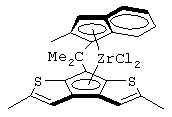
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (7,20 мл, 18,00 ммоль) к раствору 3,93 г (2-метил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана (Мм=392,66, 90,4% (ГХ-МС), 8,15 ммоль, н-BuLi:лиганд 2:1, исходя из лиганда чистотой 90,4%) в 30 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С и 30 мин при комнатной температуре с образованием темно-коричневой суспензии. Полученную суспензию снова охлаждают до -20° С и добавляют суспензию 1,91 г ZrCl4 (Мм=233,03, 8,20 ммоль, ZrCl4:лиганд 1:1, исходя из лиганда чистотой 90,4%) в 50 мл пентана, предварительно охлажденную до -20° С. Реакционную смесь выдерживают при -20° С 1 час, затем дают ей нагреться до комнатной температуры и перемешивают в течение ночи. Полученную оранжевую бледно-коричневую суспензию упаривают в вакууме, и остаток промывают эфиром, получают оранжевый порошок (5,32 г), который анализируют с помощью спектроскопии 1H ЯМР в CD2Cl2. Данные 1H ЯМР показывают присутствие желаемого катализатора вместе с не идентифицированным аддуктом координации (вероятно ZrCl4(Et2O)2 или LiCl (Et2O)). Порошок очень быстро промывают 15 мл 4 н. НСl, затем водой (30 мл), затем EtOH (20 мл) и, наконец, Et2O. После сушки выделяют 3,50 г чистого катализатора в виде оранжевого порошка. Выход чистого продукта 77,7%.
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 1,20 (с, 3Н, Si-СН3); 1,35 (с, 3Н, Si-СН3); 2,39 (д, 3Н, J=0,59 Гц, СН3); 2,45 (д, 3Н, J=1,2 Гц, СН3); 2,62 (д, 3Н, J=1,2 Гц, СН3); 6,66 (кв, 1Н, J=1,2 Гц, СН); 6,81 (уш.с, 1Н, СН); 6,87 (ддд, 1Н, J=0,98 Гц, J=6,65 Гц, J=9,0 Гц, СН); 7,21 (ддд, 1Н, J=0,98 Гц, J=6,65 Гц, J=8,61 Гц, СН); 7,45 (дт, 1Н, J=0,98 Гц, J=8,61 Гц, СН); 7,73 (дкв, 1Н, J=0,98 Гц, J=9,0 Гц, СН).
ПРИМЕР 12
Синтез хлор-(2-метил-4-фенил-1-инденил)диметилсилана
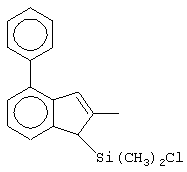
Добавляют по каплям при 0° С 2,5 М раствор н-BuLi в гексане (4,85 мл, 12,12 ммоль) к раствору 2,50 г 2-метил-4-фенилиндена (Boulder Scientific Company, Мм=206,29, 12,12 ммоль, H-BuLi:2-Me-4-Ph-Ind 1:1) в 30 мл эфира. Полученную смесь перемешивают еще 2 часа при комнатной температуре с образованием оранжевого раствора. Полученный раствор снова охлаждают до 0° С и медленно добавляют к раствору 1,58 мл дихлордиметилсилана (Aldrich, Мм=129,06, d=1,064, 13,03 ммоль, Me2SiCl2:2-Me-4-Ph-Ind 1,08:1) в 20 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 1 часа. Конечную соломенно-желтую суспензию концентрируют в вакууме, остаток экстрагируют 50 мл толуола. Экстракт сушат в вакууме, получают 3,36 г соломенно-желтого твердого вещества, которое характеризуют с помощью ГХ-МС и спектров 1Н ЯМР. Выход 92,8%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 0,24 (с, 3Н, Si-СН3); 0,48 (с, 3Н, Si-СН3); 2,31 (д, 3Н, J=0,78 Гц, СН3); 3,70 (уш. с, 1Н, СН); 6,85 (м, 1Н, J=0,78 Гц, СН); 7,19-7,59 (м, 8Н, Ar).
m/z (%): 300 (26) [M++2], 299 (18) [M++1], 298 (72) [М+], 205 (23), 204 (45), 203 (28), 202 (32), 189 (15), 165 (13), 95 (35), 93 (100).
Синтез (2-метил-4-фенил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
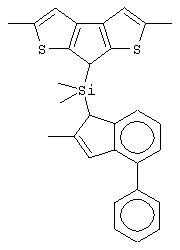
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (2,72 мл, 6,80 ммоль) к раствору 1,40 г 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена (Мм=206,32, 90,7%, 6,15 ммоль, н-BuLi:MeTh2Cp 1,1:1) в 30 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С с образованием в итоге темно-коричневой суспензии. Полученную суспензию снова охлаждают до -20° С, и к ней медленно добавляют раствор 1,90 г хлор(2-метил-4-фенил-1-инденил)диметилсилана (Мм=298,89, 6,37 ммоль, (2-Me-4-Ph-l-Ind)SiMe2Cl:MeTh2Cp 1,04:1) в 20 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 2 часа. Полученный темный (почти черный) раствор концентрируют в вакууме, остаток экстрагируют 50 мл толуола, получают маслянистый продукт, обрабатывают при 30° С при перемешивании 30 мл пентана. После перемешивания в течение 15 мин образуется порошкообразный твердый продукт, который отфильтровывают. После сушки в вакууме выделяют 2,03 г коричневого продукта.
Чистота (ГХ-МС) 83,8%. Выход чистого продукта 59,0%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): -0,35 (с, 3Н, Si-СН3); -0,32 (с, 3Н, Si-СН3); 2,23 (д, 3Н, J=0,78 Гц, СН3); 2,55 (уш.с, 3Н, СН3); 2,58 (уш.с, 3Н, СН3); 3,96 (с, 1Н, СН); 4,04 (с, 1Н, СН); 6,82 (кв, 1Н, J=0,78 Гц, СН); 6,86 (кв, 1H, J=1,17 Гц, СН); 6,88 (кв, 1H, J=1,17 Гц, СН); 7,13-7,59 (м, 8Н, Ar).
m/z (%): 469 (10) [M++1], 468 (24) [М+], 264 (28), 263 (100), 248 (14), 247 (21), 235 (20), 205 (13), 203 (16), 190 (10), 59 (14).
Синтез диметилсилил{(2-метил-4-фенил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-28
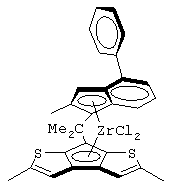
Раствор 2,58 г (5,5 ммоль) (2-метил-4-фенил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана в 40 мл эфира обрабатывают при -70° С 70 мл 1,6 М раствора н-BuLi (11,2 ммоль). Реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении, и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,28 г (5,5 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в продолжение ночи, отфильтровывают желтый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,65 г (48%). Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 13
Синтез (2-метил-1-инденил)-7-(циклопента-[1,2-b:4,3-b’]-дитиофен)диметилсилана
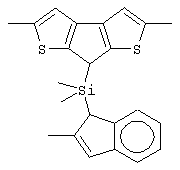
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (1,50 мл, 3,75 ммоль) к раствору 1,29 г 7Н-циклопента[1,2-b:4,3-b’]дитиофена (Мм=178,28, чистота, оцененная с помощью 1H ЯМР, составляет ~50 мас.%, 3,62 ммоль, н-BuLi:Th2Cp 1,04:1) в 20 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С с образованием темно-коричневой суспензии. Полученную суспензию снова охлаждают до -20° С и добавляют раствор 0,96 г хлор(2-метил-1-инденил)диметилсилана (83,6% (ГХ-МС), Мм=222,79, 3,62 ммоль, (2-Me-1-Ind)SiMe2Cl:Th2Cp 1:1) в 5 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и перемешивают 2 часа. Полученную черную суспензию концентрируют в вакууме, и липкий остаток экстрагируют 30 мл толуола для удаления образовавшегося LiCl. Экстракт сушат в вакууме, получают 2,26 г черного масла, которое анализируют с помощью спектроскопии 1Н ЯМР. В качестве побочных продуктов присутствовали также исходный хлор(2-метил-1-инденил)-диметилсилан, гексаэтилсилоксан, поступающий с предыдущих стадий, и смолы, но попытки очистить целевой лиганд были неудачны из-за высокой растворимости смеси в неполярном растворителе, таком как пентан. Сырой продукт затем используют в том виде, как он есть, на следующей стадии без дополнительной очистки.
Спектр 1H ЯМР (CDCl3, δ , м.д.): -0,36 (с, 3Н, Si-СН3); -0,35 (с, 3Н, Si-СН3); 2,26 (д, 3Н, J=0,98 Гц, СН3); 3,89 (с, 1Н, СН); 4,15 (с, 1Н, СН); 6,69-7,52 (м, 9Н, Ar).
Синтез диметилсилил{(2-метил-1-инденил)-7-(циклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-36
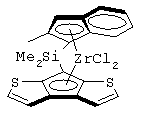
При -20° С добавляют 2,5 М раствор н-BuLi в гексане (5,00 мл, 12,50 ммоль) к раствору 2,26 г (2-метил-1-инденил)-7-(циклопента[1,2-b:4,3-b’]дитиофен)диметилсилана (Мм=364,61, 6,20 ммоль, н-BuLi:лиганд 2,02:1, исходя из лиганда чистотой 100%) в 20 мл эфира. Полученную смесь перемешивают еще 1 час при 0° С с образованием в итоге коричневой суспензии. Эту суспензию снова охлаждают до -20° С и добавляют суспензию 1,44 г ZrCl4 (Мм=233,03, 6,20 ммоль, ZrCl4:лиганд 1:1, исходя из лиганда чистотой 100%) в 30 мл пентана, предварительно охлажденного до -20° С. Реакционную смесь выдерживают при -20° С 1 час, затем дают медленно нагреться до комнатной температуры и перемешивают в течение 3 часов. Полученную коричневую суспензию упаривают в вакууме, остаток экстрагируют 30 мл толуола: экстракт, содержащий преимущественно смолы, отбрасывают, тогда как коричневый остаток (3,33 г) сушат и промывают 20 мл эфира. Данные спектроскопии 1H ЯМР в CD2Cl2 для остатка из эфира показывают присутствие желаемого катализатора вместе с не идентифицированным аддуктом координации (вероятно ZrCl4 (Et2O)2 или LiCl(Et2O)) и небольшим количеством смол. Коричневый порошок (2,28 г) снова очень быстро промывают 20 мл CH2Cl2, затем EtOH (10 мл) и, наконец, Et2O (15 мл). После сушки выделяют 0,44 г катализатора в виде светло-коричневого порошка. Выход 13,5%.
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 1,25 (с, 3Н, Si-СН3); 1,41 (с, 3Н, Si-СН3); 2,38 (уш.с, 3Н, СН3); 6,82-7,79 (м, 9Н, Ar).
ПРИМЕР 14
Синтез хлордиметил-(2-фенил-1-инденил)силана
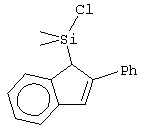
Раствор 0,96 г (5,0 ммоль) 2-фенилиндена в 30 мл Et2O обрабатывают при -70° С 3,13 мл (5,0 ммоль) 1,6 М раствора н-BuLi. После добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают 50 мин. Затем снова охлаждают до -70° С и обрабатывают раствором 0,65 г (5,0 ммоль) Me2SiCl2 в 10 мл эфира. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную смесь фильтруют для удаления LiCl, и растворитель удаляют при пониженном давлении. Сырой продукт используют на следующей стадии без дополнительной очистки.
Спектр 1H ЯМР (C6D6, δ , м.д.): 7,90-7,10 (м, 9Н, СН); 6,95 (с, 1Н, СН); 4,15 (с, 1Н, СН); -0,02 (с, 3Н, Si-СН3); -0,20 (с, 3Н, Si-СН3).
Синтез диметилсилил{(2-фенил-1-инденил)-7-(2,5-диметилциклопента [1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-31
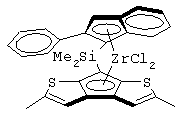
Суспензию 1,03 г (5,0 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 30 мл эфира обрабатывают при -70° С 3,13 мл 1,6 М раствора н-BuLi (5,0 ммоль). После добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают при этой температуре еще в течение 50 мин. Затем смесь снова охлаждают до -70° С и добавляют эфирный раствор (10 мл) хлордиметил-(2-фенил-1-инденил)силана с предыдущей стадии. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Лиганд (2-фенил-1-инденил)-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}диметилсилан характеризуют с помощью спектроскопии 1H ЯМР.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,70-7,25 (м, 9Н, СН); 7,20 (с, 1Н, СН); 6,90 (м, 2Н, СН); 4,60 (с, 1Н, СН); 3,70 (с, 1Н, СН); 2,65 (с, 3Н, СН3); 2,60 (с, 3Н, СН3); -0,44 (с, 3Н, Si-СН3); -0,66 (с, 3Н, Si-СН3).
Лиганд не выделяют: его раствор обрабатывают при -70° С 7,0 мл 1,6 М раствора н-BuLi (11,2 ммоль). Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,28 г (5,5 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, отфильтровывают красный осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,64 г (53% из расчета на Me2Th).
ПРИМЕР 15
Синтез (2-метил-1-инденил)-7-(2,5-диэтилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
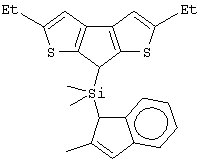
Суспензию 1,17 г (5,0 ммоль) 2,5-диэтил-7Н-циклопента-[1,2-b:4,3-b’] дитиофена в 75 мл эфира обрабатывают при -70° С 3,13 мл (5,0 ммоль) 1,6 М раствора н-BuLi. После добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час при этой температуре. Затем смесь снова охлаждают до -70° С и добавляют раствор 1,11 г (5,0 ммоль) хлор(2-метил-1-инденил)диметилсилана в 10 мл эфира. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Лиганд (2-метил-1-инденил)-7-(2,5-диэтилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилан не выделяют, а используют в растворе для синтеза катализатора (см. ниже).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,55 (д, 1Н, СН); 7,44 (д, 1Н, СН); 7,28 (м, 1Н, СН); 7,15 (м, 1Н, СН); 6,98 (м, 1Н, СН); 6,96 (м, 1Н, СН); 6,70 (м, 1Н, СН); 4,10 (с, 1Н, СН); 3,94 (с, 1Н, СН); 2,98 (м, 4Н, СН2); 2,31 (с, 3Н, СН3); 1,43 (т, 3Н, СН3); 1,41 (т, 3Н, СН3), -0,30 (с, 3Н, Si-СН3); -0,31 (с, 3Н, Si-СН3).
Синтез диметилсилил{(2-метил-1-инденил)-7-(2,5-диэтилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-34
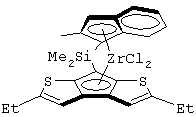
Раствор лиганда с предыдущей стадии обрабатывают при -70° С 7,0 мл (11,2 ммоль) 1,6 М раствора H-BuLi. Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении, и полученную таким образом дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 0,75 г (3,2 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, отфильтровывают желтовато-красный осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,52 г (52% относительно Et2Th).
ПРИМЕР 16
Синтез (2-метил-1-инденил)-7-(2,5-дифенилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
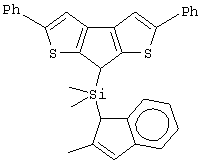
Раствор 1,32 г (4,0 ммоль) 2,5-дифенил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 30 мл эфира обрабатывают при -70° С 2,50 мл (4,0 ммоль) 1,6 М раствора н-BuLi. После добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают еще 50 мин при этой температуре. Затем смесь снова охлаждают до -70° С и добавляют раствор 0,90 г (4,0 ммоль) хлордиметил(2-метил-1-инденил)силана в 10 мл эфира. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Лиганд (2-метил-1-инденил)-7-(2,5-дифенилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилан не выделяют, а используют в растворе для синтеза катализатора.
Синтез диметилсилил{(2-метил-1-инденил)-7-(2,5-дифенилциклопента [1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-35
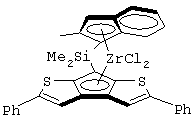
Раствор лиганда с предыдущей стадии обрабатывают при -70° С 5,6 мл (9,0 ммоль) 1,6 М раствора н-BuLi. Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают еще 1 час. После охлаждения до -70° С добавляют 1,05 г (4,5 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, отфильтровывают фиолетовый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,27 г (47% относительно Ph2Th).
ПРИМЕР 17
Синтез 3,6,6-триметилфульвена
Раствор 2-метил-1,3-циклопентадиена (125 г, 1,56 моль) в 1,2 л этанола обрабатывают при низкой температуре 126 мл (1,72 моль) ацетона и 142 мл (1,72 моль) пирролидина. Полученный раствор выдерживают при температуре ниже комнатной в течение ночи. Затем реакционную смесь нейтрализуют 10% водным раствором Н3РO4, экстрагируют гексаном (3× 150 мл) и промывают водой до нейтрального значения рН. Органическую фазу отделяют, сушат МgSO4 и концентрируют. Остаток перегоняют при 70° С/60 мм рт.ст. Выход 112,6 г (60%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 6,53 (дд, 1Н, СН); 6,35 (дд, 1Н, СН); 6,20 (м, 1Н, СН); 2,17 (с, 3Н, СН3); 2,16 (с, 3Н, СН3); 2,09 (с, 3Н, СН3).
Синтез 3-изопропил-1-метил-1,3-циклопентадиена
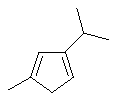
Раствор 24 г (0,2 моль) 3,6,6-триметилфульвена в 100 мл эфира добавляют при -78° С и в атмосфере азота к раствору 7,59 г (0,2 моль) алюмогидрида лития в 200 мл эфира. Реакционной смеси дают нагреться до комнатной температуры, перемешивают 2 часа и затем обрабатывают 10% водным раствором NH4Cl. Органическую фазу собирают, промывают водой, сушат с помощью MgSO4 и концентрируют. Остаток перегоняют при 63° С/50 мм рт.ст. Выход 15,88 г (65%). Названное целевое соединение характеризуют с помощью 1H ЯМР.
Синтез 1-метил-3-изопропил-6,6-диметилфульвена
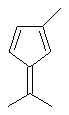
К суспензии 12,8 г (0,32 моль) гидроксида натрия в 200 мл сухого ТГФ при низкой температуре добавляют 3-изопропил-1-метил-1,3-циклопентадиен (39 г, 0,32 моль). После перемешивания в течение 30 мин реакционную смесь обрабатывают 23,8 мл (0,32 моль) ацетона. Полученный раствор выдерживают при температуре ниже комнатной в течение ночи. Затем полученную смесь нейтрализуют 10% водным раствором Н3РO4, экстрагируют гексаном (3× 100 мл) и промывают водой до нейтрального значения рН. Органическую фазу отделяют, сушат с помощью МgSO4 и концентрируют. Остаток перегоняют при 80° С/10 мм рт.ст. Выход 25, 96 г (50%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 6,21 (м, 1Н, СН); 6,05 (д, 1Н, СН); 2,67 (м, 1Н, СН); 2,24 (с, 3Н, СН3); 2,21 (с, 3Н, СН3); 2,20 (с, 3Н, СН3); 1,26 (с, 3Н, СН3); 1,28 (с, 3Н, СН3).
Синтез 2,2-(2-метил-4-изопропилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)пропана
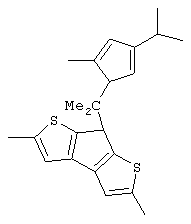
При -70° С добавляют 1,6 М раствор н-BuLi (6,25 мл, 10 ммоль) к суспензии 2,06 г (10 ммоль) 2,5-диметил-7Н-циклопента [1,2-b:4,3-b]дитиофена в 100 мл эфира. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают еще 50 мин при той же температуре. Полученную реакционную смесь обрабатывают при -70° С раствором 0,74 г (5 ммоль) 1-метил-3-изопропил-6,6-диметилфульвена, затем дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную смесь выливают в 100 мл 10%-ного водного раствора NH4Cl и экстрагируют гексаном (2× 50 мл). Органическую фазу собирают, промывают водой, сушат MgSO4 и упаривают. Остаток припускают через колонку, наполненную SiO2, используя в качестве элюента гексан. Полученный раствор сушат, получают кристаллический продукт. Выход 1,5 г (41% из расчета на исходный MeTh2Cp).
Синтез изопропилиден{(2-метил-4-изопропилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}-цирконийдихлорида С-17
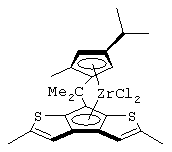
Суспензию 1,11 г (3 ммоль) 2,2-(2-метил-4-изопропил-1-циклопентадиенил)-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}пропана в 10 мл эфира и 50 мл гексана обрабатывают при -70° С 3,8 мл 1,6 М раствора н-BuLi (6,1 ммоль). По окончании добавления смеси дают нагреться до 0° С и добавляют 0,75 г (3,2 ммоль) ZrCl4. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем отфильтровывают полученный желтый осадок, промывают дважды эфиром и, наконец, перекристаллизовывают из CH2Cl2.
Выход 1,43 г (90%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 6,88 (м, 1Н, СН); 6,80 (м, 1Н, СН); 6,10 (д, 1Н, СН); 5,58 (д, 1Н, СН); 2,78 (м, 1Н, СН); 2,58 (м, 3Н, СН3); 2,56 (д, 3Н, СН3); 2,40 (с, 3Н, СН3); 2,18 (с, 3Н, СН3); 1,96 (с, 3Н, СН3); 1,14 (д, 3Н, СН3); 1,08 (д, 3Н, СН3).
ПРИМЕР 18
Синтез 1,3-диметил-1,3-циклопентадиена
Раствор 25 г (0,26 моль) 3-метил-1-циклопентен-1-она в 100 мл эфира добавляют при -78° С и в атмосфере азота к раствору метиллития в 200 мл эфира, предварительно приготовленного из 5,76 г (0,83 моль) лития и 26 мл (0,42 моль) йодметана. Реакционную смесь перемешивают 4 часа и затем обрабатывают 10% водным раствором NH4C1. Органическую фазу собирают, промывают водой, сушат МgSO4 и концентрируют. Остаток перегоняют при 42° С/100 мм рт.ст. Выход 7,3 г (30%).
Спектр 1H ЯМР (CD3COCD3, δ , м.д.): 5,98 (м, 1Н, СН); 5,75 (м, 1Н, СН); 2,80 (м, 2Н, CH2); 2,02 (д, 3Н, СН3); 1,90 (д, 3Н, СН3).
Синтез лиганда осуществляют путем сочетания литиевой соли предшественника MeTh2Cp с хлор-(2,4-диметилциклопентадиенил)-диметилсиланом, предварительно полученным из литиевой соли 1,3-диметил-1,3-циклопентадиена и Me2SiCl2.
Синтез диметилсилил{(2,4-диметилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-18
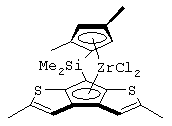
Суспензию 1,07 г (3 ммоль) (2,4-диметилциклопентадиенил)-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}диметилсилана в 20 мл эфира обрабатывают при -70° С 4,1 мл 1,6 М раствора н-BuLi (6,5 ммоль). После добавления реакционной смеси дают нагреться до 0° С и добавляют 0,75 г (3,2 ммоль) ZrCl4. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем отфильтровывают полученный желтый осадок, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2.
Выход 1,35 г (87%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 6,93 (м, 1Н, СН); 6,87 (м, 1Н, СН); 6,80-6,70 (м, 1Н, СН); 6,25 (т, 1Н, СН); 2,59 (д, 3Н, СН3); 2,56 (д, 3Н, СН3); 2,18 (с, 3Н, СН3); 2,11 (с, 3Н, СН3); 1,03 (с, 3Н, Si-СН3); 0,84 (с, 3Н, Si-СН3).
ПРИМЕР 19
Синтез диметилсилил{(3-трет-бутилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-4
Синтез осуществляют по методике, описанной в примере 18, с использованием 3-трет-бутил-1,3-циклопентадиена вместо 1,3-диметил-1,3-циклопентадиена. Продукт характеризуют с помощью спектроскопии ЯМР.
ПРИМЕР 20
Синтез изопропилиден{(тетраметилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-5
Синтез осуществляют по методике, описанной в примере 17, с использованием 1,2,3,4,6,6-эзаметилфульвена вместо 1-метил-3-изопропил-6,6-диметилфульвена. Продукт характеризуют с помощью спектроскопии ЯМР.
ПРИМЕР 21
Синтез диметилсилил{(3-триметилсилилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофена)
Синтез лиганда осуществляют путем сочетания литиевой соли предшественника MeTh2Cp с хлор-(3-триметилсилилциклопентадиенил)диметилсиланом, предварительно полученным из литиевой соли триметилсилил-1,3-циклопентадиена и Me2SiCl2.
Синтез диметилсилил{(3-триметилсилилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-9
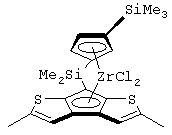
Суспензию 1,20 г (3 ммоль) (3-триметилсилил-1-циклопентадиенил)-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}диметилсилана в 20 мл эфира обрабатывают при -70° С 4,1 мл 1,6 М раствора н-BuLi (6,5 ммоль). После добавления реакционной смеси дают нагреться до 0° С и добавляют 0,75 г (3,2 ммоль) ZrCl4. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем отфильтровывают полученный желтый осадок, промывают дважды эфиром, сушат и перекристаллизовывают из СН2Сl2.
Выход 1,17 г (70%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 6,91 (м, 1Н, СН); 6,88 (м, 1Н, СН); 6,78 (м, 1Н, СН); 6,08 (т, 1Н, СН); 5,83 (т, 1Н, СН); 2,59 (д, 3Н, СН3); 2,57 (д, 3Н, СН3); 0,91 (с, 3Н, Si-СН3); 0,89 (с, 3Н, Si-СН3); 0,20 (с, 9Н, Si (СН3)3).
ПРИМЕР 22
Синтез 1-метил-3-фенил-1,3-циклопентадиена
Раствор 25 г (0,26 моль) 3-метил-2-циклопентен-1-она в 100 мл эфира добавляют при -78° С и в атмосфере аргона к раствору фениллития в 200 мл эфира, предварительно приготовленному из 5,76 г (0,83 моль) лития и 44 мл (0,42 моль) бромбензола. Реакционную смесь перемешивают 4 часа и затем обрабатывают 10% водным раствором NH4Cl. Органическую фазу собирают, промывают водой, сушат MgSO4 и концентрируют. Остаток перегоняют при 54° С/1 мм рт.ст. Выход 24,37 г (60%).
Спектр 1H ЯМР (CD3COCD3, δ , м.д.): 7,60-7,10 (м, 5Н, СН); 6,80 (д, 1Н, СН); 6,00 (м, 1Н, СН); 3,00 (с, 2Н, СН2); 1,98 (кв, 3Н, СН3).
Синтез 1-метил-3-фенил-6,6-диметилфульвена
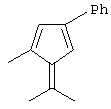
Раствор 1-метил-3-фенил-1,3-циклопентадиена (15,62 г, 0,1 моль) в 100 мл этанола обрабатывают при низкой температуре 8,6 мл (0,12 моль) ацетона и 9,7 мл (0,12 моль) пирролидина. Полученный раствор выдерживают при температуре ниже комнатной в течение ночи. Затем полученную смесь нейтрализуют 10% водным раствором Н3РO4 , экстрагируют гексаном (3× 50 мл) и промывают водой до нейтрального значения рН. Органическую фазу отделяют, сушат с помощью MgSO4 и концентрируют. Остаток перегоняют при 85° С/10 мм рт.ст. Выход 5,89 г (30%). Названное целевое соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез лиганда осуществляют по методике, описанной в примере 17, с использованием 1-метил-3-фенил-6,6-диметилфульвена вместо 1-метил-3-изопропил-6,6-диметилфульвена и 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофена вместо MeTh2Cp.
Синтез изопропилиден{(2-метил-4-фенилциклопентадиенил)-7-(2,5-триметилсилилциклопента[1,2-b:4,3-b’]дитиофен)}-цирконийдихлорида С-14
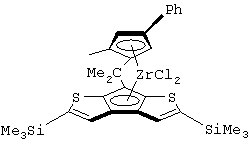
Суспензию 2,0 г (3,85 ммоль) 2,2-(2-диметил-4-фенилциклопентадиенил)-7-(2,5-триметилсилилциклопента[1,2-b:4,3-b’]дитиофен)пропана в 50 мл эфира обрабатывают при -70° С 4,8 мл 1,6 М раствора н-BuLi (7,71 ммоль). После добавления реакционной смеси дают нагреться до 0° С и добавляют 0,90 г (3,85 ммоль) ZrCl4. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем отфильтровывают полученный коричневый осадок, промывают дважды эфиром, сушат и перекристаллизовывают из СН2Сl2. Выход 1,82 г (70%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 7,34 (с, 2Н, СН); 7,32-7,12 (м, 5Н, СН); 6,62 (д, 1Н, СН); 6,26 (д, 1Н, СН); 2,50 (с, 3Н, СН3); 2,30 (с, 3Н, СН3); 2,10 (с, 3Н, СН3); 0,36 (с, 9Н, Si (СН3)3); 0,32 (с, 9Н, Si(СН3)3).
ПРИМЕР 23
Синтез изопропилиден{(2-метил-4-фенилциклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}-цирконийдихлорида С-11
Синтез осуществляют по методике, описанной в примере 22, с использованием 2,5-диметилциклопента[1,2-b:4,3-b’]дитиофена вместо 2,5-триметилсилилциклопента[1,2-b:4,3-b’]дитиофена. Продукт характеризуют с помощью спектроскопии ЯМР.
ПРИМЕР 24
Синтез изопропилиден{(4-фенилциклопентадиенил)-7-(2,5-диметилциклопента [1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-19
Синтез осуществляют по методике, описанной в примере 22, с использованием 3-фенил-6,6-диметилфульвена вместо 1-метил-3-фенил-6,6-диметилфульвена. Продукт характеризуют с помощью спектроскопии ЯМР.
ПРИМЕР 25
Синтез 1,3-диметил-1,3-циклопентадиена осуществляют как описано в примере 18.
Синтез 1,3,6,6-тетраметилфульвена
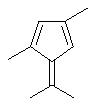
Раствор 1,3-диметил-1,3-циклопентадиена (9,42 г, 0,1 моль) в 100 мл этанола обрабатывают при низкой температуре 8,6 мл (0,12 моль) ацетона и 9,7 мл (0,12 моль) пирролидина. Полученный раствор выдерживают при температуре ниже комнатной в течение ночи. Затем реакционную смесь нейтрализуют 10% водным раствором Н3РO4, экстрагируют гексаном (3× 50 мл) и промывают водой до нейтрального значения рН. Органическую фазу отделяют, сушат над MgSO4 и концентрируют. Остаток перегоняют при 63° С/20 мм рт.ст. Выход 6,7 г (50%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 6,08 (м, 1Н, СН); 6,03 (м, 1Н, СН); 2,23 (д, 3Н, СН3); 2,17 (с, 3Н, СН3); 2,16 (с, 3Н, СН3); 1,99 (с, 3Н, СН3).
Синтез лиганда осуществляют по методике, описанной в примере 17, используя 1,3,6,6-тетраметилфульвен вместо 1-метил-3-изопропил-6,6-диметилфульвена и 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофен вместо MeTh2Cp.
Синтез изопропилиден{(2,4-диметилциклопентадиенил)-7-(2,5-триметилсилилциклопента[1,2-b:4,3-b’]дитиофен)}-цирконийдихлорида С-15
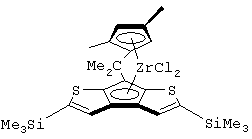
Суспензию 2,19 г (4,8 ммоль) 2,2-(2,4-диметил-1-циклопентадиенил)-7-(2,5-триметилсилилциклопента[1,2-b:4,3-b’]дитиофен)}пропана в 50 мл эфира обрабатывают при -70° С 6,0 мл 1,6 М раствора н-BuLi (9,6 ммоль). По окончании добавления реакционной смеси дают нагреться до 0° С и добавляют 1,12 г (4,8 ммоль) ZrCl4. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем отфильтровывают полученный красно-коричневый осадок, промывают дважды эфиром и, наконец, перекристаллизовывают из CH2Cl2. Выход 2,07 г (70%). Названное целевое соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 26
Синтез 3-хлор-2-метил-2-бутеналя
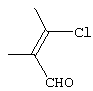
Добавляют при 0° С 1,3 моль (120 мл) РОСl3 к 1,6 моль (120 мл) ДМФА. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем смесь снова охлаждают до 0° С и обрабатывают 1 моль (90 мл) 2-бутанона. Полученной реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем ее выливают в смесь льда и воды, добавляют ацетат натрия и экстрагируют СНСl3 (3× 150 мл). Органическую фазу отделяют, промывают водой до нейтрального значения рН, сушат над МgSO4 и упаривают досуха. Остаток перегоняют в вакууме, т.кип. 45° С/10 мм рт.ст. Выход 73 г (62%).
Синтез 4,5-диметил-2-тиофенэтилкарбоксилата
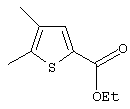
Добавляют при 0° С этил-2-меркаптоацетат (0,2 моль, 24 г) к раствору этилата натрия (0,21 моль, 14,3 г) в 150 мл этанола, и полученную смесь перемешивают при той же температуре 30 мин. Затем добавляют 3-хлор-2-метил-2-бутеналь (0,2 моль, 23,7 г) и продолжают перемешивание в течение ночи. Полученный продукт разбавляют в 100 мл воды, органическую фазу собирают, и водный слой экстрагируют СН2Сl2 (2× 150 мл). Объединенные органические слои сушат над MgSO4, упаривают досуха и остаток перегоняют в вакууме. Выход 22,48 г (61%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,52 (с, 1Н, СН); 4,32 (кв, 2Н, ОСН2); 2,35 (с, 3Н, СН3); 2,12 (с, 3Н, СН3); 1,35 (т, 3Н, СН3).
Синтез 4,5-диметил-2-тиофенкарбоновой кислоты
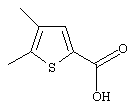
Добавляют 4,5-диметил-2-тиофенэтилкарбоксилат (0,122 моль, 22,48 г) к 30%-ному раствору гидроксида натрия в 100 мл этанола, и полученную смесь кипятят с обратным холодильником 2 часа. Затем смесь разбавляют водой, подкисляют и фильтруют. Осадок сушат под Р2О5. Выход 15,6 г (82%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,60 (с, 1Н, СН); 2,42 (с, 3Н, СН3); 2,17 (с, 3Н, СН3).
Синтез 2,3-диметилтиофена
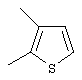
4,5-Диметил-2-тиофенкарбоновую кислоту (0,58 моль, 90 г), полученную как описано выше, нагревают до 180° С, пока не прекратиться выделение диоксида углерода. Продукт собирают и перегоняют, т.кип. 140° С. Выход 30 г (46%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,02 (д, 1Н, СН); 6,82 (д, 1Н, СН); 2,42 (с, 3Н, СН3); 2,20 (с, 3Н, СН3).
Синтез 2,3,5-триметил-5,6-дигидро-4Н-циклопента [b] тиофен-4-она
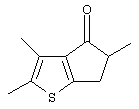
Раствор 10 г (0,07 моль) Р2О5 в 100 мл метансульфокислоты (1,54 моль) нагревают при 80° С при перемешивании. Добавляют смесь 2,3-диметилтиофена (0,27 моль, 30 г) и метакриловой кислоты (0,35 моль) в 20 мл СН2Сl2, и полученную реакционную смесь перемешивают при той же температуре 1,5 часа. Затем смесь выливают в смесь льда и воды и интенсивно перемешивают. Водный слой экстрагируют СН2Сl2 (3× 50 мл), органические слои собирают, промывают 10% водным раствором карбоната натрия до нейтрального значения рН и водой. Затем органическую фазу отделяют, сушат над MgSO4, упаривают досуха и перегоняют в вакууме, т.кип. 110° С/1 мм рт.ст. Выход 10 г (20%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 3,35 (дд, 1Н, СН2); 2,98 (кв.д, 1Н, СН); 2,66 (дд, 1Н, CH2); 2,35 (с, 3Н, СН3); 2,25 (с, 3Н, СН3); 1,52 (с, 3Н, СН3).
Синтез 2,3,5-триметил-6Н-циклопента[b]тиофена
(или 2,3,5-триметил-1-тиопенталена
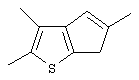
Раствор 2,3,5-триметил-5,6-дигидро-4Н-циклопента[b]тиофен-4-она (11 г, 61 ммоль) в 100 мл эфира медленно добавляют к раствору LiAlH4 (1,16 г, 30 ммоль) в 100 мл эфира и перемешивают в течение ночи. Полученную суспензию выливают в смесь льда и воды, органический слой отделяют, тогда как водный слой экстрагируют эфиром (3× 50 мл). Объединенные органические слои промывают водой, сушат над МgSO4 и упаривают досуха. Полученный таким образом 2,3,5-триметил-5,6-дигидро-4Н-циклопента[b]тиофен-4-ол растворяют в 100 мл бензола, добавляют 1 г п-толуолсульфокислоты и кипятят с обратным холодильником 10 мин. Реакционную смесь охлаждают до комнатной температуры и обрабатывают насыщенным водным раствором Nа2СО3. Органическую фазу выделяют, сушат над MgSO4 и упаривают досуха. Выход 8 г (80% из расчета на исходный кетон).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 6,44 (м, 1Н, СН); 3,05 (с, 2Н, СН2); 2,45 (с, 3Н, СН3); 2,20 (с, 3Н, СН3); 2,15 (с, 3Н, СН3).
Синтез 6Н-6-(2,3,5-триметилциклопента[b]тиофен)-хлордиметилсилана
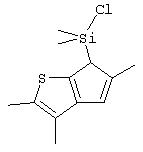
Раствор 1,28 г (5 ммоль) 2,3,5-триметил-6Н-циклопента[b] -тиофена в 40 мл Et2O обрабатывают при -70°С 3,13 мл (5 ммоль) 1,6 М раствора н-BuLi. После добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем снова охлаждают до -70° С и обрабатывают раствором 1,30 г (10 ммоль) Me2SiCl2 в 10 мл эфира. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную реакционную смесь фильтруют для удаления LiCl, и растворитель удаляют при пониженном давлении. Сырой продукт используют в том виде, как он есть, на следующей стадии без дополнительной очистки.
Спектр 1H ЯМР (C6D6, δ , м.д.): 6,30 (с, 1Н, СН); 3,25 (с, 1Н, СН); 2,20 (с, 3Н, СН3); 2,10 (с, 3Н, СН3), 1,90 (с, 3Н, СН3); 0,30 (с, 3Н, Si-СН3); -0,10 (с, 3Н, Si-СН3).
Синтез диметилсилил{6-(2,3,5-триметилциклопента[b]тиофен)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}-цирконийдихлорида С-27
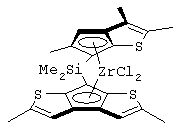
Суспензию 0,9 г (4,4 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 20 мл эфира обрабатывают при -70° С 2,75 мл 1,6 М раствора н-BuLi (4,4 ммоль). После добавления полученной смеси дают нагреться до комнатной температуры и перемешивают еще 50 мин при этой температуре. Затем смесь снова охлаждают до -70° С и добавляют к эфирному раствору (10 мл) 6Н-6-(2,3,5-триметилциклопента[b]тиофен)хлордиметилсилана, полученного на предыдущей стадии. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученный таким образом лиганд 6-{(2,3,5-триметилциклопента[b]тиофен)}-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}-диметилсилан характеризуют с помощью спектроскопии 1H ЯМР.
Спектр 1H ЯМР (C6D6, δ , м.д.): 6,70 (м, 2Н, СН); 6,50 (с, 1Н, СН); 4,40 (с, 1Н, СН); 4,10 (с, 1Н, СН); 2,39 (м, 3Н, СН3); 2,37 (д, 6Н, СН3); 2,25 (с, 3Н, СН3); 2,14 (с, 3Н, СН3); 0,18 (с, 3Н, Si-СН3); 0,07 (с, 3Н, Si-СН3).
Лиганд не выделяют: его раствор обрабатывают при -70° С 5,60 мл (9,0 ммоль) 1,6 М раствора н-BuLi. Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении, и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,17 г (5 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, отфильтровывают желтый осадок, промывают дважды эфиром, сушат и перекристаллизовывают из CH2Cl2. Выход 1,62 г (60% относительно Me2Th). Названное целевое соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 27
Синтез 3-хлор-2-метил-3-фенил-2-пропеналя
Добавляют при 0° С 1,2 моль (110 мл) РОСl3 к 2,8 моль (216 мл) ДМФА (избыток ДМФА используется в качестве растворителя). По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем смесь снова охлаждают до 0° С и обрабатывают 1 моль (134 г) пропиофенона. Полученной реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем смесь выливают в смесь льда и воды, добавляют ацетат натрия и экстрагируют СНСl3 (3× 150 мл). Органическую фазу отделяют, промывают водой до нейтрального значения рН, сушат над МgSO4 и упаривают досуха. Остаток перегоняют в вакууме, т.кип. 120° С/10 мм рт.ст. Выход 163 г (90%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 9,52 (с, 1Н, СНО); 7,45 (м, 5Н, СН); 2,12 (с, 3Н, СН3).
Синтез этилового эфира 4-метил-5-фенил-2-тиофенкарбоновой кислоты
Добавляют при 0° С этил-2-меркаптоацетат (0,9 моль, 100 мл) к раствору этилата натрия (1 моль, 68 г) в 500 мл этанола, и полученную смесь перемешивают при той же температуре 30 мин. Затем добавляют 3-хлор-2-метил-3-фенил-2-пропеналь (0,9 моль, 163 г) и продолжают перемешивание в течение ночи. Полученный продукт разбавляют в 1,5 л воды, органический слой собирают, и водный слой экстрагируют CH2Cl2 (4× 150 мл). Объединенные органические слои сушат над МgSO4, упаривают досуха и остаток используют на следующей стадии без дополнительной очистки. Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез 4-метил-5-фенил-2-тиофенкарбоновой кислоты
Добавляют полученный на предыдущей стадии этиловый эфир 4-метил-5-фенил-2-тиофенкарбоновой кислоты к 30%-ному раствору гидроксида натрия в 1 л этанола, и полученную смесь кипятят с обратным холодильником 2 часа. Затем смесь разбавляют водой и экстрагируют 200 мл бензола. Водную фазу выделяют, подкисляют и смесь фильтруют. Осадок сушат под Р2O5. Выход 127 г (65% относительно 3-хлор-2-метил-3-фенил-2-пропеналя).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,75 (с, 1Н, СН); 7,50-7,40 (м, 5Н, СН); 2,37 (с, 3Н, СН3).
Синтез 3-метил-2-фенилтиофена
4-Метил-5-фенил-2-тиофенкарбоновую кислоту (127 г, 0,58 моль), полученную как описано выше, нагревают при 220-230° С, пока не прекратится выделение диоксида углерода. Продукт собирают и перегоняют, т.кип. 120° С/10 мм рт.ст. Выход 30,3 г (30%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,60 (д, 2Н, СН); 7,48 (т, 2Н, СН); 7,35 (т, 1Н, СН); 7,25 (д, 1Н, СН); 6,98 (д, 1Н, СН), 2,39 (с, 3Н, СН3).
Синтез 2-формил-4-метил-5-фенилтиофена
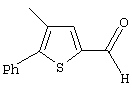
Добавляют при 0° С 0,35 моль (32 мл) РОСl3 к 1,0 моль (77 мл) ДМФА (избыток ДМФА используется в качестве растворителя). По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем смесь снова охлаждают до 0° С и обрабатывают 3-метил-2-фенилтиофеном (60 г, 0,35 моль). Полученной реакционной смеси дают нагреться до комнатной температуры, и после перемешивания при этой же температуре в течение 12 час нагревают при 80° С 2 дня. Затем смесь выливают в смесь льда и воды, добавляют ацетат натрия и экстрагируют СНСl3 (3× 150 мл). Органическую фазу отделяют, промывают водой до нейтрального значения рН, сушат над МgSO4 и упаривают досуха. Остаток кристаллизуют. Выход 60,2 г (85%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 9,88 (с, 1Н, СНО); 7,62 (с, 1Н, СН); 7,55-7,40 (м, 5Н, СН); 2,39 (с, 3Н, СН3).
Синтез 2-метил-3-(4-метил-5-фенил-2-тиенил)акриловой кислоты
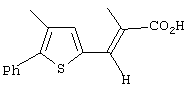
Смесь 2-формил-4-метил-5-фенилтиофена (20,2 г, 0,1 моль) и этил-2-бромпропионата (0,12 моль, 15,5 мл) добавляют к суспензии Zn (7 г, 0,1 моль) в 150 мл бензола с каталитическим количеством НgВr2. Полученную смесь кипятят с обратным холодильником при перемешивании, пока все количество Zn не растворится, и затем растворяют в воде. Органический слой выделяют, промывают 10% водным раствором НСl, сушат над МgSO4 и упаривают досуха. Остаток, соответствующий 2-метил-3-(4-метил-5-фенил-2-тиенил)этилакрилату, используют без дополнительной очистки в синтезе соответствующей кислоты. Фактически остаток добавляют к 30%-ному водному раствору гидроксида натрия в 100 мл этанола и кипятят с обратным холодильником 2 часа. Полученную реакционную смесь разбавляют в воде, подкисляют и фильтруют. Осадок сушат под Р2O5. Выход 16,7 г (65%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,90 (с, 1Н, СН); 7,50-7,30 (м, 5Н, СН); 7,20 (с, 1Н, СН); 2,40 (с, 3Н, СН3); 2,20 (с, 3Н, СН3).
Синтез 2-метил-3-(4-метил-5-фенил-2-тиенил)-2-пропановой кислоты
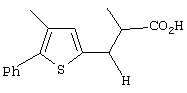
Целевое соединение 2-метил-3-(4-метил-5-фенил-2-тиенил)-2-пропановую кислоту получают электрохимическим восстановлением 2-метил-3-(4-метил-5-фенил-2-тиенил)акриловой кислоты. Выход ~ 100%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,50-7,30 (м, 5Н, СН); 6,70 (с, 1Н, СН); 3,30 (дд, 1Н, СН); 2,90 (м, 2Н, СН2); 2,30 (с, 3Н, СН3); 1,30 (д, 3Н, СН3).
Синтез 3,5-диметил-2-фенил-5,6-дигидро-4Н-циклопента[b]тиофен-4-она
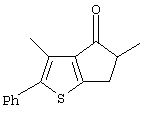
Раствор 3 г P2O5 (21 ммоль) в 30 мл метансульфоновой кислоты (0,46 моль) нагревают при 80° С при перемешивании. Добавляют раствор 2-метил-3-(4-метил-5-фенил-2-тиенил)-2-пропановой кислоты (65 ммоль, 16,9 г) в 20 мл СН2Сl2, и полученную смесь перемешивают при той же температуре 1,5 часа. Затем смесь выливают в смесь льда и воды и интенсивно перемешивают. Водный слой экстрагируют СН2Сl2 (3× 50 мл), органические слои собирают, промывают 10% водным раствором карбоната натрия до нейтрального значения рН и, наконец, водой. Далее органическую фазу выделяют, сушат над MgSO4 и упаривают досуха. Выход 6,3 г (40%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,50-7,40 (м, 5Н, СН); 3,40 (дд, 1Н, СН2); 3,05 (м, 1Н, СН); 2,80 (дд, 1Н, СН2); 2,55 (с, 3Н, СН3); 1,40 (д, 3Н, СН3).
Синтез 3,5-диметил-2-фенил-6Н-циклопента[b]тиофена (или 3,5-диметил-2-фенил-1-тиопенталена)
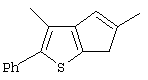
Раствор 3,5-диметил-2-фенил-5,6-дигидро-4Н-циклопента[b]-тиофен-4-она (6,3 г, 26 ммоль) в 75 мл эфира медленно добавляют к раствору LiAlH4 (0,5 г, 13 ммоль) в 50 мл эфира и перемешивают в течение ночи. Полученную суспензию выливают в смесь льда и воды, органический слой выделяют, тогда как водный слой экстрагируют эфиром (3× 50 мл). Объединенные органические слои промывают водой, сушат над МgSO4 и упаривают досуха.
Полученный таким образом 3,5-диметил-2-фенил-5,6-дигидро-4Н-циклопента[b]-тиофен-4-ол растворяют в бензоле, добавляют 1 г п-толуолсульфоновой кислоты и кипятят с обратным холодильником 10 мин. Затем реакционную смесь охлаждают до комнатной температуры и обрабатывают насыщенным водным раствором Na2СО3. Органическую фазу выделяют, сушат над MgSO4 и упаривают досуха. Выход 4,66 г (80% из расчета на исходный кетон).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,50-7,40 (м, 5Н, СН); 6,50 (кв, 1Н, СН); 3,20 (д, 1Н, CH2); 2,60 (дд, 1Н, CH2); 2,20 (с, 3Н, СН3); 1, 66 (с, 3Н, СН3).
Синтез 6Н-6-(2-фенил-3,5-диметилциклопента[b]тиофен)-хлордиметилсилана
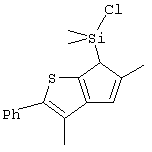
Раствор 1,70 г (7,5 ммоль) 3,5-диметил-2-фенил-6Н-циклопента[b]тиофена в 40 мл Et2O обрабатывают при -70° С 5,0 мл (8 ммоль) 1,6 М раствора н-BuLi. После добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем смесь снова охлаждают до -70° С и обрабатывают раствором 1,30 г (10 ммоль) Me2SiCl2 в 10 мл эфира. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную реакционную смесь фильтруют для удаления LiCl, и растворитель удаляют при пониженном давлении. Сырой продукт используют на следующей стадии в том виде, как он есть, без дополнительной очистки.
Спектр 1H ЯМР (C6D6, δ , м.д.): 7,60-7,20 (м, 5Н, СН); 6,50 (м, 1Н, СН); 3,40 (с, 1Н, СН); 2,84 (с, 3Н, СН3); 2,21 (с, 3Н, СН3); 0,39 (с, 3Н, Si-СН3); 0,12 (с, 3Н, Si-СН3).
Синтез диметилсилил{6-(2-фенил-3,5-диметилциклопента[b]-тиофен)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}
цирконийдихлорида С-29
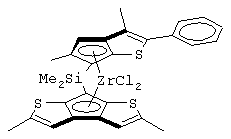
Суспензию 1,3 г (6,3 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 30 мл эфира обрабатывают при -70° С 4,0 мл 1,6 М раствора н-BuLi (6,4 ммоль). После добавления полученной смеси дают нагреться до комнатной температуры и перемешивают при этой температуре еще 50 мин. Затем смесь снова охлаждают до -70° С и добавляют эфирный раствор (10 мл) 6Н-6-(3,5-диметил-2-фенилциклопента[b]тиофен)хлордиметилсилана, полученного на предыдущей стадии. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученный таким образом лиганд 6-{(3,5-диметил-2-фенилциклопента[b]-тиофен)}-7-{[2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}-диметилсилан характеризуют с помощью спектроскопии 1H ЯМР.
Спектр 1H ЯМР (C6D6, δ , м.д.): 7,60-7,10 (м, 5Н, СН); 6,85 (с, 1Н, СН); 6,80 (с, 1Н, СН); 6,50 (м, 1Н, СН); 4,37 (с, 1Н, СН); 4,10 (с, 1Н, СН); 2,38 (д, 3Н, СН3); 2,37 (д, 3Н, СН3); 2,36 (с, 3Н, СН3); 2,12 (с, 3Н, СН3); -0,05 (с, 3Н, Si-СН3); -0,16 (с, 3Н, Si-СН3).
Лиганд не выделяют: его раствор обрабатывают при -70° С 8,2 мл (13,1 ммоль) 1,6 М раствора н-BuLi. Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении, и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,51 г (6,5 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, отфильтровывают желто-коричневый осадок, промывают дважды эфиром, сушат и перекристаллизовывают из СН2Сl2. Выход 2,30 г (56% относительно Me2Th). Названное целевое соединение характеризуют с помощью спектроскопии 1H ЯМР.
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 7,44-7,36 (м, 5Н, СН); 6,86 (кв, 1Н, J=1,24 Гц, СН); 6,73 (кв, 1H, J=1,24 Гц, СН); 6,56 (уш.с, 1H, СН); 2,63 (д, 3Н, J=1,24 Гц, СН3); 2,50 (д, 3Н, J=1,24 Гц, СН3); 2,35 (с, 3Н, СН3); 2,20 (с, 3Н, СН3); 1,22 (с, 3Н, Si-СН3); 1,14 (с, 3Н, Si-СН3).
ПРИМЕР 28
Синтез 3-хлор-2,3-дифенил-2-пропеналя
К 0,3 моль (23,5 мл) ДМФА при 0° С добавляют 0,25 моль (23 мл) РОСl3. После добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем смесь снова охлаждают до 0° С и обрабатывают 0,1 моль (19,6 г) деоксибензоина. Полученную реакционную смесь оставляют нагреваться до комнатной температуры, перемешивают 12 часов при той же температуре и затем нагревают при 100° С в течение 2 часов. Наконец, смесь выливают в смесь льда и 10%-ного раствора ацетата натрия в воде. Маслянистый осадок отфильтровывают и промывают холодным метанолом и гексаном. Остаток кристаллизуют. Выход 18,5 г (76%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 9,70 (с, 1H, СНО); 7,60-7,20 (м, 10Н, СН).
Синтез этилового эфира 4,5-дифенил-2-тиофенкарбоновой кислоты
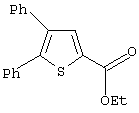
К раствору этилата натрия (42 ммоль, 2,86 г) в 50 мл этанола при 0° С добавляют этил-2-меркаптоацетат (40 ммоль, 4,4 мл), и полученную смесь перемешивают при той же температуре в течение 30 мин. Затем добавляют 3-хлор-2,3-дифенил-2-пропеналь (37 ммоль, 9,0 г) и перемешивание продолжают в течение ночи. Полученную оранжевую суспензию нагревают при 50° С в течение 2 час, затем охлаждают до комнатной температуры и разбавляют 100 мл воды. Полученный таким образом красный раствор, содержащий осадок, экстрагируют Et2O (3× 50 мл). Объединенные органические слои промывают смесью NН4Сl/вода, сушат над МgSO4 и упаривают досуха. Твердый остаток перекристаллизовывают из гексана. Выход 9,2 г (81%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,40-7,30 (м, 11Н, СН); 4,40 (кв, 2H, СН2); 1,40 (т, 3Н, СН3).
Синтез 4,5-дифенил-2-тиофенкарбоновой кислоты
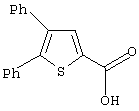
К 30%-ному раствору гидроксида натрия в 20 мл этанола добавляют этиловый эфир 4,5-дифенил-2-тиофенкарбоновой кислоты (3,6 г, 12 ммоль) с предыдущей стадии, и полученную смесь кипятят с обратным холодильником 2 часа. Затем смесь разбавляют водой, водную фазу подкисляют. Полученный белый осадок отфильтровывают и сушат при 80° С. Выход 3,16 г (94%).
Спектр 1H ЯМР ((CD3)2SO, δ , м.д.): 7,80 (с, 1Н, СН); 7,40-7,30 (м, 10Н, СН).
Синтез 2,3-дифенилтиофена
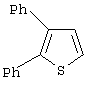
4,5-Дифенил-2-тиофенкарбоновую кислоту (0,1 моль, 28 г), полученную как описано выше, нагревают до 220-230° С, пока не прекратиться выделение диоксида углерода. Продукт разбавляют водой и экстрагируют 100 мл бензола. Органическую фазу сушат над МgSO4 и упаривают, получают кристаллический продукт. Выход 22,45 г (95%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,35 (д, 1Н, СН); 7,34-7,26 (м, 10Н, СН); 7,19 (д, 1Н, СН).
Синтез 2-формил-4,5-дифенилтиофена
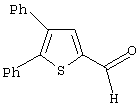
К раствору 2,3-дифенилтиофена (18 г, 76 ммоль) в ДМФА (18 мл, 0,23 моль) при 0° С добавляют РОСl3 (80 ммоль, 7,32 мл). После добавления реакционной смеси дают нагреться до комнатной температуры и кипятят с обратным холодильником 3 часа. Затем смесь охлаждают до комнатной температуры и выливают в смесь льда и 10%-ного раствора гидроксида натрия в воде. Маслянистый осадок отфильтровывают и промывают холодным метанолом и гексаном. Остаток кристаллизуют. Выход 15 г (75%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 9,95 (с, 1Н, СНО); 7,82 (с, 1Н, СН); 7,40-7,25 (м, 10Н, СН).
Синтез 2-метил-3-(4,5-дифенил-2-тиенил)этилакрилата
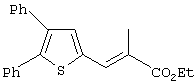
Смесь 2-формил-4,5-дифенилтиофена (14,8 г, 56 ммоль) и этил-2-бромпропионата (60 ммоль, 7,8 мл) добавляют к суспензии Zn (4,25 г, 65 ммоль) в 100 мл бензола с каталитическим количеством I2. Полученную смесь кипятят с обратным холодильником при перемешивании, пока не растворится все количество цинка, и затем растворяют в воде. Органический слой отделяют, промывают 10% водным раствором НСl, сушат над MgSO4 и упаривают досуха. Остаток растворяют в 50 мл бензола и кипятят с обратным холодильником с 0,5 г п-толуолсульфокислоты 1 час. Затем реакционную смесь охлаждают до комнатной температуры и обрабатывают насыщенным водным раствором NaCO3. Органическую фазу отделяют, сушат над MgSO4 и упаривают досуха. Выход 17,6 г (90%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,90 (с, 1Н, СН); 7,40-7,25 (м, 11Н, СН); 4,30 (кв, 2Н, CH2); 2,30 (д, 3Н, СН3); 1,40 (т, 3Н, СН3).
Синтез 2-метил-3-(4,5-дифенил-2-тиенил)акриловой кислоты
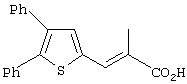
К 30%-ному раствору гидроксида натрия в 300 мл этанола добавляют 2-метил-3-(4,5-дифенил-2-тиенил)этилакрилат (18,9 г, 54 ммоль) с предыдущей стадии, и полученную смесь кипятят с обратным холодильником 2 часа. Затем смесь разбавляют водой, подкисляют и фильтруют. Осадок сушат под Р2O5. Выход 12,1 г (70%).
Спектр 1H ЯМР ((CD3)2SO, δ , м.д.): 7,85 (д, 1Н, СН); 7,55 (с, 1Н, СН); 7,40-7,20 (м, 10Н, СН); 2,15 (с, 3Н, СН3).
Синтез 2-метил-3-(4,5-дифенил-2-тиенил)-2-пропановой кислоты
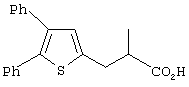
Требуемую 2-метил-3-(4,5-дифенил-2-тиенил)-2-пропановую кислоту получают электрохимическим восстановлением 2-метил-3-(4,5-дифенил-2-тиенил)акриловой кислоты. Выход ~ 100%.
Спектр 1H ЯМР ((CD3)2SO, δ , м.д.): 7,30-7,15 (м,10Н, СН); 6,90 (с, 1Н, СН); 3,10 (дд, 1Н, СН); 2,70 (м, 2Н, CH2); 1,10 (д, 3Н, СН3).
Синтез 5-метил-2,3-дифенил-5,6-дигидро-4Н-циклопента[b]-тиофен-4-она
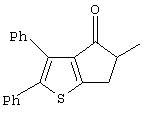
Раствор P2O5 (2,6 г, 18 ммоль) в 15 мл метансульфоновой кислоты (0,23 моль) нагревают при 80° С при перемешивании. Добавляют раствор 2-метил-3-(4,5-дифенил-2-тиенил)-2-пропановой кислоты (18 ммоль, 6,0 г) в 20 мл CH2Cl2, и полученную реакционную смесь перемешивают при той же температуре 15 мин. Затем смесь выливают в смесь льда и воды и энергично перемешивают. Водный слой экстрагируют CH2Cl2 (3× 50 мл), органические слои собирают, промывают 10% водным раствором карбоната натрия до нейтрального значения рН, затем промывают водой. Органическую фазу выделяют, сушат над МgSO4 и упаривают досуха. Остаток пропускают через наполненную силикагелем 60 колонку, используя в качестве элюента смесь гексан/этилацетат 5/1. После упаривания красной фракции получают кристаллический продукт. Выход 2,18 г (40%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,40-7,20 (м, 10Н, СН); 3,50 (дд, 1Н, СН2); 3,05 (м, 1Н, СН); 2,85 (дд, 1Н, СН2); 1,40 (д, 3Н, СН3).
Синтез 5-метил-2,3-дифенил-6Н-циклопента[b]тиофена (или 5-метил-2,3-дифенил-1-тиопенталена)
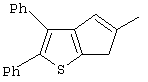
Раствор 5-метил-2,3-дифенил-5,6-дигидро-4Н-циклопента[b]-тиофен-4-она (6,08 г, 20 ммоль) в 75 мл эфира медленно добавляют к раствору LiAlH4 (0,38 г, 10 ммоль) в 50 мл эфира и перемешивают в течение ночи. Полученную суспензию выливают в смесь льда и воды, органический слой отделяют, водный слой экстрагируют эфиром (3× 50 мл). Объединенные органические слои промывают водой, сушат над MgSO4 и упаривают досуха. Полученный таким образом 5-метил-2,3-дифенил-5,6-дигидро-4Н-циклопента[b]тиофен-4-ол растворяют в 100 мл бензола, добавляют 1 г п-толуолсульфокислоты и кипятят с обратным холодильником 10 мин. Реакционную смесь охлаждают до комнатной температуры и обрабатывают насыщенным водным раствором Na2CO3. Органическую фазу отделяют, сушат над MgSO4 и упаривают досуха, получают кристаллическое вещество. Выход 3,46 г (60% относительно исходного кетона).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,40-7,20 (м, 10Н, СН); 6,48 (кв, 1Н, СН); 3,24 (с, 2Н, СН2); 2,24 (д, 3Н, СН3).
Синтез 6Н-6-(5-метил-2,3-дифенилциклопента[b]-тиофен)хлордиметилсилана
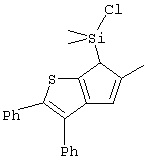
Раствор 1,28 г (4,4 ммоль) 5-метил-2,3-дифенил-6Н-циклопента [b] тиофена в 40 мл Et2O при -70° С обрабатывают 3,5 мл (5,6 ммоль) 1,6 М раствора н-BuLi. После добавления смеси дают нагреться до комнатной температуры и перемешивают в течение 1 часа. Затем реакционную смесь снова охлаждают до -70°С и обрабатывают раствором 1,30 г (10 ммоль) Me2SiCl2 в 10 мл эфира. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную реакционную смесь фильтруют, чтобы удалить LiCl, и растворитель удаляют при пониженном давлении. Сырой продукт используют на следующей стадии без дополнительной очистки.
Спектр 1H ЯМР (C6D6, δ , м.д.): 7,60-7,00 (м, 10Н, СН); 6,56 (м, 1Н, СН); 3,40 (с, 1Н, СН); 2,13 (с, 3Н, СН3); 0,37 (с, 3Н, Si-СН3); 0,15 (с, 3Н, Si-СН3).
Синтез диметилсилил{6-(5-метил-2,3-дифенилциклопента[b]-тиофен)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}-
цирконийдихлорида С-30
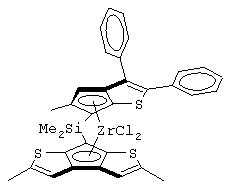
Суспензию 0,9 г (4,4 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 30 мл эфира обрабатывают при -70° С 3,0 мл 1,6 М раствора н-BuLi (4,8 ммоль). После добавления полученной смеси дают нагреться до комнатной температуры и перемешивают при этой температуре еще 50 минут. Затем смесь снова охлаждают до -70° С и добавляют эфирный раствор (10 мл) 6Н-6-(5-метил-2,3-дифенилциклопента[b]тиофен)хлордиметилсилана с предыдущей стадии. Смесь оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Полученный таким образом лиганд 6-{(5-метил-2,3-дифенилциклопента[b]тиофен)}-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}диметилсилан характеризуют с помощью спектроскопии 1H ЯМР.
Спектр 1H ЯМР (C6D6, δ , м.д.): 7,60-7,00 (м, 10Н, СН); 6,85 (м, 1Н, СН); 6,80 (м, 1Н, СН); 6,60 (м, 1Н, СН); 4,35 (с, 1Н, СН); 4,10 (с, 1Н, СН); 2,40 (д, 3Н, СН3); 2,38 (д, 3Н, СН3); 2,05 (с, 3Н, СН3); 0,05 (с, 3Н, Si-СН3); -0,20 (с, 3Н, Si-СН3).
Лиганд не выделяют: его раствор обрабатывают при -70° С 5,6 мл 1,6 М раствора н-BuLi (9,0 ммоль). Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,17 г (5,0 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, желтый осадок отфильтровывают, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,46 г (47% относительно Me2Th).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 7,39-7,24 (м, 10Н, СН); 6,88 (кв, 1Н, J=1,17 Гц, СН); 6,76 (кв, 1H, J=1,17 Гц, СН), 6,59 (уш.с, 1H, СН); 2,63 (д, 3Н, J=1,17 Гц, СН3); 2,51 (д, 3Н, J=1,17 Гц, СН3); 2,35 (с, 3Н, СН3); 1,25 (с, 3Н, Si-СН3); 1,16 (с, 3Н, Si-СН3).
ПРИМЕР 29
Синтез 3-хлор-2-фенил-2-бутеналя
К 0,45 моль (35 мл) ДМФА при 0° С добавляют 0,375 моль (35 мл) РОСl3. После добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем смесь снова охлаждают до 0°С и осторожно обрабатывают 0,15 моль (20,1 г) фенилацетона. Полученную реакционную смесь перемешивают при той же температуре в течение 1 часа. Затем смесь выливают в смесь льда и воды, добавляют ацетат натрия и экстрагируют СНСl3 (3× 50 мл). Органическую фазу отделяют, промывают водой до нейтрального значения рН, сушат над МgSO4 и осторожно упаривают досуха. Остаток перегоняют в вакууме, т.кип. 90-110° С/0,21 мм рт.ст. Выход 10 г (37%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 10,50 (с, 1Н, СНО); 7,40-7,00 (м, 5Н, СН); 2,20 (с, 3Н, СН3).
Синтез этилового эфира 5-метил-4-фенил-2-тиофенкарбоновой кислоты
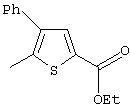
К раствору этилата натрия (46 ммоль, 3,13 г) в 50 мл этанола при 0° С добавляют этил-2-меркаптоацетат (45,8 ммоль, 5 мл), и полученную смесь перемешивают при той же температуре 30 мин. Затем добавляют 3-хлор-2-фенил-2-бутеналь (45,8 ммоль, 8,27 г) и перемешивание продолжают в течение ночи. Полученный продукт кипятят с обратным холодильником 2 часа, затем охлаждают до комнатной температуры и разбавляют 100 мл воды. Органический слой отделяют, водный слой экстрагируют CH2Cl2 (3× 50 мл). Объединенные органические слои сушат над МgSO4 и упаривают досуха. Остаток используют на следующей стадии без дополнительной очистки. Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез 5-метил-4-фенил-2-тиофенкарбоновой кислоты
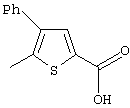
К 30%-ному раствору гидроксида натрия в 100 мл этанола добавляют 5-метил-4-фенил-2-тиофенэтилкарбоксилат с предыдущей стадии, и полученную смесь кипятят с обратным холодильником 2 часа. Затем смесь разбавляют водой и экстрагируют 50 мл бензола. Водную фазу выделяют, подкисляют и смесь фильтруют. Полученный осадок сушат под P2O5. Выход 9,5 г (95% относительно 3-хлор-2-фенил-2-бутеналя).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 12,00 (с, 1Н, СООН); 7,90 (с, 1Н, СН); 7,50-7,40 (м, 5Н, СН); 2,55 (с, 3Н, СН3).
Синтез 2-метил-3-фенилтиофена
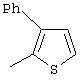
5-Метил-4-фенил-2-тиофенкарбоновую кислоту (0,25 моль, 54 г), полученную как описано выше, нагревают до 220-230° С, пока не прекратиться выделение диоксида углерода. Продукт собирают и перегоняют, т.кип. 117° С/10 мм рт.ст. Выход 30 г (70%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,45 (м, 5Н, СН); 7,15 (д, 1Н, СН); 7,10 (д, 1Н, СН); 2,55 (с, 3Н, СН3).
Синтез 2-формил-5-метил-4-фенилтиофена
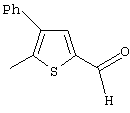
К 0,5 моль (39 мл) ДМФА при 0°С добавляют 0,166 моль (15 мл) РОСl3. После добавления смеси дают нагреться до комнатной температуры и перемешивают в течение 1 часа. Затем смесь снова охлаждают до 0° С и обрабатывают 2-метил-3-фенилтиофеном (29 г, 0,166 моль). Полученной реакционной смеси дают нагреться до комнатной температуры и после перемешивания в течение 12 часов при этой же температуре смесь нагревают при 80° С в течение 2 дней. Затем смесь выливают в смесь льда и воды и добавляют ацетат натрия. Полученный таким образов осадок отфильтровывают, промывают водой и затем гексаном. Желтый порошок сушат в вакууме. Выход 27,7 г (83%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 9,88 (с, 1Н, СНО); 7,70 (с, 1Н, СН); 7,55-7,40 (м, 5Н, СН); 2,55 (с, 3Н, СН3).
Синтез 2-метил-3-(5-метил-4-фенил-2-тиенил)этилакрилата
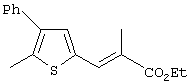
Смесь 2-формил-5-метил-4-фенилтиофена (27,6 г, 0,136 моль) и этил-2-бромпропионата (0,14 моль, 18,2 мл) добавляют к суспензии Zn (9,8 г, 0,15 моль) в 250 мл бензола с каталитическим количеством I2. Полученную смесь кипятят с обратным холодильником при перемешивании, пока не растворится все количество цинка, и затем растворяют в воде. Органический слой отделяют, промывают 10% водным раствором НСl, сушат над МgSO4 и упаривают досуха. Остаток используют в синтезе соответствующей кислоты без дополнительной очистки. Названное соединение характеризуют с помощью спектроскопии 1H ЯМР.
Синтез 2-метил-3-(5-метил-4-фенил-2-тиенил)акриловой кислоты
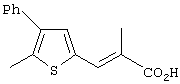
К 30%-ному водному раствору гидроксида натрия в 200 мл этанола добавляют 2-метил-3-(5-метил-4-фенил-2-тиенил)этилакрилат с предыдущей стадии и кипятят с обратным холодильником 2 часа. Полученную реакционную смесь разбавляют водой, подкисляют и фильтруют. Осадок сушат над Р2O5. Выход 26,0 г (74% относительно 2-формил-5-метил-4-фенилтиофена).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,90 (с, 1Н, СН); 7,50-7,30 (м, 5Н, СН); 7,25 (д, 1Н, СН); 2,60 (с, 3Н, СН3); 2,25 (с, 3Н, СН3).
Синтез 2-метил-3-(5-метил-4-фенил-2-тиенил)пропановой кислоты
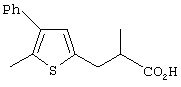
Требуемую 2-метил-3-(5-метил-4-фенил-2-тиенил)-2-пропановую кислоту получают электрохимическим восстановлением 2-метил-3-(5-метил-4-фенил-2-тиенил)акриловой кислоты. Выход ~ 100%.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,50-7,20 (м, 5Н, СН); 6,80 (с, 1Н, СН); 3,25 (дд, 1Н, СН); 2,85 (м, 2Н, СН2); 2,50 (с, 3Н, СН3); 1,30 (д, 3Н, СН3).
Синтез 2,5-диметил-3-фенил-5,6-дигидро-4Н-циклопента[b]-тиофен-4-она
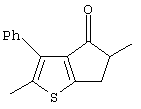
Раствор 3 г P2O5 (21 ммоль) в 30 мл метансульфоновой кислоты (0,46 моля) нагревают при перемешивании при 80° С. Добавляют раствор 2-метил-3-(5-метил-4-фенил-2-тиенил)-2-пропановой кислоты (65 ммоль, 16,9 г) в 20 мл CH2Cl2 и полученную реакционную смесь перемешивают при той же температуре 1,5 часа. Затем смесь выливают в смесь льда и воды и энергично перемешивают. Водный слой экстрагируют СН2Сl2 (3× 50 мл), органические слои собирают, промывают 10% водным раствором карбоната натрия до нейтрального значения рН, затем промывают водой. Органические слои собирают, сушат над MgSO4 и упаривают досуха. Остаток пропускают через наполненную силикагелем 60 колонку, используя в качестве элюента смесь гексан/этилацетат 3/1. После упаривания красной фракции получают маслянистый продукт. Выход 4,4 г (28%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,50-7,40 (м, 5Н, СН); 3,42 (дд, 1Н, СН2); 3,02 (м, 1Н, СН); 2,79 (дд, 1Н, СН2); 2,50 (с, 3Н, СН3); 1,35 (д, 3Н, СН3).
Синтез 2,5-диметил-3-фенил-6Н-циклопента[b]тиофена (или 2,5-диметил-3-фенил-1-тиопенталена)
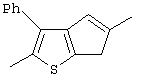
Раствор 2,5-диметил-3-фенил-5,6-дигидро-4Н-циклопента[b]-тиофен-4-она (4,4 г, 18 ммоль) в 50 мл эфира медленно добавляют к раствору LiAlH4 (0,35 г, 9 ммоль) в 50 мл эфира и перемешивают в течение ночи. Полученную суспензию выливают в смесь льда и воды, органический слой отделяют, водный слой экстрагируют эфиром (3× 50 мл). Объединенные органические слои промывают водой, сушат над МgSO4 и упаривают досуха. Полученный таким образом 2,5-диметил-3-фенил-5,6-дигидро-4Н-циклопента[b]тиофен-4-ол растворяют в 100 мл бензола, добавляют 1 г п-толуолсульфокислоты и кипятят с обратным холодильником 10 мин. Реакционную смесь охлаждают до комнатной температуры и обрабатывают насыщенным водным раствором Nа2СО3. Органическую фазу отделяют, сушат над MgSO4 и упаривают досуха. Остаток пропускают через наполненную Аl2O3 колонку, используя в качестве элюента гексан. После упаривания желтой фракции получают кристаллический продукт. Выход 1,5 г (37% относительно исходного кетона).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,50-7,40 (м, 5Н, СН); 6,52 (кв, 1Н, СН); 3,24 (д, 1Н, CH2); 2,60 (д, 1Н, СН2); 2,24 (дд, 3Н, СН3); 1,66 (с, 3Н, СН3).
Синтез 6Н-6-(2,5-диметил-3-фенилциклопента[b]тиофен)-хлордиметилсилана
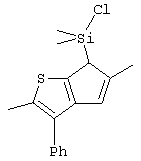
Раствор 1,20 г (5,3 ммоль) 2,5-диметил-3-фенил-6Н-циклопента[b]тиофена в 40 мл Et2O при -70° С обрабатывают 3,3 мл (5,3 ммоль) 1,6 М раствора н-BuLi. После добавления смеси дают нагреться до комнатной температуры и перемешивают 1 час. Затем реакционную смесь снова охлаждают до -70° С и обрабатывают раствором 1,30 г (10,1 ммоль) Me2SiCl2 в 10 мл эфира. По окончании добавления смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную реакционную смесь фильтруют, чтобы удалить LiCl, и растворитель удаляют при пониженном давлении. Сырой продукт используют на следующей стадии в том виде, как он есть, без дополнительной очистки.
Спектр 1H ЯМР (C6D6, δ , м.д.): 7,45-7,20 (м, 5Н, СН); 6,50 (м, 1Н, СН); 3,40 (с, 1Н, СН); 2,38 (с, 3Н, СН3); 2,12 (с, 3Н, СН3); 0,36 (с, 3Н, Si-СН3); 0,11 (с, 3Н, Si-СН3).
Синтез диметилсилил{6-(2,5-диметил-3-фенилциклопента[b]-тиофен)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}-
цирконийдихлорида С-33
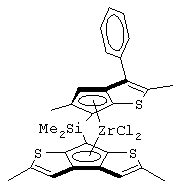
Суспензию 0,9 г (4,4 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]-дитиофена в 30 мл эфира обрабатывают при -70° С 3,0 мл 1,6 М раствора н-BuLi (4,8 ммоль). После добавления полученной смеси дают нагреться до комнатной температуры и перемешивают при этой температуре еще 50 минут. Затем смесь охлаждают до -70° С и добавляют эфирный раствор (10 мл) 6Н-6-(2,5-диметил-3-фенилциклопента[b]тиофен)хлордиметилсилана с предыдущей стадии. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученный таким образом лиганд 6-{(2,5-диметил-3-фенилциклопента[b]тиофен)}-7-{(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}диметилсилан характеризуют с помощью спектроскопии 1H ЯМР.
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,45-7,20 (м, 5Н, СН); 6,85 (с, 1Н, СН); 6,80 (м, 1Н, СН); 6,60 (м, 1Н, СН); 4,30 (с, 1Н, СН); 4,00 (с, 1Н, СН); 2,70 (д, 3Н, СН3); 2,65 (д, 3Н, СН3); 2,60 (с, 3Н, СН3); 2,30 (с, 3Н, СН3); -0,18 (с, 3Н, Si-СН3); -0,30 (с, 3Н, Si-СН3).
Лиганд не выделяют: его раствор обрабатывают при -70° С 5,6 мл 1,6 М раствора н-BuLi (9,0 ммоль). Затем реакционной смеси дают нагреться до комнатной температуры и перемешивают 1 час. Растворитель удаляют при пониженном давлении и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,17 г (5,0 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, желтый осадок отфильтровывают, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,68 г (59% относительно Me2Th). Требуемое соединение характеризуют с помощью спектроскопии 1H ЯМР.
ПРИМЕР 30
Синтез 1-бром-2-(3-инденил)этана
К раствору индена (18,6 г, Мм=116,16, 0,16 моль) в 300 мл эфира при 0° С добавляют 1,6 М раствор н-BuLi в гексане (100 мл, 0,16 моль). Полученной суспензии дают нагреться до комнатной температуры и перемешивают при той же температуре 4 часа. Суспензию индениллития снова охлаждают до -50° С и затем добавляют раствор 1,2-дибромэтана (0,24 моль, 21 мл) в 50 мл эфира. Реакционной смеси дают медленно нагреться до комнатной температуры и перемешивают в течение ночи. Затем смесь обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу выделяют, упаривают досуха и перегоняют в вакууме, т.кип. 110° С/0,5 мм рт.ст. Выход 21,6 г (60%). Названное соединение характеризуют с помощью спектроскопии ЯМР.
Синтез 1,2-(3-инденил)-7-(2,5-диметилциклопента[1,2-b]:[4,3-b’]дитиофен)этана
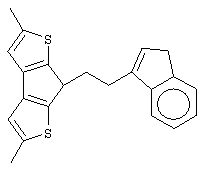
Раствор 2,5-диметилциклопента[1,2-b:4,3-b’]дитиофена (1,03 г, 5 ммоль) в 50 мл ТГФ обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (3,1 мл, 5 ммоль). Полученную смесь перемешивают еще 45 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают раствором 1-бром-2-(3-инденил)этана (1,12 г, 5 ммоль) в 25 мл ТГФ. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу отделяют, и растворители удаляют. Остаток пропускают через наполненную силикагелем колонку с использованием в качестве элюента гексана. Выход 1,26 г (72%). Названное соединение характеризуют с помощью спектроскопии ЯМР.
Синтез этилиден{(1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}цирконийдихлорида С-37
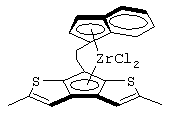
Раствор 1,2-(3-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)этана (1,26 г, 3,62 ммоль) в 15 мл эфира и 60 мл гексана обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (4,7 мл, 7,5 ммоль). Полученную суспензию перемешивают еще 2 часа при комнатной температуре, затем снова охлаждают до -70° С и добавляют ZrCl4 (0,94 г, 4 ммоль). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Темно-оранжевый осадок отфильтровывают, дважды промывают эфиром, сушат и затем перекристаллизовывают из CH2Cl2. Выход 0,92 г (50%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 7,70 (дд, 1Н, СН); 7,45 (дд, 1Н, СН); 7,20 (м, 1Н, СН); 7,10 (м, 1Н, СН); 6,75 (кв, 1Н, СН); 6,60 (кв, 1Н, СН); 6,55 (дд, 1Н, СН); 6,40 (д, 1Н, СН); 3,95-3,80 (м, 2Н, СН2); 3,65-3,55 (м, 2Н, СН2); 2,60 (д, 3Н, СН3); 2,45 (д, 3Н, СН3).
ПРИМЕР 31
Синтез хлор(4,7-диметил-1-инденил)диметилсилана
Предшественник 4,7-диметилинден получают по стандартной методике (опубликованной в Tetrahedron, 51, (1995), 4347).
К раствору 4,7-диметилиндена (14,42 г, Мм=144,22, 0,1 моль) в 200 мл гексана и 50 мл ТГФ добавляют при 0° С 1,6 М раствор н-BuLi в гексане (62,5 мл, 0,1 моль). Полученной суспензии дают нагреться до комнатной температуры и перемешивают при той же температуре 4 часа. Суспензию индениллития снова охлаждают до -50° С и затем добавляют раствор дихлордиметилсилана (0,2 моль, 24 мл) в 50 мл ТГФ. Полученной суспензии дают нагреться до комнатной температуры и перемешивают в течение ночи. Отфильтровывают хлорид лития, фильтрат упаривают досуха и перегоняют в вакууме, т.кип. 98° С/0,5 мм рт.ст. Выход 16,5 г (70%). Названное соединение характеризуют с помощью спектроскопии ЯМР.
Синтез (4,7-диметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
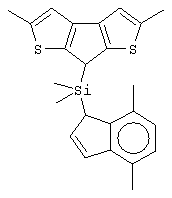
Раствор 2,5-диметилциклопента[ 1,2-b:4,3-b’]дитиофена (1,90 г, 9,2 ммоль) в 50 мл эфира обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (5,8 мл, 9,2 ммоль). Полученную смесь перемешивают еще 45 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают раствором хлор(4,7-диметил-1-инденил)-диметилсилана (2,18 г, 9,2 ммоль) в 10 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу отделяют и растворители удаляют. Остаток перекристаллизовывают из гексана. Выход 3,67 г (98%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 7,07 (дд, 1Н, СН); 7,05 (д, 1Н, СН); 6,95 (д, 1Н, СН); 6,90 (м, 2Н, СН); 6,60 (дд, 1Н, СН); 4,00 (с, 1Н, СН); 3,85 (с, 1Н, СН); 2,64 (с, 3Н, СН3); 2,60 (с, 3Н, СН3); 2,50 (с, 3Н, СН3); 2,40 (с, 3Н, СН3); -0,20 (с, 3Н, Si-СН3); -0,40 (с, 3Н, Si-СН3).
Синтез диметилсилил{(4,7-диметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-38
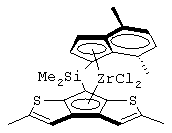
Раствор (4,7-диметил-1-инденил)-7-(2,5-диметилциклопента-[1,2-b:4,3-b’]дитиофен)диметилсилана (3,67 г, 9,02 ммоль) в 15 мл эфира и 50 мл гексана обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (12,5 мл, 20 ммоль). Полученную суспензию перемешивают еще 2 часа при комнатной температуре, затем охлаждают до -70° С и добавляют ZrCl4 (2,52 г, 10,8 ммоль). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Желтый осадок отфильтровывают, дважды промывают эфиром, сушат и затем перекристаллизовывают из СН2Сl2. Выход 3,57 г (70%). Названное соединение характеризуют с помощью спектроскопии ЯМР.
ПРИМЕР 32
Синтез хлор(2,4,7-триметил-1-инденил)диметилсилана
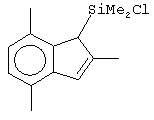
Предшественник 2,4,7-триметилинден получают по стандартной методике (опубликована в Европейской патентной заявке 0693506).
К раствору 2,4,7-триметилиндена (9,5 г. Мм=158,24, 60 ммоль) в 200 мл гексана и 50 мл ТГФ добавляют при 0° С 1,6 М раствор н-BuLi в гексане (37,5 мл, 60 ммоль). Полученной суспензии дают нагреться до комнатной температуры и перемешивают при той же температуре 4 часа. Суспензию индениллития снова охлаждают до -50° С и затем добавляют раствор дихлордиметилсилана (90 ммоль, 11 мл) в 50 мл ТГФ. Полученной суспензии дают нагреться до комнатной температуры и перемешивают в течение ночи. Отфильтровывают осадок хлорида лития, фильтрат упаривают досуха и перегоняют в вакууме, т.кип. 110° С/0,5 мм рт.ст. Выход 10,1 г (67%).
Спектр 1H ЯМР (C6D6, δ , м.д.): 6,75 (д, 1Н, СН); 6,60 (д, 1Н, СН); 6,30 (с, 1Н, СН); 3,25 (с, 1Н, СН); 2,25 (с, 3Н, СН3); 2,15 (с, 3Н, СН3); 2,10 (с, 3Н, СН3); -0,05 (с, 3Н, Si-СН3); -0,02 (с, 3Н, Si-СН3).
Синтез (2,4,7-триметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
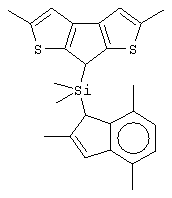
Раствор 2,5-диметилциклопента[1,2-b:4,3-b’]дитиофена (1,65 г, 8 ммоль) в 50 мл эфира обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (5,0 мл, 8 ммоль). Полученную смесь перемешивают еще 45 мин при 0° С, затем снова охлаждают до -70° С и обрабатывают раствором хлор(2,4,7-триметил-1-инденил)диметилсилана (2,0 г, 8 ммоль) в 20 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу отделяют и растворители удаляют. Выход 3,36 г (~ 100%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 6,90 (д, 1Н, СН); 6,80 (д, 1Н, СН); 6,70 (м, 2Н, СН); 6,65 (м, 1Н, СН); 4,15 (с, 1Н, СН); 4,00 (с, 1Н, CH); 2,65 (с, 3Н, СН3); 2,63 (с, 3Н, СН3); 2,55 (с, 3Н, СН3); 2,50 (с, 3Н, СН3); 2,20 (с, 3Н, СН3); -0,15 (с, 3Н, Si-СН3), -0,30 (с, 3Н, Si-СН3).
Синтез диметилсилил{(2,4,7-триметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b: 4, 3-b’]дитиофен)}цирконийдихлорида С-39
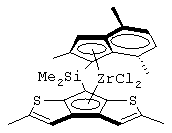
Раствор (2,4,7-триметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана (3,36 г, 8,0 ммоль) в 80 мл эфира обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (12,5 мл, 20 ммоль). Полученную суспензию перемешивают еще 2 час при комнатной температуре, затем охлаждают до -70° С и добавляют ZrCl4 (2,34 г, 10 ммоль). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Оранжевый осадок отфильтровывают, дважды промывают эфиром, сушат и затем перекристаллизовывают из CH2Cl2. Выход 2,73 г (59%). Названное соединение характеризуют с помощью спектроскопии ЯМР.
ПРИМЕР 33
Синтез 2-метил-2,3-дигидро-1Н-циклопента[а]нафталин-1-она
К суспензии АlСl3 (85 г, 0,635 моль) в 200 мл СН2Сl2 при 0° С добавляют сначала нафталин (32,5 г, 0,254 моль) и затем раствор 2-метилакрилоилхлорида (26,5 г, 0,254 моль) в 50 мл СН2Сl2. Реакционную смесь перемешивают 30 мин при 0° С, затем 2 час при комнатной температуре и, наконец, выливают в смесь льда и воды. Темный органический слой отделяют, водный слой экстрагируют СНСl3 (3× 150 мл). Органические слои собирают, промывают смесью карбонат калия/вода до нейтрального значения рН, сушат над MgSO4, упаривают досуха. Выход 28,09 г (56%). Названное соединение характеризуют с помощью спектроскопии ЯМР.
Синтез 2-метил-2,3-дигидро-1Н-циклопента[а]нафталин-1-ола
Раствор 2-метил-2,3-дигидро-1Н-циклопента[а]нафталин-1-она (29,09 г, 0,143 моль) в 100 мл ТГФ медленно добавляют к суспензии LiAlH4 (2,18 г, 58 ммоль) в 200 мл эфира и кипятят с обратным холодильником в течение 2 часов при перемешивании. Затем реакционную смесь переносят в стакан объемом 2 л и при непрерывном перемешивании медленно гидролизуют, добавляя по каплям 10% водный раствор НСl до рН 5. Органический слой отделяют, водный слой экстрагируют эфиром (3× 100 мл). Органические слои собирают, промывают смесью карбонат калия/вода до нейтрального значения рН, сушат над MgSO4 и упаривают досуха. Полученный таким образом продукт в виде смеси двух изомеров используют на следующей стадии без дополнительной очистки.
Синтез 2-метил-3Н-циклопента[а]нафталина
Смесь 2-метил-2,3-дигидро-1Н-циклопента[а]нафталин-1-ола (полученного как описано выше) и 1 г п-толуолсульфокислоты в 200 мл бензола кипятят с обратным холодильником 1 час. Затем реакционную смесь охлаждают до комнатной температуры и обрабатывают насыщенным водным раствором Na2CO3. Органическую фазу отделяют, сушат над МgSO4 и упаривают досуха. Выход 14,2 г (55% относительно исходного 2-метил-2,3-дигидро-1H-циклопента[а]нафталин-1-она).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 8,10-7,40 (м, 6Н, СН); 7,10 (с, 1Н, СН); 3,50 (с, 2Н, CH2); 2,33 (с, 3Н, СН3).
Синтез хлор(2-метил-3Н-циклопента[а]нафталин-3-ил)-диметилсилана
К раствору 2-метил-3Н-циклопента[а]нафталина (2,14 г, Мм=180,25, 11,9 ммоль) в 50 мл эфира при -50° С добавляют 1,6 М раствор н-BuLi в гексане (7,5 мл, 12 ммоль). Полученной суспензии дают нагреться до комнатной температуры и перемешивают при этой температуре 45 мин. Затем литиевую суспензию охлаждают до -70°С и добавляют дихлордиметилсилан (18 ммоль, 2,2 мл). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Осадок хлорида лития отфильтровывают, фильтрат упаривают досуха и сушат в вакууме. Выход 3,14 г (97%).
Спектр 1H ЯМР (C6D6, δ , м.д.): 8,10 (д, 1Н, СН); 7,85 (д, 1Н, СН); 7,50-7,40 (м, 4Н, СН); 7,10 (с, 1Н, СН); 3,55 (с, 1Н, СН); 2,30 (с, 3Н, СН3); 0,22 (с, 3Н, Si-СН3); -0,05 (с, 3Н, Si-СН3).
Синтез (2-метилциклопента[а]нафталин-3-ил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
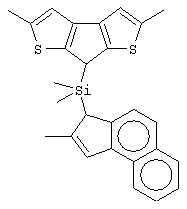
Раствор 2,5-диметилциклопента[1,2-b:4,3-b’]дитиофена (2,37 г, 11,5 ммоль) в 75 мл эфира обрабатывают при -70° С 1,6 М раствором н-BuLi в гексане (7,5 мл, 12 ммоль). Полученную смесь перемешивают 45 мин при 0° С, затем снова охлаждают до -70° С и добавляют раствор хлор(2-метил-3Н-циклопента[а]нафталин-3-ил)диметилсилана (3,14 г, 11,5 ммоль) в 25 мл эфира. Реакционной смеси дают нагреться до комнатной температуры и затем обрабатывают насыщенным водным раствором NH4Cl. Органическую фазу отделяют, растворители удаляют. Остаток пропускают через короткую колонку, наполненную силикагелем, используя в качестве элюента гексан. Выход 4,07 г (80%).
Спектр 1H ЯМР (CDCl3, δ , м.д.): 8,20 (д, 1Н, СН); 8,00 (д, 1Н, СН); 7,70-7,40 (м, 4Н, СН); 6,95 (д, 2Н, СН); 6,80 (с, 1Н, СН); 4,20 (с, 1Н, СН); 4,15 (с, 1Н, СН); 2,66 (с, 6Н, СН3); 2, 45 (с, 3Н, СН3); -0,27 (с, 3Н, Si-СН3), -0,29 (с, 3Н, Si-СН3).
Синтез диметилсилил{(2-метилциклопента[а]нафталин)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида С-40
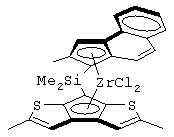
Раствор (2-метилциклопента[а]нафталин-3-ил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана (4,07 г, 9,2 ммоль) в 100 мл эфира обрабатывают при -70° С 1,6 М раствором н-BuLi (15 мл, 24 ммоль). Полученную суспензию перемешивают 2 час при комнатной температуре, затем охлаждают до -70° С и добавляют ZrCl4 (2,79 г, 12 ммоль). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Оранжевый осадок отфильтровывают, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из СН2Сl2. Выход 2,77 г (50%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 8,00 (д, 1Н, СН); 7,70 (д, 1Н, СН); 7,60 (д, 1Н, СН); 7,55 (т, 1Н, СН); 7,48 (т, 1Н, СН); 7,25 (с, 1Н, СН); 7,15 (д, 1Н, СН); 6,78 (кв, 1Н, СН); 6,65 (кв, 1Н, СН); 2,60 (д, 3Н, СН3); 2,55 (д, 3Н, СН3); 2,40 (д, 3Н, СН3); 1,32 (с, 3Н, Si-СН3); 1,18 (с, 3Н, Si-СН3).
ПРИМЕР 34
Синтез диметилсилил{(1-инденил)-7-(2,5-дитриметилсилилциклопента[1,2-b:4,3-b’]-дитиофен)}
цирконийдихлорида С-13
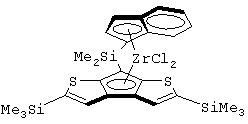
Синтез лиганда С-13 проводят сочетанием литиевой соли 2,5-дитриметилсилил-7Н-циклопента[1,2-b:4,3-b’]дитиофена с хлор(1-инденил)диметилсиланом.
Суспензию 1,48 г (3 ммоль) (1-инденил)-7-{(2,5-дитриметилсилилциклопента[1,2-b:4,3-b’]дитиофен)}диметилсилана в 50 мл эфира обрабатывают при -70° С 4,1 мл 1,6 М раствора н-BuLi (6,5 ммоль). После добавления реакционной смеси дают нагреться до 0° С и добавляют 0,75 г ZrCl4 (3,2 ммоль). Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Затем полученный красный осадок отфильтровывают, промывают дважды эфиром, сушат и, наконец, перекристаллизовывают из CH2Cl2. Выход 1,38 г (70%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 7,90-6,90 (м, 7Н, СН); 6,10 (м, 1Н, СН); 1,40 (с, 3Н, Si-СН3); 1,10 (с, 3Н, Si-СН3); 0,41 (с, 9Н, Si (СН3)3); 0,20 (с, 9Н, Si (СН3)3).
ПРИМЕР 35
Синтез (3-метил-4-триметилсилил-1-циклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
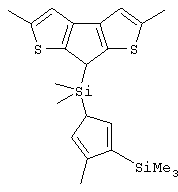
Раствор 1,03 г (5,0 ммоль) 2,5-диметил-7Н-циклопента-[1,2-b:4,3-b’]дитиофена в 40 мл эфира обрабатывают при -70° С 3,13 мл 1,6 М раствора н-BuLi (5,0 ммоль). После добавления полученной смеси дают нагреться до комнатной температуры и перемешивают при этой температуре еще 1 час. Затем смесь снова охлаждают до -70° С и добавляют раствор 1,22 г (5 ммоль) хлордиметил(3-метил-4-триметилсилил-1-циклопентадиенил)силана в 10 мл эфира. Полученной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученный таким образом лиганд (3-метил-4-триметилсилил-1-циклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилан не выделяют, а используют в виде раствора при синтезе катализатора (см. ниже).
Синтез диметилсилил{(3-метил-4-триметилсилил-1-циклопентадиенил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]-дитиофен)}-
цирконийдихлорида [С-32]
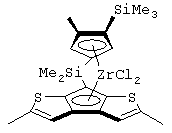
Раствор лиганда с предыдущей стадии обрабатывают при
-70° С 6,3 мл 1,6 М раствора н-BuLi (10,1 ммоль). После добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают при этой температуре еще 50 мин. Растворитель удаляют при пониженном давлении, и полученную дилитиевую соль суспендируют в гексане. После охлаждения до -70° С добавляют 1,17 г (5,0 ммоль) ZrCl4. Реакционную смесь перемешивают при комнатной температуре в течение ночи, желтый осадок отфильтровывают, промывают дважды гексаном, сушат и, наконец, перекристаллизовывают из Et2O. Выход 0,90 г (31%).
Спектр 1H ЯМР (CD2Cl2, δ , м.д.): 6,95 (кв, 1Н, J=1,17 Гц, СН); 6,91 (кв, 1Н, J=1,17 Гц, СН); 5,77 (д, 1Н, J=2,74 Гц, СН); 5,68 (д, 1Н, J=2,74 Гц, СН); 2,62 (кв, 3Н, J=1,17 Гц, СН3); 2,59 (кв, 3Н, J-1,17 Гц, СН3); 2,22 (с, 3Н, СН3); 0,91 (с, 3Н, Si-СН3); 0,89 (с, 3Н, Si-СН3); 0,22 (с, 9Н, Si-(СН3)3).
ПРИМЕР 36
Синтез 2,5-диметил-7Н-тиено[1,2-b:4,3-b’]циклопента[b]-тиофен-7-ил)(диметил)(2,3,4,5-тетраметил-2,4-циклопентадиенил)силана
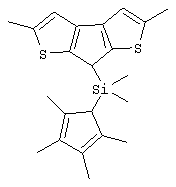
Раствор 1,03 г (5 ммоль) 2,5-диметилциклопентадиен[1,2-b:4,3-b’] дитиофена в 20 мл эфира обрабатывали при -70° С, пользуясь 3,13 мл 1,6 М BuLi (5 ммоль). Получающуюся в результате смесь перемешивали еще в течение 30 мин при 0° С, еще раз охлаждали до -70° С, а после этого обрабатывали, пользуясь 1,07 г (5 ммоль) (2,3,4,5-тетраметил-2,4-циклопентадиенил)диметилхлорсилана в 10 мл эфира. Смеси давали возможность разогреться до комнатной температуры, продукт начинал выпадать в осадок. Его отфильтровывали, промывали водой и высушивали. Выход 0,84 г (50%).
1H ЯМР (CDCl3, 20° С, δ , м.д.): 6,78 (с, 2Н); 3,90 (с, 1Н); 3,35 (уш.с, 1Н); 2,58 (с, 6Н); 2,03 (с, 6Н); 1,86 (с, 6Н); -0,38 (с, 6Н).
Получение 2,5-диметил-7Н-тиено[1,2-b:4,3-b’]циклопента[b]тиофен-7-ил)(диметил)(2,3,4,5-тетраметил-2,4-циклопентадиенил) силандихлорциркония [Me2Si(Me4Cp)(7-MeTh2-Cp)ZrCl2]
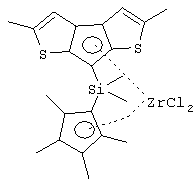
Суспензию 0,8 г (2,1 ммоль) 2,5-диметил-7Н-тиено[3’,2’:3,4]циклопента[b]тиофен-7-ил)(диметил)(2,3,4,5-тетраметил-2,4-циклопентадиенил) силана в 20 мл эфира обрабатывали, пользуясь 2,6 мл (4,2 ммоль) 1,6 М BuLi, при -70° С. Смеси давали возможность разогреться до комнатной температуры, в осадок выпадала желтоватая дилитиевая соль. Соль два раза промывали эфиром, а после этого при -50° С обрабатывали суспензией 0,48 г (2,1 ммоль) ZrCl4 в 20 мл эфира. Реакционную смесь перемешивали при температуре образования флегмы в течение 3 часов, после этого желтоватый осадок отфильтровывали, два раза промывали эфиром, высушивали и затем перекристаллизовывали из CH2Cl2. Выход 1,02 г (90%). Указанное в заголовке соединение анализировали, пользуясь методом 1Н ЯМР.
1H ЯМР (CDCl3, 20° С, δ , м.д.): 6,85 (кв, 2Н); 2,56 (д, 6Н); 2,02 (с, 6Н); 1,97 (с, 6Н); 1,05 (с, 6Н).
Пример 37
Синтез (2,5-диметил-7Н-тиено[1,2-b:4,3-b’]циклопента[b]-тиофен-7-ил)(9Н-9-флуоренил)диметилсилана
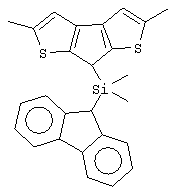
Раствор 1,03 г (5 ммоль) 2,5-диметилциклопентадиен[1,2-b:4,3-b’]дитиофена в 20 мл эфира обрабатывали при -70° С, пользуясь 3,13 мл 1,6 М BuLi (5 ммоль). Получающуюся в результате смесь перемешивали еще в течение 30 мин при 0° С, еще раз охлаждали до -70° С, а после этого обрабатывали, пользуясь 1,29 г (5 ммоль) диметил(9Н-9-флуоренил)хлорсилана в 10 мл эфира. Смеси давали возможность разогреться до комнатной температуры, после этого ее обрабатывали насыщенным водным раствором NH4Cl. Органическую фазу отделяли, растворитель удаляли, а остаток перекристаллизовывали из гексана. Выход 1,28 г (60%). Указанное в заголовке соединение анализировали, пользуясь методом спектроскопии 1H ЯМР.
1H ЯМР (CDCl3, 20° С, δ , м.д.): 7,92 (д, 2Н); 7,56 (дд, 2Н); 7,40 (тд, 2Н); 7,32 (тд, 2Н); 6,92 (д, 1Н); 4,30 (с, 2Н); 4,10 (с, 2Н); 2,55 (д, 3Н); -0,50 (с, 6Н).
Получение (2,5-диметил-7Н-тиено[1,2-b:4,3-b’]циклопента[b]-тиофен-7-ил)(9Н-9-флуоренил)диметилсилилдихлорциркония [Me2Si(Flu)(7-MeTh2-Cp)ZrCl2]
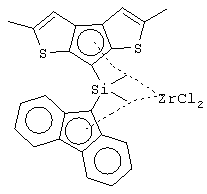
Суспензию 1,28 г (3,0 ммоль) (2,5-диметил-7Н-тиено[3’,2’:3,4]циклопента[b]тиофен-7-ил)(9Н-9-флуоренил)диметилсилана в 20 мл эфира обрабатывали, пользуясь
3,75 мл (6,0 ммоль) 1,6 М BuLi при -70° С. Смеси давали возможность разогреться до комнатной температуры, в осадок выпадала желтоватая дилитиевая соль. Соль два раза промывали эфиром и высушивали. Таким образом полученную соль суспендировали в 20 мл CH2Cl2 при -70° С, а после этого обрабатывали, пользуясь 0,7 г (3 ммоль) ZrCl4 при той же самой температуре. Реакционной смеси давали возможность разогреться до комнатной температуры, после этого ее перемешивали при температуре образования флегмы в течение 1 часа. Красный кристаллический осадок отфильтровывали, два раза промывали, пользуясь СН2Сl2, и высушивали. Выход 1,31 г (90%).
1H ЯМР (CDCl3, 20° С, δ , м.д.): 7,98 (д, 2Н); 7,66 (дд, 2Н); 7,48 (тд, 2Н); 7,41 (тд, 2Н); 6,97 (д, 1Н); 4,50 (с, 1Н); 4,30 (с, 2Н); 2,58 (д, 3Н); -0,50 (с, 6Н).
Пример 38
Синтез диметилсилил{(2,4,7-триметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофена)}
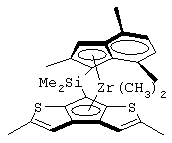
Диметилсилил{(2,4,7-триметил-1-инденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдиметил получали в соответствии со следующей методикой: лиганд - [3-(2,4,7-триметилинденил)][7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)]диметилсилан - получали так, как это описывается в примере 32 из WO 01/47939. 30,40 г данного лиганда (72,26 ммоль) и 170 мл безводного ТГФ загружали в атмосфере азота в цилиндрический стеклянный реактор, оснащенный магнитной мешалкой. Таким образом полученный коричневый раствор охлаждали и выдерживали при 0° С в то время, пока через капельную воронку по каплям добавляли 58,4 мл 2,5 М раствора н-BuLi в гексане (146 ммоль). По завершении добавления темно-коричневый раствор перемешивали в течение 1 часа при комнатной температуре, после этого охлаждали до -50° С, а затем к нему добавляли 48,6 мл 3,05 М раствора MeLi в диэтоксиметане (148,2 ммоль). В пробирке Шленка в 170 мл толуола суспендировали 16,84 г ZrCl4 (72,26 ммоль). Обе смеси выдерживали при -50° С и к раствору дианиона лиганда быстро добавляли суспензию ZrCl4. По завершении добавления реакционной смеси давали возможность достичь комнатной температуры, и ее перемешивали еще в течение часа. Получали желто-зеленую суспензию. Анализ по методу 1H ЯМР продемонстрировал полную конверсию с получением указанного в заголовке комплекса. Все летучие компоненты удаляли при пониженном давлении, а полученный сыпучий коричневый порошок суспендировали в 100 мл Et2O. После перемешивания в течение нескольких минут суспензию отфильтровывали через фритту G4. Твердую фазу на фритте после этого два раза промывали, пользуясь Et2О (до тех пор, пока растворитель для промывания из коричневого не становился желтым), затем высушивали в вакууме и, наконец, экстрагировали на фритте теплым толуолом (60° С) до тех пор, пока раствор после фильтрования из желтого не становился бесцветным (приблизительно 650 мл толуола). Экстракт высушивали при пониженном давлении до получения 28,6 г желтого порошка, который, как продемонстрировал метод 1H ЯМР, представлял собой указанный в заголовке комплекс, не содержащий примесей. Выход, рассчитанный по лиганду, составлял 73,3%.
1H ЯМР (CD2Cl2, комн. температура), м.д.: -2,09 (с, 3Н); -0,79 (с, 3Н); 1,01 (с, 3Н); 1,04 (с, 3Н); 2,38 (с, 3Н); 2,39 (с, 3Н); 2,43 (д, 3Н, J=1,37 Гц); 2,52 (с, 3Н); 2,57 (д, 3Н, J=1,37 Гц); 6,61 (дкв, 1Н, J=7,04 Гц, J=0,78 Гц); 6,81 (кв, 1H, J=1,37 Гц); 6,85 (дкв, 1H, J=7,04 Гц, J=0,78 Гц); 6,87 (kb, 1H, J=1,37 Гц); 6,91 (с, 1Н).
Пример 39
Синтез диметилсиландиил{1-(2-метил-4,6-диизопропилинденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида (А-5)
а) Синтез хлор(2-метил-4,6-диизопропил-1-инденил)диметилсилана
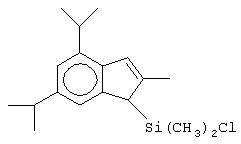
К раствору 2,93 г 2-метил-4,6-диизопропил-1-индена (13,67 ммоль) в 30 мл Et2O при 0° С по каплям добавляли 2,3 М раствор HexLi в гексане (6,00 мл, 13,80 ммоль). По завершении добавления получающейся в результате белой суспензии давали возможность разогреться до комнатной температуры, и ее перемешивали в течение 1 часа. Раствор Me2SiCl2 (99%, 1,68 мл, d=1,064, 13,68 ммоль) в 10 мл Et2O при 0° С добавляли к предварительно охлажденному до 0° С раствору литиевой соли. Реакционной смеси давали возможность разогреться до комнатной температуры, и ее перемешивали в течение 3 часов с получением в заключение белой суспензии. Растворители удаляли в вакууме, а остаток экстрагировали для удаления LiCl, пользуясь 30 мл толуола. Фильтрат доводили до сухого состояния в вакууме при 40° С и получали 3,33 г густого оранжевого масла. Выход неочищенного продукта 79,4%.
1H ЯМР (δ , м.д., CDCl3): 0,12 (с, 3Н, Si-СН3); 0,45 (с, 3Н, Si-СН3); 1,28-1,36 (м, 12Н, СН3); 2,31 (м, 3Н, СН3); 2,97 (м, 1Н, J=7,43 Гц, СН); 3,24 (м, 1Н, J=7,43 Гц, СН); 3,58 (с, 1Н, СН); 6,77 (м, 1Н, Ср-Н); 7,01 (уш.м, 1Н, Аr); 7,20 (уш.м, 1Н, Аr).
b) Синтез 1-(2-метил-4,6-диизопропилинденил)-7-(2,5-диметил-циклопента[1,2-b:4,3-b’]дитиофен)диметилсилана
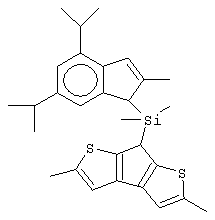
К суспензии 1,41 г 2,5-диметил-7Н-циклопента[1,2-b:4,3-b’]дитиофена (6,83 ммоль) в 30 мл Et2O при 0° С по каплям добавляли 2,3 М раствор HexLi в гексане (3,00 мл, 6,90 ммоль). Получающийся в результате коричневый раствор перемешивали при 0° С в течение 1 часа, а после этого при этой же температуре добавляли раствор 2,10 г хлор(2-метил-4,6-диизопропил-1-инденил)диметилсилана (6,84 ммоль) в 20 мл Et2O. Реакционной смеси после этого давали возможность разогреться до комнатной температуры, и ее перемешивали в течение 3 часов с получением в заключение коричневой суспензии. Растворители выпаривали при пониженном давлении, а остаток экстрагировали, пользуясь 30 мл толуола. Экстракт высушивали в вакууме и получали 2,79 г липкой темно-коричневой твердой фазы, которую анализировали по методу спектроскопии 1H ЯМР. Метод продемонстрировал присутствие ожидаемого лиганда, выход неочищенного продукта 68,5%. На следующей далее стадии продукт использовали как таковой без дополнительной очистки.
1H ЯМР (δ , м.д., CD2Cl2): -0,34 (с, 3Н, Si-СН3); -0,32 (с, Si-СН3); 1,27-1,39 (м, 12Н, СН3); 2,60 (с, 3Н, СН3); 2,62 (с, 3Н, СН3); 2,67 (с, 3Н, СН3); 2,96 (м, 1Н, J=7,24 Гц, СН); 3,29 (м, 1Н, J=7,24 Гц, СН); 3,87 (с, 1Н, СН); 4,04 (с, 1Н, СН); 6,82 (уш.с, 1Н, Ср-Н); 6,92 (м, 1Н, СН); 6,94 (м, 1Н, СН); 7,03 (уш.с, 1Н, Аr); 7,23 (уш.с, 1Н, Ar).
с) Синтез диметилсиландиил{1-(2-метил-4,6-диизопропилинденил)-7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)}цирконийдихлорида (А-5)
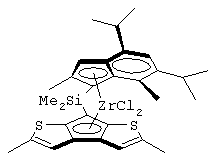
К раствору 2,79 г 1-(2-метил-4,6-диизопропилинденил) -7-(2,5-диметилциклопента[1,2-b:4,3-b’]дитиофен)диметилсилана (5,85 ммоль) в 30 мл Et2O при 0° С по каплям добавляли 2,3 М раствор HexLi (5,1 мл, 11,73 ммоль). По завершении добавления получающийся в результате коричневый раствор перемешивали в течение 1 часа при комнатной температуре. После этого его еще раз охлаждали до 0° С для добавления предварительно охлажденной до 0°С суспензии 1,36 г ZrCl4 (5,83 ммоль) в 15 мл толуола. Реакционной смеси после этого давали возможность разогреться до комнатной температуры, и ее перемешивали в течение 16 часов с получением в заключение светло-коричневой суспензии. Растворители удаляли в вакууме, а неочищенный остаток обрабатывали, пользуясь 25 мл толуола. Полученную суспензию отфильтровывали: фильтрат удаляли, тогда как остаток высушивали и получали 2,25 г оранжевого порошка, которым в результате и был указанный в заголовке комплекс, выход 60,6% с LiCl. 0,9 г данного порошка обрабатывали смесью 10 мл толуола и 2 мл изобутанола и перемешивали в течение 15 мин при комнатной температуре. После этого смесь отфильтровывали: фильтрат, содержащий LiCl и побочные продукты, обусловленные протеканием разложения, удаляли, тогда как остаток концентрировали в вакууме с получением 0,5 г оранжевого порошка, не содержащего LiCl. Согласно результатам анализа методом 1H ЯМР данный порошок был чистым комплексом.
1H ЯМР (δ , м.д., CD2Cl2): 1,18 (с, 3Н, Si-СН3); 1,34 (с, 3Н, Si-СН3); 1,19 (д, 3Н, J=6,85 Гц, СН3); 1,20 (д, 3Н, J=6, 85 Гц, СН3); 1,26 (д, 3Н, J=6,85 Гц, СН3); 1,35 (д, 3Н, J=6,85 Гц, СН3); 2,39 (с, 3Н, СН3); 2,42 (д, 3Н, J=1,17 Гц, СН3); 2,61 (д, 3Н, J=1,17 Гц, СН3); 2,82 (м, 1Н, J=6,85 Гц, СН); 3,06 (м, 1Н, J=6,85 Гц, СН); 6,64 (кв, 1H, J=1,17 Гц, СН); 6,78 (кв, 1H, J=1,17 Гц, СН); 6,80 (с, 1H, Ср-Н); 6,93 (уш.т., 1H, Ar, H5); 7,32 (уш. кв, 1H, Ar, H7).
ПРИМЕРЫ ПОЛИМЕРИЗАЦИИ
Общие методики
Сокатализатор метилалюмоксан (МАО) является коммерческим продуктом, который используется так, как он получен (Witco AG, 10 мас./об.% толуольный раствор, 1,7 М по А1). Каталитическую смесь готовят путем растворения желаемого количества металлоцена с необходимым количеством раствора МАО с получением раствора, который перед введением в автоклав перемешивают 10 мин при комнатной температуре.
Примеры полимеризации 1-33: полимеризация пропилена
В автоклав объемом 1 л из нержавеющей стали с рубашкой, снабженный магнитной мешалкой и сосудом объемом 35 мл из нержавеющей стали, соединенный с термостатом для контроля температуры, предварительно очищенный промывкой раствором Al(i-Вu)3 в гексане и высушенный при 50° С током пропена, загружают при комнатной температуре 1 ммоль А1(i-Вu)3 (в виде 1 М раствора в гексане) и 290 г пропилена. Автоклав затем термостатируют при температуре на 2°С ниже температуры полимеризации, после чего в автоклав с помощью азота под давлением через сосуд из нержавеющей стали вводят толуольный раствор, содержащий смесь катализатор/сокатализатор, температуру быстро поднимают до температуры полимеризации и проводят полимеризацию при постоянной температуре в течение 1 часа. После сброса не прореагировавшего мономера и охлаждения реактора до комнатной температуры полимер сушат при пониженном давлении при 60° С.
Условия полимеризации и данные, характеризующие полученные полимеры, представлены в таблице 1.
Пример полимеризации 34: сополимеризация пропилена/бутена
Проводя операции, как и в случае гомополимеризации пропилена, в автоклав объемом 1 л из нержавеющей стали с рубашкой, термостатированный при 58° С, загружают при комнатной температуре 1 ммоль Аl(i-Вu)3 (в виде 1 М раствора в гексане), 160 г пропилена и 154 г 1-бутена, чтобы иметь в жидкой фазе соотношение 50/50 мол.%, и затем в автоклав с помощью азота под давлением через сосуд из нержавеющей стали вводят толуольный раствор, содержащий смесь катализатор/сокатализатор (0,5 мг С-20, 0,45 ммоль МАО в 3 мл толуола), температуру быстро поднимают до температуры полимеризации, и проводят полимеризацию при постоянной температуре в течение 1 часа. Выделяют 26,5 г по существу аморфного полимера, который имеет Х.В. 2,29 дл/г, Тс -13,6° С и содержание бутена 27,7 мас.% (22,3 мол.%).
Пример полимеризации 35: сополимеризация пропилена/этилена
Проводя операции, как и в случае гомополимеризации пропилена, в автоклав объемом 1 л из нержавеющей стали с рубашкой, термостатированный при 58° С, загружают при комнатной температуре 1 ммоль Аl(i-Вu)3 (в виде 1 М раствора в гексане), пропилен и этилен так, чтобы иметь состав жидкой фазы 288 г пропилена и 1,5 г этилена (0,42 мас.%), затем в автоклав через сосуд из нержавеющей стали с помощью азота под давлением вводят толуольный раствор, содержащий смесь катализатор/сокатализатор (0,3 мг С-20, 0,27 ммоль МАО в 3 мл толуола), температуру быстро поднимают до температуры полимеризации, и проводят полимеризацию при постоянной температуре и постоянном давлении (25 бар-изб.) за счет подачи этилена (суммарное поглощение 13,3 г) в течение 1 часа. Выделяют 52,7 г по существу аморфного полимера, который имеет Х.В. 1,23 дл/г и содержание этилена 6,3 мас.% (9,2 мол.%).
Пример полимеризации 36: полимеризация пропилена с катализатором на носителе
а) Приготовление катализатора на носителе
Оборудование, используемое для нанесения катализатора, представляет собой стеклянный цилиндрический сосуд с механическим перемешиванием для обеспечения хорошего смешения между носителем и каталитическим раствором в процессе пропитки. В сосуд загружают 6 г пористого полиэтилена, имеющего Х.В. 21 дл/г, средний размер частиц 386 мкм и пористость 50,9% VN (1,07 см3/г), и механически суспендируют в токе азота. Каталитический раствор готовят путем растворения 24 мг катализатора С-20 в 12 мл раствора МАО (WITCO, 100 г/л в толуоле). Из-за ограниченной пористости носителя жидкость добавляют к твердому веществу по каплям, пока не будет достигнуто состояние начального смачивания. В этой точке растворитель упаривают в вакууме. Все операции проводят при комнатной температуре. Затем к носителю постепенно добавляют каталитический раствор. Конечный катализатор представляет собой розово-фиолетовое свободно текучее твердое вещество следующего состава: Аl 5,0 мас.% и Zr 0,0705 мас.% (мольное отношение Al/Zr 240).
б) Полимеризация
Используют реактор из нержавеющей стали объемом 4 л, снабженный лопастной турбинной магнитной мешалкой, индикатором давления, индикатором температуры и термостатирующей рубашкой. Периодическую полимеризацию осуществляют по следующей методике. При 30° С загружают 1200 г жидкого мономера, а затем 3 мл раствора TIBA 100 г/л в гексане, используемого в качестве акцептора. Реакцию полимеризации начинают путем введения в автоклав при 30° С с помощью азота под давлением 305 мг катализатора, затем температуру повышают до 60° С за 10 мин и поддерживают ее 1 час. Полимеризацию останавливают продувкой и охлаждением реактора. Никакой существенной забивки реактора не наблюдается. Полученный продукт собирают и сушат в печи, продуваемой азотом, при 70° С в течение 3 часов. Получают 360 г полимера с хорошей морфологией, который имеет Х.В. 1,94 дл/г. Активность катализатора составляет 1,2 кг ПП/г кат. на носителе в час, что соответствует 1,7 т ПП/г Zr· чac.
в) Физические свойства
К полимеру добавляют вспомогательные вещества в соответствии со стабилизирующей рецептурой и готовят из него пеллеты по следующим технологическим параметрам:
Стабилизирующая рецептура
Компоненты кг
Стеарат кальция М 0,0500
FLI-PP 99,7500
IRGANOX B215/ANOX BB 021 0,1500
Вазелиновое масло ОВ 30 0,0500
Одношнековый экструдер: MACGI диаметром 14 мм
Температура цилиндра: 230° С
Температура сырья: 230° С
Скорость шнека: 50 об/мин
Полученные таким способом пеллеты подвергают прямому прессованию с помощью пресса Carver.
Прямое прессование:
Толщина пластин образца:
Мутность, блеск, DMTA: 1,0 мм
Испытания на разрыв: 2,0 мм
Испытания на ударопрочность с надрезом по Изоду: 3,2 мм
Температура прессования пластин образца: 200° С
Время предварительного нагрева (без пресса): 5 мин
Время прессования: 5 мин
Давление: 14 бар
Охлаждение: быстрое, баня лед/вода
Пластины после прямого прессования перед испытаниями хранят при комнатной температуре, по меньшей мере, 48 часов.
Измерения ДСК: получены с помощью калориметра Mettler в соответствии со следующей методикой:
Первая стадия: Нагревание образца от -120 до 200° С при 20° С/мин;
Кристаллизация: Охлаждение образца от 200 до -120° С при 20° С/мин;
Вторая стадия: Нагревание образца от -120 до 200° С при 20° С/мин.
Физические свойства полимера представлены в таблице 2.
Примеры полимеризации 37-59: полимеризация этилен/пропилен
Этилен-пропиленовые смеси готовят в стальном цилиндре объемом 5 л, заполненном двумя газами в количествах, достаточно малых для предупреждения их конденсации. Состав газовой смеси в цилиндре контролируют с помощью ГХ. Сополимеризацию проводят при 50° С в стеклянном реакторе объемом 250 мл, снабженном механической мешалкой, шариковым холодильником, термометром и трубками для подачи мономеров и находящемся в термостатируемой бане. Вначале в продутый азотом реактор вводят 100 мл толуола и 3,5 ммоль раствора TIOAO. При температуре полимеризации азот удаляют и заменяют смесью сомономеров со скоростью потока 1,5 л/мин. При достижении равновесного давления (1,1 атм суммарного давления) добавляют 3,5 мкмоль металлоцена, растворенного в 5 мл гексана в присутствии 35 мкмоль TIOA (35 мкл 1 М раствора), для начала полимеризации. Во время реакции температуру поддерживают в пределах 50±1° С. Через 15 мин реакцию полимеризации останавливают добавлением 1 мл метанола, и сополимер осаждают 300 мл метанола, подкисленного НСl, фильтруют, промывают и сушат в течение ночи в вакууме при 50° С. Результаты полимеризации и константы сополимеризации r1 и r2 сополимеров этилен/пропилен представлены в таблицах 3, 4 и 5.
Пример полимеризации 60: полимеризация этилена с катализатором на носителе
Используют реактор из нержавеющей стали объемом 4 л, снабженный лопастной турбинной магнитной мешалкой, индикатором давления, индикатором температуры, линией подачи мономера с термическим массовым расходомером для измерения поглощения этилена и термостатирующей рубашкой. Периодическую полимеризацию осуществляют по следующей методике. В реактор при 30° С загружают 1600 мл жидкого пропана, а затем 2,5 ммоль триизо-бутилалюминия в качестве акцептора. Давление в автоклаве повышают за счет парциального давления этилена в 5 бар. Полимеризацию начинают путем введения в автоклав с помощью азота под давлением 162 мг катализатора, приготовленного в примере 37 а). Стадию полимеризации проводят при 30° С в течение 30 мин. Через этот промежуток времени температуру в реакторе повышают до 75° С, а парциальное давление этилена также увеличивают до 10 бар. Полимеризацию останавливают через 2 час путем продувки и охлаждения реактора. Выгруженный полимер сушат в печи, продуваемой азотом, при 70° С в течение 3 часов. Получают 90 г полимера с характеристической вязкостью 4,29 дл/г и температурой плавления 141,4° С.
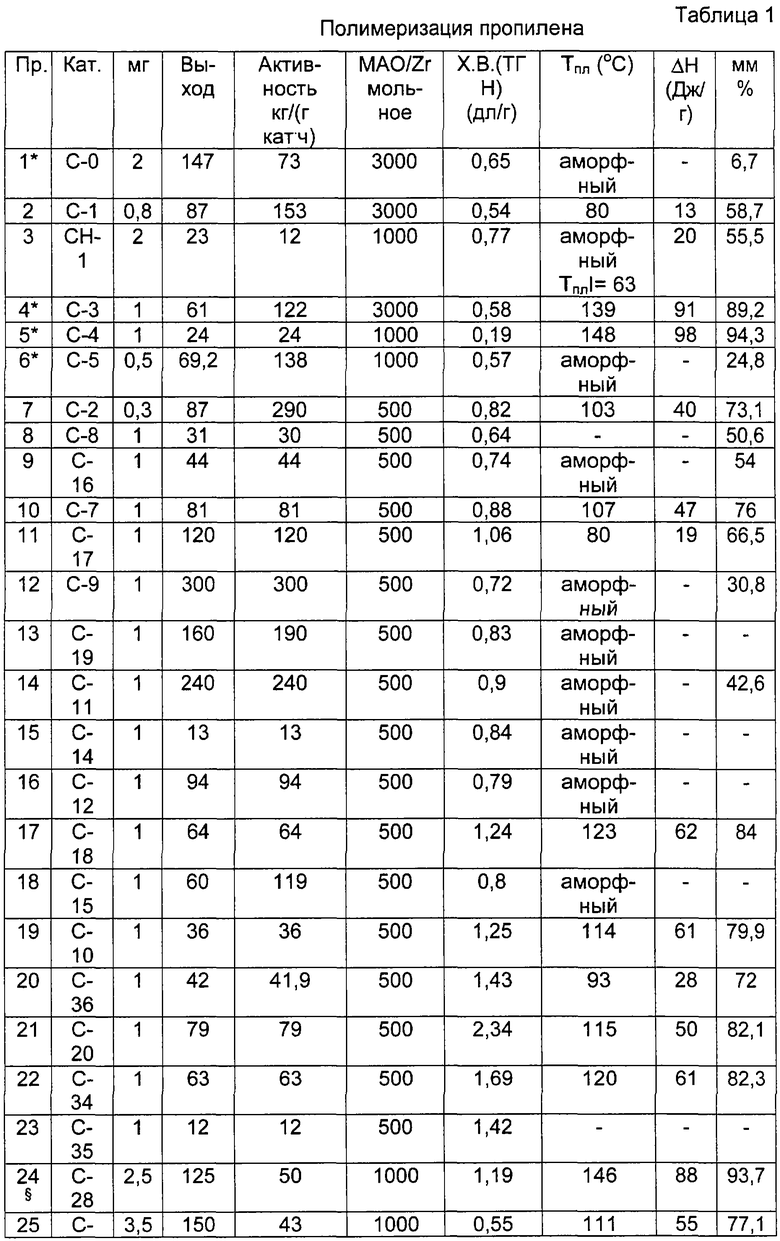
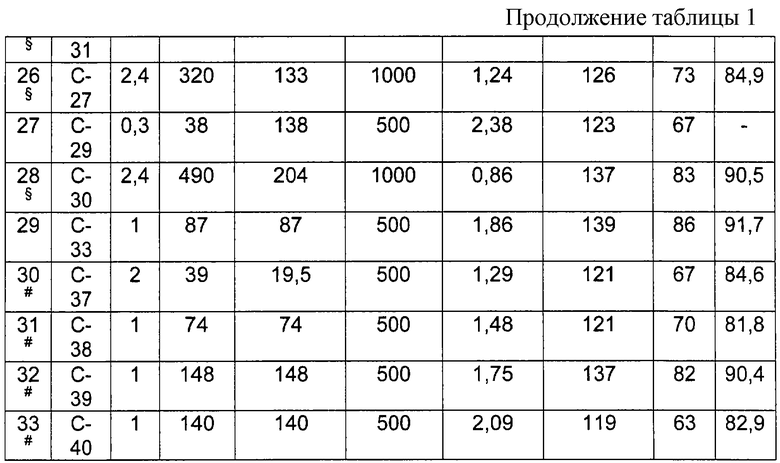
Полимеризацию осуществляют при 60° С. Для примеров 2 и 6 время полимеризации составляет 0,75 час; для примера 14 время полимеризации составляет 0,85 час; для примера 27 время равно 0,92 час; для всех других примеров время полимеризации составляет 1 час.
* - Сравнительный пример
# - Полимеризацию проводят в реакторе на 2 л с 620 г жидкого пропилена;
§ - Полимеризацию проводят в реакторе на 4 л со 1200 г жидкого пропилена.
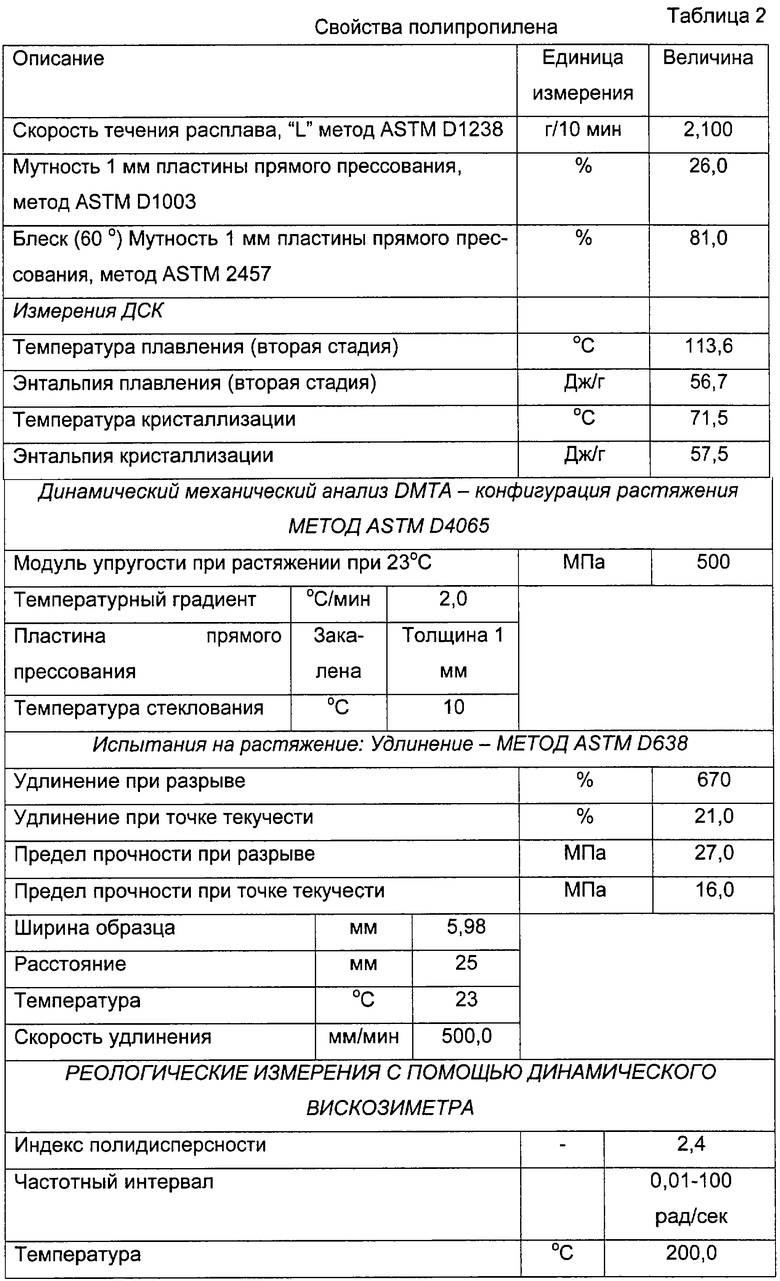
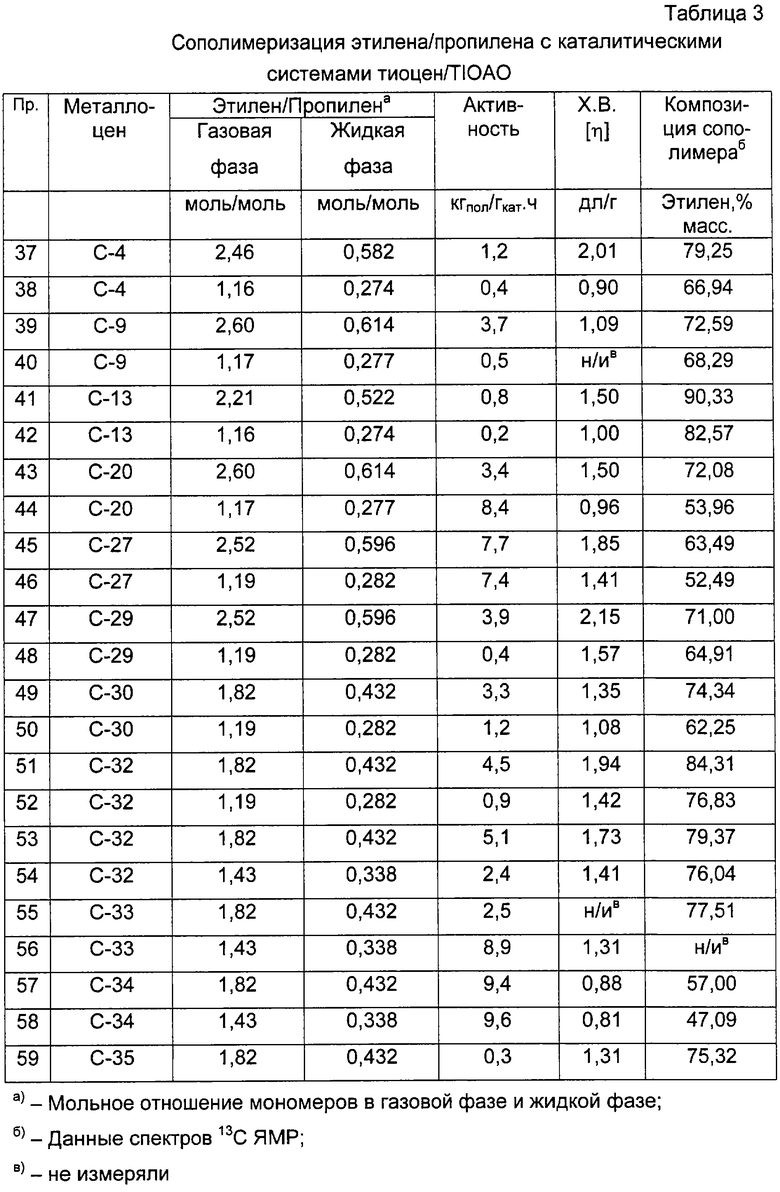
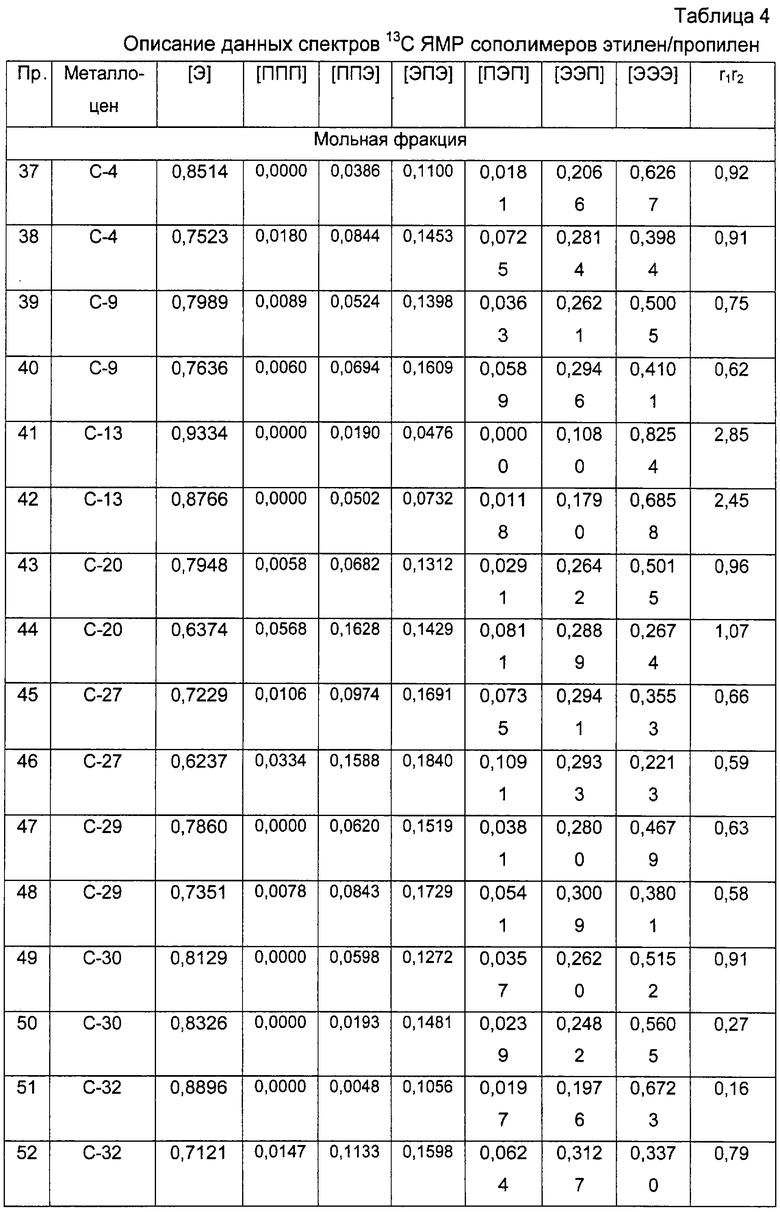
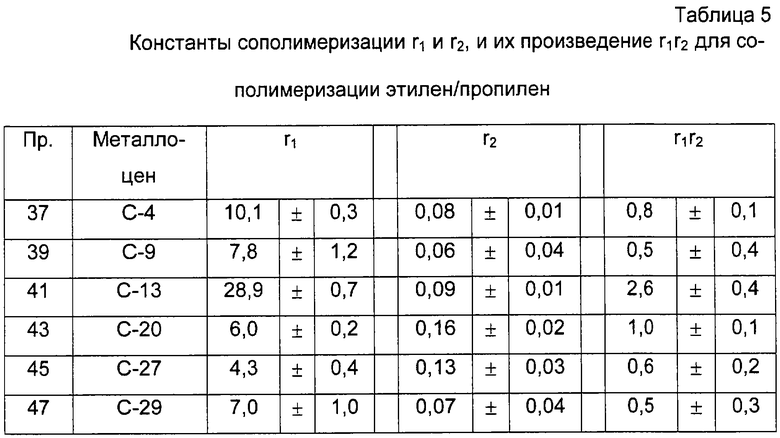
Пример полимеризации 60: полимеризация этилена
Полимеризацию этилена в стандартных условиях проводили в стеклянном автоклаве объемом 200 мл, оснащенном магнитной мешалкой, индикатором температуры и линией подачи для этилена. Автоклав очищали и продували этиленом при 35° С. При комнатной температуре вводили 140 мл гептана. Каталитическую систему получали отдельно в 10 мл гептана в результате последовательного ввода алюминийалкила - МАО или ТIOА/вода (Al/H2O 2,1) - и по истечении 5 минут перемешивания металлоцена, растворенного в толуоле (наименьшее количество из возможных). По истечении 5 минут перемешивания раствор вводили в автоклав в потоке этилена. Реактор закрывали, температуру увеличивали до 80° С и доводили давление этилена до 5,0 бар избыточного давления. Полное давление выдерживали постоянным в результате подачи этилена. По истечении периода времени, указанного в таблице 1, полимеризацию прекращали, проводя охлаждение, дегазацию реактора и ввод 1 мл этанола. Полученный полимер промывали кислым метанолом, после этого метанолом и высушивали в печи при 60° С в вакууме.
Условия полимеризации и характеристические данные для полученного полимера приведены в таблице 6.
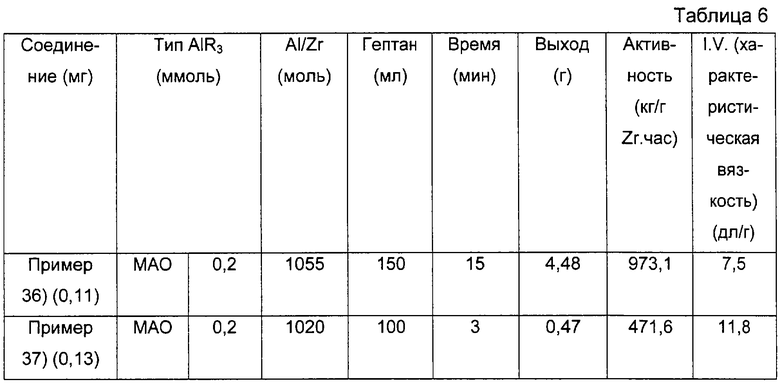
Описывается металлоценовое соединение общей формулы (I): LGZMXp, где L представляет собой двухвалентную группу; Z представляет собой остаток формулы (II), где R3 и R4 выбраны из атома водорода и углеводородных групп; А и В выбраны из S или СН, причем, если A - S, то В - CH, и если B - S, то А - СН; G представляет собой остаток формулы (III), где R6, R7, R8 и R9 выбраны из атома водорода и углеводородных групп; М представляет собой атом переходного металла: цирконий или гафний; Х выбран из атома галогена или C1-С6алкила; р является целым числом от 0 до 3. Описаны также лиганды для этих металлоценов и способы их получения. Металлоцены I используются в катализаторах при проведении способа получения полимеров альфа-олефинов, особенно пропиленовых полимеров с низкой степенью кристалличности. 9 с. и 7 з.п. ф-лы, 6 табл.
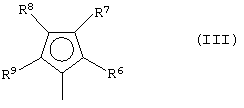

где L представляет собой двухвалентную группу, соединяющую мостиковой связью фрагменты G и Z, выбранную из CR1R2, SiR1R2 и (СН2)2, R1 и R2, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из атома водорода или C1-С6-алкильного радикала;
Z представляет собой фрагмент формулы (II)
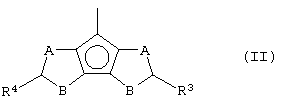
где R3 и R4, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из C1-С6-алкильного радикала, фенильного или триметилсилильного радикала;
А и В выбраны из атома серы (S) или СН при условии, что, если А представляет собой атом S, то В представляет собой СН, или если В представляет собой атом S, то А представляет собой СН, и где кольца, содержащие А и В, имеют двойную связь в допустимом положении;
G представляет собой фрагмент, выбранный из фрагментов формул (III), (IVа) и (IVd)
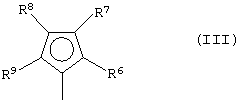
где R6 и R8 одинаковы и представляют собой водород или С1-С6-алкильный радикал;
R7 и R9, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из водорода, C1-С6-алкильного, триметилсилильного или фенильного радикала;
и R6 и R7 и R8 и R9 могут образовывать два ароматических кольца, содержащих 6 атомов углерода,
при условии, что R7 отличается от R8, и когда R7 представляет собой трет-бутильный радикал, R8 не является атомом водорода;
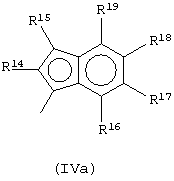
где R15 представляет собой водород;
R14, R16, R17 и R19, которые могут быть одинаковыми или могут отличаться друг от друга, выбраны из водорода, C1-С6-алкильного, триметилсилильного или фенильного радикала;
R18 представляет собой водород или вместе с R19 может образовывать бензольное кольцо,
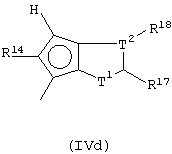
где 5-членное кольцо, образованное с участием Т1 и Т2, имеет двойные связи в каждом из допустимых положений, так что оно является ароматическим по своей природе;
Т2 представляет собой атом углерода;
Т1 представляет собой атом серы,
R14, R17 и R18, одинаковые или отличающиеся друг от друга, представляют собой водород, C1-С6-алкильный или фенильный радикал,
М представляет собой цирконий или гафний,
X, которые могут быть одинаковыми или разными, выбраны из атома галогена или C1-С6-алкильного радикала, и
р равен 2;
при этом исключены изопропилиден(3-триметилсилилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, диметилсиландиил(3-триметилсилилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, изопропилиден(3-этилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, диметилсиландиил(3-этилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, изопропилиден(3-н-бутилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, диметилсиландиил(3-н-бутилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, изопропилиден(3-метилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, диметилсиландиил(3-метилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид, изопропилиден(3-изопропилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид и диметилсиландиил(3-изопропилциклопентадиенил)(7-циклопентадитиофен)цирконий дихлорид.
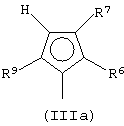
где R6 представляет собой водород;
R9 выбран из атома водорода, C1-С6-алкильного радикала;
R7 выбран из фенильной группы и группы QR11R12R13, где Q представляет собой атом Si; a R11, R12 и R13 представляют собой C1-С6-алкильный радикал.

где L принимает значения, определенные в п.1;
Z’ представляет собой остаток формулы (VI)
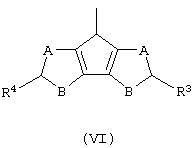
и его изомеры по двойной связи;
где А, В, R3 и R4 принимают значения, определенные в п.1;
двойные связи находятся в любом из допустимых положений;
G’ представляет собой остаток, выбранный из остатков формул (VII), (VIIa) и (VIId)
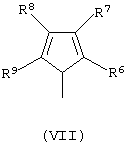
и его изомер по двойной связи,
где R6, R7, R8 и R9 принимают значения, определенные в п.1,
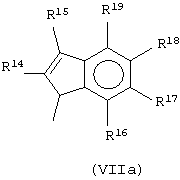
и его изомер по двойной связи,
где R14, R15, R16, R17, R18 и R19 принимают значения, определенные в п.1,
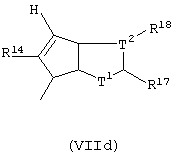
где 5-членное кольцо, образованное с участием Т1 и Т2, имеет двойные связи в каждом из допустимых положений, так что оно является ароматическим по своей природе;
Т1, Т2, R14, R17 и R18 принимают значения, определенные в п.1.

где G’, Z’ и L определены в п.4;
включающий следующие стадии:
а) контактирование соединения формулы (VIII) с основанием, выбранным из группы, включающей металлические натрий и калий, гидроксиды натрия и калия и литийорганическое соединение, где мольное отношение между соединением формулы (VIII) и указанным основанием составляет, по меньшей мере, 1:1;
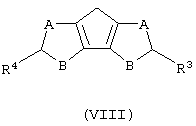
где А, В, R3 и R4 определены как указано в п.1;
b) контактирование соответствующего анионного остатка формулы (VIII) с соединением формулы (IX)
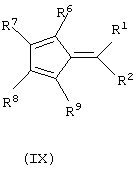
где R1, R2, R6, R7, R8 и R9 определены как указано в п.1,
а затем обработка полученного продукта протонирующим агентом.

где L, G’ и Z’ определены как указано в п.4,
включающий следующие стадии:
a) контактирование соединения формулы (VIII) с основанием, выбранным из группы, включающей металлические натрий и калий, гидроксиды натрия и калия и литийорганическое соединение, где мольное отношение между соединением формулы (VIII) и указанным основанием составляет, по меньшей мере, 1:1;
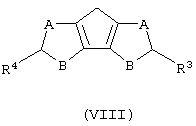
где А, В, R3 и R4 определены как указано в п.1;
b) контактирование полученных анионных соединений формулы (VIII) с соединением формулы (IX)
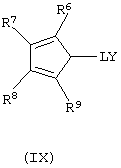
где L, R6, R7, R8 и R9 определены как указано в п.1;
Y представляет собой галогеновый радикал, выбранный из группы, состоящей из хлора, брома и иода.
(A) металлоценового соединения формулы (I)

где L, Z, М, X, G и р определены как указано в п.1; и
(B) алюмоксана.
- триады (мм), удовлетворяющие соотношению: 55<мм<85;
- энтальпия плавления (ΔН) между 5 и 70 Дж/г;
- мутность (ASTM 2457) от 15 до 30%;
- блеск (60°С) (ASTM 2457) от 60 до 95%;
- модуль упругости при растяжении (ASTM D4065) от 1000 до 200 МПа;
- относительное удлинение при разрыве (ASTM D4065) от 300 до 900%;
- предел прочности при разрыве (ASTM D638) от 10 до 40%.
- энтальпия плавления <70 Дж/г;
- триады (мм), удовлетворяющие соотношению: 30<мм<85.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| EWEN JOHN A et al | |||
| J.Amer.Chem.Soc | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |

