Изобретение относится к области пептидной и фармацевтической химии, конкретно к способу получения метилового эфира O-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина формулы:
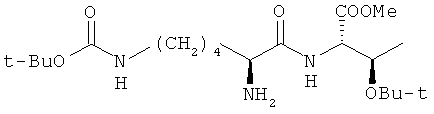
Этот защищенный дипептид является ключевым полупродуктом синтеза циклического октапептида октреотида, являющегося синтетическим аналогом соматостатина, обладает сходным с ним фармакологическими эффектами, но значительно большей продолжительностью действия. Октреотид взаимодействует со специфическими рецепторами соматостатина в различных тканях (в т.ч. в ЦНС и ЖКТ), подавляет патологически повышенную секрецию гормонов и пептидов, продуцируемых в гастроэнтеропанкреатической эндокринной системе.
Синтез этого дипептида в литературе не описан. Имеются данные, что он может быть получен в результате реакции метилового эфира О-трет-бутил-L-треонина с N-гидроксисукцинимидным эфиром Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина [1] и последующим снятием бензилоксикарбонильной защиты. Однако конкретная методика получения, пригодная для успешного синтеза дипептида в этом первоисточнике, отсутствует.
Задачей данного изобретения является разработка простого, пригодного для полупромышленного использования способа получения защищенного дипептида - метилового эфира O-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина.
Поставленная техническая задача достигается следующим способом получения метилового эфира O-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина жидкофазным методом, включающим:
а) получение пентафторфенилового эфира Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина взаимодействием Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина с пентафторфенолом в присутствии дициклогексилкарбодиимида и в среде этилацетата при 5-10°C
б) отделение фильтрованием дициклогексилмочевины;
в) конденсацию полученного на стадии а) пентафторфенилового эфира с метиловым эфиром O-трет-бутил-L-треонила в присутствии N-метилморфолина в среде этилацетата:
г) удаление кислых и основных примесей из реакционной массы;
д) гидрирование водородом полученного реакционного продукта в растворе метанола в присутствии катализатора гидроокиси палладия;
е) очистку полученного целевого продукта - защищенного дипептида с помощью щавелевой кислоты
ж) выделение целевого продукта с помощью бикарбоната натрия
Таким образом, сущность заявленного защищенного дипептида заключается в следующем.
Осуществляют реакцию Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина и пентафторфенола в присутствии дициклогексилкарбодиимида в среде этилацетата и температуре 5-10°C приводит к образованию пентафторфенилового эфира Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина. Раствор полученного соединения после отделения фильтрованием дициклогексилмочевины используют для реакции конденсации с метиловым эфиром O-трет-бутил-L-треонина с использованием N-метилморфолина в качестве акцептора выделяющегося пентафторфенола. После фильтрования соли пентафторфенола и N-метилморфолина реакционную массу обрабатывают, например, насыщенным раствором бикарбоната натрия (для удаления кислых примесей), 5% раствором, например, лимонной кислоты (для удаления основных примесей), органическую фазу упаривают и получают метиловый эфир O-трет-бутил-N-(Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизил)-L-треонина без дальнейшей очистки гидрируют в растворе метилового спирта с использованием гидроокиси палладия в качестве катализатора. Технический метиловый эфир O-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина для очистки переводят в оксалат, промывают его серным эфиром для удаления примесей и выделяют свободное основание (целевой продукт) с использованием бикарбоната натрия. Полученный продукт представляет собой белое кристаллическое соединение с содержанием основного вещества более 96% по анализу ВЭЖХ.
Для удаления кислых и основных примесей, а также выделения целевого продукта в способе используют традиционные вещества в синтезе пептидов.
Экспериментальная часть.
Аналитическую ВЭЖХ проводили на хроматографе фирмы Shimadzu. Колонка: Grom-Sil 12J ODS-4HE, 5 µm, 250*4,6 mm. Условия: градиент с 20% B до 40% B за 20 минут, A - вода+фосфатный буфер pH 3, B - ацетонитрил.
Описываемый способ иллюстрируется следующими примерами, не ограничивающими его.
Пример 1. Получают метиловый эфир O-трет-бутил-N-(Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизил)-L-треонина.
В 6 л 3-горлой колбе, снабженной механической мешалкой, растворяют 447 г (1,17 моль) Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина и 216 г (1,17 моль) пентафторфенола в 2 л этилацетата, охлаждают на ледяной бане до температуры 5°C, после чего прикапывают раствор 243 г (1,18 моль) дициклогексилкарбодиимида в 0,6 л этилацетата с такой скоростью, чтобы температурара не превышала 10°C. После прибавления реакционную массу перемешивают 2 часа при 10°C. Фильтруют выпавшую мочевину, промывают ее на фильтре 200 мл и объединяют промывные воды с органической фазой. Органическую фазу охлаждают до 5°C и при перемешивании прикапывают 223 г (1,18 моль) метилового эфира O-трет-бутил-L-треонина в 1,5 л этилацетата при температуре ниже 10°C. Перемешивают реакционную массу при этой же температуре 30 мин, после чего добавляют 120 г (1,19 моль) N-метилморфолина. Перемешивают 2 часа при комнатной температуре и оставляют реакционную массу при температуре 5°С на 12 часов. Кристаллическую соль фильтруют, промывают 200 мл ЭА. Объединенную органическую фазу промывают последовательно 500 мл воды, 2 раза по 500 мл насыщенного раствора бикарбоната натрия, 4 раза по 250 мл 5% раствором лимонной кислоты и 500 мл воды. Этилацетат упаривают на роторном испарителе при температуре не выше 40°C. Получают 608 г (94,2%) желтоватого густого масла, которое используют на следующей стадии без очистки.
Пример 2. Получают целевой продукт - метиловый эфир O-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина.
В 6 л одногорлой колбе растворяют 532 г (0,96 моль) метилового эфира O-трет-бутил-N-(Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизил)-L-треонина в 3 л метилового спирта и осуществляют стадию гидрирования, добавляя 8 г 30% водной суспензии гидроокиси палладия и пропуская ток водорода до исчезновения по ВЭЖХ исходного продукта (20-40 часов). Фильтруют катализатор, упаривают метиловый спирт на роторном испарителе, остаток растворяют в 2 л этилацетата и промывают 2 раза по 0,5 л воды. Сушат органическую фазу сульфатом натрия и упаривают досуха. Маслообразный остаток растворяют в 2 л серного эфира и при перемешивании быстро присыпают 88 г щавелевой кислоты. Выпавший белый осадок через 30 мин фильтруют и промывают на фильтре 200 мл серного эфира. Сушат на воздухе до испарения большей части эфира. Далее осуществляют выделение целевого продукта. Растворяют в 1 л воды 150 г бикарбоната натрия, прибавляют 1,5 л этилацетата и при перемешивании присыпают полученный оксалат дипептида. Перемешивают 30-60 мин до полного растворения. Отделяют органическую фазу, промывают 300 мл воды и сушат сульфатом натрия. Растворитель отгоняют на роторном испарителе. Через некоторое время полученное масло кристаллизуется. Получают 310 г (77,3%) белого кристаллического вещества. Анализ ВЭЖХ показал содержание основного вещества 96,4%.
Как видно из вышеприведенных примеров, описанный способ пригоден для получения метилового эфира O-трет-бутил-N-(Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизил)-L-треонина с достаточно высоким выходом в полупромышленном масштабе. Кроме того очистка с использованием щавелевой кислоты позволяет получать продукт высокой степени чистоты и не прибегать к более трудоемким и дорогим методам, как, например, препаративная ВЭЖХ.
Целевой продукт может быть использован, в частности, для получения октреотида или его аналогов либо иных пептидов, содержащих фрагмент L-лизил-L-треонина.
Источники информации, принятые во внимание при оформлении заявки
1. J. Chem. Soc. Perkin Tr.1 N6, p.989 (1986).
Изобретение относится к области пептидной и фармацевтической химии, конкретно к способу получения защищенного метилового эфира O-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина. Сущность изобретения заключается в том, что синтез осуществляется жидкофазным методом путем конденсации пентафторфенилового эфира Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина с метиловым эфиром О-трет-бутил-L-треонина в присутствии N-метилморфолина в среде этилацетата и последующего гидрирования полученного соединения водородом в метиловом спирте с использованием гидроокиси палладия в качестве катализатора. Очистку целевого защищенного дипептида осуществляют через соль с щавелевой кислотой. Исходный пентафторфениловый эфир получают в результате реакции Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина с пентафторфенолом и используют далее без выделения. Заявленный способ позволяет получить целевой продукт с выходом более 96% и высокой степенью чистоты. Полученный защищенный дипептид может быть использован, в частности, для получения октреотида или его аналогов либо иных пептидов, содержащих фрагмент L-лизил-L-треонина.
Способ получения метилового эфира О-трет-бутил-N-(Nε-трет-бутоксикарбонил-L-лизил)-L-треонина:
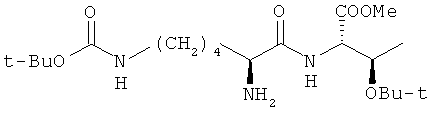
жидкофазным методом, включающий:
а) получение пентафторфенилового эфира Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина взаимодействием Nα-бензилоксикарбонил-Nε-трет-бутоксикарбонил-L-лизина с пентафторфенолом в присутствии дициклогексилкарбодиимида и в среде этилацетата при 5-10°С;
б) отделение фильтрованием дициклогексилмочевины;
в) конденсацию полученного на стадии а) пентафторфенилового эфира с метиловым эфиром О-трет-бутил-L-треонила в присутствии N-метилморфолина в среде этилацетата:
г) удаление кислых и основных примесей из реакционной массы;
д) гидрирование водородом полученного реакционного продукта в растворе метанола в присутствии катализатора гидроокиси палладия;
е) очистку полученного целевого продукта - защищенного дипептида с помощью щавелевой кислоты;
ж) выделение целевого продукта.
| Felix Arthur M | |||
| et al | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| International Journal of Peptide & Protein Research | |||
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

